
Submitted:
11 April 2024
Posted:
15 April 2024
You are already at the latest version
Abstract
Abstract Octreotide is the first synthetic peptide hormone, consisted of eight amino acids, that mimics the activity of somatostatin, a natural hormone in the body. During the past decades, advanced instrumentation and crystallographic software established X-ray Powder Diffraction (XRPD) as a valuable tool for extracting structural information of biological macromolecules. The latter was demonstrated by the successful structural determination of octreotide at a remarkably high d-spacing resolution (1.87 Å) (PDB code: 6vc1). This study focuses on the response of octreotide to different humidity levels and temperatures, with a particular focus on the stability of the polycrystalline sample. XRPD measurements were accomplished employing an Anton Paar MHC-trans humidity-temperature chamber installed within a laboratory X’Pert Pro diffractometer (Malvern Panalytical). The chamber is employed to control and maintain precise humidity and temperature levels of samples during XRPD data collection. Pawley analysis of the collected data sets revealed that the octreotide polycrystalline sample is remarkably stable and no structural transitions were observed. The compound retains its orthorhombic symmetry (space group: P212121, a = 18.57744(4) Å, b = 30.17338(6) Å, c = 39.70590(9) Å, d~ 2.35 Å). However, a characteristic structural evolution in terms of lattice parameters and volume of the unit-cell is reported mainly upon controlled relative humidity variation. In addition, an improvement of signal-to-noise ratio in the XRPD data under a cycle of dehydration/rehydration is reported. These results underline the importance of considering the impact of environmental factors, as humidity and temperature, in the context of structure-based drug design, thereby contributing to the development of more effective and stable pharmaceutical products.
Keywords:
humidity variation
; temperature variation
; X-ray crystallography
; peptides
; polymorphism
; octreotide
; drug stability
; in situ X-ray powder diffraction
1. Introduction
Structural biology plays a pivotal role in advancing the understanding of biological structures and functions offering invaluable insights into the molecular intricacies that underlie life processes. Crystallization and the preservation of peptide and protein crystallinity represent a subtle interplay, where the interaction of various factors can significantly impact the outcome (Ducruix & Giegé, 1999; Asherie, 2004; Giegé, 2013; Govada & Chayen, 2019). Among these factors, environmental conditions emerge as critical factors, with relative humidity (rH) and temperature standing out. The balance between hydration and dehydration can influence crystal size, morphology and diffraction quality (Hageman, 1988; Hudaverdyan et al., 2006). Moreover, the water content within macromolecular crystals can impact the conformational stability of the molecules, adding an extra layer of complexity to the relationship between rH and crystal structure. Temperature is another important variable as temperature fluctuations can affect the thermal motion of atoms within the crystal lattice, potentially impacting the integrity of the crystalline order (Zellnitz et al., 2015; Rosenberger et al., 1993; Budayova-Spano et al., 2007). Insights from this type of research not only have implications for fundamental biological understanding but also hold promise for the development of novel therapeutics and biotechnological applications, as peptide and protein crystals, with their advantages, are increasingly utilized in creating drug carriers and excipients that exhibit prolonged action and improved physicochemical properties, allowing for the modification of the drug's performance properties.
1.1. Structural Characterization of Peptide-Based Drugs via XRPD
In the framework of assessing the stability of polycrystalline peptide-based pharmaceuticals, XRPD is the most appropriate method in gaining insights into the crystalline integrity of a molecule. This involves the identification of potential polymorphism and the detection of conformational alterations at the unit-cell level (Norrman et al., 2006; Margiolaki & Wright, 2008; Karavassili et al., 2017; Margiolaki, 2019; Spiliopoulou et al., 2020; Triandafillidis et al., 2023). Stability studies aim to establish optimal storage conditions for a drug substance, incorporating tests for thermal stability and, if relevant, sensitivity to moisture. Consequently, conducting in situ XRPD measurements under controlled temperature and humidity conditions emerges as a potent tool for studying structural modifications of biological macromolecules. In general, temperature variations can induce polymorphic transitions or degradation, while the sorption and desorption of water can alter the structure and properties of a substance (Rupley & Careri, 1991; Fenimore et al., 2004; Hudaverdyan et al., 2006; Zellnitz et al., 2015; Atakisi et al., 2018; Trampari et al., 2018; Logotheti et al., 2019). The synergistic implementation of XRPD and a humidity-temperature chamber enables real-time observation of structural adaptation through controlled changes. This approach, involving the evaluation of diffraction patterns and their subsequent analysis, leads to important conclusions about the structural evolution of the molecule.
1.2. The Pharmaceutical Peptide Octreotide
In recent years, peptides have emerged as a distinctive class of therapeutic agents owing to their unique biochemical characteristics and therapeutic potential. While the initial focus in therapeutic peptides centred on mimicking natural hormones, the trends in discovery and development have evolved. To date, there is a shift from merely replicating hormones or peptides derived from nature to the rational design of peptides with specific and desirable biochemical and physiological activities. This evolution has been facilitated by significant advances in structural chemistry and biology and the production of polycrystalline drugs is increasingly desirable (Wang et al., 2022).
In this study, the polycrystalline sample of octreotide, an eight-amino-acid peptide and the initial synthetic analogue of somatostatin (SS), is investigated thoroughly. Octreotide is currently commercially available as a suspension of amorphous counterparts, suitable for subcutaneous or intravenous injection in the management and treatment of diseases as acromegaly and thyrotrophinomas (Pohl et al., 1995; Fili et al., 2019; Spiliopoulou et al., 2021). However, there is a prominent interest in investigating a microcrystalline form of the product. The latter could potentially offer improved absorption, distribution, metabolism and excretion (ADME) characteristics, along with prolonged action and stability.
Building upon the successful crystallization and structural characterization of the peptide achieved a few years ago using high-resolution synchrotron XRPD data (Fili et al., 2019; Spiliopoulou et al., 2021), this study aims to further explore the stability of the polycrystalline form upon variation of its environmental conditions. The main objective is to comprehensively characterize its potential as an enhanced pharmaceutical formulation, reinforcing the proposal for crystalline drugs for this peptide. Simultaneously, this study demonstrates the feasibility of such investigations in biological molecules as part of Structure Based Drug Design (SBDD) (Kuntz, 1992).
1.3. In Situ XRPD Measurements upon Controlled Relative Humidity and Temperature Variation
To successfully conduct the targeted experiments, the precise control of both rH and temperature was crucial. For this purpose, the MHC-trans humidity-temperature chamber (Multi-sample Humidity Chamber TRANsmission) from Anton Paar was installed within the laboratory X’Pert Pro diffractometer (Malvern Panalytical). This chamber is well-suited for conducting in situ XRPD measurements, enabling the observation of structural changes under carefully controlled temperature and humidity conditions. The system employs two interconnected control units to manage sample temperature and rH, ensuring accuracy while preventing condensation. The chamber accommodates up to 8 samples simultaneously, maintaining uniform conditions and allows each sample to be positioned in the X-ray beam for XRPD data collection. This setup is not only time-efficient but also ensures accurate data collection while preserving optimal experimental conditions and it is the most suitable for the stress testing of substances (Trampari et al., 2018; Logotheti et al., 2019).
2. Materials and Methods
2.1. Crystallization
The octreotide peptide, supplied in the form of a lyophilized acetate salt powder by the pharmaceutical company CBL Patras, underwent the crystallization procedure using the evaporation method reported earlier (Pohl et al., 1995; Fili et al., 2019; Spiliopoulou et al., 2021). In particular, samples with a concentration of 217 mg ml -1, were produced via the dilution of octreotide acetate powder in 0.2 M oxalic acid. The dilution process took place directly within the wells of a 24-well crystallization plate, which was subsequently sealed with Parafilm to prevent sample dehydration. Two to three pinholes were carefully created in the film to facilitate gradual evaporation. The plate underwent incubation at 322.95 K with stirring at 120 rpm until the complete dilution of octreotide powder in the solvent. Subsequently, the stirring rate was reduced to 80 rpm until the solvent was nearly evaporated. Each sample was then re-dissolved in 95 μl of ddH2O and incubated at 298 K without stirring until almost complete water evaporation. This final step was repeated several times using 47.5 μl of ddH2O until a white precipitate became visible. Following this, the samples were preserved using 47.5 μl of ddH2O until no further increase in precipitate was observed. After each step, the wells were resealed with Parafilm and pinholes were reintroduced. The resulting precipitate from each sample was transferred to an Eppendorf tube, where 250 μl of ddH2O was added before storage at 298 K. Samples were kept sealed in Eppendorf tubes and stored at RT prior to XRPD measurements. To verify the crystallinity of the produced precipitate, observation under optical microscopy was conducted (Figure 1).
2.2. Laboratory and Synchrotron XRPD Data Collection
Laboratory XRPD data were initially collected using a laboratory X’Pert Pro diffractometer (Malvern Panalytical) with focusing Kα1 geometry to measure samples loaded into 1.0 mm diameter borosilicate glass capillaries and identify the presence of the orthorhombic symmetry of the octreotide crystals (space group: P212121, a = 18.608 (2) Å, b = 30.254 (3) Å, c = 39.794 (6) Å). On the incident-beam side, a focusing X-ray mirror for Cu radiation (λ = 1.540598 Å) was paired with a 0.5° anti-scatter slit, a 0.04 rad Soller slit, a 10 mm mask and a 1/2° divergence slit. On the diffracted-beam side, the configuration included a 0.04 rad Soller slit and a PIXcel1D detector with anti-scatter shielding. 2θ scans were initiated with a starting angle of 0°, which was possible without any harm to the detector due to a beam-stop mounted on the anti-scatter device of the X-ray mirror.
In order to extract more accurate unit-cell dimensions, data with enhanced angular resolution were collected under ambient conditions at the high-resolution powder diffraction beamline, ID22 [λ = 1.3007899(8) Å, RT] (Fitch et al., 2023), of the European Synchrotron Radiation Facility (ESRF) in Grenoble. All samples were loaded into 1.0 mm diameter borosilicate glass capillaries and centrifuged in order to enhance crystal packing. Excess mother liquor was removed and the capillaries were sealed with silicone vacuum grease to prevent dehydration. The capillaries were affixed to the diffractometer and rotated at a speed of 1031 revolutions per minute to ensure sufficient powder averaging. The process involved the utilization of an automated sample-changing robot for both loading and unloading the samples. This was achieved by placing the capillaries into self-centering magnetic bases. Multiple scans were collected for each sample position and the capillaries were periodically translated axially to expose new sections of the sample that hadn't been affected by the synchrotron beam. The effects of radiation damage were observed through noticeable changes in unit-cell parameters, decrease of the diffraction signal and gradual peak broadening (Basso et al., 2005; Margiolaki & Wright, 2008; Fitch et al., 2023). These effects could be tracked by comparing the profiles measured in each of the thirteen detector channels during a single scan as well as across subsequent scans. The initial scans, taken right after the sample was translated to expose fresh material, were combined with the scans of each sample position that didn’t exhibit detectable degradation to enhance counting statistics, whereas the rest were mainly used to monitor the changes in unit-cell parameters with increasing sample exposure time to the X-ray beam. The collected patterns were typically indexed using the HighScore Plus software (Degen et al., 2014) and the fitted positions of at least the first twenty reflections of the XRPD profiles. In order to extract accurate unit-cell parameter values and to analyze peak shape and background coefficients without the use of a structural model, Pawley refinement (Pawley, 1981) processes were performed verifying the orthorhombic symmetry (space group: P212121, a = 18.57744(4) Å, b = 30.17338(6) Å, c = 39.70590(9) Å).
In situ laboratory XRPD data were collected using the transmission temperature and humidity chamber MHC-trans Anton Paar. In this case, an elliptical focusing X-ray mirror for Cu radiation (λ = 1.540598 Å) was used on the incident-beam side with 1/4° divergence slit (~ 1mm thickness), 0.04 rad Soller slits and 1/4° anti-scatter slit. On the diffracted-beam side, a PIXcel1D detector was employed with a 7.5 mm anti-scatter slit and a Soller slit of 0.04°. The detector was used in one-dimensional data-collection mode. The low 2θ angle part of the diffraction profile generally contains the strongest reflections. The slits were used to reduce the scattering close to the direct beam, enhancing signal-to-noise ratio. 2θ scans were performed within a range of 2–30°.
Protein crystalline precipitates stored in Eppendorf Tubes, were centrifuged and excess mother liquor was removed. Polycrystalline specimens for in situ XRPD measurements were loaded into thin Kapton-foil holders, in order to reduce background contribution and were placed on a multiple position sample holder (Figure 2). Excess mother liquor was removed and samples were sealed with silicone vacuum grease to prevent dehydration.
In order to examine the structural modifications in the polycrystalline samples upon rH and temperature variation as well as the effect of the variation rate on that process, different cycles of experiments were conducted following different approaches. A total of 3 cycles of XRPD data collection were performed to study the response of the peptide to varying rH levels via subsequent dehydration and rehydration processes. The aim was to test the peptide's resilience within a rH range, from 95% to 30%, at a constant temperature of 294.15 K. Each step involved a 60-minute waiting time before collecting each scan to allow for the sample to equilibrate. For each rH level, 10 scans were collected, with each scan lasting approximately for 2 minutes. To obtain a more detailed understanding of changes across a broad range of rH levels, specific steps of transition from one humidity level to the next were strategically selected. The transition steps were 5% from 95% to 70% rH and 2% from 70% to the lowest rH limit (Table 1). The samples used in this study were produced from the same crystallization experiment and were introduced into the chamber sequentially for XRPD measurements. Measurements were repeated on fresh samples in order to verify reproducibility of the results.
In order to investigate the effect of temperature variation on octreotide at distinct rH levels, 6 subsequent cycles of XRPD data collection were performed. In these cycles, the sample underwent an increase/decrease in temperature from 294.15 K to 318.15 K. This was conducted for six selected humidity levels: 95%, 85%, 75%, 65%, 55% and 45%. Intermediate waiting times of 60 minutes followed each transition from one humidity level to the next (Table 2).
Finally, all XRPD profiles were analysed using the Pawley method (Pawley, 1981) implemented in the software HighScore Plus (Degen et al., 2014) in order to obtain refined values of the unit-cell parameters and to characterize the peak shape and background coefficients.
3. Results
Following the crystallization of octreotide, the orthorhombic symmetry (space group P212121) was identified via XRPD data collection in capillary mode employing the laboratory diffractometer X’Pert Pro (Malvern Panalytical) and the ESRF beamline ID22. No intermixture of crystalline phases was observed. Pawley analysis of the laboratory XRPD data yielded satisfactory results (Figure 3) [space group: P212121, a = 18.608 (2) Å, b = 30.254 (3) Å, c = 39.794 (6) Å] with typical agreement factors of Rwp = 4.8158 % and χ2 = 2.161. Pawley analysis of the synchrotron data was satisfactory providing precise lattice parameters [a = 18.57744(4) Å, b = 30.17338(6) Å, c = 39.70590(9) Å] and typical agreement factors of Rwp = 5.39 % and χ2 = 1.29 (Figure 4). The data acquired for the octreotide polycrystalline samples extended to an enhanced d-spacing resolution of approximately 2.35 Å.
Data analysis of the diffraction patterns at all rH and temperature levels revealed the stability of the orthorhombic crystal symmetry and there was no phase transition even at the lowest rH level of 30%, at which unit-cell parameters were determined via Pawley analysis [a = 18.66 (1), b = 28.21 (2), c = 39.78 (3) Å] and agreement factors of Rwp = 2.095 % and χ2 = 2.48697.
3.1. The Effect of rH Variation on Octreotide at Ambient Temperature
Data analysis unveiled a distinct evolution of the unit-cell during subsequent dehydration and rehydration processes, as evidenced by the surface plots of the XRPD patterns, depicting a consistent modification in diffraction peak positions (Figure 5). It is noteworthy that despite the dehydration process, minimal shifts of the diffraction peaks to lower angles were observed, indicating subtle variation of the unit-cell parameters which are reversible upon rehydration. Specifically, during gradual dehydration, the orthorhombic a and c axes progressively increase by an absolute value of 0.51% and 0.27%, respectively, in the cycle with a lower relative humidity (rH) level of 30%, while the b axis significantly decreases by an absolute value of 6.43% (Figure 6). Detailed values of the modifications of unit-cell parameters for each experimental cycle are provided in Table 3. At an rH level of <75%, a sudden modification in the unit-cell occurs (Figure 7), particularly evident in the b axis, which rapidly decreases (Figure 6).
Throughout this process, crystallinity is maintained and upon subsequent rehydration, the sample recovers to its initial state. Notably, data quality improves in terms of signal-to-noise ratio under a cycle of dehydration/rehydration (Figure 8). The 60-minute waiting time interval appears satisfactory for sample adaptation.
3.2. The Effect of Temperature on Octreotide at Selected rH Levels
Surface plots of XRPD data of octreotide upon temperature variation at the selected humidity levels of 95%, 85%, 75%, 65%, 55% and 45%, reveal the absence of considerable diffraction peak shifts at 95% and 85% rH (Figure 9). This observation is evident for both the heating and cooling processes between 294.15 K and 318.15 K. At 75% and 65% rH, subtle peak shifts and intensity modifications are observed. However, with respect to the results obtained upon rH variation at ambient temperature, it is apparent that temperature has a less pronounced effect on the unit-cell dimensions. In this case, the evolution of the unit-cell parameters (Figure 10) indicate that the a, b and c axes remain relatively stable, except for the 75% and 65% rH levels, where there is a noticeable decrease primarily in the b and c axes, by an absolute value of 7.30% and 2.57%, respectively, observed at 65% rH (Table 4). These specific rH levels appear critical for the changes in peak positions and the subtle modifications observed in the diffraction patterns.
In summary, the results described above indicate that the crystal structure is significantly affected by gradual dehydration while it is a reversible process upon rehydration to the initial rH level of 95% (Figure 6, Table 3). Temperature variation has a minor effect to the crystal structure with respect to rH (Figure 10, Table 4). The diffraction signal remains satisfying at all conditions, indicating that the samples retain their crystallinity. The crystal structure is stable with a 60-minute waiting time interval to adapt at each condition, which suggests that the system reaches an equilibrium state.
4. Discussion
The integration of a controlled humidity and temperature chamber into the laboratory X-ray diffraction system has significantly enhanced our capacity to investigate the impact of environmental conditions on biomacromolecular polycrystalline specimens. In this study, our primary objective was to evaluate the resilience of the pharmaceutical peptide octreotide, under varying environmental conditions, spanning temperatures from 294.15 K to 318.15 K and rH levels from 95% to 30%. Despite notable variations in lattice parameters, the orthorhombic symmetry of octreotide remained unaltered throughout these measurements. Moreover, the peptide's crystalline structure was retained, demonstrating its ability to revert to its initial state upon rehydration. These findings not only highlight the robustness of our controlled methodology in assessing the impact of environmental fluctuations on pharmaceutical peptides but also establish a novel threshold for humidity change tolerance, contrasting previous studies indicating crystallinity loss at 60% rH (Abraham, 2010).
Furthermore, the observed stability of the octreotide peptide holds promise for advancing the development of more effective medications targeting severe medical conditions as cancerous syndromes and acromegaly. This investigation serves as a catalyst for further exploration into the potential formulation of microcrystalline pharmaceuticals, leveraging their inherent stability and physicochemical attributes. The reliability and precision of data obtained via laboratory diffraction systems underscore the efficacy of XRPD as a technique for monitoring the crystalline characteristics of macromolecular pharmaceuticals, including peptide and protein-based drugs. The scalability of this methodology to an industrial level could optimize production processes and storage conditions, thereby expediting quality control measures for pharmaceutical products and enhancing the ADME profile of drugs (Lipinski et al., 1997; Prueksaritanont & Tang, 2012).
Moreover, the increasing prominence of peptides as active pharmaceutical ingredients (APIs) underscores their diverse biological activities and therapeutic applications (Kumar et al., 2022). Incorporating peptides and proteins into pharmaceutical formulations offers advantages as high target specificity, low toxicity and precise modulation of biological pathways (Lau & Dunn, 2018; Bashir et al., 2023). These biomolecules also exhibit lower immunogenicity compared to small molecule drugs and can serve as scaffolds for designing novel therapeutic agents with enhanced efficacy and selectivity (Fosgerau & Hoffmann, 2015; Muheem et al., 2016). However, despite their promising properties, the storage of peptides and proteins poses significant challenges, including susceptibility to degradation, aggregation and denaturation under various storage conditions as temperature fluctuations, pH changes, humidity and light exposure (Manning et al., 2010; Nugrahadi et al., 2023).
To address these challenges, the utilization of crystalline suspensions emerges as a promising strategy to enhance the pharmacokinetic properties of peptide-based pharmaceuticals. Crystalline suspensions offer improved stability, solubility and bioavailability compared to amorphous formulations. Furthermore, controlled crystallization techniques can optimize ADME profiles (Mirza et al., 2009; Khadka et al., 2014). Overall, these findings highlight the potential of in situ studies with humidity chambers in conjunction with XRPD not only in structure-based drug design but also in meeting the criteria for drug stability outlined by global regulatory bodies as the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) and the Food and Drug Administration (FDA) (Guidance for Industry #5 - Drug Stability Guidelines).
Relative humidity is crucial in pharmaceutical manufacturing processes and packaging, influencing the efficacy and stability of drugs. Low rH in a production line may desiccate drugs, while excessively high rH levels may promote microbial growth. Stress testing, which includes evaluation of the effects of temperature, humidity, oxidation and photolysis on drug substances, is a standard process in pharmaceutical development. Understanding the behaviour of pharmaceutical compounds under varying humidity conditions is crucial for ensuring product stability and efficacy.
Given the potential impact of unexpected humidity fluctuations on pharmaceutical products, the findings from rH experiments can provide valuable insights into product behaviour and stability. These insights are essential for optimizing production and post-production processes to maintain the physicochemical and biochemical characteristics of drugs, including their ADME profiles.
Finally, further experiments are currently ongoing to enhance the resolution limits of collected XRPD profiles and verify the reproducibility of observations. Towards this goal, the exceptionally significant improvement in the quality of synchrotron XRPD data following the upgrade of ESRF to a fourth-generation circular accelerator, along with the utilization of a new data processing algorithm at beamline ID22, creates new expectations for more efficient analysis of XRPD diffraction data and for more precise structure determination with enhanced atomic resolution (Fitch et al., 2023). These measurements will contribute to the identification of optimal dehydration protocols and provide a deeper understanding of the structural changes induced by humidity variations, ultimately advancing pharmaceutical research and development efforts.
In conclusion, the experimental approach presented herein lays the groundwork for standardizing investigations into structural changes of primary polycrystalline samples under varying humidity and temperature conditions, offering insights into crucial biological phenomena.
5. Conclusions
The exceptional stability exhibited by the pharmaceutical peptide octreotide across diverse humidity and temperature conditions presents promising avenues for drug optimization in treating severe diseases. These findings contribute to the understanding of pharmaceutical formulations and underscore the potential of leveraging their unique stability and biochemical/physicochemical properties for therapeutic purposes. Additionally, the utilization of high-quality data from X-ray diffraction laboratory diffractometers enhances the credibility of the XRPD method for in situ assessment of macromolecular crystals, establishing a recognized practice for studying structural changes during variations in environmental conditions.
The successful crystallization of octreotide has yielded highly pure and stable crystalline material suitable for further characterization studies. High crystal yield and adequate precipitate are critical for conducting a series of XRPD measurements, facilitating the determination of crystal properties and their response to environmental factors. XRPD emerges as a powerful technique for in situ studies of crystalline materials, providing real-time information on crystal structure and phase transitions under varying environmental conditions.
Utilizing XRPD as a tool for product quality control ensures the consistency and reproducibility of pharmaceutical formulations by monitoring the crystallinity and structural integrity of active pharmaceutical ingredients (APIs) and excipients. This quality control process is vital for meeting regulatory standards and ensuring the efficacy, safety and stability of pharmaceutical products. In conclusion, the integration of XRPD into pharmaceutical research and development practices holds promise for advancing drug optimization, formulation and quality assurance processes, thereby contributing to the enhancement of healthcare outcomes and patient well-being.
Funding
This research work was supported by the Hellenic Foundation for Research and Innovation (HFRI) under the ‘First Call for HFRI Research Projects to support Faculty members and researchers and the procurement of high-cost research equipment grant’ (project No. 3051 to Irene Margiolaki).
Acknowledgments
We kindly acknowledge Malvern Panalytical for the provision of instrumentation as well as software and technical support and the ESRF for provision of beamtime at the ID22 beamline. In addition, we would like to thank CBL Patras for the provision of octreotide acetate, transfer of knowledge and expertise as well as financial support.
References
- Abraham, J. (2010). Journal of Pharmaceutical Sciences, 99, 3, 1500-1515.
- Asherie, N. (2004). Methods 34, 266–272.
- Atakisi, H., Moreau, D. W. & Thorne, R. E. (2018). Acta Crystallogr D Struct Biol 74, 264–278.
- Bashir, S., Fitaihi, R. & Abdelhakim, H. E. (2023). European Journal of Pharmaceutical Sciences 182, 106374.
- Basso, S., Fitch, A. N., Fox, G. C., Margiolaki, I. & Wright, J. P. (2005). Acta Crystallogr D Biol Crystallogr 61, 1612–1625.
- Budayova-Spano, M., Dauvergne, F., Audiffren, M., Bactivelane, T. & Cusack, S. (2007). Acta Crystallogr D Biol Crystallogr 63, 339–347.
- Degen, T., Sadki, M., Bron, E., König, U. & Nénert, G. (2014). Powder Diffr. 29, S13–S18.
- Ducruix, A. & Giegé, R. (1999). Crystallization of Nucleic Acids and Proteins: A Practical Approach Oxford University Press.
- Fenimore, P. W., Frauenfelder, H., McMahon, B. H. & Young, R. D. (2004). Proc. Natl. Acad. Sci. U.S.A. 101, 14408–14413.
- Fili, S., Valmas, A., Spiliopoulou, M., Kontou, P., Fitch, A., Beckers, D., Degen, T., Barlos, K., Barlos, K. K., Karavassili, F. & Margiolaki, I. (2019). Acta Crystallogr B Struct Sci Cryst Eng Mater 75, 611–620.
- Fitch, A., Dejoie, C., Covacci, E., Confalonieri, G., Grendal, O., Claustre, L., Guillou, P., Kieffer, J., De Nolf, W., Petitdemange, S., Ruat, M. & Watier, Y. (2023). J Synchrotron Rad 30, 1003–1012.
- Fosgerau, K. & Hoffmann, T. (2015). Drug Discovery Today 20, 122–128.
- Giegé, R. (2013). The FEBS Journal 280, 6456–6497.
- Govada, L. & Chayen, N. (2019). Crystals 9, 106.
- Hageman, M. J. (1988). Drug Development and Industrial Pharmacy 14, 2047–2070.
- Hudaverdyan, T. G., Kachalova, G. S. & Bartunik, H. D. (2006). Crystallogr. Rep. 51, 519–524.
- Karavassili, F., Valmas, A., Fili, S., Georgiou, C. & Margiolaki, I. (2017). Biomolecules 7, 63.
- Khadka, P., Ro, J., Kim, H., Kim, I., Kim, J. T., Kim, H., Cho, J. M., Yun, G. & Lee, J. (2014). Asian Journal of Pharmaceutical Sciences 9, 304–316.
- Kumar, V., Bansal, V., Madhavan, A., Kumar, M., Sindhu, R., Awasthi, M. K., Binod, P. & Saran, S. (2022). Bioengineered 13, 4309–4327.
- Kuntz, I. D. (1992). Science 257, 1078–1082.
- Lau, J. L. & Dunn, M. K. (2018). Bioorganic & Medicinal Chemistry 26, 2700–2707.
- Lipinski, C. A., Lombardo, F., Dominy, B. W. & Feeney, P. J. (1997). Advanced Drug Delivery Reviews 23, 3–25.
- Logotheti, S., Valmas, A., Trampari, S., Fili, S., Saslis, S., Spiliopoulou, M., Beckers, D., Degen, T., Nénert, G., Fitch, A. N., Karavassili, F. & Margiolaki, I. (2019). J Appl Crystallogr 52, 816–827.
- Manning, M. C., Chou, D. K., Murphy, B. M., Payne, R. W. & Katayama, D. S. (2010). Pharm Res 27, 544–575.
- Margiolaki, I. (2019). International Tables of Crystallography- Volume H: Powder Diffraction, chapter 7.1, 718-736, 2019.
- Margiolaki, I. & Wright, J. P. (2008). Acta Crystallogr A Found Crystallogr 64, 169–180.
- Mirza, S., Miroshnyk, I., Heinämäki, J., Antikainen, O., Rantanen, J., Vuorela, P., Vuorela, H. & Yliruusi, J. (2009). AAPS PharmSciTech 10, 113–119.
- Muheem, A., Shakeel, F., Jahangir, M. A., Anwar, M., Mallick, N., Jain, G. K., Warsi, M. H. & Ahmad, F. J. (2016). Saudi Pharmaceutical Journal 24, 413–428.
- Norrman, M., Ståhl, K., Schluckebier, G. & Al-Karadaghi, S. (2006). J Appl Crystallogr 39, 391–400.
- Nugrahadi, P. P., Hinrichs, W. L. J., Frijlink, H. W., Schöneich, C. & Avanti, C. (2023). Pharmaceutics 15, 935.
- Pawley, G. S. (1981). J. Appl. Crystallogr. 14, 357–361.
- Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C. & Ferrin, T. E. (2004). J. Comput. Chem. 25, 1605–1612.
- Pohl, E., Heine, A., Sheldrick, G. M., Dauter, Z., Schneider, T., Wilson, K. S. & Kallen, J. (1995). Acta Crystallogr. D Biol. Crystallogr. 51, 60–68.
- Prueksaritanont, T. & Tang, C. (2012). AAPS J. 14, 410–419.
- Rosenberger, F., Howard, S. B., Sowers, J. W. & Nyce, T. A. (1993). Journal of Crystal Growth 129, 1–12.
- Rupley, J. A. & Careri, G. (1991). Advances in Protein Chemistry, 41, 37–172.
- Spiliopoulou, M., Karavassili, F., Triandafillidis, D.-P., Valmas, A., Fili, S., Kosinas, C., Barlos, K., Barlos, K. K., Morin, M., Reinle-Schmitt, M. L., Gozzo, F. & Margiolaki, I. (2021). Acta Crystallogr. A Found. Adv. 77, 186–195.
- Spiliopoulou, M., Valmas, A., Triandafillidis, D.-P., Kosinas, C., Fitch, A., Karavassili, F. & Margiolaki, I. (2020). Crystals 10, 54.
- Trampari, S., Valmas, A., Logotheti, S., Saslis, S., Fili, S., Spiliopoulou, M., Beckers, D., Degen, T., Nénert, G., Fitch, A. N., Calamiotou, M., Karavassili, F. & Margiolaki, I. (2018). J Appl Crystallogr 51, 1671–1683.
- Triandafillidis, D. P., Karavassili, F., Spiliopoulou, M., Valmas, A., Athanasiadou, M., Nikolaras, G., Fili, S., Kontou, P., Bowler, M. W., Chasapis, C. T., Von Dreele, R. B., Fitch, A. N. & Margiolaki, I. (2023). Acta Crystallogr. D Biol. Crystallogr. 79, 374–386.
- Wang, L., Wang, N., Zhang, W., Cheng, X., Yan, Z., Shao, G., Wang, X., Wang, R. & Fu, C. (2022). Sig. Transduct. Target Ther. 7, 48.
- Zellnitz, S., Narygina, O., Resch, C., Schroettner, H. & Urbanetz, N. A. (2015). International Journal of Pharmaceutics 489, 170–176.
Figure 1.
Optical microscopy images of the polycrystalline octreotide sample.
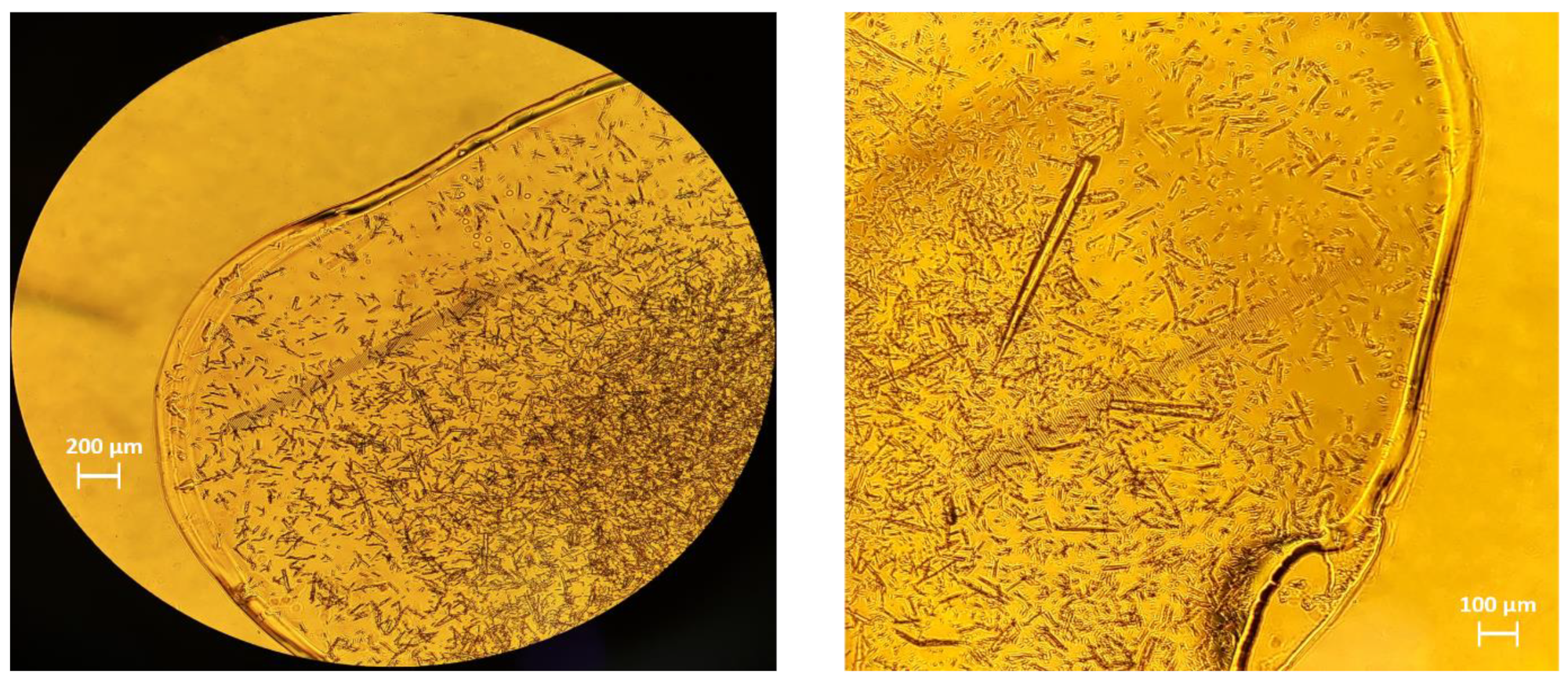
Figure 2.
(a) Empty Kapton foil holder (Left) and Kapton foil holder filled with the polycrystalline octreotide sample (Right). (b) View of the interior of the humidity chamber containing the multiple position sample holder. (c) The configuration of the X’Pert Pro diffractometer equipped with the MHC-trans humidity and temperature chamber for in situ XRPD data collection in transmission mode.
Figure 2.
(a) Empty Kapton foil holder (Left) and Kapton foil holder filled with the polycrystalline octreotide sample (Right). (b) View of the interior of the humidity chamber containing the multiple position sample holder. (c) The configuration of the X’Pert Pro diffractometer equipped with the MHC-trans humidity and temperature chamber for in situ XRPD data collection in transmission mode.

Figure 3.
Pawley fits of XRPD data of polycrystalline octreotide at ambient conditions (capillary mode) and selected rH levels (95%, 70%, 60%, 40%, 30%). The data extend up to ~ 2.35 Å resolution. They were collected employing a laboratory X-ray powder diffractometer (X’Pert Pro, Malvern Panalytical) equipped with an Anton Paar MHC-trans humidity-temperature chamber [λ = 1.540598 Å, RT]. In each panel, the black and red lines represent the experimental data and the calculated profiles, respectively, while the blue line corresponds to the difference between the experimental and calculated profiles. The vertical bars indicate the Bragg reflections compatible with this space group (P212121, lattice parameters at ambient conditions: a = 18.608 (2) Å, b = 30.254 (3) Å, c = 39.794 (6) Å).
Figure 3.
Pawley fits of XRPD data of polycrystalline octreotide at ambient conditions (capillary mode) and selected rH levels (95%, 70%, 60%, 40%, 30%). The data extend up to ~ 2.35 Å resolution. They were collected employing a laboratory X-ray powder diffractometer (X’Pert Pro, Malvern Panalytical) equipped with an Anton Paar MHC-trans humidity-temperature chamber [λ = 1.540598 Å, RT]. In each panel, the black and red lines represent the experimental data and the calculated profiles, respectively, while the blue line corresponds to the difference between the experimental and calculated profiles. The vertical bars indicate the Bragg reflections compatible with this space group (P212121, lattice parameters at ambient conditions: a = 18.608 (2) Å, b = 30.254 (3) Å, c = 39.794 (6) Å).
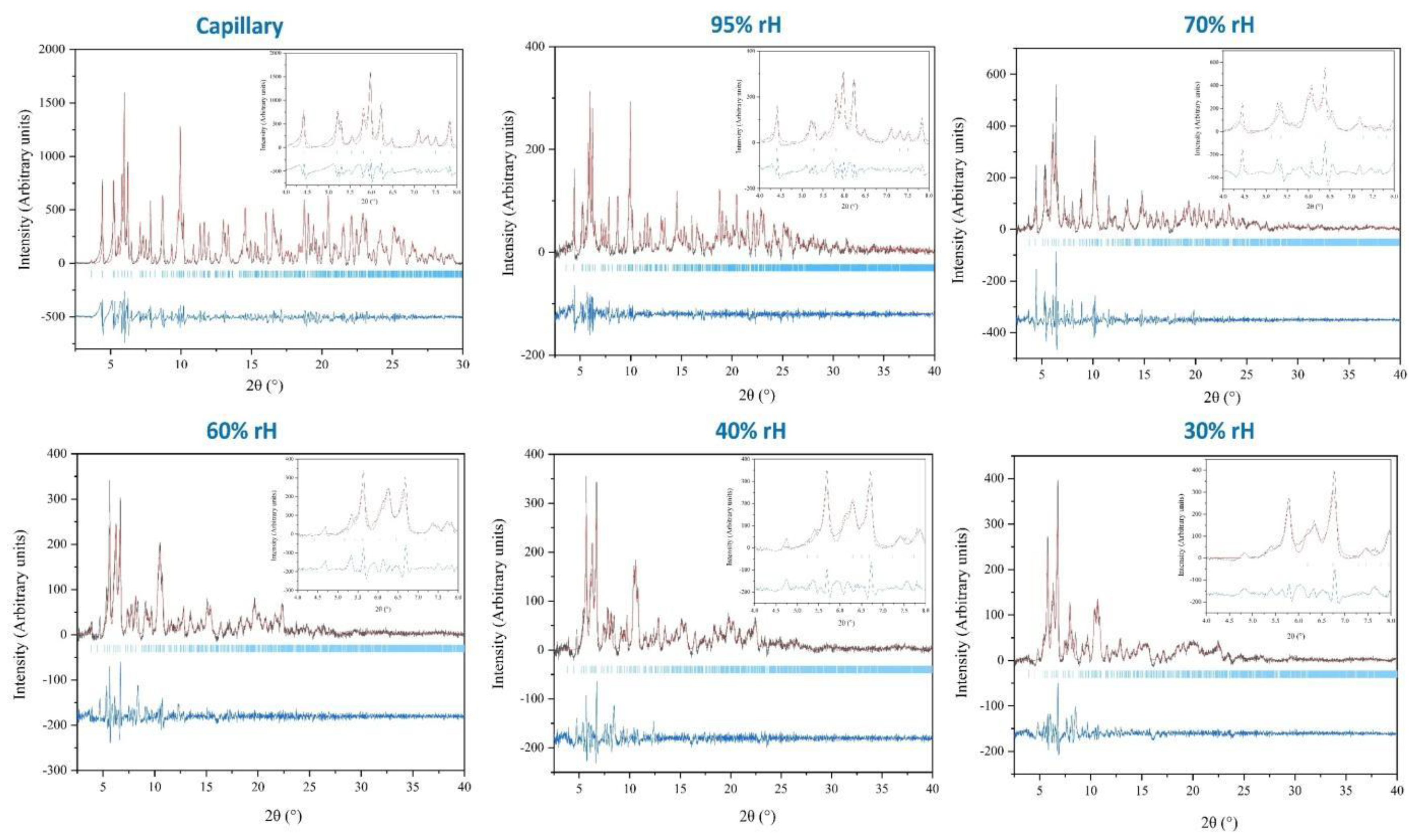
Figure 4.
Upper panel: Pawley fit of XRPD synchrotron data of Octreotide. The data were collected on ID22 at ESRF and extend up to ~ 2.35 Å resolution [λ = 1.3007899(8) Å, RT]. The black, red and lower green lines represent the experimental data, the calculated pattern and the difference between the experimental and calculated profiles, respectively. The orange vertical bars correspond to Bragg reflections compatible with this space group (P212121, a = 18.57744(4) Å, b = 30.17338(6) Å, c = 39.70590(9) Å). To highlight the enhanced d-spacing resolution, the profile was systematically multiplied by factors of 5 and 16 as indicated in the figure. Lower panel: Magnification of the 2θ range from 4.3° to 5.6°, emphasizing the enhanced angular resolution of the diffraction pattern. The background intensity has been subtracted for clarity.
Figure 4.
Upper panel: Pawley fit of XRPD synchrotron data of Octreotide. The data were collected on ID22 at ESRF and extend up to ~ 2.35 Å resolution [λ = 1.3007899(8) Å, RT]. The black, red and lower green lines represent the experimental data, the calculated pattern and the difference between the experimental and calculated profiles, respectively. The orange vertical bars correspond to Bragg reflections compatible with this space group (P212121, a = 18.57744(4) Å, b = 30.17338(6) Å, c = 39.70590(9) Å). To highlight the enhanced d-spacing resolution, the profile was systematically multiplied by factors of 5 and 16 as indicated in the figure. Lower panel: Magnification of the 2θ range from 4.3° to 5.6°, emphasizing the enhanced angular resolution of the diffraction pattern. The background intensity has been subtracted for clarity.
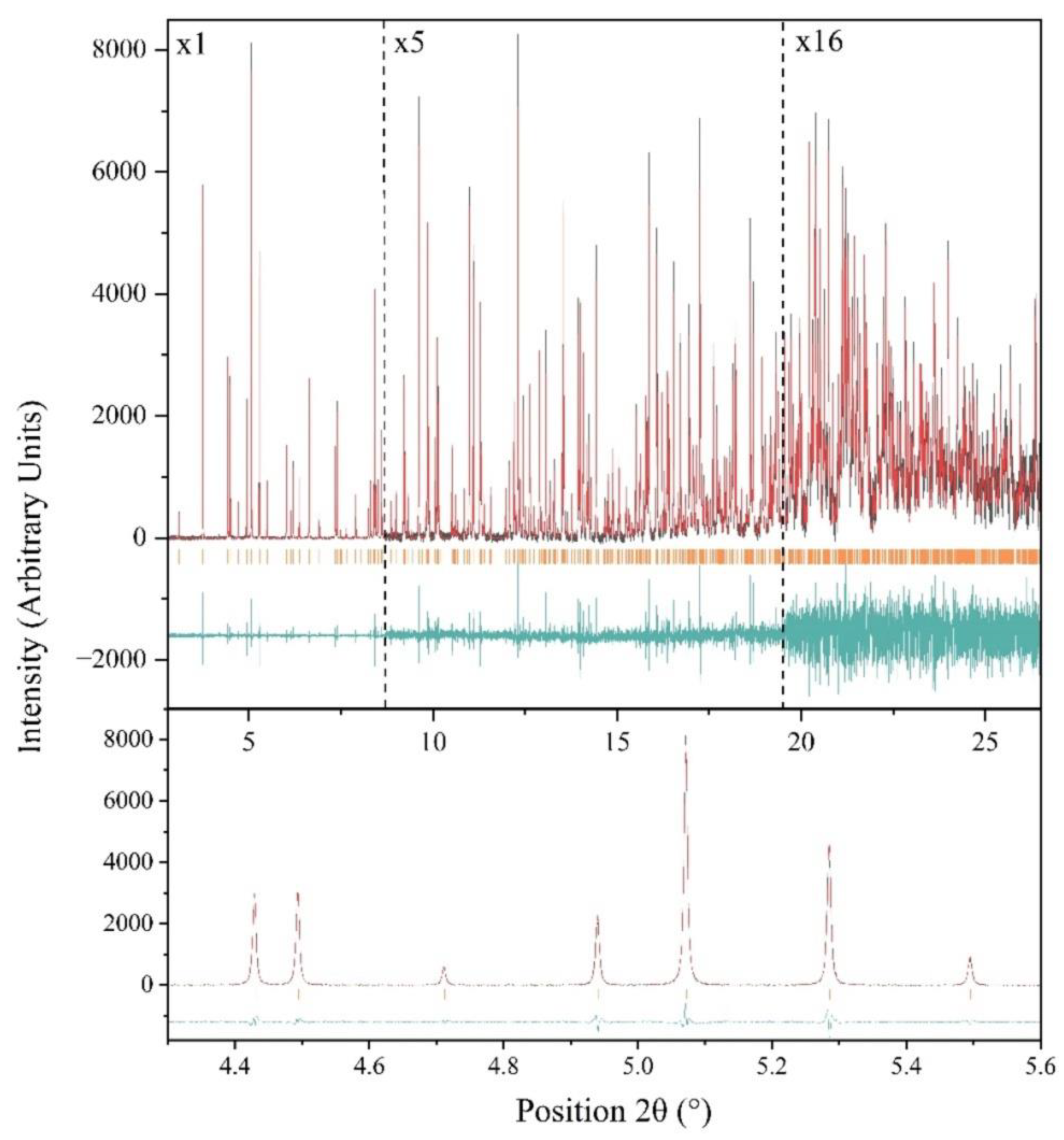
Figure 5.
Surface plots of laboratory XRPD data of the octreotide polycrystalline precipitate upon gradual dehydration/rehydration cycles from 95% to 60% rH (left), 95% to 40% rH (middle) and 95% to 30% (right). Alterations of the peak positions and intensities are evident upon gradual dehydration and rehydration cycles. Significant peak shifts become evident upon dehydration, particularly below 70% rH. Upon rehydration and above 75% rH, the sample effectively recovers to its initial state.
Figure 5.
Surface plots of laboratory XRPD data of the octreotide polycrystalline precipitate upon gradual dehydration/rehydration cycles from 95% to 60% rH (left), 95% to 40% rH (middle) and 95% to 30% (right). Alterations of the peak positions and intensities are evident upon gradual dehydration and rehydration cycles. Significant peak shifts become evident upon dehydration, particularly below 70% rH. Upon rehydration and above 75% rH, the sample effectively recovers to its initial state.
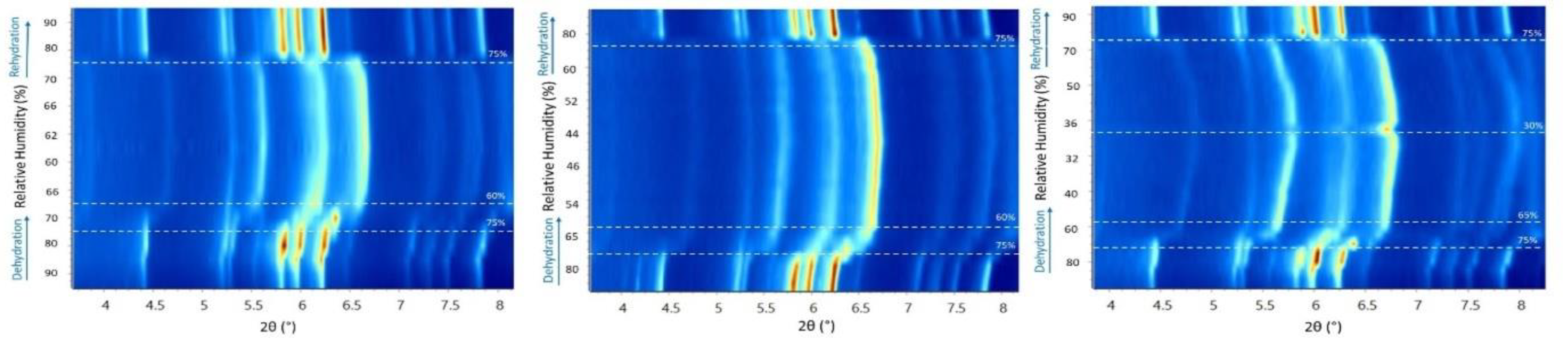
Figure 6.
Evolution of normalized unit-cell parameters upon gradual dehydration and rehydration of the octreotide polycrystalline sample from 95% to 60 % rH (upper panel), 40 % rH (middle panel) and 30 % rH (lower panel). Purple, red, green and blue symbols correspond to the extracted parameters of the unit-cell volume V, the a axis, the b axis and the c axis respectively. The lines are guides to the eye.
Figure 6.
Evolution of normalized unit-cell parameters upon gradual dehydration and rehydration of the octreotide polycrystalline sample from 95% to 60 % rH (upper panel), 40 % rH (middle panel) and 30 % rH (lower panel). Purple, red, green and blue symbols correspond to the extracted parameters of the unit-cell volume V, the a axis, the b axis and the c axis respectively. The lines are guides to the eye.
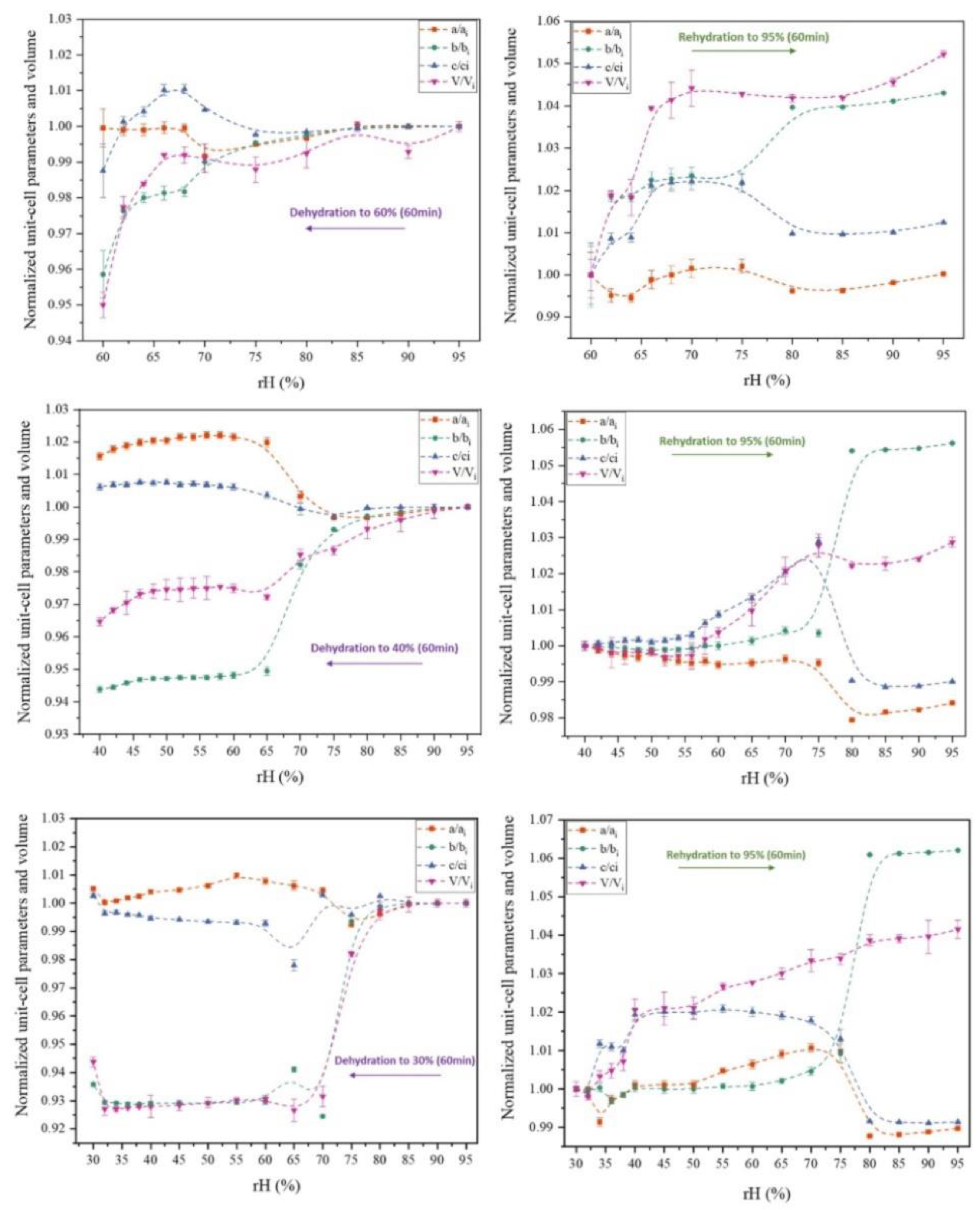
Figure 7.
Magnification of the laboratory XRPD data in the 4-8° 2θ range revealing significant peak shifts at 75% and 70% rH (RT), along with the subsequent recovery of the sample after rehydration.
Figure 7.
Magnification of the laboratory XRPD data in the 4-8° 2θ range revealing significant peak shifts at 75% and 70% rH (RT), along with the subsequent recovery of the sample after rehydration.
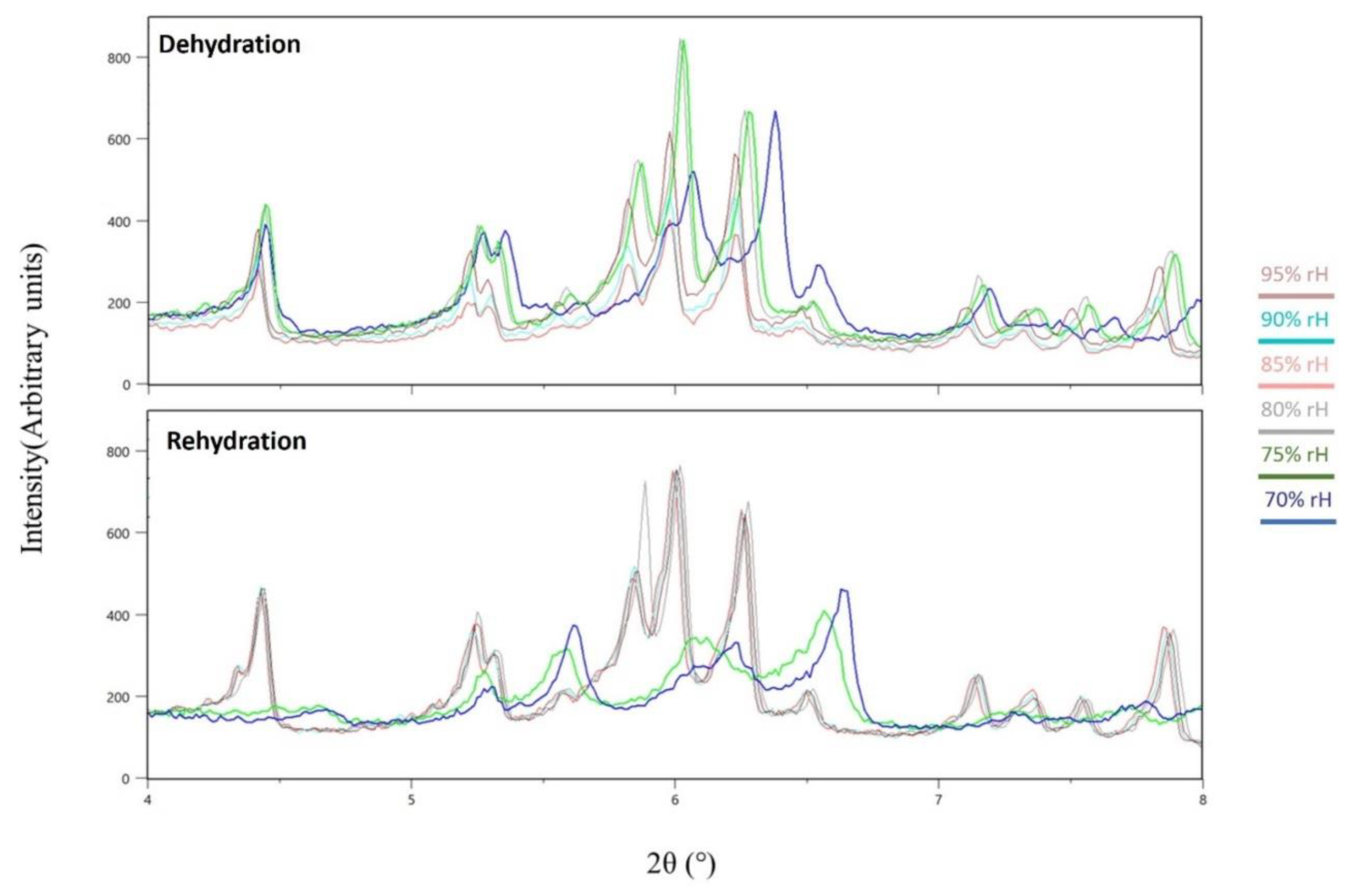
Figure 8.
Following a complete dehydration and rehydration cycle, a comparative view of XRPD data at 95% rH reveals a pronounced improvement of the XRPD data in terms of signal-to-noise ratio.
Figure 8.
Following a complete dehydration and rehydration cycle, a comparative view of XRPD data at 95% rH reveals a pronounced improvement of the XRPD data in terms of signal-to-noise ratio.
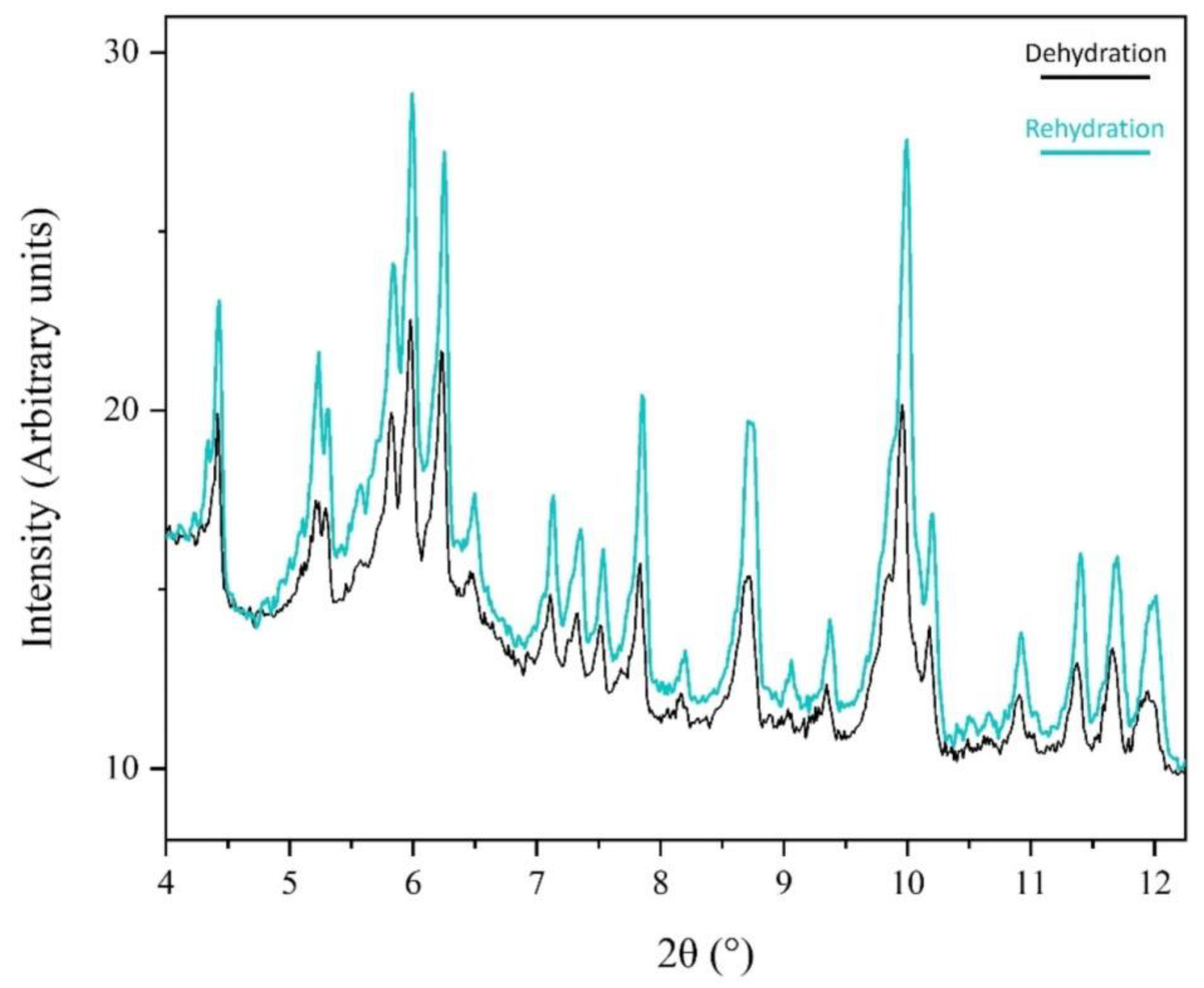
Figure 9.
Surface plots of XRPD data of octreotide polycrystalline precipitate upon gradual heating/cooling cycles at specific rH levels. A noticeable shift in the diffraction peak positions and intensities are observed at 75% and 65% rH. The latter observation may be attributed more to the effect of humidity than temperature. In addition, the XRPD data collected upon rH variation at ambient temperature described above, indicate that at rH lower than 75 % the sample exhibits a slight alteration in terms of unit-cell dimensions, yet crystallinity is maintained. The latter suggests that temperature does not significantly impact the structural integrity of the polycrystalline peptide sample.
Figure 9.
Surface plots of XRPD data of octreotide polycrystalline precipitate upon gradual heating/cooling cycles at specific rH levels. A noticeable shift in the diffraction peak positions and intensities are observed at 75% and 65% rH. The latter observation may be attributed more to the effect of humidity than temperature. In addition, the XRPD data collected upon rH variation at ambient temperature described above, indicate that at rH lower than 75 % the sample exhibits a slight alteration in terms of unit-cell dimensions, yet crystallinity is maintained. The latter suggests that temperature does not significantly impact the structural integrity of the polycrystalline peptide sample.
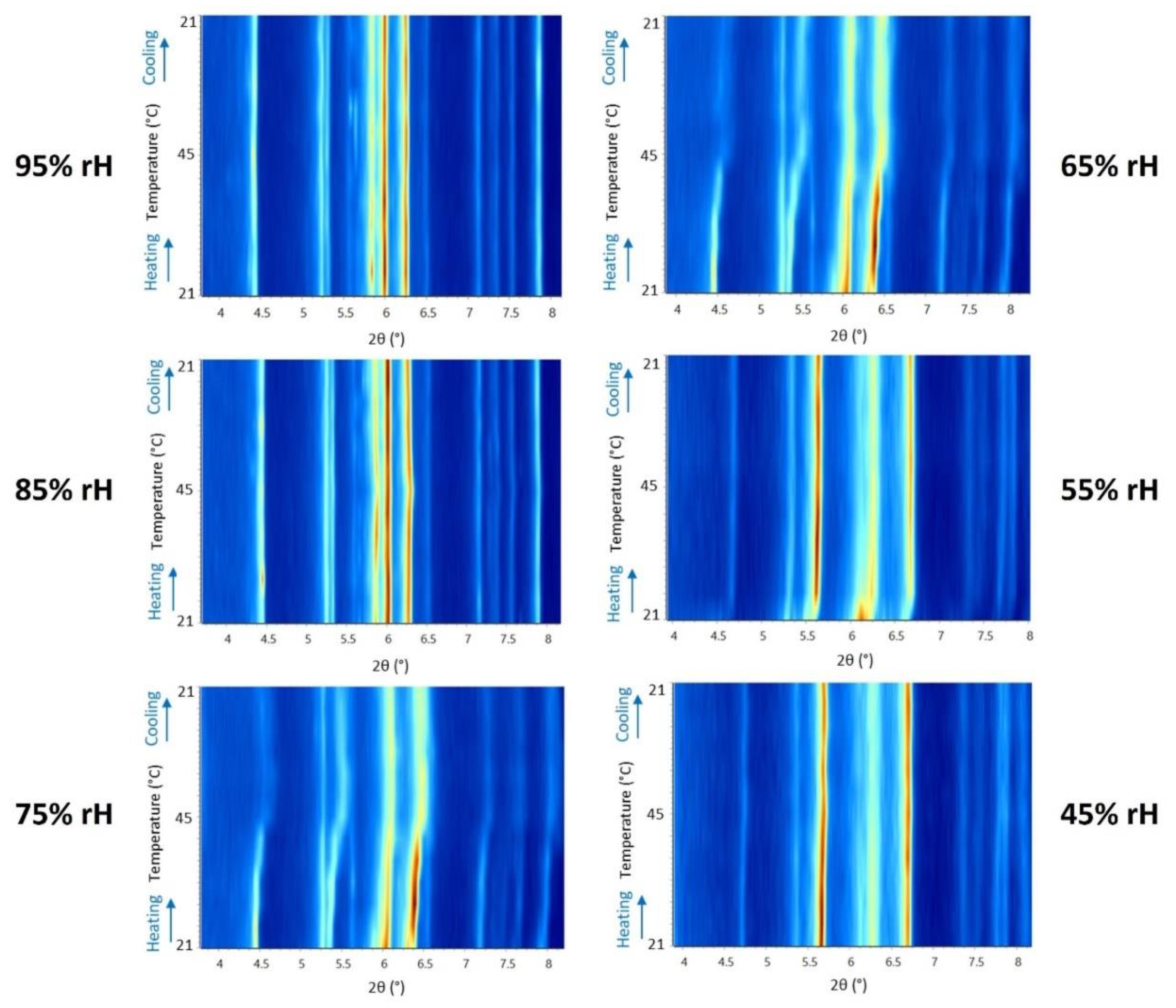
Figure 10.
Evolution of normalized unit-cell parameters upon gradual heating and cooling cycles of the octreotide polycrystalline sample from 294.15 K to 318.15 K at selected rH levels. Turquoise, pink, dark blue, light blue, brown and green symbols correspond to the rH levels of 95 %, 85 %, 75 %, 65 %, 55 % and 45 % respectively. The lines are guides to the eye.
Figure 10.
Evolution of normalized unit-cell parameters upon gradual heating and cooling cycles of the octreotide polycrystalline sample from 294.15 K to 318.15 K at selected rH levels. Turquoise, pink, dark blue, light blue, brown and green symbols correspond to the rH levels of 95 %, 85 %, 75 %, 65 %, 55 % and 45 % respectively. The lines are guides to the eye.
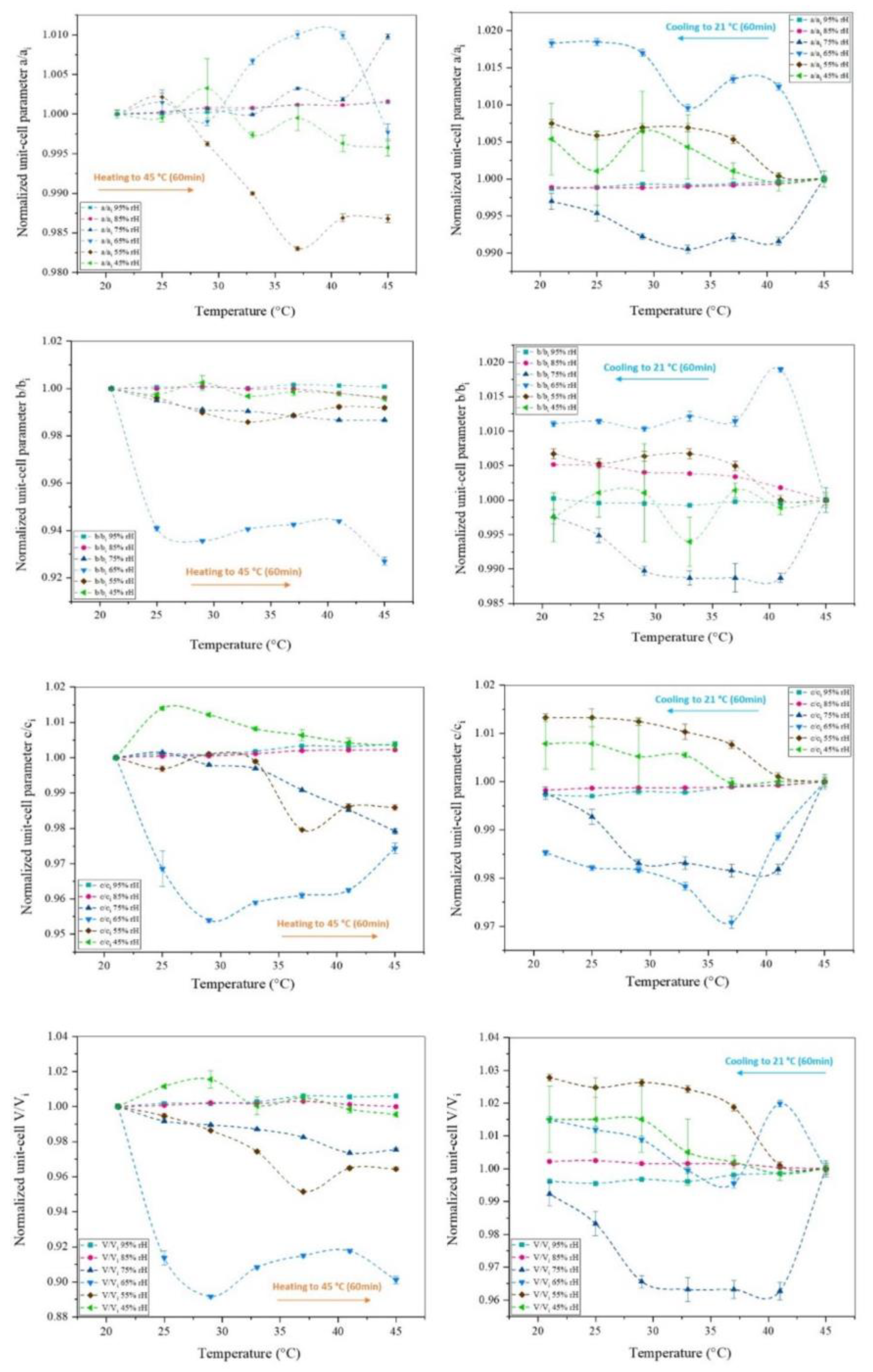

Table 1.
Data collection parameters of in situ XRPD measurements upon controlled rH variation.
| Cycle | Initial rH levels (%) | Final rH levels (%) | Step (% rH) | Temperature (K) | Waiting time | Scans /level |
|---|---|---|---|---|---|---|
| 1 | 95 | 70 | 5 | 294.15 | 60 minutes | 10 |
| 70 | 60 | 2 | 294.15 | 60 minutes | 10 | |
| 2 | 95 | 70 | 5 | 294.15 | 60 minutes | 10 |
| 70 | 40 | 2 | 294.15 | 60 minutes | 10 | |
| 3 | 95 | 70 | 5 | 294.15 | 60 minutes | 10 |
| 70 | 30 | 2 | 294.15 | 60 minutes | 10 |
Table 2.
Data collection parameters of in situ XRPD measurements upon temperature variation at selected rH levels.
Table 2.
Data collection parameters of in situ XRPD measurements upon temperature variation at selected rH levels.
| Cycle | Initial temperature (K) | Final temperature (K) | Step (K) | rH level (%) | Waiting time | Scans /level |
|---|---|---|---|---|---|---|
| 1 | 294.15 | 318.15 | 4 | 95 | 60 minutes | 10 |
| 2 | 294.15 | 318.15 | 4 | 85 | 60 minutes | 10 |
| 3 | 294.15 | 318.15 | 4 | 75 | 60 minutes | 10 |
| 4 | 294.15 | 318.15 | 4 | 65 | 60 minutes | 10 |
| 5 | 294.15 | 318.15 | 4 | 55 | 60 minutes | 10 |
| 6 | 294.15 | 318.15 | 4 | 45 | 60 minutes | 10 |
Table 3.
Modifications of unit-cell parameters of octreotide upon gradual dehydration. Delta variations were calculated as Δ(xi-xf)/xi % between the initial (xi) and final (xf) values, extracted for the orthorhombic polymorph.
Table 3.
Modifications of unit-cell parameters of octreotide upon gradual dehydration. Delta variations were calculated as Δ(xi-xf)/xi % between the initial (xi) and final (xf) values, extracted for the orthorhombic polymorph.
| 95 - 60% rH | 95 - 40% rH | 95 - 30% rH | |
|---|---|---|---|
| Δa/ai (%) | 0.04 | -1.56 | -0.51 |
| Δb/bi (%) | 4.14 | 5.61 | 6.43 |
| Δc/ci (%) | 1.24 | -0.61 | -0.27 |
| ΔV/Vi (%) | 5.00 | 3.53 | 5.63 |
Table 4.
Modifications of unit-cell parameters of octreotide upon gradual heating in each of the selected rH levels. Delta variations were calculated as Δ(xi-xf)/xi % between the initial (xi) and final (xf) values, extracted for the orthorhombic polymorph.
Table 4.
Modifications of unit-cell parameters of octreotide upon gradual heating in each of the selected rH levels. Delta variations were calculated as Δ(xi-xf)/xi % between the initial (xi) and final (xf) values, extracted for the orthorhombic polymorph.
| 294.15 - 318.15 K | 95% rH | 85% rH | 75% rH | 65% rH | 55% rH | 45% rH |
|---|---|---|---|---|---|---|
| Δa/ai (%) | -0.15 | -0.16 | -0.98 | 0.23 | 1.32 | 0.42 |
| Δb/bi (%) | -0.08 | 0.38 | 1.33 | 7.30 | 0.81 | 0.42 |
| Δc/ci (%) | -0.38 | -0.22 | 2.08 | 2.57 | 1.41 | -0.34 |
| ΔV/Vi (%) | -0.61 | 0.00 | 2.46 | 9.89 | 3.56 | 0.45 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



