
Submitted:
18 April 2024
Posted:
19 April 2024
You are already at the latest version
Abstract
Spinal muscular atrophy is a neuromuscular genetic condition associated with progressive muscle weakness and atrophy. Nusinersen is an antisense oligonucleotide therapy approved for the treatment of 5q spinal muscular atrophy in pediatric and adult patients. The objective of this clinical case series is to describe the efficacy and safety of nusinersen in treating spinal muscular atrophy in 20 pediatric and 18 adult patients across six treatment centers in Kuwait. Functional motor assessments (Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders, Hammersmith Functional Motor Scale Expanded, and Revised Upper Limb Module) were used to assess changes in motor function following nusinersen treatment. Safety assessment involved clinical monitoring of adverse events. The results demonstrate clinically meaningful or considerable improvement in motor performance for nearly all patients, lasting over 4 years in some cases. 70% of patients in the pediatric cohort and 72% of patients in the adult cohort achieved clinically meaningful improvement in motor function following nusinersen treatment. Additionally, nusinersen was well-tolerated in both cohorts. These findings add to the growing body of evidence relating to the clinical efficacy and safety of nusinersen.
Keywords:
spinal muscular atrophy
; neuromuscular disease
; nusinersen
; antisense oligonucleotide
; pediatrics
1. Introduction
Spinal muscular atrophy (SMA) is a progressive neuromuscular disorder and the most common genetic cause of infant mortality [1]. The global incidence of SMA is 1 in 10,000 live births [2]. However, the incidence of SMA in the Middle East is reportedly 1–20 in 10,000 live births [3,4,5,6]. It is thought that higher rates of consanguinity in the region may account for the increased incidence and carrier frequency [2,7]. A recent study in Saudi Arabia reported an SMA carrier frequency of 2.6%, which is higher than the reported global frequency of 1.25–2% [8].
SMA is an autosomal recessive disease characterized by muscle atrophy caused by insufficient production of survival motor neuron (SMN) protein [9]. This results in degradation of motor neurons in the anterior horn of the spinal cord [10]. SMA is of five types: 0 to IV in decreasing order of severity and increasing order of typical age of onset [11]. Around 60% of patients with SMA have the severe, infantile-onset type I form, associated with the inability to sit independently, loss of motor skills, progressive weakness, failure to thrive, scoliosis, dysphagia, and breathing problems [12]. SMA type II is associated with onset between 6 and 18 months, developmental delays, and loss of motor skills, with the ability to sit independently when placed [10]. Both SMA types I and II are associated with substantial impairment and mortality in the context of natural history of disease [13,14]. In adults, SMA type III (associated with onset between 18 months and 18 years and independent ambulation) or IV (associated with onset after 18 years and normal ambulation) can limit mobility and cause weakness and fatigue [15]. However, there is thought to be a degree of heterogeneity in terms of clinical presentations and disease manifestations [16].
Insufficient SMN protein production arises from homozygous deletion or a pathogenic mutation of the SMN1 gene located on chromosome 5q13 [17]. This results in complete reliance on the similar SMN2 gene. However, only around 10% of SMN2 transcripts generate functional SMN protein [18]. It is thought that disease severity correlates with the number of copies of SMN2, with fewer gene copies associated with more severe phenotypes [19,20].
Nusinersen is an antisense oligonucleotide approved for the treatment 5q SMA in pediatric and adult patients [21]. It increases production of SMN protein in the central nervous system by altering splicing of SMN2 to promote exon 7 inclusion, thus improving motor neuron survival [22,23]. Studies have demonstrated that nusinersen improves motor function in SMA and increases survival rates in type I patients [7,22,24]. Nusinersen is also effective in treating symptoms of SMA in adults, and real-world data is increasingly providing an understanding of its potential benefits and tolerability in adult SMA [21,25].
The aim of this case series is to describe the clinical efficacy and safety of nusinersen in 38 patients with SMA (20 pediatric and 18 adults) across six treatment centers in Kuwait.
2. Materials and Methods
2.1. Study Design and Participants
In this multicenter case series, we describe 20 pediatric and 18 adult patients with SMA who received nusinersen at Al Sabah, Al Adan, Al Jahra, Al Farwaniya, Mubarak, and Ibn Sina Hospitals in Kuwait (treatment initiation period between July 2016 and February 2022). We discuss the clinical efficacy and safety of nusinersen within these patient groups. All participants had a diagnosis of 5q SMA confirmed via genetic documentation. SMA type classification was not used to exclude patients. The age ranges of patients were 2 to 12 years in the pediatric cohort and 18 to 55 years in the adult cohort, and 14 of the 38 included patients were female.
2.2. Nusinersen Treatment
All patients received nusinersen via intrathecal injection as per current standard therapy recommendations [26]. In the loading phase, four 12 mg doses were administered (first three doses in 14-day intervals and the fourth dose 30 days after the third). Subsequently, 12 mg maintenance doses were administered every 4 months. For the purpose of this case series study, the day of administration of the first loading dose was considered the first day of treatment initiation for each patient.
2.3. Functional and Safety Assessments
Among the pediatric cohort of 20 patients, motor function was evaluated in 14 patients via the Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP-INTEND) at baseline and several points after initiation of nusinersen treatment. The CHOP-INTEND is a validated 16-item scale, with a maximum score of 4 for each checklist component [27]. For the remaining six patients in the pediatric cohort (all with SMA types II or III), gross motor function was evaluated via the Hammersmith Functional Motor Scale Expanded (HFMSE) tool, which consists of 33 items scored 0, 1, or 2, giving a maximum score of 66 [28]. Within the adult cohort, the Revised Upper Limb Module (RULM) assessment was used as the vast majority of patients were non-ambulant at the time of presentation. This is also a validated tool used in SMA types II and III, with 19 scorable items mostly graded using a 3-point system and a maximum score of 37 for each upper limb [29]. Safety assessment involved clinical monitoring of adverse events and complications.
2.4. Data Analysis
Linear trendlines (moving average models) were used to illustrate changes in functional motor outcomes for each patient. Changes in motor performance for pediatric patients 1–7 and patients 8–14 are visualized on separate graphs for clarity. The same applies for adult patients 21–29 and 30–38. Due to the retrospective nature of this study, there are some inconsistencies in the data reported, such as missing baseline functional assessment scores for some patients. Clinically meaningful improvement in functional motor performance was defined as an increase of ≥ 4 points, ≥ 3 points, and ≥ 2 points in CHOP-INTEND, HFMSE, and RULM score, respectively [30,31,32,33]. Considerable improvement in functional motor performance was defined as any increase in CHOP-INTEND, HFMSE, or RULM score ˂ 4 points, ˂ 3 points, and ˂ 2 points, respectively. Clinical stabilization was defined as maintenance of CHOP-INTEND, HFMSE, or RULM score, and motor decline was defined as any decrease in CHOP-INTEND, HFMSE, or RULM score.
3. Results
3.1. Patient Characteristics
A total of 20 pediatric and 18 adult patients with SMA were included in this case series study. Patient characteristics are summarized in Table 1 and Table 2. Among the pediatric cohort, 45% of patients were female (Figure 1). 40% were diagnosed were type I SMA, 40% with type II SMA, and 20% with type III SMA (Figure 2). 10% had one copy of SMN2, 50% had two copies, and 10% had three copies (Figure 3). 50% had a confirmed family history of SMA (Figure 4).
Among the adult cohort, 28% of patients were female (Figure 5). 11% were diagnosed with type II SMA and 89% were diagnosed with type III SMA (Figure 6). 17% had two copies of SMN2, 44% had three copies, and 11% had four copies (Figure 7). 72% had a confirmed family history of SMA (Figure 8) and 94% of cases were associated with parental consanguinity (Figure 9).
3.2. Functional Motor Studies
In the pediatric cohort, motor function was evaluated in 14 patients via CHOP-INTEND assessment at baseline and several points following treatment initiation (Figure 10 and Figure 11). In 13 patients, CHOP-INTEND assessments demonstrated a significant or considerable improvement in functional motor performance after treatment initiation compared with baseline, and one patient was clinically stabilized. Note that fluctuations were observed in the motor performance of patients 6, 7, and 14 over time. This may be attributed to these patients missing doses or receiving delayed doses due to national travel restrictions imposed in Kuwait following the COVID-19 pandemic. Within this subgroup of patients, the average increase in CHOP-INTEND from the earliest recorded assessment point to the latest was 13.7. 71% of these patients experienced a clinically meaningful improvement in motor performance (≥ 4 CHOP-INTEND points) following nusinersen treatment, and no patients experienced motor decline associated with SMA progression.
For six patients in the pediatric cohort, motor function was evaluated via HFMSE assessment (Figure 12). Four patients in this subgroup experienced a significant improvement in functional motor performance, whereas one patient experienced a considerable improvement. Patient 15 had a low baseline HFMSE score and exhibited stabilization over 50 months of treatment. Within this subgroup, the average increase in HFMSE from the earliest to the latest assessment point was 3.5. 67% of patients in this subgroup experienced a clinically meaningful improvement in motor performance (≥ 3 HFMSE points) following nusinersen treatment, and no patients experienced motor decline associated with SMA progression. Overall, in the pediatric cohort (i.e., patients 1-20), 70% of patients experienced a clinically meaningful improvement in motor function following nusinersen treatment, and 20% of patients experienced a considerable improvement in motor function (Figure 13).
For the adult SMA cohort, RULM assessment was used to evaluate changes in motor performance following treatment initiation (Figure 14 and Figure 15). Of the 18 patients in the adult cohort, 13 experienced clinically meaningful improvement in functional motor performance, three experienced considerable improvement, and one was clinically stabilized. Just one patient with a high baseline RULM score exhibited a slight decline in motor performance associated with SMA disease progression. This is thought to be related to the patient missing or receiving delayed doses of nusinersen due to national travel restrictions implemented in Kuwait following the COVID-19 pandemic. Within the adult cohort, the average increase in RULM score was 7.3 from the earliest to the latest assessment point. 72% of patients in the adult cohort experienced a clinically meaningful improvement in motor performance (≥ 2 RULM points) following nusinersen treatment, and 17% of patients experienced a considerable improvement (Figure 16).
3.3. Clinical Presentations and Motor Milestones
11 patients of the 20 in the pediatric cohort were diagnosed with SMA and initiated nusinersen treatment in infancy. 10 of these patients exhibited significant or considerable motor function improvement, and six were capable of sitting independently. Nine patients in the pediatric cohort were diagnosed after the age of 12 months. Of them, three were entirely wheelchair bound, two were limited to assisted walking, and four were capable of unassisted walking.
Among the adult SMA cohort of 18 patients, 14 were non-ambulant at the time of initial presentation. All patients responded well to nusinersen therapy, with no deterioration in motor function observed.
3.4. Safety Outcomes
From the pediatric cohort, three patients were admitted for chest infections on more than one occasion. One patient developed severe scoliosis and underwent corrective surgery. Six other patients also developed mild or moderate scoliosis. After 2 years of nusinersen therapy, one patient experienced mild elevations in prothrombin time and activated partial thromboplastin time (17.9 and 37.0 seconds, respectively). This resolved naturally within 3 months. In relation to drug administration, one patient occasionally experienced post-injection headaches and another experienced fibrous tissue formation at the site of lumbar puncture. The development of scoliosis among these patients is thought to be a consequence of disease activity as opposed to a side effect of nusinersen. All other adverse safety events were transient and were considered mild.
Among the adult SMA patient cohort, nusinersen was very well-tolerated. The only adverse safety event was the development of mild restrictive lung disease in one patient, which was possibly unrelated to nusinersen.
4. Discussion
In this retrospective, multicenter study in Kuwait, we investigated the effects of nusinersen in 38 patients with SMA. There were 20 patients in the pediatric cohort, eight of whom were diagnosed with SMA type I, eight of whom were diagnosed with SMA type II, and four of whom were diagnosed with SMA type III. In the adult cohort, two patients were diagnosed with SMA type II and 16 were diagnosed with SMA type III. This is the first retrospective clinical case series of nusinersen for SMA in Kuwait, and its findings add to the growing body of efficacy and safety data pertaining to antisense oligonucleotide therapy.
In the pediatric cohort, 70% of patients achieved clinically meaningful improvement in motor function following nusinersen treatment. Nearly all patients experienced either considerable or significant improvement in motor function as assessed by CHOP-INTEND or HFMSE. Two exceptions were patients with low baseline functional motor scores, who were clinically stabilized and showed no further motor deterioration. Three other patients experienced fluctuations in motor performance over time, which is thought to relate to missed doses and interruptions in physical therapy due to national travel restrictions imposed following the COVID-19 pandemic. In the adult cohort, 72% of patients achieved clinically meaningful improvement in motor function following nusinersen treatment. Nearly all patients in the adult cohort experienced either significant or considerable improvements in functional motor performance as assessed by RULM. Two exceptions were patients with very high baseline RULM scores, in whom the ceiling effect was observed.
These findings demonstrate improved motor function in patients with SMA following nusinersen treatment in real-world clinical settings in Kuwait. The findings are congruent with results from the ENDEAR and CHERISH phase III clinical trials, which investigated the efficacy and safety of nusinersen in SMA in 121 infants and 126 children, respectively [23,24]. In the SHINE open-label extension study, nusinersen demonstrated improvement or stabilization in motor function measures for up to nearly 6 years [34]. Furthermore, a 2021 meta-analysis that included observational data from pediatric and adult patients with SMA types II and III also reported that nusinersen provided a favorable benefit in motor performance over a 10–14-month period [35]. In terms of safety, nusinersen was well-tolerated within both cohorts in this study, which is consistent with recently published observational studies [21,36,37]. In addition, the generally improved functional performance of patients treated with nusinersen is thought to be associated with enhanced function and/or survival of motor neurons caused by increased SMN protein production.
It has previously been demonstrated that the pharmacology of nusinersen is consistent with its intended mechanism of action, with increases in the amount of functional SMN2 messenger RNA transcript and SMN protein having been observed [12]. Therefore, the drug reaches its target tissues and promotes exon 7 inclusion during gene splicing in the central nervous system. It is also thought that the number of SMN2 gene copies affects SMA phenotype, with fewer copies associated with more severe disease [20,38]. Indeed, similar associations between SMA type and SMN2 copy number were noted within the cohorts of patients in this study.
There are a few limitations associated with this case series. Firstly, baseline functional motor data were unavailable for some patients who initiated nusinersen treatment outside of Kuwait. For these patients, we report the earliest recorded functional motor score obtained a few months after therapy was initiated. Had baseline functional scores been available for all patients, even greater average improvements in motor performance may have been observed. Additionally, clinical measures solely relied on assessments of motor function, and as such, the benefits of nusinersen in non-motor domains were not captured. Despite these limitations, the study is meaningful as it is the first to report on the clinical efficacy and safety of nusinersen in patients with SMA in Kuwait.
Overall, this case series describes promising clinical outcomes following nusinersen therapy in SMA. This is particularly relevant to Kuwait, a country with higher-than-average levels of consanguinity and disease incidence. Further clinical data generation and research is recommended in the country to identify novel protocols for optimal disease management and improved patient outcomes.
5. Conclusions
This case series demonstrates that nusinersen is generally well-tolerated in pediatric and adult patients with SMA in Kuwait. Most patients experienced a clinically meaningful improvement in functional motor performance following treatment initiation, with benefits sustained for over 4 years in eight patients. Further large-scale studies are warranted to confirm the findings reported.
Author Contributions
Conceptualization, A.T.; data curation, A.T., M.Z., W.K., L.B., G.A.I.E., M.E.A.A., D.S., A.A., E.E.A.E., O.S., F.A., V.M., and N.S.; writing—original draft preparation, A.T.; writing—review and editing, M.Z., W.K., L.B., G.A.I.E., M.E.A.A., D.S., A.A., E.E.A.E., O.S., F.A., V.M., and N.S.; visualization, A.T.; project administration, A.T. All authors have read and agreed to the published version of the manuscript.
Funding
Medical writing support and article processing charges were funded by Biogen.
Institutional Review Board Statement
This study was approved by the Kuwait Ministry of Health on 18 February 2024.
Informed Consent Statement
All reported patient data is anonymized. Patients or their families consented to the use of their data in this publication.
Data Availability Statement
The data supporting the findings of this study are available within the article.
Acknowledgments
The authors acknowledge the pediatric/adult neurology teams and physiotherapy teams at Al Sabah, Al Adan, Al Jahra, Al Farwaniya, Mubarak, and Ibn Sina Hospitals in Kuwait for their contributions. We also thank the patients and their families for their support. Medical writing was provided by Luqman Khan of Connect Communications, Dubai, UAE and funded by Biogen.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Arnold, W.D.; Kassar, D.; Kissel, J.T. Spinal muscular atrophy: diagnosis and management in a new therapeutic era. Muscle Nerve 2015, 51, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Verhaart, I.E.C.; Robertson, A.; Wilson, I.J.; Aartsma-Rus, A.; Cameron, S.; Jones, C.C.; Cook, S.F.; Lochmuller, H. Prevalence, incidence and carrier frequency of 5q-linked spinal muscular atrophy - a literature review. Orphanet J Rare Dis 2017, 12, 124. [Google Scholar] [CrossRef] [PubMed]
- Al-Rajeh, S.; Bademosi, O.; Gascon, G.G.; Stumpf, D. Werdnig Hoffman’s disease (spinal muscular atrophy type I): A clinical study of 25 Saudi nationals in Al-Khobar. Ann Saudi Med 1992, 12, 67–71. [Google Scholar] [CrossRef] [PubMed]
- al Rajeh, S.; Bademosi, O.; Ismail, H.; Awada, A.; Dawodu, A.; al-Freihi, H.; Assuhaimi, S.; Borollosi, M.; al-Shammasi, S. A community survey of neurological disorders in Saudi Arabia: the Thugbah study. Neuroepidemiology 1993, 12, 164–178. [Google Scholar] [CrossRef] [PubMed]
- Rajab, A.; Bappal, B.; Al-Shaikh, H.; Al-Khusaibi, S.; Mohammed, A.J. Common autosomal recessive diseases in Oman derived from a hospital-based registry. Community Genet 2005, 8, 27–30. [Google Scholar] [CrossRef] [PubMed]
- al-Gazali, L.I.; Dawodu, A.H.; Sabarinathan, K.; Varghese, M. The profile of major congenital abnormalities in the United Arab Emirates (UAE) population. J Med Genet 1995, 32, 7–13. [Google Scholar] [CrossRef]
- Ali, H.G.; Ibrahim, K.; Elsaid, M.F.; Mohamed, R.B.; Abeidah, M.I.A.; Al Rawwas, A.O.; Elshafey, K.; Almulla, H.; El-Akouri, K.; Almulla, M.; et al. Gene therapy for spinal muscular atrophy: the Qatari experience. Gene Ther 2021, 28, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Al Jumah, M.; Al Rajeh, S.; Eyaid, W.; Al-Jedai, A.; Al Mudaiheem, H.; Al Shehri, A.; Hussein, M.; Al Abdulkareem, I. Spinal muscular atrophy carrier frequency in Saudi Arabia. Mol Genet Genomic Med 2022, 10, e2049. [Google Scholar] [CrossRef] [PubMed]
- Iyer, C.C.; McGovern, V.L.; Murray, J.D.; Gombash, S.E.; Zaworski, P.G.; Foust, K.D.; Janssen, P.M.; Burghes, A.H. Low levels of Survival Motor Neuron protein are sufficient for normal muscle function in the SMNDelta7 mouse model of SMA. Hum Mol Genet 2015, 24, 6160–6173. [Google Scholar] [CrossRef]
- Prior, T.W.; Leach, M.E.; Finanger, E. Spinal Muscular Atrophy. In GeneReviews((R)), Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; Seattle (WA), 1993.
- Kolb, S.J.; Kissel, J.T. Spinal Muscular Atrophy. Neurol Clin 2015, 33, 831–846. [Google Scholar] [CrossRef]
- Finkel, R.S.; Chiriboga, C.A.; Vajsar, J.; Day, J.W.; Montes, J.; De Vivo, D.C.; Yamashita, M.; Rigo, F.; Hung, G.; Schneider, E.; et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study. Lancet 2016, 388, 3017–3026. [Google Scholar] [CrossRef] [PubMed]
- D‘Amico, A.; Mercuri, E.; Tiziano, F.D.; Bertini, E. Spinal muscular atrophy. Orphanet Journal of Rare Diseases 2011, 6, 71. [Google Scholar] [CrossRef] [PubMed]
- Finkel, R.S.; McDermott, M.P.; Kaufmann, P.; Darras, B.T.; Chung, W.K.; Sproule, D.M.; Kang, P.B.; Foley, A.R.; Yang, M.L.; Martens, W.B.; et al. Observational study of spinal muscular atrophy type I and implications for clinical trials. Neurology 2014, 83, 810–817. [Google Scholar] [CrossRef] [PubMed]
- Piepers, S.; van den Berg, L.H.; Brugman, F.; Scheffer, H.; Ruiterkamp-Versteeg, M.; van Engelen, B.G.; Faber, C.G.; de Visser, M.; van der Pol, W.L.; Wokke, J.H. A natural history study of late onset spinal muscular atrophy types 3b and 4. J Neurol 2008, 255, 1400–1404. [Google Scholar] [CrossRef]
- Wan, H.W.Y.; Carey, K.A.; D‘Silva, A.; Vucic, S.; Kiernan, M.C.; Kasparian, N.A.; Farrar, M.A. Health, wellbeing and lived experiences of adults with SMA: a scoping systematic review. Orphanet J Rare Dis 2020, 15, 70. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, S.; Burglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Benichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M.; et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995, 80, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Aslesh, T.; Yokota, T. Restoring SMN Expression: An Overview of the Therapeutic Developments for the Treatment of Spinal Muscular Atrophy. Cells 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, S.; Burlet, P.; Liu, Q.; Bertrandy, S.; Clermont, O.; Munnich, A.; Dreyfuss, G.; Melki, J. Correlation between severity and SMN protein level in spinal muscular atrophy. Nat Genet 1997, 16, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Feldkotter, M.; Schwarzer, V.; Wirth, R.; Wienker, T.F.; Wirth, B. Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am J Hum Genet 2002, 70, 358–368. [Google Scholar] [CrossRef]
- Lam, K.; Wu, A. Clinical Outcome of Adult Spinal Muscular Atrophy Patients Treated with Nusinersen: A Case Series Review. Perm J 2020, 25, 1. [Google Scholar] [CrossRef]
- Albrechtsen, S.S.; Born, A.P.; Boesen, M.S. Nusinersen treatment of spinal muscular atrophy - a systematic review. Dan Med J 2020, 67. [Google Scholar]
- Finkel, R.S.; Mercuri, E.; Darras, B.T.; Connolly, A.M.; Kuntz, N.L.; Kirschner, J.; Chiriboga, C.A.; Saito, K.; Servais, L.; Tizzano, E.; et al. Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N Engl J Med 2017, 377, 1723–1732. [Google Scholar] [CrossRef] [PubMed]
- Mercuri, E.; Darras, B.T.; Chiriboga, C.A.; Day, J.W.; Campbell, C.; Connolly, A.M.; Iannaccone, S.T.; Kirschner, J.; Kuntz, N.L.; Saito, K.; et al. Nusinersen versus Sham Control in Later-Onset Spinal Muscular Atrophy. N Engl J Med 2018, 378, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Cintas, P. Current treatments of spinal muscular atrophy in adults. Rev Neurol (Paris) 2023, 179, 106–113. [Google Scholar] [CrossRef] [PubMed]
- MacCannell, D.; Berger, Z.; East, L.; Mercuri, E.; Kirschner, J.; Muntoni, F.; Farrar, M.A.; Peng, J.; Zhou, J.; Nestorov, I.; et al. Population pharmacokinetics-based recommendations for a single delayed or missed dose of nusinersen. Neuromuscul Disord 2021, 31, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Glanzman, A.M.; Mazzone, E.; Main, M.; Pelliccioni, M.; Wood, J.; Swoboda, K.J.; Scott, C.; Pane, M.; Messina, S.; Bertini, E.; et al. The Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND): test development and reliability. Neuromuscul Disord 2010, 20, 155–161. [Google Scholar] [CrossRef] [PubMed]
- O‘Hagen, J.M.; Glanzman, A.M.; McDermott, M.P.; Ryan, P.A.; Flickinger, J.; Quigley, J.; Riley, S.; Sanborn, E.; Irvine, C.; Martens, W.B.; et al. An expanded version of the Hammersmith Functional Motor Scale for SMA II and III patients. Neuromuscul Disord 2007, 17, 693–697. [Google Scholar] [CrossRef] [PubMed]
- Mazzone, E.S.; Mayhew, A.; Montes, J.; Ramsey, D.; Fanelli, L.; Young, S.D.; Salazar, R.; De Sanctis, R.; Pasternak, A.; Glanzman, A.; et al. Revised upper limb module for spinal muscular atrophy: Development of a new module. Muscle Nerve 2017, 55, 869–874. [Google Scholar] [CrossRef]
- Lusakowska, A.; Wojcik, A.; Fraczek, A.; Aragon-Gawinska, K.; Potulska-Chromik, A.; Baranowski, P.; Nowak, R.; Rosiak, G.; Milczarek, K.; Konecki, D.; et al. Long-term nusinersen treatment across a wide spectrum of spinal muscular atrophy severity: a real-world experience. Orphanet J Rare Dis 2023, 18, 230. [Google Scholar] [CrossRef]
- Pera, M.C.; Coratti, G.; Forcina, N.; Mazzone, E.S.; Scoto, M.; Montes, J.; Pasternak, A.; Mayhew, A.; Messina, S.; Sframeli, M.; et al. Content validity and clinical meaningfulness of the HFMSE in spinal muscular atrophy. BMC Neurol 2017, 17, 39. [Google Scholar] [CrossRef]
- Pera, M.C.; Coratti, G.; Mazzone, E.S.; Montes, J.; Scoto, M.; De Sanctis, R.; Main, M.; Mayhew, A.; Muni Lofra, R.; Dunaway Young, S.; et al. Revised upper limb module for spinal muscular atrophy: 12 month changes. Muscle Nerve 2019, 59, 426–430. [Google Scholar] [CrossRef] [PubMed]
- Stolte, B.; Bois, J.M.; Bolz, S.; Kizina, K.; Totzeck, A.; Schlag, M.; Kleinschnitz, C.; Hagenacker, T. Minimal clinically important differences in functional motor scores in adults with spinal muscular atrophy. Eur J Neurol 2020, 27, 2586–2594. [Google Scholar] [CrossRef] [PubMed]
- A Study for Participants With Spinal Muscular Atrophy (SMA) Who Previously Participated in Nusinersen (ISIS 396443) Investigational Studies (SHINE). Available online: https://clinicaltrials.gov/ct2/show/NCT02594124.
- Coratti, G.; Cutrona, C.; Pera, M.C.; Bovis, F.; Ponzano, M.; Chieppa, F.; Antonaci, L.; Sansone, V.; Finkel, R.; Pane, M.; et al. Motor function in type 2 and 3 SMA patients treated with Nusinersen: a critical review and meta-analysis. Orphanet J Rare Dis 2021, 16, 430. [Google Scholar] [CrossRef] [PubMed]
- Hagenacker, T.; Wurster, C.D.; Gunther, R.; Schreiber-Katz, O.; Osmanovic, A.; Petri, S.; Weiler, M.; Ziegler, A.; Kuttler, J.; Koch, J.C.; et al. Nusinersen in adults with 5q spinal muscular atrophy: a non-interventional, multicentre, observational cohort study. Lancet Neurol 2020, 19, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Krupa, D.; Czech, M.; Chudzynska, E.; Kon, B.; Kostera-Pruszczyk, A. Real World Evidence on the Effectiveness of Nusinersen within the National Program to Treat Spinal Muscular Atrophy in Poland. Healthcare (Basel) 2023, 11. [Google Scholar] [CrossRef]
- Zhang, Y.H.; Zhang, Y.Q.; Zhu, B.S.; He, J.; Wang, L.; Tang, X.H.; Guo, J.J.; Jin, C.C.; Chen, H.; Zhang, J.; et al. [Association of copy number of SMN1 and SMN2 with clinical phenotypes in children with spinal muscular atrophy]. Zhongguo Dang Dai Er Ke Za Zhi 2019, 21, 239–243. [Google Scholar] [CrossRef]
Figure 1.
Gender proportion of patients in the pediatric cohort.
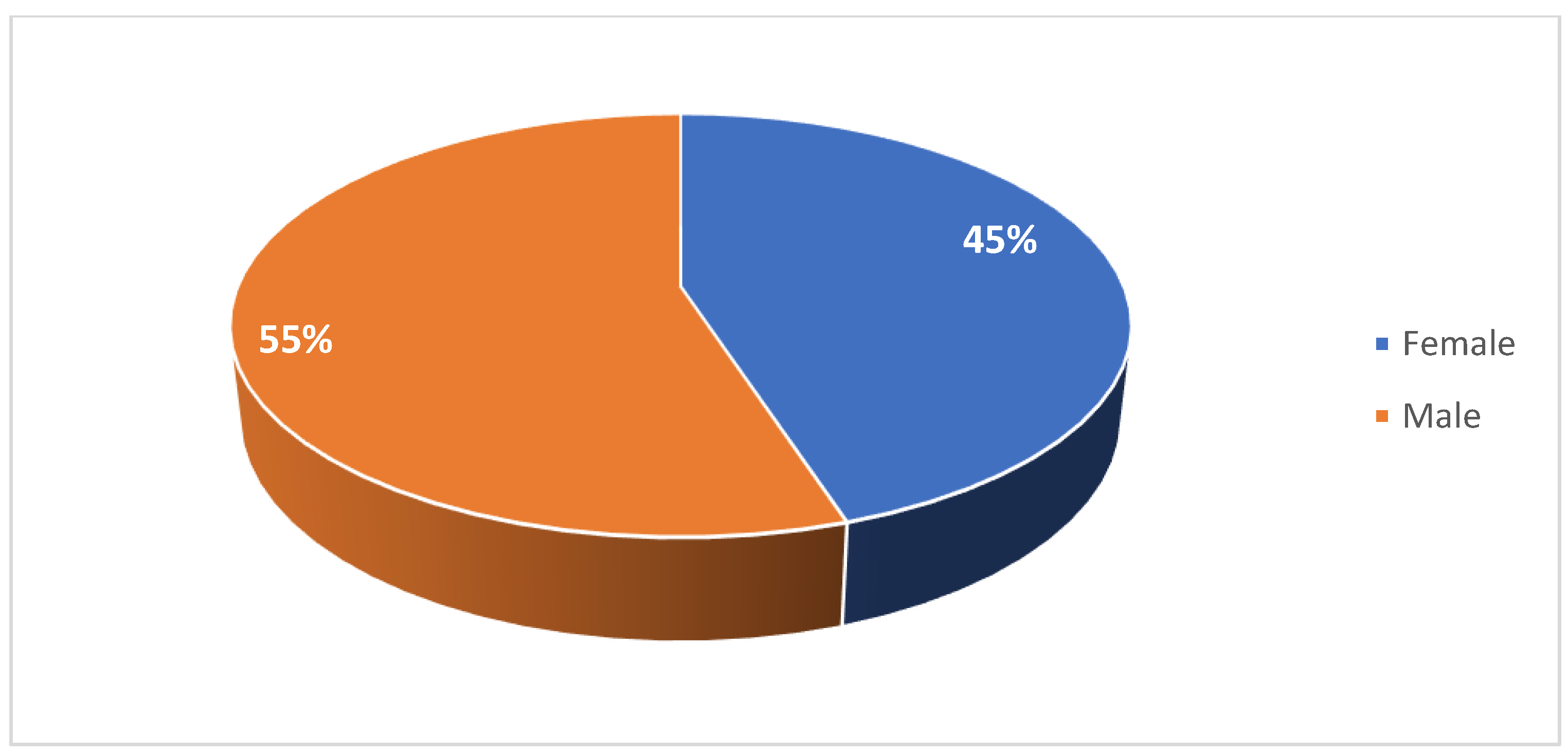
Figure 2.
Distribution of patients in the pediatric cohort by SMA type.
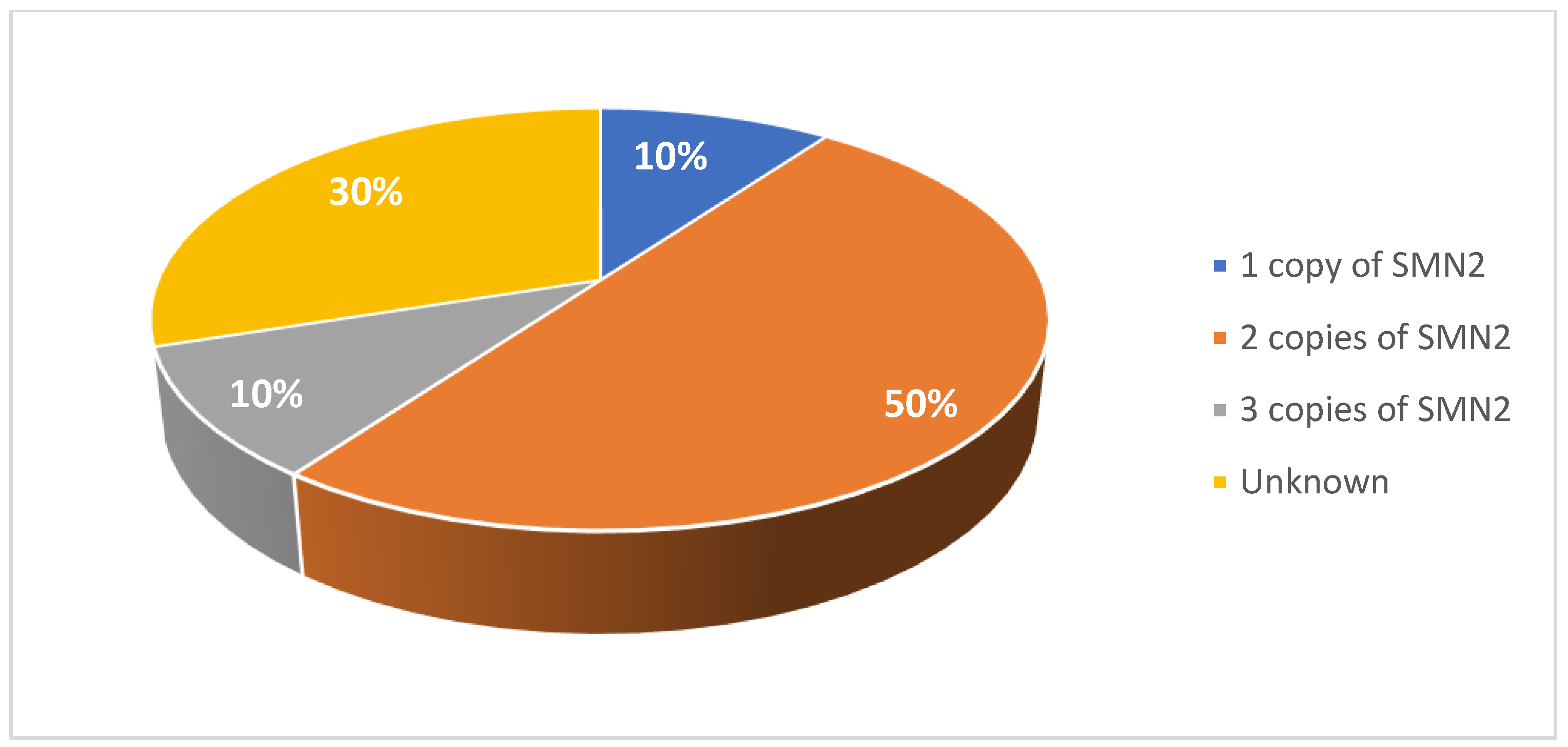
Figure 3.
Number of SMN2 gene copies in patients in the pediatric cohort.
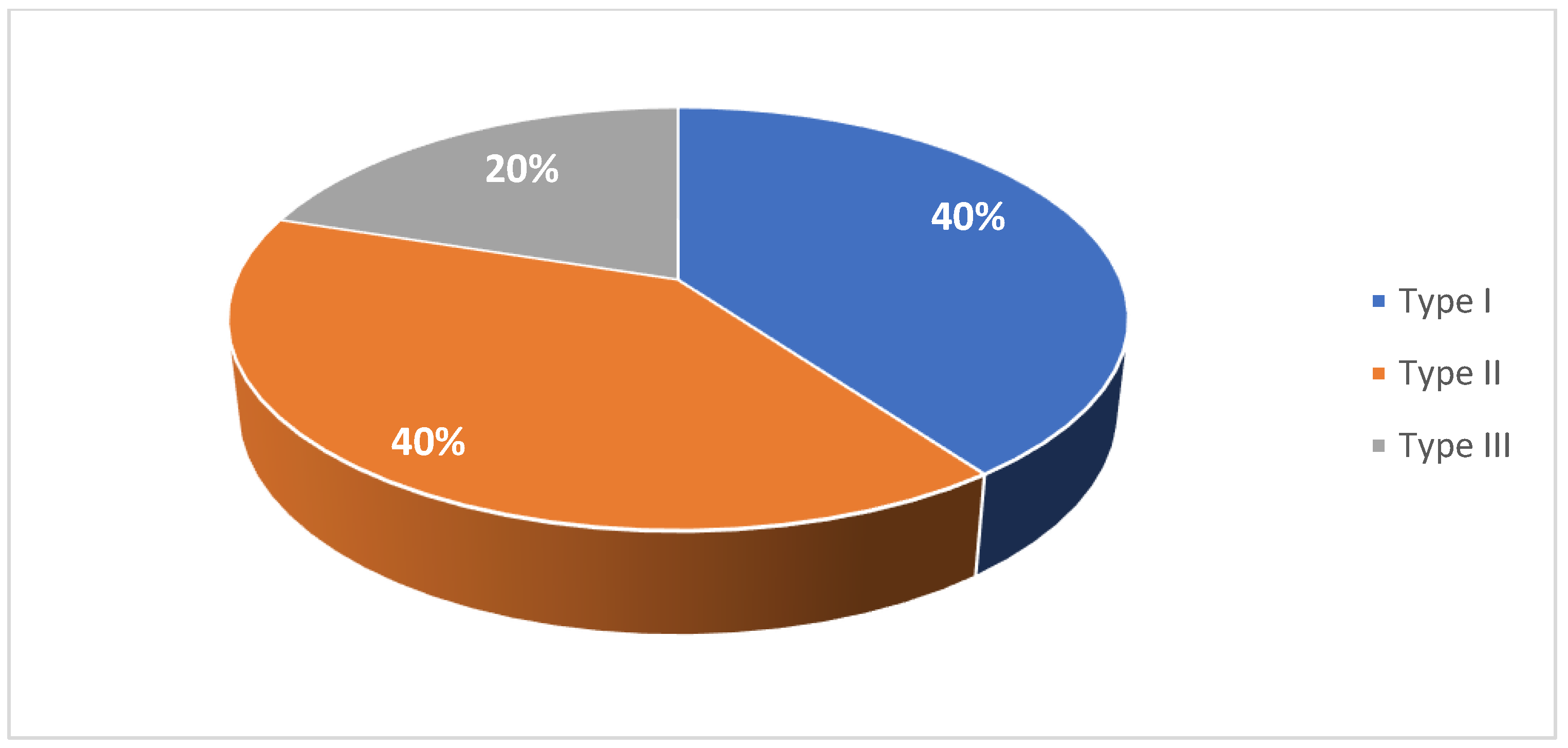
Figure 4.
Proportion of patients with a family history of SMA in the pediatric cohort.
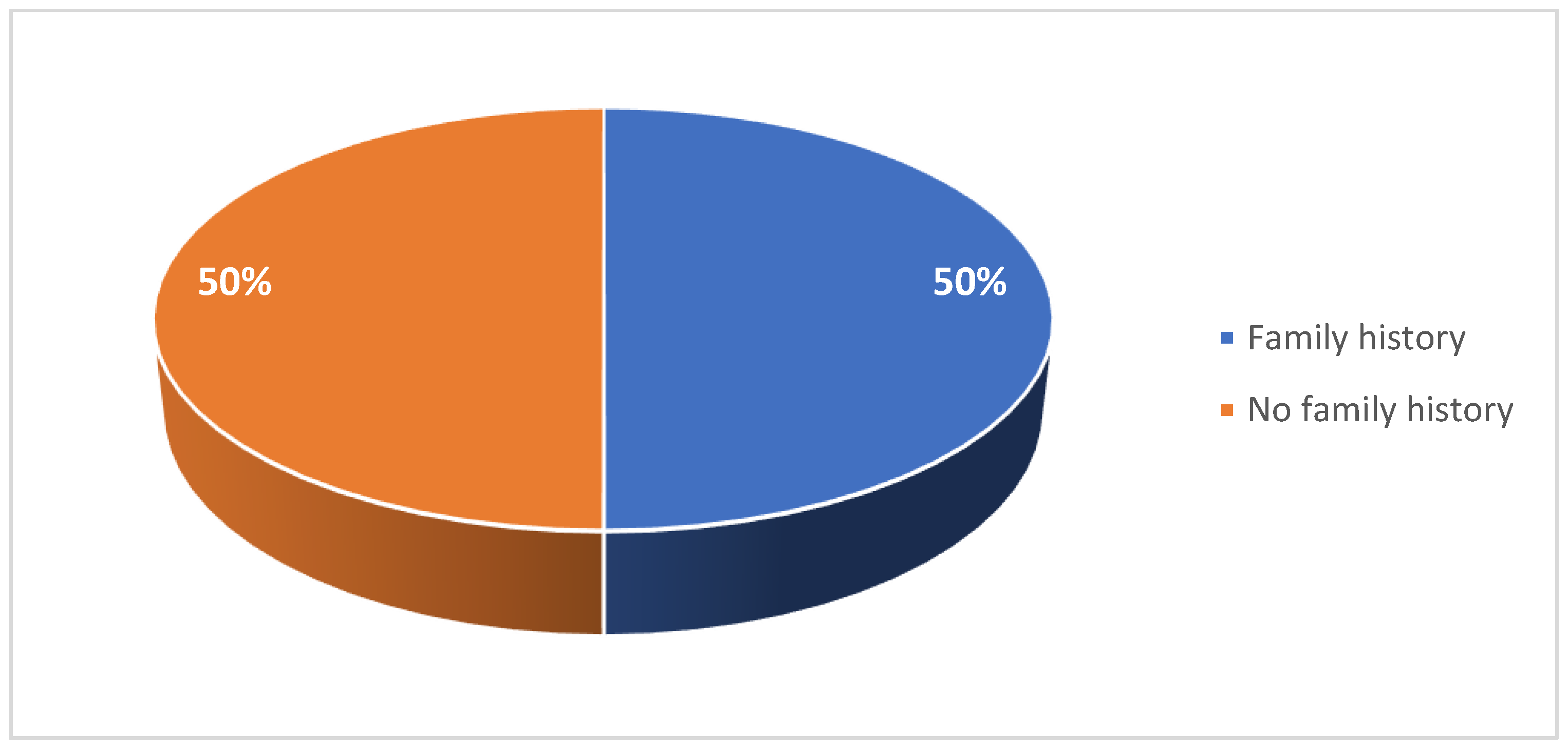
Figure 5.
Gender proportion of patients in the adult cohort.
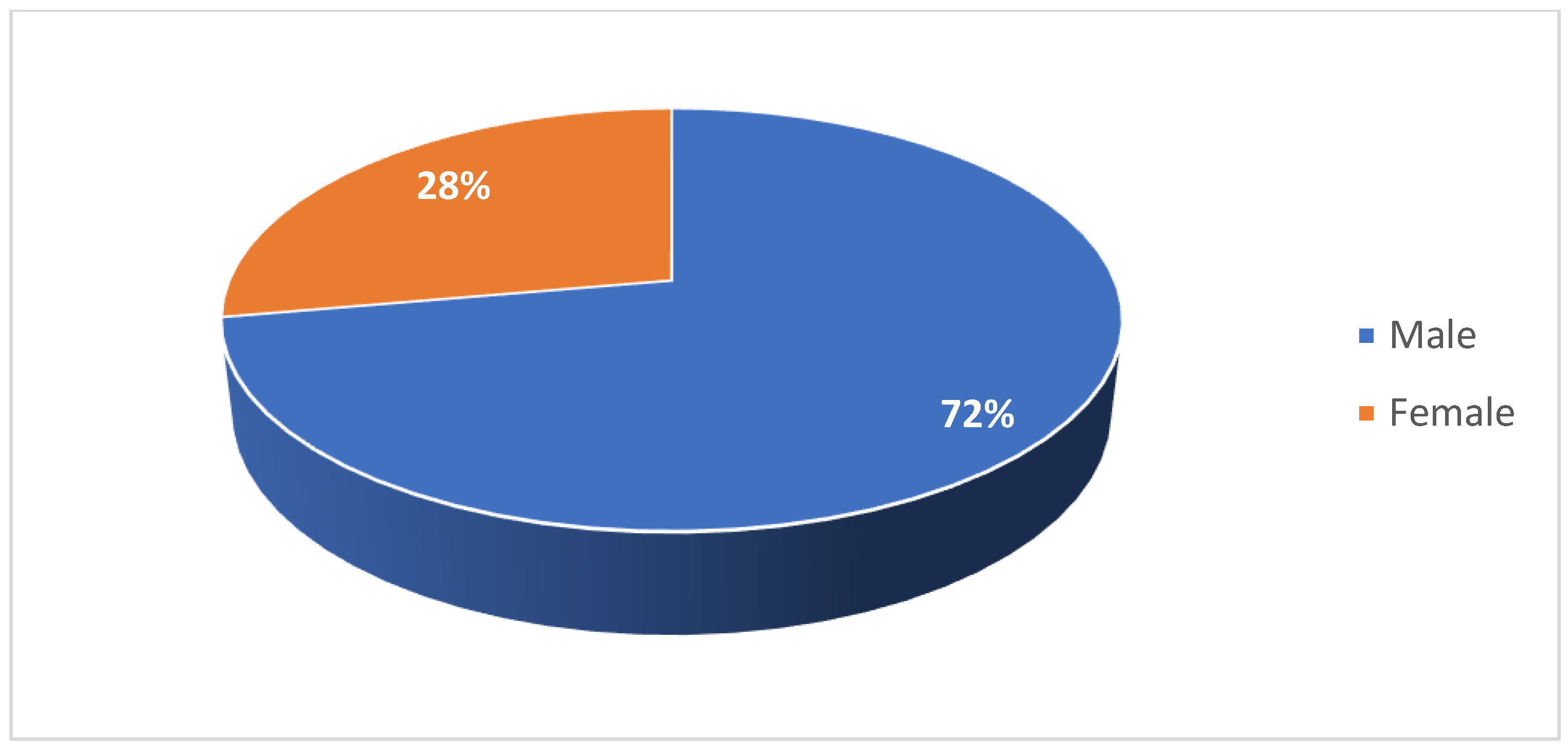
Figure 6.
Distribution of patients in the adult cohort by SMA type.
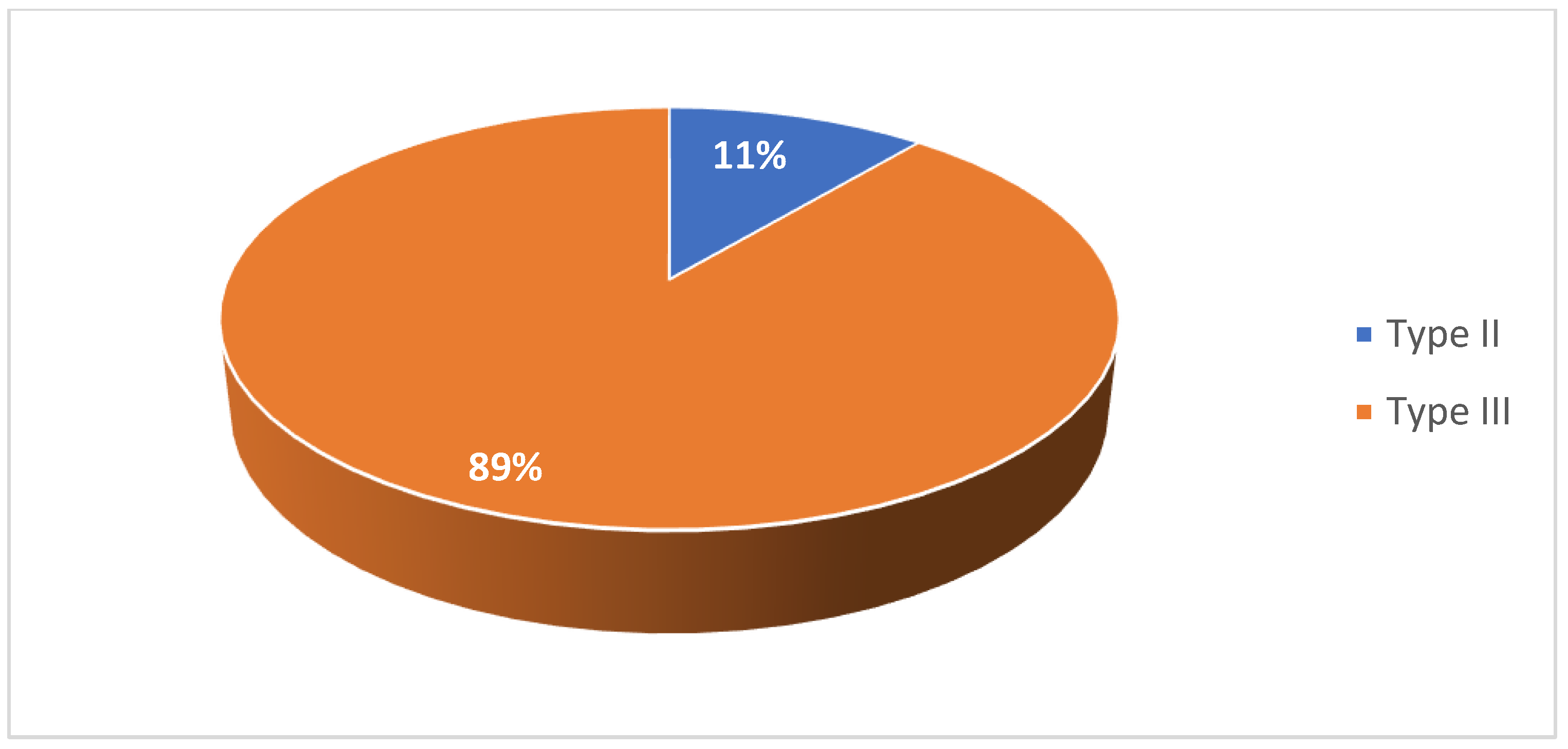
Figure 7.
Number of SMN2 gene copies in patients in the adult cohort.
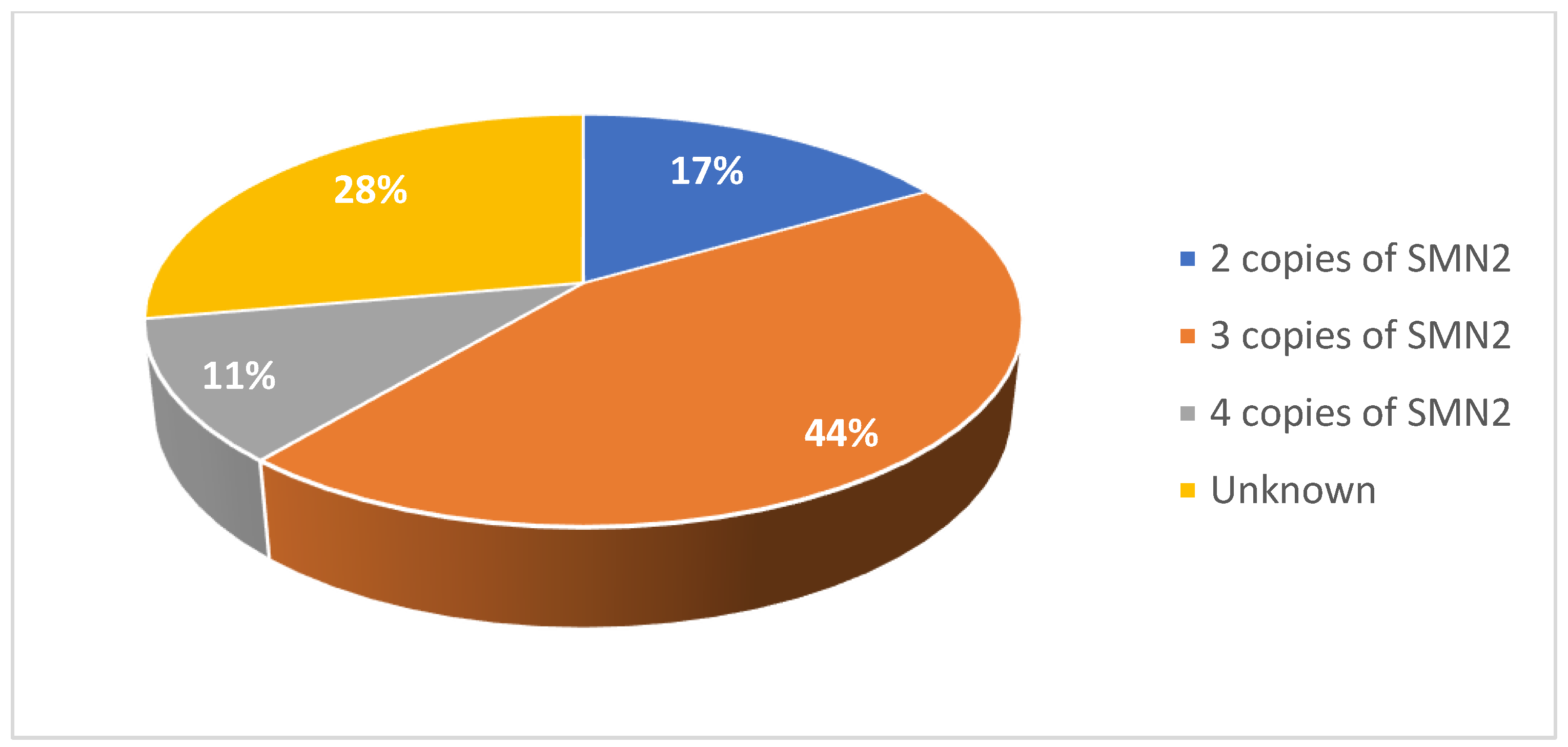
Figure 8.
Proportion of patients with a family history of SMA in the adult cohort.
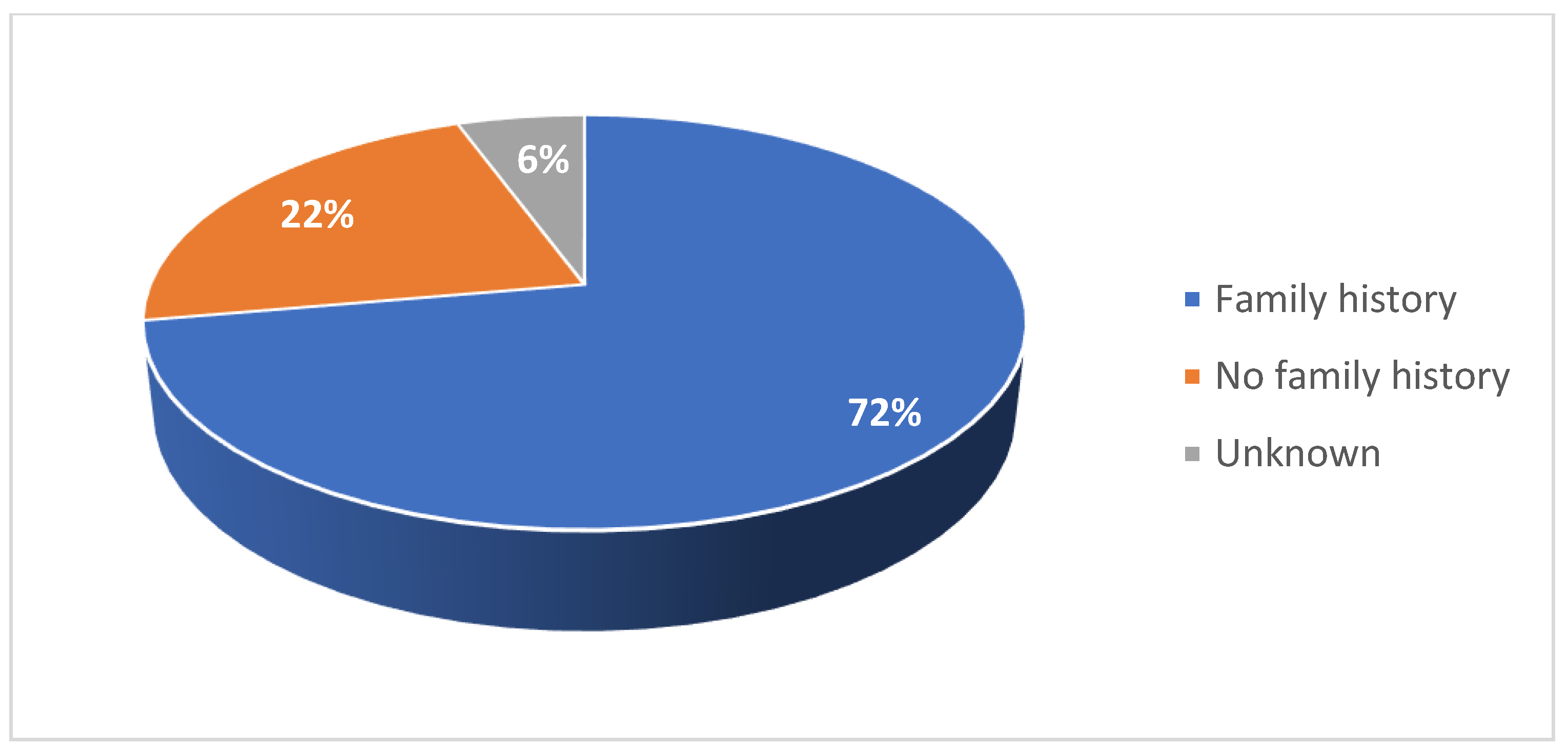
Figure 9.
Proportion of patients with parental consanguinity in the adult cohort.
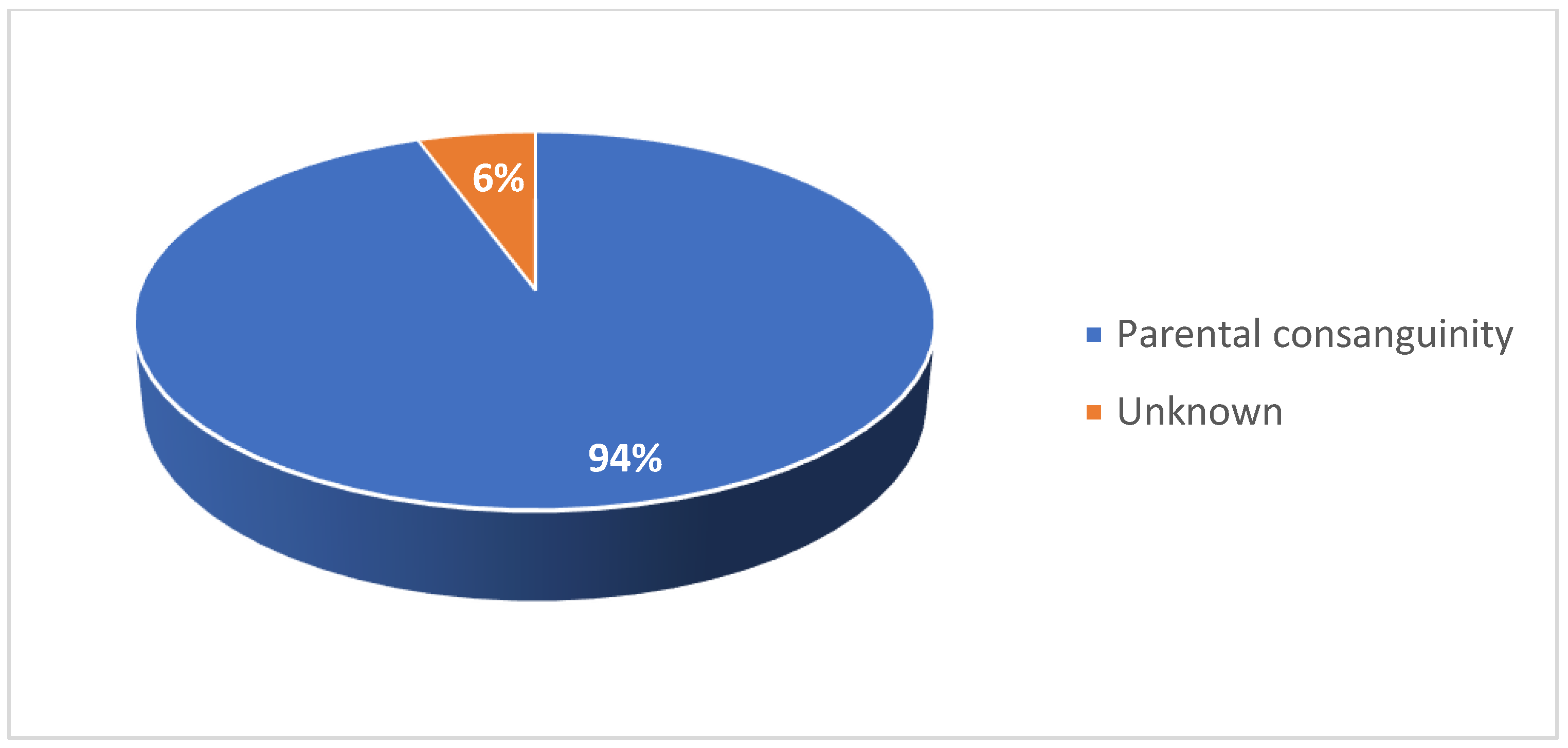
Figure 10.
CHOP-INTEND scores for patients 1–7 after nusinersen treatment initiation CHOP-INTEND, Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders.
Figure 10.
CHOP-INTEND scores for patients 1–7 after nusinersen treatment initiation CHOP-INTEND, Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders.
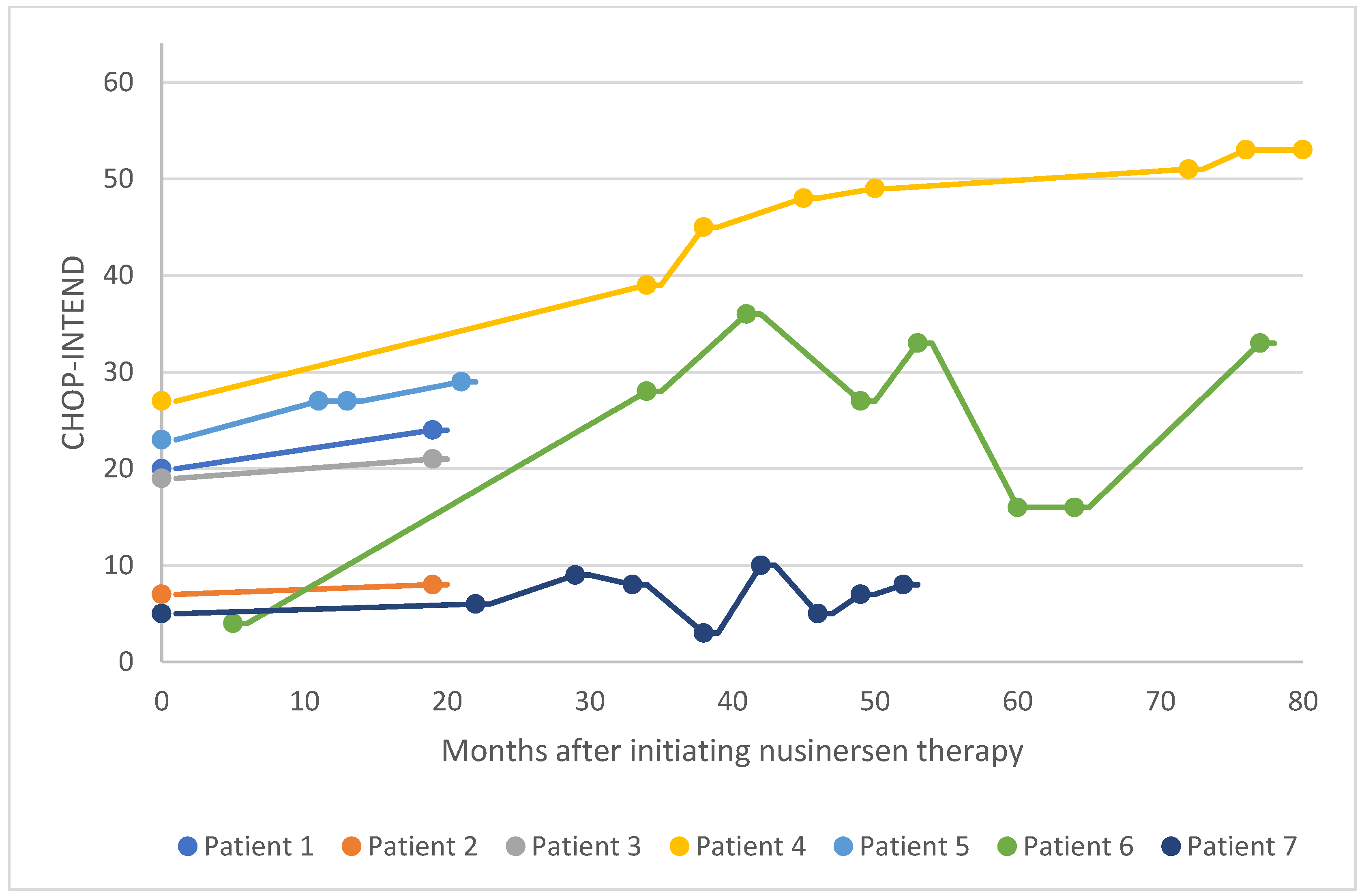
Figure 11.
CHOP-INTEND scores for patients 8–14 after nusinersen treatment initiation CHOP-INTEND, Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders.
Figure 11.
CHOP-INTEND scores for patients 8–14 after nusinersen treatment initiation CHOP-INTEND, Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders.
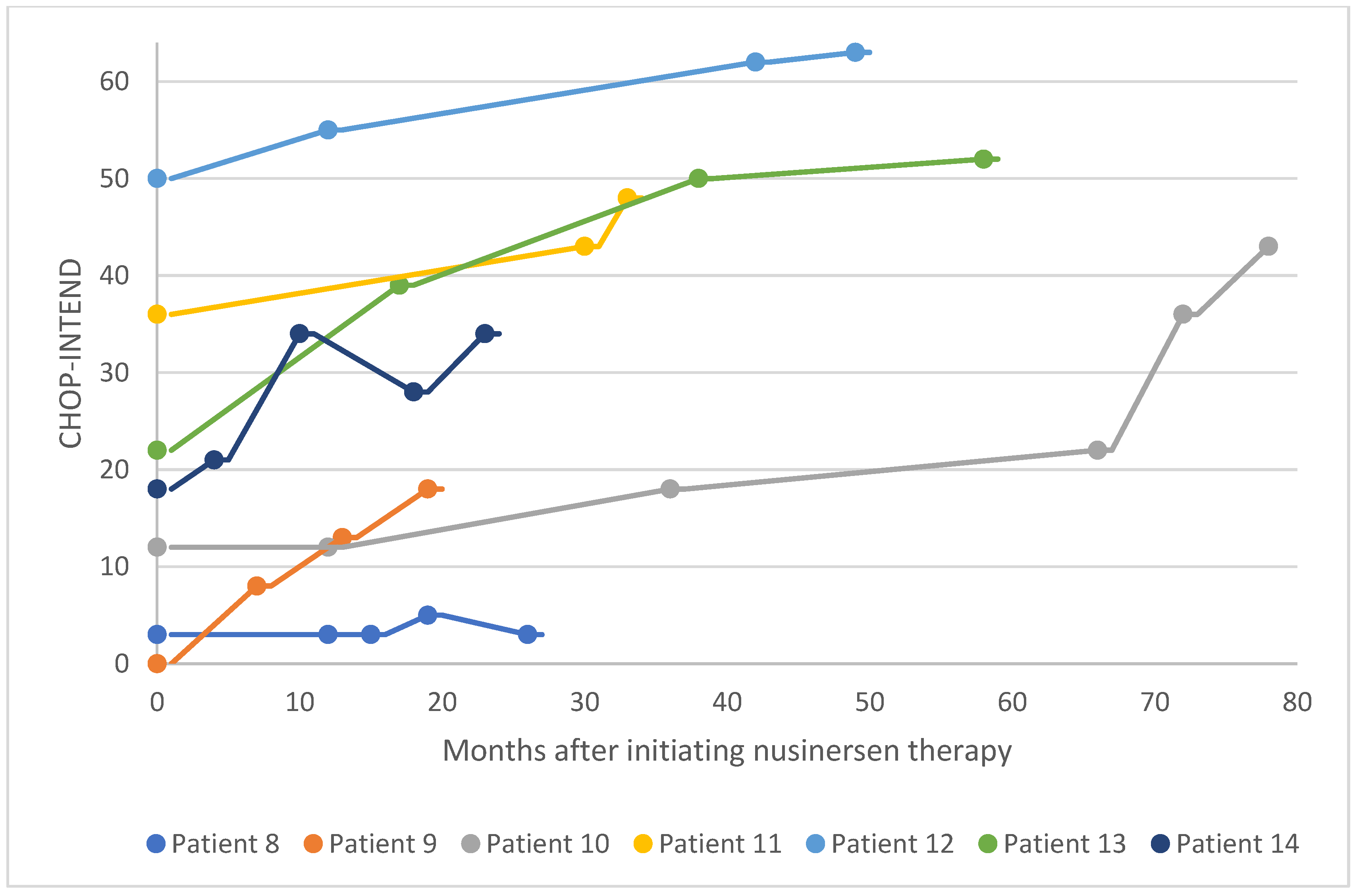
Figure 12.
HFMSE scores for patients 15–20 after nusinersen treatment initiation HFMSE, Hammersmith Functional Motor Scale Expanded.
Figure 12.
HFMSE scores for patients 15–20 after nusinersen treatment initiation HFMSE, Hammersmith Functional Motor Scale Expanded.
Figure 13.
Motor function response to nusinersen treatment among pediatric patients 1–20.
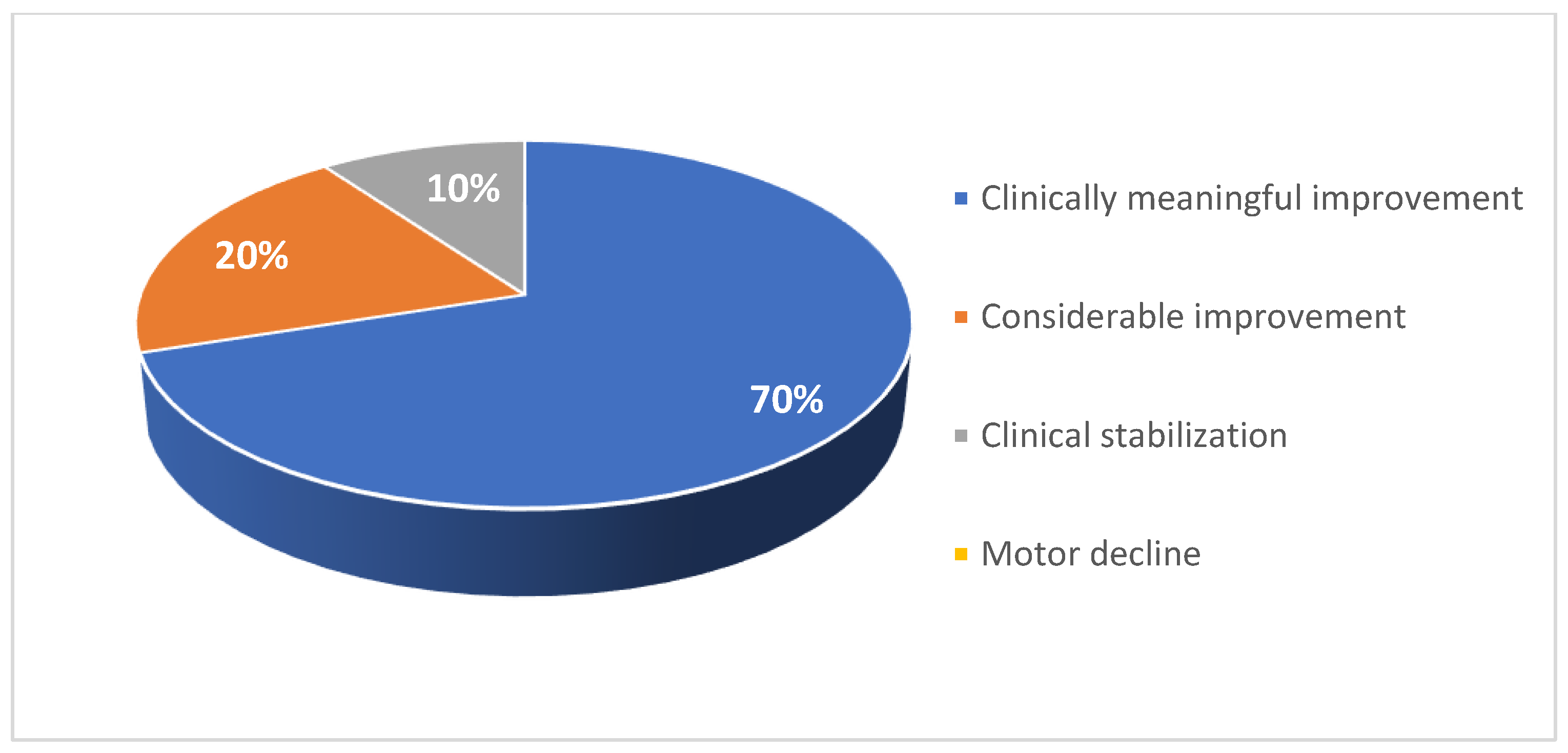
Figure 14.
Upper limb RULM scores for patients 21–29 after nusinersen treatment initiation. RULM, Revised Upper Limb Module.
Figure 14.
Upper limb RULM scores for patients 21–29 after nusinersen treatment initiation. RULM, Revised Upper Limb Module.

Figure 15.
Upper limb RULM scores for patients 30–38 after nusinersen treatment initiation. RULM, Revised Upper Limb Module.
Figure 15.
Upper limb RULM scores for patients 30–38 after nusinersen treatment initiation. RULM, Revised Upper Limb Module.
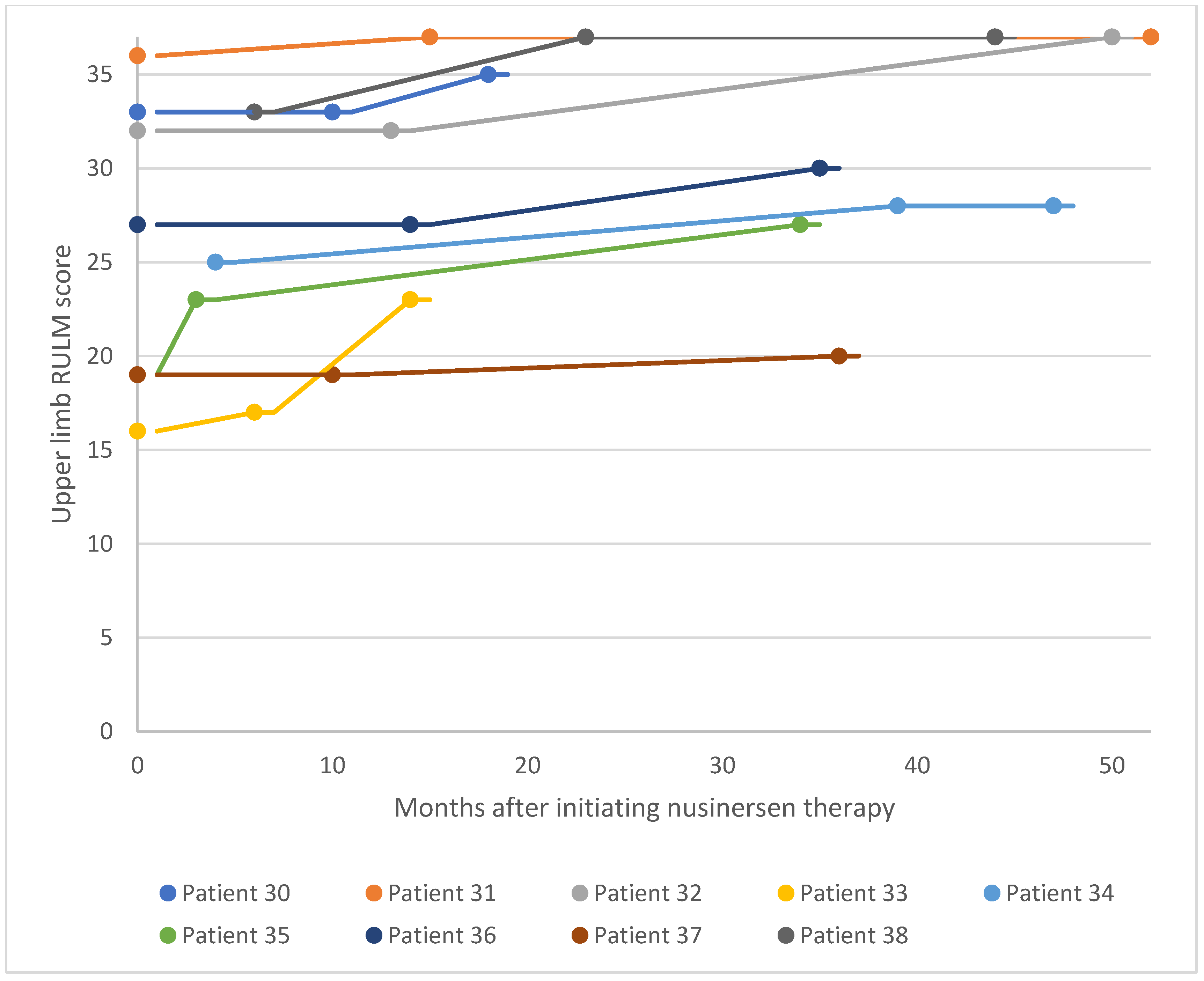
Figure 16.
Motor function response to nusinersen treatment among adult patients 21–38.
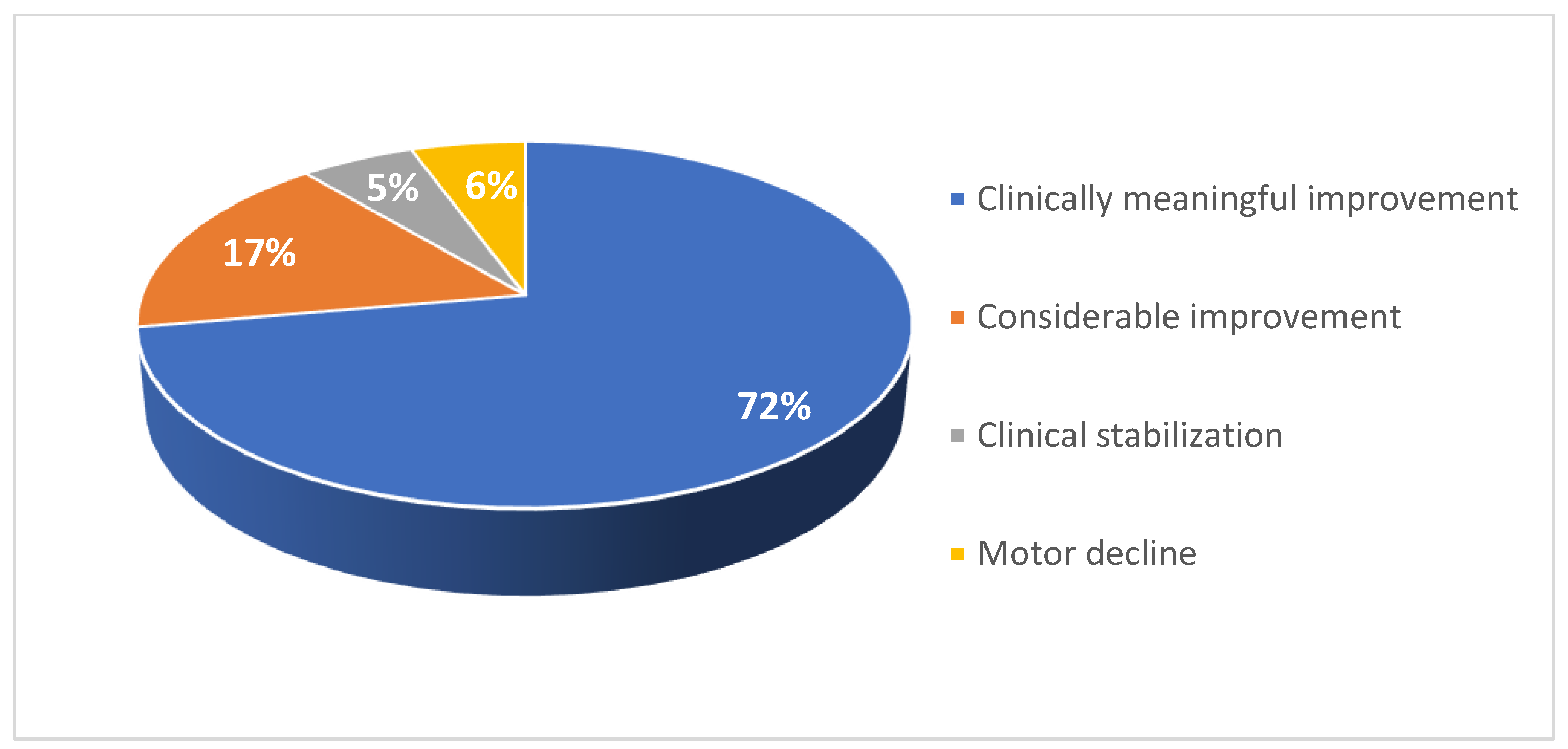

Table 1.
Patient characteristics of the pediatric SMA cohort.
| Case number | Gender | SMA type | Number of SMN2 gene copies | Family history of SMA |
|---|---|---|---|---|
| 1 | Female | I | 1 | (+) |
| 2 | Male | II | 1 | (+) |
| 3 | Female | II | 2 | (+) |
| 4 | Female | I | 2 | (+) |
| 5 | Female | I | 2 | (+) |
| 6 | Male | I | 2 | (-) |
| 7 | Male | I | 2 | (-) |
| 8 | Female | I | 2 | (-) |
| 9 | Female | I | Unknown | (-) |
| 10 | Male | II | Unknown | (-) |
| 11 | Male | II | 2 | (-) |
| 12 | Female | II | Unknown | (-) |
| 13 | Male | II | Unknown | (-) |
| 14 | Male | I | 2 | (-) |
| 15 | Male | II | 2 | (-) |
| 16 | Female | III | Unknown | (+) |
| 17 | Male | II | 2 | (+) |
| 18 | Male | III | 3 | (+) |
| 19 | Female | III | Unknown | (+) |
| 20 | Male | III | 3 | (+) |
SMA, spinal muscular atrophy.
Table 2.
Patient characteristics of the adult SMA cohort.
| Case number | Gender | SMA type | Number of SMN2 gene copies | Family history of SMA | Parental consanguinity |
|---|---|---|---|---|---|
| 21 | Male | III | 3 | (+) | (+) |
| 22 | Male | III | Unknown | (+) | (+) |
| 23 | Male | III | 4 | (+) | (+) |
| 24 | Male | III | 2 | (+) | (+) |
| 25 | Female | III | 3 | (+) | (+) |
| 26 | Female | III | 4 | (-) | (+) |
| 27 | Male | III | Unknown | (-) | (+) |
| 28 | Female | III | Unknown | (+) | (+) |
| 29 | Male | III | 2 | Unknown | Unknown |
| 30 | Male | III | Unknown | (+) | (+) |
| 31 | Male | III | 3 | (-) | (+) |
| 32 | Male | III | 3 | (+) | (+) |
| 33 | Male | III | Unknown | (+) | (+) |
| 34 | Male | III | 2 | (+) | (+) |
| 35 | Male | III | 3 | (+) | (+) |
| 36 | Male | II | 3 | (+) | (+) |
| 37 | Female | III | 3 | (+) | (+) |
| 38 | Female | II | 3 | (-) | (+) |
SMA, spinal muscular atrophy.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



