
Submitted:
19 April 2024
Posted:
22 April 2024
You are already at the latest version
Abstract
Maternal obesity and over/undernutrition can have a long-lasting impact on offspring health during critical periods in the first 1000 days of life. Children born to mothers with obesity have reduced immune responses to stimuli which increase susceptibility to infections. Recently, maternal western-style diets (WSD), high in fat and simple sugars, have been associated with skewing neonatal immune cell development, and recent evidence suggests that dysregulation of innate immunity in early life has long-term consequences on metabolic diseases and behavioral disorders in later life. Several factors contribute to abnormal innate immune tolerance or trained immunity, including changes in gut microbiota, metabolites, and epigenetic modifications. Critical knowledge gaps remain regarding the mechanisms whereby these factors impact fetal and postnatal immune cell development, especially in precursor stem cells in bone marrow and fetal liver. Components of the maternal microbiota that are transferred from mothers consuming a WSD to their offspring are understudied and identifying cause and effect on neonatal innate and adaptive immune development needs to be refined. Tools including single-cell RNA-sequencing, epigenetic analysis, and spatial location of specific immune cells in liver and bone marrow are critical for understanding immune system programming. Considering the vital role of immune function plays in offspring health, it will be important to understand how maternal diets can control developmental programming of innate and adaptive immunity.
Keywords:
maternal obesity
; fetal-maternal health
; innate immune regulation
; microbiome
; metabolites
; epigenetics
; multi-omics
1. Introduction
Obesity is a world-wide epidemic that impacts human health at all ages, including pregnant women and their offspring, with socioeconomic consequences across generations in both developed and developing countries. While difficult to approximate, pan-global studies have estimated that about 500 million people are currently obese, and that number is expected to reach over 1 billion by 2030 [1,2]. With the rising global incidence of obesity, especially in children and women of reproductive age, it is important to understand how obesity and poor maternal diet modify neonatal outcomes, particularly in the first 1000 days from conception to 2 years of age. Critical developmental windows in a child’s life can have lifelong impacts on the structure and function of organs and tissues, including developmental programming of the immune system, which plays a significant role in disease susceptibility [3,4,5]. Maternal diet, particularly a western-style diet (WSD), high in fat and simple sugars, has been shown in animal models to drive developmental programming of the immune system, beginning in utero [6,7]. The underlying causes and mechanisms for this remodeling, its impact on future disease pathways, and the critical events that drive developmental programming in the immune system are just beginning to be investigated. The purpose of this review is to explore how exposure to excess maternal fuels during fetal life can influence innate immune cell development and responses, microbial dysbiosis, metabolic effects from metabolites, and epigenetic modifications. How these changes affect the development of mechanisms underlying long-lived chronic inflammatory diseases such as metabolic dysfunction-associated steatotic liver disease (MASLD; formerly NAFLD) including microglial and liver function in the offspring, are reviewed. Possible interventions in pre-clinical models of disease are also discussed.
2. Immune Cell Development: Hematopoiesis
From embryo to neonate, the developing fetus undergoes many changes to its immune system. Central to this process, hematopoiesis undergoes significant changes in the bone and liver during fetal development to yield all blood cells which are derived from a group of self-renewing, multilineage hematopoietic stem and progenitor cells (HSPCs) [8]. However, the notion that there is a single progenitor cell has been replaced by a layered concept in which the various immune cells arise from predecessor cells in three broadly defined waves (Figure 1) [9,10]. The first cells observed in an embryo are not HSPCs but rather erythrocytes that supply oxygen to the fetal yolk sac [11]. These erythrocytes differ from their adult cousins in that they are nucleated and express embryonic hemoglobin [12]. In addition to these embryonic red blood cells, primitive macrophages develop within the yolk sac that are distinct from circulating macrophages. These cells travel to the fetal liver (establishing a resident population of macrophages and mast cells) before migrating to the brain as microglial cells or the lung as alveolar macrophages [13,14]. This is usually considered the first wave (termed “primitive”), which begins at about 4-weeks postconception, as these primitive macrophages are the first immune cells to emerge from the yolk sac [15]. The second wave, beginning around 6-weeks postconception, comes in the form of erythro-myeloid progenitors which are generated from hemogenic endothelial cells [16]. These progenitors have the potential to differentiate into macrophages, monocytes, granulocytes, and mast cells [17,18,19]. Finally, around 10 to 12-weeks postconception, the third wave, known as definitive hematopoiesis, occurs when adult-type HSPCs emerge from the aorta gonad mesonephros region and seed the fetal liver, followed shortly thereafter by colonization of the fetal bone marrow [20]. It has been shown that this seeding of the bone marrow by HSPCs happens as early as embryonic day 15.5 in mice models [21]. These cells have the potential to generate the full spectrum of immune cells needed for proper functions throughout life [22].
In addition to myeloid cells, common lymphoid progenitor cells go through a series of developments outside of the bone marrow during the maturation of a fetus [23]. Lymphoid cells are a diverse group of immune cells from the adaptive responses of T and B cells to their innate partners including natural killer (NK) cells and innate lymphoid cells. Lymphoid progenitors derived from progenitor cells in the yolk sac expand in the fetal bone marrow where they account for ~40% of the total progenitor pool [24,25]. Primary development of these tissue resident immune cells happens early in fetal gestation and once seeded into tissues, changes to “central” hematopoiesis and has little effect on the peripheral systems. Interestingly, the organs required for normal development (bone marrow and thymus) are not fully formed at this time. Despite this, cells with a similar genetic profile as lymphoid-myeloid progenitors are found within the fetal liver suggesting that the pathways for the development of T/B cells are present at this stage [26]. Development of T-cell receptor subtypes undergo increasing diversity as development proceeds starting with invariant γδ T cells and proceeding through the more complex αβ T cells [27]. While B cells are traditionally associated with adaptive immunity where they produce antibodies in response to specific pathogens, they also play an important role in the innate immune system. Human B cell development is a dynamic lifelong process that starts in utero in the bone marrow at around 6-weeks post-conception [28]. B cell-mediated innate immunity is the body's immediate, nonspecific defense mechanism against pathogens. B cells can interact with gut microbiota, which play a crucial role in metabolic health [29]. Naïve B cells also show lineage divergence depending on when the cells are seeded. Tissue resident B-1 cells can only be differentiated from fetal bone marrow or fetal liver; this contrasts with conventional B-2 cells which are derived only from adult bone marrow [30].
3. Impact of Maternal Obesity/WSD on Maternal and Placental Immune Function
Normal pregnancy proceeds with a large change in maternal immune function and a highly regulated inflammatory response is required for proper development of the fetus [31]. During early pregnancy, the yolk sac is responsible for protecting the fetus from environmental stressors until the placenta is fully developed at about 12 weeks of gestation [32]. The placenta plays a crucial role by providing oxygen, nutrients, and immune protection to the developing fetus, as well as serving as an endocrine organ that regulates maternal-fetal communication and maternal physiology. The predominate immune cells in the placenta are the uterine NK (uNK) cells and macrophages (Hofbauer cells) [33,34,35]. Unlike normal NK cells which are cytotoxic, uNK cells are generally responsible for angiogenesis in the placenta and maternal immune tolerance towards the fetus [36]. Additionally, Hofbauer cells play a role in morphogenesis and homeostasis and display an M2 phenotype (related to tissue repair and renewal for the growing fetus) by affecting the trophoblast cells [37]. Proper regulation of these cells is crucial for normal development of the fetus.
Maternal obesity can have many negative influences on the regulation of immunity and inflammation over the course of gestation [38]. Pregravid obesity impacts the systemic maternal immune system, e.g., reducing the numbers of NK T cells and plasmacytoid dendritic cells (DCs) [39,40]. Additionally, changes to the adaptive immune cells have been observed. While the total number of CD4+ T cells remained constant, the number of naïve CD4+ T cells was reported to increase in plasma across gestation in obese pregnancies [39,40]. As for CD8+ T cells, studies on maternal obesity suggest an increase in their numbers from mid to late pregnancy, possibly as a response to the growing demands of the maternal-fetal interface and the need to maintain immune surveillance [39,40]. A majority of studies demonstrate that pregnant women with obesity exhibit increased inflammation and circulating inflammatory cytokines compared with normal-weight pregnant women, and factors including fuel overload, insulin resistance, and excess gestational weight gain contribute to this inflammation [41,42,43,44]. Extracellular vesicles (EVs) also play critical roles in promoting maternal immune tolerance by acting as placental fragments that the immune system may rely upon [45,46]. Further, EVs can contribute to gestational diabetes mellitus (GDM) by altering metabolism and inflammation through proteins and circulating miRNAs predicted to affect liver and pancreatic beta cells [47,48,49].
3.1. Maternal Obesity Changes Placental Immunity
Whether maternal obesity leads to heightened cytokine transport across the placenta, thus directly inducing inflammation in the fetus, is an open question. Nevertheless, excess maternal adiposity has been reported to alter the composition and function of maternal monocytes in peripheral blood, as well as macrophages found in placental and maternal adipose tissues during late gestation, toward a proinflammatory phenotype [41,50]. A recent study showed that Hofbauer cells and microglia share a similar developmental lineage and correlations in gene dysregulation were observed between the populations during maternal obesity [51]. This suggests that the readily abundant cells in the placenta provide insight into the developmental function of neonatal microglia. Higher maternal BMI has also been correlated with increased numbers of B cells during pregnancy [40]. Furthermore, pregnant women with obesity exhibit increased activation of uNK cells promoting TNF- production [52,53]. The normal cellular population of the placenta is also altered under obese conditions; an increased accumulation of CD68+ macrophages and a reduced number of M1 (proinflammatory) polarized macrophages have been reported [54,55]. Instead of inflammation, the accumulating cells in the placenta are hyporesponsive to TLR stimulation which is associated with systemic and adipose tissue inflammation in obese pregnancies [56,57]. These cells at the maternal-fetal interface thus become excessively tolerant to stimuli and the tolerance is then imparted onto the fetus by shared developmental lineages.
3.2. Cord Blood Changes in Offspring from Obese Pregnancies
Changes during pregnancy can have a profound impact on the immune cells and inflammatory cytokines supplied to the fetus and the developmental trajectory of the neonate (Figure 2). In cord blood from obese pregnancies, an increase of NK T cells and plasmacytoid DCs with a corresponding drop in CD34+ stems cells, eosinophils, and regulatory T cells (Tregs) have been shown [58]. This drop in hematopoietic stem cells may alter the host’s ability to combat a wide range of insults to the immune system and instead skew it towards predetermined remodeling. Indeed, Sureshchandra et al. found that memory T cells within cord blood from obese pregnancies were increased in number; however, the CD4+ T cells had functional defects and blunted responses to stimuli [59]. This diminished ability of T helper cells impacts their ability to produce IL-4 as it is negatively correlated with increasing maternal BMI [56]. Paradoxically, unusually high levels of inflammatory mediators such as TNF- and C-reactive protein have been observed in the cord blood from obese pregnancies [60,61]. All these changes lead to blunted responses in fetal immune cells, decreased placental blood flow/angiogenesis, and increased fatty acid uptake with a corresponding increase in fetal liver mass in obese gravida [62]. Further, ex vivo studies have shown that monocytes in cord blood from obese pregnancies exhibit increased susceptibility and severity to infections which arose from aberrant monocyte responses caused in part by reduced nuclear translocation of NF-B [7,54]. These studies showed that reduced availability of proinflammatory gene promoters led to decreased inflammatory mediators such as IL-6, IL-10, and IL-12. Taken together, maternal obesity has the capacity to change both relative immune cell populations in the pregnant woman and in her fetus to an immune-tolerant phenotype that can impact inflammation pathways postnatally.
3.3. Maternal Obesity Impacts Adaptive and Innate Immunity in the Neonate
In normal human pregnancy, the fetal immune system has a helper T (Th) 2 phenotype to prevent alloimmune responses against the mother [63]. This is facilitated by a rare B cell subpopulation (regulatory B cells) and primarily by cytokines IL-10, IL-35, and TGF- [64]. The immune tolerance function of these cells acts by inhibiting Th1 cell activation, Th17 differentiation, and maintenance of Tregs [65]. This phenotype remains after delivery, with the infant’s immune system still biased towards a Th2 phenotype, presenting low IL-2 and IFN-γ [66]. The low production of such cytokines can be due to functional differences in DCs which may prevent the differentiation of naïve CD4+ T cells towards Th1. However, after multiple pathogenic encounters and in a time- and age-dependent manner, a switch towards Th1 polarization occurs. This promotes a proinflammatory profile with important consequences to the offspring’s long-term health, such as the prevention of childhood autoimmune diseases including asthma, allergies, atopy, as well as type 1 diabetes, all with strong adaptive immune system interactions. These diseases are more prevalent in children born to mothers with obesity, thereby linking early immune education to disease risk [67,68,69,70,71,72]. Additionally, Odaka et al. found that mice born from dams on a high-fat diet (HFD) had increased IgE production [73]. Since IgE is one of the main effectors of allergy, maternal obesity may explain the increasing prevalence of childhood allergies. Finally, these changes may have a lasting impact on the offspring through dysregulated hematopoiesis and as noted earlier, is paramount to healthy immune system development and long-term health.
4. Impact of Maternal Obesity on the Fetal Liver and Offspring Immunity
In animal models, maternal diet-induced obesity has been shown to modify fetal liver and immune system development and function [74,75]. Studies in rodents and nonhuman primates (NHP), in addition to human neonates, link maternal obesity with increased neonatal liver fat [76,77,78]. Further, exposure to a maternal WSD can increase the risk of the pediatric form of MASLD in adolescent youth [79]. Maternal factors that impact fetal immune system population dynamics and programming are a complex interplay involving pregravid obesity, dietary patterns, and inflammation. Increased inflammation is thought to be a driving force towards MASLD [80,81,82]. MASLD is a chronic, progressive condition affecting about 37% of the global population and is strongly associated with features of the metabolic syndrome, including obesity and type 2 diabetes [83]. Thus, the advanced form of the disease can progress to its active inflammatory form, metabolic dysfunction-associated steatohepatitis (MASH) and both the innate and adaptive immune systems contribute to the progression [84]. Liver resident Kupffer cells respond to nutrient overload by releasing chemokines CCL1, CCL2, and CCL5 to recruit monocytes and promote their polarization into M1 proinflammatory macrophages [85]. Chemically blocking these mediators has been shown to reduce hepatic fibrosis in human trials [86]. However, inflammation from the innate immune system is not the only driving force for increased complications in the liver, as there is an influx of T and B cells into the liver forming ectopic lymphoid structures [87]. During hepatic steatosis in mice, CD4+ T cells will produce IFN- from Th1 cells through the expression of T-bet, further increasing inflammation [88]. Studies using IFN--deficient mice have shown decreased hepatic steatosis and fibrosis when compared to wild-type mice [89]. Similar findings have been corroborated in humans through clinical observations in both adults and children [90,91], suggesting that animal studies on the developmental effects of MASLD are translational to humans.
4.1. Causal Factors for Maternal WSD Remodeling of Fetal and Neonatal Immunity
Maternal WSD can induce a proinflammatory response in fetal HSPCs and macrophages from NHPs [92]. Several adaptive programs have also been described in macrophages, including priming, tolerance, and trained immunity [93,94]. Specific pathways and mechanisms differ between the various adaptive programs in innate immunity, involving epigenetic, transcriptional, and metabolic reprogramming. Innate immune cells can undergo any of these functional adaptive programs, but the exact definition depends on the context under which the cells are studied and the nature of the stimulus used before and after to demonstrate an activated state of programmed immunity or inactivated state of immune tolerance to secondary stimuli [95,96]. Depending on their inflammatory polarization, macrophages in the liver either promote resolution from inflammation and prevent progression to fibrosis or remodel the tissue and provoke fibrosis. [97,98,99,100,101,102]. Monocytes from offspring from obese pregnancies may have an impaired capacity to respond to infection [103]. Their state of immune tolerance may affect their ability to expand in number, leading to cellular exhaustion in resident tissues in response to secondary challenges such as infection or a WSD. Given that immune tolerance can lead to downregulation of inflammatory and restorative pathways in the liver, pathogenic immune tolerance may have broad consequences on increasing programming effects through susceptibility, initiation, or progression of fibrosis across a variety of tissues.
An example of this can be found with IL-6 signaling as it is both a regulator of T cells and increased during obesity or maternal infection. Xu et al. found that IL-6Rα knockout mice, when fed an HFD, displayed reduced inflammation and increased insulin sensitivity during the first 8 weeks on an HFD compared with control mice [104]. However, the disease phenotype returned to control levels at 16 weeks of age due to a switch to enhanced IL-6-trans-signalling which utilizes the soluble form of the IL-6 receptor. Further, Lim et al. found that IL-6, induced by a mild maternal infection, caused epigenetic imprinting in the fetal intestine that has a long-term impact on immune responses to enteric pathogens and tissue inflammation in adulthood [105]. These data clearly illustrate that the timing is critical for proper immune regulation and the duration of effects may be greater when applied during fetal development.
Fetal and neonatal T and B cells are not spared from changes to normal effector functions due to external stimuli. CD4+ and CD8+ T cells respond to antigens in HFDs to drive MASH and metabolic syndrome [88,106]. These cells are recruited into the liver by IFN- and promote insulin resistance and glucose metabolism during HFD feeding [107]. Disruption of the gut microbiota-B cell axis may also contribute to obesity-related inflammation and metabolic dysfunction. B cells have also been implicated in the pathogenesis of MASLD, particularly in its more severe form MASH, as they are shown to be pro-fibrogenic in the form of plasma cells secreting IgA [108]. These are formed by proinflammatory mediators which stimulate hepatic stellate cells into producing retinoic acid which is detected by the B cells [109]. The adaptive arm of immunity can also be linked back to innate immunity. Independent of T cells, B-1 cells can secrete IgM natural antibodies which can react with endogenous antigens such as oxidized phospholipids [110]. These changes both increase the susceptibility to MASLD and drive its progression forward.
5. Microglia Activation by a Maternal WSD
Microglia are the resident immune cells of the brain and have similar functions to macrophages such as immune defense and brain maintenance [111]. This group of self-renewing cells originate from the fetal yolk sac and any adverse effects on this population at birth can cause long-term consequences on brain function [112]. Many associations have been made between inflammation and neuropsychiatric disorders including ADHD, autism, schizophrenia, and eating disorders [113]. Mattei et al. found that maternal immune activation with Poly(I:C) led to a downregulation of genes involved in the inflammatory response, microglial development, and phagocytosis in adult offspring [114]. However, genes involved in migration of microglial cells were upregulated, and these transcriptional changes could be reversed with treatment of minocycline, an antibiotic used as a possible treatment for schizophrenia [115]. Studies in an NHP model showed that offspring from dams with WSD-induced obesity have an inflammatory phenotype in the brain that includes increased microglial cell counts [116]. This study showed that while pre-pregnancy adiposity was positively correlated with increased microglial cell counts, these counts were also negatively correlated with maternal WSD. The exact cause of these relationships is not entirely clear. However, further research by this group found that IL-12 was increased in the dams due to maternal WSD which caused neurodevelopmental disorders in the offspring [117]. Another group confirmed the previous findings of decreased microglial cell counts due to maternal WSD but also showed that the deceased cell count did not persist one year postnatally [118]. This would suggest that neurodevelopmental trajectory and immune response is heavily dictated by early life events. Additionally, this activation is accompanied with the release of proinflammatory cytokines, e.g., TNF-, IL-1, and IL-6, which have been linked to neuronal loss, brain damage, and other neurodegenerative diseases [119]. For example, induction of the inflammasome through NLRP3 causes increased neuroinflammation and cognitive impairment and knocking out its receptor IL-1R1 can restore these deficiencies back to control levels [120]. This increased inflammation damages brain tissue and can alter the proper functioning of other resident cells. IL-6 was also found to be a critical regulator of microglial activation and it effects were abrogated either by knockout mice or neutralizing antibodies or increased by maternal IL-6 administration [121]. Lastly, IL-17A was also shown to increase microglial activation through direct administration to the fetal mouse brain [122].
6. Maternal Obesity Influences the Fetal Microbiome and Immune Development
Maternal diet alters the microbiome of both the mother and the offspring to regulate many processes including gene expression, immune tolerance, and metabolism [123,124,125,126]. The maternal microbiome has been studied in great detail recently and studies have shown that differences in maternal diet have a profound impact on the diversity and taxa of the maternal and fetal microbiota [127,128,129,130,131,132,133,134]. Individual bacteria and their components such as endotoxins have been linked with changes in gut and circulating inflammatory cells, and these changes have been implicated in many chronic western diseases [123,135]. Data suggest that gross changes in abundances of obesity-enriched microbial genes are positively correlated with blood glucose levels in pregnant women with obesity and GDM and can be controlled by maternal diet, but the functionality of these changes remains unknown [136,137]. A portion of the maternal microbiome is vertically transmitted to the neonate, and early-life microbiota of the infant depends in part on the mode of delivery [138]. The process of neonatal colonization, whether it begins in utero, and the mechanisms involved are complex and not completely understood. Commensal microbes make their way to the infant through vertical transmission from the mother and their diversity is affected by perinatal conditions [139,140]. Research has shown that 72% of the neonate gut microbiome comes from the mother, but this number is substantially reduced to 25% in cases of recent maternal antibiotic use [141]. Infants exposed to antibiotics obtained antimicrobial resistant species mostly from the environment rather than through vertical transmission. This shows the potential dangers that can arise with nosocomial infections and antibiotic exposure in neonates. The hygiene hypothesis suggests that reduced diversity of early-life microbial exposure is associated with an elevated risk of allergic diseases later in life; however, studies have also linked susceptibility to allergic disorders to immune regulatory pathways triggered by some commensal microbes [142]. Soderborg et al. showed that when microbes from 2-week-old infants born to mothers with obesity are transferred to germ-free mice, this imbalance disrupts myeloid cells (macrophage precursors) derived from the bone marrow and provokes obesity and a leaky gut leading to MASLD after a WSD challenge [143]. This metabolic rewiring of maturing macrophages is likely accompanied by epigenetic reprogramming that causes reduced bacterial phagocytosis and prevents liver inflammation. This may profoundly disrupt the establishment of the host-microbe symbiosis and suggests that there is a direct impact on offspring’s stem cells, which regulate immune development.
Concepts on the origins of developmental immune programming and how early life interactions can shape lifelong alterations and disease susceptibility through immune tolerance or trained immunity of the innate immune system have been reviewed by Hong and Medzhitov [144]. The first 1000 days of life are a critical period for proper development of a healthy gut, dominated by a diverse group of infant-type bifidobacteria including Bifidobacterium longum, B. bifidum, and B. breve [145,146,147]. These species can metabolize human milk oligosaccharides that are abundant in breast milk and thus have a metabolic advantage in early colonization [148]. Breastfeeding has been correlated with reduced disorders such as pathogen infection, autoimmune disorders, diabetes, and obesity [149,150]. Likewise, the acquisition of founder strains in the Enterobacteriaceae family in human newborns plays an essential role in development of normal neonatal immunity and susceptibility to metabolic diseases in later life. Enterobacteriaceae, as well as being a facilitative anaerobe, is a producer of lipopolysaccharide (LPS) important for trained innate and adaptive immunity and is critical for normal immune tolerance [150]. Maternal diet can also shift the community of commensals [133,135]. An HFD can increase the number of anaerobes such as Bacteroidetes and obese individuals have a high Bacteroidetes to Firmicutes ratio; however, this is not a universal finding [151]. Table 1 lists key studies exploring the nature between the microbiome and disordered offspring immunity.
The microbiome can also have a wide range of effects on shaping and training the host immune system [68,157,158]. Normal colonization of microbiota has been shown to assist in the proper development of CD8+ T cell immunity through the priming of monocytes and macrophages which release TNF that signals conventional DCs to produce the CD8+ T cell target IL-12p40 [159]. They can also promote early innate effectors of unconventional T cells including mucosal-associated invariant T (MAIT) cells, NK cells, and γδ T cells [160]. Furthermore, commensals are necessary for promoting immune tolerance and creating proper immune defenses in the gut. Peripherally induced Tregs (pTregs) can support tolerance in the gut through RORγt while also abrogating Th17 responses [161,162]. Murine studies have shown that early in life (2-3 weeks old) is a critical window for pTreg generation and failure to generate Tregs can lead to altered immune homeostasis and increased inflammatory intestinal diseases [163]. These alterations to immune signaling contribute to immunologic and metabolic disorders which are normally prevented through proper priming of the immune system. This priming can occur as early as the second trimester; memory T cells were found in fetal lymph nodes, which can be activated by Staphylococcus and Lactobacillus [164]. Further, it has been shown that germ-free mice have compromised CD4+ T cell and Treg development, suggesting that microbial interaction is necessary for early immune system development [165]. Finally, serotonin was shown to be highly enriched during the first 3 weeks of life in the luminal contents from the gut of specific pathogen-free neonatal mice and is driven by the gut commensal microbiota to directly alter cell differentiation into Tregs [166].
Microbes can also directly influence the differentiation and effect of bone marrow HSPCs as the fetus develops. Studies have shown that mice lacking proper commensal microbes during development have low HSPC counts and defective myelopoiesis which resulted in impaired resistance to bacterial infections [167,168]. Recolonization of these mice with a healthy microbiota can rescue this phenotype via metabolites, microbial components, and nutritional support [169,170,171]. However, there is a complex interplay at work as not all stimuli provide the same outcome. An example of this was found in mice given different bacterial components and those supplemented with LPS had an increased pathogen killing capacity from myeloid cells compared with those given bacterial DNA instead [172]. The heterogeneity coincides with other studies that have found differences among monocytes in both mice and humans in that inflammatory environmental cues can influence the resulting population of monocytes [173,174]. These differences have lasting effects on both the myeloid cells as well as HSPCs [175]. Luo et al. found an overall decreased capacity for hematopoiesis with an altered microbiome brought on by HFD feeding in adult mice, and these changes were carried over by fecal transplantation of microbes from the HFD group to normal mice [176]. On the other hand, most studies of early life LPS result in dampening of innate immunity in monocytes in offspring from LPS-treated dams compared with offspring from control pregnancies, suggesting an impaired capacity to respond to infection. In infants born to mothers with chorioamnionitis for example, monocytes have impaired production of inflammatory cytokines and lower expression of genes involved in antigen presentation and adaptive immunity [177,178]. Taken together, altered microbiomes can have a profound effect both on the proper development and maintenance of immunity during critical periods of fetal development.
7. Metabolites Regulate Fetal Metabolism and Immune Development
As noted earlier, infant-type Bifidobacteria species are well suited for colonizing the infant gut due to their ability to metabolize breast milk [145]. They produce a myriad of metabolites including various types of short-chain fatty acids (SCFAs) and aromatic amino acids which can directly regulate and alter host pathways (Table 2) [179,180,181,182]. The most common SCFAs in the gut are acetate, propionate, and butyrate which are able to influence host processes through different mechanisms [183]. Butyrate is the preferred energy source for gut epithelial cells and can directly modulate genes by acting as histone deacetylases (HDACs) [184]. HDACs have been shown to suppress tumor growth in cancerous cells and have anti-inflammatory effects in healthy cells [185,186,187]. Moreover, butyrate supplementation in mammalian models has been shown to elevate beneficial gut microbiota, improve intestinal barrier function, and promote fat mobilization and utilization, resulting in the attenuation of hepatic steatosis through STAT3 signaling and HDACs [188,189,190]. Acetate and propionate are activators of G protein-coupled receptors and thus promote lipid and glucose metabolism in white adipose tissue [191,192]. Chen et al. found that maternal supplementation with propionate ameliorated intrauterine growth restriction by downregulating glucose and lipid metabolism caused by hypoxia [193].
Another class of metabolites important for regulating inflammation are derived from the essential amino acid tryptophan (Table 2). Tryptophan regulation and utilization are important during pregnancy and are essential for proper fetal growth [194]. Tryptophan metabolites are derived from gut metabolism and are altered in obesity; higher amounts of the tryptophan product kynurenine and lower amounts of serotonin are associated with higher BMI and visceral fat mass [195,196]. Gut commensals metabolize dietary tryptophan to produce metabolites capable of modulating the host immune system, thereby linking the gut microbiota with nutrition, metabolism, and the innate immune response. The indole derivatives tryptamine and indole-3-acetate were shown to suppress the inflammatory responses of macrophages and reduce lipid production in hepatocytes [197]. Other tryptophan metabolites, e.g., indole-3-ethanol, indole-3-pyruvate, and indole-3-aldehyde, can prevent gut permeability by maintaining tight junctions [198]. Finally, indole-3-carbinol can decrease adipogenesis, thermogenesis, and inflammation by rescuing key transcription factors that become dysregulated by HFD feeding [199]. Central to most of these pathways is the aryl hydrocarbon receptor (AHR); many tryptophan metabolites are AHR ligands [200]. AHR signals the transcription factors NRF2 and NF-κB and has been shown to play a role in the progression of MASLD [201]. Ghiboub et al. reviewed the ongoing research into therapeutic treatments using tryptophan including direct supplementation, microbiota-derived supplementation, and AHR ligand activation [202].
8. Role of Epigenetic Programming in Immune Cells from Offspring Exposed to WSD
Epigenetic changes due to maternal diet/obesity can occur throughout neonatal tissues and cells with lasting impact on neonatal development and beyond. Recent studies have shown that cord blood DNA methylation in neonates born to mothers with obesity correlated with maternal triglycerides and childhood adiposity [208]. In NHP studies, both HSPCs and bone marrow-derived macrophages (BMDM) from maternal WSD-exposed 3-year-old offspring were epigenetically modified to a more proinflammatory signature [6], despite weaning at 7 months to a normal chow diet. More open chromatin (higher transcription) in regions promoting glycolysis and more closed chromatin in regions relating to oxidative phosphorylation were observed in fetal mononuclear cells, suggestive of immune cell chromatin remodeling by exposure to maternal WSD. These progeny cells were then able to transform into macrophages with an increase in genes that mark early development of proinflammatory macrophages such as FOS/JUN, NF-κB, C/EBPβ, and STAT6 [6]. These findings indicate that maternal WSD led to a classic shift, increasing glycolytic metabolism that favors the proinflammatory M1 cell type [6]. This effect may also be further modified by the tissue microenvironment which affect their proper function [209,210]. Suter et al. showed that in NHPs, maternal WSD can alter the expression of NPAS2 (regulator of circadian genes in peripheral organs) in the offspring liver through histone modification at the promoter regions of genes favoring liver lipid deposition [211,212].
Maternal overnutrition also causes epigenetic changes within the hypothalamus of the fetal and postnatal brain [213,214,215]. Mouse studies showed that maternal WSD alters the arcuate nucleus region that controls NPY and AgRP and the appetite suppression neurons of POMC and CART [216]. Maternal overnutrition can increase the expression of NPY/AgRP while reducing POMC in the adult offspring’s brain [217,218]. Overall, this led to less sensitivity to the appetite suppression hormones leptin and insulin [217,219,220,221]. Specifically, hypermethylation occurs at the enhancer and promoter regions for POMC [222,223]. Interestingly, hypermethylation of the enhancer regions (but not the promoter) persisted into adulthood showing the prolonged programming effects of overnutrition [222]. Modulation of genes can also occur though HDACs. Suter et al. showed in the NHP model that a maternal WSD increased acetylation of histone H3 through the downregulation of the lysine deacetylase SIRT1 in the liver [224]. A study in mice showed that in response to an HFD, Hdac5 and Hdac8 levels were increased in the hypothalamus, suggesting that they also play a role in gene expression under different metabolic conditions [225]. These alterations to the environment can alter the local hypothalamic region and, coupled with the proinflammatory response of activated microglia and astrocytes, suggests that metabolic reprogramming during gestation will drive microglia innate immune memory to shape their immune reactivity leading to abnormalities of the brain in areas of both appetite stimulation and suppression [226,227].
9. Conclusions and Future Directions
As outlined in this review, due to maternal factors, the developing immune system in offspring goes through many changes, both functionally and superficially in development, beginning with the fetus, to the neonatal period and beyond (Figure 3). However, many challenges remain as the interpretation and translation of these findings are hampered by our relatively limited mechanistic understanding of immune programming. As single-cell ‘omic technologies are maturing and more high-throughput data analyses are conducted, more information is being gathered on different cell populations, especially immune cell types, rare cells types, disease state cells, and the whole spectrum of cells in between that lead to complex interactions within tissues [228]. The signals underlying development of HSPCs, specifically in the fetal liver and bone marrow under the influence of maternal WSD, and the timing and distribution of cells is of critical importance. Tools with single-cell resolution for analyses of the transcriptome, epigenetic changes, splice variants, surface protein expression, and spatial location will enable us to uncover their interactions. Analyzing these types of datasets in tandem will allow us to discover novel interactions. Additionally, despite evidence that T and B cells can be derived from the yolk sac in mice, no definitive proof of this has been reported in humans as fate-mapping in adults from their developmental origins is not yet possible [23]. Which HSPC populations function primarily during fetal development and which populations are lifelong will be key to understanding how early factors influence disease development.
Maternal proinflammatory dietary factors including high fat/high sugar can alter the microbiome of both the mother and their offspring and the metabolites these microbes produce. However, the critical roles of a complex microbiome in influencing HSPCs and fetal programming/immunity are needed. The exact composition of the human maternal microbiota that is transferred to their offspring is vastly different between individuals. Therefore, protocols and mouse models mimicking the human condition for discovery of these relationships during critical windows of development need to be refined. Alterations in the maternal microbiome can lead to functional changes in offspring immune cell populations; this research will generate a diverse array of datasets including transcriptomics, proteomics, metabolomics, and integrated together, meta-omics, to create a functional understanding of the processes of fetal immune development. This information can better inform us of the treatments needed and pre/probiotic usage during pregnancy for healthier outcomes.
Lastly, questions remain as to how to correct the immune system especially if we medically supplement with an external source. Studies have sought to use maternal diets as an intervention for altered immunological programming and neural defects in offspring [229]. Will it be possible in the future to introduce targeted genetically modified immune cells as therapy for metabolically rewiring the immune system [230,231]? Given the increasing numbers of children and adolescents suffering from metabolic and behavioral disorders, and the role that inflammation may play in triggering the origins of disease, understanding the relationship between poor maternal diets and developmental programming of the immune system during early life may play a crucial role in maintaining the delicate balance between health and disease in the 21st century.
Author Contributions
Conceptualization, B.N. and J.F.; writing—original draft preparation, B.N.; writing—review and editing, B.N. and J.F.; visualization, B.N.; supervision, J.F.; project administration, J.F.; funding acquisition, J.F. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Institute of Diabetes and Digestive and Kidney Diseases grants R01DK121951 (J.F.) and R01DK128416 (J.F.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable
Acknowledgments
All figures were created with BioRender.com.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Finucane, M.M.; Stevens, G.A.; Cowan, M.J.; Danaei, G.; Lin, J.K.; Paciorek, C.J.; Singh, G.M.; Gutierrez, H.R.; Lu, Y.; Bahalim, A.N.; et al. National, regional, and global trends in body-mass index since 1980: systematic analysis of health examination surveys and epidemiological studies with 960 country-years and 9.1 million participants. Lancet 2011, 377, 557–567. [Google Scholar] [CrossRef] [PubMed]
- Kelly, T.; Yang, W.; Chen, C.S.; Reynolds, K.; He, J. Global burden of obesity in 2005 and projections to 2030. Int J Obes (Lond) 2008, 32, 1431–1437. [Google Scholar] [CrossRef] [PubMed]
- Brines, J.; Rigourd, V.; Billeaud, C. The First 1000 Days of Infant. Healthcare (Basel) 2022, 10. [Google Scholar] [CrossRef]
- Likhar, A.; Patil, M.S. Importance of Maternal Nutrition in the First 1,000 Days of Life and Its Effects on Child Development: A Narrative Review. Cureus 2022, 14, e30083. [Google Scholar] [CrossRef] [PubMed]
- Simione, M.; Moreno-Galarraga, L.; Perkins, M.; Price, S.N.; Luo, M.; Kotelchuck, M.; Blake-Lamb, T.L.; Taveras, E.M. Effects of the First 1000 Days Program, a systems-change intervention, on obesity risk factors during pregnancy. BMC Pregnancy Childbirth 2021, 21, 729. [Google Scholar] [CrossRef] [PubMed]
- Nash, M.J.; Dobrinskikh, E.; Soderborg, T.K.; Janssen, R.C.; Takahashi, D.L.; Dean, T.A.; Varlamov, O.; Hennebold, J.D.; Gannon, M.; Aagaard, K.M.; et al. Maternal diet alters long-term innate immune cell memory in fetal and juvenile hematopoietic stem and progenitor cells in nonhuman primate offspring. Cell Rep 2023, 42, 112393. [Google Scholar] [CrossRef] [PubMed]
- Sureshchandra, S.; Doratt, B.M.; Mendza, N.; Varlamov, O.; Rincon, M.; Marshall, N.E.; Messaoudi, I. Maternal obesity blunts antimicrobial responses in fetal monocytes. Elife 2023, 12. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Yoshimoto, M. Multiple waves of fetal-derived immune cells constitute adult immune system. Immunol Rev 2023, 315, 11–30. [Google Scholar] [CrossRef] [PubMed]
- Mass, E.; Gentek, R. Fetal-Derived Immune Cells at the Roots of Lifelong Pathophysiology. Front Cell Dev Biol 2021, 9, 648313. [Google Scholar] [CrossRef]
- Donald, K.; Finlay, B.B. Early-life interactions between the microbiota and immune system: impact on immune system development and atopic disease. Nat Rev Immunol 2023, 23, 735–748. [Google Scholar] [CrossRef]
- Palis, J.; Yoder, M.C. Yolk-sac hematopoiesis: the first blood cells of mouse and man. Exp Hematol 2001, 29, 927–936. [Google Scholar] [CrossRef]
- Brittain, T. Molecular aspects of embryonic hemoglobin function. Mol Aspects Med 2002, 23, 293–342. [Google Scholar] [CrossRef] [PubMed]
- Chia, S.L.; Kapoor, S.; Carvalho, C.; Bajenoff, M.; Gentek, R. Mast cell ontogeny: From fetal development to life-long health and disease. Immunol Rev 2023, 315, 31–53. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [PubMed]
- Tober, J.; Koniski, A.; McGrath, K.E.; Vemishetti, R.; Emerson, R.; de Mesy-Bentley, K.K.; Waugh, R.; Palis, J. The megakaryocyte lineage originates from hemangioblast precursors and is an integral component both of primitive and of definitive hematopoiesis. Blood 2007, 109, 1433–1441. [Google Scholar] [CrossRef] [PubMed]
- McGrath, K.E.; Frame, J.M.; Fegan, K.H.; Bowen, J.R.; Conway, S.J.; Catherman, S.C.; Kingsley, P.D.; Koniski, A.D.; Palis, J. Distinct Sources of Hematopoietic Progenitors Emerge before HSCs and Provide Functional Blood Cells in the Mammalian Embryo. Cell Rep 2015, 11, 1892–1904. [Google Scholar] [CrossRef]
- Gomez Perdiguero, E.; Klapproth, K.; Schulz, C.; Busch, K.; Azzoni, E.; Crozet, L.; Garner, H.; Trouillet, C.; de Bruijn, M.F.; Geissmann, F.; et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 2015, 518, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Hoeffel, G.; Chen, J.; Lavin, Y.; Low, D.; Almeida, F.F.; See, P.; Beaudin, A.E.; Lum, J.; Low, I.; Forsberg, E.C.; et al. C-Myb(+) erythro-myeloid progenitor-derived fetal monocytes give rise to adult tissue-resident macrophages. Immunity 2015, 42, 665–678. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Liu, S.; Xu, J.; Zhang, X.; Han, D.; Liu, J.; Xia, M.; Yi, L.; Shen, Q.; Xu, S.; et al. Adult Connective Tissue-Resident Mast Cells Originate from Late Erythro-Myeloid Progenitors. Immunity 2018, 49, 640–653e645. [Google Scholar] [CrossRef]
- Calvanese, V.; Mikkola, H.K.A. The genesis of human hematopoietic stem cells. Blood 2023, 142, 519–532. [Google Scholar] [CrossRef]
- Hall, T.; Sriram, P.; McKinney-Freeman, S. Murine Fetal Bone Marrow HSPCs Undergo a Dramatic Shift in Frequency at Birth. Blood 2019, 134, 2471–2471. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, F. The evolving views of hematopoiesis: from embryo to adulthood and from in vivo to in vitro. J Genet Genomics 2023. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Wijanarko, K.; Liani, O.; Strumila, K.; Ng, E.S.; Elefanty, A.G.; Stanley, E.G. Lymphoid cell development from fetal hematopoietic progenitors and human pluripotent stem cells. Immunol Rev 2023, 315, 154–170. [Google Scholar] [CrossRef]
- O'Byrne, S.; Elliott, N.; Rice, S.; Buck, G.; Fordham, N.; Garnett, C.; Godfrey, L.; Crump, N.T.; Wright, G.; Inglott, S.; et al. Discovery of a CD10-negative B-progenitor in human fetal life identifies unique ontogeny-related developmental programs. Blood 2019, 134, 1059–1071. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, M.; Porayette, P.; Glosson, N.L.; Conway, S.J.; Carlesso, N.; Cardoso, A.A.; Kaplan, M.H.; Yoder, M.C. Autonomous murine T-cell progenitor production in the extra-embryonic yolk sac before HSC emergence. Blood 2012, 119, 5706–5714. [Google Scholar] [CrossRef] [PubMed]
- Luis, T.C.; Luc, S.; Mizukami, T.; Boukarabila, H.; Thongjuea, S.; Woll, P.S.; Azzoni, E.; Giustacchini, A.; Lutteropp, M.; Bouriez-Jones, T.; et al. Initial seeding of the embryonic thymus by immune-restricted lympho-myeloid progenitors. Nat Immunol 2016, 17, 1424–1435. [Google Scholar] [CrossRef] [PubMed]
- Krangel, M.S. Mechanics of T cell receptor gene rearrangement. Curr Opin Immunol 2009, 21, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Jackson, T.R.; Ling, R.E.; Roy, A. The Origin of B-cells: Human Fetal B Cell Development and Implications for the Pathogenesis of Childhood Acute Lymphoblastic Leukemia. Front Immunol 2021, 12, 637975. [Google Scholar] [CrossRef]
- Pabst, O.; Nowosad, C.R. B cells and the intestinal microbiome in time, space and place. Semin Immunol 2023, 69, 101806. [Google Scholar] [CrossRef]
- Dorshkind, K.; Crooks, G. Layered immune system development in mice and humans. Immunol Rev 2023, 315, 5–10. [Google Scholar] [CrossRef]
- Racicot, K.; Kwon, J.Y.; Aldo, P.; Silasi, M.; Mor, G. Understanding the complexity of the immune system during pregnancy. Am J Reprod Immunol 2014, 72, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Ander, S.E.; Diamond, M.S.; Coyne, C.B. Immune responses at the maternal-fetal interface. Sci Immunol 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Moffett-King, A. Natural killer cells and pregnancy. Nat Rev Immunol 2002, 2, 656–663. [Google Scholar] [CrossRef]
- Nagamatsu, T.; Schust, D.J. The contribution of macrophages to normal and pathological pregnancies. Am J Reprod Immunol 2010, 63, 460–471. [Google Scholar] [CrossRef] [PubMed]
- Suryawanshi, H.; Morozov, P.; Straus, A.; Sahasrabudhe, N.; Max, K.E.A.; Garzia, A.; Kustagi, M.; Tuschl, T.; Williams, Z. A single-cell survey of the human first-trimester placenta and decidua. Sci Adv 2018, 4, eaau4788. [Google Scholar] [CrossRef]
- Burke, S.D.; Barrette, V.F.; Gravel, J.; Carter, A.L.; Hatta, K.; Zhang, J.; Chen, Z.; Leno-Duran, E.; Bianco, J.; Leonard, S.; et al. Uterine NK cells, spiral artery modification and the regulation of blood pressure during mouse pregnancy. Am J Reprod Immunol 2010, 63, 472–481. [Google Scholar] [CrossRef]
- Yao, Y.; Xu, X.H.; Jin, L. Macrophage Polarization in Physiological and Pathological Pregnancy. Front Immunol 2019, 10, 792. [Google Scholar] [CrossRef]
- Wanaditya, G.K.; Putra, I.W.A.; Aryana, M.B.D.; Mulyana, R.S. Obesity in Pregnant Women and Its Impact on Maternal and Neonatal Morbidity. European Journal of Medical and Health Sciences 2023, 5, 17–21. [Google Scholar] [CrossRef]
- Sureshchandra, S.; Marshall, N.E.; Wilson, R.M.; Barr, T.; Rais, M.; Purnell, J.Q.; Thornburg, K.L.; Messaoudi, I. Inflammatory Determinants of Pregravid Obesity in Placenta and Peripheral Blood. Frontiers in Physiology 2018, 9. [Google Scholar] [CrossRef]
- Sen, S.; Iyer, C.; Klebenov, D.; Histed, A.; Aviles, J.A.; Meydani, S.N. Obesity impairs cell-mediated immunity during the second trimester of pregnancy. American Journal of Obstetrics and Gynecology 2013, 208, 139–e131.e138. [Google Scholar] [CrossRef]
- Bravo-Flores, E.; Mancilla-Herrera, I.; Espino, Y.S.S.; Ortiz-Ramirez, M.; Flores-Rueda, V.; Ibarguengoitia-Ochoa, F.; Ibanez, C.A.; Zambrano, E.; Solis-Paredes, M.; Perichart-Perera, O.; et al. Macrophage Populations in Visceral Adipose Tissue from Pregnant Women: Potential Role of Obesity in Maternal Inflammation. Int J Mol Sci 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Pendeloski, K.P.T.; Ono, E.; Torloni, M.R.; Mattar, R.; Daher, S. Maternal obesity and inflammatory mediators: A controversial association. Am J Reprod Immunol 2017, 77. [Google Scholar] [CrossRef] [PubMed]
- Tinius, R.A.; Blankenship, M.M.; Furgal, K.E.; Cade, W.T.; Pearson, K.J.; Rowland, N.S.; Pearson, R.C.; Hoover, D.L.; Maples, J.M. Metabolic flexibility is impaired in women who are pregnant and overweight/obese and related to insulin resistance and inflammation. Metabolism 2020, 104, 154142. [Google Scholar] [CrossRef] [PubMed]
- Pantham, P.; Aye, I.L.; Powell, T.L. Inflammation in maternal obesity and gestational diabetes mellitus. Placenta 2015, 36, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Paul, N.; Sultana, Z.; Fisher, J.J.; Maiti, K.; Smith, R. Extracellular vesicles- crucial players in human pregnancy. Placenta 2023, 140, 30–38. [Google Scholar] [CrossRef]
- Gercel-Taylor, C.; O'Connor, S.M.; Lam, G.K.; Taylor, D.D. Shed membrane fragment modulation of CD3-zeta during pregnancy: link with induction of apoptosis. J Reprod Immunol 2002, 56, 29–44. [Google Scholar] [CrossRef] [PubMed]
- Gillet, V.; Ouellet, A.; Stepanov, Y.; Rodosthenous, R.S.; Croft, E.K.; Brennan, K.; Abdelouahab, N.; Baccarelli, A.; Takser, L. miRNA Profiles in Extracellular Vesicles From Serum Early in Pregnancies Complicated by Gestational Diabetes Mellitus. J Clin Endocrinol Metab 2019, 104, 5157–5169. [Google Scholar] [CrossRef] [PubMed]
- Kandzija, N.; Zhang, W.; Motta-Mejia, C.; Mhlomi, V.; McGowan-Downey, J.; James, T.; Cerdeira, A.S.; Tannetta, D.; Sargent, I.; Redman, C.W.; et al. Placental extracellular vesicles express active dipeptidyl peptidase IV; levels are increased in gestational diabetes mellitus. J Extracell Vesicles 2019, 8, 1617000. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Wang, S.; Huang, X.; Chen, P.; Deng, L.; Li, S.; Lin, S.; Wang, Z.; Liu, B. Plasma Exosomal miRNAs Associated With Metabolism as Early Predictor of Gestational Diabetes Mellitus. Diabetes 2022, 71, 2272–2283. [Google Scholar] [CrossRef]
- Sacks, G.P.; Studena, K.; Sargent, K.; Redman, C.W. Normal pregnancy and preeclampsia both produce inflammatory changes in peripheral blood leukocytes akin to those of sepsis. Am J Obstet Gynecol 1998, 179, 80–86. [Google Scholar] [CrossRef]
- Batorsky, R.; Ceasrine, A.M.; Shook, L.L.; Kislal, S.; Bordt, E.A.; Devlin, B.A.; Perlis, R.H.; Slonim, D.K.; Bilbo, S.D.; Edlow, A.G. Hofbauer cells and fetal brain microglia share transcriptional profiles and responses to maternal diet-induced obesity. bioRxiv 2023. [Google Scholar] [CrossRef] [PubMed]
- Castellana, B.; Perdu, S.; Kim, Y.; Chan, K.; Atif, J.; Marziali, M.; Beristain, A.G. Maternal obesity alters uterine NK activity through a functional KIR2DL1/S1 imbalance. Immunol Cell Biol 2018, 96, 805–819. [Google Scholar] [CrossRef] [PubMed]
- Fukui, A.; Funamizu, A.; Yokota, M.; Yamada, K.; Nakamua, R.; Fukuhara, R.; Kimura, H.; Mizunuma, H. Uterine and circulating natural killer cells and their roles in women with recurrent pregnancy loss, implantation failure and preeclampsia. J Reprod Immunol 2011, 90, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Cifuentes-Zuniga, F.; Arroyo-Jousse, V.; Soto-Carrasco, G.; Casanello, P.; Uauy, R.; Krause, B.J.; Castro-Rodriguez, J.A. IL-10 expression in macrophages from neonates born from obese mothers is suppressed by IL-4 and LPS/INFgamma. J Cell Physiol 2017, 232, 3693–3701. [Google Scholar] [CrossRef] [PubMed]
- Challier, J.C.; Basu, S.; Bintein, T.; Minium, J.; Hotmire, K.; Catalano, P.M.; Hauguel-de Mouzon, S. Obesity in pregnancy stimulates macrophage accumulation and inflammation in the placenta. Placenta 2008, 29, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.M.; Marshall, N.E.; Jeske, D.R.; Purnell, J.Q.; Thornburg, K.; Messaoudi, I. Maternal obesity alters immune cell frequencies and responses in umbilical cord blood samples. Pediatr Allergy Immunol 2015, 26, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Haghiac, M.; Surace, P.; Challier, J.C.; Guerre-Millo, M.; Singh, K.; Waters, T.; Minium, J.; Presley, L.; Catalano, P.M.; et al. Pregravid obesity associates with increased maternal endotoxemia and metabolic inflammation. Obesity (Silver Spring) 2011, 19, 476–482. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Espinosa, L.O.; Montiel-Cervantes, L.A.; Guerra-Marquez, A.; Penaflor-Juarez, K.; Reyes-Maldonado, E.; Vela-Ojeda, J. Maternal obesity associated with increase in natural killer T cells and CD8+ regulatory T cells in cord blood units. Transfusion 2016, 56, 1075–1081. [Google Scholar] [CrossRef] [PubMed]
- Sureshchandra, S.; Mendoza, N.; Jankeel, A.; Wilson, R.M.; Marshall, N.E.; Messaoudi, I. Phenotypic and Epigenetic Adaptations of Cord Blood CD4+ T Cells to Maternal Obesity. Front Immunol 2021, 12, 617592. [Google Scholar] [CrossRef]
- Dosch, N.C.; Guslits, E.F.; Weber, M.B.; Murray, S.E.; Ha, B.; Coe, C.L.; Auger, A.P.; Kling, P.J. Maternal Obesity Affects Inflammatory and Iron Indices in Umbilical Cord Blood. J Pediatr 2016, 172, 20–28. [Google Scholar] [CrossRef]
- McCloskey, K.; Ponsonby, A.L.; Collier, F.; Allen, K.; Tang, M.L.K.; Carlin, J.B.; Saffery, R.; Skilton, M.R.; Cheung, M.; Ranganathan, S.; et al. The association between higher maternal pre-pregnancy body mass index and increased birth weight, adiposity and inflammation in the newborn. Pediatr Obes 2018, 13, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Sureshchandra, S.; Marshall, N.E.; Messaoudi, I. Impact of pregravid obesity on maternal and fetal immunity: Fertile grounds for reprogramming. J Leukoc Biol 2019, 106, 1035–1050. [Google Scholar] [CrossRef] [PubMed]
- Esteve-Sole, A.; Luo, Y.; Vlagea, A.; Deya-Martinez, A.; Yague, J.; Plaza-Martin, A.M.; Juan, M.; Alsina, L. B Regulatory Cells: Players in Pregnancy and Early Life. Int J Mol Sci 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Rosser, E.C.; Mauri, C. Regulatory B cells: origin, phenotype, and function. Immunity 2015, 42, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Flores-Borja, F.; Bosma, A.; Ng, D.; Reddy, V.; Ehrenstein, M.R.; Isenberg, D.A.; Mauri, C. CD19+CD24hiCD38hi B cells maintain regulatory T cells while limiting TH1 and TH17 differentiation. Sci Transl Med 2013, 5, 173ra123. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.M.; Messaoudi, I. The impact of maternal obesity during pregnancy on offspring immunity. Mol Cell Endocrinol 2015, 418 Pt 2, 134–142. [Google Scholar] [CrossRef]
- Abenavoli, L.; Scarpellini, E.; Colica, C.; Boccuto, L.; Salehi, B.; Sharifi-Rad, J.; Aiello, V.; Romano, B.; De Lorenzo, A.; Izzo, A.A.; et al. Gut Microbiota and Obesity: A Role for Probiotics. Nutrients 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, H.M.; Bettini, M.L. Early-life microbiota-immune homeostasis. Front Immunol 2023, 14, 1266876. [Google Scholar] [CrossRef] [PubMed]
- Denizli, M.; Capitano, M.L.; Kua, K.L. Maternal obesity and the impact of associated early-life inflammation on long-term health of offspring. Front Cell Infect Microbiol 2022, 12, 940937. [Google Scholar] [CrossRef]
- Dumas, O.; Varraso, R.; Gillman, M.W.; Field, A.E.; Camargo, C.A., Jr. Longitudinal study of maternal body mass index, gestational weight gain, and offspring asthma. Allergy 2016, 71, 1295–1304. [Google Scholar] [CrossRef]
- Harpsoe, M.C.; Basit, S.; Bager, P.; Wohlfahrt, J.; Benn, C.S.; Nohr, E.A.; Linneberg, A.; Jess, T. Maternal obesity, gestational weight gain, and risk of asthma and atopic disease in offspring: a study within the Danish National Birth Cohort. J Allergy Clin Immunol 2013, 131, 1033–1040. [Google Scholar] [CrossRef] [PubMed]
- Lindell, N.; Carlsson, A.; Josefsson, A.; Samuelsson, U. Maternal obesity as a risk factor for early childhood type 1 diabetes: a nationwide, prospective, population-based case-control study. Diabetologia 2018, 61, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Odaka, Y.; Nakano, M.; Tanaka, T.; Kaburagi, T.; Yoshino, H.; Sato-Mito, N.; Sato, K. The influence of a high-fat dietary environment in the fetal period on postnatal metabolic and immune function. Obesity (Silver Spring) 2010, 18, 1688–1694. [Google Scholar] [CrossRef] [PubMed]
- Alfaradhi, M.Z.; Fernandez-Twinn, D.S.; Martin-Gronert, M.S.; Musial, B.; Fowden, A.; Ozanne, S.E. Oxidative stress and altered lipid homeostasis in the programming of offspring fatty liver by maternal obesity. Am J Physiol Regul Integr Comp Physiol 2014, 307, R26–34. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, J.M.; Gomez-Lopez, J.; Medina-Bravo, P.; Juarez-Sanchez, F.; Contreras-Ramos, A.; Galicia-Esquivel, M.; Sanchez-Urbina, R.; Klunder-Klunder, M. Maternal obesity increases oxidative stress in the newborn. Obesity (Silver Spring) 2015, 23, 1650–1654. [Google Scholar] [CrossRef] [PubMed]
- Brumbaugh, D.E.; Friedman, J.E. Developmental origins of nonalcoholic fatty liver disease. Pediatr Res 2014, 75, 140–147. [Google Scholar] [CrossRef] [PubMed]
- McCurdy, C.E.; Bishop, J.M.; Williams, S.M.; Grayson, B.E.; Smith, M.S.; Friedman, J.E.; Grove, K.L. Maternal high-fat diet triggers lipotoxicity in the fetal livers of nonhuman primates. J Clin Invest 2009, 119, 323–335. [Google Scholar] [CrossRef]
- Mouralidarane, A.; Soeda, J.; Visconti-Pugmire, C.; Samuelsson, A.M.; Pombo, J.; Maragkoudaki, X.; Butt, A.; Saraswati, R.; Novelli, M.; Fusai, G.; et al. Maternal obesity programs offspring nonalcoholic fatty liver disease by innate immune dysfunction in mice. Hepatology 2013, 58, 128–138. [Google Scholar] [CrossRef]
- Cohen, C.C.; Francis, E.C.; Perng, W.; Sauder, K.A.; Scherzinger, A.; Sundaram, S.S.; Shankar, K.; Dabelea, D. Exposure to maternal fuels during pregnancy and offspring hepatic fat in early childhood: The healthy start study. Pediatr Obes 2022, 17, e12902. [Google Scholar] [CrossRef]
- Luci, C.; Bourinet, M.; Leclere, P.S.; Anty, R.; Gual, P. Chronic Inflammation in Non-Alcoholic Steatohepatitis: Molecular Mechanisms and Therapeutic Strategies. Front Endocrinol (Lausanne) 2020, 11, 597648. [Google Scholar] [CrossRef]
- Platek, A.E.; Szymanska, A. Metabolic dysfunction-associated steatotic liver disease as a cardiovascular risk factor. Clin Exp Hepatol 2023, 9, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, N.; Doskey, L.C.; Malhi, H. The Role of Endoplasmic Reticulum in Lipotoxicity during Metabolic Dysfunction-Associated Steatotic Liver Disease (MASLD) Pathogenesis. Am J Pathol 2023, 193, 1887–1899. [Google Scholar] [CrossRef]
- Chan, W.K.; Chuah, K.H.; Rajaram, R.B.; Lim, L.L.; Ratnasingam, J.; Vethakkan, S.R. Metabolic Dysfunction-Associated Steatotic Liver Disease (MASLD): A State-of-the-Art Review. J Obes Metab Syndr 2023, 32, 197–213. [Google Scholar] [CrossRef] [PubMed]
- Sutti, S.; Albano, E. Adaptive immunity: an emerging player in the progression of NAFLD. Nat Rev Gastroenterol Hepatol 2020, 17, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Tacke, F. Targeting hepatic macrophages to treat liver diseases. J Hepatol 2017, 66, 1300–1312. [Google Scholar] [CrossRef]
- Friedman, S.L.; Ratziu, V.; Harrison, S.A.; Abdelmalek, M.F.; Aithal, G.P.; Caballeria, J.; Francque, S.; Farrell, G.; Kowdley, K.V.; Craxi, A.; et al. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology 2018, 67, 1754–1767. [Google Scholar] [CrossRef] [PubMed]
- Pitzalis, C.; Jones, G.W.; Bombardieri, M.; Jones, S.A. Ectopic lymphoid-like structures in infection, cancer and autoimmunity. Nat Rev Immunol 2014, 14, 447–462. [Google Scholar] [CrossRef] [PubMed]
- Sutti, S.; Jindal, A.; Locatelli, I.; Vacchiano, M.; Gigliotti, L.; Bozzola, C.; Albano, E. Adaptive immune responses triggered by oxidative stress contribute to hepatic inflammation in NASH. Hepatology 2014, 59, 886–897. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.Y.; Takahara, T.; Kawai, K.; Fujino, M.; Sugiyama, T.; Tsuneyama, K.; Tsukada, K.; Nakae, S.; Zhong, L.; Li, X.K. IFN-gamma deficiency attenuates hepatic inflammation and fibrosis in a steatohepatitis model induced by a methionine- and choline-deficient high-fat diet. Am J Physiol Gastrointest Liver Physiol 2013, 305, G891–899. [Google Scholar] [CrossRef]
- Ferreyra Solari, N.E.; Inzaugarat, M.E.; Baz, P.; De Matteo, E.; Lezama, C.; Galoppo, M.; Galoppo, C.; Chernavsky, A.C. The role of innate cells is coupled to a Th1-polarized immune response in pediatric nonalcoholic steatohepatitis. J Clin Immunol 2012, 32, 611–621. [Google Scholar] [CrossRef]
- Inzaugarat, M.E.; Ferreyra Solari, N.E.; Billordo, L.A.; Abecasis, R.; Gadano, A.C.; Chernavsky, A.C. Altered phenotype and functionality of circulating immune cells characterize adult patients with nonalcoholic steatohepatitis. J Clin Immunol 2011, 31, 1120–1130. [Google Scholar] [CrossRef] [PubMed]
- Sureshchandra, S.; Chan, C.N.; Robino, J.J.; Parmelee, L.K.; Nash, M.J.; Wesolowski, S.R.; Pietras, E.M.; Friedman, J.E.; Takahashi, D.; Shen, W.; et al. Maternal Western-style diet remodels the transcriptional landscape of fetal hematopoietic stem and progenitor cells in rhesus macaques. Stem Cell Reports 2022, 17, 2595–2609. [Google Scholar] [CrossRef]
- Iwasaki, A.; Medzhitov, R. Control of adaptive immunity by the innate immune system. Nat Immunol 2015, 16, 343–353. [Google Scholar] [CrossRef]
- Netea, M.G.; Joosten, L.A.; Latz, E.; Mills, K.H.; Natoli, G.; Stunnenberg, H.G.; O'Neill, L.A.; Xavier, R.J. Trained immunity: A program of innate immune memory in health and disease. Science 2016, 352, aaf1098. [Google Scholar] [CrossRef]
- Kucuksezer, U.C.; Ozdemir, C.; Akdis, M.; Akdis, C.A. Influence of Innate Immunity on Immune Tolerance. Acta Med Acad 2020, 49, 164–180. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.; Weis, S.; Netea, M.G.; Wetzker, R. Remembering Pathogen Dose: Long-Term Adaptation in Innate Immunity. Trends Immunol 2018, 39, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Pellicoro, A.; Ramachandran, P.; Iredale, J.P.; Fallowfield, J.A. Liver fibrosis and repair: immune regulation of wound healing in a solid organ. Nat Rev Immunol 2014, 14, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, P.; Iredale, J.P. Macrophages: central regulators of hepatic fibrogenesis and fibrosis resolution. J. Hepatol. 2012, 56, 1417–1419. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, M.A.; MacKinnon, A.C.; Ramachandran, P.; Dhaliwal, K.; Duffin, R.; Phythian-Adams, A.T.; van Rooijen, N.; Haslett, C.; Howie, S.E.; Simpson, A.J.; et al. Ly6Chi monocytes direct alternatively activated profibrotic macrophage regulation of lung fibrosis. Am J Respir Crit Care Med 2011, 184, 569–581. [Google Scholar] [CrossRef]
- Li, M.; Riddle, S.; Zhang, H.; D'Alessandro, A.; Flockton, A.; Serkova, N.J.; Hansen, K.C.; Moldvan, R.; McKeon, B.A.; Frid, M.; et al. Metabolic reprogramming regulates the proliferative and inflammatory phenotype of adventitial fibroblasts in pulmonary hypertension through the transcriptional co-repressor C-terminal binding protein-1. Circulation 2016, 134, 1105–1121. [Google Scholar] [CrossRef]
- El Kasmi, K.C.; Stenmark, K.R. Contribution of metabolic reprogramming to macrophage plasticity and function. Semin Immunol 2015, 27, 267–275. [Google Scholar] [CrossRef] [PubMed]
- El Kasmi, K.C.; Pugliese, S.C.; Riddle, S.R.; Poth, J.M.; Anderson, A.L.; Frid, M.G.; Li, M.; Pullamsetti, S.S.; Savai, R.; Nagel, M.A.; et al. Adventitial fibroblasts induce a distinct proinflammatory/profibrotic macrophage phenotype in pulmonary hypertension. J. Immunol. 2014, 193, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Sureshchandra, S.; Marshall, N.E.; Mendoza, N.; Jankeel, A.; Zulu, M.Z.; Messaoudi, I. Functional and genomic adaptations of blood monocytes to pregravid obesity during pregnancy. iScience 2021, 24, 102690. [Google Scholar] [CrossRef]
- Xu, E.; Pereira, M.M.A.; Karakasilioti, I.; Theurich, S.; Al-Maarri, M.; Rappl, G.; Waisman, A.; Wunderlich, F.T.; Bruning, J.C. Temporal and tissue-specific requirements for T-lymphocyte IL-6 signalling in obesity-associated inflammation and insulin resistance. Nat Commun 2017, 8, 14803. [Google Scholar] [CrossRef] [PubMed]
- Lim, A.I.; McFadden, T.; Link, V.M.; Han, S.J.; Karlsson, R.M.; Stacy, A.; Farley, T.K.; Lima-Junior, D.S.; Harrison, O.J.; Desai, J.V.; et al. Prenatal maternal infection promotes tissue-specific immunity and inflammation in offspring. Science 2021, 373. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.J.; Adili, A.; Piotrowitz, K.; Abdullah, Z.; Boege, Y.; Stemmer, K.; Ringelhan, M.; Simonavicius, N.; Egger, M.; Wohlleber, D.; et al. Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell 2014, 26, 549–564. [Google Scholar] [CrossRef]
- Ghazarian, M.; Revelo, X.S.; Nohr, M.K.; Luck, H.; Zeng, K.; Lei, H.; Tsai, S.; Schroer, S.A.; Park, Y.J.; Chng, M.H.Y.; et al. Type I Interferon Responses Drive Intrahepatic T cells to Promote Metabolic Syndrome. Sci Immunol 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Grohmann, M.; Wiede, F.; Dodd, G.T.; Gurzov, E.N.; Ooi, G.J.; Butt, T.; Rasmiena, A.A.; Kaur, S.; Gulati, T.; Goh, P.K.; et al. Obesity Drives STAT-1-Dependent NASH and STAT-3-Dependent HCC. Cell 2018, 175, 1289–1306 e1220. [Google Scholar] [CrossRef]
- Thapa, M.; Chinnadurai, R.; Velazquez, V.M.; Tedesco, D.; Elrod, E.; Han, J.H.; Sharma, P.; Ibegbu, C.; Gewirtz, A.; Anania, F.; et al. Liver fibrosis occurs through dysregulation of MyD88-dependent innate B-cell activity. Hepatology 2015, 61, 2067–2079. [Google Scholar] [CrossRef]
- Tsiantoulas, D.; Sage, A.P.; Mallat, Z.; Binder, C.J. Targeting B cells in atherosclerosis: closing the gap from bench to bedside. Arterioscler Thromb Vasc Biol 2015, 35, 296–302. [Google Scholar] [CrossRef]
- Magalhaes, M.S.; Potter, H.G.; Ahlback, A.; Gentek, R. Developmental programming of macrophages by early life adversity. Int Rev Cell Mol Biol 2022, 368, 213–259. [Google Scholar] [CrossRef] [PubMed]
- Saijo, K.; Glass, C.K. Microglial cell origin and phenotypes in health and disease. Nat Rev Immunol 2011, 11, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Rivera, H.M.; Christiansen, K.J.; Sullivan, E.L. The role of maternal obesity in the risk of neuropsychiatric disorders. Front Neurosci 2015, 9, 194. [Google Scholar] [CrossRef] [PubMed]
- Mattei, D.; Ivanov, A.; Ferrai, C.; Jordan, P.; Guneykaya, D.; Buonfiglioli, A.; Schaafsma, W.; Przanowski, P.; Deuther-Conrad, W.; Brust, P.; et al. Maternal immune activation results in complex microglial transcriptome signature in the adult offspring that is reversed by minocycline treatment. Transl Psychiatry 2017, 7, e1120. [Google Scholar] [CrossRef]
- Solmi, M.; Veronese, N.; Thapa, N.; Facchini, S.; Stubbs, B.; Fornaro, M.; Carvalho, A.F.; Correll, C.U. Systematic review and meta-analysis of the efficacy and safety of minocycline in schizophrenia. CNS Spectr 2017, 22, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.A.; Mitchell, A.J.; Selby, M.; Fair, D.A.; Gustafsson, H.C.; Sullivan, E.L. Maternal diet and obesity shape offspring central and peripheral inflammatory outcomes in juvenile non-human primates. Brain Behav Immun 2022, 102, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, A.J.; Khambadkone, S.G.; Dunn, G.; Bagley, J.; Tamashiro, K.L.K.; Fair, D.; Gustafsson, H.; Sullivan, E.L. Maternal Western-style diet reduces social engagement and increases idiosyncratic behavior in Japanese macaque offspring. Brain Behav Immun 2022, 105, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Papadakis, S.; Thompson, J.R.; Feczko, E.; Miranda-Dominguez, O.; Dunn, G.A.; Selby, M.; Mitchell, A.J.; Sullivan, E.L.; Fair, D.A. Perinatal Western-style diet exposure associated with decreased microglial counts throughout the arcuate nucleus of the hypothalamus in Japanese macaques. J Neurophysiol 2024, 131, 241–260. [Google Scholar] [CrossRef] [PubMed]
- Subhramanyam, C.S.; Wang, C.; Hu, Q.; Dheen, S.T. Microglia-mediated neuroinflammation in neurodegenerative diseases. Semin Cell Dev Biol 2019, 94, 112–120. [Google Scholar] [CrossRef]
- Guo, D.H.; Yamamoto, M.; Hernandez, C.M.; Khodadadi, H.; Baban, B.; Stranahan, A.M. Visceral adipose NLRP3 impairs cognition in obesity via IL-1R1 on CX3CR1+ cells. J Clin Invest 2020, 130, 1961–1976. [Google Scholar] [CrossRef]
- Smith, S.E.; Li, J.; Garbett, K.; Mirnics, K.; Patterson, P.H. Maternal immune activation alters fetal brain development through interleukin-6. J Neurosci 2007, 27, 10695–10702. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Tome, S.; Takei, Y. Intraventricular IL-17A administration activates microglia and alters their localization in the mouse embryo cerebral cortex. Mol Brain 2020, 13, 93. [Google Scholar] [CrossRef] [PubMed]
- Aron-Wisnewsky, J.; Vigliotti, C.; Witjes, J.; Le, P.; Holleboom, A.G.; Verheij, J.; Nieuwdorp, M.; Clement, K. Gut microbiota and human NAFLD: disentangling microbial signatures from metabolic disorders. Nat Rev Gastroenterol Hepatol 2020, 17, 279–297. [Google Scholar] [CrossRef] [PubMed]
- Conway, J.; N, A.D. Ageing of the gut microbiome: Potential influences on immune senescence and inflammageing. Ageing Res Rev 2021, 68, 101323. [Google Scholar] [CrossRef]
- Nichols, R.G.; Davenport, E.R. The relationship between the gut microbiome and host gene expression: a review. Hum Genet 2021, 140, 747–760. [Google Scholar] [CrossRef]
- Cox, T.O.; Lundgren, P.; Nath, K.; Thaiss, C.A. Metabolic control by the microbiome. Genome Med 2022, 14, 80. [Google Scholar] [CrossRef] [PubMed]
- Human Microbiome Project, C. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ley, R.E.; Hamady, M.; Fraser-Liggett, C.M.; Knight, R.; Gordon, J.I. The human microbiome project. Nature 2007, 449, 804–810. [Google Scholar] [CrossRef]
- Ley, R.E.; Backhed, F.; Turnbaugh, P.; Lozupone, C.A.; Knight, R.D.; Gordon, J.I. Obesity alters gut microbial ecology. Proc Natl Acad Sci U S A 2005, 102, 11070–11075. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef]
- Wilson, A.S.; Koller, K.R.; Ramaboli, M.C.; Nesengani, L.T.; Ocvirk, S.; Chen, C.; Flanagan, C.A.; Sapp, F.R.; Merritt, Z.T.; Bhatti, F.; et al. Diet and the Human Gut Microbiome: An International Review. Dig Dis Sci 2020, 65, 723–740. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.K.; Chang, H.W.; Yan, D.; Lee, K.M.; Ucmak, D.; Wong, K.; Abrouk, M.; Farahnik, B.; Nakamura, M.; Zhu, T.H.; et al. Influence of diet on the gut microbiome and implications for human health. J Transl Med 2017, 15, 73. [Google Scholar] [CrossRef]
- Koren, O.; Konnikova, L.; Brodin, P.; Mysorekar, I.U.; Collado, M.C. The maternal gut microbiome in pregnancy: implications for the developing immune system. Nat Rev Gastroenterol Hepatol 2024, 21, 35–45. [Google Scholar] [CrossRef]
- Jennison, E.; Byrne, C.D. The role of the gut microbiome and diet in the pathogenesis of non-alcoholic fatty liver disease. Clin Mol Hepatol 2021, 27, 22–43. [Google Scholar] [CrossRef] [PubMed]
- Kuang, Y.S.; Lu, J.H.; Li, S.H.; Li, J.H.; Yuan, M.Y.; He, J.R.; Chen, N.N.; Xiao, W.Q.; Shen, S.Y.; Qiu, L.; et al. Connections between the human gut microbiome and gestational diabetes mellitus. Gigascience 2017, 6, 1–12. [Google Scholar] [CrossRef]
- Sugino, K.Y.; Hernandez, T.L.; Barbour, L.A.; Kofonow, J.M.; Frank, D.N.; Friedman, J.E. A maternal higher-complex carbohydrate diet increases bifidobacteria and alters early life acquisition of the infant microbiome in women with gestational diabetes mellitus. Front Endocrinol (Lausanne) 2022, 13, 921464. [Google Scholar] [CrossRef] [PubMed]
- Miko, E.; Csaszar, A.; Bodis, J.; Kovacs, K. The Maternal-Fetal Gut Microbiota Axis: Physiological Changes, Dietary Influence, and Modulation Possibilities. Life (Basel) 2022, 12. [Google Scholar] [CrossRef] [PubMed]
- Yassour, M.; Vatanen, T.; Siljander, H.; Hamalainen, A.M.; Harkonen, T.; Ryhanen, S.J.; Franzosa, E.A.; Vlamakis, H.; Huttenhower, C.; Gevers, D.; et al. Natural history of the infant gut microbiome and impact of antibiotic treatment on bacterial strain diversity and stability. Sci Transl Med 2016, 8, 343ra381. [Google Scholar] [CrossRef]
- Wang, S.; Ryan, C.A.; Boyaval, P.; Dempsey, E.M.; Ross, R.P.; Stanton, C. Maternal Vertical Transmission Affecting Early-life Microbiota Development. Trends Microbiol 2020, 28, 28–45. [Google Scholar] [CrossRef]
- Li, W.; Tapiainen, T.; Brinkac, L.; Lorenzi, H.A.; Moncera, K.; Tejesvi, M.V.; Salo, J.; Nelson, K.E. Vertical Transmission of Gut Microbiome and Antimicrobial Resistance Genes in Infants Exposed to Antibiotics at Birth. J Infect Dis 2021, 224, 1236–1246. [Google Scholar] [CrossRef] [PubMed]
- Guarner, F.; Bourdet-Sicard, R.; Brandtzaeg, P.; Gill, H.S.; McGuirk, P.; van Eden, W.; Versalovic, J.; Weinstock, J.V.; Rook, G.A. Mechanisms of disease: the hygiene hypothesis revisited. Nat Clin Pract Gastroenterol Hepatol 2006, 3, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Soderborg, T.K.; Clark, S.E.; Mulligan, C.E.; Janssen, R.C.; Babcock, L.; Ir, D.; Young, B.; Krebs, N.; Lemas, D.J.; Johnson, L.K.; et al. The gut microbiota in infants of obese mothers increases inflammation and susceptibility to NAFLD. Nat Commun 2018, 9, 4462. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.Y.; Medzhitov, R. On developmental programming of the immune system. Trends Immunol 2023, 44, 877–889. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Lin, Y.; Zhang, H.; Wang, G.; Zhao, J.; Zhang, H.; Chen, W. Intestinal 'Infant-Type' Bifidobacteria Mediate Immune System Development in the First 1000 Days of Life. Nutrients 2022, 14. [Google Scholar] [CrossRef] [PubMed]
- Turroni, F.; Peano, C.; Pass, D.A.; Foroni, E.; Severgnini, M.; Claesson, M.J.; Kerr, C.; Hourihane, J.; Murray, D.; Fuligni, F.; et al. Diversity of bifidobacteria within the infant gut microbiota. PLoS One 2012, 7, e36957. [Google Scholar] [CrossRef] [PubMed]
- Turroni, F.; van Sinderen, D.; Ventura, M. Genomics and ecological overview of the genus Bifidobacterium. Int J Food Microbiol 2011, 149, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Asakuma, S.; Hatakeyama, E.; Urashima, T.; Yoshida, E.; Katayama, T.; Yamamoto, K.; Kumagai, H.; Ashida, H.; Hirose, J.; Kitaoka, M. Physiology of consumption of human milk oligosaccharides by infant gut-associated bifidobacteria. J Biol Chem 2011, 286, 34583–34592. [Google Scholar] [CrossRef] [PubMed]
- Russell, J.T.; Roesch, L.F.W.; Ordberg, M.; Ilonen, J.; Atkinson, M.A.; Schatz, D.A.; Triplett, E.W.; Ludvigsson, J. Genetic risk for autoimmunity is associated with distinct changes in the human gut microbiome. Nat Commun 2019, 10, 3621. [Google Scholar] [CrossRef]
- Vatanen, T.; Kostic, A.D.; d'Hennezel, E.; Siljander, H.; Franzosa, E.A.; Yassour, M.; Kolde, R.; Vlamakis, H.; Arthur, T.D.; Hamalainen, A.M.; et al. Variation in Microbiome LPS Immunogenicity Contributes to Autoimmunity in Humans. Cell 2016, 165, 842–853. [Google Scholar] [CrossRef]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Microbial ecology: human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef] [PubMed]
- Weaver, L.K.; Minichino, D.; Biswas, C.; Chu, N.; Lee, J.J.; Bittinger, K.; Albeituni, S.; Nichols, K.E.; Behrens, E.M. Microbiota-dependent signals are required to sustain TLR-mediated immune responses. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Soderborg, T.K.; Carpenter, C.M.; Janssen, R.C.; Weir, T.L.; Robertson, C.E.; Ir, D.; Young, B.E.; Krebs, N.F.; Hernandez, T.L.; Barbour, L.A.; et al. Gestational Diabetes Is Uniquely Associated With Altered Early Seeding of the Infant Gut Microbiota. Front Endocrinol (Lausanne) 2020, 11, 603021. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Qin, Y.; Chen, M.; Zhang, Y.; Wang, X.; Dong, T.; Chen, G.; Sun, X.; Lu, T.; White, R.A., 3rd; et al. Gestational diabetes mellitus is associated with the neonatal gut microbiota and metabolome. BMC Med 2021, 19, 120. [Google Scholar] [CrossRef] [PubMed]
- Pinto, Y.; Frishman, S.; Turjeman, S.; Eshel, A.; Nuriel-Ohayon, M.; Shrossel, O.; Ziv, O.; Walters, W.; Parsonnet, J.; Ley, C.; et al. Gestational diabetes is driven by microbiota-induced inflammation months before diagnosis. Gut 2023, 72, 918–928. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.M.; Reinke, S.N.; Mousavi-Derazmahalleh, M.; Garssen, J.; Jenmalm, M.C.; Srinivasjois, R.; Silva, D.; Keelan, J.; Prescott, S.L.; Palmer, D.J.; et al. Maternal prebiotic supplementation during pregnancy and lactation modifies the microbiome and short chain fatty acid profile of both mother and infant. Clin Nutr 2024, 43, 969–980. [Google Scholar] [CrossRef] [PubMed]
- Barman, P.K.; Goodridge, H.S. Microbial Sensing by Hematopoietic Stem and Progenitor Cells. Stem Cells 2022, 40, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Paucar Iza, Y.A.; Brown, C.C. Early life imprinting of intestinal immune tolerance and tissue homeostasis. Immunol Rev 2024. [Google Scholar] [CrossRef] [PubMed]
- Kohler, A.; Delbauve, S.; Smout, J.; Torres, D.; Flamand, V. Very early-life exposure to microbiota-induced TNF drives the maturation of neonatal pre-cDC1. Gut 2021, 70, 511–521. [Google Scholar] [CrossRef]
- Constantinides, M.G.; Belkaid, Y. Early-life imprinting of unconventional T cells and tissue homeostasis. Science 2021, 374, eabf0095. [Google Scholar] [CrossRef]
- Sefik, E.; Geva-Zatorsky, N.; Oh, S.; Konnikova, L.; Zemmour, D.; McGuire, A.M.; Burzyn, D.; Ortiz-Lopez, A.; Lobera, M.; Yang, J.; et al. MUCOSAL IMMUNOLOGY. Individual intestinal symbionts induce a distinct population of RORgamma(+) regulatory T cells. Science 2015, 349, 993–997. [Google Scholar] [CrossRef] [PubMed]
- Eshleman, E.M.; Shao, T.Y.; Woo, V.; Rice, T.; Engleman, L.; Didriksen, B.J.; Whitt, J.; Haslam, D.B.; Way, S.S.; Alenghat, T. Intestinal epithelial HDAC3 and MHC class II coordinate microbiota-specific immunity. J Clin Invest 2023, 133. [Google Scholar] [CrossRef] [PubMed]
- Al Nabhani, Z.; Dulauroy, S.; Marques, R.; Cousu, C.; Al Bounny, S.; Dejardin, F.; Sparwasser, T.; Berard, M.; Cerf-Bensussan, N.; Eberl, G. A Weaning Reaction to Microbiota Is Required for Resistance to Immunopathologies in the Adult. Immunity 2019, 50, 1276–1288 e1275. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Lai, G.C.; Yao, L.J.; Aung, T.T.; Shental, N.; Rotter-Maskowitz, A.; Shepherdson, E.; Singh, G.S.N.; Pai, R.; Shanti, A.; et al. Microbial exposure during early human development primes fetal immune cells. Cell 2021, 184, 3394–3409 e3320. [Google Scholar] [CrossRef]
- Hu, M.; Eviston, D.; Hsu, P.; Marino, E.; Chidgey, A.; Santner-Nanan, B.; Wong, K.; Richards, J.L.; Yap, Y.A.; Collier, F.; et al. Decreased maternal serum acetate and impaired fetal thymic and regulatory T cell development in preeclampsia. Nat Commun 2019, 10, 3031. [Google Scholar] [CrossRef] [PubMed]
- Sanidad, K.Z.; Rager, S.L.; Carrow, H.C.; Ananthanarayanan, A.; Callaghan, R.; Hart, L.R.; Li, T.; Ravisankar, P.; Brown, J.A.; Amir, M.; et al. Gut bacteria-derived serotonin promotes immune tolerance in early life. Sci Immunol 2024, 9, eadj4775. [Google Scholar] [CrossRef] [PubMed]
- Khosravi, A.; Yanez, A.; Price, J.G.; Chow, A.; Merad, M.; Goodridge, H.S.; Mazmanian, S.K. Gut microbiota promote hematopoiesis to control bacterial infection. Cell Host Microbe 2014, 15, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Josefsdottir, K.S.; Baldridge, M.T.; Kadmon, C.S.; King, K.Y. Antibiotics impair murine hematopoiesis by depleting the intestinal microbiota. Blood 2017, 129, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Staffas, A.; Burgos da Silva, M.; Slingerland, A.E.; Lazrak, A.; Bare, C.J.; Holman, C.D.; Docampo, M.D.; Shono, Y.; Durham, B.; Pickard, A.J.; et al. Nutritional Support from the Intestinal Microbiota Improves Hematopoietic Reconstitution after Bone Marrow Transplantation in Mice. Cell Host Microbe 2018, 23, 447–457 e444. [Google Scholar] [CrossRef]
- Trompette, A.; Gollwitzer, E.S.; Yadava, K.; Sichelstiel, A.K.; Sprenger, N.; Ngom-Bru, C.; Blanchard, C.; Junt, T.; Nicod, L.P.; Harris, N.L.; et al. Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat Med 2014, 20, 159–166. [Google Scholar] [CrossRef]
- Clarke, T.B.; Davis, K.M.; Lysenko, E.S.; Zhou, A.Y.; Yu, Y.; Weiser, J.N. Recognition of peptidoglycan from the microbiota by Nod1 enhances systemic innate immunity. Nat Med 2010, 16, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Yanez, A.; Coetzee, S.G.; Olsson, A.; Muench, D.E.; Berman, B.P.; Hazelett, D.J.; Salomonis, N.; Grimes, H.L.; Goodridge, H.S. Granulocyte-Monocyte Progenitors and Monocyte-Dendritic Cell Progenitors Independently Produce Functionally Distinct Monocytes. Immunity 2017, 47, 890–902 e894. [Google Scholar] [CrossRef] [PubMed]
- Menezes, S.; Melandri, D.; Anselmi, G.; Perchet, T.; Loschko, J.; Dubrot, J.; Patel, R.; Gautier, E.L.; Hugues, S.; Longhi, M.P.; et al. The Heterogeneity of Ly6C(hi) Monocytes Controls Their Differentiation into iNOS(+) Macrophages or Monocyte-Derived Dendritic Cells. Immunity 2016, 45, 1205–1218. [Google Scholar] [CrossRef] [PubMed]
- Villani, A.C.; Satija, R.; Reynolds, G.; Sarkizova, S.; Shekhar, K.; Fletcher, J.; Griesbeck, M.; Butler, A.; Zheng, S.; Lazo, S.; et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 2017, 356. [Google Scholar] [CrossRef] [PubMed]
- Jonscher, K.R.; Abrams, J.; Friedman, J.E. Maternal Diet Alters Trained Immunity in the Pathogenesis of Pediatric NAFLD. J Cell Immunol 2020, 2, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Chen, G.L.; Hannemann, N.; Ipseiz, N.; Kronke, G.; Bauerle, T.; Munos, L.; Wirtz, S.; Schett, G.; Bozec, A. Microbiota from Obese Mice Regulate Hematopoietic Stem Cell Differentiation by Altering the Bone Niche. Cell Metab 2015, 22, 886–894. [Google Scholar] [CrossRef] [PubMed]
- Bermick, J.; Schaller, M. Chorioamnionitis exposure dampens the preterm monocyte response to subsequent challenges. Immunol Cell Biol 2018, 96, 789–791. [Google Scholar] [CrossRef] [PubMed]
- de Jong, E.; Hancock, D.G.; Wells, C.; Richmond, P.; Simmer, K.; Burgner, D.; Strunk, T.; Currie, A.J. Exposure to chorioamnionitis alters the monocyte transcriptional response to the neonatal pathogen Staphylococcus epidermidis. Immunol Cell Biol 2018, 96, 792–804. [Google Scholar] [CrossRef] [PubMed]
- Rios-Covian, D.; Ruas-Madiedo, P.; Margolles, A.; Gueimonde, M.; de Los Reyes-Gavilan, C.G.; Salazar, N. Intestinal Short Chain Fatty Acids and their Link with Diet and Human Health. Front Microbiol 2016, 7, 185. [Google Scholar] [CrossRef]
- Koh, A.; De Vadder, F.; Kovatcheva-Datchary, P.; Backhed, F. From Dietary Fiber to Host Physiology: Short-Chain Fatty Acids as Key Bacterial Metabolites. Cell 2016, 165, 1332–1345. [Google Scholar] [CrossRef]
- Hsu, C.N.; Tain, Y.L. Developmental Programming and Reprogramming of Hypertension and Kidney Disease: Impact of Tryptophan Metabolism. Int J Mol Sci 2020, 21. [Google Scholar] [CrossRef]
- Patil, N.Y.; Friedman, J.E.; Joshi, A.D. Role of Hepatic Aryl Hydrocarbon Receptor in Non-Alcoholic Fatty Liver Disease. Receptors (Basel) 2023, 2, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.H.; Pomare, E.W.; Branch, W.J.; Naylor, C.P.; Macfarlane, G.T. Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut 1987, 28, 1221–1227. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, R.W. Histone-deacetylase inhibitors: novel drugs for the treatment of cancer. Nat Rev Drug Discov 2002, 1, 287–299. [Google Scholar] [CrossRef]
- Chang, P.V.; Hao, L.; Offermanns, S.; Medzhitov, R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 2247–2252. [Google Scholar] [CrossRef]
- Flint, H.J.; Scott, K.P.; Louis, P.; Duncan, S.H. The role of the gut microbiota in nutrition and health. Nat Rev Gastroenterol Hepatol 2012, 9, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Thangaraju, M.; Prasad, P.D.; Martin, P.M.; Lambert, N.A.; Boettger, T.; Offermanns, S.; Ganapathy, V. Blockade of dendritic cell development by bacterial fermentation products butyrate and propionate through a transporter (Slc5a8)-dependent inhibition of histone deacetylases. J Biol Chem 2010, 285, 27601–27608. [Google Scholar] [CrossRef] [PubMed]
- Beisner, J.; Filipe Rosa, L.; Kaden-Volynets, V.; Stolzer, I.; Gunther, C.; Bischoff, S.C. Prebiotic Inulin and Sodium Butyrate Attenuate Obesity-Induced Intestinal Barrier Dysfunction by Induction of Antimicrobial Peptides. Front Immunol 2021, 12, 678360. [Google Scholar] [CrossRef]
- Fang, W.; Xue, H.; Chen, X.; Chen, K.; Ling, W. Supplementation with Sodium Butyrate Modulates the Composition of the Gut Microbiota and Ameliorates High-Fat Diet-Induced Obesity in Mice. J Nutr 2019, 149, 747–754. [Google Scholar] [CrossRef]
- Zhou, D.; Pan, Q.; Xin, F.Z.; Zhang, R.N.; He, C.X.; Chen, G.Y.; Liu, C.; Chen, Y.W.; Fan, J.G. Sodium butyrate attenuates high-fat diet-induced steatohepatitis in mice by improving gut microbiota and gastrointestinal barrier. World J Gastroenterol 2017, 23, 60–75. [Google Scholar] [CrossRef]
- Kimura, I.; Ozawa, K.; Inoue, D.; Imamura, T.; Kimura, K.; Maeda, T.; Terasawa, K.; Kashihara, D.; Hirano, K.; Tani, T.; et al. The gut microbiota suppresses insulin-mediated fat accumulation via the short-chain fatty acid receptor GPR43. Nat Commun 2013, 4, 1829. [Google Scholar] [CrossRef] [PubMed]
- Le Poul, E.; Loison, C.; Struyf, S.; Springael, J.Y.; Lannoy, V.; Decobecq, M.E.; Brezillon, S.; Dupriez, V.; Vassart, G.; Van Damme, J.; et al. Functional characterization of human receptors for short chain fatty acids and their role in polymorphonuclear cell activation. J Biol Chem 2003, 278, 25481–25489. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Wang, Y.Y.; Li, S.P.; Zhao, H.M.; Jiang, F.J.; Wu, Y.X.; Tong, Y.; Pang, Q.F. Maternal propionate supplementation ameliorates glucose and lipid metabolic disturbance in hypoxia-induced fetal growth restriction. Food Funct 2022, 13, 10724–10736. [Google Scholar] [CrossRef] [PubMed]
- Badawy, A.A. Tryptophan metabolism, disposition and utilization in pregnancy. Biosci Rep 2015, 35. [Google Scholar] [CrossRef] [PubMed]
- Cussotto, S.; Delgado, I.; Anesi, A.; Dexpert, S.; Aubert, A.; Beau, C.; Forestier, D.; Ledaguenel, P.; Magne, E.; Mattivi, F.; et al. Tryptophan Metabolic Pathways Are Altered in Obesity and Are Associated With Systemic Inflammation. Front Immunol 2020, 11, 557. [Google Scholar] [CrossRef] [PubMed]
- Lischka, J.; Schanzer, A.; Baumgartner, M.; de Gier, C.; Greber-Platzer, S.; Zeyda, M. Tryptophan Metabolism Is Associated with BMI and Adipose Tissue Mass and Linked to Metabolic Disease in Pediatric Obesity. Nutrients 2022, 14. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, S.; Ding, Y.; Saedi, N.; Choi, M.; Sridharan, G.V.; Sherr, D.H.; Yarmush, M.L.; Alaniz, R.C.; Jayaraman, A.; Lee, K. Gut Microbiota-Derived Tryptophan Metabolites Modulate Inflammatory Response in Hepatocytes and Macrophages. Cell Rep 2018, 23, 1099–1111. [Google Scholar] [CrossRef] [PubMed]
- Scott, S.A.; Fu, J.; Chang, P.V. Microbial tryptophan metabolites regulate gut barrier function via the aryl hydrocarbon receptor. Proc. Natl. Acad. Sci. U. S. A. 2020, 117, 19376–19387. [Google Scholar] [CrossRef]
- Choi, Y.; Kim, Y.; Park, S.; Lee, K.W.; Park, T. Indole-3-carbinol prevents diet-induced obesity through modulation of multiple genes related to adipogenesis, thermogenesis or inflammation in the visceral adipose tissue of mice. J Nutr Biochem 2012, 23, 1732–1739. [Google Scholar] [CrossRef]
- Shinde, R.; McGaha, T.L. The Aryl Hydrocarbon Receptor: Connecting Immunity to the Microenvironment. Trends Immunol 2018, 39, 1005–1020. [Google Scholar] [CrossRef]
- Bock, K.W. Aryl hydrocarbon receptor (AHR), integrating energy metabolism and microbial or obesity-mediated inflammation. Biochem Pharmacol 2021, 184, 114346. [Google Scholar] [CrossRef] [PubMed]
- Ghiboub, M.; Verburgt, C.M.; Sovran, B.; Benninga, M.A.; de Jonge, W.J.; Van Limbergen, J.E. Nutritional Therapy to Modulate Tryptophan Metabolism and Aryl Hydrocarbon-Receptor Signaling Activation in Human Diseases. Nutrients 2020, 12. [Google Scholar] [CrossRef]
- den Besten, G.; van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.J.; Bakker, B.M. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J Lipid Res 2013, 54, 2325–2340. [Google Scholar] [CrossRef]
- Frost, G.; Sleeth, M.L.; Sahuri-Arisoylu, M.; Lizarbe, B.; Cerdan, S.; Brody, L.; Anastasovska, J.; Ghourab, S.; Hankir, M.; Zhang, S.; et al. The short-chain fatty acid acetate reduces appetite via a central homeostatic mechanism. Nat Commun 2014, 5, 3611. [Google Scholar] [CrossRef]
- Laursen, M.F.; Sakanaka, M.; von Burg, N.; Morbe, U.; Andersen, D.; Moll, J.M.; Pekmez, C.T.; Rivollier, A.; Michaelsen, K.F.; Molgaard, C.; et al. Bifidobacterium species associated with breastfeeding produce aromatic lactic acids in the infant gut. Nat Microbiol 2021, 6, 1367–1382. [Google Scholar] [CrossRef]
- Wang, J.P.; Yoo, J.S.; Lee, J.H.; Jang, H.D.; Kim, H.J.; Shin, S.O.; Seong, S.I.; Kim, I.H. Effects of phenyllactic acid on growth performance, nutrient digestibility, microbial shedding, and blood profile in pigs. J Anim Sci 2009, 87, 3235–3243. [Google Scholar] [CrossRef]
- Zugasti, O.; Bose, N.; Squiban, B.; Belougne, J.; Kurz, C.L.; Schroeder, F.C.; Pujol, N.; Ewbank, J.J. Activation of a G protein-coupled receptor by its endogenous ligand triggers the innate immune response of Caenorhabditis elegans. Nat Immunol 2014, 15, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Waldrop, S.W.; Niemiec, S.; Wood, C.; Gyllenhammer, L.E.; Jansson, T.; Friedman, J.E.; Tryggestad, J.B.; Borengasser, S.J.; Davidson, E.J.; Yang, I.V.; et al. Cord blood DNA methylation of immune and lipid metabolism genes is associated with maternal triglycerides and child adiposity. Obesity (Silver Spring) 2024, 32, 187–199. [Google Scholar] [CrossRef]
- van der Heijden, C.; Noz, M.P.; Joosten, L.A.B.; Netea, M.G.; Riksen, N.P.; Keating, S.T. Epigenetics and Trained Immunity. Antioxid Redox Signal 2018, 29, 1023–1040. [Google Scholar] [CrossRef] [PubMed]
- Fanucchi, S.; Dominguez-Andres, J.; Joosten, L.A.B.; Netea, M.G.; Mhlanga, M.M. The Intersection of Epigenetics and Metabolism in Trained Immunity. Immunity 2021, 54, 32–43. [Google Scholar] [CrossRef]
- Suter, M.; Bocock, P.; Showalter, L.; Hu, M.; Shope, C.; McKnight, R.; Grove, K.; Lane, R.; Aagaard-Tillery, K. Epigenomics: maternal high-fat diet exposure in utero disrupts peripheral circadian gene expression in nonhuman primates. FASEB J 2011, 25, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Suter, M.A.; Takahashi, D.; Grove, K.L.; Aagaard, K.M. Postweaning exposure to a high-fat diet is associated with alterations to the hepatic histone code in Japanese macaques. Pediatr Res 2013, 74, 252–258. [Google Scholar] [CrossRef]
- Harmancioglu, B.; Kabaran, S. Maternal high fat diets: impacts on offspring obesity and epigenetic hypothalamic programming. Front Genet 2023, 14, 1158089. [Google Scholar] [CrossRef] [PubMed]
- Borengasser, S.J.; Zhong, Y.; Kang, P.; Lindsey, F.; Ronis, M.J.; Badger, T.M.; Gomez-Acevedo, H.; Shankar, K. Maternal obesity enhances white adipose tissue differentiation and alters genome-scale DNA methylation in male rat offspring. Endocrinology 2013, 154, 4113–4125. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, C.M.; Segovia, S.A.; Vickers, M.H. Experimental Models of Maternal Obesity and Neuroendocrine Programming of Metabolic Disorders in Offspring. Front Endocrinol (Lausanne) 2017, 8, 245. [Google Scholar] [CrossRef] [PubMed]
- Ross, M.G.; Desai, M. Developmental programming of appetite/satiety. Ann Nutr Metab 2014, 64 Suppl 1, 36–44. [Google Scholar] [CrossRef]
- Chen, H.; Simar, D.; Morris, M.J. Hypothalamic neuroendocrine circuitry is programmed by maternal obesity: interaction with postnatal nutritional environment. PLoS One 2009, 4, e6259. [Google Scholar] [CrossRef] [PubMed]
- Vickers, M.H.; Breier, B.H.; Cutfield, W.S.; Hofman, P.L.; Gluckman, P.D. Fetal origins of hyperphagia, obesity, and hypertension and postnatal amplification by hypercaloric nutrition. Am J Physiol Endocrinol Metab 2000, 279, E83–87. [Google Scholar] [CrossRef] [PubMed]
- Desai, M.; Han, G.; Ross, M.G. Programmed hyperphagia in offspring of obese dams: Altered expression of hypothalamic nutrient sensors, neurogenic factors and epigenetic modulators. Appetite 2016, 99, 193–199. [Google Scholar] [CrossRef]
- Sardinha, F.L.; Telles, M.M.; Albuquerque, K.T.; Oyama, L.M.; Guimaraes, P.A.; Santos, O.F.; Ribeiro, E.B. Gender difference in the effect of intrauterine malnutrition on the central anorexigenic action of insulin in adult rats. Nutrition 2006, 22, 1152–1161. [Google Scholar] [CrossRef]
- Galjaard, S.; Devlieger, R.; Van Assche, F.A. Fetal growth and developmental programming. J Perinat Med 2013, 41, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Gali Ramamoorthy, T.; Allen, T.J.; Davies, A.; Harno, E.; Sefton, C.; Murgatroyd, C.; White, A. Maternal overnutrition programs epigenetic changes in the regulatory regions of hypothalamic Pomc in the offspring of rats. Int J Obes (Lond) 2018, 42, 1431–1444. [Google Scholar] [CrossRef] [PubMed]
- Plagemann, A.; Harder, T.; Brunn, M.; Harder, A.; Roepke, K.; Wittrock-Staar, M.; Ziska, T.; Schellong, K.; Rodekamp, E.; Melchior, K.; et al. Hypothalamic proopiomelanocortin promoter methylation becomes altered by early overfeeding: an epigenetic model of obesity and the metabolic syndrome. J Physiol 2009, 587, 4963–4976. [Google Scholar] [CrossRef]
- Suter, M.A.; Chen, A.; Burdine, M.S.; Choudhury, M.; Harris, R.A.; Lane, R.H.; Friedman, J.E.; Grove, K.L.; Tackett, A.J.; Aagaard, K.M. A maternal high-fat diet modulates fetal SIRT1 histone and protein deacetylase activity in nonhuman primates. FASEB J 2012, 26, 5106–5114. [Google Scholar] [CrossRef] [PubMed]
- Funato, H.; Oda, S.; Yokofujita, J.; Igarashi, H.; Kuroda, M. Fasting and high-fat diet alter histone deacetylase expression in the medial hypothalamus. PLoS One 2011, 6, e18950. [Google Scholar] [CrossRef] [PubMed]
- Dalvi, P.S.; Chalmers, J.A.; Luo, V.; Han, D.Y.; Wellhauser, L.; Liu, Y.; Tran, D.Q.; Castel, J.; Luquet, S.; Wheeler, M.B.; et al. High fat induces acute and chronic inflammation in the hypothalamus: effect of high-fat diet, palmitate and TNF-alpha on appetite-regulating NPY neurons. Int J Obes (Lond) 2017, 41, 149–158. [Google Scholar] [CrossRef]
- Grayson, B.E.; Levasseur, P.R.; Williams, S.M.; Smith, M.S.; Marks, D.L.; Grove, K.L. Changes in melanocortin expression and inflammatory pathways in fetal offspring of nonhuman primates fed a high-fat diet. Endocrinology 2010, 151, 1622–1632. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Chen, W.; Jin, H.; Chen, X. A Review of Single-Cell RNA-Seq Annotation, Integration, and Cell-Cell Communication. Cells 2023, 12. [Google Scholar] [CrossRef]
- Chen, B.; de Launoit, E.; Renier, N.; Schneeberger, M. Maternal nutritional programming shapes the cerebral landscape. Trends Endocrinol Metab 2023. [Google Scholar] [CrossRef]
- Wu, R.; Prachyathipsakul, T.; Zhuang, J.; Liu, H.; Han, Y.; Liu, B.; Gong, S.; Qiu, J.; Wong, S.; Ribbe, A.; et al. Conferring liver selectivity to a thyromimetic using a novel nanoparticle increases therapeutic efficacy in a diet-induced obesity animal model. PNAS Nexus 2023, 2, pgad252. [Google Scholar] [CrossRef]
- Ayyangar, U.; Karkhanis, A.; Tay, H.; Afandi, A.F.B.; Bhattacharjee, O.; Ks, L.; Lee, S.H.; Chan, J.; Raghavan, S. Metabolic rewiring of macrophages by epidermal-derived lactate promotes sterile inflammation in the murine skin. EMBO J 2024, 43, 1113–1134. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Simplified overview of fetal immune cell development. Immune cells develop in three overlapping waves: firstly, primitive macrophages emerge from the yolk sac, secondly, erythro-myeloid progenitors (EMP) form in the fetal liver. During the second and third trimesters, fully functioning hematopoietic stem and progenitor cells (HSPC) seed the fetal liver and migrate from the liver to the fetal bone marrow. The relative contribution of each wave is plotted against fetal gestational age in the number of weeks postconception (wpc) until birth. T and B cells require further development through primary and secondary lymphoid organs starting from the common lymphoid progenitor (CLP) in the bone marrow (simplified here). Circulating leukocytes are replenished from this central reservoir in the bone marrow.
Figure 1.
Simplified overview of fetal immune cell development. Immune cells develop in three overlapping waves: firstly, primitive macrophages emerge from the yolk sac, secondly, erythro-myeloid progenitors (EMP) form in the fetal liver. During the second and third trimesters, fully functioning hematopoietic stem and progenitor cells (HSPC) seed the fetal liver and migrate from the liver to the fetal bone marrow. The relative contribution of each wave is plotted against fetal gestational age in the number of weeks postconception (wpc) until birth. T and B cells require further development through primary and secondary lymphoid organs starting from the common lymphoid progenitor (CLP) in the bone marrow (simplified here). Circulating leukocytes are replenished from this central reservoir in the bone marrow.
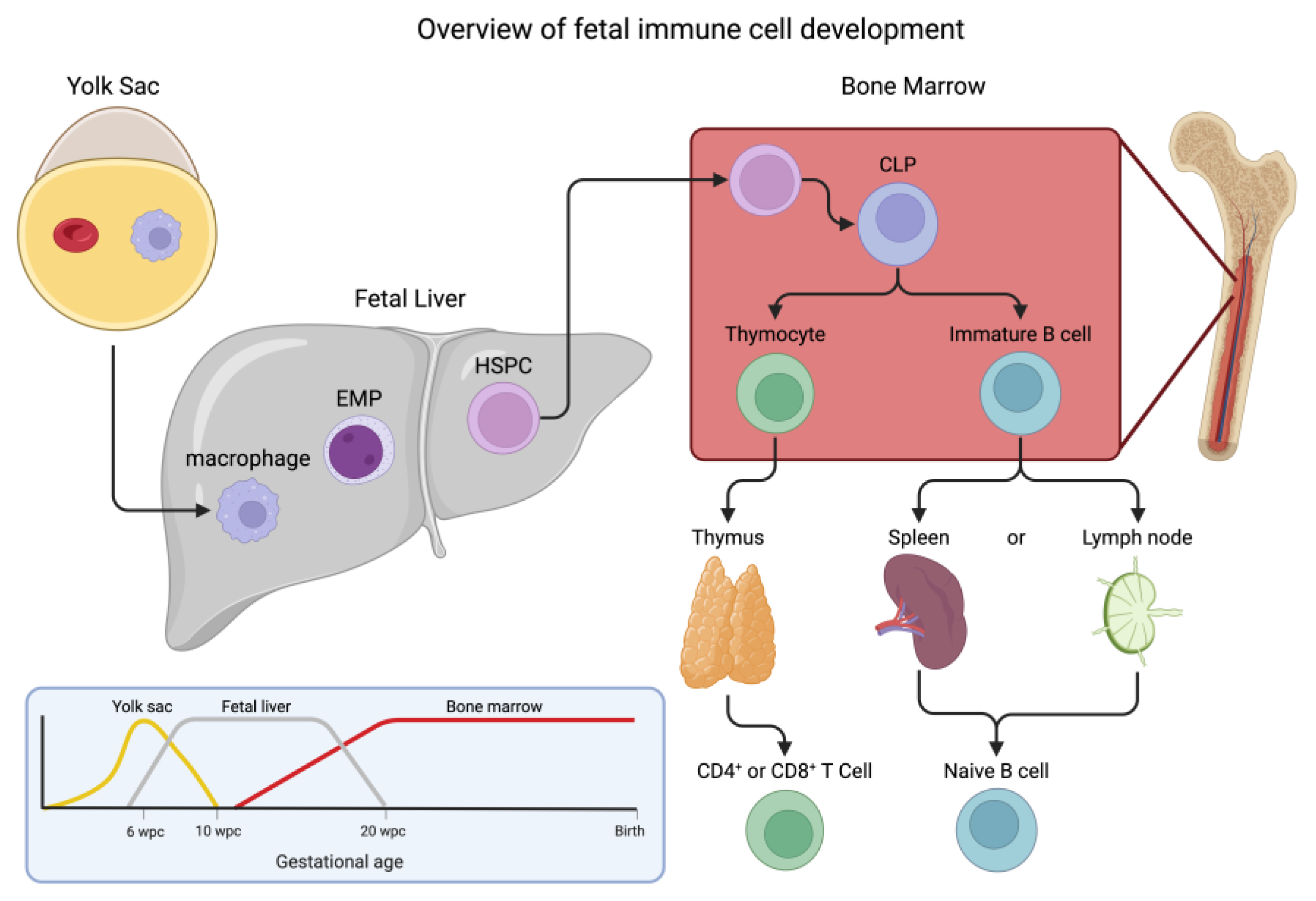
Figure 2.
Changes to immune system effectors in the mother, fetus, and neonate due to maternal obesity. Maternal obesity is accompanied by increased inflammation and abnormal uterine natural killer (NK) cell activation. These changes alter the blood supply to the fetus by reducing fetal pluripotent CD34+ cells and regulatory capabilities and altering macrophage polarization. The neonate born to a mother with obesity may have imbalanced T helper subtypes, and increased circulating IgE, and altered hematopoiesis leading to an increased chance of childhood allergies and autoimmune diseases. Bregs, B regulatory cells; NK, natural killer; NKT, NK T cells; pDCs, plasmacytoid dendritic cells; Th, T helper; Tregs, T regulatory cells.
Figure 2.
Changes to immune system effectors in the mother, fetus, and neonate due to maternal obesity. Maternal obesity is accompanied by increased inflammation and abnormal uterine natural killer (NK) cell activation. These changes alter the blood supply to the fetus by reducing fetal pluripotent CD34+ cells and regulatory capabilities and altering macrophage polarization. The neonate born to a mother with obesity may have imbalanced T helper subtypes, and increased circulating IgE, and altered hematopoiesis leading to an increased chance of childhood allergies and autoimmune diseases. Bregs, B regulatory cells; NK, natural killer; NKT, NK T cells; pDCs, plasmacytoid dendritic cells; Th, T helper; Tregs, T regulatory cells.
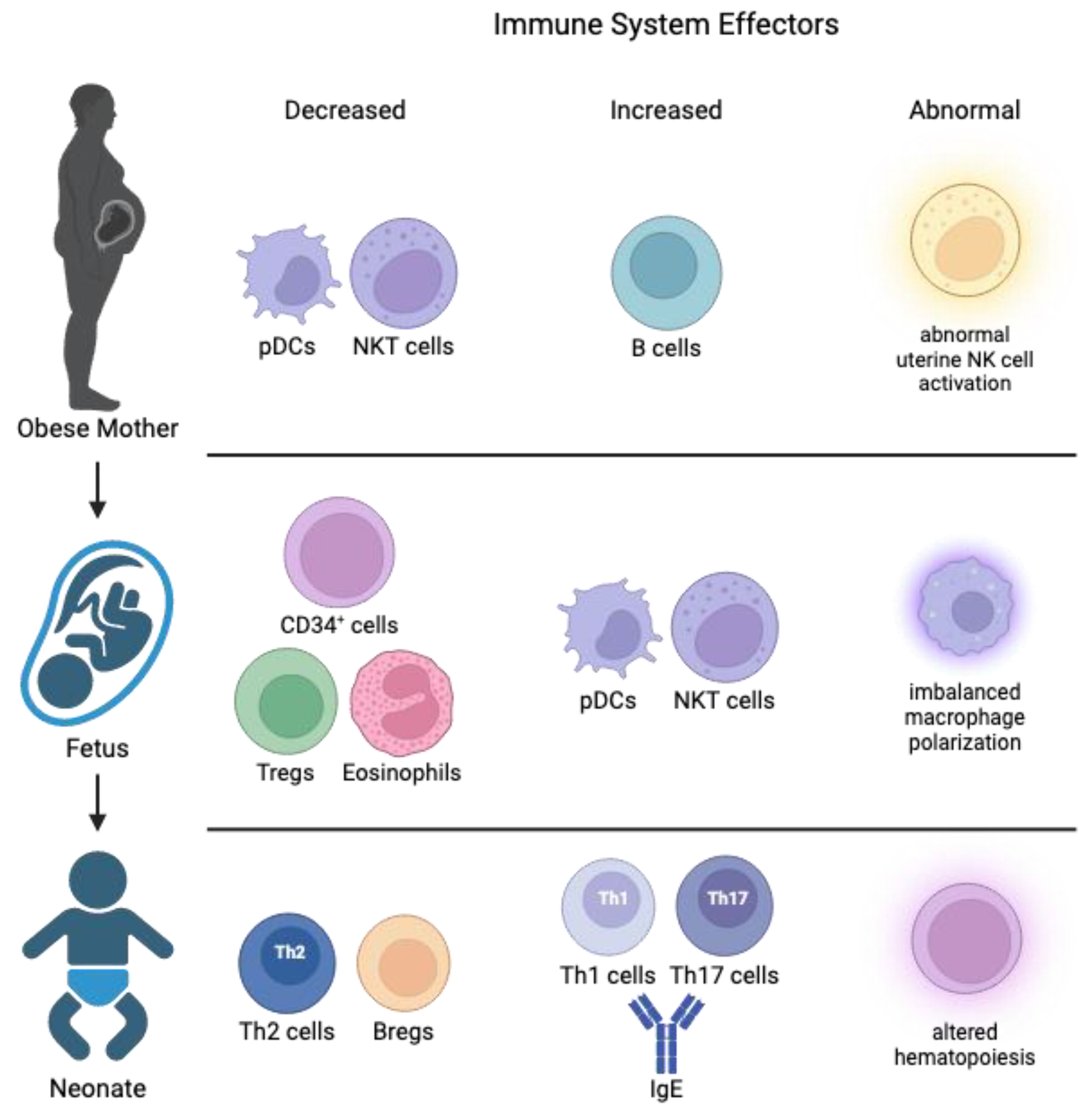
Figure 3.
Maternal factors underlie developmental programming and immune cell development. Several maternal factors brought on by a maternal WSD include increased immune activation, altered microbiome, and changes to bacterial metabolite composition. These exposures underlie dysregulated hematopoiesis, excessive immune tolerance, epigenetic modifications, and disordered infant microbiome. The outcomes affect postnatal developmental programming in organs and tissues with sustained and long-term metabolic and neurodevelopmental effects on future health.
Figure 3.
Maternal factors underlie developmental programming and immune cell development. Several maternal factors brought on by a maternal WSD include increased immune activation, altered microbiome, and changes to bacterial metabolite composition. These exposures underlie dysregulated hematopoiesis, excessive immune tolerance, epigenetic modifications, and disordered infant microbiome. The outcomes affect postnatal developmental programming in organs and tissues with sustained and long-term metabolic and neurodevelopmental effects on future health.
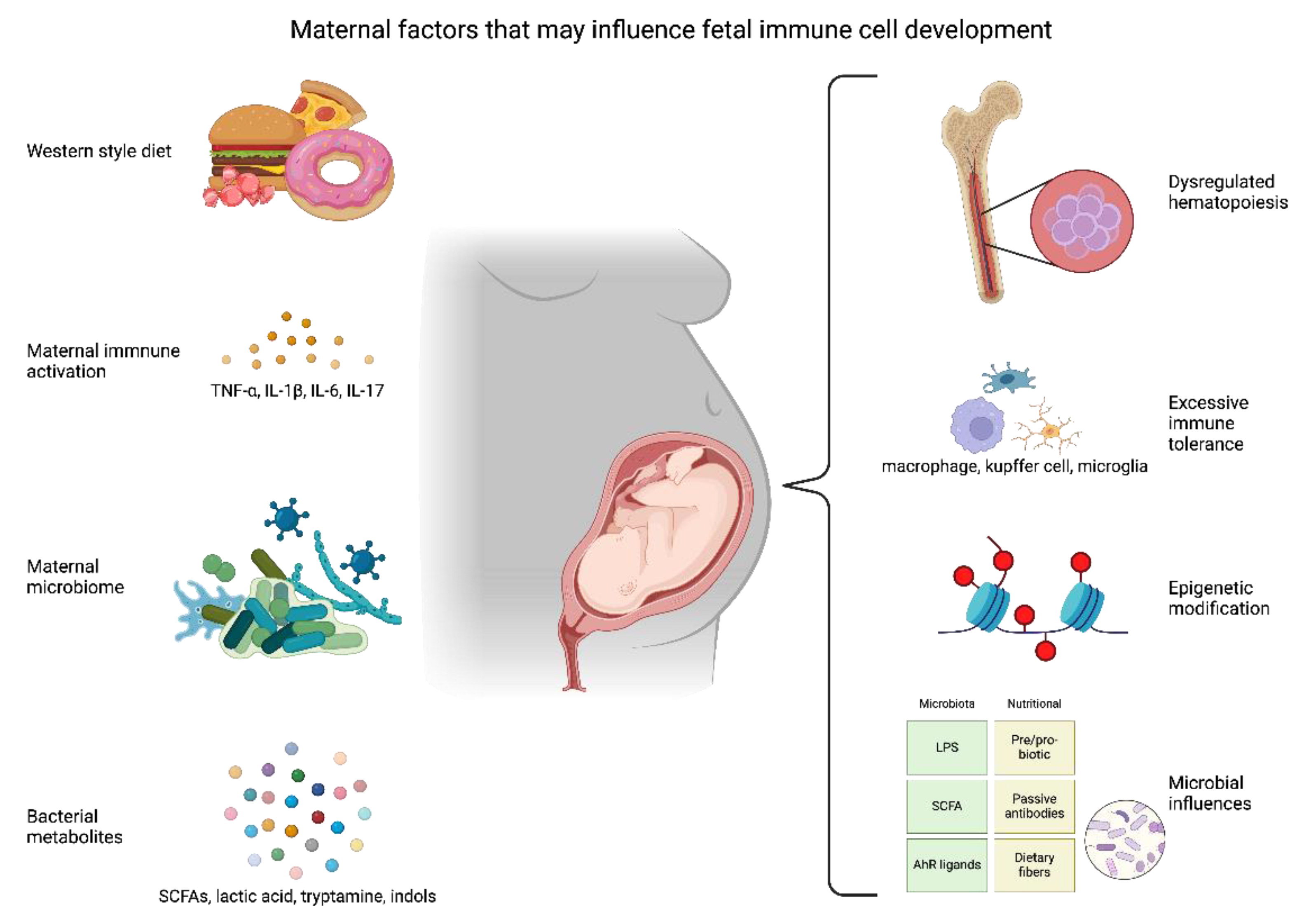

Table 1.
Maternal obesity impacts commensal microbiota and immunity in the offspring.
| Subjects (year) | Physiological outcomes | Programming effects | Reference |
| 222 human infants and NOD mice (2016) |
|
|
[150] |
| Germ-free mice colonized with microbes of infants from obese or NW mothers (2018) |
|
|
[143] |
| 17,055 neonates from the general population (2019) |
|
|
[149] |
| Antibiotic-treated and germ-free mice (2019) |
|
|
[152] |
| 46 full-term neonates from obese mother with and without GDM (2020) |
|
|
[153] |
| 418 mothers and their neonates (2021) |
|
|
[154] |
| 394 pregnant women in the first trimester and germ-free mice (2023) |
|
|
[155] |
| 65 mother-infant pairs, half receiving a prebiotic (2024) |
|
|
[156] |
GDM, gestational diabetes mellitus; LPS, lipopolysaccharide; SCFA, short-chain fatty acid; T1D, type 1 diabetes.
Table 2.
Common metabolites from commensal microbiota, their mechanism of action, and the effect on the host.
Table 2.
Common metabolites from commensal microbiota, their mechanism of action, and the effect on the host.
| Metabolite | Mechanism | Effect | Reference |
|---|---|---|---|
| Acetate | FFAR2 and acetyl-CoA carboxylase | Promoted lipid metabolism and reduced appetite | [203,204] |
| Propionate | FFAR2 and FFAR3 | Promoted glucose metabolism | [203] |
| Butyrate | FFAR3 | Anti-inflammatory effects and promoted lipid metabolism | [203] |
| Indolelactic Acid | AHR and HCA3 | Improved gut health and immune responses | [205] |
| Pheyllactic Acid | Mineral utilization | Increased lymphoid cell count without infection | [206] |
| 4-Hydroxyphenyllactic Acid | DCAR-1 | Triggered increased innate immune responses | [207] |
| Tryptamine | AHR | Suppressed inflammation | [197] |
| Indole-3-acetate | AHR | Suppressed inflammation | [197] |
| Indole-3-ethanol | MyoIIA, ezrin | Maintained gut permeability | [198] |
| Indole-3-pyruvate | MyoIIA, ezrin | Maintained gut permeability | [198] |
| Indole-3-aldehye | MyoIIA, ezrin | Maintained gut permeability | [198] |
| Indole-3-carbinol | UCPs, PPAR, sirtuin-1, leptin, aP2 | Decreased adipogenesis, thermogenesis, and inflammation | [199] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



