
Submitted:
15 April 2024
Posted:
17 April 2024
You are already at the latest version
Abstract
“Pecorino” is a typical semi-hard cheese obtained with raw or heat-treated sheep milk using procedures to valorize the raw material's chemical and microbiological properties. In the present study, using a high throughput method of 16S rRNA gene sequencing, we assessed the evolution of the microbiome composition from milk to Pecorino cheese in artisanal processes using milk from Comisana and Lacaune sheep breeds. The comparative analysis of the bacterial com-munity composition revealed significant differences in the presence and abundance of specific taxa in the milk microbiome of the Comisana and Lacaune breeds. NGS analysis also revealed differences in the curd microbiome related to dairy farming practices, which have a relevant effect on the final structure of the Pecorino-cheese microbiome.
Keywords:
Comisana breed
; Lacaune breed
; sheep milk
; microbiome
; Next-Generation Sequencing (NGS)
; Pecorino cheese
1. Introduction
Fermented foods have been a significant part of the human diet since prehistoric times [1-2]. These foods benefit consumers through nutritional content, high digestibility, and microbial stability and represent the means of storage of humanity's oldest foods [3]. Fermented foods are characterized by microorganisms, which define the product's organoleptic characteristics and provide beneficial components such as probiotics and antioxidant and anti-pathogenic compounds [4-5]. They may also contain prebiotics that promote beneficial bacteria growth and, therefore, can modulate the host microbiota [6].
Among fermented foods, cheese represents a key component of the human diet, and its consumption is increasing worldwide [7-8-9]. Pecorino cheese is commonly referred to as a variety of hard and semi-hard cheese obtained exclusively with raw or heat-treated (temperature comprised 45-48°C) sheep’s milk by traditional procedures [10-11].
Italy is well known for producing many “Pecorino” and other sheep milk cheeses [12-13-14]. Among them, Pecorino Romano is one of the most important Italian DOP cheeses, producing more than 32,6 tons in 2022 [15]. Besides these PDO cheeses, many Italian Pecorino-like no PDO cheeses are manufactured in small artisanal farms following traditional methods. These artisanal products are appreciated for their distinctive traits linked to the production environment and milk’s microbial biodiversity.
Raw-milk artisanal cheeses convey ideas of tradition and culture, mainly for countries such as France and Italy [16], to such an extent that cheese tourism is seen as a possible development perspective in rural, mountain, and natural remote areas [17]. Moreover, raw milk cheeses have been associated with a complex profile of volatile acids and highly sensorial attributes, conferring unique organoleptic properties [18] compared to processed cheeses, which show a less intense flavor and ripen more quickly [19-20].
Although the organoleptic quality of artisanal cheeses produced using raw milk and natural curd is superior to the most widespread pasteurized milk cheese, these products may pose a threat to the consumer’s health and, therefore, their safety should be carefully assessed to protect the producer and consumer interests [21-22]. Thus, monitoring microbiota composition and its evolution during fermentation and ripening is crucial for obtaining products with optimal sensory properties and safety characteristics [23]. Much effort has been put into investigating the raw milk microbial communities to improve cheese production and safety due to their importance for the world's population [24-25].
In recent years, High-Throughput Sequencing (HTS) of 16 rDNA gene amplicons has been widely used to investigate the evolution of the microbiome during the fermentation process [26]. This method overcomes the limitations of culture methods and permits the study of the microbial community profile and the taxonomic evolution during space and time in dairy products [19; 27-31].
Several factors, including animal breed and farming practice, can affect the structure of the milk microbiome [32-33].
The main aims of this study were to assess the microbiota diversity in raw milk, curd, and Pecorino-like cheese of two different sheep breeds, Comisana and Lacaune, and the evolution of these microbiomes during the cheesemaking process, using high-throughput sequencing of the 16S rRNA gene.
2. Materials and Methods
2.1. Samples Collection
Milk, curd, and cheese samples were collected from two dairy farms of the Amaseno Valley in the Province of Frosinone (Central Italy): one raising the Comisana sheep breed (CSB) and the other the Lacaune sheep breed (LSB). Each farm used raw milk and native starter cultures in the Pecorino Romano-like cheesemaking process. Two independent bulk milk samples of 500 mL and about 50 g of curd and middle-aged cheese (10-20 days) were sampled for each farm. All samples were transported immediately, at 4°C, to the laboratory. Curd and cheese samples were divided into several aliquots (about 0.5 g each). Milk was pretreated to reduce fat content, and total cells (somatic and bacterial) were recovered by centrifugation, as Luziatelli et al. described [34]. All materials were stored at -20°C until DNA extraction.
2.2. DNA Extraction and Purification
Total DNA was extracted from raw milk, curd, and cheese samples using a commercially available kit (DNeasy Blood & Tissue kit, Qiagen) with the following modifications. The cellular pellet, obtained from pretreated milk samples stored at − 20°C, was resuspended in 5 mL of saline phosphate buffer (PBS) containing 10μL of 0.5 M EDTA pH 8.0 and centrifuged at 10,000 rpm for 10 min at 4°C. The resulting pellet was resuspended in 5 mL of saline buffer, amended with 1% Triton X-100, and incubated for 2 hours at 37°C to lysate the somatic cells. The suspension was then centrifuged at 10,000 for 10 min at 4°C. The bacterial pellet obtained was resuspended in 180μL of enzymatic lysis buffer and treated as described in the DNeasy Blood & Tissue kit instructions.
For purifying total DNA from curd and cheese, samples (100 and 250 mg, respectively) were homogenized and treated as described for milk samples.
2.3. DNA Quantification
The DNA was quantified using the Qubit® fluorometer 3.0 with the Qubit™ dsDNA HS Assay Kit (Thermo Fisher Scientific, Rodano [MI], Italy).
2.4. Next-Generation Sequencing and Data Analysis
The metagenomic was amplified for the V3-V4 region of the 16S rRNA gene using the primers pairs 343F (5’-TAC GGR AGG CAG CAG-3’) and 802R (5’-TACNVGGGTWT CTA ATC C-3’).
The sequencing was performed on the Illumina MiSeq version 3 sequencing platform system in 300-nucleotide (nt) paired-end mode.
Run statistics were determined using CLC Genomics Workbench 12 (Qiagen GmbH, Hilden, Germany). The Illumina-generated reads were demultiplexed, quality filtered, and analyzed using the “Quantitative Insights Into Microbial Ecology” (QIIME) pipeline [35]. Operational Taxonomic Units (OTUs) were assigned to the reads using an open reference approach with the UCLUST algorithm [36] against the SILVA database release 138.1 (https://www.arb-silva.de/) clustered at 97% identity [37].
For the microbiome definition, the OTU data obtained from each sample's replicate were combined and used to describe the most abundant bacteria in raw milk, curd, and cheese samples. The resulting number of OTUs was converted into percentage abundance in Microsoft Excel (Microsoft Corp., Redmond, WA) and used for comparative analysis.
3. Results
This work compares the microbiome evolution from milk to cheese in Pecorino Romano-like cheesemaking processes using milk from Comisana (CSB) and Lacaune (LSB) sheep breeds.
NGS analysis of the 16S V3-V4 region generated reads between 28584 (curd_1; CSB) and 56259 (cheese_1; CSB; Table S1). Approximately 58-85% of raw reads per sample passed the merging, trimming, and chimera filtering steps and were analyzed with QIIME2 [35] (Table S1).
The comparative analysis of the different microbiomes indicated that the number of OTUs per sample differed in the two sheep breeds. As shown in Table S1, in the CBS microbiomes, the number of OTUs increased from 50 (milk) to 207 (curd), while in LSB microbiomes, the OTU number varied between 66 (milk) and 165 (curd). Differences in the OTUs abundance were also observed in the corresponding cheese samples: 19 (CSB) - 54 (LSB; Table S1). In Table S1, the taxonomic assignment of each OTU is reported using the BLAST search against the SILVA database.
The rarefaction plots of 16S rRNA datasets showed that all curves reached a plateau (Figure 1), indicating that the datasets represent the microbial community well.
3.1. Sheep Milk Microbiota
The comparative analysis of the milk microbiomes showed that the CSB and LSB bacterial communities clustered separately (Figure 2, Panel A), with the principal coordinate 1 accounting for 100% of the total variance. The Venn diagram constructed using the OTUs taxonomy data (Figure 2, Panel B) revealed the presence of 25 OTUs shared in both milk samples. This core population represented 50% of the total CSB milk microbiome and 38% of the entire LSB milk microbial community (Figure 2, Panel B).
As shown in Table S3, the OTUs comprised in the milk core microbiome were affiliated to 6 phylum and 19 families. About two-thirds of these 25 OTUs (16) belonged to Proteobacteria with Pseudomonadaceae (4 OTUs ID 332, 333, 338, and 340) and Xanthomonadaceae (3 OTUs ID 347, 348, and 350) as more abundant families. About 25% of the core OTUs were equally distributed between Actinobacteria (3 OTUs) and Firmicutes (3 OTUs), with Streptococcaceae (OTUs ID 156 and 159) as the more abundant family in the latter taxa.
We used a Principal Component Analysis (PCA) of the two datasets at family (Figure 3, Panel A) and OTU (Figure 3, Panel B) level to evaluate the main differences in the milk microbiome of the two sheep breeds. This analysis revealed the presence of 4 families (Pseudomonadaceae, Xanthomonadaceae, Enterobacteriaceae, and Streptococcaceae), whose abundance significantly varied among the two milk samples (Figure 3, Panel A). In the CSB milk samples, about 90% of the total OTUs were equally distributed between Pseudomonadaceae and Xanthomonadaceae. In contrast, the OTUs affiliated with these two families in the LSB milk samples were about 84% of the total reads. Pseudomonadaceae-affiliated OTUs represented approximately 60% of the total OTUs (Table S2). The PCA analysis also revealed that Enterobacteriaceae and Streptococcaceae were differentially abundant in the milk samples of the two sheep breeds (Figure 3, Panel A). Enterobacteriaceae were about 8-fold higher in LSB vs CSB, whereas Streptococcaceae were 7.7-fold more abundant in CSB than in LSB milk samples (Table S2).
The analysis at the OTU level (Figure 3, Panel B) revealed that the differences at a family level were associated with the relative abundance of five different OTUs. The OTU 348 and 350, affiliated to Xanthomonadaceae (Stenotrophomonas spp.), and OTU 159, belonging to Streptococcaceae (Streptococcus sp.), were more abundant in the CSB microbiome. The OTU ID 340 and 307, belonging to Pseudomonadaceae (Pseudomonas veronii) and Enterobacteriaceae (unknown species), respectively, were more abundant in the LSB milk microbiota (Figure 3, Panel B).
3.2. Curd Microbiota
The PCA analysis constructed with all CSB and LSB datasets (raw milk, curd, and Pecorino-like cheese) revealed that the differences between the two-curd microbiota were broader than those observed in a pairwise comparison between milk and cheese samples (Figure 4).
As reported in Table S1, the number of OTUs in the raw curd samples was about 4.1 and 2.5 times higher than in the corresponding CSB and LSB milk samples.
The Venn diagram constructed using the OTUs taxonomy data revealed the presence of 67 OTUs shared in both curd samples, representing 32.4 and 34.3% of the total curd microbiome in CSB and LSB samples, respectively (Figure 5, Panel A). The shared OTUs, affiliated with 37 different families, represented 93.4 (CSB) and 43.6% (LSB) of the total curd OTUs (Table S4).
The PCA analysis at the OTU level indicated that the main differences between the curd microbiome datasets were associated with 9 OTUs, 2 of which (OTU 79 and 271) were absent in the curd core microbiome. OTU ID number 315 (Serratia sp.) and 337 (Pseudomonas fragi) were the most abundant in the CSB curd samples (Figure 5), representing about 4.3 and 80.5% of the total OTUs, respectively (Table S2). The OTUs ID number 79 (Flavobacterium frigidarium), 156 (Lactococcus sp.), 271 (Comamonas sp.), 324 (Acinetobacter sp.), 326 (Acinetobacter johsonii), 328 (Enhydrobacter sp.) and 340 (P. veronii) were highly represented in LSB curd samples with F. frigidarium (OTU 79) as the most abundant OTU (37.6%; Figure 5 and Table S2).
3.3. Cheese Microbiota
Data reported in Table S1 indicated a 2.8-fold difference in the OTUs of the two cheese microbiomes (19 CSB vs. 54 LSB). In the two datasets, the number of OTUs whose abundance was higher than 1% was 5 (CSB) and 8 (LSB), respectively. The relative abundance of these OTUs was 99 (CSB) and 92% (LSB), respectively.
The PCA analysis indicated that the differences between the CSB and LSB milk datasets were comparable with those at the cheese level (Figure 4).
3.3.1. Influence of the Milk Microbiome
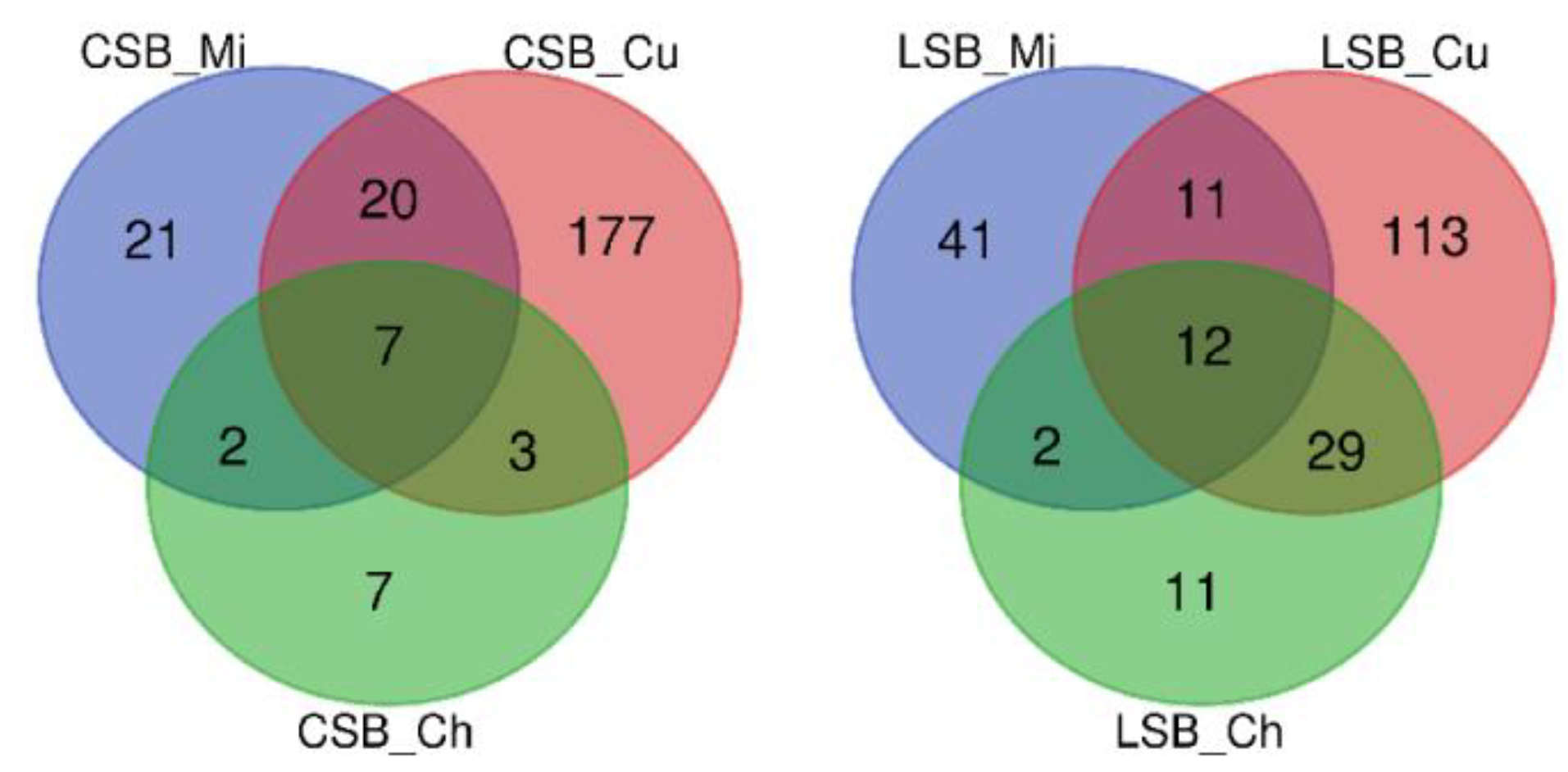
We used the OTUs taxonomy datasets to construct the Venn diagrams reported in Figure 6 to describe the microbiome evolution during cheesemaking. This analysis revealed the presence of 9 (CSB) and 14 (LSB) shared OTUs between milk and cheese. About 80% of these OTUs were affiliated with Proteobacteria and Firmicutes, and their most abundant taxa in milk (Pseudomonas veronii-affiliated OTU 340; Table S5, Panel A) and cheese (Lactococcus sp.-affiliated OTU 156; Table S5, Panel B).
Analyzing the relative abundance of the shared OTUs between milk and cheese microbiomes in the CSB datasets, we identified 4 OTUs whose abundance increased in cheese, 4 OTUs whose abundance was higher in milk, and 1 OTU whose relative abundance remained constant during the cheesemaking process (Table S5).
In the LSB datasets, the abundance of 4 of the shared OTUs increased from milk to cheese, 5 OTUs were more abundant in the milk microbiome, and 3 OTUs remained constant in their relative abundance in the two microbiomes (Table S5).
In detail, the relative abundance of P. veronii OTU 340 significantly decreased from 43.8% (milk) to 0.02%(cheese) in CSB and from 58.9 (milk) to 0.04 (cheese) in LSB datasets. In contrast, the relative abundance of Lactococcus sp. OTU 156 increased 66-fold in the LSB cheese (from 1.02 to 67.2% of the total OTUs) and 542-fold in the CSB samples (from 0.1 to 54.2% of the total OTUs; Table S5).
The analysis of the LAB population showed that 4 OTUs related to 4 different genera (Lactobacillus, Lactococcus, Streptococcus, and Leuconostoc) were shared between milk and the corresponding cheese (Table S5). Notably, 3 OTUs were part of the milk core microbiome (Lactobacillus zeae OTU 153, Lactococcus sp. OTU 156, and Streptococcus sp. OTU 159; Table S3). The OTU affiliated with the Leuconostoc genus differed in the two datasets: ID 155 in CSB and ID 154 in the LSB.
Except for OTU 159 in CSB milk samples, the other OTUs belonging to the LAB category increased during cheesemaking (Table S5).
The Venn diagrams also show the presence of 7 and 11 unique OTUs in Pecorino-like cheese from CSB and LSB, respectively (Figure 6).
As shown in Table S6, Panel A, in CSB Pecorino-like cheese, most of them (4 out of 7) were affiliated to Lactobacillaceae (2 OTUs), Streptococcaceae (1 OTU), and an unclassified family of the Lactobacillales order (1 OTU). The remaining OTUs were affiliated with three families: Bifidobacteriaceae, Clostridiaceae (Clostridium perfringens), and Enterobacteriaceae (Rahnella aquatilis).
In the LSB cheese sample, the unique 11 OTUs belonged to 10 families, among which the most represented was Enterobacteriaceae (Citrobacter sp.; 2 OTUs; Table S6, Panel B).
The remaining OTUs were associated with nine different taxa: 1) Micrococcaceae (Rhotia sp.); 2) Pasteurellaceae (Mannheimia sp.); 3) Pseudoaltermonadaceae (Pseudoalteromonas sp.); 4) Lactobacillaceae (Lactobacillus paralimentarus); 5) Streptococcaceae (Lactococcus gavieae); 6) unclassified species belonging to (6) Caronobacteriaceae, (7) Dermabacteraceae, (8) Flavobacteriales and (9) Lactobacillales (Table S6, Panel B).
3.3.2. Influence of the Curd Microbiome
The OTUs shared between curd and cheese can be divided into two groups: those common to milk, curd, and cheese (described above) and those shared only between curd and cheese. To analyze the potential role of the latter group of OTUs (3 in CSB and 29 in LSB datasets; Figure 6), we arbitrarily clustered these OTUs into four categories as described by Secchi et al. [39]: LAB and other probiotics (LAB), spoilage (SP), pathogenic (P) and other bacteria (O). In CSB samples, one of the shared OTUs (ID 127) belonged to the pathogenic/spoilage category, and two (ID 315 and 337) to the spoilage category (Table S7).
The LSB curd and cheese samples also revealed the OTU 127, affiliated with Staphylococcus (Table S7). Its abundance decreased about 6-fold (from 0.06 to 0.01% of the total OTUs) from curd to cheese in the CSB samples and only 20% in the corresponding LSB samples (from 1.54 to 1.32% of the total OTUs).
The abundance of the shared SP-taxa decreased in the CSB samples from 84.81% (curd) to 39.55% (cheese) of the total OTUs. Interestingly, the trend of these two spoilage-related OTUs followed a different pattern: the abundance of the OTU 337 (P. fragi) decreased about 20-fold (from 65 to 3% of the total OTUs), whereas the abundance of the OTU 315 (Serratia sp.) increased approximately 10-fold (from 3 to 29% of the total OTUs).
The abundance of OTU 126, affiliated with Macrococcus sp. (putative pathogenic/spoilage taxon), increased about 4-fold (from 0.05 to 0.20% of the total OTUs) in LSB samples during the cheesemaking process. In the same samples, the SP category comprised 15 shared OTUs belonging to 4 phyla: Proteobacteria, which was the most representative phylum (10 OTUs); Bacteroidetes (2 OTUs), Firmicutes (2 OTUs) and Actinobacteria (1 OUT; Table S7). Most of the taxa included psychrotrophic environmental bacteria, generally present in the soil, water, and air [40]. The total abundance of these SP-OTUs decreased about 25-fold (from 63.9 to 2.6% of the total OTUs) during the LSB cheesemaking process. In particular, OTU 79, affiliated with F. frigidarium, represented about two-thirds of the total SP-related OTUs and decreased about 125-fold (from 37.6 to 0.3% of the total OTUs) from curd to cheese samples (Table S7).
In the LSB samples, OTUs belonging to the LAB and O categories were also revealed (Table S7). The LAB comprised 9 OTUs belonging to 5 families and nine species. Half of these OTUs were affiliated with Lactobacillus (2 OTUs) and Streptococcus (3 OTUs) species. During the cheesemaking process, the total abundance of the LAB-related OTUs increased about 10-fold (from 2.2 to 25.8% of the total OTUs). In particular, OTU 145 and 155, affiliated with Lactobacillus sp. and Leuconostoc mesenteroides, increased about 12- and 23-fold, respectively (Table S7). The total abundance of the bacteria belonging to the O category (OTUs 23, 122, and 285) decreased about 3.4-fold from 1.44 to 0.42% of the total OTUs during the cheesemaking process.
4. Discussion
The main aim of this study was to assess the effect of the diversity of the milk microbiome of two different sheep breeds, Comisana and Lacaune, on the microbial community of artisanal Pecorino Romano-like cheese. The analysis was carried out on samples collected at various stages of the cheesemaking process (milk, curd, and mid-ripening cheese) using high-throughput sequencing of the 16S rRNA gene.
The rarefaction curves reported in Figure 1 indicated that the 16S rRNA datasets represented the bacterial community's complexity well.
To gain information about the fingerprints of the microbiome of Comisana and Lacaune milk, we combined OTU data from replicate samples. We used the resulting datasets for Venn and PCA analysis. The comparative analysis datasets through the Venn diagram allowed us to identify common (core microbiome) and unique (accessory microbiome) OTUs occurring in the two milk samples. PCA analysis allowed us to identify the OTUs whose abundance profile varied between the two microbiome datasets. A similar approach was used to evaluate the contribution of the milk and curd microbiome to the cheese microbial community. The Venn diagram (Figure 2, Panel A) showed that the Comisana and Lacaune milk datasets shared 25 OTUs, representing about 97.5% and 96.1% of the total reads, respectively (Table S3).
The PCA analysis indicated that Comisana and Lacaune milk microbiomes were markedly distinct and that differences in the two datasets were due to the relative abundance of the shared taxa (Figure 3). In both milk microbiomes, 44 and 59% of the total reads were associated with the P. veronii-affiliated OTU 340, representing more than 97% of the total reads belonging to Pseudomonadaceae. These results on the occurrence of specific taxa in samples collected from farms of the same geographic area supported the hypothesis that the environment shapes the milk microbiota [41].
P. veronii is a non-pathogenic environmental microorganism originally isolated from mineral water. It is known for its ability to degrade aromatic compounds [42-44]. The presence of P. veronii in the milk microbiome has already been reported for buffalo and other mammals [34; 45-46] but has yet to be observed in the milk microbiome of different sheep breeds, such as Assaf dairy ewes [47]
The PCA analysis also revealed differences in the abundance of OTUs affiliated with Xanthomonadaceae (OTU 348 and 350). In CSB milk samples, these OTUs were about 2-fold higher than in LSB milk datasets, indicating that the breed and farming environments potentially influence the presence of these taxa in the milk microbiome. The relative taxa belonged to Stenotrophomonas, a genus whose members are known components of the core milk microbiome of goats [48] and cows [49]. Stenotrophomonas comprises psychrotrophic and proteolytic strains, which can be involved in bovine mastitis [50] and raw milk spoilage [51]. Notably, the OTUs 348 and 350 were not affiliated with Stenotrophomonas maltophilia, a pathogen reported to be associated with human respiratory infections [52-54].
Another significant difference between the two milk microbiomes was the abundance of Enterobacteriaceae and Streptococcaceae families and their representative OTUs (ID 307, 315, and 159; Figure 3). The OTU 307 represented more than 98% of the total Enterobacteriaceae-affiliated reads in both milk microbiomes, while Enterobacteriaceae-affiliated OTU 315 was detected only in the LSB milk datasets. The OTU 159 represented more than 97% of the total reads belonging to Streptococcaceae in the CSB milk samples and only 4% of the Streptococcaceae-affiliated reads in the LSB microbiome. The differential abundance of OTUs of the core microbiome underlines the effect of the dairy farming practice on the composition of the milk microbial community.
The core members of Comisana and Lacaune microbiomes reported in this work show differences with the Assaf dairy ewes microbiome described by Esteban-Blanco et al. [47]. These authors reported that the milk microbiome of healthy Assaf sheep comprised five dominant genera: Corynebacterium, Escherichia/Shigella, Lactobacillus, Staphylococcus, and Streptococcus [47]. In contrast, we identified 22 different genera that were shared in the microbiomes of Comisana and Lacaune. Three of them, Lactobacillus, Streptococcus, and Corynebacterium, occurred in the microbiome of all three sheep breeds. We detected the presence of an OTU affiliated with Enterobacteriaceae but not belonging to the Escherichia/Shigella phylogroup. Staphylococcus-affiliated OTUs were observed in the Assaf and Lacaune milk microbiomes but were absent in the Comisana milk. These data suggested that the Staphylococcus genus is not part of the core microbiome of the sheep milk.
PCA analysis of different datasets revealed that the Comisana and Lacaune raw milk coagulation curd possessed a distinct complex microbiome (Figure 4). As shown in Table S1, the total OTUs significantly increased from milk to curd in both samples, indicating that the combination of rennet and cheese starter used in the two cheesemaking processes significantly affected the biodiversity of the curd microbiome. In the CSB datasets, the number of OTUs in the curd samples was 4.1-fold higher than in the corresponding milk (207 vs. 50 OTUs; Table S1), while in the LSB datasets, this number increased about 2.5-fold (265 vs. 66; Table S1). About 46 (23 out of 50 OTUs) and 65% (43 out of 66 OTUs) of the total OTUs occurring in CSB and LSB milk were not present in the corresponding curd (Figure 5). The comparative analysis of milk and curd microbiomes revealed that the Xanthomonadaceae-associated OTUs (ID 347, 348, and 350) of the core milk microbiome were drastically reduced or disappeared after the thermal treatment (Table S2). The same analysis also revealed that the most representative OTUs of the core milk microbiome affiliated with Pseudomonodaceae (ID 340) and Enterobacteriaceae (ID 307) disappeared during the cheesemaking process. In contrast, OTU ID 159 (Streptococcoccus sp.) decreased 25-fold in CSB curd and increased about 8.2-fold in LSB curd compared to the corresponding milk samples.
Data reported in Figure 5, Panel A also indicated the presence of 44 OTUs shared only between the two curd datasets, whose presence could be due to environmental contamination. The relative abundance of most of these shared OTUs was below 0.1% (36 OTUs in the CSB curd dataset and 30 OTUs in the LSB dataset), and only a few of them (1 OTU in the CSB curd dataset and 6 OTUs in the LSB dataset) were present in the corresponding mid-ripened cheese at a relative abundance higher than 0.1% (Table S1).
The PCA analysis of the curd datasets, which explains over 99% of the total variance (Figure 5, Panel B), indicated that the significant differences between the two curd microbiomes were due to the abundance of 8 shared OTUs and two 2 OTUs (ID 79 and 271) that were present only in LSB samples. The latter were identified as F. frigidarium (OTU 79) and Comamonas sp. (OTU 271), two environmental taxa whose presence was reported in artisan Mongolian sheep cheese by Guo et al. [55].
Two out of the eight shared OTUs belong to Serratia (ID 315) and Pseudomonas (ID 337) genera, and their presence could be related to environmental contamination since these microorganisms are ubiquitous in water, soil, and other environments [56-58]. Both genera include species involved in food spoilage often associated with dairy products that were recognized as resident microbiota of food processing plants for their ability to produce biofilms resistant to cleaning procedures [26; 59-61].
Furthermore, Ruta et al. [62] reported the presence of Serratia and Pseudomonas in Pecorino Siciliano curds samples collected in 5 different farms. In both cheese ripening processes, the abundance of the P. fragi-associated OTU 337 significantly decreased (25-fold in CSB samples) or disappeared (in LSB samples; Table S2). This effect can be related to the environmental changes associated with Pecorino-like cheese production (high salinity and low pH), which inhibit this taxon's growth and survival [63-64]. Comparing the microbiome pattern of curd and the corresponding cheese, we observed a different trend in the abundance of Serratia-associated OTU 315. In CSB samples, this increased about 8.5-fold from curd to cheese, whereas in LSB samples, its abundance decreased up to 0.03% of the total OTUs. Members of the Serratia genus are commonly isolated from cheese. Todaro et al. [65], analyzing the effect of the salting technologies on the cheese microbiome, reported the presence of Serratia in different PDO Pecorino cheeses. These authors suggested that the survival of unwanted bacteria, including Serratia, is inversely correlated to the abundance of LAB. Our data indicated that the Lactobacillales-affiliated OTUs represented more than 92% of the total OTUs in LSB cheese samples, in which we observed a low level of Serratia. Meanwhile, Serratia represented about one-third of the total cheese microbiome in CSB cheese samples, in which Lactobacillales were only 58% of the total OTUs. Both Lactobacillales and Serratia are known to produce bacteriocins active against Gram-negative bacteria, including Escherichia coli and Pseudomonas [66-70].
Moreover, bacteriocins produced by LAB can be active against Serratia, which can be valuable in the cheesemaking sector to reduce the development of these unwanted spoilage microorganisms. A more detailed analysis of the Lactobacilalles-affiliated OTUs indicated a strong effect of the cheesemaking process on the number and abundance of these taxa. No OTU related to Carnobacteriaceae and Enterococcaceae was present in CSB cheese samples, while in the LSB cheese samples, they represented about 0.43% and 0.81% of the entire microbiome, respectively. Members of the Streptococcus (St) and Lactobacillus (Lb) genera were differentially represented in the two cheeses. Taxa belonging to these genera were more abundant in LSB (3.80%, St; 8.90%, Lb) than CSB (0.02%, St; 0.3, Lb) cheese samples.
The comparative analysis of the two Pecorino cheese microbiomes revealed that the main differences were related to 5 OTUs: 3 LAB-affiliated OTUs (Lactobacillus sp. OTU 145, L. mesenteroides OTU 155 and Lactococcus sp. OTU 156) and two environmental contaminants (Serratia sp. OTU 315 and P. fragi OTU 337). The relative abundance of these taxa is 96 (CSB) and 85% (LSB) of the total OTUs, respectively (Table S8). Only two were present in milk and the corresponding cheese at a detectable level (OTU ID 155 and 156 in CSB samples; OTU 156 and 315 in LSB samples). Interestingly, OTU 156, corresponding to the LAB involved in the acidification process, was 10-fold more abundant in LSB (1.02% of the total OTUs) than in CSB (0.1% of the total OTUs) milk samples. Despite the data reported in Table S10 indicating that growth rates of Lactococcus-affiliated OTU 156, from milk to curd, were similar in the two datasets, the different initial concentrations of this taxa in the raw CSB and LSB milk affected the acidification process generating environmental conditions that in LSB samples favored the development of natural non-starter lactic acid bacteria (NSLAB; L. mesenteroides affiliated OTU 155) and the containment of Serratia and Pseudomonas contaminants.
These data indicate that the structure and composition of Lacaune sheep breed microbiota are valuable in an artisanal process to obtain Pecorino-like cheese with a higher concentration of NSLAB (L. mesenteroides), which can have a positive effect on flavor development, and a lower concentration of spoilage bacteria (Serratia sp. and P. fragi).
The presence of unique OTUs in both cheese samples can be related to taxa (e.g., Lactobacillales and Clostridiales) whose relative abundance falls below the detectable limit in the milk and curd microbiomes. Based on our results, establishing the origin of these taxa (milk or curd) is impossible. Still, it is worth mentioning that taken together, they represent only a minor part of the entire cheese microbiome: 1.5% in LSB and 1.7% in CSB (Table S6).
5. Conclusions
In conclusion, our study revealed differences in the milk microbiome of Comisana and Lacaune. These differences were associated with the relative abundance of starter and non-starter LAB, which were important for the organoleptic and safety properties of artisanal cheeses.
The profiling of Comisana and Lacaune milk microbiomes allowed us to determine the effect of the environment on the milk microbiome, identify the set of genera that form the sheep milk core microbiome, and evaluate the impact of changes in core OTU abundance on the cheesemaking.
Our data underline the importance of Next-Generation Sequencing as a valuable tool for developing a fermentation process that valorizes the autochthonous microbiota and increases the safety of artisanal products.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1. Summary of NGS data used in this study. Table S2. CBS and LSB milk, curd, and cheese microbiome. Table S3. Core microbiome of CBS and LSB milk. Table S4. Core microbiome of CBS and LSB curd. Table S5. ID and taxonomic affiliation of the OTUs shared between milk and cheese samples. Table S6. ID and taxonomic affiliation of the unique OTUs in cheese samples. Table S7. ID, taxonomic affiliation, and microbial categories of curd-cheese shared OTUs. Table S8. ID and taxonomic affiliation of cheese-shared OTUs.
Author Contributions
FL and MR contributed to the conception and design of the study and wrote the first draft of the manuscript. FL, RAJ, FM, VM, and MR contributed to defining the methodology. FL and MR contributed to software analysis and data validation. FL, RAJ, FM, VM, and MR contributed to the investigation. FL and MR wrote and edited the final version of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This study was partially funded by LazioInnova, Gruppi di Ricerca-2020, prot. A0375-2020- 36613.
Data Availability Statement
The original contributions presented in the study are included in the article; further inquiries can be directed to the corresponding author.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Tamang, J.P.; Cotter, P.D.; Endo, A.; Han, N.S.; Kort, R.; Liu, S.Q.; et al. Fermented foods in a global age: east meets West. Compr. Rev. Food Sci. Food Saf. 2020, 19, 184–217. [Google Scholar] [CrossRef] [PubMed]
- Chilton, S.N.; Burton, J.P.; Reid, G. Inclusion of fermented foods in food guides around the world. Nutrients 2015, 7, 390–404. [Google Scholar] [CrossRef]
- Marco, M.L.; Heeney, D.; Binda, S.; Cifelli, C.J.; Cotter, P.D.; Foligné, B.; et al. Health benefits of fermented foods: microbiota and beyond. Curr. Opin. Biotechnol. 2017, 44, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Kok, C.R.; Hutkins, R. Yogurt and other fermented foods as sources of health-promoting bacteria. Nutr. Rev. 2018, 76, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Leeuwendaal, N.K.; Stanton, C.; O'Toole, P.W.; Beresford, T.P. Fermented foods, health and the gut microbiome. Nutrients 2022, 14, 1527. [Google Scholar] [CrossRef] [PubMed]
- Vinderola, G.; Cotter, P.D.; Freitas, M.; Gueimonde, M.; Holscher, H.D.; Ruas-Madiedo, P.; Salminen, S.; Swanson, K.S.; Sanders, M.E.; Cifelli, C.J. Fermented foods: a perspective on their role in delivering biotics. Front. Microbiol. 2023, 14, 1196239. [Google Scholar] [CrossRef]
- Shiby, V.K.; Mishra, H.N. Fermented milks and milk products as functional foods—a review. Crit. Rev. Food Sci. Nutr. 2013, 53, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Dadousis, C.; Pegolo, S.; Rosa, G.J.M.; Gianola, D.; Bittante, G.; Cecchinato, A. Pathway-based genome-wide association analysis of milk coagulation properties, curd firmness, cheese yield, and curd nutrient recovery in dairy cattle. J. Dairy Sci. 2017, 100, 1223–1231. [Google Scholar] [CrossRef]
- Dairy and Dairy Products (Chapter 7). in OECD/FAO (2021), OECD-FAO Agricultural Outlook 2021-2030, OECD Publishing, Paris. [CrossRef]
- Gobbetti, M.; Di Cagno, R. Chapter 32 - Extra-Hard Varieties. In Cheese, 4th ed.; McSweeney, P.L.H., Fox, P.F., Cotter, P.D., Everett, D.W., Eds.; Academic Press, Cambridge, MA, USA, 2017, pp. 809–828, ISBN 9780124170124.
- Gobbetti, M.; Di Cagno, R. Extra-hard Varieties. In Encyclopedia of Dairy Sciences, 3rd ed.; McSweeney, P.L.H., McNamara, J.P., Eds; Academic Press, Cambridge, MA, USA, 2022; Volume 3, pp. 172–195, ISBN 9780128187678.
- Caridi, A.; Micari, P.; Caparra, P.; Curari, A.; Sarullo, V. Ripening and seasonal changes in microbial groups and in physico-chemical properties of the ewes’ cheese Pecorino del Poro. Int. Dairy J. 2003, 13, 191–200. [Google Scholar] [CrossRef]
- Di Cagno, R.; Banks, J.; Sheehan, L.; Fox, P.F.; Brechany, E.Y.; Corsetti, A.; Gobbetti, M. Comparison of the microbiological, compositional, biochemical, volatile profile and sensory characteristics of three Italian PDO ewes’ milk cheeses. Int. Dairy J. 2003, 13, 961–972. [Google Scholar] [CrossRef]
- Coda, R.; Brechany, E.; De Angelis, M.; De Candia, S.; Di Cagno, R.; Gobbetti, M. Comparison of the compositional, microbiological, biochemical, and volatile profile characteristics of nine Italian ewes' milk cheeses. J. Dairy Sci. 2006, 89, 4126–4143. [Google Scholar] [CrossRef]
- Ozbun, T. Production volume of Pecorino Romano PDO in Italy 2012-2022. (February 13, 2024) https://www.statista.com/statistics/551472/pecorino-romano-pdo-production-volume-in-italy/.
- Carloni, E.; Petruzzelli, A.; Amagliani, G.; Brandi, G.; Caverni, F.; Mangili, P.; Tonucci, F. Effect of farm characteristics and practices on hygienic quality of ovine raw milk used for artisan cheese production in central Italy. Anim. Sci J. 2016, 87, 591–599. [Google Scholar] [CrossRef]
- Fusté-Forné, F. Developing cheese tourism: a local-based perspective from Valle de Roncal (Navarra, Spain). J. Ethn. Foods. 2020, 7. [Google Scholar] [CrossRef]
- Montel, M.C.; Buchin, S.; Mallet, A.; Delbes-Paus, C.; Vuitton, D.A.; Desmasures, N.; Berthier, F. Traditional cheeses: rich and diverse microbiota with associated benefits. Int. J. Food Micro. 2014, 177, 136–154. [Google Scholar] [CrossRef] [PubMed]
- Fuka, M.M.; Wallisch, S.; Engel, M.; Welzl, G.; Havranek, J.; Schloter, M. Dynamics of bacterial communities during the ripening process of different croatian cheese types derived from raw ewe's milk cheeses. PLOS ONE. 2013, 8, e80734. [Google Scholar] [CrossRef] [PubMed]
- Hattem, H.E.; Taleb, A.T.; Manal, A.N.; Hanaa, S.S. Effect of pasteurization and season on milk composition and ripening of Ras cheese. J. Brew. Distilling. 2012, 3, 15–22. [Google Scholar] [CrossRef]
- Pasquali, F.; Valero, A.; Possas, A.; Lucchi, A.; Crippa, C.; Gambi, L.; Manfreda, G.; De Cesare, A. Occurrence of foodborne pathogens in Italian soft artisanal cheeses displaying different intra- and inter-batch variability of physicochemical and microbiological parameters. Front. Microbiol. 2022, 959648. [Google Scholar] [CrossRef] [PubMed]
- Condoleo, R.; Palumbo, R.; Mezher, Z.; Bucchini, L.; Taylor, R.A. Microbial risk assessment of Escherichia coli shiga-toxin producers (STEC) in raw sheep’s milk cheeses in Italy. Food Control 2022, 137, 108951. [Google Scholar] [CrossRef]
- Pinto, U.M.; De Dea, L.J. Editorial: Community series in microbiological safety and quality aspects of fermented dairy products, volume II. Front. Microbiol. 2023, 14, 1182373. [Google Scholar] [CrossRef]
- Yuan, H.; Han, S.; Zhang, S.; Xue, Y.; Zhang, Y.; Lu, H.; Wang, S. Microbial properties of raw milk throughout the year and their relationships to quality parameters. Foods 2022, 1, 3077. [Google Scholar] [CrossRef]
- Bettera, L.; Dreier, M.; Schmidt, R.S.; Gatti, M.; Berthoud, H.; Bachmann, H.P. Selective enrichment of the raw milk microbiota in cheese production: Concept of a natural adjunct milk culture. Front. Microbiol. 2023, 14, 1154508. [Google Scholar] [CrossRef] [PubMed]
- De Filippis, F.; La Storia, A.; Villani, F.; Ercolini, D. Exploring the sources of bacterial spoilers in beefsteaks by culture-independent high-throughput sequencing. PLoS One 2013, 8, e70222. [Google Scholar] [CrossRef] [PubMed]
- Lusk, T.S.; Ottesen, A.R.; White, J.R.; Allard, M.W.; Brown, E.W.; Kase, J.A. Characterization of microflora in Latin-style cheeses by next-generation sequencing technology. BMC microbiol. 2012, 12, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ercolini, D. High-throughput sequencing and metagenomics: moving forward in the culture-independent analysis of food microbial ecology. Appl. Environ. Microbiol. 2013, 79, 3148–3155. [Google Scholar] [CrossRef] [PubMed]
- De Pasquale, I.; Di Cagno, R.; Buchin, S.; De Angelis, M.; Gobbetti, M. Microbial ecology dynamics reveal a succession in the core microbiota involved in the ripening of pasta filata Caciocavallo Pugliese cheese. Appl. Environ. Microbiol. 2014, 80, 6243–6255. [Google Scholar] [CrossRef] [PubMed]
- Kergourlay, G.; Taminiau, B.; Daube, G.; Vergès, M.C.C. Metagenomic insights into the dynamics of microbial communities in food. Int. J. Food Microbiol. 2015, 213, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Dugat-Bony, E.; Garnier, L.; Denonfoux, J.; Ferreira, S.; Sarthou, A.S.; Bonnarme, P.; Irlinger, F. Highlighting the microbial diversity of 12 French cheese varieties. Int. J. Food Microbiol. 2016, 238, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Cremonesi, P.; Ceccarani, C.; Curone, G.; Severgnini, M.; Pollera, C.; Bronzo, V.; Riva, F.; Addis, M.F.; Filipe, J.; Amadori, M.; et al. Milk microbiome diversity and bacterial group prevalence in a comparison between healthy Holstein Friesian and Rendena cows. PLoS ONE 2018, 13, e0205054. [Google Scholar] [CrossRef]
- Esteban-Blanco, C.; Gutiérrez-Gil, B.; Puente-Sánchez, F.; Marina, H.; Tamames, J.; Acedo, A.; Arranz, J.J. Microbiota characterization of sheep milk and its association with somatic cell count using 16s rRNA gene sequencing. J. Anim. Breed Genet. 2020, 137, 73–83. [Google Scholar] [CrossRef]
- Luziatelli, F.; Melini, F.; Ficca, A.G.; Melini, V.; Nardilli, F.; Ruzzi, M. Core microbiome and bacterial diversity of the Italian Mediterranean river buffalo milk. Appl. Microbiol. Biotechnol. 2023, 107, 1875–1886. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C.; Flyvbjerg, H. Error filtering, pair assembly and error correction for next-generation sequencing reads. Bioinform. 2015, 31.21, 3476–3482. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, P.; Parfrey, L.W.; Yarza, P.; Gerken, J.; Pruesse, E.; Quast, C.; Schweer, T.; Peplies, J.; Ludwig, W.; Glöckner, F.O. The SILVA and "All-species Living Tree Project (LTP)" taxonomic frameworks. Opens external link in new window. Nucl. Acids Res. 2014, 42, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Hammer, O.; Harper, D.A.T.; Ryan, P.D. Paleontological Statistics (PAST) 2.06. University of Oslo, Oslo, Norway. 2015, Retrieved, 24.
- Secchi, G.; Amalfitano, N.; Carafa, I.; Franciosi, E.; Gallo, L.; Schiavon, S.; Sturaro, E.; Tagliapietra, F.; Bittante, G. Milk metagenomics and cheese-making properties as affected by indoor farming and summer highland grazing. J. Dairy Sci. 2023, 106, 96–116. [Google Scholar] [CrossRef]
- Yuan, L.; Sadiq, F.A.; Burmølle, M.; Wang, N.I.; He, G. Insights into psychrotrophic bacteria in raw milk: a review. J. Food Prot., 2019, 82, 1148–1159. [Google Scholar] [CrossRef]
- Esteban-Blanco, C.; Gutiérrez-Gil, B.; Marina, H.; Pelayo, R.; Suárez-Vega, A.; Acedo, A.; Arranz, J.-J. The Milk microbiota of the Spanish Churra sheep breed: new insights into the complexity of the milk microbiome of dairy species. Animals 2020, 10, 1463. [Google Scholar] [CrossRef]
- Elomari, M.; Coroler, L.; Hoste, B.; Gillis, M.; Izard, D.; Leclerc, H. DNA relatedness among Pseudomonas strains isolated from natural mineral waters and proposal of Pseudomonas veronii sp. nov. Int. J. Syst. Bacteriol. 1996, 4, 1138–1144. [Google Scholar] [CrossRef] [PubMed]
- Nam, I.H.; Chang, Y.S.; Hong, H.B.; et al. A novel catabolic activity of Pseudomonas veronii in biotransformation of pentachlorophenol. Appl. Microbiol. Biotechnol. 2003, 62, 284–290. [Google Scholar] [CrossRef]
- Onaca, C.; Kieninger, M.; Engesser, K.H.; Altenbuchner, J. Degradation of alkyl methyl ketones by Pseudomonas veronii MEK700. J. Bacteriol. 2007, 10, 3759–3767. [Google Scholar] [CrossRef]
- Meng, L.; Liu, H.; Dong, L.; Zheng, N.; Xing, M.; Zhang, Y.; Zhao, S.; Wang, J. Identification and proteolytic activity quantification of Pseudomonas spp. isolated from different raw milks at storage temperatures. J. Dairy Sci., 2018, 101, 2897–2905. [Google Scholar]
- Kamal-Eldin, A.; Ayyash, M.; Sobti, B.; Nagy, P. Camel Milk. In Encyclopedia of Dairy Sciences, 3rd ed.; McSweeney, P.L.H., John, P. McNamara, J.P., Eds.; Academic Press, Cambridge, MA, USA, 2022; Volume 5, pp. 504–513, ISBN 9780128187678.
- Oikonomou, G.; Addis, M.F.; Chassard, C.; Nader-Macias, M.E.F.; Grant, I.; Delbès, C.; Bogni, C.I.; Le Loir, Y.; Even, S. Milk microbiota: what are we exactly talking about? Front. Microbiol. 2020, 11, 60. [Google Scholar] [CrossRef]
- McInnis, E.A.; Kalanetra, K.M.; Mills, D.A.; Maga, E.A. Analysis of raw goat milk microbiota: Impact of stage of lactation and lysozyme on microbial diversity. Food Microbiol. 2015, 46, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Derakhshani, H.; Fehr, K.B.; Sepehri, S.; Francoz, D.; De Buck, J.; Barkema, H.W.; Plaizier, J.C.; Khafipour, E. Invited review: Microbiota of the bovine udder: contributing factors and potential implications for udder health and mastitis susceptibility. J. Dairy Sci. 2018, 101, 10605–10625. [Google Scholar] [CrossRef]
- Kuehn, J.S.; Gorden, P.J.; Munro, D.; Rong, R.; Dong, Q.; et al. Bacterial community profiling of milk samples as a means to understand culture-negative bovine clinical mastitis. PLoS ONE 2013, 8, e61959. [Google Scholar] [CrossRef]
- Zeinhom, M.M.A.; Hassan, G.M.; Salem, H.A.M.; Corke, H. Prevalence and survival of Stenotrophomonas species in milk and dairy products in Egypt. Foodborne Pathog. Dis., 2021, 18, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Barchitta, M.; Cipresso, R.; Giaquinta, L.; Romeo, M.A.; Denaro, C.; Pennisi, C.; Agodi, A. Acquisition and spread of Acinetobacter baumannii and S. maltophilia in intensive care patients. Int. J. Hyg. Environ. Health. 2009, 212, 330–337. [Google Scholar] [CrossRef] [PubMed]
- De Vrankrijker, A.M.; Wolfs, T.F.; Van der Ent, C.K. Challenging and emerging pathogens in cystic fibrosis. Paediatr. Respir. Rev. 2010, 11, 246–254. [Google Scholar] [CrossRef]
- Brooke, J.S. Stenotrophomonas maltophilia: an emerging global opportunistic pathogen. Clin. Microbiol Rev. 2012, 25, 2–41. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Xu, W.L.; Li, C.D.; Wang, F.C.; Guo, Y.S.; Ya, M. Determination of the microbial community of traditional Mongolian cheese by using culture-dependent and independent methods. Food Sci. Nutr. 2022, 2, 828–837. [Google Scholar] [CrossRef]
- Lyerly, D.M.; Kreger, A.S. Importance of Serratia protease in the pathogenesis of experimental Serratia marcescens pneumonia. Infect. Immun. 1983, 40, 113–119. [Google Scholar] [CrossRef]
- Abreo, E.; Altier, N. Pangenome of Serratia marcescens strains from nosocomial and environmental origins reveals different populations and the links between them. Sci. Rep. 2019, 9, 46. [Google Scholar] [CrossRef]
- Crone, S.; Vives-Flórez, M.; Kvich, L.; Saunders, A.M.; Malone, M.; Nicolaisen, M.H.; Martínez-García, E.; Rojas-Acosta, C.; Catalina Gomez-Puerto, M.; Calum, H.; Whiteley, M.; Kolter, R.; Bjarnsholt, T. The environmental occurrence of Pseudomonas aeruginosa. APMIS 2020, 128, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Endres, C.M.; Castro, I.M.S.; Trevisol, L.D.; Severo, J.M.; Mann, M.B.; Varela, A.P.M.; Frazzon, A.P.G.; Mayer, F.Q.; Frazzon, J. Molecular characterization of the bacterial communities present in sheep's milk and cheese produced in South Brazilian Region via 16S rRNA gene metabarcoding sequencing. LWT 2021, 147, 111579. [Google Scholar] [CrossRef]
- Stellato, G.; De Filippis, F.; La Storia, A.; Ercolini, D. Coexistence of lactic acid bacteria and potential ppoilage microbiota in a dairy processing environment. Appl. Environ. Microbiol. 2015, 22, 7893–7904. [Google Scholar] [CrossRef] [PubMed]
- Stellato, G.; La Storia, A.; Cirillo, T.; Ercolini, D. Bacterial biogeographical patterns in a cooking centre for hospital foodservice. Int. J. Food Microbiol. 2015, 193, 99–108. [Google Scholar] [CrossRef]
- Ruta, S.; Murray, M.; Kampff, Z.; McDonnell, B.; Lugli, G.A.; Ventura, M.; Todaro, M.; Settanni, L.; van Sinderen, D.; Mahony, J. Microbial Ecology of Pecorino Siciliano PDO Cheese Production Systems. Fermentation 2023, 9, 620. [Google Scholar] [CrossRef]
- Wolfe, B.E.; Button, J.E.; Santarelli, M.; Dutton, R.J. Cheese rind communities provide tractable systems for in situ and in vitro studies of microbial diversity. Cell 2014, 158, 422–433. [Google Scholar] [CrossRef]
- Tirloni, E.; Bernardi, C.; Stella, S. Pseudomonas spp.: are food grade organic acids efficient against these spoilage microorganisms in fresh cheeses? Foods 2021, 4, 891. [Google Scholar] [CrossRef] [PubMed]
- Todaro, M.; Francesca, N.; Reale, S.; Moschetti, G.; Vitale, F.; Settanni, L. Effect of different salting technologies on the chemical and microbiological characteristics of PDO Pecorino Siciliano cheese. Eur. Food Res. Technol., 2011, 233, 931–940. [Google Scholar] [CrossRef]
- Foulds, J.D.; Shemin, D. Properties and characteristics of a bacteriocin from Serratia marcescens. J. Bacteriol. 1969, 99, 655–660. [Google Scholar] [CrossRef]
- Enfedaque, J.; Ferrer, S.; Guasch, J.F.; Tomás, J.; Regué, M. Bacteriocin 28b from Serratia marcescens N28b: identification of Escherichia coli surface components involved in bacteriocin binding and translocation. Can. J. Microbiol. 1996, 42, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Kuo, P.A.; Kuo, C.H.; Lai, Y.K.; Graumann, P.L.; Tu, J. Phosphate limitation induces the intergeneric inhibition of Pseudomonas aeruginosa by Serratia marcescens isolated from paper machines. FEMS Microbiol. Ecol. 2013, 84, 577–587. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Sheoran, P.; Gupta, A.; Yadav, J.; Tiwari, S.K. Antibacterial property of bacteriocin produced by Lactobacillus plantarum LD4 isolated from a fermented food. Ann. Microbiol. 2016, 66, 1431–1440. [Google Scholar] [CrossRef]
- Younas, S.; Mazhar, B.; Liaqat, I.; Ali, S.; Tahir, H.M.; Ali, N.M. Bacteriocin production by Lactobacilli and their role as antibacterial tool against common pathogens. J. Oleo Sci. 2022, 4, 541–550. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Rarefaction curves for the samples of the five different farms. LSB = Lacaune Sheep Breed, CSB = Comisana Sheep Breed, Mi = milk samples, Cu = curd samples, Ch = cheese samples.
Figure 1.
Rarefaction curves for the samples of the five different farms. LSB = Lacaune Sheep Breed, CSB = Comisana Sheep Breed, Mi = milk samples, Cu = curd samples, Ch = cheese samples.
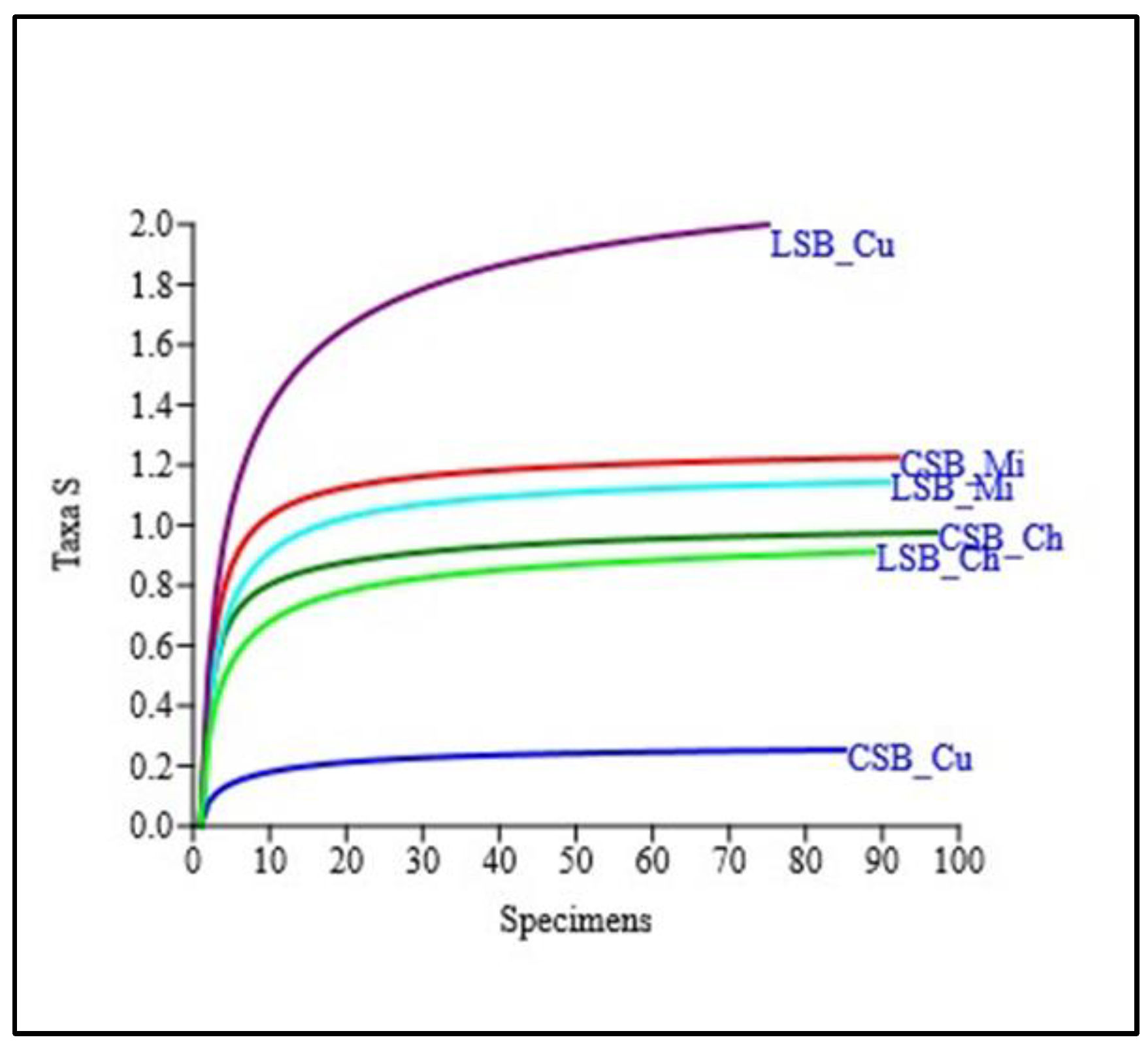
Figure 2.
Comparative analysis of milk microbiomes. (A) Score plot showing the principal components (PCO) 1 and 2 calculated with the abundance matrix of the CSB and LSB milk microbiota. Each symbol represents one microbiota. The plot model explains 100% of the total data variance. (B) Venn diagram showing the number of shared and unique OTUs (≥ 2 reads) in the CSB and LSB milk microbiota.
Figure 2.
Comparative analysis of milk microbiomes. (A) Score plot showing the principal components (PCO) 1 and 2 calculated with the abundance matrix of the CSB and LSB milk microbiota. Each symbol represents one microbiota. The plot model explains 100% of the total data variance. (B) Venn diagram showing the number of shared and unique OTUs (≥ 2 reads) in the CSB and LSB milk microbiota.
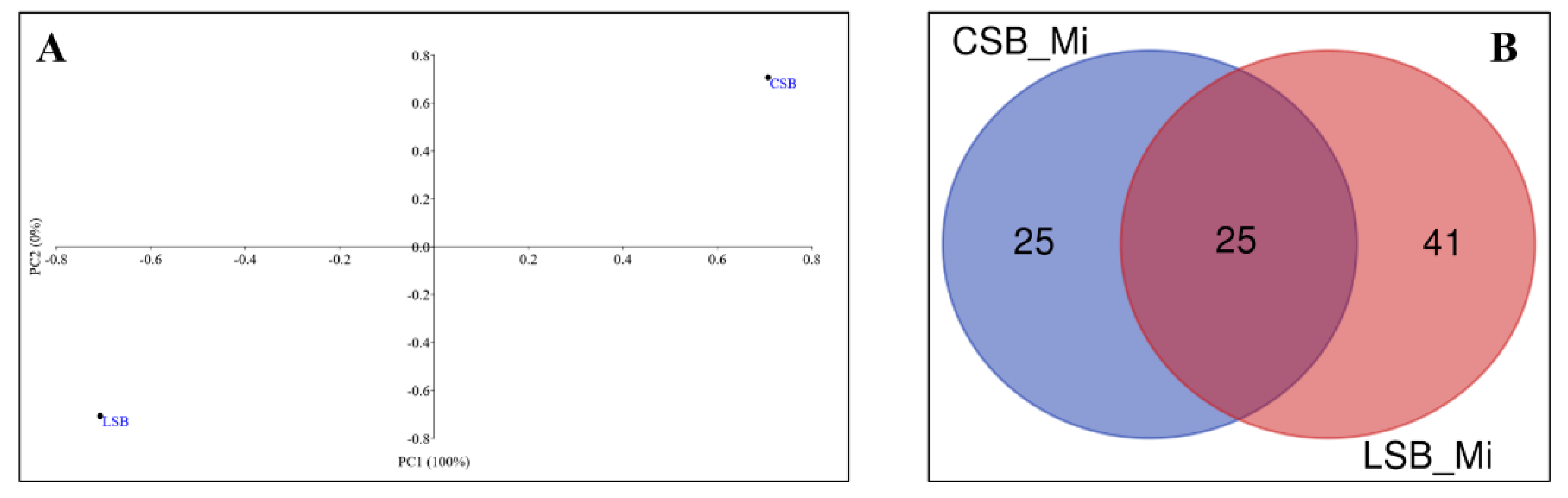
Figure 3.
Principal component analysis (PCA) of taxa occurring in the CSB and LSB milk microbiota based on the relative abundance distribution at family (A) and OTU (B) levels.
Figure 3.
Principal component analysis (PCA) of taxa occurring in the CSB and LSB milk microbiota based on the relative abundance distribution at family (A) and OTU (B) levels.
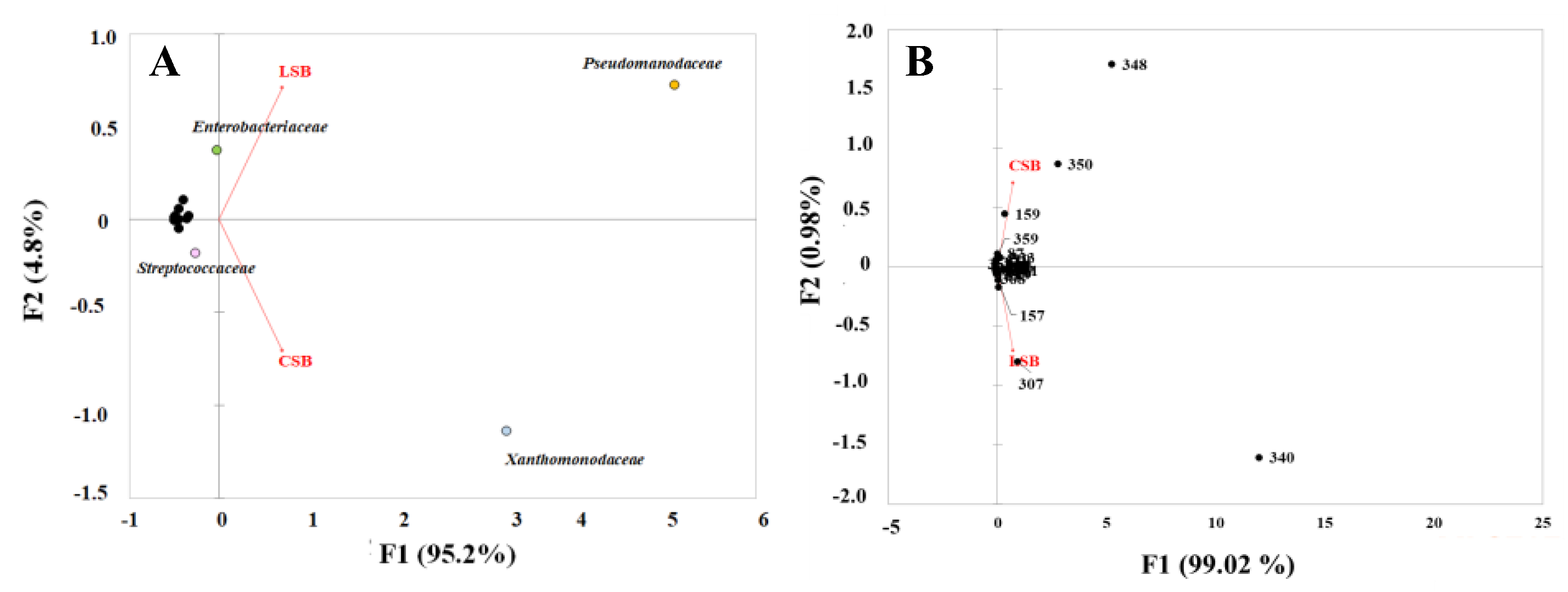
Figure 4.
Principal component analysis of taxa occurring in the CSB (blue symbols) and LSB (red symbols) in the milk (circle), curd (square), and cheese (inverted triangle) microbiota based on the relative abundance distribution at the family level.
Figure 4.
Principal component analysis of taxa occurring in the CSB (blue symbols) and LSB (red symbols) in the milk (circle), curd (square), and cheese (inverted triangle) microbiota based on the relative abundance distribution at the family level.
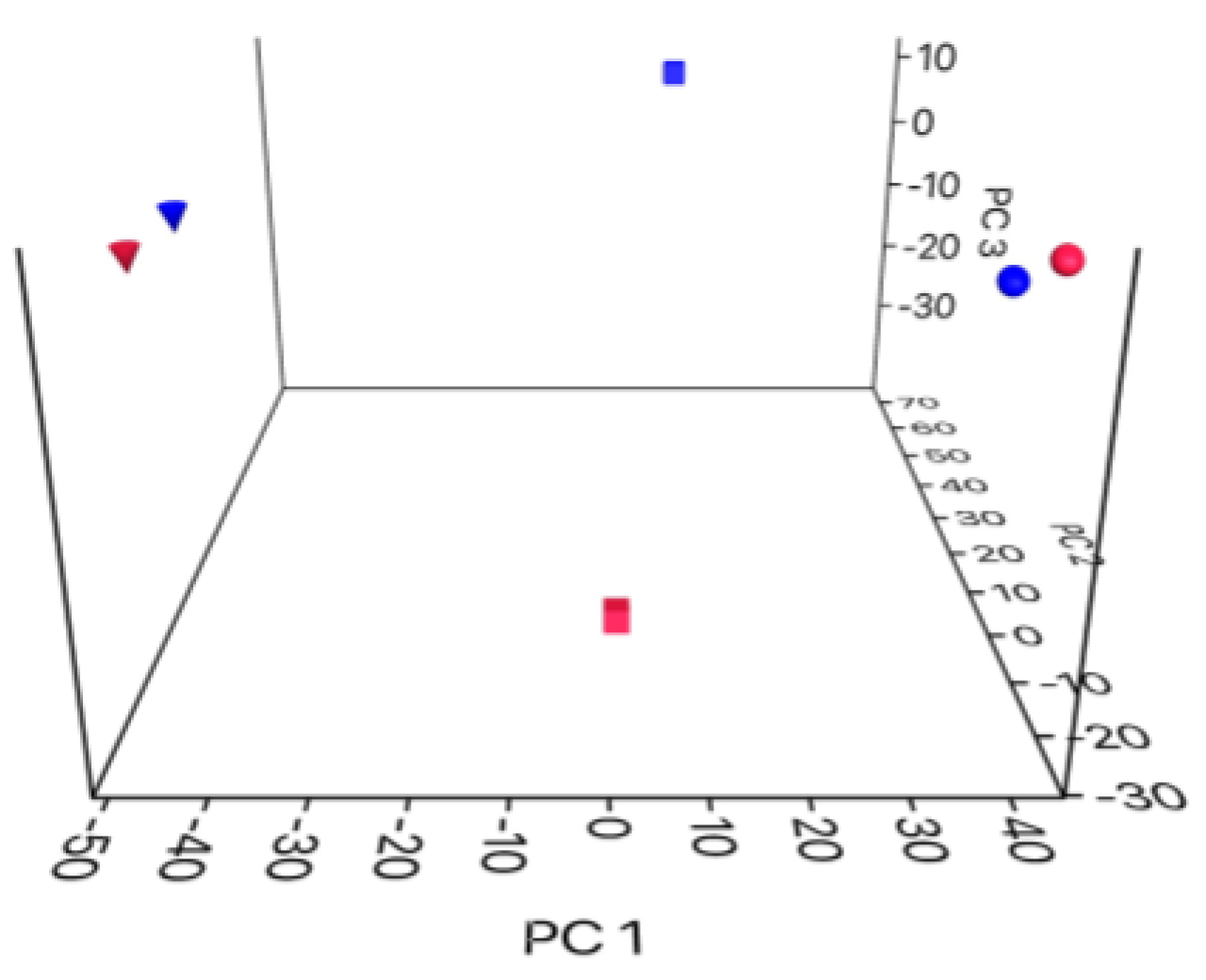
Figure 5.
Comparative analysis of curd microbiomes. (A) The Venn diagram shows the number of shared and unique OTUs (≥ 2 reads) in the CSB and LSB milk and curd microbiota. (B) Principal component analysis (PCA) of taxa occurring in the CSB and LSB curd microbiota is based on the relative abundance distribution at the OTU level.
Figure 5.
Comparative analysis of curd microbiomes. (A) The Venn diagram shows the number of shared and unique OTUs (≥ 2 reads) in the CSB and LSB milk and curd microbiota. (B) Principal component analysis (PCA) of taxa occurring in the CSB and LSB curd microbiota is based on the relative abundance distribution at the OTU level.
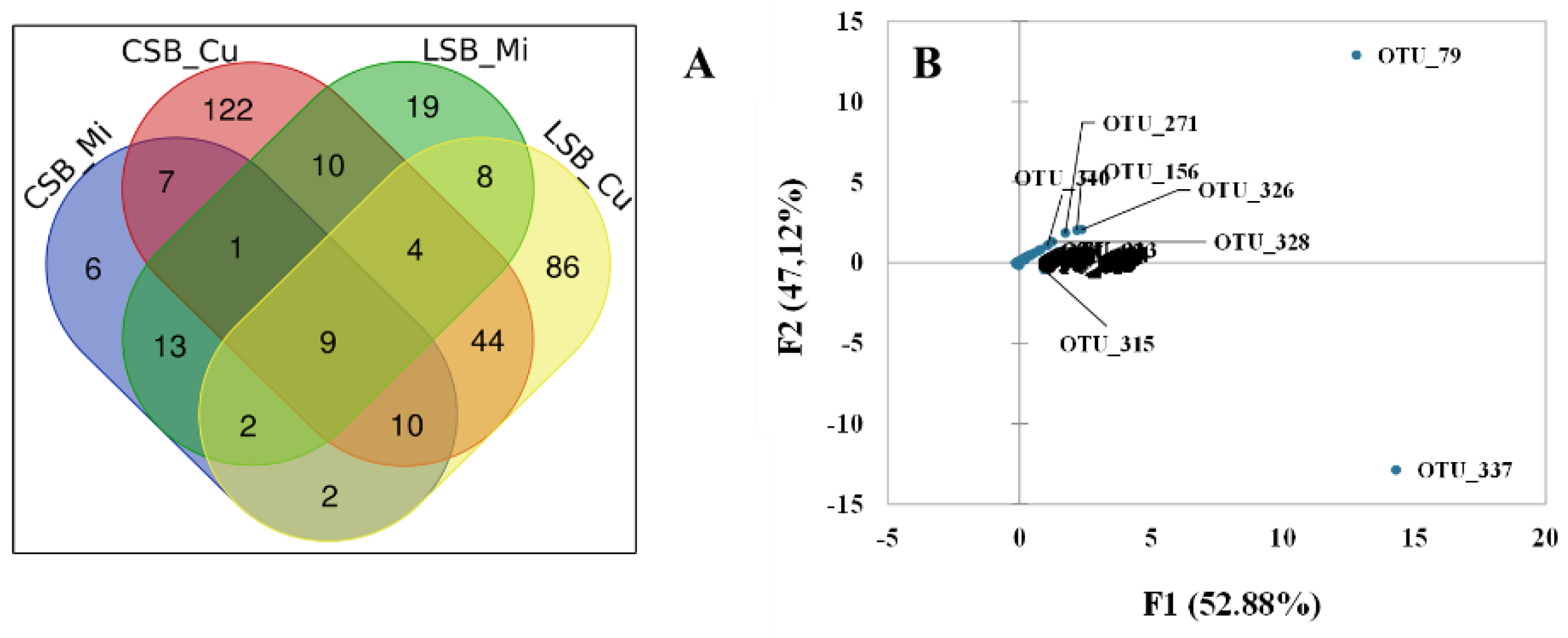
Figure 6.
The Venn diagrams show the number of shared and unique OTUs (≥ 2 reads) in the milk, curd, and cheese microbiota from CSB (Left Panel) and LSB (Right Panel) cheesemaking processes.
Figure 6.
The Venn diagrams show the number of shared and unique OTUs (≥ 2 reads) in the milk, curd, and cheese microbiota from CSB (Left Panel) and LSB (Right Panel) cheesemaking processes.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



