
Submitted:
15 April 2024
Posted:
15 April 2024
You are already at the latest version
Abstract
A method for accurately determining the chemical composition of deposits at the bottom of pores during the electrocoloring (e-coloring) of aluminum anodic oxide (AAO) layers in tin-based solutions is developed. The aluminum samples were AC e-colored after DC sulfuric anodization. Free-standing, tin e-colored aluminum oxide film was obtained by selective dissolution of the metallic aluminum from the AAO in copper chloride solution to access the deposit directly at the bottom of the pore. This allowed us to take XPS analysis directly from the deposits at pore bottoms without any interference from the base material or insulating barrier layer. The results revealed the presence of a mixture of tin oxide and metal in the deposits, which were richer in oxide content. Furthermore, a cyclic voltammetry experiment mimicking real polarization conditions during AC conditions was optimized and used to gain a deeper understanding of the electrochemical reactions that occur during AC electrocoloring. The comparison of CV results in tin-free and tin-containing electrolytes indicated that the tin deposited during a cathodic cycle is oxidized in the anodic cycle. The formation of tin based deposits radically changed the CV behavior. The XPS and cyclic voltammetry results consistently show that the deposits formed during e-coloring comprised a mixture of metallic and oxidic tin species richer in oxide content.
Keywords:
Aluminum
; anodization
; electrocoloring
; tin
; XPS:
1. Introduction
Under ambient conditions, aluminum metal already has a very thin oxide film on its surface. However, the corrosion resistance of this oxide film needs to be improved since it is thin, inhomogeneous, and incoherent. [1,2,3,4,5]. With the anodic oxidation process, a thick, homogenous, and protective oxide film can be grown on the aluminum surface with higher hardness, corrosion, and abrasion resistance. If the anodic aluminum oxide (AAO) film is formed in acidic electrolytes, the morphology of the resulting film will be highly porous. The thin, dense, and dielectric barrier layer is the first structure that forms on the surface, and it builds the basis for the growth of the porous layer [4,5,6]. The mechanism of pore formation is still a matter of discussion [2,3,4,5,6,7]. The morphology (pore dimensions and shape) of the AAO layer changes with electrolyte type, applied voltage, current density, temperature, and process time [3,5].
Sulfuric acid is the most widely used electrolyte for the production of AAO layers because of its low cost, speed, lower energy consumption, and suitability of pore structure for further coloring. In addition, the porous nature of AAO layers makes them suitable for coloring using different methods that find extensive use in architectural and decorative applications. AAO layers can be colored with organic, inorganic dyes, or by integral coloring. However, the most preferred coloring method is electrolytic (e-coloring) since it has an extended service life compared to the other methods while preserving the metallic texture of the substrate even after the coloring process. [7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25]
Electrolytic coloring of AAO is generally carried out in an acidic solution that contains metal ions such as Ni, Co, Sn, and Cu. During the process of coloring, species are deposited in the AAO pores using AC/DC or pulse polarization. Today, the most common e-coloring electrolytes are tin-based, and is realized by applying 10-20 V AC voltage on sulfuric acid anodized aluminum alloys. With this process, all shades of bronze to black colors can be obtained [8,9]. The mechanism of the electrolytic coloring of AAO and the chemical nature of the deposit in the pores has yet to be fully established [11,12,13,14,15,16,17,18,19,20,21,22,23,24,25]. Many studies in the literature are devoted to understanding the nature of the deposits that form in the AAO by using different e-coloring electrolytes and characterization techniques. In a variety of studies, the nature of the deposit is claimed to be colloidal or nano-sized metallic particles [8,9,13,14,15,19,20,21,22,24,25]; there are also a limited number of studies that claim the presence of a mixture of metallic and oxidic species in the pores [10,11]. We found only two studies on the industrially most widely used tin-based e-coloring electrolytes. Cohen et.al. analyzed the deposits by Mössbauer spectroscopy and concluded that metal deposition in the pores occurs on a resistive oxide surface [23]. Jagminas et al. used Cu, Ni, Co, and Sn-based electrolytes and characterized the deposits with FT-IR studies. Their results showed that Me(OH)x-type structures were formed in the pore base [18].
As presented above, the characterization results obtained from different sources in the literature contradict each other. Since the nature of products within the pores defines the mechanism of coloring, a sound method is needed to determine the nature of deposits in the pores. Additionally, considering the high magnitude of positive and negative voltages applied and the possible local chemistry changes within the restricted volumes of pores during e-coloring, a discrete electrochemical method that can simulate the complex conditions during AC e-coloring is also required. In the literature, we found no electrochemical studies conducted under AC e-coloring conditions. Accordingly, the study aims to develop a sound method to determine the chemical nature of the deposits formed at pore bottoms and a reliable electrochemical method that can simulate the conditions during AC-coloring and intercorrelate the results to clarify the mechanism of e-coloring in tin-based electrolytes.
2. Materials and Methods
2.1. Preparation and Characterization of Free-Standing AAO Layers
Before anodization, high, purity aluminum sheets (99.999%, 0.7 mm thick) were annealed at 450 °C for 3h to obtain the ordered nanostructure after two-step anodization [26,27]. Annealed samples (20 mm x 80mm in size) were then electropolished in 80% phosphoric +20% sulfuric acid solution at a constant voltage of 17 V for 5min at 70 °C. First, anodization was carried out by applying 16V DC at 19 °C in a 170 g/l H2SO4 solution in a conventional two-electrode cell with an aluminum counter electrode for 30 minutes. After dissolving this layer in chromic acid solution, a second anodization step was performed under the same conditions. In these experiments, only one face of the aluminum was exposed to the anodization solution by masking the other surface with cellulosic lacquer. During this step, the anodization time was increased to obtain an AAO thickness of 40 µm. Increasing AAO thickness is crucial to obtaining a free-standing AAO layer after the selective dissolution of remnant metallic aluminum below the AAO layer.
Electro-coloring was carried out in an Sn-based solution consisting of 190g/l SnSO4, 18g/l H2SO4, and sulfosalicylic acid as a stabilizer. The coloring time was kept between 10 and 12 minutes at an AC of 17 V. After the coloring process, samples were sealed in hot water to protect Sn species deposited at the bottom of the pores during the selective dissolution of metallic aluminum substrate. Self-standing AAO layers were produced by the selective dissolution of metallic aluminum in 3% CuCl2 solution after dissolving the lacquer layer in acetone, covering the metallic Al. By stripping, it became possible to obtain free-standing films ready to be analyzed by different techniques without the interfering effects of the base metal.
The structure and chemistry of the self-standing films were characterized by FE-SEM (JEOL JSM_7000F field emission gun scanning electron microscope).
The XPS investigations were conducted using a Thermo Scientific K-Alpha X-Ray Photoelectron Spectrometer with a monochromatized Al K-alpha X-ray source. The binding energy scale was calibrated with respect to C1s (285 eV). Before the analyses, the barrier layer of free-standing film is removed with argon sputtering. A flood gun was also used to avoid valence state-changing problems associated with Ar etching. Casa XPS software is used to deconvolute the spectra. Gaussian/Lorentzian functions and Shirley background were used during the XPS spectra deconvolution. The area ratios of doublets and the energy difference between the doublets were kept constant during deconvolution.
2.2. Electrochemical Investigation Methodology for the Determination of the Coloring Mechanism of AAO in Sn-Based Electro-Coloring Solutions
2.2.1. Preparation of AAO Films for e-Coloring
In these experiments, the most commonly anodized aluminum alloy (AA6063-T6) was used as the substrate to determine the e-coloring mechanism in Sn-based solutions. Their composition is specified in Table 1.
These alloys were anodized after alkaline etching in a sodium hydroxide solution (10 %NaOH, 60 oC, 3 mins.) followed by nitric acid-based neutralization. The AAO films with 11±1 µm thicknesses were grown by applying 16V DC at 19 °C in a 170 g/l H2SO4 aqueous solution in a conventional two-electrode cell with an aluminum counter electrode. In all e-coloring experiments, the anodization parameters were kept constant.
2.2.2. CV-Controlled E-Coloring Experiments
In the industrial practice, E-coloring of AAO in Sn-based solutions is realized by the application of 10-20 V of AC voltage (50 Hz) in an acidic solution consisting of 12g/l SnSO4, 18g/l H2SO4 and stabilizer at room temperature. Accordingly, very high scan rates and high potentials are required to simulate these parameters with a CV method. The high-rate CV experiment potential limit of the available potentiostat is +/-10V, and the scan rate that can obtain reliable data collection results is 100 V/s. The potential limit of 10 V is sufficient for e-coloring [15,21]. On the other hand, in the case of frequency, a 10-fold decreased frequency can be obtained compared to 50 Hz. Thus, checking the possibility of e-coloring using these limit parameters became necessary.
Accordingly, the first experiments were conducted to determine the limits of scan rate for e-coloring. A series of experiments with scan rates 100 V/s and in the potential range of +/-10V were realized in a three-electrode cell consisting of an Ag/AgCl reference electrode (Ag/Ag/Cl, +0.197 V/NHE), a graphite counter electrode, and the anodized aluminum working electrode.
After determining the suitability of the scan rate and potential range for e-coloring, time (cycle) dependent CV experiments were conducted in Sn-free and Sn-containing acidic solutions. Cycle-dependent changes in the morphology of the AAO layers were investigated by FEG SEM.
3. Results and Discussion
3.1. Chemical Characterization of Tin Deposits in Free-Standing AAO Films
XPS analysis of free-standing e-colored AAO films obtained after stripping the metallic aluminum from their backside and sputtering the barrier layer covering the deposits was conducted. SEM images taken from the backside of the films after XPS analysis revealed the presence of Sn-based deposits that show themselves as brighter spots (Figure 1)
The major peaks in the XPS survey were related to different binding states of Al, O, C, S, and Sn. The occurrence of sulfur in AAO is related to the use of sulfuric acid as the anodization electrolyte [1,5]. Figure 2 presents high-resolution scans of Al, Sn, O, and S.
As expected, the position of the Al 2p peak at 74.6 eV is consistent [34,35] with the binding energy of Al2O3 (Figure 2a).
The deconvolution of the S2s peak revealed the presence of elemental (0) and (+3) valent S species with binding energies of 162.1 and 163.7 eV, respectively. These binding energy values are also consistent with the literature [28,29] (Figure 2b).
The deconvolution 1Os spectra of the sample comprised three different peaks centered at 530.3eV, 531.5eV, and 532.8eV (Figure 2c). The O1s line at 530.3eV corresponded to the tin oxide species, and the others are oxygen bound to aluminum oxides and sulfur, respectively [28,30,31].
The deconvoluted 3d region of Sn peaks of samples revealed the presence of metallic tin and tin oxide species in the deposits (Figure 2d). After the deconvolution, two separate peak groups with Sn3d5/2 binding energy at 486.7, 485 eV and Sn3d3/2 binding energy at 495.23eV, 493.53eV, were observed. The peaks at 493.5 and 485 eV represent 3d3/2 and 3d5/2 of metallic tin [39,40]. The binding energies Sn+2 and Sn+4 have binding energies very close to each other (0.5 eV) that fall into the detection sensitivity range of 0.6 eV of the instrument (3d5/2 for +2 and +4 valent Sn: 486.3±0.6 eV) [30,31,32,33] that makes a differentiation between them very difficult. Despite this difficulty, it is not wrong to say that the tin species in the pores consisted of a mixture of metallic and oxidic tin species.
These results indicated the formation of tin oxide species in the deposits at AAO pore bottoms. In the following sections of the study, the mechanism behind the formation of these deposits will be investigated using a CV method compatible with the AC e-coloration process.
3.2. Electrochemical Analysis of e-Coloring Process with High Scan Rate CV
In this section, studies are presented for optimizing a CV method compatible with AC e-coloring, followed by studies in tin-free and tin-containing electrolytes using optimized CV parameters.
3.2.1. Determination of the Suitability of Potential and Scan Rate Limits for e-Coloring
To control the possibility of e-coloring within voltage and scan rate limits of the potentiostat, CV measurements of anodized samples in the potential range of +/-10V and with 100V/s scan rate for 10 min (2500 cycles) were realized. After this experiment, the typical brown color of anodized aluminum in tin-containing electrolytes was obtained. SEM cross-section of the same sample indicated the deposition of tin species in the pores of AAO (Figure 3). Accordingly, the +/-10 V range and 100 V/s scan rate are used for further CV experiments.
3.2.2. CV Experiments in Tin-Free and Tin-Containing Electrolytes
CV experiments were conducted first in the base e-coloring electrolyte free from tin ions to determine the contribution of tin ions in the electrolyte. Before detailing the CV behavior, the meanings of the potentials and currents observed during cathodic and anodic polarization need to be explained to understand the behavior of AAO layers while cycling. In Figure 4, the critical potentials are marked from 1 to 6. The CV is presented on an E-logi scale to show potential positions more precisely.
The cycling starts from point 1 towards the cathodic direction, giving very low cathodic currents limited by the presence of the barrier layer till point 2. This potential is called cathodic breakdown potential, indicating the breakdown of the barrier layer, followed by vigorous hydrogen evolution at the metal-oxide interface [33,34,35,36] that leads to the alkalinization of the electrolyte in the pores in which AAO is chemically unstable. Breakdown of the barrier layer depends on the amount of charge build-up during cathodic cycling [37,38], showing a strong dependence on scan rate, the defect density of the AAO, and electrolyte properties. Lower scan rates lead to the earlier breakdown of the barrier layer since the charge build-up is time-dependent. Thus, a sufficiently high scan rate is required to prevent total dissolution of the barrier layer due to local alkalinization and further build-up of porous AAO in the anodic cycle [38]. Upon reversal of scan direction (point 3), the cathodic current decays, showing a hysteresis behavior, indicating ongoing activity of the cathodic reaction till point 4. The potential at point 4 is the corrosion potential of the aluminum in the alkalinized pore [39,40,41,42]. Starting from point 4, a low anodic current starts to flow until point 5, at which AAO formation and OER start. The charge build-up on AAO is released upon reversing the potential towards the cathodic direction, indicating a capacitive behavior [43,44]. After releasing the anodic charge, the cathodic charging of the AAO layers starts at point 6 and continues till the breakdown potential.
The possible chemical and electrochemical reactions at different potential regions are listed below.
- At Point 2 after cathodic breakdown till point 4;
The electrochemical cathodic reactions (reactions 1,2) followed lead to alkalinization that induces the chemical dissolution of Al and Al2O3 by producing aluminate ions.
- At 4 Point for till Point 5
At point 4 Al metal is not protected at pore bottoms thus corrosion starts at the mixed potential of the anodic (reaction 5) and cathodic (reaction 6) half-cell reactions in aluminate rich pore bottom electrolyte. Followed by active dissolution and passivation of Al (reaction7)
- At Point 5
Oxygen evolution reaction (OER) and further anodic oxidation starts.
- At Point 6
The discharge of anodic charge on the dielectric AAO and initiation of cathodic charge build up starts.
3.2.3. Variation of CV Behavior with Cycling in Tin Free Electrolytes
Cycle-dependent E vs i and E vs logi relations are presented in Figure 5a and b. The breakdown shifts to more negative potentials (-4 to -5.30V) with cycling, indicating the requirement of more charge build-up for breakdown related to the change in the barrier layer properties of the AAO layer that forms at the potential range between 4 and 5. The SEM images of the AAO layer (Figure 6) after cycling indicate that a new AAO layer is formed during the anodic cycles. The possibility of AAO growth with AC polarization has already been shown in several studies [45,46,47,48,49]. The new AAO layer's morphology differs from that of the original anodization electrolyte. The AAO is more porous and irregular and possesses larger pore diameters than those formed in lower pH anodization electrolytes (Figure 6). This new oxide morphology can be attributed to changes in pH in the pores and/or precipitation of aluminum hydroxides within the pores [34,35,36,45] during cathodic cycling that dehydrates during anodic polarization. The shift of breakdown potentials can be related to the thicker barrier layer of the new AAO that forms in higher pH solutions. Another indication of increased pore bottom pH is the magnitude of cathodic currents after breakdown. The cathodic maximum currents decrease after 20 cycles from -145 to -57 mA/cm2), which can be related to the lower activity of hydrogen ions due to the alkalinization (Figure 5a).
The potential at point 3 is only observed after cathodic breakdown, indicating the corrosion behavior of aluminum. The potential values (-1.57±0.2V) lie close to the corrosion potential of aluminum metal in alkaline solutions [35,39,41,50,51]. After point 4, the variation of anodic currents is negligible due to the well-known diode behavior of aluminum [38,46]. The onset of anodization potential (point 5) did not vary with cycling. However, the increase in cycling decreases the maximum anodic currents from 17 to 13 mA/cm2, which can also be attributed to the increased thickness of AAO and higher pH within the pores. The discharge potentials at point 6 (+5.7 V± 0.1V) did not vary significantly, indicating similar capacitive behavior of the layers after cycling.
3.2.4. Tin Ion Containing Electrolytes
The CV behavior during the first 20 cycles is similar to that of a solution free from tin ions. However, after 20 cycles, cathodic (peak A) and anodic (peak B) peaks appear, and the CV behavior changes completely (Figure 7a,b).
At the 20th cycle, after the realization of the cathodic breakdown and hydrogen evolution, the deposition of tin becomes apparent at point A. The tin's deposition potentials at point A are comparable with the electrodeposition potential of tin in alkaline electrolytes containing stabilizers [52,53]. After the deposition of tin in the pores, the potential at point 4 also starts to change in a positive direction towards the corrosion potential of tin in alkaline electrolytes [53,54,55]. With the change of potential towards anodic direction, oxidation of deposited tin starts to commence at point B [54,55,56,57,58,59]. After the oxidation of metallic tin, a very low anodic current is recorded even beyond the anodic oxidation potential (oxygen evolution reaction) (point5), similar to the anodic behavior of tin in alkaline solutions anodized at high anodic potentials. All of these observations indicated that after the deposition of tin in the pores, the tin deposits govern the electrochemical behavior. Another support for this observation is the lack of new AAO growth (Figure 8) contrary to the ones subjected to cyclic polarization in tin-free electrolytes. Upon reversal of the potential, the capacitive behavior of the system also changed dramatically towards more positive potentials, indicating higher charge accumulation ability that aroused from the presence of tin oxide species in the pores [60,61]. Interestingly, no sign of cathodic reduction is observed upon reaching the reduction potential of tin ions during cathodic cycling. That can be attributed to the non-conductive nature of tin oxide species in the pores or a thin AAO barrier layer below them. The potential (point 2) at which cathodic reduction starts also switches to higher cathodic potentials after the build-up of tin oxide species in the pores. The charge transfer reactions through the barrier layer strongly depend on their thickness; if the oxide layer thickness is lower than approx. 10 nm [36] charge transfer is realized at the oxide solution interface. However, when the thickness is higher than 10 nm, the charge transfer reaction occurs via proton transfer at the metal-oxide interface. Assuming the presence of a thinner barrier layer at point 2, allowing charge transfer at the oxide solution interface in the presence of tin ions is not wrong because of their hindering effect on oxide growth at point 5 (Figure 8). Charge transfer through a thinner barrier layer is expected to occur at lower cathodic potentials, which means the charge will be readily available to reduce hydrogen ions. The delay of initiation of HER in the presence of a tin oxide-covered barrier layer can be explained by the conversion of nonconductive +4 tin oxide species towards a conductive nature by conversion into a sub-stoichiometric conductive oxide [53] during cathodic polarization or by the difficulty of charge transport through relatively thick tin oxide layer. The shift of cathodic breakdown potential to more negative values with cycling and thickening of the tin oxide layer may indicate that the second mechanism better explains this shift. The observation of lower cathodic currents at point 2 is due to the well-known high overvoltage of HER on tin oxides [52,54]. With the increase in cycle numbers beyond 20, the reduction and oxidation current increases (points A and B) and stabilizes after 50 cycles.
In the presence of tin ions in the solution the possible changes in the chemical and electrochemical reactions with respect to tin free electrolytes can be summarized as follows:
- At point 2 after cathodic breakdown till point A, rates of reaction 1 and 2 decreased because of high over potential of HER on tin oxides. The rates of chemical dissolution of AAO layer (reaction 3) and Al (reaction 4) at the pore bottom is also decrease because of limited alkalinization.
- At Point A;
- At Point B;
- At point 5
Reactions 8 and 9 take place at a lower rate compared to tin-free electrolytes because of the presence of tin oxides at the pore bottom.
When the deposit chemistry determination and cycling voltammetry results are evaluated together, it is concluded that the pores of AAO in tin-based e-coloring solutions are a mixture of metallic and oxidic tin species. These results indicate that the semiconductor nature of the deposited tin oxide species will also contribute to the coloring of AAO by absorption and explain different color tints observed while using different e-coloring electrolytes.
4. Conclusions
The analysis of free-standing e-colored AAO with XPS showed that the pore deposits are a mixture of tin and tin oxides.
It is determined that cyclic voltammetry in the +/-10V potential range and 100V/s scan rate can simulate the e-coloring process using AC voltage.
Five critical potentials, the cathodic breakdown, corrosion potential, anodic oxidation potential, and discharge potential, are present in the CV of AAO in tin-free e-coloring electrolytes. It has been determined that after cathodic breakdown, alkalinization of the electrolyte at the bottoms is realized, which is supported by the appearance of corrosion potential during switching from cathodic to anodic cycle. Under the conditions utilized, an AAO layer with a more porous and irregular nature is formed.
Significant differences exist between the CV behavior of AAO in tin ion-bearing electrolytes and tin-free electrolytes. Additional peaks related to the reduction of tin ions and oxidation of metallic peaks appeared after 20 cycles, indicating the deposition of metallic tin during the cathodic cycle and its oxidation during the anodic cycle.
After the deposition of tin species in the pores, the cathodic breakdown potential shifted toward a more cathodic potential, the corrosion potential switched toward the corrosion potential of tin in alkaline solutions, and the anodic oxidation and discharge potentials shifted toward more positive values. Related currents decreased.
The presence of tin ions in the electrolyte hindered the growth of a new AAO layer during anodic polarization.
The results of CV experiments supported the findings of XPS analysis, which indicated the presence of a tin oxide-rich tin metal and oxide mixture in the AAO pores.
The methodology used in this study can be applied to other e-coloring processes to understand the nature of deposits in the AAO.
Further work is required to clarify the chemical and electrochemical reactions occurring at different regions of anodic and cathodic polarization during cycling.
Author Contributions
Pinar Afsin: Methodology, Conceptualization; Investigation; Visualization; Writing – original draft: Validation. Can Akyil: Review & editing; Resources, Funding. Kursat Kazmanli: Methodology, Resources; Review &Editing Mustafa Urgen: Conceptualization; Methodology; Project Resources; Supervision; Writing – review & editing; Validation.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Sheasby, P. G.; Pinner R. Anodizing of aluminum, in The Surface Treatment and Finishing of Aluminum and its Alloys, UK: Finishing Publications Ltd. 2001. Vol. 1., 330-420.
- Schwirn, K.; Lee, W.; Hillebrand, R.; Steinhart, M.; Nielsch, K.; Gösele, U. Self-Ordered Anodic Aluminum Oxide Formed by H2SO4 Hard Anodization. ACS Nano 2008, 2, 302–310. [CrossRef]
- Lee, K.; Tang, Y.; Ouyang, M. Self-Ordered, Controlled Structure Nanoporous Membranes Using Constant Current Anodization. Nano Lett 2008, 8, 4624–4629. [CrossRef]
- Sulka, G.D.; Parkoła, K.G. Temperature Influence on Well-Ordered Nanopore Structures Grown by Anodization of Aluminium in Sulphuric Acid. Electrochim Acta, 2007, 52, 1880–1888. [CrossRef]
- Thompson, G.E.; Wood, G.C. Anodic Films on Aluminium. In; Treatise on Material Science and Technology, Editor J.C. Scully. Elsevier,1983, Volume 23, pp. 205–3. [CrossRef]
- Diggle, J.W.; Downie, T.C.; Goulding, C.W. Anodic Oxide Films On Aluminum; Chem. Rev. 1969, 69, 3, 365–405. [CrossRef]
- Jani, A.M.; Losic, D.; Voelcker, N.H. Nanoporous Anodic Aluminium Oxide: Advances in Surface Engineering and Emerging Applications. Prog Mater Sci., 2013, 58, 636–704. [CrossRef]
- Nozari Nezhad, M.; Kolahi, A.; KazemZad, M.; Saiedifar, M. Electrolytic Coloring of Anodized Aluminum by Copper. In Proceedings of the Advanced Materials Research, 2014, 829, 381–385.
- Liang, K.; Liang, C.H.; Wang, H. Structure and Distribution of Electrodeposits on Anodic Aluminium Films by Electrolytic Colouring in Zinc Sulphate Solution. Transactions of the Institute of Metal Finishing 2007, 85, 159–161. [CrossRef]
- Doughty, A.S.; Thompson G.E., Wood, G.C., Investigation of the electrolytic Coloring of Porous Anodic Films on Aluminum using Electron Microscopy. Transactions of the Institute of Metal Finishing 1975, 53, 33-35.
- Nahar, N.M.; Mo, G.H.; Ignatiev, A. Development of an Al203-Co Selective Absorber For Solar Collectors. Thin Solid Films 1989, 172, 19-25. [CrossRef]
- Suzer, S.S.; Kadirgan, F.; Sohmen, H.M.S.; Wetherilt, A.J.; Tureture, I.E. Spectroscopic Characterization of Al O-Ni Selective Absorbers for Solar Collectors; Solar Energy Materials and Solar Cells. 1998, 52; 55-60.
- Hakimizad, A.; Raeissi, K.; Ashrafizadeh, F. Characterization of Aluminum Anodized Layers Modified in Sulfuric and Phosphoric Acid Baths and Their Effect on Conventional Electrolytic Coloring. Surf Coat Technol 2012, 206, 2438–2445. [CrossRef]
- De Graeve, I.; Laha, P.; Goossens, V.; Furneaux, R.; Verwimp, D.; Stijns, E.; Terryn, H. Colour Simulation and Prediction of Complex Nano-Structured Metal Oxide Films. Test Case: Analysis and Modeling of Electro-Coloured Anodized Aluminium. Surf Coat Technol 2011, 205, 4349–4354. [CrossRef]
- Santiago, S.; Fernandez, A.M. Electrolytic Nickel Impregnation of Porous Anodic Aluminum Oxide Films Using AC Voltage as Solar Selective Absorber. In Proceedings of the Energy Procedia; Elsevier Ltd, 2014; Vol. 57, pp. 2733–2742. [CrossRef]
- Sheasby, P. G., Pinner R. Anodizing in Architecture in The Surface Treatment and Finishing of Aluminum and its Alloys, UK: Finishing Publications Ltd. 2001. Vol. 2., 700-740.
- Lin, C.-F.; Hebert, K.R. Change Produced by Cathodic Polarization in the Electrical Conduction Behavior of Surface Films on Aluminum Electrochemical Systems; J. Electrochem. Soc. 1994; 141; 105-110.
- Jagminas, A.; Niaura, G.; Kuzmarskyte, J.; Butkiene, R. Surface-Enhanced Raman Scattering Effect for Copper Oxygenous Compounds Array within the Alumina Template Pores Synthesized by AC Deposition from Cu(II) Acetate Solution. Appl Surf Sci 2004, 225, 302–308. [CrossRef]
- Salmi, J.; Bonino, J.-P.; Bes, R.S. Nickel Pigmented Anodized Aluminium as Solar Selective Absorbers J. of Materials Science, 2000, 35, 1347-1351.
- Goad, D.G.W.; Moskovits, M. Colloidal Metal in Aluminum-Oxide. J Appl Phys 1978, 49, 2929–2934. [CrossRef]
- Tsangaraki-Kaplanoglou, I.; Theohari, S.; Dimogerontakis, T.; Kallithrakas-Kontos, N.; Wang, Y.M.; Kuo, H.H. (Harry); Kia, S. An Investigation of Electrolytic Coloring Process of Anodized Aluminum Coatings. Surf Coat Technol 2006, 201, 2749–2759. [CrossRef]
- Akolkar, R.; Wang, Y.M.; Kuo, H.H. Kinetics of the Electrolytic Coloring Process on Anodized Aluminum. J Appl Electrochem 2007, 37, 291–296. [CrossRef]
- Cohen, R.L.; Raub, C.J.; Muramaki, T. The State of Tin in Tin-Anodized Aluminum. J. Electrochem. Soc. 1978 ,125 34. [CrossRef]
- Zemanová, M., Chovancová, M. & Krivošík, P. A New Approach to Nickel Electrolytic Colouring of Anodised Aluminium. Chem. Pap. 2009 63, 62–70. [CrossRef]
- Shaffei, M.F.; Abd El-Rehim, S.S.; Shaaban, N.A.; Huisen, H.S. Electrolytic Coloring of Anodic Aluminum for Selective Solar Absorbing Films: Use of Additives Promoting Color Depth and Rate. Renewable Energy, 2001, 23, 489-495. [CrossRef]
- Bouchama, L.; Azzouz, N.; Boukmouche, N.; Chopart, J.P.; Daltin, A.L.; Bouznit, Y. Enhancing Aluminum Corrosion Resistance by Two-Step Anodizing Process. Surf Coat Technol 2013, 235, 676–684. [CrossRef]
- Han, X.Y.; Shen, W.Z. Improved Two-Step Anodization Technique for Ordered Porous Anodic Aluminum Membranes. Journal of Electroanalytical Chemistry 2011, 655, 56–64. [CrossRef]
- St. Smart, R.C.; Skinner, W.M.; Gerson, A.R. XPS of Sulphide Mineral Surfaces: Metal-Deficient, Polysulphides, Defects and Elemental Sulphur. Surface and Interface Analysis 1999; 28;.101-105.
- Lee, A.F.; Wilson, K.; Goldoni, A.; Larciprete, R.; Lizzit, S. A Fast XPS Study of Sulphate Promoted Propene Decomposition over Pt{1 1 1}. Surface Science, 2002, 553, 140-148.
- Zähr, J.; Oswald, S.; Türpe, M.; Ullrich, H.J.; Füssel, U. Characterisation of Oxide and Hydroxide Layers on Technical Aluminum Materials Using XPS. In Proceedings of the Vacuum; March 14 2012; Vol. 86, pp. 1216–1219. [CrossRef]
- Thomas, B.; Skariah, B. Spray Deposited Mg-Doped SnO2 Thin Film LPG Sensor: XPS and EDX Analysis in Relation to Deposition Temperature and Doping. J Alloys Compd. 2015, 625, 231–240. [CrossRef]
- Oswald, S. X -Ray Photoelectron Spectroscopy in Analysis of Surfaces Update Based on the Original Article by Steffen Oswald, Encyclopedia of Analytical Chemistry , John Wiley & Sons, Ltd. 2000, pp. 19-21.
- Nisancioglu, K.; Holtan, H. Cathodic Polarization of Commercially Pure Aluminium. Corros Sci, 1979, 19, 537-552. [CrossRef]
- Bunker, B.C.; Nelson, G.C.; Zavadil, K.R.; Barbour, J.C.; Wall, F.D.; Sullivan, J.P.; Windisch, C.F.; Engelhardt, M.H.; Baer, D.R. Hydration of Passive Oxide Films on Aluminum. J of Physical Chemistry B 2002, 106, 4705–4713. [CrossRef]
- Adhikari, S.; Hebert, K.R. Participation of Aluminum Hydride in the Anodic Dissolution of Aluminum in Alkaline Solutions. J. Electrochem. Soc. 2008, 155, C189. [CrossRef]
- Seo, Jong & Lee, Dong Nyung. Assessment of Proton Transport in Amorphous Aluminum Oxide by Cathodic Polarization. J of Electrochem. Soc 2003, 150, 329-335. [CrossRef]
- Hassel, A.W.; Lohrengel, M.M. Initial Stages of Cathodic Breakdown of Thin Anodic Aluminium Oxide Films. Electrochim Acta, 1995, 40, 433-437.
- Gasco Owens, A.; Veys-Renaux, D.; Rocca, E. Reverse Scan Polarization of Anodic Aluminum Oxide until Detachment in Sulfuric Acid: Mechanisms and Morphologies. Electrochim Acta 2022, 435. [CrossRef]
- Zhang, J.; Klasky, M.; Letellier, B.C. The Aluminum Chemistry and Corrosion in Alkaline Solutions. Journal of Nuclear Materials, 2009, 384, 175–189.
- Thompson, G. E; Wood, G.C., The Effect of Alternating Voltage on Aluminum Electrodes. Corros Sci., 1978 18, 721-746. [CrossRef]
- Adhikari, S.; Hebert, K.R. Factors Controlling the Time Evolution of the Corrosion Potential of Aluminum in Alkaline Solutions. Corros Sci. 2008, 50, 1414–1421. [CrossRef]
- Ching-Feng, L.; Porter, M.D. Lin, Surface Films Produced by Cathodic Polarization of Aluminum. J. Electrochem. Soc. 1994, 141, 96-104.
- Hong, C.; Chu, L.; Lai, W.; Chiang, A.S.; Fang, W. Implementation of a New Capacitive Touch Sensor Using the Nanoporous Anodic Aluminum Oxide (Np-AAO) Structure. IEEE Sens J 2011, 11, 3409–3416. [CrossRef]
- Marsal, L.F.; Vojkuvka, L.; Formentin, P.; Pallarés, J.; Ferré-Borrull, J. Fabrication and Optical Characterization of Nanoporous Alumina Films Annealed at Different Temperatures. Opt Mater (Amst) 2009, 31, 860–864. [CrossRef]
- Sacchi, F.; Paolini, G. A Study of AC Anodizing of Aluminium in Sulphuric Acid Solutions. Transactions of the IMF 1964, 42, 298–311. [CrossRef]
- Segawa, H.; Okano, H.; Wada, K.; Inoue, S. Fabrication of Alumina Films with Laminated Structures by AC Anodization. Sci Technol Adv Mater 2014, 15. [CrossRef]
- Segawa, H.; Okano, H.; Wada, K.; Inoue, S.; Byun, I. Synthesis of Laminated Alumina Films by AC Oxidation. J Electrochem Soc 2013, 160, D240–D245. [CrossRef]
- Kape, J.M.; Further Development in the AC Anodizing of Aluminum in Sulfuric Acid Electrolytes. Transactions of the Institute of Metal Finishing, 1985, 63, 90-97.
- Balasubramanian, V.; John, S.; Shenoi, B.A. Influence of Addition Agents for A.C: Anodizing in Sulphuric Acid Electrolytes. Surface Technology, 1983, 19, 293-303. [CrossRef]
- Pyun, SI; Moon, SM. Corrosion Mechanism of Pure Aluminium in Aqueous Alkaline Solution. J. Solid State Electrochem. 2000, 4, 267-272.
- Tran, T.T.M.; Tribollet, B.; Sutter, E.M.M. New Insights into the Cathodic Dissolution of Aluminium Using Electrochemical Methods. Electrochim Acta 2016, 216, 58–67. [CrossRef]
- Broggi, R.L.; De Oliveira, G.M.; Barbosa, L.L.; Pallone, E.M.J.A.; Carlos, I.A. Study of an Alkaline Bath for Tin Deposition in the Presence of Sorbitol and Physical and Morphological Characterization of Tin Film. J Appl Electrochem 2006, 36, 403–409. [CrossRef]
- Kwaśniewski, D.; Grdeń, M. Electrochemical Behaviour of Tin in Alkaline Electrolyte. Electrochem Commun 2015, 61, 125–128. [CrossRef]
- Ammar, I.A.; Darwish, S.; Khalil, M.W.; Galal, A. Potentiodynamic and Cyclic Voltametric Studies on the Passivity of Tin in Neutral Phosphate Buffer. Materwiss Werksttech 1985, 16, 194–203. [CrossRef]
- Brunetti, V.; López Teijelo, M. Oxide/Hydroxide Films on Tin. II: Characterization of the Anodic Growth in Alkaline Solutions. J Electroanalytical Chemistry 2008, 613, 16–22. [CrossRef]
- Kapusta, S.D.; Hackerman, N. Anodic Passivation of Tin in Slightly Alkaline Solutions. Electrochim Acta 1979; 25, 1625-1639. [CrossRef]
- Tunold, R.; Broli, A. Anodic And Cathodic Behaviour of Tin In Acidic Sulphate Solutions. Corros Sci 1973, 13, 361-373. [CrossRef]
- Akyil, C.; Akdas, G.; Afsin, P.; Ürgen, M. Freestanding SnO2 Films Produced with Anodic Polarization in Acidic Media Containing Colloidal Tin Hydroxides. Mater Chem Phys 2019, 221. [CrossRef]
- Er, D.; Avcı, B.; Ürgen, M. Electrocatalytic Performance of Interconnected Self-Standing Tin Nanowire Network Produced by AAO Template Method for Electrochemical CO2 Reduction. ChemElectroChem 2023, 10. [CrossRef]
- Zaraska, L.; Gawlak, K.; Wiercigroch, E.; Malek, K.; Kozieł, M.; Andrzejczuk, M.; Marzec, M.M.; Jarosz, M.; Brzózka, A.; Sulka, G.D. The Effect of Anodizing Potential and Annealing Conditions on the Morphology, Composition and Photoelectrochemical Activity of Porous Anodic Tin Oxide Films. Electrochim Acta 2019, 319, 18–30. [CrossRef]
- Zaraska, L.; Gawlak, K.; Gurgul, M.; Dziurka, M.; Nowak, M.; Gilek, D.; Sulka, G.D. Influence of Anodizing Conditions on Generation of Internal Cracks in Anodic Porous Tin Oxide Films Grown in NaOH Electrolyte. Appl Surf Sci 2018, 439, 672–680. [CrossRef]
Figure 1.
FEG-SEM images of the back side of the free-standing AAO layer after sputtering.

Figure 2.
XPS spectra of freestanding AAO film e-colored in tin based electrolyte (a) Al 2p, (b) S2s, (c) O1s, (d) Sn 3d.
Figure 2.
XPS spectra of freestanding AAO film e-colored in tin based electrolyte (a) Al 2p, (b) S2s, (c) O1s, (d) Sn 3d.
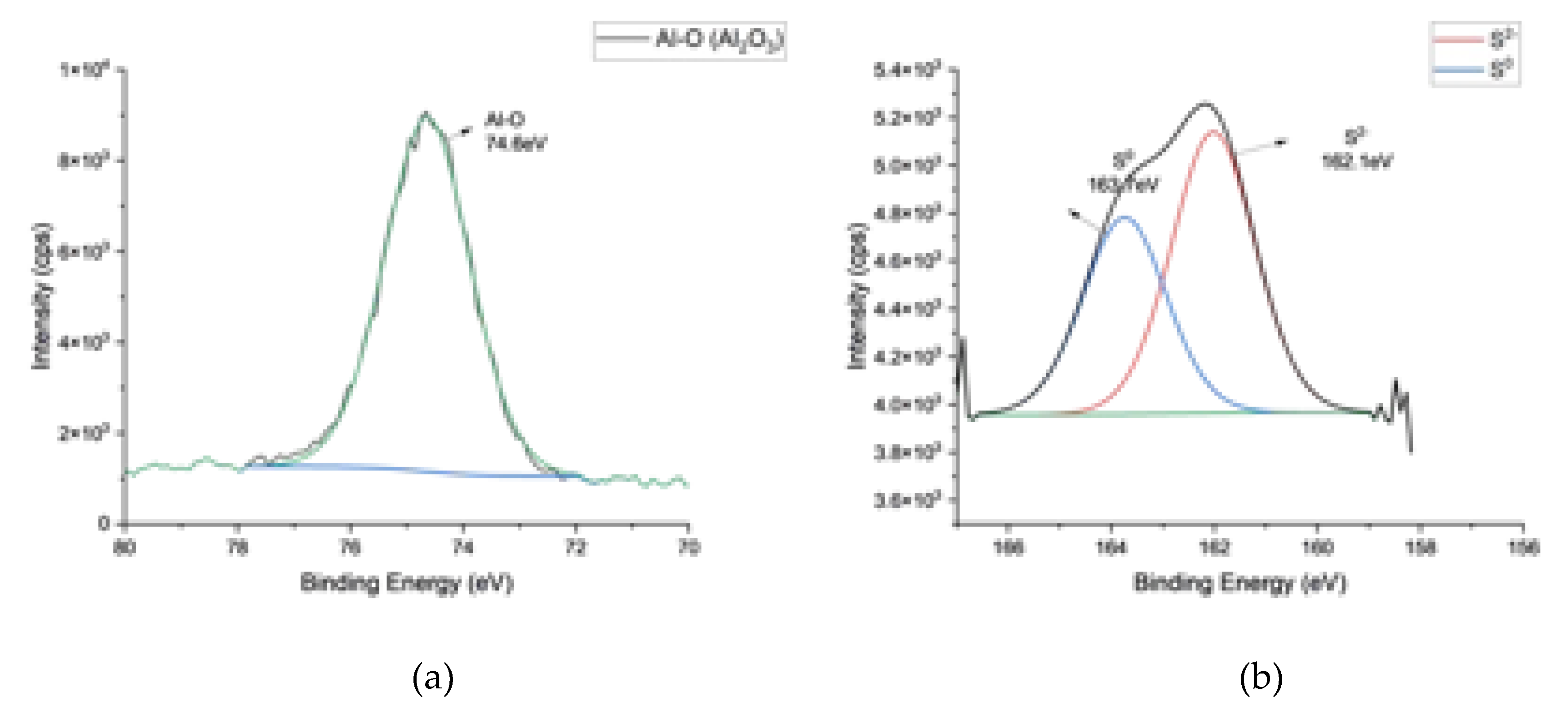
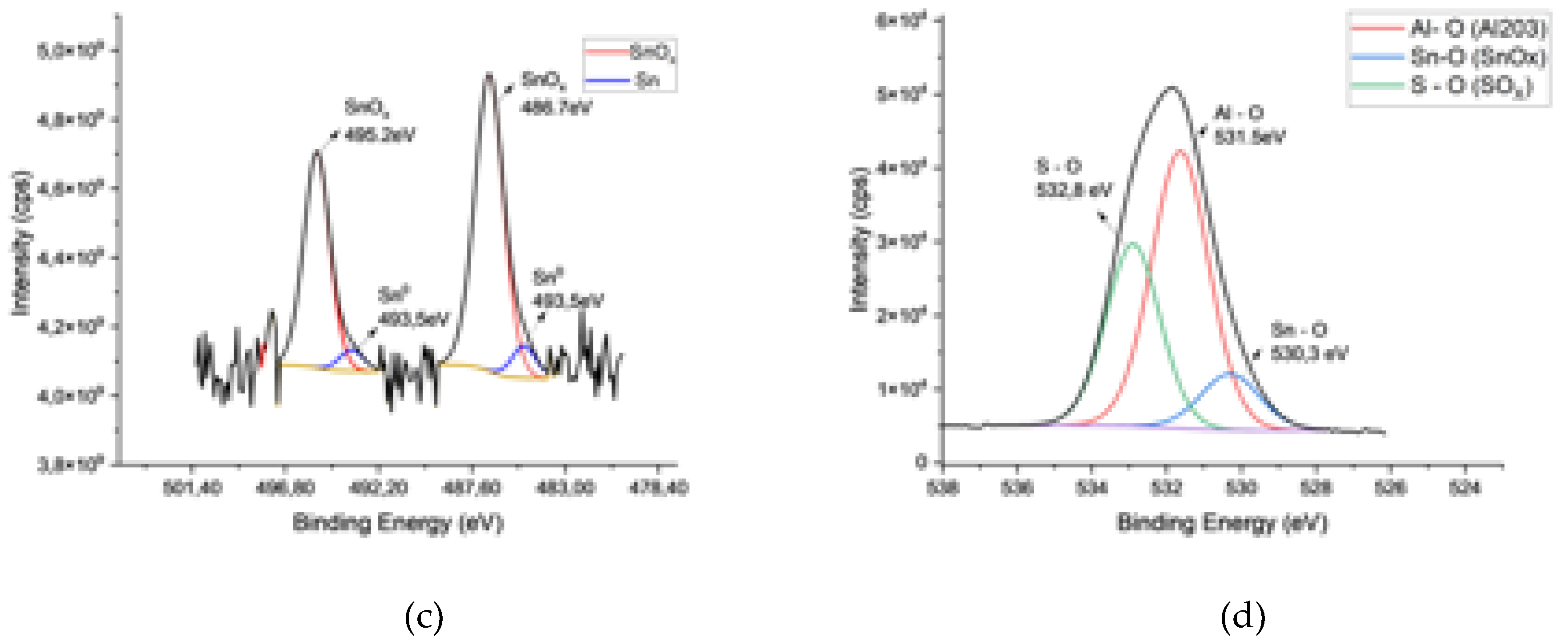
Figure 3.
Cross-section image of tin e-colored samples produced by using potential range +/-10V and 100V/s scan rate for 10 min (2500 cycles), (a) 20K magnification, (b)100K magnification.
Figure 3.
Cross-section image of tin e-colored samples produced by using potential range +/-10V and 100V/s scan rate for 10 min (2500 cycles), (a) 20K magnification, (b)100K magnification.

Figure 4.
Critical potentials recorded during CV in Tin free electrolyte.
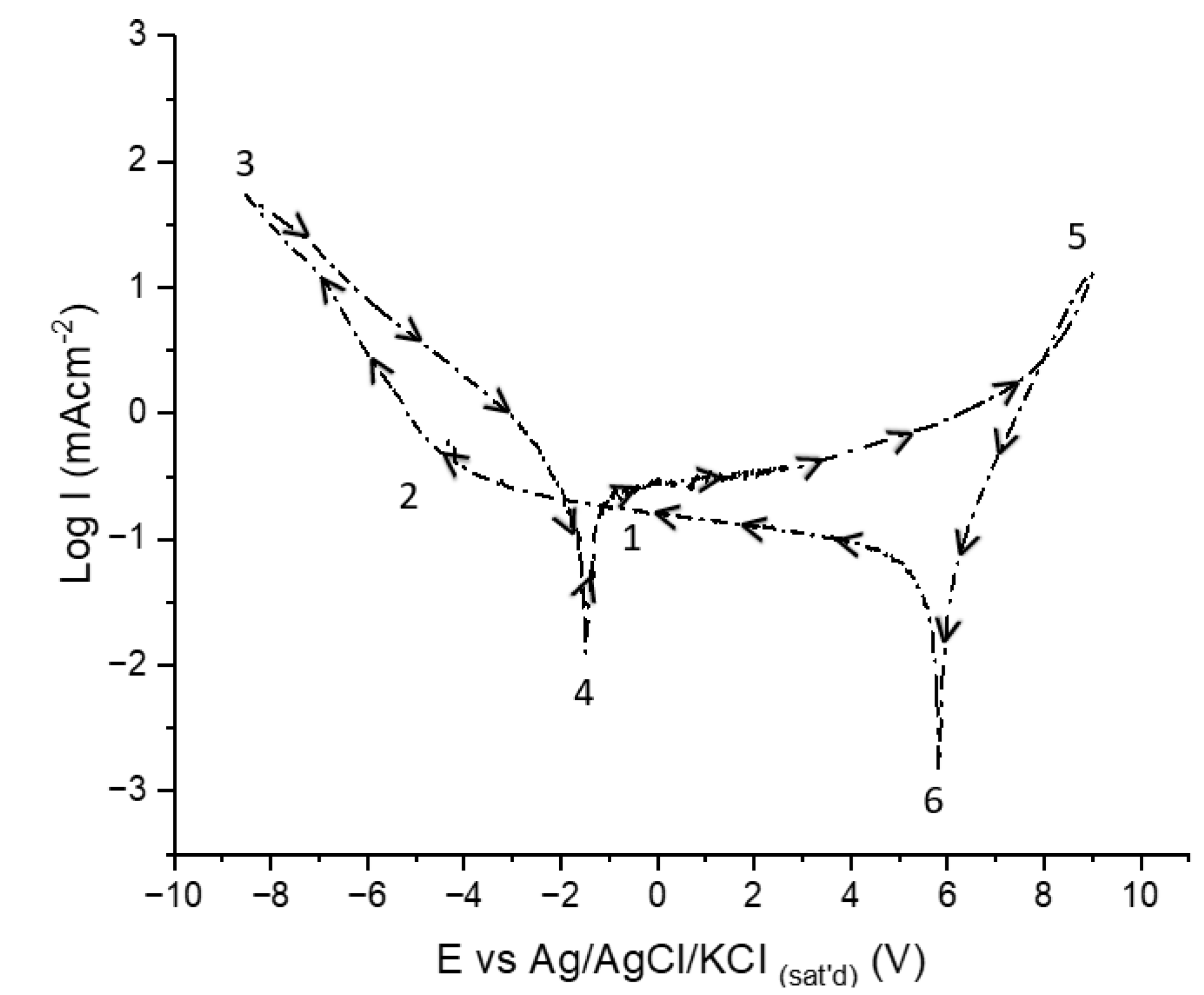
Figure 5.
Cyclic voltammograms obtained in the potential range of +/-10V and 100 V/s in tin Free solution; presented in (a) E-Logi and (b) E-i scales.
Figure 5.
Cyclic voltammograms obtained in the potential range of +/-10V and 100 V/s in tin Free solution; presented in (a) E-Logi and (b) E-i scales.
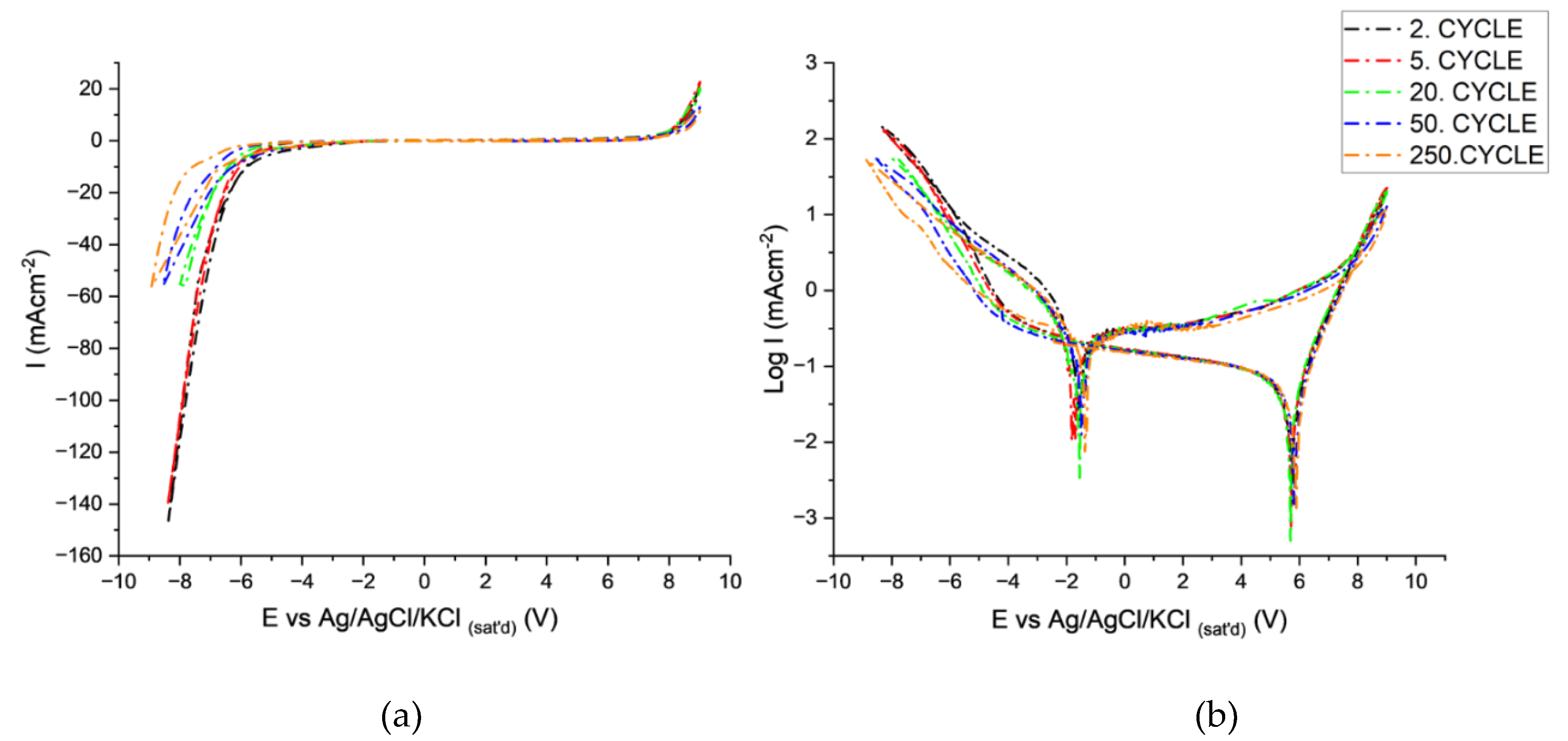
Figure 6.
SEM image of newly created AAO layer (a) after 1000 cycles , (b) after 2500 cycles in tin free electrolyte.
Figure 6.
SEM image of newly created AAO layer (a) after 1000 cycles , (b) after 2500 cycles in tin free electrolyte.

Figure 7.
Cyclic voltammograms obtained in the potential range of +/-10V and 100 V/s presented in (a) E-i and (b) E-logi scales in tin-containing solution.
Figure 7.
Cyclic voltammograms obtained in the potential range of +/-10V and 100 V/s presented in (a) E-i and (b) E-logi scales in tin-containing solution.
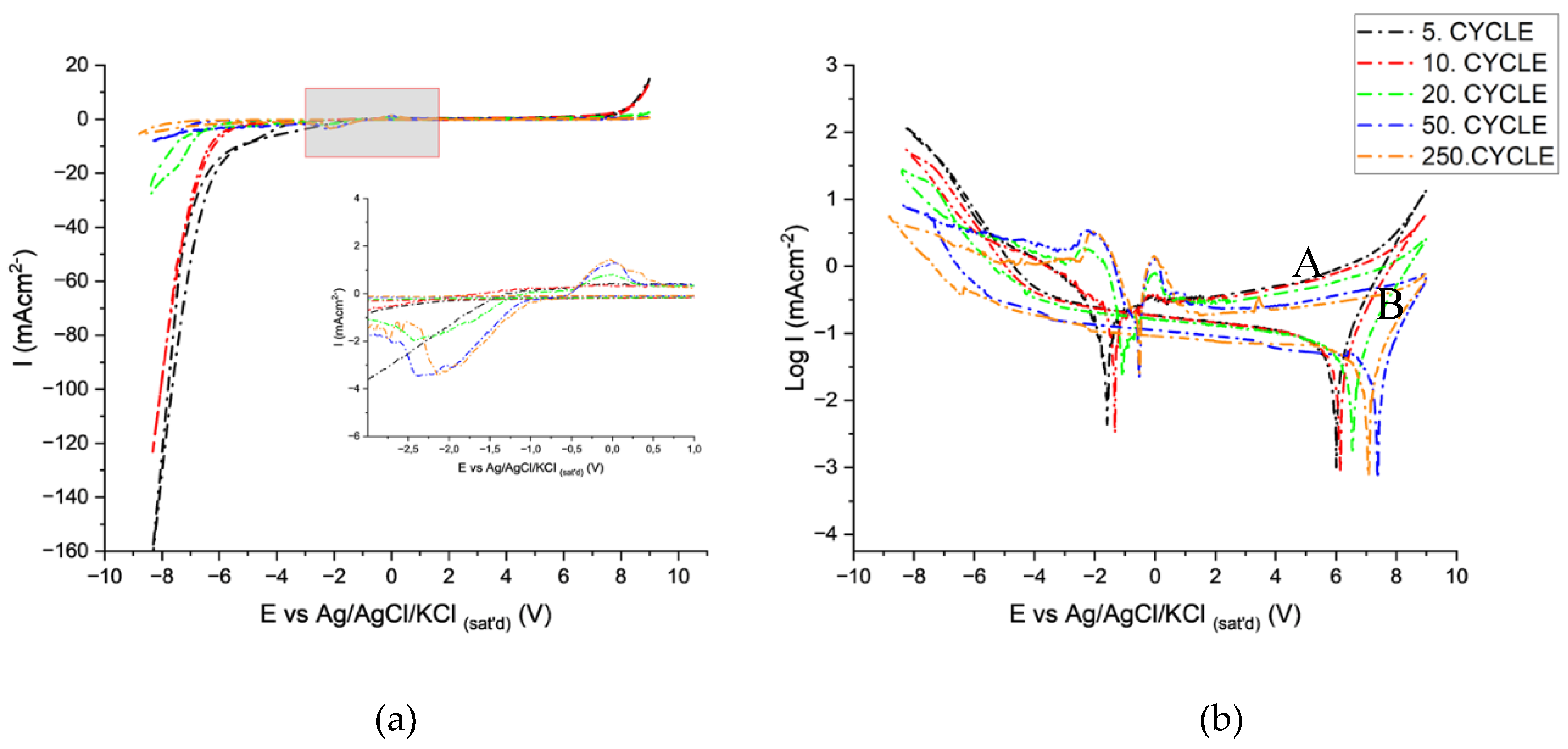
Figure 8.
SEM cross section images of AAO after CV experiments in tin based solution.
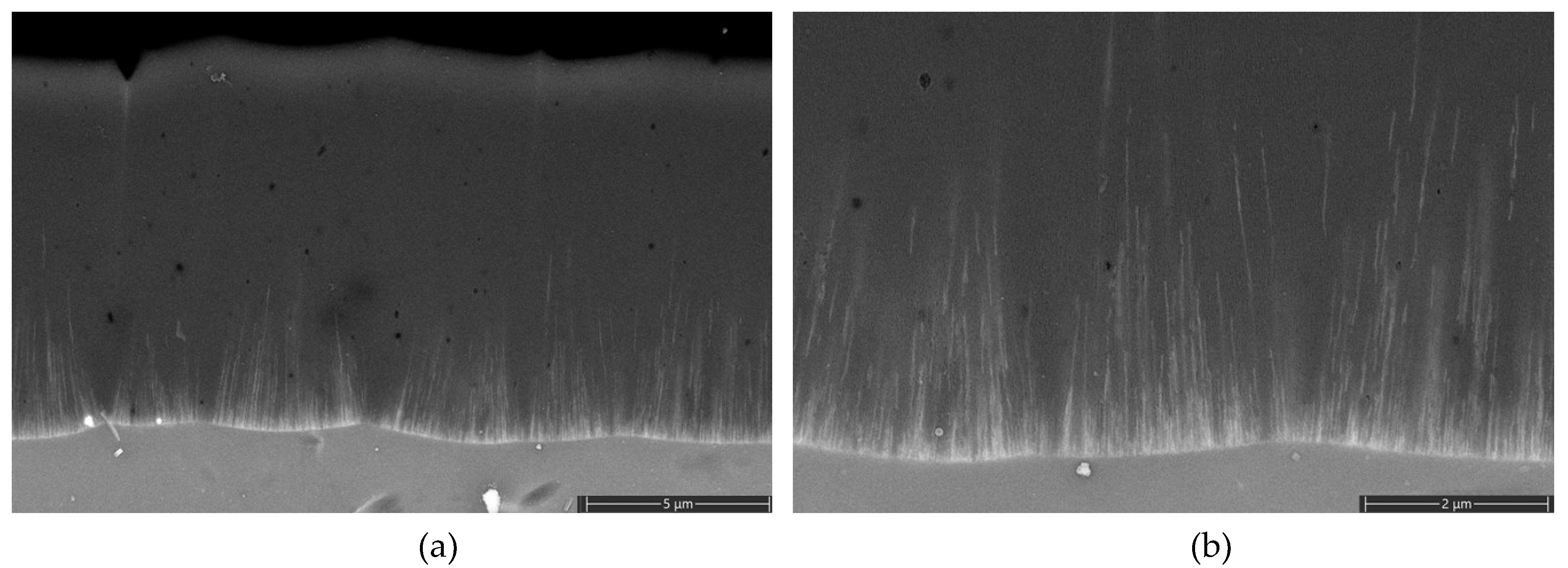

Table 1.
Composition of AA6063-T6 used in experiments.
| Alloying Element | Si | Fe | Cu | Mg | Cr | Zn | Mn | Al |
|---|---|---|---|---|---|---|---|---|
| % | 0.41 | 0.11 | 0.05 | 0.51 | <0.10 | <10 | <10 | Balance |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



