
Submitted:
17 April 2024
Posted:
18 April 2024
You are already at the latest version
Abstract
Alzheimer´s disease (AD) is characterized by the brain deposition of senile plaques composed by amyloid-β peptides (Aβ) that increase inflammation. An endogenous peptide derived from the insulin-like growth factor (IGF)-I, glycine-proline-glutamate (GPE) has IGF-I-sensitizing and neuroprotective actions. Here, we examined the effects of GPE on Aβ levels and hippocampal inflammation generated by the intracerebroventricular infusion of Aβ25-35 during 2 weeks (300 pmol/day) in ovariectomized rats and the signaling-related pathways and levels of Aβ-degrading enzymes associated with these GPE-related effects. GPE prevented the Aβ-induced increase in the phosphorylation of p38 mitogen-activated protein kinase and the reduction in activation of signal transducer and activator of transcription 3, insulin receptor substrate-1 and Akt, as well as on interleukin (IL)-2 and IL-13 levels in the hippocampus. The functionality of somatostatin, measured as the percentage of inhibition of adenylate cyclase activity and the levels of insulin-degrading enzyme were also preserved by GPE co-treatment. These findings indicate that GPE co-administration may protect from Aβ insult by changing hippocampal cytokine content and somatostatin functionality through regulation of leptin- and IGF-I-signaling pathways that could influence the reduction in Aβ levels through modulation of levels and/or activity of Aβ proteases.
Keywords:
Alzheimer´s disease
; cytokines
; Gly-Pro-Glu
; IGF-I signaling
; inflammation
1. Introduction
Alzheimer’s disease (AD) is an irreversible pathology that predominantly affects individuals over the age of 65 and is influenced by many factors that contribute to its onset and progression. These include the accumulation of intracellular neurofibrillary tangles and the presence of extracellular deposits of amyloid fibrils at the core of senile plaques, which are associated with neuronal death and a decline in cognitive function [1]. One of the main components of these plaques is amyloid β-peptide (Aβ), produced from the amyloid precursor protein (APP) by sequential enzymatic alternative processing, and is considered to be a key factor in the pathogenesis of the disease [2].
Inflammation is another important factor contributing to the pathogenesis of AD through the activation of microglia and astrocytes, leading to the secretion of pro-inflammatory cytokines [3]. This dysregulation of interleukins and chemokines in the brain causes neurodegeneration through modulation of several signaling pathways, most notably nuclear factor kappa B (NFκB) [4]. One of the factors that affect the generation of an inflammatory milieu is the increased production and deposition of Aβ peptides that activate microglia and the subsequent production of cytokines that further enhance Aβ synthesis [5], a vicious circle that leads to neuronal death and pathological changes in astrocytes that impair Aβ clearance [6].
Chronic infusion of Aβ peptides is an experimental approach to AD as it induces hippocampal Aβ deposition associated with neuronal death, deficits in synaptic plasticity and learning, as well as changes in the inflammatory milieu similar to those seen in AD [7,8]. In particular, the neurotoxic fragment Aβ25-35 fragment has a more pronounced deleterious effect than Aβ1-42 [9], as it is associated with the key domain for aggregation [10].
Neurodegenerative diseases are a serious health concern worldwide, with a high incidence due to increasing life expectancy and the lack of restorative treatments. Therapies based on the use of different proteins have emerged as a possible strategy due to their high specificity and activity on different biological targets [11]. Several endogenous peptides have anti-apoptotic and neuroprotective properties in the central nervous system and among which, glycine-proline-glutamate (GPE) a natural peptide cleaved from the N-terminus of insulin-like growth factor I (IGF)-I, is a protective agent in brain injury [12] and has shown neuroprotective capabilities in experimental models of AD [13,14].
GPE and its analogues have anti-inflammatory properties, which is one of their most important effects, since inflammation favors aggregation processes and decreases the efficiency of glial cells in the processes of clearance of Aβ aggregates [6]. In this sense, IGF-I is involved in Aβ clearance [15] and also activates the Akt pathway, as does GPE [16]. A de-crease in IGF-I sensitivity increases Aβ toxicity, while activation of its intracellular pathway is associated with an increase in the synthesis and activity of Aβ-degrading enzymes [17], such as insulin-degrading enzyme (IDE).
These aforementioned data suggest that GPE may be useful in AD. However, there is little information on the efficacy of GPE on the possible protective effect against the inflammatory environment generated by continuous infusion of Aβ25-35 and its relation-ship with changes in activation of the Akt pathway. Therefore, we analyzed the activation of some signaling pathways involved in the alterations of the inflammatory environment in the hippocampus by studying several pro- and anti-inflammatory cytokines after Aβ infusion in the presence and absence of peripherally administered GPE. As the expression of Aβ-degrading enzymes is related to changes in the Akt pathway, we studied its activation, as well as the leptin signaling that can modulate it and others related to the expression of certain cytokines [18,19]. Finally, since somatostatin (SRIF) modulates the action of Aβ proteases [20], we studied the functionality of this neuropeptide after Aβ infusion and the effect of GPE therapy.
2. Results
2.1. GPE Reduces Hippocampal Aβ25-35 Levels and Activation of Inflammatory Pathways After Aβ25-35 Infusion
Aβ25-35 infusion increased its levels in the hippocampus and this augment was partially blocked by co-administration of GPE. GPE treatment of control rats did not alter Aβ25-35 levels (Figure 1A). Phosphorylation of p38 mitogenactivated protein kinase (p38MAPK) was increased in Aβ25-35-treated rats and GPE prevented the changes induced by Aβ (Figure 1B). Phosphorylation of NFκB at Ser536 is essential for the inhibition of NFκB responses, thereby counteracting inflammatory processes [21]. Infusion of Aβ25-35 reduced NFκB phosphorylation at Ser536 and co-administration of GPE prevented the changes induced by Aβ25-35, whereas GPE had no effect on NFκB in control rats (Figure 1C).
2.2. GPE Partially Counteracts the Inhibitory Effects of Aβ25-35 on the Activation of Leptin Signaling
Chronic infusion of Aβ25-35 reduced serum leptin levels and co-administration of GPE prevented the effects of this toxic fragment on circulating leptin concentrations, whereas administration of GPE to control rats had no effect (Figure 1D). Phosphorylation of signal transducer and activator of transcription 3 (STAT3) at Tyr 705 was reduced in both Aβ25-35 and Aβ25-35 plus GPE-treated rats (Figure 1E), and reduction in phosphorylation of STAT3 at Ser 727 induced by Aβ25-35 was avoided by GPE co-administration (Figure 1F).
2.3. Aβ25-35-Induced Downregulation of IGF-I-Related Signalling is Prevented by GPE Treatment
Serum IGF-I levels were not modified by Aβ25-35 infusion and GPE increased IGF-I when administered to Aβ25-35 and control rats (Figure 2A). Hippocampal IGF-I levels did not change in any of the experimental groups (Figure 2B). Phosphorylation of the IGF-I receptor (IGF-IR) at specific tyrosine residues is a critical step in the activation ofof this signaling pathway [22]. Phosphorylation of IGF-IR on Tyr1131 was decreased in Aβ25-35 treated rats treated, with no changes in the other groups studied (Figure 2C). Phosphorylation of insulin receptor substrate (IRS)-1 on Tyr residues was reduced in Aβ25-35- and Aβ25-35 plus GPE-treated groups (Figure 2D). Phosphorylation of IRS-1 at Ser 636 residue inhibits the activation of downstream targets [23]. Aβ25-35 infusion increased phosphorylation at this residue and GPE co-administration prevented this increase (Figure 2E). Finally, Akt phosphorylation at Thr308 was reduced after Aβ25-35 infusion and GPE co-administration prevented this decrease, whereas GPE had no effect in control rats (Figure 2F).
2.4. Effects of Aβ25-35 and GPE on Serum and Hippocampal Cytokine Content
Circulating levels of interferon-γ (IFN-γ) were increased in Aβ25-35-treated rats (Figure 3A), interleukin (IL)-2 was unchanged (Figure 3C), IL-13 was decreased after Aβ25-35 infusion (Figure 3E), and concentrations of IL-17A were increased in Aβ25-35- and Aβ25-35 plus GPE-treated rats (Figure 3G). Hippocampal concentrations of IFN-γ were augmented in Aβ25-35-treated rats (Figure 3B), IL-2 levels were reduced after Aβ25-35 infusion and co-administration of GPE- prevented this reduction (Figure 3D), IL-13 levels were reduced after Aβ25-35 infusion and GPE increased IL-13 levels compared to control rats (Figure 3F) and IL-17A concentrations were augmented in Aβ25-35-treated rats and co-administration of GPE prevented this increase (Figure 3H).
2.5. Aβ25-35 and GPE Are Involved in Modulating the Activity of AC and the Levels of an Aβ-Degrading Enzyme
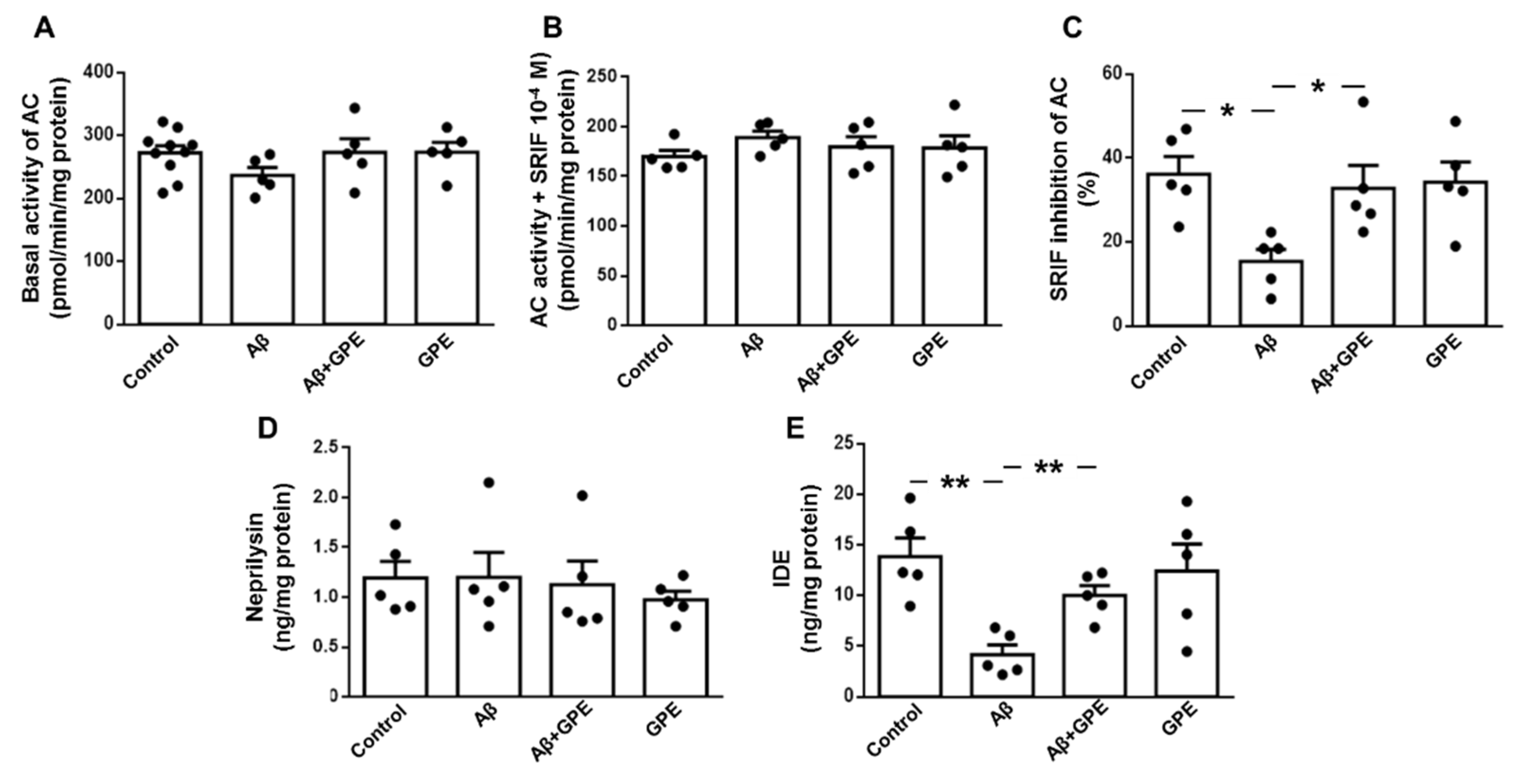
SRIF stimulates the activity and levels of Aβ-degrading enzymes [24,25]. We therefore investigated the functionality of this neurotransmitter. As SRIF receptors are coupled to adenylate cyclase (AC) in an inhibitory manner, we examined basal AC activity, and SRIF-mediated inhibition in membrane fraction from hippocampus. No differences in basal and inhibited AC activitiy were observed between the experimental groups (Figure 4A and 4B, respectively). However, the capacity of SRIF to inhibit basal AC activity was significantly lower in the Aβ25-35-treated group, without changes in the other groups (Figure 4C).
Hippocampal neprilysin levels were similar in all experimental groups (Figure 4D). IDE concentrations were reduced in Aβ25-35-treated rats and co-administration of GPE prevented the effects of Aβ infusion. GPE alone had no effect in the control group (Figure 4E).
Figure 4.
Effects of Aβ25-35 and GPE co-administration on basal adenylyl cyclase (AC) activity (pmol/min/mg protein) as well as on somatostatin (SRIF)-mediated inhibition of AC activity in hippocampal membranes (A and B, respectively), percentage of SRIF inhibition of AC activity (C), levels of neprilysin (D) and insulin-degrading enzyme (IDE) (E) in ovariectomized (Ovx) rats (control), Ovx rats treated with β-amyloid 25-35 peptide (Aβ), Ovx rats treated with Aβ25-35 plus GPE (Aβ + GPE) and Ovx rats treated with GPE (GPE). Data are expressed as mean ± SEM. N = 5. * p < 0.05, ** p < 0.01.
Figure 4.
Effects of Aβ25-35 and GPE co-administration on basal adenylyl cyclase (AC) activity (pmol/min/mg protein) as well as on somatostatin (SRIF)-mediated inhibition of AC activity in hippocampal membranes (A and B, respectively), percentage of SRIF inhibition of AC activity (C), levels of neprilysin (D) and insulin-degrading enzyme (IDE) (E) in ovariectomized (Ovx) rats (control), Ovx rats treated with β-amyloid 25-35 peptide (Aβ), Ovx rats treated with Aβ25-35 plus GPE (Aβ + GPE) and Ovx rats treated with GPE (GPE). Data are expressed as mean ± SEM. N = 5. * p < 0.05, ** p < 0.01.
2.6. GPE does not Alter the Aβ25-35-Induced Decrease in Leptin and IGF Signaling in Neuronal Cultures
2.7. GPE Co-Administration Modifies Aβ25-35-Induced Changes in Glial Cell Signaling and Cytokine Secretion
Aβ25-35 decreased phosphorylation of STAT3 and IRS1 and IDE levels in glial cells (Figure 5D, 5E and 5F, respectively). Co-administration of GPE partially prevented changes in STAT3 and IRS1 phosphorylation (Figure 5D and 5F) and fully prevented Aβ25-35-induced IDE reduction.(Figure 5F).
Levels of IFN-γ in culture media were unaffected by Aβ-25-35 or GPE co-administration (Figure 6A). Aβ25-35 induced a reduction of IL-2 levels that was prevented by co-administration of GPE (Figure 6B). Concentrations of IL-13 were reduced after addition of Aβ25-35 and GPE had no effect (Figure 6C). Finally, Aβ-25-35 augmented IL-17A levels in culture media and GPE co-administration partially prevented these changes (Figure 6D).
2.8. Aβ25-35 Content Shows an Inverse Relation to IL-2, SRIF Functionality and IDE
Hippocampal concentrations of Aβ-25-35 were inversely correlated with hippocampal IL-2 content (Figure 7A) and the capacity of SRIF to inhibit AC activity, a measure of SRIF action (Figure 7B). Levels of Aβ25-35 did not show a relationship with neprilysin (Figure 7C), but did show a negative association with IDE (Figure 7D).
2.9. Correlation of Aβ25-35, SRIF Functionality and Aβ-Degrading Enzymes with Phosphorylation of Signaling Targets and Cytokine Levels in the Hippocampus
Linear regression analyses showed a direct correlation of Aβ25-35 levels with phosphorylation of the pro-inflammatory signaling targets, IFN-γ and IL-17A, and an inverse relationship with phosphorylation of the leptin- and IGF-I-signaling targets, IL-2 and IL-13. In contrast, the percentage inhibition of AC by SRIF and IDE concentrations exhibited an inverse relationship with phosphorylation of pro-inflammatory targets IFN-γ and IL-17A, and a positive correlation with leptin- and IGF-I-intracellular targets, IL-2 and IL-13 (Table 1). No correlations were found between neprilysin and the above-mentioned markers (data not shown).
3. Discussion
Extracellular plaque-like deposits within the hippocampus lead to cognitive impairment and cause inflammation as Aβ protofibrils activate microglia, triggering an inflammatory response and the release of neurotoxic cytokines [26]. This study was designed to analyze the effect of a neuroprotective agent derived from IGF-I, the tripeptide GPE, on the changes in the inflammatory environment of the hippocampus and its possible relationship with the activation of various signaling pathways related to these processes. Here, we report that GPE blocks most of the changes in cytokine content in the hippocampus induced by continuous infusion of Aβ and that this effect may be mediated by preserving the activation of leptin- and IGF-I-related signaling pathways. In addition, we show that the decrease in IDE after Aβ insult is blocked by co-administration of GPE, contributing to the reduction in hippocampal Aβ levels.
Our experimental model is a chronic infusion of Aβ, which induces some of the major changes seen in AD patients, such as cognitive deficits [27] and increased brain inflammation [28]. The Aβ25-35 fragment was chosen because it is proposed to be the functional domain of Aβ responsible for its neurotoxic properties and it is also present in the brain of AD patients [10]. We chose the experimental model of female rats after ovariectomy, as estrogens diminish Aβ toxicity [29] and most women who suffer from AD are elderly and their estrogen levels have already dropped [30]. In addition, inhibition of estradiol synthesis affects hippocampal synaptic plasticity only in females [31].
Our data shows an increase in the activation of pro-inflammatory signaling targets after Aβ infusion. There was an augment in p38MAPK phosphorylation and a reduction in the Ser residue of NFκB, that activates this molecule. As we have found in this study, it has previously been reported that activation of these targets increases the levels of IFN-γ and IL-17A [32], while decreasing the content of the anti-inflammatory IL-13 [33]. A striking finding was the decrease in hippocampal levels of IL-2, a cytokine classically associated with an inflammatory profile. This finding may be related to the decrease in STAT-3 activation, since phosphorylation of this target increases the levels of this interleukin and its subsequent signaling [34,35]. One of the mechanisms that may influence STAT-3 phosphorylation is Aβ itself, as it is a negative allosteric modulator of the leptin receptor [36], with consequent decreased activation of downstream targets.
Co-administration of GPE was able to modify most of the Aβ-induced changes in signaling pathways and inflammation. Hence, systemic administration of GPE reduces p38MAPK activation [37] and suppresses the NFκB inflammatory pathway in experimental models of neurodegenerative disease [38]. The effects of GPE mimic those exerted by IGF-I, increasing Akt activation [16], although it does not bind to IGF-IR. Activation of the Akt pathway may be favored by the increase in leptin signaling after GPE co-administration, as it was reported in other situations [39], and the increase in serum leptin levels may explain, at least in part, the activation in its signaling and subsequent phosphorylation of IGF-I-related targets. In this way, disruption of leptin signaling in a mouse model of AD reduces Akt in parallel with upregulation of suppressor of cytokine signaling 3 (SOCS3) in the hippocampus [40], and we have demonstrated that central infusion of leptin reduced the association of SOCS3 with IGF-IR, increasing its phosphorylation and activation of downstream targets [41].
A role for reactive glia in neuronal damage and recovery has been reported. Treatment with GPE suppresses microglial proliferation and prevents the loss of astrocytes after injury [42]. Our “in vitro” experiments seem to demonstrate that glial cells are involved in the modifications of cytokine levels in the hippocampus, both after Aβ administration and co-treatment with GPE, which partially or totally restores the levels of cytokines affected by Aβ infusion. Aβ activates astrocytes, inducing an increase in GFAP, vimentin and pro-inflammatory cytokines, whereas GPE normalizes GFAP, vimentin and cytokine profile [43,44]. Hence, GPE binds to astrocytes and reduces brain inflammation [45].
Not only the changes in the activation of signaling pathways studied here modify cytokine levels, but these factors can also regulate signaling themselves. For example, IL-2 synergizes with IGF-I in processes related to memory enhancement in experimental animals and promotes Akt activation in homeostatic processes of proliferation [46]. IL-13 also has anti-apoptotic and proliferative effects in different tissues modulating the pathways analyzed here. Antiapoptotic effects of this interleukin have been described through activation of the Akt pathway [47] and proliferative effects by increasing STAT-3 phosphorylation [48]. Therefore, among the multiple activities associated with the pathological conditions of AD [49], we may speculate that GPE may be prevent/reverse Aβ damage through changes in interrelated signaling pathways and cytokine profiles, thereby enhancing its beneficial actions on this disease.
This study shows that the deleterious effects of Aβ on SRIF functionality are blocked by GPE. Although the regulatory mechanisms of SRIF tone are partially unknown, both our previous results [13] and new data included in this study suggest that activation of leptin and IGF-I signaling may be involved in the protective effect of GPE on this neurotransmitter. Leptin may be involved in the preservation of SRIF cells against Aβ effects as this adipokine protects against Aβ-induced cell death through a STAT3-dependent mechanism [50]. IGF-I-related signaling may promote SRIF synthesis, as Akt activation promotes CREB phosphorylation, which induces the expression of SRIF and its receptors [51].
One of the most striking findings was the reduction in Aβ levels when GPE was co-administered. In this way, the increase in SRIFergic tone may modulate the expression of Aβ-degrading proteases [52]. Here we found an increase in hippocampal IDE levels, with no differences in neprilysin content. Nevertheless, as the activity of neprilysin is regulated by SRIF [53], the increased functionality of this neuropeptide suggests an active role for this protease in the decrease of Aβ levels. IDE may also be regulated by phosphatidylinositol 3-kinase (PI3K) activation, as factors that augment Akt phosphorylation may rise IDE expression and synthesis [54].
We cannot rule out additional factors mediating the effects of GPE on Aβ levels. This tripeptide can be metabolized to cycloprolylglycine, another important metabolite of IGF-I [55]. This dipeptide improves memory and reduces Aβ plaque load in double transgenic mice APP/presenilin-1 (PS1) [56]. Leptin signaling may also be involved in the depletion of Aβ in the hippocampus, as has been reported in diabetic rats subjected to high-intensity interval training, which showed an increase in leptin receptor, Janus kinase 2 (JAK2) and STAT3, and a concomitant reduction in glycogen synthase kinase 3β (GSK3β), neurofibrillary tangles and Aβ levels [57].
Several interleukins may also be involved in the decrease in Aβ content, particularly IL-2 and IL-13, which increase after co-administration of GPE. For example, a decrease in IL-2 levels has been found in hippocampal biopsies from patients with AD. Furthermore, in the hippocampus of APP/PS1 transgenic mice, IL-2 administration induces the activation and regrouping of astrocytes around amyloid plaques, decrease Aβ content and improves synaptic plasticity [58]. Central infusion of IL-13 ameliorated cognitive deficits via degradation and clearance of intra- and extraneuronal Aβ peptides in APP23 mice, by modulating Aβ-degrading proteases [59]. The decrease in the hippocampal content of IL-17A levels after GPE co-treatment may also be related to diminished Aβ levels. Thus, this interleukin promotes AD progression in the APP/PS1 mouse model by increasing neuroinflammation through the NFκB pathway and Aβ deposition [60].
It is clear that more research is needed to better understand the role of changes in the activation of signaling pathways and their relationship with inflammatory markers in experimental models of this disease. Further “in vitro” studies could provide additional information on the effects of these cytokines in relation to changes in the activation of these signaling targets and enzymes involved in Aβ degradation, as well as the cell populations involved in these actions. Another aspect to take into account is the lack of memory testing in this study and the relationship with changes in peripheral inflammation. Our results showed inflammatory changes in the circulation, although they were more pronounced in the hippocampus. Some studies have shown an association between the increase in serum cytokines and the progressive decline in spatial memory after Aβ infusion [61]. In relation to this finding, there are also reports showing the association between biomarkers of inflammation and the degree of dementia in AD patients [62].
In summary, our results show that GPE activates signaling pathways that modulate the inflammatory milieu. These changes may increase the levels of one of the key Aβ-degrading enzymes, with a subsequent decrease in amyloid burden, one of the major hallmarks of this neurodegenerative disease. Given the limited success in the development of therapies for AD, GPE could be a successful tool to reduce one of the main factors affecting the development of this disease, and therefore represent a possible future perspective for the treatment of this disease.
4. Materials and Methods
4.1. Materials
All chemicals were purchased from Merck (Darmstadt, Germany) unless otherwise noted. Osmotic minipumps were from Alzet (Palo Alto, CA, USA).
4.2. Preparation of Aβ25-35
Aβ25-35 peptide was prepared according to the method reported by Burgos-Ramos et al. [63]. This fragment was dissolved in 1% acetic acid, following the manufacturer’s instructions. One day before the implantation, osmotic minipumps were connected and filled with 200 μl of Aβ25-35 solution and primed in 0.9% saline solution at 37 °C overnight [64].
4.3. Animals and Experimental Design
This study was approved by the Ethics Committee of the Universidad de Alcalá de Henares (SAF 2010–22277, Ministerio de Ciencia y Tecnología) and complied with Royal Decree 1201/2005 (Boletín Oficial del Estado, BOE No. 252) pertaining to the protection of experimental animals and with the European Communities Council Directive (86/609/EEC). Female Wistar rats, weighing 250–280 g, supplied by Harlan Laboratories Models S.L. (Barcelona, Spain), were housed in groups of 2 rats per cage on a 12 h light/dark cycle with free access to water and food and were allowed one week of acclimatization before the start of the experiments. Care was taken to use the minimum number of animals.
Twenty female Wistar rats of 8 weeks of age were bilaterally ovariectomized under anesthesia (0.02 mL of ketamine/100 g body weight and 0.04 mL of xylazine/100 g body weight) as previously reported [43]. Three weeks after ovariectomy, the animals were distributed into four groups. In the first group, a cannula attached to an osmotic minipump was implanted in the right cerebral ventricle (-0.3 mm anteroposterior, 1.1 mm lateral) and Aβ25-35 was infused for 14 days (300 pmol/day, infusion rate 0.5 μL/h) as described [65]. In a second group, Aβ25-35 was infused at the same time and dose, and three intraperitoneal injections of GPE (300 μg, dissolved in isotonic saline) were administered at 0, 6 and 12 days. Another group received GPE alone, as described for the previous group. Control rats received vehicle by the same administration routes. On day 14, the rats were sacrificed, the serum was stored at -80ºC and the brain was dissected on ice to obtain the hippocampus [66].
4.5. Tissue Homogenization and Protein Quantification
For immunodetection of Aβ25-35, phosphorylated (p) Thr308Akt, Akt, IDE, IFN-γ, IGF-I, IGF-IR, IL-2, IL-13, IL-17A, pSer636-IRS1, pTyr-IRS1, IRS1, pThr180/Tyr182-p38MAPK, p38MAPK, neprilysin, pSer536-NFkB, NFκB, pSer727STAT3, pTyr705STAT3 and STAT3, hippocampus was homogenized on ice in 400 μL of lysis buffer (Merck). Lysates were frozen 12 hours at -80ºC and then, centrifuged at 12,000X g for 5 min at 4ºC. Supernatants were stored at -80ºC until assayed. Protein levels were determined by the Bradford method (Bio-Rad Laboratories, Madrid, Spain).
4.6. ELISAs
4.6.1. Aβ25-35
Hippocampal levels of Aβ25-35 were determined using an ELISA kit from Blue Gene Biotech (China), with a monoclonal capture antibody against Aβ25-35 and another detection antibody conjugated to horseradish peroxidase (HRP). After 60 min incubation at 37°C, the wells were washed and incubated with a substrate and the absorbance was read at 450 nm.
4.6.2. Aβ-Degrading Enzymes
Neprilysin levels in the hippocampus were measured using an ELISA from Cusabio (Wuhan, China). Homogenates were incubated with a capture antibody for 120 min at 37°C. Once samples were removed, a biotin-antibody was added. After 60 min, HRP-avidin and a substrate were added until the color developed.
Levels of IDE were assessed using a kit from Cloud-Clone Corp. (Houston, TX, USA). After incubating the homogenates for 120 min with a biotin conjugated-IDE antibody, an avidin-HRP complex was added, incubated for 90 min at 37°C and subsequently washed. Substrate solution was added until a blue color developed.
4.6.3. IGF-I
Serum and hippocampal IGF-I concentrations were analyzed using an ELISA kit from R&D Systems (Minneapolis, MN, USA). Serum and homogenates were incubated with a monoclonal anti-IGF-I capture antibody for 120 min at 25ºC. After washing, conjugate was added and incubated for 120 min. Wells were washed again and incubated with a substrate solution for 30 min and the absorbance read at 450 nm.
4.6.4. Phosphorylation of IGF-I Receptor
The assay (Cell Signaling Technology, Danvers, MA, USA) detects levels of IGF-I receptor protein when phosphorylated at Tyr1131 residue. Homogenates were incubated for 120 min at 37ºC in a plate coated with the pTyr1131-IGF-I antibody. After washing, a detection antibody was added and incubated at 37ºC for 60 min. Afterwards, the plate was washed again and an HRP-linked secondary antibody was added and incubated 37ºC for 30 min. Finally, after washing, the substrate was added and the absorbance read at 450 nm.
4.6.5. Leptin
Serum leptin levels were measured using a kit from Merck. Standards, controls and samples were added together with a capture antibody, to a plate coated with a capture antibody. After 120 min of incubation, the plate was washed and the enzyme was added and incubated for 30 min. After washing, the substrate was added until the development of a blue color and then read at 450 nm.
The intra- and inter-assay coefficients of variation were lower than 10% for all assays.
4.7. Multiplexed Bead Immunoassays
Phosphorylated and total levels of Akt, IRS1, p38MAPK, NFκB and STAT3 in the hippocampus as well as concentrations of IFN-γ, IL-2, IL-13 and IL-17A in serum and hippocampus were measured using multiplexed bead immunoassays (Bio-Rad Laboratories and Merck) following the manufacturer’s recommendations. Beads conjugated to antibodies and serum or homogenates (25 μL each) were incubated, and antibody conjugated to biotin was added and incubated. Then, beads were incubated with streptavidin–phycoerythrin. At least 50 beads per variable were examined in the Bio-Plex suspension array system 200 (Bio-Rad Laboratories). Raw data (median fluorescence intensity, MFI) were evaluated using Bio-Plex Manager Software 6.2 (Bio-Rad Laboratories). The intra- and inter-assay coefficients of variation were lower than 10%.
4.8. Adenylyl Cyclase Assay
Membranes from the hippocampus were prepared as previously described [67]. Adenylyl cyclase activity was measured in membranes from hippocampus (0.06 mg/mL) incubated with 1.5 mM ATP, 5 mM MgSO4, 10 mM GTP, an ATP-regenerating system, 1 mM 3-isobutyl-1-methylxanthine, 0.1 mM phenylmethylsulphonyl fluoride, 1 mg/mL bacitracin, 1 mM EDTA, and 10-4 M SRIF. After a 15 min incubation at 30ºC, the reaction was stopped by heating. After cooling, 0.2 mL of an alumina slurry (0.75 g/mL in Tris/HCl buffer, pH 7.4) was added and the suspension was centrifuged. The supernatant was employed for the assay of cyclic AMP [68].
4.9. Cell Cultures and Treatments
4.9.1. Culture of Rat Hippocampal Neurons
Cultures were performed as reported [43]. Briefly, pregnant Sprague-Dawley rats were sacrificed and 18-day rat embryos collected. Hippocampi were dissected in Neurobasal medium (Gibco-Invitrogen, Madrid, Spain) containing 10% of fetal bovine serum (FBS, Gibco-Invitrogen). The cell suspension was centrifuged for 10 min at 600X g. The pellet was resuspended in fresh medium, and the cells were plated at a density of 5×106 cells/dish in poly-D-lysine 100 mm Petri dishes. After 10 days of culture, the neurons were treated for 24 h with 1 μM Aβ25-35 alone or in combination with 100 μM GPE for 24 h. We measured the phosphorylated and total levels of STAT3 and IRS-1 and IDE concentrations in the lysates by a multiplexed bead immunoassay and an ELISA, respectively.
4.9.2. Mixed Glial Culture
For this culture, 3–5-day old Sprague-Dawley rats were used. Briefly, the rats were sacrificed and hippocampi were dissected by pipetting in Dulbecco’s Modified Eagle Medium (DMEM)/F12 medium (Thermo Fisher, Madrid, Spain) supplemented with 20% of FBS. Then, the cells were filtered using a 40 μm cell strainer and centrifuged for 8 min at 900X g. Finally, the cells were seeded in DMEM/F12 with 20% FBS at a density of 5×106 cells/dish in 100 mm Petri dishes and cultured at 37°C in humidified 5%CO2/95% air. Once confluence was achieved after 7-10 days, glial cells were treated with DMEM/F12 with 10% FBS alone (basal condition), with 1 μm Aβ25-35 alone and with 1 μM Aβ25-35 plus 100 μM GPE for 24 h. In cell lysates from glial cultures, we determined phosphorylated and total levels of STAT3 and IRS-1 and IDE content and in the extracellular culture media, we measured IFN-γ, IL-2, IL-13 and IL-17A concentrations by a multiplexed bead immunoassay.
4.10. Statistical Analysis
Data are summarized as mean ± SEM. The analysis of all data was carried out using one-way ANOVA followed by Bonferroni’s post hoc tests. Relationships between variables were performed by linear regression analysis. Values were considered significantly different when the p value was less than 0.05. Analyses were performed using Statview software (Statview 5.01, SAS Institute, Cary, NC, USA) and graphs were generated using GraphPad Prism 8 (San Diego, CA, USA) software.
Author Contributions
Conceptualization, L.M.F., M.G.L. and V.B.; methodology, M.R.-P., E.B.-R., and S.C.; validation, L.M.F. and J.A..; formal analysis, E.B.-R. and V.B.; investigation, M.R.-P., E. A.-F. and V.B..; writing—original draft preparation, E.B.-R., M.G.L., J.A. and V.B.; writing—review and editing L.M.F., E.B.-R., M.G.L., E.A.-F., and V.B.; funding acquisition, L.M.F., E.B.-R., M.R.-P., J.A., and V.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Junta de Comunidades de Castilla-La Mancha (JCCM) (SBPLY/23/180225/000030), Ministerio de Ciencia e Innovación (FIS-PI19/00166 and FIS-PI22/01820) funded by MCIN/AEI/10.13039/501100011033 and PDC2022-133809-I00; Community of Madrid ref CAM-P2022/BMD-7230 and by “ERDF A way of making Europe”, by the “European Union”, (BFU 2017-82565-C2-1 and PID2021-122653OB-I00) funded by MICIN/AEI/10.13039/501100011033 and by “ERDF A way of making Europe”, by the “European Union”) and the Centro de Investigación Biomédica en Red Fisiopatología de Obesidad y Nutrición (CIBEROBN), Instituto Carlos III. S.C. was supported by CIBEROBN.
Institutional Review Board Statement
This study was approved by the Ethics Committee of the Universidad de Alcalá de Henares (SAF 2010–22277, Ministerio de Ciencia y Tecnología) and complied with Royal Decree 1201/2005 (Boletín Oficial del Estado, BOE No. 252) pertaining to the protection of experimental animals and with the European Communities Council Directive (86/609/EEC).
Informed Consent Statement
Not applicable.
Data Availability Statement
All relevant data are included within the manuscript.
Acknowledgments
The authors would like to thank Dr. Julie A. Chowen for the critical review of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Abbreviations
| Aβ | Amyloid-β peptide |
| AC | Adenylate cyclase |
| AD | Alzheimer´s disease |
| Akt | Protein kinase B |
| ANOVA | Analysis of variance |
| APP | Amyloid precursor protein |
| AU | Absorbance units |
| DMEM | Dulbecco’s modified Eagle medium |
| ELISA | Enzyme-linked immunosorbent assay |
| FBS | Fetal bovine serum |
| GFAP | Glial fibrillary acidic protein |
| GPE | Glycine-proline-glutamate |
| GSK3β | Glycogen synthase kinase 3β |
| HRP | Horseradish peroxidase |
| IDE | Insulin-degrading enzyme |
| IFN-γ | Interferon-γ |
| IGF-I | Insulin-like growth factor I |
| IGF-IR | IGF-I receptor |
| IL | Interleukin |
| IRS1 | Insulin receptor substrate 1 |
| JAK2 | Janus kinase 2 |
| MFI | Median fluorescent intensity |
| NFκB | Nuclear factor kappa B |
| Ovx | Ovariectomized |
| p | Phosphorylated |
| PI3K | Phosphatidylinositol 3-kinase |
| PS1 | Presenilin-1 |
| p38MAPK | p38 mitogen-activated protein kinase |
| SOCS3 | Suppressor of cytokine signaling 3 |
| SRIF | Somatostatin |
| STAT3 | Signal transducer and activator of transcription 3 |
References
- Ludewig, S.; Korte, M. Novel insights into the physiological function of the APP (Gene) family and its proteolytic fragments in synaptic plasticity. Front. Mol. Neurosci. 2017, 9, 161. [CrossRef]
- Li, R.; Li, Y.; Zuo, H.; Pei, G.; Huang, S.; Hou, Y. Alzheimer’s amyloid-beta accelerates cell senescence and suppresses SIRT1 in human neural stem cells. Biomolecules 2024, 14, 189. [CrossRef]
- Fornari Laurindo, L.; Aparecido Dias, J.; Cressoni Araújo, A.; Torres Pomini, K.; Machado Galhardi, C.; Rucco Penteado Detregiachi, C.; Santos de Argollo Haber, L.; Donizeti Roque, D.; Dib Bechara, M.; Vialogo Marques de Castro, M.; et al. Immunological dimensions of neuroinflammation and microglial activation: exploring innovative immunomodulatory approaches to mitigate neuroinflammatory progression. Front. Immunol. 2024, 14, 1305933. [CrossRef]
- Merighi, S.; Nigro, M.; Travagli, A.; Gessi, S. Microglia and Alzheimer’s disease. Int. J. Mol. Sci. 2022, 23, 12990. [CrossRef]
- Wang, H.; Sun, M.; Li, W.; Liu, X.; Zhu, M.; Qin, H. Biomarkers associated with the pathogenesis of Alzheimer’s disease. Front. Cell. Neurosci. 2023, 17, 1279046. [CrossRef]
- Kim, J.; Yoo, I.D.; Lim, J.; Moon, J.S. Pathological phenotypes of astrocytes in Alzheimer’s disease. Exp. Mol. Med. 2024, 56, 95-99. [CrossRef]
- Salman, M.; Akram, M.; Shahrukh, M.; Ishrat, T.; Parvez, S. Effects of pramipexole on beta-amyloid(1-42) memory deficits and evaluation of oxidative stress and mitochondrial function markers in the hippocampus of Wistar rat. Neurotoxicology 2022, 92, 91-101. [CrossRef]
- Fekete, C.; Vastagh, C.; Dénes, Á.; Hrabovszky, E.; Nyiri, G.; Kalló, I.; Liposits, Z.; Sárvári, M. Chronic amyloid beta oligomer infusion evokes sustained inflammation and microglial changes in the rat hippocampus via NLRP3. Neuroscience 2019, 405, 35-46. [CrossRef]
- Aguado-Llera, D.; Arilla-Ferreiro, E.; Campos-Barros, A.; Puebla-Jiménez, L.; Barrios V. Protective effects of insulin-like growth factor-I on the somatostatinergic system in the temporal cortex of beta-amyloid-treated rats. J. Neurochem. 2005, 92, 607-615. [CrossRef]
- Kubo, T.; Nishimura, S.; Kumagae, Y.; Kaneko, I. In vivo conversion of racemized beta-amyloid ([D-Ser 26]A beta 1-40) to truncated and toxic fragments ([D-Ser 26]A beta 25-35/40) and fragment presence in the brains of Alzheimer’s patients. J. Neurosci. Res. 2002, 70, 474-483. [CrossRef]
- Pirhaghi, M.; Mamashli, F.; Moosavi-Movahedi, F.; Arghavani, P.; Amiri, A.; Davaeil, B.; Mohammad-Zaheri, M.; Mousavi-Jarrahi, Z.; Sharma, D.; Langel, Ü.; et al. Cell-penetrating peptides: promising therapeutics and drug-delivery systems for neurodegenerative diseases. Mol. Pharm. 2024. [CrossRef]
- Guan, J.; Thomas, G.B.; Lin, H.; Mathai, S.; Bachelor, D.C.; George, S.; Gluckman, P.D. Neuroprotective effects of the N-terminal tripeptide of insulin-like growth factor-1, glycine-proline-glutamate (GPE) following intravenous infusion in hypoxic-ischemic adult rats. Neuropharmacology 2004, 47, 892-903. [CrossRef]
- Burgos-Ramos, E.; Martos-Moreno, G.A.; López, M.G.; Herranz, R.; Aguado-Llera, D.; Egea, J.; Frechilla, D.; Cenarruzabeitia, E.; León, R.; Arilla-Ferreiro, E.; et al. The N-terminal tripeptide of insulin-like growth factor-I protects against beta-amyloid-induced somatostatin depletion by calcium and glycogen synthase kinase 3 beta modulation. J. Neurochem. 2009, 109, 360-370. [CrossRef]
- Silva-Reis, S.C.; Sampaio-Dias, I.E.; Costa, V.M.; Correia, X.C.; Costa-Almeida, H.F.; García-Mera, X.; Rodríguez-Borges, J.E. Concise overview of glypromate neuropeptide research: from chemistry to pharmacological applications in neurosciences. ACS Chem. Neurosci. 2023, 14, 554-572. [CrossRef]
- Herrero-Labrador, R.; Trueba-Saiz, A.; Martinez-Rachadell, L.; Fernandez de Sevilla, M.E.; Zegarra-Valdivia, J.A.; Pignatelli, J.; Diaz-Pacheco, S.; Fernandez, A.M.; Torres Aleman, I. Circulating insulin-like growth factor I is involved in the effect of high fat diet on peripheral amyloid beta clearance. Int. J. Mol. Sci. 2020, 21, 9675. [CrossRef]
- Almengló, C.; Devesa, P.; Devesa, J.; Arce, V.M. GPE promotes the proliferation and migration of mouse embryonic neural stem cells and their progeny in vitro. Int. J. Mol. Sci. 2017, 18, 1280. [CrossRef]
- Messier, C.; Teutenberg, K. The role of insulin, insulin growth factor, and insulin-degrading enzyme in brain aging and Alzheimer’s disease. Neural Plast. 2005, 12, 311-328. [CrossRef]
- Yook, J.S.; Rakwal, R.; Shibato, J.; Takahashi, K.; Koizumi, H.; Shima, T.; Ikemoto, M.J.; Oharomari, L.K.; McEwen, B.S.; Soya, H. Leptin in hippocampus mediates benefits of mild exercise by an antioxidant on neurogenesis and memory. Proc. Natl. Acad. Sci. USA 2019, 116, 10988-10993. [CrossRef]
- Al-Kuraishy, H.M.; Al-Gareeb, A.I.; Alsayegh, A.A.; Hakami, Z.H.; Khamjan, N.A.; Saad, H.M.; Batiha, G.E.; De Waard, M. A potential link between visceral obesity and risk of Alzheimer’s disease. Neurochem. Res. 2023, 48, 745-766. [CrossRef]
- Tundo, G.R.; Di Muzio, E.; Ciaccio, C.; Sbardella, D.; Di Pierro, D.; Polticelli, F.; Coletta, M.; Marini, S. Multiple allosteric sites are involved in the modulation of insulin-degrading-enzyme activity by somatostatin. FEBS J. 2016, 283, 3755-3770. [CrossRef]
- Christian, F.; Smith, E.L.; Carmody, R.J. The regulation of NF-κB subunits by phosphorylation. Cells 2016, 5, 12. [CrossRef]
- Grønborg, M.; Wulff, B.S.; Rasmussen, J.S.; Kjeldsen, T.; Gammeltoft S. Structure-function relationship of the insulin-like growth factor-I receptor tyrosine kinase. J. Biol. Chem. 1993, 268, 23435-23440. PMID: 7693688.
- Tzatsos, A. Raptor binds the SAIN (Shc and IRS-1 NPXY binding) domain of insulin receptor substrate-1 (IRS-1) and regulates the phosphorylation of IRS-1 at Ser-636/639 by mTOR. J. Biol. Chem. 2009, 284, 22525-22534. [CrossRef]
- Saito, T.; Iwata, N.; Tsubuki, S.; Takaki, Y.; Takano, J.; Huang, S.M.; Suemoto T.; Higuchi M.; Saido T.C. Somatostatin regulates brain amyloid beta peptide Abeta42 through modulation of proteolytic degradation. Nat. Med. 2005, 11, 434-439. [CrossRef]
- Sandoval, K.; Umbaugh, D.; House, A.; Crider, A.; Witt, K. Somatostatin receptor subtype-4 regulates mRNA expression of amyloid-beta degrading enzymes and microglia mediators of phagocytosis in brains of 3xTg-AD mice. Neurochem. Res. 2019, 44, 2670-2680. [CrossRef]
- Weggen, S.; Rogers, M.; Eriksen, J. NSAIDs: small molecules for prevention of Alzheimer’s disease or precursors for future drug development? Trends Pharmacol. Sci. 2007, 28, 536-543. [CrossRef]
- Campolongo, P.; Ratano, P.; Ciotti, M.T.; Florenzano, F.; Nori, S.L.; Marolda, R.; Palmery, M.; Rinaldi, A.M.; Zona, C.; Possenti, R.; et al. Systemic administration of substance P recovers beta amyloid-induced cognitive deficits in rat: involvement of Kv potassium channels. PLoS One 2013, 8, e78036. [CrossRef]
- Tang, L.; Xiang, Q.; Xiang, J.; Zhang, Y.; Li, J. Tripterygium glycoside ameliorates neuroinflammation in a mouse model of Abeta25-35-induced Alzheimer’s disease by inhibiting the phosphorylation of IkappaBalpha and p38. Bioengineered 2021, 12, 8540-8554. [CrossRef]
- Napolitano, M.; Costa, L.; Piacentini, R.; Grassi, C.; Lanzone, A.; Gulino, A. 17β-estradiol protects cerebellar granule cells against β-amyloid-induced toxicity via the apoptotic mitochondrial pathway. Neurosci. Lett. 2014, 561, 134-139. [CrossRef]
- Lopez-Lee, C.; Torres, E.R.S.; Carling, G.; Gan, L. Mechanisms of sex differences in Alzheimer’s disease. Neuron 2024, S0896-6273(24)00050-3. [CrossRef]
- Brandt, N.; Vierk, R.; Rune, G.M. Sexual dimorphism in estrogen-induced synaptogenesis in the adult hippocampus. Int. J. Dev. Biol. 2013, 57, 351-356. [CrossRef]
- Fei, X.; Zhang, P.Y.; Zhang, X.; Zhang, G.Q.; Bao, W.P.; Zhang, Y.Y.; Zhang, M.; Zhou, X. IL-17A monoclonal antibody partly reverses the glucocorticoids insensitivity in mice exposed to Ozonec. Inflammation 2017, 40, 788-797. [CrossRef]
- Yuan, R.; Wang, L.; Deng, Z.H.; Yang, M.M.; Zhao, Y.; Hu, J.; Zhang, Y.; Li, Y.; Liu, M.; Liu, S.F., et al. Protective effects of mesenchymal stem cells against central nervous system injury in heat stroke. Curr. Stem Cell. Res. Ther. 2023, 18, 401-409. [CrossRef]
- Oliva, A.A. Jr.; Kang, Y.; Sanchez-Molano, J.; Furones, C.; Atkins, C.M. STAT3 signaling after traumatic brain injury. J. Neurochem. 2012, 120, 710-720. [CrossRef]
- Espírito-Santo, S.A.; Nunes-Tavares, N.; Mendonça, H.R.; Serfaty, C.A.; Sholl-Franco, A.; Campello-Costa, P. Intravitreal Interleukin-2 modifies retinal excitatory circuits and retinocollicular innervation. Exp. Eye Res. 2021, 204, 108442. [CrossRef]
- Cecon, E.; Lhomme, T.; Maurice, T.; Luka, M.; Chen, M.; Silva, A.; Wauman, J.; Zabeau, L.; Tavernier, J.; Prévot, V.; et al. Amyloid beta peptide is an endogenous negative allosteric modulator of leptin receptor. Neuroendocrinology 2021, 111, 370-387. [CrossRef]
- Arora, T.; Caviedes, P.; Sharma, S.K. Effects of a tripeptide on mitogen-activated protein kinase and glycogen synthase kinase activation in a cell line derived from the foetal hippocampus of a trisomy 16 mouse: an animal model of Down syndrome. Neurotox. Res. 2020, 37, 714-723. [CrossRef]
- Minelli, A.; Conte, C.; Cacciatore, I.; Cornacchia, C.; Pinnen, F. Molecular mechanism underlying the cerebral effect of Gly-Pro-Glu tripeptide bound to L-dopa in a Parkinson’s animal model. Amino Acids 2012, 43, 1359-1367. [CrossRef]
- Park, S.; Hong, S.M.; Sung, S.R.; Jung, H.K. Long-term effects of central leptin and resistin on body weight, insulin resistance, and beta-cell function and mass by the modulation of hypothalamic leptin and insulin signaling. Endocrinology 2008, 149, 445-454. [CrossRef]
- King, A.; Brain, A.; Hanson, K.; Dittmann, J.; Vickers, J.; Fernandez-Martos, C. Disruption of leptin signalling in a mouse model of Alzheimer’s disease. Metab. Brain Dis. 2018, 33, 1097-1110. [CrossRef]
- Barrios, V.; Frago, L.M.; Canelles, S.; Guerra-Cantera, S.; Arilla-Ferreiro, E.; Chowen, J.A.; Argente, J. Leptin modulates the response of brown adipose tissue to negative energy balance: implication of the GH/IGF-I axis. Int. J. Mol. Sci. 2021, 22, 2827. [CrossRef]
- Maragakis, N.J.; Rothstein, J.D.; Mechanisms of disease: astrocytes in neurodegenerative disease. Nat. Clin. Pract. Neurol. 2006, 2, 679-689. [CrossRef]
- Guan, J.; Gluckman, P.D. IGF-1 derived small neuropeptides and analogues: a novel strategy for the development of pharmaceuticals for neurological conditions. Br. J. Pharmacol. 2009, 157, 881-891. [CrossRef]
- Aguado-Llera, D.; Canelles, S.; Fernández-Mendívil, C.; Frago, L.M.; Argente, J.; Arilla-Ferreiro, E.; López, M.G.; Barrios, V. Improvement in inflammation is associated with the protective effect of Gly-Pro-Glu and cycloprolylglycine against Aβ-induced depletion of the hippocampal somatostatinergic system. Neuropharmacology 2019, 151, 112-126. [CrossRef]
- Svedin, P.; Guan, J.; Mathai, S.; Zhang, R.; Wang, X.; Gustavsson, M.; Hagberg, H.; Mallard, C. Delayed peripheral administration of a GPE analogue induces astrogliosis and angiogenesis and reduces inflammation and brain injury following hypoxia-ischemia in the neonatal rat. Dev. Neurosci. 2007, 29, 393-402. [CrossRef]
- Shapiro, M.R.; Peters, L.D.; Brown, M.E.; Cabello-Kindelan, C.; Posgai, A.L.; Bayer, A.L.; Brusko, T.M. Insulin-like growth factor-1 synergizes with IL-2 to induce homeostatic proliferation of regulatory T cells. J. Immunol. 2023, 211, 1108-1122. [CrossRef]
- Relic, B.; Guicheux, J.; Mezin, F.; Lubberts, E.; Togninalli, D.; Garcia, I.; van den Berg, W.B.; Guerne, P.A. IL-4 and IL-13, but not IL-10, protect human synoviocytes from apoptosis. J. Immunol. 2001, 166, 2775-2782. [CrossRef]
- Marella, S.; Sharma, A.; Ganesan, V.; Ferrer-Torres, D.; Krempski, J.W.; Idelman, G.; Clark, S.; Nasiri, Z.; Vanoni, S.; Zeng, C.; et al. IL-13-induced STAT3-dependent signaling networks regulate esophageal epithelial proliferation in eosinophilic esophagitis. J. Allergy Clin. Immunol. 2023, 152, 1550-1568. [CrossRef]
- Turkez, H.; Cacciatore, I.; Marinelli, L.; Fornasari, E.; Aslan, M.E.; Cadirci, K.; Kahraman, C.Y.; Caglar, O.; Tatar, A.; Di Biase, G.; et al. Glycyl-L-prolyl-L-glutamate pseudotripeptides for treatment of Alzheimer’s disease. Biomolecules 2021, 11, 126. [CrossRef]
- Doherty, G.H.; Beccano-Kelly, D.; Yan, S.D.; Gunn-Moore, F.J.; Harvey, J. Leptin prevents hippocampal synaptic disruption and neuronal cell death induced by amyloid beta. Neurobiol. Aging. 2013, 34, 226-237. [CrossRef]
- Gonzalez, G.A.; Montminy, M.R. Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell 1989, 59, 675-680. [CrossRef]
- Sandoval, K.; Umbaugh, D.; House, A.; Crider, A.; Witt, K. Somatostatin receptor subtype-4 regulates mRNA expression of amyloid-beta degrading enzymes and microglia mediators of phagocytosis in brains of 3xTg-AD mice. Neurochem. Res. 2019, 44, 2670-2680. [CrossRef]
- Burgos-Ramos, E.; Hervás-Aguilar, A.; Aguado-Llera, D.; Puebla-Jiménez, L.; Hernández-Pinto, A.M.; Barrios, V.; Arilla-Ferreiro, E. Somatostatin and Alzheimer’s disease. Mol. Cell. Endocrinol. 2008, 286, 104-111. [CrossRef]
- El Sayed, N.S.; Kandil, E.A.; Ghoneum, M.H. Enhancement of insulin/PI3K/Akt signaling pathway and modulation of gut microbiome by probiotics fermentation technology, a kefir grain product, in sporadic Alzheimer’s disease model in mice. Front. Pharmacol. 2021, 12, 666502. [CrossRef]
- Guan. J.; Harris. P.; Brimble, M.; Lei, Y.; Lu, J.; Yang, Y.; Gunn, A.J. The role for IGF-1-derived small neuropeptides as a therapeutic target for neurological disorders. Expert Opin. Ther. Targets 2015, 19, 785-793. [CrossRef]
- Arora, T.; Sharma, S.K. Cyclic glycine-proline improves memory and reduces amyloid plaque load in APP/PS1 transgenic mouse model of Alzheimer’s disease. Int. J. Alzheimers Dis. 2023, 2023, 1753791. [CrossRef]
- Rezaei, M.H.; Madadizadeh, E.; Aminaei, M.; Abbaspoor, M.; Schierbauer, J.; Moser, O.; Khoramipour, K.; Chamari, K. Leptin signaling could mediate hippocampal decumulation of beta-amyloid and tau induced by high-intensity interval training in rats with type 2 diabetes. Cell. Mol. Neurobiol. 2023, 43, 3465-3478. [CrossRef]
- Alves, S.; Churlaud, G.; Audrain, M.; Michaelsen-Preusse, K.; Fol, R.; Souchet, B.; Braudeau, J.; Korte, M.; Klatzmann, D.; Cartier, N. Interleukin-2 improves amyloid pathology, synaptic failure and memory in Alzheimer’s disease mice. Brain 2017, 140, 826-842. [CrossRef]
- Kawahara, K.; Suenobu, M.; Yoshida, A.; Koga, K.; Hyodo, A.; Ohtsuka, H.; Kuniyasu, A.; Tamamaki, N.; Sugimoto, Y.; Nakayama, H. Intracerebral microinjection of interleukin-4/interleukin-13 reduces beta-amyloid accumulation in the ipsilateral side and improves cognitive deficits in young amyloid precursor protein 23 mice. Neuroscience 2012, 207, 243-260. [CrossRef]
- Cao, M.; Liu, J.; Zhang, X.; Wang, Y.; Hou Y.; Song, Q.; Cui, Y.; Zhao, Y.; Wang, P. IL-17A promotes the progression of Alzheimer’s disease in APP/PS1 mice. Immun. Ageing 2023, 20, 74. [CrossRef]
- Shallie, O.F.; Dalle, E.; Mabandla, M.V. Memory decline correlates with increased plasma cytokines in amyloid-beta (1-42) rat model of Alzheimer’s disease. Neurobiol. Learn. Mem. 2020, 169, 107187. [CrossRef]
- Foley, K.E.; Winder, Z.; Sudduth, T.L.; Martin, B.J.; Nelson, P.T.; Jicha, G.A.; Harp, J.P.; Weekman, E.M.; Wilcock, D.M. Alzheimer’s disease and inflammatory biomarkers positively correlate in plasma in the UK-ADRC cohort. Alzheimers Dement. 2024, 20, 1374-1386. [CrossRef]
- Burgos-Ramos, E.; Hervás-Aguilar, A.; Puebla-Jiménez, L.; Boyano-Adánez, M.C.; Arilla-Ferreiro, E. Chronic but not acute in-tracerebroventricular administration of amyloid beta-peptide (25-35) decreases somatostatin content, adenylate cyclase activity, somatostatin-induced inhibition of adenylate cyclase activity, and adenylate cyclase I levels in the rat hippocampus. J. Neurosci. Res. 2007, 85, 433-442. [CrossRef]
- Dao, A.T.; Zagaar, M.A.; Levine, A.T.; Salim, S.; Eriksen, J.L.; Alkadhi, K.A. Treadmill exercise prevents learning and memory impairment in Alzheimer’s disease-like pathology. Curr. Alzheimer Res. 2013, 10, 507-515. [CrossRef]
- Nag, S.; Yee, B.K.; Tang, F. Reduction in somatostatin and substance P levels and choline acetyltransferase activity in the cortex and hippocampus of the rat after chronic intracerebroventricular infusion of beta-amyloid (1-40). Brain Res. Bull. 1999, 50, 251-262. [CrossRef]
- Glowinski, J.; Iversen, L.L. Regional studies of catecholamines in the rat brain. I. The disposition of [3H]norepinephrine, [3H]dopamine and [3H]dopa in various regions of the brain. J. Neurochem. 1966, 13, 655-669. [CrossRef]
- Reubi, J.C.; Perrin, M.H.; Rivier, J.E.; Vale, W. High affinity binding sites for a somatostatin-28 analog in rat brain. Life Sci. 1981, 28, 2191-2198. [CrossRef]
- Gilman, A.G. A protein binding assay for adenosine 3´:5´-cyclic monophosphate. Proc. Natl. Acad. Sci. USA 1970, 67, 305-312. [CrossRef]
Figure 1.
Effects of Aβ25-35 and GPE co-administration on hippocampal Aβ25-35 levels and phosphorylation of pro-inflammatory and leptin signaling targets. Levels of (A) Aβ25-35, relative protein levels of (B) p38 mitogen activated protein kinase (pMAPK) phosphorylated (p) at Thr180 and Tyr182 (pThr180Tyr182p38MAPK) and (C) nuclear factor kappa B (NFκB) phosphorylated at Ser536 (pSer536NFkB), (D) serum leptin levels and relative protein levels of (E) signal transducer and activator of transcription 3 (STAT3) phosphorylated at Tyr705 (pTyr705STAT3) and (F) STAT3 phosphorylated at Ser727 (pSer727STAT3) in ovariectomized (Ovx) rats (control), Ovx rats treated with β-amyloid 25-35 peptide (Aβ), Ovx rats treated with Aβ25-35 plus GPE (Aβ + GPE) and Ovx rats treated with GPE (GPE). Data are expressed as mean ± SEM. N = 5. MFI, median fluorescent intensity * p < 0.05, ** p < 0.01.
Figure 1.
Effects of Aβ25-35 and GPE co-administration on hippocampal Aβ25-35 levels and phosphorylation of pro-inflammatory and leptin signaling targets. Levels of (A) Aβ25-35, relative protein levels of (B) p38 mitogen activated protein kinase (pMAPK) phosphorylated (p) at Thr180 and Tyr182 (pThr180Tyr182p38MAPK) and (C) nuclear factor kappa B (NFκB) phosphorylated at Ser536 (pSer536NFkB), (D) serum leptin levels and relative protein levels of (E) signal transducer and activator of transcription 3 (STAT3) phosphorylated at Tyr705 (pTyr705STAT3) and (F) STAT3 phosphorylated at Ser727 (pSer727STAT3) in ovariectomized (Ovx) rats (control), Ovx rats treated with β-amyloid 25-35 peptide (Aβ), Ovx rats treated with Aβ25-35 plus GPE (Aβ + GPE) and Ovx rats treated with GPE (GPE). Data are expressed as mean ± SEM. N = 5. MFI, median fluorescent intensity * p < 0.05, ** p < 0.01.
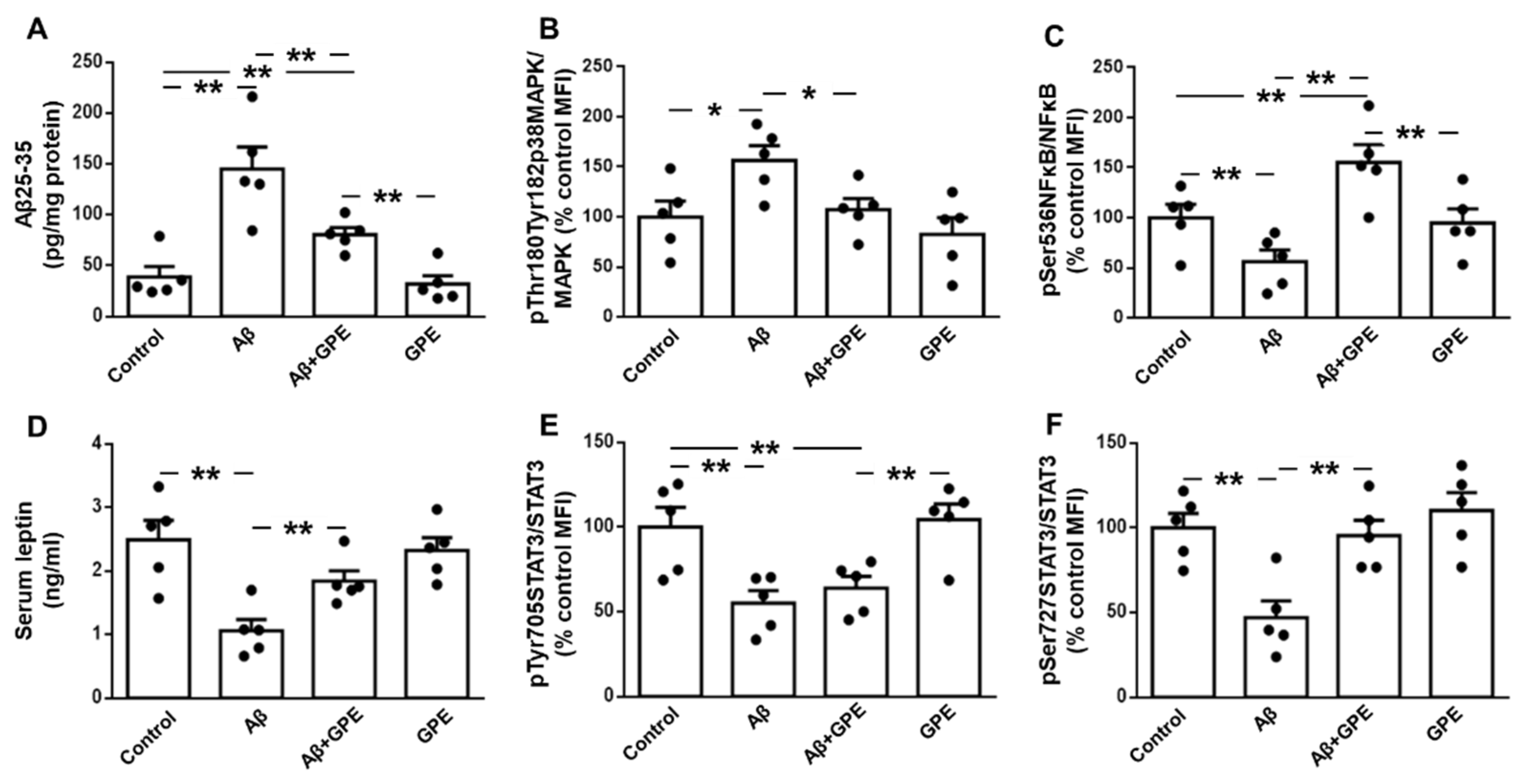
Figure 2.
Effects of Aβ25-35 and GPE co-administration on IGF-I levels and IGF-I-related signaling targets. Serum (A) and (B) hippocampal levels of IGF-I and relative protein levels of (C) insulin-like growth factor-I receptor (IGF-IR) phosphorylated at Tyr1131 (pTyr1131IGF-IR), (D) insulin receptor substrate 1 (IRS1) phosphorylated at Tyr residues (pTyrIRS1), (E) IRS1 phosphorylated at Ser636 (pSer636IRS1) and (F) Akt phosphorylated at Thr308 (pThr308Akt) in ovariectomized (Ovx) rats (control), Ovx rats treated with β-amyloid 25-35 peptide (Aβ), Ovx rats treated with Aβ25-35 plus GPE (Aβ + GPE) and Ovx rats treated with GPE (GPE). Data are expressed as mean ± SEM. N = 5. AU, absorbance units, MFI, median fluorescent intensity * p < 0.05, ** p < 0.01, *** p < 0.001.
Figure 2.
Effects of Aβ25-35 and GPE co-administration on IGF-I levels and IGF-I-related signaling targets. Serum (A) and (B) hippocampal levels of IGF-I and relative protein levels of (C) insulin-like growth factor-I receptor (IGF-IR) phosphorylated at Tyr1131 (pTyr1131IGF-IR), (D) insulin receptor substrate 1 (IRS1) phosphorylated at Tyr residues (pTyrIRS1), (E) IRS1 phosphorylated at Ser636 (pSer636IRS1) and (F) Akt phosphorylated at Thr308 (pThr308Akt) in ovariectomized (Ovx) rats (control), Ovx rats treated with β-amyloid 25-35 peptide (Aβ), Ovx rats treated with Aβ25-35 plus GPE (Aβ + GPE) and Ovx rats treated with GPE (GPE). Data are expressed as mean ± SEM. N = 5. AU, absorbance units, MFI, median fluorescent intensity * p < 0.05, ** p < 0.01, *** p < 0.001.
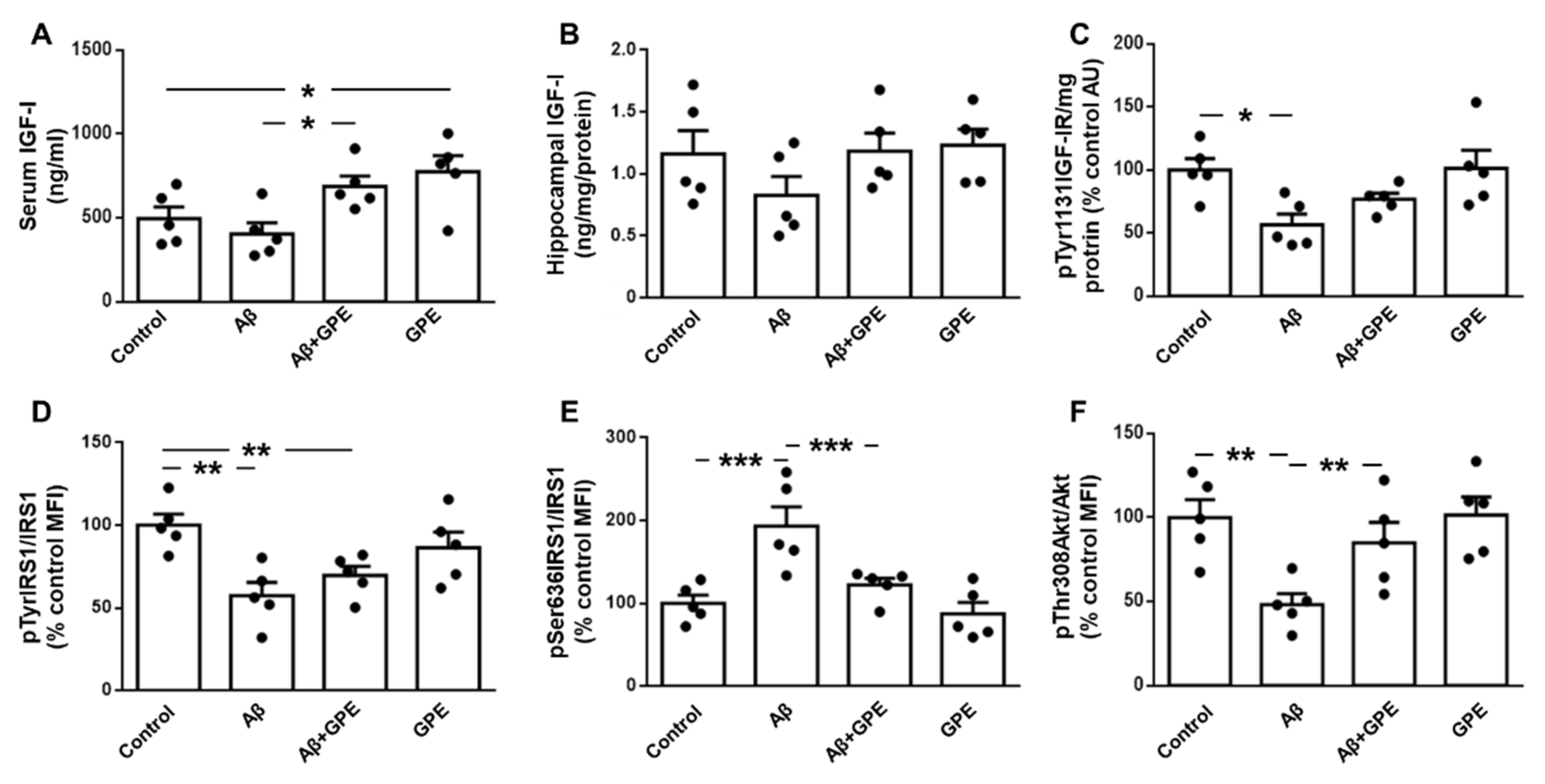
Figure 3.
Effects of Aβ25-35 and GPE co-administration on serum and hippocampal cytokine levels. Serum levels of interferon (IFN)-γ (A), interleukin (IL)-2 (C), IL-13 (E) and IL-17A (G) and hippocampal concentrations of IFN-γ (B), IL-2 (D), IL-13 (F) and IL-17A (H) in ovariectomized (Ovx) rats (control), Ovx rats treated with β-amyloid 25-35 peptide (Aβ), Ovx rats treated with Aβ25-35 plus GPE (Aβ + GPE) and Ovx rats treated with GPE (GPE). Data are expressed as mean ± SEM. N = 5. * p < 0.05, ** p < 0.01.
Figure 3.
Effects of Aβ25-35 and GPE co-administration on serum and hippocampal cytokine levels. Serum levels of interferon (IFN)-γ (A), interleukin (IL)-2 (C), IL-13 (E) and IL-17A (G) and hippocampal concentrations of IFN-γ (B), IL-2 (D), IL-13 (F) and IL-17A (H) in ovariectomized (Ovx) rats (control), Ovx rats treated with β-amyloid 25-35 peptide (Aβ), Ovx rats treated with Aβ25-35 plus GPE (Aβ + GPE) and Ovx rats treated with GPE (GPE). Data are expressed as mean ± SEM. N = 5. * p < 0.05, ** p < 0.01.
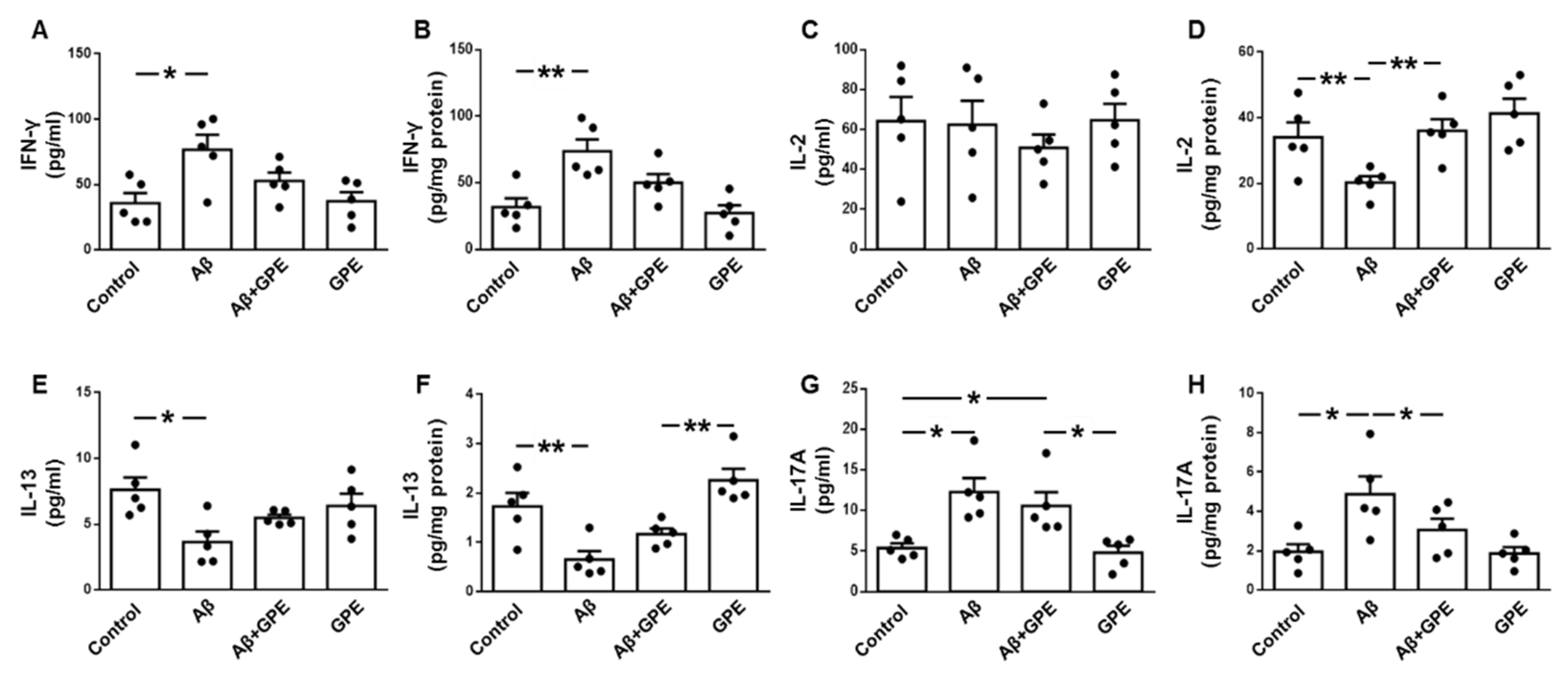
Figure 5.
Effects of Aβ25-35 and GPE co-administration on phosphorylation of signaling targets and levels of insulin-degrading enzyme (IDE) in neuronal and glial cultures. Relative protein levels in neuronal and glial cultures of (A and D, respectively) of signal transducer and activator of transcription 3 (STAT3) phosphorylated (p) at Ser727 (pSer727STAT3), (B and E, respectively) insulin receptor substrate 1 (IRS1) phosphorylated at Tyr residues (pTyrIRS1) and protein concentrations (C and F, respectively) insulin-degrading enzyme (IDE). Data are expressed as mean ± SEM. N = 5. * p < 0.05.
Figure 5.
Effects of Aβ25-35 and GPE co-administration on phosphorylation of signaling targets and levels of insulin-degrading enzyme (IDE) in neuronal and glial cultures. Relative protein levels in neuronal and glial cultures of (A and D, respectively) of signal transducer and activator of transcription 3 (STAT3) phosphorylated (p) at Ser727 (pSer727STAT3), (B and E, respectively) insulin receptor substrate 1 (IRS1) phosphorylated at Tyr residues (pTyrIRS1) and protein concentrations (C and F, respectively) insulin-degrading enzyme (IDE). Data are expressed as mean ± SEM. N = 5. * p < 0.05.
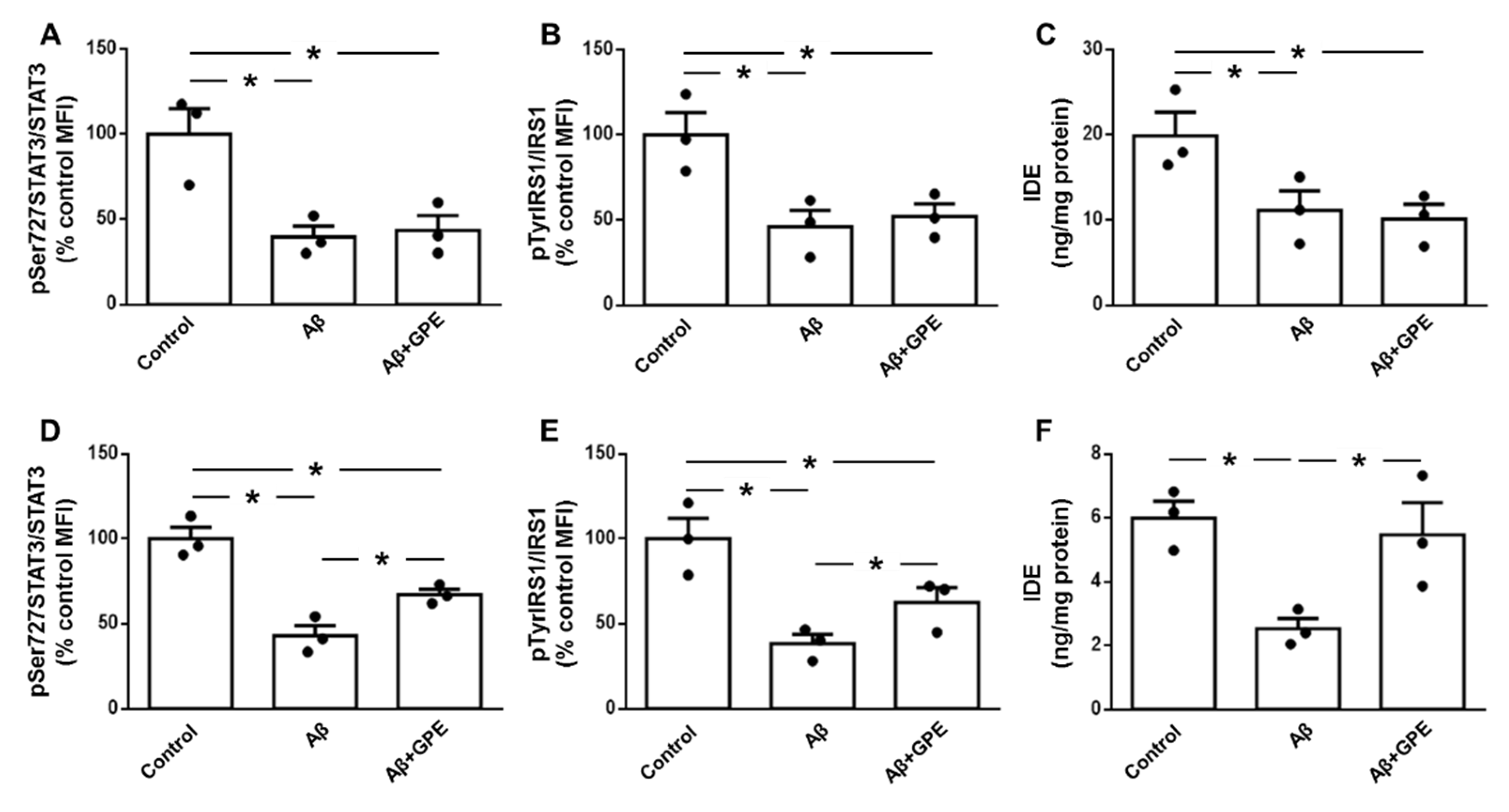
Figure 6.
Effects of Aβ25-35 and GPE co-administration on interleukin secretion in glial cultures. Protein levels in culture media of interferon (IFN)-γ (A), interleukin (IL)-2 (B), IL-13 (C) and IL-17A (D). Data are expressed as mean ± SEM. N = 5. * p < 0.05.
Figure 6.
Effects of Aβ25-35 and GPE co-administration on interleukin secretion in glial cultures. Protein levels in culture media of interferon (IFN)-γ (A), interleukin (IL)-2 (B), IL-13 (C) and IL-17A (D). Data are expressed as mean ± SEM. N = 5. * p < 0.05.
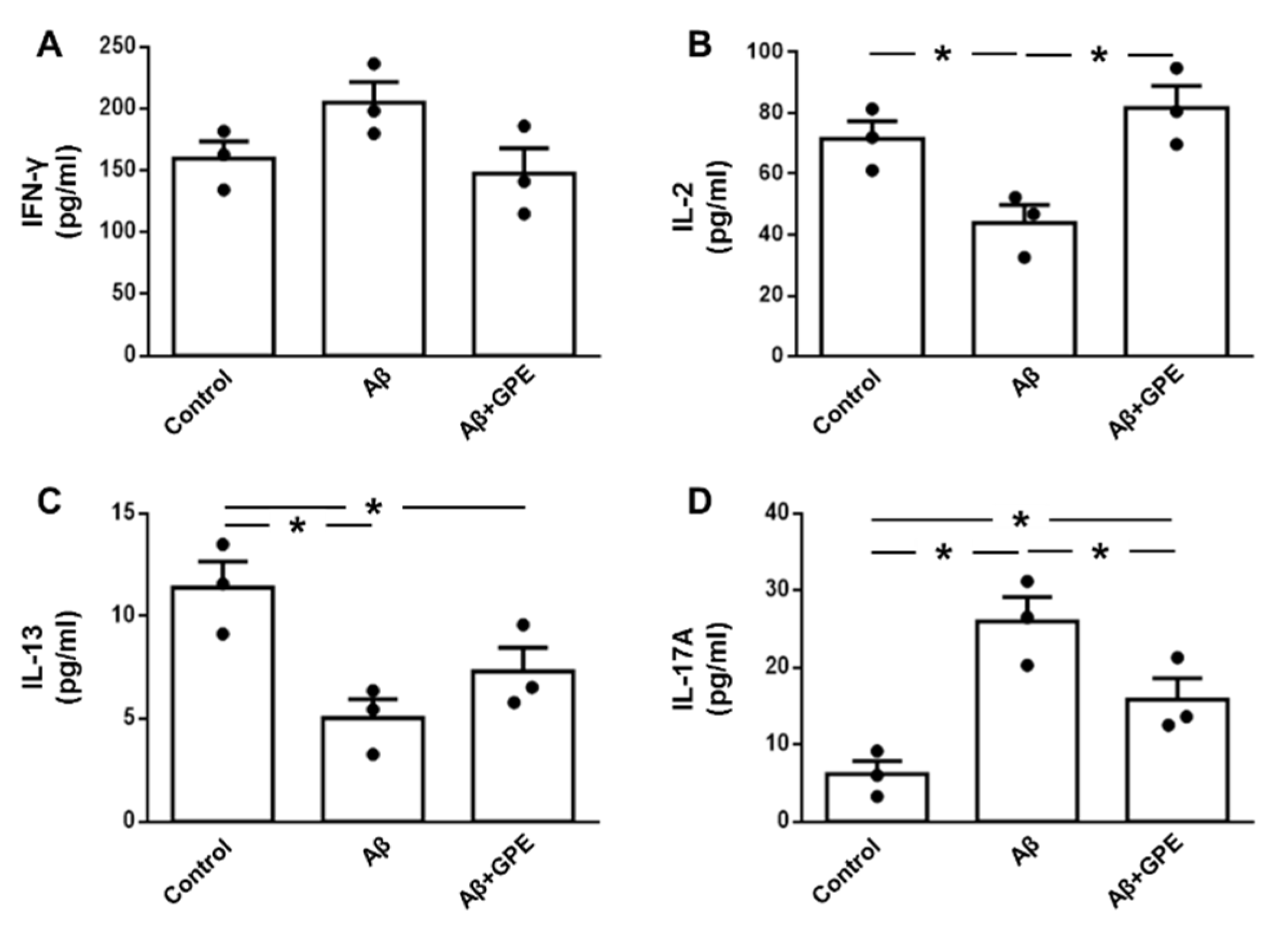
Figure 7.
Correlation of Aβ25-35 with (A) interleukin (IL)-2 content, (B) percentage of inhibition of adenylate cyclase (AC) activity, (C) neprilysin and (D) insulin-degrading enzyme (IDE) levels in the hippocampus. Correlation coefficients (r) and p values are represented for each analysis. NS, non-significant.
Figure 7.
Correlation of Aβ25-35 with (A) interleukin (IL)-2 content, (B) percentage of inhibition of adenylate cyclase (AC) activity, (C) neprilysin and (D) insulin-degrading enzyme (IDE) levels in the hippocampus. Correlation coefficients (r) and p values are represented for each analysis. NS, non-significant.
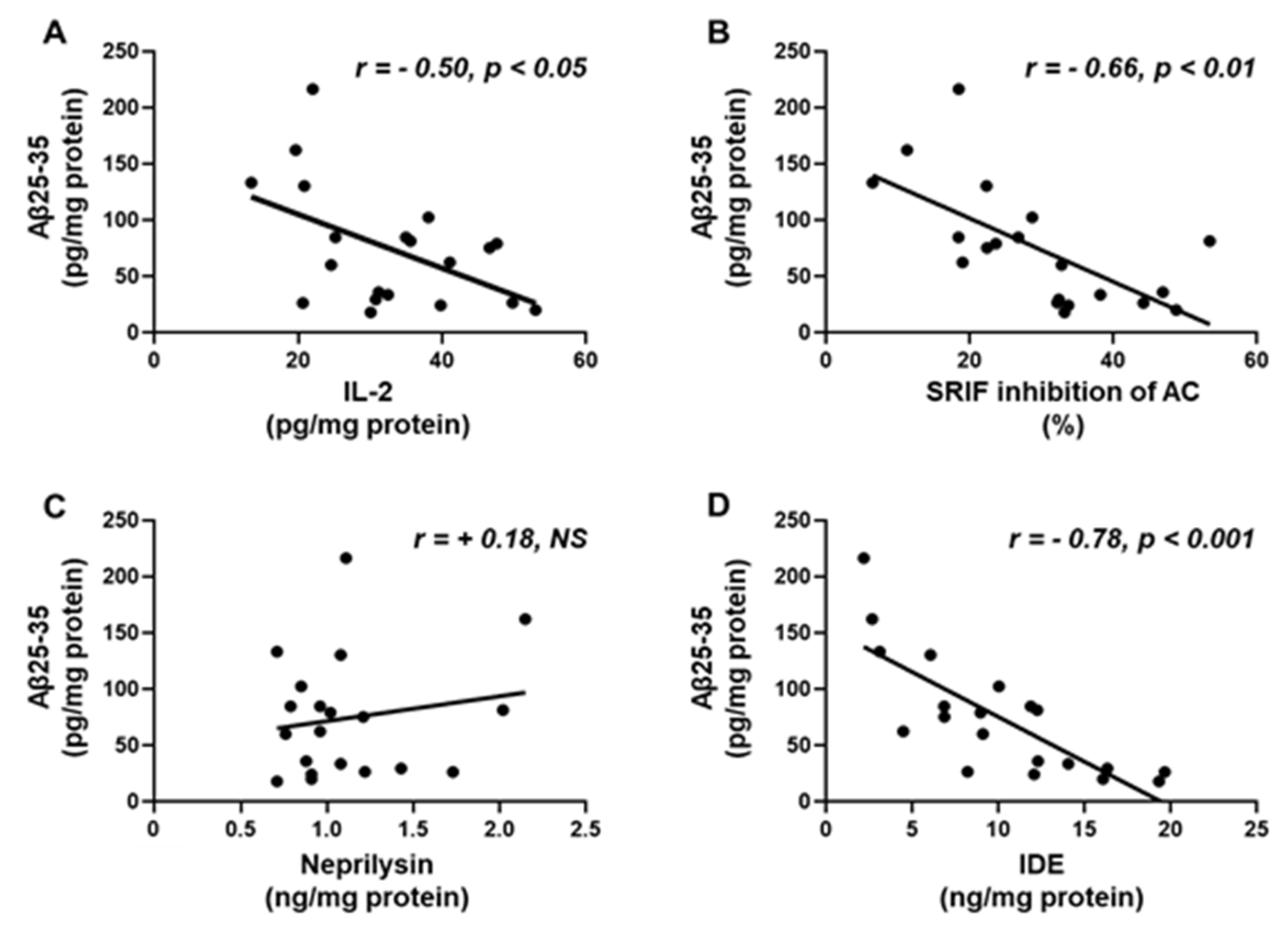

Table 1.
Correlation between Aβ25-35 levels, SRIF inhibition of AC activity and IDE levels with phosphorylation of intracellular signaling targets and cytokine content in the hippocampus.
Table 1.
Correlation between Aβ25-35 levels, SRIF inhibition of AC activity and IDE levels with phosphorylation of intracellular signaling targets and cytokine content in the hippocampus.
| Aβ25-35 (pg/mg) | SRIF inhibition AC (%) | IDE (ng/mg) | |
|---|---|---|---|
| r p | r p | r p | |
| p-p38MAPK/MAPK (%) | + 0.53 * | - 0.59 ** | - 0.45 * |
| pSerNFκB/NFκB (%) | - 0.40 NS | + 0.49 * | + 0.38 NS |
| pTyrSTAT3/STAT3 (%) | - 0.74 *** | + 0.57 ** | + 0.60 ** |
| pSerSTAT3/STAT3 (%) | - 0.76 *** | + 0.65 ** | + 0.71 *** |
| pTyrIGF-IR/mg protein | - 0.63 ** | + 0.61 ** | + 0.53 * |
| pTyrIRS1/IRS1 (%) | - 0.61 ** | + 0.41 NS | + 0.42 NS |
| pSerIRS1/IRS1 (%) | + 0.86 *** | - 0.67 ** | - 0.72 *** |
| pThrAkt/Akt (%) | - 0.66 ** | + 0.62 ** | + 0.55 * |
| IFN-γ (pg/mg) | + 0.80 *** | - 0.60 ** | - 0.72 *** |
| IL-2 (pg/mg) | - 0.50 * | + 0.37 NS | + 0.70 *** |
| IL-13 (pg/mg) | - 0.78 *** | + 0.51 * | + 0.69 *** |
| IL-17A (pg/mg) | + 0.60 ** | - 0.54 * | - 0.59 ** |
AC, adenylate cyclase; pThrAkt, Akt phosphorylated (p) at Thr308; IFN-γ, interferon-γ; pTyrIGF-IR, insulin-like growth factor-I receptor (IGF-IR) phosphorylated (p) at Tyr1131; IL, interleukin; pTyrIRS1, insulin receptor substrate 1 (IRS1) phosphorylated at Tyr residues (pTyrIRS1), pSer636IRS1, IRS1 phosphorylated at Ser636; p-p38MAPK/MAPK, p38 mitogen activated protein kinase (pMAPK) phosphorylated at Thr180 and Tyr182; pSerNFκB, nuclear factor kappa B (NFκB) phosphorylated at Ser536; SRIF, somatostatin; pTyrSTAT3, signal transducer and activator of transcription 3 (STAT3) phosphorylated at Tyr705; pSerSTAT3, STAT3 phosphorylated at Ser727. Correlation coefficients (r) and p values are provided for each analysis. N = 5. NS, non-significant. * p < 0.05, ** p < 0.01, *** p < 0.001.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



