
Submitted:
16 April 2024
Posted:
17 April 2024
You are already at the latest version
Abstract
We have developed a family of N-alkyl-nitrobenzamides that exhibit promising antitubercular ac-tivities. These compounds are a simplification of the structure of known inhibitors of DprE1, an essential Mtb enzyme and an emergent anti-TB target. The compounds were obtained by a simple synthetic methodology and their antitubercular, cyto-toxicity and stability were studied. The most active compounds were the 3,5-dinitro and the 3-nitro-5-trifluoromethyl derivatives. Lipophilicity was shown to be a main parameter modulating activity within each family. The most active compound presented MIC of 16 ng/mL and had in-termediate lipophilicities. The activity was also studied in a macrophage model of infection. In this model the compounds with chain lengths of 6 and 12 carbon atoms presented the best results, ex-hibiting activity profiles comparable to isoniazid. Docking studies performed suggested that the fit of our highly active compounds to the DprE1 binding pocket was very similar to the literature compounds. Furthermore, the metrics used in the analysis were able to distinguish between the families of the highly and least active compounds. Assessment of susceptibility over multiple species and computational together with the structure of the compounds are complementary information that supports DprE1 as a likely target of action for this type of compounds.
Keywords:
Tuberculosis
; nitrobenzamides
; DprE
; mycobacteria
1. Introduction
Tuberculosis is a respiratory tract disease caused by Mycobacterium tuberculosis species complex that spread between people through aerosols containing as little as 1-3 bacilli [1]. Although TB mostly affects the lungs, it may adversely impact the brain, kidneys, or spine. Despite being a treatable disease, tuberculosis still remains one of the leading causes of death globally, being the second most common single infectious agent-related cause of death (ranking above HIV/AIDS and only second to COVID-19 in 2022) [2,3,4]. The COVID-19 pandemic has reversed years of global progress in tackling tuberculosis [5] and, for the first time in over a decade, TB deaths had increased, and only now are starting to return to pre-pandemic levels, according to the World Health Organization’s 2023 Global TB report [4].
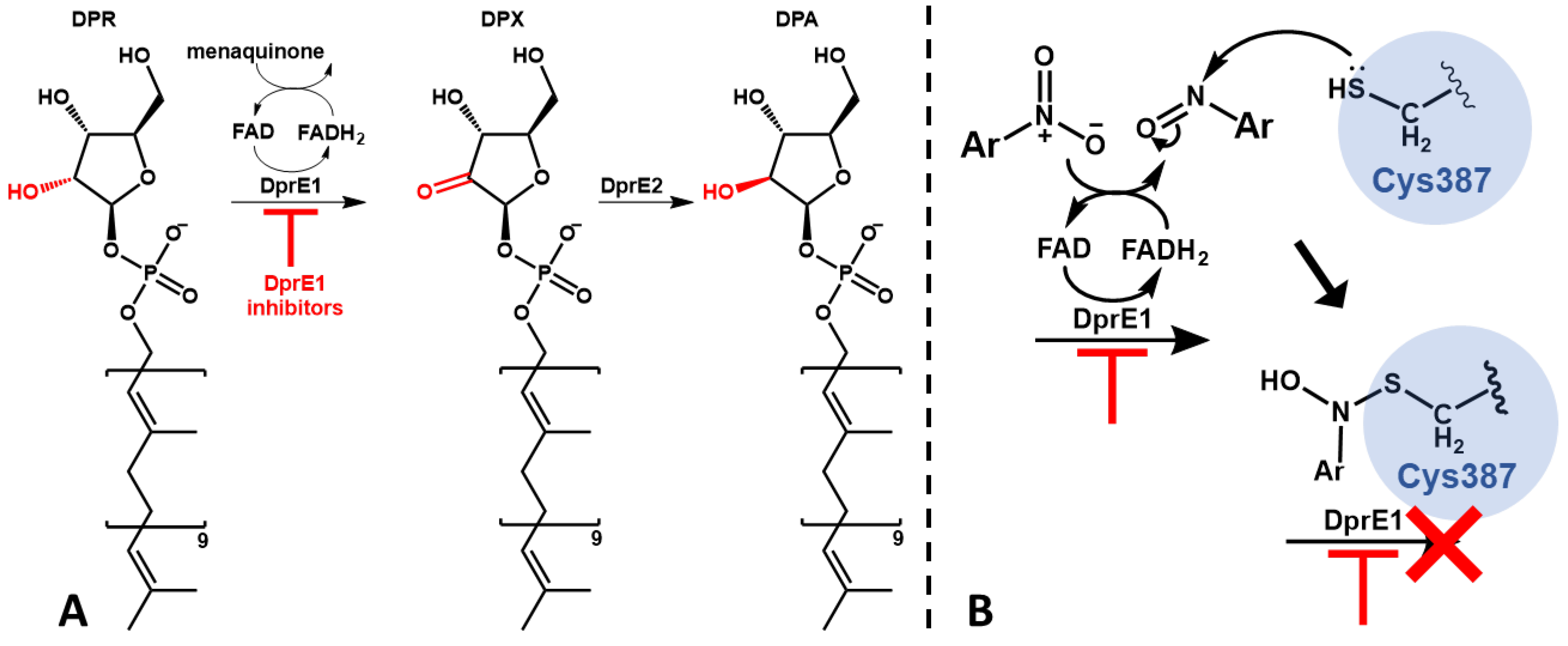
Most cases of tuberculosis can be treated and the spread of infection can be prevented with prompt diagnosis and six months of first-line antibiotic treatment [6]. However the increasing appearance of multidrug-resistant (MDR) and extensive drug-resistant (XDR) TB makes clear the need for new drugs that can be effective and used in shorter therapeutic regimens [7]. Numerous novel therapeutic targets have been established in recent years with the effort to develop new effective anti-TB therapeutics, many of which targeting essential proteins implicated in the synthesis of the cell wall components [8,9]. One relevant example is DprE1, considered one of the promising targets for the development of new anti-TB drugs [10]. DprE1 is a component of the DprE1-DprE2 complex, an heterodimeric protein that catalyzes the epimerization of decaprenylphosphoryl-D-ribose (DPR) to decaprenylphosphoryl-D-arabinose (DPA) (Figure 1A) [11]. DPA is a vital precursor in the production of both lipoarabinomannan and arabinogalactan, essential in cell wall biosynthesis [12,13]. One of the reasons that makes DprE1 so promising is its intracellular location, since by being located in the periplasmatic space of the mycobacteria the only barrier to the action of its inhibitors is the cell wall [14].
Inhibitors of DprE1 are generally subdivided into covalent and non-covalent inhibitors. The mechanism of covalent inhibition is based on the formation of a non-reversible covalent bond between the inhibitor and the cysteine 387 residue (Cys387) of DprE1. For this to happen, covalent inhibitors make use of an aromatic nitro moiety that, upon interaction with the active site of DprE1, undergoes reduction of the nitro group to nitroso, which then forms a covalent adduct with Cys387 irreversibly hindering the protein’s function (Figure 1B) [15,16]. This residue is profoundly conserved in mycobacteria, aside from in Mycobacterium avium and Mycobacterium aureum where cysteine is replaced by alanine and serine, respectively [17,18].
Figure 1.
Schematic representation of the DprE1-DprE2 complex function and its inhibition; (A) – Epimerization of DPR to DPA; (B) - Mechanism of activation and DprE1 inhibition of nitroaromatic suicide inhibitors.
Figure 1.
Schematic representation of the DprE1-DprE2 complex function and its inhibition; (A) – Epimerization of DPR to DPA; (B) - Mechanism of activation and DprE1 inhibition of nitroaromatic suicide inhibitors.
Our research group has studied benzoic acid and its derivatives for their antimycobacterial potential, specifically against Mtb H37Rv strain, attempting to develop alkyl esters as prodrugs capable of increasing the weak acids’ antitubercular activity [19,20]. We provided proof that mycobacteria may quickly hydrolyze a range of organic acid esters [21,22] and that esters demonstrate greater in vitro activity than that of the corresponding free organic acids [23,24], indicating that they are appropriate prodrugs for the substances, helping the molecules enter the cells and releasing the free acid.
In previous work we found that alkyl esters of 3,5-dinitrobenzoic acid showed very relevant antitubercular activities [25]. The activity was much higher than anticipated by Zhang et al. [19] based on the pka of the liberated free acid, indicating that the presence of the nitro groups might be important for the mechanism of action. Our inability to correlate the activity with ester hydrolysis rates led us to hypothesize that these compounds might act directly as active drugs rather than prodrugs. If the compounds could be acting as drugs, then a series of isosteres could be explored for activity.
Building on this, after exploring the potential of nitrobenzoates and nitrothiobenzoates [26], we decided to explore the antimycobacterial potential of bioisosteric amide analogues. Only two 3,5-dinitrobenzamides with linear alkyl chains were reported in the literature as having antimycobacterial activity [27] but they were somehow disregarded, as their activity was lower when compared with other more complex derivatives containing a terminal aromatic moiety (such as in the DNB1 or DNB2). The reason for that is probably because the authors unfortunately only explored the derivatives with C6 and C16 N-alkyl groups. Because we knew from our previous work with nitro containing esters that the alkyl chain could be optimized for activity by modifying the chain length of the compounds [26], we decided to apply the same approach to the N-alkyl benzamide analogues with the objective of improving their antimycobacterial efficacy.
Our initial efforts focused on synthesizing a series of nitrobenzamides with N-alkyl chains ranging from four to sixteen carbon atoms and evaluating their antitubercular activities. Encouraged by the results obtained on the activity of these compounds, we extended our study to include their performance in a macrophage infection model. Given the fears associated with nitroaromatic compounds in drug development [28] the cytotoxicity of the compounds here reported was assessed using human macrophages. We also decided to study the stability of the compounds on buffer, plasma and in a mycobacterial homogenate. Buffer stability studies were used to evaluate the chemical stability of the compounds at pH 7.4 and plasma stability studies served to evaluate if the compounds exhibit stability that allows the determination of activity in vivo. Stability in mycobacterial homogenate was examined to determine the effect of mycobacterial enzymes on the compounds. Since we had no access to purified DprE1 to directly test the inhibition of purified DprE1, we evaluated the effects of our compounds on various mycobacteria species known to have differing responses to DprE1 inhibitors. In parallel, computational docking studies were performed to analyze how our compounds fit within the DprE1 binding pocket, compared to the DNB1 and DNB2, known inhibitors of DprE1. These studies supported the potential of DprE1 as a possible target for our compounds, and lead to the discovery of new DNBs with antitubercular activities comparable to established benchmarks like DNB1.
3. Results
3.1. Compound Library and Antitubercular Activity
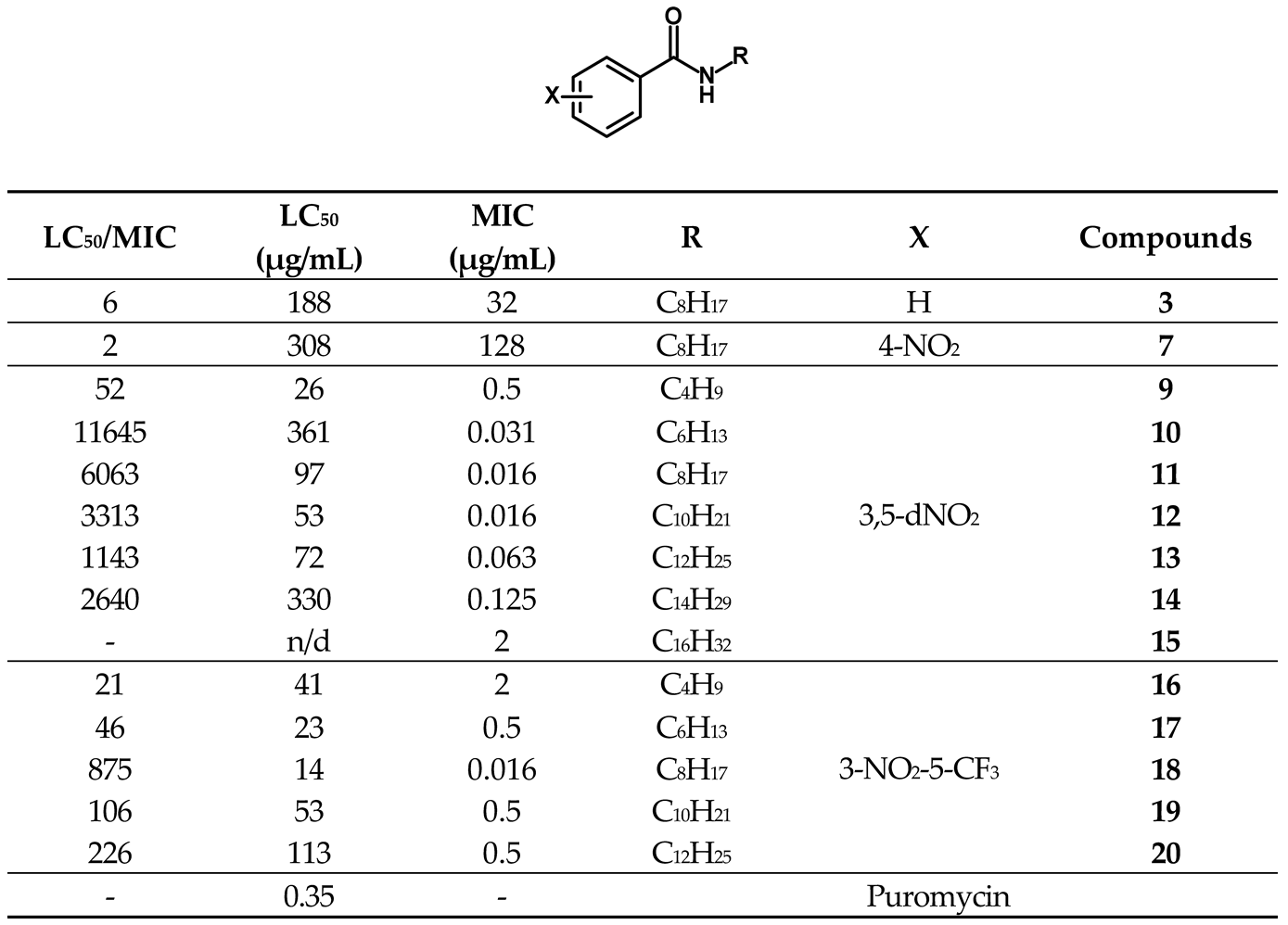
A library comprising of four series of benzamides, with 4-NO2, 3,5-NO2, 3-NO2-5-CF3 and H- substitution in the aromatic ring and N-alkyl groups of varying length, was obtained following a standard synthetic approach. The objective was to explore the effect of nitro substitution on the activity of the compounds and at the same time, study the effect of the N-alkyl group length on the activity of each series. The compounds obtained as well as their corresponding antitubercular activity, determined as minimal inhibitory concentration (MIC) and minimal bactericidal concentration (MBC) are presented in Table 1.
The first observation is that the compounds with a nitro substituent in the 3-position of the aromatic ring presented activities significantly higher than the other compounds. Not only the 3,5-dinitro and 3-NO2-5-CF3 series presented activities significantly higher than the other two series but also the most active compounds in the series containing a 3-nitro substitution (11, 12 and 18) presented MIC ca 2000 times lower than the most active compounds in each the other two series (3 and 6).
The library contains compounds with very different lipophilicities, as predicted octanol-water partition coefficient (logP) ranged from 2.21 to 8.59. When we analyzed the library as a whole, no correlation between the logP value and the antitubercular activity was found. However, when we look at each series independently, a different pattern emerged, as in all the series the more active compounds are the ones with intermediate lipophilicity (and number of carbons in the alkyl chain between 6-10). Figure 2 represents the correlation between logP and MIC values for the 3,5-dinitro substituted benzamides. A polynomial correlation between the logP and the antimycobacterial activity can be identified, indicating that quite possibly this is indeed a main parameter modulating activity.
3.2. Susceptibility Assessment of Multiple Mycobacterial Species
The difference in activity found between the series of compounds tested could be indicative that the two more active series are most likely acting by a diverse mode of action to the remainder of the compounds. Our N-alkyl-3,5-dinitrobenzamides can be considered a simplification of the classic antitubercular amide DNB1 [30], containing a simple lipophilic alkyl group instead of a more complex substituent in the amide nitrogen. Similarly, the N-alkyl-3-nitro-5-trifluoromethylbenzamides are in turn a simplification of the active derivative A1 (itself a bioisostere of DNB1) [31] or a simplification of CT319 [32]. Both A1 and CT319 are active 3-nitro-5-trifluoromethylbenzamide derivatives that contain a trifluoromethyl group in the 5-position instead of a nitro group. Those three compounds act on DprE1 [30,31,32]. Although not definite, a simple approach to test if DprE1 could be a possible target for these compounds consists in challenging a range of bacterial strains with our compounds and known DprE1 inhibitors [17,33]. Since the different bacterial strains possess diverse sensitivities to DprE1 inhibition, a bioactivity profile of action could be established. Hence, the susceptibility of M. tuberculosis, M. bovis BCG, M. avium and M. smegmatis was assessed, and compared to DNB1 and DNB2, known DprE1 inhibitors [30,34]. The results are presented on Table 2. Compounds from a previous study [26] were also tested together with the amides, which are the compounds O9 – O15, comprising the ester analogues of compounds 9-15.
The first part of Table 2 shows the bioactivity profile of DprE1 inhibitors when compared with two control drugs isoniazid (INH) and para-aminosalicylic acid (PAS). INH is highly active against Mtb and M. bovis, and moderately active against M. smegmatis and M. avium. PAS only presented high activity against Mtb and M. bovis. The known DprE1 inhibitors DNB1 and DNB2 possessed high activity against Mtb and M. bovis, moderate activity against M. smegmatis (MIC of 0.5 µg/mL) and a lower activity against M. avium (MIC of 32 and 64 respectively). The results obtained with the DprE1 inhibitors are in accordance with the literature. Indeed, M. avium does not have the codon that codes for Cys387 in DprE1, with cysteine being replaced by alanine [17]. This mutation confers natural resistance to the DprE1 inhibitors. The NfnB nitroreductase is overexpressed in M. smegmatis, which may cause the drug to become inactive due to the reduction of the nitro group to an amino group [18,35]. The other two species, M. bovis BCG, and M. tuberculosis H37Rv, are equally susceptible to DprE1 inhibitors [36].
The second part of Table 2 shows the activities of compounds 3-15 against the same mycobacteria. Comparing the profile of DprE1 inhibitors DNB1 and DNB2 with the most active 3,5 dinitro substituted derivatives (compounds 11 and 12) we can observe similar effects: while the MIC values for M. bovis are the same or slightly increased when compared to the Mtb values, the susceptibility of M. smegmatis was decreased and M. avium was shown to be completely resistant. Furthermore, this profile is not observed for amides 3 and 7 that present different structures. Compound 3 does not present a nitro substituent and compound 7 presents a single nitro substituent but in the 4-position of the aromatic ring.
Finally, we also determined in the present study the activity of esters O9-O15, structurally similar to the class of more active amides found in our studies (9-15). All compounds whose identification starts with an O are ester isosteres of the corresponding N-alkyl-3,5-dinitrobenzamides with the same number. Although the activity of the esters is lower than the corresponding amides, two different patterns can also be observed. The more active esters (O11 and O12) present a similar activity for Mtb and M. bovis, no significantly different activity for M. smegmatis, and a complete loss of activity against M. avium. The less active esters (compounds O9 and O15) do not show the same marked differences between the different mycobacteria species studied.
3.3. Cytotoxicity Evaluation in Human Macrophages
Given that aromatic compounds containing nitro groups are associated to adverse effects, namely due to their toxicity, genotoxicity, mutagenicity and/or carcinogenicity [37,38], the cytotoxicity of the compounds under study was assessed on the THP-1 human monocytic cell line (ATCC56 TIB202). Although there are no guidelines for admissible values, cytotoxicity can be estimated by determining the lethal concentration for 50% of the cell population, LC50 [39]. The lower the LC50 value, the lower the lethal concentration of the compound used, hence, higher toxicity to eukaryotic cells. Furthermore, the ratio between the LC50 and MIC values (LC50/MIC), was determined, in order to better sort the compounds according to their toxicity [39]. This ratio also serves as an indicator of a compound's selectivity between mycobacterial and eukaryotic cells, with a higher LC50/MIC ratio suggesting greater potential interest in the compound.

Table 3.
Results obtained from the cytotoxicity assays, represented by LC50 values (μg/mL), the MIC values (μg/mL) and the ratio LC50/MIC.
Table 3.
Results obtained from the cytotoxicity assays, represented by LC50 values (μg/mL), the MIC values (μg/mL) and the ratio LC50/MIC.
 |
The amides under study have LC50 values between 14 and 360 μg/mL. However, since the LC50 values obtained for such compounds correspond to concentrations much higher than their MIC values, it results in high values of LC50/MIC, or good selectivity to the mycobacterial targets. For example, for compound 11, LC50/MIC = 6063, which means that the LC50 value of this compound is 6063 times higher than its MIC value. Hence, the results here reported show that there is a large difference between the concentration of compound that impacts the mycobacteria (MIC) and the one that impacts human cells (LC50).
3.3. Biological Activity in Macrophage Model of Infection
Since the compounds under study showed promising antitubercular activities, their activity in an infection model was studied, namely by evaluating their antimycobacterial activity in THP-1 human macrophages infected with M. tuberculosis H37Rv as a model of mycobacterial infection. To assess the mycobacterial intracellular survival/killing, bacteria that survive the treatment were recovered from infected treated macrophages, plated on solid medium and CFU counts were assessed on days 0, 1, 3, 5 and 7 post infection. The results obtained are presented in Figure 3.
Figure 3 shows the effects of the compounds on the intracellular killing in infected macrophages relatively to the control (non-treated culture). INH was used as positive control. At the dose tested (0.15 μg/mL), not all compounds that showed good activity in vitro confirmed the activity in the macrophage assay. The compounds that presented higher activity both in vitro and ex vivo in infected macrophages were compounds 10, 12 and 13. Compound 18, the most active compound in the 3-nitro-5-trifluoromethyl benzamide series did not show good activity in macrophages. The same can be said about compound 11 that was one of compounds that showed higher activity in vitro. The compound that had the best results in both assays was compound 12.
3.4. Stability Studies
In previous studies, the compounds under study were esters and an attempt at establishing a correlation between antimicrobial activity and their stability in multiple biological media was made. However, since esters were prone to hydrolysis, it was difficult to state that the compounds were not acting as prodrugs of weak acids that indeed are known for light antimycobacterial activity [19,40]. The amide analogues that are now here discussed emerged as analogues with potentially higher resistance to hydrolysis, hence, their stability was also assessed.
For this, phosphate buffer saline PBS, human plasma, and M. smegmatis homogenate were used and all the compounds were incubated with the media. The detection of the respective free acid allowed the determination of the pseudo first order rate of the hydrolysis (for the compounds that decomposed faster) and the percentage of decomposition at 72 hours (for the most stable compounds). The results are presented in Table 4. No other products other than the free acid resulting from hydrolysis were detected in the incubations. The stability of the compounds was initially tested on PBS pH 7.4 and no degradation was observed over two weeks indicating that all the degradation observed in human plasma and mycobacterial homogenate is mainly enzymatic.
These results show that the amides are highly resistant to hydrolysis in both media assayed, with only the shorter chained derivatives being hydrolyzed. Such effect is in accordance with what was observed for the corresponding esters [26], where the shorter chain derivatives showed the most hydrolysis. Nevertheless, short chain N-alkyl amides were more easily hydrolyzed in mycobacterial homogenate than in plasma in our incubation conditions. Since the mycobacterial homogenate is richer in enzymes than the plasma due to being made from the whole mycobacterial cell [21], it is probably richer in amidases and thus can perform more easily the hydrolysis of short chain N-alkylamides. Our more active compounds were not easily hydrolyzed by the mycobacterial homogenate.
3.5. Computational Studies
Considering the structure of our nitro containing compounds, the difference in activity observed, the structural similarity with known DprE1 inhibitor, and the results obtained in activity against different mycobacterial species presented, DprE1 is the most attractive target for the compounds presented in this study. With the aim of further exploring this possibility, a docking-based computational study was performed, where the position of our compounds within the binding pocket of DprE1 was studied and compared with that of DNB1 and DNB2. The 4P8L PDB entry was chosen for this purpose since it is the only structure with a completely resolved DprE1 enzyme. In this study, 5 docking runs were performed using 3 scoring functions (AutoDock, Vina and Vinardo), and their top scoring pose was selected. Next, the poses obtained were analyzed in relation to their distance to the mechanistically relevant locations for DprE1 inhibition, the cysteine 387 residue and the FAD cofactor. Using these poses, the distance of the nitro groups to both the cysteine 387 (NDCys387) and the FAD (NDFAD) were calculated and since some compounds do not contain a nitro group, the center of the aromatic moiety was also considered (CDCys387 and CDFAD). This analysis allows to study the fit of the compounds in the binding pocket in a way that is less dependent on arbitrary interactions and more relevant to the expected mechanism of inhibition.
To exemplify this analysis, we selected compound DNB1 (gray), compound 11 (green) and compound 18 (pink) and the overlap of their best docking poses is present in Figure 4A. These compounds were selected as representatives of their respective families, the 3,5-dinitro- substituted and 3-nitro-5-(trifluoromethyl)-substituted respectively, since all compounds within these families have similar distances to both the cysteine residue and the FAD cofactor. Since, according to the mechanism of action of DNBs (Figure 1B), the nitro group must firstly be activated by the FADH2 and then quickly be attacked by the sulfur atom of the cysteine residue, these moieties can the considered a pre-reactive triad (Figure 4B). As Figure 4A shows, all the selected compounds are in the proper position to form this triad.
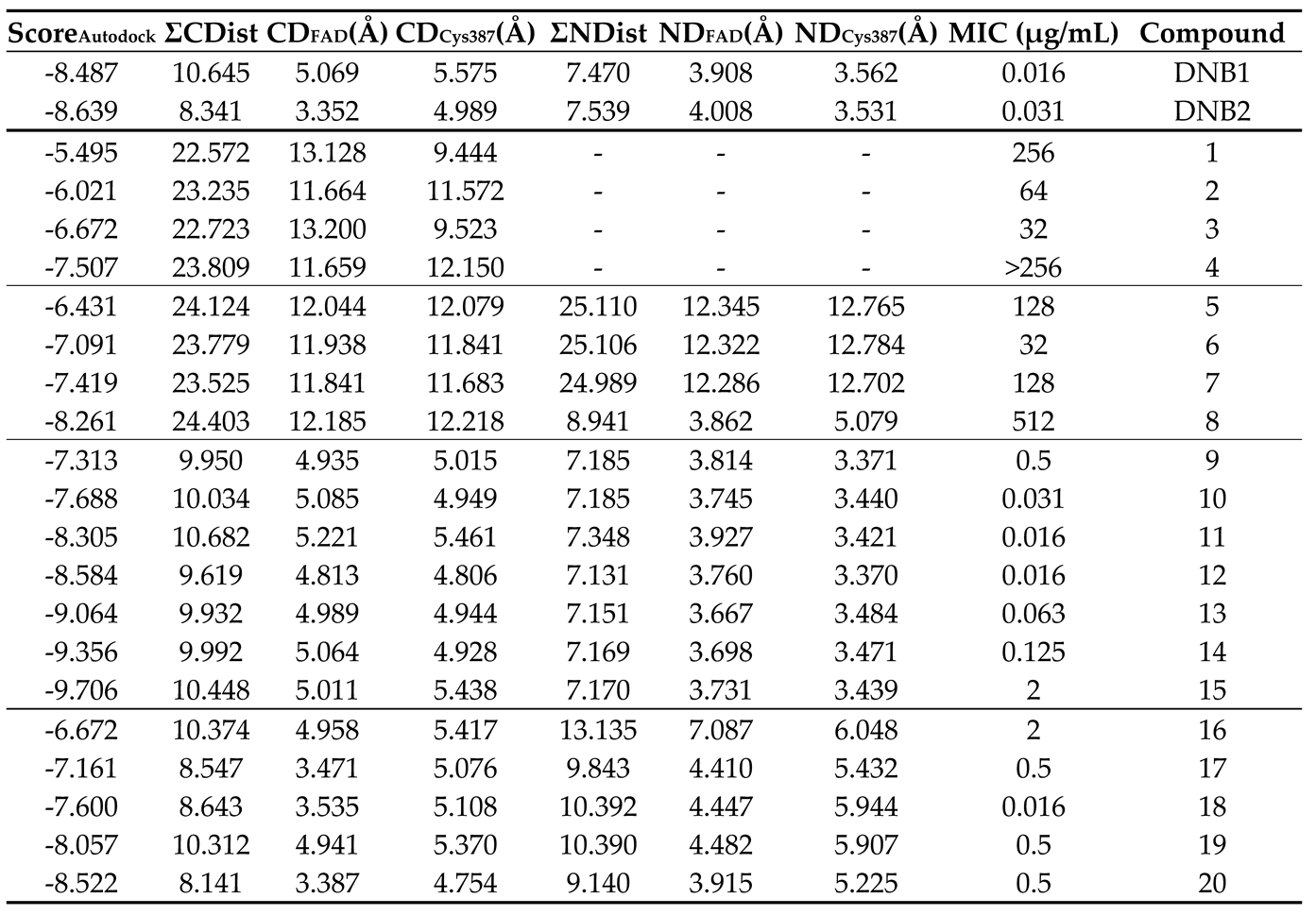
The results that led to the “best fit” of the compounds to the binding pocket, which corresponds to the poses with shorter NDCys387, NDFAD, CDCys387 and/or CDFAD distances, are present in Table 5.
The sum of the distances to cysteine 387 and FAD was included (both ΣNDist and ΣCDist) as an easier way to retrieve some conclusions of their spatial position in the binding pocket of DprE1. Firstly, we can observe that, in general, within the same family of compounds, the distances measured are similar, indicating that the variation of alkyl chain length does not significantly impact the fit of the compounds to the binding pocket, even though their score values indicate a tendency to increase with the increasing chain length. Furthermore, we can see that both ΣNDist and ΣCDist can be used to distinguish the most active families of compounds from the least active families. In example, compounds 5-8, which have higher MIC values (512 - 32 µg/mL), have higher ΣNDist (25.110 – 8.941 Å) and ΣCDist (~24 Å) than the family of compounds 9-15 that are considerably more active (MIC values of 2 - 0.016 µg/mL), for which these distances were notably lower (ΣNDist ~ 7 and ΣCDist ~ 10 Å). Moreover, the most active family of compounds has similar values of ΣNDist and ΣCDist when compared to DNB1 and DNB2.
3. Discussion
The benzamides presented here were easily obtained by reacting the acid chloride with the corresponding amide. Detailed synthesis procedures yields and results not presented in the manuscript are available in the Supplementary Material.
The large difference in activity observed between compounds 9-20 versus others suggests distinct mechanisms of action. The in vitro activity of these benzamides is primarily centered on the presence of a nitro group at the 3-position of the benzene ring. Substitution of this nitro group or its relocation to the 4-position significantly diminishes activity, echoing findings by Christophe et al. [30].
The fact that most of the activity was maintained by substitution of the nitro group in the position 5 of the aromatic ring by a trifluoromethyl group is also in accordance with the literature [31,41]. In an interesting study, Li et al. developed nitrobenzamide compounds by simplifying the structure of PBTZ169 [31]. They found that substituting the nitro group with a CF3 group slightly reduced the compounds' activity. However, the overall activity remained comparable to the original nitro derivatives, suggesting that a nitro group could be replaced with a CF3 group to address pharmacokinetic or toxicological concerns.
Lipophilicity is also important, but it only modulates the activity of each series with N-alkyl chain lengths of 6-10 carbons being optimal. Since our computational studies indicate that the lengths of the alkyl chain only interfere slightly with the position of the inhibitor in the binding pocket of DprE1, it seems that an optimum lipophilicity of the compounds is mainly needed to allow the compounds to access the periplasmatic space where DprE1 is located.
The relationship between logP and antimycobacterial activity in the 3,5-dinitro substituted derivatives indicates lipophilicity as a critical factor (Figure 2). Considering mycobacterial cell wall lipidic rich composition, increasing lipophilicity may enhance membrane permeation however it also reduces water solubility, posing challenges in biological assays, especially for compounds with longer than ten carbon chain. Therefore, it is conceivable that longer alkyl chains have the capacity to exhibit comparable or even higher intrinsic DprE1 inhibition, but their solubility prevents its expression leading to the parabolic behavior observed in Figure 2.
To support our hypothesis that our compounds inhibit DprE1, we evaluated their effectiveness against M. tuberculosis, M. bovis BCG, and M. avium and compared these results with those of known DprE1 inhibitors, DNB1 and DNB2. Taking in consideration the limitation that M. avium is naturally resistant to most antibiotics [24], we can indeed observe that the amides 11 and 12 showed high activity in M. bovis and M. tuberculosis, that are naturally sensitive to DprE1 inhibitors, however, the activity is completely lost in M. avium, which has a mutation in the DprE1 enzyme, having an alanine residue in the place of Cys387. This cysteine is fundamental for the mode of action of covalent DprE1 inhibitors as it is its thiol group that reacts with the nitroso group of the activated inhibitor (Figure 1). Similar results were presented by the benzothiazinone BTZ043 which was tested by Caroline et al. [17], where BTZ043 showed high activity in M. tuberculosis, that, was lost in M. avium. The NfnB nitroreductase is overexpressed in M. smegmatis, which may cause the drug to become inactive due to the reduction of a nitro group to amine, which is unable to react with the cysteine residue. The fact that our more active 3,5-dinitrobenzamides presented activities over the four mycobacterial species that follow the described pattern of DprE1 sensitivity provided important clues that support the hypothesis of the disubstituted derivatives acting as DprE1 inhibitors. The remaining compounds tested did not present a similar spectrum of activity and presented intrinsically low activities against Mtb indicating that they either do not inhibit DprE1 or are unable to reach the periplasmic space where DprE1 is located [12,42].
One important aspect of any new antitubercular drug is its activity over dormant inactive cells [43] We did not test our compounds in any model suitable to assess its activity over non-replicating M. tuberculosis as the probable mechanism of action does not support a relevant activity over dormant cells, however this hypothesis should be kept open.
The cytotoxicity assessments indicated that despite their nitroaromatic nature, the compounds exhibit low toxicity relative to their antimycobacterial activity. This finding was particularly pronounced in compounds with MIC values below 0.5 µg/mL, where the effective concentrations were substantially lower than the cytotoxic levels, suggesting potential safety in therapeutic contexts. However, in the future, other toxicity studies (in silico, in vitro and in vivo) must be performed in parallel with the advancement of this family of compounds.
Since no significant impact on macrophage viability was observed at the MIC concentrations used, an infection model with Mtb infected macrophages was challenged with the most active compounds. Figure 3 allows to easily distinguish the compounds that lead to intracellular killing of mycobacteria (decreasing the CFU count number) to the ones that do not. For compounds 10, 12 and 13 it was observed pronounced intracellular mycobacterial death, although for compound 12 a diminished antitubercular activity was achieved. Interestingly, compound 11 did not show activity, despite being a compound with one of the lowest MIC values. Since compound 12 also showed a diminished effect when compared to compounds 10 and 13, it is possible that this loss of activity is due to the length of the alkyl group, or its associated physical properties, that, in this experimental setup, were detrimental to antitubercular activity, contrary to the in vitro experimental setup.
From Table 2, is clear that amide isosteres of the 3,5-dinitrobenzoates have better activity over M. tuberculosis than the esters. In previous work, ester analogues of the amides under study were associated with some susceptibility to hydrolysis [25]. However, here it was shown that the generality of amide derivatives was stable in all biological media assessed, and the most active compounds, both in vitro and in the macrophage model, were very stable.
Finally, computational studies were performed aimed at assessing DprE1 as a possible target of action for the most active families of compounds, namely the 3,5-dinitro and 3-nitro-5-trifluoromethyl substituted derivatives. By comparing our compounds with the literature compounds DNB1 and DNB2, we can observe that, while the score values do not accurately allow to distinguish highly active (MIC ≤ 2 µg/mL) from less active (MIC > 2 µg/mL) compounds, the distances to the cysteine 387 residue and the FAD cofactor, both to the aromatic ring or the nitro group, allowed to make this distinction.
The similarity in distance measurements between our highly active compounds and established DprE1 inhibitors suggests a similar fit within the DprE1 binding pocket, reinforcing DprE1 as a suitable target for these compounds. Metrics such as ΣNDist and ΣCDist help distinguish between highly active and less active compound families, providing a valuable tool for future research and identifying potential new inhibitors.
It is important to note that these distance metrics do not vary significantly with different alkyl chain lengths because of the enzyme's broad binding pocket, which can accommodate large, branched chains like those in the natural substrate of DprE1. While this confirms the structural fit is crucial for inhibitory activity, it complicates the ability to rank compounds within families based on effectiveness.
Regarding the correlation between score values and MIC levels, no direct relationship was observed. Instead, score values increased linearly with alkyl chain length and logP values, indicating that differences in MIC are likely due to solubility and absorption challenges rather than binding affinity to DprE1.
4. Materials and Methods
Materials. Balanced salt solution, phosphate-buffered saline (PBS), Dulbecco’s modified Eagle’s medium (DMEM), and L-glutamine were purchased from Invitrogen. Sodium dodecyl sulphate (SDS), Triton X-100, benzoic acid, 4-nitrobenzoic acid, 3,5-dinitrobenzoic acid, 3-nitro-5-(trifluoromethyl)benzoic acid, n-butylamine, n-hexylamine, n-octylamine, n-decylamine, n-dodecylamine, n-tetradecylamine, n-hexadecylamine and trypan blue were purchased from Sigma-Aldrich Quimica SA. Middlebrook 7H10 agar was purchased from Difco (BD Difco, Franklin Lakes, NJ, USA). Microwell tissue culture plates were purchased from Nunc. All amides presented were synthesized according to the procedures described in this paper. Compounds were prepared in stock solutions of 40 mg/ml in dimethyl sulfoxide (DMSO – AppliChem Panreac). Isoniazid (Sigma-Aldrich) is a first line antibiotic against tuberculosis and was used as a positive control for M. tuberculosis killing.
Bacterial strains and cell lines. Bacteria broth culture medium Middlebrook 7H9 and solid culture medium Middlebrook 7H10 were purchased from Difco (BD Difco, Franklin Lakes, NJ, USA). All Mycobacterium spp. were cultivated in Middlebrook’s 7H9 medium supplemented with 10% OADC (oleic acid, albumin, dextrose, catalase) enrichment (BD Difco, Franklin Lakes, NJ, USA), 0.02% glycerol, and 0.05% tyloxapol (Merck, KGaA) incubated at 37 °C until exponential growth phase was achieved. M. tuberculosis H37Rv (ATCC 27294), M. avium (DSM44156) and M. bovis BCG (CIP 105050) were used for MIC evaluation. M. smegmatis (ATCC607, mc2 155) was used for homogenate preparation and for MIC determination.
Synthesis. 1. Acyl chloride synthesis: A solution of the chosen benzoic acid derivative in thionyl chloride (3 mL per mmol of acid) was refluxed for 5 h, leading to the formation of the desired acyl chloride. The excess thionyl chloride was removed by low pressure evaporation. The product was used without further purification. 2. General protocol to Amide synthesis: A solution of the appropriate acyl chloride (1 eq.) in dichloromethane was added dropwise to a solution of corresponding amine and triethylamine (1.5 eq.) in dichloromethane at 0 °C. When the reaction was complete (as assessed by TLC using hexane:ethyl acetate, 5:1 to 1:1, or ethyl acetate as eluent) the reaction mixture was filtered and the filtrate washed successively with 10 mL of distilled water and with 15 mL of saturated sodium bicarbonate solution. The dichloromethane solution was subsequently dried, and the solvent evaporated. The residue was purified by column chromatography (silica gel 60) using hexane: ethyl acetate, 5:1 to 1:1, or ethyl acetate as eluent. All compounds were characterized by 13C NMR, 1H NMR, IR and HRMS. The purity of the compounds was further tested by HPLC and TLC. Synthesis specifications, yields and structural data for compounds 11, 12 and 18 is described below. Data for the rest of the compounds is available in the Supplementary Material.
Synthesis of N-octyl-3,5-dinitrobenzamide (11). Following the described general procedure, 6 mmol (0,837 mL) of 3,5-dinitrobenzoyl chloride were dissolved in DCM (2,5 mL) and added to a solution in DCM (2,5 mL) of 9 mmol (1,480 mL) of n-octylamine and 6 mmol (0,833 mL) of triethylamine. N-octyl-3,5-dinitrobenzamide - yellow solid; Yield 43%; 1H NMR (300 MHz, Chloroform-d) δ 9.18 (t, J = 2.1 Hz, 1H), 8.95 (d, J = 2.1 Hz, 2H), 6.37 (s, 1H), 3.54 (td, J = 7.3, 5.7 Hz, 2H), 1.79 – 1.61 (m, 2H), 1.50 – 1.19 (m, 10H), 0.90 (t, J = 7.3 Hz, 3H). 13C RMN (300 MHz, Chloroform-d) δ 162.79 (C7), 120.95 (C4), 138.20 (C1), 127.17 (C2 and C6), 148.64 (C3 and C5), 40.89 (C9), 31.76 (C10), 29.44 29.23 29.16 26.98 (C11-C15), 14.05 (C16). Infra-red (IR) - (n, cm-1) - 1720,43 (C=O). HRMS (ESI+): m/z calculated for C15H21N3O5: 323.34446, found: 324.1562 (M+H+). Purity by HPLC 99,5%.
Synthesis of N-decyl-3,5-dinitrobenzamide (12). Following the described general procedure, 6 mmol (0,837 mL) of 3,5-dinitrobenzoyl chloride were dissolved in DCM (2,5 mL) and added to a solution in DCM (2,5 mL) of 9 mmol (1,776 mL) of n-decylamine and 6 mmol (0,833 mL) of triethylamine. N-decyl-3,5-dinitrobenzamide - yellow solid; Yield 41%; 1H NMR (300 MHz, Chloroform-d) δ 9.17 (t, J = 2.0 Hz, 1H), 8.96 (d, J = 2.1 Hz, 2H), 6.47 (s, 1H), 3.54 (td, J = 7.3, 5.7 Hz, 2H), 1.78 – 1.63 (m, 2H), 1.49 – 1.18 (m, 14H), 0.89 (t, J = 7.3 Hz, 3H). 13C RMN (300 MHz, Chloroform-d) δ 162.79 (C7), 120.94 (C4), 138.20 (C1), 127.18 (C2 and C6), 148.64 (C3 and C5), 40.90 (C9), 31.86 (C10), 29.52 29.44 29.28 26.98 22.65 (C11-C17), 14.08 (C18). Infra-red (IR) - (n, cm-1) - 1720,43 (C=O). HRMS (ESI+): m/z calculated for C17H25N3O5: 351.39762, found: 352.1869 (M+H+). Purity by HPLC 99,5%.
Synthesis of N-octyl-3-nitro-5-(trifluoromethyl)benzamide (18). Following the described general procedure, 6 mmol (0,967 mL) of 3-nitro-5-(trifluoromethyl)benzoyl chloride were dissolved in DCM (2,5 mL) and added to a solution in DCM (2,5 mL) of 9 mmol (1,480 mL) of n-octylamine and 6 mmol (0,833 mL) of triethylamine. N-octyl-3-nitro-5-(trifluoromethyl)benzamide - yellow solid; Yield 35%; 1H NMR (300 MHz, Chloroform-d) δ 8.76 (br. s, 1H), 8.60 (br. s, 1H), 8.41 (br. s, 1H), 6.40 (s, 1H), 3.50 (td, J = 7.3, 5.7 Hz, 2H), 1.75 – 1.55 (m, 2H), 1.48 – 1.16 (m, 10H), 0.88 (t, J = 7.3 Hz, 3H). 13C RMN (300 MHz, Chloroform-d) δ 163.77 (C7), 120.64 (C4), 137.63 (C1), 132.90 (q, JCF = 34.7 Hz, C5), 124.75 (C6), 129.97 (C2), 123.04 (CF3), 148.36 (C3), 40.77 (C9), 31.76 (C10), 29.44 29.23 29.16 26.97 22.60 (C11-С15), 14.03 (C16). Infra-red (IR) - (n, cm-1) - 1720,43 (C=O). HRMS (ESI+): m/z calculated for C16H21F3N2O3: 346.34482, found: 347.1496 (M+H+). Purity by HPLC 98,7%.
Compound characterization. 1H NMR and 13C-NMR spectra were recorded on a Bruker Ultra-Shield 300 MHz spectrometer, in the indicated solvent; chemical shifts are reported in parts per million (ppm), relative to tetramethylsilane (TMS). The spectra were referenced to the solvent peak and coupling constants (J) are quoted in hertz (Hz). The IR spectra were recorded on a 400 FTIR Nicolet Impact spectrometer between 4000 and 400 cm-1. High-resolution mass spectra (HRMS) were obtained on a Bruker Impact II quadrupole time-of-flight mass spectrometer (Bruker Daltoniks), and m/z values are reported in Daltons.
HPLC analysis: HPLC determinations were performed in a HPLC Hitachi LaChrom Ultra system comprising of two Hitachi L-2160U pumps, a Hitachi UV-L-2400U detector, a Hitachi L-2200U auto sampler, a Hitachi L-2300 column oven, a Merck-Hitachi D-2500 integrator and a Merck RP-8 column. The eluant was a mixture of acetonitrile (60% to 80%) and aqueous phosphate buffer with 5% of KH2PO4/H3PO4 0.025M (40% to 20%). The flow rate was 1 mL min-1 and the wavelength was set to 230 nm. All quantifications were evaluated using calibration curves from stock solutions. Purity by HPLC was determined using a Merck RP-8 column % 50% ACN in water for compounds 1, 2, 3, 5, 7, 9, 10, 11, 16, 17, 18 and 70% acetonitrile in water for the other compounds.
Mycobacterial homogenate preparation: A crude whole mycobacterial homogenate was prepared according to reference [21]. A culture of exponentially growing M. smegmatis ATCC607 variant mc2 155 with an O.D. 600 nm of 0.8–1.0 was harvested by centrifugation at T = 4 °C for 10 min, washed and re-suspended in pH = 7.4 phosphate buffer saline PBS (25 mL for each 750 mL of the initial growing broth). The bacterial homogenate was prepared using an ultrasound probe with a sequence of five cycles of 2 min each. The homogenate was afterwards divided in 1 mL portions and kept at −80 °C until use. Total protein concentration was 1.4 mg mL−1.
Stability studies. Conditions of incubations and preparation of samples: In all stability assays, the initial concentration of the compound under study was 5 × 10−4 M. All incubations were carried out at pH 7.4 and 37 °C under agitation using PBS as diluting agent. The benzamides were added from 2.5 × 10−2 M acetonitrile stock solutions. After incubation, aliquots of 50 μL were taken into vials processed as described below, injected into the HPLC and analysed for quantification of the corresponding acid and remaining benzamide. All quantifications were performed using calibration curves. Stability in buffer. 1600 µL pH 7.4 phosphate buffer saline PBS, 360 µL ACN and 40 µL a stock solution of the compound in ACN were mixed in a 2 mL vial. The solution was incubated at 37 °C and 50 µL aliquots were removed, mixed with 450 µL of ACN:H2O 1:1 and analysed by HPLC. Plasma stability. Pooled human plasma (640 µL), pH 7.4 phosphate buffered saline (160 µL) and 16 µL of a 2.5 × 10−2 M stock solution of the compound in ACN were mixed in a vial. The suspensions were incubated at 37 °C and 50 µL aliquots were removed, mixed with 450 µL of ACN:ZnSO4 1% 1:1 and centrifuged for 10 min at 15,000 rpm. The supernatant was then removed and analysed by HPLC. Homogenate stability. 948 µL pH 7.4 phosphate buffer, 32 µL of M. smegmatis homogenate and 20 µL of a 2.5 × 10−2 M stock solution of the compound in ACN were mixed in a vial. The solution was incubated at 37 °C and 50 µL aliquots were removed, mixed with 450 µL of ACN:H2O 1:1 and centrifuged for 10 min at 15,000 rpm. The supernatant was then removed and analysed by HPLC.
Determination of the Minimum Inhibitory Concentration (MIC) and Minimum Bactericidal Concentrations (MBC): The MICs were determined by the broth microdilution method in 96-well plates. Briefly, M. tuberculosis bacterial cultures in exponential growth phase were collected by centrifugation, washed with PBS and re-suspended in fresh culture medium. Clumps of bacteria were removed by ultrasonic treatment of the bacteria suspension in an ultrasonic water bath for 5 min followed by a low-speed centrifugation (500 x g) for 2 min. Single cell suspension was verified by microscopy. The microplates containing a bacterial suspension corresponding to approximately 105 colony-forming units per ml were incubated with the selected concentrations of the compounds. Every other day, the optical density of the wells was measured in a Tecan M200 spectrophotometer, following 30 s of orbital agitation. These values were used to produce the growth curves. At the 10th day of incubation, the MIC was determined, corresponding to the concentration with no visible turbidity. Optical density measurements were taken until the 15th day of incubation. The MBC values were determined using an established methodology [44]. Briefly, following MIC determination, the bacterial samples were recovered from the MIC test microplates and plated in 7H10 + OADC solid medium. The MBC was determined following 3 weeks of incubation, corresponding to the concentration of compound that produced no colonies on the solid medium. Bacteria treated with DMSO solvent at the same proportions as present during the compound tests were used as a control. Isoniazid was used as a positive control for bacteria killing and assay validation following EUCAST guidelines (MIC = [0.03, 0.12] μg/mL). All MIC and MBC results presented comprise the mode value of a minimum of triplicate experiments.
Determination of macrophage viability after treatment with compounds: THP-1 Human monocytic cell line THP-1 (ATCC TIB202) was used to determine the effect of the compounds on cell viability. The cells were grown in RPMI 1640 (BD Difco, Franklin Lakes, NJ, USA) supplemented with 10 % fetal bovine serum (FBS; BD Difco, Franklin Lakes, NJ, USA), 10 mM HEPES (BD Difco, Franklin Lakes, NJ, USA), 1 mM sodium pyruvate, and maintained at 37 ºC with 5 % CO2. Differentiation of THP-1 monocytes into macrophages was induced for 48 h with 20 nM phorbol 12-myristate 13-acetate (PMA) following a 24 h resting period without PMA. Differentiated macrophages in 96-well plates, 5 × 104 cells per well, were treated with the compounds. After three days of treatment, cell viability was determined using PrestoBlue (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s indications. Briefly, the cells were washed with PBS and incubated with PrestoBlue 10 % (v/v) in cell culture medium. After 4 h of incubation the fluorescence of each well was measured in a Tecan M200 spectrophotometer (Em: 560 nm/Ex: 590 nm). Viability was calculated relatively to non-treated cells. Cells treated with DMSO solvent at the same proportions were used as a control. Puromycin was used as a positive control for cell death.
Macrophage Infection and intracellular bacteria killing following compounds treatment: Before infection, M. tuberculosis was cultivated for seven days at 37 °C and 5% CO2 until the exponential growth phase was reached. Bacterial suspensions were centrifuged and washed in phosphate-buffered saline (PBS) and resuspended in macrophage culture medium without antibiotics. Clumps of bacteria in the suspension were disrupted using an ultrasonic bath treatment for 5 min and removed using centrifugation at a low speed of 500× g for 1 min. The obtained single-cell suspension was verified through fluorescence microscopy and quantified by measuring the optical density at 600 nm. Then, the infection was performed with a multiplicity of infection (MOI) of 1 bacterium per macrophage for 3 h at 37 °C and 5% CO2. Following this incubation period, the cells were washed with PBS and the compounds (0.15 μg/mL) were added in fresh, complete medium. DMSO solvent was used as a negative control and isoniazid (0.15 μg/mL) was used as a positive control for bacteria killing. At the specified time-points, macrophages were disrupted using 0.05% Igepal and the resulting bacterial suspension was serial-diluted and plated in 7H10 OADC agar plates. Micro-colonies were counted following approximately 2 weeks of incubation at 37 °C.
Molecular docking: 3D models of the compounds under study were built and optimized (MMFF94forcefield) using the open-source cheminformatics toolkit RDK [45]. Meeko library (https://github.com/forlilab/Meeko) was used to obtain the corresponding PDBQT files, allowing all available torsions to rotate freely. The receptor structure was retrieved from the RCSB Protein Data Bank entry 4P8L (PDB: 4P8L), correspondent to the crystal structure of M. tuberculosis DprE1 in complex with the non-covalent inhibitor Ty36c, and only the atoms pertaining to the chain A of the protein were considered. A PDBQT file of the receptor was obtained using AutoDockTools [46], keeping only the polar hydrogen atoms. Molecular docking simulations were performed using AutoDock Vina 1.2.5 [47,48]. Two search spaces of 22 Å3 and 30 Å3 were used, centered on the position of the ligand from the crystallographic structure. For each compound, 5 independent docking runs were performed to increase sampling, using 3 scoring functions for pose selection: Vina, Vinardo and AutoDock. All poses were then sorted according to each of the scoring functions, selecting the lowest-energy conformations for each docking run and each compound. The coordinates of the 5 top poses for each compound were further used to calculate the distance from the N atom of their nitro groups and the center of their aromatic ring moiety to two atoms of the receptor: the S atom in the cysteine 387 residue and the N atom in the center portion of the isoalloxazine ring of the FAD cofactor that is in the binding pocket region (Figure 4B).
5. Conclusions
The synthesis of a library of nitro-substituted alkyl benzamides allowed to enhance the understanding of the role of alkyl groups in the antitubercular activity of the compounds.
The results here discussed show that high antitubercular activities are obtained with di-substitution of the aromatic moiety as well as an alkyl chain length of 8 to 10 carbon atoms. Regarding the alkyl chain length, a parabolic influence on activity was observed for the most active family of compounds. Also, we demonstrated that such compounds have good profiles of activity in infection models, comparable to INH, a known antitubercular drug.
Although not the definite proof, the similarity of structure with known inhibitors, the assessment of susceptibility of multiple species and the computational studies are complementary information that supports DprE1 as a likely target of action for this type of compounds. The computational studies also indicate that a good fit in the binding pocket is associated with a good activity, but the physical properties of compounds, such as logP must also be taken into account, especially for their impact in solubility and their capacity for crossing the cell wall (but not the membrane) into the cell periplasm where DprE1 is located [14].
The current study demonstrates that adding a simple alkyl group of optimal chain length as the N-substituent of the DNB class of compounds leads to simple compounds with good activity. This finding paves the way for the creation of new derivatives that benefit from the added flexibility provided by alkyl substituents.
6. Patents
The current work led to European Patent Application EP22199251. (EPO). Luis Filipe Vicente Constantino, João Pedro Almeida Pais, Tiago Alexandre Duarte Delgado, Olha Antoniuk, Elsa Maria Ribeiro Dos Santos Anes, David Alexandre Rodrigues Pires. (2022). Benzoic acid derivatives, methods and uses thereof.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Synthesis specifications and characterization of compounds.
Author Contributions
Data curation, J.P.P., D.P., E.A., P.J.C. and L.C.; formal analysis, J.P.P., D.P., P.J.C., E.A. and L.C.; funding acquisition, L.C., P.J.C. and J.P.P.; investigation, J.P.P., O.A., T.D., A.F. and D.P.; methodology, J.P.P., E.A., P.J.C. and L.C.; project administration, L.C.; supervision, L.C.; writing—original draft, J.P.P. and L.C.; writing—review and editing, J.P.P., E.A., P.J.C. and L.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Fundação para a Ciência e Tecnologia (FCT), Grant EXPL/SAU-INF/1097/2021. It also received financial support from FCT (via ImedULisboa) from projects UIDB/04138/2020 UIDP/04138/2020 and CP iMed IDEA2023 (EA/LC). Computational studies were funded by strategic projects UIDB/04046/2020−UIDP/04046/2020 (BioISI) and also by the European Union (TWIN2PIPSA, GA 101079147). The views and opinions expressed are, however, those of the author(s) only and do not necessarily reflect those of the European Union or European Research Executive Agency (REA). Neither the European Union nor the granting authority can be held responsible for them.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
Data available within the article.
Acknowledgments
The authors acknowledge the financial support from FCT and Portugal 2020 to the Portuguese Mass Spectrometry Network (Rede Nacional de Espectrometria de Massa—RNEM; LISBOA-01-0145-FEDER-402-022125. FCT is also acknowledged for the Individual Call to Scientific Employment Stimulus contract 2021.00381.CEECIND (P.J.C.) and thank FCT for a doctoral grant SFRH/BD/146447/2019 (A.F.).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Cambier, C.J.; Falkow, S.; Ramakrishnan, L. Host Evasion and Exploitation Schemes of Mycobacterium Tuberculosis. Cell 2014, 159, 1497–1509. [Google Scholar] [CrossRef]
- Gill, C.M.; Dolan, L.; Piggott, L.M.; McLaughlin, A.M. New Developments in Tuberculosis Diagnosis and Treatment. Breathe 2022, 18, 210149. [Google Scholar] [CrossRef]
- Lange, C.; Aarnoutse, R.; Chesov, D.; van Crevel, R.; Gillespie, S.H.; Grobbel, H.P.; Kalsdorf, B.; Kontsevaya, I.; van Laarhoven, A.; Nishiguchi, T.; et al. Perspective for Precision Medicine for Tuberculosis. Front. Immunol. 2020, 11, 566608. [Google Scholar] [CrossRef]
- Global Tuberculosis Report 2023; World Health Organization: Geneva, Switzerland, 2023.
- Pai, M.; Kasaeva, T.; Swaminathan, S. COVID-19’s Devastating Effect on Tuberculosis Care—A Path to Recovery. N. Engl. J. Med. 2022, 386, 1490–1493. [Google Scholar] [CrossRef] [PubMed]
- Global Tuberculosis Report 2022; World Health Organization: Geneva, Switzerland, 2022.
- Blondiaux, N.; Moune, M.; Desroses, M.; Frita, R.; Flipo, M.; Mathys, V.; Soetaert, K.; Kiass, M.; Delorme, V.; Djaout, K.; et al. Reversion of Antibiotic Resistance in Mycobacterium Tuberculosis by Spiroisoxazoline SMARt-420. Science 2017, 355, 1206–1211. [Google Scholar] [CrossRef] [PubMed]
- Shetye, G.S.; Franzblau, S.G.; Cho, S. New Tuberculosis Drug Targets, Their Inhibitors, and Potential Therapeutic Impact. Transl. Res. 2020, 220, 68–97. [Google Scholar] [CrossRef] [PubMed]
- Riccardi, G.; Pasca, M.R.; Chiarelli, L.R.; Manina, G.; Mattevi, A.; Binda, C. The DprE1 Enzyme, One of the Most Vulnerable Targets of Mycobacterium Tuberculosis. Appl. Microbiol. Biotechnol. 2013, 97, 8841–8848. [Google Scholar] [CrossRef]
- Imran, M.; Alshrari, A.S.; Thabet, H.K.; Abida; Afroz Bakht, M. Synthetic Molecules as DprE1 Inhibitors: A Patent Review. Expert Opin. Ther. Pat. 2021, 31, 759–772. [Google Scholar] [CrossRef]
- Gawad, J.; Bonde, C. Decaprenyl-Phosphoryl-Ribose 2′-Epimerase (DprE1): Challenging Target for Antitubercular Drug Discovery. Chem. Cent. J. 2018, 12, 1–12. [Google Scholar] [CrossRef]
- Amado, P.S.M.; Woodley, C.; Cristiano, M.L.S.; O’Neill, P.M. Recent Advances of DprE1 Inhibitors against Mycobacterium Tuberculosis: Computational Analysis of Physicochemical and ADMET Properties. ACS Omega 2022, 7, 40659–40681. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.; Soni, A.; Tanwar, O.; Bhadane, R.; Besra, G.S.; Kawathekar, N. DprE1 Inhibitors: Enduring Aspirations for Future Antituberculosis Drug Discovery. ChemMedChem 2023, 18, e202300099. [Google Scholar] [CrossRef] [PubMed]
- Brecik, M.; Centárová, I.; Mukherjee, R.; Kolly, G.S.; Huszár, S.; Bobovská, A.; Kilacsková, E.; Mokošová, V.; Svetlíková, Z.; Šarkan, M.; et al. DprE1 Is a Vulnerable Tuberculosis Drug Target Due to Its Cell Wall Localization. ACS Chem. Biol. 2015, 10, 1631–1636. [Google Scholar] [CrossRef] [PubMed]
- Richter, A.; Rudolph, I.; Möllmann, U.; Voigt, K.; Chung, C.W.; Singh, O.M.P.; Rees, M.; Mendoza-Losana, A.; Bates, R.; Ballell, L.; et al. Novel Insight into the Reaction of Nitro, Nitroso and Hydroxylamino Benzothiazinones and of Benzoxacinones with Mycobacterium Tuberculosis DprE1. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Neres, J.; Pojer, F.; Molteni, E.; Chiarelli, L.R.; Dhar, N.; Boy-Röttger, S.; Buroni, S.; Fullam, E.; Degiacomi, G.; Lucarelli, A.P.; et al. Structural Basis for Benzothiazinone-Mediated Killing of Mycobacterium Tuberculosis. Sci. Transl. Med. 2012, 4, 150ra121. [Google Scholar] [CrossRef]
- Foo, C.S.Y.; Lechartier, B.; Kolly, G.S.; Boy-Röttger, S.; Neres, J.; Rybniker, J.; Lupien, A.; Sala, C.; Piton, J.; Cole, S.T. Characterization of DprE1-Mediated Benzothiazinone Resistance in Mycobacterium Tuberculosis. Antimicrob. Agents Chemother. 2016, 60, 6451–6459. [Google Scholar] [CrossRef] [PubMed]
- de Jesus Lopes Ribeiro, A.L.; Degiacomi, G.; Ewann, F.; Buroni, S.; Incandela, M.L.; Chiarelli, L.R.; Mori, G.; Kim, J.; Contreras-Dominguez, M.; Park, Y.S.; et al. Analogous Mechanisms of Resistance to Benzothiazinones and Dinitrobenzamides in Mycobacterium Smegmatis. PLoS ONE 2011, 6, e26675. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, H.; Sun, Z. Susceptibility of Mycobacterium Tuberculosis to Weak Acids. J. Antimicrob. Chemother. 2003, 52, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Gu, P.; Constantino, L.; Zhang, Y. Enhancement of the Antituberculosis Activity of Weak Acids by Inhibitors of Energy Metabolism but Not by Anaerobiosis Suggests That Weak Acids Act Differently from the Front-Line Tuberculosis Drug Pyrazinamide. J. Med. Microbiol. 2008, 57, 1129–1134. [Google Scholar] [CrossRef] [PubMed]
- Valente, E.; Simões, M.F.; Testa, B.; Constantino, L. Development of a Method to Investigate the Hydrolysis of Xenobiotic Esters by a Mycobacterium Smegmatis Homogenate. J. Microbiol. Methods 2011, 85, 98–102. [Google Scholar] [CrossRef]
- Valente, E.; Testa, B.; Constantino, L. Activation of Benzoate Model Prodrugs by Mycobacteria. Comparison with Mammalian Plasma and Liver Hydrolysis. Eur. J. Pharm. Sci. 2021, 162, 105831. [Google Scholar] [CrossRef]
- Simões, M.F.; Valente, E.; Gómez, M.J.R.; Anes, E.; Constantino, L. Lipophilic Pyrazinoic Acid Amide and Ester Prodrugs Stability, Activation and Activity against M. Tuberculosis. Eur. J. Pharm. Sci. 2009, 37, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.; Valente, E.; Simões, M.F.; Carmo, N.; Testa, B.; Constantino, L.; Anes, E. Esters of Pyrazinoic Acid Are Active against Pyrazinamide-Resistant Strains of Mycobacterium Tuberculosis and Other Naturally Resistant Mycobacteria In Vitro and Ex Vivo within Macrophages. Antimicrob. Agents Chemother. 2015, 59, 7693–7699. [Google Scholar] [CrossRef] [PubMed]
- Pais, J.P.; Magalhães, M.; Antoniuk, O.; Barbosa, I.; Freire, R.; Pires, D.; Valente, E.; Testa, B.; Anes, E.; Constantino, L. Benzoic Acid Derivatives as Prodrugs for the Treatment of Tuberculosis. Pharmaceuticals 2022, 15, 1118. [Google Scholar] [CrossRef] [PubMed]
- Pais, J.P.; Antoniuk, O.; Freire, R.; Pires, D.; Valente, E.; Anes, E.; Constantino, L. Nitrobenzoates and Nitrothiobenzoates with Activity against M. Tuberculosis. Microorganisms 2023, 11, 969. [Google Scholar] [CrossRef] [PubMed]
- Munagala, G.; Yempalla, K.R.; Aithagani, S.K.; Kalia, N.P.; Ali, F.; Ali, I.; Rajput, V.S.; Rani, C.; Chib, R.; Mehra, R.; et al. Synthesis and Biological Evaluation of Substituted N-Alkylphenyl-3,5-Dinitrobenzamide Analogs as Anti-TB Agents. Medchemcomm 2014, 5, 521–527. [Google Scholar] [CrossRef]
- Nepali, K.; Lee, H.Y.; Liou, J.P. Nitro-Group-Containing Drugs. J. Med. Chem. 2019, 62, 2851–2893. [Google Scholar] [CrossRef] [PubMed]
- Tetko, I.V.; Gasteiger, J.; Todeschini, R.; Mauri, A.; Livingstone, D.; Ertl, P.; Palyulin, V.A.; Radchenko, E.V.; Zefirov, N.S.; Makarenko, A.S.; et al. Virtual Computational Chemistry Laboratory—Design and Description. J. Comput. Aided. Mol. Des. 2005, 19, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Christophe, T.; Jackson, M.; Hee, K.J.; Fenistein, D.; Contreras-Dominguez, M.; Kim, J.; Genovesio, A.; Carralot, J.P.; Ewann, F.; Kim, E.H.; et al. High Content Screening Identifies Decaprenyl-Phosphoribose 2′ Epimerase as a Target for Intracellular Antimycobacterial Inhibitors. PLoS Pathog. 2009, 5, e1000645. [Google Scholar] [CrossRef]
- Li, L.; Lv, K.; Yang, Y.; Sun, J.; Tao, Z.; Wang, A.; Wang, B.; Wang, H.; Geng, Y.; Liu, M.; et al. Identification of N-Benzyl 3,5-Dinitrobenzamides Derived from PBTZ169 as Antitubercular Agents. ACS Med. Chem. Lett. 2018, 9, 741–745. [Google Scholar] [CrossRef]
- Trefzer, C.; Rengifo-Gonzalez, M.; Hinner, M.J.; Schneider, P.; Makarov, V.; Cole, S.T.; Johnsson, K. Benzothiazinones: Prodrugs That Covalently Modify the Decaprenylphosphoryl-β-D-Ribose 2′-Epimerase DprE1 of Mycobacterium Tuberculosis. J. Am. Chem. Soc. 2010, 132, 13663–13665. [Google Scholar] [CrossRef]
- Makarov, V.; Manina, G.; Mikusova, K.; Möllmann, U.; Ryabova, O.; Saint-Joanis, B.; Dhar, N.; Pasca, M.R.; Buroni, S.; Lucarelli, A.P.; et al. Benzothiazinones Kill Mycobacterium Tuberculosis by Blocking Arabinan Synthesis. Science 2009, 324, 801–804. [Google Scholar] [CrossRef] [PubMed]
- Favrot, L.; Ronning, D.R. Targeting the Mycobacterial Envelope for Tuberculosis Drug Development. Expert Rev. Anti. Infect. Ther. 2012, 10, 1023. [Google Scholar] [CrossRef] [PubMed]
- Manina, G.; Bellinzoni, M.; Pasca, M.R.; Neres, J.; Milano, A.; De Jesus Lopes Ribeiro, A.L.; Buroni, S.; Škovierová, H.; Dianišková, P.; Mikušová, K.; et al. Biological and Structural Characterization of the Mycobacterium Smegmatis Nitroreductase NfnB, and Its Role in Benzothiazinone Resistance. Mol. Microbiol. 2010, 77, 1172–1185. [Google Scholar] [CrossRef]
- Neres, J.; Hartkoorn, R.C.; Chiarelli, L.R.; Gadupudi, R.; Pasca, M.R.; Mori, G.; Venturelli, A.; Savina, S.; Makarov, V.; Kolly, G.S.; et al. 2-Carboxyquinoxalines Kill Mycobacterium Tuberculosis through Noncovalent Inhibition of DprE1. ACS Chem. Biol. 2015, 10, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Maeda, T.; Nakamura, R.; Kadokami, K.; Ogawa, H.I. Relationship between Mutagenicity and Reactivity or Biodegradability for Nitroaromatic Compounds. Environ. Toxicol. Chem. 2007, 26, 237–241. [Google Scholar] [CrossRef]
- Purohit, V.; Basu, A.K. Mutagenicity of Nitroaromatic Compounds. Chem. Res. Toxicol. 2000, 13, 673–692. [Google Scholar] [CrossRef]
- Polaquini, C.; Torrezan, G.; Santos, V.; Nazaré, A.; Campos, D.; Almeida, L.; Silva, I.; Ferreira, H.; Pavan, F.; Duque, C.; et al. Antibacterial and Antitubercular Activities of Cinnamylideneacetophenones. Molecules 2017, 22, 1685. [Google Scholar] [CrossRef] [PubMed]
- Wade, M.M.; Zhang, Y. Effects of Weak Acids, UV and Proton Motive Force Inhibitors on Pyrazinamide Activity against Mycobacterium Tuberculosis In Vitro. J. Antimicrob. Chemother. 2006, 58, 936–941. [Google Scholar] [CrossRef]
- Karabanovich, G.; Dušek, J.; Savková, K.; Pavliš, O.; Pávková, I.; Korábečný, J.; Kučera, T.; Kočová Vlčková, H.; Huszár, S.; Konyariková, Z.; et al. Development of 3,5-Dinitrophenyl-Containing 1,2,4-Triazoles and Their Trifluoromethyl Analogues as Highly Efficient Antitubercular Agents Inhibiting Decaprenylphosphoryl-β-d-Ribofuranose 2′-Oxidase. J. Med. Chem. 2019, 62, 8115–8139. [Google Scholar] [CrossRef]
- Chikhale, R.V.; Barmade, M.A.; Murumkar, P.R.; Yadav, M.R. Overview of the Development of DprE1 Inhibitors for Combating the Menace of Tuberculosis. J. Med. Chem. 2018, 61, 8563–8593. [Google Scholar] [CrossRef]
- Egorova, A.; Salina, E.G.; Makarov, V. Targeting Non-Replicating Mycobacterium Tuberculosis and Latent Infection: Alternatives and Perspectives (Mini-Review). Int. J. Mol. Sci. 2021, 22, 13317. [Google Scholar] [CrossRef] [PubMed]
- Santos, N.C.d.S.; Scodro, R.B.d.L.; Sampiron, E.G.; Ieque, A.L.; Carvalho, H.C.d.; Santos, T.d.S.; Ghiraldi Lopes, L.D.; Campanerut-Sá, P.A.Z.; Siqueira, V.L.D.; Caleffi-Ferracioli, K.R.; et al. Minimum Bactericidal Concentration Techniques in Mycobacterium Tuberculosis: A Systematic Review. Microb. Drug Resist. 2020, 26, 752–765. [Google Scholar] [CrossRef] [PubMed]
- Landrum, G. RDKit: Open-Source Cheminformatics. Release 2014.03.1 2015.
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
Figure 2.
Graphical representation of the correlation between logP and log(1/MIC) values for the N-alkyl-3,5-dinitrobenzamide series (compounds 9 to 15).
Figure 2.
Graphical representation of the correlation between logP and log(1/MIC) values for the N-alkyl-3,5-dinitrobenzamide series (compounds 9 to 15).
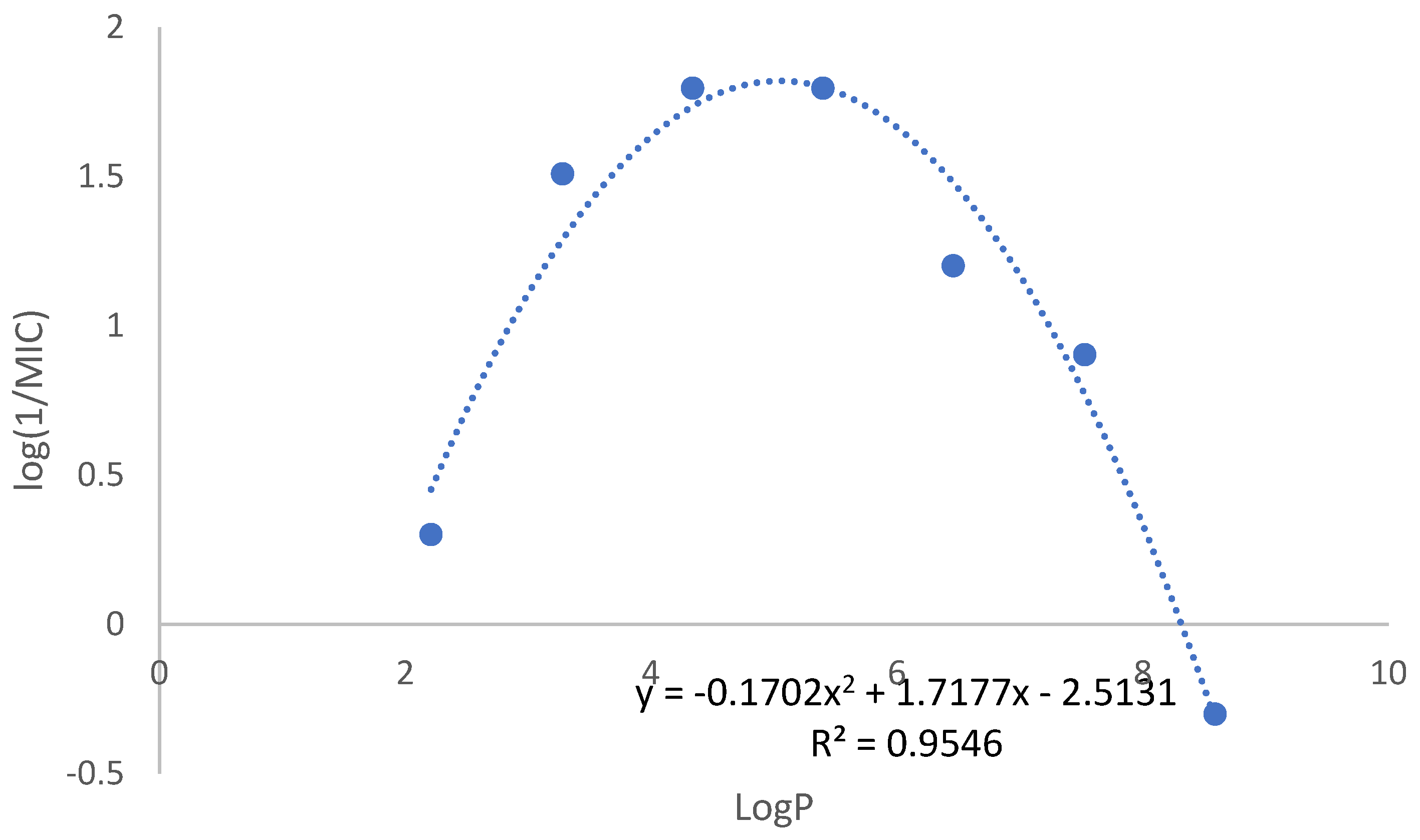
Figure 3.
Biological activity of the most active amides under study, in a macrophage model of infection. The results presented are the average of triplicate experiments and the error bars report a standard deviation (σ). Compounds and isoniazid (INH) were tested at 0.15 μg/mL.
Figure 3.
Biological activity of the most active amides under study, in a macrophage model of infection. The results presented are the average of triplicate experiments and the error bars report a standard deviation (σ). Compounds and isoniazid (INH) were tested at 0.15 μg/mL.
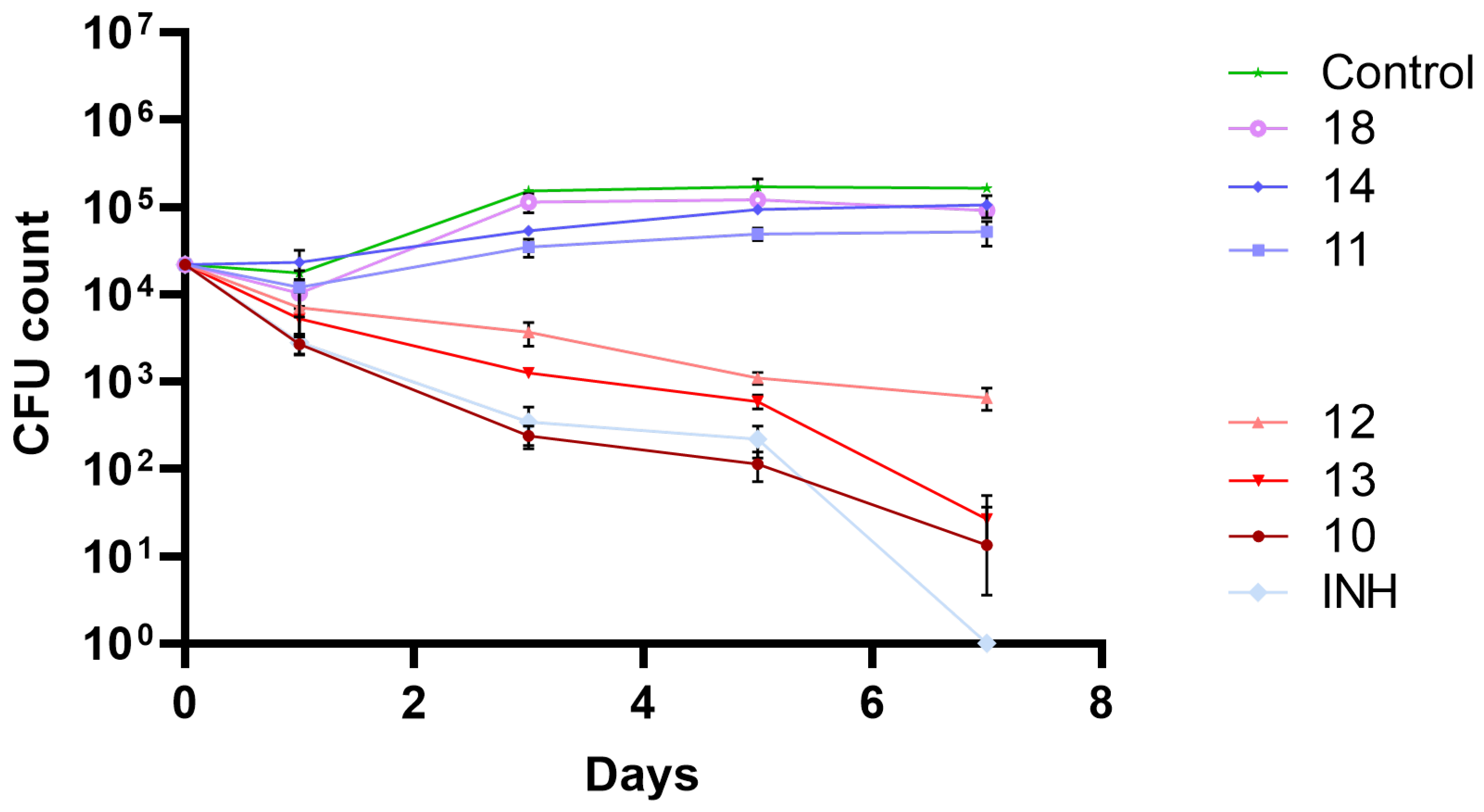
Figure 4.
Representation of the best binding poses obtained by docking computational study, between a PDB crystal structure (PDB: 4P8L) and: (A) - The compounds DNB1 (gray), 11 (green) and 18 (pink); (B) – Distance between compound 11 (green) and both the cysteine residue (Cys387) and the FAD cofactor (FADH2).
Figure 4.
Representation of the best binding poses obtained by docking computational study, between a PDB crystal structure (PDB: 4P8L) and: (A) - The compounds DNB1 (gray), 11 (green) and 18 (pink); (B) – Distance between compound 11 (green) and both the cysteine residue (Cys387) and the FAD cofactor (FADH2).
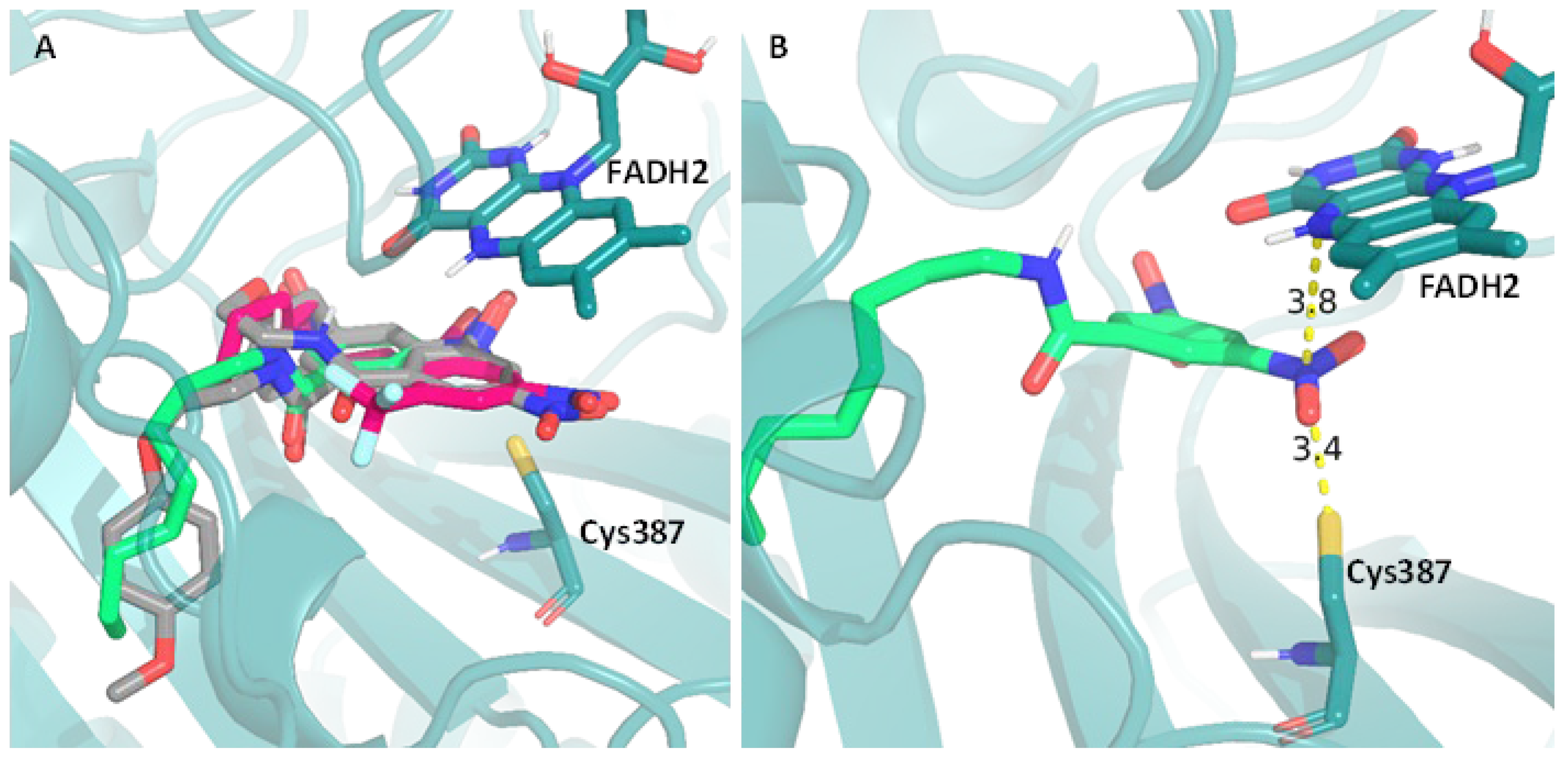
Table 1.
Library of compounds under study and their corresponding predicted logP values, as well as their antitubercular activity against M. tuberculosis expressed as MIC and MBC values, the mode value of triplicate experiments.
Table 1.
Library of compounds under study and their corresponding predicted logP values, as well as their antitubercular activity against M. tuberculosis expressed as MIC and MBC values, the mode value of triplicate experiments.
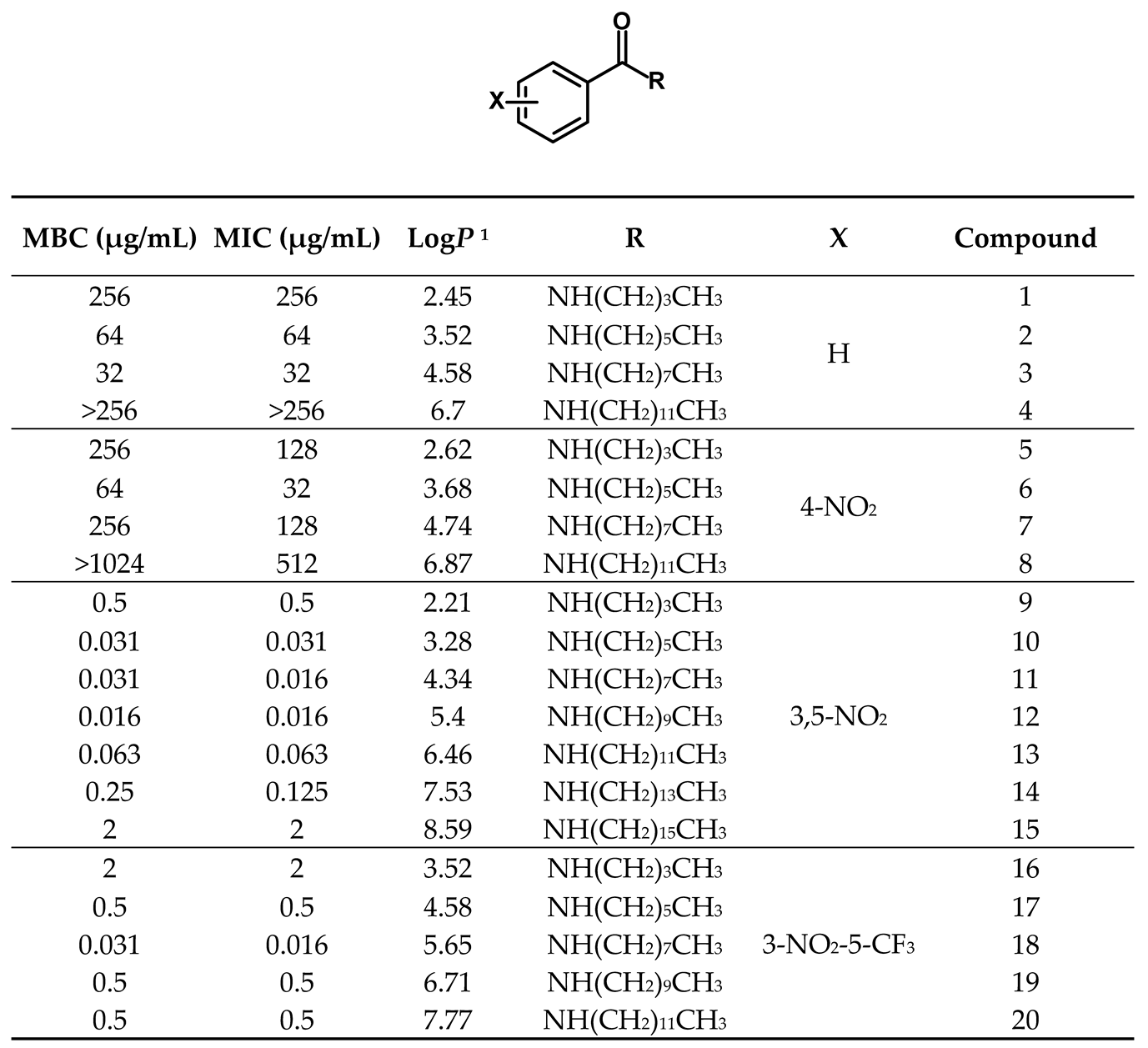 |
1 LogP values were predicted by the software ALOGPS 2.1 [29].
Table 2.
Susceptibility of M. tuberculosis, M. bovis BCG, M. avium and M. smegmatis to synthesized and control compounds, expressed as MIC (µg/mL) and MBC (µg/mL) values. Compounds DNB1, DNB2, PAS and INH are literature compounds, tested as internal standards.
Table 2.
Susceptibility of M. tuberculosis, M. bovis BCG, M. avium and M. smegmatis to synthesized and control compounds, expressed as MIC (µg/mL) and MBC (µg/mL) values. Compounds DNB1, DNB2, PAS and INH are literature compounds, tested as internal standards.
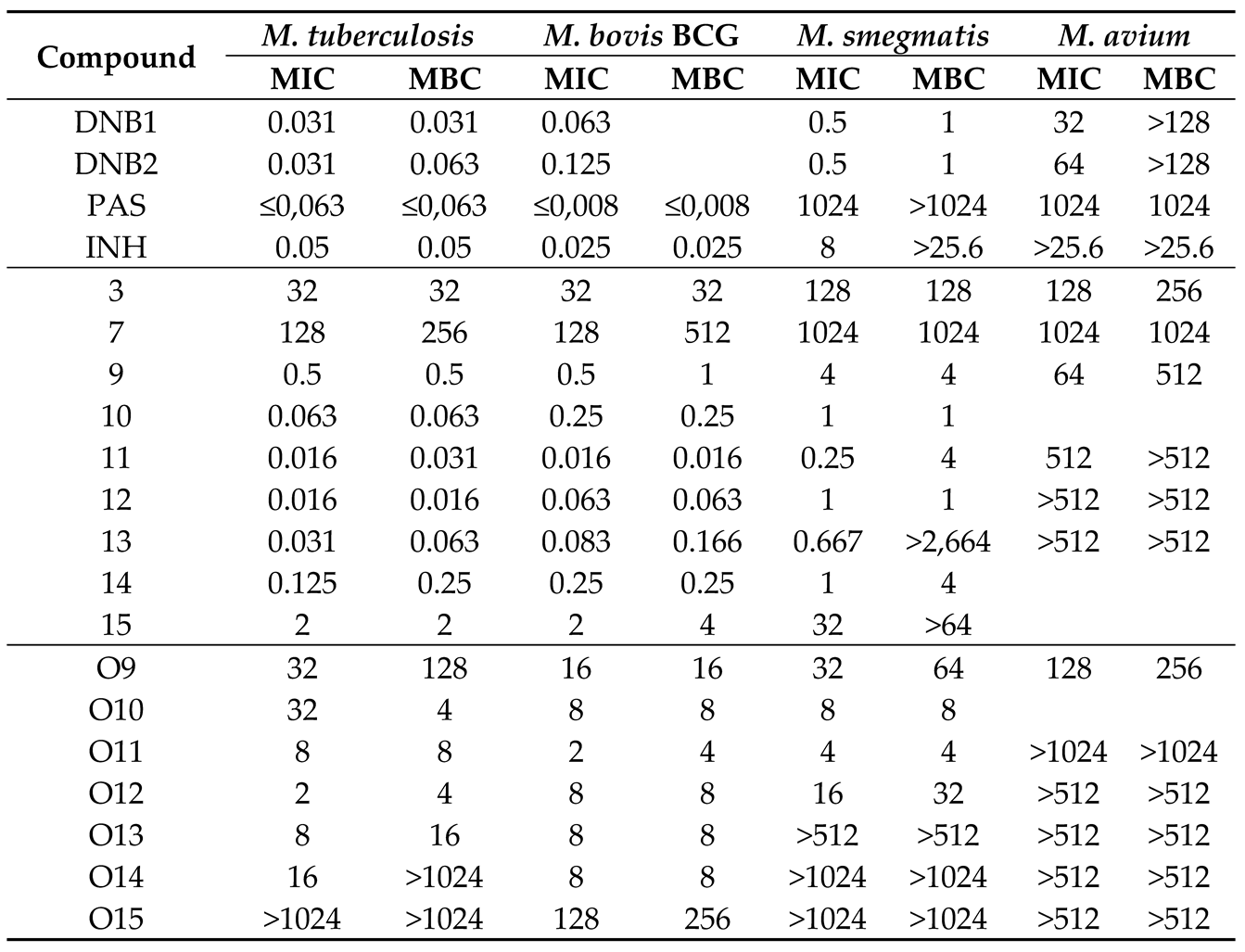 |
Table 4.
Results obtained from the stability assays in human plasma and mycobacterial homogenate. ND refers to the Kobs that could not be accurately calculated as the hydrolysis rate was too slow. %(72h) or %(48h) represents the percentage of hydrolysis after a 72- or 48-hour incubation period, respectively, and allows for a comparison of relative stability.
Table 4.
Results obtained from the stability assays in human plasma and mycobacterial homogenate. ND refers to the Kobs that could not be accurately calculated as the hydrolysis rate was too slow. %(72h) or %(48h) represents the percentage of hydrolysis after a 72- or 48-hour incubation period, respectively, and allows for a comparison of relative stability.
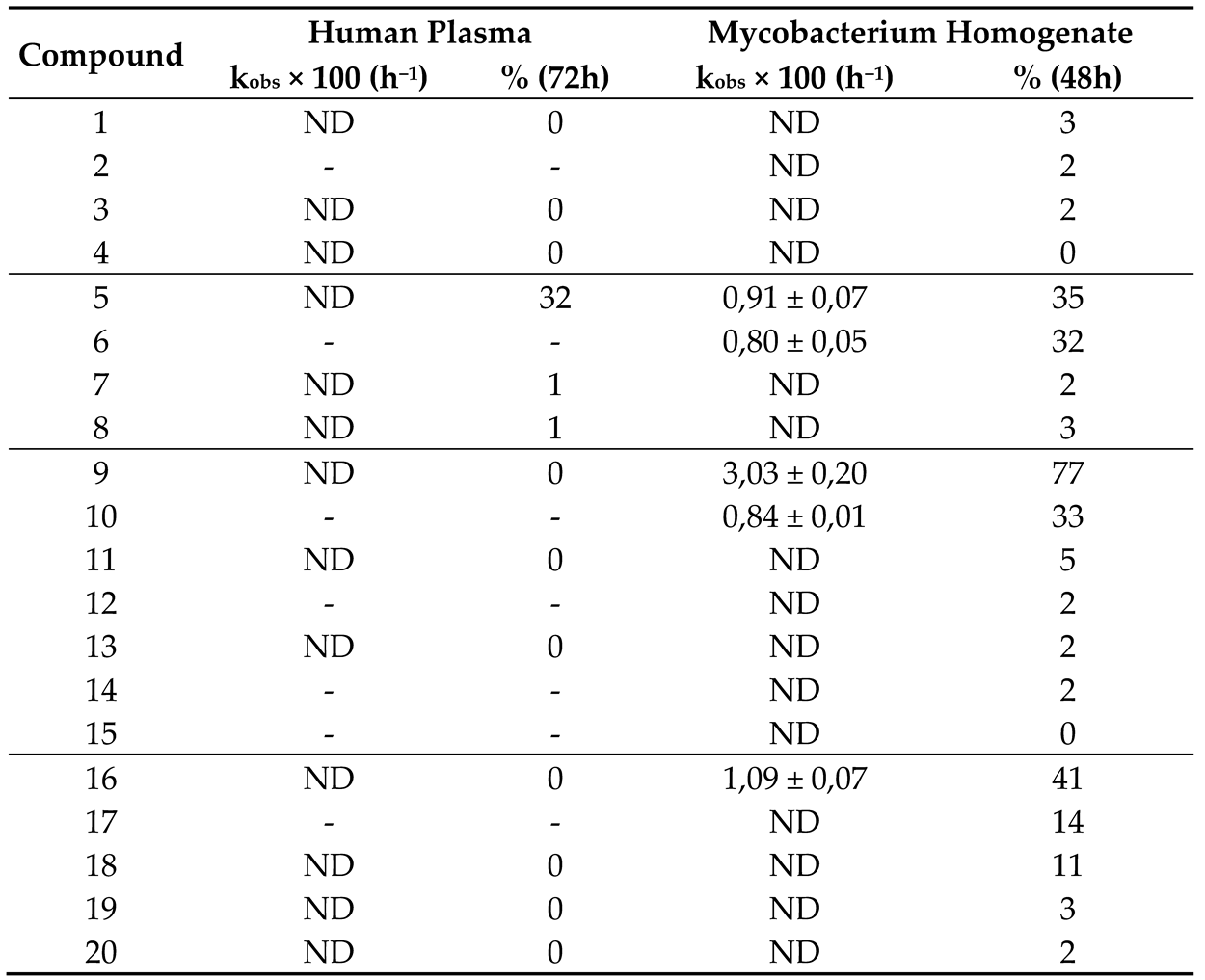 |
ND (not determined); - (not assayed).
Table 5.
Values of the distances obtained from the analysis of the best binding poses (AutoDock) of the compounds in study and their correspondent scores (kcal/mol) and MIC (µg/mL) values. NDCys387 and NDFAD refers to the distance between the nitro group to the cysteine 387 and FAD residues, respectively. Similarly, CDCys387 and CDFAD refers to the distance between the center of the aromatic moiety to the cysteine 387 and FAD residues, respectively. ΣNDist and ΣCDist refer to the sum of the distances to the nitro group and the aromatic center, respectively.
Table 5.
Values of the distances obtained from the analysis of the best binding poses (AutoDock) of the compounds in study and their correspondent scores (kcal/mol) and MIC (µg/mL) values. NDCys387 and NDFAD refers to the distance between the nitro group to the cysteine 387 and FAD residues, respectively. Similarly, CDCys387 and CDFAD refers to the distance between the center of the aromatic moiety to the cysteine 387 and FAD residues, respectively. ΣNDist and ΣCDist refer to the sum of the distances to the nitro group and the aromatic center, respectively.
 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



