
Submitted:
10 April 2024
Posted:
11 April 2024
You are already at the latest version
Abstract
Angiostrongylus cantonensis, a zoonotic parasite, can invade the human central nervous system (CNS) and cause acute eosinophilic meningitis or eosinophilic meningoencephalitis. Mice infected with A. cantonensis show elevated levels of pro-inflammatory cytokines, plasminogen activators, and matrix metalloproteinase-9, resulting in disruption of the blood-brain barrier (BBB) and immune cell infiltration into the CNS. Caveolin-1 (Cav-1) regulates the permeability of the BBB, which affects immune cells and cerebrospinal fluid. This intricate interaction ultimately fuels the progression of brain damage and edema. This study aims to investigate the regulatory role of Cav-1 in the pathogenesis of meningoencephalitis induced by A. cantonensis infection. We investigate pathological alterations by triphenyl-tetrazolium chloride, brain water content, BBB permeability, Western blot analysis, and gelatin zymography in BALB/c mice after A. cantonensis. The study evaluates the critical role of Cav-1 regulation through the TLR4 / MyD88 signaling pathway, modulates tight junction proteins, influences BBB permeability, and contributes to brain damage in A. cantonensis-induced meningoencephalitis.
Keywords:
Angiostrongylus cantonensis
; Caveolae-1
; blood-brain barrier permeability
; cerebral edema
1. Introduction
Angiostrongylus cantonensis is a kind of zoonotic parasite [1], A. cantonensis larvae can invade the human central nervous system and cause acute eosinophilic meningitis, or eosinophilic meningoencephalitis, in the most severe infection, increased brain pressure, increased cerebrospinal fluid pressure, coma and death [2], pathological mechanisms causing brain damage include physical destruction of tissues by larval migration [3], and immune responses induced by A. cantonensis infection include hippocampus and brain. In the parenchyma, astrocytes, microglia and neurones undergo apoptosis or necroptosis and demyelination, loss of Purkinje cells in the cerebellum and blood-brain barrier (BBB) destroy [10] and other pathological phenomena [4,5,6,7,8,9].
The neurovascular unit is made up of cells including endothelial cells, pericytes and vascular smooth muscle cells, and glial cells. Stable and proper interaction between all components of the neurovascular unit is essential for the stability and function of the central nervous system. It is essential that if one of the members is out of balance, it will cause the appearance of the disease [11]. Caveolin (Cav) and cholesterol collaborate to shape and maintain the architecture of caveolae. Cav-1 adopts a hairpin-shaped configuration within the cellular membrane, with the N- and C-terminals extending into the cytoplasm. These terminals facilitate binding to various receptors and numerous message-transmitting molecules through the caveolin scaffold domain. However, the impact of these interactions on downstream cell signaling pathways, whether positive or negative, depends on the specific cell type, stimuli, and induced cellular signaling pathways [12,13,14,15]. Cav-1 participates in the mechanism of regulation of physiology and pathology through a complex information transmission pathway system, such as endocytosis, cholesterol regulation of transport, lipid metabolism, transcellular trafficking, and immune response [16,17,18]. Cav-1 is present in endothelial cells, astrocytes, and neurons in the neurovascular unit of the brain, especially abundant in endothelial cells [19,20,21,22]. Cav-1 changes BBB permeability of the BBB by regulating the endocytosis of vascular endothelial cells and intercellular tight junction proteins and adherent junction proteins. The degradation of the extracellular matrix in the basal layer, transmembrane proteins, and tight junction proteins between endothelial cells will lead to the destruction of the BBB structure, resulting in the leakage of immune cells,, red blood cells and fluid from the blood vessels to the central nervous system [11,16,17,18,23]. Cav-1 participates in the physiological and pathological mechanisms of many brain lesions and brain edema processes, including the regulation of immune responses and BBB communication. Therefore, the role of Cav-1 in the regulation of BBB damage and permeability after brain injury remains controversial.
In mice infected with A. cantonensis, it can cause eosinophilic meningitis, brain injury, cerebral hemorrhage, and cerebral edema. There are still many places that remain unclear. This study clarifies that Cav -1 plays a role in the promotion to inflammation and damage of the BBB in hemorrhagic brain injury caused by A. cantonensis-induced eosinophilic meningoencephalitis and cerebral edema, increasing the permeability of the BBB or inhibiting the immune response to protect the brain and reduce damage.
2. Materials and Methods
2.1. Collection of Third-stage Larvae (AcL3) of A. cantonensis
After infected rats with A. cantonensis, the feces will contain first-stage larvae (AcL1) of A. cantonensis. These AcL1 larvae will be collected from feces to infect water snails (Biomphalaria glabrata). After 60 days of infection, the AcL1 larvae will mature into third- stage larvae (AcL3). The tissue of the water snails was homogenized in a ratio of 1:30 (tissue: artificial pepsin digestion solution containing pepsin from Sigma, USA) using a digestive juice grinder. The mixture was then evenly stirred and digested in a 37 ° C incubator for 1 hour. The lower layer of sediment was carefully collected using a dropper, transferred to a glass dish, and observed under a dissecting microscope to identify and collect the AcL3. AcL3 were then quantified, and every 30 larvae were grouped into a 1.5 ml microcentrifuge tube for future feeding experiments with mice.
2.2. Induction of Animal Models of Eosinophilic Meningitis
Sixty-three BALB/c mice were randomly assigned into 7 groups, each consisting of 9 mice. These groups included the non-infected control group, the group of infection with A. cantonensis, and the group infection with A. cantonensis and treated with Methyl-β-cyclodextrin (MβCD). In the groups infected with A. cantonensis and treated with MβCD, each mouse received 30 AcL3. Mice in the A. cantonensis infection group were sacrificed on days 5, 10, 15, 20, and 25 after infection for time course experiments. The non-infected control group received an equivalent volume of deionized water on day 0, as did the infected groups, and were euthanized on day 25. In the MβCD treatment group, MβCD (Merck, USA) was administered intraperitoneally at a dose of 100 mg / kg / day from day 0 of A. cantonensis infection to day 24, with euthanasia of mice on day 25 after infection.
2.3. Cerebral Edema Determination
Mice were anesthetized with isoflurane and sacrificed by decapitation. The brains were removed and washed with phosphate buffered saline (PBS). The appearance of the brain was monitored for hemorrhage and infection by A. cantonensis, and then the brain tissue was placed in cold PBS for ten minutes and then placed in a brain tissue slice mold to cut into brain slices with a thickness of 1 mm, infiltrated with 2% TTC (2, 3, 5-triphenyl-tetrazolium chloride solution) at 37 ° C and kept away from staining. It was carried out for 30 minutes in a light environment and then the brain tissue was fixed with 10% formaldehyde for 24 hours. After scanning, the volume of the brain was calculated with an image processing system (AIS software, Imaging research INC, Canada).
2.4. Measurement of Brain Water Content
The water content of the brain was measured using the wet dry weight method, and then the mice were sacrificed to remove brain tissue and weighed as wet weight. Brain tissue was fully dried in a constant temperature oven at 80 ° C for 24 hours and weighed several times to obtain a stable dry weight (average weighing error <0.002g). The percentage of brain water content was calculated as follows: brain water content = (wet weight - dry weight) / wet weight × 100%.
2.5. Measurement of BBB Permeability
The permeability of the BBB was examined by an in vivo permeability assay. The 40kDa FITC-dextran (6 mg / ml, 4 mg / kg body weight, Sigma) was injected into the tail vein. The control group was injected with normal saline. After 30 minutes, the mice were anesthetized with isoflurane and CSF was sucked through the dura mater with a capillary tube. Excitation: 490 and emission: 520 nm were used to measure absorbance on a spectrophotometer (Hitachi U4500, Japan) and the 40 kDa FITC-dextran concentration in the CSF was calculated using the standard curve.
2.6. Western blot
The protein detection method followed the procedures described in previous research conducted in our laboratory [8]. In summary, the sample was centrifuged at 12,000 g for 10 minutes at 4 ° C and the resulting supernatant was collected and quantified. The freshly extracted proteins were then loaded onto the SDS electrophoresis page for electrophoresis. After electrophoresis, the proteins on the page were transferred onto a PVDF membrane. A primary antibody was used followed by a horseradish peroxidase (HRP) conjugated secondary antibody for the reaction, which facilitates protein detection of proteins through enhanced chemiluminescence (ECL). Subsequently, the protein bands were analyzed using a cold-light imager (iBright CL750 imaging system, USA).
2.7. Gelatin zymography
Brain tissue protein was loaded onto 7.5% (mass/volume) SDS-polyacrylamide gel electrophoresis after copolymerization with 0.1% gelatin (Sigma, USA). The stacking gels were 4% (mass/volume) polyacrylamide. Electrophoresis was carried out in running buffer (25 mM Tris, 250 mM glycine, 1% SDS) at room temperature at 110 V for 1 h. The gel was washed twice at room temperature for 30 min each in 2.5% Triton X-100 and then twice with double-distilled H2O for 10 min each. The gel was incubated in reaction buffer (50 mM Tris-HCl, pH 7.5, containing 200 mM NaCl, 10 mM CaCl2, 0.02% Brij-35, 0.01% NaN3) at 37 ° C for 18 h. The gel was stained with 0.25% Coomassie Brilliant Blue R-250 (Sigma, USA) for 1 hour and destained in 15% methanol / 7.5% acetic acid.
2.8. Statistical analysis
The test results obtained by different groups were statistically analyzed by Kruskal-Wallis, and multiple comparisons were made by Dunn's multiple comparison. The results are expressed as mean±standard deviation (means±SD), and p<0.05 means statistically significant.
3. Results
3.1. Alterations in the Permeability of the BBB Result in Cerebral Edema and Haemorrhage
Following A. cantonensis infection in mice, a progressive increase in brain hemorrhage was consistently observed, as well as swelling and congestion of blood vessels throughout the course of the infection. In particular, five-stage larvae of A. cantonensis were detected at the base of the skull on days 20-25 after infection (Figure 1). Subsequent evaluation of brain volume and water content revealed a positive correlation with duration of infection, showing a significant increase on day 10 (Figure 2a). TTC staining revealed a 12% increase in brain volume and an 8% increase in brain water content on day 25 after infection (Figure 2b). Furthermore, the concentration of 40 kDa FITC-dextran was examined in the CSF, revealing a positive correlation with the duration of infection and a substantial increase from day 10 to 25 after infection (Figure 3a). The correlation analysis between the concentration of 40 kDa FITC-dextran in the CSF and the volume of the brain showed a significant positive correlation (r2=0.791, p<0.05) (Figure 3b). These findings suggest that A. cantonensis infection induces alterations in the permeability of the BBB, leading to changes in the permeability of the BBB and subsequently causing cerebral edema and hemorrhage.
3.2. Cav-1 Through the TLR4 / MyD88 Signaling Pathway Modifies BBB Permeability
In the brains of mice infected with A. cantonensis, there was a significant increase in Cav-1 and p-Cav-1 levels on day 10 after infection, with a subsequent increase observed as infection progressed (Figure 4). The analysis correlating of 40 kDa FITC-dextran with the Cav-1 protein revealed a positive association (r2=0.722, p<0.05, Figure 5). Furthermore, previous findings indicated a positive correlation between concentration of 40 kDa FITC-dextran in the CSF and brain volume. Investigation of the Cav-1 regulatory pathway of Cav-1 revealed a significant elevation in the TLR4 and MyD88 signaling pathways in the brains of mice infected with A. cantonensis on days 10 and 15 after infection (Figure 6), which is in agreement with the observed increase in Cav-1 and p-Cav-1 proteins during the same time frame. This study proposes that Cav-1 regulates BBB permeability through the TLR4 / MyD88 signaling pathway.
3.3. Treatment with Caveolae / Cav-1 Specific Inhibitor MβCD
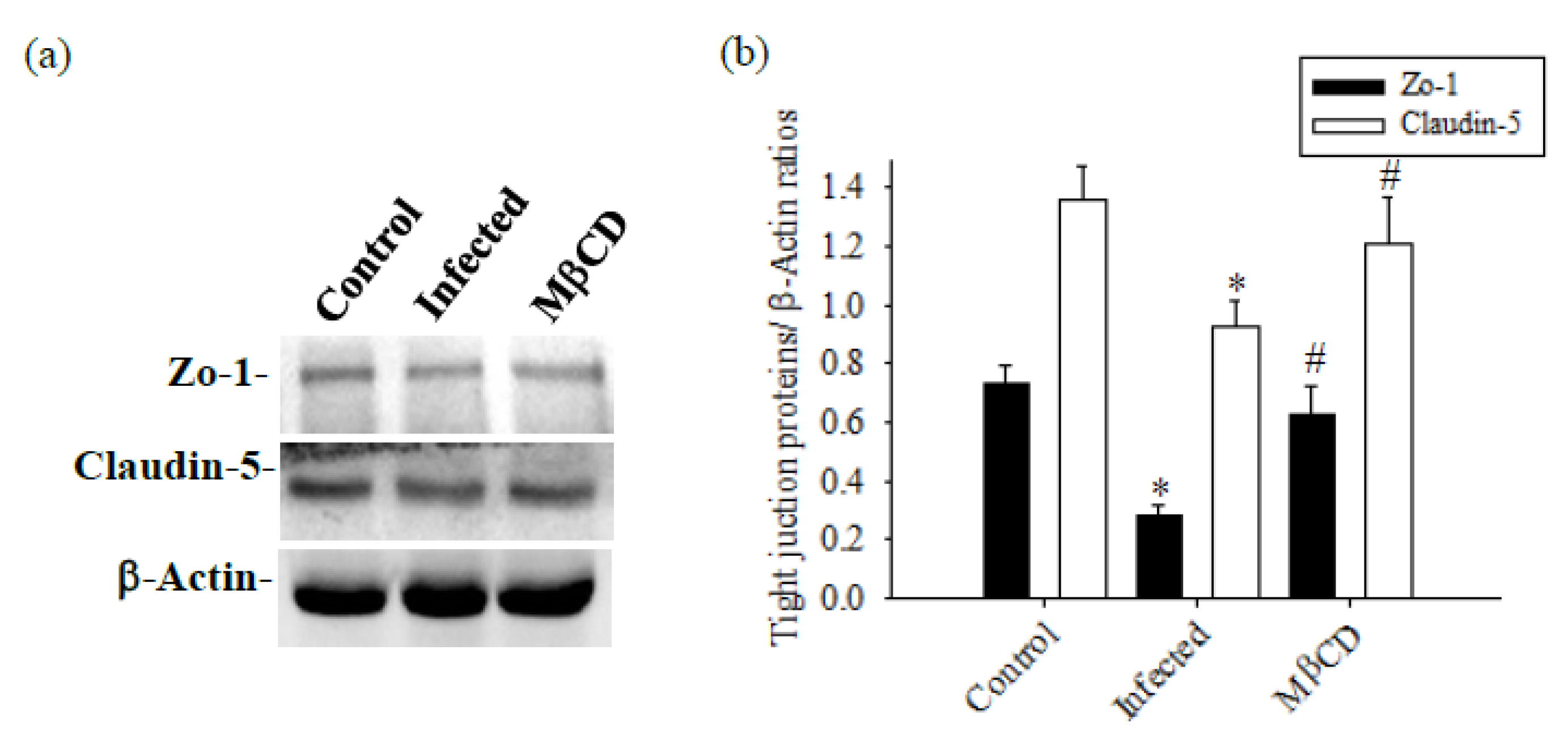
Mice infected with A. cantonensis received treatment with the specific Caveolae / Cav-1 inhibitor MβCD, Cav-1, p-Cav-1, TLR4 and MyD88 expression levels were significantly decreased in MβCD treatment group compared to the infected group (Figure 7, p<0.05). Concurrently, MβCD treatment significantly reduced the activity of MMP-9, an enzyme known to damage the BBB, in both CSF and brain tissue (Figure 7b). The 40 kDa FITC-dextran in CSF, indicative of the permeability of the BBB, showed a significant increase in the infected group and a significant decrease in the MβCD treatment group compared to the infected group (Figure 7c, p<0.05). Similar trends were observed in changes in brain volume, determined by TTC staining, with a significant reduction observed in the infected group following MβCD treatment compared to the infected group (Figure 7d, p<0.05). Subsequently, the levels of Zo-1 and claudin-5, crucial tight junction proteins in the brain, increased significantly in MβCD treatment group compared to the infected group (p<0.05), although still slightly lower than in the control group (Figure 8).
4. Discussion
Cerebral edema frequently manifests in response to various triggers, such as infection, stroke, and traumatic brain injury. Multiple factors influence the development of brain edema, including activation of inflammatory pathways, disruption of the blood-brain barrier (BBB), dysregulation of ion channels, and changes in osmotic gradients. Increasing experimental and clinical evidence underscores the link between BBB dysfunction and various severe central nervous system (CNS) disorders, including multiple sclerosis, stroke, brain tumors, epilepsy, and Alzheimer's disease [24]. In an animal model of traumatic brain injury, an observed increase of approximately 6% in brain water content has been documented [25]. Furthermore, the neuropathological changes resulting from Toxoplasma gondii infection lead to localized vascular edema, contributing to an overall increase of approximately 1% in the water content [26]. In A. cantonensis causing eosinophilic meningitis, the group infected with A. cantonensis, compared to the control group, exhibited a 12% increase in brain volume and an 8% increase in brain water content. This resulted in cerebral hemorrhage, brain edema, and inflammatory responses involving cellular factors. Changes in cerebrospinal fluid (CSF) composition, including total protein, white blood cells, and eosinophils, were correlated with BBB permeability [2,10,27,28,29,30]. Interesting, in the context of acute focal cerebral ischemia, significant areas of brain tissue necrosis appear as white regions in TTC staining [31]. However, when mice are infected with A. cantonensis, causing eosinophilic meningitis, TTC staining to measure brain volume does not reveal significant areas of necrosis of brain tissue. This suggests that although there is substantial infiltration of immune cells in the subarachnoid space and ventricles, brain tissue exhibits demyelination [32]. The extent of brain injury may not reach the necrotic state or areas of damaged brain tissue near the subarachnoid space may be minimal, making it difficult to observe. More research is needed to explore these intriguing findings.
Caveolin-1 is present in various components of the cerebral neurovascular unit, including endothelial cells, astrocytes, and neurons. In particular, Cav-1 is particularly abundant in endothelial cells, where it plays a crucial role in the regulation of BBB permeability by influencing endothelial transcytosis and modulation of tight junction proteins [19,20,21,22]. In conditions such as ischemia and lipopolysaccharide-induced injury, Cav-1 expression increases early, contributing to a decrease in claudin-5 and occludin, resulting in the breakdown of the BBB. Subsequent up-regulation of Cav-1 leads to degradation of tight junction proteins (claudin-5 and occludin), exacerbating damage to the BBB and increasing permeability [17,33]. In hypoxic brain edema, Cav-1 forms membrane invaginations, facilitating claudin-5 endocytosis and subsequent autophagy, ultimately increasing BBB permeability and causing cerebral edema [34]. In ischemic brain injury, tissue-type plasminogen activator (tPA) treatment activates matrix metalloproteinase-9 (MMP-9) directly in vascular endothelial cells or indirectly through Cav-1, promoting MMP-9 activation and subsequent degradation of tight junction proteins. This process leads to the breakdown of the BBB and increased permeability [35,36]. Interestingly, tPA, through Cav-1, also inhibits MMP-9 activity to protect the BBB and reduce damage [37]. In a previous study, infection with A. cantonensis in mice resulted in increased expression of plasminogen activators and MMP-9, leading to damage to the BBB, degradation of the extracellular matrix (ECM), and infiltration of immune cells, leading to eosinophilic meningitis [10,29,30]. To investigate the involvement of Cav-1 in the regulation of BBB permeability, we evaluated MMP activity and expression, as well as BBB permeability. The expression of the Cav-1 protein correlates positively with brain edema, BBB permeability, and MMP-9 activity in infection-induced meningoencephalitis with A. cantonensis. To further elucidate the role of Cav-1 in the regulation of BBB permeability, we employed methyl-β-cyclodextrin (MβCD), a cholesterol depleting agent that disrupts caveolae by inhibiting Cav-1 activity [38]. Treatment with MβCD, which prevents caveola formation, resulted in a Cav-1-induced decrease in meningoencephalitis in A. cantonensis infection, leading to a decrease in MMP-9 activity in the brain and CSF, as well as degradation of tight junction proteins (Zo-1 and claudin-5), thus mitigating damage to the BBB. Cav-1 / MMP-9 signaling cascades play a critical role in mediating the damage to the BBB that causes cerebral hemorrhage and brain edema during cerebral ischemia-meningoencephalitis induced by A. cantonensis infection.
Traumatic brain injury improves pro-inflammatory responses, neuronal loss, and long-term behavioral deficits, which Cavs can regulate cytokine production to regulate neuronal and glial survival signaling [39]. In Cav-1 knockout mice, there is a reduction in heme oxygenase-1 (HO-1), macrophage inflammatory protein 2, MMP-9, and COX-2 during hemorrhagic stroke, leading to decreased immune cell infiltration and mitigated hemin-induced neuronal toxicity, ultimately improving brain damage [40]. It is interesting that Cav-1 plays a neuroprotective role by modulating cytokine expression and thus mitigating neuroinflammation. Cav-1 signaling through the Cav-1 / VEGF pathway promotes both neural regeneration and vascular formation, while simultaneously reducing apoptosis. This multifaceted approach appears to alleviate brain damage caused by oxidative stress [37]. Cav-1, by binding to eNOS, inhibits the activation of eNOS, reducing the production of toxic NO. Furthermore, it suppresses the activation of MMP-9, reducing the degradation of tight junction proteins such as claudin-5 and occludin in the BBB. This attenuation mitigates the disruption of the BBB and the consequent extracellular brain edema. Cav-1 also regulates inflammatory responses in the vascular system by modulating the expression of receptors for inflammatory molecules such as TNF-α, thus reducing the ability of endothelial cells to maintain the integrity of the inner layer [22,41,42,43,44]. Consistent with previous studies, NF-κB and pro-inflammatory factors, including tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), IL-5, HO-1 and (peroxisome proliferator activated receptor) PPAR-γ also participate in the regulatory mechanisms governing A. cantonensis-induced disruption of the BBB, leakage of immune cells into the subarachnoid space, and subsequent eosinophilic meningitis and brain damage [28,29,45]. In A. cantonensis infection, the expression of HO-1 and COX-2 in the mouse brain increases, indicating their participation in regulation and their association with MMP-9 [45]. Our investigation of the role of Cav-1 in A. cantonensis-induced eosinophilic meningoencephalitis, which leads to brain damage, cerebral hemorrhage, and cerebral edema, revealed an increase in Cav-1 levels in the brains of infected mice. This increase was correlated with increased BBB permeability and inflammation, suggesting that Cav-1 may contribute to inflammation and damage to the BBB by increasing the production of pro-inflammatory cytokines.
Cav-1 interacts with Toll-like receptor 4 (TLR4), activating downstream inflammatory signals and exacerbating neuroinflammation and BBB damage [46]. Activation of the TLR4 / MyD88 signaling pathway through MAPK, ERK1 / 2, and JNK to regulate NF-κB and AP-1, thus facilitating upregulation of pro-inflammatory cytokines [46,47,48]. In the pathogenesis of neuroinflammatory-related diseases, activated astrocytes under inflammatory conditions, through the TLR4 / MyD88 signaling pathway, regulate the expression of pro-inflammatory molecules via NF-κB, MAPK, and Jak1 / Stat1 pathways, influencing the inflammatory responses of surrounding cells [49,50]. Furthermore, activation of the TLR4 / MyD88 signaling pathway regulates the expression of VCAM-1 and ICAM-1 in endothelial cells, facilitating the passage of immune cells through the BBB, but also leading to breakdown of the BBB. Inhibition of TLR4 can reduce damage to the BBB [51]. In cerebral pathologies such as ischemic brain injury, cerebral hemorrhage, and cerebral edema, increased expression of Cav-1 contributes to increased BBB permeability, exacerbating the symptoms of brain injury. After ischemic stroke, cortical neuronal death, and hypoxia-induced astrocyte apoptosis, Cav-1, through the Ras / Ras / ERK signaling pathway, protects against hypoxia-induced astrocyte apoptosis. Furthermore, through the Src kinase and ERK 1/2 signaling pathways, it prevents ischemic cell death in neurons [52,53]. After cerebral ischemia, miR-199a-5p regulates Cav-1 expression, leading to increased expression of BDNF and VEGF, promoting neural stem cell differentiation and endogenous neurogenesis [54]. Therefore, in neuroinflammatory conditions, TLR4 through the MyD88 / NF-κB signaling pathway, regulates the expression of pro-inflammatory factors, leading to increased BBB permeability and neuronal cell damage. Previous investigations have revealed that the infection-induced increase in MMP-9 induced by A. cantonensis is governed by the NF-kB or MAPK / ERK / AP-1 pathways. NF-κB and AP-1, as ubiquitous transcription factors, interact with DNA to regulate the expression of inflammatory genes, thus modulating inflammation [55,56]. Consequently, it is speculated that in A. cantonensis infected mice, leading to eosinophilic meningoencephalitis, MβCD can negatively regulate Cav-1, subsequently degrading tight junction proteins of the TLR4 / MyD88 signaling pathway, thus altering the permeability of the BBB and ultimately contributing to cerebral hemorrhage and cerebral edema. It is hypothesized that infection with A. cantonensis activates the TLR4 / MyD88 signaling pathway through Cav-1 phosphorylation, leading to increased activity of the NF-kB or MAPK / ERK / AP-1 signaling pathway, subsequently increasing MMP-9 activity, resulting in damage to the BBB, increased permeability, and inflammatory responses.
Caveolae / caveolins in endothelial cells have emerged as a crucial pathway for the invasion of pathogenic microorganisms into the body. Caveolae, through nonclathrin-regulated macropinocytosis, provide a pathway for the cellular internalization of numerous molecules. Unlike clathrin-mediated endocytosis, caveolae play a distinct role in the entry of various pathogens such as viruses, bacteria, parasites, and toxins into cells. Through the caveolae mechanism, pathogens invading host cells can escape lysosomal fusion and escape host cell lysosomal killing mechanisms [57,58,59]. In the context of HIV infection, PPARα and PPARγ, regulated by Cav-1, modulate the ERK and Akt signaling pathways to reduce MMP-9 expression, thus mitigating damage to the BBB [60,61]. In parasitic infections, host macrophages use caveolae to engulf Leishmania chagasi. This process delays the fusion of L. chagasi with lysosomes, resulting in increased intracellular survival of L. chagasi [62]. Trichomonas vaginalis, in addition to its interaction with glycosaminoglycans on the surface of the host cell and its specific binding to heparan sulfate on proteoglycans, also enters host cells through caveolae-regulated endocytosis dependent on host cell cholesterol. This process occurs independently of clathrin-mediated endocytosis, using lipid-raft-mediated endocytosis [63]. Unlike the invasion mechanisms of protozoans, during infection by the nematode A. cantonensis, an increase in Cav-1 expression in the brain and through the TLR4 / MyD88 signaling pathway to activate MMP-9 leads to degradation of tight junction proteins (Zo-1 and claudin-5). This modulation alters the permeability of the BBB, resulting in infiltration of immune cells and brain edema.
5. Conclusions
The crucial role of Cav-1 in A. cantonensis-induced eosinophilic meningoencephalitis involves regulating MMP-9 expression via the TLR4 / MyD88 signaling pathway, leading to degradation of BBB and contributing to cerebral complications, hemorrhage and edema.
6. Patents
Author Contributions
Conceptualization, A.-C.C. and K.-M.C.; methodology, S.-C.L. and K.-M.C.; investigation, C.-Y.L.; writing initial draft preparation, K.-M.C. and S.-C.L.; writing - review and editing, A.-C.C. and K.-M.C.; supervision, K.-M.C. project administration, K.-M.C. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by the Chung-Shan Medical University Hospital (CSH-2023-C-043).
Institutional Review Board Statement
The animal care and handling protocols were approved by the Animal Center Animal Centre Agreement of the Chung-Shan Medical University (IACUC approval number 2573).
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained in the article.
Conflicts of Interest
The authors have no conflicts of interest to declare. Sponsors had no role in the design of the study, in the collection, analysis or interpretation of the data, in the writing of the manuscript or in the decision to publish the results.
References
- Alicata, J.E. Biology and distribution of the rat lungworm, Angiostrongylus cantonensis, and its relationship to eosinophilic meningoencephalitis and other neurological disorders of man and animals. Adv Parasitol. 1965, 223–248. [Google Scholar]
- Tsai, H.C.; Chen, Y.S.; Yen, C.M. Human Parasitic Meningitis Caused by Angiostrongylus cantonensis Infection in Taiwan. Hawaii J Med Public Health. 2013, 72, 26–27. [Google Scholar]
- Finsterer, J.; Auer, H. Parasitoses of the human central nervous system. J Helminthol. 2013, 87, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.M.; Lee, H.H.; Lai, S.C.; Hsu, L.S.; Wang, C.J.; Liu, J.Y. Apoptosis in meningoencephalitis of Angiostrongylus cantonensis-infected mice. Exp Parasitol. 2008, 119, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Mengying, Z.; Yiyue, X.; Tong, P.; Yue, H.; Limpanont, Y.; Ping, H.; Okanurak, K.; Yanqi, W.; Dekumyoy, P.; Hongli, Z.; Watthanakulpanich, D.; Zhongdao, W.; Zhi, W.; Zhiyue, L. Apoptosis and necroptosis of mouse hippocampal and parenchymal astrocytes, microglia and neurons caused by Angiostrongylus cantonensis infection. Parasit Vectors. 2017, 10, 611. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.Y.; Chen, K.M.; Lan, K.P.; Lee, H.H.; Lai, S.C. Alterations of myelin proteins in inflammatory demyelination of BALB/c mice caused by Angiostrongylus cantonensis. Vet Parasitol. 2010, 171, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, K. Mechanism of parasite killing by eosinophils in parasitic infections. Nihon Rinsho. 1993, 51, 657–663. [Google Scholar] [PubMed]
- Chen, K.M.; Lee, H.H.; Lu, K.H.; Tseng, Y.K.; Hsu, L.S.; Chou, H.L.; Lai, S.C. Association of matrix metalloproteinase-9 and Purkinje cell degeneration in mouse cerebellum caused by Angiostrongylus cantonensis. Int J Parasitol. 2004, 34, 1147–1156. [Google Scholar] [CrossRef] [PubMed]
- Lai, S.C.; Lu, C.Y.; Shyu, L.Y.; Chen, K.M. Angiostrongylus cantonensis infection induces MMP-9 and causes tight junction protein disruption associated with Purkinje cell degeneration. Parasitol Res. 2020, 119, 3433–3441. [Google Scholar] [CrossRef]
- Chen, K.M.; Liu, J.Y.; Lai, S.C.; Hsu, L.S.; Lee, H.H. Association of plasminogen activators and matrix metalloproteinase-9 proteolytic cascade with blood-CNS barrier damage of angiostrongyliasis. Int J Exp Pathol. 2006, 87, 113–119. [Google Scholar] [CrossRef]
- Villabona-Rueda, A.; Erice, C.; Pardo, C.A.; Stins, M.F. The Evolving Concept of the Blood Brain Barrier (BBB): From a Single Static Barrier to a Heterogeneous and Dynamic Relay Center. Front Cell Neurosci. 2019, 13, 405. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Huang, Q.; Zeng, L.; Hu, Z.; Tang, X. Caveolin-1 and MLRs: A potential target for neuronal growth and neuroplasticity after ischemic stroke. Int J Med Sci. 2019, 16, 1492–1503. [Google Scholar] [CrossRef] [PubMed]
- Lamaze, C.; Tardif, N.; Dewulf, M.; Vassilopoulos, S.; Blouin, C.M. The caveolae dress code: structure and signaling. Curr Opin Cell Biol. 2017, 47, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Ikezu, T.; Ueda, H.; Trapp, B.D.; Nishiyama, K.; Sha, J.F.; Volonte, D.; Galbiati, F.; Byrd, A. L.; Bassell, G.; Serizawa, H.; Lane, W.S.; Lisanti, M.P.; Okamoto, T. Affinitypurification and characterization of caveolins from the brain differential expression of caveolin-1, -2, and -3 in brain endothelial and astroglial cell types. Brain Res.
- de Almeida, C.J.G. Caveolin-1 and Caveolin-2 Can Be Antagonistic Partners in Inflammation and Beyond. Front Immunol. 2017, 8, 1530. [Google Scholar] [CrossRef]
- Liu, P.; Rudick, M.; Anderson, R.G. Multiple functions of caveolin-1. J Biol Chem. 2002, 277, 41295–41298. [Google Scholar] [CrossRef] [PubMed]
- Nag, S.; Manias, J.L.; Stewart, D.J. Expression of endothelial phosphorylated caveolin-1 is increased in brain injury. Neuropathol Appl Neurobiol. 2009, 35, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Pang, L.; Yang, S.; Dai, W.; Wu, S.; Kong, J. Role of caveolin-1 in human organ function and disease: friend or foe? Carcinogenesis. 2022, 43, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Knowland, D.; Arac, A.; Sekiguchi, K.J.; Hsu, M.; Lutz, S.E.; Perrino, J.; Steinberg, G.K.; Barres, B.A.; Nimmerjahn, A.; Agalliu, D. Stepwise recruitment of transcellular and paracellular pathways underlies blood-brain barrier breakdown in stroke. Neuron. 2014, 82, 603–617. [Google Scholar] [CrossRef] [PubMed]
- Blochet, C.; Buscemi, L.; Clément, T.; Gehri, S.; Badaut, J.; Hirt, L. Involvement of caveolin-1 in neurovascular unit remodeling after stroke: Effects on neovascularization and astrogliosis. J Cereb Blood Flow Metab. 2020, 40, 163–176. [Google Scholar] [CrossRef]
- Badaut, J.; Ajao, D.O.; Sorensen, D.W.; Fukuda, A.M.; Pellerin, L. Caveolin expression changes in the neurovascular unit after juvenile traumatic brain injury: signs of blood-brain barrier healing? Neuroscience. 2015, 285, 215–226. [Google Scholar] [CrossRef]
- Gu, Y.; Zheng, G.; Xu, M.; Li, Y.; Chen, X.; Zhu, W.; Tong, Y.; Chung, S.K.; Liu, K.J.; Shen, J. Cav-1 regulates nitric oxide-mediated matrix metalloproteinases activity and blood-brain barrier permeability in focal cerebral ischemia and reperfusion injury. J Neurochem. 2012, 120, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.; Kahle, M.P.; Bix, G.J. Perlecan and the blood-brain barrier: beneficial proteolysis? Front Pharmacol. 2012, 3, 55. [Google Scholar] [CrossRef] [PubMed]
- Weiss, N.; Miller, F.; Cazaubon, S.; Couraud, P.O. The blood-brain barrier in brain homeostasis and neurological diseases. Biochim Biophys Acta. 2009, 1788, 842–857. [Google Scholar] [CrossRef] [PubMed]
- Kimbler, D.E.; Shields, J.; Yanasak, N.; Vender, J.R.; Dhandapani, K.M. Activation of P2X7 promotes cerebral edema and neurological injury after traumatic brain injury in mice. PLoS One. 2012, 7, e41229. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, M.A.; Babcock, I.W.; Royo, M.A.; Sibley, L.A.; Kelly, A.G.; Harris, T.H. Vascular Endothelial Growth Factor-C Treatment Enhances Cerebrospinal Fluid Outflow during Toxoplasma gondii Brain Infection but Does Not Improve Cerebral Edema. Am J Pathol. 2024, 194, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.D.; Tsai, L.Y.; Chen, C.H.; Wang, J.J.; Hsiao, J.K.; Yen, C.M. Blood-brain barrier dysfunction occurring in mice infected with Angiostrongylus cantonensis. Acta Trop. 2006, 97, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.M.; Lai, S.C. Biochemical and pathological evaluation of albendazole/thalidomide co-therapy against eosinophilic meningitis or meningoencephalitis induced by Angiostrongylus cantonensis. J Antimicrob Chemother. 2007, 59, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Chiu, P.S.; Lai, S.C. Matrix metalloproteinase-9 leads to claudin-5 degradation via the NF-κB pathway in BALB/c mice with eosinophilic meningoencephalitis caused by Angiostrongylus cantonensis. PLoS One. [CrossRef]
- Chiu, P.S.; Lai, S.C. Matrix metalloproteinase-9 leads to blood-brain barrier leakage in mice with eosinophilic meningoencephalitis caused by Angiostrongylus cantonensis. Acta Trop. 2014, 140, 140,141–50. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Huang, S.S.; Lin, S.M.; Lin, M.J.; Chu, Y.C.; Chih, C.L.; Tsai, M.J.; Lin, H.C.; Huang, W.C.; Tsai, S.K. The neuroprotective effect of glial cell line-derived neurotrophic factor in fibrin glue against chronic focal cerebral ischemia in conscious rats. Brain Res. 2005, 1033, 28–33. [Google Scholar] [CrossRef]
- Lin, K.Y.; Lai, S.C. Induction of 2',3'-cyclic nucleotide 3'-phosphodiesterase in demyelination of BALB/c mice caused by Angiostrongylus cantonensis. J Comp Pathol. 2009, 141, 248–253. [Google Scholar] [CrossRef]
- Liu, W.Y.; Wang, Z.B.; Wang, Y.; Tong, L.C.; Li, Y.; Wei, X.; Luan, P.; Li, L. Increasing the Permeability of the Blood-brain Barrier in Three Different Models in vivo. CNS Neurosci Ther. 2015, 21, 21,568–74. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Wang, X.; Wan, B.; Wang, D.; Li, M.; Cheng, K.; Luo, Q.; Wang, D.; Lu, Y.; Zhu, L. Cav-1 accelerates hypoxia-induced endothelial dysfunction in high-altitude cerebral edema. Cell Commun Signal. 2022, 20, 160. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Sun, Y.; Xu, J.; Liu, W. Cav-1 mediates tissue plasminogen activator-induced MMP-9 up-regulation in cultured brain microvascular endothelial cells. J Neurochem. 2015, 132, 132,724–30. [Google Scholar] [CrossRef]
- Madaro, L.; Antonangeli, F.; Favia, A.; Esposito, B.; Biamonte, F.; Bouché, M.; Ziparo, E.; Sica, G.; Filippini, A.; D'Alessio, A. Knock down of Cav-1 affects morphological and functional hallmarks of human endothelial cells. J Cell Biochem. 2013, 114, 1843–1851. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Zhong, W.; Hu, Z.; Tang, X. A review of the role of cav-1 in neuropathology and neural recovery after ischemic stroke. J Neuroinflammation. 2018, 15, 348. [Google Scholar] [CrossRef] [PubMed]
- Hua, H.; Munk, S.; Whiteside, C.I. Endothelin-1 activates mesangial cell ERK1/2 via EGF-receptor transactivation and caveolin-1 interaction. Am J Physiol Renal Physiol. 2003, 284, 284,F303–12. [Google Scholar] [CrossRef] [PubMed]
- Niesman, I.R.; Schilling, J.M.; Shapiro, L.A.; Kellerhals, S.E.; Bonds, J.A.; Kleschevnikov, A.M.; Cui, W.; Voong, A.; Krajewski, S.; Ali, S.S.; Roth, D.M.; Patel, H.H.; Patel, P.M; Head, B.P. Traumatic brain injury enhances neuroinflammation and lesion volume in caveolin deficient mice. J Neuroinflammation. 2014, 11, 39. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.F.; Chen, S.F.; Lee, T.S.; Lee, H.F. Chen, S.F.; Shyue, S.K. Cav-1 deletion reduces early brain injury after experimental intracerebral hemorrhage. Am J Pathol. 2011, 178, 178,1749–61. [Google Scholar] [CrossRef] [PubMed]
- D'Alessio, A.; Al-Lamki, R.S.; Bradley, J.R.; Pober, J.S. Caveolae participate in tumor necrosis factor receptor 1 signaling and internalization in a human endothelial cell line. Am J Pathol. 2005, 166, 1273–1282. [Google Scholar] [CrossRef]
- Frank, P.G. Endothelial caveolae and Cav-1 as key regulators of atherosclerosis. Am J Pathol. 2010, 177, 544–546. [Google Scholar] [CrossRef]
- Fernández-Hernando, C.; Yu, J.; Suárez, Y.; Rahner, C.; Dávalos, A.; Lasunción, M.A.; Sessa, W.C. Genetic evidence supporting a critical role of endothelial Cav-1 during the progression of atherosclerosis. Cell Metab. 2009, 10, 10,48–54. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.H.; Kim, H.S.; Park, M.S.; Lee, E.B.; Lee, J.K.; Kim, J.T.; Kim, J.H.; Lee, M.C.; Lee, H.J.; Cho, K.H. Overexpression of Cav-1 attenuates brain edema by inhibiting tight junction degradation. Oncotarget. 2016, 7, 67857–67867. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.M.; Lan, K.P.; Lai, S.C. Heme oxygenase-1 modulates brain inflammation and apoptosis in mice with angiostrongyliasis. Parasitol Int. 2022, 87, 102528. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Tian, J.; Liu, Y.; Ye, Z.; Xu, M.; Huang, R.; Song, X. TLR4-Myd88 pathway upregulated caveolin-1 expression contributes to coronary artery spasm. Vascul Pharmacol. 2022, 142, 106947. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kim, H.; Lee, J.H.; Hwangbo, C. Toll-like receptor 4 (TLR4): new insight immune and aging. Immun Ageing. 2023, 20, 67. [Google Scholar] [CrossRef] [PubMed]
- Deguine, J.; Barton, G.M. MyD88: a central player in innate immune signaling. F1000Prime Rep. 2014, 6, 97. [Google Scholar] [CrossRef] [PubMed]
- Lei, C.; Li, Y.; Zhu, X.; Li, H.; Chang, X. HMGB1/TLR4 induces autophagy and promotes neuroinflammation after intracerebral hemorrhage. Brain Res. 2022, 9, 148003. [Google Scholar] [CrossRef] [PubMed]
- Gorina, R.; Font-Nieves, M.; Márquez-Kisinousky, L.; Santalucia, T.; Planas, A.M. Astrocyte TLR4 activation induces a proinflammatory environment through the interplay between MyD88-dependent NFkappaB signaling, MAPK, and Jak1/Stat1 pathways. Glia. 2011, 59, 242–255. [Google Scholar] [CrossRef]
- Bhaskar, S.; Sudhakaran, P.R.; Helen, A. Quercetin attenuates atherosclerotic inflammation and adhesion molecule expression by modulating TLR-NF-κB signaling pathway. Cell Immunol. 2016, 310, 131–140. [Google Scholar] [CrossRef]
- Xu, L.; Wang, L.; Wen, Z.; Wu, L.; Jiang, Y.; Yang, L.; Xiao, L.; Xie, Y.; Ma, M.; Zhu, W.; Ye, R.; Liu, X. Caveolin-1 is a checkpoint regulator in hypoxia-induced astrocyte apoptosis via Ras/Raf/ERK pathway. Am J Physiol Cell Physiol. 2016, 310, C903–C910. [Google Scholar] [CrossRef]
- Head, B.P.; Patel, H.H.; Tsutsumi, Y.M.; Hu, Y.; Mejia, T.; Mora, R.C.; Insel, P.A.; Roth, D.M.; Drummond, J.C.; Patel, P.M. Caveolin-1 expression is essential for N-methyl-D-aspartate receptor-mediated Src and extracellular signal-regulated kinase 1/2 activation and protection of primary neurons from ischemic cell death. FASEB J. 2008, 22, 828–840. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.Q.; Jiang, W.F.; Zheng, X.T.; Li, L.; Fang, Y.; Yang, Y.; Hu, X.W.; Chu, L.S. MiR-199a-5p enhances neuronal differentiation of neural stem cells and promotes neurogenesis by targeting Cav-1 after cerebral ischemia. CNS Neurosci Ther. 2023, 29, 3967–3979. [Google Scholar] [CrossRef] [PubMed]
- Lan, K.P.; Wang, C.J.; Hsu, J.D.; Chen, K.M.; Lai, S.C.; Lee, H.H. Induced eosinophilia and proliferation in Angiostrongylus cantonensis-infected mouse brain are associated with the induction of JAK/STAT1, IAP/NF-kappaB and MEKK1/JNK signals. J Helminthol. 2004, 78, 78,311–7. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.H.; Shiow, S.J.; Chung, H.C.; Huang, C.Y.; Lin, C.L.; Hsu, J.D.; Shyu, L.Y.; Wang, C.J. Development of brain injury in mice by Angiostrongylus cantonensis infection is associated with the induction of transcription factor NF-kappaB, nuclear protooncogenes, and protein tyrosine phosphorylation. Exp Parasitol. 2000, 95, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.S.; Abraham, S.N. Co-option of endocytic functions of cellular caveolae by pathogens. Immunology. 2001, 102, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Norkin, L.C. Caveolae in the uptake and targeting of infectious agents and secreted toxins. Adv Drug Deliv Rev. 2001, 49, 301–315. [Google Scholar] [CrossRef] [PubMed]
- Sieczkarski, S.B.; Whittaker, G.R. Influenza virus can enter and infect cells in the absence of clathrin-mediated endocytosis. J Virol. 2002, 76, 10455–10464. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Wen, Z.; Gao, W.; Lin, Z.; Zhong, J.; Jiu, Y. Multifaceted Functions of Host Cell Caveolae/Cav-1 in Virus Infections. Viruses. 2020, 12, 487. [Google Scholar] [CrossRef]
- Huang, W.; András, I.E.; Rha, G.B.; Hennig, B.; Toborek, M. PPARα and PPARγ protect against HIV-1-induced MMP-9 overexpression via caveolae-associated ERK and Akt signaling. FASEB J. 2011, 25, 3979–3988. [Google Scholar] [CrossRef]
- Rodríguez, N.E.; Gaur, U.; Wilson, M.E. Role of caveolae in Leishmania chagasi phagocytosis and intracellular survival in macrophages. Cell Microbiol. 2006, 8, 1106–1120. [Google Scholar] [CrossRef]
- Rai, A.K.; Johnson, P.J. Trichomonas vaginalis extracellular vesicles are internalized by host cells using proteoglycans and caveolin-dependent endocytosis. Proc Natl Acad Sci U S A. 2019, 116, 21354–21360. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Cranial base hemorrhage in mice infected with A. cantonensis (a) control, (b) day 5, (c) day 10, (d) day 15, (e) day 20, (f) day 25. After infected mice with A. cantonensis, the cranial base of the brain began to have a slight bleeding on day 10, and bleeding and expansion and congestion became more obvious on days 15 to 25, and the fifth stage of A. cantonensis could be found on days 20 and 25 (arrows).
Figure 1.
Cranial base hemorrhage in mice infected with A. cantonensis (a) control, (b) day 5, (c) day 10, (d) day 15, (e) day 20, (f) day 25. After infected mice with A. cantonensis, the cranial base of the brain began to have a slight bleeding on day 10, and bleeding and expansion and congestion became more obvious on days 15 to 25, and the fifth stage of A. cantonensis could be found on days 20 and 25 (arrows).
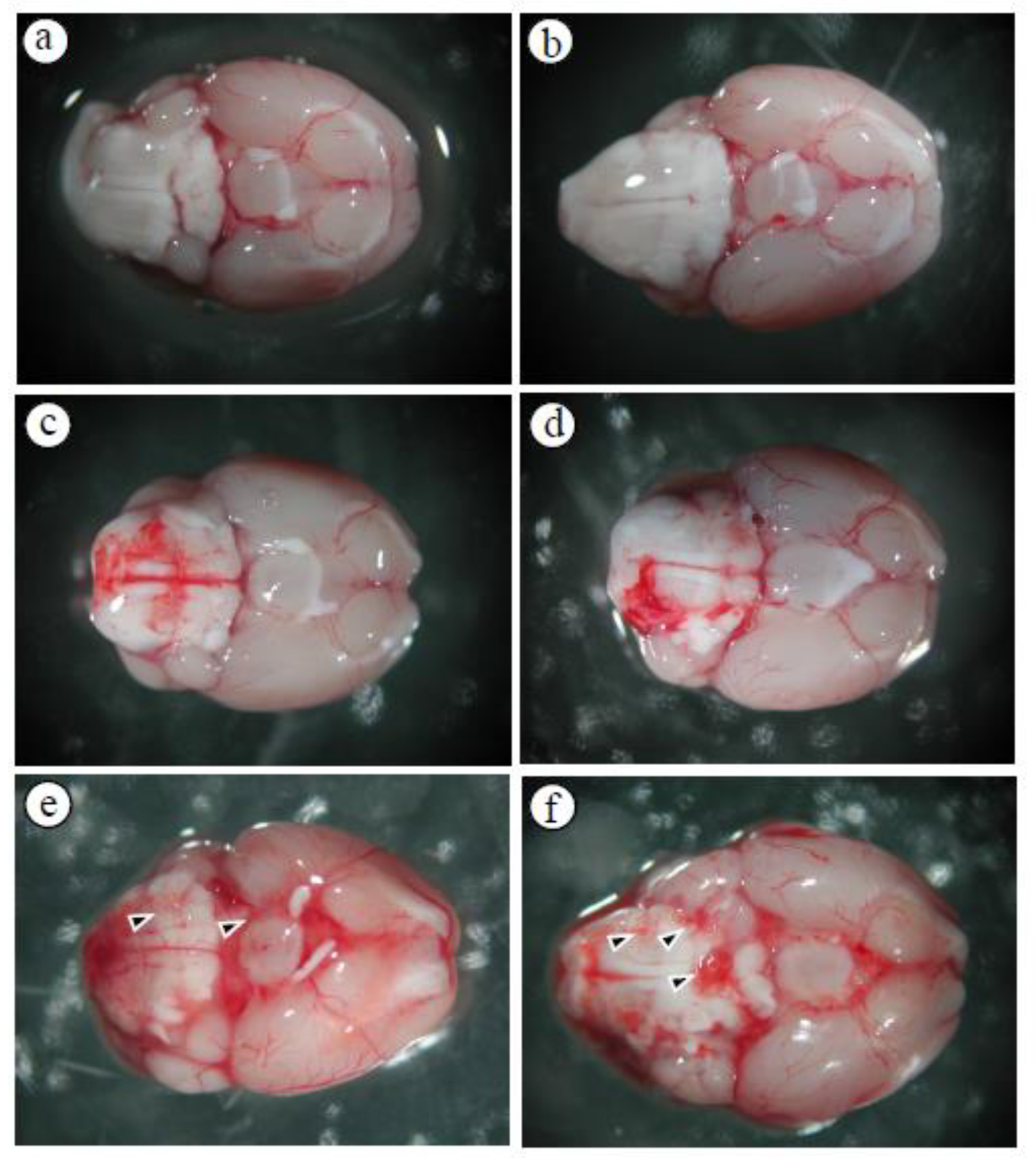
Figure 2.
Changes in brain volume and edema. (a) Brain slices were stained with TTC, brain volume of the brain was calculated by the image processing system (AIS software, Imaging research INC, Canada) and (VDay-Vcontrol)/ Vcontrol x100% was used to calculate the percentage of increase in cerebellar volume after infection with A. cantonensis, the area was found to increase and edema on days 10 to 25 after infection with A. cantonensis. (b) As measured by water content, cerebral edema increased significantly on days 15 to 25 after infection with A. cantonensis. Data for 3 mice in each group are expressed as Mean±SD, and * indicates a significant difference compared to the control group (p<0.05).
Figure 2.
Changes in brain volume and edema. (a) Brain slices were stained with TTC, brain volume of the brain was calculated by the image processing system (AIS software, Imaging research INC, Canada) and (VDay-Vcontrol)/ Vcontrol x100% was used to calculate the percentage of increase in cerebellar volume after infection with A. cantonensis, the area was found to increase and edema on days 10 to 25 after infection with A. cantonensis. (b) As measured by water content, cerebral edema increased significantly on days 15 to 25 after infection with A. cantonensis. Data for 3 mice in each group are expressed as Mean±SD, and * indicates a significant difference compared to the control group (p<0.05).
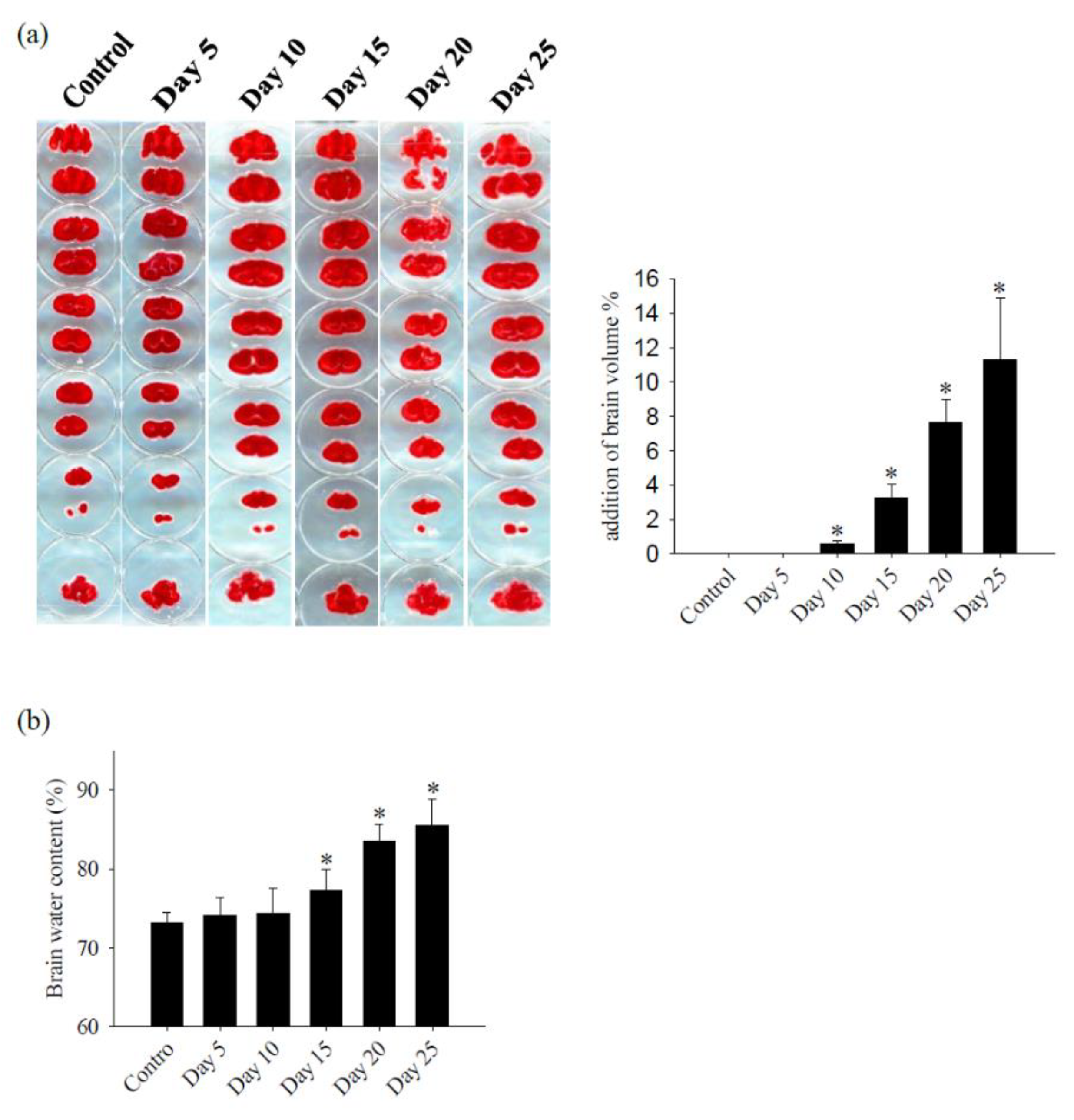
Figure 3.
Correlation analysis between 40 kDa FITC dextran concentration and brain edema. (a)The concentration of 40 kDa FITC-dextran in the CSF of mice infected with A. cantonensis on days 10 to 25 increased significantly. Data for 3 mice in each group are expressed as Mean±SD, and * indicates a significant difference compared to the control group. (p<0.05). (b) The concentration of 40 kDa FITC-dextran in the CSF and brain edema after mice were infected with A. cantonensis (n=3, r2=0.791, p<0.05).
Figure 3.
Correlation analysis between 40 kDa FITC dextran concentration and brain edema. (a)The concentration of 40 kDa FITC-dextran in the CSF of mice infected with A. cantonensis on days 10 to 25 increased significantly. Data for 3 mice in each group are expressed as Mean±SD, and * indicates a significant difference compared to the control group. (p<0.05). (b) The concentration of 40 kDa FITC-dextran in the CSF and brain edema after mice were infected with A. cantonensis (n=3, r2=0.791, p<0.05).
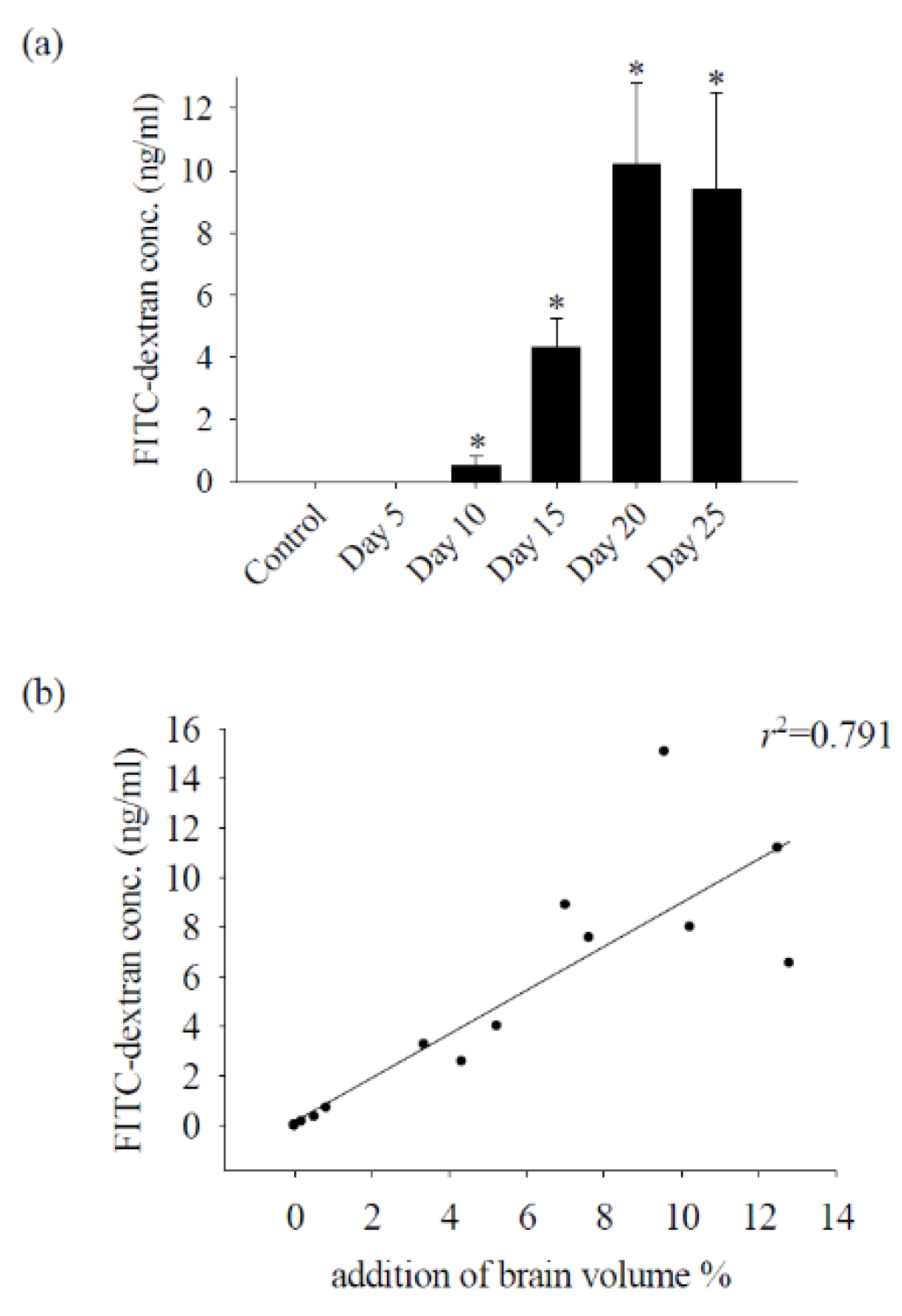
Figure 4.
Expression of the Cav-1 and p-Cav-1 proteins in the brains of mice infected with A. cantonensis. (a) Western blot detection of p-Cav-1 protein expression, the bands quantification of p-Cav-1 expression by the iBright CL750 imaging system increases significantly on days 10 to 25 after infection. (b) Western blot detection of p-Cav-1 protein expression, the bands quantification of p-Cav-1 expression by the iBright CL750 imaging system increases significantly on days 10 to 25 after infection. β-Actin is the loading control. Data for 3 mice in each group are expressed as Mean±SD, and * indicates a significant difference compared to the control group (p<0.05).
Figure 4.
Expression of the Cav-1 and p-Cav-1 proteins in the brains of mice infected with A. cantonensis. (a) Western blot detection of p-Cav-1 protein expression, the bands quantification of p-Cav-1 expression by the iBright CL750 imaging system increases significantly on days 10 to 25 after infection. (b) Western blot detection of p-Cav-1 protein expression, the bands quantification of p-Cav-1 expression by the iBright CL750 imaging system increases significantly on days 10 to 25 after infection. β-Actin is the loading control. Data for 3 mice in each group are expressed as Mean±SD, and * indicates a significant difference compared to the control group (p<0.05).
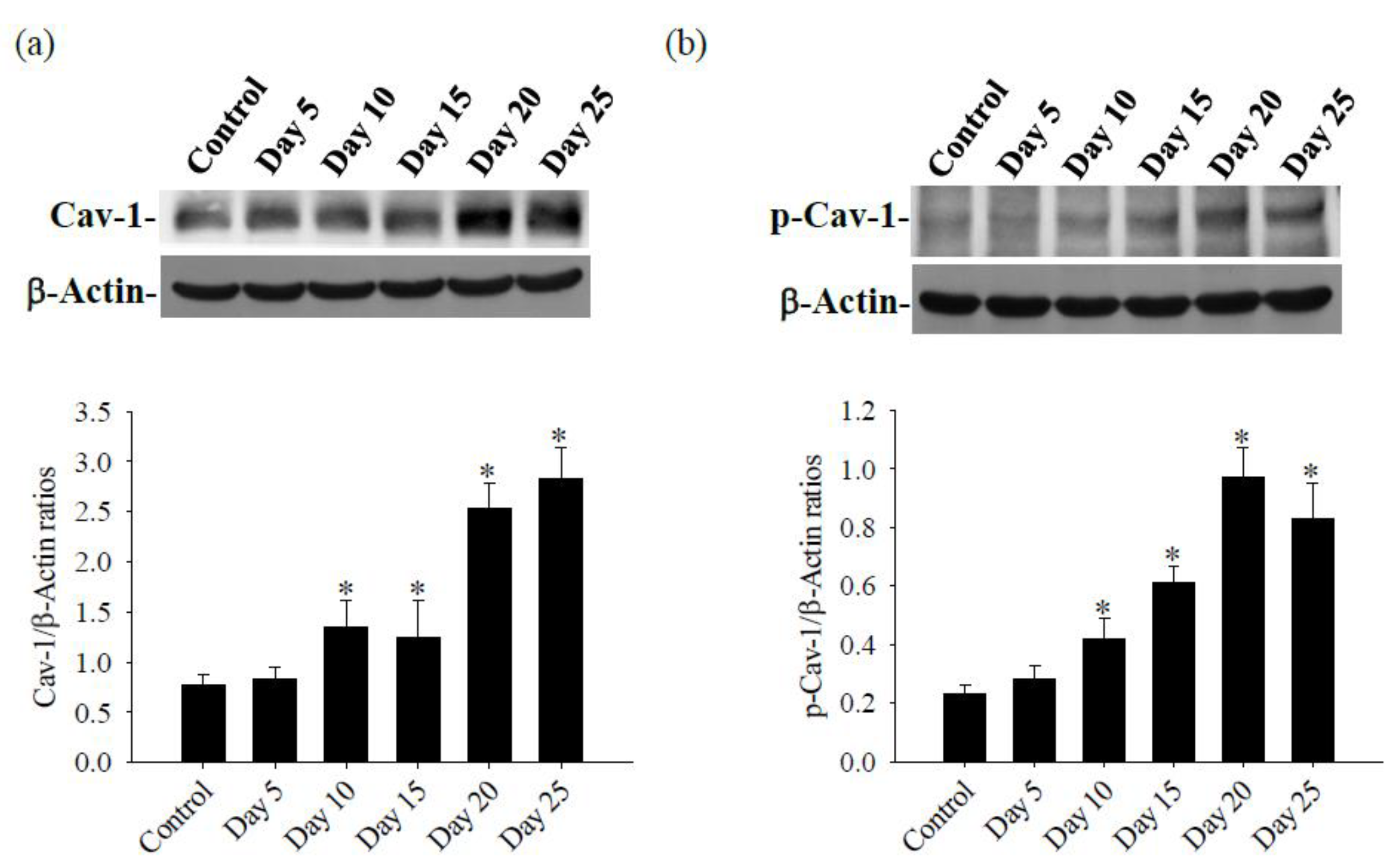
Figure 5.
Correlation analysis between 40 kDa FITC dextran concentration and Cav-1 protein. The 40 kDa FITC dextran concentration in CSF and Cav-1 protein expression in brain (n=3, r2=0.722, p<0.05) were significantly correlated.
Figure 5.
Correlation analysis between 40 kDa FITC dextran concentration and Cav-1 protein. The 40 kDa FITC dextran concentration in CSF and Cav-1 protein expression in brain (n=3, r2=0.722, p<0.05) were significantly correlated.
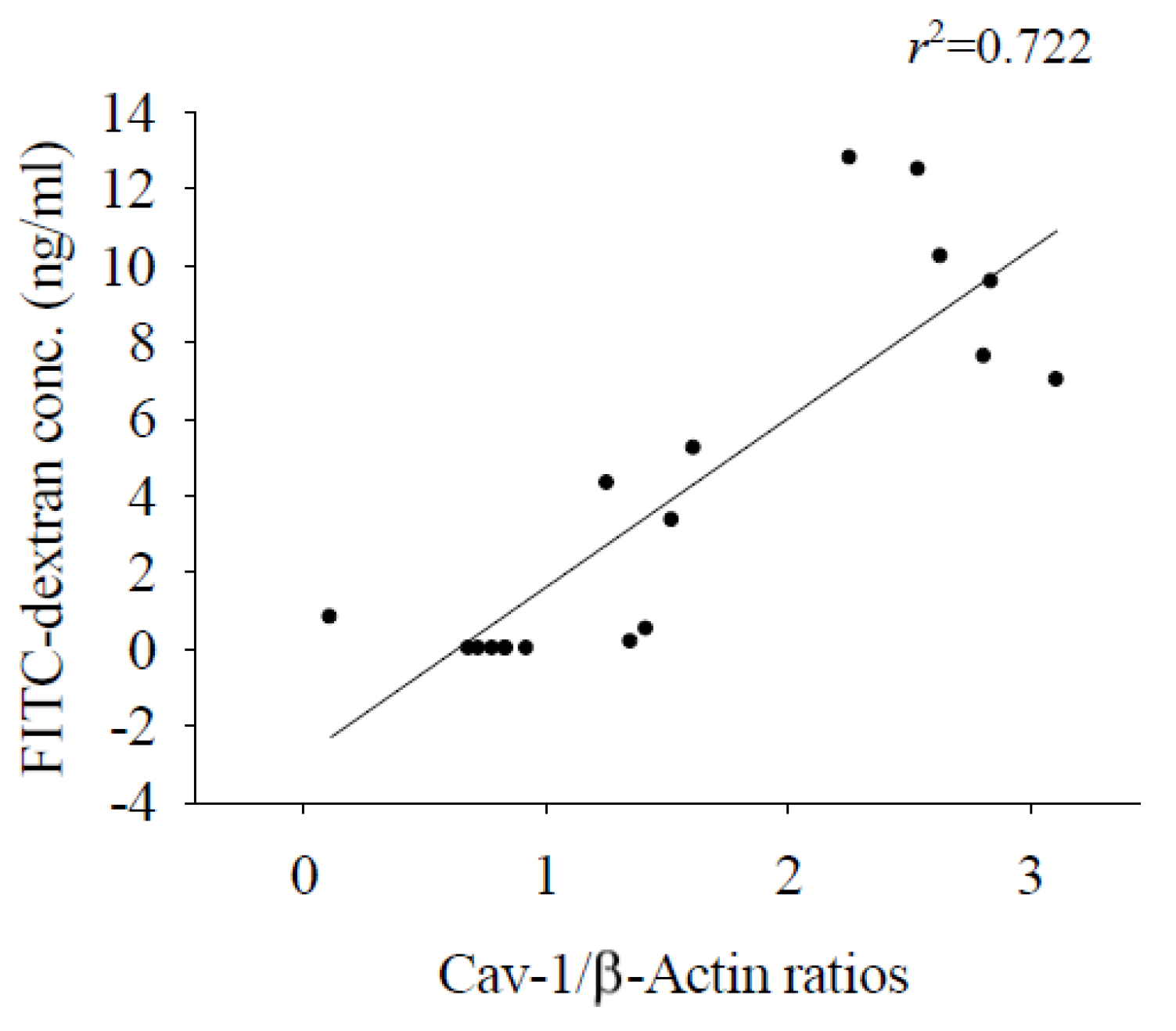
Figure 6.
Expression of TLR4 and MyD88 proteins in the brains of mice infected with A. cantonensis. Western blot to detect the expression of the TLR4 and MyD88 protein, the band quantified by the iBright CL750 imaging system, the expression of TLR4 increased significantly on days 10 to 25 after infection, and the expression of MyD88 expression increased significantly on days 15 to 25 after infection. β-Actin is the loading control. Data for 3 mice in each group are expressed as Mean±SD, and * indicates a significant difference compared to the control group (p<0.05).
Figure 6.
Expression of TLR4 and MyD88 proteins in the brains of mice infected with A. cantonensis. Western blot to detect the expression of the TLR4 and MyD88 protein, the band quantified by the iBright CL750 imaging system, the expression of TLR4 increased significantly on days 10 to 25 after infection, and the expression of MyD88 expression increased significantly on days 15 to 25 after infection. β-Actin is the loading control. Data for 3 mice in each group are expressed as Mean±SD, and * indicates a significant difference compared to the control group (p<0.05).
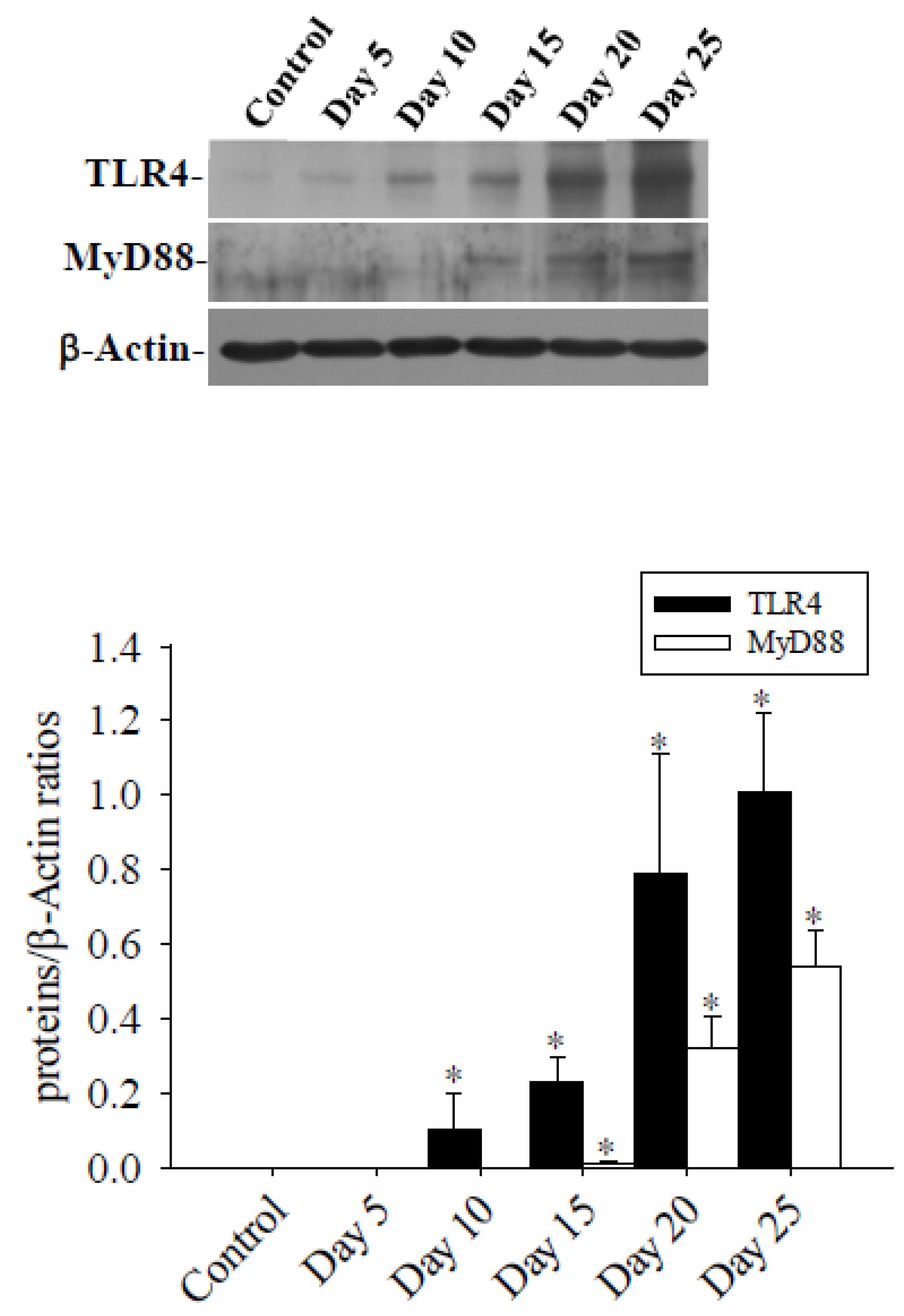
Figure 7.
The Caveolae / Cav-1 specific inhibitor MβCD was treated with mice infected with A. cantonensis. (a) The levels of Cav-1, p-Cav-1, TLR4, and MyD88 proteins exhibited significant increases in the infection group compared to the control group, but significantly decreased in the MβCD treatment group compared to the infection group. β-Actin is the loading control. (b) In the brain and CSF of MMP-9 activation measured by zymography, MMP-9 activity increased significantly on day 15 and reached a peak on days 15 and 20. MMP-9 activity decreased significantly in the group treated with MβCD treatment. (c) The concentration in the CSF increased significantly in the infected group and decreased significantly in the group treated with MβCD treatment. (d) Changes in brain volume observed with TTC stain. Brain volume increased significantly and decreased significantly in the group treated with MβCD treatment. Data from 3 mice in each group are expressed as Mean±SD, * indicates a significant difference compared to the infected group with the control group (p<0.05), # indicates a significant difference compared to the MβCD treatment group with the infected group (p<0.05).
Figure 7.
The Caveolae / Cav-1 specific inhibitor MβCD was treated with mice infected with A. cantonensis. (a) The levels of Cav-1, p-Cav-1, TLR4, and MyD88 proteins exhibited significant increases in the infection group compared to the control group, but significantly decreased in the MβCD treatment group compared to the infection group. β-Actin is the loading control. (b) In the brain and CSF of MMP-9 activation measured by zymography, MMP-9 activity increased significantly on day 15 and reached a peak on days 15 and 20. MMP-9 activity decreased significantly in the group treated with MβCD treatment. (c) The concentration in the CSF increased significantly in the infected group and decreased significantly in the group treated with MβCD treatment. (d) Changes in brain volume observed with TTC stain. Brain volume increased significantly and decreased significantly in the group treated with MβCD treatment. Data from 3 mice in each group are expressed as Mean±SD, * indicates a significant difference compared to the infected group with the control group (p<0.05), # indicates a significant difference compared to the MβCD treatment group with the infected group (p<0.05).
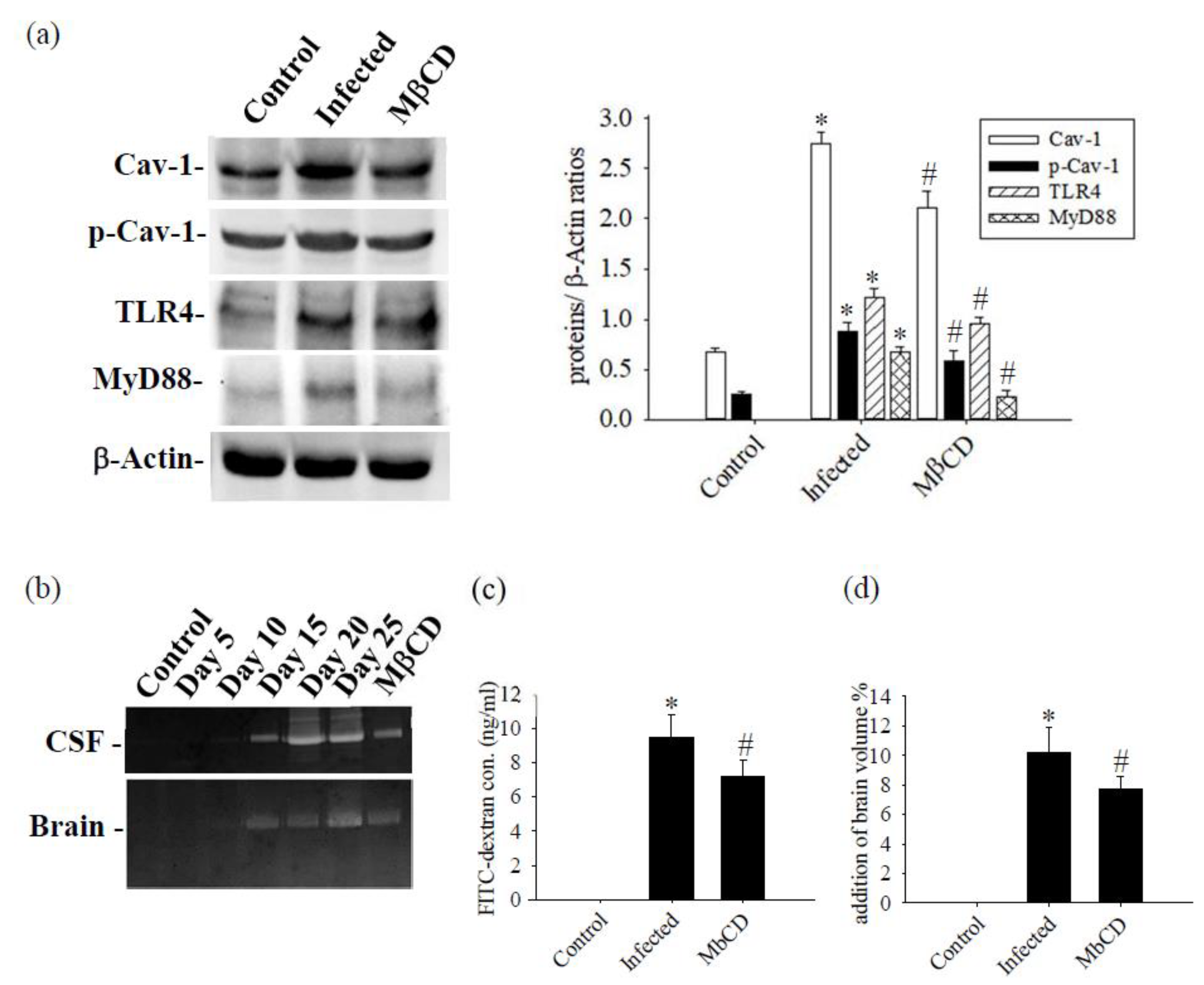
Figure 8.
The expression of tight junction proteins was treated with MβCD in mice infected with A. cantonensis. (a) Compared to the infected group, the expression of the Zo-1 and claudin-5 protein was significantly reduced when treated with MβCD treatment. β-Actin is the loading control. (b) The bands quantified by the iBright CL750 imaging system, the expression of the Zo-1 and claudin-5 proteins showed a significant increase with MβCD treatment. Data from 3 mice in each group are expressed as Mean±SD, * indicates a significant difference compared to the infected group with the control group (p<0.05), # indicates a significant difference compared to the MβCD treatment group with the infected group (p<0.05).
Figure 8.
The expression of tight junction proteins was treated with MβCD in mice infected with A. cantonensis. (a) Compared to the infected group, the expression of the Zo-1 and claudin-5 protein was significantly reduced when treated with MβCD treatment. β-Actin is the loading control. (b) The bands quantified by the iBright CL750 imaging system, the expression of the Zo-1 and claudin-5 proteins showed a significant increase with MβCD treatment. Data from 3 mice in each group are expressed as Mean±SD, * indicates a significant difference compared to the infected group with the control group (p<0.05), # indicates a significant difference compared to the MβCD treatment group with the infected group (p<0.05).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



