
Submitted:
17 April 2024
Posted:
18 April 2024
You are already at the latest version
Abstract
The present study aimed to determine the genetic diversity of isolates of Mycobacterium tuberculosis (Mtb) from presumed drug resistant tuberculosis patients from several states of Brazil. The isolates had been submitted to conventional drug susceptibility testing for first- and second-line drugs. Multidrug-resistant (MDR-TB) (54.8%) was the most frequent phenotypic resistance profile, in addition to an important high frequency of pre-extensively resistant (p-XDR-TB) (9.2%). Using whole-genome sequencing (WGS), we characterized 298 Mtb isolates from Brazil. Besides analysis of genotype distribution and possible correlations between molecular and clinical data, we determined the performance of an in-house WGS pipeline with other online pipelines for Mtb lineages and drug resistance profile definition. Sub-lineage 4.3 (52%) was the most frequent genotype and the genomic approach revealed a p-XDR-TB level of 22.5%. We detected 20 novel mutations in three resistance genes, and six of these were observed in eight phenotypically resistant isolates. Cluster analysis of 170 isolates showed that 43.5% of the TB patients belonged to 24 genomic clusters, suggesting considerable ongoing transmission of DR-TB, including two interstate transmissions. The in-house WGS pipeline showed the best overall performance in drug-resistance prediction, presenting the best accuracy values to five of nine drugs tested. Significant associations were observed between suffering from fatal disease and genotypic p-XDR-TB (p = 0.03) and either phenotypic (p = 0.006) and genotypic (p = 0.0007) ethambutol resistance. The use of WGS analysis improved our understanding of the population structure of MTBC in Brazil, genetic and clinical data correlations and demonstrating its utility for surveillance efforts for spread of DR-TB, hopefully helping to avoid the emergence of even more resistant strains and to reduce TB incidence and mortality rates.
Keywords:
Mycobacterium tuberculosis
; Drug Resistance
; Whole Genome Sequencing
; Genetic Diversity
; Brazil
; Novel Mutations
1. Introduction
Tuberculosis (TB) accounted for 1.4 million of deaths and an estimated 10.6 million of new cases in 2023, and was the leading cause of death from a single infectious agent worldwide until COVID-19 pandemic [1]. The emergence of drug resistant (DR) TB is a global threat that hinders successful TB treatment. TB multidrug resistant (MDR-TB), defined as simultaneous resistance to rifampicin (RIF) and isoniazid (INH), results in a worse prognosis, prolonged TB treatment with second-line drugs that are more toxic, more expensive and with the possibility of evolving to pre-extensive drug resistant (p-XDR-TB). The latter is defined as strains that fulfil the definition of multidrug resistant or rifampicin resistant (MDR/RR-TB) plus resistance to any fluoroquinolone, and subsequently can evolve to extensive drug resistant (XDR-TB), defined as strains p-XDR-TB plus resistance to bedaquiline and/or linezolid.
Brazil is one of the thirty countries with highest TB burden, and although the incidence rate decreased until 2014, during the period of 2015-2019 a notified increased incidence of 34.3 to 37.4 cases/100.000 inhabitants was observed [2]. In addition, due to the COVID-19 pandemic, it is estimated that after a decade of decline, TB mortality has increased in Brazil and globally [3]. An additional concern for TB control in Brazil is the considerable number of DR-TB patients; in 2018 around 2.500 MDR/RR-TB cases were estimated [4], including an increase of MDR-TB among patients that had not previously been treated in Rio de Janeiro State [5].
Rapid DR-TB detection and epidemiological surveillance, as well as knowledge about the genetic diversity of isolates of the Mycobacterium tuberculosis complex (MTBC) in different settings are factors that may contribute to DR-TB elimination. In this scenario, molecular tools have become important and quite recently, next generation sequencing (NGS) made it possible to quickly characterize the whole genome of MTBC strains, enabling both identification of resistance-related genetic variants and lineages identification involved in ongoing transmission [6,7]. Culture-based phenotypic drug susceptibility test (DST), although still the current gold standard, has limitations due to the slow growth rate of the MTBC organisms. Thus, molecular methods for drug resistance prediction are being steadily introduced as a routine in low TB incidence countries [8].
WGS is a promising tool and an approach for DR/MDR-TB detection, since it provides detailed sequence information from different genomic regions, thus enabling drug resistance prediction [9]. However, the high amount of sequencing data generated by WGS created the challenge to develop bioinformatics tools to translate the data into information of clinical and laboratory interest [8]. To permit the use of WGS by professionals with little or no bioinformatics skills, user friendly tools for analysis and interpretation of WGS data have been developed and implemented, permitting accessibility and broad implementation of NGS-based approaches [10,11,12,13]. Nonetheless, due to the complexity of large scale data analyses, some bioinformatics command line skills are still required and sometimes, user a friendly graphical interface is not available [14,15].
Due to global variability of prevalence of MTBC lineages and evidence of differential association with drug resistance [16,17], it is evident that combined detection of both characteristics in different countries and regions may interfere with performance and management of DR-TB detection. Therefore, genomic information of MTBC strains together with conventional phenotypic drug susceptibility testing (DST) and clinical outcome is urgently needed. In addition, evaluation of the different available tools for extraction of DST profile from WGS became important to evaluate the regional differences in DR-TB surveillance and to understand local TB transmission.
Here we conducted a genetic diversity study on genomes from a large collection of Mtb isolates from several states of Brazil that mostly had phenotypic DST for primary and secondary drugs. WGS data were evaluated by an in-house WGS pipeline and different online available pipelines, including Mykrobe Predictor, KvarQ, TB Profiler version 0.3.4 and the more recent TB Profiler 5.0 to predict drug susceptibility and the in-house WGS pipeline, KvarQ, TB Profiler 5.0 and RD-Analyzer to predict the genotype (TB lineages).
2. Results
2.1. Study Samples, Patient Data and Phenotypic Drug Susceptibility Testing
Among 298 Mtb isolates from presumed DR-TB patients with high quality WGS data, 294 had conventional phenotypic DST data for at least one drug. Phenotypic DST resulted in 64 (21.8%) pan-susceptible; 22 (7.5%) isoniazid monoresistant (IMR); nine (3.1%) rifampicin monoresistant (RMR); one (0.3%) ethambutol monoresistant (EMR); 10 (3.4%) poly-resistant (Poly-R); 161 (54.8%) MDR and 27(9.2%) p-XDR (Supplementary Table S1).
Among 170 patients for which we had identification, 112 had sociodemographic, clinical, radiographic and treatment outcome data available (Supplementary Table S2). Among them, 65 (57.5%) were male, seven (6.3%) were smokers, eight (7.1%) were illicit drug users, 16 (14.2%) had diabetes, 12 (10.7%) were alcohol users and seven (6.3%) were HIV infected. In relation to chest radiographic images, 72 patients (64.3%) had bilateral cavitary disease, 15 (13.4%) unilateral and 16 (14.3%) had no image available. Regarding treatment outcome, 50 (44.6%) cured or had treatment completion, 32 (28.6%) died due to TB, 16 (14.3%) were lost to follow-up, five (4.5%) were still under treatment, five (4.5%) died from another cause and four (3.6%) patients had no clinical data available.
2.2. Novel Mutations, In Silico Drug Susceptibility and Resistance Prediction Using Different Pipelines
Considering the in-house WGS pipeline, the 298 sequenced genomes were classified as follows: 80 (26.8%) pan-susceptible; 11 (3.7%) IMR; 4 (1.3%) RMR; 1 (0.3%) EMR; 1 (0.3%) SMR; 10 (3.4%) Poly-R; 124 (41.6%) MDR and 67 (22.5%) p-XDR. The nature and frequencies of the mutations detected with this pipeline are presented in Figure 1 and in Supplementary Table S3.
Using the 294 genomes that had DST available for at least one drug, we compared the performance for drug resistance inference of the five predictive drug resistance pipelines against phenotypic DST (three different pipelines and two versions of TB Profiler). The in-house WGS pipeline showed slightly better overall results when considering sensitivity, specificity, positive and negative predictive values, and accuracy when comparing with Mykrobe Predictor, both versions of TB Profiler and kvarQ (Table 1). For prediction of resistance to RIF and INH, in all tools, the sensitivity was higher than 80% but the specificity was lower than 95%. Regarding the prediction to the other first line drugs, the in-house WGS pipeline showed sensitivity higher than 80% to EMB, while high specificity to STR was observed using the in-house WGS pipeline and Mykrobe Predictor. With respect to second line drugs, highest sensitivity to FQ ofloxacin (OFL) was obtained using TB Profiler, and over 90% specificity to AMK, KAN and CAP was observed in all pipelines. Highest sensitivity to PZA was obtained in the most updated version of TB Profiler (68%).
The agreement among phenotypic DST and the in-house WGS pipeline, KvarQ, Mykrobe Predictor, TB Profiler 0.3.4 and TB Profiler 5.0 was assessed for all drugs considering resistance and susceptibility prediction (regardless mutation type in pipelines). The highest agreement level among the six methods was found in RIF (k = 0.89), and the worst agreement level was in PZA (k = 0.32). An excellent level of agreement was also detected to INH, AMK, KAN, CAP (Supplementary Table S4).
Using TB Profiler 5.0, that is able to detect and report unique mutations, i.e. not yet reported, 20 novel mutations in three resistance genes (katG, pncA and ethA) were identified, which six were present in eight phenotypically resistant isolates (gene-drug related) as shown in Table 2. Unfortunately, ethionamide susceptibility had not been phenotypically characterized in the present study. Mostly isolates with novel mutations were phenotypically MDR (68.7%) and genotypically p-XDR (46.8%). All mutations observed using TB Profiler 5.0 are shown in Supplementary Table S5.
2.3. Genomic Diversity, Phylogenetic Analysis and Lineage Classification Using Different Pipelines
Among the 298 genomes analyzed by the in-house WGS pipeline, 99% belonged to Lineage 4, of which 155 (52%) to sub-lineage 4.3 [LAM], 42 (14.1%) to sub-lineage 4.10 [PGG3], 40 (13.4%) to sub-lineage 4.1.2 [Haarlem], 35 (11.7%) to sub-lineage 4.1.1 [X], 17 (5.7%) to sub-lineage 4.4 [Vietnam], two (0.6%) had not been sub-lineage detected and four (1.3%) demonstrated a mixed classification of L4.3/L4.1.1 (n = 1), L4.3/L4.1.2 (n = 1), L4.3/L4.10 (n = 1) and L4.1.1/L4.4 (n = 1), indicating mixed infections or contaminations; three (1%) were classified as Lineage 1.
A phylogenetic tree (Figure 2) based on 38,563 genome-wide SNPs was constructed using 293 genomes. Five genomes with high count of mixed SNP calls were excluded (four with mixed sub-lineage classification and one L4.1.2 [Haarlem]). The topology was congruent with different lineage and sub-lineages classifications assigned by all SNP-based pipelines as previously described [33,34,35].
Upon comparing SNP-based lineage classification pipelines (in-house WGS pipeline, kvarQ-barcode-Coll14 and TB Profiler) and a RD-based analysis pipeline (RD-Analyzer), the L4.3 [LAM] (and higher resolutions classifications when available, e.g., L4.3.2, L4.3.3, L4.3.4, L4.3.4.1, L4.3.4.2, L4.3.4.2.1) was predominant with all tools. Classification results and lineage proportions are presented in Supplementary Figure S1.
Some discordant genotypic classifications were encountered and described in the Supplementary Figure S2, mainly among RD-based pipeline analyzes and SNP-based pipelines. Nevertheless, the free-marginal kappa coefficient obtained presented an excellent agreement level (k = 0.89 [95%CI: 0.86-0.93]) and the overall agreement was 92.06%.
With respect to the sample origin (Figure 3), 212 (71.2%) were isolates from patients residents from the Southeast region including 173 (58,1%) isolates from Rio de Janeiro and 39 (13.1%) from São Paulo; 46 (15.4%) were from the Midwest region, including 42 (14,1%) from the Distrito Federal, two (0,7%) from Goiás and two (0,7%) from Mato Grosso; 15 (4.9%) were form the northeast including six (2%) from Pernambuco, four (1,3%) from Ceará, three (1%) from Maranhão, one (0,3%) from Piauí and one (0,3%) from Sergipe; nine 9 (3%) were from the South Region including six (2%) from Santa Catarina and three (1%) from Paraná and finally, six (2%) were from the North Region, including three (1%) from Amazonas, two (0,7%) from Tocantins and one (0,3%) from Acre. From eight (2.7%) isolates we had not data about state origin.
2.4. Genomic Clusters Analysis
Among the 170 isolates with patient identification available, 74 (43.5%) belonged to 24 genomic clusters with sizes ranging from 2 to 10 isolates, therefore characterized by a clustering rate of 0.294. The most frequent genotypic resistance profiles in this sampling was MDR-TB (53.5%), followed by p-XDR-TB (32.9%) and a similar distribution was observed among the clustered population, composed by 63.5% MDR-TB and 33.7% p-XDR-TB isolates (Figure 4). Importantly, a significantly association between being part of a cluster and having an MDR-TB genotype was observed (Chi-square = 5.2512; p=0.02).
Upon analysis of genotypes of these 170 isolates using the in-house WGS pipeline, 95 (55.8%) belonged to sub-lineage 4.3 [LAM], 26 (15.2%) were 4.1.2 [Haarlem], 20 (11.7%) were 4.10 [PGG3], 18 (10.5%) were 4.1.1 [X], nine (5.3%) were 4.4 [Vietnam], one (0.6%) was classified as Lineage 1, and one (0.6%) as Lineage 4 without sub-lineage detection. Among the isolates in clusters, L4.3 LAM also was observed most frequently (n = 39; 52.7%).
When concentrating on the origin of these samples, 138 were from Rio de Janeiro (81.1%), six from Pernambuco (3.5%;) and Santa Catarina (3.5%) each, four from Ceará (2.3%), three from Amazonas (1.7%), Maranhão (1.7%) and Paraná (1.7%) each, two from Minas Gerais (1.1%) and Goiás (1.1%) each and one from Tocantins (0.6%), Acre (0.6%) and Mato Grosso (0.6%) each. An even more pronounced amount of the clustered isolates (95.9%) were from Rio de Janeiro while only three isolates were from other states (Ceará [GC8], Goiás and Tocantins [GC15]). The latter explains why we observed two genomic clusters with isolates belonging to patients from different states, GC8 with one from Ceará and three from Rio de Janeiro, and GC15 with one from Goiás and one from Tocantins.
2.5. Evolution of Drug Susceptibility Patterns in Patients with Multiple Isolates of Mycobacterium Tuberculosis
Included were 133 genomes from 112 patients from whom clinical and epidemiologic data were available and from which 15 had been followed-up (Table 3 and Supplementary Table S6). These 15 patients had at least two isolates with WGS resulting in a total of 36 genomes: two patients with four isolates, two patients with three isolates and 11 patients with two isolates each. Only two patients presented a change in phenotypical drug resistance: one was susceptible to AMK and changed to resistant without changing the mutational profile (patient P39). Another was phenotypically susceptible to KAN and CAP and changed to resistant (patient P64) but again, without changing the mutational profile.
Regarding the mutational profile changes of those 15 patients, seven patients (two with three isolates each and five with two isolates each) had their genotypic drug resistance profile changed (Table 3). The other eight patients presented no change in the genomes of their respective isolates (genetic distance = 0 SNPs) (Supplementary Table S6).
Emergence of resistance mutations for FQ was observed in five patients in gyrA gene (including P28 = S91P [T→C], P27 = A90V [C→T], P63 = D94H [G→C], P10 = D94A [A→C], P39 = D94G [A→G] / A90V [C→T]); one patient cured from disease while the other four died of tuberculosis. Two patients had an EMB mutation resistance emergence in embB gene (P54 = G406D [G→A] and P35 = Q497R [A→G]); the first was cured while the second patient abandoned treatment with no further information obtained.
2.6. Treatment Outcome, Risk Factors and Lineage Associations with Genotypic and Phenotypic Resistance
The association of TB treatment outcome and genotypic and phenotypic drug resistance was evaluated. Genotypic p-XDR-TB was significantly associated with mortality (Chi-square = 4.6231; p = 0.03). Both phenotypic resistance (Chi-square = 7.3445; p = 0.006) and genotypic resistance to EMB (Fisher’s exact test p = 0.0007) were significantly associated with mortality. Another significant association was EMB genotypic resistance (Chi-square = 24.364; p = 0.00001) with the p-XDR-TB genotype. All isolates with phenotypic EMB available are summarized in Supplementary Table S7. In short, the table shown EMB test available isolates divided in four groups: EMB genotypically susceptible and resistant and phenotypically susceptible and resistant; each of these groups was also divided in two groups: cured and deceased.
Comparing patients who had any risk factors to develop TB (HIV, diabetes, alcoholism, smoking and/or illicit drugs abuse) (Supplementary Table S2) among the two outcome groups, deceased and cured, a significant association between presence of any risk factor and deceased outcome was observed (Chi-square = 5.7672; p = 0.01).
Upon comparing of the frequency of the presence drug resistance related mutations in Mtb Isolates classified as L4.3 (LAM) when compared to those of other sub-lineages, a statistically significant lower frequency of mutation related with resistance to STR (Chi-square = 5.3496; p = 0.02), CAP (Chi-square = 6.4498; p = 0.01), ETH (Chi-square = 5.7636; p = 0.01) and AMK (Fisher’s exact test p = 0.001) were associated with LAM sub-lineage in comparison to non-LAM isolates.
3. Discussion
In this study we performed a genome-based characterization of the genetic diversity of mainly drug resistant isolates of Mtb in different regions in Brazil. The analyzed Mtb strains predominantly belonged to the sub-lineage 4.3 (LAM) (52%). This is in accordance with previous studies performed in various Brazilian regions in both susceptible and DR-TB samples [18,19,20,21,22,23,24]. L4.3 was predominant in the South, North, Southeast and Midwest region. In the Northeast, this predominance by L4.3 was not observed but the number of samples from this region was low. Three isolates (from São Paulo, Rio de Janeiro and Distrito Federal) belonged to Lineage 1, known to be restricted mostly to Eastern Africa and South and East of Asia [16], but apparently described with a certain frequency in the Northern Brazil region [24,25,26], probably imported trough slave trade from East Africa [27]. Although L1 is usually not associated with DR, two of three isolates in this study presented some resistance (one IMR and one Poly-R). This might be due to sampling bias as the CRPHF mainly receives samples from patients suspicious for being DR, either due to treatment failure, treatment abandonment, relapse TB or contact with TB resistant patients.
The four pipelines presented a high agreement level (k = 0.89), with almost complete agreement among SNP-based pipelines, but showing divergences mainly in lineage classifications by RD-Analyzer. The Mtb isolates with high counts of mixed SNP calls may be indicative of mixed infection as described by others [28,29,30]. Analysis among five isolates with a high proportion of mixed SNP calls, showed four mixed classifications by the in-house WGS pipeline only, while TB Profiler and KvarQ detected only one of the two lineages present in these isolates. The RD-Analyzer had some difficulties to classify lineage in comparison with SNP-based tools and presented the most divergences among all tools tested, including 31 isolates with mixed classification and six unidentified despite good genome quality and low mixed SNP call counts.
The majority of clusters were identified in Rio de Janeiro, and although sampling from this state was clearly over represented in our setting, this may represent higher levels of recent infection and drug resistant TB transmission, differing of TB cases due to reactivation of latent infection [31,32]. In this Brazilian State, an increase of primary MDR-TB has been described as associated with low TB control performance [5]. Studies in other Brazilian States also shown important ongoing transmission of MDR-TB, as observed in Sao Paulo [33,34] and Santa Catarina [35] and also, in a genome-based study [36], in Rio Grande do Sul. In the latter, as well as in our study, a significant association between clustering and genotypic MDR-TB and significant ongoing transmission in p-XDR-TB resistance profiles was observed, with practically half of the patients infected with genotypic p-XDR-TB (44.6%; n = 25) in clusters, reflecting an alarming scenario of insufficient DR-TB control [36].
In addition to clusters of isolates derived from residents of particular states, we also observed clusters suggestive of interstate transmission of particular genotypes that seem to have been circulating in the country for a considerable time. Examples are GC8 (three patients from Rio de Janeiro and one from Ceará) and GC15 (one from Goiás and one from Tocantins) presenting genetic distances ≤12 SNPs. Furthermore, we observed among isolates of unidentified patients, that were not included in clustering analysis, one isolate from Distrito Federal (isolated in the year 2007) that had genetic distance of ≤12 SNPs to the GC4 isolates that were from Rio de Janeiro (2008-2012), and also one isolate from São Paulo (2004) and one isolate from Distrito Federal (2007) that had genetic distance of 11 SNPs between them. These could point to other two interstate transmission events, requiring further investigation.
From 15 patients, we had several isolates collected during and after treatment. In most of these cases, the infecting lineage strain during follow up of the patients was the same and only in one patient (P35), reinfection with another strain and lineage was observed. In a retrospective cohort study from China [37], a MIRU-VNTR-based recurrence definition observed a lower frequency of reinfection (n = 21; 36.2%) in comparison to relapse (n = 37; 63.8%). In a population-based study from Malawi [38] using WGS, lower frequency of reinfection (n = 20; 14.3%) compared with relapse (n = 55; 39.5%) was also observed; 64 (46%) of recurrent patients remained unclassified, showing how challenging it remains to classify such cases.
We observed emergence of FQ resistance conferring mutations among one third of the patients that were followed-up during treatment. These bacterial populations had likely been selected due non-compliance of drug treatment [39,40]. In addition, emergence of phenotypic resistance to AMK in patient P39, and phenotypic resistance to KAN and CAP in patient P64 was observed, but without detection of known mutations in rrs and eis. This could be caused by not yet known resistant conferring mutations to those drugs and/or by efflux pump activity, not investigated here. Such pumps have been described to be induced by sub-inhibitory levels of antibiotics such as FQ, RIF and INH in inappropriate treatment regimens [41]. Additionally, the presence of a minor undetected population of drug resistant bacteria, so-called heteroresistance, can also explain differences between culture and in silico based drug susceptibility outcome [42].
Another observation was the emergence of the G406D mutation in embB in patient P54 (Table 3), however, a mixed call was detected in this codon and may indicate selection of the mutant population within a single strain, probably due to inappropriate treatment. Patient P35 presented emergence of the Q497R mutation in embB, and in addition a shift of RIF resistance genotype, initially presenting Q432P mutation in rpoB and posteriorly the S450L mutation. Besides that, in the first isolate the sub-lineage detected was L4.1.1 (X) and in the second was L4.3 (LAM), and also a genomic difference of 484 SNPs was observed between both, indicating a TB recurrence by reinfection [38].
We observed a statistically significant lower frequency of resistance related mutations to STR, CAP, ETH and AMK in strains belonging to sub-lineage 4.3 (LAM) in comparison with non-LAM sub-lineages. Lower frequency of mutations related to resistance in some lineages has been described, including lower frequency of S315T mutation in katG in LAM spoligofamily when compared to Haarlem [43]. Another study showed a higher mutation rate in Mtb strains of lineage 2 and as such, a faster emergence of resistance conferring mutations in strains of lineage 2, accompanied by development of DR-TB [44].
Upon comparing among patient treatment outcome, risk factors for TB development, phenotypic and genotypic resistance and Mtb lineage, we observed a significant association of EMB phenotypic (Chi-square p = 0.006) and genotypic (Fisher’s exact test p = 0.0007) resistance with TB mortality. EMB resistance has been described as a risk factor for mortality, mainly when associated with MDR pattern [43]. In our study, genotypic p-XDR-TB was significantly associated with EMB genotypic resistance, while genotypic p-XDR-TB profile was significantly associated with higher morbidity (Chi-square p = 0.03). In addition, the presence of any of the five risk factors (HIV, diabetes, smoking cigarette, alcohol and/or illicit drug use) was also significantly associated with morbidity but independent of genotypic or phenotypic EMB resistance. The increased risk of mortality in MDR-TB and p-XDR-TB patients has been described [46,47] and is related with increasing level of drug resistances, whereas accumulating of even more factors increases the probability of an unfavorable outcome [48,49]; EMB resistance could be an important stage of resistance accumulation in Mtb.
We observed a similar proportion of main drug resistance conferring mutations, frequencies and genes affected detected by all pipelines tested and this is in agreement with other studies [50,51,52,53,54].
Unprecedented was the detection of 20 novel mutations in 32 strains using TB Profiler 5.0, including a novel frameshift mutation katG_c.2070delC in an isolate that is likely associated with INH resistance, considering the impact this may have on the catalase-peroxidase structure and activity, accompanied by phenotypic resistance against INH. The same isolate also presented the mutation ahpC_c.-81C>T, classified in WHO catalogue as with uncertain significance, but may be acting in synergy with the newly described to confer resistance; this needs to be better investigated. This isolate was misclassified as genetically susceptible to INH by all pipelines but TB Profiler, and the same was observed in other isolates with the mutation ahpC_c.-81C>T, highlighting the importance of using updated pipelines.
Only four of 32 isolates were fully phenotypically characterized for the nine drugs available in this study. Unfortunately, thirteen of the isolates in which we detected a novel mutation were not phenotypically characterized for the drug of interest, making it impossible to establish the genetic and phenotypic correlation. Among the four isolates with the frameshift mutation pncA_c.193_200dupTCCTCGTC, two had no PZA DST available and two were PZA resistant (these two were from the same patient); the mutation pncA_c.305dupC was observed in one resistant and one with DST not available; finally, the mutations pncA_c.452dupT and pncA_c.75_79delCGCGC were both presents in resistant isolates, all suggesting resistance association. On the other hand, the frameshift mutation pncA_c.443_444dupGC was detected in both susceptible and resistant isolates, making it difficult to interpret. Other isolates observed with novel frameshift mutations in pncA and katG genes and phenotypically susceptible could be explained by an acquired low-level resistance, under the antibiotic concentration threshold used here, or due to some laboratorial error in phenotypic resistance determination. Phenotypic susceptible isolates harboring confident resistance mutations have been reported in other studies [55,56].
Among ten other mutations we detected that were absent in the WHO catalogue (Supplementary Table S5), eight [57,58,59,60,61,62,63,64] in twelve isolates had not yet reported in Brazil [63,65], four (katG_p.Leu634Phe, pncA_p.Asp63His, pncA_p.Gly23Val, pncA_c.521_522insT) were present in phenotypically resistant isolates in our study. Another mutation associated with resistance was detected for the first time in Brazil, rpoB_p.Ile491Phe, a mutation outside of the 81 bp hotspot of the rpoB gene, what make it undetectable by commercial assays such as GeneXpert MTB/RIF and MTBDRplus [66,67]. In Eswatini, for example, the presence of this mutation is a significant problem because of its prevalence of >60% in MDR strains [68]. In our study, this mutation was observed in one isolate genotypically and phenotypically MDR, that also carried the rpoB_p.Ser493Leu mutation, an uncertain variant according to WHO. However, their combination can be acting in synergy because the former is a borderline resistance mutation [69].
The comparison among the pipelines evaluating their performance through sensitivity, specificity, accuracy, positive and negative predictive values calculation for drug resistance prediction, using phenotypic resistance results as a reference, showed a better overall performance of resistance prediction by the in-house WGS pipeline. For all pipelines, the sensitivity of prediction of resistance to RIF and INH was higher than 80% but the specificity was lower than 95% and therefore below that of the WHO recommendations [70]. High positive predictive value-PPV (>80%) was observed in all pipelines used for detection of resistance conferring mutations for RIF, INH, OFL, AMK and similar to that described by others [71,72]. Lower PPV was observed for detection of resistance mutations whose action mechanisms are more complex and genetic bases of resistance is less understood, such as resistance to PZA, KAN and CAP. Unreliable PZA resistance prediction has been reported in various studies [25,71,72,73] and in order to enhance PZA resistance prediction, the following strategies have been described: (i) improving the mutation library of the pipelines mainly by including detection of indels in pncA, (ii) the “non-wild type sequence” approach, that consist in interpret any non-synonymous mutations or indels in pncA as genotypic PZA-resistant strains, (iii) besides manually checking of the sequence reads [74,75]. Not surprisingly, the best sensitivity performance for PZA resistance prediction was achieved by TB Profiler 5.0, the most up-to-date pipeline for detection of DR-associated mutations, accounting with the largest PZA-mutations catalogue among all pipelines used and able to detect novel mutations, including fourteen novel indels observed in this study.
Another important finding in our study is that, compared to conventional DST, more than twice as many cases were classified as p-XDR-TB using WGS. This difference in proportion could be beyond the accuracy of genomic tests to second-line resistance detection. However, one should take into account that the criteria for performing phenotypic DST tests to second line drugs in Brazil are causing the underestimates of the detection of resistance to such drugs. These results highlight the importance of using WGS for epidemiologic surveillance and control of DR/MDR/p-XDR-TB, as these discrepancies would not be detected and reported without it use.
Without any doubt, a major limitation of this study is that phenotypic tests for second line drugs were not done in all samples. Among the 161 isolates classified as phenotipically MDR-TB only 22 were tested to all second line drugs and only 10 were tested for nine drugs; one of 10 Poly-R-TB and six of the 27 p-XDR-TB were submitted to the full DST. This further emphasizes the importance of using molecular methods for TB diagnosis and comprehensive resistance analyzes. Another limitation is the small sample size from regions outside Rio de Janeiro, Distrito Federal and Sao Paulo States, therefore not representing the national scenario of DR-TB in Brazil.
In conclusion, the evaluated pipelines for prediction of MTBC lineage and drug resistance work well in the Brazilian sampling studied here and our data favor the use of WGS in cases without conventional DST. Although the in-house WGS pipeline performed slightly better in general, all tools performed well in predicting DR for RIF, INH, AMK, KAN and CAP. Importantly they allowed detection of p-XDR-TB strains that otherwise would probably have been unreported. Analysis of isolates of DR-TB patients that were sampled sometimes years apart, demonstrated that several accumulated drug resistance mutations showing that resistance evolution occurs by acquisition of mutations in the same strain, probably due to noncompliance of treatment. Phylogenetic analysis and lineage characterization contribute to better understand the MTBC genotypes spreading in the population, so WGS improves the knowledge of MTBC population structure and evolution and offers rapid and reliable assessment of resistance related mutations allowing faster access to effective treatment. A surprisingly low level of reinfection was observed in an area with high TB incidence and, even in patients with DR and prolonged treatment. We believe that these findings highlight the importance of the need for active surveillance throughout the national territory in order to avoid further aggravation of the TB scenario in Brazil.
4. Materials and Methods
4.1. Sample Collections and Phenotypic Drug Resistance
We used a convenience sample for this study of 298 Mtb clinical isolates available at the National Tuberculosis Reference Center Professor Hélio Fraga (CRPHF; Rio de Janeiro, Brazil). The sample set included isolates of patients from 16 different Brazilian States with presumed DR-TB due to a history of treatment failure, treatment abandonment, relapse TB or contact with DR-TB patients. The isolates were therefore submitted to phenotypic drug susceptibility testing (DST).
DST was performed to the first line drugs RIF, INH, PZA, EMB and to the second line drugs STR, OFL, AMK KAN and CAP, using the liquid MB/BacT system (Organon Teknika Corp., Durham, NC, USA). The following cut-off values were used: RIF (1.0 mg/L), INH (0.1 mg/L), EMB (5.0 mg/L), PZA (100.0 mg/L), STR (1.0 mg/L), OFX (2.0 mg/L), AMK (1.0 mg/L), KAN (4.0 mg/L) and CAP (2.5 mg/L).
4.2. Whole Genome Sequencing and in-House Pipeline
The Mtb genomic DNA was extracted as described previously [76]. To perform WGS, sequencing libraries were prepared as described previously [77], and sequenced on an Illumina platform at Division of Infectious Diseases and Environmental Health of the Norwegian Institute of Public Health. To describe the MTBC genetic diversity and genotypic resistance profile, a Single Nucleotide Polymorphism (SNP)-based Mtb lineage and sub-lineage, and resistance related genes (11 coding genes and four promoter regions) was assigned first using an in-house pipeline developed at the Swiss Tropical and Public Health Institute (Swiss TPH) as described before [78], hereafter called in-house WGS pipeline. In brief, FASTQ files were processed with Trimmmomatic v 0.33 (SLIDINGWINDOW:5:20) [79] to remove Illumina adaptors and trim low quality reads. Overlapping reads were then merged with SeqPrep v 1.2. Duplicated reads were marked by the MarkDuplicates module of Picard v 2.9.1. The resulting reads were mapped to a reconstructed ancestral sequence of MTBC as described previously [80] using the BWA-MEM v 0.7.13 algorithm. Pysam v 0.9.0 was used to exclude reads with alignment score lower than (0.93*read_length)-(read_length*4*0.07)): this corresponds to more than seven miss-matches per 100 bp. SNPs were called with Samtools v 1.2 mpileup and VarScan v 2.4.1. SNPs annotation used was that of Mycobacterium tuberculosis H37Rv reference strain (NC_000962.3). Repetitive regions of the genome as PE/PPE/PGRS were excluded as described before [81].
4.3. Phylogenetic Analysis
A maximum likelihood phylogenetic tree using 100 bootstraps was constructed using an alignment containing all variable positions from all quality filtered genomes with ≥15x average coverage using MEGAX v 10.2 [82], and the resulting tree was rooted using M. canettii (Genbank accession number: NC_019950.1). The tree was visualized using iTOL v 6.7 [83].
4.4. Lineage Classification Pipelines Comparisons
Mtb lineages classification by the in-house WGS pipeline was compared with three online available pipelines: TB Profiler version 5.0 [84], KvarQ version 0.12.2 [10] but using the SNP-barcode used elsewhere [11], and RD-Analyzer version 1.01 - a region of difference based analyzer of Mtb from sequence reads [85].
4.5. Genotypic Resistance Detection and Pipelines Evaluation
In order to identify the drug resistance related variants among the included Mtb genomes, we used the in-house WGS pipeline and the command line versions of the pipelines Mykrobe Predictor version 0.3.3 [12], TB Profiler version 0.3.4 [73], KvarQ version 0.12.2 [10] and TB Profiler version 5.0 (https://github.com/jodyphelan/TBProfiler), a recent pipeline version that accounts for the updated WHO catalogue of mutations conferring resistance (https://www.who.int/publications/i/item/9789240082410).
4.6. Cluster Analysis and Recent Transmission
Genomic clusters were delineated using the tool snp-dists v0.7.0 (https://github.com/tseemann/snp-dists). A SNP-based distance matrix was built using the alignment of 170 high quality genomes which not presented mixed calls. A genomic cluster was defined when two or more isolates presented genomes with ≤12 SNPs of difference, which encompasses not only linked cases such as household contacts (genetic distance from zero to five SNPs), but also related cases (five to 12 SNPs). These thresholds are considered as representing very likely recent transmissions event, especially in settings using isolates from chronically infected patients and DR-TB [86,87,88]. To estimate recent transmission, 170 genomes, corresponding to one genome per patient appropriately identified (patient ID), were used. Samples without patient identification and/or duplicated ID were excluded of this analysis. The reconstructed maximum likelihood tree using 100 bootstraps was visualized in GrapeTree [89]. Clustering rate was calculated using the following formula (nc – c)/n, where nc = the total isolates in cluster, c = the number of clusters and n = the total number of isolates.
4.7. Patient data
Patient clinical data available, included TB treatment outcome, diabetes, alcohol use, illicit drug, cigarette smoking, HIV coinfection status and sex, were obtained from the "Sistema de Informação de Agravos de Notificação" (SINAN) and "Sistema de Informação de Tratamentos Especiais de Tuberculose" (SITETB), two national databases for disease surveillance. The project was approved by Research Ethics Committee of Federal University of Rio de Janeiro (CAEE 10126919.2.0000.5257).
4.8. Statistical Analyses
For prediction of drug resistance, we calculated sensitivity, specificity, accuracy and positive and negative predictive values using the statistical software R (Version 3.6.1). First and second-line anti-TB drugs prediction were evaluated for the four pipelines, using MB/BacT DST as the reference standard.
Free-marginal Kappa Randolph’s statistics [90] with a 95% confidence interval was used to determine agreement among Mtb lineage classification by the four pipelines. The same procedure was followed to determine agreement among phenotypic drug resistance detected by MB/BacT and the genotypic drug resistance detected by the four pipelines, using four levels of agreement: <0.40 (poor), 0.40-0.59 (fair), 0.60-0.80 (moderate/good) and >0.80 (excellent).
Pearson’s chi-square test was used to determine associations among treatment outcome, risk factors, clusters, lineage, genotypic and phenotypic drug resistance using the chisq.test function in the statistical software R (Version 3.6.1). In case the expected value is less than 5, Fisher’s exact test was calculated using the fisher.test function in R.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Lineage classification of 298 genomes isolates using four pipelines; Figure S2: Venn diagram for the four pipelines used in lineage classification of the 298 genomes isolates; Table S1: Phenotypic profile of 294 isolates included in the study; Table S2: Clinical data of 112 patients obtained from SITETB and SINAN database; Table S3: Mutations resistance related detected by the in-house WGS pipeline in 298 isolates; Table S4: Agreement among DST phenotypic test, in-house WGS pipeline, KvarQ, Mykrobe Predictor, TB Profiler version 0.3.4 and 5.0 according free-marginal kappa coefficient. Table S5: Mutations resistance-related detected by TB Profiler version 5.0 in 298 isolates. Table S6: Chronical patients without changes in genotypic profile mutation resistance. Table S7: Genotypic and phenotypic profiles of the 82 patients with cured and deceased treatment outcome.
Author Contributions
L.S.E., P.N.S. and A.L.K. contributed to conceptualization; all authors contributed to methodology; L.S.E, D.B., P.N.S., A.L.K., V.E. and S.G. contributed to formal analysis; all authors contributed to investigation; A.L.K., P.N.S., M.L.R., V.E. and S.G. contributed to resources; L.S.E., P.N.S., A.L.K., L.L.G., M.B. and F.C.O.F. contributed to data curation; L.S.E contributed to writing—original draft preparation, L.S.E., D.B., R.S., P.N.S., A.L.K., M.L.R., V.E. and S.G. contributed to writing—review and editing; P.N.S., A.L.K., M.L.R., and S.G. contributed to supervision; L.S.E. contributed to project administration; P.N.S. and A.L.K. contributed to funding acquisition. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Coordination for the Improvement of Higher Education Personnel (CAPES/MEC/Brazil) and to the National Council for Scientific and Technological Development (CNPq/MCTI/Brazil). Leonardo Souza Esteves was supported by doctorate and sandwich doctorate scholarship 888881.134437/2016-01 (CAPES/Brazil-UFRJ/PPG Clínica Médica, Coordinator: José R. Lapa e Silva), and postdoctoral fellowship by Programa Fiocruz de Fomento à Inovação (INOVA/Fiocruz).
Institutional Review Board Statement
The project was approved by Research Ethics Committee of Federal University of Rio de Janeiro (CAEE 10126919.2.0000.5257).
Acknowledgments
The authors would like to thank to sciCORE from University of Basel for helping in computational processing, the staff from Centro de Referência Professor Hélio Fraga (CRPHF/Brazil/RJ) for helping in samples processing and culture management, Laboratory of Molecular Biology Applied to Mycobacteria at IOC/FIOCRUZ and Laboratory of Infection Biology and Molecular Epidemiology at Swiss TPH and Norwegian Institute of Public Health for the infrastructure.
Conflicts of Interest
The authors declare that they have no potential conflicts of interest.
References
- WHO Global Tuberculosis Report 2023; Geneve, 2023; ISBN 9789240083851.
- Brazil Epidemiological Report - Tuberculosis 2021; 2021.
- WHO Global Tuberculosis Report 2021; Geneva, 2021.
- WHO Global Tuberculosis Report 2019; Geneva, 2019.
- Bhering, M.; Kritski, A. Short Communication Trends in Primary Multidrug-Resistant Tuberculosis in the State of Rio de Janeiro : A Retrospective Study Conducted during 2000-2019. Rev. Soc. Bras. Med. Trop. 2021, 54, 12–15. [Google Scholar] [CrossRef] [PubMed]
- Tagliani, E.; Anthony, R.; Kohl, T.A.; De Neeling, A.; Nikolayevskyy, V.; Ködmön, C.; Maurer, F.P.; Niemann, S.; Van Soolingen, D.; Van Der Werf, M.J.; et al. Use of a Whole Genome Sequencingbased Approach for Mycobacterium Tuberculosis Surveillance in Europe in 2017-2019: An ECDC Pilot Study. Eur. Respir. J. 2021, 57. [Google Scholar] [CrossRef]
- Walker, T.M.; Choisy, M.; Dedicoat, M.; Drennan, P.G.; Wyllie, D.; Yang-Turner, F.; Crook, D.W.; Robinson, E.R.; Walker, A.S.; Smith, E.G.; et al. Mycobacterium Tuberculosis Transmission in Birmingham, UK, 2009–19: An Observational Study. Lancet Reg. Heal. - Eur. 2022, 17, 100361. [Google Scholar] [CrossRef] [PubMed]
- Meehan, C.J.; Goig, G.A.; Kohl, T.A.; Verboven, L.; Dippenaar, A.; Ezewudo, M.; Farhat, M.R.; Guthrie, J.L.; Laukens, K.; Miotto, P.; et al. Whole Genome Sequencing of Mycobacterium Tuberculosis: Current Standards and Open Issues. Nat. Rev. Microbiol. 2019, 1. [Google Scholar] [CrossRef]
- Papaventsis, D.; Casali, N.; Kontsevaya, I.; Drobniewski, F.; Cirillo, D.M.; Nikolayevskyy, V. Whole Genome Sequencing of Mycobacterium Tuberculosis for Detection of Drug Resistance: A Systematic Review. Clin. Microbiol. Infect. 2017, 23, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Steiner, A.; Stucki, D.; Coscolla, M.; Borrell, S.; Gagneux, S. KvarQ: Targeted and Direct Variant Calling from Fastq Reads of Bacterial Genomes. BMC Genomics 2014, 15, 881. [Google Scholar] [CrossRef] [PubMed]
- Coll, F.; McNerney, R.; Guerra-Assunção, J.A.; Glynn, J.R.; Perdigão, J.; Viveiros, M.; Portugal, I.; Pain, A.; Martin, N.; Clark, T.G. A Robust SNP Barcode for Typing Mycobacterium Tuberculosis Complex Strains. Nat. Commun. 2014, 5, 4–8. [Google Scholar] [CrossRef]
- Bradley, P.; Gordon, N.C.; Walker, T.M.; Dunn, L.; Heys, S.; Huang, B.; Earle, S.; Pankhurst, L.J.; Anson, L.; De Cesare, M.; et al. Rapid Antibiotic-Resistance Predictions from Genome Sequence Data for Staphylococcus Aureus and Mycobacterium Tuberculosis. Nat. Commun. 2015, 6, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Verboven, L.; Phelan, J.; Heupink, T.H.; Van Rie, A. TBProfiler for Automated Calling of the Association with Drug Resistance of Variants in Mycobacterium Tuberculosis. PLoS One 2022, 17, 1–15. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Freedom in Bioinformatics. Front. Genet. 2014, 5, 1–2. [Google Scholar] [CrossRef]
- Smith, D.R. Buying in to Bioinformatics: An Introduction to Commercial Sequence Analysis Software. Brief. Bioinform. 2014, 16, 700–709. [Google Scholar] [CrossRef] [PubMed]
- Coscolla, M.; Gagneux, S. Consequences of Genomic Diversity in Mycobacterium Tuberculosis. Semin. Immunol. 2014, 26, 431–444. [Google Scholar] [CrossRef] [PubMed]
- Oppong, Y.E.A.; Phelan, J.; Perdigão, J.; Machado, D.; Miranda, A.; Viveiros, M.; Clark, T.G.; Hibberd, M.L. Genome-Wide Analysis of Mycobacterium Tuberculosis Polymorphisms Reveals Lineage- Specific Associations with Drug Resistance. 2019, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Salvato, R.S.; Schiefelbein, S.; Barcellos, R.B.; Praetzel, B.M.; Anusca, I.S.; Esteves, L.S.; Halon, M.L.; Unis, G.; Dias, C.F.; Miranda, S.S.; et al. Molecular Characterisation of Multidrug-Resistant Mycobacterium Tuberculosis Isolates from a High-Burden Tuberculosis State in Brazil. Epidemiol. Infect. 2019, 147. [Google Scholar] [CrossRef] [PubMed]
- Verza, M.; Scheffer, M.C.; Salvato, R.S.; Schorner, M.A.; Barazzetti, F.H.; Machado, H.D.M.; Medeiros, T.F.; Rovaris, D.B.; Viveiros, M. Genomic Epidemiology of Mycobacterium Tuberculosis in Santa Catarina, Southern Brazil. Sci. Rep. 2020, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Esteves, L.S.; Dalla Costa, E.R.; Vasconcellos, S.E.G.; Vargas, A.; Ferreira Junior, S.L.M.; Halon, M.L.; Ribeiro, M.O.; Rodenbusch, R.; Gomes, H.M.; Suffys, P.N.; et al. Genetic Diversity of Mycobacterium Tuberculosis Isoniazid Monoresistant and Multidrug-Resistant in Rio Grande Do Sul, a Tuberculosis High-Burden State in Brazil. Tuberculosis 2018, 110. [Google Scholar] [CrossRef] [PubMed]
- Gomes, H.M.; Elias, A.R.; Oelemann, M.A.C.; Pereira, M.A. da S.; Montes, F.F.O.; Marsico, A.G.; Kritski, A.L.; Filho, L. dos A.; Caldas, P.C.; Possuelo, L.G.; et al. Spoligotypes of Mycobacterium Tuberculosis Complex Isolates from Patients Residents of 11 States of Brazil. Infect. Genet. Evol. 2012, 12, 649–656. [Google Scholar] [CrossRef] [PubMed]
- Vasconcellos, S.E.G.; Acosta, C.C.; Gomes, L.L.; Conceição, E.C.; Lima, K.V.; De Araujo, M.I.; De Lourdes Leite, M.; Tannure, F.; De Souza Caldas, P.C.; Gomes, H.M.; et al. Strain Classification of Mycobacterium Tuberculosis Isolates in Brazil Based on Genotypes Obtained by Spoligotyping, Mycobacterial Interspersed Repetitive Unit Typing and the Presence of Large Sequence and Single Nucleotide Polymorphism. PLoS One 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Dantas, N.G.T.; Suffys, P.N.; Carvalho, W. da S.; Gomes, H.M.; de Almeida, I.N.; de Assis, L.J.; Augusto, C.J.; Gomgnimbou, M.K.; Refregier, G.; Sola, C.; et al. Genetic Diversity and Molecular Epidemiology of Multidrug-Resistant Mycobacterium Tuberculosis in Minas Gerais State, Brazil. BMC Infect. Dis. 2015, 15, 1–11. [Google Scholar] [CrossRef]
- Conceição, E.C.; Rastogi, N.; Couvin, D.; Lopes, M.L.; Furlaneto, I.P.; Gomes, H.M.; Vasconcellos, S.E.G.; Suffys, P.N.; Schneider, M.P.C.; de Sousa, M.S.; et al. Genetic Diversity of Mycobacterium Tuberculosis from Pará, Brazil, Reveals a Higher Frequency of Ancestral Strains than Previously Reported in South America. Infect. Genet. Evol. 2017, 56, 62–72. [Google Scholar] [CrossRef]
- Guimarães, A.E.D.S.; Sharma, A.; Furlaneto, I.P.; Rutaihwa, L.; Cardoso, J.F.; da Conceição, M.L.; Spinassé, L.B.; Machado, E.; Lopes, M.L.; Duarte, R.S.; et al. Evaluation of Drug Susceptibility Profile of Mycobacterium Tuberculosis Lineage 1 from Brazil Based on Whole Genome Sequencing and Phenotypic Methods. Mem. Inst. Oswaldo Cruz 2020, 115, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Conceição, E.C.; Refregier, G.; Gomes, H.M.; Olessa-Daragon, X.; Coll, F.; Ratovonirina, N.H.; Rasolofo-Razanamparany, V.; Lopes, M.L.; van Soolingen, D.; Rutaihwa, L.; et al. Mycobacterium Tuberculosis Lineage 1 Genetic Diversity in Pará, Brazil, Suggests Common Ancestry with East-African Isolates Potentially Linked to Historical Slave Trade. Infect. Genet. Evol. 2019, 73, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Menardo, F.; Gagneux, S.; Rutaihwa, L.K.; Zwyer, M.; Borrell, S.; Comas, I.; Conceição, E.C.; Coscolla, M.; Cox, H.; Joloba, M.; et al. Local Adaptation in Populations of Mycobacterium Tuberculosis Endemic to the Indian Ocean Rim. F1000Research 2021, 10, 1–24. [Google Scholar] [CrossRef]
- Sobkowiak, B.; Glynn, J.R.; Houben, R.M.G.J.; Mallard, K.; Phelan, J.E.; Guerra-assunção, J.A.; Banda, L.; Mzembe, T.; Viveiros, M.; Mcnerney, R.; et al. Identifying Mixed Mycobacterium Tuberculosis Infections from Whole Genome Sequence Data. 2018, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Eyre, D.W.; Cule, M.L.; Griffiths, D.; Crook, D.W.; Peto, T.E.A.; Walker, A.S.; Wilson, D.J. Detection of Mixed Infection from Bacterial Whole Genome Sequence Data Allows Assessment of Its Role in Clostridium Difficile Transmission. PLoS Comput. Biol. 2013, 9. [Google Scholar] [CrossRef] [PubMed]
- Mallard, K.; McNerney, R.; Crampin, A.C.; Houben, R.; Ndlovu, R.; Munthali, L.; Warren, R.M.; French, N.; Glynn, J.R. Molecular Detection of Mixed Infections of Mycobacterium Tuberculosis Strains in Sputum Samples from Patients in Karonga District, Malawi. J. Clin. Microbiol. 2010, 48, 4512–4518. [Google Scholar] [CrossRef]
- Perdigão, J.; Gomes, P.; Miranda, A.; Maltez, F.; Machado, D.; Silva, C.; Phelan, J.E.; Brum, L.; Campino, S.; Couto, I.; et al. Using Genomics to Understand the Origin and Dispersion of Multidrug and Extensively Drug Resistant Tuberculosis in Portugal. Sci. Rep. 2020, 10, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Wyllie, D.H.; Davidson, J.A.; Grace Smith, E.; Rathod, P.; Crook, D.W.; Peto, T.E.A.; Robinson, E.; Walker, T.; Campbell, C. A Quantitative Evaluation of MIRU-VNTR Typing Against Whole-Genome Sequencing for Identifying Mycobacterium Tuberculosis Transmission: A Prospective Observational Cohort Study. EBioMedicine 2018, 34, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Ferrazoli, L.; Palaci, M.; Marques, L.R.M.; Jamal, L.F.; Afiune, J.B.; Chimara, E.; Martins, M.C.; Da Silva Telles, M.A.; Oliveira, C.A.F.; Palhares, M.C.; et al. Transmission of Tuberculosis in an Endemic Urban Setting in Brazil. Int. J. Tuberc. Lung Dis. 2000, 4, 18–25. [Google Scholar]
- Telles, M.A.S.; Ferrazoli, L.; Waldman, E.A.; Giampaglia, C.M.S.; Martins, M.C.; Ueki, S.Y.M.; Chimara, E.; Silva, C.A.; Cruz, V.; Waldman, C.C.S.; et al. A Population-Based Study of Drug Resistance and Transmission of Tuberculosis in an Urban Community. Int. J. Tuberc. Lung Dis. 2005, 9, 970–976. [Google Scholar]
- Nogueira, C.L.; Prim, R.I.; Senna, S.G.; Rovaris, D.B.; Maurici, R.; Rossetti, M.L.; Couvin, D.; Rastogi, N.; Bazzo, M.L. First Insight into the Molecular Epidemiology of Mycobacterium Tuberculosis in Santa Catarina, Southern Brazil. Tuberculosis 2016, 97, 57–64. [Google Scholar] [CrossRef]
- Salvato, R.; Regina, E.; Costa, D.; Júlia, A.; Hee, S.; Laura, M.; Bones, R.; Unis, G.; Fontoura, C.; Viveiros, M.; et al. Infection, Genetics and Evolution First Insights into Circulating XDR and Pre-XDR Mycobacterium Tuberculosis in Southern Brazil. Infect. Genet. Evol. 2020, 78, 104127. [Google Scholar] [CrossRef] [PubMed]
- Zong, Z.; Huo, F.; Shi, J.; Jing, W.; Ma, Y.; Liang, Q.; Jiang, G.; Dai, G.; Huang, H.; Pang, Y. Relapse versus Reinfection of Recurrent Tuberculosis Patients in a National Tuberculosis Specialized Hospital in Beijing, China. Front. Microbiol. 2018, 9, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Guerra-Assunção, J.A.; Houben, R.M.G.J.; Crampin, A.C.; Mzembe, T.; Mallard, K.; Coll, F.; Khan, P.; Banda, L.; Chiwaya, A.; Pereira, R.P.A.; et al. Recurrence Due to Relapse or Reinfection with Mycobacterium Tuberculosis: A Whole-Genome Sequencing Approach in a Large, Population-Based Cohort with a High HIV Infection Prevalence and Active Follow-Up. J. Infect. Dis. 2015, 211, 1154–1163. [Google Scholar] [CrossRef] [PubMed]
- Hughes, D.; Andersson, D.I. Evolutionary Trajectories to Antibiotic Resistance. Annu. Rev. Microbiol. 2017, 71, 579–596. [Google Scholar] [CrossRef] [PubMed]
- Anley, D.T.; Akalu, T.Y.; Dessie, A.M.; Anteneh, R.M.; Zemene, M.A.; Bayih, W.A.; Solomon, Y.; Gebeyehu, N.A.; Kassie, G.A.; Mengstie, M.A.; et al. Prognostication of Treatment Non-Compliance among Patients with Multidrug-Resistant Tuberculosis in the Course of Their Follow-up: A Logistic Regression–based Machine Learning Algorithm. Front. Digit. Heal. 2023, 5, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kanji, A.; Hasan, R.; Zhang, Y.; Shi, W.; Imtiaz, K.; Iqbal, K.; Shafiq, S.; Hasan, Z. Increased Expression of Efflux Pump Genes in Extensively Drug-Resistant Isolates of Mycobacterium Tuberculosis. Int. J. Mycobacteriology 2016, 5, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Rigouts, L.; Miotto, P.; Schats, M.; Lempens, P.; Cabibbe, A.M.; Galbiati, S.; Lampasona, V.; de Rijk, P.; Cirillo, D.M.; de Jong, B.C. Fluoroquinolone Heteroresistance in Mycobacterium Tuberculosis: Detection by Genotypic and Phenotypic Assays in Experimentally Mixed Populations. Sci. Rep. 2019, 9, 1–8. [Google Scholar] [CrossRef]
- Dalla Costa, E.R.; Ribeiro, M.O.; Silva, M.S.; Arnold, L.S.; Rostirolla, D.C.; Cafrune, P.I.; Espinoza, R.C.; Palaci, M.; Telles, M.A.; Ritacco, V.; et al. Correlations of Mutations in KatG, OxyR-AhpC and InhA Genes and in Vitro Susceptibility in Mycobacterium Tuberculosis Clinical Strains Segregated by Spoligotype Families from Tuberculosis Prevalent Countries in South America. BMC Microbiol. 2009, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ford, C.B.; Shah, R.R.; Maeda, M.K.; Gagneux, S.; Murray, M.B.; Cohen, T.; Johnston, J.C.; Gardy, J.; Lipsitch, M.; Fortune, S.M. Mycobacterium Tuberculosis Mutation Rate Estimates from Different Lineages Predict Substantial Differences in the Emergence of Drug-Resistant Tuberculosis. Nat. Genet. 2013, 45, 784–790. [Google Scholar] [CrossRef]
- Gayoso, R.; Dalcolmo, M.; Ueleres, J.; Barreira, D. Predictors of Mortality in Multidrug-Resistant Centers, 2005 to 2012. Brazilian J. Infect. Dis. 2018, 22, 305–310. [Google Scholar] [CrossRef]
- Zürcher, K.; Reichmuth, M.L.; Ballif, M.; Loiseau, C.; Borrell, S.; Reinhard, M.; Skrivankova, V.; Hömke, R.; Sander, P.; Avihingsanon, A.; et al. Articles Mortality from Drug-Resistant Tuberculosis in High-Burden Countries Comparing Routine Drug Susceptibility Testing with Whole-Genome Sequencing : A Multicentre Cohort Study. 2021, 320–330. [Google Scholar] [CrossRef]
- Araujo, L.G.; Garcia, M.T.; Zaccarioto, T.R.; MOretti, M.L.; Levy, C.E.; Resende, M.R. Clinical Outcomes and Molecular Characterization of Drug-Resistant Tuberculosis in Pre- and Extensively Drug-Resistant Disease Based on Line Probe Assays. 2021, 5, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Mitnick, C.D.; Franke, M.F.; Rich, M.L.; Viru, F.A.A.; Appleton, S.C.; Atwood, S.S.; Bayona, J.N.; Bonilla, C.A.; Chalco, K.; Fraser, H.S.F.; et al. Aggressive Regimens for Multidrug-Resistant Tuberculosis Decrease All-Cause Mortality. 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Peters, L.; Olson, L.; Khu, D.T.K.; Linnros, S.; Le, N.K.; Hanberger, H.; Hoang, N.T.B.; Tran, D.M.; Larsson, M. Multiple Antibiotic Resistance as a Risk Factor for Mortality and Prolonged Hospital Stay: A Cohort Study among Neonatal Intensive Care Patients with Hospital-Acquired Infections Caused by Gram-Negative Bacteria in Vietnam. PLoS One 2019, 14. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.S. Mutations Found in Mycobacterium Tuberculosis. 2018, 103, 1034–1045. [Google Scholar] [CrossRef]
- Matsui, T.; Maíra, J.; Pinhata, W.; Christiane, M.; Brandão, A.P.; Ferrazoli, L.; Leão, S.C.; Viana-niero, C. Frequency of First and Second-Line Drug Resistance-Associated Mutations among Resistant Mycobacterium Tuberculosis Clinical Isolates from São Paulo, Brazil. 2020, 115, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Daoqun, L.; Yuzhu, S.; Cheng-lin, Z.; Xiaofei, L.; Xueshan, X. Journal of Infection and Public Health Screening Mutations in Drug-Resistant Mycobacterium Tuberculosis Strains in Yunnan, China. J. Infect. Public Health 2017, 10, 630–636. [Google Scholar] [CrossRef]
- Dokht, A.; Etemad, N.; Hashemzadeh, M. Journal of Global Antimicrobial Resistance Frequency of Rrs and RpsL Mutations in Streptomycin-Resistant Mycobacterium Tuberculosis Isolates from Iranian Patients. Integr. Med. Res. 2017, 9, 51–56. [Google Scholar] [CrossRef]
- Shi, D.; Li, L.; Zhao, Y.; Jia, Q.; Li, H.; Coulter, C.; Jin, Q.; Zhu, G. Characteristics of EmbB Mutations in Multidrug-Resistant Mycobacterium Tuberculosis Isolates in Henan, China. 2011, 2240–2247. [Google Scholar] [CrossRef]
- Chen, Y.; Takiff, H.E.; Gao, Q. Phenotypic Instability of Mycobacterium Tuberculosis Strains Harbouring Clinically Prevalent Drug-Resistant Mutations. The Lancet Microbe 2023, 4, e292. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Feng, S.; Chen, Y.; Yu, M.; Wei, W.; Tian, G.B. Antimicrobial Susceptibility Testing in Clinical Mycobacterium Tuberculosis Isolates. The Lancet Microbe 2023, 4, e68. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Lei, B.; Musser, J.M.; Tu, S. Isoniazid Activation Defects in Recombinant Mycobacterium Tuberculosis Catalase-Peroxidase (KatG) Mutants Evident in InhA Inhibitor Production. Antimicrob. Agents Chemother. 2003, 47, 670–675. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.J.; Thibert, L.; Sanchez, T.; Heifets, L.; Zhang, Y. PncA Mutations as a Major Mechanism of Pyrazinamide Resistance in Mycobacterium Tuberculosis: Spread of a Monoresistant Strain in Quebec, Canada. Antimicrob. Agents Chemother. 2000, 44, 528–532. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.W.; Lee, J.-M.; Jung, K.-S. Characterization of PncA Mutations of Pyrazinamide-Resistant Mycobacterium Tuberculosis in Korea. J Korean Med Sci 2001, 16, 537–543. [Google Scholar] [CrossRef]
- Li, K.; Yang, Z.; Gu, J.; Luo, M.; Deng, J.; Chen, Y. Characterization of PncA Mutations and Prediction of PZA Resistance in Mycobacterium Tuberculosis Clinical Isolates From Chongqing, China. Front. Microbiol. 2021, 11, 1–10. [Google Scholar] [CrossRef]
- Scorpio, A.; Lindholm-Levy, P.; Heifets, L.; Gilman, R.; Siddiqi, S.; Cynamon, M.; Zhang, Y. Characterization of PncA Mutations in Pyrazinamide-Resistant Mycobacterium Tuberculosis. Antimicrob. Agents Chemother. 1997, 41, 540–543. [Google Scholar] [CrossRef] [PubMed]
- Marttila, H.J.; Marjamäki, M.; Vyshnevskaya, E.; Vyshnevskiy, B.I.; Otten, T.F.; Vasilyef, A. V.; Viljanen, M.K. PncA Mutations in Pyrazinamide-Resistant Mycobacterium Tuberculosis Isolates from Northwestern Russia. Antimicrob. Agents Chemother. 1999, 43, 1764–1766. [Google Scholar] [CrossRef]
- Morlock, G.P.; Metchock, B.; Sikes, D.; Crawford, J.T.; Cooksey, R.C. EthA, InhA, and KatG Loci of Ethionamide-Resistant Clinical Mycobacterium Tuberculosis Isolates. Antimicrob. Agents Chemother. 2003, 47, 3799–3805. [Google Scholar] [CrossRef]
- Al-Ghafli, H.; Kohl, T.A.; Merker, M.; Varghese, B.; Halees, A.; Niemann, S.; Al-Hajoj, S. Drug-Resistance Profiling and Transmission Dynamics of Multidrug-Resistant Mycobacterium Tuberculosis in Saudi Arabia Revealed by Whole Genome Sequencing. Infect. Drug Resist. 2018, 11, 2219–2229. [Google Scholar] [CrossRef]
- Valim, A.R.M.; Rossetti, M.L.R.; Ribeiro, M.O.; Zaha, A. Mutations in the RpoB Gene of Multidrug-Resistant Mycobacterium Tuberculosis Isolates from Brazil. J. Clin. Microbiol. 2000, 38, 3119–3122. [Google Scholar] [CrossRef] [PubMed]
- Sanches-Padilha, E.; Merker, M.; Beckert, P.; Jochims, F.; Dlamini, T.; Kahn, P.; Bonnet, M.; Niemann, S. Detection of Drug-Resistant Tuberculosis by Xpert MTB/RIF in Swaziland. N. Engl. J. Med. 2015, 372, 1179–1181. [Google Scholar] [CrossRef] [PubMed]
- Makhado, N.A.; Matabane, E.; Faccin, M.; Pinçon, C.; Jouet, A.; Boutachkourt, F.; Goeminne, L.; Gaudin, C.; Maphalala, G.; Beckert, P.; et al. Outbreak of Multidrug-Resistant Tuberculosis in South Africa Undetected by WHO-Endorsed Commercial Tests: An Observational Study. Lancet Infect. Dis. 2018, 18, 1350–1359. [Google Scholar] [CrossRef] [PubMed]
- Beckert, P.; Sanchez-Padilla, E.; Merker, M.; Dreyer, V.; Kohl, T.A.; Utpatel, C.; Köser, C.U.; Barilar, I.; Ismail, N.; Omar, S.V.; et al. MDR M. Tuberculosis Outbreak Clone in Eswatini Missed by Xpert Has Elevated Bedaquiline Resistance Dated to the Pre-Treatment Era. Genome Med. 2020, 12, 1–11. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization Technical Report on Critical Concentrations for Drug Susceptibility Testing of Isoniazid and the Rifamycins (Rifampicin, Rifabutin and Rifapentine); 2021; ISBN 9789240017283.
- Denkinger, C.M.; Kik, S. V; Cirillo, M.; Casenghi, M.; Shinnick, T.; Weyer, K.; Gilpin, C.; Boehme, C.C.; Schito, M.; Kimerling, M.; et al. Defining the Needs for Next Generation Assays for Tuberculosis. 2015, 211, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Macedo, R.; Nunes, A.; Portugal, I.; Duarte, S.; Vieira, L.; Gomes, J.P. Dissecting Whole-Genome Sequencing-Based Online Tools for Predicting Resistance in Mycobacterium Tuberculosis: Can We Use Them for Clinical Decision Guidance? Tuberculosis 2018, 110, 44–51. [Google Scholar] [CrossRef] [PubMed]
- van Beek, J.; Haanperä, M.; Smit, P.W.; Mentula, S.; Soini, H. Evaluation of Whole Genome Sequencing and Software Tools for Drug Susceptibility Testing of Mycobacterium Tuberculosis. Clin. Microbiol. Infect. 2019, 25, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Coll, F.; McNerney, R.; Preston, M.D.; Guerra-Assunção, J.A.; Warry, A.; Hill-Cawthorne, G.; Mallard, K.; Nair, M.; Miranda, A.; Alves, A.; et al. Rapid Determination of Anti-Tuberculosis Drug Resistance from Whole-Genome Sequences. Genome Med. 2015, 7, 51. [Google Scholar] [CrossRef]
- Coll, F.; Phelan, J.; Hill-Cawthorne, G.A.; Nair, M.B.; Mallard, K.; Ali, S.; Abdallah, A.M.; Alghamdi, S.; Alsomali, M.; Ahmed, A.O.; et al. Genome-Wide Analysis of Multi- and Extensively Drug-Resistant Mycobacterium Tuberculosis. Nat. Genet. 2018, 50, 307–316. [Google Scholar] [CrossRef]
- Iwamoto, T.; Murase, Y.; Yoshida, S.; Aono, A.; Kuroda, M.; Sekizuka, T.; Yamashita, A.; Kato, K.; Takii, T.; Arikawa, K.; et al. Overcoming the Pitfalls of Automatic Interpretation of Whole Genome Sequencing Data by Online Tools for the Prediction of Pyrazinamide Resistance in Mycobacterium Tuberculosis. PLoS One 2019, 14, 1–13. [Google Scholar] [CrossRef]
- Embden, J.D.A. van.; Cave, M.D.; Crawford, J.T.; Dale, J.W.; Eisenach, K.D.; Gicquel, B.; Hermans, P.; Martin, C.; Mcadam, R.; Shinnick, T.M.; et al. Strain Identification of Mycobacterium Tuberculosis by DNA Fingerprinting : Recommendations for a Standardized Methodology. 1993, 31, 406–409. [Google Scholar] [CrossRef] [PubMed]
- Eldholm, V.; Pettersson, J.H.O.; Brynildsrud, O.B.; Kitchen, A.; Rasmussen, E.M.; Lillebaek, T.; Rønning, J.O.; Crudu, V.; Mengshoel, A.T.; Debech, N.; et al. Armed Conflict and Population Displacement as Drivers of the Evolution and Dispersal of Mycobacterium Tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 13881–13886. [Google Scholar] [CrossRef]
- Menardo, F.; Loiseau, C.; Brites, D.; Coscolla, M.; Gygli, S.M.; Rutaihwa, L.K.; Trauner, A.; Beisel, C.; Borrell, S.; Gagneux, S. Treemmer: A Tool to Reduce Large Phylogenetic Datasets with Minimal Loss of Diversity. BMC Bioinformatics 2018, 19, 164. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Comas, I.; Chakravartti, J.; Small, P.M.; Galagan, J.; Niemann, S.; Kremer, K.; Ernst, J.D.; Gagneux, S. Human T Cell Epitopes of Mycobacterium Tuberculosis Are Evolutionarily Hyperconserved. Nat. Genet. 2010, 42, 498–503. [Google Scholar] [CrossRef]
- Stucki, D.; Brites, D.; Jeljeli, L.; Coscolla, M.; Qingyun; Liu, Andrej Trauner, Lukas Fenner, Liliana Rutaihwa, Sonia Borrell, T.; Luo, Qian Gao, Midori Kato-Maeda, Marie Ballif, Matthias Egger, R.; Macedo, Helmi Mardassi, Milagros Moreno, Griselda Tudo Vilanova11, J.; Fyfe, Maria Globan, Jackson Thomas, Frances Jamieson, J.L.; Guthrie, Adwoa Asante-Poku, Dorothy Yeboah-Manu, E.W.; et al. Mycobacterium Tuberculosis Lineage 4 Comprises Globally Distributed and Geographically Restricted Sublineages. 2016, 48, 1535–1543. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (ITOL) v4: Recent Updates and New Developments. Nucleic Acids Res. 2019, 47, 256–259. [Google Scholar] [CrossRef]
- Phelan, J.E.; O’Sullivan, D.M.; Machado, D.; Ramos, J.; Oppong, Y.E.A.; Campino, S.; O’Grady, J.; McNerney, R.; Hibberd, M.L.; Viveiros, M.; et al. Integrating Informatics Tools and Portable Sequencing Technology for Rapid Detection of Resistance to Anti-Tuberculous Drugs. Genome Med. 2019, 11, 1–7. [Google Scholar] [CrossRef]
- Faksri, K.; Xia, E.; Tan, J.H.; Teo, Y.Y.; Ong, R.T.H. In Silico Region of Difference (RD) Analysis of Mycobacterium Tuberculosis Complex from Sequence Reads Using RD-Analyzer. BMC Genomics 2016, 17, 1–10. [Google Scholar] [CrossRef]
- Walker, T.M.; Ip, C.L.C.; Harrell, R.H.; Evans, J.T.; Kapatai, G.; Dedicoat, M.J.; Eyre, D.W.; Wilson, D.J.; Hawkey, P.M.; Crook, D.W.; et al. Whole-Genome Sequencing to Delineate Mycobacterium Tuberculosis Outbreaks : A Retrospective Observational Study. Lancet Infect. Dis. 2013, 13, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Comas, I. Genomic Epidemiology of Tuberculosis. Adv. Exp. Med. Biol. 2017, 1019, 79–93. [Google Scholar] [CrossRef] [PubMed]
- Merker, M.; Kohl, T.A.; Niemann, S.; Supply, P. The Evolution of Strain Typing in the Mycobacterium Tuberculosis Complex; 2017; Vol. 1019; ISBN 9783319643717.
- Zhou, Z.; Alikhan, N.F.; Sergeant, M.J.; Luhmann, N.; Vaz, C.; Francisco, A.P.; Carriço, J.A.; Achtman, M. Grapetree: Visualization of Core Genomic Relationships among 100,000 Bacterial Pathogens. Genome Res. 2018, 28, 1395–1404. [Google Scholar] [CrossRef]
- Warrens, M.J. Inequalities between Multi-Rater Kappas. 2010, 271–286. [Google Scholar] [CrossRef]
Figure 1.
Proportion of resistance related mutations of 298 isolates included in the study based on in-house WGS pipeline analysis. Three most frequent mutations found are highlighted and the remaining mutations are shown in Supplementary table S3. RIF = rifampicin, INH = isoniazid, PZA = pyrazinamide, EMB = ethambutol, STR = streptomycin, FQ = fluoroquinolone, AMK, KAN, CAP = amikacin, kanamycin, capreomycin, ETH = ethionamide.
Figure 1.
Proportion of resistance related mutations of 298 isolates included in the study based on in-house WGS pipeline analysis. Three most frequent mutations found are highlighted and the remaining mutations are shown in Supplementary table S3. RIF = rifampicin, INH = isoniazid, PZA = pyrazinamide, EMB = ethambutol, STR = streptomycin, FQ = fluoroquinolone, AMK, KAN, CAP = amikacin, kanamycin, capreomycin, ETH = ethionamide.
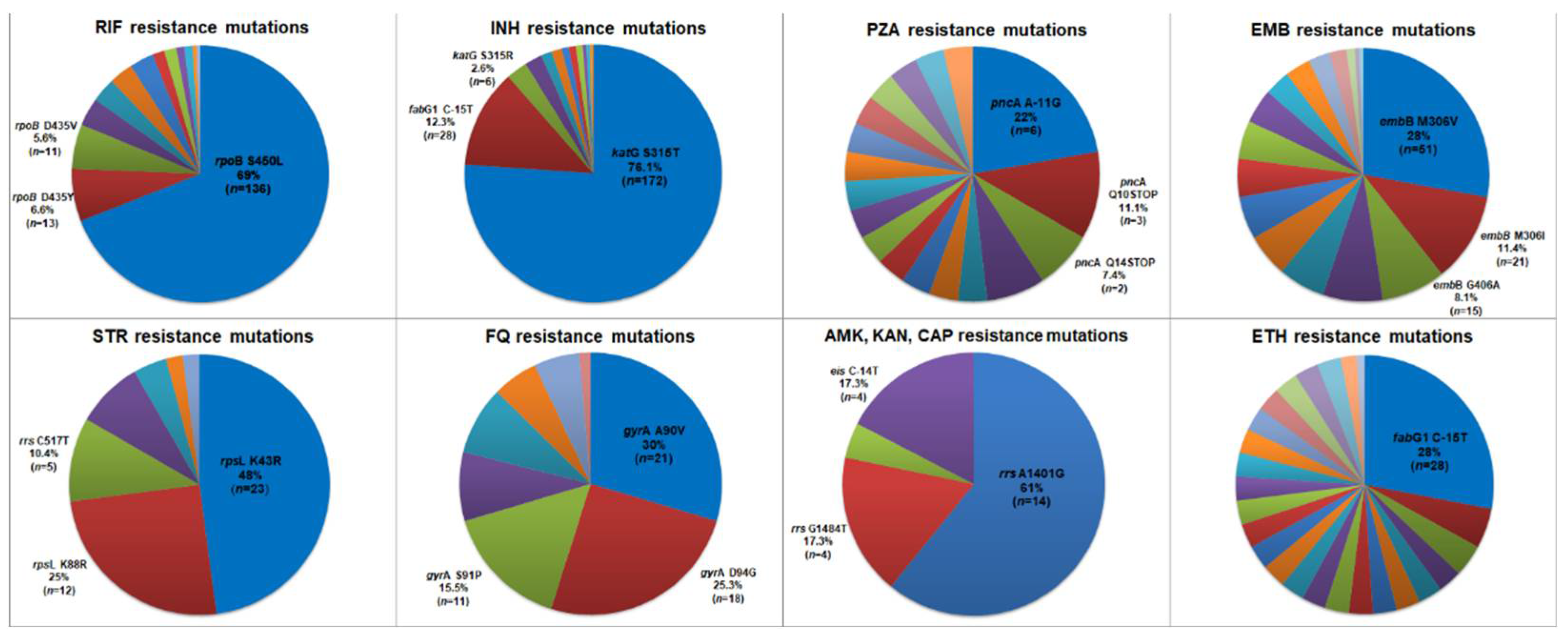
Figure 2.
Maximum likelihood phylogenetic tree for the 293 M. tuberculosis isolates constructed based on 38,563 SNPs rooted with M. canettii. Presented from the outside to the inside: SNP-based lineage classification by the in-house WGS pipeline; Genotypic resistance profile (circle); and Phenotypic resistance profile (square). Tips (triangle) are shown colored according to the genomic cluster from 170 patients with identification (see legend). GC = Genomic Cluster; NC = non-clustered. Tree branch scale represents the number of nucleotide substitution per site. .
Figure 2.
Maximum likelihood phylogenetic tree for the 293 M. tuberculosis isolates constructed based on 38,563 SNPs rooted with M. canettii. Presented from the outside to the inside: SNP-based lineage classification by the in-house WGS pipeline; Genotypic resistance profile (circle); and Phenotypic resistance profile (square). Tips (triangle) are shown colored according to the genomic cluster from 170 patients with identification (see legend). GC = Genomic Cluster; NC = non-clustered. Tree branch scale represents the number of nucleotide substitution per site. .
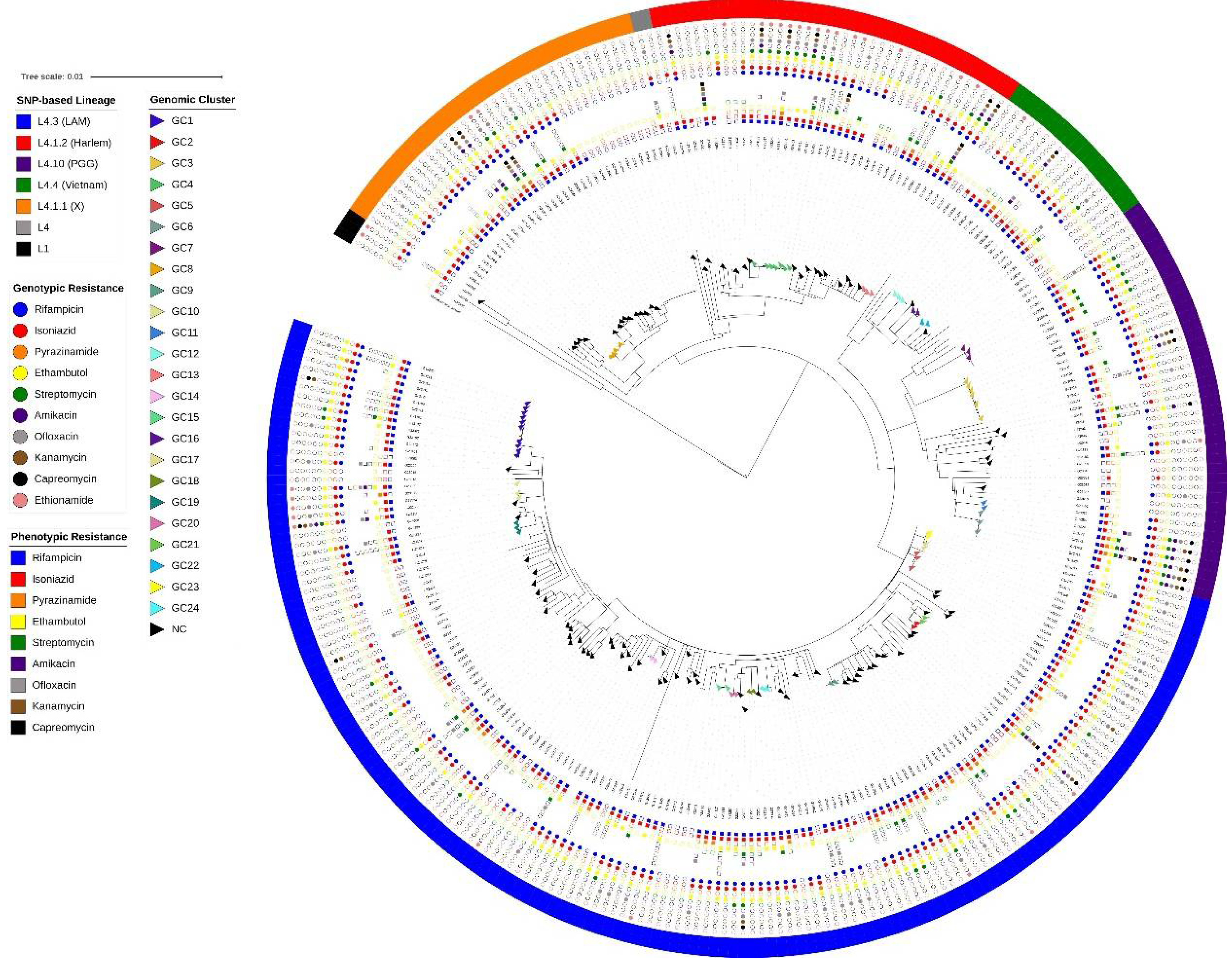
Figure 3.
Geographic proportion distribution of Mycobacterium tuberculosis lineages classified by in-house WGS pipeline of 290 isolates. Eight isolates (three L4.3 [LAM], two L4.10 [PGG3], two L4.1.1 [X] and one L4.1.2 [Haarlem]) have no data about geographic origin.
Figure 3.
Geographic proportion distribution of Mycobacterium tuberculosis lineages classified by in-house WGS pipeline of 290 isolates. Eight isolates (three L4.3 [LAM], two L4.10 [PGG3], two L4.1.1 [X] and one L4.1.2 [Haarlem]) have no data about geographic origin.
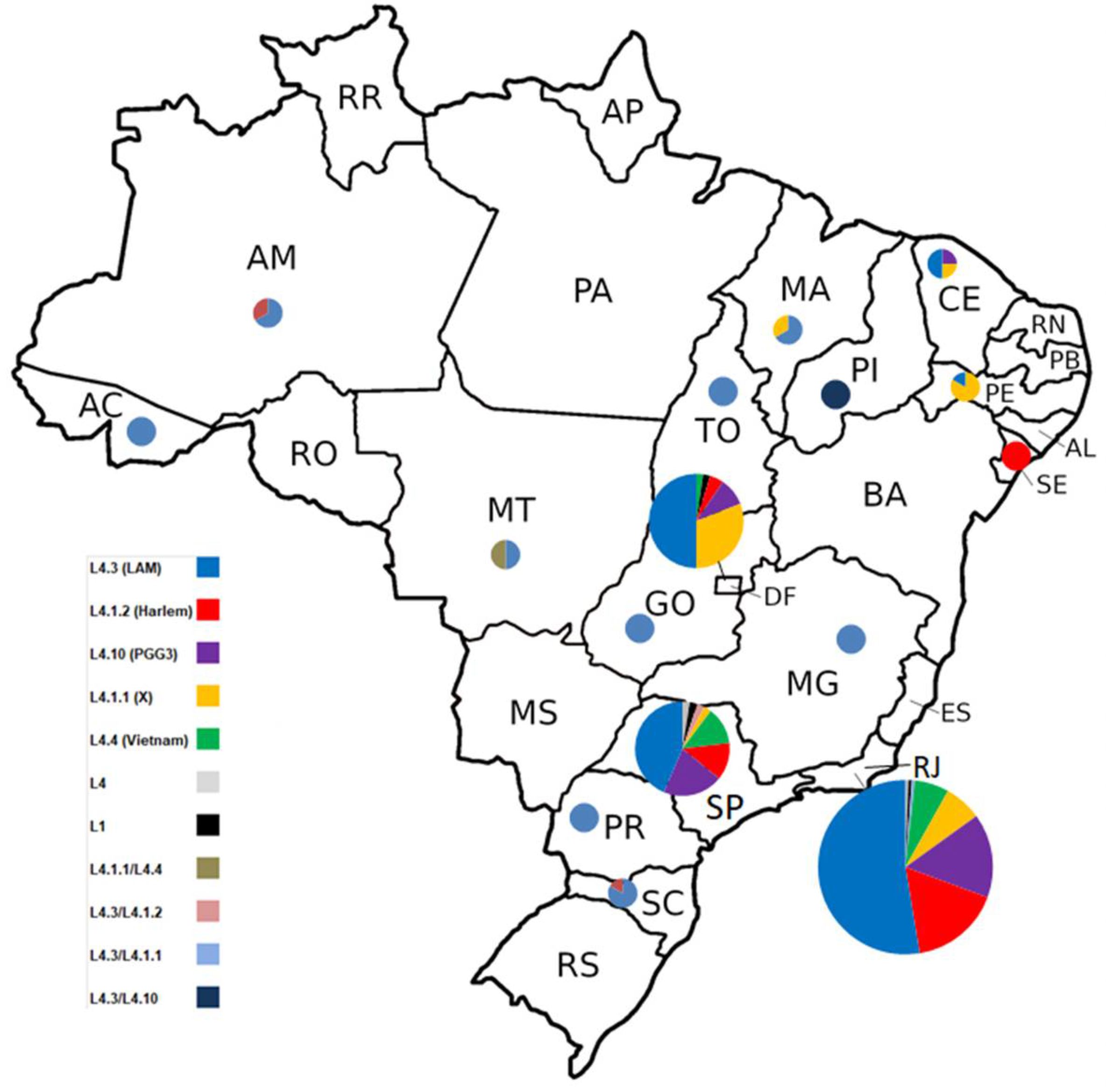
Figure 4.
Radial tree of the 170 isolates used for clustering analysis. Tips were colored according to genotypic resistance profile. Pie charts in the tips represent two or more isolates with no SNPs differences among genomes. NC = non-clustered. The dashed line represents the L1 isolate with greater distance in comparison to the rest of the isolates. Tree branch scale represents the number of nucleotide substitution per site. .
Figure 4.
Radial tree of the 170 isolates used for clustering analysis. Tips were colored according to genotypic resistance profile. Pie charts in the tips represent two or more isolates with no SNPs differences among genomes. NC = non-clustered. The dashed line represents the L1 isolate with greater distance in comparison to the rest of the isolates. Tree branch scale represents the number of nucleotide substitution per site. .
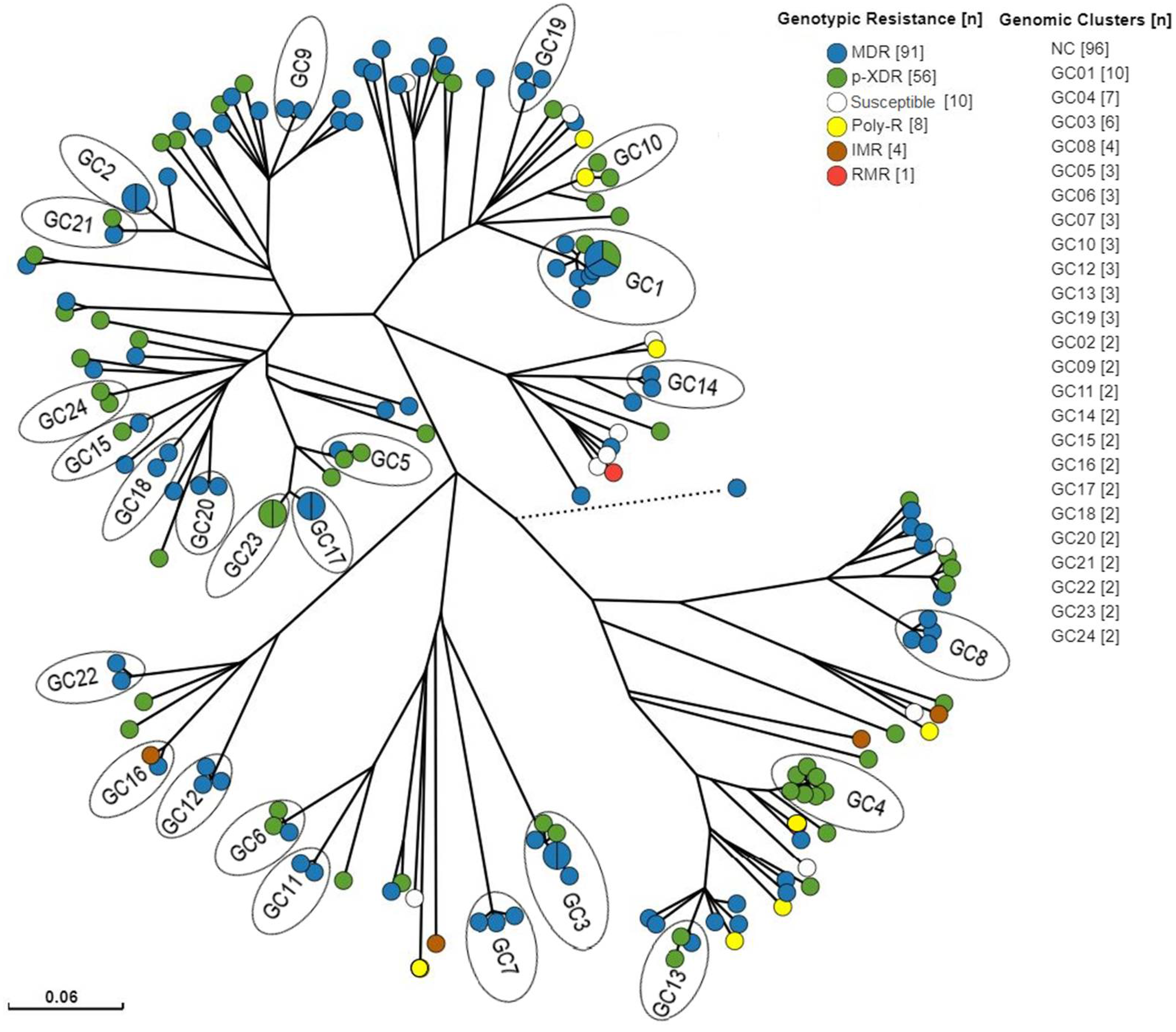

Table 1.
Evaluation of WGS-based drug susceptibility prediction based on the in-house WGS pipeline, KvarQ, Mykrobe Predictor, TB Profiler and TB Profiler 5.0 presenting sensitivity, specificity, positive and negative predictive values, and accuracy.
Table 1.
Evaluation of WGS-based drug susceptibility prediction based on the in-house WGS pipeline, KvarQ, Mykrobe Predictor, TB Profiler and TB Profiler 5.0 presenting sensitivity, specificity, positive and negative predictive values, and accuracy.
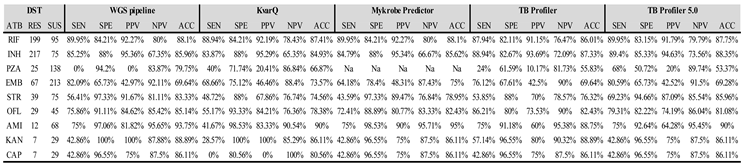
ATB = Antibiotic, RES = Resistant, SUS = Susceptible, SEN = Sensitivity, SPE = Specificity, PPV = Positive Predictive Value, NPV = Negative Predictive Value, ACC = Accuracy.
Table 2.
Novel mutations observed in this study using TB Profiler 5.0.
| n Isolates | Drug | DST | Novel Mutation | Phenotypic Profile | Genotypic Profile╪ |
|---|---|---|---|---|---|
| 1 | INH | R | katG_c.2070delC¹ | MDR | p-XDR |
| 1 | INH | S | katG_c.1141dupG² | RMR | IMR |
| 1 | ETH | NA | ethA_c.306_307delCA | MDR | p-XDR |
| 1 | ETH | NA | ethA_c.-382_*857del | p-XDR | p-XDR |
| 1 | ETH | NA | ethA_c.40dupA | MDR | MDR |
| 4 | ETH | NA | ethA_c.851dupC | MDR | 2 MDR/2 p-XDR |
| 4 | PZA | 2R/2NA | pncA_c.193_200dupTCCTCGTC | MDR | MDR |
| 1 | PZA | NA | pncA_c.289_293dupGGTGC | MDR | MDR |
| 1 | PZA | NA | pncA_c.502delA | Poly-R | Poly-R |
| 1 | PZA | R | pncA_c.452dupT | MDR | p-XDR |
| 2 | PZA | R | pncA_c.75_79delCGCGC | p-XDR | p-XDR |
| 4 | PZA | R/2S/NA | pncA_c.443_444dupGC³ | MDR | 2 MDR/2 p-XDR |
| 1 | PZA | S | pncA_c.117_124delGGACTACC | MDR | p-XDR |
| 1 | PZA | S | pncA_c.300delC | MDR | Poly-R |
| 2 | PZA | R/NA | pncA_c.305dupC | p-XDR/Poly-R | p-XDR |
| 1 | PZA | S | pncA_c.329_338delACGAGAACGG | p-XDR | Poly-R |
| 1 | PZA | S | pncA_c.-3449_*7353del | p-XDR | p-XDR |
| 1 | PZA | S | pncA_c.423_424delGA | p-XDR | p-XDR |
| 2 | PZA | S | pncA_c.454_455insT | MDR | MDR |
| 1 | PZA | S | pncA_c.527dupG | MDR | MDR |
╪ = Genotypic resistance profile classified according to TB Profiler 5.0 ¹Genotypic isoniazid resistance profile = ahpC_c.-81C>T, katG_c.2070delC ²Genotypic isoniazid resistance profile = ahpC_c.-48G>A, katG_c.1141dupG ³Genotypic pyrazinamide resistance profile = pncA_c.443_444dupGC, pncA_p.Phe81Val. Isolate PZA phenotypically susceptible.
Table 3.
Chronic TB patients with changes in genotypic profile mutation and phenotypic resistance.
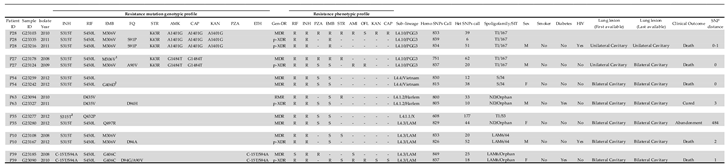
1In addition to the mutation M306V, there was a gap in codons 405 and 406, which resulted in flag G→T E405D, G→C E405D, G→T G406C, G→A G406S, G→A G406D, G→C G406A. 2Mixed SNP call. ND = not determined. Homo SNPs Call = all bases detected in a same genome position are the same. Het SNP Call = bases detected in a same genome position are different.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



