
Submitted:
19 April 2024
Posted:
22 April 2024
You are already at the latest version
Abstract
Acute lymphoblastic leukaemia is currently treated with bacterial L-Asparaginase; however, its side effects bring up the special need for improved and efficient novel enzymes development. Previously, we obtained low anti-asparaginase antibody production and high serum enzyme half-life in mice with the P40S/S206C mutant; however, its specific activity was significantly reduced. Thus, our aim was to test single mutants, S206C and P40S, for in vitro and in vivo assays. Our results showed that the drop in specific activity was caused by P40S substitution. Besides, our single mutants were highly stable in biological environment simulation, unlike double mutant P40S/S206C. The in vitro cell viability assay demonstrated that mutant enzymes have higher cytotoxic effect than WT on T-cells derived ALL and on some solid cancer cell lines. The in vivo assays were performed in mice to identify toxicological effects, to evoke immunological response and to study the enzymes pharmacokinetics. From these tests, none of the enzymes was toxic; however, S206C evoked lower physiological changes and immune responses. In relation to pharmacokinetic profile, S206C has two-fold higher activity than WT and P40S two hours after injection. In conclusion, we present bioengineered E. coli asparaginases with high specific enzyme activity and lesser side effects.
Keywords:
E. coli asparaginase
; bioengineer
; leukaemia
; nonclinical assays
1. Introduction
Pharmacological, toxicological and pharmacokinetics preclinical studies are essential for any developing biopharmaceutical and must follow the guidelines of the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) Document S6, S6 Preclinical Safety Evaluation of Biotechnology-Derived Pharmaceuticals, from 1997 [1]. In 2010, a specific guidance called Nonclinical Evaluation for Anticancer Pharmaceuticals was released [2]. According to the document, a successful nonclinical experiment should consider important details as pharmacologic properties of the drug, non-toxic doses for future extrapolation to humans and a toxicological profile to identify damage to specific organs and reversible/irreversible effects. In addition, in 2012 an S6 Addendum to Preclinical Safety Evaluation of Biotechnology-Derived Pharmaceuticals was published with complementary, useful and updated recommendations for in vivo assays.
In this context, well detailed nonclinical trials for a novel asparaginase biopharmaceutical are critical for future successful clinical trials. Actually, L-asparaginase enzyme for bacteria, commonly known as ASNase, is worldwide used to treat blood cancer, such as well succeeded therapy for acute lymphoblastic leukaemia (ALL) in children and adolescent. First approved FDA enzymes were ASNases from Escherichia coli (EcA) and Erwinia chrysanthemi (ErA) in 1978 and 2011, respectively. However, side effects related to their use, mainly on adult patients, such as neurotoxicity, pancreatitis, hypersensitivity or even anaphylactic shock [3] compel the need for improved versions of this enzyme. In this sense, preclinical toxicological and immune assays are crucial to go further with the clinical trials. Studies with different ASNases on several biomodels are reported on literature under different treatment schemes [4,5,6,7,8]. In this sense, herein we proposed a detailed study to complement a previous reported study with double mutant P40S/S206C [9] and its separated mutations (P40S and S206C) to better characterize the specific effect of them on animals.
Several studies are being carried out looking for better catalytic properties of ASNase to overcome side effects, including prospection of new ASNase sources or development of biobetters [4,10,11,12]. Protein bioengineering has emerged as an excellent tool that gives researchers the possibility to create new proteins/enzymes with desired profile, such as improved shelf-live and serum half-life stability, better substrate affinity/selectivity and low immunogenicity [13]. In this context, our research group has been focused on protein bioengineering techniques to create ASNase-biobetters. Appling this approach, several E. coli ASNases clones were developed and, after in vitro and in vivo assays, it was suggested that the P40S/S206C double mutant could be an interesting ALL treatment option as it presented improved plasma half-life and induced low anti-ASNase antibodies formation [9]. However, the P40S/S206C double mutant enzyme lost significant specific activity to asparagine hydrolysis and presented high relative glutaminase activity. These features motivated a deeper investigation of the double mutant ASNase P40S/S206C, exploring the individual mutation contributions to its improved resistance, stability, and immune system evasion.
Thus, our goals with this study are to characterize and evaluate biological in vitro activity of single-point mutation ASNase P40S and S206C in comparison with the double mutant from Rodrigues’ work (P40S/S206C). Also, to define whether the encouraging results come from the double mutation interaction or if we could rescue specific activity with a single point mutation, while preserving increased serum half-life and low antibodies production. Additionally, published studies of cytotoxicity assays with mutant ASNases on solid tumours are limited. Hence, we evaluated the potential action of these mutants on immortalized adherent cancer cells viability. Finally, to accomplish a detailed in vivo assay with multiple doses scheme, it was accessed toxicological analysis and measured hypersensitivity and inflammatory responses of these proteoforms. These nonclinical studies are essential to researcher groups for designing and performing a safe and effective treatment scheme for clinical studies in humans.
Our results indicate that mutation P40S was detrimental to enzyme activity and isolated S206C mutant rescued WT ability to hydrolyse asparagine. Nonetheless, both single mutants presented activity against ALL cell line. Interestingly, single mutants presented different behaviours related to activity against solid tumour cell lines, induction of antibody titres and resulted in specific physiologic alterations related to biopharmaceutical toxicity. Overall, the S206C mutant seems to be the best option, but each proteoform contains different features and can be more adequate for different situations and/or patients, reinforcing the importance of generating options of ASNase versions for future personalized therapeutic application.
2. Results
2.1. Biochemical Characterization of ASNases
The asparaginase gene from E. coli (ansB gene), inserted on vector pET15b, as well as its mutants, were properly expressed and the proteoforms successfully purified (Figure S1), a purity percentage above 95% was accepted to continue with the experiments (Figure S2).
The results for asparaginase (ASNase activity) and for glutaminase (GLNase activity) activities are presented on Figure 1 and summarized on Table 1. It was observed that ASNase specific activity was higher for native enzyme, called herein as wild type WT, and S206C, 76.2 and 74.9 U/mg, respectively, than for P40S (60.8 U/mg) and double mutant P40S/S206C (52.9 U/mg) enzymes. As previously mentioned, P40S/S206C protein was also developed in our laboratory and found that it lost around 30% to 40% of the activity compared to the WT enzyme, as reported by Rodrigues (2020) [9]. This may be an issue in leukaemia treatment since more enzyme mass is needed to achieve the therapeutic effect.
Regarding to GLNase activity, reported to be the main reason of adverse effects caused by the asparaginase therapy [12,14], a decrease was observed for all mutant enzymes (less than 2 U/mg), with the lowest value of 1.3 U/mg observed for P40S protein (Table 1). All the other mutants presented similar values. However, when correlating with ASNase activity, double mutant P40S/S206C enzyme has the higher percentage of intrinsic relative GLNase activity (3.3%), followed by WT, S206C and P40S proteins, with 2.4%, 2.3% and 2.1%, respectively (Table 1).
2.1.1. ASNases Stability on Human Serum
Aiming to simulate a physiological environment, the activity of WT, S206C, P40S and P40S/S206C enzymes was tested for in vitro stability in presence of human serum (HS) prior to the nonclinical assay to select the enzymes that would be evaluated in vivo for toxicity and immune hypersensitivity under our proposed scheme on Item 4.4.2. Therefore, ASNase activity was quantified during 96 hours of incubation with HS and statistical differences were observed from 72 to 96 hours. As shown in Table 2, P40S/S206C enzyme considerably lost the activity both in PBS 1x (80% lower) and HS (56% lower) after 96 hours; its activity started to decrease after 72 hours of incubation (Figure S3). In contrast, WT, S206C and P40S enzymes only lost activity in PBS 1x after 96 hours. Also, from Table 2 the highest stability on HS was observed for enzymes S206C, followed by P40S, WT and P40S/S206C, with residual activities of 83%, 76%, 70% and 44%, respectively. This means that the double mutant conserved only half of the ASNase activity of the single mutant S206C; indeed, even in PBS 1x the activity was 2-fold higher for S206C, WT and P40S proteins comparing to the P40S/S206C residual activity.
Our results show that the single mutants S206C and P40S conserved high catalytic activity, closer to WT, and also high stability in human serum.
2.2. Kinetic Parameters of ASNases
Figure 2 shows the kinetic profiles of commercial Leuginase®, WT and single mutant ASNases possessing a non-allosteric Michaelis-Menten behaviour. The kinetic parameters are summarized in Table 3 and from these results we concluded that WT and S206C enzymes had lower KM than commercial Leuginase® and P40S proteoform. Nonetheless, Vmax was similar for all the ASNases tested.
2.3. Nonclinical Studies of ASNases
2.3.1. In Vitro Assays
2.3.1.1. Blood Cancer Cell Lines Cytotoxicity
The Table 4 shows the cytotoxicity results of WT ASNase and its mutant proteoforms against T (MOLT-4) and B (REH) cells precursor derived ALL. P40S enzyme has significantly higher anti-tumour effect on T cells (MOLT-4) than WT. For REH cells, IC50 values were similar for WT and mutants.
2.3.1.2. Solid Tumour Cell Lines Cytotoxicity
From the MTT assay results, shown in Table 5, in relation to WT protein, S206C enzyme had a significant lower IC50 on Caco-2, PANC-1 and U-87 MG cell lines, while P40S enzyme is higher cytotoxic on PANC-1 and U-87 MG. However, on ovarian cancer cell line (SK-OV-3), S206C mutant had less effect and consequently higher IC50 than WT and P40S ASNases. On the other hand, P40S enzyme has the highest cytotoxic effect on glioblastoma cells (U-87 MG) with significantly lower IC50 compared to WT and S206C. Our results confirm that the mutant enzymes are biologically active as WT on these solid cancer cell lines; indeed, they even have higher cytotoxic effect against some cell lines despite their mutations. On the other hand, for breast cancer line MDA-MB-231, P40S ASNase has lower cytotoxic potential. Taken together, the results highlight the importance of the development of ASNase diversity options, since specific cytotoxic effect is observed for enzymes with the same biochemical function – deplete asparagine, with different consequences to tumour cell line viability.
2.3.2. In Vivo Assays
Following the guidelines of S6 for anticancer pharmaceuticals and after to characterize the enzymes, test their catalytic potential, stability and confirm their anti-tumour activity on leukaemic and some solid cancer cells in vitro, we performed the nonclinical assay in mice. Results of treatment scheme described for toxicity analysis are presented as follow, as well as immunogenic responses and pharmacokinetic profile (Item 4.4.2).
2.3.2.1. Organ Toxicity Analysis after ASNase Exposure
For toxicity tests, animals’ health must be controlled all over the experiments to avoid unnecessary suffering. Thus, body weight was considered as an indicator of wellness, which showed an increasing pattern in all groups throughout the experiment with no significant difference among them (Figure 3A).
Regarding the body temperature, no variations of normal temperature were detected on days 0 and 14 after single doses of 1050 U/Kg of ASNase. However, on day 23, after the challenge dose (fivefold higher), a decrease in body temperature was observed in all groups, except for the control (group treated only with buffer). Mice treated with P40S and S206C enzymes had the major drop of body temperature around 30 minutes after the injection, while it happened around 45 minutes for the group treated with the WT enzyme. For all the groups, body temperature was recovered 2 hours after treatment. Indeed, only minor significant differences were found between body temperatures of control group against all enzyme treated groups (Figure 3B).
Blood biometry was evaluated after the challenge dose on day 23. For all groups, control, and ASNase treated WT, S206C and P40S, ordinary counts of red and white blood cells were detected, except for haematocrit and granulocytes. Even though they are within normal ranges, the highest values observed for both parameters were resulted from animal group treated with P40S enzyme, followed by groups treated with S206C and WT enzymes (Figure 4A,B). On the other hand, platelet concentration analysis (Figure 4C) exhibited significant differences between control and all enzyme treated groups, but also between WT and S206C. The results showed a less damage in platelet count when the mutant proteoform S206C is applied in comparison to other ASNases.
In addition, the average weight of some organs from mice groups treated is presented in Table 6. Animals treated with enzymes, both WT and mutants, had significant reduced pancreas mass and, except for the S206C protein treated group, increased liver when compared with the control group. Also, histopathological results indicated that morphology and architecture of organs was preserved in the animals of the control group, within normal parameters. On the other hand, all groups treated with ASNases presented mild secondary histological alterations. The most frequent findings were microvesicular steatosis of liver, acute tubular necrosis of kidneys and mild intercellular edema of heart; other tissues were conserved, and no damage was found. Pictures from the WT group, S206C and P40S are shown on Figure S4 and no major differences were found among WT and mutant enzymes exposed groups.
2.3.2.2. Immunogenic and Inflammatory Potential of ASNases
The expression of immunoglobulins IgG and IgE anti-ASNase due to the enzyme administration, commonly related with hypersensitivity, were quantified as well as Platelet-activating factor (PAF) and Monocyte chemoattractant protein (MCP-4), mediators of several leukocyte functions, inflammation, sepsis, and anaphylaxis [15,16]. S206C mutant enzyme induced less humoral response throughout the experiment, since IgG and IgE levels were lower than WT and P40S treated groups, especially on the fourth week. Indeed, P40S enzyme treated animal group had the highest IgE titre of all groups since the second week, unlike IgG that increased on the fourth week (Figure 5). Regarding PAF and MCP-4, animals injected with P40S enzyme showed the lowest concentration of both compounds and the highest values were from mice treated with WT (PAF) and S206C (MCP-4) enzymes, respectively (Figure 6).
During this assay, body weight and temperature after each injection of ASNase was also controlled. As expected, all animals gained weight normally throughout the entire experiment, no variations of temperature were observed after each injection and at the end of the assay no alterations were detected on blood biometry. All these results highlight that the treatments brought low impact on animals' health.
2.3.2.3. Pharmacokinetics of ASNases
Finally, asparaginase activity was measured on mice plasma at 2, 6, 12 and 24 hours after one single dose injection of 1,050 U/Kg of each ASNase, WT and mutants (as described in Item 4.4.2). As expected, the maximum enzyme activity was detected 2 hours after the injection, in all cases, and the minor after 24 hours. No significant differences were found between all enzymes from 6 hours until 24 hours, except after two hours of the injection when S206C residual plasma activity was significantly higher than WT and P40S (Figure 7).
3. Discussion
Considering the ALL-BFM-IC 2009 protocol that recommends an ASNase dose of 5,000 U/m2 every third day, on days 12, 15, 18, 21, 24, 27, 30, 33 (8 doses) during induction phase, an equivalent scheme in mouse should involve around 5 doses within a 24 days-treatment. Mice used in these experiments aged 6 – 8-week-old, with an average weight of 20 grams and an estimated body surface area of 0.007 m2 [17], which correlate with a human adolescent of 13 years-old [18]. Thus, the animal equivalent dose in mouse should be around 1,900 U/Kg [19]. However, we decided to use a three concentrated doses scheme, considering that our enzymes formulations did not have stabilizers and osmolytes. Therefore, doses of 1,050 and 5,250 U/Kg were administered to the animals on days 0, 14 and 23 to investigate whether these novel mutants evoke toxic responses in healthy and immunocompetent animals. In this context, discomfort or pain, animal weight and corporal temperature were analysed, as well as organ’s weight, morphology, and histology [5,20,21]. Animal weight loss is associated with toxicity or discomfort since it can be caused by lack of appetite due to pain/allergies. Although, it can also result from behavioural factors such as stress, lack of adaptation or fear, metabolic changes such as malabsorption syndrome or increased physical activity [22]. Indeed, to guarantee animal’s welfare, in the case of losses equal to or greater than 20% of body weight and lack of water consumption, the humane endpoint should be performed [22,23]. Our results suggest no progressive chronic diseases or acute toxicity due to the enzyme administration, since until the end of the experiment significant animal weight loss was not observed in any of the tested groups, they all grew normally.
Regarding the analysis of corporal temperature over two hours after each enzymatic formulation injection (days 0, 14 and 23) the first two doses were harmless, and no variation was observed. In contrast, the temperature decreased at the last challenge dose within each group. In general terms, the maximum loss of corporal temperature was 1.9 °C for the WT group, 2.8 °C for the S206C group and 2.6 °C for P40S group. Some authors consider body temperature drop as the first symptom of hypersensitivity reaction [5,21]. In that context, except for the control group, all animals had significant drop in body temperature within the first hour after injection; however, temperature was recovered soon after. This means that even at higher doses than the ALL-BFM-IC 2009 protocol recommends these mutant enzymes were not lethal. This could be considered as an induced hypothermia for an initial anaesthetic effect due to the introduction of the “foreign body”, the enzymatic solution, instead of toxicity effect [24]. Under standard conditions, mice have normal temperatures between 35.5 and 37.5 °C and mild hypothermia range from 32 to 35°C [25]. Hence, temperature values recorded after the last dose indicate a transient mild hypothermia for around 2 hours. Like weight loss, body temperature drop can be considered as a symptom of animal's discomfort; however, it can also be related to a compensation and protection system [25]. Hypothermia has neuroprotective effects in ischemia or encephalopathy in animal models and human [26,27,28,29,30]. In the case of ischemia, it can reduce the cerebral metabolic rate and preserve energy [31], reduce the release of free radicals [32], reduce the formation of cerebral edema and stabilize membranes [33], and also reduce the rate of apoptosis [27,34,35].
Concerning the blood biometry results, the decrease observed on platelets count may be explained by anti-asparaginase antibodies production. Bougie and collaborators (2010) demonstrated that transient thrombocytopenia can be induced by the presence of anti-drug antibodies. Therefore, anti-asparaginase antibodies may cause thrombocytopenia, with the worst scenario observed in WT group followed for P40S enzyme, although no animal suffered from spontaneous bleeding. Morowski and collaborators (2013) observed that reductions of 70 to 80% of the normal number of platelets do not prevent the correct formation of thrombi in case of injuries or ruptures of veins or small arterioles and that even small amounts of platelets (approximately 3%) are effective in maintaining homeostasis without the presence of spontaneous bleeding [36]. Based on this, all animals were capable to live without any bleeding complications; even more considering the decreased platelet condition is transitory due to enzyme administration. Our results show that S206C proteoform has the lower impact on platelet count, which can be an advantage to patients with ALL. Also, it is important to mention that higher haematocrit (HCT) percentage on P40S treated animals may be due to dehydration or shock caused by the mutant itself, unlike WT or S206C enzymes effect. In addition, the elevated concentration of granulocytes observed upon P40S ASNase administration may be correlated with enzyme digestion and consequently antigen presentation to the immune system, in agreement with the higher IgG and IgE expression observed.
Michael and colleagues had gathered regulatory guidelines and data from several pharmaceutical companies located in Europe, North America and Japan, regarding toxicity on organs to identify it in rodent. Accordingly, the organs analysed in this work are considered relevant to evaluate drug toxicity. Liver for instance, that reflects physiologic and metabolic distresses, is suitable to identify hepatocellular hypertrophy that matches with histopathological changes, peroxisome proliferation and lipidosis. Also, liver has little animal-to-animal variations and has a primordial function as it is the animal’s primary detoxification organ. Kidneys are frequently a target of toxicity and could reflect acute injuries. Thymus and spleen are essential as they are important indicators of immune toxicities, physiological discomfort, stress and histopathological changes may perfectly correlate with organ weight changes. Also, heart could be valuable by its limited inter-animal variability and its sensitivity to identify toxicity [37]. In this context, results found in these experiments indicate minor hepatotoxicity, microvesicular steatosis that can result in increased fat accumulation in liver cells, in all enzyme treated groups and an increased organ size, except for the S206C group. This may be caused by an impairment of liver metabolic functions that includes defective fatty acid oxidations, enhanced lipogenesis, irregular triglyceride secretion or increased absorptions of fat acids from the diet [38,39]. Hepatic failure, as platelet decrease, may be related with anti-asparaginase antibodies such as IgG and IgE, which also may form and accumulate immune complexes and could be responsible for the increased organ size on WT and P40S enzymes treated groups, unlike S206C exposed group. It is possible that the animals treated with S206C enzyme did not showed an increased organ size due to less recognition of mutant enzyme from host antibodies, as also seen for the double mutant P40S/S206C [9]. This could mean less accumulation of immune complexes from blood to hepatocytes through the portal vein and, therefore, no liver impairment was produced. Instead, S206C mutation could give the enzyme the ability to escape from hepatocytes and liver macrophages/monocytes from degradation/clearance circumventing its binding to pattern recognition receptors (PRR) as pathogen associated molecular pattern (PAMP) or damage-associated molecular patterns (DAMP). In this sense, avoiding phagocytosis and degradation by lysosomal proteases may prevent S206C liver bioaccumulation or an exacerbated immune response, while maintaining its blood asparaginase activity higher than the other enzymes after injection [40,41].
Rathod et al. (2019) identify that ASNase binds mainly to basophils and B cells, and in less amount to neutrophils and macrophages upon two sensitization doses of 10 mg of E. coli ASNase and challenged with 100 mg on day 24 on 8-week-old female C57BL/6 mice [21]. This scheme of treatment was used to stimulate a humoral hypersensitivity by immune complexes sensitization and a re-exposure with the antigen with the challenge dose, likewise our assay. The authors also found that ASNase binding to immune cells could be either free (cell-associated IgE) or by anti-ASNase IgG and IgE immune complexes, mainly to basophils, which express both high affinity IgE receptor FcεRI and low-affinity IgG receptor, FcγRIII. Thus, our results show that anti-ASNase IgG mediated hypersensitivity after the challenge dose was significantly higher on WT and P40S enzymes treated groups on the fourth week, unlike S206C enzyme, which induces the lowest titre of both anti-ASNase IgG and IgE throughout the experiment. Indeed, IgE concentration in mice on WT and S206C enzymes treated groups slightly increased from the second to the fourth week, unlike P40S exposed animals that effectively produced high and increasing titre of anti-ASNase IgE on both measurements.
Hypersensitivity reactions developed to ASNase are mainly related to induction of CD4+ T cells rather than B-cells [21]. Therefore, MCP-4 chemokine was also quantified as it preferentially attracts T cells, monocytes, and eosinophils. In addition, it is considered homolog to human chymase because both share high substrate specificity, similar tissue distribution and functional serglycin-binding properties [42,43]. Our results showed the highest MCP-4 increase in S206C group, as previously reported by Medjene et al (2020). This chymase increase observed can be protective, since it had a potent anti-inflammatory effect in mice with renal ischemia reperfusion injury and bacterial infection, by controlling neutrophil extravasation activation and, consequently, limiting their contribution to the possible associated injuries toward the pathogen/antigen response [42,44]. Also, recently MCP-4 was inversely correlated with IgE levels [42]. Indeed, P40S enzyme treated group had the highest titre of anti-ASNase IgE and the lowest MCP-4 concentration. The same correlation is true for mice treated with S206C enzyme – high MCP-4 concentration and low IgE titre. Relative to PAF expression, anti-ASNase IgE induces the release of PAF from granulocytes upon re-exposure to the enzyme on day 23 and anaphylaxis reactions may occur. In contrast, if PAF receptor antagonist is used, hypersensitive reactions are considerably reduced [21]. In this context, significant increased concentrations of PAF were found in the blood of animals treated with WT enzyme, confirming the advantage of S206C mutant, but in this case, also P40S mutant.
Concerning the pharmacokinetic data collected, enzyme activity of S206C treated group after two hours of the single dose was significantly higher than WT and P40S exposed groups, likely due to less protease inactivation of the enzyme by asparaginyl endopeptidase (AEP or legumain) or cathepsin B (CTSB), as previously described [9,41,45,46,47]. Also, as related by Rathod et al. (2019), under the scheme of sensitization, macrophages clearance of ASNase is likely to occur within the following hours after the injection, due to macrophages number increasing up to 10% every hour [21]. This result was already expected since it was reported that P40S/S206C mutations may protect and camouflage the enzyme from recognition and degradation by AEP and CTSB [9].
Previous studies indicate that even at relatively low concentration, around of 0.1 U/mL, ASNase is capable to fully deplete physiological concentrations of asparagine (Asn) within seconds in blood plasma and central nervous system [7,48,49,50]. Here, all proteoforms were therapeutically effective until 12 hours after injection. Also, Horvath et al. (2019) found that mice treated with three doses of 1,000 U/Kg of commercial ASNase had a decrease of Asn concentration to half (22 µM approximately) after 24 hours of administration and reaches its lowest level after 48 hours (6 µM) [7]. In mice, the normal range of Asn is from 40 to 50 µM, as they had reported. Based on that, our scheme results are relatively close to those obtained in Horvath et al. (2019) experiments using the same dose as this work. It is important to highlight central role of monocyte/macrophages (phagocytic cells) on the liver, spleen, and bone marrow, which promote ASNase degradation and clearance by cathepsin B activity, with ASNase pharmacokinetics therefore reducing its blood and bone marrow niche half-life. Thus, ASNase local rapid degradation specifically in the bone marrow niche mediated by CTSB may explain a resistance mechanism of leukaemic cells to escape from apoptosis [41]. So, we can hypothesize a reduced affinity of S206C to surface receptors of phagocytic cells or a decrease on lysosomal process of the proteoform and its epitope exposure, which may attenuate immune and allergic response, as observed in S206C group data together.
Finally, relative to cytotoxicity on solid tumour cells, S206C proteoform presents lower IC50 values, which mean higher cytotoxic effect on Caco-2, PANC-1 and U-87 MG than WT enzyme; however, on SK-OV-3 cell line S206C is less effective. On the other hand, P40S has higher anti-cancer activity on U-87 MG and PANC-1, only defeated for WT on MDA-MB-231 cell line. Indeed, previous results from literature described the IC50 against Caco-2 of 68.28 U for ASNase from Pseudonomas aeruginosa [51], 30 U for ASNase from Pyrococcus furiosus [14] and 5 U/mL from Pyrococcus abyssi compared to 0.41 U and 7.1 U/mL as here identified. On the other hand, cytotoxicity on PANC-1 is not well known. A study using commercial E. coli ASNase Spectrila® identified IC50 around 0.11 U/mL [52], higher efficient when comparing to 3.3 and 2.7 U/mL of our WT and S206C enzymes. Nonetheless, S206C mutant showed improved immunological response than E coli ASNase (WT). Indeed, ASNase sensitivity in PANC-1 is correlated with a decrease in purine synthesis pathway and with Gln starvation due to GLNase co-activity. Also, the resistance mechanism is likely because glutamine synthetase gene over-expression, instead of asparagine synthetase gene [52]. Thus, to further investigate glutaminase activity of the mutants, we cultured PANC-1 cells without and with the addition of Gln and increasing concentrations of ASNases (WT, S206C and P40S). However, no significant differences on IC50 were found, suggesting low or absent glutaminase activity of the optimized and bioengineered ansB gene (data not shown) compared with formulated recombinant ASNase Spectrila®. This may explain the differences on IC50 for Spectrila® [52] and the mutants presented here. Regarding glioblastoma cells, a work from Karpel-Massler et al. (2016) on different glioblastoma cells treated with recombinant L-asparaginase from E. coli from Sigma Aldrich effectively showed that these tumour cell lines are sensitive to ASNase, with IC50 between 0.1 and 1.55 U/mL [53], while we obtained 1.2 and 0.8 U/mL for S206C and P40S enzymes, respectively. In fact, they demonstrated the increased rate of intrinsic and extrinsic apoptosis under ASNase treatment in vitro and its enhanced inhibition of glioblastoma cells implanted into SCID SHO mice.
In conclusion, we present the novel S206C mutant ASNase from E. coli that shows improved characteristics compared to native enzyme and recovers specific activity loss from double mutant P40S/S206C. This novel mutant induces lower humoral immunological response, lower allergenic and higher protective inflammatory reaction when compared with the WT enzyme. Additionally, it has the highest enzyme activity two hours after the single dose according to the pharmacokinetic profile. Considering that macrophage recognition and clearance is higher in this period, it suggests less immune stimulation and possibly no hepatic bioaccumulation of immune complex. Furthermore, regarding to cytotoxicity on solid tumours, S206C showed higher cytotoxic effect mainly on Caco-2, PANC-1 and U-87 MG cells than WT. Finally, we propose S206C as a promising option for WT enzyme that could improve the patient´s quality of life and treatment.
4. Materials and Methods
4.1. Enzyme Production and Purification
4.1.1. Gene and Vector Information
Native E. coli asparaginase type II enzyme (WT), encoded by ansB gene (UNIPROT P00805), was synthesized with optimized codon usage by GenScript (Piscataway, New Jersey, USA), and used as template to obtain the following novel mutants: S206C and P40S, as well as P40S/S206C used as a reference [9]. Briefly, site-directed mutation was performed with the QuikChange - Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, California, USA), following supplier instructions. WT and mutants were inserted into NdeI and BamHI restriction sites of pET15b expression vector and cloned in E. coli BL21 (DE3) (Novagen-Merck-Millipore, Burlington, Massachusetts, USA).
4.1.2. Bacterial Culture
Transformed bacteria were cultured in solid lysogenic broth medium (LB) added with carbenicillin (tryptone 10 g/L, yeast extract 5 g/L, NaCl 5 g/L, carbenicillin 50 μg/mL). Then, clones were grown in 400 mL of liquid LB and carbenicillin at 37°C and 250 rpm overnight. Afterward, cells from pre-inoculum were diluted into 1 litre of fresh medium to an initial OD600 of 0.2 and after reaching OD600 of 0.7 – 0.8, protein production was induced by the addition of isopropyl β-D-1-thiogalactopyranoside (IPTG 1 mM) for 22 hours at 37°C. Subsequently, cells were harvested by centrifugation at 5,000 g-force for 20 minutes at 4°C and submitted to osmotic shock to obtain periplasmic fractions [9].
4.1.3. Protein Purification
Osmotic shock of collected cells was performed as following: one gram of wet cells was resuspended in 15 mL of ice-cold hyperosmotic buffer (50 mM Tris-HCl pH 8.0, 500 mM sucrose, 0.5 mM ethylenediaminetetraacetic acid-EDTA) and centrifuged at 5,000 g-force for 30 minutes at 4°C. Next, the supernatant was washed out and cells were resuspended in 0.5 mM phenylmethylsulfonyl fluoride (PMSF) and ultra-pure water (5 mL per gram of wet cell) and centrifuged at 7,500 g-force for 20 minutes at 4°C. The resultant supernatant is the periplasmic fraction which was filtered through a 0.2 µm membrane and diluted in 50 mM Tris-HCl pH 8.8 buffer for further purification steps.
First, buffered periplasmic fraction was loaded onto a weak anion exchange chromatography column (HiTrap™ DEAE FF 5 mL, GE Healthcare Life Sciences, Chicago, Illinois, USA) pre-equilibrated with start buffer (50 mM Tris-HCl pH 8.8), and proteins were eluted by an increasing gradient from 0 to 100% NaCl buffer (0–500 mM NaCl in Tris-HCl 50 mM buffer, pH 8.8) in 10 column volumes (cv) at a flow of 2 mL/minute on AKTATM Start (GE Healthcare Life Sciences). All fractions containing the enzyme were pooled and concentrated on Amicon Ultra centrifugal filter 10 kDa MWCO (Millipore Merck KGaA, Darmstadt, Germany), and submitted to size exclusion chromatography (SEC, column Superdex 200 Increase 10/300 GL, GE Life Sciences) on AKTATM Purifier (GE Healthcare Life Sciences). ASNase was eluted using 50 mM Tris HCl and 100 mM glycine buffer, pH 7.4 with a flow rate of 1 mL/min on 5 cv. The purity of the collected fractions was estimated by SDS-PAGE (14%), stained with Coomassie Blue R-250. Finally, pure fractions were pooled and sterilized by filtration through a sterile MILLEX-GV 0.22 μm pore membrane in laminar flow cabinet. The protein concentration of pure enzyme was determined by absorbance at 280 nm, using the molar extinction coefficient of ε= 23,505 M-1cm-1 and molecular weight (MW) of 34.5 kDa.
4.2. Enzymes’ Biochemical Characterization
Asparaginase and glutaminase activities were determined using the Nessler´s reagent (Merck-Millipore). The specific activity is expressed as U (1 unit corresponds to 1 μmole of ammonia released per minute) per milligram of pure protein. Briefly, the enzymes were diluted from 0.000013 to 0.00026 mg and incubated for 10 minutes at 37 °C with Tris HCl 50 mM buffer, pH 8.8 and L-Asn 44 mM. Likewise, for glutaminase activity, enzymes were diluted from 0.00013 to 0.0026 mg and mixed with Tris HCl 50 mM buffer, pH 8.8 and L-glutamine (Gln) 44 mM and incubated under the same conditions. Following, 37 µL of the reaction volume were diluted in water with 20 µL of TCA 1.5 M to stop the enzyme activity and finally added of 37 µL of Nessler´s reagent; and the absorbance was analysed at 436 nm in a microplate reader SpectraMax M2 (Molecular Devices, California, USA). Blank controls were performed with all reagents except substrate (Asn and Gln) and negative controls without enzyme. Higher absorbance values were discounted from sample absorbances.
The µmole of NH3 released was calculate by interpolation of absorbance values of reactions to a standard curve of known concentrations of ammonium sulphate ((NH4)2SO4) vs absorbance at 436nm. Assay was performed in technical triplicate and analytic triplicate. Finally, the values of µmoles of NH3 per minute were plotted against the milligrams of enzyme and the slope of the linear regression curve equation corresponds to the specific activity of the enzyme (U/mg). Statistical analysis was performed with one-way ANOVA and Tukey’s multiple comparisons test on Prism 9.0 (Graphpad).
4.2.1. Enzymes’ Stability on Human Serum
Ten micrograms of each ASNase proteoform were incubated with sterile phosphate buffer saline (PBS) 1X, pH 7.4 and 10% of human serum (HS) (Invitrogen, Waltham, MA, USA) at 37 °C for 96 hours. Control samples without HS were incubated at the same conditions. Asparaginase activity was measured at points 0, 24, 48, 72 and 96 hours using the Nessler´s reagent. Assay was performed with technical triplicate and analytic duplicate. Statistical analysis was performed by two-way ANOVA and Dunnett’s multiple comparisons test on Prism 9.0 (Graphpad).
4.3. Kinetics Parameters Determination for ASNases Studied
Kinetic profile of all enzymes was evaluated following the coupled enzymatic reaction to glutamic-oxaloacetic transaminase (GOT G2751 Sigma) and malic dehydrogenase (MDH M2634 Sigma), in which for one mole of L-aspartate produced after L-asparagine hydrolysis, one equivalent mole of NADH is oxidized to NAD+, which decrease in absorbance was spectrophotometrically measured at 340 nm and 37 °C [54,55] . Thus, 80 ηM of each ASNase (WT or mutants) was mixed with Tris HCl 100 mM buffer, pH 7.4, 0.4 mM of α-ketoglutarate, 0.4 mM of NADH, 5 U/mL of GOT, 5 U/mL of MDH and with 2.5 to 600 µM of Asn for linear rates. Also, commercial asparaginase Leuginase® (Beijing SL Pharmaceuticals) was used for comparison. Then, the level of NADH continuous consumption in µmole was calculated based on absorbance readings at 340 nm using the Lambert-Beer law (ε=6.22 mM-1cm-1). The consumed µmoles of NADH were plotted against time (minutes) and the slope corresponds to the initial velocity (V0, µmoles of L-aspartate/minute). Kinetic parameters were determined on Prism 9 (Graphpad) using V0 values and Asn concentration for determining KM and maximal velocity (Vmax) with non-linear regression fit. Blank and negative control reactions were performed with no substrate or enzyme addition, respectively, and the highest value was discounted of experimental absorbance. Assay was executed in technical quadruplicate and analytic duplicate.
4.4. Nonclinical Evaluation of ASNases
4.4.1. In Vitro Assays
4.4.1.1. Blood Cancer Cell Lines Cytotoxic Effect of ASNases
MOLT-4 cell line was used as ALL model derived from T-cells and REH as ALL pre-B cells-type. For both cell lines, 2.0 x 10^4 cells/well were treated for 72 hours with increasing ASNase doses ranging from 0.01 to 1 U/mL in clear Nunc 96-Well Flat Bottom plates (Thermo Scientific™ Denmark). Positive controls were performed by adding buffer and negative controls with 20% of Dimethyl Sulfoxide (DMSO). The cells were cultured in Advance RPMI 1640 medium supplemented with 10% of foetal bovine serum (FBS), 1% of penicillin/streptomycin 100x (P/S 1X), 0.01 M of HEPES, 1 mM pyruvate and 2.5 g/L glucose solution (Gibco Taiwan) at 37°C and 5% of CO2. After the incubation time, 0.5 mg/mL of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (Invitrogen™ Canada) was added to each sample and further incubated for 3 hours. Finally, 150 µL of a solution with 10% SDS and 0.01 M of HCl was added, incubated for 16 hours to dissolve the formazan crystals and absorbance was read at 570 nm. Inhibitory concentration (IC50) was calculated on Prism 9.0 (Graphpad) by Log (inhibitor) vs. normalized response and statistical analyses of different IC50 within cell lines were performed by ordinary one-way ANOVA and Tukey’s multiple comparisons test. Assay was performed with technical triplicate and analytic duplicate.
4.4.1.2. Solid Tumour Cell Lines Cytotoxic Effect of ASNases
Cytotoxic effect of ASNase WT and mutants was evaluated on immortalized adherent cancer cells of breast (MDA-MB-231), colon (Caco-2), ovarian (SK-OV-3), pancreas (PANC-1) and glioblastoma (U-87 MG). The culture medium for MDA- MB-231 and SK-OV-3 was Advance RPMI 1640 supplemented with 10% of FBS, 1% of GlutaMAX 100x and P/S 1x (Gibco). For Caco-2, PANC-1 and U-87 MG cells DMEM High glucose, 10% of FBS, 1% of MEM non-essential amino acids (NEAA) 100x and P/S 1x (Gibco) was used. All cell lines were cultured at 37°C and 5% of CO2 at passage between 10 and 20, except for Caco-2, that was used between passages 30 to 38. In all cases, cells were seeded in clear Nunc 96-Well Flat Bottom plates (Thermo Scientific™) with 1.0 x 10^4 cells/well and treated with 0 to 20 U/mL of each ASNase (WT, S206C and P40S) for 72 hours. Following, 0.5 mg/mL of MTT was added to each well and the plates incubated for more 3 hours under the same conditions. Next, the media was discarded and finally 200 µL of DMSO added under agitation, until the crystals were dissolved. The absorbance was measured at 570 and 620 nm for reference; all assays were performed with technical and analytic triplicate. Inhibitory concentration (IC50) was calculated on Prism 9.0 (Graphpad) by Log (inhibitor) vs. normalized response and statistical analyses of different IC50 within separated cell lines were performed by ordinary one-way ANOVA and Tukey’s multiple comparisons test.
4.4.2. In Vivo Assay
Male Balb/c SPF mice from 6- to 8-week-old were used as models in this study. Animal care was constant, changing cages twice a week, applying weight control between enzyme administrations and checking the parameters of the Grimace scale in mice, following guidance from the National Center for Replacement, Refinement & Reduction of Animals in Research NC3RS, on days of the injections. Animals had a 12-hour light/12-hour dark cycle, food and water ad libitum and were maintained on rooms with relative humidity at 45 to 65% and temperature 20-24 °C. In fact, they were properly monitored all over the assay, and at the beginning of each experiment they underwent a two-week adaptation period to reduce the stress of transport and get used to the new experimentation room, where they normally fed, and no strange behaviour was ever observed.
The in vivo assay comprised three sets of independent experiments; the first one was to access toxicological effects after multiple doses, including a final challenge dose (three animals per group), as explained in Figure 8A, in which body weight, corporal temperature, blood count and general animal health conditions were evaluated. Body temperature was measured by infrared digital thermometer NX-2000 for up to two hours after the enzyme injection, on the administration days 0, 14 and 23 to monitor animal health and, if necessary, perform a humane endpoint. Animal weight (that was controlled throughout the entire experiment) and body temperature measurements (after each injection) were statistically analysed by two-way ANOVA, α=0.05 and Tuckey´s multiple comparison test on Prism 9 (Graphpad).
In the second set, the antigenic and allergenic potential of each enzyme was investigated by means of IgG, IgE, PAF and MCP-4 quantifications in plasma (five animals per group – Figure 8B). The third set of experiment aimed at the pharmacokinetics profile of the enzymes after a single dose as shown in Figure 8C (five animals per group). ASNase doses were administered via intraperitoneal. Each set of experiments comprised four groups, namely control group (treated only with 50 mM Tris HCl and 100 mM glycine buffer, pH 7.4), WT group, S206C group and P40S group.
This project was approved by the Ethical Committee for Animal Experimentation of the School of Pharmaceutical Sciences of the Universidade de São Paulo (protocol number CEUA/FCF 036.2020-P605).
4.4.2.1. Organ Toxicity Analysis
After 2.5 hours of the challenge dose injection of each ASNase, animals were anesthetized with isoflurane, exsanguinated, and blood sample was collected for blood biometry. Immediately after, they were heart perfused with 100 mL of ice-cold PBS 1x to remove all remaining blood and next fixed with 200 mL of ice-cold paraformaldehyde (PFA) 4% pH 6.9. Subsequently, various organs were harvested, rinsed with saline solution 0.9% and weighted, including heart, liver, kidney, thymus, spleen and pancreas. Next, organs were fixed for 48 hours in buffered formalin, dehydrated in graded concentrations of ethanol and embedded in paraffin, cut 3 to 5 microns thick with a microtome, and finally stained with haematoxylin-eosin (H&E) for visualization in light microscope by a professional unaware of the treatment. Statistical analysis of organ’s weights was performed at Prism 9.0 (GraphPad) using Multiple t-test and the two-stage linear step-up procedure of Benjamini, Krieger and Yekutieli, with Q = 1%.
4.4.2.2. Immunogenic and Inflammatory Potential
Blood samples were collected on days 7 and 23 in Eppendorf tubes containing EDTA 15 mg/mL, centrifuged at 8,000 g-force, 4 °C for 3 minutes and the plasma was separated and stored at -80 °C until analysis. The specific anti-ASNase IgG immunoglobulin quantification was performed by the indirect ELISA assay. Briefly, 96 well microplate F-bottom HIGH BINDING Greiner bio-one was coated with 0.6 µg/mL of asparaginase (WT or mutants) previously diluted in PBS 1x, packed in foil and incubated overnight at 4°C. The enzyme solution (antigen solution) was removed, and the plate washed with wash solution WS (PBS 1x with 0.5% tween 20). Then, the well bottom was sealed by adding 200 µL of PBS 1x solution supplemented with 1% of bovine serum albumin (BSA) for 90 minutes at 100 rpm, 37 °C and the plate packed in cling film. The wells were washed and the plasma diluted in work solution, PBS 1x with 10% FBS was added and incubated for 90 minutes at 100 rpm and 37 °C; wells were washed with WS. Next, 100 µL of secondary antibody (Anti-mouse IgG, horseradish peroxidase linked whole antibody from sheep, NA931V, GE Healthcare Life Science, UK) diluted 1:3,000 in working solution was added, and incubated for an additional 90 minutes, and then washed twice with WS and once with PBS 1x. After that, for the colorimetric reaction, 100 µL of 1% o-phenylenediamine (Lot#SLBT7214 Sigma, USA) solution in 50 mM phosphate citrate buffer, pH 5.5 and 0.03% hydrogen peroxide were added per well and the plates incubated at 37 °C under agitation for 15 minutes, protected from light. Finally, the reaction was stopped by adding 100 µL H2SO4 2 M and absorbance was read at 492 nm [15]. Specific anti-ASNase IgE (Mouse IgE ELISA kit Sigma RAB0799-1KT USA), PAF (PAF ELISA KIT CK-bio-16762, EZ Assays, Florida-USA) and MCP-4 (CCL13/MCP-4 ELISA KIT CK-bio-16613, EZ Assays, Florida-USA) quantifications were performed as the manufacturer’s instructions. Statistical analysis of IgG and IgE were performed by two-way ANOVA and Tukey’s multiple comparisons test and PAF and MCP-4 data were analysed with an ordinary one-way ANOVA and Tukey’s multiple comparison test on Prism 9.0 (GraphPad).
4.4.2.3. Pharmacokinetics Profile of ASNases
As defined in Figure 8C, blood was collected in Eppendorf tubes containing EDTA at 15 mg/mL, centrifuged at 8,000 g-force, 4 °C for 3 minutes and plasma was separated and stored at -80 °C until analysis. Asparaginase activity was measured as described by Cecconello et al. (2020). Briefly, 20 µL of plasma were diluted with 180 µL of a 2 mM aspartic acid β-hydroxamate (AHA) solution dissolved in 15 mM Tris HCl buffer, pH 7.3, supplemented with 0.015% (w/v) BSA, and incubated for 30 min at 37 °C. Next, 50 µL of the resulting supernatant were added to a new tube to react with 200 µL of oxine reagent, formed by one part of 2% 8-hydroxyquinoline dissolved in absolute ethanol (w/v) and three parts of 1 M Na2CO3. Afterward, the tube was heat at 95 °C for 1 minute, cooled for 10 minutes and finally absorbance was measured at 690 nm [56]. A standard curve was constructed from 0 to 2 U/mL under the same reaction conditions to interpolate the absorbance values obtained experimentally from mice plasma samples. Statistical analysis of asparaginase activity was performed at Prism 9.0 (GraphPad) using Multiple t-test and the two-stage linear step-up procedure of Benjamini, Krieger and Yekutieli, with Q = 1%.
Supplementary Materials
The following supporting information can be downloaded at: the website of this paper posted on Preprints.org.
Author Contributions
Individual contribution is detailed as follow: Conceptualization, G.R.L. and G.M.; methodology, G.R.L. and G.M.; formal analysis, G.R.L., J.M.C. and G.M.; experimentation, G.R.L., T.C.S., J.M.C., M.G.F., J.C.E. and M.G.M.; resources, G.M. and K.M.R..; writing—original draft preparation, G.R.L.; writing—review and editing, C.R.Y., K.M.R., T.C.S. and G.M.; supervision, G.M. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by State of São Paulo Research Foundation (FAPESP) grants 2018/15104-0, 2022/02456-0. G.M. received a Productivity Fellowship from the Brazilian National Counsel of Technological and Scientific Development (CNPq 306060/2022-1). G.R.L. and M.G.F. received fellowship from the Coordination of Improvement of Higher Education Personnel (CAPES-Brazil, finance code: 001). T.C.S. is supported by ‘chamada universal‘ from MCTIC/CNPq 28/2018 (423532/2018-9).
Institutional Review Board Statement
The animal study protocol was approved by the Ethical Committee for Animal Experimentation of the School of Pharmaceutical Sciences of the Universidade de São Paulo (protocol number CEUA/FCF 036.2020-P605).
Data Availability Statement
All data supporting the results can be found within the manuscript.
Acknowledgments
The authors would like to thank Dr Iris Munhoz Costa which contributed with experimental advice mainly related to in vitro data, as well as Mona Belaid for her invaluable collaboration at King´s College London.
Conflicts of Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- European Medicines Agency. ICH S6 (R1) Preclinical safety evaluation of biotechnology-derived pharmaceuticals - Scientific guideline | European Medicines Agency 1997. https://www.ema.europa.eu/en/ich-s6-r1-preclinical-safety-evaluation-biotechnology-derived-pharmaceuticals-scientific-guideline (accessed April 14, 2024).
- European Medicines Agency. ICH S9 Non-clinical evaluation for anticancer pharmaceuticals - Scientific guideline | European Medicines Agency 2010. https://www.ema.europa.eu/en/ich-s9-non-clinical-evaluation-anticancer-pharmaceuticals-scientific-guideline (accessed April 14, 2024).
- Costa-Silva TA, Costa IM, Biasoto HP, Lima GM, Silva C, Pessoa A, et al. Critical overview of the main features and techniques used for the evaluation of the clinical applicability of L-asparaginase as a biopharmaceutical to treat blood cancer. Blood Rev 2020;43. [CrossRef]
- Sengupta S, Biswas M, Gandhi K, Gota V, Sonawane A. Pre-Clinical Evaluation of Novel L-Asparaginase Mutants for the Treatment of Acute Lymphoblastic Leukemia. Blood 2021;138:4919–4919. [CrossRef]
- Fernandez CA, Smith C, Karol SE, Ramsey LB, Liu C, Pui CH, et al. Effect of premedications in a murine model of asparaginase hypersensitivity. J Pharmacol Exp Ther 2015;352:541–51. [CrossRef]
- Suresh SA, Ethiraj S, Rajnish KN. Toxicity Analysis of Recombinant L-asparaginase I and II in Zebrafish. Indian J Microbiol 2020;60:535. [CrossRef]
- Horvath TD, Chan WK, Pontikos MA, Martin LA, Du D, Tan L, et al. Assessment of l-Asparaginase Pharmacodynamics in Mouse Models of Cancer. Metabolites 2019;9. [CrossRef]
- Sengupta S, Biswas M, Gandhi KA, Gupta SK, Gera PB, Gota V, et al. Preclinical evaluation of engineered L-asparaginase variants to improve the treatment of Acute Lymphoblastic Leukemia. Transl Oncol 2024;43. [CrossRef]
- Rodrigues MAD, Pimenta M V., Costa IM, Zenatti PP, Migita NA, Yunes JA, et al. Influence of lysosomal protease sensitivity in the immunogenicity of the antitumor biopharmaceutical asparaginase. Biochem Pharmacol 2020;182:114230. [CrossRef]
- Ardalan N, Sepahi AA, Khavari-Nejad RA. Development of Escherichia coli asparaginase II for the Treatment of Acute Lymphocytic Leukemia: In Silico Reduction of asparaginase II Side Effects by a Novel Mutant (V27F). Asian Pac J Cancer Prev 2021;22:1137. [CrossRef]
- Munhoz Costa I, Schultz L, De Araujo B, Pedra B, Silva M, Leite M, et al. Recombinant L-asparaginase 1 from Saccharomyces cerevisiae: an allosteric enzyme with antineoplastic activity OPEN 2016. [CrossRef]
- Husain I, Sharma A, Kumar S, Malik F. Purification and Characterization of Glutaminase Free Asparaginase from Enterobacter cloacae: In-Vitro Evaluation of Cytotoxic Potential against Human Myeloid Leukemia HL-60 Cells. PLoS One 2016;11. [CrossRef]
- Brumano LP, da Silva FVS, Costa-Silva TA, Apolinário AC, Santos JHPM, Kleingesinds EK, et al. Development of L-asparaginase biobetters: Current research status and review of the desirable quality profiles. Front Bioeng Biotechnol 2019;6. [CrossRef]
- Saeed H, Hemida A, El-Nikhely N, Abdel-Fattah M, Shalaby M, Hussein A, et al. Highly efficient Pyrococcus furiosus recombinant L-asparaginase with no glutaminase activity: Expression, purification, functional characterization, and cytotoxicity on THP-1, A549 and Caco-2 cell lines. Int J Biol Macromol 2020;156:812–28. [CrossRef]
- Breland UM, Michelsen AE, Skjelland M, Folkersen L, Krohg-Sørensen K, Russell D, et al. Raised MCP-4 levels in symptomatic carotid atherosclerosis: An inflammatory link between platelet and monocyte activation. Cardiovasc Res 2010;86:265–73. [CrossRef]
- Horkheimer I, Schuster DP. THE ROLE OF PLATELET ACTIVATING FACTOR IN SEPSIS: A BENCH-TO-BEDSIDE REVIEW ADVANCES. ADVANCES IN SEPSIS 2002;2.
- Jacob S, Nair AB, Morsy MA. Dose Conversion Between Animals and Humans: A Practical Solution. Indian Journal of Pharmaceutical Education and Research 2022;56:600–7. [CrossRef]
- Wang S, Lai X, Deng Y, Song Y. Correlation between mouse age and human age in anti-tumor research: Significance and method establishment. Life Sci 2020;242:117242. [CrossRef]
- Nair A, Jacob S. A simple practice guide for dose conversion between animals and human. J Basic Clin Pharm 2016;7:27. [CrossRef]
- Silvânia M. P. Neves, Flavia de Moura Prates Ong, Lívia Duarte Rodrigues, Renata Alves dos Santos, Renata Spalutto Fontes, Roseni de Oliveira Santana. Manual de cuidados e procedimentos com animais de laboratório do Biotério de Produção e Experimentação da FCF-IQ/USP. São Paulo: 2013. [CrossRef]
- Rathod S, Ramsey M, Relling M V., Finkelman FD, Fernandez CA. Hypersensitivity reactions to asparaginase in mice are mediated by anti-asparaginase IgE and IgG and the immunoglobulin receptors FceRI and FcγRIII. Haematologica 2019;104:319–29. [CrossRef]
- Talbot SR, Biernot S, Bleich A, van Dijk RM, Ernst L, Häger C, et al. Defining body-weight reduction as a humane endpoint: a critical appraisal. Lab Anim 2020;54:99–110. [CrossRef]
- Morton DB, Griffiths PH. Guidelines on the recognition of pain, distress and discomfort in experimental animals and an hypothesis for assessment. Vet Rec 1985;116:431–6. [CrossRef]
- Gavin HE, Satchell KJF. Surface hypothermia predicts murine mortality in the intragastric Vibrio vulnificus infection model. BMC Microbiol 2017. [CrossRef]
- Vaquero J, Bélanger M, James L, Herrero R, Desjardins P, Côté J, et al. Mild Hypothermia Attenuates Liver Injury and Improves Survival in Mice With Acetaminophen Toxicity 2007. [CrossRef]
- Bernard SA, Gray TW, Buist MD, Jones BM, Silvester W, Gutteridge G, et al. Treatment of Comatose Survivors of Out-of-Hospital Cardiac Arrest with Induced Hypothermia. New England Journal of Medicine 2002;346:557–63. [CrossRef]
- Holzer M. Mild Therapeutic Hypothermia to Improve the Neurologic Outcome after Cardiac Arrest. New England Journal of Medicine 2002;346:549–56. [CrossRef]
- Eicher DJ, Wagner CL, Katikaneni LP, Hulsey TC, Bass WT, Kaufman DA, et al. Moderate hypothermia in neonatal encephalopathy: Efficacy outcomes. Pediatr Neurol 2005;32:11–7. [CrossRef]
- Shankaran S, Laptook AR, Ehrenkranz RA, Tyson JE, McDonald SA, Donovan EF, et al. Whole-Body Hypothermia for Neonates with Hypoxic–Ischemic Encephalopathy. New England Journal of Medicine 2005;353:1574–84. [CrossRef]
- Stewart CR, Landseadel JP, Gurka MJ, Fairchild KD. Hypothermia increases interleukin-6 and interleukin-10 in juvenile endotoxemic mice $watermark-text $watermark-text $watermark-text. Pediatr Crit Care Med 2010;11:109–16. [CrossRef]
- Laptook AR, Corbett RJT, Sterett R, Garcia D, Tollefsbol G. Quantitative relationship between brain temperature and energy utilization rate measured in Vivo using 31P and 1H magnetic resonance spectroscopy. Pediatr Res 1995;38:919–25. [CrossRef]
- Globus MY -T, Alonso O, Dietrich WD, Busto R, Ginsberg MD. Glutamate Release and Free Radical Production Following Brain Injury: Effects of Posttraumatic Hypothermia. J Neurochem 1995;65:1704–11. [CrossRef]
- Gunn AJ, Gunn TR, De Haan HH, Williams CE, Gluckman PD. Dramatic neuronal rescue with prolonged selective head cooling after ischemia in fetal lambs. Journal of Clinical Investigation 1997;99:248–56. [CrossRef]
- Ginsberg MD, Belayev L. Biological and molecular mechanisms of hypothermic neuroprotection. Therapeutic Hypothermia, CRC Press; 2004, p. 85–141. [CrossRef]
- Wei H, Yin M, Lu Y, Yang Y, Li B, Liao X-X, et al. Mild hypothermia improves neurological outcome in mice after cardiopulmonary resuscitation through Silent Information Regulator 1-actviated autophagy. Cell Death Discov 2019;5:129. [CrossRef]
- Morowski M, Vögtle T, Kraft P, Kleinschnitz C, Stoll G, Nieswandt B. Only severe thrombocytopenia results in bleeding and defective thrombus formation in mice. Blood 2013;121:4938–47. [CrossRef]
- Michael B, Yano B, Sellers RS, Perry R, Morton D, Roome N, et al. Evaluation of Organ Weights for Rodent and Non-Rodent Toxicity Studies: A Review of Regulatory Guidelines and a Survey of Current Practices. Http://DxDoiOrg/101080/01926230701595292 2017;35:742–50. [CrossRef]
- Koo SH. Nonalcoholic fatty liver disease: molecular mechanisms for the hepatic steatosis. Clin Mol Hepatol 2013;19:210–5. [CrossRef]
- Alex S, Boss A, Heerschap A, Kersten S. Exercise training improves liver steatosis in mice. Nutr Metab (Lond) 2015;12:1–11. [CrossRef]
- Robinson MW, Harmon C, O’Farrelly C. Liver immunology and its role in inflammation and homeostasis. Cellular & Molecular Immunology 2016 13:3 2016;13:267–76. [CrossRef]
- Van Der Meer LT, Terry SYA, Van Ingen Schenau DS, Andree KC, Franssen GM, Roeleveld DM, et al. In vivo imaging of antileukemic drug asparaginase reveals a rapid macrophage-mediated clearance from the bone marrow. Journal of Nuclear Medicine 2017;58:214–20. [CrossRef]
- Céspedes N, Donnelly EL, Lowder C, Hansten G, Wagers D, Briggs AM, et al. Mast Cell Chymase/Mcpt4 Suppresses the Host Immune Response to Plasmodium yoelii, Limits Malaria-Associated Disruption of Intestinal Barrier Integrity and Reduces Parasite Transmission to Anopheles stephensi. Front Immunol 2022;13. [CrossRef]
- Pejler G, Rönnberg E, Waern I, Wernersson S. Mast cell proteases: multifaceted regulators of inflammatory disease. Blood 2010;115:4981–90. [CrossRef]
- Madjene LC, Danelli L, Dahdah A, Vibhushan S, Bex-Coudrat J, Pacreau E, et al. Mast cell chymase protects against acute ischemic kidney injury by limiting neutrophil hyperactivation and recruitment. Kidney Int 2020;97:516–27. [CrossRef]
- Holland M, Castro F V., Alexander S, Smith D, Liu J, Walker M, et al. RAC2, AEP, and ICAM1 expression are associated with CNS disease in a mouse model of pre-B childhood acute lymphoblastic leukemia. Blood 2011;118:638–49. [CrossRef]
- Patel N, Krishnan S, Offman MN, Krol M, Moss CX, Leighton C, et al. A dyad of lymphoblastic lysosomal cysteine proteases degrades the antileukemic drug L-asparaginase. Journal of Clinical Investigation 2009;119:1964–73. [CrossRef]
- Qi Q, Obianyo O, Du Y, Fu H, Li S, Ye K. Blockade of Asparagine Endopeptidase Inhibits Cancer Metastasis. J Med Chem 2017;60:7244–55. [CrossRef]
- Riccardi R, Holcenberg S, Glaubiger D, Wood J, Poplack D. L-asparaginase pharmacokinetics and asparagine levels in cerebrospinal fluid of rhesus monkeys and humans . Cancer Res 1981;11:4554–8.
- Asselin B, Rizzari C. Asparaginase pharmacokinetics and implications of therapeutic drug monitoring. Leuk Lymphoma 2015;56:2273. [CrossRef]
- Purwaha P, Lorenzi PL, Silva LP, Hawke DH, Weinstein JN. Targeted metabolomic analysis of amino acid response to L-asparaginase in adherent cells. Metabolomics 2014;10:909–19. [CrossRef]
- Saeed H, Hemida A, Abdel-Fattah M, Eldoksh A, Shalaby M, Nematalla H, et al. Pseudomonas aeruginosa recombinant L-asparaginase: Large scale production, purification, and cytotoxicity on THP-1, MDA-MB-231, A549, Caco2 and HCT-116 cell lines. Protein Expr Purif 2021;181:105820. [CrossRef]
- Blachier J, Cleret A, Guerin N, Gil C, Fanjat JM, Tavernier F, et al. L-asparaginase anti-tumor activity in pancreatic cancer is dependent on its glutaminase activity and resistance is mediated by glutamine synthetase. Exp Cell Res 2023;426:113568. [CrossRef]
- Karpel-Massler G, Ramani D, Shu C, Halatsch ME, Westhoff MA, Bruce JN, et al. Metabolic reprogramming of glioblastoma cells by L-asparaginase sensitizes for apoptosis in vitro and in vivo. Oncotarget 2016;7:33512. [CrossRef]
- Schalk AM, Lavie A. Structural and Kinetic Characterization of Guinea Pig l-Asparaginase Type III. Biochemistry 2014;53:2318. [CrossRef]
- Munhoz Costa I, Custódio Moura D, Meira Lima G, Pessoa A, Oresco dos Santos C, de Oliveira MA, et al. Engineered asparaginase from Erwinia chrysanthemi enhances asparagine hydrolase activity and diminishes enzyme immunoreactivity - a new promise to treat acute lymphoblastic leukemia. Journal of Chemical Technology and Biotechnology 2022;97:228–39. [CrossRef]
- Cecconello DK, Rechenmacher C, Werlang I, Zenatti PP, Yunes JA, Alegretti AP, et al. Implementation of the asparaginase activity assessment technique for clinical use: experience of a Brazilian Center. Sci Rep 2020;10:21481. [CrossRef]
Figure 1.
(A) Asparaginase activity of WT, S206C, P40S and P40S/S206C enzymes. The enzyme specific activity is represented as the slope of the linear regression equation of plotted µmole NH3/min against the milligrams of enzyme (U/mg). n=3. (B) Glutaminase activity of WT, S206C, P40S and P40S/S206C enzymes. The slope value corresponds to the glutaminase specific activity in U/mg. As well as asparaginase activity, µmole of NH3 released was quantified by Nessler´s reagent, n=3.
Figure 1.
(A) Asparaginase activity of WT, S206C, P40S and P40S/S206C enzymes. The enzyme specific activity is represented as the slope of the linear regression equation of plotted µmole NH3/min against the milligrams of enzyme (U/mg). n=3. (B) Glutaminase activity of WT, S206C, P40S and P40S/S206C enzymes. The slope value corresponds to the glutaminase specific activity in U/mg. As well as asparaginase activity, µmole of NH3 released was quantified by Nessler´s reagent, n=3.

Figure 2.
Kinetic profile of a commercial asparaginase Leuginase® (A), and the enzymes produced on our laboratory, WT (B), S206C (C) and P40S (D) ASNases. The µmoles of L-aspartate/minute released after asparaginase hydrolysis of asparagine was quantified by the coupled enzymatic reaction, where for each mole of L-aspartate produced after L-asparagine hydrolysis one mole of NADH is oxidized to NAD+ which decrease in absorbance was continuously measured at 340 nm and 37°C. The analysis was performed on Prism 9 for Enzyme Kinetics - Substrate vs. Velocity with Michaelis-Menten equation.
Figure 2.
Kinetic profile of a commercial asparaginase Leuginase® (A), and the enzymes produced on our laboratory, WT (B), S206C (C) and P40S (D) ASNases. The µmoles of L-aspartate/minute released after asparaginase hydrolysis of asparagine was quantified by the coupled enzymatic reaction, where for each mole of L-aspartate produced after L-asparagine hydrolysis one mole of NADH is oxidized to NAD+ which decrease in absorbance was continuously measured at 340 nm and 37°C. The analysis was performed on Prism 9 for Enzyme Kinetics - Substrate vs. Velocity with Michaelis-Menten equation.
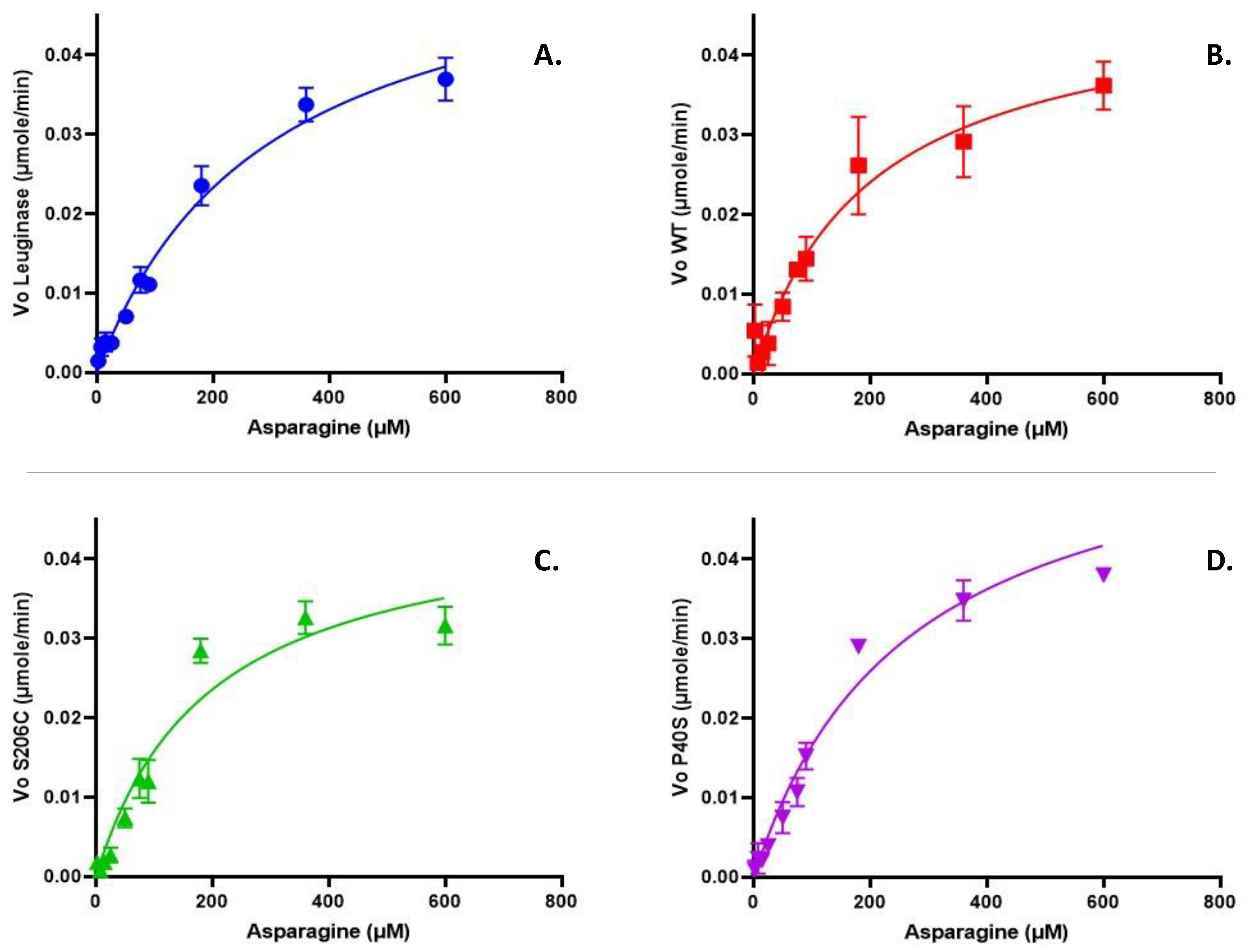
Figure 3.
Animal health and wellness monitoring. (A) Weight measured on days 0, 3, 5, 10, 14, 18 and 23 of each group (Control, and ASNase treated WT, S206C and P40S) under a scheme of two doses of 1,050 U/Kg on days 0 and 14 and a final dose of 5,250 U/Kg on day 23. Standard deviation is shown by vertical bar, n=3. No significant differences were found between groups or days of treatment. (B) Body temperature values for each enzyme treated group measured for two hours after the challenge injection of 5,250 U/Kg. Standard deviation is shown by vertical bar. n=3. Statistical differences relative to control - *: p value <0.05.
Figure 3.
Animal health and wellness monitoring. (A) Weight measured on days 0, 3, 5, 10, 14, 18 and 23 of each group (Control, and ASNase treated WT, S206C and P40S) under a scheme of two doses of 1,050 U/Kg on days 0 and 14 and a final dose of 5,250 U/Kg on day 23. Standard deviation is shown by vertical bar, n=3. No significant differences were found between groups or days of treatment. (B) Body temperature values for each enzyme treated group measured for two hours after the challenge injection of 5,250 U/Kg. Standard deviation is shown by vertical bar. n=3. Statistical differences relative to control - *: p value <0.05.
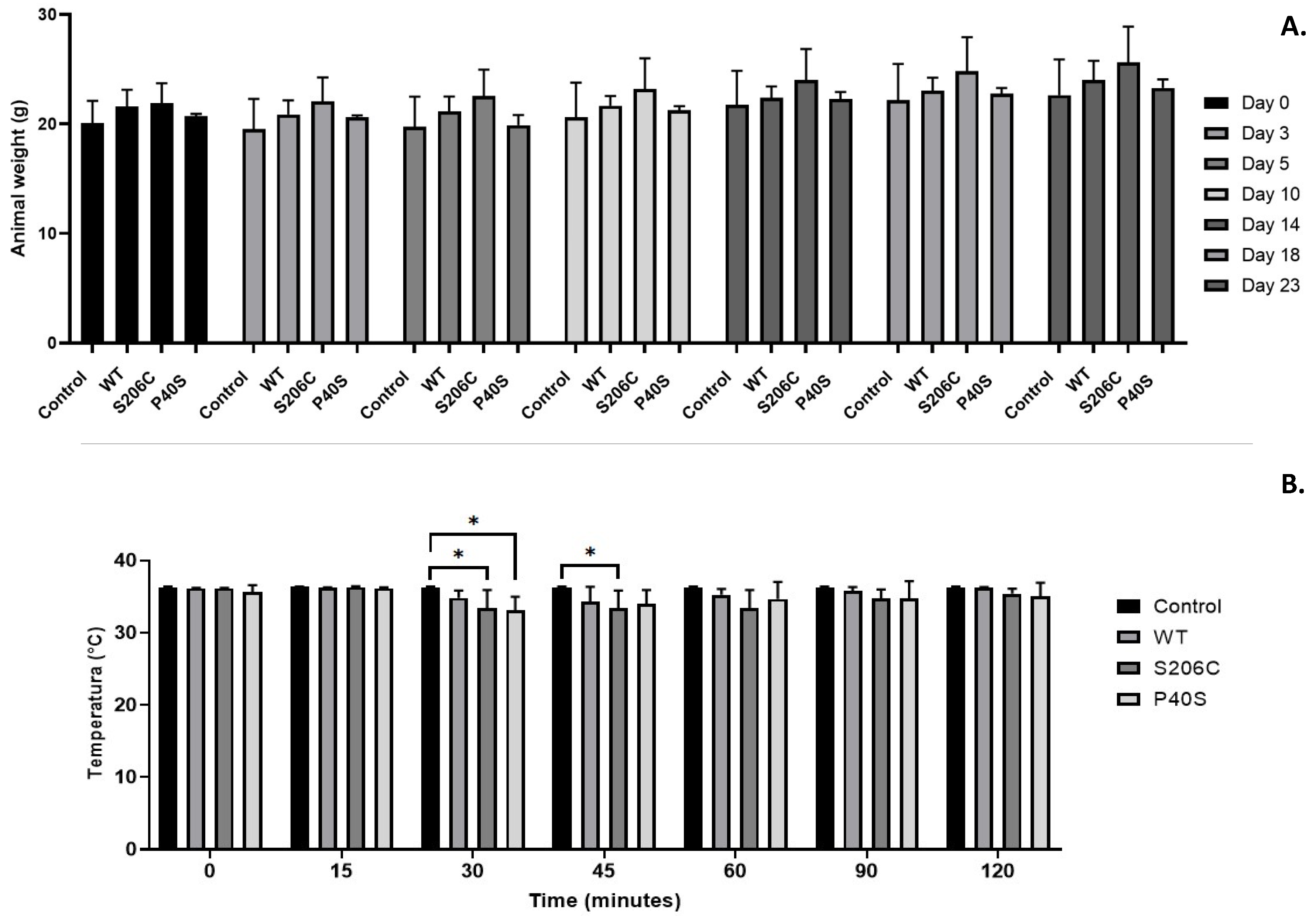
Figure 4.
Blood biometry after challenge injection. (A) Haematocrit percentage, (B) granulocytes concentration and (C) platelets count for all experimental groups. Standard deviation is shown by vertical bar, n=3, *: p value <0.05, **: p value <0.005, ***: p value <0.0005. No statistically significant differences were observed on haematocrit percentage or granulocytes comparing the groups.
Figure 4.
Blood biometry after challenge injection. (A) Haematocrit percentage, (B) granulocytes concentration and (C) platelets count for all experimental groups. Standard deviation is shown by vertical bar, n=3, *: p value <0.05, **: p value <0.005, ***: p value <0.0005. No statistically significant differences were observed on haematocrit percentage or granulocytes comparing the groups.
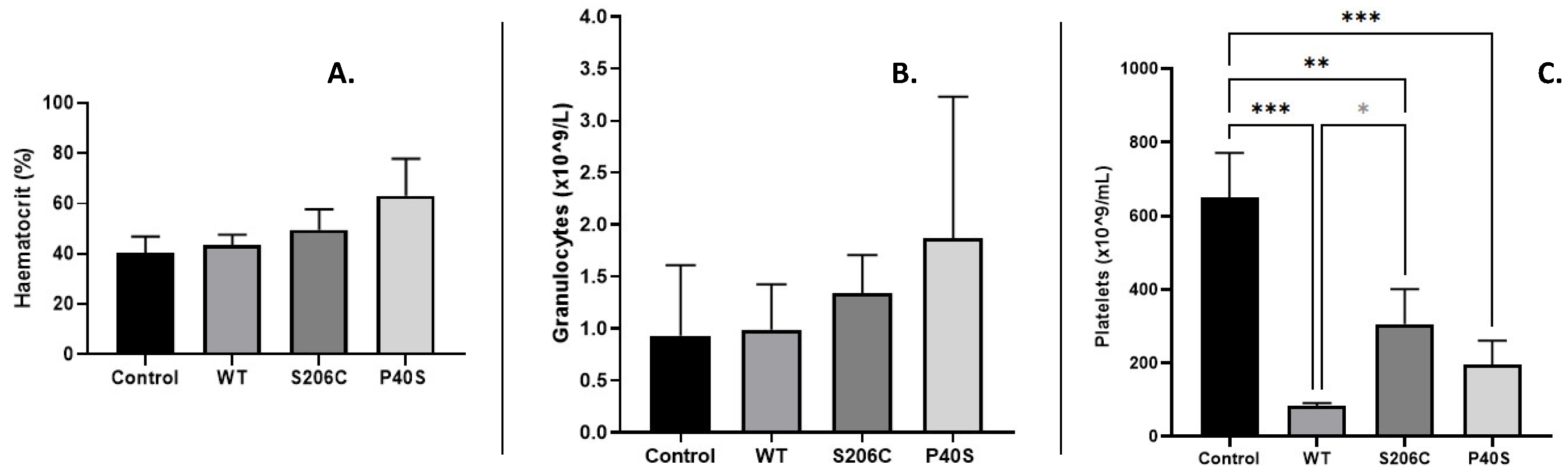
Figure 5.
Antibody anti-ASNase quantification by ELISA of (A) IgG and (B) IgE after treatment scheme detailed in Figure 1B. Standard deviation is shown by vertical bar, n=5, ***: p value <0.0005, ****: p value <0.00005.
Figure 5.
Antibody anti-ASNase quantification by ELISA of (A) IgG and (B) IgE after treatment scheme detailed in Figure 1B. Standard deviation is shown by vertical bar, n=5, ***: p value <0.0005, ****: p value <0.00005.
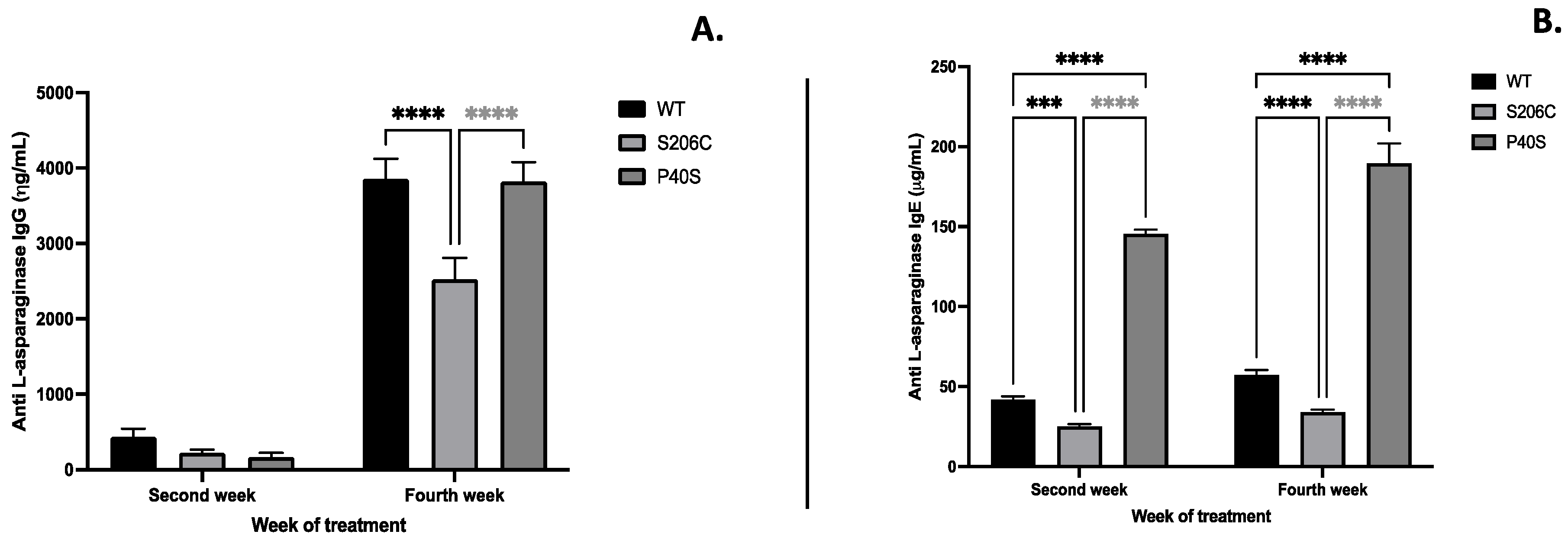
Figure 6.
(A) PAF and (B) MCP-4 quantification by ELISA on the fourth week after treatment scheme detailed in Figure 1B. Standard deviation is shown by vertical bar, n=5, *: p value <0.05, **: p value <0.005, ***: p value <0.0005.
Figure 6.
(A) PAF and (B) MCP-4 quantification by ELISA on the fourth week after treatment scheme detailed in Figure 1B. Standard deviation is shown by vertical bar, n=5, *: p value <0.05, **: p value <0.005, ***: p value <0.0005.
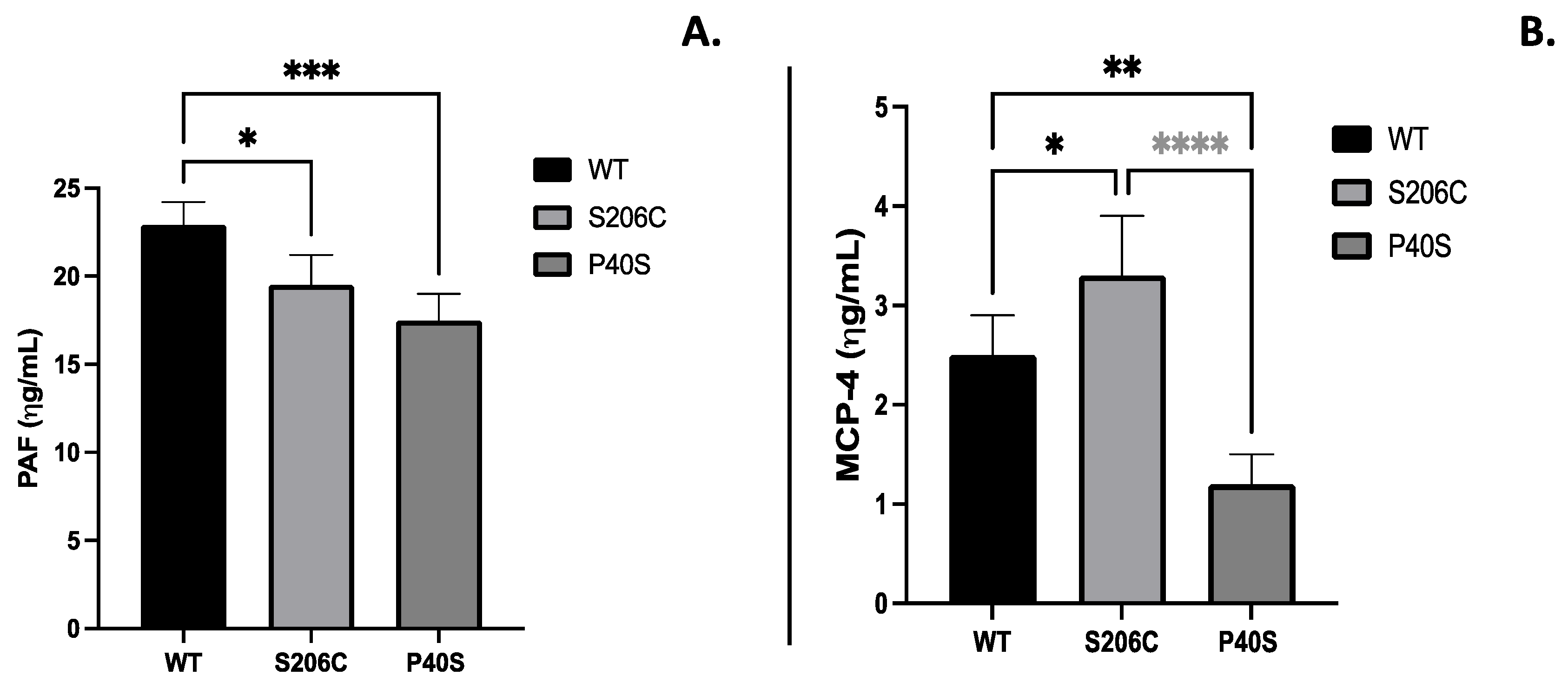
Figure 7.
Asparaginase activity measured in mice plasma after 2, 6, 12 and 24 hours of one single injection dose of 1050 U/Kg of each ASNase, WT and mutants, quantified by oxine method. Standard deviation is shown by vertical bar, n=5, ***: p value <0.0005, ****: p value <0.00005.
Figure 7.
Asparaginase activity measured in mice plasma after 2, 6, 12 and 24 hours of one single injection dose of 1050 U/Kg of each ASNase, WT and mutants, quantified by oxine method. Standard deviation is shown by vertical bar, n=5, ***: p value <0.0005, ****: p value <0.00005.
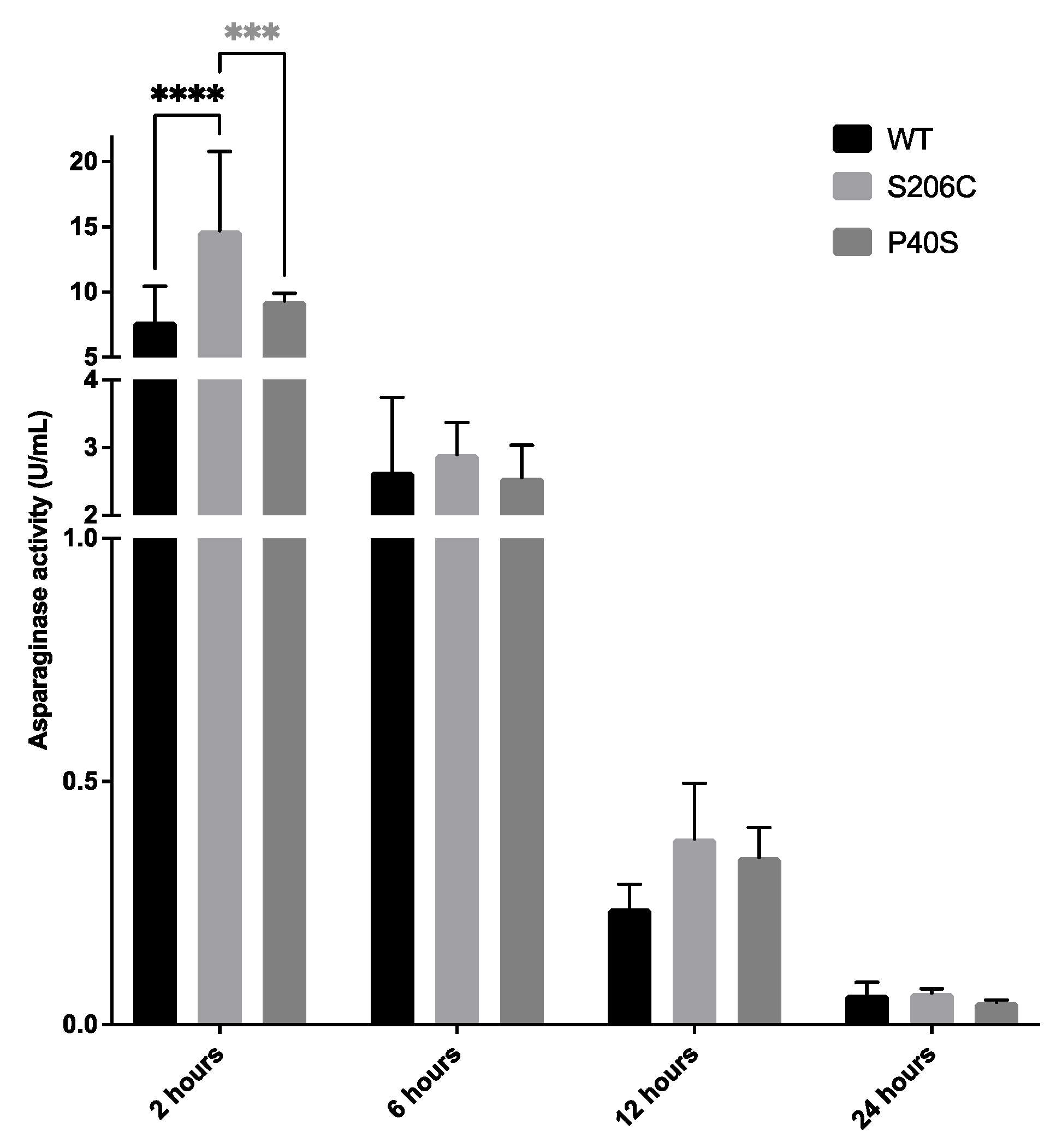
Figure 8.
Flowchart of experimental design for in vivo studies. (A): Multi-organs toxicological effects of animal exposure to multiple doses of ASNase (WT and mutants) for 23 days experimentation, with continuous weight control on days 0,3,5,10,14,18 and 23. Temperature was measured for two hours after each ASNase injection on days 0, 14 and 23. On day 23, blood sample was collected to biometry analysis and organs were collected and treated with haematoxylin and eosin (H&E stain). (B): Antigenic and Allergenic potential evaluation of each proteoform after two sensitization doses per IgG, IgE, PAF and MCP-4 plasmatic quantification. (C): Pharmacokinetics profile of ASNase (WT and mutants) for 24 hours, with enzyme activity detection at 2,6,12 and 24 hours after injection. Asparaginase activity was measured by oxine method in plasma.
Figure 8.
Flowchart of experimental design for in vivo studies. (A): Multi-organs toxicological effects of animal exposure to multiple doses of ASNase (WT and mutants) for 23 days experimentation, with continuous weight control on days 0,3,5,10,14,18 and 23. Temperature was measured for two hours after each ASNase injection on days 0, 14 and 23. On day 23, blood sample was collected to biometry analysis and organs were collected and treated with haematoxylin and eosin (H&E stain). (B): Antigenic and Allergenic potential evaluation of each proteoform after two sensitization doses per IgG, IgE, PAF and MCP-4 plasmatic quantification. (C): Pharmacokinetics profile of ASNase (WT and mutants) for 24 hours, with enzyme activity detection at 2,6,12 and 24 hours after injection. Asparaginase activity was measured by oxine method in plasma.
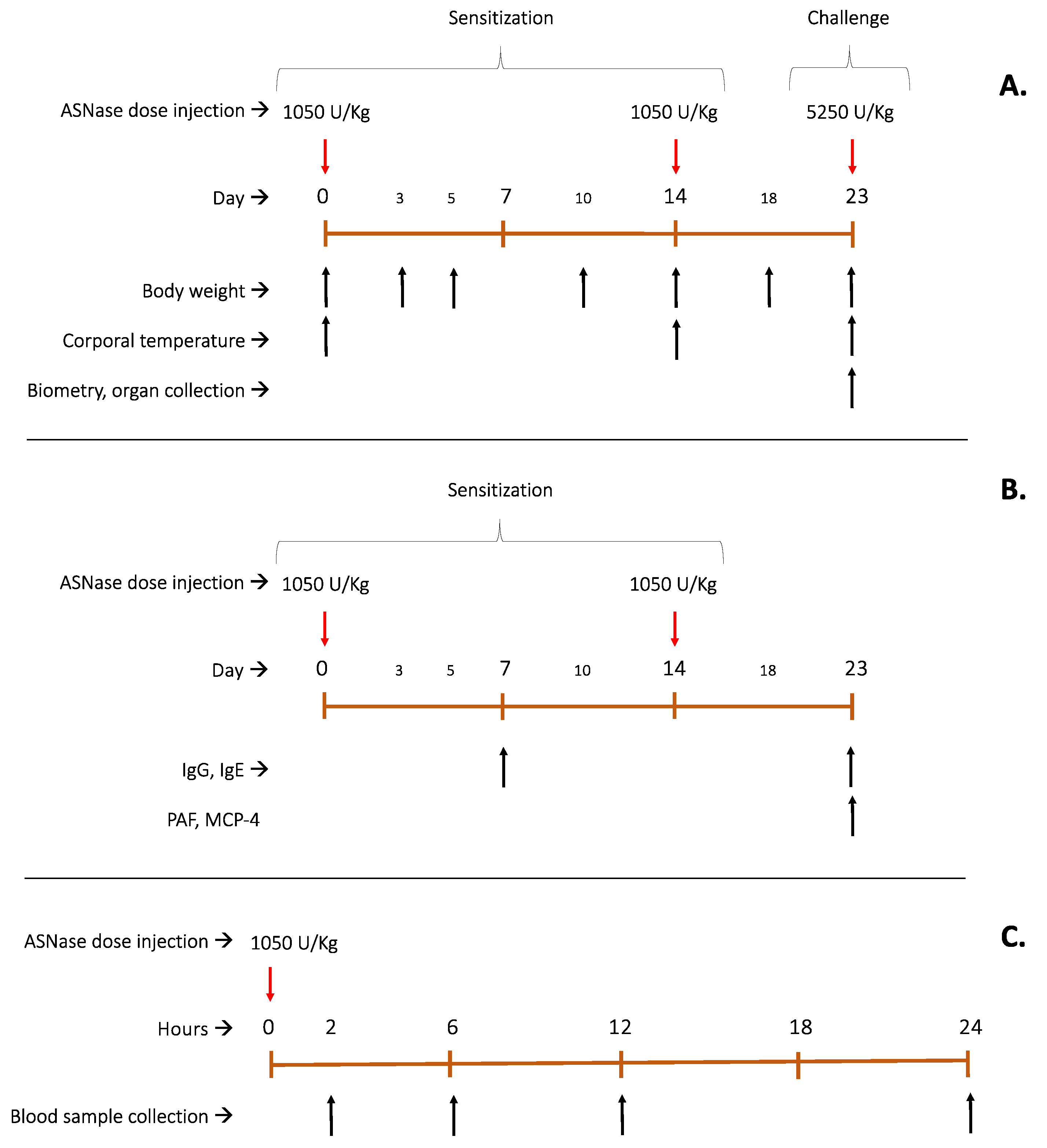

Table 1.
Specific activity for L-asparagine and L-glutamine of WT, S206C, P40S and P40S/S206C enzymes and their relative glutaminase activity percentage. Statistical differences with WT- **: p value <0.005; ***: p value <0.0005, ****: p value <0.00005. Statistical differences with S206C- ##: p value <0.005; ###: p value <0.0005, ####: p value <0.00005. Statistical differences with P40S- ++: p value <0.005.
Table 1.
Specific activity for L-asparagine and L-glutamine of WT, S206C, P40S and P40S/S206C enzymes and their relative glutaminase activity percentage. Statistical differences with WT- **: p value <0.005; ***: p value <0.0005, ****: p value <0.00005. Statistical differences with S206C- ##: p value <0.005; ###: p value <0.0005, ####: p value <0.00005. Statistical differences with P40S- ++: p value <0.005.
| Enzyme | ASNase activity (U/mg) | GLNase activity (U/mg) |
Relative GLNase activity % |
|---|---|---|---|
| WT | 76.22 ±0.82 | 1.79 ±0.09 | 2.35 |
| S206C | 74.86 ±0.32 | 1.73 ±0.04 | 2.31 |
| P40S | 60.82 ±0.72***### | 1.27 ±0.02**## | 2.09 |
| P40S/S206C | 52.88 ±1.12****####++ | 1.76 ±0.05++ | 3.33 |
Table 2.
Asparaginase activity of WT, S206C, P40S and P40S/S206C enzymes incubated with PBS 1x or 10% of human serum (HS) at 0 and 96 hours and the percentage of residual activity. n=2. Statistical differences from 0 hours to 96 hours within each row - *: p value <0.05; **: p value <0.005, ***: p value <0.0005.
Table 2.
Asparaginase activity of WT, S206C, P40S and P40S/S206C enzymes incubated with PBS 1x or 10% of human serum (HS) at 0 and 96 hours and the percentage of residual activity. n=2. Statistical differences from 0 hours to 96 hours within each row - *: p value <0.05; **: p value <0.005, ***: p value <0.0005.
| Enzyme |
ASNase activity (0 hours) |
ASNase activity (96 hours) |
Residual activity |
|---|---|---|---|
| WT-PBS 1x | 88.1 ±19.8 | 51.4 ±10.1** | 58.3% |
| WT-HS | 87.6 ±14.8 | 61.7 ±13.2 | 70.4% |
| S206C-PBS 1x | 74.7 ±5.0 | 43.9 ±14.8* | 58.8% |
| S206C-HS | 78.0 ±3.3 | 64.8 ±13.2 | 83.2% |
| P40S-PBS 1x | 69.4 ±2.5 | 28.0 ±10.8** | 40.3% |
| P40S-HS | 69.5 ±2.0 | 53.1 ±15.1 | 76.4% |
| P40S/S206C-PBS 1x | 61.1 ±1.6 | 12.1 ±5.6*** | 19.7% |
| P40S/S206C-HS | 62.8 ±1.7 | 27.6 ±9.4* | 44.0% |
Table 3.
Kinetic parameters obtained for commercial Leuginase®, and WT, S206C and P40S ASNases. The analysis was performed on Prism 9 for Enzyme Kinetics - Substrate vs. Velocity with non-linear regression - Michaelis-Menten equation fit. n=2. Statistical differences with Leuginase - *: p value <0.05. Statistical differences with S206C- #: p value <0.05.
Table 3.
Kinetic parameters obtained for commercial Leuginase®, and WT, S206C and P40S ASNases. The analysis was performed on Prism 9 for Enzyme Kinetics - Substrate vs. Velocity with non-linear regression - Michaelis-Menten equation fit. n=2. Statistical differences with Leuginase - *: p value <0.05. Statistical differences with S206C- #: p value <0.05.
| Leuginase | WT | S206C | P40S | ||
|---|---|---|---|---|---|
| Kinetic parameters | Vmax (µmole/min) |
0.05728 ±0.0063 | 0.04787 ±0.0073 | 0.04647 ±0.0065 | 0.06001 ±0.0055 |
| KM (µM) |
291.3 ±23.6 | 197.6 ±17.9* | 194.0 ±12.5* | 262.7.2 ±9.8# | |
| Goodness of fit | R2 | 0.9744 | 0.9203 | 0.9362 | 0.9389 |
Table 4.
IC50 (U/mL) for different blood cancer cell lines (MTT assay) when treated for 72 hours with ASNases WT, S206C and P40S. n=2, Statistical differences with WT - *: p value <0.05. No significant differences were observed between mutants.
Table 4.
IC50 (U/mL) for different blood cancer cell lines (MTT assay) when treated for 72 hours with ASNases WT, S206C and P40S. n=2, Statistical differences with WT - *: p value <0.05. No significant differences were observed between mutants.
| Cell line | ASNase type | ||
|---|---|---|---|
| WT | S206C | P40S | |
| MOLT-4 | 0.112 ±0.020 | 0.076 ±0.012 | 0.045 ±0.013* |
| REH | 0.114 ±0.019 | 0.149 ±0.031 | 0.114 ±0.002 |
Table 5.
IC50 (U/mL) for different solid cancer cell lines (MTT assay) treated for 72 hours with ASNases WT, S206C and P40S. n=3. Statistical differences with WT - *: p value <0.05; **: p value <0.005; ***: p value <0.0005. Statistical differences with S206C- #: p value <0.05; ##: p value <0.005.
Table 5.
IC50 (U/mL) for different solid cancer cell lines (MTT assay) treated for 72 hours with ASNases WT, S206C and P40S. n=3. Statistical differences with WT - *: p value <0.05; **: p value <0.005; ***: p value <0.0005. Statistical differences with S206C- #: p value <0.05; ##: p value <0.005.
| CELL LINE | ASNASE TYPE | ||
|---|---|---|---|
| WT | S206C | P40S | |
| MDA-MB-231 | 5.9 ±0.075 | 6.3 ±0.434 | 7.5 ±0.584**# |
| Caco-2 | 9.2 ±0.522 | 7.1±0.249* | 9.0 ±1.074# |
| SK-OV-3 | 8.5 ±1.344 | 11.9 ±0.43* | 9.4 ±1.013 |
| PANC-1 | 3.3 ±0.166 | 2.7 ±0.075** | 2.9 ±0.201* |
| U-87 MG | 1.5 ±0.107 | 1.2 ±0.099* | 0.8 ±0.098***## |
Table 6.
Average weight of organs collected after 23 days of treatment with multiple doses of ASNase (groups WT and mutants) and control group, n=3. Statistical difference with control group - *: p value <0.05. No statistical significance was identified between WT and mutants.
Table 6.
Average weight of organs collected after 23 days of treatment with multiple doses of ASNase (groups WT and mutants) and control group, n=3. Statistical difference with control group - *: p value <0.05. No statistical significance was identified between WT and mutants.
| Organ | Weight (mg) | |||
|---|---|---|---|---|
| Control | WT | S206C | P40S | |
| Heart | 147.1 ±7.3 | 162.8 ±4.0 | 151.9 ±3.7 | 153.8 ±11.5 |
| Spleen | 99.1 ±18.0 | 100.2 ±2.3 | 94.3 ±10.3 | 91.7 ±3.2 |
| Liver | 1529.2 ±38.2 | 1846.7* ±50.1 | 1686.7 ±163.7 | 1827.0* ±34.7 |
| Kidney | 317.5 ±27.0 | 291.9 ±1.1 | 264.8 ±7.2 | 251.5 ±12.5 |
| Pancreas | 940.0 ±2.9 | 774.8* ±21.3 | 732.2* ±30.2 | 713.3* ±38.1 |
| Thymus | 85.6 ±13.9 | 99.4 ±11.7 | 57.6 ±2.0 | 84.7 ±7.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



