Submitted:
19 April 2024
Posted:
19 April 2024
You are already at the latest version
Abstract
Asymmetric lithium salts, such as Lithium (difluoromethanesulfonyl)(trifluoromethanesulfonyl)imide (LiDFTFSI), have been demonstrated to surpass traditional symmetric lithium salts with improved Li+ conductivity and the capacity to generate stable solid electrolyte interphase (SEI) while maintaining compatibility with aluminum (Al0) current collector. However, the intrinsic reductive mechanism through which LiDFTFSI influences battery performance remains unclear and under debate. Herein, the detailed SEI reactions of LiDFTFSI-based electrolytes were investigated by combining density functional theory and molecular dynamics, aiming to clarify the formation process and atomic structure of SEI. Our results show that asymmetric DFTFSI– weakens the interaction between carbonate solvents and Li+, and substantially alter the solvation structure, exhibiting a well-balanced coordination capacity to bis(trifluoromethanesulfonyl)imide (TFSI–). Nanoseconds hybrid molecular dynamics simulation further reveals that the preferential decomposition of LiDFTFSI produces sufficient LiF and Li2O to facilitate a robust SEI. Moreover, the abundant F– generated from LiDFTFSI decomposition accumulates on the Al surface and subsequently combines with Al3+ from the current collector to form AlF3, potentially inhibiting corrosion of the current collector. Overall, these findings elucidate how LiDFTFSI regulates the solvation sheath and SEI structure, advancing the development of high-performance electrolytes compatible with current collector.
Keywords:
Lithium Metal Battery
; Electrolyte Solvation Structure
; Battery Interphase
; LiDFTFSI
; Hybrid Molecular Dynamics
1. Introduction
As the demand for sustainable energy storage grows, developing and innovating high-energy-density batteries has become one of the most important research areas in the field of energy storage and a driving force behind the development of new energy sources [1,2,3,4]. Lithium metal batteries (LMBs), which serve as the most promising next-generation energy storage devices due to their ultrahigh battery capacity and compatibility with existing battery manufacturing equipment, have currently been drawing significant research interests all around the world. LMBs possess a specific capacity of 3860 mAh g−1 and a low electrochemical potential of -3.04 V (vs. standard hydrogen electrode, SHE), showing advantages in terms of energy density compared to those of lithium-ion batteries (LIBs) [5,6,7,8,9,10]. Significant challenges, including uncontrollable lithium dendrite growth and the formation of an unstable solid electrolyte interphase (SEI) during charging and discharging cycles. These issues can be attributed to the high reactivity of lithium metal and inevitable side reactions between the electrolyte and the anode [11,12,13,14,15]. These issues compromise the cycle life and performance of LMBs and, more critically, pose serious safety threaten during use [16].
To address these concerns, significant efforts have been devoted in designing advanced lithium anode structures [17,18,19], constructing artificial SEIs [20,21] and optimizing electrolytes [22,23]. Among various strategies, electrolyte engineering is one of the most effective and feasible approaches for regulating the homogenous and stable formation of an SEI. Electrolyte engineering mainly includes (1) adjusting the electrolyte formula based on the existing components, such as by using high-concentration electrolytes or additives, and (2) molecular design based on existing electrolyte components. Increasing the salt concentrations facilitate the sacrificial reduction of aions and limit the coordination of the solvent with lithium ions (Li+), thus enabling inorganic dominated SEI growth [24,25]. Furthermore, additives (fluoroethylene carbonate [26,27], LiNO3 [28,29], and saccharin sodium [30]) enhance the stable formation of SEIs by accelerating decomposition and reducing side reactions between other electrolyte components and the electrode, thereby improving battery performance. LiF is crucial for forming highly stable SEIs, prompting the introduction of various fluorine-containing components to increase its content. For example, 1,4-dimethoxybutane [31] promotes both ionic conductivity and enriched LiF formation in the SEI, thus inhibiting Li dendrite growth by facilitating uniform Li deposition [32].
Various electrolytes exist, but lithium salts in LMBs are notably limited. Lithium hexafluorophosphate (LiPF6) is widely used commercially owing to its good performance and affordable price. LiPF6 features a suitable Li+ transportation property and chemical/electrochemical stability, along with an effective passivation effect on the aluminum (Al0) current collector. However, the chemical and thermodynamic stability of LiPF6, especially toward Al0, is insufficient. PF6– rapidly decomposes at elevated temperatures and is hydrolytically sensitive, limiting its use in advanced batteries [33]. Alternatives to LiPF6, like LiBOB and LiDFOB, offer better thermal stability and can passivate Al foil, yet their low conductivity and solubility fail to satisfy battery system requirements [34]. Lithium salts like LiTFSI, with high charge delocalization, improve conductivity and SEI uniformity but severely corrode the Al0 current collector. Recently, Zhang et al. developed a novel asymmetric lithium salt, difluoromethanesulfonyl)(trifluoromethanesulfonyl)imide (LiDFTFSI), aiming to counteract the shuttling effect of polysulfide intermediates in lithium-sulfur batteries through anion modification. [35]. Altering the -CF3 group to the weaker acidic -CF2H enhances Li+ conductivity by limiting anionic mobility through hydrogen bonding and promotes stable SEI formation via enhanced anion reduction. More importantly, this straightforward modification by disrupting functional group symmetry substantially improves the compatibility with the Al0 current collector. Qiao et al. proposed the formation of sufficient AlF3 from LiDFTFSI-based electrolyte can passivate aluminum foil even at 4.2 V (verse Li/Li+), which explains its greater compatibility with the Al0 current collector compared to LiTFSI [36]. Nevertheless, the underlying reductive reactions of DFTFSI–, SEI formation and collector passivation mechanisms remain unclear. Elucidating the elementary reaction mechanisms helps to establish design criteria from a chemical perspective, enabling rational electrolyte design.
In this work, we employed multiscale simulation to investigate the physical and chemical properties of electrolytes and underlying reaction mechanisms, which provided insights to understand SEI formation and its structure evolution. Specifically, the unique solvation structure of DFTFSI– and Li+ was studied and compared with that of carbonate solvents in an electrolyte. To study electrolyte reduction and the SEI formation mechanism, the hybrid ab initio and reactive force field molecular dynamics (HAIR) method [37], which combines ab initio molecular dynamics (AIMD) with reactive force field molecular dynamics (RMD) simulations, was employed to improve computing efficiency while retaining accuracy. To compare the electrolyte decomposition products with the experimental results, an X-ray photoelectron spectroscopy (XPS) simulation of the SEI structure was performed. Additionally, we explored the passivation mechanism of DFTFSI– on the Al0 current collector using accurate density functional theory (DFT) calculations to elucidate the thermodynamic reductions of DFTFSI– and TFSI– on the Al electrode. This study not only bridges the knowledge gap regarding LiDFTFSI behavior at the atomic level but also offers new perspectives for designing innovative lithium salts to advance the commercialization of LMBs.
2. Computational Details
2.1. Classical Molecular Dynamics (cMD) Simulations
cMD simulations were carried out using GROMACS (version 2020.3) [38] to simulate the bulk electrolytes. The workflow was as follows: First, model optimization was performed, followed by a 2 ns canonical (NVT) simulation at 300 K. Subsequently, a 2 ns isothermal-isobaric (NPT) simulation was conducted at 300 K and 1.0 bar pressure using a Nose‒Hoover thermostat. After reaching equilibrium, a final 2 ns NVT simulation was carried out. Additionally, a custom code was utilized to extract and statistically analyze the coordination, with a cutoff radius of 2.7 Å used to determine the coordination of Li+ with anions or solvent molecules.
Based on the experimental electrolyte formula (1 M LiDFTFSI in a mixture of ethylene carbonate (EC) and ethyl methyl carbonate (EMC)), a simulation box with size of 4 × 4 × 4 nm3 was built, consisting of 40 Li+, 40 DFTFSI–, 160 EC, and 240 EMC. To compare, the 1 M LiTFSI EC/EMC electrolyte simulation was built with a box size of 4 × 4 × 4 nm3, consisting of 40 Li+, 40 TFSI–, 160 EC, and 240 EMC.
2.2. AIMD and RMD Simulations
The AIMD simulation was carried out using VASP (version 5.4.4) with the Projected Augmented Wave (PAW) method [39] and a plane wave basis set. The method is density functional theory (DFT) with generalized gradient approximations (GGA) of Perdew-Burke-Ernzerhof (PBE) functional [40]. The energy cutoff for the plane-wave basis expansion was set at 400 eV, and the convergence threshold energy for the self-consistent calculation and geometry relaxation was less than 1×10-5 eV. These MD simulations used only the gamma point of the Brillouin zone with no consideration of symmetry. A dispersion correction, the DFT-D3 method with Becke-Jonson damping [41], was included in the calculations.
The RMD simulation was carried out using LAMMPS (version 12 Dec 2018). The time step for the RMD simulation was set to 0.25 fs, and the NVT ensemble simulation was conducted at 300 K.
The workflow was as follows: first, a 50 ps AIMD simulation was used to explore the chemical reactions occurring in the electrolyte system during the initial stage, followed by a HAIR simulation that alternated between AIMD and RMD to accelerate the reaction. Each lap of the AIMD simulation was 0.5 ps, and the RMD simulation was accelerated by 10-fold. As the simulation proceeded, the electrochemical reaction gradually decreased, and mass transport dominated, so 100-fold acceleration was used to study deeper reactions and extend the simulation scale beyond 2.5 ns. The diagram of the workflow is shown in Figure S1.
To investigate electrolyte decomposition, the box size was set to 10.5 × 10.5 × 26.5 Å3, consisting of 1 LiDFTFSI, 4 EC and 6 EMC. Six layers (3 × 3) of Li (100) were set at the bottom of the box to simulate Li anode. During the simulation, the bottom two layers were fixed to represent the bulk anode. To relax this electrolyte/anode interface, geometry optimization was carried out. The optimized geometries are shown in Figure S2.
3. Results and Discussion
3.1. Electrolyte Solvation Structure Analysis
cMD simulations were carried out to investigate the specified solvation structure caused by the asymmetric structure on the LiDFTFSI-EC/EMC. The solvation structure of an electrolyte is determined by its molecular structure, which can be distinguished into three categories [32,42]: (1) solvents surrounding Li+ (SSL), (2) Li+-anion single pairs (LASPs) and (3) Li+-anion clusters (LACs). Strong Li+-solvent interaction should be avoided, but it inhibits desolvation and triggers solvent cointercalation during Li+ reduction while charging [43]. The comparisons of sovlation structures between LiDFTFSI and LiTFSI are as shown in Figure 1a andFigure S3a. Based on Li+ coordination with oxygen, the proportions of SSL, LASP, and LAC in the LiDFTFSI system are 35%, 27.5%, and 37.5%, respectively, while in the LiTFSI system, these are 47.5%, 27.5%, and 25%, respectively. Thus, the proportion of SSL in the LiDFTFSI system is much lower than that in the LiTFSI system. The significant increase in the proportion of LAC solvation structures suggests a shift in the solvation structure from SSL to LASP and LAC.
In the LiDFTFSI system, a greater number of anions penetrate the solvation sheath and contribute to the first solvation sheath of Li+. To understand such an effect, DFT calculations were used to obtain the electrostatic potential (ESP) energy surface of TFSI– and DFTFSI–. In Figure 1b, the (-CF2H) substituent of DFTFSI– differs significantly from the (-CF3) substituent of TFSI–, resulting in a less negative charge and enhancing anion-Li+ interaction. Meanwhile, DFTFSI– facilitates hydrogen bonds with solvent molecules, in turn, enhancing ionic conductivity. A considerable amount of DFTFSI– enters the first solvation sheath, displacing solvent molecules and enriching the solvation structure with DFTFSI–. Table S1 provides additional statistics on Li+ coordination. Compared to the LiTFSI system, the LiDFTFSI system exhibits a higher average anion coordination number and a lower average solvent coordination number. This anion-rich solvation structure can facilitate salt preferential decomposition, resulting in the formation of an inorganic-rich SEI interface.
The radial distribution function (RDF) provides further insights into the detailed structure features of anions and solvent molecules within the solvation sheath. As shown in Figure 1c, Li+-O(DFTFSI–), Li+-O(EC) and Li+-O(EMC) peaks at 0.208, 0.216 and 0.212 nm, respectively, indicating that DFTFSI– has strong coordination with Li+, followed by EMC and EC, further confirming that DFTFSI– penetrates into the first solvation sheath. In LiTFSI, Li+-O(TFSI–), Li+-O(EC) and Li+-O(EMC) peaks at 0.212, 0.212 and 0.210 nm, respectively. Compared to LiDFTFSI, the anion peak exhibit a systematical right-shift, and EMC has larger coordination than TFSI– and EC (Figure S3b). The coordination number of Li+-O(DFTFSI–) increases compared to that of Li+-O(TFSI–), and the coordination number distribution plot also shows a decrease in the coordination number of Li+-O(EC) in the LiDFTFSI system. These observations indicate that DFTFSI– weakens the interaction between EC and Li+ and enters the solvation sheath, thereby modifying the solvation structure, consistent with previous report [44].
3.2. Underlying mechanism of electrolyte reduction and SEI formation
Hybrid molecular dynamics simulations were carried out to investigate the decomposition and the initial SEI formation of LiDFTFSI-EC/EMC electrolyte. Electrolyte reduction is crucial for SEI formation at the lithium metal anode, a 2.86 ns HAIR simulation using the model shown in Figure S2b to reveal the detailed SEI reaction. As shown in Figure 2, DFTFSI– decomposed within 7 ps AIMD simulation. The detailed reactions are as follows: the N‒S bond on ‒CF2H side breaks at 0.19 ps (Figure 2a), indicating that the asymmetric substituent increases the reactivity. Subsequently, the C‒F bonds on ‒CF2H quickly break at 1.21 ps due to the formation of LiF as reduced by Li0. As shown in Figure 2a, the S‒‒O bond breaks at 1.51 ps, along with Li2O formation. After extended simulation, the dehydrogenation reaction occurs at 72.60 ps, leading to LiH formation, consistent with experimental report. These results demonstrate that introducing hydrogen atoms to the asymmetric group increases the salt reactivity, and produces ionic conductive LiH and mechanically stable LiF.
On the other hand, the ‒CF3 group detached from the salt at 3.20 ps through C‒S bond cleavage, similar to the previous report [37]. Deep reduction of DFTFSI–, followed by S‒O, N‒S, and C‒F cleavages, contributed to the inorganic components of the SEI (Figure 2b). The breaking of the N‒S bond occurred at 5.26 ps, and all F atoms were released from DFTFSI– at 6.88 ps (Figure 2b). This detailed mechanism demonstrated that asymmetric DFTFSI– can rapidly react with Li0 and generate products, including LiF, Li2O, LiH, and Li3N, which can potentially improve the ionic conductivity, regulate the mechanical properties of the SEI and inhibit dendrite formation [6]. The decomposition mechanism of DFTFSI– revealed by AIMD simulation supports the experimental hypothesis, as illustrated inFigure 2c and Figure S4.
The solvent decomposition mechanism is crucial. AIMD simulations indicate that EC is reduced during charging, contributing to SEI formation. EC starts to decompose within 50 ps, and three different initial pathways can be distinguished based on the reactive trajectories. At 2.51 ps, in a Li+-rich environment, EC undergoes a one-electron reduction, breaking its C‒O bond and transforming from a ring to a chain structure, forming OC2H4OCO–. Subsequently, OC2H4OCO– follows two pathways: it accepts another electron to form OC2H4O2–, releasing one CO at 4.00 ps (Figure 3a), and releases CO2 while forming the free radical anion •C2H4O–, consistenting with previous DFT [45]. At 61.21 ps, •C2H4O– is further reduced by Li0 and forms Li2O along with one electron transfer (Figure 3b). In addition to one-electron reduction, two-electron reduction was also observed in the simulation. EC underwent a two-electron reduction to form CO32– and C2H4, as previously observed in experiment. Further reductions occurred at 50.60 and 56.50 ps, resulting in the formation of Li2O from CO32– (Figure 3c).
The decomposition of EMC is slower than EC and generally falls into two categories. As shown in Figure 3d, the C‒O bond breaks with Li2O at 50.6 ps, and then the C‒O bond breaks, leading to the formation of CO and CH3O– at 55.69 ps. The other reaction path involves the initial detachment of the ‒OCH3 group from the carbon chain via one-electron reduction, followed by the breaking of the C‒O bond in the remaining ‒OCOCH2CH3 species, yielding CO and C2H5O– (Figure 3e). EMC decomposition did not occur in the first 50 ps of the AIMD simulation but began in the early stages of the HAIR simulation, revealing similar reaction mechanism.
After initial decomposition, deeper reactions continue, forming the main products of the SEI. The products and the primitive SEI formed after 2.86 ns of HAIR simulation are shown in Figures S5 and S6. The inner SEI layer, close to the anode surface, predominantly contains inorganic compounds like Li2O, LiF, etc. [46,47]. These inorganic compounds improve mechanical properties, inhibit lithium dendrite growth, decrease electrolyte consumption, and promote a uniform and stable SEI, thereby potentially enhancing battery cycle performance. Instead, the outer SEI layer, close to the electrolyte, consists mainly of carbon-oxygen organic compounds from solvent decomposition, consistent with previous experiment [35,36]. To further analyze the products and atomic structure of the SEI, the XPS spectra of C 1 s, O 1 s and F 1 s are simulated, as shown in Figure 4. C atoms exhibit a complex chemical environment characterized by various bond types, including C‒O, C‒C, C‒H, and C=O bonds (Figure 4a), primarily arising from the decomposition of solvents into RC‒O (e.g., CH3OLi, C2H5OLi). In Figure 4b, O atoms are either bonded to C, forming C‒O and C=O bonds, which confirm the presence of carbon-oxygen groups and correlate with the C 1 s XPS spectrum, or exist as Li2O. The schematic diagram of DFTFSI– and solvent decomposition shows that F atoms bond with Li+ to form LiF (Figure 4c). The theoretical XPS predictions well agree with the experimental results, demonstrating the reliability of the simulations.
3.3. DFTFSI–Passivated Al
In addition to regulating the anode interphase, LiDFTFSI, as the reactive active component, can remarkably suppress the dissolution of the Al0 current collector at high potentials (>4.2 V versus Li/Li+) in comparison with LiTFSI. The Al0 current collector maintains good stability within the battery, because Al foil naturally forms a protective Al2O3 layer [52]. However, when the voltage exceeds the threshold, Al2O3 can be destroyed in the organic solution, exposing fresh Al0. This leads to progressive corrosion of the Al0 current collector. The asymmetric structure of LiDFTFSI and the high reactivity of the ‒CF2H group rapidly dissociate to form AlF3, which then protects the anode. To test such hypothesis, we examined the defluorination process on the ‒CF2 group on the Al metal surface, as shown in Figure 5. DFT calculations show that the Gibbs free energy change (ΔG) for this process is -1.64 eV, compared to -0.94 eV for LiTFSI. The results indicate that LiDFTFSI defluorinates more readily on the Al electrode, facilitating the subsequent formation of AlF3, well validating our hypothesis. Therefore, the protective mechanism of LiDFTFSI on Al foil can be described as follows: LiDFTFSI diffuses and adsorbs onto the Al foil, decomposes, and the resulting F ions react with Al3+ to form an AlF3 passivation layer. This layer prevents further exposure of the Al metal and suppresses interfacial side reactions.
4. Conclusions
To summarize, we conducted atomic simulations to investigate how the asymmetric sulfonyl imide salt (LiDFTFSI) influences the microscopic solvation structure and the underlying interfacial reaction mechanisms in LMBs. According to cMD simulations, the presence of the asymmetric functional group (‒CF2H) increased the proportion of the LAC solvation structure in the LiDFTFSI-EC/EMC system compared to the LiTFSI-based electrolyte, while substantially decreasing the portion of the SSL solvation structure. cMD simulation also suggests that DFTFSI– has a greater coordination capacity with Li+, thereby triggering salt-prone interfacial reactions. HAIR simulation was employed to investigate the initial interface reactions and the detailed SEI generation process of the LiDFTFSI-based electrolyte. A homogeneous and stable SEI layer relying on the substantial production of Li2O and LiF was formed as a result of the preferred decomposition of LiDFTFSI. The reliability of the HAIR and XPS simulations was confirmed by comparing theoretical XPS simulations of the SEI with actual XPS spectra.
Furthermore, LiDFTFSI tends to defluorinate on the Al surface, facilitating the formation of a passivation layer that suppresses corrosion of the Al0 current collector, providing a clear explanation to the perplex experimental observation. This study clearly clarify the solvation structure and interface chemistry of electrolytes, providing new insights in the design of new electrolytes to enhance the performance of LMBs.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org., Figure S1: The workflow diagram for simulating interfacial reactions; Figure S2: Atomic molecular geometries for anions and solvents and the initial model for the 1 M LiDFTFSI-EC/EMC system; Figure S3: Examples of three solvation structures, the RDF and the CN of the LiTFSI-EC/EMC electrolyte; Figure S4: The experimentally speculated reduction mechanism for LiDFTFSI; Figure S5: The molecular geometries of the final products in the SEI obtained from the 2.8 ns HAIR simulation; Figure S6: Composition and structure of the SEI in the 1 M LiDFTFSI-EC/EMC system for the simulations; Table S1: Li+ coordination in the LiDFTFSI-based and LiTFSI-based electrolytes.
Author Contributions
Conceptualization, S.F. and T.C.; validation, T.Y., L.B. and T.C.; formal analysis, S.F.; writing—original draft preparation, S.F.; writing—review and editing, S.F., Y.L. and T.C.; supervision, T.C.; project administration, T.C.; funding acquisition, T.C. All authors have read and agreed to the published version of the manuscript. S.F. and T.Y. contributed equally to this work.
Data Availability Statement
The data presented in this study are available upon request from the corresponding author.
Acknowledgments
T.C. acknowledges support from the National Key Research and Development Program of China (Grant No. 2022YFB2502200), Suzhou Key Laboratory of Functional Nano & Soft Materials, the Collaborative Innovation Center of Suzhou Nano Science & Technology, the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), the 111 Project, Joint International Research Laboratory of Carbon-Based Functional Materials and Devices, the National Natural Science Foundation of China (22173066), and the Natural Science Foundation of Jiangsu Province (BK20230065). Y.L. acknowledges support from the National Natural Science Foundation of China (22303058) and the Natural Science Foundation of Jiangsu Province (BK20230475).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Winter, M.; Barnett, B.; Xu, K. Before Li ion batteries. Chem. Rev. 2018, 118, 11433-11456. [CrossRef]
- Larcher, D.; Tarascon, J.M. Towards greener and more sustainable batteries for electrical energy storage. Nat. Chem. 2015, 7, 19-29. [CrossRef]
- Lin, D.; Liu, Y.; Cui, Y. Reviving the lithium metal anode for high-energy batteries. Nat. Nanotechnol. 2017, 12, 194-206. [CrossRef]
- Dunn, B.; Kamath, H.; Tarascon, J.-M. Electrical energy storage for the grid: a battery of choices. Science 2011, 334, 928-935. [CrossRef]
- Wang, H.; Yu, Z.; Kong, X.; Kim, S.C.; Boyle, D.T.; Qin, J.; Bao, Z.; Cui, Y. Liquid electrolyte: The nexus of practical lithium metal batteries. Joule 2022, 6, 588-616. [CrossRef]
- Li, M.; Wang, C.; Davey, K.; Li, J.; Li, G.; Zhang, S.; Mao, J.; Guo, Z. Recent progress in electrolyte design for advanced lithium metal batteries. SmartMat 2023, 4, e1185. [CrossRef]
- Hobold, G.M.; Lopez, J.; Guo, R.; Minafra, N.; Banerjee, A.; Shirley Meng, Y.; Shao-Horn, Y.; Gallant, B.M. Moving beyond 99.9% coulombic efficiency for lithium anodes in liquid electrolytes. Nat. Energy 2021, 6, 951-960. [CrossRef]
- Liu, J.; Bao, Z.; Cui, Y.; Dufek, E.J.; Goodenough, J.B.; Khalifah, P.; Li, Q.; Liaw, B.Y.; Liu, P.; Manthiram, A.; et al. Pathways for practical high-energy long-cycling lithium metal batteries. Nat. Energy 2019, 4, 180-186. [CrossRef]
- Nanda, S.; Gupta, A.; Manthiram, A. Anode-Free Full Cells: A Pathway to High-energy density lithium-metal batteries. Adv. Energy Mater. 2021, 11, 2000804.
- Jie, Y.; Ren, X.; Cao, R.; Cai, W.; Jiao, S. Advanced liquid electrolytes for rechargeable Li metal batteries. Adv. Funct. Mater. 2020, 30, 1910777. [CrossRef]
- Jagger, B.; Pasta, M. Solid electrolyte interphases in lithium metal batteries. Joule 2023, 7, 2228-2244. [CrossRef]
- Qi, M.; Xie, L.; Han, Q.; Zhu, L.; Chen, L.; Cao, X. An overview of the key challenges and strategies for lithium metal anodes. J. Energy Storage 2022, 47, 103641. [CrossRef]
- Cheng, X.-B.; Zhang, R.; Zhao, C.-Z.; Zhang, Q. Toward safe lithium metal anode in rechargeable batteries: A review. Chem. Rev. 2017, 117. [CrossRef]
- Wang, Q.; Yao, Z.; Zhao, C.; Verhallen, T.; Tabor, D.P.; Liu, M.; Ooms, F.; Kang, F.; Aspuru-Guzik, A.; Hu, Y.-S.; et al. Interface chemistry of an amide electrolyte for highly reversible lithium metal batteries. Nat. Commun. 2020, 11, 4188. [CrossRef]
- Zhai, P.; Liu, L.; Gu, X.; Wang, T.; Gong, Y. Interface engineering for lithium metal anodes in liquid electrolyte. Adv. Energy Mater. 2020, 10, 2001257. [CrossRef]
- Liu, B.; Zhang, J.-G.; Xu, W. Advancing lithium metal batteries. Joule 2018, 2, 833-845. [CrossRef]
- Liu, S.; Ma, Y.; Zhou, Z.; Lou, S.; Huo, H.; Zuo, P.; Wang, J.; Du, C.; Yin, G.; Gao, Y. Inducing uniform lithium nucleation by integrated lithium-rich li-in anode with lithiophilic 3D framework. Energy Storage Mater. 2020, 33, 423-431. [CrossRef]
- Wang, X.; Zeng, W.; Hong, L.; Xu, W.; Yang, H.; Wang, F.; Duan, H.; Tang, M.; Jiang, H. Stress-driven lithium dendrite growth mechanism and dendrite mitigation by electroplating on soft substrates. Nat. Energy 2018, 3, 227-235. [CrossRef]
- Wang, T.; Zhai, P.; Legut, D.; Wang, L.; Liu, X.; Li, B.; Dong, C.; Fan, Y.; Gong, Y.; Zhang, Q. S-doped graphene-regional nucleation mechanism for dendrite-free lithium metal anodes. Adv. Energy Mater. 2019, 9, 1804000.
- Zhao, J.; Liao, L.; Shi, F.; Lei, T.; Chen, G.; Pei, A.; Sun, J.; Yan, K.; Zhou, G.; Xie, J.; et al. Surface fluorination of reactive battery anode materials for enhanced stability. J. Am. Chem. Soc. 2017, 139, 11550-11558. [CrossRef]
- Liu, Y.; Lin, D.; Yuen, P.Y.; Liu, K.; Xie, J.; Dauskardt, R.H.; Cui, Y. An artificial solid electrolyte interphase with high Li-ion conductivity, mechanical strength, and flexibility for stable lithium metal anodes. Adv. Mater. 2017, 29, 1605531.
- Zheng, J.; Engelhard, M.H.; Mei, D.; Jiao, S.; Polzin, B.J.; Zhang, J.-G.; Xu, W. Electrolyte additive enabled fast charging and stable cycling lithium metal batteries. Nat. Energy 2017, 2, 17012. [CrossRef]
- Weber, R.; Genovese, M.; Louli, A.J.; Hames, S.; Martin, C.; Hill, I.G.; Dahn, J.R. Long cycle life and dendrite-free lithium morphology in anode-free lithium pouch cells enabled by a dual-salt liquid electrolyte. Nat. Energy 2019, 4, 683-689. [CrossRef]
- Pham, T.D.; Bin Faheem, A.; Lee, K.-K. Design of a LiF-rich solid electrolyte interphase layer through highly concentrated LiFSI–THF electrolyte for stable lithium metal batteries. Small 2021, 17, 2103375.
- Jiao, S.; Ren, X.; Cao, R.; Engelhard, M.H.; Liu, Y.; Hu, D.; Mei, D.; Zheng, J.; Zhao, W.; Li, Q.; et al. Stable cycling of high-voltage lithium metal batteries in ether electrolytes. Nat. Energy 2018, 3, 739-746. [CrossRef]
- Zhang, X.-Q.; Cheng, X.-B.; Chen, X.; Yan, C.; Zhang, Q. Fluoroethylene carbonate additives to render uniform Li deposits in lithium metal batteries. Adv. Funct. Mater. 2017, 27, 1605989. [CrossRef]
- Hou, T.; Yang, G.; Rajput, N.N.; Self, J.; Park, S.-W.; Nanda, J.; Persson, K.A. The influence of FEC on the solvation structure and reduction reaction of LiPF6/EC electrolytes and its implication for solid electrolyte interphase formation. Nano Energy 2019, 64, 103881.
- Wang, X.; Wang, S.; Wang, H.; Tu, W.; Zhao, Y.; Li, S.; Liu, Q.; Wu, J.; Fu, Y.; Han, C.; et al. Hybrid electrolyte with dual-anion-aggregated solvation sheath for stabilizing high-voltage lithium-metal batteries. Adv. Mater. 2021, 33, 2007945.
- Fu, J.; Ji, X.; Chen, J.; Chen, L.; Fan, X.; Mu, D.; Wang, C. Lithium nitrate regulated sulfone electrolytes for lithium metal batteries. Angew. Chem. Int. Ed. 2020, 59, 22194-22201. [CrossRef]
- Yang, F.; Wang, P.; Huang, Q.; Luo, J.; Hu, R.; Huang, Q.; Mao, C.; Yang, L.; Liang, G.; Li, Y.; et al. Saccharin sodium coupling fluorinated solvent enabled stable interface for high-Vvoltage Li-metal batteries. Small 2024, 2311961.
- Yu, Z.; Wang, H.; Kong, X.; Huang, W.; Tsao, Y.; Mackanic, D.G.; Wang, K.; Wang, X.; Huang, W.; Choudhury, S.; et al. Molecular design for electrolyte solvents enabling energy-dense and long-cycling lithium metal batteries. Nat. Energy 2020, 5, 526-533. [CrossRef]
- Yu, Z.; Rudnicki, P.E.; Zhang, Z.; Huang, Z.; Celik, H.; Oyakhire, S.T.; Chen, Y.; Kong, X.; Kim, S.C.; Xiao, X.; et al. Rational solvent molecule tuning for high-performance lithium metal battery electrolytes. Nat. Energy 2022, 7, 94-106. [CrossRef]
- Xu, K. Nonaqueous liquid electrolytes for lithium-based rechargeable batteries. Chem. Rev. 2004, 104, 4303-4418. [CrossRef]
- Wang, S.; Qiu, W.; Guan, Y.; Yu, B.; Zhao, H.; Liu, W. Electrochemical characteristics of LiMxFe1−xPO4 cathode with LiBOB based electrolytes. Electrochim. Acta 2007, 52, 4907-4910. [CrossRef]
- Zhang, H.; Oteo, U.; Judez, X.; Eshetu, G.G.; Martinez-Ibañez, M.; Carrasco, J.; Li, C.; Armand, M. Designer anion enabling solid-state lithium-sulfur batteries. Joule 2019, 3, 1689-1702. [CrossRef]
- Qiao, L.; Oteo, U.; Martinez-Ibañez, M.; Santiago, A.; Cid, R.; Sanchez-Diez, E.; Lobato, E.; Meabe, L.; Armand, M.; Zhang, H. Stable non-corrosive sulfonimide salt for 4-V-class lithium metal batteries. Nat. Mater. 2022, 21, 455-462. [CrossRef]
- Liu, Y.; Yu, P.; Wu, Y.; Yang, H.; Xie, M.; Huai, L.; Goddard, W.A., III; Cheng, T. The DFT-ReaxFF hybrid reactive dynamics method with application to the reductive decomposition reaction of the TFSI and DOL electrolyte at a lithium-metal anode surface. J. Phys. Chem. Lett. 2021, 12, 1300-1306.
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1-2, 19-25. [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758-1775. [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. B 1996, 77, 3865-3868. [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456-1465. [CrossRef]
- Qi, F.; Yu, P.; Zhou, Q.; Liu, Y.; Sun, Q.; Ma, B.; Ren, X.; Cheng, T. Preferential decomposition of the major anion in a dual-salt electrolyte facilitates the formation of organic-inorganic composite solid electrolyte interphase. J. Chem. Phys. 2023, 158, 104704. [CrossRef]
- Chen, Y.; He, Q.; Zhao, Y.; Zhou, W.; Xiao, P.; Gao, P.; Tavajohi, N.; Tu, J.; Li, B.; He, X.; et al. Breaking solvation dominance of ethylene carbonate via molecular charge engineering enables lower temperature battery. Nat. Commun. 2023, 14, 8326. [CrossRef]
- Cheng, H.; Sun, Q.; Li, L.; Zou, Y.; Wang, Y.; Cai, T.; Zhao, F.; Liu, G.; Ma, Z.; Wahyudi, W.; et al. Emerging era of electrolyte solvation structure and interfacial model in batteries. ACS Energy Lett. 2022, 7, 490-513. [CrossRef]
- Liu, Y.; Wu, Y.; Sun, Q.; Ma, B.; Yu, P.; Xu, L.; Xie, M.; Yang, H.; Cheng, T. Formation of linear oligomers in solid electrolyte interphase via two-electron reduction of ethylene carbonate. Adv. Theory Simul. 2022, 5, 2100612. [CrossRef]
- Zhao, Q.; Stalin, S.; Archer, L.A. Stabilizing metal battery anodes through the design of solid electrolyte interphases. Joule 2021, 5, 1119-1142. [CrossRef]
- Gong, C.; Pu, S.D.; Gao, X.; Yang, S.; Liu, J.; Ning, Z.; Rees, G.J.; Capone, I.; Pi, L.; Liu, B.; et al. Revealing the role of fluoride-rich battery electrode interphases by operando transmission electron microscopy. Adv. Energy Mater. 2021, 11, 2003118. [CrossRef]
- Stephan, A.M.; Prem Kumar, T.; Thomas, S.; Bongiovanni, R.; Nair, J.R.; Angulakshmi, N.; Pollicino, A. Ca3(PO4)2-incorporated poly(ethylene oxide)-based nanocomposite electrolytes for lithium batteries. Part II. Interfacial properties investigated by XPS and a.c. impedance studies. J. Appl. Polym. Sci. 2012, 124, 3255-3263.
- Schulz, N.; Hausbrand, R.; Dimesso, L.; Jaegermann, W. XPS-Surface Analysis of SEI layers on Li-ion cathodes: Part I. Investigation of initial surface chemistry. J. Electrochem. Soc. 2018, 165, A819.
- Rustomji, C.S.; Yang, Y.; Kim, T.K.; Mac, J.; Kim, Y.J.; Caldwell, E.; Chung, H.; Meng, Y.S. Liquefied gas electrolytes for electrochemical energy storage devices. Science 2017, 356, eaal4263. [CrossRef]
- Fang, D.; He, F.; Xie, J.; Xue, L. Calibration of binding energy positions with C1s for XPS results. Journal of Wuhan University of Technology-Mater. Sci. Ed. 2020, 35, 711-718. [CrossRef]
- Yang, S.; Zhong, J.; Li, S.; Li, B. Revisiting aluminum current collector in lithium-ion batteries: Corrosion and countermeasures. J. Energy Chem. 2024, 89, 610-634. [CrossRef]
Figure 1.
(a) Statistical diagrams of the proportion of solvation structures after cMD simulation and examples of three solvation structures of the LiDFTFSI-EC/EMC electrolyte. Color codes: lithium: purple, oxygen: red, carbon: gray, fluorine: cyan, sulfur: yellow, nitrogen: blue, hydrogen: white. (b) ESP energy diagrams of DFTFSI– and TFSI–. (c) RDF, g(r), and CN, n(r) of Li+-O.
Figure 1.
(a) Statistical diagrams of the proportion of solvation structures after cMD simulation and examples of three solvation structures of the LiDFTFSI-EC/EMC electrolyte. Color codes: lithium: purple, oxygen: red, carbon: gray, fluorine: cyan, sulfur: yellow, nitrogen: blue, hydrogen: white. (b) ESP energy diagrams of DFTFSI– and TFSI–. (c) RDF, g(r), and CN, n(r) of Li+-O.
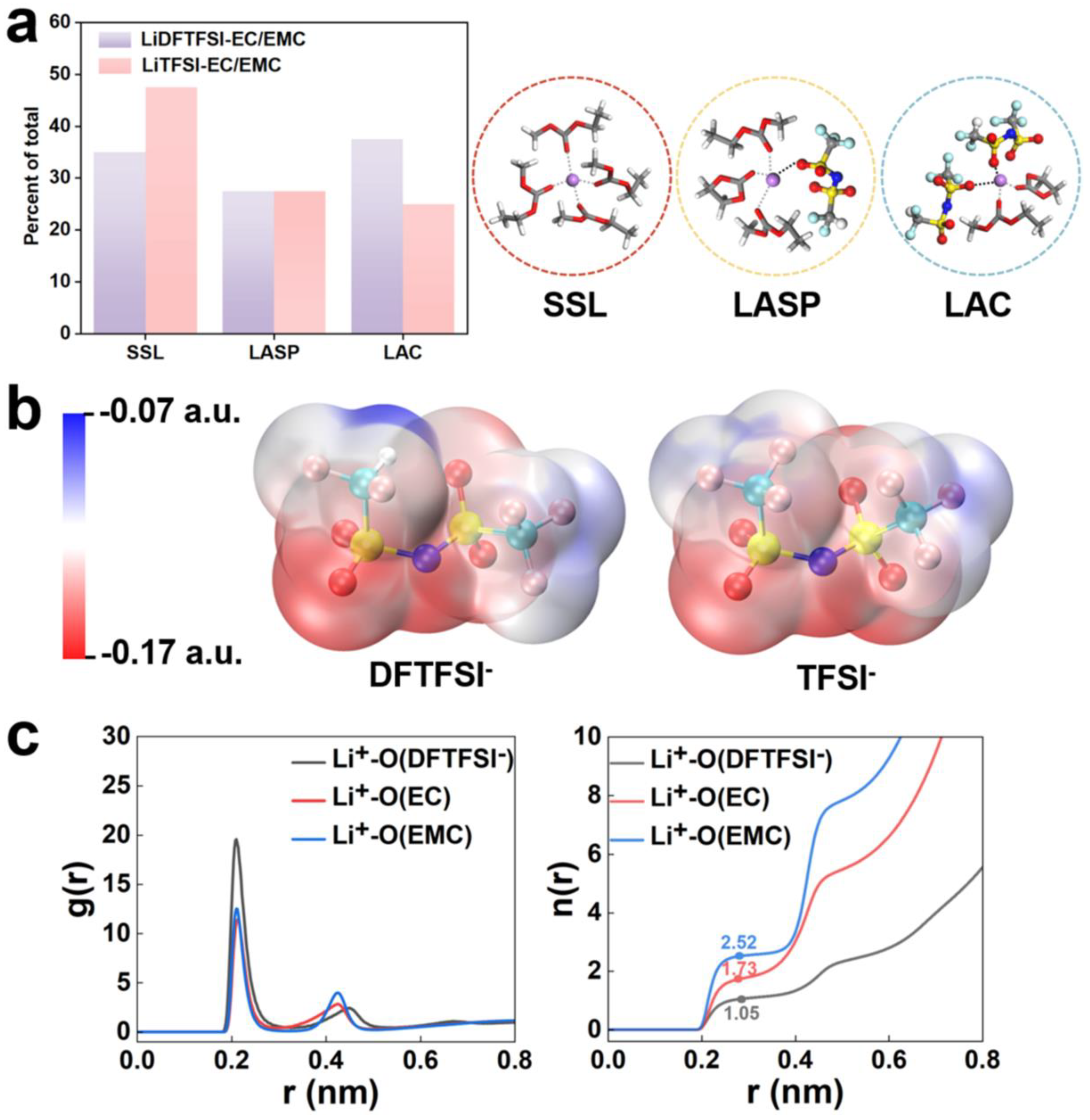
Figure 2.
Sequence of DFTFSI decompositions obtained from AIMD and HAIR simulations for the 1 M LiDFTFSI-EC/EMC system. (a) The decompositions of the side of the ‒CF2H group. (b) The decompositions of the side of the ‒CF3 group. (c) DFTFSI– reaction pathway obtained from simulations. The color codes are the same as those in Figure 1a.
Figure 2.
Sequence of DFTFSI decompositions obtained from AIMD and HAIR simulations for the 1 M LiDFTFSI-EC/EMC system. (a) The decompositions of the side of the ‒CF2H group. (b) The decompositions of the side of the ‒CF3 group. (c) DFTFSI– reaction pathway obtained from simulations. The color codes are the same as those in Figure 1a.

Figure 3.
Decomposition pathways of EC and EMC for the 1 M LiDFTFSI-EC/EMC system obtained from AIMD and HAIR simulations between 0 and 61.21 ps. (a) Pathway 1 of EC, (b) pathway 2 of EC, (c) pathway 3 of EC, (d) pathway 1 of EMC and (e) pathway 2 of EMC. The color codes are the same as those in Figure 1a.
Figure 3.
Decomposition pathways of EC and EMC for the 1 M LiDFTFSI-EC/EMC system obtained from AIMD and HAIR simulations between 0 and 61.21 ps. (a) Pathway 1 of EC, (b) pathway 2 of EC, (c) pathway 3 of EC, (d) pathway 1 of EMC and (e) pathway 2 of EMC. The color codes are the same as those in Figure 1a.
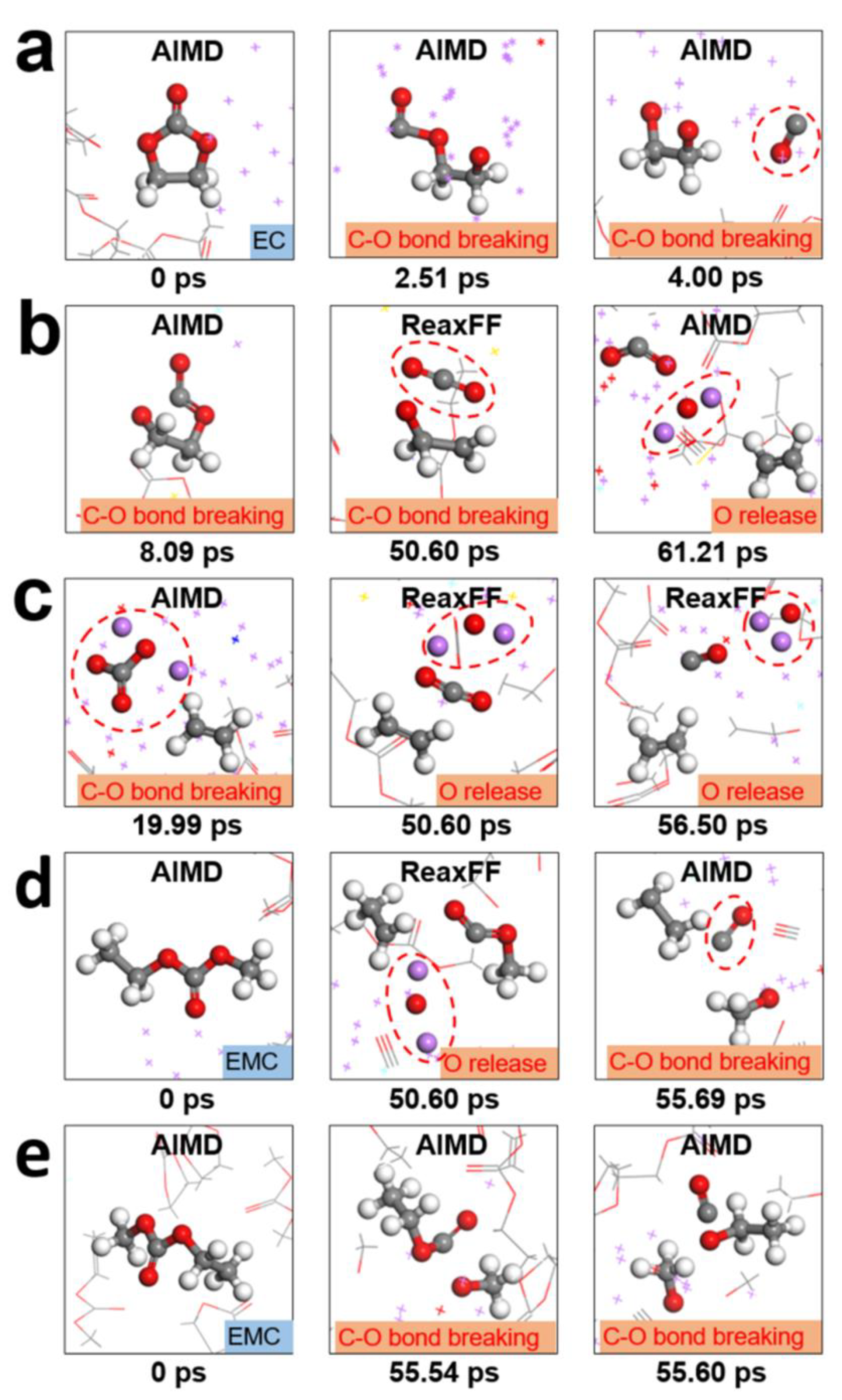
Figure 4.
XPS of the LiDFTFSI-EC/EMC system. (a) C 1 s, (b) O 1 s and (c) F 1 s. The experimental binding energy shifts in C 1 s, O 1 s and F 1 s are marked with red dots [48,49,50,51].
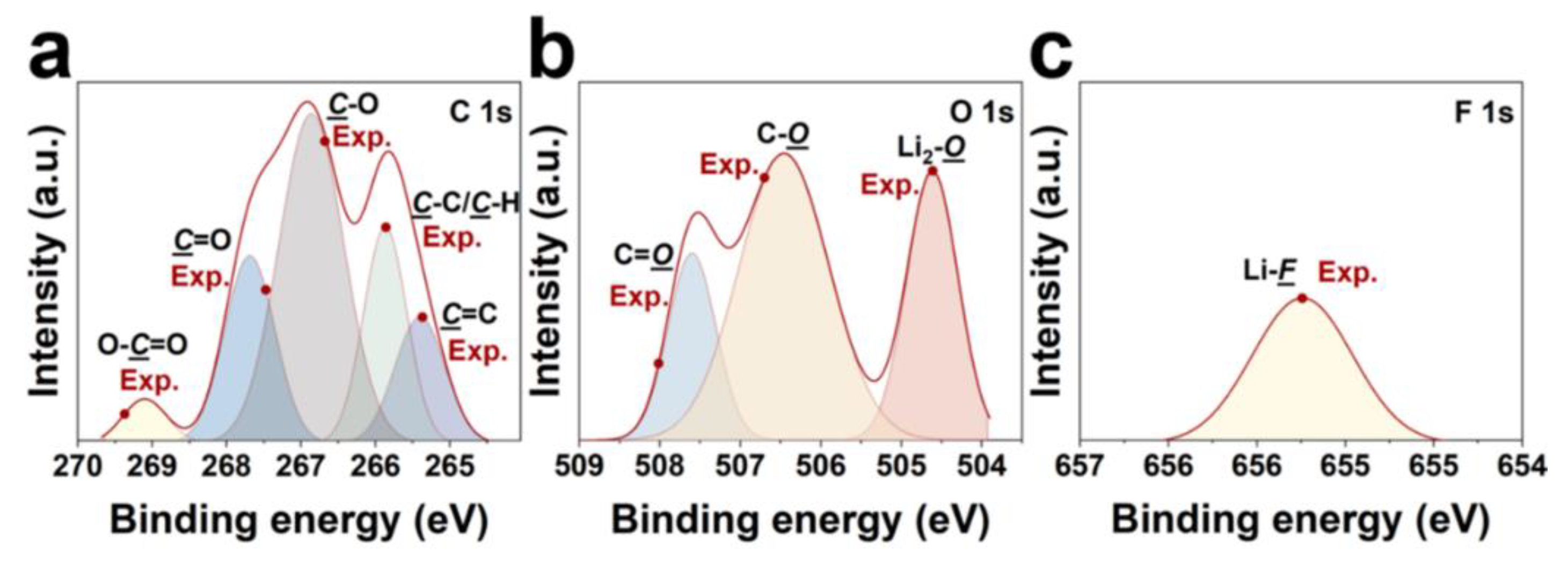
Figure 5.
LiDFTFSI defluorination achieves passivating Al. (a) ΔG of defluorination of lithium salt on the Al (111) surface and (b) schematic diagram of LiDFTFSI passivating Al. Color codes are the same as in Figure 1a.
Figure 5.
LiDFTFSI defluorination achieves passivating Al. (a) ΔG of defluorination of lithium salt on the Al (111) surface and (b) schematic diagram of LiDFTFSI passivating Al. Color codes are the same as in Figure 1a.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



