
Submitted:
17 April 2024
Posted:
18 April 2024
You are already at the latest version
Abstract
Stroke represents one of the neurological diseases most responsible for death and permanent disability in the world. Different factors, such as thrombus, emboli and atherosclerosis, take part in the intricate pathophysiology of stroke. Comprehending the molecular processes involved in this mechanism is crucial to developing new, specific and efficient treatments. Some common mechanisms are excitotoxicity and calcium overload, oxidative stress, and neuroinflammation. Furthermore, non-coding RNAs (ncRNAs) are critical in pathophysiology and recovery after cerebral ischaemia. ncRNAs, particularly microRNAs, and the long non-coding RNAs (lncRNAs) are essential for angiogenesis and neuroprotection, and they have been suggested to be therapeutic, diagnostic and prognostic tools in cerebrovascular diseases, including stroke. This review summarizes the intricate molecular mechanisms underlying ischemic and hemorrhagic stroke and delves into the function of miRNAs in the development of brain damage. Furthermore, we will analyze new perspectives on treatment based on molecular mechanisms in addition to traditional stroke therapies.
Keywords:
Stroke
; Ischemic
; hemorrhagic
; pathophysiology
; Long non-coding RNAs
; MicroRNAs
; Stroke therapeutics
1. Introduction
Stroke is characterized by focal neurological signs or symptoms with a vascular cause that endures at least 24h; brain MRI or CT in baseline conditions and brain CT with contrast medium must verify the presence of ischemic lesions after 48–72 h. In the occidental part of the world, stroke is the third highest cause of death and the most common motive of invalidity. It affects 15 million people per year in the world; one-third of them die, and the other 5 million are permanently physically challenged [1]. Ethnicity is correlated with the incidence of stroke; for example, in the United States, the hazard of pathology is increased in Hispanic and Black people than in Caucasians, as reported by multiple studies [1]. Stroke incidence is about 62.8 per 100,000 in men, while women have a stroke incidence of 59 per 100k; this suggests that this disease affects fewer women than men. However, this detection concerns only young subjects; the American Heart Association recently recommended that women over 75 are more affected than men of the same age [2].
Furthermore, the probability of stroke is proportional to age, and nearly three-quarters of all strokes take place in patients older than 64 years [1], even though 33% of the cases of strokes occur in younger persons, proposing that this pathology does not affect just older people. Regarding the pathophysiology, we can identify two principal categories of stroke that are utterly different: haemorrhage and ischemia. Haemorrhage is represented by blood in the cerebral parenchyma, which can accumulate and press on the adjacent parenchyma.
In contrast, insufficient blood flow and inability to satisfy the requirement of oxygen and nourishing substances of the cerebral tissue define ischemia. Cerebral ischemia is caused by a clot that interrupts blood flow to the brain and represents 87% of all stroke cases [3]. TOAST classification distinguishes five different subtypes [4]: (1) extensive artery atherosclerosis (LAAS); (2) cardioembolic infarct (CEI); (3) lacunar infarct (LAC); (4) stroke of other determined etiology (ODE); (5) stroke of undetermined etiology (UDE). Most of the strokes with ischemic origin are caused by embolism with cardiac genesis and by atherosclerosis affecting the large artery. A small or large artery thrombus provokes approximately 45% of ischemic strokes [5,6], whereas cardioembolic stroke accounts for 14–30% of all strokes [7,8]. The lacunar is another important subtype (15–25% of all ischemic strokes) [9,10]. Therefore, cardiac embolism, occlusion of small vessels, and atherosclerosis of the cerebral circulation can be the cause of ischemic stroke. Among all of them, cardioembolic stroke is essential for two principal reasons: first, cardiac embolism provokes the most severe strokes [11], and secondarily, although developing better therapies for dyslipidemia and arterial hypertension, embolism with cardiac origin represents a rising source of stroke in wealthy nations, for example, Canada [12].
Various molecular mechanisms are involved in stroke, some of which are shared by both ischemic and hemorrhagic stroke, such as inflammation and oxidative stress. In addition, excitotoxicity and calcium overload are involved in ischemic stroke [13,14]. Since the pathophysiology involves diverse mechanisms, strategies have continuously evolved to target these causal mechanisms for more promising stroke therapies. Therefore, the review focuses on the pathophysiology, molecular basis, and potential therapeutic strategies for stroke, both established and newly evolving.
2. Ischemic Stroke Pathophysiology
Ischemic stroke occurs due to occlusion of cerebral arteries due to a travelling embolus, such as a cardiogenic embolus, an artery-to-artery embolus, or vascular stenosis of the artery itself. This occlusion interrupts cerebral blood flow and, consequently, significant functional and neurological impairment. Most of the strokes with ischemic origin are caused by atherosclerosis affecting the large artery and by embolism with cardiac genesis. Ischemic and hemorrhagic strokes manifest in neurological deficits due to various pathophysiologic mechanisms (Figure 1).
2.1. Atherothrombotic Stroke
Atherothrombotic stroke is the most common. Atherosclerosis is described as a reduction of calibre or hardening of the arteries and usually affects medium and large arteries. The process begins with damage to the internal coating of an artery (the endothelium). The damage can occur secondly from physical stress, such as arterial hypertension. Damage to the arteries can also be caused by excess cholesterol in the blood or high blood sugar, which is consequently inflammation mediated by the immune system. Grassi, cholesterol, platelets, cellular debris, and calcium gather in the walls of damaged arteries, stimulating the creation and accumulation of other types of cells. The plaque accumulates fat inside with connective tissue around [15]. The resulting deposits are highly thrombogenic because they hinder blood flow and exercise high cutting stresses on the vessel wall [16]. The plaque thickens the artery wall, narrowing the vessel. Blood flow is decreased by narrowing, reducing oxygen supply to the part of the body that the artery serves. One of the major causes of ischemic stroke is atherosclerosis of the carotid artery, with artery-to-artery embolism being the central stroke mechanism in patients with atherosclerosis of the carotid artery [17]. Another biophysical mechanism of obstruction is the development of a local thrombus within a cerebral artery [18]. As a response to vessel injury or atherosclerotic lesions, there is thrombosis or the formation of a blood clot [19]. The coagulation cascade is activated, resulting in the aggregation of platelets and the transformation of prothrombin into a fibrin clot.
2.2. Embolic Stroke
In contrast to a thrombus, an embolus represents a travelling or migrating source of obstruction, thus originating from a distant site. An embolus can be arterial, cardiac, from the peripheral circulation, aortic, or from an unknown source. Cardiac embolism represents an increasing percentage of cerebral ischemia, and it will probably rise mainly in the future. This kind of stroke is caused by an occlusion of a cerebral artery from an embolus formed in the heart. The most severe type of ischemic stroke is considered cardioembolic stroke, and emboli larger in size correspond with higher severity in outcome and injury. From all possible cardiac origins, we have different risks of causing an ischemic stroke; in fact, some of them have a medium risk of provoking embolism, and others have a high risk instead. It is necessary to identify at least one cardiac cause of embolism to establish that cardioembolism produced cerebral ischemia. The most common cause of cardioembolic strokes seems to be the low cardiac output and blood stasis associated with atrial fibrillation. Other high-risk cardiac conditions include valvular heart disease, acute myocardial infarction, bacterial endocarditis, and dilated cardiomyopathy [20]. Interestingly, a hypercoagulable state has proven to be associated with the novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), potentially increasing the risk of embolic strokes in COVID-19 patients [21].
3. Molecular Mechanisms o the Ischemic Stroke Pathophysiology
The onset of ischemia is followed by a cascade of events producing or activating different components, disrupting the homeostatic balance within the brain and leading to cellular death. Among these, excitotoxicity, oxidative stress, and the inflammatory response play vital roles.
3.1. Excitotoxicity and Calcium Overload
A cascade of events producing or activating different components follows the onset of ischemia, which leads to cellular death. After cell death, stressors such as organelle swelling, plasma membrane disruption, and cellular content leak are responsible for neuronal damage [22]. Inflammation, the release of excitatory amino acids, increased intracellular calcium levels, and the production of prostaglandins, leukotrienes, and reactive oxygen species are all critical processes that contribute to the pathological picture of stroke and are potential targets of interest in therapeutic research [23]. In the early stages of ischemic injury, excitotoxicity and calcium build-up are closely associated. In response to the ischemia-triggered energy deficit and failure of ion pumps and reuptake mechanisms, excitatory amino acids, mainly the neurotransmitter glutamate, are released and accumulate in the neuronal extracellular space. Glutamate overload and prolonged glutamate receptors NMDA and AMPA activation lead to the influx and increased intracellular calcium concentration [24]. Significant shifts in ion homeostasis are secondary to AMPA receptor overstimulation, accounting for the increased concentrations of intracellular sodium and chloride [25]. This ion imbalance triggers water influx into the neurons, leading to cell lysis and tissue oedema [26].
Furthermore, increased intracellular calcium promotes calcium release from the mitochondria and other cellular stores and mitochondrial dysfunction [27], emphasizing the complex role of calcium in excitotoxicity and the various mechanisms that contribute to calcium dysregulation. In addition to glutamate receptors, other potential targets in ischemic stroke injury could be channels and ion pumps involved in calcium dysregulation during ischemia, such as the acid-sensing ion channels and Na/Ca2+ exchanger [28]. Besides its role in calcium overload and excitotoxicity, mitochondrial dysfunction leads to swelling, mitochondrial permeability, and membrane collapse, activating apoptotic and oxidative stress pathways.
3.2. Oxidative Stress
Brain ischemia and, to a more significant extent, brain reperfusion are also associated with producing free oxygen radicals. Calcium influx and overload activate calcium-dependent enzymes such as nitric oxide synthase (NOS), which leads to injury and cellular damage through the formation of reactive oxygen species (ROS), such as peroxynitrite (ONOO-) [29]. Mitochondria, long believed to be the primary source of ROS during ischemia, plays a significant role in ROS generation during reperfusion injury. Swelling and collapse of the mitochondria due to mitochondrial dysfunction involves activating pro-apoptotic cascades and releasing free oxygen radicals [30]. A key contributor to ROS production is the enzyme NADPH oxidase, which has recently been shown to generate the majority of superoxide anion in ischemia and NMDA receptor activation compared to the mitochondria [31].
3.3. Neuroinflammation
After ischemic stroke, the brain insult results in apoptosis and necrosis, all of which drive an inflammatory response; this response is called neuroinflammation and is characterized by the participation of numerous cytotypes, first with the activation of resident glial cells and then with the infiltration of leukocytes, monocytes and other immune cells in the brain and the releasing of inflammatory agents. After infiltration into the lesion, dynamic immune cascades occur almost simultaneously to induce beneficial and detrimental effects after stroke onset and continue in the later phases. This immune system is summarized in Table 1.
3.3.1. Roles of Cytokines in Cerebral Ischemia
Cytokines are immunomodulating agents that play a significant role in cell activation, proliferation, and differentiation. Almost every nucleated cell can produce cytokines to modulate the interaction of immune cells such as T cells, B cells, and monocytes/macrophages, thereby arranging immune responses [32,33]. Many processes in the brain involve proinflammatory cytokines that can trigger neurons, glia, and endothelial cells directly. Because of this complex and multiphasic pathway, they may induce further damage or increase cellular viability.
TNF-α
One of the proteins most involved in an inflammatory and immune process is TNF-α, in the past called differentiation factor or cachectin.
This cytokine can be produced by monocytes, mast cells, T cells, macrophages, keratinocytes, neutrophils, and fibroblasts. TNF-α has two different forms: a soluble one, which is biologically active form (sTNF-α) made by the tumor necrosis factor-alpha converting enzyme (TACE), and a transmembrane one (tmTNF-α), which modulates inflammation locally via cell-cell interactions [34]. The soluble form operates systemically by promoting macrophages' cytotoxic and phagocytic action and stimulating the production of other cytokines, such as IL-6 and IL-1. TNF works not only systemically but also locally in the brain. TNF-α acts through receptors (TNFR-1 and TNFR-2), which have different affinity for TNF-α and degree of glycosylation. sTNF-α is correlated with TNFR1, while tmTNF-α with TNFR-2 and TNFR-1. Growing evidence suggests that TNF-α plays a crucial role in the pathophysiology of stroke and has both a neuroprotective and a neurotoxic result in the ischemic brain [32,33]. Proinflammatory mediators, such as interleukin-1 (IL-1), TNF-α, and IL-6, contribute to developing harmful postischemic inflammation in the brain [24,25,26]. Increasing evidence from studies [32,33], using both preclinical animal models and human samples picked up from patients affected by ischemic stroke, suggests that TNF-α, a pleiotropic and powerful proinflammatory cytokine, is involved in the development of injuries in the ischemic brain.
Furthermore, this cytokine is overexpressed in the brain after both transient [36] and permanent [35] middle cerebral artery occlusion [MCAO]. TNF-α is one of the first cytokines to emerge in the context of the inflammatory response to ischemic brain injury and contributes to stimulating the cascade of other inflammatory components in both blood serum and the cerebrospinal fluid [37,38]. Indeed, TNF-α's appearance in the brain following the post-ischemic damage is early because it has an initial peak in the first hours [1-3 hours] and a second one after more than 24-36 hours [37,38]. In the literature, some reports indicate that TNF-α represents a valuable indicator for evaluating prognosis and an accurate parameter for determining the beginning of the inflammatory response [39]. In stroke patients, as soon as 6–12 hours after the onset of symptoms [40] can be seen a rising concentration of TNF-α. An increase in TNF-α concentration within 24 and 48 hrs following a stroke has also been shown; the slight decrease, which occurs within 72 and 144 hrs after a stroke, correlates with clinical improvement in patients during the acute phase of ischemic stroke [41]. Furthermore, TNF-α is also involved in the neuroprotective process against ischemic brain damage [42]. This cytokine seems to play a bivalent role in the brain's inflammatory responses that follow ischemia because it plays an immunosuppressive role during the chronic phase and a proinflammatory role through the acute phase of the inflammatory response in the CNS.
IL-1β
The IL-1 family includes at least three proteins, IL-1α, IL-1β, and IL-1ra, that are the products of separate genes sharing a significant homology and are implicated in the pathogenesis of many human diseases, including stroke. IL-1β is a principal proinflammatory and immunoregulatory cytokine able to influence almost all cell types. IL-1β seems to be the central IL-1 agonist induced in the brain after responding to local insults (e.g., trauma, stroke) or systemic ones (e.g., infection, injury) within one hour during experimental cerebral ischemic brain injury. IL-1β is produced following the formation of an inflammasome, such as monocytes and macrophage/microglia [43]. After ischemic stroke, IL-1β can turn on the nuclear factor (NF)-κB through the activation of TLRs; after this process, NF-κB can transactivate genes correlated with cytokines, chemokines, and other proinflammatory agents [44].
After ischemic stroke, the microglia will be switched to the proinflammatory phenotype called M1, able to express IL-1β, which is a proinflammatory cytokine with neurotoxic effects. Besides, IL-1β can interact with the endothelium and increase leukocyte adherence, promoting oedema formation [45]. IL-1β knockout mice have significantly decreased brain injury induced by MCAO [46]. Furthermore, brain injury increased when IL-1β was administered to rats [47]. Multiple studies report that inhibiting the IL-1 receptor 1, which binds to both IL-1α and IL-1β and is detected in various cytotypes, reduces the area of the brain damaged by ischemia, preserving neurological functions [48]. Therefore, this evidence suggests considering IL-1βa as a crucial contributor to ischemic brain damage. IL- 1β, when tied up with its related receptor, the IL-1 receptor (IL-1R), provokes IL-1R-dependent increase in NF-κB pathways. However, if the levels of IL-1β are increased above a specific cutoff, it can promote the expression of the IL-1 receptor antagonist [IL-1Ra]. This balance between IL-1β and its antagonist, the IL-1Ra, is more critical than just IL-1β because of its global effect and role [49]. Thus, this balance might be a good predictor for a patient's outcome following an ischemic stroke. However, just a few clinical studies have used their level as stroke biomarkers. IL-1β levels were mainly correlated with poor long-term functional outcomes in the study [50]; on the other hand, IL-1Ra levels seemed to predict post-stroke infection development [51].
IL-6
Various cytotypes, including microglial cells, leukocytes, astrocytes, and endothelial cells, can produce IL-6 in response to brain injury. It stimulates hepatocytes to synthesize acute phase proteins (APPs), primarily fibrinogen and CRP (C-reactive protein). IL-6 activates APPs and involves the phosphorylation of the NF-IL-6 transcription factor, which can enhance the transcription of numerous genes [52]. The production of IL-6 requires IL-1 and TNF, which stimulate endothelial cells, fibroblasts, and keratinocytes, thereby increasing the expression of IL-6. IL-6 is a proinflammatory cytokine with several crucial beneficial and harmful functions to CNS cells. Various other molecules, such as IL-1, interleukine-4 (IL-4), prostaglandins, and TNF-α, can trigger and modulate the production of this cytokine, suggesting that the process of expression of various cytokines is intricately linked to the inflammatory cascade. This complexity presents a fascinating challenge for further research in this field.
In recent years, many pieces of research have been made to explain the role of interleukins in the aetiology and development of stroke. Although IL-6 is a cytokine with a proinflammatory role, it has been proposed that it plays an essential function in cerebral ischemia not only as a mediator of the inflammatory development in the acute stage of stroke but also as a neurotrophic element during the late phase of the progress of cerebral ischemia [53]. It has been verified that IL-6 is known as an essential inflammatory marker in stroke; several studies proved a meaningful increase in the concentration of IL-6 in serum, which took place within a few hours following the onset of ischemia and lasted for up to 90 days after the stroke [54]. Thus, these studies show that IL-6 concentration increases during an ischemic stroke, while in physiological conditions, IL-6 cerebral expression is modest [54].
Conversely, the administration of recombinant human IL-6 has shown significant reductions in ischemic damage in a rat model of stroke [54]. Moreover, Sotgiu et al. [40] have reported a negative correlation between the measure of cerebral infarction and the level of IL-6. From this, the authors have concluded that in the intricate network of inflammation that occurs during an ischemic stroke, IL-6 is not a neurotoxic factor but a neuroprotective one. This compelling evidence suggests a potential bivalent role of IL-6 in the ischemic brain, inspiring further exploration into its therapeutic applications.
IFN-γ
The IFN group cytokines can be divided into two types. Type I IFNs represent the largest class and comprise the IFN-α, -β, -ε, -κ, and -ω, which share remarkable sequence homology and are produced by most cytotypes. IFN-γ is a unique member of the type II IFN and has a crucial role in stimulating and modulating an array of immune responses [55]. Mostly monocytes, macrophages, natural killer [NK] cells, T cells, dendritic cells, and B-lymphocytes secrete IFN-γ. It is a significant regulator of immune function and offers a robust first-line defense against invading pathogens.
Furthermore, IFN-γ has many biological actions, including regulating multiple aspects of the immune responses and promoting antigen presentation via upregulating class I and class II major histocompatibility complex [MHC] molecules on the surface of macrophages and T cells. The heterodimeric receptor (IFN-γR) on the cell's surface mediates the cellular response to IFN- γ, activating downstream signal transduction cascades, ultimately regulating gene expression. IFN-γ, when bound to its related receptor, can switch a variety of downstream signalling pathways on, in particular, the Janus kinase (JAK)/signal transducer and activator of transcription (STAT) [56]. The potential influence of IFN-γ on atherogenesis and ischemic stroke is a compelling area of research, with a growing body of evidence reporting its greater expression in atherosclerotic lesions [57]. This suggests its crucial function in the process of atherogenesis and its potential role in the pathogenesis of ischemic stroke, underscoring the need for further investigation.
Following an ischemic brain insult, MHC class II specific CD4 cells will be activated and may effortlessly penetrate the CNS through the blood-brain barrier [BBB] [58]. Therefore, microglia have the chance to retain and further arouse CD4 cells already primed to differentiate into either T helper 1 [TH1] cells, able to produce proinflammatory cytokines such as IL-6, IFN-γ, and TNF-α, or into T helper 2 [TH2] cells producing cytokines capable of supporting antibody-mediated responses [IL-4, IL-5, IL-10, lL-13] [59]. The role of IFN-γ in the polarization of microglia is a crucial aspect of its function. TH1 cells produce proinflammatory IFN-γ cytokines that can turn microglia into M1 phenotype; this cytotype is responsible for a proinflammatory reaction and can produce oxidative metabolites and proinflammatory cytokines, highlighting the significant impact of IFN-γ on immune response.
Anti-Inflammatory Cytokines
The unevenness between inflammatory and anti-inflammatory responses seems critical in brain damage after ischemia [60].
IL-10 is an anti-inflammatory cytokine encoded by the IL10 gene on chromosome 1, and it is mainly produced by monocytes and, secondary, by TH2 lymphocytes, mastocytes, CD4 CD25 Foxp3 regulatory T cells, and by a specific subset of activated T cells and B cells [60]. IL-10 plays a crucial role in the pathogenesis of stroke by repressing the overexpression of proinflammatory cytokines. These data are validated by a study that showed a potential role of IL-10 in reducing the infarct area in normal mice [60]; on the other hand, IL-10-deficient mice have a larger lesion size after MCAO [61].
TGF-β is a pleiotropic growth factor with three isoforms binding the same receptors, specifically TGF-β1, TGF-β2, and TGF-β3, which is implicated in neuronal survival and brain tissue repair after brain injury.
Studies have shown [62] that TGF-β exerts a suppressive action against neutrophils and astrocytes, capable of producing inflammatory cytokines with a harmful effect during the inflammatory response to cerebral ischemic injury. After ischemic stroke, TGF-β, provided by activated M2 phenotype macrophages, is anti-inflammatory and contributes to healing after brain injury [63]. TGF-β reduces the potentially damaging effects associated with activated microglia by inhibiting microglial activation. Therefore, TGF-β appears to have a neuroprotective and anti-inflammatory role in ischemic stroke.
IL-4, a cytokine that can regulate various immune and inflammatory responses, plays a vital role during TH2 cell differentiation [64]. It can also polarise macrophages/microglia toward the anti-inflammatory M2 phenotype [65]. M2 macrophages/microglia express anti-inflammatory mediators and produce various neurotrophic factors that support the resolution of inflammation, mediated by increased trophic input, and the enhancement of both phagocytosis and proteolysis of dead, diseased cells/proteins. This ultimately promotes tissue restoration [66]. Therefore, IL-4 may have a neuroprotective action, encouraging tissue repair and potentially playing a therapeutic role.
3.3.2. Recruitment of Inflammatory Cells in Ischemic Brain Injury
Microglia
Microglia are part of the resident innate immune cells of the brain and represent 5–20 % of the glial population. This cytotype is activated after ischemic stroke, and morphological and phenotypical changes can be observed [67].
Resident microglia cells are among the first cells to respond to brain injury because they are immediately activated within a few minutes after the ischemic brain insult and tend to augment their count in the days after, reaching the climax ten days after transient focal cerebral ischemia [68]. On the other hand, blood-derived macrophages are generally recruited with an unavoidable delay. They commence appearing in the injured brain tissue on day 4, reaching peak numbers on day 7, and decrease after that [68].
After ischemic brain injury, microglia become activated, withdraw their ramifications, and assume an amoeboid morphology identical to activated macrophages. These changes contribute to the acquisition of macrophage-like skills, including phagocytosis, cytokine production, antigen presentation, and the production of matrix metalloproteinases that may damage the BBB, increasing its permeability. These events may encourage early infiltration of circulating leukocytes into the brain, which is consequently exposed to systemic inflammatory agents that worsen the ischemic damage. Activated microglia have two phenotypes: classically activated [M1] and alternatively activated [M2] [69]. The M1 microglia play a proinflammatory role and thus secrete cytokines and oxidative metabolites such as IL-1β, TNF, IL-6, and nitric oxide [70]; on the contrary, M2 microglia contribute to recovery after brain injury. This one also expresses anti-inflammatory mediators, such as IL-10 and IL-4, producing neurotrophic factors that prevent inflammation and promote recovery.
During ischemic stroke, the M2 phenotype is predominant in local microglia and newly recruited macrophages at earlier phases. The M1 phenotype increases gradually in peri-infarct areas. Thus, neurons in ischemic regions induce changes in the M2 phenotype in microglia and macrophages [70]. Reflecting on the opposite roles of microglia phenotypes in ischemic stroke, developing a therapeutic strategy to suppress the morphological modification and encourage the benefits of microglia is crucial.
Astrocytes
Like microglia, astrocytes are the resident cells of the brain with a fundamental role in maintaining homeostasis in the central nervous system. They actively modulate ion and water balance, release neurotrophic agents and scavenging transmitters released during synapses, shuttle metabolites, and waste products; astrocytes are also involved in BBB formation [71]. In normal physiological conditions, excessive glutamate is taken up by astrocytes from extracellular space and converted to glutamine for neuronal new utilization. Still, during brain injury, the extent of damage to the astrocytes might negatively influence their ability to glutamate uptake [71]. After ischemia, cytokines produced by neurons and glial cells conduct the astrocyte's reactivity hyperplasia. Astrocyte's proliferation ends in the expression of inflammatory agents such as monocyte chemotactic protein-1, IL-1β [72], glial fibrillary acidic protein [GFAP], nestin, and vimentin that can cause reactive gliosis and scar formation [73].
After a stroke, because of the failure of the Na+ and K+ pump, astrocytes enlarge; this fact provokes high intracerebral pressure and less cerebral perfusion [74]. Reactive astrocytes liberate matrix metalloproteinase two, leading to matrix protein degradation [75]. Reactive astrocytes also result in inhibitory conditions by inducing ephrin-A5 at the lesion center, interfering with axonal sprouting [76].
Neutrophils
Aside from microglia and blood-derived macrophages, neutrophils are one of the most significant leukocytes infiltrating the ischemic brain. They come out early, between thirty minutes and a few hours following ischemic injury, reaching a peak within the first three days and gradually declining over time [77]. Further, neutrophil infiltration rises on day 1, spikes on day 3, and begins to decrease but is detectable through days 7 and 15 after cerebral ischemia. Neutrophils express different endothelial adhesion molecules within 15 min post-ischemia [78]. Neutrophils have already encompassed cerebral vessels after six to 8 h of the stroke and immediately start infiltration [79]. The significance of neutrophils in the pathogenesis of ischemic brain damage is the effect of a complex role exerted by this immune cytotype through multiple possible mechanisms, such as the restriction of cerebral blood flow [CBF] by vessel plugging or releasing vasoconstrictive agents, excessive production of the hydrolytic enzyme, ROS and proinflammatory factors [80]. Furthermore, neutrophils are also involved in ischemic injury, as well as MMP-9, the matrix metalloproteinase most closely implicated in cerebral injury during ischemia. It is a protease able to degrade the basal lamina and encourage the disruption of BBB after cerebral parenchymal damage, causing oedema and hemorrhagic transformation of ischemic stroke [81].
Dendritic Cells (DCs)
DCs are the professional antigen-presenting cells (APCs) that express MHC II to promote T-cell activation [83]. However, DCs are usually not found in the brain parenchyma [83]. The role of DCs is well documented in animal models of ischemic stroke. After inducing transient MCAO in mice, DCs accumulate in the ischemic hemisphere at 24 h. When these DCs were further specified, the DCs in the core area of the infarct were from the periphery, whereas resident DCs mainly occupied the penumbra [84]. However, the role of DCs needs to be clearly described in humans. In acute ischemic stroke patients, significantly reduced numbers of circulating myeloid DC precursors (mDCPs) and plasmacytoid DC precursors (pDCPs) were observed, which recovered after a few days. This transient decrease of circulating DCPs may result from their recruitment from the blood into the ischemic brain. The patients with lower DCPs show bigger stroke lesion sizes and co-localization of myeloid DCs with T-cells around cerebral vessels in the infarcted area, indicating that antigen-mediated T-cell activation and long-lasting immune responses have occurred in the brain [85].
T lymphocytes
Unlike neutrophils, T lymphocytes play a crucial role in the later stages of ischemic brain injury. These leukocytes infiltrate the peripheral zone surrounding the lesion by day three, sparing the centre and often in proximity to blood vessels. Their numbers increase on the third day, peak on the seventh day, and decline during the next seven days [86]. The growing body of research is focused on unravelling the function of different T cell subtypes in the complex pathophysiology of ischemic stroke.
Three main groups of T cells can be distinguished based on their function: cytotoxic T cells [CD8+], helper T cells [Th, CD4+], and regulatory T cells [Tregs]. Each subtype has distinct markers on the cell surface and unique cytokine secretion profiles, providing valuable insights into T cells' diverse nature and function. Notably, studies on cerebral I/R in T-cell-deficient mice have shown that CD4+ and CD8+ T cells play a crucial role in brain inflammatory and thrombogenic responses, brain damage, and neurological symptomatology associated with experimental stroke by operating on a shared pathway [87].
Tregs, a significant CD4+ T-cell subtype, represent 10% of all CD4+ T-cells and express CD25 on the surface and the transcription factor Foxp3. Researchers are exploring the potential of modulating the activity of these cells due to their neuroprotective activity. Several studies [88,89] suggest that Treg lymphocytes and their major cytokine IL-10 represent the main neuroprotective factors from inflammatory postischemic brain damage. They appear to counteract the damaging production of inflammatory cytokines, such as IFN-γ and TNF-α, and to inhibit the activation and infiltration of other immune cytotypes, such as microglia and leukocytes, that promote the amplification of the brain's inflammatory reaction following ischemia. Moreover, immunodepletion of Treg, mediated by CD-25 specific antibody, was associated with more serious neurological symptomatology at day seven after MCAO-induced ischemia in mice [89] and exacerbated tissue loss. However, Treg cell therapy is already under study, including strategies such as isolation and purification, a period for ex vivo expansion, and the use of its anergic properties.
Populations of T lymphocytes also contain a small subset, γδ T cells, a unique and conserved population of lymphocytes that is about 5% of T cells. γδ T cells express a peerless T cell receptor [TCR], usually composed of α and β glycoprotein chains, made by one γ chain and one δ chain. Even this cytotype seems to be involved in the pathogenesis of ischemic stroke as shown by experimental studies where it was observed a significant decrease in infarct extent in TCR-γδ knockout mice; the same result was noticed in mice treated with TCR-γδ specific antibody [90]; this evidence may propose new prospective therapies to modulate the brain's inflammatory response following ischemia.
Inside the circulating CD4+ T cell population, a subset of cells lacking the costimulatory molecule CD28 on their surface. This unusual subset of helper cells is called CD4+CD28 null, a sparse population in most healthy subjects; indeed, it represents 0.1% - 2.5% of peripheral blood total CD4+ T-lymphocytes. Once activated, they are proinflammatory, producing large amounts of IFN-γ and TNF-α [91,92]. Furthermore, they have a harmful effect by expressing cytotoxic molecules usually produced by TCD8+ lymphocytes and NK cells, not by TCD4+ lymphocytes, such as granzyme A, granzyme B, and perforin. Further essential properties of these cells are represented by the fact that they are immune to apoptotic cell death, unresponsive to suppression by natural regulatory T cells, and also incapable of providing signals required for antibody production by B lymphocytes due to the lack of CD40 molecule expression caused by the absence of CD28 receptor [93]. Nowik et al. [94] assessed whether these cells might play a pathogenic function during an ischemic stroke with an atherosclerotic background, showing that CD4+CD28null lymphocytes are involved in processes able to increase the risk of acute ischemic stroke, rejecting that their appearance is just a consequence of stroke. The authors showed that the percentage of CD4+CD28null lymphocytes was not significantly different between the two groups [a group in the acute phase of ischemic stroke and another one with no story of ischemic stroke but with either hypertension or diabetes] but was significantly higher than in controls. This difference may suggest a possible connection between these cells and the other risk factors in accelerating plaque destabilization and atherosclerotic development. Moreover, this study also proved that the intensity of neurological damage, evaluated by the presence or increase of an ischemic lesion observed in a brain CT scan performed within 24 hours from the stroke onset, is not associated with the percentage of lymphocytes.
This finding contrasts with the hypothesis that an increase in these cells follows the cerebral ischemic insult, suggesting that their rise occurs before stroke onset. Another study conducted by Z.G. Nadareishvili et al. [95] investigated whether CD4+CD28null lymphocytes rose in patients with death after acute ischemic stroke or recurrent stroke. In this study, they evaluated the percentage of CD4+CD28null lymphocytes in the peripheral blood of one-hundred-six patients during the first 48 hours after symptoms began, conducting a follow-up up to one year after the onset of stroke. Authors found out that a higher percentage of these cells is correlated with a higher risk of stroke recurrence or death, supposing that an increased rate of CD4+CD28null lymphocytes could represent a biomarker for recurrent stroke and death. These results could be explained through a combination of multiple factors, such as the fact that CD4+CD28null cells may provoke first-ever and recurrent stroke damaging endothelium and brain tissue by inflammation, or probably this subset of T lymphocytes could be associated with defective adaptive immunity and, consequently, considered as a marker of poor general health.
Tuttolomondo et al. performed a study [96] to examine the peripheral frequency of CD28 null cells in subjects with acute ischemic stroke concerning the TOAST diagnostic subtype and to appraise their correlation with scores of clinical severity of the acute ischemic stroke and their predictive function in the diagnosis of acute ischemic stroke and in determining the diagnostic subtype. Ninety-eight consecutive subjects with a diagnosis of ischemic stroke were enrolled in the research; as controls were enrolled, 66 hospitalized patients without a diagnosis of acute ischemic stroke. Authors have recounted that subjects with acute ischemic stroke had a meaningfully higher peripheral frequency of CD4+ cells and CD28 null cells related to control subjects without acute ischemic stroke. Besides, subjects affected by cardioembolic stroke had a significantly higher peripheral rate of CD4+ cells and CD28 null cells than subjects with other TOAST subtypes. They also reported a relevant correlation between CD28 null cell outer percentage and some markers of stroke severity, such as the NIHSS and the SSS score, used to assess the type and degree of neurological deficit during the acute phase. The results of this study hypothesize that a higher percentage of peripheral CD4CD28 null cells might be associated with a more profound brain injury.
In humans, a dominant part of natural killer (NK) and T cell target recognition depends on the monitoring of human leukocyte antigen (HLA) class I molecules by killer immunoglobulin-like receptors (KIRs). Recent evidence [97] suggests the T cell implication in the acute complication of atherosclerosis, implying a possible function of KIR genetic background in regulating inflammatory cell entanglement during the acute cardiovascular event. Based on this, Tuttolomondo et al. performed research [98] to increase the knowledge about the immunological genetic background of acute ischemic stroke susceptibility in correlation with the frequency of the KIR genes and HLA alleles. Between November 2013 and February 2016, 116 consecutive patients with acute ischemic stroke were recruited. As healthy controls, 66 subjects were enrolled without acute ischemic stroke. Patients affected by acute ischemic stroke and control patients were genotyped for the presence of KIR genes, and of the three main KIR ligand groups, HLA-C1, HLA-C2, and HLA-Bw4, both HLA-B and HLA-A loci. Authors found in subjects with ischemic stroke a higher frequency of activating "proinflammatory" KIR genes, which could justify the immunoinflammatory activation during the acute phase of the stroke.
Indeed, the previous finding [96] of an increased percentage of CD28null cells in the peripheral blood of subjects with ischemic stroke may also be related to the higher prevalence of KIR gene activators such as 2S2 and 2DS4. Authors concluded that an increased degree of cytokine and cell-mediated (NK cells or T cell subset such as CD4+CD28null cells) inflammation could also depend on KIR genes responsible for expressing activator KIR receptors on these inflammatory cytotypes.
B-Cells
B-cells can also contribute to neuroprotection after the ischemic stroke, depending on IL-10 [178,179]. Unlike T-cells, B-cells did not show improvements against inflammation and brain injury when MCAO was induced in B-cell-deficient mice [32]. B-cell-deficient μMT/ mice showed larger infarcts and a higher mortality rate after 48 h of MCAO compared to wild-type mice. After 48 h, accumulation of leukocytes such as neutrophils, T-cells, microglia, and macrophages that produced IFN γ and TNF α was observed in the ischemic hemisphere of μMT/ mice. Adoptive transfer of IL-10 -/- B cells to μMT/ mice did not show a reduction in infarct volume after MCAO, showing that B-cells can restore the beneficial outcome, including inhibition of proinflammatory cytokine release from T-cells after stroke through IL-10 production [99].
Similarly, the adoptive transfer of naïve B-cells from wild-type mice to MCAO-induced mice generated smaller infarcts at 3 and 7 days. The same experiment with IL-10 deficient B-cells did not show similar protection. Using whole-brain volumetric serial two-photon tomography, diapedesis of B-cells was observed in distant areas from the injury area, such as the cerebral cortex, dentate gyrus, olfactory areas, and hypothalamus, where motor and cognitive abilities are governed, to promote long-term recovery. These observations show that B-cell depletion impacted post-stroke neurogenesis and cognitive functions [100]. Another study showed that B-cells showed delayed infiltration in the lesion around seven weeks after the onset. During that period, B-cells were closely associated with T-cells and CD11c-expressing cells, potentially DCs, for antigen transfer to induce isotype switching. Some CD19+ B cells co-expressed CD138, indicating these cells are plasma cells. After seven weeks of stroke in mice, an increased level of IgG (IgG1 and IgG2b), IgA, and IgM was observed in the lesion area. B-cell-deficient μMT -/- mice did not show IgG in the lesion area and cognitive deficit after stroke, even though the infarct size and T-cell infiltration were similar to wild-type mice. This indicates that the antibodies produced by B-cells mediate cognitive ability after stroke. When mice were treated with CD20 antibodies to ablate B-cells, cognitive deficits and IgG expression after stroke were also prevented [101].
However, Schuhmann et al. showed that depleting B-cells pharmacologically using anti-CD20 24 h before the MCAO in mice or using B-cell-deficient JHD -/- mice that do not have circulating B-cells did not affect the stroke lesion size, number of neutrophils and monocytes, and levels of TNF α and IL-1β at day 1 and 3 post-MCAO [102].
3.3.3. Neuroimmune Crosstalk in the Pathogenesis of Ischemic Stroke
A tight connection exists between the central nervous and immune systems through complex communication networks. The immune system monitors the brain functioning and reacts when cerebral homeostasis is altered because of injuries or diseases. Stroke promotes strong phlogosis involving the production of cytokines (p.e. TNF-α) by various cytotypes in the brain, including human neurons, activated glial and endothelial cells, with consequent blood-brain barrier detriment and infiltration of numerous types of leukocytes after a determined gap of time. These cytotypes can play a neuro-damaging or neuroprotective role, and the severity of the cerebral damage is strictly correlated with the balance between these two possible parts.
The relations between the various cytotypes of the immune system during the acute phase of ischemic stroke is an elaborate mechanism controlled by several factors. The ischemic stroke is a complex pathology, and essential factors, such as the ischemic lesion's harshness, the stroke's location, age, and comorbidities, can affect the interaction and the equilibrium between the cytotypes in the necrotic cerebral parenchyma. A
ll these factors can influence the local cytokine secretion, critical in modulating the interactions between the immune cells. For example, severe ischemic damage and a raised proinflammatory milieu with massive IL-6 and TNF-α release may cause an increased neutrophil recall in necrotic cerebral tissue.
Neutrophils are considered damaging since compelling evidence correlates this cytotype with blood-brain barrier breakdown and brain injury. An increased blood neutrophil count is also associated with larger infarct areas in subjects affected by cerebral ischemia [87]. Furthermore, the role of microglia and macrophages/monocytes during cerebral ischemia depends on the M1/M2 polarisation status. Specific cytokines in the local milieu (i.e., M1: IFN-γ, M2: IL-10, TGF-β) influence the polarization of one of the two phenotypes.
The prevalence of the M1 phenotype is correlated with more severe ischemic damage, hypoxia-inducible factor-1 (HIF-1) activation, and increased anaerobic glycolysis. Polarization of the microglia to the M1 phenotype and the resulting increased production of IL-23 facilitates the recruitment and stimulation of γδ T cells, a subset of unconventional innate T cells with a diverse T cell receptor that could play a detrimental function during acute ischemic stroke. Rising evidence reinforces that γδ T cells are pathogenic in testing cerebral ischemia/reperfusion models by secreting IL-17 and stimulating phlogosis [103].
On the other hand, the prevalence of a local anti-inflammatory milieu stimulated by the secretion of IL-10 and TGF-β facilitates the polarisation of the microglia to the anti-inflammatory M2 phenotype, which has a neuroprotective function. Furthermore, releasing these anti-inflammatory cytokines promotes the recruitment of regulatory lymphocytes that play immunomodulatory and immunosuppressor functions in the injured cerebral parenchyma. Indeed, consistent evidence supports the beneficial functions of Tregs in an experimental cerebral ischemia model [87]. While partially understood, the puzzle of the immune system's interaction during ischemic stroke remains a complex and intriguing study area. The known role of the cytokine environment is just the tip of the iceberg, as these interactions involve intercellular crosstalks by mechanisms yet to be fully comprehended. This complexity underscores the need for further research and piques our curiosity, driving us to delve deeper into understanding the immune response to stroke. The equilibrium between neuroprotective and neurodegenerative actions, a crucial aspect of this response, is influenced by several factors, reflecting the heterogeneous nature of the ischemic stroke.
4. Hemorrhagic Stroke Pathophysiology
A hemorrhagic stroke happens less frequently than an ischemic stroke but has a high morbidity and mortality rate (about 40%) [104]. Hemorrhagic stroke primarily occurs due to blood vessel rupture and extravasation of blood in either the brain parenchyma and ventricles, called intracerebral haemorrhage (ICH), or the subarachnoid space, called subarachnoid haemorrhage (SAH). ICH can also happen after the ischemic stroke and is associated with hematoma expansion, oedema, and intraventricular haemorrhage. The leading causes of bleeding include hypertension, the use of anticoagulants and thrombolytic agents, and head injury [105]. Other causes of hemorrhagic stroke and neurologic impairment and disability include cerebral aneurysms. Described as ballooning or pouching of the vessel wall, a fully developed and untreated aneurysm continues to grow and expand until it ruptures, leading to a hemorrhage.
4.1. Brain Injuries after Intracerebral Hemorrhage
The bleeding after ICH causes the disruption of the brain architecture within a few hours, and this damage due to the primary injury is nearly impossible to prevent [106]. During the first day of ICH, many patients suffer from hematoma expansion and increased hemorrhagic volume, which are the critical factors determining the clinical outcome post-ICH [107]. The secondary injury results from intra-parenchymal hematoma, which causes multiple detrimental events, leading to various neurological deficits [108]. Various blood components activate cytotoxic, excitotoxic, oxidative, and inflammatory pathways [109]. The significant contributors to secondary brain injury post-ICH are thrombin, iron, and hemoglobin from the hematoma [110].
4.2. Oxidative Stress and Hemorrhagic Stroke
Oxidative stress plays a predominant role in secondary brain injury after ICH. Oxidative stress is described as the disruption of the balance between the production of ROS and the capability of scavenging the former by the physiological antioxidant machinery of cells [111,112]. Superoxide radicals (O2−), hydrogen peroxide (H2O2), and hydroxyl radicals (OH) constitute the primary forms of ROS [112]. Oxidative stress contributes to the growth and progression of perihematomal edema in brain hemorrhage patients [113]. ROS damages the central nervous system through cell death and structural damage, especially blood-brain barrier disruption. Apoptosis releases excess free radicals, which induces lipid and nucleic acid peroxidation through various pathways [114]. Furthermore, H2O2 can alter mitochondrial function and upregulate the expression of pro-apoptotic genes, ultimately inducing apoptosis following ICH [115].
4.3. Neuroinflammation in Hemorrhagic Stroke
As already mentioned above, intraparenchymal blood activates various inflammatory pathways. Recently, it has been shown that these immune pathways are very similar in ischemic and hemorrhagic stroke [89]. Hemorrhagic stroke can initiate inflammatory responses, induce cerebral edema, and disrupt BBB with neurotoxic materials such as thrombin, fibrin, and erythrocytes [116]. The accumulation of erythrocytes can initiate inflammation by activating Toll-like receptor 4 (TLR4) in the microglia to release TNF α and IL-1β [117]. Thus, local microglia and astrocytes are the first cells to respond to the ICH. Once activated, they promote the influx of circulating macrophages. Subsequently, various inflammatory cytokines are upregulated, further activating lymphocytes and ultimately deteriorating ICH-induced injury by disrupting BBB [118]. NLR family, a pyrin domain containing three (NLRP3) inflammasomes, induces inflammation via caspase-1 and interleukin (IL)-1β post-ICH [87]. Similarly, TLR4 activates ICH's inflammatory pathway and neuronal death (Fei et al., 2019). Another critical player in the ICH inflammatory processes is the damage-associated molecular patterns (DAMPs), such as high mobility group box 1 (HMGB1), a non-histone nuclear protein that plays an essential proinflammatory action once released into the extracellular space [114]. The secreted HMGB1 interacts with TLR-2 and TLR-4 to trigger inflammation. HMGB1 secreted by monocytes and macrophages was observed to interact with TLR2, TLR4, and TLR5 to upregulate nuclear factor kappa-light-chain-enhancer of activated B-cells (NF-kB). In studies involving animal models of ICH, HMGB1 levels increased in peri-hematoma regions post-ICH [119]. Furthermore, an increase in the serum levels of HMGB1 is observed in patients with ICH, which significantly correlates with stroke severity [120]. Thus, the expression level of HMGB1 in plasma and CSF can be related to SAH outcomes. As the inflammation proceeds, leukocytes are recruited via adhesion molecules to secrete diverse chemokines and cytokines to induce endothelial cell death, recruit more immune cells, and damage the tight junctions in the BBB [116]. Microglia and macrophages phagocytose erythrocytes and degraded debris after the injury [121]. T-cells can be detrimental, but Tregs can be neuroprotective in a rat model of hemorrhagic stroke, as mentioned for ischemic stroke [122].
5. New Epigenetic Players in Stroke Pathogenesis: From Non-Coding RNAs to Exosomal Non-Coding RNAs
MiRNAs are essential in modulationg gene expression in all types of cells. Binding to messenger RNA (mRNA) can cause cleavage or inhibit translation into protein. The process of miRNA biosynthesis is a complex biological process, which involves the transcription of primary miRNA (pri-miRNA) followed by processing with the nuclear endonuclease Drosha to create pre-miRNA. Once transported to the cytoplasm, the enzyme Dicer processes the pre-miRNA to form a short double-stranded RNA sequence. One of the chains is degraded, leaving behind the mature miRNA.
The mature miRNA then binds to Ago2, forming RNA-induced gene silencing complexes (RISC) that enable it to interact with mRNA targets to cause degradation or translational repression. Notably, a single miRNA can regulate the expression of hundreds of mRNAs, and vice versa, an mRNA can present various sequences to interact with multiple miRNAs [123].
An alternative biosynthetic pathway for miRNAs exists, where some miRNAs are produced from specific introns called "mirtrons." Mirtrons are of the size required to synthesize pre-miRNAs, eliminating the need for Drosha endonuclease directly. This pathway adds another layer of complexity to the miRNA biogenesis process. After pre-miRNAs are synthesized, they are transported to the cytoplasm, where they undergo processing by the Dicer enzyme. The Dicer enzyme cuts the pre-miRNAs to produce short double-stranded RNA sequences that are 19-25 nucleotides long. These double-stranded RNA molecules are then loaded into the RNA-induced silencing complex (RISC), which leads the RISC to the target messenger RNAs (mRNAs), influencing the mRNA translation.
To conclude, the biosynthetic pathway of miRNAs is a complex multi-step mechanism involving multiple steps and pathways. The alternative pathway through mirtrons adds another layer of complexity to the process and highlights the diversity of miRNA biogenesis [124].
5.1. Mechanism of Action
Typically, the miRNA binds to a complementary sequence in the 3'UTR region of the target mRNA. However, new findings indicate that binding can occur in the 5'UTR region. The "seed region," spanning nucleotides 2 to 8 of a mature miRNA, is crucial for recognizing the target mRNA [125].
It is crucial to note that a miRNA can regulate the expression of multiple mRNAs. Similarly, an mRNA can have several sequences interacting with different miRNAs simultaneously. Additionally, it is essential to consider that miRNAs are not immediately degraded after interacting with a single target. Therefore, they can demodulate other mRNAs as well [126].
Identifying miRNA target genes is a complex process that requires careful consideration. Several elements can affect the accuracy of the identification process, such as the accessibility of the target sequence and the secondary structure. Despite these challenges, researchers continue to work towards identifying miRNA targets with greater precision than the mRNA [127].
The role of different miRNAs in ischemic stroke’s pathogenesis is summarized in Table 2.
5.2. Circulating miRNA as a Biomarker
Given the current situation, it is critical to find an examination that is both highly sensitive and effective while also being budget-friendly and non-intrusive to identify and predict the prognosis of haemorrhagic or ischemic stroke and its subtypes. Biomarkers can shed light on these diseases' underlying mechanisms. They may be proteins, nucleic acids, or metabolites and can provide valuable information on a given disease's risk, diagnosis, severity, and prognosis through their quantification [128].
Physicians use brain imaging to determine the most appropriate treatment for stroke patients. However, this method has certain limitations, such as high costs, contraindications, and the need for expert radiologists to interpret the results. Additionally, access to brain imaging services is often limited. As a result, researchers are working hard to develop more reliable tools to help healthcare providers manage stroke patients effectively. Unlike other common diseases like diabetes or acute myocardial infarction, there is no specific or sensitive biomarker available to aid in the diagnosis and treatment of stroke. This fact is mainly due to the complexity and heterogeneity of the disease, as well as the impact of the blood-brain barrier on the circulation of biomarkers [129].
5.3. Exosomes Biogenesis
Extracellular vesicles (EVs) are a new promising study area. These small or large lipid bilayer membrane particles are released from all living cells into the extracellular environment. Exosomes, the smallest type of EVs, have an average diameter ranging between 30 and 150 nm and have been the most studied. It has been discovered that EVs play an active role in different biological mechanisms, including intracellular homeostasis and intercellular communication. These vesicles may hold the key to finding a reliable biomarker for stroke diagnosis and treatment. [130,131].
Exosomes are derived from endosomal structures that originate through endocytosis of invaginated endosomes from the plasma membrane. [132]
Those tiny vesicles are released from cells and carry necessary molecular signals to other cells, which can affect the recipient cells in various ways. These vesicles can be loaded with specific cargo, and their release can be regulated by environmental and cellular cues such as stress, inflammation, or cell-cycle events. Exosomes travel through the circulation system to reach their target cells, where they can bind to the cell surfaces and get absorbed through specific mechanisms. Recent studies suggest that the uptake of exosomes can be regulated and that certain classes of exosomes may contain specific targeting molecules that lead to some degree of specificity towards specific recipient tissues. [133,134].
Exosomes can also undergo transcytosis, allowing them to cross the blood-brain barrier gaining access to the central nervous system (CNS). [135].
5.4. Exosomes in Brain Injury
Cells release exosomes into biofluids such as cerebrospinal fluid (CSF). These vesicles are enriched with tetra-spanin proteins (CD81 & CD63), Alix (a regulator of the endosomal trafficking pathway), and chaperone protein HSP70. However, the volume of exosomes varies according to the cell origin and the pathological and physiological conditions.
Various studies have shown that exosomes carry cargoes of proteins, RNAs, and lipids such as miRNAs and mRNAs. Through centrifugation and other procedures, it is possible to isolate exosomes from bio-fluids and the supernatant of cells that have been cultured in an exosome-free medium. Proteomic and RNA analyses have shown that exosomes are a rich source of biomolecules with potential applications in diagnostics and therapeutics. [136,137].
Various cell types can release exosomes in the CNS during brain injury and may play a significant role in post-stroke brain remodelling. [138].
The information of neural death in ischaemic brain is communicated through exosomes to other cells in the brain. Because of this, miRNAs in exosomes play a crucial role in inter-cellular communication [139].
Astrocytes release exosomes under both physiological and pathological conditions. These exosomes contain various biological molecules such as DNA, miRNA, and proteins, but their composition varies depending on the stimuli. Under normal conditions, astrocyte-derived exosomes contain neuroprotective and neurotrophic elements and molecules that help in neurite outgrowth, synaptic transmission, and neuronal survival. Numerous studies suggest that astrocytes are activated during cerebral ischemia and secrete exosomes to protect the central nervous system. Pei et al. demonstrated that astrocyte-derived exosomes inhibit autophagy and enhance neuronal viability in an ischemic stroke model [140].
Moreover, astrocyte-derived exosomes also contain miRNAs, such as miR-34c and miR-361, that protect neurons and prevent nerve damage after cerebral ischemia [141,142].
The detection of stroke biomarkers in the bloodstream is challenging because of the blood-brain barrier (BBB), which acts as an interface between the central nervous system and the peripheral circulation. As a result, biomarkers from cerebrospinal fluid (CSF) have difficulty transitioning into the bloodstream. However, exosomes have shown potential to cross the BBB, making them a promising candidate for being measured in the blood. Some authors have recently explored the use of exosomes as stroke biomarkers. Patients who have had an ischemic stroke show different cargo exosomes than those of healthy controls. miRNAs, the most investigated exosome content, could provide valuable information.
5.5. miRNA as a Biomarker in Clinical Practice
The importance of having a tool for a rapid diagnosis of stroke represents a medical need since the diagnosis of stroke is remarkably time-consuming, being mainly dependent upon examination by a clinical care provider and using various neuroimaging techniques. The functions of miRNA were initially identified in original tissue samples. Most studies focused on the correlation between miRNAs in brain tissues and underlying mechanisms of stroke, including cellular apoptosis, neuroinflammation, and oxidative stress. [143,144,145,146].
Tan et al. conducted a study comparing miRNAs' expression in healthy individuals and those diagnosed with cerebral ischemia. They used microarray analysis of blood specimens and selective TaqMan Quantitative polymerase chain reaction (qPCR) of miRNAs to evaluate miRNA expression. The study found that miR-25, miR-125b-2, miR-125b-627, miR-125b-27a, miR-125b-488, and miR-145 are significant biomarkers for the diagnosis and treatment of stroke. These miRNAs may be relevant in developing effective treatments for cerebral ischemia [147].
In their study, Wang et al. analyzed circulating miRNAs in plasma from patients diagnosed with acute stroke and non-stroke diseases. They aimed to determine whether such miRNAs could be used as biomarkers for diagnosing and treating stroke [148].
Several miRNAs have been identified as potential biomarkers for diagnosing certain conditions. These include miR-106b-5p, miR-126, miR-30a, miR-4306, let-7b27, and let-7b28. Studies have shown that high levels of miR-130a in serum contribute to serious brain oedema and poor prognosis after acute intracerebral haemorrhage (ICH) [149].
5.6. miRNA as a Biomarker in Acute Ischemic Stroke
Among the several miRNAs considered, miR-101a-3p emerges as the most promising ischemic stroke biomarker for further future evaluation. The levels of expression of miR-101a- 3p increase at early time intervals, 0.5 and 3 h, after permanent ischemia induction and at a later time interval (nine hours) after transient ischemia. No changes were detected after hemorrhagic stroke [152].
According to research by Liu et al., miRNA can play both beneficial and harmful roles. Their research discovered that suppressing the expression of miR-155 can enhance the proliferation, migration, and tube formation ability of human brain micro-vessel endothelial cells by reducing cellular apoptosis and reactive oxygen species (ROS) production [153].
Researchers found that inhibiting miR-27b could alleviate neurological deficits by suppressing neuroinflammation and reducing cell death. Additionally, miR-155 and miR-124 were reported to play a role in macrophage polarization [154].
miR-134 can facilitate the remodelling of neuronal structures by inhibiting the translation of Limk1-mRNA, a protein kinase that affects dendritic spine development [155].
Some miRNA types can exert multiple functions, such as miR-181a, which can improve cellular survival by suppressing inflammation responses in monocytes and macrophages [156].
Moon's report demonstrated that inhibiting miR-181a reduced neuronal apoptosis induced by forebrain ischemia [157].
5.7. miRNA in Hemorrhagic Stroke
Studies have shown that specific miRNAs (microRNAs) can be used as biomarkers to differentiate between subarachnoid haemorrhage (SAH) patients with delayed or non-delayed cerebral infarction and controls. Similarly, a specific panel of miRNAs in cerebrospinal fluid can distinguish SAH patients from controls and differentiate between SAH patients with vasospasm and those without it. In addition, clinical studies have identified that serum miR-130a or a panel of blood-specific miRNAs can differentiate between intracerebral haemorrhage (ICH) patients and controls. Moreover, plasma miR-29c and miR-122 can help distinguish between the hematoma and non-hematoma enlargement groups of ICH patients [145]
The miRNA levels can be increased or decreased to treat or prevent hemorrhagic stroke. In an experimental model of ICH, injecting a miR-130a inhibitor into the right lateral ventricle before ICH induction in male rats reduces the expression of miR-130a, decreases brain oedema, and improves neurological function [150].
A miR-367 mimic can increase miR-367 levels, inhibit the inflammatory response, and decrease brain oedema and neurological injury in mice with ICH [158].
Overexpression of miR-223 through a miR-223 mimic injection in ICH mice reduces brain edema and improves neurological functions while inhibiting the inflammatory response [159].
5.8. miRNA as Target Therapy: The of Role miRNA Mimics
MiRNA-based therapeutics hold significant potential in combating various diseases and can be broadly categorized into two types - miRNA mimics and miRNA inhibitors.
MiRNA mimics can help compensate for the loss of specific miRNAs that are downregulated and correlate with disease progression. In contrast, miRNA inhibitors (or antimiRs) can suppress the over-expressed miRNAs that contribute to disease pathogenesis. To imitate miRNA precursors, miRNA mimics are designed as synthetic short double-stranded oligonucleotides. Once introduced into cells, these oligonucleotides can be recognized and processed by miRNA biogenesis machinery. To ensure effectiveness, miRNA mimics are constructed with one "guide strand" and one fully or partially complementary "passenger strand," which play a crucial role in regulating gene expression [160,161].
5.9. miRNA as Target Therapy: The Role of AntagomiRNAs
Numerous studies have proved the therapeutic benefits of intravenous infusion of antagomiR in cardiovascular pathologies. A promising new technique called "miRNA masking" has been developed to improve the specificity of AMO (anti-MIRNA-oligonucleotides). This technique selectively inhibits the binding of a particular miRNA to a target mRNA without interfering with other targets [162].
Although AMOs have been found to be dose-dependent and require high doses for effective inhibition, a new strategy called "miRNA sponges" promises to overcome this limitation. These RNA sequences present multiple binding sites for miRNAs and can be designed to interact with all members of a miRNA cluster, achieving inhibition of an entire functional class. These observations provide hope for a possible use of these molecules in treating hypercholesterolemia [163,164,165].
5.10. The Role of Angiogenesis as Potential Target
Angiogenesis is a natural process in which new blood vessels are formed from existing ones. While the growth of blood vessels is tightly regulated in adult brains under normal conditions, research from both human and animal stroke models indicates that neovascularization occurs in the adult brain after cerebral ischemia [166,167].
Studies have shown that the develop of new blood vessels, known as post-ischemic angiogenesis, is critical in restoring blood flow and neuronal metabolism following a stroke. This cumulative evidence highlights the importance of angiogenesis as a potential therapeutic target in stroke recovery [168].
For instance, when a miR-107 mimic was used, it lessened the ischemic brain infarction and increased the number of capillaries in the penumbral area, possibly boosting the endothelial VEGF165/164 levels, which promoted angiogenesis [169].
Post-stroke angiogenesis contributes to improved neurological recovery by promoting tissue repair, vascular remodelling, and plasticity in both stroke patients and animal stroke models (cit1 e cit2) [170,171].
Another findings by Sun P. et al. show that several miRNAs play essential roles in regulating post-stroke angiogenesis, including miR-107 and miR-150. They demonstrated that deleting the miR-15a/16-1 cluster in the endothelium had a potent pro-angiogenic effect following reperfusion of the ischemic brain. This fact led to significant improvements in long-term neurological recovery after ischemic stroke. This study confirmed the existence of vascular remodelling during the prolonged recovery phase and showed that endothelium-targeted deletion of the miR-15a/16-1 cluster promoted revascularization and angiogenesis in the peri-infarct brain areas following ischemic stroke. Therefore, endothelial miR-15a/16-1 is a promising pharmacological target for improving post-stroke neurological recovery by enhancing cerebral angiogenesis [172].
5.11. The Role of Synapitc Plasticity as Possible Target
The results of a recent study by Xin, Wang, and their colleagues have uncovered an exciting new development in stroke research. Their research shows that miR-17-92 enriched exosomes can promote neural plasticity and improve recovery in rats post-transient middle cerebral artery occlusion. The downstream effector proteins in these exosomes can promote neurite remodelling, axonal growth, and cell proliferation in primary cortical neurons, leading to enhanced functional recovery. Moreover, when miR-133b enriched exosomes were administered to a stroke model, further exosomes were released from glial cells and astrocytes, providing trophic support to axons and ultimately leading to increased neuronal plasticity and stress protection. The study highlights the significant therapeutic benefits of neuroprotective miRNA-enriched exosomes, which can improve neuroprotection and recovery under oxidative stress conditions. These findings are truly remarkable and offer hope for future stroke treatments [173,174].
5.12. The Role of Post Stroke Inflammation
In a groundbreaking study by Xu et al. in 2017, a promising mechanism to combat post-stroke inflammation was discovered. The researchers identified the suppression of Toll-like receptor 4 (TLR4) by miR-1906, which triggers the proinflammatory cascade in the brain after a stroke. By administering miRNA-1906 agomir to rats, the team found that TLR4 protein levels were significantly reduced despite the presence of TLR4 mRNA, indicating that miRNA-1906 suppresses TLR4 translation rather than TLR4 mRNA degradation. Consequently, the downstream signalling pathways that activate proinflammatory genes are inhibited, reducing post-stroke inflammation, infarct volume, and peri-infarct tissue damage. Xu et al.'s findings offer a promising avenue for developing targeted therapies for post-stroke inflammation, which could improve patient outcomes and quality of life [175].
5.13. miRNA Involved in Neuroprotection
The highly expressed miR-204-5p (2075-fold), miR-125b- 5p (108-fold), miR-9-5p (299-fold), miR-338-3p (146-fold), miR-187-3p (21-fold), and miR-9-3p (42-fold) in CSF were associated with the regulation of matrix metalloproteinase-9 (MMP-9), interleukin (IL)-1b, IL-6, occludin, and selectin E, among others. miRNA profiles in CSF were physiologically contiguous with profiles in brain extracellular fluid. However, only a small number of studies have been reviewed. This area of stroke research would benefit from more studies focussing on the function of miRNAs in CSF of patients with stroke [176].
miRNA 21, miR-99a, and miR-497 have been found to reduce ischemic volume and protect neuronal cells from apoptosis, thus improving neurological functions in rats and in vitro models of ischemic stroke [177,178].
Except for the functions mentioned above, over-expression of miR-424 and miR-let-7c-5p could also suppress the activation of microglia in cerebral ischemia [179].
Overelease of miR-132, miR-126, miR-103, and miR-367 can lead to a reduction in neurobehavioral and neuropathological changes by improving BBB integrity, suppressing neuroinflammation, and reducing neuronal apoptosis in hemorrhagic stroke [158,180,181,182].
Additionally, excess miR-210 could promote angiogenesis and neurogenesis in mouse brains and help repair the injured brain tissues [183].
5.14. Future Perspectives
In the future, pre-miRNAs and anti-sense RNAs can be synthesized and administered intravenously to target the abnormal DNA translational control, which can either increase or decrease miRNA levels. However, extensive research is needed to establish the specificity and sensitivity of such treatments and potential side effects and uptake/elimination mechanisms. Additionally, using engineered exosomes and microvesicles that can deliver therapeutic miRNAs to the site of brain injury could open up new avenues for stroke therapy. Although some studies have shown promising results, proof-of-concept studies are still required to determine whether such therapies can improve stroke outcomes.
6. Molecular Mechanisms and Therapies in Stroke: Update on Recent Developments
In the last years, researchers’ aim was a better comprehension of the molecular mechanisms underlying ischemic stroke and the consequent cerebral damage. Understanding different pathways could lead to developing new therapies apart from the classic thrombolysis, improving the chances of a better neurological outcome.
6.1. Inflammation
At the beginning of the ischemic process, inflammation contributes to the disruption of the blood-brain barrier (BBB), causing oedema; on the other hand, at a later time, it contributes to fixing the damaged cerebral tissue [184].
Following an ischemic stroke, microglia become M1 or M2-activated macrophages. M2 ones have an anti-inflammatory function promoting the production factors such as insulin-like growth factor 1 (IGF-1) and brain-derived neurotrophic factor (BDNF). M1 macrophages have a crucial function in the release of multiple factors either involved in the development of further inflammation or toxic for the neuronal cells, such as NO, ROS, IL-6, TNF-α and IL-1B [185].
IL-1, especially IL-1β, promotes a further production of IL-6 and TNF-α, triggering the other cytotypes involved in inflammation; all this process leads to further injury to the cerebral tissue compromising the neurological outcome [186].
In addition, inflammation promotes the overexpression of adhesion molecules, such as E-selectin and ICAM-1, that let dendritic cells, astrocytes and lymphocytes reach the cerebral tissue affected by ischaemia [187]; furthermore, this amount of cells interferes with blood circulation [188].
During phlogosis, it is possible to observe an increased production of matrix-metalloproteases (MMPs) capable of harming the BBB with consequent oedema and cerebral injury; their expression is, in fact, correlated to the entity of cerebral damage and the successive chance of bleeding [189].
Clausen et al. observed that etanercept, a monoclonal antibody against TNF-α, improved the neurological outcome in mice affected by induced ischemic stroke, not the extent of the infarcted area yet [190].
cTfRMAb-TNFR, capable of moving TNFR crosswise the BBB, was able to improve the neurological injury and decrease the size of the ischemic cerebral portion [191].
6.2. Excitotoxicity
Ischemia leads to a lack of oxygen causing an increased production of glutamate and the consequent activation of Na+/Ca2+ channels associated with N-methyl-D-aspartate receptors (NMDARs) [196] provoking excessive burden of Ca 2+ in cytosol and mitochondria [197]. This process is called excitotoxicity and, by kainite receptors, NMDARs and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid [198], promotes the release of free radicals and the activation of lipases, kinases and proteases [199] resulting in apoptosis [200].
On the other hand, it seems that peculiar NMDARs in the synaptic space containing the GluN2A subunit have a protective role in reducing oxidative stress and, consequently, excitotoxicity [201].
The Tat-NR2B9c peptide is able to modulate the NMDARs with a potential therapeutic role in ischemic stroke. In fact, multiple studies in animal models showed that this molecule decreased the ischemic cerebral area with a better neurological outcome [202,203,204]; ZL006 and IC87201 are two little molecules capable of modulating NMDARs and showed similar results in ischemic stroke [205,206].
Hong et al. studied the possible use of Neu2000, an NMDARs antagonist, demonstrating a beneficial role in ischemic stroke [207].
Unfortunately, all these molecules which antagonise the NMDARs pathway do not have a clinical application because of the normal functions of these receptors and, consequently, all the side effects, such as cardiovascular and neuropsychiatric diseases [208].
6.3. BBB Alterations and Matrix Metalloproteases (MMPs
In animal and human stroke models, an increased release of MMPs was observed [208], promoting BBB disruption, cerebral oedema formation, and further inflammation [210]. Barr et al. reported that MMP9 has a crucial role in damaging the BBB, increasing its permeability [211]; similarly, Chelluboina et al. observed increased levels of MMP12 and its silencing was correlated with a better neurological prognosis [212]. The role of MMP2 in ischemic stroke is still unclear, but it seems to interact with VEGF to repair the BBB [213].
Since their crucial role in BBB disruption, MMPs’ repression may have a role in ischemic stroke therapy. Hydrogen sulfide and the triggering of the vagus nerve seem capable of inhibiting MMP9, protecting the BBB [214,215]; in addition, even the administration of norcantharidin suppressed this metalloproteinase protecting the BBB [216]. Michalski et al. observed a correlation between high-pressure oxygen and BBB integrity, possibly modulating MMP2 [217].
6.4. Inflammasome
Inflammasomes are formed by a group of proteins [218] and take part in inflammation during ischemic stroke causing apoptosis [219]. NLRP3 inflammasome seems to participate in cerebral inflammation modulating microglia [220] by the NF-κB pathway [221]; on this basis, NLRP3 inflammasome may represent a therapeutic target in cerebral ischemia decreasing inflammation [222]. Wang et al. reported that genistein, a phytoestrogen, can restrain the formation of this inflammasome reducing the neurological deficit in mice models [223]; similarly, MCC950, capable of blocking NLRP3, reduced caspase-3-mediated cell death and decreased the cerebral ischemic areas [224].
6.5. Chemokines
Chemokines are little proteins with signalling functions crucial during neuroinflammation in cerebral ischemia. These proteins, such as microglial response factor-1, cytokine-induced neutrophil chemoattractant and fractalkine, are stimulated by all the proinflammatory cytokines [225]. During ischemic stroke, Chemokine-chemokine ligand 2 (CCL2) and its receptor, CCR2, are overexpressed and promote the infiltration of immune cells in the ischemic area leading to further inflammation [226,227,228]. Takami et al., reported a detrimental effect of the administration of CCL3 during ischemic stroke in animal models [229].
6.6. Hypoxia-Inducible Factor (HIF)
HIF is a protein produced during ischemia able to stimulate transcription and is formed by HIF-1α and HIF-1β [232]; HIF-1 promotes the switch to anaerobic cellular metabolism, supporting cells during hypoxia [233]. Ziello et al. reported that ROS stimulate the release of HIF-1α [234]; consequently, HIF-1 needs an oxidative environment to work. On the contrary, several studies showed that HIF-1α could preserve brain cells during ischemia: for example, Kim et al. observed that decreasing HIF-1α in animal ischemic models resulted in a worse neurological dysfunction [235]; in addition, HIF-1α seems to preserve the neuron’s integrity during oxidative stress [236].
6.7. Cell-Based Therapies
Stem cells seem a promising therapeutic option since they participate in neuroplasticity [237]. Neural stem cells (NSC) can turn into different cerebral cytotypes, such as neurons, glial cells, astrocytes and oligodendrocytes [238] and can develop new cells over time [239]; on this basis, Baker et al. reported an improved neurological outcome in animal ischemic stroke models after NSC therapy [240]. Furthermore, Zhang et al. reported the possibility of combination therapy with NSC, brain-derived neurotrophic factors and vascular endothelial growth factors [241].
6.8. Drug-Based Therapies
At the moment, tissue Plasminogen Activator (tPA) is the only therapy approved for ischemic stroke, but it has a short therapy window, several contraindications and does not play a role in neuroplasticity [246]; due to this, only a tiny percentage of patients can benefit from this treatment.
Fibrinogen-depleting agents derive from viper venom and could represent a possible alternative to tPA [247]; these molecules can improve blood perfusion in ischemic areas by clearing blood clots [248].
Gamma-aminobutyric receptor agonists (GABA) agonists, such as diazepam and midazolam, seem to reduce neurological symptoms and protect brain cells from inflammation during ischaemia [249].
Inhibiting calcium and sodium channels could find a role in the treatment of ischemic stroke; amlodipine, a Ca2+ channel antagonist, decreased the chance of ischemic stroke in patients affected by essential hypertension by 13.5% [250], in addition, Frank et al. reported that blocking sodium channels interferes with cellular depolarization, making brain cells less excitable during ischemic stroke [247].
As said above, excitotoxicity is mainly mediated by glutamate receptors and has a detrimental effect on cerebral cells [251]; since these receptors are ubiquitous, researchers focus on blocking the NMDAR pathways rather than the receptors, causing fewer side effects [252].
DM199 is a synthetic form of human tissue kallikrein-1 (KLK) that is able to improve blood circulation and promote the development of new vessels without the contraindications and side effects of tPA or thrombectomy; in China, this type of treatment is already used for acute cerebral ischaemia (Kailikang®) after several studies and a large trial.
To conclude, cofilin inhibitors could be a potential treatment in ICH; cofilin is a protein with a crucial role in cytoskeleton formation and has a pathological function in different neurological pathologies [253] promoting inflammation and cellular injury. Alaqel et al. showed that inhibiting cofilin in ICH animal model improved the neurological outcome [254].
7. Conclusions
It is widely accepted that the consequences of a stroke are rarely contained within the CNS and are instead characterized by both the influx and efflux of cytokines, cells, and fluid that can significantly influence stroke outcomes. Neuroinflammation, oxidative stress, and excitotoxicity are among the protagonists of the interactions within this complex mechanism. In addition, miRNAs play crucial roles in stroke pathogenesis. Several studies reported that these molecules have critical roles in different stages of stroke and have diagnostic, prognostic and therapeutic potential in brain diseases.
Moreover, exosomes have been regarded as the new materials with prominent inter-cellular roles in mediating neuro-restorative events following strokes and neural injuries. It is, therefore, clear how the detailed understanding of these molecules, which are the basis of the pathogenesis of stroke, is necessary for new therapeutic perspectives, which are crucial considering that classic treatments such as tPA only have a limited therapeutic window. Regarding the limitations of the tPA, DM199 seems to be a promising therapy that is much more versatile for patients, as it significantly increases the recovery rate in affected individuals. Other targets for treatment have also been identified, such as GABA receptors, glutamate receptors, sodium and calcium channel blockers, and fibrinogen-depleting agents, all of which, when altered or targeted, reduce symptoms and improve functional outcomes. Stem cell research focuses mainly on post-stroke recovery, including neural stem cells, HSC stem cells, and HUBC-MSC stem cells. Overall, stroke therapeutic research is on the path to developing safe and efficient therapies for stroke, researchers have performed a tremendous amount of work in unravelling potential molecular targets and generated avenues for stroke researchers to try these agents in preclinical models of different types of strokes.
References
- McKay. Judith; World Health Organization; Mensah, George A; Mendis, Shanthi; Greenlund, Kurt the atlas of heart disease and stroke 2004.
- Mozaffarian D, Benjamin EJ, Go AS, et al. Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation 2016; 133(4): E38-e360.
- Lynch JR, Blessing R, White WD, Grocott HP, Newman MF, Laskowitz DT. Novel diagnostic test for acute stroke. Stroke 2004; 35(1): 57-63.
- Lin HJ, Wolf PA, Kelly-Hayes M, Beiser AS, Kase CS, Benjamin EJ, D’Agostino RB. Stroke severity in atrial fibrillation. The Framingham Study. Stroke. 1996;27:1760–1764.
- Bogiatzi C, Hackam DG, McLeod AI, Spence JD. Secular trends in ischemic stroke subtypes and stroke risk factors. Stroke. 2014;45:3208–3213. [CrossRef]
- Candeias SM, Gaipl US. The immune system in cancer prevention, development and therapy. Anticancer Agents Med Chem 2016;16:101-7.
- Odendall C, Kagan JC. Activation and pathogenic manipulation of the sensors of the innate immune system. 2: Microbes Infect2017;19, 2017.
- Amantea D, Micieli G, Tassorelli C, Cuartero MI, Ballesteros I, Certo M, Moro MA, Lizasoain I, Bagetta G. Rational modulation of the innate immune system for neuroprotection in ischemic stroke. 1: Front Neurosci 2015;9, 2015.
- den Haan JMM, Arens R, van Zelm MC. The activation of the adaptive immune system: cross-talk between antigen-presenting cells, T cells and B cells. Immunology Letters 2014;162:103-12.
- Kanai M, Kubo H, Kitamura Y, Izawa KP, Ono K, Ando H, Nozoe M, Mase K, Shimada S. Difference in autonomic nervous activity in different subtypes of non-cardioembolic ischemic stroke. 1: Int J Cardiol 2015;201, 2015.
- Moskowitz MA, Lo EH, Iadecola C. The science of stroke: mechanisms in search of treatments. Neuron 2010;67:181-98.
- Lucas DR, Newhouse JP. The toxic effect of sodium L-glutamate on the inner layers of the retina. AMA Arch Ophthalmol 1957;58:193-201.
- Campbell BCV, Khatri P, 2020. Stroke. Lancet Lond. Engl. 396, 129–142. 10. 1016.
- Kuriakose D, Xiao Z, 2020. Pathophysiology and Treatment of Stroke: Present Status and Future Perspectives. Int. J. Mol. Sci. 21, 7609. 10. 3: [PubMed, 3390.
- Ramel D, et al., 2019. Immune and Smooth Muscle Cells Interactions in Atherosclerosis: How to Target a Breaking Bad Dialogue? Front. Pharmacol. 10, 1276. 10.3389/fphar.2019. 3: [PubMed, 0127.
- Libby P, et al., 2019. Atherosclerosis. Nat. Rev. Dis. Primer 5, 1–18. 10. 1038.
- Woo HG, et al., 2020. Atherosclerotic plaque locations may be related to different ischemic lesion patterns. BMC Neurol. 20, 288. 10. 3: [PubMed, 1186.
- Weisel JW, Litvinov RI, 2021. Visualizing thrombosis to improve thrombus resolution. Res. Pract. Thromb. Haemost. 5, 38–50. 10.1002/rth2. 3: [PubMed, 1246.
- Ashorobi D, Ameer MA, Fernandez R, 2022. S: Thrombosis, in.
- Carlo Domenico Maida, Rosario Luca Norrito, Mario Daidone, Antonino Tuttolomondo, Antonio Pinto. Neuroinflammatory Mechanisms in Ischemic Stroke: Focus on Cardioembolic Stroke, Background, and Therapeutic Approaches.Int J Mol Sci. 2020 Sep 4;21(18):6454. [CrossRef]
- Nalbandian A, et al., 2021. Post-acute COVID-19 syndrome. Nat. Med. 27, 601–615. 10. 3: s41591-021-01283-z [PubMed, 1038.
- Şekerdağ E, Solaroğlu I, Gürsoy-Özdemir Y, 2018. Cell Death Mechanisms in Stroke and Novel Molecular and Cellular Treatment Options. Curr. Neuropharmacol. 16, 1396–1415. 10. 2: [PubMed, 2174.
- Jurcau A, Ardelean AI, 2022. Oxidative Stress in Ischemia/Reperfusion Injuries following Acute Ischemic Stroke. Biomedicines 10, 574. 10. 3: [PubMed, 3390.
- Belov Kirdajova D, Kriska J, Tureckova J, Anderova M, 2020. Ischemia-Triggered Glutamate Excitotoxicity From the Perspective of Glial Cells. Front. Cell. Neurosci. 14, 51. 10.3389/ fncel.2020. 3: [PubMed, 0005.
- Sun Y, et al., 2019. Phased Treatment Strategies for Cerebral Ischemia Based on Glutamate Receptors. Front. Cell. Neurosci. 13, 168. 10.3389/fncel.2019. 3: [PubMed, 0016.
- Suzuki H, Kawakita F, Asada R, 2022. Neuroelectric Mechanisms of Delayed Cerebral Ischemia after Aneurysmal Subarachnoid Hemorrhage. Int. J. Mol. Sci. 23, 3102. 10. 3: [PubMed, 3390.
- Ludhiadch A, et al., 2022. Role of Calcium Homeostasis in Ischemic Stroke: A Review. CNS Neurol. Disord. Drug Targets 21, 52–61. 10. 3: [PubMed, 2174.
- hen Z, et al., 2022. Glutamate excitotoxicity: Potential therapeutic target for ischemic stroke. Biomed. Pharmacother. 151, 113125. 10.1016/j.biopha.2022. 3: [PubMed, 1131.
- Wu M-Y, et al., 2018. Current Mechanistic Concepts in Ischemia and Reperfusion Injury. Cell. Physiol. Biochem. 46, 1650–1667. 10. 2: [PubMed, 1159.
- Andrabi SS, Parvez S, Tabassum H, 2020. Ischemic stroke and mitochondria: mechanisms and targets. Protoplasma 257, 335–343. 10. 3: [PubMed, 1007.
- Ferrari F, Gorini A, Hoyer S, Villa RF, 2018. Glutamate metabolism in cerebral mitochondria after ischemia and post-ischemic recovery during aging: relationships with brain energy metabolism. J. Neurochem. 146, 416–428. 10.1111/jnc. 2: [PubMed, 1446.
- Yilmaz G, Arumugam TV, Stokes KY, et al. Role of T lymphocytes and interferon-g in ischemic stroke. Circulation 2006; 113: 2105-2112.
- Lambertsen KL, Gregersen R, Meldgaard M, et al. A role for interferon-g in focal cerebral ischemia in mice. 9: J Neuropathol Exp Neurol 2004; 63, 2004.
- Stone MJ, Hayward JA, Huang C. Mechanisms of regulation of the chemokine-receptor network. Int J Mol Sci. 2017;18:342–375. [CrossRef]
- Liu T, Clark RK, McDonnell PC, et al. Tumor necrosis factor-α expression in ischemic neurons. Stroke1994; 5: 1481-1488.
- Wang X, Yue TL, Barone FC, et al. Concomitant cortical expression of TNF-α and IL-1 α mRNAs follows early response gene expression in transient focal ischemia. Mol Chem Neuropathol 1994; 23: 103-114.
- Murakami Y, Saito K, Hara A, et al. Increases in tumor necrosis factor-alpha following transient global cerebral ischemia do not contribute to neuron death in mouse hippocampus. J Neurochem 2005; 93: 1616-1622.
- Offner H, Subramanian S, Parker SM, et al. Experimental stroke induces massive, rapid activation of the peripheral immune system. J Cereb Blood Flow Metab 2006; 26: 654-665.
- Huţanu A, Iancu M, Bălaşa R, et al. Predicting the functional outcome of ischemic stroke patients in Romania based on plasma CRP, sTNFR-1, D-Dimers, NGAL and NSE measured using a biochip array. ActaPharmacol Sin. 2018;39:1228–1236. [CrossRef]
- Sotgiu S, Zanda B, Marchetti B, et al. Inflammatory biomarkers in blood of patients with acute brain ischemia. Eur J Neurol. 2006;13:505–513. [CrossRef]
- Nayak AR, Kashyap SR, Kabra D, et al. Time course of inflammatory cytokines in acute ischemic stroke patients and their relation to inter-alfa trypsin inhibitor heavy chain 4 and outcome. Ann Indian Acad Neurol. 2012;15:181–185. [CrossRef]
- Barger SW, Horster D, Furukawa K, et al. Tumor necrosis factors alpha and beta protect neurons against amyloid beta-peptide toxicity: evidence for involvement of a kappa B-binding factor and attenuation of peroxide and Ca2+ accumulation. Proc Natl Acad Sci U S A 1996; 92: 9328- 9332.
- Satoh T, Otsuka A, Contassot E, French LE. The inflammasome and IL-1beta: implications for the treatment of inflammatory diseases. Immunotherapy 2015;7:243-54.
- Liao Z, Xiao HT, Zhang Y, Tong RS, Zhang LJ, Bian Y, He X. IL-1β: a key modulator in asthmatic airway smooth muscle hyper-reactivity. Expert Rev Respir Med 2015;9:429-36.
- del Zoppo, GJ. Inflammation and the neurovascular unit in the setting of focal cerebral ischemia. Neuroscience 2009;158:972-82.
- Boutin H, LeFeuvre RA, Horai R, et al. Role of IL-1alpha and IL- 1beta in ischemic brain damage. 5: J Neurosci 2001; 21, 2001.
- Yamasaki Y, Matsuura N, Shozuhara H, et al. Interleukin-1 as a pathogenetic mediator of ischemic brain damage in rats. Stroke 1995; 26: 676-680.
- Basu A, Lazovic J, Krady JK, et al. Interleukin-1 and the interleukin-1 type 1 receptor are essential for the progressive neurodegeneration that ensues subsequent to a mild hypoxic/ischemic injury. J Cereb Blood Flow Metab 2005; 25: 17- 29.
- Rothwell, N. Interleukin-1 and neuronal injury: mechanisms, modification, and therapeutic potential. Brain Behav Immun 2003;17:152-7.
- Protopsaltis J, Kokkoris S, Korantzopoulos P, Milionis HJ, Karzi E, Anastasopoulou A, Filioti K, Antonopoulos S, Melidonis A, Giannoulis G. Prediction of long-term functional outcome in patients with acute ischemic non-embolic stroke. Atherosclerosis 2009;203:228-35.
- Tanzi P, Cain K, Kalil A, Zierath D, Savos A, Gee JM, Shibata D, Hadwin J, Carter K, Becker K. Post-stroke infection: a role for IL-1ra? Neurocrit Care 2011;14:244-52.
- Erta M, Quintana A, Hidalgo J. Interleukin-6, a major cytokine in the central nervous system. Int J Biol Sci. 2012;8:1254–1266. [CrossRef]
- Suzuki S, Tanaka K, Suzuki N. Ambivalent aspects of interleukin-6 in cerebral ischemia: inflammatory versus neurotrophic aspects. J Cereb Blood Flow Metab. 2009;29:464–479. [CrossRef]
- Loddick SA, Turnbull AV, Rothwell NJ. Cerebral interleukin-6 is neuroprotective during permanent focal cerebral ischemia in the rat. J Cereb Blood Flow Metab 1998; 18: 176-179.
- Yu XH, Jiang N, Zheng XL, Cayabyab FS, Tang ZB, Tang CK. Interleukin-17A in lipid metabolism and atherosclerosis. Clin Chim Acta 2014;431:33-9.
- Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defences. Annu Rev Immunol 2014;32:513-45.
- Folsom AR, Gottesman RF, Appiah D, Shahar E, Mosley TH. Plasma d-Dimer and incident ischemic stroke and coronary heart disease: the atherosclerosis risk in communities study. 1: Stroke 2016;47, 2016.
- Schroeter M, Jander S, Witte OW, Stoll G. Local immune responses in the rat cerebral cortex after middle cerebral artery occlusion. J Neuroimmunol 1994;55:195-203.
- Mosmann TR, Sad S. The expanding universe of T-cell subsets: Th1, Th2 and more. Immunol Today 1996;17:138-46.
- O’Garra A, Vieira PL, Vieira P, Goldfeld AE. IL-10–producing and naturally occurring CD4+ Tregs: limiting collateral damage. J Clin Invest 2004; 114: 1372-1378.
- Grilli M, Barbieri I, Basudev H, et al. Interleukin-10 modulates neuronal threshold of vul- nerability to ischaemic damage. Eur J Neurosci 2000; 12: 2265-2272.
- Gross CE1, Bednar MM, Howard DB, Sporn MB. Transforming growth factor-beta 1 reduces infarct size after experimental cerebral ischemia in a rabbit model. Stroke 1993; 24(4): 558-562.
- Nair MG, Guild KJ, Artis D. Novel effector molecules in type 2 inflammation: lessons drawn from helminth infection and allergy. J Immunol 2006;177:1393-9.
- Cekanaviciute E, Buckwalter MS. Astrocytes: integrative regulators of neuroinflammation in stroke and other neurological diseases.Neurotherapeutics 2016;13:685-701.
- Lin J, Kakkar V, Lu X. Essential roles of Toll-like receptors in atherosclerosis. Curr Med Chem 2016;23:431-54.
- Rietdijk CD, Van Wezel RJA, Garssen J, Kraneveld AD. Neuronal toll-like receptors and neuro-immunity in Parkinson’s disease, Alzheimer’s disease and stroke. Neuroimmunol Neuroinflammation 2016;3:27.
- Guruswamy R, ElAli A. Complex roles of microglial cells in ischemic stroke pathobiology: new insights and future directions. International journal of molecular sciences. 2017;18(3). [CrossRef]
- Schilling M, Besselmann M, Müller M. Predominant phagocytic activity of resident microglia over hematogenous macrophages following transient focal cerebral ischemia: an investigation using green fluorescent protein transgenic bone marrow chimeric mice. 2: Exp Neurol 2005; 196(2), 2005.
- Geissmann F, Gordon S, Hume DA, Mowat AM, Randolph GJ. Unravelling mononuclear phagocyte heterogeneity. Nat Rev Immunol 2010;10:453-60.
- Hu X, Li P, Guo Y, Wang H, Leak RK, Chen S, Gao Y, Chen J. Microglia/ macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke 2012;43:3063-70.
- Bylicky MA, Mueller GP, Day RM. Mechanisms of endogenous neuroprotective effects of astrocytes in brain injury. Oxid Med Cell Longev. 2018;2018:6501031.
- Hennessy E, Griffin ÉW, Cunningham C. Astrocytes are primed by chronic neurodegeneration to produce exaggerated chemokine and cell infiltration responses to acute stimulation with the cytokines IL-1β and TNF-α. J Neurosci. 2015;35(22):8411–22.
- Wang H, Song G, Chuang H, Chiu C, Abdelmaksoud A, Ye Y, et al. Portrait of glial scar in neurological diseases. Int J Immunopathol Pharmacol. 2018;31: 2058738418801406.
- Sykova, E. Glial diffusion barriers during aging and pathological states. Prog Brain Res. 3: 2001;132, 2001. [Google Scholar]
- Rempe RG, Hartz AMS, Bauer B. Matrix metalloproteinases in the brain and blood-brain barrier: versatile breakers and makers. J Cereb Blood Flow Metab. 2016;36(9):1481–507.
- Overman JJ, Clarkson AN, Wanner IB, Overman WT, Eckstein I, Maguire JL, et al. A role for ephrin-A5 in axonal sprouting, recovery, and activity- dependent plasticity after stroke. Proc Natl Acad Sci U S A. 2012;109(33): E2230–9.
- Jickling GC, Liu D, Ander BP, Stamova B, Zhan X, Sharp FR. Targeting neutrophils in ischemic stroke: translational insights from experimental studies. J Cereb Blood Flow Metab. 2015;35(6):888–901.
- Weston RM, Jones NM, Jarrott B, Callaway JK. Inflammatory cell infiltration after endothelin-1-induced cerebral ischemia: histochemical and myeloperoxidase correlation with temporal changes in brain injury. J Cereb Blood Flow Metab. 2007;27(1):100–14.
- Perez-de-Puig I, Miró-Mur F, Ferrer-Ferrer M, Gelpi E, Pedragosa J, Justicia C, et al. Neutrophil recruitment to the brain in mouse and human ischemic stroke. Acta Neuropathol. 2015;129(2):239–57.
- Connolly ES Jr, Winfree CJ, Springer TA, et al. Cerebral protection in homozygous null ICAM-1 mice after middle cerebral artery occlusion. Role of neutrophil adhesion in the pathogenesis of stroke. J Clin Invest 1996; 97: 209-216.
- Castellanos M, Leira R, Serena J, et al. Plasma metalloproteinase-9 concentration predicts hemorrhagic transformation in acute ischemic stroke. Stroke 2003; 34: 40-46.
- Gokhan S, Ozhasenekler A, Mansur Durgun H, Akil E, Ustundag M, Orak M. Neutrophil lymphocyte ratios in stroke subtypes and transient ischemic attack. Eur Rev Med Pharmacol Sci. 2013;17(5):653–7.
- Jian,Z. ;Liu,R.;Zhu,X.;Smerin,D.;Zhong,Y.;Gu,L.;Fang,W.;Xiong,X.TheInvolvementandTherapyTargetofImmuneCells After Ischemic Stroke. Front. Immunol. 2019, 10, 2167.
- Felger,J. C.;Abe,T.;Kaunzner,U.W.;GottfriedBlackmore,A.;GalToth,J.;McEwen,B.S.;Iadecola,C.;Bulloch,K.Braindendriticcells in ischemic stroke: Time course, activation state, and origin. Brain Behav. Immun. 2010, 24, 724–737.
- Yilmaz,A. ;Fuchs,T.;Dietel,B.;Altendorf,R.;Cicha,I.;Stumpf,C.;Schellinger,P.D.;Blümcke,I.;Schwab,S.;Daniel,W.G.;etal.Transient decrease in circulating dendritic cell precursors after acute stroke: Potential recruitment into the brain. Clin. Sci. 2009, 118, 147–157.
- Feng Y, Liao S, Wei C, Jia D, Wood K, Liu Q, et al. Infiltration and persistence of lymphocytes during late-stage cerebral ischemia in middle cerebral artery occlusion and photothrombotic stroke models. J Neuroinflammation. 2017; 14(1):248.
- Yun Hwa Choi, Collin Laaker, Martin Hsu, Peter Cismaru, Matyas Sandor, Zsuzsanna Fabry 2 4.Molecular Mechanisms of Neuroimmune Crosstalk in the Pathogenesis of Stroke.Int J Mol Sci. 2021 Aug 31;22(17):9486. [CrossRef]
- Sakaguchi S, Ono M, Setoguchi R, et al. Foxp3 CD25 CD4 natural regulatory T cells in dominant self tolerance and autoimmune disease. Immunol Rev 2006; 212: 8-27.
- Liesz A, Suri-Payer E, Veltkamp C. Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat Med 2009; 15: 138 -139.
- Shibata K, Yamada H, Hara H, et al. Resident V δ1+γδ T cells control early infiltration of neutrophils after Escherichia coli infection via IL-17 production. 4: J Immunol 2007; 178, 2007.
- Snyder MR, Nakajima T, Leibson PJ, et al. Stimulatory killer Ig- like receptors modulate T cell activation through DAP12- dependent and DAP12-independent mechanisms. 3: J Immunol 2004;173, 2004.
- Zal B, Kaski JC, Arno G, et al. Baboonian, Heat-shock protein 60-reactive CD4+CD28null T cells in patients with acute coronary syndromes. Circulation 2004; 109: 1230-1235.
- Weyand CM, Brandes JC, Schmidt D, et al. Functional properties of CD4þ CD28- T cells in the aging immune system. Mech Ageing Dev 1998; 102: 131-147.
- Nowik M, Nowacki P, Grabarek J, et al. Can we talk about CD4+CD28- lymphocytes as a risk factor for ischemic stroke. Eur Neurol 2007; 58(1): 26-33.
- Nadareishvili ZG, Li H, Wright V, et al. Elevated proinflammatory CD4+CD28– lymphocytes and stroke recurrence and death. Neurology 2004; 63: 1446-1451.
- Tuttolomondo A, Pecoraro R, Casuccio A, Di Raimondo D, Buttà C, Clemente G, Della Corte V, Guggino G, Arnao V, Maida C, Simonetta I, Maugeri R, Squadrito R, Pinto A.Peripheral Frequency of CD4+ CD28- Cells in Acute Ischemic Stroke: Relationship With Stroke Subtype and Severity Markers. Medicine (Baltimore). 2015 May;94(20):e813. [CrossRef]
- Nakajima T, Goek O, Zhang X, Kopecky SL, Frye RL, Goronzy JJ, Weyand CM. De novo expression of killer immunoglobulin-like receptors and signalling proteins regulate the cytotoxic function of CD4 T cells in acute coronary syndromes. Circ Res. 1: 2003;93(2), 2003.
- Tuttolomondo A, Domenico Di Raimondo 2, Rosaria Pecoraro 3 4, Alessandra Casuccio 5, Danilo Di Bona 6, Anna Aiello 7, Giulia Accardi 7, Valentina Arnao 8, Giuseppe Clemente 2, Vittoriano Della Corte 9, Carlo Maida 2, Irene Simonetta 2, Calogero Caruso 7, Rosario Squatrito 3, Antonio Pinto 2, KIRIIND (KIR Infectious and Inflammatory Diseases) Collaborative Group. HLA and Killer Cell Immunoglobulin-Like Receptor (KIRs) Genotyping in Patients With Acute Ischemic Stroke. J Neuroinflammation. 2019 Apr 17;16(1):88. [CrossRef]
- Ren, X.; Akiyoshi, K.; Dziennis, S.; Vandenbark, A.A.; Herson, P.S.; Hurn, P.D.; Offner, H. Regulatory B cells limit CNS inflammation and neurologic deficits in murine experimental stroke. J. Neurosci. 2011, 31, 8556–8563. [Google Scholar] [CrossRef] [PubMed]
- Ortega,S. B.;Torres,V.O.;Latchney,S.E.;Whoolery,C.W.;Noorbhai,I.Z.;Poinsatte,K.;Selvaraj,U.M.;Benson,M.A.;Meeuwissen, A.J.M.; Plautz, E.J.; et al. B cells migrate into remote brain areas and support neurogenesis and functional recovery after focal stroke in mice. Proc. Natl. Acad. Sci. USA 2020, 117, 4983.
- Doyle,K. P.;Quach,L.N.;Solé,M.;Axtell,R.C.;Nguyen,T.V.V.;SolerLlavina,G.J.;Jurado,S.;Han,J.;Steinman,L.;Longo,F.M.; et al. B-lymphocyte-mediated delayed cognitive impairment following stroke. J. Neurosci. 2015, 35, 2133–214.
- Schuhmann,M. K.;Langhauser,F.;Kraft,P.;Kleinschnitz,C.Bcellsdonothaveamajorpathophysiologicroleinacuteischemic stroke in mice. J. Neuroinflamm. 2017, 14, 112.
- Gelderblom,M. ;Gallizioli,M.;Ludewig,P.;Thom,V.;Arunachalam,P.;Rissiek,B.;Bernreuther,C.;Glatzel,M.; Korn, T.; Arumugam, T.V.; et al. IL-23 (Interleukin-23)–Producing Conventional Dendritic Cells Control the Detrimental IL-17 (Interleukin-17) Response in Stroke. Stroke 2018, 49, 155–164.
- Rincon,F. ;Mayer,S.A.TheepidemiologyofintracerebralhemorrhageintheUnitedStatesfrom1979to2008.Neurocrit.Care 2013, 19, 95–102.
- Aronowski J, Zhao X, 2011. Molecular pathophysiology of cerebral hemorrhage: secondary brain injury. Stroke 42, 1781–1786. 10.1161/STROKEAHA.110. 2: [PubMed, 5967.
- Tschoe C, et al., 2020. Neuroinflammation after Intracerebral Hemorrhage and Potential Therapeutic Targets. J. Stroke 22, 29–46. 10.5853/jos.2019. 3: [PubMed, 0223.
- Li Z, et al., 2020. Hematoma Expansion in Intracerebral Hemorrhage: An Update on Prediction and Treatment. Front. Neurol. 11, 702. 10.3389/fneur.2020. 3: [PubMed, 0070.
- Xiao A, et al., 2021. GDF11 alleviates secondary brain injury after intracerebral hemorrhage via attenuating mitochondrial dynamic abnormality and dysfunction. Sci. Rep. 11, 3974. 10. 3: s41598-021-83545-x [PubMed, 1038.
- Madangarli N, Bonsack F, Dasari R, Sukumari–Ramesh S, 2019.
- Blood Components and Neurotoxicity. Brain Sci. 9, 316. 10. 3: [PubMed, 3390.
- Zhang R, et al., 2020. Intracerebral hemorrhage in translational research. Brain Hemorrhages 1, 13– 18. 10.1016/j.hest.2020.02.
- Yao Z, Bai Q, Wang G, 2021. Mechanisms of Oxidative Stress and Therapeutic Targets following Intracerebral Hemorrhage. Oxid. Med. Cell. Longev. 2021, 8815441. 10. 3: [PubMed, 1155.
- Zhang Y, et al., 2022. Oxidative Stress Following Intracerebral Hemorrhage: From Molecular Mechanisms to Therapeutic Targets. Front. Immunol. 13, 847246. 10.3389/fimmu.2022. 3: [PubMed, 8472.
- Kim M, et al., 2021. Reactive Oxygen Species Scavenger in Acute Intracerebral Hemorrhage Patients. Stroke 52, 1172–1181. 10.1161/STROKEAHA.120. 3: [PubMed, 0322.
- Yan H, et al., 2021. Ferroptosis: mechanisms and links with diseases. Signal Transduct. Target. Ther. 6, 1–16. 10. 3: [PubMed, 1038.
- Higgins GC, Devenish RJ, Beart PM, Nagley P, 2012. Transitory phases of autophagic death and programmed necrosis during superoxide-induced neuronal cell death. Free Radic. Biol. Med. 53, 1960–1967. 10.1016/j.freeradbiomed.2012.08. 2: [PubMed, 2298.
- Fang,Y. ;Gao,S.;Wang,X.;Cao,Y.;Lu,J.;Chen,S.;Lenahan,C.;Zhang,J.H.;Shao,A.;Zhang,J.ProgrammedCellDeathsandPotential Crosstalk with Blood-Brain Barrier Dysfunction after Hemorrhagic Stroke. Front. Cell. Neurosci. 2020, 14, 68.
- Luo, C.; Yao, X.; Li, J.; He, B.; Liu, Q.; Ren, H.; Liang, F.; Li, M.; Lin, H.; Peng, J.; et al. Paravascular pathways contribute to vasculitis and neuroinflammation after subarachnoid hemorrhage independently of glymphatic control. Cell Death Dis. 2016, 7, e2160 [CrossRef] [PubMed]. [Google Scholar] [CrossRef]
- Zille M, et al., 2022. Novel targets, treatments, and advanced models for intracerebral haemorrhage. eBioMedicine 76. 10.1016/j.ebiom.2022. 1038.
- Lei Chunyan, et al., 2015. Activation of the high-mobility group box 1 protein-receptor for advanced glycation end-products signaling pathway in rats during neurogenesis after intracerebral hemorrhage. Stroke 46, 500–506. 10.1161/STROKEAHA.114. 2: [PubMed, 0068.
- Zhou Y, et al., 2010. Elevation of high-mobility group protein box-1 in serum correlates with severity of acute intracerebral hemorrhage. Mediators Inflamm. 2010, 142458. 10. 2: [PubMed, 1155.
- Lan,X. ;Han,X.;Liu,X.;Wang,J.Inflammatoryresponsesafterintracerebralhemorrhage:Fromcellularfunctiontotherapeutic targets. J. Cereb. Blood Flow Metab. 2019, 39, 184–186.
- Quan,W. ;Zhang,Z.;Li,P.;Tian,Q.;Huang,J.;Qian,Y.;Gao,C.;Su,W.;Wang,Z.;Zhang,J.;etal.RoleofRegulatoryTcellsin Atorvastatin Induced Absorption of Chronic Subdural Hematoma in Rats. Aging Dis. 2019, 10, 992–1002.
- Lee Y, Ahn C, Han J, Choi H et al. The nuclear RNAse III Drosha initiates microRNA processing. Nature 2003; 425: 415-419. Yi R, Qin Y, Macara IG, Cullen B. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev. 2003. [Google Scholar]
- Okamura K, Hagen J, Duan H, Tyler DM et al. The mirtron pathway generates microR- NA-class regulatory RNAs in Drosophila. Cell 2007; 130: 89-100.
- Brennecke J, Stark A, Russell RB, Cohen SM. Principles of microRNA-target recognition. PLoS Biol. 2005; 3: e85.
- Hutvagner G, Zamore PD. A microRNA in a multiple-turnover RNAi enzyme complex. Sci- ence 2002; 297: 2056-2060.
- Didiano D, Hobert O. Perfect seed pairing is not a generally reliable predictor for mirNA- target interactions. Nat. Struct. Mol. Biol. 2006; 13: 849-851.
- Kamtchum-Tatuene, J. , & Jickling, G. C. ( 21(4), 344–368. [CrossRef] [PubMed]
- Dagonnier, M.; Donnan, G.A.; Davis, S.M.; Dewey, H.M.; Howells, D.W. Acute Stroke Biomarkers: Are We There Yet? Front. Neurol. 2021, 12, 619721. [Google Scholar] [CrossRef] [PubMed]
- Reymond, S.; Vujic ́, T.; Sanchez, J.C. Neurovascular Unit-Derived Extracellular Vesicles: From Their Physiopathological Roles to Their Clinical Applications in Acute Brain Injuries. Biomedicines 2022, 10, 2147. [Google Scholar] [CrossRef] [PubMed]
- Yokoi, A.; Ochiya, T. Exosomes and extracellular vesicles: Rethinking the essential values in cancer biology. Semin. Cancer Biol. 2021, 74, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Jella KK, Nasti TH, Li Z, Malla SR, Buchwald ZS, and Khan MK (2018). Exosomes, their biogenesis and role in inter-cellular communication, tumor microenvironment and cancer immunotherapy. 6.
- Gurunathan S, Kang MH, and Kim JH (2021). A comprehensive review on factors influences biogenesis, functions, therapeutic and clinical implications of exosomes. Int. J. Nanomedicine 16, 1281–1312. 3: [PubMed, 3362.
- Murphy DE, de Jong OG, Brouwer M, Wood MJ, Lavieu G, Schiffelers RM, and Vader P (2019). Extracellular vesicle-based therapeutics: natural versus engineered targeting and trafficking. Exp. Mol. Med 51, 1–12.
- Ramirez SH, Andrews AM, Paul D, and Pachter JS (2018). Extracellular vesicles: mediators and biomarkers of pathology along CNS barriers. Fluids Barriers CNS 15, 19. [PubMed: 29960602].
- Fruhbeis C, et al., Extracellular vesicles as mediators of neuron-glia communication. Front. 1: CellNeurosci, 2013; 7, 2013.
- György B, Hung ME, Breakefield XO, Leonard JN, Therapeutic applications of extracellular vesicles: clinical promise and open questions, Annu. Rev. Pharmacol. Toxicol 55 (2015) 439– 464. 2: [PubMed, 2529.
- Gharbi, T.; Zhang, Z.; Yang, G.Y. The Function of Astrocyte Mediated Extracellular Vesicles in Central Nervous System Diseases. Front. Cell Dev. Biol. 2020, 8, 568889. [Google Scholar] [CrossRef] [PubMed]
- Valadi, H. , Ekström, K., Bossios, A., Sjöstrand, M., Lee, J. J., & Lötvall, J. O. (2007). Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nature Cell Biology, 9(6), 654. [CrossRef]
- Pei, X.; Li, Y.; Zhu, L.; Zhou, Z. Astrocyte-derived exosomes suppress autophagy and ameliorate neuronal damage in experimental ischemic stroke. Exp. Cell Res. 2019, 382, 111474. [Google Scholar] [CrossRef]
- Bu, X.; Li, D.; Wang, F.; Sun, Q.; Zhang, Z. Protective role of astrocyte-derived exosomal microRNA-361 in cerebral ischemic reperfusion injury by regulating the AMPK/mTOR signaling pathway and targeting CTSB. Neuropsychiatr. Dis. Treat. 2020, 16, 1863–1877. [Google Scholar] [CrossRef]
- Wu, W.; Liu, J.; Yang, C.; Xu, Z.; Huang, J.; Lin, J. Astrocyte-derived exosome-transported microRNA-34c is neuroprotective against cerebral ischemia/reperfusion injury via TLR7 and the NF-kappaB/MAPK pathways. Brain Res. Bull. 2020, 163, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Zhang L, Dong LY, Li YJ, Hong Z, Wei WS. The microRNA miR-181c controls microglia-mediated neuronal apoptosis by suppressing tumor necrosis factor. J Neuroinflammation. 2: 2012; 9, 2012.
- Wen Y, Zhang X, Dong L, Zhao J, Zhang C, Zhu C. Acetyl- britannilactone modulates microRNA-155-mediated inflammatory response in ischemic cerebral tissues. Mol Med. 1: 2015;21, 2015.
- Martinez B, Peplow PV. Blood microRNAs as potential diagnostic markers for hemorrhagic stroke. Neural Regen Res. 1: 2017;12(1), 2017.
- Banerjee S, Xie N, Cui H, Tan Z, Yang S, Icyuz M, Abraham E, Liu G. MicroRNA let-7c regulates macrophage polarization. J Immunol. 6: 2013;190(12), 2013.
- Tan KS, Armugam A, Sepramaniam S, Lim KY, Setyowati KD, Wang CW, Jeyaseelan K. Expression profile of Micro- RNAs in young stroke patients. PLoS One. e: 2009;4(11), 2009.
- Wang W, Sun G, Zhang L, Shi L, Zeng Y. Circulating micro- RNAs as novel potential biomarkers for early diagnosis of acute stroke in humans. J Stroke Cerebrovasc Dis. 2: 2014; 23(10), 2014.
- LongG, WangF,LiH,YinZ,SandipC,LouY,WangY,Chen C, Wang DW. Circulating miR-30a, miR-126 and let-7b as biomarker for ischemic stroke in humans. BMC Neurol. 1: 2013;13, 2013.
- Wang MD, Wang Y, Xia YP, Dai JW, Gao L, Wang SQ, et al. High serum MiR-130a levels are associated with severe perihematomal edema and predict adverse outcome in acute ICH. Mol Neurobiol. 1: 2016; 53(2), 2016.
- WangJ, ZhuY, JinF, TangL, HeZ, HeZ. Differential expression of circulating microRNAs in blood and haematoma samples from patients with intracerebral haemorrhage. J Int Med Res. 4: 2016;44(3), 2016.
- Bejleri, J.; Jirström, E.; Donovan, P.; Williams, D.J.; Pfeiffer, S. Diagnostic and Prognostic Circulating MicroRNA in Acute Stroke: A Systematic and Bioinformatic Analysis of Current Evidence. J. Stroke 2021, 23, 162–182 [CrossRef] [PubMed]. [Google Scholar] [CrossRef] [PubMed]
- LiuY, PanQ, ZhaoY, HeC, BiK, ChenY, ZhaoB, ChenY, Ma X. MicroRNA-155 regulates ROS production, no generation, apoptosis and multiple functions of human brain microvessel endothelial cells under physiological and pathological conditions. J Cell Biochem. 2: 2015;116(12), 2015.
- Yin M, Chen Z, Ouyang Y, Zhang H, Wan Z, Wang H, Wu W, Yin X. Thrombin-induced, TNFR-dependent miR-181c downregulation promotes MLL1 and NF-kB target gene expression in human microglia. J Neuroinflammation. 1: 2017;14(1), 2017.
- Schratt GM, Tuebing F, Nigh EA, Kane CG, Sabatini ME, Kiebler M, Greenberg ME. A brain-specific microRNA regulates dendritic spine development. Nature. 2: 2006;439(7074), 2006.
- Xie W, Li M, Xu N, Lv Q, Huang N, He J, Zhang Y. Mir-181a regulates inflammation responses in monocytes and macro- phages. PLoS One. e: 2013;8, 2013.
- Moon JM, Xu L, Giffard RG. Inhibition of microRNA-181 reduces forebrain ischemia-induced neuronal loss. J Cereb Blood Flow Metab. 1: 2013;33(12), 2013.
- Yuan B, Shen H, Lin L, Su T, Zhong L, Yang Z (2015) MicroRNA367 negatively regulates the inflammatory response of microglia by targeting IRAK4 in intracerebral hemorrhage. 2: J Neuroinflammation 12.
- Yang Z, Zhong L, Xian R, Yuan B (2015) MicroRNA-223 regulates inflammation and brain injury via feedback to NLRP3 inflammasome after intracerebral hemorrhage. Mol Immunol 65:267-276.
- van Rooij E, Marshall WS and Olson EN. Toward microRNA-based therapeutics for heart disease: the sense in anti-sense. 9: Circ Res 2008; 103, 2008.
- Latronico MV and Condorelli, G. Therapeutic use of microRNAs in myocardial diseases. 1: Curr Heart Fail Rep 2011; 8, 2011. [Google Scholar]
- Krützfeldt J, Rajewsky N, Braich R, Rajeev KG, et al. Silencing of microRNAs in vivo with 'antagomirs'. 6: Nature 2005; 438, 2005.
- Elmén J, Lindow M, Silahtaroglu A, Bak M, et al. Antagonism of microRNA-122 in mice by systemically administered LNA-antimiR leads to up-regulation of a large set of predicted target mRNAs in the liver. 1: Nucleic Acids Res 2008; 36, 2008.
- Elmén J, Lindow M, Schütz S, Lawrence M, et al. LNA-mediated microRNA silencing in non- human primates. 8: Nature 2008; 452, 2008.
- Ebert MS, Neilson JR, Sharp PA. MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nat Methods. 7: 2007; 4, 2007.
- Hayashi T, Noshita N, Sugawara T and Chan PH. Temporal profile of angiogenesis and expression of related genes in the brain after ischemia. J Cereb Blood Flow Metab. 2003;23:166–80. 1: [PubMed, 1257.
- Marti HJ, Bernaudin M, Bellail A, Schoch H, Euler M, Petit E and Risau W. Hypoxia-induced vascular endothelial growth factor expression precedes neovascularization after cerebral ischemia. Am J Pathol. 2000;156:965–76. 1: [PubMed, 1070.
- Krupinski J, Kaluza J, Kumar P, Kumar S and Wang JM. Role of angiogenesis in patients with cerebral ischemic stroke. Stroke. 1994;25:1794–8. 7: [PubMed, 7521.
- Li Y, Mao L, Gao Y, Baral S, Zhou Y and Hu B. MicroRNA-107 contributes to post-stroke angiogenesis by targeting Dicer-1. Sci Rep. 2015;5:13316. 2: [PubMed, 2629.
- Wang J, Shi Y, Zhang L, Zhang F, Hu X, Zhang W, Leak RK, Gao Y, Chen L and Chen J. Omega-3 polyunsaturated fatty acids enhance cerebral angiogenesis and provide long-term protection after stroke. Neurobiol Dis. 2014;68:91–103. 2: [PubMed, 2479.
- Hoffmann CJ, Harms U, Rex A, Szulzewsky F, Wolf SA, Grittner U, Lattig-Tunnemann G, Sendtner M, Kettenmann H, Dirnagl U, Endres M and Harms C. Vascular signal transducer and activator of transcription-3 promotes angiogenesis and neuroplasticity long-term after stroke. Circulation. 2015;131:1772–82. 2: [PubMed, 2579.
- Sun P, Zhang K, Hassan SH, Zhang X, Tang X, Pu H, Stetler RA, Chen J, Yin KJ. Endothelium-Targeted Deletion of microRNA-15a/16-1 Promotes Poststroke Angiogenesis and Improves Long-Term Neurological Recovery. Circ Res. 2020 Apr 10;126(8):1040-1057. [CrossRef] [PubMed]
- Xin, H. , Katakowski, M., Wang, F., Qian, J.-Y., Liu, X. S., Ali, M. M.,... Chopp, M. (2017). MicroRNA-17–92 cluster in exosomes enhance neuroplasticity and functional recovery after stroke in rats. Stroke, 48(3), 747–753. [CrossRef]
- Xin, H. , Wang, F., Li, Y., Lu, Q.-E., Cheung, W. L., Zhang, Y. I.,... Chopp, M. (2017). Secondary release of exosomes from astrocytes contributes to the increase in neural plasticity and improvement of functional recovery after stroke in rats treated with exosomes harvested from microRNA 133b-overexpressing multipotent mesen- chymal stromal cells. Cell Transplantation, 26(2), 243–257. https:// doi.org/10. 3727. [Google Scholar]
- Xu, X. , Wen, Z., Zhao, N., Xu, X., Wang, F., Gao, J.,... Liu, X. (2017). MicroRNA-1906, a novel regulator of toll-like receptor 4, amelio- rates ischemic injury after experimental stroke in mice. Journal of Neuroscience, 37(43), 10498–10515. [CrossRef]
- Iwuchukwu I, Nguyen D, Sulaiman W. MicroRNA Profile in cerebrospinal fluid and plasma of patients with spontaneous intracerebral hemorrhage. CNS Neurosci Ther. 2016;22(12): 1015–1018.
- TaoZ, ZhaoH,WangR,LiuP,YanF,ZhangC,JiX,LuoY. Neuroprotective effect of microRNA-99a against focal cerebral ischemia-reperfusion injury in mice. J Neurol Sci. 1: 2015; 355(1–2), 2015.
- Yin KJ, Deng Z, Huang H, Hamblin M, Xie C, Zhang J, Chen YE. miR-497 regulates neuronal death in mouse brain after transient focal cerebral ischemia. Neurobiol Dis. 1: 2010;38(1), 2010.
- ZhaoH,WangJ,GaoL,WangR,LiuX,GaoZ,TaoZ,XuC, Song J, Ji X, Luo Y. MiRNA-424 protects against permanent focal cerebral ischemia injury in mice involving suppressing microglia activation. Stroke. 1: 2013;44, 2013.
- ZhangY,HanB,HeY,LiD,MaX,LiuQ,HaoJ.MicroRNA- 132 attenuates neurobehavioral and neuropathological changes associated with intracerebral hemorrhage in mice. Neurochem Int. 1: 2017;107, 2017.
- KongF,ZhouJ,ZhouW,GuoY,LiG,YangL.Protectiverole of microRNA-126 in intracerebral hemorrhage. Mol Med Rep. 1: 2017;15(3), 2017.
- Wang LM, Xie Y, Xu LL, Ye RD, Liu XF. Target-regulated caveolin-1 by miR-103 improves neurological deficits follow- ing subarachnoid hemorrhage. Chin J Geriatr Heart Brain Ves- sel Dis. 5: 2016;18(5), 2016.
- C ZengL,HeX,WangY,TangY,ZhengC,CaiH,LiuJ, Wang Y, Fu Y, Yang GY. MicroRNA-210 overexpression induces angiogenesis and neurogenesis in the normal adult mouse brain. Gene Ther. 3: 2014;21(1), 2014.
- Candelario-Jalil E, Dijkhuizen RM, Magnus T, 2022. Neuroinflammation, Stroke, Blood-Brain Barrier Dysfunction, and Imaging Modalities. Stroke 53, 1473–1486. 10.1161/STROKEAHA.122. 0369.
- Jayaraj RL, Azimullah S, Beiram R, Jalal FY, Rosenberg GA, 2019. Neuroinflammation: friend and foe for ischemic stroke. J. Neuroinflammation 16, 142. 10.1186/s12974-019-1516-2; Hanisch U-K, Kettenmann H, 2007. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 10, 1387–1394. [Google Scholar]
- Luheshi NM, et al., 2011. Interleukin-1α expression precedes IL-1β after ischemic brain injury and is localised to areas of focal neuronal loss and penumbral tissues. J. Neuroinflammation 8, 186. 10.1186/1742-2094-8-186; Boutin H, et al., 2001. Role of IL-1α and IL-1β in Ischemic Brain Damage. J. Neurosci. 21, 5528–5534. 10.1523/JNEUROSCI.21-15-05528.
- Finger CE, et al., 2022. Age-related immune alterations and cerebrovascular inflammation. Mol. Psychiatry 27, 803–818. 10. 1038.
- Chen R, et al., 2021. New Insight into Neutrophils: A Potential Therapeutic Target for Cerebral Ischemia. Front. Immunol.
- Solár P, Zamani A, Lakatosová K, Joukal M, 2022. The blood–brain barrier and the neurovascular unit in subarachnoid hemorrhage: molecular events and potential treatments. Fluids Barriers CNS 19, 29. 10. 1186.
- Clausen, B. H. et al. Systemically administered anti-TNF therapy ameliorates functional outcomes after focal cerebral ischemia. J. Neuroinflammation. 2014; 11. [Google Scholar]
- Sumbria, R. K. , Boado, R. J. & Pardridge, W. M. Brain protection from stroke with intravenous TNF alpha decoy receptor-Trojan horse fusion protein. J. Cereb. Blood Flow. Metab. 1933; 32. [Google Scholar]
- Liguz-Lecznar, M. , Zakrzewska, R. & Kossut, M. Inhibition of Tnf-alpha R1 signaling can rescue functional cortical plasticity impaired in early poststroke period. Neurobiol. Aging 36, 2877–2884 / Works, M. G., Koenig, J. B. & Sapolsky, R. M. Soluble TNF receptor 1-secreting ex vivo-derived dendritic cells reduce injury after stroke. J. Cereb. Blood Flow. Metab. 1376; 33. [Google Scholar]
- Scheinfeld, N. A comprehensive review and evaluation of the side effects of the tumor necrosis factor-alpha blockers etanercept, in!iximab, and adalimumab. J. Dermatol. Treat. 15.
- Loddick, S. A. & Rothwell, N. J. Neuroprotective effects of human recombinant interleukin-1 receptor antagonist in focal cerebral ischaemia in the rat. J. Cereb. Blood Flow. Metab. 16, 932–940 / Nawashiro, H., Martin, D. & Hallenbeck, J. M. Neuroprotective effects of TNF binding protein in focal cerebral ischemia. Brain Res.
- Emsley, H. C. A. et al. A randomised phase II study of interleukin-1 receptor antagonist in acute stroke patients. J. Neurol. Neurosurg. 1366; 76. [Google Scholar]
- Willard, S. S. & Koochekpour, S. Glutamate, glutamate receptors, and downstream signaling pathways. Int. J. Biol. Sci. 9.
- George, P. M. & Steinberg, G. K. Novel Stroke Therapeutics: Unraveling Stroke Pathophysiology And Its Impact On Clinical Treatments. 87.
- Moskowitz, M. A. , Lo, E. H. & Iadecola, C. The science of stroke: mechanisms in search of treatments. 67.
- Mazala, D. A. G. , Grange, R. W. & Chin, E. R. The role of proteases in excitation contraction coupling failure in muscular dystrophy. Am. J. Physiol. Cell. Physiol.
- Casas, A. I. et al. Calcium-dependent blood-brain barrier breakdown by NOX5 limits postreperfusion bene"t in stroke. J. Clin. Invest. 129, 1772–1778 / Liu, F. et al. Mitochondria in Ischemic Stroke: New Insight and Implications. Aging Dis. 9.
- Zhu, J. et al. Up regulation of GluN2A-containing NMDA receptor protects cultured cortical neuron cells from oxidative stress. 0097; 4. [Google Scholar]
- Teves, L. M. , Cui, H. & Tymianski, M. Efficacy of the PSD95 inhibitor Tat-NR2B9c in mice requires dose translation between species. J. Cereb. Blood Flow. Metab. 36.
- Soriano, F. X. et al. Speci"c targeting of pro-death NMDA receptor signals with differing reliance on the NR2B PDZ Ligand. J. Neurosci. 1069; 28. [Google Scholar]
- Chen, Y. et al. Tat-NR2B9c prevents excitotoxic neuronal superoxide production. J. Cereb. Blood Flow. Metab. 35.
- Luo, C.-X. et al. Interaction of nNOS with PSD-95 negatively controls regenerative repair after stroke. J. Neurosci. 1353; 34. [Google Scholar]
- Lai, T. W. , Zhang, S. & Wang, Y. T. Excitotoxicity and stroke: Identifying novel targets for neuroprotection. Prog. Neurobiol.
- Hong, J. M. et al. Safety and optimal neuroprotection of neu2000 in acute Ischemic stroke with recanalisation: study protocol for a randomised, doubleblinded, placebo-controlled, phase-II trial. 19.
- Lai, T. W. , Shyu, W.-C. & Wang, Y. T. Stroke intervention pathways: NMDA receptors and beyond. Trends Mol. Med. 17.
- Rosenberg, G. A. & Yang, Y. Vasogenic edema due to tight junction disruption by matrix metalloproteinases in cerebral ischemia. Neurosurg. Focus. 22.
- Bao Dang, Q. et al. High-density lipoproteins limit neutrophil-induced damage to the blood-brain barrier in vitro. J. Cereb. Blood Flow. Metab. 33.
- Barr, T. L. et al. Blood-brain barrier disruption in humans is independently associated with increased matrix metalloproteinase-9. 41.
- Chelluboina, B. et al. Matrix metalloproteinase-12 induces blood-brain barrier damage after focal cerebral ischemia. 3523; 46. [Google Scholar]
- Shen, Y. et al. Inhibition of HIF-1 alpha reduced blood brain barrier damage by regulating MMP-2 and VEGF during acute cerebral ischemia. Front. Cell. Neurosci. 12.
- Yang, Y. et al. Non-invasive vagus nerve stimulation reduces blood-brain barrier disruption in a rat model of ischemic stroke. Brain Stimul. 11.
- Liu, H. et al. Hydrogen sul"de attenuates tissue plasminogen activator-induced cerebral hemorrhage following experimental stroke. Transl. Stroke Res. 7.
- Khan, I. S. et al. Intraarterial administration of norcantharidin attenuates ischemic stroke damage in rodents when given at the time of reperfusion: novel uses of endovascular capabilities. J. Neurosurg.
- Michalski, D. et al. Early outcome and blood-brain barrier integrity after coadministered thrombolysis and hyperbaric oxygenation in experimental stroke. Exp. Transl. Stroke Med. 3.
- Mohamed, I. N. , Ishrat, T., Fagan, S. C. & El-Remessy, A. B. Role of inflammasome activation in the pathophysiology of vascular diseases of the neurovascular unit. Antioxid. Redox Signal. 1188; 22. [Google Scholar]
- Fann, D. Y.-W. et al. Pathogenesis of acute stroke and the role of inflammasomes. Ageing Res. Rev. 12.
- Yang, F. et al. NLRP3 deficiency ameliorates neurovascular damage in experimental ischemic stroke. J. Cereb. Blood Flow. Metab. 34.
- Chi, W. et al. HMGB1 promotes the activation of NLRP3 and caspase-8 inflammasomes via NF-kappa B pathway in acute glaucoma. J. Neuroinflammation. 12.
- Hong, P. et al. NLRP3 inflammasome as a potential treatment in ischemic stroke concomitant with diabetes. J. Neuroinflammation.
- Wang, S. Q. et al. Genistein attenuates acute cerebral ischemic damage by inhibiting the NLRP3 inflammasome in reproductively senescent mice. Front. Aging Neurosci.
- Ye, X. C. et al. Purinergic 2X7 receptor/NLRP3 pathway triggers neuronal apoptosis after ischemic stroke in the mouse. Exp. Neurol.
- Mennicken, F. , Maki, R., de Souza, E. B. & Quirion, R. Chemokines and chemokine receptors in the CNS: a possible role in neuroinflammation and patterning. Trends Pharmacol. Sci. 20.
- Fang, W. et al. CCR2-dependent monocytes/macrophages exacerbate acute brain injury but promote functional recovery after ischemic stroke in mice. 3530; 8. [Google Scholar]
- Guo, Y.-Q. et al. Expression of CCL2 and CCR2 in the hippocampus and the interventional roles of propofol in rat cerebral ischemia/reperfusion. Exp. Ther. Med. 8.
- Dimitrijevic, O. B. , Stamatovic, S. M., Keep, R. F. & Andjelkovic, A. V. Absence of the chemokine receptor CCR2 protects against cerebral Ischemia/reperfusion injury in mice. 1345; 38. [Google Scholar]
- Takami, S. et al. Chemokine receptor antagonist peptide, viral MIP-II, protects the brain against focal cerebral ischemia in mice. J. Cereb. Blood Flow. Metab. 1430; 21. [Google Scholar]
- Hammond, M. D. et al. CCR2(+) Ly6C(hi) inflammatory monocyte recruitment exacerbates acute disability following intracerebral hemorrhage. J. Neurosci. 3901; 34. [Google Scholar]
- Wattananit, S. et al. Monocyte-derived macrophages contribute to spontaneous long-term functional recovery after stroke in mice. J. Neurosci. 4182; 36. [Google Scholar]
- Hou, Y. , Wang, J. & Feng, J. The neuroprotective effects of curcumin are associated with the regulation of the reciprocal function between autophagy and HIF-1α in cerebral ischemia-reperfusion injury. Drug Des., Dev. Ther. 1135; 13. [Google Scholar]
- Semenza, G. L. Regulation of oxygen homeostasis by hypoxia-inducible factor 1. 24.
- Ziello, J. E. , Jovin, I. S. & Huang, Y. Hypoxia-Inducible Factor (HIF)-1 regulatory pathway and its potential for therapeutic intervention in malignancy and ischemia. Yale J. Biol. Med. 80.
- Kim, J. W. , Tchernyshyov, I., Semenza, G. L. & Dang, C. V. HIF-1 mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 3.
- Hu, C. J. et al. Differential regulation of the transcriptional activities of hypoxia-inducible factor 1 alpha (HIF-1 alpha) and HIF-2 alpha in stem cells. Mol. Cell. Biol. 3514; 26. [Google Scholar]
- Chrostek, et al., 2019. Efficacy of stem cell-based therapies for stroke. Brain Res. 1722, 146362. 10.1016/j.brainres.2019. 1463.
- Bergström T, Forsberg-Nilsson K, 2012. Neural stem cells: Brain building blocks and beyond. Ups. J. Med. Sci. 117, 132–142. 10.3109/03009734.2012. 6650.
- Boese AC, et al., 2018. Neural stem cell therapy for subacute and chronic ischemic stroke. Stem Cell Res. Ther. 9, 154. 10. 1186.
- Baker EW, Kinder HA, West FD, 2019. Neural stem cell therapy for stroke: A multimechanistic approach to restoring neurological function. Brain Behav. 9, e01214. 10.1002/brb3. 1214.
- Zhang G-L, Zhu Z-H, Wang Y-Z, 2019. Neural stem cell transplantation therapy for brain ischemic stroke: Review and perspectives. World J. Stem Cells 11, 817–830. 10.4252/wjsc.v11.i10.
- Rascón-Ramírez FJ, et al., 2021. Are We Ready for Cell Therapy to Treat Stroke? Front. Cell Dev. Biol.
- Modi J, Menzie-Suderam J, Xu H, Trujillo P, Medley K, Marshall ML, Tao R, Prentice H, Wu J-Y, 2020. Mode of action of granulocyte-colony stimulating factor (G-CSF) as a novel therapy for stroke in a mouse model. J. Biomed. Sci. 27, 19. 10. 1186.
- Chen K-H, et al., 2020. Human Umbilical Cord–Derived Mesenchymal Stem Cell Therapy Effectively Protected the Brain Architecture and Neurological Function in Rat After Acute Traumatic Brain Injury. Cell Transplant. 29, 0963689720929313. 10. 1177.
- Liu G, David BT, Trawczynski M, Fessler RG, 2020. Advances in Pluripotent Stem Cells: History, Mechanisms, Technologies, and Applications. Stem Cell Rev. Rep. 16, 3–32. 10. 1007.
- Pan Y, Shi G, 2021. Silver Jubilee of Stroke Thrombolysis With Alteplase: Evolution of the Therapeutic Window. Front. Neurol.
- Frank D, et al., 2022. The Development of Novel Drug Treatments for Stroke Patients: A Review. Int. J. Mol. Sci. 23, 5796. 10. 3390.
- Hao Z, et al., 2012. Fibrinogen depleting agents for acute ischaemic stroke. Cochrane Database Syst. Rev CD000091. 10.1002/14651858.CD000091.
- Liu J, Zhang J, Wang LN. Gamma aminobutyric acid (GABA) receptor agonists for acute stroke. Cochrane Database Syst Rev. 2018 Oct 30;10(10):CD009622. [CrossRef] [PubMed]
- Rothwell PM, et al., 2010. Effects of beta blockers and calcium-channel blockers on within-individual variability in blood pressure and risk of stroke. Lancet Neurol. 9, 469–480. 10. 1016.
- Lai TW, Zhang S, Wang YT, 2014. Excitotoxicity and stroke: Identifying novel targets for neuroprotection. Prog. Neurobiol., 2013 Pangu Meeting on Neurobiology of Stroke and CNS Injury: Progresses and Perspectives of Future 115, 157–188. [CrossRef]
- Wu QJ, Tymianski M, 2018. Targeting NMDA receptors in stroke: new hope in neuroprotection. Mol. Brain 11, 15. 10. 1186.
- Alhadidi Q, et al., 2018. Cofilin Knockdown Attenuates Hemorrhagic Brain Injury-induced Oxidative Stress and Microglial Activation in Mice. Neuroscience 383, 33–45. 10.1016/j.neuroscience.2018.04.
- Alaqel SI, et al., 2022. Synthesis and Development of a Novel First-in-Class Cofilin Inhibitor for Neuroinflammation in Hemorrhagic Brain Injury. ACS Chem. Neurosci. 10.1021/acschemneuro. 0001.
- Rahmati M, Ferns GA, Mobarra N. The lower expression of circulating miR-210 and elevated serum levels of HIF-1α in ischemic stroke; Possible markers for diagnosis and disease prediction. J Clin Lab Anal. 2021 Dec;35(12):e24073. [CrossRef] [PubMed]
Figure 1.
A summary of the pathophysiology involved in ischemic stroke. A) Excitotoxicity in ischemic stroke, in which excessive glutamate is released, and both synaptic and extra synaptic NMDARs are involved; B) Neuroinflammation and BBB breakdown in ischemic stroke; C) Oxidative stress, which is mainly characterized by ROS production and mitochondrial dysfunction that involves Ca2+influx into mitochondria ad MPTP in ischemic stroke; D) Cell death signalling pathways, which manly involves autophagy, apoptosis and necroptosis in ischemic stroke.
Figure 1.
A summary of the pathophysiology involved in ischemic stroke. A) Excitotoxicity in ischemic stroke, in which excessive glutamate is released, and both synaptic and extra synaptic NMDARs are involved; B) Neuroinflammation and BBB breakdown in ischemic stroke; C) Oxidative stress, which is mainly characterized by ROS production and mitochondrial dysfunction that involves Ca2+influx into mitochondria ad MPTP in ischemic stroke; D) Cell death signalling pathways, which manly involves autophagy, apoptosis and necroptosis in ischemic stroke.
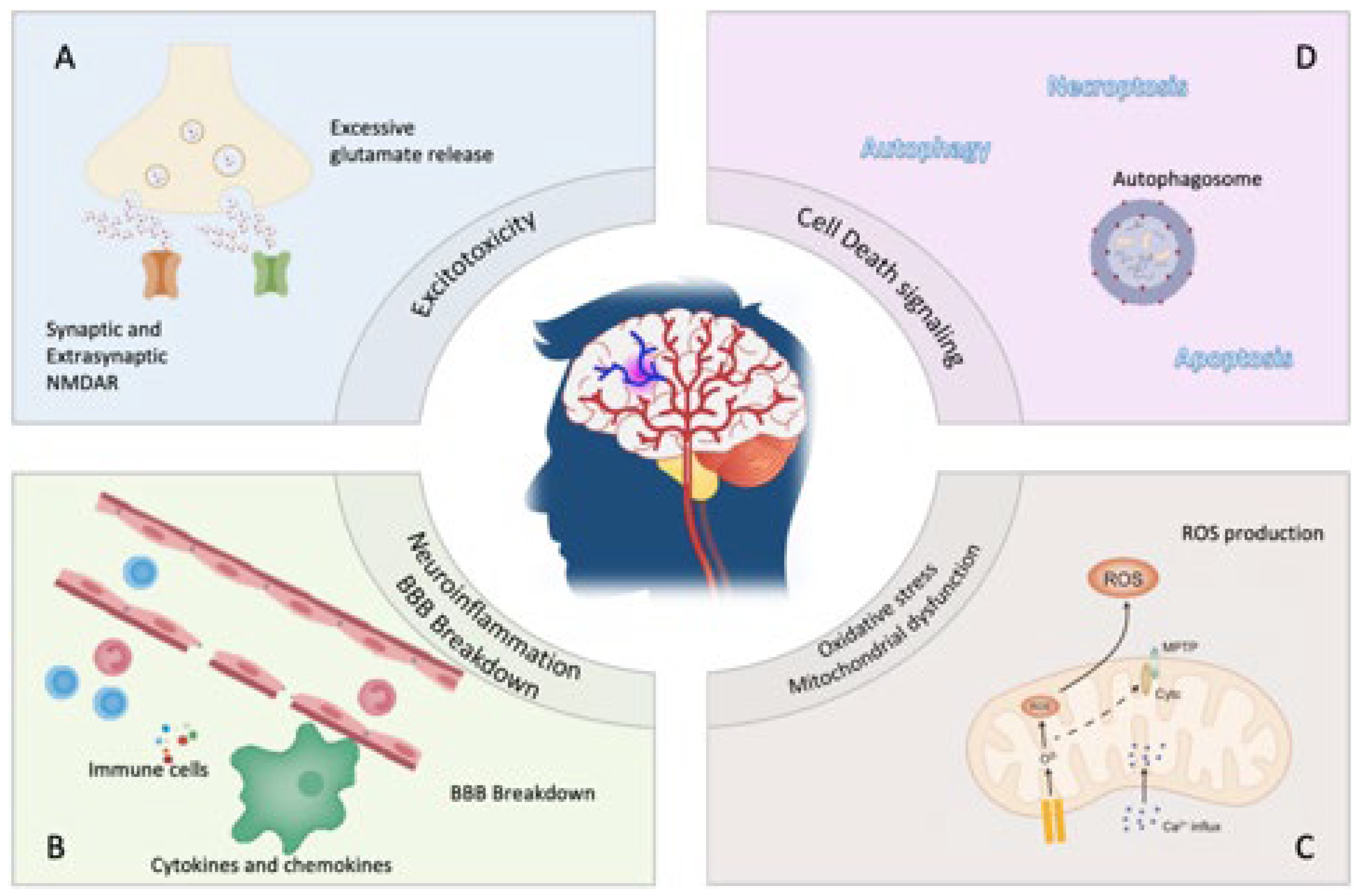

Table 1.
A table summarizing the temporal trend, the actions of each immune cell after stroke and the produced cytokines and chemokines.
Table 1.
A table summarizing the temporal trend, the actions of each immune cell after stroke and the produced cytokines and chemokines.
| Immune Cells | Temporal Trend | Produced Cytokines/ Chemokines | Action(s) |
|---|---|---|---|
| Neutrophils | Accumulate after 3h, peak at day 1–3, and dissipate over 7 days | Elastases MMP-9 IL-1, VEGF ROS, MMP-9 Annexin-1 Resolvins Protectins |
Cerebral edema, BBB destruction, and neuronal death Degradation of DAMP signaling and vascular remodeling Cerebral angiogenesis Microglia migration toward the infarct core after 1 day Decrease neutrophil migration and pro-inflammatory cytokine release |
| Mast cells | Significant increase after 24h | Histamine Heparin Vasoactive agents Chymase MMP-2, 9 |
Destruct BBB, increase vascular permeability, leukocyte recruitment, cerebral edema, destroy tight junctions, and disrupt hemostasis |
| Monocyte/ Macrophage | Shown as early as 3 h, peak at day 3, and become anti-inflammatory at day 7 |
TNF-α IL-1β IL-10, 23 TGF-β PDGF CD302, 163, 206 Fibronectin 1 Arginase 1 |
Augment immune responses IL-17a production from T-cells Tissue repair and wound healing |
| 4NK cells | 3 h, peak at 12 h, and remain elevated at least 4 days | IFN-γ IL-17a, 6, 12, 1β TNF-α ROS |
Augment immune responses and development of cerebral infarction |
|
CD4-/CD8- T-cells |
1–3 days | TNF-α | Augment immune responses |
| CD8+ T-cells | Detected as early as 3 h and stay for about 30 days | Perforin Fas ligand |
Neurotoxicity and augment immune responses |
|
CD4+ T-cells (Th1 and Th17) CD4+ T-cells (Th2) |
Shown at 24 h and stay for about 30 days | IL-2, 12, 17, 21, 22, 23 TNF-α IFN-γ IL-4, 5, 6, 10, 13 |
Augment immune responses Immunosuppression |
| Tregs | Shown after several days and stays for about 30 days |
IL-10 IL-17 (in certain conditions) |
Suppress astrogliosis, regulate astrocyte neurotoxicity, and functional recovery Inhibit CD4+ T-cell proliferation |
| B-cells | Delayed appearance after weeks of onset | IL-10 | Neuroprotection |
Table 2.
This table provides an overview of studies that have analyzed the role of miRNAs in the pathogenesis of ischemic and hemorrhagic stroke.
Table 2.
This table provides an overview of studies that have analyzed the role of miRNAs in the pathogenesis of ischemic and hemorrhagic stroke.
| Authors | Stroke type | MIRNA involved |
MIRNA profile | Role |
|---|---|---|---|---|
|
Iwuchukwu et al. [176] |
Hemorragic | Panel: miRNA 204-5p + miRNA 9-5p + miRNA-338-3p | Lower | Target: MMP-9 Elevated MMP -> Increased damage during acute phase of ICH |
|
Tan et al. [147] |
Ischemic | miRNA 126 miRNA 130 |
Increased Increased |
Endotelial cell / CV functions Angiogenesis |
|
Wang et al. [151] |
Hemorragic | miRNA-126 miRNA 21-5p |
Lower Lower |
Endotelial cell / CV functions Protective role against ischemia induced apoptosis |
|
Wen et al. [144] |
Ischemic | miRNA-124 | Increased with MCAO (middle cerebral artery occlusion) | miR-155 can exert both pro- and antiinflammatory effects by targeting different mediators of inflammatory signaling, such as SHIP1, SOCS1, SMAD2 and TAB2 |
|
Zhang et al. [143] |
Post-ischemic neuronal damage | miRNA-181c | Lower | miRNA-181 suppress TNF-a expression |
|
Rahmati et al. [255] |
Ischemic | miRNA-210 + HIF1-a | Lower | HIF-1° induce miRNA—210: could prevent apoptosis, protect stem cell survivance, induce angiogenesis |
|
Tao Z. et al. [177] |
Ischemic | miRNA 99a | Lower | MiR-99a prevented apoptosis and blocked cell cycle progression in neuro-2a cel |
|
Yin et al. [178] |
Ischemic | miRNA-497 | Increased | miR-497 promotes ischemic neuronal death by negatively regulating anti-apoptotic proteins, bcl-2 and bcl-w |
|
Zhao et al. [179] |
Ischemic | miRNA-424 | Lower | Expression prevented ischemic brain injury through a mechanism involving suppressing microglia activation |
|
Yuah et al. [158] |
Hemorragic | miRNA-367 | Lower | miR-367 was a crucial regulator of TLRs downstream NF-κB signaling by direct targeting IRAK4 |
|
Moon et al. [157] |
Ischemic | miRNA-181 | Increased in infarct core; decreased in penumbra after focal ischemia |
miR-181 was also shown to sensitize glioblastoma cells to apoptosis by reducing Bcl-2 |
|
Li Y. et al. [169] |
Ischemic (MCAO) | miRNA-107 | Increased | might regulate post-stroke angiogenesis and therefore serve as a therapeutic target. |
|
Sun et al. [172] |
Ischemic | microRNA-15a/16–1 | Increased | represses pro angiogenic factors VEGFA, FGF2, and their receptors VEGFR2 and FGFR1 |
|
Yang et al. [159] |
Hemorragic | microRNA-223 | Lower |
Could downregulate NLRP3 to inhibit inflammation and brain edema |
|
Xin et al. [174] |
Ischemic (MCAO) | microRNA-133 | Lower | overexpressing MSCs further stimulate and increase exosomes release from astrocytes, possibly by downregulating the RABEPK expression |
|
Xu et al. [175] |
Ischemic | microRNA-1906 | Increased in glial cell Decreased in neurons |
Abolishment of TLR4 protein expression; could ameliorate brain injury in experimental stroke |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



