
Submitted:
16 April 2024
Posted:
16 April 2024
You are already at the latest version
Abstract
Cholesterol, a crucial component of cell membranes, influences various biological processes, including membrane trafficking, signal transduction, and host-pathogen interactions. Disruptions in cholesterol homeostasis have been linked to congenital and acquired conditions, including neurodegenerative disorders such as Alzheimer’s disease (AD). Previous research from our group has demonstrated that herpes simplex virus type I (HSV-1) induces an AD-like phenotype in several cell models of infection. This study explores the interplay between cholesterol and HSV-1-induced neurodegeneration. The impact of cholesterol was determined by modulating its levels with methyl-beta-cyclodextrin (MβCD) using the neuroblastoma cell lines SK-N-MC and N2a. We have found that HSV-1 infection triggers the intracellular accumulation of cholesterol in structures resembling endolysosomal/autophagic compartments, a process reversible upon MβCD treatment. Moreover, MβCD exhibits inhibitory effects at various stages of HSV-1 infec-tion, underscoring the importance of cellular cholesterol levels not only in the viral entry pro-cess but also in subsequent post-entry stages. MβCD also alleviated several features of AD-like neurodegeneration induced by viral infection, including lysosomal impairment and intracellu-lar accumulation of amyloid-beta peptide (Aβ) and phosphorylated tau. In conclusion, these findings highlight the connection between cholesterol, neurodegeneration, and HSV-1 infection, providing valuable insights into the underlying mechanisms of AD.
Keywords:
Alzheimer’s disease
; HSV-1
; cholesterol
; neuroblastoma cells
; methyl-beta-cyclodextrin
; infection
; neurodegeneration
; lysosomal alterations
; beta-amyloid
; hyperphosphorylated tau
1. Introduction
Cholesterol is an essential lipidic component of cell membranes that regulates their structure and fluidity, and serves as a precursor of diverse compounds, such as steroid hormones, oxysterol, and bile acids. Cholesterol is involved in a wide range of biological processes, including membrane trafficking, signal transduction, host-pathogen interactions, and the immune function. Thus, the maintenance of cholesterol homeostasis is essential for proper functioning of cells and its disruption has been related to several human congenital (Nieman-Pick type C disease (NPC), Familial hypercholesterolemia) and acquired (cardiovascular disorders, neurodegenerative processes, and certain types of cancer) diseases [1,2,3]. In this context, cellular cholesterol arises as a crossroad in cell pathophysiology which could lead to the discovery of new molecular mechanisms involved in disease progression or even serve as therapeutic targets.
Cholesterol is derived from both dietary sources and de novo biosynthesis, primarily occurring in the liver and the brain. The central nervous system (CNS) contains approximately 20-25% of total cholesterol, highlighting its crucial role in neuronal function. Most of CNS cholesterol resides in myelin, and is essential for synaptogenesis, synaptic transmission, and plasticity [2]. Cholesterol homeostasis and metabolism have been extensively studied in the context of neurodegenerative diseases such as Alzheimer’s disease (AD) (reviewed in [4])—the most common form of dementia. Although the main hallmarks of AD—senile plaques mainly composed of beta-amyloid (Aβ) peptides and neurofibrillary tangles composed of hyperphosphorylated tau protein—have been known for more than a century [5], the molecular mechanisms involved in disease progression remain obscure. Recently, cholesterol, along with other genetic and non-genetic factors, have emerged as a potential contributor to the sporadic forms of the disease [6].
On the other hand, previous reports have accounted for the ability of certain viruses to induce a metabolic reprogramming in host cells and interfere with cholesterol homeostasis [7,8]. For many years, our lab has focused on studying the links between AD and herpes simplex virus type I (HSV-1) infection. HSV-1 is a highly prevalent, ubiquitous, and neurotropic DNA virus that, apart from infecting epithelial cells and causing mucosal lesions, can also reach the nervous system and establish latent infections [9]. We have characterized several in vitro models using different cell lines—murine and human neuroblastoma cells and human neural precursors—where HSV-1 infection was able to induce an AD-like phenotype. This phenotype is marked by the inhibition of Aβ secretion, intracellular accumulation of Aβ and hyperphosphorylated tau protein, and alterations in autophagy-lysosomal pathway [10,11,12,13,14]. These results contribute to the growing evidence supporting the infectious hypothesis of AD and the potential role of HSV-1 in neurodegeneration, initially proposed at the end of the 20th century [15,16]. Recent studies point that susceptibility to HSV-1 could be aggravated by the APOE4 genotype, suggesting a shared risk factor for infection and AD-like neurodegeneration [17].
Genetic expression studies in our cell model of infection and oxidative stress identified the lysosomal pathway and cholesterol homeostasis as the main pathways altered [14]. Neurons are particularly vulnerable to disruption of the lysosome system since they live long without cell division. Defects in autophagy and endocytosis may thus play a significant role in neurodegenerative diseases, including AD, as suggested by previous reports [18,19]. In line with this, we have described that HSV-1 infection is able to alter both lysosomal load and lysosome activity. Moreover, we found that deficiency in LAMP2—a lysosome membrane protein involved in fusion of autophagosomes with lysosomes and cholesterol trafficking—leads to a significant reduction of viral infection and attenuates the neurodegeneration markers induced by the virus [20].
Overall, we aim to keep deciphering the connections between HSV-1 infection and AD-like neurodegeneration. In this work, considering that cholesterol dysregulation has been associated with markers of AD neurodegeneration [4] and proposed to play multiple roles in the HSV-1 replicative cycle [21], we focus on the potential role of cholesterol as a bridge between HSV-1 and AD neurodegeneration. Using an in vitro approach to modulate cholesterol levels in neuroblastoma cells, we have observed that HSV-1 triggers intracellular cholesterol accumulation. Moreover, lowering cholesterol levels mitigates infection and ameliorates certain aspects of neurodegeneration, including beta-amyloid accumulation, tau hyperphosphorylation and lysosomal alterations within infected cells. These results point to a pivotal role of cellular cholesterol in the AD-like phenotype induced by HSV-1 in neuronal cells.
2. Materials and Methods
Cell lines and culture medium. SK-N-MC cells (HTB-10), initially described as neuroblastoma and afterwards catalogued as part of the Ewing’s sarcoma tumour family [22], were obtained from the American Type Culture Collection (ATCC). They were cultured as a monolayer in minimal Eagle’s medium (MEM) supplemented with 10% heat-inactivated foetal calf serum, 1 mM sodium pyruvate, nonessential amino acids, 2 mM glutamine, and 50 μg/ml gentamycin. Murine neuroblastoma cell line N2a was supplied by Paul Saftig’s lab [23]. N2a cells were cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% foetal calf serum, 2 mM glutamine, nonessential amino acids, 1 mM sodium pyruvate and 50 μg/ml gentamycin. Both cell lines were cultured at 37°C in a humidified 5% CO2 atmosphere.
HSV-1 infection. When cell cultures reached 70-75% confluence, they were infected with wild-type HSV-1 strain KOS 1.1 at a multiplicity of infection (moi) of 10 plaque-forming units per cell (pfu/cell). The virus was obtained, propagated, and purified from Vero cells, as described in [24]. Two different procedures were followed. In the first procedure, cells were incubated with the viral solution for 1 hour at 37°C. Subsequently, the unbound virus was removed, and the cells were further incubated in culture medium at 37°C until collection. In the second procedure, cells were incubated with the viral solution for 2 hours at 37°C. After viral adsorption, cells were inactivated for two minutes in citrate buffer pH3 and then, culture medium was added. Control samples (mock) were incubated in virus-free suspensions. Viral titres in cell lysates (intracellular viral titre) and cell culture supernatants (extracellular viral titre) were determined by plaque assay. Finally, to modify cholesterol levels, 1.5-3 mM methyl-beta-cyclodextrin (MβCD), 25 μg/ml U18666A and 15 μg/ml cholesterol were used.
Viral DNA quantification. DNA was purified by using the QIAamp® DNA Blood Kit (QIAGEN). The amount of HSV-1 DNA was quantified by real-time quantitative PCR with a CFX-384 Real-Time PCR System (BioRad) using a custom designed TaqMan assay specific for the US12 viral gene (forward primer: 5’-CGTACGCGATGAGATCAATAAAAGG-3’; reverse primer: 5’-GCTCCGGGTCGTGCA-3’; TaqMan probe: 5’-AGGCGGCCAGAACC-3’). Viral DNA content was normalized in terms of human genomic DNA, quantified with a predesigned TaqMan assay specific for the 18S (Hs9999991_s1; Applied Bio-systems). The quantification data were expressed as viral DNA copy number per ng of genomic DNA.
Cell viability assay. To assess cell viability upon different treatments, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction to formazan colorimetric assay was performed. Briefly, cells were culured in 96-well plates and 0.5 mg/ml MTT (Sigma) was added. After 2 hours of incubation with MTT at 37°C, 100 μl of solubilisation buffer (20% SDS, 50% N, N-dimethyl-formamide, pH 4.7) was added, followed by an overnight incubation at 37°C, protected from light. The next day, formazan absorbance was measured at 550 nm in an ELISA Microplate Reader model 680 (BioRad).
Cholesterol quantification. Intracellular cholesterol levels were quantified using the enzymatic fluorogenic assay Amplex Red Cholesterol Assay Kit (Invitrogen) following the provider’s instructions. For the experiments, 10 μg of protein lysates were analysed. Fluorescence was measured in a plate fluorimeter at 560 nm excitation and 590 nm emission wavelengths.
Immunofluorescence analysis and filipin staining. Cells grown on coverslips were fixed in 4% paraformaldehyde (PFA) and permeabilized with blocking solution (2% horse or foetal calf serum, 0.2% Triton X-100 in phosphate buffer saline [PBS] pH 7.4). Then, coverslips were incubated with primary antibodies and with Alexa Fluor-coupled secondary antibodies diluted in blocking solution (Table 1). Finally, cells were counterstained with 4,6-diamino-2-phenylindole (DAPI) (Merck) in PBS and mounted on microscope slides using Mowiol medium (Sigma-Aldrich). The overall procedure was performed at room temperature (RT) and samples were protected from light.
For filipin staining, 4% PFA-fixed cells were incubated for 30 minutes with 50 μg/ml filipin and 1 μM TO-PRO-3, a compound that binds to nuclear DNA, in PBS before mounting with Mowiol medium.
Sample visualization was performed in a FRET inverted microscope Axiovert200 (Zeiss) coupled to a monochrome ccd camera and in a LSM 900 laser scanning confocal microscope (Zeiss) coupled to a vertical Axio Imager 2 vertical microscope (Zeiss). Immunofluorescence images were obtained using Metamorph or ZEN Blue 3.4 imaging software and processed with Adobe Photoshop.
Cell lysates and Western blot analysis. Lysates were obtained by incubating cell samples in a preparation containing proteases (CompleteTM, Mini, EDTA-free Protease Inhibitor Cocktail, Roche) and phosphatases inhibitors (PhosSTOPTM, Roche) in the radioimmunoprecipitation assay (RIPA) buffer (10 mM Tris-HCl pH 7.5, 50 mM NaCl, 1% Nonidet P-40, 0.2% sodium deoxycholate, 0.1% sodium dodecyl sulfate [SDS], 1 mM EDTA). Before Western blot analysis, protein concentration in cell lysates was determined by Bicinchoninic acid assay (BCA, Pierce) following the provider’s instructions. Protein separation was performed using Laemmli discontinuous SDS-polyacrylamide gel electrophoresis. After electrophoresis and transfer to a nitrocellulose membrane, membranes were blocked with PBS-3% BSA-0.2% Tween 20 or PBS-5% nonfat milk-0.2% Tween 20 solution. Incubations with primary and secondary antibodies (Table 1) diluted in dilution buffer (PBS-1% BSA-0.05%Tween 20 or PBS-1% nonfat milk -0.05% Tween 20) were performed at RT. Secondary antibodies were coupled to peroxidase and washing steps were performed in PBS-0.05% Tween 20. Finally, detection by enhanced chemiluminescence was carried out using ECL Western blotting detection reagents (Amersham Biosciences) according to the manufacturer’s instructions.
Quantification of lysosome load. The lysosome load was determined using the acidotropic probe LysoTracker Red DND-99 (LTR), which freely pass through cell membranes and typically concentrate in acidic organelles. As a control, cells were exposed to Bafilomycin A1 (0.1 μM). Before the end of infection or treatments, cells were exposed to 0.5 μM LTR for 1 hour at 37°C in culture medium and then washed with PBS. Then, cells were lysed with RIPA buffer and centrifuged at 13,000 g for 10 minutes. The protein concentration of the lysates was quantified by the BCA method, and fluorescence was recorded using a FLUOstar OPTIMA microplate reader (BMG LABTECH) (excitation wavelength 560 nm and emission wavelength 590 nm).
Cathepsin activity assays. Cathepsin activity assays were conducted following a previously described protocol with minor modifications [25]. Briefly, cells were lysed in a buffer containing 50 mM sodium acetate (pH 5.5), 0.1 M NaCl, 1 mM EDTA, and 0.2% Triton X-100. Lysates were clarified by centrifugation and immediately used for determination of proteolytic activity. 20-50 μg of protein from cell lysates were incubated for 30 minutes in the presence of the following fluorogenic substrates (all from Enzo Life Sciences): Z-VVR-fluorophore 7-amino-4-methyl-coumarin (AMC) (P-199; most sensitive substrate for cathepsin S; 20 mM), and the Cathepsin D/E fluorogenic substrate Mca-GKPILFFRLK (Dnp)-D-Arg-NH2 (P-145; 10 mM). The AMC released was quantified using a microtiter plate reader (Tecan Trading AG) with excitation at 360 nm and emission at 430 nm (Z-VVR-AMC), or excitation at 320 nm and emission at 400 nm for Cathepsin D and E fluorogenic substrates.
Statistical Analysis. Differences between groups were analysed pairwise using the two-tailed student t-test or the one sample t-test, in the case of data expressed as relative values. Significance levels were denoted at p < 0.05 (*), p < 0.01 (**), and p < 0.001 (***). Statistical analyses were performed using Microsoft Excel and GraphPad Software online resources (https://www.graphpad.com/quickcalcs/oneSampleT1/).
3. Results
3.1.1. HSV-1 Infection Increases Intracellular Cholesterol Levels
Previous studies conducted by our research group identified cholesterol homeostasis as one of the most disrupted processes in cell models of HSV-1 infection [14]. Our first aim was to determine cholesterol levels in infected SK-N-MC and N2a cells using a fluorometric method. To validate this approach, we employed two cholesterol modulators known to elevate cellular cholesterol levels: U18666A and water-soluble cholesterol (Figure 1A). Upon exposure to HSV-1, both cell lines also exhibited a significant increase in cholesterol levels (Figure 1B).
Another well-established method for visualizing cellular cholesterol through fluorescence microscopy involves the use of filipin staining. Filipin is a naturally fluorescent polyene antibiotic that selectively binds to cholesterol and not esterified sterols. We employed this method to detect free cholesterol in our cell models. In uninfected cells, cholesterol predominantly localizes to the plasma membrane. However, HSV-1 infection induced a substantial accumulation of intracellular cholesterol (Figure 1C). As a positive control, cells were treated with U18666A, a compound that specifically inhibits cholesterol transport out of late endosomes/lysosomes, leading to cholesterol accumulation within endolysosomal compartments. Intriguingly, the staining pattern of HSV-1-infected cells closely resembled that observed in U18666A-treated cells (Figure 1C). Additionally, the rounded morphology of filipin-positive structures suggested a vesicular nature, implying that HSV-1 infection may trigger the accumulation of cholesterol within endolysosomal compartments. These results raise the question of in which cellular compartments cholesterol accumulates in HSV-1-infected cells. With this goal in mind, the distribution pattern of filipin was examined through confocal analysis. When SK-N-MC cells were exposed to HSV-1, filipin staining colocalized with CD222 (a late endosome marker) and LC3 (an autophagosome marker), but no significant colocalization of filipin with early endosomal (EEA1) or lysosomal markers (LAMP2) was observed (Figure 1D). Taken together, these findings suggest that cholesterol accumulates in late endosomal/autophagic compartments. The absence of colocalization of filipin with lysosome markers may indicate inefficient fusion between late endosomes/autophagosomes and lysosomes caused by HSV-1 infection, as previously described by our group in human neuroblastoma cells [11].
3.1.2. MβCD Reverts the Cholesterol Accumulation Induced by HSV-1
To investigate the role of cellular cholesterol levels on the AD-like phenotype induced by HSV-1, the initial step involved optimizing the agent used to modulate these levels. Methyl-β-cyclodextrin (MβCD) is a compound that extracts cholesterol from cellular membranes, thereby reducing its levels. Initially, the impact of MβCD on cellular viability of SK-N-MC and N2a cells was examined. The results indicated that MβCD did not affect cellular viability at concentrations up to 2 mM in SK-N-MC cells and 3 mM in N2a cells (Figure 2A).
To verify the effectiveness of reducing cholesterol levels with MβCD, the amount of cholesterol present in SK-N-MC and N2a cells infected with HSV-1 and subjected to increasing concentrations of the drug was quantified. The results confirmed the increase in cholesterol levels induced by the infection, and revealed a dose-dependent decrease in cholesterol levels across all tested conditions with MβCD in both cell lines. Specifically, infected cells treated with concentrations of 1.5 - 2 mM in SK-N-MC cells and 2.5 - 3 mM in N2a cells, showed a significant reduction in cholesterol levels, reaching levels comparable to those observed in uninfected and untreated cells (Figure 2B). Based on these findings, concentrations of 1.5 - 2 mM of MβCD were selected for treatments of SK-N-MC cells, while 2.5 - 3 mM was chosen for treatments of N2a cells in subsequent experiments.
With the aim of assessing whether MβCD treatment could reduce cholesterol accumulation in lysosomal compartments, filipin labeling experiments were performed. SK-N-MC and N2a cells were treated with the U18666A compound followed by MβCD treatment for this purpose. Filipin visualization through fluorescence microscopy revealed that U18666A induced intracellular cholesterol accumulation. Interestingly, MβCD resulted in a significant decrease in filipin labeling in U18666A-treated cells (Figure 2C).
After optimizing the treatment conditions with MβCD, the next step was to investigate whether MβCD could reduce intracellular cholesterol accumulation induced by HSV-1. Both uninfected and HSV-1-infected SK-N-MC and N2a cells treated with MβCD were labeled with filipin. Analysis by fluorescence microscopy revealed that MβCD treatment led to a substantial reduction in cholesterol in the plasma membrane of uninfected cells. Moreover, the previously observed virus-induced intracellular cholesterol accumulation was confirmed and a reduction in the intensity of intracellular filipin labeling was noted with MβCD treatment in infected cells (Figure 2D). These findings indicate that MβCD treatment results in a reduction of intracellular cholesterol accumulation in infected neuroblastoma cells.
3.1.3. MβCD Inhibits the Efficiency of HSV-1 Infection
To explore the relevance of cholesterol accumulation induced by HSV-1, the effect of reducing cholesterol levels using the drug MβCD on various aspects related to the efficiency of infection was analyzed. Initially, we aimed to determine whether the decrease in cholesterol levels affected viral protein expression. Western Blot analysis targeting the ICP4 protein (an immediately early protein essential for viral transcription and replication) and glycoprotein C (gC; a “true” late viral protein belonging to the lipid envelope) revealed a significant decrease in viral protein levels induced by MβCD in both SK-N-MC (Figure 3A) and N2a (Figure 3B) cells. Then, quantitative PCR experiments demonstrated that MβCD induced a reduction in viral DNA quantity in both cell lines, indicating its impact on the viral DNA replication process (Figure 3C). Finally, to evaluate the effect of cholesterol level reduction on the formation of infectious viral particles, we analyzed the viral titer in culture supernatants and cell lysates of MβCD-treated SK-N-MC and N2a cells. The results showed that MβCD completely blocked the production of both extracellular and intracellular infectious viral particles (Figure 3D). Collectively, these findings confirm the inhibitory effect of reduced cellular cholesterol levels on HSV-1 infection.
3.1.4. Cholesterol Plays a Role in Post-Entry Stages of HSV-1 Infection
Several studies have identified cholesterol as essential for HSV-1 entry [21,26]. To validate this effect in neuroblastoma cells, MβCD treatment was administered either 1 hour before (-1 hpi) or after viral adsorption (0 hpi). The impact on viral entry was assessed by monitoring the levels of viral protein ICP4 and the number of HSV-1 infected cells at early stages of infection using immunoblotting and immunofluorescence assays with a specific antibody for ICP4. ICP4 is encoded by an immediate-early gene, with expression initiating prior to HSV-1 DNA replication and accumulating within the nucleus of infected cells. Assessing the ICP4 levels at early infection times serves as an indirect measure of viral entry.
Treatment with MβCD one hour before viral adsorption led to a strong reduction in both ICP4 levels (Figure 4A) and the number of HSV-1-infected cells at 5 hpi (Figure 4B). Conversely, the addition of MβCD after viral adsorption did not result in any changes in total ICP4 levels or the percentage of infected cells (Figure 4A and Figure 4B). These findings highlight the significance of cellular cholesterol levels in the viral entry process in SK-N-MC and N2a neuroblastoma cells.
To assess whether cell cholesterol also influences post-entry phases of HSV-1 infection, SK-N-MC and N2a cells were exposed to HSV-1 for 2 hours at 37°C to allow viral entry. Non-internalized viral particles were subsequently inactivated by treatment with citrate buffer. The infected cells were then cultured for 5 or 18 hours in the presence of MβCD. The viral inactivation method involving incubation with citrate buffer at pH 3, allows the removal of all extracellular virus after adsorption, thereby ensuring that any effects induced by MβCD, added to the culture medium after viral inactivation, impact processes subsequent to viral entry. Levels of ICP4 (at 5 hpi) and gC (at 18 hpi) were analyzed using Western Blot. Data analysis revealed that the levels of ICP4 remained unchanged in the presence of MβCD under both experimental conditions (with or without citrate buffer), indicating the absence of significant cholesterol-related impacts on viral entry (Figure 4C). The late protein gC serves as a robust marker at later stages of infection. Interestingly, no changes in gC levels were observed upon extracellular virus inactivation, suggesting that the reduction in gC levels induced by MβCD remains consistent, irrespective of the virus inactivation state (Figure 4D).
In summary, these findings strongly indicate that cholesterol plays a pivotal role in processes subsequent to the viral entry phase. In the following experiments aimed at assessing the role of cholesterol accumulation in the onset of HSV-1-induced AD-like phenotype, MβCD will be administered after viral adsorption to analyze post-entry effects of the drug.
3.1.5. MβCD Reverts the Lysosomal Alterations Induced by HSV-1
To investigate the significance of cholesterol accumulation in AD-like alterations induced by HSV-1, we initially assessed its relevance in various aspects related to HSV-1-induced lysosomal dysfunction. Previous studies from our group demonstrated that HSV-1 infection leads to an increased lysosomal content in SK-N-MC cells [14]. Consequently, we examined the impact of cholesterol reduction on lysosome load in this cell line using the fluorescent acidotropic probe LysoTracker Red (LTR).
The LTR fluorescence assay was validated with bafilomycin A1, a potent vacuolar H+-ATPase inhibitor known to neutralize lysosomal pH and induce a decrease in LTR fluorescence levels (Figure 5A). Quantification of LTR fluorescence levels confirmed a significant increase in lysosomal content provoked by HSV-1. However, when the effects of MβCD were examined, a substantial decrease in LTR fluorescence levels was observed in infected cells, reaching values similar to those observed in untreated and uninfected cells (Figure 5B).
To monitor the effect of cholesterol modulation with MβCD on lysosomal functionality in the SK-N-MC cell line in the presence and absence of HSV-1, the enzymatic activity of lysosomal cathepsins D/E and S was quantified using fluorogenic substrates specific for these proteases. Previous experiments from our group revealed that HSV-1 infection decreased the activity of these hydrolases [14]. To investigate whether MβCD could reverse this effect, both uninfected and infected SK-N-MC cells were treated with MβCD. As a control, cells were exposed to bafilomycin A1, which induces a substantial decrease in cathepsin activities, validating the efficacy of the assay (Figure 5C). Enzymatic activity analysis confirmed the inhibition of cathepsin D/E and S induced by the virus. MβCD treatment did not show any effects in uninfected cells. In contrast, MβCD resulted in an increase in cathepsin enzymatic activity in HSV-1-infected cells, reaching levels similar to those of uninfected control cells (Figure 5D).
In summary, treatment with MβCD is capable of reversing the increase in lysosomal burden and the inhibition of cathepsin activity caused by HSV-1 infection. These data suggest a role of cholesterol in the lysosomal alterations observed in infected cells.
3.1.6. MβCD Attenuates the Neurodegeneration Markers Induced by HSV-1
Our group has previously demonstrated that HSV-1 and HSV-2 infection lead to intracellular accumulation of Aβ peptides and hyperphosphorylated tau [10,12,27]. Thus, our next aim was to assess the impact of cholesterol on these processes. As shown in Figure 1D, our results suggest that cholesterol accumulates in late endosomal/autophagic compartments in infected cells. Moreover, our previous research has shown that Aβ peptides accumulate in these same compartments in infected human neuroblastoma cells [12]. With this background, we conducted an analysis of the colocalization of cholesterol, Aβ and phosphorylated tau (using two antibodies that specifically recognize two phosphorylated epitopes of tau protein present in AD brains: Thr205 and Ser422) in SK-N-MC cells exposed to HSV-1. Confocal microscopy analysis revealed colocalization of both isoforms of Aβ (Figure 6A) and phosphorylated tau (Figure 6B) with filipin staining in infected cells. These data suggest that the accumulation of cholesterol in endolysosomal/autophagic compartments may play a role in the intracellular accumulation of Aβ and phosphorylated tau.
To investigate the impact of cholesterol on Aβ accumulation, we examined Aβ levels in MβCD-treated N2a cells exposed to HSV-1 using confocal microscopy and ELISA assays. These experiments were conducted in N2a cells instead of SK-N-MC cells because the former secrete higher levels of Aβ, and the ELISA analysis results are more consistent and reproducible. Immunofluorescence assays revealed a decrease in the number of Aβ-positive cells in HSV-1-infected cells treated with MβCD (Figure 6C). In addition, ELISA assays were performed to determine changes in secreted Aβ levels resulting from the reduction in cholesterol amount. HSV-1 infection significantly inhibited Aβ secretion, consistent with previous findings from our group in SK-N-MC cells [12]. In contrast to the results of immunofluorescence experiments, extracellular Aβ levels remained unchanged in MβCD-treated cells (Figure 6D).
Finally, to assess whether MβCD treatment affects the levels of phosphorylated tau protein, immunological analyses were performed in SK-N-MC cells. We found that the number of phosphorylated tau-positive cells was considerably lower in infected cells treated with MβCD, consistent with the data obtained in Aβ experiments (Figure 6E). Western blot assays also confirmed that HSV-1 induces an increase in levels of phosphorylated tau protein at the Ser422 epitope. Interestingly, we observed that the MβCD treatment leads to a decrease in levels of phosphorylated tau (Figure 6F). These findings suggest that the reduction of cholesterol levels partially reverses the accumulation of phosphorylated tau induced by the virus.
In summary, our results suggest that intracellular cholesterol accumulation may contribute to the development of an AD-like phenotype induced by HSV-1 in neuroblastoma cells.
4. Discussion
HSV-1 is recognized as a risk factor in sporadic AD, as it triggers the appearance of characteristic neurodegeneration markers of the disease. Previous reports have indicated a potential link between cholesterol and both AD and viral infections [28,29]. Therefore, we hypothesized that cholesterol could mediate the interactions between HSV-1 infection and neurodegeneration. In this context, previous findings from our laboratory have implicated genes related to cholesterol metabolism and autophagy-lysosome pathway in the process of HSV-1 infection and AD pathogenesis [20,30,31]. Thus, we aimed to assess the impact of cellular cholesterol on both infection and neurodegeneration induced by HSV-1 in murine and human neuroblastoma cell cultures.
The fact that viruses are able to hijack the metabolic machinery of host cells paves the way for identifying potential targets to fight against viral infections and their deleterious effects. Virtually, any cellular pathway can be altered during viral infection [32], however, lipid and cholesterol metabolism are underscored among them, given their pleiotropic nature and their role as a central node in cell physiology (reviewed in [33]). In this work, we confirm that changes in cholesterol homeostasis are one of the alterations induced by HSV-1 in our in vitro models. Indeed, we observed an increase in cholesterol levels upon infection along with changes in its intracellular distribution pattern. To date, there are few reports linking herpes virus infections with alterations in cholesterol homeostasis. HSV-1 infection has been shown to impair cholesterol metabolism resulting in the accumulation of cholesteryl esters [34], and to alter cholesterol trafficking in human arterial smooth muscle cells [35]. To our knowledge, this is the first study to describe the accumulation of cholesterol in endolysosomal/autophagic compartments as a consequence of HSV-1 infection. This accumulation points to a likely connection between lysosomal dysfunction and alterations in cholesterol homeostasis during HSV-1 infection, supporting previous findings from our research group [14].
To investigate the role of cholesterol accumulation, we evaluated the effects of MβCD treatment on HSV-1 infection and neurodegeneration. Cyclodextrins (CDs) are commonly used compounds to rapidly deplete cells of cholesterol. They have been tested for clinical purposes in certain Lysosome Storage Disorders (LSDs), such as NPC, and certain neuro-degenerative diseases (reviewed in [36]). NPC is a severe congenital disease caused by mutations in the NPC1 and NPC2 genes, which are involved in cholesterol transport within the lysosome [37]. Commonalities between LSDs with central nervous system manifestations and AD, together with increasing evidence regarding the pivotal role of endolysosomal and autophagic dysfunction in AD, have led some authors to propose AD as a LSD itself [18].
First of all, we established the optimal conditions for MβCD treatment in our experiments to restore cholesterol levels upon infection without compromising cell viability. It is noteworthy that the degree of cholesterol reduction induced by cyclodextrins may vary significantly across different cell types, even under similar concentrations and incubation times. For instance, previous studies have shown that exposure to low concentrations of MβCD for a short duration increased cellular cholesterol in T lymphocytes from young subjects, an effect reversed by extending the exposure time [38]. These observations emphasize the importance of assessing the impact of MβCD on each specific cell line and experimental conditions. In our neuroblastoma cell-based HSV-1 infection models, an 18-hour treatment with MβCD, initiated after viral adsorption, appears to be the optimal strategy for reversing virus-induced intracellular cholesterol accumulation. However, this approach has some drawbacks when studying the role of intracellular cholesterol in the AD-like phenotype induced by HSV-1. Firstly, it significantly affects the infection process. Secondly, the cholesterol reduction takes place not only in endolysosomal organelles but also in all cellular membranes, raising the possibility of unwanted side effects in non-lysosomal membranes. These limitations should be considered when interpreting the experimental results.
To explore the involvement of cholesterol in the HSV-1 viral cycle in neuroblastoma cells, we evaluated various stages of the infection process. As an initial step, we observed a significant inhibition of viral entry with MβCD treatment. These findings align with previous reports emphasizing the importance of cholesterol in the HSV-1 infection process, particularly in virus entry [26,39]. Furthermore, we deepened into these analyses and confirmed the participation of cholesterol in processes subsequent to viral entry phase by adding MβCD after inactivation of non-internalized viral particles, once viral adsorption was completed. Notably, MβCD had a more pronounced effect on the formation of infectious viral particles than on the levels of viral DNA or proteins, with viral particles becoming undetectable in the presence of MβCD. These findings suggest that cholesterol may play a role in later stages of infection such as virion maturation, formation of infectious particles, or virus release from the cell. Thus, we successfully replicated the results obtained by [21], supporting our experimental hypothesis by ensuring the plausible implication of cholesterol in post entry effects of viral infection, particularly those associated with AD-like neurodegeneration.
Our group has previously shown that HSV-1 impairs two processes closely associated with neurodegeneration: tau phosphorylation and amyloid-beta precursor protein proteolytic processing [10,12]. In this report, we describe the accumulation of hyperphosphorylated tau and Aβ in compartments of the autophagy-lysosome pathway in infected cells. Considering the accumulation of both cholesterol and AD-like neurodegeneration markers in these compartments, it is tempting to speculate that neurodegeneration observed in HSV-1-infected cells could be linked to cholesterol accumulation. According to this hypothesis, MβCD completely restored changes related to lysosomal dysfunction, including lysosome load and cathepsin function in infected neuroblastoma cells. In this regard, MβCD is referred as a potential treatment for LSDs and its role restoring autophagy flux and reducing cholesterol accumulation has been confirmed in vitro [37] and in vivo [40]. Moreover, several studies have revealed that inhibition or loss of cathepsins triggers lysosomal dysfunction, leading to intracellular cholesterol and Aβ accumulation, suggesting their involvement in the appearance of neurodegeneration markers [41]. Finally, reduced lysosomal enzyme activity has been associated with AD in various reports (reviewed in [18]), underscoring the importance of cathepsin activity in maintaining lysosomal function and preventing neurodegenerative processes.
In line with these findings, MβCD also ameliorated the alterations in phosphorylated tau and Aβ levels observed in our models. A decrease in the number of cells showing accumulation of Aβ and phosphorylated tau was observed in MβCD-treated neuroblastoma cells exposed to HSV-1. Consistent with this, quantification of intracellular phosphorylated tau revealed that MβCD treatment leads to a decrease in levels of phosphorylated tau, partially reversing the effects of HSV-1 infection. In this regard, alterations in cholesterol homeostasis have been associated with the accumulation of hyperphosphorylated tau and intracellular Aβ [42,43], and cyclodextrin treatment reduced neuroinflammation and cognitive deficits in a transgenic AD mouse model [44]. The decrease in intracellular Aβ and phosphorylated tau upon MβCD treatment described in this work could be explained by several mechanisms. First, a reduction in the number of cells in which the infection successfully progresses could be behind the alleviation of be-ta-amyloid and tau pathology. Second, this effect could be caused by the restoration of lysosomal function, enabling more efficient degradation of protein aberrant forms, or providing cellular protection against HSV-1 infection.
In conclusion, whereas it is unlikely that efficient treatments for neurodegeneration will consist of single-target strategies, these basic research approaches are useful to progressively elucidate the complex scenarios and molecular networks involved in AD and its links with different environmental risk factors such as HSV-1 infection. Although further analyses are required to confirm these hypotheses, our proposal and the results obtained support the relevance of cholesterol in viral infection and its impact on neurodegeneration, which shed some light on this likely bridge connecting HSV-1 with AD.
Author Contributions
Conceptualization, J.A. and M.J.B.; methodology, J.A. and M.J.B.; validation, J.A and M.J.B.; formal analysis, J.A.; investigation, J.A., B.I., A.Z., I.S., V.M., J.T. and H.K..; resources, J.A and M.J.B.; data curation, J.A.; writing—original draft preparation, B.S. and J.A.; writing—review and editing, B.S., J.A and M.J.B.; visualization, J.A.; supervision, J.A and M.J.B.; project administration, J.A and M.J.B.; funding acquisition, M.J.B.. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by the Spanish Ministerio de Ciencia e Innovación (PID2020-113921RB-I00). Blanca Salgado is recipient of a Predoctoral Research Contract from the Spanish Ministerio de Universidades (FPU21/06616).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
Institutional grants from the Fundación Ramón Areces and Banco de Santander to the CBMSO are acknowledged.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Luo, J.; Yang, H.; Song, B.L. Mechanisms and Regulation of Cholesterol Homeostasis. Nat. Rev. Mol. Cell Biol. 2019, 21, 225–245. [CrossRef]
- Ho, W.Y.; Hartmann, H.; Ling, S. Central Nervous System Cholesterol Metabolism in Health and Disease. IUBMB Life 2022, 74, 826–841. [CrossRef]
- Cardoso, D.; Perucha, E. Cholesterol Metabolism: A New Molecular Switch to Control Inflammation. Clin. Sci. 2021, 135, 1389–1408. [CrossRef]
- Hu, Z.L.; Yuan, Y.Q.; Tong, Z.; Liao, M.Q.; Yuan, S.L.; Jian, Y.; Yang, J.L.; Liu, W.F. Reexamining the Causes and Effects of Cholesterol Deposition in the Brains of Patients with Alzheimer’s Disease. Mol. Neurobiol. 2023, 1, 1–17. [CrossRef]
- Stelzmann, R.A.; Norman Schnitzlein, H.; Reed Murtagh, F. An English Translation of Alzheimer’s 1907 Paper, “Über Eine Eigenartige Erkankung Der Hirnrinde.” Clin. Anat. 1995, 8, 429–431. [CrossRef]
- Breijyeh, Z.; Karaman, R.; Muñoz-Torrero, D.; Dembinski, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules 2020, 25, 5789. [CrossRef]
- Huang, P.; Wang, X.; Lei, M.; Ma, Y.; Chen, H.; Sun, J.; Hu, Y.; Shi, J. Metabolomics Profiles Reveal New Insights of Herpes Simplex Virus Type 1 Infection. Int. J. Mol. Sci. 2023, 24, 1521–1521. [CrossRef]
- Lange, P.; Lagunoff, M.; Tarakanova, V. Chewing the Fat: The Conserved Ability of DNA Viruses to Hijack Cellular Lipid Metabolism. Viruses 2019, 11, 119. [CrossRef]
- Marcocci, M.E.; Napoletani, G.; Protto, V.; Kolesova, O.; Piacentini, R.; Li Puma, D.D.; Lomonte, P.; Grassi, C.; Palamara, A.T.; De Chiara, G. Herpes Simplex Virus-1 in the Brain: The Dark Side of a Sneaky Infection. Trends Microbiol. 2020, 28, 808–820. [CrossRef]
- Álvarez, G.; Aldudo, J.; Alonso, M.; Santana, S.; Valdivieso, F. Herpes Simplex Virus Type 1 Induces Nuclear Accumulation of Hyperphosphorylated Tau in Neuronal Cells. J. Neurosci. Res. 2012, 90, 1020–1029. [CrossRef]
- Santana, S.; Bullido, M.J.; Recuero, M.; Valdivieso, F.; Aldudo, J. Herpes Simplex Virus Type i Induces an Incomplete Autophagic Response in Human Neuroblastoma Cells. J. Alzheimers Dis. 2012, 30, 815–831. [CrossRef]
- Santana, S.; Recuero, M.; Bullido, M.J.; Valdivieso, F.; Aldudo, J. Herpes Simplex Virus Type I Induces the Accumulation of Intracellular β-Amyloid in Autophagic Compartments and the Inhibition of the Non-Amyloidogenic Pathway in Human Neuroblastoma Cells. Neurobiol. Aging 2012, 33, 430.e19-430.e33. [CrossRef]
- Salgado, B.; Sastre, I.; Bullido, M.J.; Aldudo, J. Herpes Simplex Virus Type 1 Induces AD-like Neurodegeneration Markers in Human Progenitor and Differentiated ReNcell VM Cells. Microorganisms 2023, 11, 1205. [CrossRef]
- Kristen, H.; Sastre, I.; Muñoz-Galdeano, T.; Recuero, M.; Aldudo, J.; Bullido, M.J. The Lysosome System Is Severely Impaired in a Cellular Model of Neurodegeneration Induced by HSV-1 and Oxidative Stress. Neurobiol. Aging 2018, 68, 5–17. [CrossRef]
- Ball, M.J. “Limbic Predilection in Alzheimer Dementia: Is Reactivated Herpesvirus Involved?” Can. J. Neurol. Sci. 1982, 9, 303–306. [CrossRef]
- Jamieson, G.A.; Maitland, N.J.; Wilcock, G.K.; Craske, J.; Itzhaki, R.F. Latent Herpes Simplex Virus Type 1 in Normal and Alzheimer’s Disease Brains. J. Med. Virol. 1991, 33, 224–227. [CrossRef]
- Linard, M.; Letenneur, L.; Garrigue, I.; Doize, A.; Dartigues, J.F.; Helmer, C. Interaction between APOE4 and Herpes Simplex Virus Type 1 in Alzheimer’s Disease. Alzheimers Dement. 2020, 16, 200–208. [CrossRef]
- Whyte, L.S.; Lau, A.A.; Hemsley, K.M.; Hopwood, J.J.; Sargeant, T.J. Endo-Lysosomal and Autophagic Dysfunction: A Driving Factor in Alzheimer’s Disease? J. Neurochem. 2017, 140, 703–717. [CrossRef]
- Malik, B.R.; Maddison, D.C.; Smith, G.A.; Peters, O.M. Autophagic and Endo-Lysosomal Dysfunction in Neurodegenerative Disease. Mol. Brain 2019, 12. [CrossRef]
- Kristen, H.; Sastre, I.; Aljama, S.; Fuentes, M.; Recuero, M.; Frank-García, A.; Martin, A.; Sanchez-Juan, P.; Lage, C.; Bullido, M.J.; et al. LAMP2 Deficiency Attenuates the Neurodegeneration Markers Induced by HSV-1 Infection. Neurochem. Int. 2021, 146, 105032. [CrossRef]
- Wudiri, G.A.; Nicola, A.V. Cellular Cholesterol Facilitates the Postentry Replication Cycle of Herpes Simplex Virus 1. J. Virol. 2017, 91, e00445-17. [CrossRef]
- Staege, M.S.; Hutter, C.; Neumann, I.; Foja, S.; Hattenhorst, U.E.; Hansen, G.; Afar, D.; Burdach, S.E.G. DNA Microarrays Reveal Relationship of Ewing Family Tumors to Both Endothelial and Fetal Neural Crest-Derived Cells and Define Novel Targets. Cancer Res. 2004, 64, 8213–8221. [CrossRef]
- Rothaug, M.; Stroobants, S.; Schweizer, M.; Peters, J.; Zunke, F.; Allerding, M.; D’Hooge, R.; Saftig, P.; Blanz, J. LAMP-2 Deficiency Leads to Hippocampal Dysfunction but Normal Clearance of Neuronal Substrates of Chaperone-Mediated Autophagy in a Mouse Model for Danon Disease. Acta Neuropathol. Commun. 2015, 3, 6. [CrossRef]
- Burgos, J.S.; Ramirez, C.; Sastre, I.; Bullido, M.J.; Valdivieso, F. Involvement of Apolipoprotein E in the Hematogenous Route of Herpes Simplex Virus Type 1 to the Central Nervous System. J. Virol. 2002, 76, 12394–12398. [CrossRef]
- Porter, K.; Nallathambi, J.; Lin, Y.; Liton, P.B. Lysosomal Basification and Decreased Autophagic Flux in Oxidatively Stressed Trabecular Meshwork Cells. Autophagy 2013, 9, 581–594. [CrossRef]
- Bender, F.C.; Whitbeck, J.C.; Ponce De Leon, M.; Lou, H.; Eisenberg, R.J.; Cohen, G.H. Specific Association of Glycoprotein B with Lipid Rafts during Herpes Simplex Virus Entry. J. Virol. 2003, 77, 9542–9552. [CrossRef]
- Kristen, H.; Santana, S.; Sastre, I.; Recuero, M.; Bullido, M.J.; Aldudo, J. Herpes Simplex Virus Type 2 Infection Induces AD-like Neurodegeneration Markers in Human Neuroblastoma Cells. Neurobiol. Aging 2015, 36, 2737–2747. [CrossRef]
- Gao, Y.; Ye, S.; Tang, Y.; Tong, W.; Sun, S. Brain Cholesterol Homeostasis and Its Association with Neurodegenerative Diseases. Neurochem. Int. 2023, 171, 105635. [CrossRef]
- Wang, Y.; Gao, L. Cholesterol: A Friend to Viruses. Int. Rev. Immunol. 2024, 1–15. [CrossRef]
- Llorente, P.; Mejías, V.; Sastre, I.; Recuero, M.; Aldudo, J.; Bullido, M.J. Matrix Metalloproteinase 14 Regulates HSV-1 Infection in Neuroblastoma Cells. Antiviral Res. 2021, 192, 105116. [CrossRef]
- Recuero, M.; Vicente, M.C.; Martínez-García, A.; Ramos, M.C.; Carmona-Saez, P.; Sastre, I.; Aldudo, J.; Vilella, E.; Frank, A.; Bullido, M.J.; et al. A Free Radical-generating System Induces the Cholesterol Biosynthesis Pathway: A Role in Alzheimer’s Disease. Aging Cell 2009, 8, 128–139. [CrossRef]
- Leblanc, P.; Vorberg, I.M. Viruses in Neurodegenerative Diseases: More than Just Suspects in Crimes. PLOS Pathog. 2022, 18, e1010670. [CrossRef]
- Omasta, B.; Tomaskova, J. Cellular Lipids—Hijacked Victims of Viruses. Viruses 2022, 14, 1896. [CrossRef]
- Hajjar, D.P.; Pomerantz, K.B.; Falcone, D.J.; Weksler, B.B.; Grant, A.J. Herpes Simplex Virus Infection in Human Arterial Cells. Implications in Arteriosclerosis. J. Clin. Invest. 1987, 80, 1317–1321. [CrossRef]
- Hsu, H.Y.; Nicholson, A.C.; Pomerantz, K.B.; Kaner, R.J.; Hajjar, D.P. Altered Cholesterol Trafficking in Herpesvirus-Infected Arterial Cells. Evidence for Viral Protein Kinase-Mediated Cholesterol Accumulation. J Biol Chem 1995, 270, 19630–19637. [CrossRef]
- Braga, S.S. Molecular Mind Games: The Medicinal Action of Cyclodextrins in Neurodegenerative Diseases. Biomolecules 2023, 13, 666. [CrossRef]
- Dai, S.; Dulcey, A.E.; Hu, X.; Wassif, C.A.; Porter, F.D.; Austin, C.P.; Ory, D.S.; Marugan, J.; Zheng, W. Methyl-β-Cyclodextrin Restores Impaired Autophagy Flux in Niemann-Pick C1-Deficient Cells through Activation of AMPK. Autophagy 2017, 13, 1435–1451. [CrossRef]
- Zidovetzki, R.; Levitan, I. Use of Cyclodextrins to Manipulate Plasma Membrane Cholesterol Content: Evidence, Misconceptions and Control Strategies. Biochim Biophys Acta 2007, 1768, 1311–1324. [CrossRef]
- Rahn, E.; Petermann, P.; Hsu, M.-J.; Rixon, F.J.; Knebel-Mörsdorf, D. Entry Pathways of Herpes Simplex Virus Type 1 into Human Keratinocytes Are Dynamin- and Cholesterol-Dependent. PLoS ONE 2011, 6, e25464. [CrossRef]
- Davidson, C.D.; Ali, N.F.; Micsenyi, M.C.; Stephney, G.; Renault, S.; Dobrenis, K.; Ory, D.S.; Vanier, M.T.; Walkley, S.U. Chronic Cyclodextrin Treatment of Murine Niemann-Pick C Disease Ameliorates Neuronal Cholesterol and Glycosphingolipid Storage and Disease Progression. PloS One 2009, 4, e6951. [CrossRef]
- Cermak, S.; Kosicek, M.; Mladenovic-Djordjevic, A.; Smiljanic, K.; Kanazir, S.; Hecimovic, S. Loss of Cathepsin B and L Leads to Lysosomal Dysfunction, NPC-Like Cholesterol Sequestration and Accumulation of the Key Alzheimer’s Proteins. PLOS ONE 2016, 11, e0167428. [CrossRef]
- Barbero-Camps, E.; Roca-Agujetas, V.; Bartolessis, I.; De Dios, C.; Fernández-Checa, J.C.; Marí, M.; Morales, A.; Hartmann, T.; Colell, A. Cholesterol Impairs Autophagy-Mediated Clearance of Amyloid Beta While Promoting Its Secretion. Autophagy 2018, 14, 1129–1154. [CrossRef]
- Van Der Kant, R.; Langness, V.F.; Herrera, C.M.; Williams, D.A.; Fong, L.K.; Leestemaker, Y.; Steenvoorden, E.; Rynearson, K.D.; Brouwers, J.F.; Helms, J.B.; et al. Cholesterol Metabolism Is a Druggable Axis That Independently Regulates Tau and Amyloid-β in iPSC-Derived Alzheimer’s Disease Neurons. Cell Stem Cell 2019, 24, 363-375.e9. [CrossRef]
- Yao, J.; Ho, D.; Calingasan, N.Y.; Pipalia, N.H.; Lin, M.T.; Beal, M.F. Neuroprotection by Cyclodextrin in Cell and Mouse Models of Alzheimer Disease. J. Exp. Med. 2012, 209, 2501–2513. [CrossRef]
Figure 1.
HSV-1 infection induces intracellular cholesterol accumulation. (A) Intracellular cholesterol levels expressed as nanograms of cholesterol per micrograms of protein in SK-N-MC and N2a neuroblastoma cells treated with U18666A and water soluble cholesterol. (B) Intracellular levels of cholesterol in cultures infected with HSV-1 at multiplicity of infection (moi) 10 for 18 hours compared to mock-infected cells. The graph data show the mean ± SEM of at least 4 independent experiments. Significance was recorded at p < 0.05 (*), p < 0.01 (**), and p < 0.001 (***). (C) Confocal microscopy images of filipin staining (green) show cholesterol distribution pattern in SK-N-MC and N2a cells infected with HSV-1 (moi 10) or treated with U18666A. TO-PRO-3-stained nuclei (red) are also shown. (D) Immunofluorescence images of SK-N-MC cells infected with HSV-1 at moi 10 were obtained with filipin staining and antibodies recognizing different markers of early (EEA1) and late endosomes (CD222), autophagosomes (LC3) and lysosomes (LAMP2). Arrowheads in the merge panels showed the colocalization of filipin with CD222 and LC3 in HSV-1-infected cells. Scale bar: 10 µm.
Figure 1.
HSV-1 infection induces intracellular cholesterol accumulation. (A) Intracellular cholesterol levels expressed as nanograms of cholesterol per micrograms of protein in SK-N-MC and N2a neuroblastoma cells treated with U18666A and water soluble cholesterol. (B) Intracellular levels of cholesterol in cultures infected with HSV-1 at multiplicity of infection (moi) 10 for 18 hours compared to mock-infected cells. The graph data show the mean ± SEM of at least 4 independent experiments. Significance was recorded at p < 0.05 (*), p < 0.01 (**), and p < 0.001 (***). (C) Confocal microscopy images of filipin staining (green) show cholesterol distribution pattern in SK-N-MC and N2a cells infected with HSV-1 (moi 10) or treated with U18666A. TO-PRO-3-stained nuclei (red) are also shown. (D) Immunofluorescence images of SK-N-MC cells infected with HSV-1 at moi 10 were obtained with filipin staining and antibodies recognizing different markers of early (EEA1) and late endosomes (CD222), autophagosomes (LC3) and lysosomes (LAMP2). Arrowheads in the merge panels showed the colocalization of filipin with CD222 and LC3 in HSV-1-infected cells. Scale bar: 10 µm.
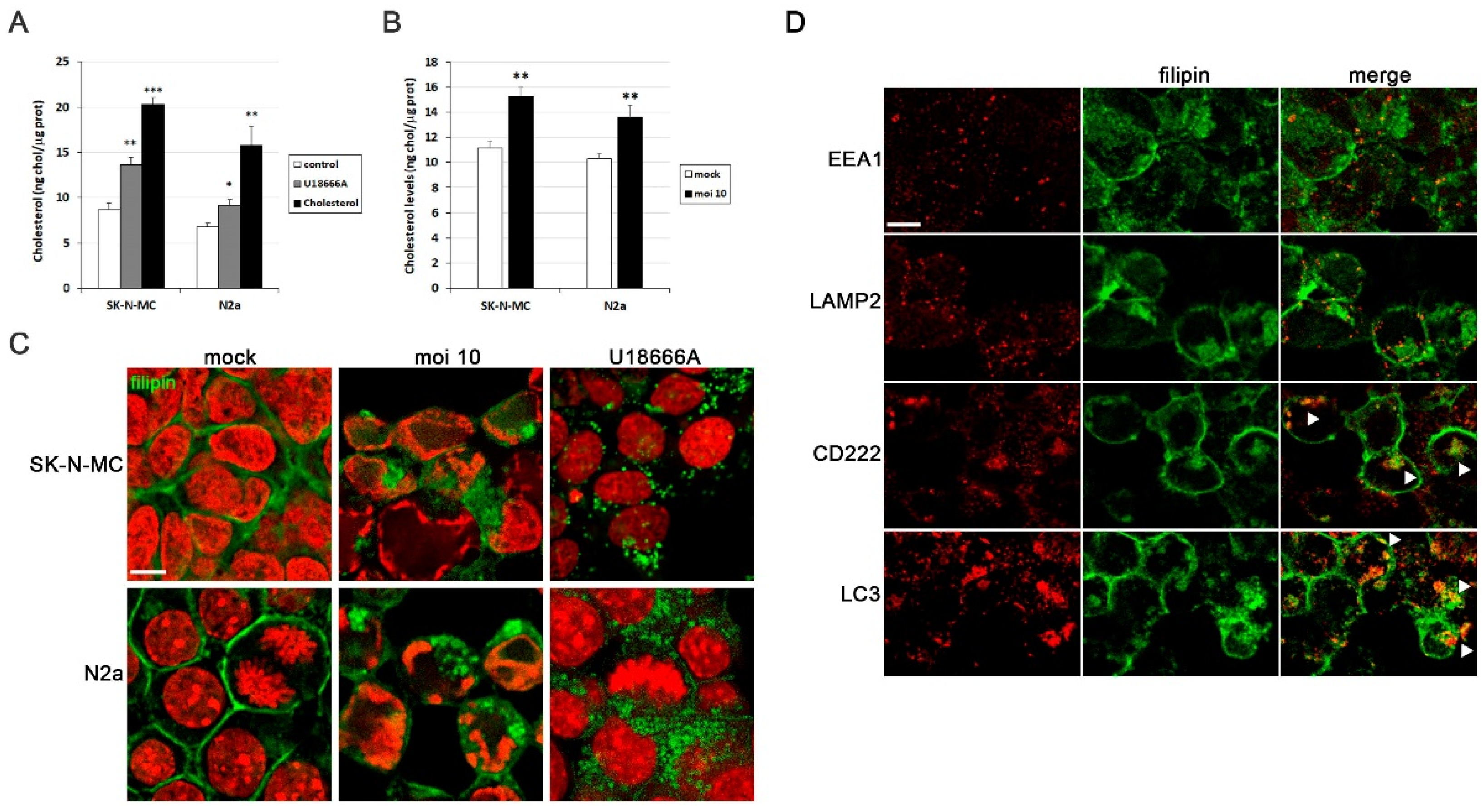
Figure 2.
MβCD reverses the cholesterol accumulation induced by HSV-1. (A) Cell viability of SK-N-MC and N2a cells subjected to 18 hours of treatment with different doses of MβCD was monitored using the MTT reduction assay. (B) Effects of different concentrations of MβCD on intracellular levels of cholesterol in infected (moi 10) and mock-infected cells. Cholesterol levels are shown as a percentage versus mock-infected and non-treated cultures. The graph data show the mean ± SEM of at least 3 independent experiments. Significance was recorded at p < 0.05 (*) and p < 0.01 (**). (C and D) The effect of MβCD on cholesterol distribution pattern in SK-N-MC and N2a cells treated with U18666A (C) or infected with HSV-1 at moi 10 for 18 hours (D) was tested. Fluorescence microscopy images show filipin staining in green and cellular nuclei stained with TO-PRO-3 in red. Scale bar: 10 µm.
Figure 2.
MβCD reverses the cholesterol accumulation induced by HSV-1. (A) Cell viability of SK-N-MC and N2a cells subjected to 18 hours of treatment with different doses of MβCD was monitored using the MTT reduction assay. (B) Effects of different concentrations of MβCD on intracellular levels of cholesterol in infected (moi 10) and mock-infected cells. Cholesterol levels are shown as a percentage versus mock-infected and non-treated cultures. The graph data show the mean ± SEM of at least 3 independent experiments. Significance was recorded at p < 0.05 (*) and p < 0.01 (**). (C and D) The effect of MβCD on cholesterol distribution pattern in SK-N-MC and N2a cells treated with U18666A (C) or infected with HSV-1 at moi 10 for 18 hours (D) was tested. Fluorescence microscopy images show filipin staining in green and cellular nuclei stained with TO-PRO-3 in red. Scale bar: 10 µm.
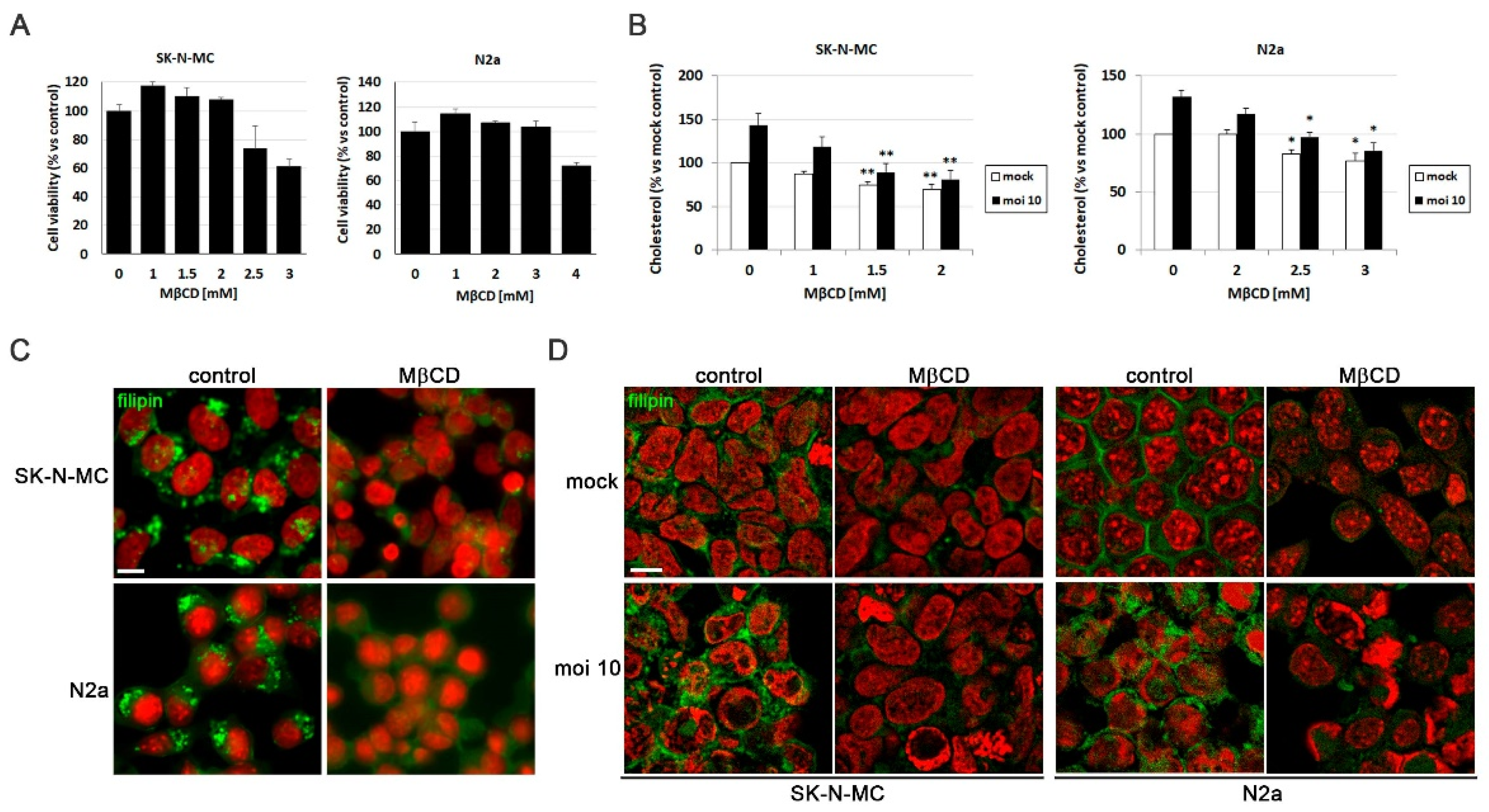
Figure 3.
MβCD inhibits the efficiency of HSV-1 infection. The impact of MβCD treatment on the levels of viral proteins ICP4 and glycoprotein C (gC) was determined through Western blot analysis in SK-N-MC (A) and N2a (B) cells exposed to HSV-1 at moi 10 for 18 hours. A tubulin blot is shown to ensure equal protein load. In all graphs, data represent the mean ± SEM of 3 experiments. (C) The effect of MβCD on viral replication was evaluated by quantifying viral DNA copies in both neuroblastoma cells lines. The graph data show the mean ± SEM of 4 independent experiments. Significance was recorded at p < 0.05 (*), p < 0.01 (**), and p < 0.001 (***). (D) Intracellular and extracellular viral titres were determined by plaque assays in SK-N-MC and N2a cells infected with HSV-1 at moi 10 for 18 hours and subsequently exposed to MβCD (2 mM for SK-N-MC cells and 3 mM for N2a cells). Data from a representative experiment are shown.
Figure 3.
MβCD inhibits the efficiency of HSV-1 infection. The impact of MβCD treatment on the levels of viral proteins ICP4 and glycoprotein C (gC) was determined through Western blot analysis in SK-N-MC (A) and N2a (B) cells exposed to HSV-1 at moi 10 for 18 hours. A tubulin blot is shown to ensure equal protein load. In all graphs, data represent the mean ± SEM of 3 experiments. (C) The effect of MβCD on viral replication was evaluated by quantifying viral DNA copies in both neuroblastoma cells lines. The graph data show the mean ± SEM of 4 independent experiments. Significance was recorded at p < 0.05 (*), p < 0.01 (**), and p < 0.001 (***). (D) Intracellular and extracellular viral titres were determined by plaque assays in SK-N-MC and N2a cells infected with HSV-1 at moi 10 for 18 hours and subsequently exposed to MβCD (2 mM for SK-N-MC cells and 3 mM for N2a cells). Data from a representative experiment are shown.
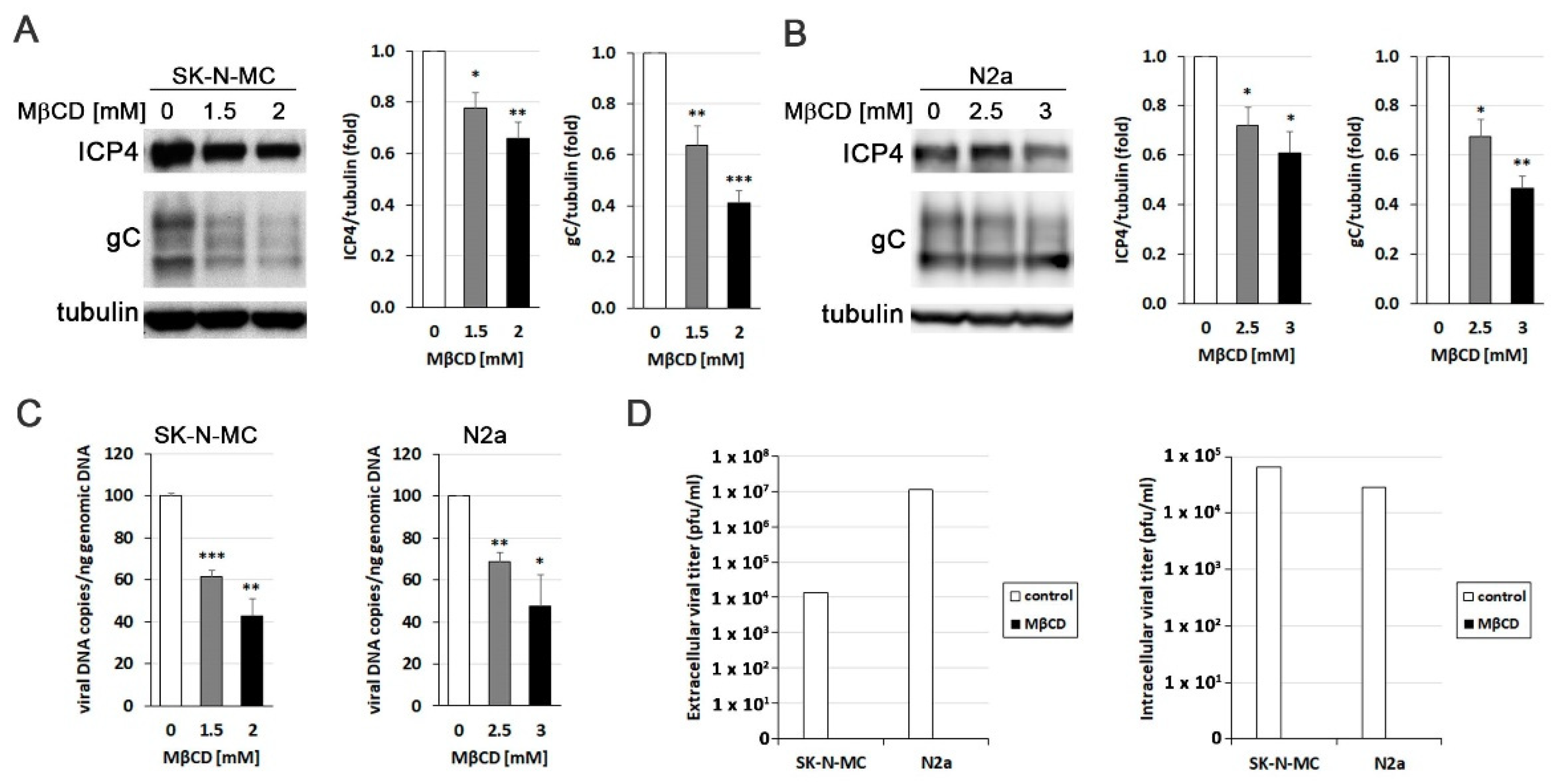
Figure 4.
MβCD affects post-entry stages of HSV-1 infection. (A) The effects of MβCD treatment at different times (1 hour before (-1 hpi) or just after (0 hpi) viral adsorption) on ICP4 levels were assessed through Western blot analysis in SK-N-MC and N2a cells infected with HSV-1 at moi 10 for 5 hours. (B) Immunofluorescence images of ICP4 in neuroblastoma cells infected with HSV-1 at moi 10 for 5 hours show the effects of MβCD added either before (-1 hpi) or after (0 hpi) viral adsorption. DAPI-stained nuclei are also shown. Scale bar: 10 µm. (C and D) SK-N-MC and N2a cells infected with HSV-1 (moi 10) were treated with citrate buffer (pH 3.0) to inactivate non-internalized virus. Cells were then treated with different concentrations of MβCD. Levels of ICP4 (C) and gC (D) viral proteins were monitored at 5 and 18 hpi respectively by Western blot analysis. In all panels, a tubulin blot is shown to ensure equal protein load, and the ratio of viral proteins to tubulin, obtained by densitometric analysis, is shown below the blots. In the Figure 4A, Figure 4C and Figure 4D, the vertical black lines indicate that the blots have been spliced. The spliced fragments come from the same original image.
Figure 4.
MβCD affects post-entry stages of HSV-1 infection. (A) The effects of MβCD treatment at different times (1 hour before (-1 hpi) or just after (0 hpi) viral adsorption) on ICP4 levels were assessed through Western blot analysis in SK-N-MC and N2a cells infected with HSV-1 at moi 10 for 5 hours. (B) Immunofluorescence images of ICP4 in neuroblastoma cells infected with HSV-1 at moi 10 for 5 hours show the effects of MβCD added either before (-1 hpi) or after (0 hpi) viral adsorption. DAPI-stained nuclei are also shown. Scale bar: 10 µm. (C and D) SK-N-MC and N2a cells infected with HSV-1 (moi 10) were treated with citrate buffer (pH 3.0) to inactivate non-internalized virus. Cells were then treated with different concentrations of MβCD. Levels of ICP4 (C) and gC (D) viral proteins were monitored at 5 and 18 hpi respectively by Western blot analysis. In all panels, a tubulin blot is shown to ensure equal protein load, and the ratio of viral proteins to tubulin, obtained by densitometric analysis, is shown below the blots. In the Figure 4A, Figure 4C and Figure 4D, the vertical black lines indicate that the blots have been spliced. The spliced fragments come from the same original image.
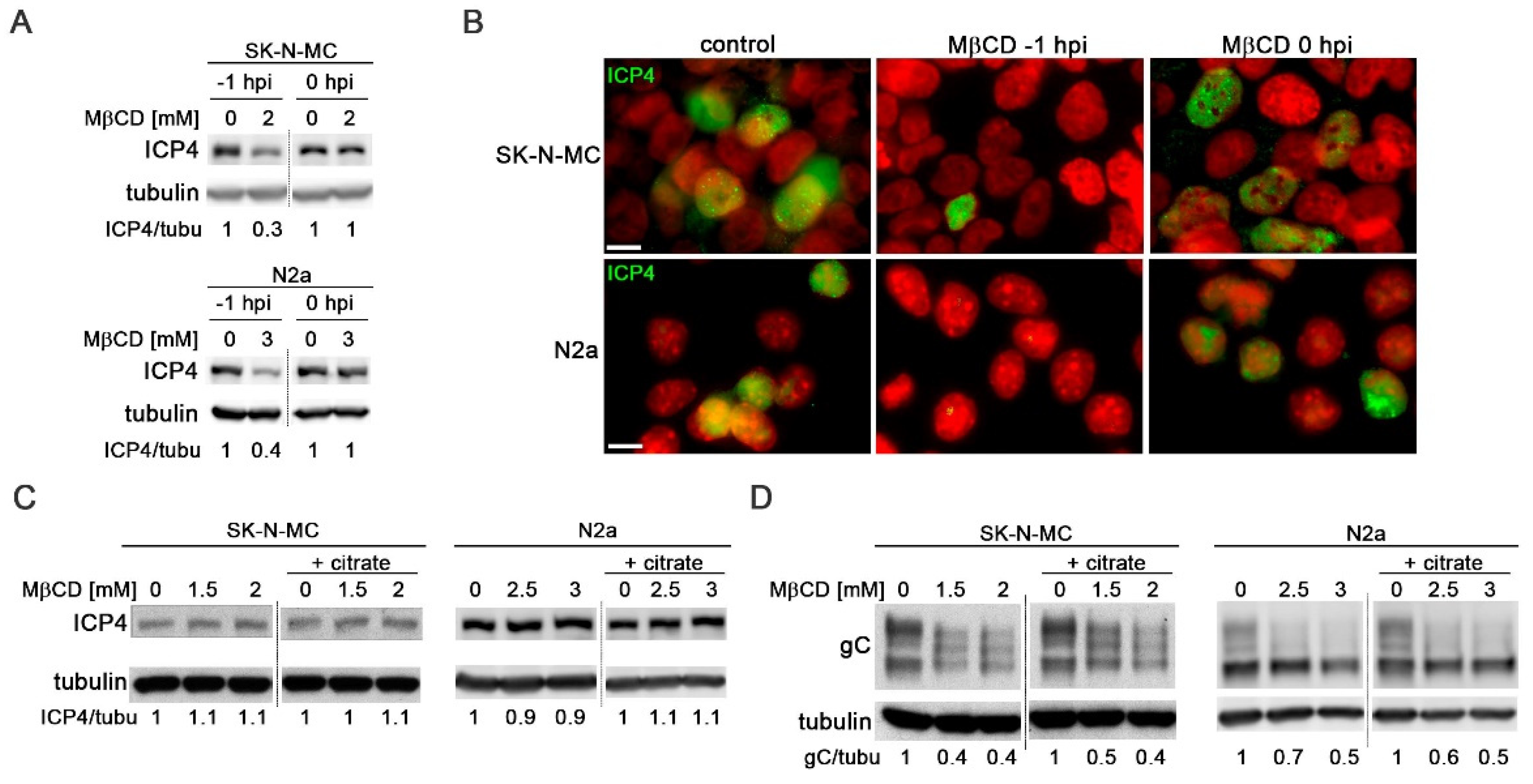
Figure 5.
MβCD treatment reverses the lysosomal alterations induced by HSV-1. (A and B) Analysis of lysosomal load by quantification of LysoTracker Red (LTR) fluorescence in SK-N-MC cells treated with 0.1 µM bafilomycin A1 (A) or infected with HSV-1 at moi 10 and treated with 2 mM MβCD for 18 hours (B). (C and D) The relative enzymatic activities of cathepsins D/E and S were quantified in SK-N-MC cells treated with 0.1 µM bafilomycin A1 (C) or infected with HSV-1 at moi 10 and treated with 2 mM MβCD for 18 hours (D). The graph data show the mean ± SEM from at least 4 independent experiments. Significance was recorded at p < 0.05 (*), p < 0.01 (**), and p < 0.001 (***).
Figure 5.
MβCD treatment reverses the lysosomal alterations induced by HSV-1. (A and B) Analysis of lysosomal load by quantification of LysoTracker Red (LTR) fluorescence in SK-N-MC cells treated with 0.1 µM bafilomycin A1 (A) or infected with HSV-1 at moi 10 and treated with 2 mM MβCD for 18 hours (B). (C and D) The relative enzymatic activities of cathepsins D/E and S were quantified in SK-N-MC cells treated with 0.1 µM bafilomycin A1 (C) or infected with HSV-1 at moi 10 and treated with 2 mM MβCD for 18 hours (D). The graph data show the mean ± SEM from at least 4 independent experiments. Significance was recorded at p < 0.05 (*), p < 0.01 (**), and p < 0.001 (***).
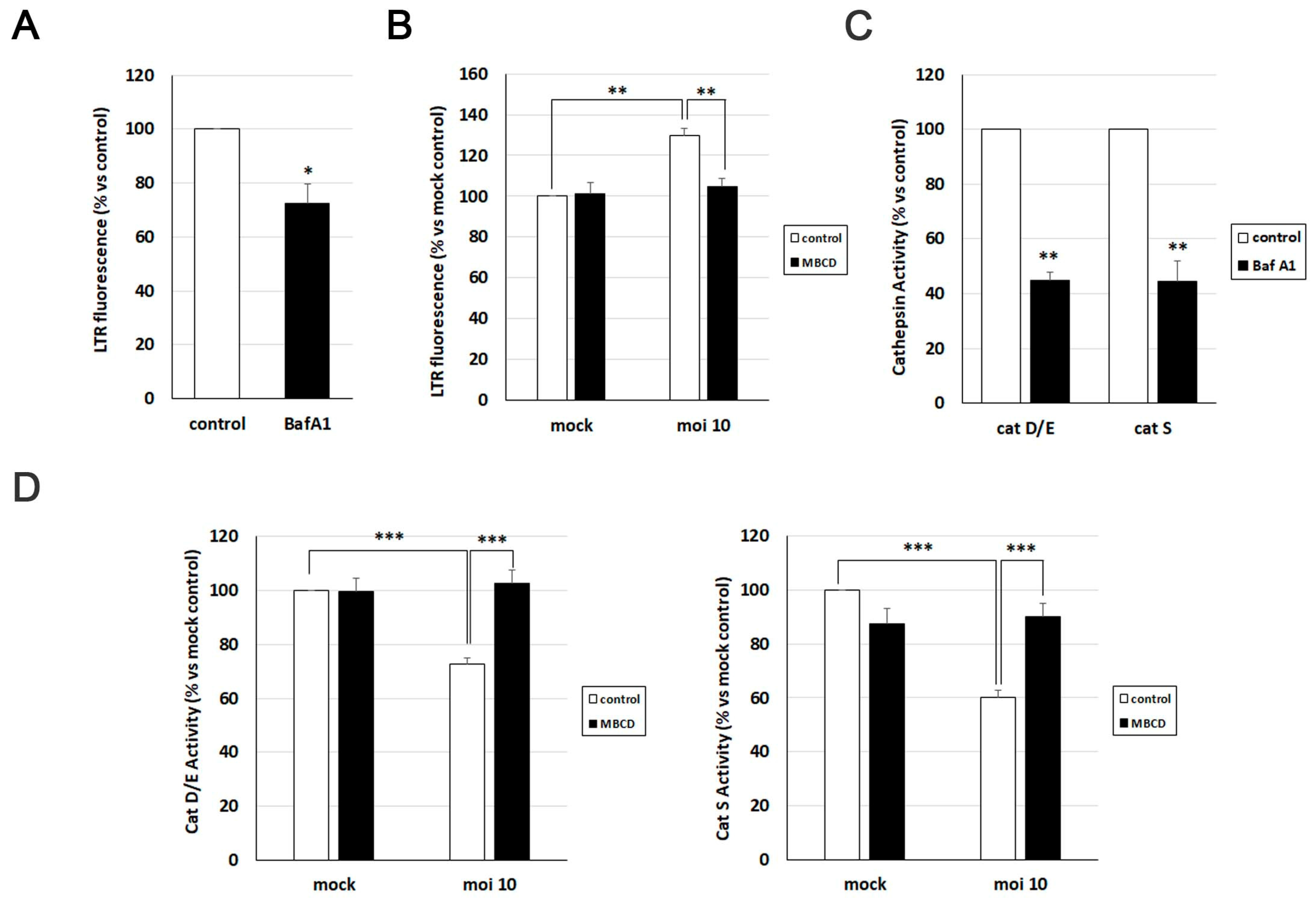
Figure 6.
MβCD attenuates the neurodegeneration markers induced by HSV-1. (A and B) Immunofluorescence images of SK-N-MC cells uninfected (mock) or infected with HSV-1 at moi 10 for 18 hours were obtained with filipin staining and antibodies recognizing Aβ peptides (A) and phosphorylated tau at epitopes Thr205 and Ser422 (B). Arrowheads in the merge panels showed the colocalization of filipin with Aβ and phosphorylated tau in HSV-1-infected cells. Scale bar: 10 µm. (C) N2a cells were infected with HSV-1 at moi 10 for 18 hours and treated with 3 mM MβCD. Immunofluorescence images show Aβ40 and Aβ42 staining in green and cellular nuclei stained with DAPI in red. Scale bar: 10 µm. (D) Quantitative analysis of secreted Aβ levels by ELISA. N2a cells were infected with HSV-1 (moi 10) for 18 hours and treated with 3 mM MβCD. The graph data show the mean ± SEM of 6 independent experiments. Significance was recorded at p < 0.001 (***). (E) SK-N-MC cells were infected with HSV-1 at moi 10 for 18 hours and treated with 2 mM MβCD. Phosphorylation of Thr205 epitope of tau was analyzed by immunofluorescence. DAPI-stained nuclei are also shown (red). Scale bar: 10 µm. (F) SK-N-MC cells were infected with HSV-1 at moi 10 for 18 hours and treated with different concentrations of MβCD. Levels of Ser422-phosphorylated tau were monitored by Western blot analysis. A tubulin blot is shown to ensure equal protein load. The ratio of phosphorylated tau to tubulin, obtained by densitometric analysis, is shown below the blots.
Figure 6.
MβCD attenuates the neurodegeneration markers induced by HSV-1. (A and B) Immunofluorescence images of SK-N-MC cells uninfected (mock) or infected with HSV-1 at moi 10 for 18 hours were obtained with filipin staining and antibodies recognizing Aβ peptides (A) and phosphorylated tau at epitopes Thr205 and Ser422 (B). Arrowheads in the merge panels showed the colocalization of filipin with Aβ and phosphorylated tau in HSV-1-infected cells. Scale bar: 10 µm. (C) N2a cells were infected with HSV-1 at moi 10 for 18 hours and treated with 3 mM MβCD. Immunofluorescence images show Aβ40 and Aβ42 staining in green and cellular nuclei stained with DAPI in red. Scale bar: 10 µm. (D) Quantitative analysis of secreted Aβ levels by ELISA. N2a cells were infected with HSV-1 (moi 10) for 18 hours and treated with 3 mM MβCD. The graph data show the mean ± SEM of 6 independent experiments. Significance was recorded at p < 0.001 (***). (E) SK-N-MC cells were infected with HSV-1 at moi 10 for 18 hours and treated with 2 mM MβCD. Phosphorylation of Thr205 epitope of tau was analyzed by immunofluorescence. DAPI-stained nuclei are also shown (red). Scale bar: 10 µm. (F) SK-N-MC cells were infected with HSV-1 at moi 10 for 18 hours and treated with different concentrations of MβCD. Levels of Ser422-phosphorylated tau were monitored by Western blot analysis. A tubulin blot is shown to ensure equal protein load. The ratio of phosphorylated tau to tubulin, obtained by densitometric analysis, is shown below the blots.
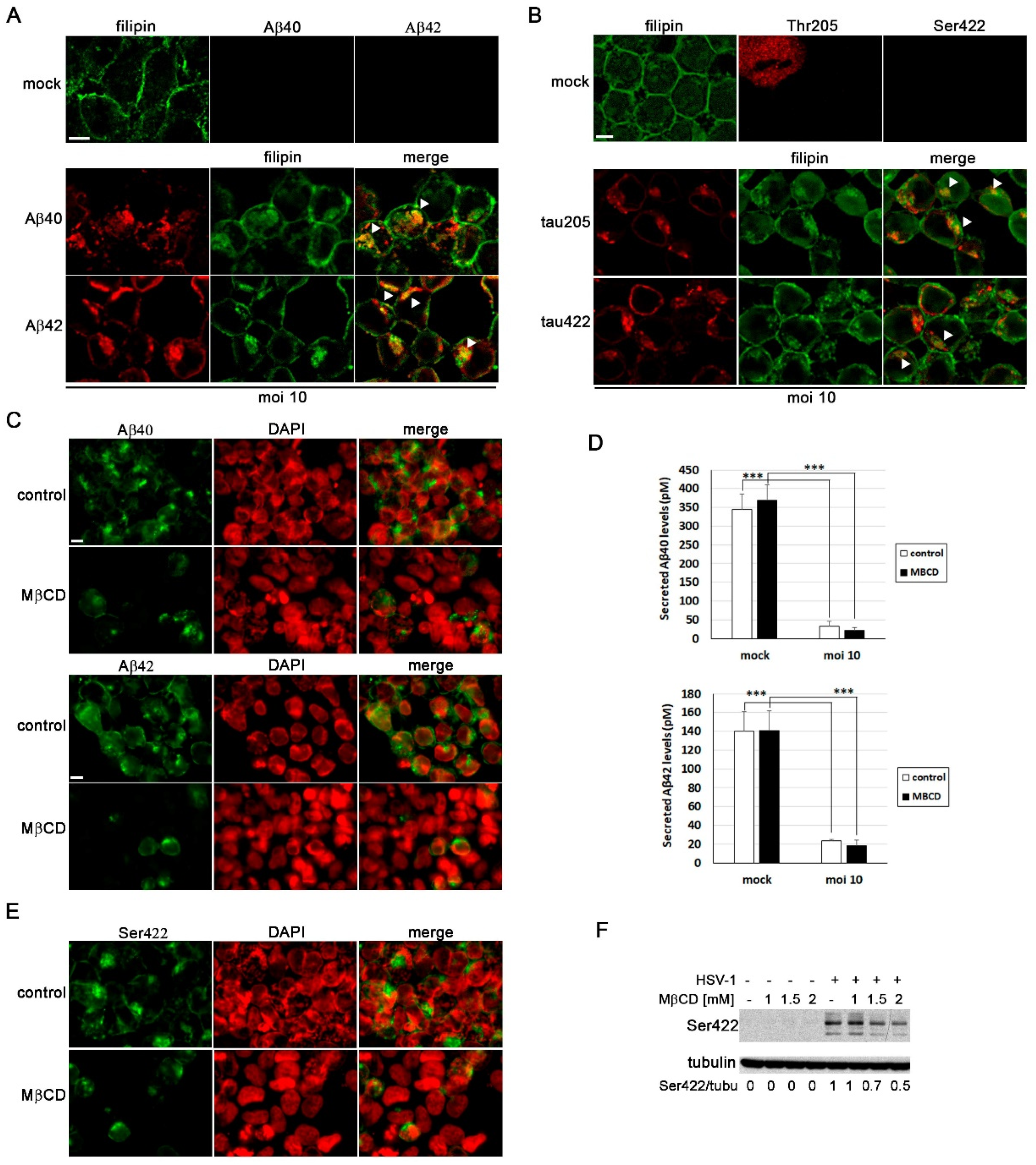

Table 1.
List of antibodies used in Western blot (WB) and immunofluorescence (IF) analysis. .
| Primary Antibodies | Species | Dilution | Reference | |
| HSV-1 infection | ICP4 | Mouse | 1/1000 (WB) 1/100 (IF) |
Abcam ab6514 |
| gC | Mouse | 1/3000 (WB) 1/300 (IF) |
Abcam ab6509 | |
| Neurodegeneration markers | Aβ40 | Rabbit | 1/100 (IF) | Invitrogen 44348A |
| Aβ42 | Rabbit | 1/100 (IF) | Invitrogen 44-344 | |
| p-Tau Thr205 | Rabbit | 1/250 (WB) 1/50 (IF) |
Invitrogen 44-738G | |
| p-Tau Ser422 | Rabbit | 1/250 (WB) 1/50 (IF) |
Invitrogen 44-764G | |
| Autophagy-lysosomal pathway | EEA1 | Mouse | 1/1000 (WB) 1/100 (IF) |
BD Biosciences 610457 |
| Human LAMP2 | Mouse | 1/1000 (WB) 1/50 (IF) |
DSHB H4B4 | |
| CD222 | Mouse | 1/100 (IF) | BioLegend 315902 | |
| LC3B | Rabbit | 1/500 (WB) 1/100 (IF) |
Sigma L7543 | |
|
Housekeeping protein |
α-Tubulin | Mouse | 1/10000 (WB) | Sigma T5168 |
| Secondary Antibodies | Species | Dilution | Reference | |
| Mouse-POD | Horse | 1/25000 (WB) | Vector PI-2000 | |
| Rabbit-POD | Goat | 1/25000 (WB) | Nordic GAR/IgG (H+L)/PO | |
| Alexa-555 anti-mouse | Goat | 1/1000 (IF) | Thermo Fisher A-21137 |
|
| Alexa-488 anti-rabbit | Donkey | 1/1000 (IF) | Thermo Fisher A-21206 |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



