Submitted:
11 April 2024
Posted:
16 April 2024
You are already at the latest version
Abstract
Immune checkpoint inhibitors (ICI) are a promising form of immunotherapy that has significantly changed the therapeutic landscape for many advanced cancers. They have shown unique clinical benefit against a broad range of tumour types and a strong overall impact on survival in studied patient populations. However, there are still many limitations holding back this immunotherapy from reaching its full potential as a possible curative option for advanced cancer patients. A great deal of research is being undertaken in the hope of driving advancements in this area, building a better understanding of the mechanisms behind immune checkpoint inhibition and ultimately developing more effective, safer, and wider-reaching agents. Taking into account the current literature on this topic, this review aims to explore in depth the basis of the use of ICIs in the treatment of advanced cancers, evaluate its efficacy and safety, consider its current limitations, and finally reflect on what the future holds for this very promising form of cancer immunotherapy.
Keywords:
Immune checkpoint inhibitor
; ipilimumab
; pembrolizumab
1. Introduction
Over the past few decades, many types of cancer immunotherapies have been studied such as monoclonal antibodies (mAb), cancer vaccines and adoptive T-cell therapies [1,2,3]. They each target different components of the immune system and have shown varying levels of promise as well as limitations [4]. However, no immunotherapy has shown as much promise in recent years as immune checkpoint inhibitors (ICI) [5]. Cancer cells can evade immunosurveillance through activation of immune checkpoint pathways, inhibiting T-cell activation and diminishing anti-tumour immune responses [6]. Immune checkpoint pathways are activated when co-inhibitory molecules on the surface of cancer cells or antigen-presenting cells (APCs) bind to T-cell surface molecules known as immune checkpoints [7]. ICIs act to block immunosuppressive signalling by inhibiting this binding, reviving anti-tumour activity and preventing cancer progression [5]. In 2011, the Food and Drug Administration (FDA) approved the first ICI for clinical use, Ipilimumab, for treatment of metastatic melanoma, an inhibitory mAb targeting CD152, cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) [8]. This marked the beginning of a new era of cancer immunotherapy and since then other ICIs have also been approved such as Pembrolizumab targeting the protein CD279, programmed cell death protein (PD-1) and Atezolizumab targeting its ligand CD274, programmed cell death-ligand 1 (PD-L1) [9]. Clinical trials have shown promising therapeutic outcomes of ICIs in several different cancers, most notably in melanoma, lung cancer, urothelial cancer and renal cancer, for which their mainstream use has been approved [10,11,12,13]. Additionally, there are hundreds of active clinical trials being undertaken to evaluate the efficacy and safety of multiple ICIs across many different cancers [2]. However, despite the success of ICIs in the studied patient population, only a fraction of cancer patients benefit from ICIs in practice [5]. The efficacy of ICI relies on the basis that there is an underlying antitumour immune response present, and the ICI is simply removing the brakes to release this function. However, the degree of antitumour immunity is variable and in fact regulated by complex factors in the tumour microenvironment (TME), the immunogenicity of the tumour and its mutational burden [14,15,16]. ICIs are also associated with a unique spectrum of immune-related adverse effects (irAEs), such as dermatological, gastrointestinal and endocrine inflammatory reactions [17]. These are some limitations that must be addressed if ICIs are to be effective in wider patient populations.
2. Immunoediting: Elimination, Equilibrium and Escape
It is well recognized the importance of the immune system in protecting the body against pathogens such as bacteria, viruses, fungi and parasites [18]. However, the idea that the immune system has extrinsic mechanisms by which it can detect cancerous cells, destroy them and prevent tumour growth is a concept that has only been universally accepted in recent decades [19]. This concept, known as cancer immunosurveillance, was first hypothesised by Burnet and Thomas in the 1950s, but little attention was given to it due to lack of strong experimental evidence [20,21]. It wasn’t until the mid-1990s that interest was renewed by experimental data seen in mice, which demonstrated the link between immunodeficiency and increased tumorigenesis [22]. In the years to follow, further experimental data from animal models combined with correlative data seen in humans firmly established the concept of cancer immunosurveillance and its role in tumour suppression [23]. However, there was more to unveil, as there was still confusion surrounding the fact that immunocompetent patients could still develop cancer. This was addressed through further research which showed that not only can the immune system protect against tumour development but can also selectively promote growth of cancer cells with reduced immunogenicity [19]. These contradicting roles of the immune system in host protection and tumour progression has led to the revised term known as cancer immunoediting. This is a dynamic process involving 3 distinct phases: elimination, equilibrium and escape, known as the 3 E’s of immunoediting (Figure 1). [23]
The elimination phase is essentially the original concept of immunosurveillance, in which the immune system detects and destroys neoplastic cells through a combination of innate and adaptive immune responses [24]. In some cases, this can represent an ideal endpoint in immunoediting, whereby all cancerous cells are destroyed [19]. Alternatively, in cases where variants of cancer cells are able to evade or withstand this process and there is incomplete destruction of cancer cells, immunoediting enters the equilibrium phase. In this phase the immune system is able to control net growth of a tumour, but a latent pool of cancer cells remains, as such it is considered a dormant phase of tumour growth and its presence is clinically inapparent [23]. If this equilibrium is maintained for entirety of an individual’s life, this can represent a second stable endpoint of cancer immunoediting. The two other possible outcomes from this midpoint are the eventual elimination by the immune system or evasion from immune responses and transition into final escape phase of immunoediting [19]. In the escape phase, the functional dormancy of cancer cells is broken, and they are able to grow in an unrestricted manner and become clinically apparent. The occurs due to either mutational changes in the cancer cells which result in variant population that is able to evade immune destruction or there is a significant degree of immunosuppression, either cancer induced or due to other causes, that allows unrestricted growth of cancer cells [22]. This process of transition from equilibrium to escape phase usually involves several distinct phases of immunoediting that eventually result in cancer cell population capable of evading the immune system. This is why this process is known as immunoediting, as it almost as if the immune system ‘edits’ the a cancer cell population over time, reducing its immunogenicity and sculpting a distinct phenotype of cancer cell from the original parent population that is able to escape the immunoediting process entirely [25].
This newly established understanding of immunoediting has formed a foundation for modern immunotherapy, in that all forms of immunotherapy act to augment this immune mediated process in some way [2]. Whether it is through augmenting the immune system and enhancing the elimination phase or blocking the methods of immune evasion employed by cancer cells and preventing the escape phase. In both cases, the aim is to pin back the dynamic process of immunoediting and prevent progression to the escape phase [25]. ICIs do exactly this by inhibiting the activation of immune checkpoints, which would ordinarily allow cancer cells to evade the immune system [26]. There are so many other mechanisms of immune evasion and therefore so much therapeutic potential of treating cancer in a similar manner to ICIs [27]. Theoretically, Immunotherapies primarily target the processes that occur in the elimination and escape phase of immunoediting, therefore it follows that the least is known about the equilibrium phase comparatively [25]. Therefore, more research is required to better understand the processes that underpin this phase and further prove its existence. This especially important considering it is likely to longest phase and that the continual interactions that are thought to occur between the immune systems and sub-clinical cancer cell population during this time, eventually results into the immunogenic phenotype that is able to escape its control [19]. A better understanding of the processes during this phase could mean more potential therapeutic avenues to explore for cancer treatment or even possibly cancer screening. If, hypothetically, these subclinical cancer populations could somehow be detected at such an early stage, this could mean earlier diagnosis and treatment, and overall better outcomes for patients.
3. The Anti-Tumour Immunity
The body has several mechanisms in place to prevent cancer formations at all stages of its development, from a genetic to cellular level [22]. The first barriers in place are a variety of intrinsic tumour suppressor mechanisms that act to repair damaged DNA or trigger cell apoptosis in instances where DNA repair fails and cell replication becomes aberrant [28]. When these barriers fail, extrinsic tumour suppressor mechanisms come in to place to prevent growth on a cellular level. The immune system is an important extrinsic mechanism that acts to identify and limit growth of cancer cells through the action of its effector leukocytes. Its role in tumour prevention can be further subdivided in to three mechanisms. It can prevent viral-induced tumours by eliminating viral infections, it can prevent prolonged inflammatory microenvironments that be conductive for tumorigenesis by prompt resolution of other infections and finally, it can directly target cancer cells in the process of immunosurveillance as discussed previously. The latter is arguably the most important extrinsic mechanisms and the bodies last line of defence from cancer progression [29].
As will all immune responses, the innate and adaptive immune cells play important and combined roles in identifying and eliminating cancer cells. Innate immune system is made up of cellular, biochemical and physical structural mechanisms, in place to respond rapidly to pathogens or cancer in a non-specific manner [30]. In general, the innate immune system can only detect limited set of antigens through pattern recognition receptors (PRR), receptors that are germ line encoded but do not undergo somatic recombination [31]. Innate immune cells such as macrophages, natural killer (NK) cells and dendritic cells all play a vital role in anti-tumour immunity [22]. The adaptive immune system offers a more antigen specific immune response through its effector B and T Lymphocytes, and their humoral mediators such as antibodies and cytokines. This outstanding range of antigen specificity is achieved through random somatic rearrangement of antigen receptor genes which lead to an almost infinite repertoire of antigen specific T-cell receptors and immunoglobulins for B-cells [31]. T-cells can be divided into two main types, CD8+ cytotoxic T lymphocytes (CTLs), which have an important role in cell mediated immunity, and CD4+ helper T-cells which influence many aspects of the immune response through its many subsets and their effector cytokines. Its two main subsets are Th1 cells, which promote cell-mediated immunity, and Th2 cells which promote humoral immunity [32] There are also other CD4+ subsets such as T-reg and TH17 cells, which contribute to the complex interplay between cancer and the immune system as discussed below [29]. B-cells control the humoral immune response, producing a variety of antigen specific immunoglobulins, vital for immunity against extracellular pathogens such as bacteria and parasites [32]. Of these two main types of adaptive immune cells, T-cells play a more dominant role in the anti-tumour immune response [29].
3.1. Role of Innate Immune system
The innate immune response is the first line of defence in the immunosurveillance process. When a tumour begins to grow, there is angiogenesis and increased blood supply to its population of cells. This invasive growth induces inflammatory signals leading to the recruitment of innate immune cells such as NK cells, macrophages and dendritic cells [33]. NK cells are a subset of leukocytes which play an important role in cell-mediated immunity, they produce pro-inflammatory cytokines such as interferon gamma (IFN-γ) and carry out cell-mediated cytotoxicity of cancer cells. They have the capacity to detect antigenic changes in transformed cells, through a limited set of germ line encoded inhibitory and activating receptors. Once activated, NK cells can kill cancer cells through release of cytolytic enzymes perforin and granzyme, and through expression of TNF-related apoptosis-inducing ligand (TRAIL) [29]. NK cells also have an additional role in facilitating dendritic cell (DC) maturation, through release of IFN- γ, into DC-1 phenotype, a specific subset of DCs responsible for presenting tumour associated antigens (TAA) to CD8+ CTLs [34]. This moves us on to the role of dendritic cells, a type of phagocyte considered to be the most potent antigen presenting cells (APC). Their main role is to present TAAs to both CD8+ and CD4+ T-cells, acting as a bridge between the innate and adaptive immune response [33]. Macrophages, another form of phagocyte, have a more complicated relationship with the anti-tumour immune response [35]. Like other immune cells, macrophages have a spectrum of subsets, each with functionally different phenotypes [35]. Factors in the microenvironment such as cytokines and differential oxygen tension determine what subsets a macrophage matures in to [36]. Classically activated (M1) macrophages and alternatively activated (M2) macrophages are two distinct phenotypes which represent extremes on either side of the subset spectrum [37]. Macrophages undergo M1 activation in response to pro-inflammatory cytokines such as INF- γ and TNF- α [35]. M1 macrophages have an important role in facilitating anti-tumour cytotoxic activity, as they produce a wide array of pro-inflammatory cytokines and chemokines, leading to recruitment of Th1 cells, CD8+ CTLs and NK cells, resulting in a robust cytotoxic response [38] [39]. In contrast, macrophages undergo M2 activation in response to Th2 cytokines (interleukin-4 (IL-4) and IL-13), IL-10 and transforming growth factor β (TGF-β). The resulting cells often promote tumour growth and metastasis by releasing an array of anti-inflammatory cytokine and chemokines, leading to recruitment of regulatory T-cells (T-reg) and Th2 cell, resulting in an immunosuppressive environment [35]. T-reg cells play an important role in preventing autoimmunity and maintaining self-tolerance, therefore in this context, they contribute to the immunosuppressive environment [29]. M2 macrophages also produce many growth factors and matrix metalloproteases, promoting angiogenesis and matrix remodelling, further facilitating tumour progression [40]. A high number of tumour associated macrophages (TAM) is often associated poor prognosis in many cancers, such as breast cancer [41]. In such cases, this is likely due to M2 macrophages representing a large proportion of the TAM population [36].
3.2. Role of Adaptive Immune system
The adaptive immune system offers a more focused and antigen specific attack against cancer cells [22]. The process starts through action of APCs, such as DCs, which present TAAs to T-cells [29]. For CD8+ T-cells, TCR stimulation alone is not enough to sustain optimal activation of naïve or memory CD8+ T-cell. Presence of co-stimulatory signals, via the CD28 receptor, and a third signal from Th1 cytokines, such as IL-2, are generally required for full activation [42]. Additionally, absence of co-inhibitory signals, through immune checkpoints such as CTLA-4, is also essential [43]. Successful activation results in the effector CD8+ CTLs, the principle mediators of cytotoxic immune response. CTLs eliminate cancer cells in a similar manner to NK cells, through action of cytolytic granules perforin and granzyme [42]. CTLs may be the principle cell behind cancer cell elimination but its success is dependent on input of several different cells and balance of many factors. CD4+ helper T-cells are more heterogenous population of T-cells, and therefore have a varied role in the anti-tumour response. Following antigen presentation, CD4+ T-cell develop into different functional phenotypes depending on the polarizing cytokines present [44]. In the presence of IL-12, they polarise into Th1 cells, a subset whose role in anti-tumour immunity have already been mentioned. This has direct influence through release pro-inflammatory cytokines such as IFN- and TNF-α, and indirectly through activation and expansion of CTLs [33]. In contrast, the other main subset, Th2 cells, impedes cell-mediated anti-tumour immunity [29]. Th2 cells develop in the presence of IL-4, and once fully activated, produce high level of IL-4, IL-5, IL-6, IL-10 and IL-13. These cytokines promote the B-cell mediated humoral response, but also induce T-cell anergy and inhibit T-cell-mediated cytotoxicity [33]. As mentioned before, CTLs require additional signals from Th1 cytokines to become fully activated, however, in cases where they receive Th2 cytokine signals instead, they become functionally inactivated and remain in a prolonged hypoactive state, known as T-cell anergy. The end product is a greatly supressed cytotoxic response [45]. There are two additional types of CD4+ T-cell which have some influence on cancer immunity, T-reg cells and Th17 cells [29]. T-reg cells harbour immunosuppressive function as their primary role is to prevent organ specific autoimmunity by identifying and eliminating self-reactive lymphocytes [46]. In setting of cancer, this immunosuppressive function promotes growth and progression [47]. T-regs cells effects are mediated though several mechanisms such as release of immunosuppressive cytokines, such as TGF-β and IL-10, and through direct cytolysis of T-cell, NK cells and DCs using perforin and granzyme [29,48]. The presence of T-regs cells in the TME has been associated with poor prognosis in many cancers, such as hepatocellular and ovarian cancer [49,50]. However, in some cases, for example in colorectal cancer, T-reg accumulation acts to supress bacteria-driven inflammation which promotes carcinogenesis [51]. Therefore, although T-regs primarily act to supress the anti-tumour immune response, they have somewhat a dual role in carcinogenesis. Finally, Th17 cells are subset of CD4+ cells which has an important role in mucosal immunity and produce contradicting effects on the anti-tumour immune response [29]. Unlike Th1 and Th2 cells, which represent terminal differentiation product of CD4+ cells, Th17 can transdifferentiate into different phenotypes [52]. For example, they can differentiate into Th1-like cells which release inflammatory cytokine and recruit CTLs, NKs and DCs, thus enhancing the anti-tumour cell mediated response [33]. On the other hand, they can also transdifferentiate into immunosuppressive T-regs, which promote tumour growth and progression [33].
In summary, on a cellular level the immune system can both impede and promote tumour growth through its many different immune cells. The objective of immunotherapies such as ICIs is to selectively enhance the cytotoxic cell-mediated immune responses responsible for fighting against cancer cells. Additionally, presence of cells such as CD8+ CTLS, Th1, NK and M1 cells can also be used a predictive biomarkers for ICI therapy, as it shows presence of an underlying anti-tumour immunity [5]. Therefore, knowledge of cellular processes underlying immunosurveillance have been useful in developing and guiding clinical use of immunotherapies such as ICIs.
4. Immune Checkpoint Pathways
Immune checkpoints are important regulators of the immune system. They act as TCR co-signalling partners, delivering either activating or inhibitory signals to T-cells upon stimulation. These signalling pathways are vital for maintaining self-tolerance as well as ensuring an adequate immune response [53]. An example of a stimulatory checkpoint protein is the previously mentioned CD28 molecule, a protein highly expressed on CD4+ and CD8+ T-cells, which initiates co-stimulatory signals when its binds to its corresponding ligands, CD80 and CD86, found on APCs [54]. There is a total of 7 other stimulatory checkpoints that have been identified, all of which act in similar way to activate and stimulate T-cell activity [53]. However, it is inhibitory immune checkpoints which have been the main subject of attention in recent years. Their co-inhibitory pathways have been shown to promote tumour formation and progression, through dampening of cancer immunosurveillance [55]. In fact, cancer cells have been shown to utilise these pathways by expression of their ligands, activating immunosuppressive pathways and allowing for immune evasion [56]. The therapeutic potential surrounding these pathways is evident and since the success of first ICI, Ipilimumab, targeting CTLA-4, many more inhibitory immune checkpoints have been identified. As of now, the immune checkpoints which have been successfully targeted with approved ICIs are CTLA-4, PD-1 and its ligand PD-L1 [5]. Other co-inhibitory immune checkpoints include lymphocyte activation gene-3 (LAG-3), T-cell immunoglobulin and mucin-domain containing-3 (TIM-3), T-cell immunoglobulin and ITIM domain (TIGIT), V-domain Ig suppressor of T-cell activation (VISTA) and many more (Figure 2). These are all emerging ICI targets, currently under clinical trial or development, with many showing promising outcomes [57]. However, before we focus on what the future holds, it is important the understand the current established ICI targets, CTLA-4 and PD-1, as ultimately it is their success and current limitations which is guiding research in to these new targets in the hope of improvement. In the next section will look more depth at the normal physiological roles of these immune checkpoints, the mechanism of their blockade by ICIs and analyse the success of this intervention in treatment of advanced cancers.
5. CTLA-4 Physiological Role
CTLA-4 is a transmembrane protein belonging to immunoglobulin superfamily, and its expression and function is intrinsically linked to T-cell activation [53] [8]. In fact, CTLA-4 is not detectable on naïve T-cell but is immediately induced and upregulated in response to TCR activation [55]. Its expression is regulated by strength of TCR signalling, whereby stronger TCR signalling induce greater expression of CTLA-4 [58]. CTLA-4 is a homologue of the co-stimulatory checkpoint CD28, and as such shares its ligands, CD80 (B7-1) and CD86 (B7-2), also known as the B7 ligands [55]. Following its expression, it begins to compete with CD28 for the B7 ligands, however, as it boasts a higher affinity and avidity for these ligands, it can effectively outcompete CD28, blocking costimulatory signals and attenuating T-cell activation [58]. Thus, it regulates the availability of B7 ligands to CD28, forming a homeostatic feedback loop in response to TCR stimulation. The net outcome following TCR stimulation is dependent on relative binding of CD28 versus CTLA-4 to the B7 ligands and can result in either T-cell activation or anergy [59]. Other than its primary cell intrinsic role in regulating T-cell activity, it also influences immunological tolerance through cell extrinsic mechanisms, mainly through action of T-reg cells [55]. Unlike effector T-cell, CTLA-4 is constitutively expressed on T-reg cells [58]. Of course, the importance of T-reg cells in maintaining tolerance has already been established and its expression of CTLA-4 is yet another component it of suppressor function. CTLA-4 expressed on T-regs act in similar way, limit availability of B7 ligands, in a cell-extrinsic manner, and blocking CD28 costimulatory activation of nearby effector T-cell through competitive inhibition [55]. Additionally, beyond just the binding of B7 ligands, CTLA-4 has also been reported to limit the overall availability of B7 ligands through trans-endocytosis of these ligands from APCs [60]. It is quite evident that CTLA-4’s function is inherently linked to the interaction of its homologue CD28 and its ligands, as its primary mechanisms all act in some way to impede this signalling. However, some evidence suggests that CTLA-4 may also have a role independent of CD28, in which it directly antagonises TCR signalling following B7-1 engagement, through its own cell intrinsic signals [61]. A proven mechanism has yet to be established and further research is needed to understand the intracellular signalling of CTLA-4. Ultimately, CTLA-4 fundamental role is to the maintain self-tolerance and as such, it primarily regulates T-cell activation at sites of T-cell priming, such as lymph nodes [55]. Its critical role in tolerance has been proven by observations seen in CTLA-4 knockout mice, which developed uncontrolled lymphoproliferative disease and fatal autoimmunity, due to unrestrained CD28 co-stimulation [62].
Considering it has such an important role in self-tolerance, it makes sense that on the opposite end of the scale, excessive CTLA-4 activity can produce an immunosuppressive environment conductive for tumorigenesis [58]. Such a concept was given more weight by pre-clinical studies done in late 1990s, which showed decreased tumour growth and improved survival in mouse models following CTLA-4 pathway blockade [63]. Of course, the next step was to develop approved anti-CTLA4 therapy that could replicate such results in human cancer patients. Initially, three anti-CTLA4 antibodies entered clinical trials: tremelimumab, its parental antibody known as CP-642,570 and Ipilimumab [8]. CP-642,570 did not make it past the first human trial and Tremelimumab showed initial promise in Phase I and II trials, but ultimately failed to meet it endpoints in Phase III trials [8] [64,65]. This left Ipilimumab, a fully humanized IgG1 mAb, that went on to achieve FDA and EMA approval for treatment of unresectable stage III and metastatic melanoma, following many successful clinical trials [8]. In the last few years, it has also been approved as part of combination therapy with Nivolumab, an anti-PD1 mAbs, for unresectable or metastatic melanoma, advanced renal cell carcinoma (RCC) and some forms of advanced colorectal carcinoma [66].
5.1. Mechanism of CTLA-4 Blockade Induced Tumour Rejection
The exact mechanism of CTLA4 blockade induced tumour rejection by ipilimumab is still not completely clear, with many distinct mechanisms thought to play a role. The primary mechanism is centred around direct blockade of CTLA4 competition for B7 ligands, allowing unrestricted CD28 co-stimulation and increasing activation of effector T-cell in response to TAAs (Figure 3) [55]. As B7 ligands are not found on cancer cells, this mainly occurs in the tumour-draining lymph nodes, where APCs cross-present TAAs to tumour reactive effector cells [55]. This end result is increased activation and activity of effector CD8+ CTLS and Th1 cells, the primary mediators of the anti-tumour immune response [55]. Additionally, within the TME, Ipilimumab acts to reactivate and expand exhausted CD8 T-cell and Th1 cells through blockade of CTLA-4 [67]. Exhaustion occurs due chronic TAA-induced TCR stimulation, resulting in persistently high CTLA-4 expression and greatly reduced activity of primed immune cells [68]. Therefore, CTLA-4 blockade removes this inhibitory control, reviving effector T-cell function [67].
Depletion of T-reg cells within the TME is the other main mechanism that is thought to contribute to CTLA-4 blockade induced tumour rejection [69]. CTLA-4 signalling plays an important role in the immunosuppressive function of T-regs, and as such blockade of such signalling removes this immunosuppressive influence that otherwise promotes tumour growth and progression [69]. This is achieved through selective depletion of intra-tumoural T-reg cells, not peripheral populations [70]. This selectivity is likely due to increased expression of CTLA-4 T-regs in the TME or, additionally, differences in activity and availability of receptor for crystallizable fragment of immunoglobulin (FcR), which is thought to mediate the mechanism of T-reg depletion. Laboratory investigations showed that binding of the Fc portion of anti-CTLA4 antibodies to FcR found on APCs were necessary for the depletion of intra-tumoural T-regs and subsequent anti-tumour effect [71].
Interestingly, a recent study has gone as far as to say that efficacy of Ipilimumab is completely independent of B7 ligands, instead solely driven by antibody mediated T-reg depletion [72]. Such a conclusion of course contradicts years of prior evidence, and although recent studies do suggest a greater role of T-reg depletion, a large body of research backs the concept of B7 ligand regulation as critical mechanism of Ipilimumab [55]. Also, there is not yet any definitive evidence of T-reg depletion in humans treated with Ipilimumab. In reality, both mechanisms could likely contribute to the efficacy of Ipilimumab to some degree, as both effector T-cells and T-regs express CTLA-4 and have roles its checkpoint signalling pathways [73]. However, the relative contribution of cell intrinsic expansion of effector T-cell function versus T-reg depletion remains unclear and as such further research is needed to better understand the weight of each mechanism. To be able to delineate the primary and more effective mechanism behind CTLA-4 blockade induce tumour rejection could potentially guide development of new anti-CTLA4 therapies, more focused on targeting these mechanisms. For example, Ipilimumab was initially developed based on the knowledge CTLA-4 blockade would enhance T-cell activity and as such was not developed to be depleting antibody and target T-regs in any way [74]. If in fact T-reg depletion played a more central role in tumour rejection in humans, then this would change the direction of future anti-CTLA4 therapies.
5.2. Efficacy Of Iplilmumab In Treatment Of Advanced Melanoma
For decades prior to the introduction to ipilimumab, there was little in improvement in the treatment available for patients with advanced or metastatic melanoma, with no significant improvement seen in overall survival (OS) of patients in this time [75]. The chemotherapeutic drug dacarbazine remained the reference single agent of choice, despite it showing no impact on survival [76]. Ipilumumab was the first immunotherapy to show improved OS in this patient population with high unmet needs, completely changing the therapeutic outlook for this cancer following its FDA approval [8]. Pre-clinical data of CTLA-4 blockade in mice showed that it could not only enhance the endogenous anti-tumour immunity against cancers, but also be used synergistically in combination with other interventions such as chemotherapy or vaccines to improve response against less immunogenic cancer [77]. Findings from Phase I trials extended these promising results, showing anti-tumour immunity in various advanced tumours, especially melanoma, and demonstrated a favourable safety and toxicity profile both alone and in combination [76].
As Ipilimumab’s clinical development progressed in to phase II trials, it continued to show promising outcomes. In a Phase II trial (MDX010-08), 72 previously untreated advanced melanoma patients were randomised to receive either ipilimumab 3mg/kg monotherapy or in combination with dacarbazine. In both cases, ipilimumab showed strong objective clinical outcomes and promising OS in patients [78]. When comparing the long-term survival data in this trial against another trial, MDX010-15, a phase 1/2 dose-ranging study in which 23 patients were treated with ipilimumab at 10 mg/kg dose every 3 weeks, a trend towards better survival with a higher dose was demonstrated. 2- year survival rate of 36% was seen in 10mg/kg dose vs 22% for 3mg/kg dose [79]. A similar trend was seen in a phase 1/2 study (CA184-022), in which 217 previously treated advanced melanoma patients underwent induction therapy with one of three doses of ipilimumab, followed by maintenance therapy. Disease control rate (DCR) was 13.7%, 26.4%, and 29.2% in the 0.3 mg/kg, 3 mg/kg, and 10 mg/kg groups, respectively. There was similar safety profile between the two higher doses, with irAE rate of 70% with 10 mg/kg and 65% with 3 mg/kg [80]. Overall, a series of phase II trials evaluated the efficacy and safety of ipilimumab in over 500 patients [81]. The results were of course very promising, but there were two key lessons derived from the findings of these trials. Firstly, irAEs were the most common toxicity associated with ipilimumab, however, they were in most cases reversible [81]. Secondly, these trials revealed the unique response pattern of ipilimumab, in which some patients showed response following apparent disease progression. In fact, objective responses were sometimes observed 6-12 month after treatment initiation [81]. Therefore, it was recognised that overall survival (OS) was the superior endpoint to reflect clinical activity of Ipilimumab and as such it was chosen as the primary endpoint for future Phase III trials [81].
Phase III trials were the last step in the journey towards FDA approval for Ipilimumab. A key phase III trial, MDX010-20, evaluated Ipilimumab treatment at 3mg/kg in previously treatment patients, comparing it against cancer vaccine gp100 alone, and in combination [12]. A significantly improved OS was observed in patients treated with Ipilimumab, with or without gp100, compared to patients receiving gp100 alone. There was almost double in 1 and 2-year survival rates with ipilimumab compared to gp100. As well as this, clinical outcomes were independent of important prognostic factors of melanoma, such as stage at presentation, baseline lactate dehydrogenase levels and age. Once again, there was high frequency of irAEs, of approximately 60%, but they were low grade and manageable in most cases [12]. The results of this study were the basis for approval of ipilimumab at 3mg/kg dose for previously treated metastatic melanoma and, in some countries, for treatment-naive metastatic melanoma [76]. A recent meta-analysis of 1861 patients across 12 clinical trials of ipilimumab in melanoma showed a favourable median OS of 11.4 months and 3-year of 22% survival across all patients [82]. It also showed that ipilimumab was more effective in improving OS and other clinical outcomes in combination with PD-1 inhibitors, such as Nivolumab. This synergistic relationship between these two ICI has therefore been utilised and approved for treatment of melanoma and other cancers [82]. In summary, ipilimumab was the first instance of success for ICI therapy as a whole, paving the way for the further research, development and clinical application of immune checkpoint blockade.
6. PD-1/PD-L1 Physiological Role
PD-1 is a transmembrane protein which, much like CTLA-4, has an important role in co-inhibitory regulation of TCR activation [83]. Although both CTLA-4 and PD-1 have similar inhibitory effects on T-cells activity, the responsible signalling mechanisms, timing of downregulation and anatomic location of immune inhibition differ significantly [58]. Unlike CTLA-4 expression, which is confined to T-cells, PD-1 is expressed more broadly in activated T-cells, B-cells and myeloid cells. Furthermore, while CTLA-4 primarily functions during the priming phase of T-cell activation within secondary lymphoid organs, PD-1 functions during the effector phase and therefore within peripheral tissue [84]. PD-1 has two ligands to which it binds to mediate its effects, PD-L1 and PD-L2, whose expression also differ in comparison to CTLA-4 ligands [55]. Whereas B7 ligands are expressed on APCs, the PDL ligands are expressed widely on leukocytes, non-hematopoietic cells and non-lymphoid tissues [84].
When PD-1 binds to its ligands in the presence of TCR or B-cell receptor activation, it transduces a direct inhibitory signal to the activating receptor (Figure 4). This co-inhibitory signal prevents phosphorylation of key signalling intermediates, inhibiting activation of T-cells or B-cells [85]. Signalling through PD-1 also heavily influences cytokine production by T-cells, inhibiting release of pro-inflammatory cytokines such as INF-γ, TNF-α and IL-2, which ordinarily act to promote T-cell activity and proliferation [86]. Additionally, PD-1 signalling also inhibits upregulation of Bcl-xl, an anti-apoptotic protein, and limits expression transcriptional factors which are important for T-cell effector function [86]. Like CTLA-4, the net effect of PD-1 co-inhibitory signalling is dependent on the extent of the opposing co-stimulatory signalling of CD28 and other stimulatory signals. These immune checkpoints all act together to form a homeostatic feedback loop, that closely regulates the degree of T-cell activation and activity [58]. Considering the widespread distribution of PD-1 and its ligands, alongside its role in inhibitory regulation, it follows that’s the inherent role of PD-1 is to maintain peripheral tolerance of effector lymphocytes [55]. Experimental evidence supporting this concept has been seen in mice studies, in which genetic deletion of Pdcd1 gene (encoding for PD-1) lead to various autoimmune pathologies [86,87]
6.1. Mechanism of PD-1/PD-L1 Blockade Induced Tumour Rejection
PD-L1 expression has been detected on many different cancer cells types, which are able utilise the PD-1 signalling pathway to evade the immune system, taking advantage of its immunosuppressive function to grow and progress [55,88]. As a result, several different ICIs have been developed targeting the PD-1 signalling pathways. Currently, Pembrolizumab, Nivolumab and Cemiplimab are the approved anti-PD-1 ICIs, and Atezolizumab, Avelumab and Durvalumab are approved anti-PD-L1 ICIs. They are collectively able to target 9 different cancer, which really highlights the significance of PD-1/PD-L1 signalling in cancer progression, and the great deal of therapeutic benefit that can be derived from blockade of these pathways [89]. PD-1 blockade acts to induce tumour rejection through reinvigoration of exhausted T-cells and overall enhancement of their effector function through blockade of the many inhibitory mechanisms of PD-1 mentioned before [86]. PD-L1 blockade is thought to induce tumour rejection in largely similar manner to anti-PD1 antibodies, due to the dominance in expression of PD-L1 compared to PD-L2 [58]. Also, considering PD-L1 is induced predominantly by Th1 cytokines whereas PD-L2 is induced by Th2 cytokines, PD-L1 blockade would produce a Th-1 skewed response that would favour anti-tumour immunity [90]. Furthermore, selective inhibition of PD-L1 blocks PD-1/PD-L1 interactions while preserving PD-1/PD-L2 interactions. This in theory could produce a more targeted response with less unwanted toxicity, as there will be partial preservation of PD-1 self-tolerance mechanisms, via PD-L2 binding. However, as of yet there is limited clinical data to prove this concept [58]. Also, PD-L1 is also known to bind to B7-1, as well as PD-1, to induce inhibitory signals to T-cells. Therefore PD-L1 blockade could also stimulate T-cell activation in a very similar manner to CTLA4 blockade, increasing availability of B7-1 ligand for CD28 co-stimulation [91]. Ultimately, more research is needed to better understand the potential multifaceted mechanisms of both anti-PD-1 and anti-PD-L1 therapies, in order to optimize their use in the many different targeted cancers.
6.2. Efficacy Of Anti-PD-1/PD-L1 Agents
6.2.1. Pembrolizumab
Of the ICIs that target the PD-1/PD-L1 signalling pathway, the PD-1 ICI Pembrolizumab and the PD-L1 ICI Atezolizumab are the most established, as such they are the best ICIs to assess in order to understand the efficacy of PD-1/PD-L1 therapies as a whole [89]. Pembrolizumab is a fully humanised mAb that received accelerated FDA approval in 2014 for treatment of unresectable or metastatic melanoma, and shortly after for non-small cell lung cancer (NSCLC) in 2015 [92]. KEYNOTE-001, a phase I dose-finding trial of melanoma patients previously treated with ipilimumab, assessed pembrolizumab given intravenously at multiple doses ranging from 1 to 10 mg/kg, either every 2 or 3 weeks [93]. The overall response rate (ORR) was highest in the dose-dense and dose-intense group at the expense of higher irAEs incidence [93]. Consequent expansion cohorts of this study explored the safety and efficacy of many pembrolizumab regimes in patients with advanced melanoma and NSCLC [92]. The KEYNOTE-001 data showed significant activity in patients with advanced melanoma, regardless of previous ipilimumab treatment. However, notably higher response rates were seen in ipilimumab-naïve patients [92]. A recent 5-year follow up this patient cohort in KEYNOTE-001 showed favourable 5-year OS rates, further proving the durable antitumour activity and tolerability of pembrolizumab in advanced melanoma [94]. A phase 3 trial, KEYNOTE-006, compared the efficacy of pembrolizumab against ipilimumab and 5-year follow up results also recently became available. After median follow-up of 57.7 months, median OS was 32.7 months in pembrolizumab groups compared to 15.9 months in the ipilimumab group. There was also a lower incidence of high-grade treatment-related AEs seen with pembrolizumab. These results extended the finding in the original trial, showing a superiority of pembrolizumab over ipilimumab in patients with advanced melanoma [95].
Pembrolizumab also showed impressive outcomes in 5-year follow of the NSCLC expansion cohort of the KEYNOTE-001 trial. Based on these results estimated OS rates was 23.2% for treatment-naive patients and 15.5% for previously treated patients, which are very favourable when compared to pre-immunotherapy OS rates of around 5%. There was also a very low incidence of treatment-related AEs [96]. A phase 2/3 trial, KEYNOTE-010, also showed improved OS with Ipilimumab when compared to Docetaxel in previously treated patients with advanced NSCLC, with long term follow-up data from this same trial extending this positive trend [97]. Overall, through many trials, Pembrolizumab has demonstrated durable responses and prolonged OS in NSCLC, especially in variants with high expression of PDL-1. Thus, this has been recognized as an important predictive biomarker of the efficacy of Pembrolizumab in this patient population [92]. Beyond melanoma and NSLC, Pembrolizumab has been approved and showed success in treatment of many cancer including head and neck cancer, gastric cancer, Hodgkin lymphoma and urothelial cancer. This really highlights the wide-reaching therapeutic potential of Pembrolizumab and PD-1 ICIs as a whole [92].
6.2.2. Atezoluzumab
Atezolizumab was the first anti-PDL-1 ICI of its kind, receiving FDA approval in 2016 for treatment of locally advanced or metastatic urothelial carcinoma (mUC) resistant to platinum-based chemotherapy. The following year, it also received accelerated approval as an initial treatment in patients who were ineligible for cisplatin chemotherapy [98]. In both cases, approval was primarily based on data from the IMvigor210 study, a phase II trial which evaluated the safety and efficacy of Atezolizumab in patients with locally advanced or mUC, regardless of PD-L1 expression. Patients were split in to 2 cohorts, cohort 1 comprised treatment-naïve patients and cohort 2 comprised of previously treated patients. The median follow-up for Cohort 1 was 29 months, and 33 months for Cohort 2. In Cohort 1, the ORR was 24% and in cohort 2 the ORR 16%, which were both very favourable when compared to the historical control ORR of 10%. Additionally, in both cohorts a higher PDL-1 expression of tumour-infiltrating lymphocytes (>5%) was associated with improved ORR. The incidence of irAEs in cohort 1 was 14% and in cohort 2 10%, with the incidence of high grade irAEs being 7% and 6% respectively [99,100].
In 2016, Atezolizumab received accelerated approval for treatment of cisplatin-resistant metastatic NSCLC, specifically associated with EGFR or ALK gene abnormalities [101]. This approval is based on results from the Phase III OAK and Phase II POPLAR studies [102,103]. The largest study, OAK, evaluated the efficacy and safety of Atezolizumab compared with docetaxel in 1,225 patients. The results showed that those treated with Atezolizumab had median OS of 13.8 months, 4.2 months longer than those treated with docetaxel chemotherapy [103]. Since then it has also been approved as an initial treatment in combination with chemotherapy for metastatic non-squamous NSCLC and extensive-stage Stage Small Cell Lung Cancer [104].
Finally, in 2019 Atezolizumab received accelerated approval for treatment of PD-L1-Positive, Metastatic Triple-Negative Breast Cancer (TNBC), in combination with chemotherapy [105]. TNBC is aggressive variant of the cancer with few treatment options, in which cancer cells lack expression of hormone receptors and do not overexpress HER2 protein [106]. The accelerated approval was based on data from the Phase III IMpassion130 study, which evaluated the efficacy and safety of Atezolizumab plus nab-paclitaxel compared with placebo plus nab-paclitaxel in unresectable locally advanced or metastatic TNBC patients [106]. The study demonstrated that the combination therapy significantly reduced the risk of cancer progression or death by 40 percent compared with nab-paclitaxel alone specifically in patients who has PDL-1 positive disease and had not received any prior chemotherapy. The combination therapy was also shown to safe and tolerable, consistent with the known individual safety profiles of each medication and with no new safety concerns identified [106]. Much like PD-1 ICIs such as Pembrolizumab, Atezolizumab has proven to be a versatile and effective therapy for many end-stage cancers and it currently under trial for several other types of cancer, such as pancreatic cancer, gastric cancer and ovarian cancer [107].
7. Adverse Effects Of Immune Checkpoint Inhibitors
Immune checkpoints have an important role in maintaining immune homeostasis and preventing autoimmunity. Therefore, although blockade of its immunosuppressive pathways by ICI’s may revive anti-tumour immunity, it can lead to potentially severe and even lethal autoimmune pathologies known as immune-related adverse events (irAEs) [108]. irAEs are clinically diverse with most organs being potentially affected such as the skin, liver, lung, pituitary, thyroid, gastrointestinal tract and even the central nervous system (Figure 5) [108]. These toxicities are often unpredictable and highly variable, however in most cases are mild and easily managed [109]. Dermatological reactions are the most common irAEs associated with ICIs, such as rash, pruritis and vitiligo, occurring in more than 40% patients with ipilimumab and in around 20% of those with PD-1/PD-L1 inhibitors [109]. Gastrointestinal irAEs are also frequently seen, commonly presenting as diarrhoea and colitis, and once again incidence of which are reported to be higher in anti-CTLA4 therapies [109]. In general, the incidence of irAEs are higher in anti-CTLA4 therapies compared to anti-PD1/PDL1 therapies, including high grade cases [58]. For example, in a phase III trial of melanoma patients the overall rate of grade ≥3 irAEs was higher in ipilimumab compared with pembrolizumab (20% vs 13%) [110]. In general, the management of mild to moderate cases is with supportive care and conservative measures, however in severe cases, corticosteroid, biological therapy or even treatment cessation may be indicated [109].
Interestingly, although irAE are unwanted side effects and can have potentially serious implications, in some cases their presence and in particular their onset can be a positive prognostic marker of ICI efficacy [111]. Among ICIs, irAE onset appears to be more strongly associated with anti-PD-1/PD-L1 therapies compared to anti-CTLA4 therapies, a link demonstrated by a growing body of research [111]. Other characteristics of irAEs such as site, severity and timing of onset have also been considered as potential clinical biomarkers, however current research is limited, and further studies are needed to understand the true implications of these parameters on ICI efficacy [111]. Overall, there is a close link between autoimmunity and anti-tumour effect elicited by ICIs, therefore it is an important area of research interest to determine whether these two aspects of ICI therapy can be uncoupled in order to maximize benefit while minimizing toxicities for patients.
8. Predictive Biomarkers
Although ICIs have transformed the treatment landscape for many patients with advanced cancers, response rates range from 15-60%, leaving a significant proportion of patients who do not derive any benefit [111]. Therefore, identifying predictive biomarkers is critical in order to delineate responders from non-responders and prevent unnecessary treatment and adverse effects of therapy, as well as improve cost-effectiveness of this often expensive therapy by only applying it to those most likely to benefit [5] [112]. Thus far, biomarkers that have been focussed on include PDL-1 overexpression, immune cell constitution of TME, neoantigens and genetic and epigenetic signatures of a tumour [113].
PDL-1 overexpression has proven to be a very accurate predictive biomarker of anti-PD-1/PD-L1 ICI efficacy, so much so that it is a vital criterion for treatment of cancers such as urothelial cancer and NSCLC with such therapies [114]. The efficacy of ICI therapy relies on underlying presence of anti-tumour immunity, thus the immune constitution within the TME is another important prognostic biomarker. For example, in responders the TME is associated with high levels of TILs, a high T-effector /Treg ratio and increased secretion of IFN-γ and other inflammatory cytokines [5]. The TME is classified into three major types based on degree of immune cell infiltration: immune desert, immune excluded and immune inflamed. Immune inflamed phenotype is characterised by infiltration of multiple immune cell subtypes and is associated with the best prognosis. Immune excluded phenotype exhibits presence of many inflammatory cells and mediators; however, there is lack of TME infiltration of accumulated T-cells. Immune desert phenotype is characterised by absence of T-cells in the TME and unsurprisingly does not respond well to ICI therapy [14]. The degree of anti-tumour immune response is also determined by the immunogenicity of a cancer, so biomarkers such the tumour mutational burden (TMB) and presence of neoantigens are also important predictors of response [115]. Furthermore, certain genetic signatures of tumour can also confer a better prognosis with ICI therapy. For example, microsatellite instability (MSI) positive colorectal cancers show a better prognosis following ICI as they are associated with greater degree of CD8+ T-cell infiltration as well as high TMB and neoantigen expression [116,117]. Finally, an example of epigenetic biomarker is DNA methylation of certain genes, for example a study showed methylation status of a single gene, FOXP3, which regulates Treg cells function, was also found to be predictive of ICI response in NSCLC patients [118].
There are many more biomarkers that have been identified but as of yet there is no single predictive model for ICI efficacy [119]. There is a great degree of heterogeneity seen in each clinical case as immune responses are not uniform across all malignancies and individuals [120]. Therefore, in reality a case by case approach is needed in which multiple potential biomarkers are utilised to best predict the efficacy of ICI-based therapy [119]. Ultimately, continued research into this area will help identify and guide clinical application of predictive biomarkers, in hope of extending the reach of ICI therapy and maximising patient benefit.
9. Future Directions
ICIs have proven to be major breakthrough in cancer immunotherapy, revealing a promising avenue for treatment of a high risk and diverse patient population. Despite the ICIs to date showing compelling clinical effectiveness is certain tumour types, the overall efficiency of ICI therapy remains unsatisfactory [57]. Therefore, going forward research goals are to optimise the current ICI therapies as well as explore additional immune checkpoints in hope of creating a more effective, safer, and wider reaching therapy.
Beyond CTLA-4 and PD-1, novel immune checkpoints are being discovered continuously and many ICIs are currently under development or clinical trial, targeting these new immune checkpoints [57]. An example of a promising new ICI target is LAG-3. LAG-3 is a co-inhibitory checkpoint which has a negative regulatory role in Th1 cells activation, proliferation and cytokine production [121]. It is widely expressed on number of different activated lymphocytes and it structurally similar to CD4, and as such MHC-II is considered it primary ligand [122]. A phase I trial of the anti-LAG-3 ICI, IMP321, conducted in patients with metastatic RCC showed minimal toxicity and increased level of circulating CD8+ T-cells which was correlated to tumour growth reduction [123]. A subsequent trial of IMP321 in metastatic breast cancer also showed favourable results, with 50% ORR compared to historical ORR of 25% [124]. Another anti-LAG-3 mAb, Relatlimab, is in an ongoing phase I clinical trial in which the efficacy as a monotherapy or in combination with Nivolumab is being evaluated in patients with various advanced malignancies including melanoma, NSCLC, and RCC [122]. This exploration into combination therapy was evoked by many preclinical studies which showed compensatory upregulation LAG-3 and other immune checkpoints in response to PD-1/PD-L1 blockade [125]. Therefore, this concomitant blockade could possibly produce a synergistic effect in inhibiting tumour growth [122]. Other promising new targets include TIM-3 which has at least 8 antagonistic mAbs currently under trial and TIGIT, with around 6 major agents in trial, as well as many more [57]. There is also more to understand when it come to the biology and signalling mechanisms of many immune checkpoints, for example the identification of ligands for the immune checkpoints VISTA and B7-H3, which would be the key to fully understanding their therapeutic potential [57].
While monotherapies have shown very promising results, more attempts are being made to design ICI-based combination therapies that target multiple non-redundant pathways in order to achieve synergistic anti-tumour effects [126]. The combination of Ipilimumab and Nivolumab were the first such combination therapy, utilising together the centrally acting CTLA-4 and peripherally acting PD-1 pathways [127]. This has in fact shown greater efficacy in cancers such as melanoma, but also a higher incidence of adverse effect. Therefore, it seems synergistic effects of combined immune checkpoint blockade also comes at the cost of greater loss of intrinsic immune-tolerance mechanisms [127]. Additionally, many novel ICIs are also under trial in combination with PD-1 inhibitors as there is convincing rationale that other immune checkpoints are upregulated following PD-1/PD-L1 blockade [57]. There is also potential of combining ICIs with other forms of immunotherapy such as adoptive T-cell therapies or cellular vaccines, which could also act via complementary mechanisms to produce synergistic effects [126]. Finally, ICIs already have a basis for combination with conventional treatments such as chemotherapeutic drugs, but now combination with radiotherapy is also being considered [128]. While radiotherapy can be immunosuppressive, it can also enhance antigenicity by triggering release of TAAs [126]. Preclinical data has also supported this theory with positive results seen in mouse models treated with radiotherapy and anti-CTLA4 therapy, and as such there are number of clinical trials exploring this potential combination [129].
Beyond identification and development of new ICI-based regimes, in order to optimise its current and future clinical use it is essential that accurate biomarkers are identified to predict for both efficacy and adverse effects [119]. Subsequently, such adverse effects need should be diagnosed and management promptly and effectively [109]. Research into these areas will help formulate, design, and improve on the current management guideline’s lines in place. Overall, there is so much left to learn and understand regarding immune checkpoints and thus continued preclinical and clinical research into all avenues is needed to guide further advancements.
10. Conclusion
ICI therapy has led to important clinical advancements and provided a new weapon against advanced cancers, giving hope to a patient population with previously dismal prognosis. The therapy has elicited durable clinical responses with significant improvements in patient survival, showing its potential as a curative option. However, the therapy is still held back many limitations such as the small proportion of responders, toxicity caused by irAEs and its significant resource implications. There are so many avenues of research being undertaken in relation to ICIs with the aim of addressing such limitations and ultimately guiding the development of new and more effective agents.
Author Contributions
writing—original draft preparation, Abdullah Younis.
Funding
This review received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
Nil.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Borghaei, H.; Smith, M.R.; Campbell, K.S. Immunotherapy of cancer. Vol. 625, European Journal of Pharmacology. 2009, 41–54.
- Zhang, H.; Chen, J. Current status and future directions of cancer immunotherapy. J. Cancer 2018, 9, 1773–1781. [Google Scholar] [CrossRef] [PubMed]
- Mellman, I.; Coukos, G.; Dranoff, G. Cancer immunotherapy comes of age. Nature 2011, 480, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Voena, C.; Chiarle, R. Advances in cancer immunology and cancer immunotherapy. Discov Med. 2016, 21, 125–33. [Google Scholar] [PubMed]
- Darvin, P.; Toor, S.M.; Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors: recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Kates, M.; Sopko, N.A.; Matsui, H.; Drake, C.G.; Hahn, N.M.; Bivalacqua, T.J. Immune checkpoint inhibitors: a new frontier in bladder cancer. World J. Urol. 2015, 34, 49–55. [Google Scholar] [CrossRef]
- Camacho, L.H. CTLA-4 blockade with ipilimumab: Biology, safety, efficacy, and future considerations. Cancer Med. 2015, 4, 661–72. [Google Scholar] [CrossRef] [PubMed]
- Barbee, M.S.; Ogunniyi, A.; Horvat, T.Z.; Dang, T.-O. Current Status and Future Directions of the Immune Checkpoint Inhibitors Ipilimumab, Pembrolizumab, and Nivolumab in Oncology. Ann. Pharmacother. 2015, 49, 907–937. [Google Scholar] [CrossRef]
- Rosenberg, J.E.; Hoffman-Censits, J.; Powles, T.; van der Heijden, M.S.; Balar, A.V.; Necchi, A.; Dawson, N.; O’Donnell, P.H.; Balmanoukian, A.; Loriot, Y.; et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet 2016, 387, 1909–1920. [Google Scholar] [CrossRef]
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Aren Frontera, O.; Melichar, B.; Choueiri, T.K.; Plimack, E.R.; Barthélémy, P.; Porta, C.; George, S.; et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2018, 378, 1277–1290. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Blankenstein, T.; Coulie, P.G.; Gilboa, E.; Jaffee, E.M. The determinants of tumour immunogenicity. Nat. Rev. Cancer 2012, 12, 307–313. [Google Scholar] [CrossRef]
- Fancello, L.; Gandini, S.; Pelicci, P.G.; Mazzarella, L. Tumor mutational burden quantification from targeted gene panels: major advancements and challenges. J. Immunother. Cancer 2019, 7, 183. [Google Scholar] [CrossRef]
- Kourie, H.R.; Klastersky, J. Immune checkpoint inhibitors side effects and management. Immunotherapy 2016, 8, 799–807. [Google Scholar] [CrossRef]
- Corthay, A. Does the immune system naturally protect against cancer? Vol. 5, Frontiers in Immunology. Frontiers Research Foundation; 2014.
- Smyth, M.J.; Dunn, G.P.; Schreiber, R.D. Cancer Immunosurveillance and Immunoediting: The Roles of Immunity in Suppressing Tumor Development and Shaping Tumor Immunogenicity. Vol. 90, Advances in Immunology. 2006, 1–50.
- Burnet, F.M. The concept of immunological surveillance. Vol. 13, Progress in experimental tumor research. Fortschritte der experimentellen Tumorforschung. Progres de la recherche experimentale des tumeurs. 1970, 1–27.
- Thomas, L. On Immunosurveillance in Human Cancer. Vol. 55, THE YALE JOURNAL OF BIOLOGY AND MEDICINE. 1982.
- Vesely, M.D.; Kershaw, M.H.; Schreiber, R.D.; Smyth, M.J. Natural Innate and Adaptive Immunity to Cancer. Annu. Rev. Immunol. 2011, 29, 235–271. [Google Scholar] [CrossRef]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The Three Es of Cancer Immunoediting. Annu. Rev. Immunol. 2004, 22, 329–360. [Google Scholar] [CrossRef]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The Immunobiology of Cancer Immunosurveillance and Immunoediting. Immunity 2004, 21, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Mittal, D.; Gubin, M.M.; Schreiber, R.D.; Smyth, M.J. New insights into cancer immunoediting and its three component phases-elimination, equilibrium and escape. Vol. 27, Current Opinion in Immunology. 2014, 16–25.
- Efremova, M.; Rieder, D.; Klepsch, V.; Charoentong, P.; Finotello, F.; Hackl, H.; Hermann-Kleiter, N.; Löwer, M.; Baier, G.; Krogsdam, A.; et al. Targeting immune checkpoints potentiates immunoediting and changes the dynamics of tumor evolution. Nat. Commun. 2018, 9, 32. [Google Scholar] [CrossRef]
- Beatty, G.L.; Gladney, W.L. Immune Escape Mechanisms as a Guide for Cancer Immunotherapy. Clin. Cancer Res. 2015, 21, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Lowe, S.W.; Cepero, E.; Evan, G. Intrinsic tumour suppression. Nature 2004, 432, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Narendra, B.L.; Reddy, K.E.; Shantikumar, S.; Ramakrishna, S. Immune system: a double-edged sword in cancer. Inflamm. Res. 2013, 62, 823–834. [Google Scholar] [CrossRef] [PubMed]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Innate Immunity. 2002. [Google Scholar]
- Flajnik, M.F.; Kasahara, M. Origin and evolution of the adaptive immune system: genetic events and selective pressures. Nat. Rev. Genet. 2009, 11, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Chaplin, D.D. Overview of the immune response. J. Allergy Clin. Immunol. 2010, 125, S3–S23. [Google Scholar] [CrossRef] [PubMed]
- Hastings, K.T. Innate and adaptive immune responses to cancer. In: Fundamentals of Cancer Prevention. Springer Berlin Heidelberg; 2005, 79–108.
- Collin, M.; Bigley, V. Human dendritic cell subsets: an update. Immunology 2018, 154, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Hao, N.-B.; Lü, M.-H.; Fan, Y.-H.; Cao, Y.-L.; Zhang, Z.-R.; Yang, S.-M. Macrophages in Tumor Microenvironments and the Progression of Tumors. J. Immunol. Res. 2012, 2012, 1–11. [Google Scholar] [CrossRef]
- Helen, J.K.; Knowles, H.J.; Harris, A.L. Macrophages and the hypoxic tumour microenvironment. Front. Biosci. 2007, 12, 4298–314. [Google Scholar] [CrossRef]
- Martinez, F.O.; Helming, L.; Gordon, S. Alternative Activation of Macrophages: An Immunologic Functional Perspective. Annu. Rev. Immunol. 2009, 27, 451–483. [Google Scholar] [CrossRef]
- Najafi, M.; Hashemi Goradel, N.; Farhood, B.; Salehi, E.; Nashtaei, M.S.; Khanlarkhani, N.; et al. Macrophage polarity in cancer: A review [Internet]. Vol. 120, Journal of Cellular Biochemistry. Wiley-Liss Inc.; 2019, 2756–65. Available from: http://www.ncbi.nlm.nih.gov/pubmed/30270458.
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef]
- Coffelt, S.B.; Hughes, R.; Lewis, C.E. Tumor-associated macrophages: Effectors of angiogenesis and tumor progression [Internet]. Vol. 1796, Biochimica et Biophysica Acta - Reviews on Cancer. 2009, 11–8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19269310.
- Laoui, D.; Movahedi, K.; Van Overmeire, E.; Van den Bossche, J.; Schouppe, E.; Mommer, C.; Nikolaou, A.; Morias, Y.; De Baetselier, P.; Van Ginderachter, J.A. Tumor-associated macrophages in breast cancer: distinct subsets, distinct functions. Int. J. Dev. Biol. 2011, 55, 861–867. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Bevan, M.J. CD8+ T Cells: Foot Soldiers of the Immune System. Immunity 2011, 35, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Scheipers, P.; Reiser, H. Role of the CTLA-4 receptor in t cell activation and immunity. Immunol. Res. 1998, 18, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Chong, M.M.W.; Littman, D.R. Plasticity of CD4+ T Cell Lineage Differentiation [Internet]. Vol. 30, Immunity. 2009 [cited 2020 Feb 10], 646–55. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19464987.
- Schwartz, R.H. T C ELL A NERGY. Annu Rev Immunol. 2003, 21, 305–34. [Google Scholar] [CrossRef] [PubMed]
- Romano, M.; Fanelli, G.; Albany, C.J.; Giganti, G.; Lombardi, G. Past, Present, and Future of Regulatory T Cell Therapy in Transplantation and Autoimmunity. Front. Immunol. 2019, 10, 43. [Google Scholar] [CrossRef] [PubMed]
- Zou, W. Regulatory T cells, tumour immunity and immunotherapy [Internet]. Vol. 6, Nature Reviews Immunology. 2006, 295–307. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16557261.
- Cao, X.; Cai, S.F.; Fehniger, T.A.; Song, J.; Collins, L.I.; Piwnica-Worms, D.R.; Ley, T.J. Granzyme B and Perforin Are Important for Regulatory T Cell-Mediated Suppression of Tumor Clearance. Immunity 2007, 27, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Curiel, T.J.; Coukos, G.; Zou, L.; Alvarez, X.; Cheng, P.; Mottram, P.; Evdemon-Hogan, M.; Conejo-Garcia, J.R.; Zhang, L.; Burow, M.; et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 2004, 10, 942–949. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Qiu, S.-J.; Fan, J.; Zhou, J.; Wang, X.-Y.; Xiao, Y.-S.; Xu, Y.; Li, Y.-W.; Tang, Z.-Y. Intratumoral Balance of Regulatory and Cytotoxic T Cells Is Associated With Prognosis of Hepatocellular Carcinoma After Resection. J. Clin. Oncol. 2007, 25, 2586–2593. [Google Scholar] [CrossRef] [PubMed]
- Frey, D.M.; Droeser, R.A.; Viehl, C.T.; Zlobec, I.; Lugli, A.; Zingg, U.; Oertli, D.; Kettelhack, C.; Terracciano, L.; Tornillo, L. High frequency of tumor-infiltrating FOXP3+ regulatory T cells predicts improved survival in mismatch repair-proficient colorectal cancer patients. Int. J. Cancer 2010, 126, 2635–2643. [Google Scholar] [CrossRef]
- Guéry, L.; Hugues, S. Th17 Cell Plasticity and Functions in Cancer Immunity. BioMed Res. Int. 2015, 2015, 1–11. [Google Scholar] [CrossRef]
- Immune Checkpoint Signaling Pathway - Creative Diagnostics [Internet]. [cited 2020 Mar 21]. Available from: https://www.creative-diagnostics.com/immune-checkpoint-signaling-pathway.htm.
- Nirschl, C.J.; Drake, C.G. Molecular Pathways: Coexpression of Immune Checkpoint Molecules: Signaling Pathways and Implications for Cancer Immunotherapy. Clin. Cancer Res. 2013, 19, 4917–4924. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef] [PubMed]
- Juneja, V.R.; McGuire, K.A.; Manguso, R.T.; LaFleur, M.W.; Collins, N.; Haining, W.N.; Freeman, G.J.; Sharpe, A.H. PD-L1 on tumor cells is sufficient for immune evasion in immunogenic tumors and inhibits CD8 T cell cytotoxicity. J. Exp. Med. 2017, 214, 895–904. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Xu, L.; Yi, M.; Yu, S.; Wu, K.; Luo, S. Novel immune checkpoint targets: moving beyond PD-1 and CTLA-4. Mol. Cancer 2019, 18, 155. [Google Scholar] [CrossRef] [PubMed]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 pathways similarities, differences, and implications of their inhibition. Vol. 39, American Journal of Clinical Oncology: Cancer Clinical Trials. Lippincott Williams and Wilkins; 2016, 98–106.
- Sansom, D.M. CD28, CTLA-4 and their ligands: Who does what and to whom? Vol. 101, Immunology. Wiley-Blackwell; 2000, 169–77.
- Qureshi, O.S.; Zheng, Y.; Nakamura, K.; Attridge, K.; Manzotti, C.; Schmidt, E.M.; Baker, J.; Jeffery, L.E.; Kaur, S.; Briggs, Z.; et al. Trans-Endocytosis of CD80 and CD86: A Molecular Basis for the Cell-Extrinsic Function of CTLA-4. Science 2011, 332, 600–603. [Google Scholar] [CrossRef] [PubMed]
- Fallarino, F.; Fields, P.E.; Gajewski, T.F. B7-1 Engagement of Cytotoxic T Lymphocyte Antigen 4 Inhibits T Cell Activation in the Absence of CD28. J. Exp. Med. 1998, 188, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, P.; Penninger, J.M.; Timms, E.; Wakeham, A.; Shahinian, A.; Lee, K.P.; Thompson, C.B.; Griesser, H.; Mak, T.W. Lymphoproliferative Disorders with Early Lethality in Mice Deficient in Ctla-4. Science 1995, 270, 985–988. [Google Scholar] [CrossRef]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of Antitumor Immunity by CTLA-4 Blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef]
- Camacho, L.H.; Antonia, S.; Sosman, J.; Kirkwood, J.M.; Gajewski, T.F.; Redman, B.; Pavlov, D.; Bulanhagui, C.; Bozon, V.A.; Gomez-Navarro, J.; et al. Phase I/II Trial of Tremelimumab in Patients With Metastatic Melanoma. J. Clin. Oncol. 2009, 27, 1075–1081. [Google Scholar] [CrossRef]
- Ribas, A.; Kefford, R.; Marshall, M.A.; Punt, C.J.A.; Haanen, J.B.; Marmol, M.; et al. Phase III randomized clinical trial comparing tremelimumab with standard-of-care chemotherapy in patients with advanced melanoma. J Clin Oncol [Internet]. 2013, 31, 616–22. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23295794.
- Rotte, A. Combination of CTLA-4 and PD-1 blockers for treatment of cancer. J. Exp. Clin. Cancer Res. 2019, 38, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.C.; Levine, J.H.; Cogdill, A.P.; Zhao, Y.; Anang, N.-A.A.S.; Andrews, M.C.; Sharma, P.; Wang, J.; Wargo, J.A.; Pe’Er, D.; et al. Distinct Cellular Mechanisms Underlie Anti-CTLA-4 and Anti-PD-1 Checkpoint Blockade. Cell 2017, 170, 1120–1133. [Google Scholar] [CrossRef]
- Wherry, E.J. T cell exhaustion. Vol. 12, Nature Immunology. 2011, 492–9.
- Simpson, T.R.; Li, F.; Montalvo-Ortiz, W.; Sepulveda, M.A.; Bergerhoff, K.; Arce, F.; Roddie, C.; Henry, J.Y.; Yagita, H.; Wolchok, J.D.; et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti–CTLA-4 therapy against melanoma. J. Exp. Med. 2013, 210, 1695–1710. [Google Scholar] [CrossRef] [PubMed]
- Selby, M.J.; Engelhardt, J.J.; Quigley, M.; Henning, K.A.; Chen, T.; Srinivasan, M.; Korman, A.J. Anti-CTLA-4 Antibodies of IgG2a Isotype Enhance Antitumor Activity through Reduction of Intratumoral Regulatory T Cells. Cancer Immunol. Res. 2013, 1, 32–42. [Google Scholar] [CrossRef]
- Bulliard, Y.; Jolicoeur, R.; Windman, M.; Rue, S.M.; Ettenberg, S.; Knee, D.A.; Wilson, N.S.; Dranoff, G.; Brogdon, J.L. Activating Fc γ receptors contribute to the antitumor activities of immunoregulatory receptor-targeting antibodies. J. Exp. Med. 2013, 210, 1685–1693. [Google Scholar] [CrossRef]
- Du, X.; Tang, F.; Liu, M.; Su, J.; Zhang, Y.; Wu, W.; Devenport, M.; A Lazarski, C.; Zhang, P.; Wang, X.; et al. A reappraisal of CTLA-4 checkpoint blockade in cancer immunotherapy. Cell Res. 2018, 28, 416–432. [Google Scholar] [CrossRef] [PubMed]
- Peggs, K.S.; Quezada, S.A.; Chambers, C.A.; Korman, A.J.; Allison, J.P. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti–CTLA-4 antibodies. J. Exp. Med. 2009, 206, 1717–1725. [Google Scholar] [CrossRef]
- Ramagopal, U.A.; Liu, W.; Garrett-Thomson, S.C.; Bonanno, J.B.; Yan, Q.; Srinivasan, M.; Wong, S.C.; Bell, A.; Mankikar, S.; Rangan, V.S.; et al. Structural basis for cancer immunotherapy by the first-in-class checkpoint inhibitor ipilimumab. Proc. Natl. Acad. Sci. 2017, 114, E4223–E4232. [Google Scholar] [CrossRef]
- Altekruse S, Kosary C, Krapcho M, gov/csr NN cancer., 2010 undefined. SEER Cancer Statistics Review, 1975–2007, National Cancer Institute. Bethesda, MD, based on November 2009 SEER data submission, posted to the.
- Tarhini, A.; Lo, E.; Minor, D.R. Releasing the Brake on the Immune System: Ipilimumab in Melanoma and Other Tumors. Cancer Biotherapy Radiopharm. 2010, 25, 601–613. [Google Scholar] [CrossRef]
- Peggs, K.S.; A Quezada, S.; Korman, A.J.; Allison, J.P. Principles and use of anti-CTLA4 antibody in human cancer immunotherapy. Curr. Opin. Immunol. 2006, 18, 206–213. [Google Scholar] [CrossRef]
- Hersh, E.M.; O’day, S.J.; Powderly, J.; Khan, K.D.; Pavlick, A.C.; Cranmer, L.D.; Samlowski, W.E.; Nichol, G.M.; Yellin, M.J.; Weber, J.S. A phase II multicenter study of ipilimumab with or without dacarbazine in chemotherapy-naïve patients with advanced melanoma. Investig. New Drugs 2010, 29, 489–498. [Google Scholar] [CrossRef]
- Hersh, E.; Weber, J.; Powderly, J.; Pavlik, A.; Nichol, G.; Yellin, M.; Cranmer, L.; Urba, W.; O'Day, S. Long-term survival of patients (pts) with advanced melanoma treated with ipilimumab with or without dacarbazine. J. Clin. Oncol. 2009, 27, 9038–9038. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Neyns, B.; Linette, G.; Negrier, S.; Lutzky, J.; Thomas, L.; Waterfield, W.; Schadendorf, D.; Smylie, M.; Guthrie, T.; et al. Ipilimumab monotherapy in patients with pretreated advanced melanoma: a randomised, double-blind, multicentre, phase 2, dose-ranging study. Lancet Oncol. 2010, 11, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Hodi, F.S.; Weber, J.S.; Allison, J.P.; Urba, W.J.; Robert, C.; O'Day, S.J.; Hoos, A.; Humphrey, R.; Berman, D.M.; et al. Development of ipilimumab: a novel immunotherapeutic approach for the treatment of advanced melanoma. Ann. New York Acad. Sci. 2013, 1291, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Schadendorf, D.; Hodi, F.S.; Robert, C.; Weber, J.S.; Margolin, K.; Hamid, O.; Patt, D.; Chen, T.-T.; Berman, D.M.; Wolchok, J.D. Pooled Analysis of Long-Term Survival Data From Phase II and Phase III Trials of Ipilimumab in Unresectable or Metastatic Melanoma. J. Clin. Oncol. 2015, 33, 1889–1894. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Sun, Q.; Zhang, X. PD-1 and its ligands are important immune checkpoints in cancer. Oncotarget 2016, 8, 2171–2186. [Google Scholar] [CrossRef] [PubMed]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and Its Ligands in Tolerance and Immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef] [PubMed]
- Yokosuka, T.; Takamatsu, M.; Kobayashi-Imanishi, W.; Hashimoto-Tane, A.; Azuma, M.; Saito, T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J. Exp. Med. 2012, 209, 1201–1217. [Google Scholar] [CrossRef]
- Riella, L.V.; Paterson, A.M.; Sharpe, A.H.; Chandraker, A. Role of the PD-1 Pathway in the Immune Response. Am. J. Transplant. 2012, 12, 2575–2587. [Google Scholar] [CrossRef]
- Nishimura, H.; Nose, M.; Hiai, H.; Minato, N.; Honjo, T. Development of Lupus-like Autoimmune Diseases by Disruption of the PD-1 Gene Encoding an ITIM Motif-Carrying Immunoreceptor. Immunity 1999, 11, 141–151. [Google Scholar] [CrossRef]
- Xiang, X.; Yu, P.-C.; Long, D.; Liao, X.-L.; Zhang, S.; You, X.-M.; Zhong, J.-H.; Li, L.-Q. Prognostic value of PD -L1 expression in patients with primary solid tumors. Oncotarget 2017, 9, 5058–5072. [Google Scholar] [CrossRef] [PubMed]
- PD-1/PD-L1 Landscape Analysis - Cancer Research Institute (CRI) [Internet]. Available from: https://www.cancerresearch.org/scientists/immuno-oncology-landscape/pd-1-pd-l1-landscape.
- Loke, P.; Allison, J.P. PD-L1 and PD-L2 are differentially regulated by Th1 and Th2 cells. Proc. Natl. Acad. Sci. 2003, 100, 5336–5341. [Google Scholar] [CrossRef] [PubMed]
- Butte, M.J.; Keir, M.E.; Phamduy, T.B.; Sharpe, A.H.; Freeman, G.J.; Sharpe, H. PD-L1 interacts specifically with B7-1 to inhibit T cell proliferation. Immunity [Internet]. 2009, 27, 111–22. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2707944%7B&%7Dtool=pmcentrez%7B&%7Drendertype=abstract.
- Khoja, L.; Butler, M.O.; Kang, S.P.; Ebbinghaus, S.; Joshua, A.M. Pembrolizumab. J. Immunother. Cancer 2015, 3, 36. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Ribas, A.; Wolchok, J.D.; Hodi, F.S.; Hamid, O.; Kefford, R.; Weber, J.S.; Joshua, A.M.; Hwu, W.-J.; Gangadhar, T.C.; et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet 2014, 384, 1109–1117. [Google Scholar] [CrossRef] [PubMed]
- Hamid, O.; Robert, C.; Daud, A.; Hodi, F.S.; Hwu, W.J.; Kefford, R.; Wolchok, J.D.; Hersey, P.; Joseph, R.; Weber, J.S.; et al. Five-year survival outcomes for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. Ann. Oncol. 2019, 30, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Ribas, A.; Schachter, J.; Arance, A.; Grob, J.-J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.M.; Lotem, M.; et al. Pembrolizumab versus ipilimumab in advanced melanoma (KEYNOTE-006): post-hoc 5-year results from an open-label, multicentre, randomised, controlled, phase 3 study. Lancet Oncol. 2019, 20, 1239–1251. [Google Scholar] [CrossRef] [PubMed]
- Garon, E.B.; Hellmann, M.D.; Rizvi, N.A.; Carcereny, E.; Leighl, N.B.; Ahn, M.-J.; Eder, J.P.; Balmanoukian, A.S.; Aggarwal, C.; Horn, L.; et al. Five-Year Overall Survival for Patients With Advanced Non‒Small-Cell Lung Cancer Treated With Pembrolizumab: Results From the Phase I KEYNOTE-001 Study. J. Clin. Oncol. 2019, 37, 2518–2527. [Google Scholar] [CrossRef]
- Herbst, R.S.; Garon, E.B.; Kim, D.-W.; Cho, B.C.; Perez-Gracia, J.L.; Han, J.-Y.; Arvis, C.D.; Majem, M.; Forster, M.D.; Monnet, I.; et al. Long-Term Outcomes and Retreatment Among Patients With Previously Treated, Programmed Death-Ligand 1‒Positive, Advanced Non‒Small-Cell Lung Cancer in the KEYNOTE-010 Study. J. Clin. Oncol. 2020, 38, 1580–1590. [Google Scholar] [CrossRef] [PubMed]
- Atezolizumab for Urothelial Carcinoma | FDA [Internet]. Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/atezolizumab-urothelial-carcinoma.
- Hoffman-Censits, J.H.; Grivas, P.; Van Der Heijden, M.S.; Dreicer, R.; Loriot, Y.; Retz, M.; Vogelzang, N.J.; Perez-Gracia, J.L.; Rezazadeh, A.; Bracarda, S.; et al. IMvigor 210, a phase II trial of atezolizumab (MPDL3280A) in platinum-treated locally advanced or metastatic urothelial carcinoma (mUC). J. Clin. Oncol. 2016, 34, 355–355. [Google Scholar] [CrossRef]
- Balar, A.V.; Galsky, M.D.; Rosenberg, J.E.; Powles, T.; Petrylak, D.P.; Bellmunt, J.; Loriot, Y.; Necchi, A.; Hoffman-Censits, J.; Perez-Gracia, J.L.; et al. Atezolizumab as first-line treatment in cisplatin-ineligible patients with locally advanced and metastatic urothelial carcinoma: A single-arm, multicentre, phase 2 trial. Lancet 2017, 389, 67–76. [Google Scholar] [CrossRef]
- Atezolizumab (TECENTRIQ) | FDA [Internet]. Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/atezolizumab-tecentriq.
- Fehrenbacher, L.; Spira, A.; Ballinger, M.; Kowanetz, M.; Vansteenkiste, J.; Mazieres, J.; Park, K.; Smith, D.; Artal-Cortes, A.; Lewanski, C.; et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): a multicentre, open-label, phase 2 randomised controlled trial. Lancet 2016, 387, 1837–1846. [Google Scholar] [CrossRef]
- Rittmeyer, A.; Barlesi, F.; Waterkamp, D.; Park, K.; Ciardiello, F.; von Pawel, J.; Gadgeel, S.M.; Hida, T.; Kowalski, D.M.; Dols, M.C.; et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. Lancet 2017, 389, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Hematology/Oncology (Cancer) Approvals & Safety Notifications | FDA [Internet]. Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/hematologyoncology-cancer-approvals-safety-notifications.
- FDA approves atezolizumab for PD-L1 positive unresectable locally advanced or metastatic triple-negative breast cancer | FDA [Internet]. Available from: https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-atezolizumab-pd-l1-positive-unresectable-locally-advanced-or-metastatic-triple-negative.
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Hegg, R.; Im, S.-A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef]
- Clinical Trials Using Atezolizumab - National Cancer Institute [Internet]. Available from: https://www.cancer.gov/about-cancer/treatment/clinical-trials/intervention/atezolizumab.
- Weinmann, S.C.; Pisetsky, D.S. Mechanisms of immune-related adverse events during the treatment of cancer with immune checkpoint inhibitors. Rheumatology 2019, 58 (Suppl. 7), vii59–vii67. [Google Scholar] [CrossRef]
- Liu, Y.-H.; Zang, X.-Y.; Wang, J.-C.; Huang, S.-S.; Xu, J.; Zhang, P. Diagnosis and Management of Immune Related Adverse Events (irAEs) in Cancer Immunotherapy. Biomed. Pharmacother. 2019, 120, 109437. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef]
- Das, S.; Johnson, D.B. Immune-related adverse events and anti-tumor efficacy of immune checkpoint inhibitors. J. Immunother. Cancer 2019, 7, 306. [Google Scholar] [CrossRef] [PubMed]
- Andrews, A. Treating with Checkpoint Inhibitors-Figure $1 Million per Patient. Am Heal drug benefits 2015, 8, 9–9. [Google Scholar]
- Arora, S.; Velichinskii, R.; Lesh, R.W.; Ali, U.; Kubiak, M.; Bansal, P.; Borghaei, H.; Edelman, M.J.; Boumber, Y. Existing and Emerging Biomarkers for Immune Checkpoint Immunotherapy in Solid Tumors. Adv. Ther. 2019, 36, 2638–2678. [Google Scholar] [CrossRef]
- Davis, A.A.; Patel, V.G. The role of PD-L1 expression as a predictive biomarker: an analysis of all US Food and Drug Administration (FDA) approvals of immune checkpoint inhibitors. J. Immunother. Cancer 2019, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, G.M.; Miller, V.; et al. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther [Internet]. 2017, 16, 2598–608. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28835386.
- Schlötterer, C.; Harr, B.; Schlo, C. Microsatellite Instability Microsatellite Instability. 2017, 1–4.
- Drescher, K.M.; Sharma, P.; Watson, P.; Gatalica, Z.; Thibodeau, S.N.; Lynch, H.T. Lymphocyte recruitment into the tumor site is altered in patients with MSI-H colon cancer. Fam. Cancer 2009, 8, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Duruisseaux, M.; Martínez-Cardús, A.; Calleja-Cervantes, M.E.; Moran, S.; de Moura, M.C.; Davalos, V.; Piñeyro, D.; Sanchez-Cespedes, M.; Girard, N.; Brevet, M.; et al. Epigenetic prediction of response to anti-PD-1 treatment in non-small-cell lung cancer: a multicentre, retrospective analysis. Lancet Respir. Med. 2018, 6, 771–781. [Google Scholar] [CrossRef]
- Havel, J.J.; Chowell, D.; Chan, T.A. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat. Rev. Cancer 2019, 19, 133–150. [Google Scholar] [CrossRef]
- Gibney, G.T.; Weiner, L.M.; Atkins, M.B. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. 2016, 17, e542–e551. [Google Scholar] [CrossRef]
- Maçon-Lemaître, L.; Triebel, F. The negative regulatory function of the lymphocyte-activation gene-3 co-receptor (CD223) on human T cells. Immunology [Internet]. 2005, 115, 170–8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15885122.
- Andrews, L.P.; Marciscano, A.E.; Drake, C.G.; Vignali, D.A.A. LAG3 (CD223) as a cancer immunotherapy target. Immunol. Rev. 2017, 276, 80–96. [Google Scholar] [CrossRef]
- Brignone, C.; Escudier, B.; Grygar, C.; Marcu, M.; Triebel, F. A phase I pharmacokinetic and biological correlative study of IMP321, a novel MHC class II agonist, in patients with advanced renal cell carcinoma. Clin Cancer Res [Internet]. 2009, 15, 6225–31. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19755389.
- Brignone, C.; Gutierrez, M.; Mefti, F.; Brain, E.; Jarcau, R.; Cvitkovic, F.; Bousetta, N.; Medioni, J.; Gligorov, J.; Grygar, C.; et al. First-line chemoimmunotherapy in metastatic breast carcinoma: combination of paclitaxel and IMP321 (LAG-3Ig) enhances immune responses and antitumor activity. J. Transl. Med. 2010, 8, 71–71. [Google Scholar] [CrossRef]
- Huang, R.-Y.; Francois, A.; McGray, A.R.; Miliotto, A.; Odunsi, K. Compensatory upregulation of PD-1, LAG-3, and CTLA-4 limits the efficacy of single-agent checkpoint blockade in metastatic ovarian cancer. OncoImmunology 2016, 6, e1249561. [Google Scholar] [CrossRef] [PubMed]
- Seliger, B. Combinatorial Approaches With Checkpoint Inhibitors to Enhance Anti-tumor Immunity. Front. Immunol. 2019, 10, 999. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [PubMed]
- Vanpouille-Box, C.; Formenti, S.C.; Demaria, S. Toward precision radiotherapy for use with immune checkpoint blockers [Internet]. Vol. 24, Clinical Cancer Research. American Association for Cancer Research Inc.; 2018, 259–65. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28751442.
- Rudqvist, N.P.; Pilones, K.A.; Lhuillier, C.; Wennerberg, E.; Sidhom, J.W.; Emerson, R.O.; et al. Radiotherapy and CTLA-4 blockade shape the tcr repertoire of tumor-infiltrating t cells. Cancer Immunol Res [Internet]. 2018, 6, 139–50. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29180535.
Figure 1.
Three phases of cancer immunoediting – diagram illustrating balance of immunological cancer populations versus less immunologic cancer population capable of cancer progression, in each phase of immunoediting.
Figure 1.
Three phases of cancer immunoediting – diagram illustrating balance of immunological cancer populations versus less immunologic cancer population capable of cancer progression, in each phase of immunoediting.
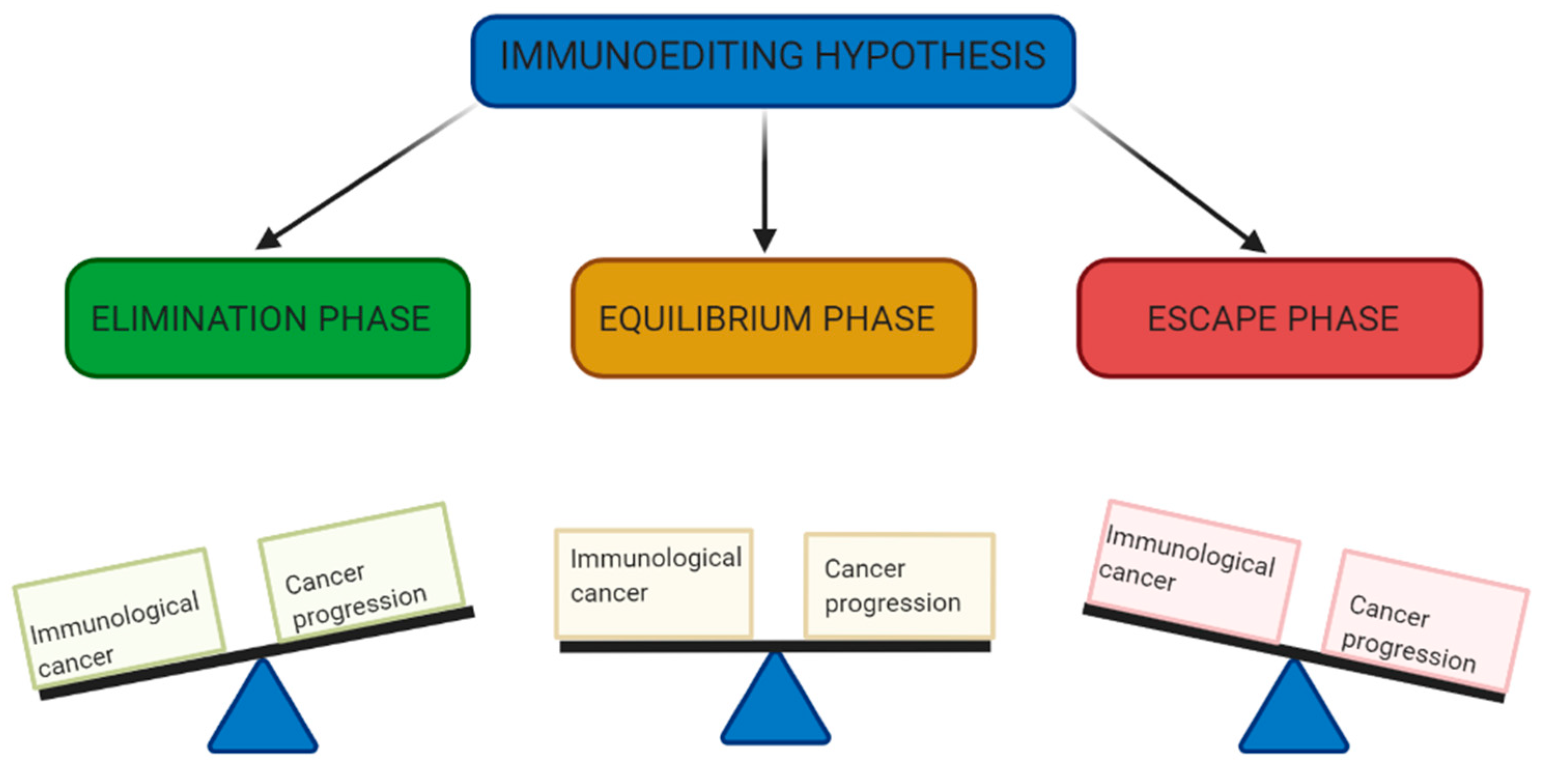
Figure 2.
Current and emerging checkpoint receptors and their respective ligands. They transmit co-inhibitory or co-stimulatory signals to TCR upon binding to their ligands.
Figure 2.
Current and emerging checkpoint receptors and their respective ligands. They transmit co-inhibitory or co-stimulatory signals to TCR upon binding to their ligands.
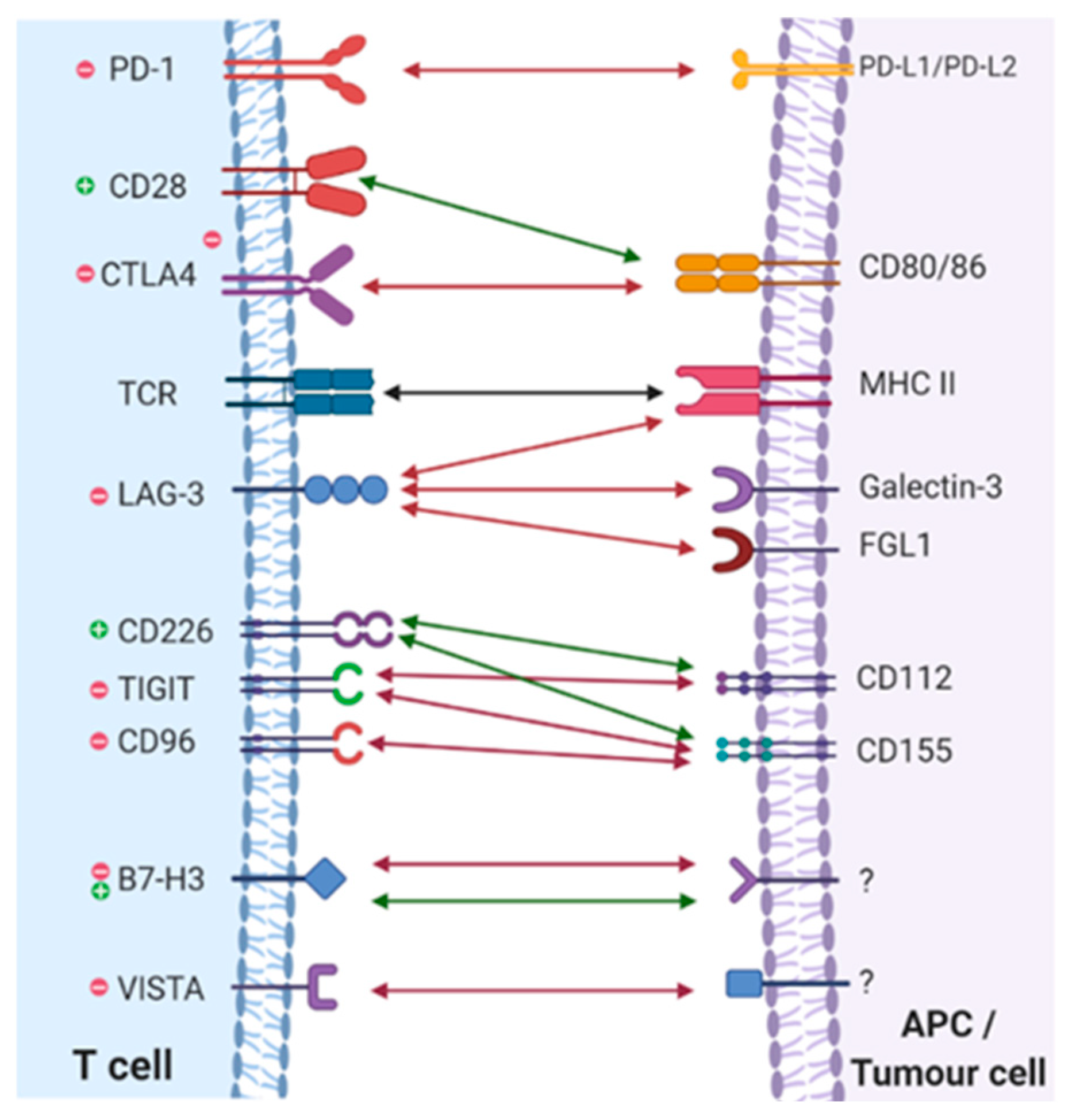
Figure 3.
CTLA4:CD80/86 binding inhibits TCR activation via competitive inhibition of CD28 co-stimulatory signalling. Anti-CTLA4 antibodies such as Ipilimumab block CTLA4:CD80/86 binding allowing unrestrained CD28 co-stimulation and increased TCR activation.
Figure 3.
CTLA4:CD80/86 binding inhibits TCR activation via competitive inhibition of CD28 co-stimulatory signalling. Anti-CTLA4 antibodies such as Ipilimumab block CTLA4:CD80/86 binding allowing unrestrained CD28 co-stimulation and increased TCR activation.
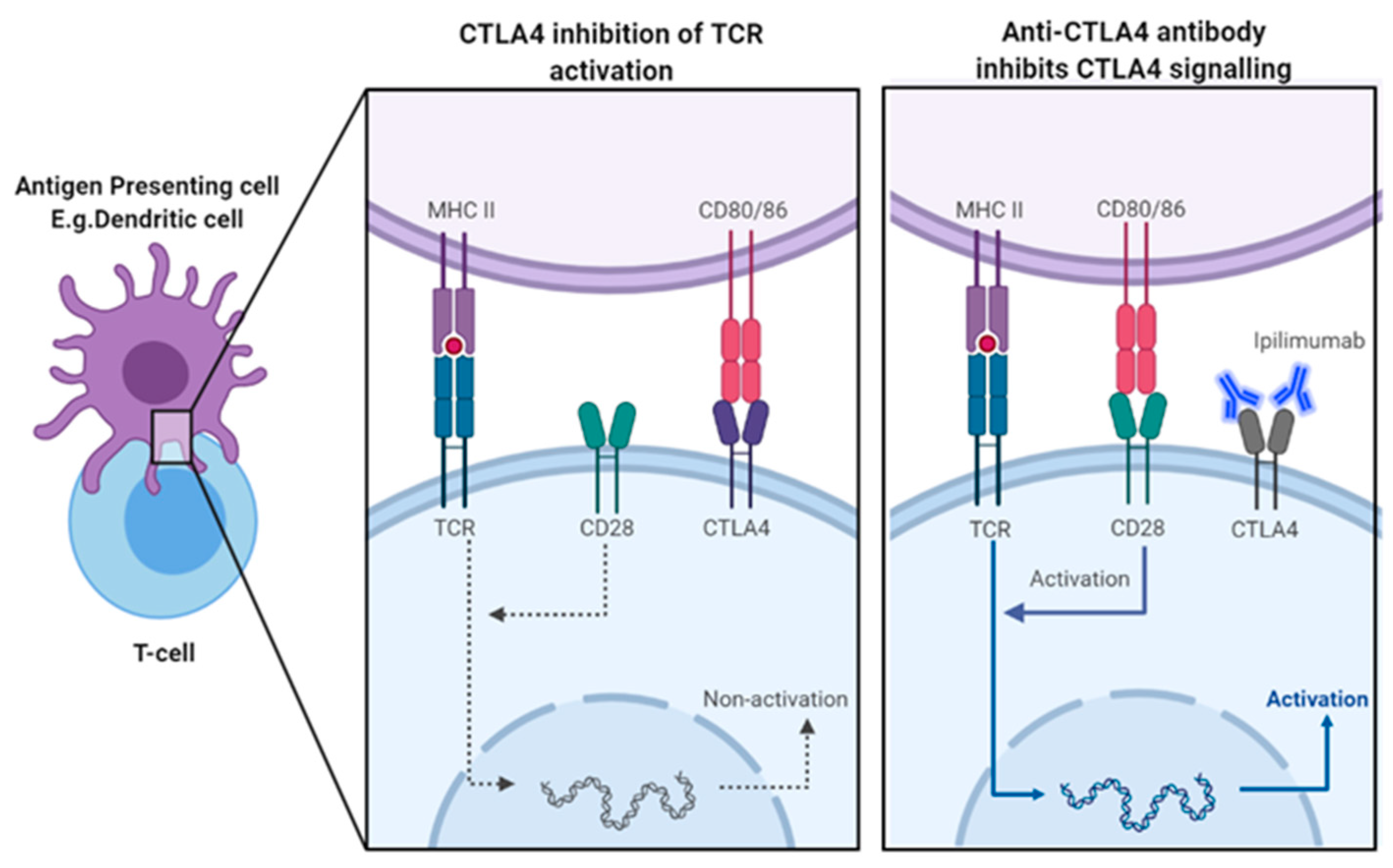
Figure 4.
Tumour cells can express PD-L1 (and PDL2, not shown) which can directly bind to inhibitory immune checkpoint PD-1 on surface of T cells. PD-1:PD-L1 binding induces co-inhibitory signalling that inhibits TCR activations, anti-PD-1 antibodies, such as Pembrolizumab, and anti-PD-L1 antibodies, such as Atezolizumab, inhibit PD:PD-L1 signalling, allowing unsrestrained TCR activation.
Figure 4.
Tumour cells can express PD-L1 (and PDL2, not shown) which can directly bind to inhibitory immune checkpoint PD-1 on surface of T cells. PD-1:PD-L1 binding induces co-inhibitory signalling that inhibits TCR activations, anti-PD-1 antibodies, such as Pembrolizumab, and anti-PD-L1 antibodies, such as Atezolizumab, inhibit PD:PD-L1 signalling, allowing unsrestrained TCR activation.
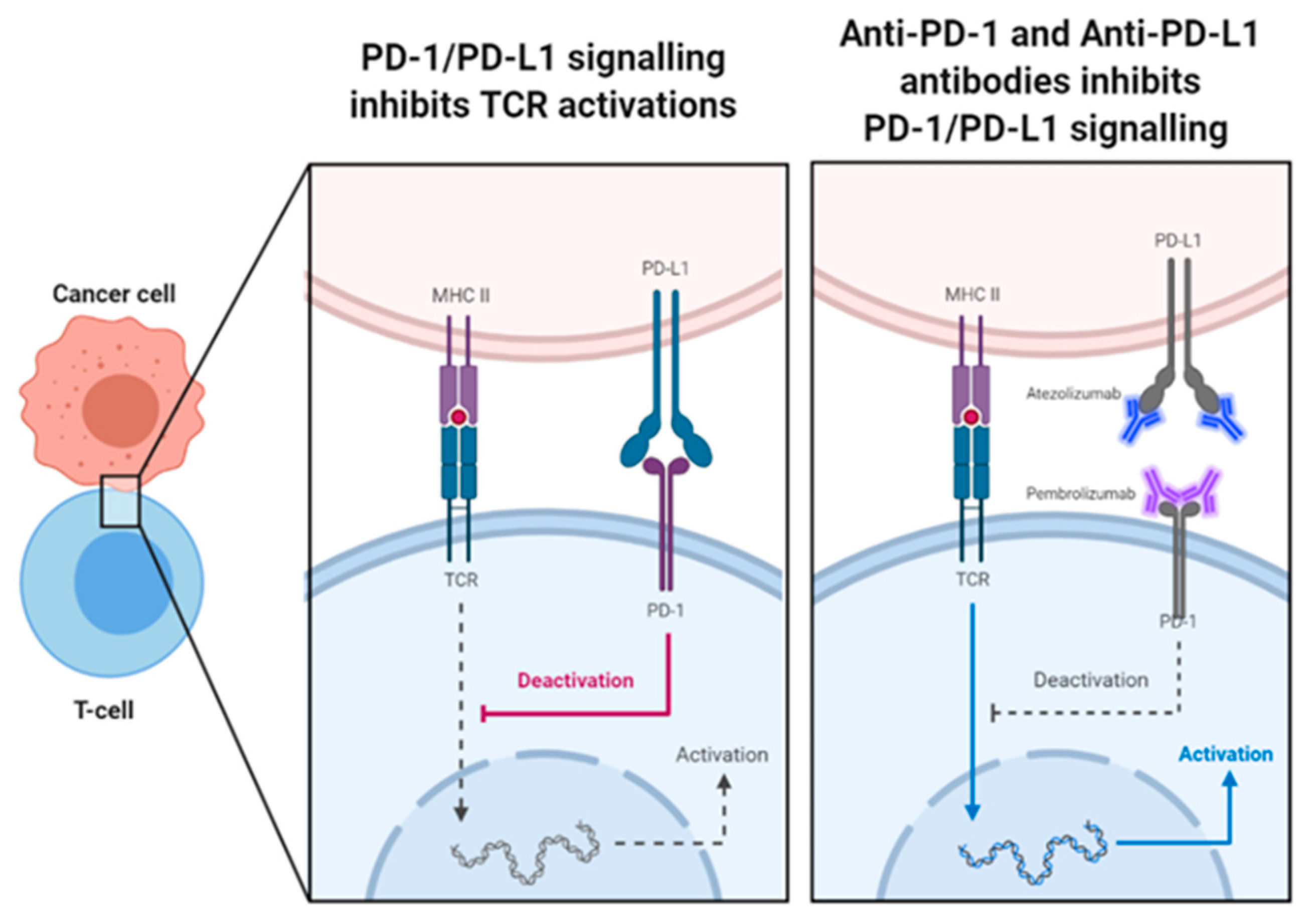
Figure 5.
Immune related adverse effects can affect almost every organ system. Examples of inflammtory pathology in each organ is shown above.
Figure 5.
Immune related adverse effects can affect almost every organ system. Examples of inflammtory pathology in each organ is shown above.
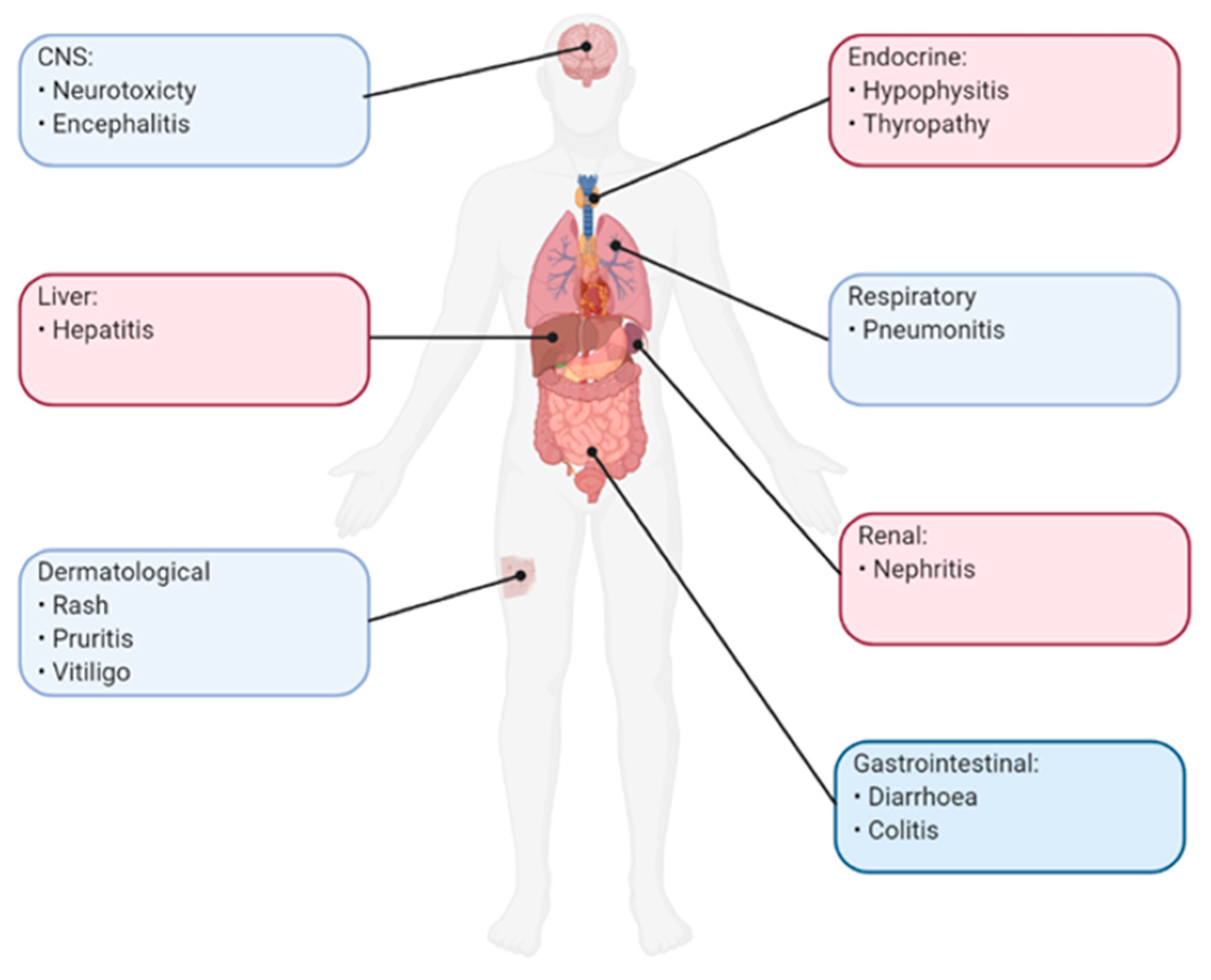
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



