
Submitted:
16 April 2024
Posted:
16 April 2024
You are already at the latest version
Abstract
Considering the worldwide impact of heart failure, it is crucial to develop approaches that can help us comprehend its root cause and make accurate predictions about its outcome. This is essential for lowering the suffering and death rates connected with this widespread illness. Cardiomyopathies frequently result from genetic factors, and the study of heart failure genetics is advancing quickly. Dilated cardiomyopathy (DCM) is the most prevalent kind of cardiomyopathy, encompassing both hereditary and nongenetic abnormalities. It is distinguished by the enlargement of the left ventricle or both ventricles, accompanied by reduced contractility. The discovery of the molecular origins and subsequent awareness of the molecular mechanism is broadening our knowledge of DCM development. Additionally, it emphasizes the complicated nature of DCM and the necessity to formulate several different strategies to address the diverse underlying factors contributing to this disease. Genetic variations that can be transmitted from one generation to another can be a significant contributor to causing family or isolated DCM. Genetic variation also plays a significant role in determining susceptibility for acquired triggers for DCM. The genetic causes of DCM can have a diverse range of phenotypic expressions. It is crucial to select the most probable patients to have advantages from genetic testing. The purpose of this research is to emphasize the significance of identifying genetic DCM, the crucial relationships between genotype and phenotype, and the usefulness of genetic testing in the therapeutic care of both those affected and their relatives. This approach is expected to gain importance once treatment is guided by genotype-specific advice and disease-modifying medications.
Keywords:
dilated cardiomyopathy
; genetics
; heart failure
; genotype - specific therapy
1. Introduction
Dilated cardiomyopathy (DCM) affects more than 0.4% of the general population and is the second most frequent cause of heart failure, accounting for around 36% of all heart failure patients, second only to coronary artery disease. The 5-year survival rate following a heart failure diagnosis is just 50%, and mortality might result from arrhythmias or the gradual worsening of heart failure [1]. While there have been some decreases in the occurrence of heart failure due to a decrease in risk factors for cardiovascular disease and more effective therapy, the economic cost associated with HF remains substantial [2]. Furthermore, there is an increasing demand to diagnose and treat HF. Approximately 25-30% of individuals diagnosed with DCM are believed to have a hereditary factor that can be identified[3]. The 2023 European Society of Cardiology guidelines for the management of cardiomyopathies characterize DCM as the presence of left ventricular or biventricular systolic dysfunction and dilatation that cannot be attributed to inappropriate loading circumstances or ischemic heart disease[4].
The exact prevalence of genetically caused DCM is unknown. The prevalence of idiopathic DCM, which refers to cases where no identified acquired cause of DCM is present, is estimated to vary from 4‰ to 0.4‰. Additionally, around 25% of individuals with idiopathic DCM have a family history of the disease].These data are obtained from predominantly from European people [3].
Genetic factors that contribute to DCM are not simply caused by individual DNA variations with significant effects, as in a Mendelian model. Instead, it is probable that several variants with minor individual effects on disease risk together contribute to the observed inheritance. Even in the presence of a recognizable cause for DCM (such as alcohol abuse or pregnancy) one cannot exclude the possibility of a significant hereditary susceptibility to the condition, which might impact its treatment [5].
Nowadays, genetic testing has become readily accessible on a large scale. It is crucial to identify individuals who need genetic testing and recognize those who are at risk within the family as a whole. Genetic testing provides benefits not only to individuals and their families, but also contributes to the expansion of scientific understanding regarding genetics and its influence on heart illness [3].
The present research specifically examines cases of DCM that in adult patients. Our objective is to assist clinicians in comprehending the clinical significance of the genetic cause of DCM, identifying individuals and their families who could benefit from genetic evaluations, acquainting them with frequently conducted genetic tests, guiding them in understanding genetic findings, and informing them about the medical importance of these findings.
2. Defining Familial Dilated Cardiomyopathy (FDCM)
DCM can be defined by a left ventricular ejection fraction (LVEF) of less than 45%, together with a left ventricular end-diastolic size that is more than 112% of the anticipated value based on age and body surface area, as stated in the 2023 ESC Guidelines for the management of cardiomyopathies [4]. The term 'idiopathic' is used only after ruling out the numerous probable reasons, several of whom can be identified using non-invasive assessment and, more commonly, cardiac MRI, with a focus on the patient's clinical history [6].
Idiopathic or familial DCM (FDCM) is diagnosed when at least two members of the family fulfil the criteria for DCM, or when the patient corresponds the criteria for DCM and has a first-degree family member who has suffered sudden cardiac death before the age of 35 [7]. Nevertheless, these criteria are ambiguous and sometimes hard to precisely establish. FDCM is typically anticipated when individuals in the proband's family have a history of heart failure or premature unexpected cardiac death without a clearly identifiable reason. If an enlargement of the left ventricle (LV) is observed without a significant decrease in LVEF is found at the evaluation of close relatives, it is advisable to consider a diagnosis of FDCM.
3. Genetics
The area of genetic testing has made significant progress in recent decades. During these years, several variations in more than 100 potential disease genes have been documented. However, this wide-ranging list has been narrowed down to less than 20 genes that have strong evidence linking them to the etiology of FDCM, as determined by systematic analysis [3]. In general, the rate of success in genetic testing is still very modest, at around 20-30%. The grading method developed by the American College of Medical Genetics and Genomics (ACMG) has proved itself as the benchmark for variant interpretation and is currently widely utilized [8]. This approach examines many parameters, such as the frequency of the variant in the overall population, the expected effect on the protein it encodes, the number of occurrences in suffering people, and its prevalence within families. The ACMG classification categorizes variations into five groups: pathogenic (Class V), likely-pathogenic (Class IV), variant of undetermined significance (VUS), Class III, likely benign (Class II), and benign (Class I). Only genetic variations that are known to cause disease or are highly predicted to cause disease should be routinely utilized for screening and making therapeutic choices.
Recently, clinicians have developed techniques to assist them in determining whether genetic testing should be pursued in patients with suspected GDCM. The Madrid Genotype Score assesses the likelihood of obtaining a meaningful genetic test outcome based on five factors: existing family history of DCM, low voltage on ECG, evidence of skeletal myopathy, lack of hypertension, and lack of left bundle branch block. When four or more of these criteria were fulfilled, a pathogenic or potentially pathogenic variation was detected in 79% of the patients [9].
FDCM exhibits genetic heterogeneity, with a substantial number of cases manifesting as monogenic disorders, while a more complex genetic architecture is present in a number of cases. The majority of monogenic types of FDCM have an autosomal dominant pattern of inheritance. The genes linked to FDCM are involved in several biological processes, such as sarcomere components, cytoskeletal and desmosomal proteins, and mitochondrial proteins, among others, as shown in Figure 1. The genetic etiology of other cardiomyopathies, such as hypertrophic cardiomyopathy, is less diverse compared to this. This indicates that FDCM might be a common endpoint for multiple pathways. [5]
Abbreviations: DSP, desmoplakin; LMNA, lamin A/C; MYHN, myosin heavy chain; MYPN, myopalladin; RBM20, RNA binding protein 20; TNNT, cardiac muscle troponin T2; TPM1, α- tropomyosin 1;SCN5A, sodium channel protein type 5
Pathogen mutations can arise in genes that code for a diverse range of proteins found in the cardiomyocyte sarcomere, cytoskeleton, and nucleus. Autosomal recessive, mitochondrial, and X-linked inheritance have also been documented.[10]. Table 1 provides a list of important genes associated with inherited DCM.
The primary genetic cause of FDCM is represented by dividing mutations in the TTN gene, which encodes the large protein called titin. These mutations are present in 10-20% of instances. Titin has a significant role in the structure and function of the sarcomeres of the adult myocardium [11].
The second most prevalent reason for FDCM and possible the most severe one consists in alterations in the LMNA gene, which encrypts the proteins Lamins A and C of the nuclear envelope. These modifications result in a severe phenotype marked by gradual heart failure, severe irregular heart rhythms, and abnormalities in the electrical conduction of the heart. These alterations lead to a severe phenotype characterized by progressive HF, abnormalities in cardiac conduction and severe arrhythmias. [3].
Furthermore, there is a notable allelic heterogeneity, where several mutations within the same gene might result in a comparable phenotype. It is also important to mention that multiple variants of the same gene can induce different morphological characteristics [11].
Several genetic variations in DCM demonstrate partial and age-related penetrance, as well as heterogenous expressivity. Penetrance refers to the percentage of individuals with a certain genetic variant who exhibit the illness. Incomplete penetrance indicates that not all individuals having the genetic variant will have the disease. Penetrance in FDCM is generally age-dependent. This means that an individual inherits the genetic mutation responsible for DCM from birth, but the symptoms of DCM usually do not appear until middle age (over 40 years old) or may not appear at all. The level of penetrance for various FDCM variations is still unclear and is being actively researched. Heterogeneous expressivity means the presence of differences in both the severity and spectrum of clinical characteristics that are observed in individuals with a certain genetic condition. For instance, if we take into account two subjects from the same family who have the exact same genetic variation, one of them may experience severe DCM, having indication of heart transplantation, while the other one may only exhibit minimal symptoms. This suggests that there is a potential for other genetic, epigenetic, or external factors, to contribute to the observed characteristics [12,13].
It is increasingly apparent that several genes can be linked to certain characteristics, such as arrhythmic forms of FDCM, with a high risk of sudden cardiac death. Identifying these alarming phenotypes is crucial and should prompt immediate consideration of genetic testing, as shown in Table 2.
4. Genetic Variants Causing DCM
Laminopathies. Lamin A/C is a protein found in the nuclear envelope. It is encoded by the LMNA gene and serves as a structural component, similar to intermediate filaments. The development of LMNA-associated DCM is characterized by the disturbance of chromatin organization during cell division and the interruption of signal transduction in non-dividing cells [14]. LMNA variations typically lead to the development of both dilated cardiomyopathy (DCM) and conduction system illness. DCM usually manifests in individuals in their forties and fifties and often necessitates the use of a pacemaker in the majority of patients. In patients with LMNA variations, the risk of sudden cardiac death is significantly higher (up to 46%) compared to controls. Additionally, a significant number of these patients need to undergo pacemaker implantation [15]. The exact prevalence of LMNA mutations as a cause of DCM is uncertain, however it is generally estimated to be between 4% and 8%. LMNA variants were found in 30% of patients with familial DCM and conduction system disease [4].
Sarcomeric protein cardiomyopathies. Genetic variations in the genes that code for sarcomeric proteins, which play a role in generating contractile force, are responsible for many types of cardiomyopathies, ranging from hypertrophic remodeling to ventricular dilatation. It is usual to observe heterogeneity in the characteristics of these conditions. Some genetic variations in the genes that encode sarcomeric proteins may cause DCM by reducing the ability of the heart to respond to normal physiological stress [16, 17]. The primary focus in relation to DCM is the sarcomeric protein called titin, which is encoded by the TTN gene. Multiple TTN mutations leading to protein truncation have been identified [18]. TTN variants may account for around 25% of familial instances of DCM, which is inherited in an autosomal-dominant manner, and 12-18% of sporadic occurrences of DCM [19]. TTN variants often do not have a direct correlation with cardiac conduction abnormalities or skeletal muscle disorders, as stated by the 2023 ESC guidelines on cardiomyopathies [4]. The manifestation of titin cardiomyopathy often resembles that of idiopathic types of DCM, with symptoms appearing between the ages of 40 and 50. The prognosis is comparable, however, those with titin cardiomyopathy may have a more favourable response to medical treatment compared to those with idiopathic dilated cardiomyopathy [20]. Genes that encode proteins involved in the sarcomeric contractile apparatus, such as TNNT2, TPM1, MYH7, MYBPC3, MYH6, TNNI3, ACTC1, , MYL3, and MYL2 have been found to contain potential disease-causing variations. These genes are responsible for producing proteins like myosin 7, myosin 6, cardiac muscle troponin T, cardiac-type myosin-binding protein C, cardiac muscle troponin I, α-tropomyosin, myosin regulatory light chain 2, myosin light chain 3, and α-cardiac actin. The prevalence of these variants follows the order: MYH7, MYH6, TNNT2, MYBPC3, TNNI3, TPM1, MYL2, MYL3, and ACTC1 [4]. The percentage of familial DCM that is caused by genetic variations in genes that code for contractile proteins might be as high as 10%, and there is significant variation in the observable characteristics (phenotypic heterogeneity). Gene variants can also be found in proteins of the Z-disc, leading to comparable phenotypes of DCM [21].
DCM-causing mutations have been found in the genes ACTN2 (which encodes α-actinin 2), NKRD1 (which encodes ankyrin repeat domain-containing protein 1), CSRP3 (which encodes muscle LIM protein), LDB3 (which encodes LIM domain binding protein 3), MYOZ2 (which encodes myozenin 2), TCAP (which encodes telethonin), and VCL (which encodes vinculin) [22, 23, 24]. European individuals with DCM were discovered to have variants in the MYPN gene, which encodes myopalladin, a significant protein linked with the Z-disc in the heart. These variants were seen at a frequency of 3-4% [25].
Myofibrillar cardiomyopathy. In addition to the sarcomere unit, intermediate filaments play a crucial role in preserving the structural stability of the myofibril, enabling the production of force, and aiding in signaling processes. Desmin is the primary intermediate filament protein found in both cardiomyocytes and skeletal myocytes. It is encoded by the DES gene. DES variants are often inherited in an autosomal dominant manner and are characterized by conduction abnormalities that can lead to syncopal episodes or sudden cardiac death [26, 27]. Previously, it was believed that changes in FLNC, which is responsible for producing filamin C, a structural protein, may impact both skeletal and cardiac muscles. However, recent study has demonstrated that these changes specifically lead to a phenotype of DCM [28]. FLNC pathogenic mutations have a high degree of penetrance and are strongly linked to an elevated risk of sudden cardiac death. The guidelines indicate class IIa for cardioverter-defibrillator implantation in the presence of a pathogenic FLNC mutation and a left ventricular ejection fraction (LVEF) of less than 45% [29].
Myofibrillar cardiomyopathy can also be caused by genetic variations in BAG3, which is responsible for producing an intracellular chaperone protein. [30] is provided.
Desmosomal cardiomyopathy. Desmosomes are proteins that play a role in the organization and operation of intercalated discs. These discs are crucial for the transfer of electrical activity between cells, as well as for mechanical and signaling activities. Genetic variations in genes that encode desmosomal proteins are commonly linked to arrhythmogenic right ventricular cardiomyopathy. Nevertheless, it is possible for left ventricular dysfunction to occur in a pattern of DCM without any involvement of the right ventricle. As a result, the exact cause of arrhythmogenic right ventricular cardiomyopathy or DCM caused by variations in genes that encode desmosomal proteins is not fully understood. The most responsible mutations for DCM appear to be found in the desmosomal genes DSP (which encodes desmoplakin) and PKP2 (which encodes plakophilin 2). The presence of harmful mutations in these genes may not be adequately acknowledged. According to one report, 13% of patients undergoing cardiac transplantation for idiopathic DCM have pathogenic variants in genes that encode desmosomal proteins [31]. Other studies have found that DSP is the fourth most common gene to have DCM-causing variants in patients with isolated DCM [32].
Tafazzin- related cardiomyopathy. TAZ variants, namely the tafazzin proteins G4.5, that regulate cardiolipin can lead to Barth syndrome in neonates. Barth syndrome is characterised by features such as dilated cardiomyopathy (DCM), short stature, lactic acidosis, and neutropenia [33,34]. Nevertheless, other variations in TAZ might lead to a milder manifestation of DCM. DTNA variants, which encode dystrobrevin- α, may result in a phenotype of X-linked LV noncompaction cardiomyopathy [35] Captur.
Additional genetic variations linked to cardiomyopathies.
Various genetic variations have been linked to DCM, each with different disease-causing pathways. In 2009, it was discovered that variations in the RBM20 gene, which produces a protein called RNA-binding protein [36], are responsible for causing DCM. This protein plays a role in modifying mRNA after it has been transcribed in spliceosomes. It is relatively unusual for individuals to experience rapid advancement towards severe heart failure by the age of 40, and there is a substantial likelihood of ventricular arrhythmias and sudden cardiac death in patients with RBM20 mutations [37]. SCN5A gene variations, which affect the α-subunit of the sodium channel in the heart, are commonly linked to Brugada syndrome or long QT syndrome. However, these variations may also contribute to an increased risk of heart failure. Individuals with SCN5A mutations may have conduction abnormalities and arrhythmias, which are characteristic of the DCM phenotype [38]. The development of cardiomyopathy linked with SCN5A variations occurs at a young age, often in the second to third decade of life [39].
Important syndromic DCMs
Mitochondrial myopathies. Mitochondrial abnormalities are linked to a wide variety of cardiomyopathies. Mitochondrial myopathies have two modes of heredity: maternal transmission, which occurs due to the segregation of mitochondria during cell division, and autosomal dominant inheritance, which is caused by certain nuclear proteins that interact with mitochondria and can lead to illness. Genetic testing for mitochondrial myopathies involves suppressing the activity of mitochondrial DNA to identify specific harmful variations. Unlike nuclear genes, the manifestation of variations in mitochondrial genes is directly related to the quantity of these variation [40].
Although there is a range of ways in which it might be presented, the more often occurring mitochondrial myopathy syndromes can exhibit distinct cardiac characteristics.
In Kearns-Sayre syndrome, a condition that often appears during adolescence and is characterized by increasing weakness of the eye muscles and degeneration of the retina, the majority of patients also experience cardiac conduction anomalies, including heart block [41].
In individuals with MELAS (mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes) syndrome, the development of heart failure is a notable feature. However, this is typically accompanied by significant and symmetrical left ventricular hypertrophy, which can manifest throughout infancy or maturity [42].
Lysosomal storage disorders. Lysosomal storage disorders are a group of diseases that cause the build-up of certain substances within cells. These disorders affect the heart as part of their development. The illness processes vary significantly in terms of their appearance, symptoms, and severity. [43,44].
Neuromuscular syndromes. Duchenne muscular dystrophy is the quintessential example of a neuromuscular illness that affects the heart. Genetic mutations in the DMD gene, which codes for the protein dystrophin, are connected to a pattern of inheritance that follows an X-linked recessive pattern. Similar to desmin, dystrophin establishes a connection between the sarcomeric proteins and the plasma membrane through the dystroglycan complex. Duchenne muscular dystrophy is characterized by the loss of cell membrane integrity and subsequent cell death and fibrofatty replacement due to muscle tension. DCM often appears during the second decade of life and consistently presents with a well-documented history of skeletal myopathy [45]. Since cardiac involvement is unavoidable if mortality from other causes is prevented, it is crucial to test for cardiomyopathy annually in adult patients [45].
Becker muscular dystrophy may have a pattern like that of Duchenne muscular dystrophy, however not all cases affect the heart and, when it does, the development of DCM tends to occur at a later age [45].
X-linked DCM), which is connected to the DMD gene, is a more serious kind of cardiomyopathy. Unlike other forms, it does not affect skeletal muscle and is characterized by early start (in the second decade of life) and fast development [46].
LMNA variants are linked to certain skeletal muscle illnesses that are inherited in an autosomal dominant manner.
Emery-Dreifuss muscular dystrophy and limb girdle muscular dystrophy type 1B both involve the progressive degeneration of skeletal muscle [47, 48]. However, the cardiac symptoms, such as DCM, atrioventricular block, and ventricular arrhythmias, are similar to those seen in diseases caused by LMNA variants, where the cardiac symptoms are more prominent [49, 50]. The phenotypic cardiac symptoms observed in Emery-Dreifuss muscular dystrophy and limb girdle muscular dystrophy type 1B are influenced by the specific position of the mutation within the LMNA gene [51, 52]. Emery-Dreifuss muscular dystrophy can also be connected to an X-linked inheritance pattern due to variations in the EMD gene, which codes for emerin, a protein found in the nuclear lamina [53].
5. Clinical Picture
The majority of patients diagnosed with DCM are adults, with the age of onset usually occurring in the third or fourth decade of life. Heart failure and palpitations frequently occur as symptoms [54]. The first symptoms might be confusing and often do not provide enough information to distinguish it from other causes of DCM. The manifestations of illness vary from those who show no symptoms and are found during screening, to individuals who have severe heart failure or life-threatening arrhythmia and unexpected cardiac death.
Indicators that raise suspicion of an underlying genetic cause include a family history of DCM, unexplained cardiac arrest or sudden cardiac death, as well as the occurrence of cardiomyopathy or arrhythmia at a young age (under 35 years). Other conditions that may be associated are peripartum cardiomyopathy, unexplained syncope, muscle disorders, and cardiomyopathy with concurrent arrhythmia [55].
DCM as the main phenotypic characteristic occurs in a significant number of patients. Subjects might experience symptoms related to heart failure, such as tiredness, breathing difficulties, orthopnea, and ankle swelling. Family members having a harmful mutation in their genes may present no symptoms or only experience mild DCM that is identified by family screening. Cardiac ventricular and atrial arrhythmias, as well as conduction disorders may develop, especially when the left ventricular ejection fraction decreases [56].
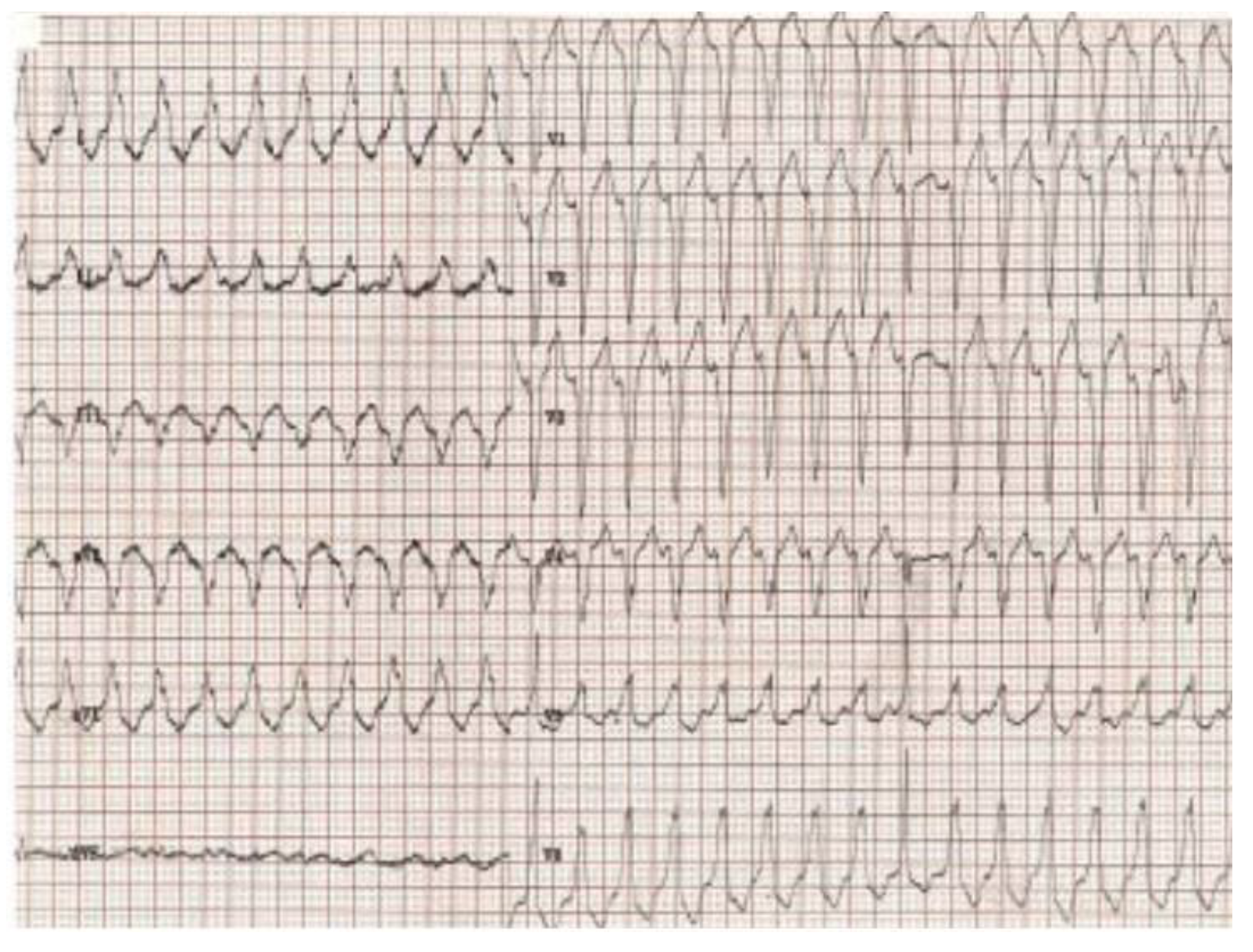
Some individuals with FDCM have early or obvious abnormalities in their cardiac conduction system. When a younger individual experiences conduction disorders, it should raise the possibility of hereditary cardiomyopathy. Electrocardiogram findings may include bradycardia, atrioventricular blocks, axis deviation, or bundle branch blocks. Figure 2 presents the ECG of an adult patient with familial dilated and arrhythmogenic right ventricular cardiomyopathy at sinus rhythm (A), with ventricular late potentials (B) and during a sustained ventricular tachycardia (C).
Figure 2A.
12-leads electrocardiogram. Sinus rhythm with recurring premature ventricular beats originating from the right ventricle. T wave inversions and S waves in all precordial leads. Potential epsilon waves in leads DII, DIII, and aVF.
Figure 2A.
12-leads electrocardiogram. Sinus rhythm with recurring premature ventricular beats originating from the right ventricle. T wave inversions and S waves in all precordial leads. Potential epsilon waves in leads DII, DIII, and aVF.
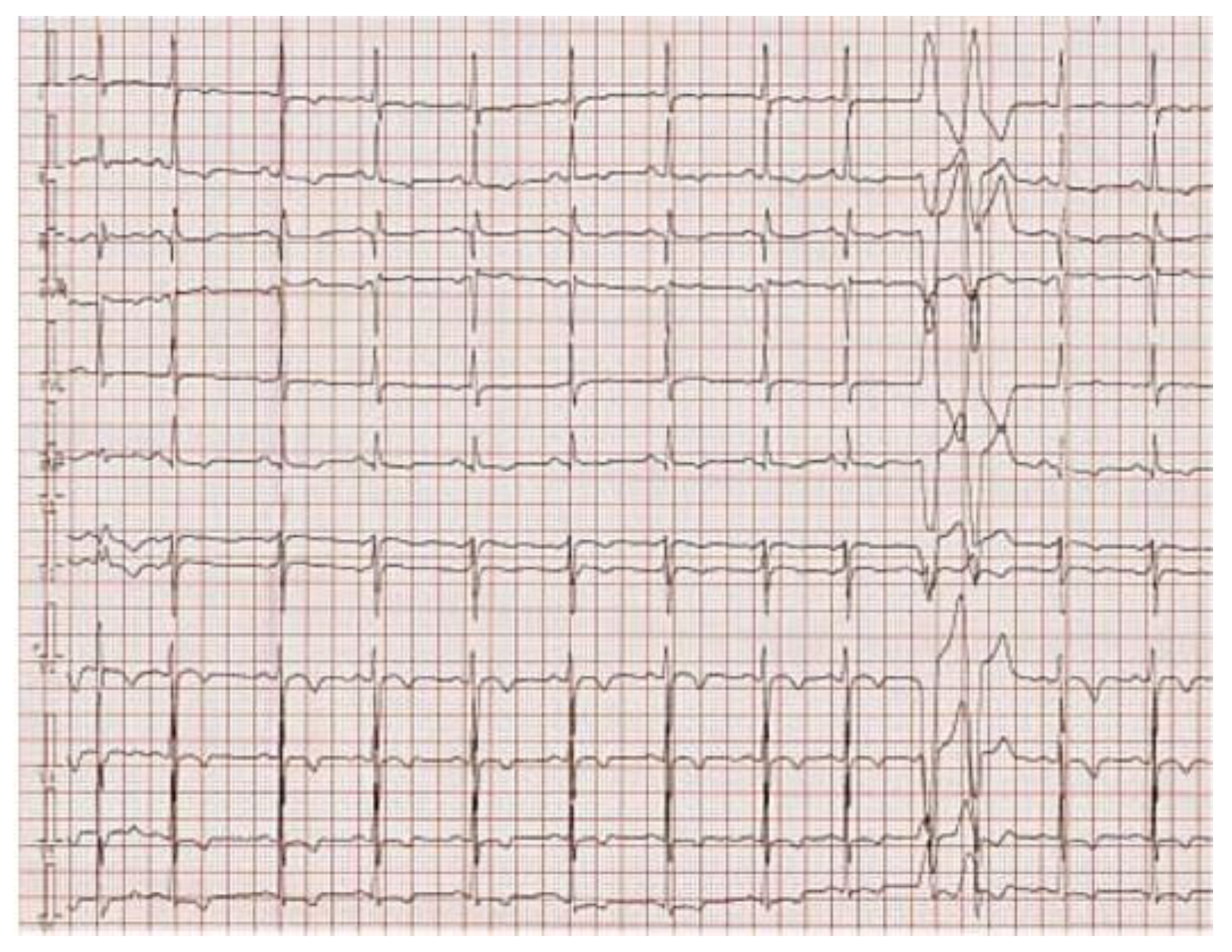
Figure 2B.
Ventricular late potentials.
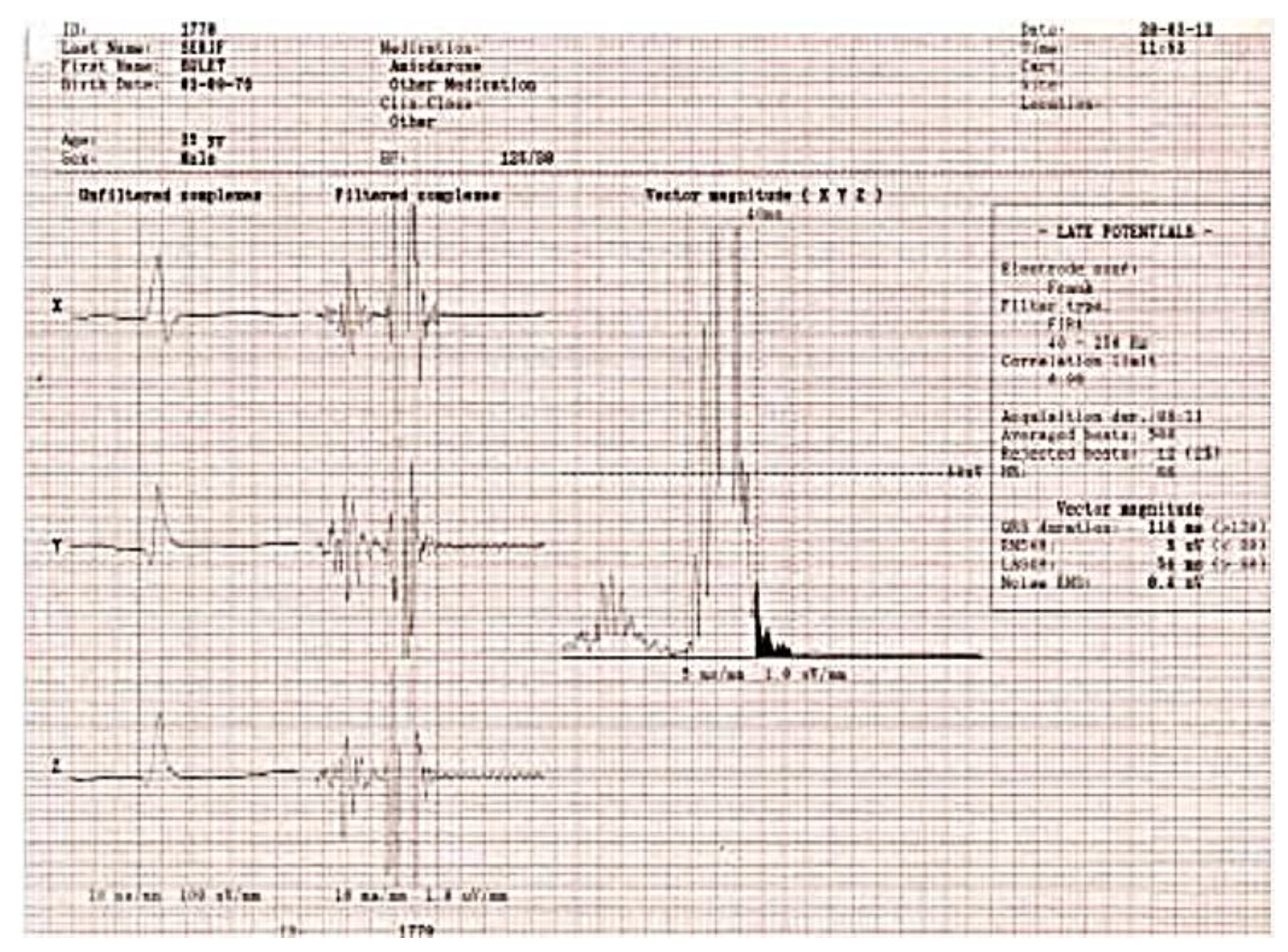
Figure 2C.
Sustained ventricular tachycardia originating in the right ventricle, with an appearance of left bundle branch block.
Figure 2C.
Sustained ventricular tachycardia originating in the right ventricle, with an appearance of left bundle branch block.
The genes LMNA and DES have a specific predilection towards early conduction disorder that may precede by decades the development of DCM. These disorders may also be linked to atrial and ventricular arrhythmias as well as skeletal muscle diseases [56].
Arrhythmogenic FDCM might present with sudden cardiac death as the initial symptom. The occurrence and intensity of cardiac arrhythmias may not be directly related to the amount of LVEF decrease. The significant similarity in genetics and physical features between arrhythmogenic DCM and arrhythmogenic right ventricular cardiomyopathy (ARVC) with left ventricular involvement has caused a lot of difficulty in diagnosis. As a result, a novel category called 'arrhythmogenic cardiomyopathy' has emerged. The genes linked to arrhythmogenic cardiomyopathy are LMNA, DSP, SCN5A, RBM20, FLNC, DES, and PLN [57].
Although once considered a rare non-genetic condition, current research indicates that up to 15% of individuals with peripartum cardiomyopathy carry harmful genetic mutations in genes related with DCM, like TTN. For women who have a genetic predisposition, pregnancy is believed to act as a trigger by causing higher stress on the circulatory system. This can lead to the manifestation of the disease at an earlier stage than it would typically occur [58, 59]. Women with a positive genotype may have a lower likelihood of experiencing a reversal of DCM after pregnancy, compared to those who have a negative genotype.
FDCM may be linked to other symptoms of a multisystem disease, such as sensori-neural deafness, skeletal myopathy, neuropathy, partial lipodystrophy, neuropathy, and other disorders. A significant number of individuals diagnosed with inherited skeletal myopathies, such as Duchenne or Becker, will experience the development of DCM as they get older. DCM can also emerge as part the clinical picture in systemic illnesses such storage or mitochondrial abnormalities. While certain genetic variations may exhibit foreseeable trends, there can be significant variability in the manifestation of diseases, the age at which they appear, and their severity among individuals who possess variants in the same gene. This variability can occur both within families and between different families. The penetrance of DCM can be affected by environmental variables, genes modulators, associated medical conditions, and lifestyle choices [60]. These are currently being studied. It is currently being recognized that genetic diversity may play a role in determining one's vulnerability to acquired types of DCM, for instance alcohol or anthracycline-induced DCM. This confuses the boundary between hereditary and acquired diseases [8,61].
6. Evaluation of the Initial Patient with Recently Diagnosed FDCM
Generally, the causes of DCM can be categorized as ischemic, toxic, metabolic, infiltrative, inflammatory, and infectious. Among them, ischemic cardiomyopathy is the most prevalent cause. A comprehensive evaluation is particularly necessary for all patients who have recently been diagnosed with DCM, including those with peripartum cardiomyopathy. This evaluation is crucial to ensure that any potentially curable or reversible causes are not overlooked. Nevertheless, the presence of a visible immediate factor such as pregnancy or chemotherapy does not rule out the possibility of a hereditary element in cardiomyopathy. Women who have a genetic predisposition to peripartum cardiomyopathy may experience a faster beginning of the disease and show symptoms at a younger age compared to other members of their family. As a result, the family's medical history at the time of diagnosis may show no signs of illness. Regularly reviewing the family history is crucial, since it may evolve to become positive over time. Genetic testing may be necessary in this situation or in women who do not exhibit improvement in heart function during the post-partum interval [3].
The diagnosis for the patient with DCM is mostly established by the analysis of clinical and imaging data. Genetic testing can be helpful in confirming the diagnosis in a small subset of individuals who have ambiguous diagnostic results. For instance, when considering a dual pathology like sarcoid disease, the existence of an alternative genetic diagnosis might help determine whether to stop further intensive and less productive diagnostic tests, such as endomyocardial biopsy for isolated cardiac sarcoid [62].
FDCM is recognized clinically when it develops in two or more closely related individuals, after ruling out other potential causes. A diagnosis of FDCM does not rely on positive genetic testing, because this typically fails to provide useful information. An exhaustive family history should encompass a three-generation genealogy [63] that investigates any instances of heart failure, arrhythmia, unexplained mortality, heart failure, or other cardiac symptoms, especially if they arose at an age younger than expected. An awareness of other symptoms, such as muscular disorders, is also necessary.
The symptoms of the family member do not necessarily have to match those of the reference patient. There are several difficulties in establishing a satisfactory heritage. The variety of illness onset and penetrance of the genetic anomaly, as well as the long period of time it takes for the disease to develop, might complicate the screening process for those who are willing to undergo it. Certain family members manifest hesitation or incapacity to undergo evaluation, while in other cases, families are limited in size and possess not enough data. Although there are difficulties, it is crucial to create a pedigree as it aids in determining the likelihood of a genetic test being positive.
It is worth mentioning that around 25% of individuals with sporadic DCM are later identified as having familial DCM after a thorough evaluation of their medical history, physical examination, electrocardiogram, and transthoracic echocardiogram of both the affected individual and their family members [64]. Therefore, it is advisable to advise genetic testing for individuals who have a positive family history or who develop apparent sporadic idiopathic DCM. Genetic testing can provide valuable information that can influence treatment decisions, predict prognosis, and enable screening of other family members.
It is advisable to carry out clinical screening for all unaffected first-degree relatives of any person who has a genetic anomaly linked to proven or suspected FDCM [56]. The screening of family members not only helps diagnose the primary case, but also often leads to the detection of the disease in persons who show no symptoms, who might benefit from clinical examination. Genealogy assessment also aids in the in the identification of family members exhibiting pertinent phenotypic characteristics, such as isolated arrhythmias or conduction anomalies, and it allows for earlier treatment and perhaps better outcomes [58]. Additionally, it can aid in the process of family planning.
Non-invasive imaging methods are essential for diagnosing and monitoring patients with cardiomyopathies. These methods include ultrasound-based techniques, cardiac magnetic resonance (CMR) imaging, computed tomography (CT), and nuclear techniques including positron emission tomography (PET) and scintigraphy [4].
Regular screening, including clinical evaluation, electrocardiogram (ECG), and transthoracic echocardiography (TTE), should begin in adulthood or earlier if there are treatment implications [63]. The frequency of screening can range from every six months to every five years, depending on the existence or not of high-risk indicators and the usual age at which the illness typically appears in the family [60, 63]. LV dilatation on TTE in asymptomatic relatives might indicate subclinical illness, incidental illness, or physiological modifications. It is difficult to differentiate between these conditions, and since there is no dependable method to identify those who may develop obvious illness, regular surveillance is required. Currently, the screening criterion in TTE continues to be the LV size and ejection fraction. However, continuing research is being conducted to explore alternative preclinical indicators. Recently, researchers have assessed global longitudinal strain of the LV and observed a decrease in patients who are genotype-positive but phenotype-negative, compared to genotype-negative controls. Yet, the exact significance of global longitudinal strain in detecting or monitoring subclinical FDCM has not been determined [5]. Myocardial deformation imaging, specifically using speckle tracking or tissue Doppler, is a more sensitive indicator than EF (ejection fraction) for detecting subtle ventricular dysfunction. This is particularly useful in cases such as genotype-positive HCM (hypertrophic cardiomyopathy), DCM, and ARVC (arrhythmogenic right ventricular cardiomyopathy) in family members. Additionally, it can assist in distinguishing between different causes of hypertrophy, such as amyloidosis, HCM, and athlete's heart. Mechanical dispersion serves as an indicator of unevenness in contraction and draws attention to subtle structural alterations that would go unnoticed by alternative methods [64-67]. Figure 3 exemplifies a patient with FDCM examined by two-dimensional (2D) transthoracic conventional echocardiography and by 2D- speckle-tracking echography, showing dilated heart chambers, severe LV dysfunction (LVEF of 30%) and a Bull's eye myocardial deformation pattern with overall segmental low peak longitudinal strain values.
Three-dimensional echocardiography accurately measures the volumes of heart chambers, but it requires a sufficient acoustic window. Contrast agents may be used to enhance the visibility of endocardium in order to show the existence of excessive muscle trabeculation, a condition known as hyper-trabeculation. Additionally, contrast agents can help rule out the presence of intracardiac thrombus [4].
CMR imaging has become the most reliable method for evaluating the size and function of the ventricles. It should be performed in all patients who have recently been diagnosed with cardiomyopathy. CMR can offer comprehensive anatomical and physiological data that surpasses that of TTE. Additionally, CMR can provide insights into cardiac tissue features, including inflammation and fibrosis, through the use of late gadolinium enhancement (LGE). CMR has the benefits of being non-invasive and not requiring an acoustic window, while also allowing for the analysis of tissue characteristics. The latter benefit is especially crucial in diagnosing non-compaction LV cardiomyopathy (Figure 4) , ARVC, myocarditis, amyloidosis, sarcoidosis, and other types of inflammatory diseases, as well as haemochromatosis. Standard initial examination should regularly include cine imaging sequences, T2-weighted sequences, pre- and post-contrast T1 mapping, and LGE. These findings should be evaluated alongside genetic results and other clinical features by experienced professionals in cardiac imaging and heart muscle disease assessment.
Regular follow-up CMR should be conducted every 2-5 years, depending on the initial severity and clinical course of the disease. This can help assess the progression of the disease and the effectiveness of therapy [4]. The existence of LGE is linked to the possibility for arrhythmias and more severe illness. Diseases connected to DSP and FLNC frequently exhibit a fibrosis pattern resembling a ring around the outer layer of the heart, known as subepicardial fibrosis. This observation may suggest the need for considering the installation of an implantable cardioverter-defibrillator (ICD). CMR has the capability to identify illness in persons who do not show any symptoms and may also be used to track the progression of the disease [68].
The existence of LGE is linked to the possibility for arrhythmias and more severe illness. Diseases connected to DSP and FLNC frequently exhibit a fibrosis pattern resembling a ring around the outer layer of the heart, known as subepicardial fibrosis, as presented in Figure 5. This observation may suggest the need for considering the installation of an implantable cardioverter-defibrillator (ICD). CMR has the capability to identify illness in persons who do not show any symptoms and may also be used to track the progression of the disease [68].
Computed tomography-based imaging is mostly employed in patients suspected of having cardiomyopathy to exclude coronary artery disease (CAD), either as an alternative diagnosis or as a comorbidity that impacts clinical symptoms and progression.
Additional functional testing and imaging may be necessary on a case-by-case basis during the evaluation process. Endomyocardial biopsy is a topic of debate and is not currently included in the usual diagnostic process for most individuals with DCM [60].
Laboratory assessment. Currently, there are no established disease-specific biomarkers for DCM. However, researchers are actively investigating this area. Despite the lack of acute myocardial ischemia, high-sensitivity blood troponins frequently show aberrant levels in both acute and chronic heart failure. While troponin testing may offer some risk assessment in cases of cardiomyopathy, it has a little role in the evaluation of suspected DCM.
Consistently high levels of serum creatinine kinase (CK) can indicate the presence of myopathies or neuromuscular illnesses such as dystrophinopathies (such as Becker muscular dystrophy or X-linked DCM), laminopathies, desminopathies, or, less commonly, a myofibrillar myopathy [68]. Patients with arrhythmogenic right ventricular cardiomyopathy (ARVC) and non-dilated left ventricular cardiomyopathy (NDLVC) may exhibit elevated levels of C-reactive protein, especially during bouts resembling myocarditis [69]. Increased concentrations of iron and ferritin in the blood, together with a high transferrin saturation, may indicate a potential diagnosis of haemochromatosis. This should prompt additional investigation into the underlying cause (primary or secondary) by genetic testing. Lactic acidosis, myoglobinuria, and leucocytopenia may indicate the presence of mitochondrial disorders [4].
Ordering genetic testing has become fairly simple, but interpreting the results can be complicated. It is recommended to have this done in a specialized clinic with multidisciplinary input, given the challenges in interpreting genetic variants in the absence of a confirmed phenotypic diagnosis.
Conducting tests on the reference patient, who is the most probable person affected, provides the greatest likelihood of identifying a harmful genetic mutation. Genetic testing is commonly performed using commercially available next-generation sequencing arrays. Choosing the appropriate array for a certain patient is crucial due to subtle variations. Moreover, as new data arises, variations may be reassessed and either promoted or lowered in their categorization. This has resulting implications for management and might potentially lead to the need for re-screening of family members. Genetic testing is valuable for both the proband and their family members since it provides crucial information about the likelihood of DCM recurring or being transmitted. Identifying a disease-causing mutation in a potential parent can also be utilized to provide information for prenatal genetics, specifically pre-implantation genetic diagnosis. This process involves in vitro fertilization with the selection or modification of embryos [70]. This is a highly invasive procedure that involves many stages, including oocyte retrieval, in vitro fertilization, genetic testing of embryos, and implantation of embryos that do not have harmful variants. It is important for all patients who have a strong diagnosis with a clearly identified disease-causing or potentially disease-causing genetic mutation to be informed about this possibility. Chorionic villus sampling and amniocentesis are alternative methods used for prenatal genetic diagnosis. Given the multitude of choices available, it is imperative that patients get comprehensive pre-test genetic counselling to guarantee they possess the necessary knowledge and assistance to make a well-informed choice. This encompasses the ability to comprehend the possible consequences of test results, examine the decision-making process in reaction to the results, and facilitate communication with other family who may be at risk [11].
7. Evaluation of the Relatives of the Initial Patient with Recently Diagnosed FDCM
A systematic methodology for the genetic assessment of FDCM is presented in Figure 6.
Genetic testing on the relatives of the proband. It is advisable to do cascade genetic testing on relatives of the patient of interest if a disease-causing variant has been identified [54 Hershberg]. Individuals who are negative for an illness but have a detected harmful or possibly harmful genetic mutation should have regular and frequent screenings. Unaffected carriers should receive counselling on the symptoms and signs of early-stage DCM. They should also undergo more frequent clinical surveillance, with check-ups scheduled every 1-3 years as recommended by the European Society of Cardiology guidelines [4] or every 3–5years according to the American Heart Association guidelines [56].
Family members who do not carry the variation might be given reassurance and released from further screening [54].
Families without an identified pathogenic variation, however still regarded susceptible to the disease, are recommended to take part in clinical screening. Typically, it is advised to undergo this procedure every two years, however the frequency may be modified according on factors such as age and risk level [13]. In addition, advancements in genetic knowledge might require families to undergo genetic re-evaluation in order to identify harmful variations.
Genetic testing holds significant potential to enhance precision medicine by providing valuable information for risk assessment and tailored treatment approaches. Currently, in DCM, there are only a limited number of genes that have been found to have pathogenic variations that could possibly impact the management of the condition. One particularly noteworthy gene is LMNA (lamin), which has been found to have variations that are strongly linked to the development of conduction disorders, atrial and ventricular arrhythmias, and sudden cardiac death. This might potentially reduce the minimum need for implanting a cardioverter-defibrillator (ICD) for primary prevention, and promote more frequent monitoring of heart rhythm [11].
Advanced remote monitoring devices in the future may be capable of using machine learning analysis to identify or exclude a DCM phenotype at an early stage. These devices would analyse both clinical and electrocardiographic features. They could be used by general practitioners or even by family members themselves. The adoption of such devices might potentially decrease the need for frequent cardiological surveillance [11].
Types of genetic testing
Genetic variant testing for candidates. Performing genetic testing for specific variants in an asymptomatic individual is advised when their first-degree relative has DCM that is linked to a gene variant known to cause disease or likely to cause disease. This type of testing is called predictive testing because it aims to predict the likelihood of developing symptoms or structural abnormalities in the individual. When certain clinical symptoms are present, such as conduction tissue disease, advanced heart block in the context of DCM, or the coexistence of skeletal muscle involvement indicated by an asymptomatic increase in creatine kinase level or a noticeable neuromuscular disorder, genetic testing is likely to provide valuable results. In such cases, it is recommended to consider targeted single-gene testing or the utilization of a syndromic cardiomyopathy gene panel [71, 72].
Gene panel testing for cardiomyopathy. Commonly used gene panels are often employed to detect genetic variations in cardiomyopathies. There is variation in the quantity and categories of genes available for sequencing across different panels. Previous functional investigations or familial segregation analysis have often identified a correlation between the genes and cardiomyopathies. Research examining a comprehensive gene panel consisting of 46 genes in a group of 766 randomly selected patients with dilated cardiomyopathy (DCM) revealed that as many as 37% of them may possess an underlying causative genetic variation. For gene panels consisting of five to ten genes, the testing output was less than 10%. However, when using a bigger panel of 46 genes, the success rate jumped to over 30%. It is important to note that this larger panel also had a high proportion of variations of unknown significance (VUSs), accounting for 60% of the results.
Sequencing of the whole exome. The use of whole-exome sequencing in clinical settings is growing due to the significant reduction in costs connected with this type of testing over time, allowing for the acquisition of information regarding the occurrence of mutations in unrelated genes. Whole-exome sequencing can aid in the discovery of new genetic variations, especially in genes that are not yet linked to illness development. This is typically done through trio testing, which involves testing the affected individual and both of their parents to establish if the genetic variant is inherited and associated with the condition. The incidence of detection of a disease-causing genetic mutation in specific, previously undetected Mendelian disorders can reach up to 25% [6].
The regular use of whole-exome sequencing is connected with significant constraints The extent of the test's coverage is restricted to the specific genes being studied. Therefore, the interpretation of the test findings must be done with caution due to potential problems, such as mistakenly identifying a mutation as disease-causing. Due to our limited understanding of the functional significance of variations in intronic or intergenic regions, whole-genome sequencing is not commonly employed in clinical settings [6].
Genetic variations are categorized into harmful, likely pathogenic, unknown importance, likely benign, or benign [11]. After undergoing a genetic test, there are two main outcomes: either an informative variation is detected, which may be classified as pathogenic or likely pathogenic, or no informative variant is detected, which can be classified as no variant, benign, likely benign, or variant of uncertain significance (VUS).
The lack of an identifiable genetic mutation does not automatically indicate the absence of a genetic cause for dilated cardiomyopathy (DCM). The potential explanations are as follows: (a) a gene associated with DCM that is not included in the gene panel used for testing, which could be due to recently discovered disease genes or the gene has not been identified yet; or (b) DCM may not have a purely monogenic cause, there may be a pattern of polygenic disease within families.
8. Genetic Risk Factors and Prognosis
There is significant interest in using genetic markers to assess prognosis and aid in risk stratification for individuals with DCM, in addition to predicting their likelihood of getting the condition. Patients who have pathogenic LMNA variations may have a higher chance of developing ventricular arrhythmias, regardless of the extent to which their LVEF is diminished. This makes them suitable candidates for the implantation of a cardioverter-defibrillator as a preventive measure against sudden cardiac death. Single nucleotide polymorphisms (SNPs) can also provide valuable information for the categorization of risk. In a large-scale genome-wide association study (GWAS), researchers found that a single nucleotide polymorphism (SNP) located on chromosomal band 5q22 was associated with a 36% higher risk of mortality in individuals diagnosed with heart failure [73].
Previous genome-wide association studies (GWAS) have found a correlation between single nucleotide polymorphisms (SNPs) and a higher likelihood of developing heart failure and experiencing cardiovascular events [74,75]. Nevertheless, further data is needed to support the regular use of genetic testing for predicting outcomes in arrhythmic DCM.
9. Management
The current approach to treating GDCM involves using guideline-directed medicinal interventions that try to prevent or decelerate the advancement of heart failure. The drugs consist of the usual combination of beta blockers, angiotensin receptor-neprilysin inhibitors, sodium-glucose contransporter-2 inhibitors, and mineralocorticoid receptor antagonists [76]. While there are no specific clinical studies focused on pharmacological treatment for heart failure in genetic DCM, all the significant trials on heart failure with decreased ejection fraction did involve patients with non-ischemic cardiomyopathy, and it is likely that some of these patients had genetic causes for their condition.
The variation in individual responsiveness to β-blockade is likely due to genetic variants in the gene encoding the β-adrenergic receptor within the population. Polymorphisms in ADRB1 and GRK5, which encode the downstream G-protein-coupled receptor kinase 5, have an impact on the response to β-blockade [77]. Additional results indicate that amino acid changes in the β-adrenergic receptor, such as Arg389Gly, might potentially impact the response to β-blockade. This effect may be due to a reduction in the activation of the renin-angiotensin-aldosterone pathway. Nevertheless, this polymorphism has not been linked to any alteration in morbidity or death in the sub-studies of the WOSCOPS trial [78] or the MERIT-HF trial [79].
Genes that encode angiotensin II and its accompanying receptors have a role in the development of heart failure and are targeted by antagonistic medication according to medical guidelines. These genes have polymorphisms that result in different responses to treatment. Evidence indicates that individuals with a homozygous DD genotype for the ACE gene, which increases angiotensin II levels, along with a polymorphism in AGTR1 (which encodes the type 1 angiotensin II receptor), may experience higher activation of the renin-angiotensin-aldosterone system and a poorer prognosis, even when treated with ACE inhibitors [80]. Research was conducted to observe individuals with congestive heart failure. It was shown that having at least one D allele of the ACE gene was linked to decreased survival rates, particularly for patients who were not receiving β-blocker medication. However, in another experiment where patients were categorized based on the existence of previously recognized harmful changes in AGTR1, no disparities in long-term mortality were found [81].
While the impact magnitude may not be significant, certain single nucleotide polymorphisms (SNPs) have been discovered that elevate the risk of angio-oedema produced by ACE inhibitors [80]. In the clinical arena, routine testing for SNPs that are linked to variable responses to neuro-hormonal blocking is not commonly done since there is a lack of clinical validity and usefulness.
The interplay between environmental variables and an individual's genetic composition is of utmost significance. Environmental variables and triggers have the potential to influence the observable characteristics of DCM in a patient who already has a genetic susceptibility for the condition. Peripartum cardiomyopathy has been linked to both a genetic susceptibility and the interaction between an underlying pathogenic variation and the physiological stress of pregnancy [58, 59]. Furthermore, the cardiotoxicity of anthracyclines and other antineoplastic drugs may be influenced by genetic predisposition [82]. Upon thorough evaluation, it is evident that environmental triggers may be linked to a poorer prognosis if there is an existing genetic susceptibility to the illness [83]. Crucially, it is vital for both DCM patients and their families to focus on addressing changeable variables that contribute to cardiovascular disease and overall health. The conventional approach to managing hypertension, quitting smoking, preventing diabetes, addressing obesity, and moderating alcohol intake continues to be essential, since additional harmful factors affecting the heart are linked to poor outcomes [83]. A primary care setting will handle the management of many of these concerns.
Pharmacological treatments for hereditary DCM do not often affect the course of the illness. A notable exception to this is the p.R222Q SCN5A mutation, which allows for the possibility of tailored treatment based on particular mechanisms. This genetic mutation leads to the development of multifocal ectopic Purkinje-related premature contractions (MEPPC), atrial arrhythmias, and DCM as a result of increased activity in the heart sodium channels. The symptoms mentioned can be efficiently managed by using a sodium channel antagonist, such as flecainide. This type of treatment is considered a gene-specific therapy and is a significant example of precision medicine in familial DCM. Additional gene-specific medicines that are currently being studied but have not yet been used in clinical practice include myosin activator medications (such as omecamtiv mecarbil) and the suppression of the mitogen-activated protein kinase signaling pathway in laminopathies [5].
Implantable cardiac defibrillator (ICD) treatment can be utilized as either primary or secondary prevention after experiencing a life-threatening arrhythmia. Current guidelines employ genetic variations such as LMNA, FLNC, and PLN to categorize individuals based on their risk for malignant arrhythmias. These guidelines suggest the use of primary prevention ICDs for patients with a higher risk, even if their left ventricular ejection fraction (LVEF) is over the usual criterion of 35% [29].
Determining a genetic anomaly is also crucial in making judgements on the appropriate timing for heart transplantation. Patients with pathogenic or possibly pathogenic LMNA mutations frequently experience a swiftly deteriorating clinical condition and require early consideration of cardiac transplantation.
Certain pathogenic variations have stricter guidelines. For instance, individuals with LMNA-associated DCM should avoid engaging in intense physical activity since it is linked to a higher risk of deterioration in heart function and death [84]. It is extremely advantageous and important to pay attention to the psychological well-being and receive genetic counselling from specialists who have been trained in this field. This should not be disregarded or underestimated.
Contraception or pregnancy planning is crucial for pre-menopausal women with DCM. Women who test positive for a certain genotype and become pregnant should have thorough echocardiographic surveillance both during and after pregnancy to identify any indications of worsening after childbirth. Peripartum cardiomyopathy has the potential to reoccur earlier and with greater severity in subsequent pregnancies. Indeed, for certain women with confirmed DCM, pregnancy may be advised against due to its exceedingly precarious nature, which poses significant risks to both the mother and the foetus [85]. When it comes to contraception for individuals with cardiomyopathy, it is recommended to use barrier techniques or long-acting contraceptive methods that contain levonorgestrel.
The overall prognosis is variable depending on the specific gene alterations. Administering treatment to individuals with DCM caused by truncating TTN variants frequently leads to the reversal of left ventricular remodeling. But, familial DCM can be a progressive condition, with a substantial number of individuals advancing to end-stage heart failure and needing cardiac transplantation [86]. Despite a reported 50% death rate at 5 years, the general prognosis seems to be becoming better. This can be attributed to improved management of cardiovascular risk factors, earlier detection of the condition, and advancements in heart failure treatment [87].
For those who have a positive genotype but do not display any physical signs of the condition, there is presently no strong evidence to support a specific approach for managing their condition. Furthermore, there are no treatments available that have been proven to prevent the development of DCM in these individuals [88] . It is crucial to provide genetic counselling and effectively control cardiovascular risk factors in this particular group.
9. Unresolved Issues and Future Directions
Over the past decade, there has been significant advancement in understanding and improving our knowledge of the genetic foundation of DCM. The objective for the next decade should be to utilize this information in order to develop specific treatments. There are some unresolved inquiries in our comprehension of the genetics of DCM. An enigma is in the very modest genetic diagnostic success rate, especially in cases of familial DCM. This presumably indicates situations with complex genetic structures that are more challenging to investigate—highly influenced by genetics, but not caused by a single gene. These theories encompass oligogenic and polygenic combinations, in which genetic susceptibility is influenced by the cumulative impact of numerous common genetic variations. This is in contrast to a dominant inheritance pattern driven by a single strong genetic component, as seen in Mendelian inheritance. These extra variations can have either additive effects, where the combined burden of these more frequent variants contributes to the disease, or non-additive interactions, where the more common variants affect the impact of a rare variant. Gene-environment interactions are expected to alter the impact of both uncommon and common variations.
10. Conclusions
DCM remains a highly complex condition with broad phenotypic heterogeneity that includes both arrhythmogenic and non-arrhythmogenic forms.
The heart failure community must advance in the identification and validation of genetic markers that indicate the risk of DCM, the progression of the disease, and the response to therapy. This progress will ultimately lead to the realization of precision medicine in heart failure, with the potential for improved outcomes for patients and their families.
Author Contributions
Conceptualization, D.-A.A, O.-M.C, D.-F.L. and M.-C.T.; methodology, I.C., D.C., A.-P.T, D.-F.L; software, I.C., M.A., B.-C.B.; validation, E.-S.B., D.C., D.-C.J. and S.-F.A.; formal analysis, I.-R.L.; investigation, D.-A.A., M.A., and I.-R.L; resources, C.O.; data curation, D.-A.A, D.-F.L.; writing—original draft preparation, D.-A.A., I.-R.L., A.-P.T; writing—B.-C.B., E.-S.B, D.-F.L.; review and editing, O.-M.C.; visualization, I.C., D.-C.J, S.-F.A.; supervision, M.-C.T, C.O.; project administration, M.-C.T.; funding acquisition, O.-M.C, D.-F.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Victor Babes University of Medicine and Pharmacy, Timisoara, Romania.
Institutional Review Board Statement
Not applicable..
Informed Consent Statement
Not applicable.
Acknowledgments
The authors thank the medical staff from the echocardiography and cardiac MRI laboratories for their technical support.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Stehlik J, Edwards LB, Kucheryavaya AY, Benden C, Christie JD, Dobbels F, et al. The Registry of the International Society for Heart and Lung Transplantation: Twenty-eighth Adult Heart Transplant Report−−2011. J Heart Lung Transplant. 2011, 30, 1078–1094. [Google Scholar] [CrossRef]
- Heidenreich, P. A. Forecasting the impact of heart failure in the United States a policy statement from the American Heart Association. Circ. Heart Fail 2013, 6, 606–619. [Google Scholar] [CrossRef] [PubMed]
- Paul C, Peters S, Perrin M, Fatkin D, Amerena J. Non-ischemic dilated cardiomyopathy: recognizing the genetic links. Intern Med J. 2023, 53, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Arbelo E, Protonotarios A, Gimeno R. J,, Arbustini E, Barriales-Villa R, et.al. 2023 ESC Guidelines for the management of cardiomyopathies: Developed by the task force on the management of cardiomyopathies of the European Society of Cardiology (ESC). European Heart Journal 2023, 44, 3503–3626. [Google Scholar]
- Bui, Q. M, Ding J, Hong K.N, Adler E.A. The Genetic Evaluation of Dilated Cardiomyopathy. Structural Heart 2023, 7, 100200. [Google Scholar] [CrossRef]
- Rosenbaum AN, Agre KE, Pereira NL. Genetics of dilated cardiomyopathy: practical implications for heart failure management. Nat Rev Cardiol. 2020, 17, 286–297. [Google Scholar] [CrossRef] [PubMed]
- Sweet, M. E. , Taylor, M. R. & Mestroni, L. Diagnosis, prevalence, and screening of familial dilated cardiomyopathy. Expert. Opin. Orphan Drugs 2015, 3, 869–876. [Google Scholar] [PubMed]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Escobar-Lopez L, Ochoa JP, Royuela A, et al. Clinical risk score to predict pathogenic genotypes in patients with dilated cardiomyopathy. J Am Coll Cardiol. 2022, 80, 1115–1126. [Google Scholar] [CrossRef]
- Jordan E, Peterson L, Ai T, et al. Evidence-based assessment of genes in dilated cardiomyopathy. Circulation. 2021, 144, 7–19. [Google Scholar] [CrossRef]
- Tayal U, Newsome S, Buchan R, et al. Phenotype and clinical outcomes of titin cardiomyopathy. J Am Coll Cardiol. 2017, 70, 2264–2274. [Google Scholar] [CrossRef] [PubMed]
- Musunuru K, Hershberger RE, Day SM, et al. Genetic testing for inherited cardiovascular diseases: a scientific statement from the American Heart Association. Circ Genom Precis Med. 2020, 13, e000067. [Google Scholar] [CrossRef] [PubMed]
- Shah RA, Asatryan B, Sharaf Dabbagh G, et al. Frequency, penetrance, and variable expressivity of dilated cardiomyopathy-associated putative pathogenic gene variants in UK Biobank Participants. Circulation. 2022, 146, 110–124. [Google Scholar] [CrossRef] [PubMed]
- Barriales-Villa R, Ochoa JP, Larranaga-Moreira JM, Salazar-Mendiguchia J, Diez-Lopez C, Restrepo-Cordoba MA, et al. Risk predictors in a Spanish cohort with cardiac laminopathies. The REDLAMINA registry. Rev Esp Cardiol (Engl Ed) 2021, 74, 216–224. [Google Scholar]
- Rootwelt-Norberg C, Christensen AH, Skjolsvik ET, Chivulescu M, Vissing CR, Bundgaard H, et al. Timing of cardioverter-defibrillator implantation in patients with cardiac laminopathies–external validation of the LMNA-risk ventricular tachyarrhythmia calculator. Heart Rhythm 2023, 20, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Vikhorev, P.G. Abnormal contractility in human heart myofibrils from patients with dilated cardiomyopathy due to mutations in TTN and contractile protein genes. Sci. Rep. 2017, 7, 14829. [Google Scholar] [CrossRef] [PubMed]
- Gramlich, M. Stress- induced dilated cardiomyopathy in a knock- in mouse model mimicking human titin- based disease. J. Mol. Cell. Cardiol. 2009, 47, 352–358. [Google Scholar] [CrossRef]
- Cannata A, Merlo M, Dal Ferro M, Barbati G, Manca P, Paldino A, et al. Association of titin variations with late-onset dilated cardiomyopathy. JAMA Cardiol 2022, 7, 371–377. [Google Scholar] [CrossRef]
- Heliö T, Elliott P, Koskenvuo JW, Gimeno JR, Tavazzi L, Tendera M, et al. ESC EORP cardiomyopathy registry: real-life practice of genetic counselling and testing in adult cardiomyopathy patients. ESC Heart Fail. 2020, 7, 3013–3021. [Google Scholar] [CrossRef]
- Vissing CR, Rasmussen TB, Dybro AM, Olesen MS, Pedersen LN, Jensen M, et al. Dilated cardiomyopathy caused by truncating titin variants: long-term outcomes, arrhythmias, response to treatment and sex differences. J Med Genet 2021, 58, 832–841. [Google Scholar] [CrossRef]
- Van Waning JI, Caliskan K, Michels M, Schinkel AFL, Hirsch A, Dalinghaus M, et al. Cardiac phenotypes, genetics, and risks in familial noncompaction cardiomyopathy. J Am Coll Cardiol 2019, 73, 1601–1611. [Google Scholar] [CrossRef] [PubMed]
- Jordan E, Peterson L, Ai T, Asatryan B, Bronicki L, Brown E, et al. Evidence-based assessment of genes in dilated cardiomyopathy. Circulation 2021, 144, 7–19. [Google Scholar] [CrossRef] [PubMed]
- James CA, Jongbloed JDH, Hershberger RE, Morales A, Judge DP, Syrris P, et al. International evidence based reappraisal of genes associated with arrhythmogenic right ventricular cardiomyopathy using the clinical genome resource framework. Circ Genom Precis Med 2021, 14, e003273. [Google Scholar] [CrossRef] [PubMed]
- Ingles J, Goldstein J, Thaxton C, Caleshu C, Corty EW, Crowley SB, et al. Evaluating the clinical validity of hypertrophic cardiomyopathy genes. Circ Genom Precis Med 2019, 12, e002460. [Google Scholar] [CrossRef] [PubMed]
- Duboscq- Bidot, L. Mutations in the Z- band protein myopalladin gene and idiopathic dilated cardiomyopathy. Cardiovasc. Res. 2008, 77, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Dalakas, M. Desmin myopathy, a skeletal myopathy with cardiomyopathy caused by mutations in the desmin gene. N. Engl. J. Med. 2000, 342, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Begay, R.L. Filamin C truncation mutations are associated with arrhythmogenic dilated cardiomyopathy and changes in the cell–cell adhesion structures. JACC Clin. Electrophysiol. 2018, 4, 504–514. [Google Scholar] [CrossRef] [PubMed]
- Gigli M, Stolfo D, Graw SL, et al. Phenotypic expression, natural history, and risk stratification of cardiomyopathy caused by filamin C truncating variants. Circulation. 2021, 144, 1600–1611. [Google Scholar] [CrossRef] [PubMed]
- Wilde, A.A.M. European Heart Rhythm Association (EHRA)/Heart Rhythm Society (HRS)/Asia Pacific Heart Rhythm Society (APHRS)/Latin American Heart Rhythm Society (LAHRS) Expert Consensus Statement on the State of Genetic Testing for Cardiac Diseases. Heart Rhythm 2022, 19, 623–629. [Google Scholar] [CrossRef]
- Domínguez F, Cuenca S, Bilinska Z, et al. Dilated cardiomyopathy due to BLC2-associated athanogene 3 (BAG3) mutations. J Am Coll Cardiol. 2018, 72, 2471. [Google Scholar] [CrossRef]
- Garcia- Pavia, P. Desmosomal protein gene mutations in patients with idiopathic dilated cardiomyopathy undergoing cardiac transplantation: a clinicopathological study. Heart 2011, 97, 1744–1752. [Google Scholar] [CrossRef]
- Klauke B, Gaertner-Rommel A, Schulz U, et al. High proportion of genetic cases inpatients with advanced cardiomyopathy including a novel homozygous Plakophilin 2-gene mutation. PLoS One. 2017, 12, e0189489. [Google Scholar]
- Bione, S. A novel X- linked gene, G4.5. is responsible for Barth syndrome. Nat. Genet. 1996, 12, 385–389. [Google Scholar] [CrossRef]
- Ichida, F. Novel gene mutations in patients with left ventricular noncompaction or barth syndrome. Circulation 2001, 103, 1256–1263. [Google Scholar] [CrossRef] [PubMed]
- Captur, G. & Nihoyannopoulos, P. Left ventricular non- compaction: genetic heterogeneity, diagnosis and clinical course. Int. J. Cardiol. 2010, 140, 145–153. [Google Scholar]
- Van den Hoogenhof MMG, Beqqali A, Amin AS, et al. RBM20 mutations induce an arrhythmogenic dilated cardiomyopathy related to disturbed calcium handling. Circulation. 2018, 138, 1330–1342. [Google Scholar] [CrossRef] [PubMed]
- Kułach A, Majewski M, Gąsior Z, Gardas R, Goscinska-Bis K, Gołba KS. Dilated cardiomyopathy with severe arrhythmias in Emery-Dreifuss muscular dystrophy. Cardiol J. 2020, 27, 93–111. [Google Scholar] [CrossRef]
- McNair WP, Sinagra G, Taylor MR, et al. SCN5A mutations associate with arrhythmic dilated cardiomyopathy and commonly localize to the voltage-sensing mechanism. J Am Coll Cardiol. 2011, 57, 2160–2168. [Google Scholar] [CrossRef] [PubMed]
- Olson, T.M. Sodium channel mutations and susceptibility to heart failure and atrial fibrillation. JAMA 2005, 293, 447–454. [Google Scholar] [CrossRef]
- Zaragoza, M. V. , Brandon, M. C., Diegoli, M., Arbustini, E. & Wallace, D. C. Mitochondrial cardiomyopathies: how to identify candidate pathogenic mutations by mitochondrial DNA sequencing, MITOMASTER and phylogeny. Eur. J. Hum. Genet. 2011, 19, 200–207. [Google Scholar]
- Brega, A. , Narula, J. & Arbustini, E. Functional, structural, and genetic mitochondrial abnormalities in myocardial diseases. J. Nucl. Cardiol. 2001, 8, 89–97. [Google Scholar] [PubMed]
- Anan, R. Cardiac involvement in mitochondrial diseases: a study on 17 patients with documented mitochondrial DNA defects. Circulation 1995, 91, 955–961. [Google Scholar] [CrossRef] [PubMed]
- Nair, V. , Belanger, E. C. & Veinot, J.P. Lysosomal storage disorders affecting the heart: a review. Cardiovasc. Pathol. 2019, 39, 12–24. [Google Scholar] [PubMed]
- Agarwal R, Paulo JA, Toepfer CN, et al. Filamin C cardiomyopathy variants cause protein and lysosome accumulation. Circ Res. 2021, 129, 751–766. [Google Scholar] [CrossRef] [PubMed]
- Kamdar, F. & Garry, D.J. Dystrophin- deficient cardiomyopathy. J. Am. Coll. Cardiol. 2016, 67, 2533–2546. [Google Scholar] [PubMed]
- Towbin, J. A. X- linked dilated cardiomyopathy -molecular- genetic evidence of linkage to the Duchenne muscular- dystrophy (dystrophin) gene at the Xp21 locus. Circulation 1993, 87, 1854–1865. [Google Scholar] [CrossRef] [PubMed]
- Bonne, G. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery- Dreifuss muscular dystrophy. Nat. Genet. 1999, 21, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Heller SA, Shih R, Kalra R, Kang PB. Emery-Dreifuss muscular dystrophy. Muscle Nerve. 2020, 61, 436–448. [Google Scholar] [CrossRef] [PubMed]
- Sanna T, Dello Russo A, Toniolo D, et al. Cardiac features of Emery-Dreifuss muscular dystrophy caused by lamin A/C gene mutations. Eur Heart J. 2003, 24, 2227–2236. [Google Scholar] [CrossRef]
- Bonne G, Leturcq F, Ben Yaou R. Emery-Dreifuss Muscular Dystrophy. 2004 Sep 29 [Updated 2019 Aug 15]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. https://www.ncbi.nlm.nih. 1436.
- Nigro, V. & Savarese, M. Genetic basis of limb- girdle muscular dystrophies: the 2014 update. Acta Myol. 2014, 33, 1–12. [Google Scholar]
- El-Battrawy I, Zhao Z, Lan H, et al. Ion Channel dysfunctions in dilated cardiomyopathy in limb-girdle muscular dystrophy. Circ Genom Precis Med. 2018, 11, e001893. [Google Scholar] [CrossRef] [PubMed]
- Emery AE, Dreifuss FE. Unusual type of benign x-linked muscular dystrophy. J Neurol Neurosurg Psychiatry. 1966, 29, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Fatkin, D. Guidelines for the diagnosis and management of familial dilated cardiomyopathy. Heart Lung Circ 2011, 20, 691–693. [Google Scholar] [CrossRef] [PubMed]
- Hershberger RE, Morales A, Siegfried JD. Clinical and genetic issues in dilated cardiomyopathy: a review for genetics professionals. Genet Med. 2010, 12, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Hershberger RE, Givertz MM, Ho CY, Judge DP, Kantor PF, McBride KL et al. Genetic evaluation of cardiomyopathy: a clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet Med 2018, 20, 899–909. [Google Scholar] [CrossRef] [PubMed]
- Peters S, Kumar S, Elliott P,Kalman JM, Fatkin D. Arrhythmic genotypes in familial dilated cardiomyopathy: implications for genetic testing and clinical management. Heart Lung Circ 2019, 28, 31–38. [Google Scholar] [CrossRef]
- Ware JS, Li J, Mazaika E, Yasso CM, DeSouza T, Cappola TP et al. Shared genetic predisposition in peripartum and dilated cardiomyopathies. N Engl J Med 2016, 374, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Van Spaendonck- Zwarts, K.Y. Peripartum cardiomyopathy as a part of familial dilated cardiomyopathy. Circulation. 2010, 121, 2169–2175. [Google Scholar] [CrossRef] [PubMed]
- Fatkin D, Johnson R, McGaughran J, Weintraub RG, Atherton JJ. Position statement on the diagnosis and management of familial dilated cardiomyopathy. Heart Lung Circ 2017, 26, 1127–1132. [Google Scholar] [CrossRef]
- Hershberger RE, Cowan J, Jordan E, Kinnamon DD. The complex and diverse genetic architecture of dilated cardiomyopathy. Circ Res 2021, 128, 1514–1532. [Google Scholar] [CrossRef]
- Dorn, GW. Genetics of common forms of heart failure. Curr Opin Cardiol. 2011, 26, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Peters S, Johnson R, Birch S, Zentner D, Hershberger RE, Fatkin D. Familial dilated cardiomyopathy. Heart Lung Circ 2020, 29, 566–574. [Google Scholar] [CrossRef] [PubMed]
- Hudson L, Morales A, Mauro AC, Whellan D, Adams KF, O’Connor CM et al. Family history of dilated cardiomyopathy among patients with heart failure from the HF-ACTION genetic ancillary study. Clin Transl Sci 2013, 6, 179–183. [Google Scholar] [CrossRef]
- Haugaa KH, Hasselberg NE, Edvardsen T. Mechanical dispersion by strain echocardiography: a predictor of ventricular arrhythmias in subjects with lamin A/C mutations. JACC Cardiovasc Imaging 2015, 8, 104–106. [Google Scholar] [CrossRef] [PubMed]
- Haugaa KH, Goebel B, Dahlslett T, Meyer K, Jung C, Lauten A, et al. Risk assessment of ventricular arrhythmias in patients with nonischemic dilated cardiomyopathy by strain echocardiography. J Am Soc Echocardiogr 2012, 25, 667–673. [Google Scholar] [CrossRef]
- Leren IS, Saberniak J, Haland TF, Edvardsen T, Haugaa KH. Combination of ECG and echocardiography for identification of arrhythmic events in early ARVC. JACC Cardiovasc Imaging 2017, 10, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Augusto JB, Eiros R, Nakou E, MouraFerreira S, Treibel TA, Captur G et al. Dilated cardiomyopathy and arrhythmogenic left ventricular cardiomyopathy: a comprehensive genotype-imaging phenotype study. Eur Heart J Cardiovasc Imaging 2020, 21, 326–336. [Google Scholar]
- Bonny A, Lellouche N, Ditah I, Hidden-Lucet F, Yitemben MT, Granger B, et al. C-reactive protein in arrhythmogenic right ventricular dysplasia/cardiomyopathy and relationship with ventricular tachycardia. Cardiol Res Pract 2010, 2010, 919783. [Google Scholar]
- Sylvius, N. In vivo and in vitro examination of the functional significances of novel lamin gene mutations in heart failure patients. J. Med. Genet. 2005, 42, 639–647. [Google Scholar] [CrossRef]
- Miller DT, Lee K, Gordon AS, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2021 update: a policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2021, 23, 1391–1398. [Google Scholar] [CrossRef]
- Miller DT, Lee K, Abul-Husn NS, et al. ACMG SF v3.1 list for reporting of secondary findings in clinical exome and genome sequencing: a policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2022, 24, 1407–1414. [Google Scholar] [CrossRef] [PubMed]
- Smith, J. G. Discovery of genetic variation on chromosome 5q22 associated with mortality in heart failure. PLOS Genet. 2016, 12, e1006034. [Google Scholar] [CrossRef] [PubMed]
- Morrison, A.C. Genomic variation associated with mortality among adults of European and African ancestry with heart failure: the Cohorts for Heart and Aging Research in Genomic Epidemiology Consortium. Circ. Cardiovasc. 2010; 3. [Google Scholar]
- Myers, V. D. Association of variants in BAG3 with cardiomyopathy outcomes in African American individuals. JAMA Cardiol. 2018, 3, 929–938. [Google Scholar] [CrossRef] [PubMed]
- Members WC, Members AAJC. 2022 AHA/ACC/HFSA guideline for the management of heart failure. J Card Fail. 2022, 28, e1–e167. [Google Scholar] [CrossRef] [PubMed]
- Cresci, S. Clinical and genetic modifiers of long-term survival in heart failure. J. Am. Coll. Cardiol. 2009, 54, 432–444. [Google Scholar] [CrossRef]
- White, H. An evaluation of the β1 adrenergic receptor Arg389Gly polymorphism in individuals at risk of coronary events: a WOSCOPS substudy. Eur. Heart J. 2002, 23, 1087–1092. [Google Scholar] [CrossRef] [PubMed]
- White, H. L. An evaluation of the β1 adrenergic receptor Arg389Gly polymorphism in individuals with heart failure: a MERIT- HF sub- study. Eur. J. Heart Fail. 2003, 5, 463–468. [Google Scholar] [PubMed]
- Pare, G. Genetic variants associated with angiotensin- converting enzyme inhibitor- associated angioedema. Pharmacogenet. Genomics 2013, 23, 470–478. [Google Scholar]
- Nelveg- Kristensen, K.E. Pharmacogenetic risk stratification in angiotensin- converting enzyme inhibitor- treated patients with congestive heart failure: a retrospective cohort study. PLOS ONE 2015, 10, 1–16. [Google Scholar]
- Scott, E. , Hasbullah, J. S., Ross, C. J. & Carleton, B.C. Reducing anthracycline- induced cardiotoxicity through pharmacogenetics. Pharmacogenomics. 2018, 19, 1147–1150. [Google Scholar]
- Hazebroek MR, Moors S, Dennert R, van den Wijngaard A, Krapels I, Hoos M et al. Prognostic relevance of gene-environment interactions in patients with dilated cardiomyopathy: applying the MOGE(S) classification. J Am Coll Cardiol 2015, 66, 1313–1323. [Google Scholar] [CrossRef] [PubMed]
- Skjølsvik ET, Hasselberg NE, Dejgaard LA, Lie ØH, Andersen K, Holm T et al. Exercise is associated with impaired left ventricular systolic function in patients with Lamin A/C genotype. J Am Heart Assoc 2020, 9, e012937. [Google Scholar] [CrossRef] [PubMed]
- Regitz-Zagrosek V, Roos-Hesselink JW, Bauersachs J, Blomström-Lundqvist C, Cífkova R, De Bonis M et al. 2018 ESC guidelines for the management of cardiovascular diseases during pregnancy: the Task Force for the Management of Cardiovascular Diseases during pregnancy of the European Society of Cardiology (ESC). Eur Heart J. 2018, 39, 3165–3241. [Google Scholar] [CrossRef] [PubMed]
- De Boer RA, Heymans S, Backs J,Carrier L, Coats AJS, Dimmeler S et al. Targeted therapies in genetic dilated and hypertrophic cardiomyopathies: from molecular mechanisms to therapeutic targets. A position paper from the Heart Failure Association (HFA) and the Working Group on Myocardial Function of the European Society of Cardiology (ESC). Eur J Heart Fail 2022, 24, 406–420. [Google Scholar]
- Merlo M, Cannatà A, Pio Loco C, Stolfo D, Barbati G, Artico J et al. Contemporary survival trends and aetiological characterization in non- ischaemic dilated cardiomyopathy. Eur J Heart Fail 2020, 22, 1111–1121. [Google Scholar] [CrossRef]
- Pinto YM, Elliott PM, Arbustini E, Adler Y, Anastasakis A, Böhm M et al. Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: a position statement of the ESC working group on myocardial and pericardial diseases. Eur Heart J 2016, 37, 1850–1858. [Google Scholar] [CrossRef]
Figure 1.
Celular proteins linked to DCM.
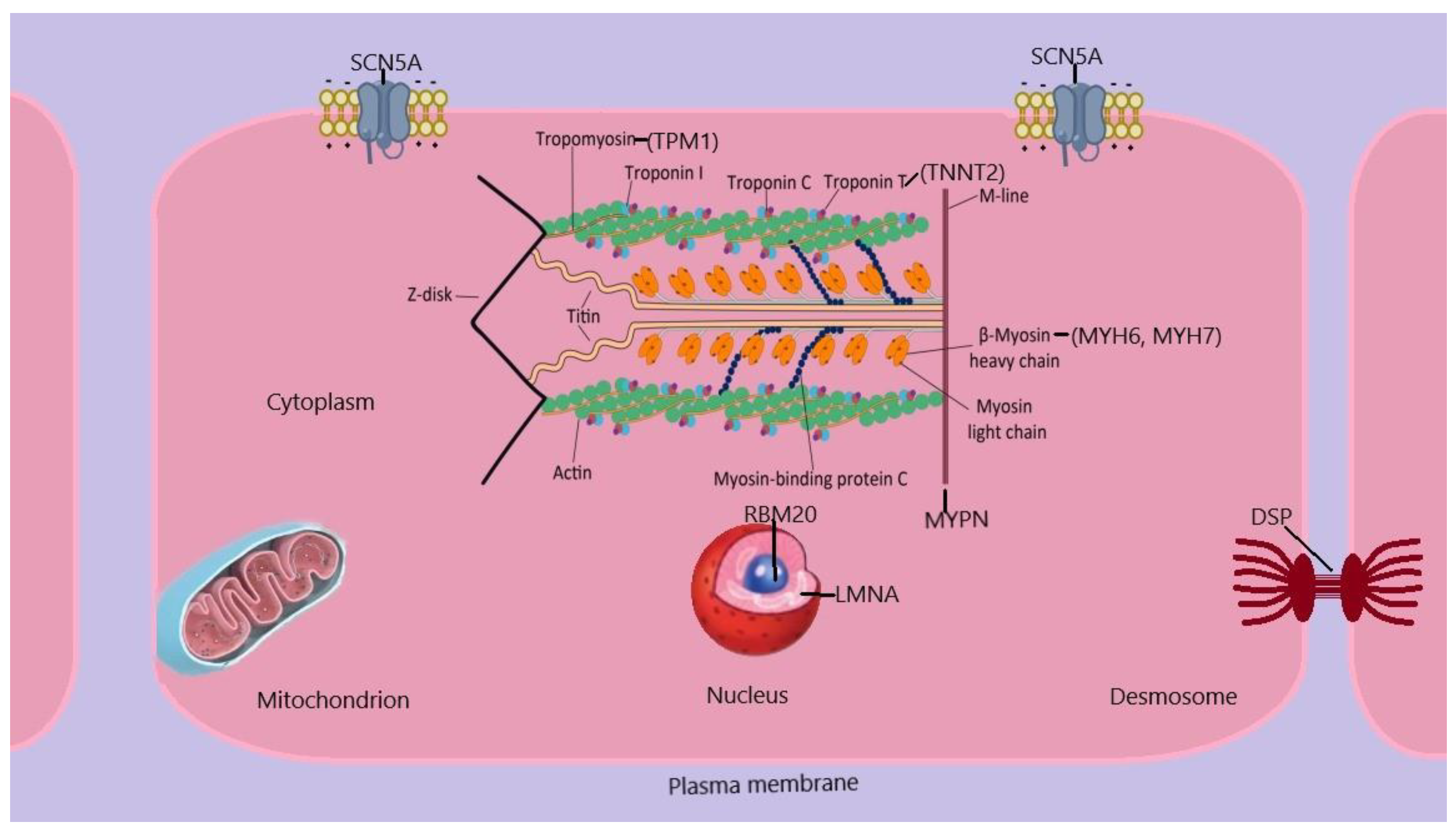
Figure 3.
Familial dilated cardiomyopathy. Two-dimensional trans-thoracic conventional echography in apical 4 chambers and parasternal short axis views. Bull's eye myocardial deformation with overall reduced segmental longitudinal peak strain patterns.
Figure 3.
Familial dilated cardiomyopathy. Two-dimensional trans-thoracic conventional echography in apical 4 chambers and parasternal short axis views. Bull's eye myocardial deformation with overall reduced segmental longitudinal peak strain patterns.
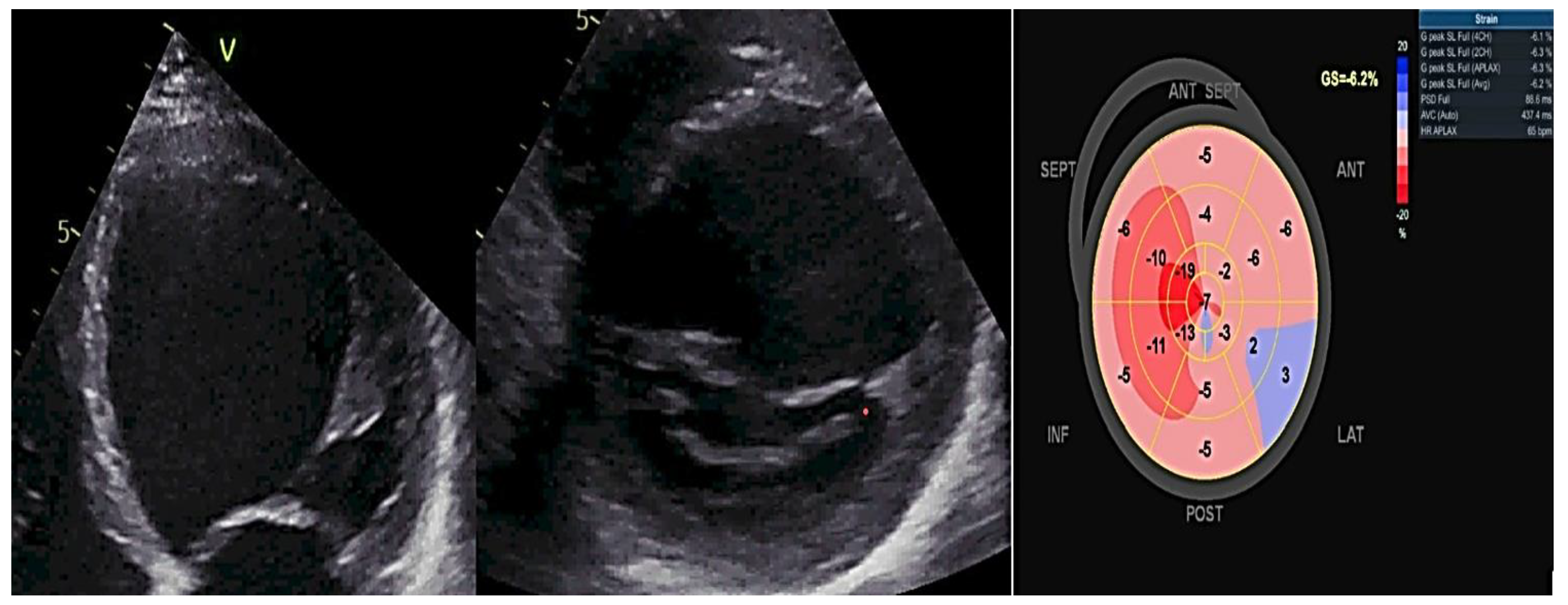
Figure 4.
Cardiac magnetic resonance images, 4 chamber (horizontal long axis) and 2 chamber (vertical long axis). Dilated left ventricle with hypertrabeculations of the antero-lateral wall; late gadolinium enhancement shows septal mid-wall fibrosis, suggestive of idiopathic dilative cardiomyopathy with left ventricular non- compaction.
Figure 4.
Cardiac magnetic resonance images, 4 chamber (horizontal long axis) and 2 chamber (vertical long axis). Dilated left ventricle with hypertrabeculations of the antero-lateral wall; late gadolinium enhancement shows septal mid-wall fibrosis, suggestive of idiopathic dilative cardiomyopathy with left ventricular non- compaction.
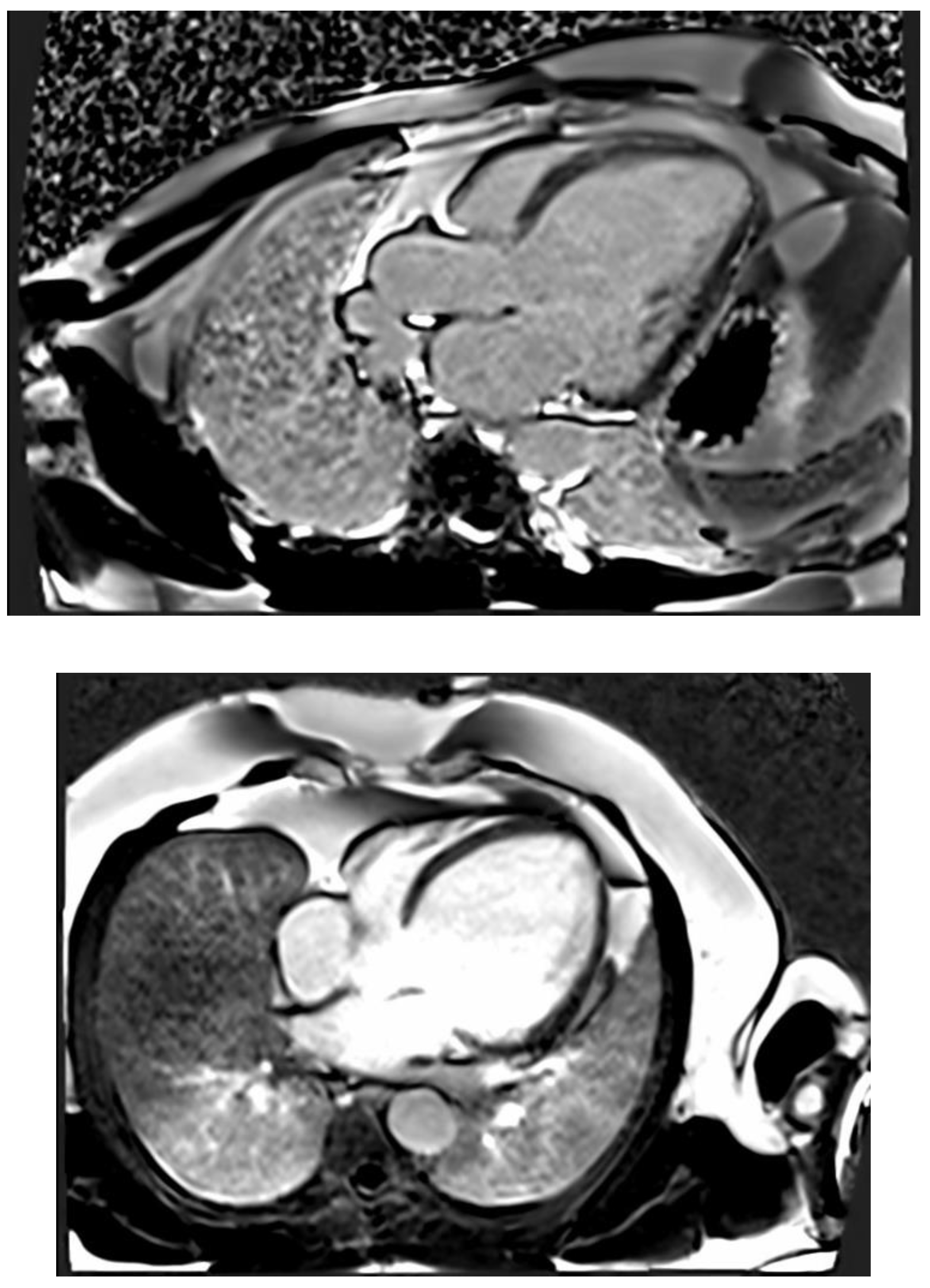
Figure 5.
Cardiac magnetic resonance image, short axis. Dilated left ventricle with thin walls; late gadolinium enhancement highlights circumferential "ring-shape" mid wall fibrosis suggestive of desmoplakin cardiomyopathy.
Figure 5.
Cardiac magnetic resonance image, short axis. Dilated left ventricle with thin walls; late gadolinium enhancement highlights circumferential "ring-shape" mid wall fibrosis suggestive of desmoplakin cardiomyopathy.
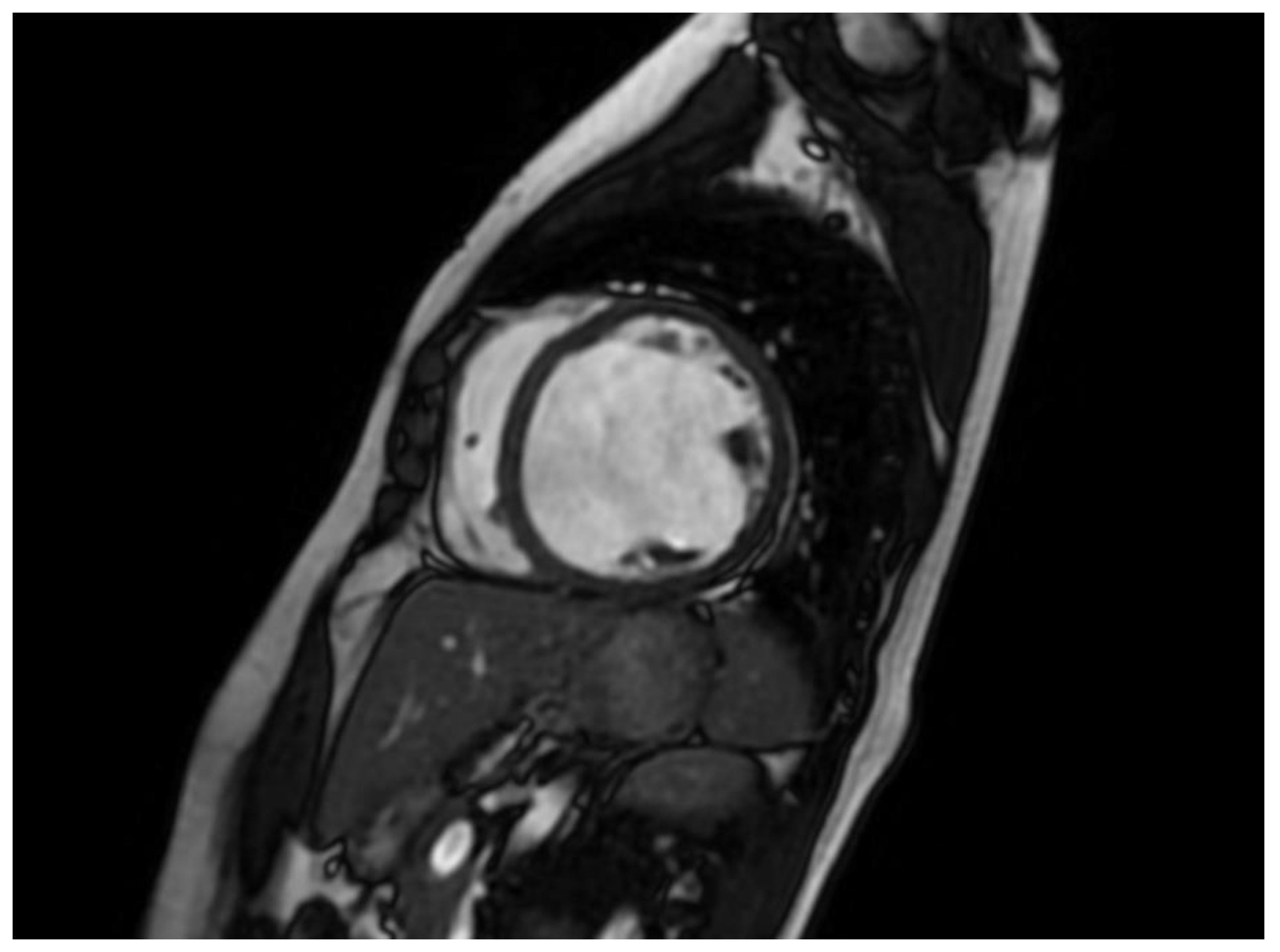
Figure 6.
A practical approach for genetic testing in dilated cardiomyopathies.
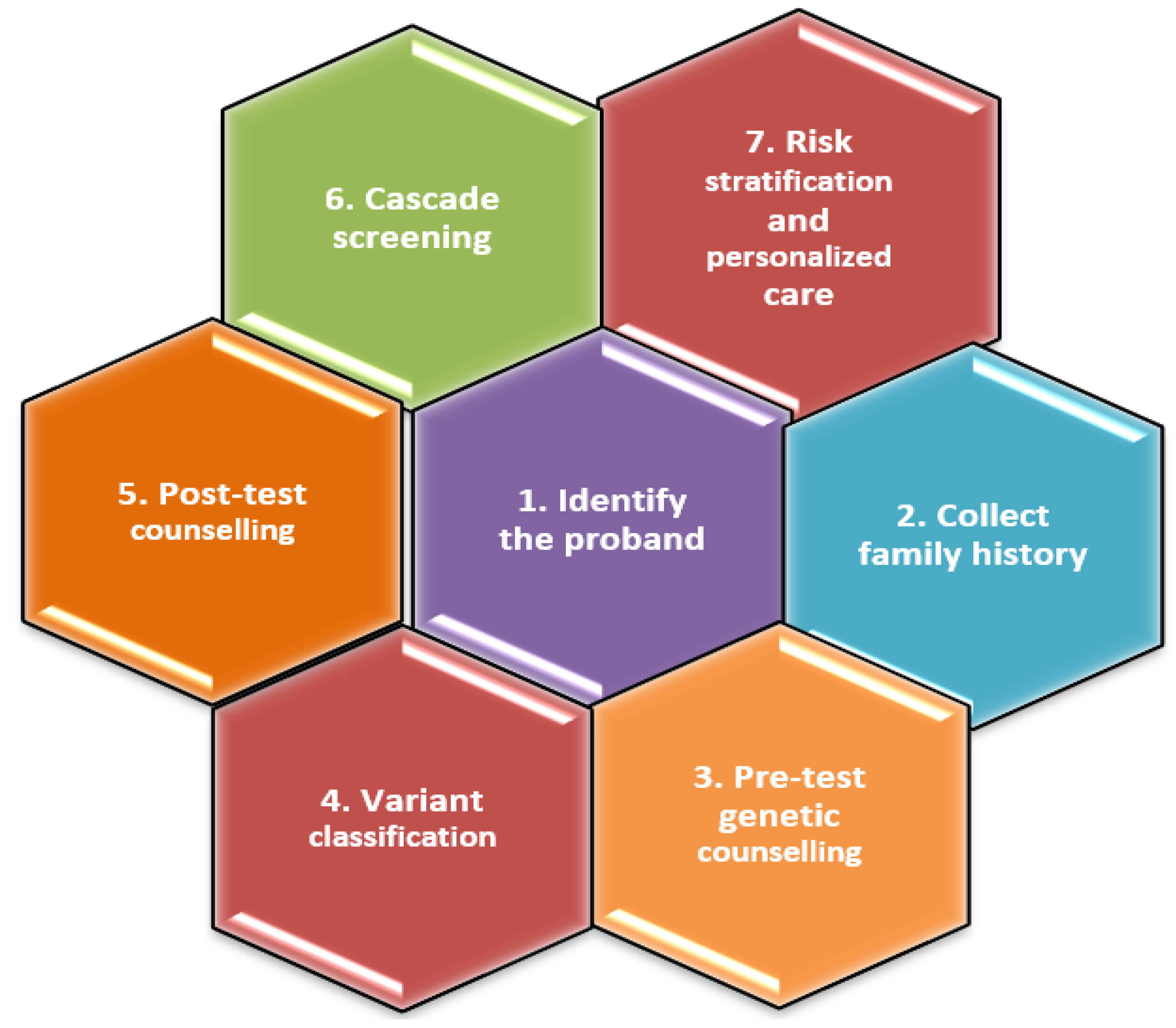

Table 1.
Genes associated with inherited DCM.
| Gene | Protein | Function | Clinical picture | |
|---|---|---|---|---|
| LMNA | Lamin A/C | Nuclear lamina | Severe DCM, with severe heart failure, High risk for cardiac death or heart transplantation Atrial and ventricular arrythmias Cardiac conduction disorders Highly penetrant Emery-Dreifuss muscular dystrophy Limb-girdle muscular dystrophy |
|
| RBM20 | RNA binding motif 20 | Nucleus RNA binding | DCM Atrial and ventricular arrhythmias Sudden cardiac death LV non-compaction CM |
|
| TTN | Titin | Sarcomere | DCM Atrial fibrillation Ventricular arrythmias LV non-compaction CM Muscular dystrophy |
|
| MYH7 | Myosin heavy chain beta 7 | Sarcomere | DCM LV non-compaction CM |
|
| TNNT2 | Cardiac troponin T2 | Sarcomere | DCM | |
| TNNC1 | Troponin C | Sarcomere | DCM | |
| BAG3 | BAG cochaperone 3 | Chaperone protein | Severe DCM Myofibrillar myopathy |
|
| DSP | Desmoplakin | Desmoplakin | Arrhythmogenic CM-left dominant Arrhythmogenic RV CM Ventricular arrhythmias |
|
| DES | Desmin | Cytoskeletal | DCM Arrhythmogenic RV CM Atrial and ventricular arrhythmias LV non-compaction CM Limb girdle muscular dystrophy |
|
| FLNC | Filamin C | Cytoskeletal | DCM Atrial and ventricular arrhythmias Myofibrillar myopathy |
|
| PLN | Phospholamban | Ca | Calcium channel regulator | DCM Arrhythmogenic right ventricular CM Atrial and ventricular arrhythmias |
| SNCSA | Voltage-gated sodium channel SA | Sodium channel regulator | DCM Atrial and ventricular arrhythmias often reversible with sodium-channel blockade Long QT syndrome Brugada syndrome |
Abbreviations: LV, left ventricular; RV, right ventricular; CM, cardiomyopathy; DCM, dilated cardiomyopathy
Table 2.
High risk genes in inherited DCM.
| High risk genes | Clinical picture | Investigations |
|---|---|---|
| LMNA RBM20 BAG3 DSP DES FLNC PLN SNCSA |
Family history of conduction disease, heart transplant or sudden cardiac death Rapid disease progression Severe heart failure Myocarditis (DSP) Sudden cardiac death |
Severely reduced LVEF Ventricular arrhythmias Conduction disorders Late gadolinium enhancement on cardiac MRI |
| Abbreviations: DCM, dilated cardiomyopathy; LVEF, left ventricular ejection fraction; MRI, magnetic resonance imaging | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



