
Submitted:
21 April 2024
Posted:
23 April 2024
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a positive sense single stranded RNA genome containing virus which has infected millions of people all over the world particularly China. The virus has been mutating rapidly enough resulting in the emergence of new variants and sub-variants which have reportedly been spread from Wuhan city of China, the epicenter of the virus, to the rest of China and all over the world. The occurrence of mutations in the viral genome especially in the viral spike protein region has resulted in the evolution of multiple variants and sub-variants which gives the virus benefit of host immune evasion and render the modern-day vaccines and therapeutics ineffective. Therefore, there is a continuous need to study the genetic characteristics and evolutionary dynamics of the SARS-CoV-2 variants. Hence, in this study, a total of 833 complete genomes of SARS-CoV-2 variants (including the wild-type or reference sequence) from Taiyuan and Wuhan cities of China were genetically characterized and studies their phylogenetic and evolutionary dynamics using phylogenetics, genetic similarity, and phylogenetic network analysis. Our results show a lot of useful information regarding the evolutionary dynamics of the viral variants, their genetic similarities/dissimilarities, and their phylogenetic relationships. It has been reported in this study that the SARS-CoV-2 variants have nine recombinant events which suggest very important information regarding the viral evolution. In addition, the phylogenetic network analysis shows the number of mutations resulting in emergence and clustering of the viral variants of both cities which reveal significant information regarding the phylodynamics of the virus. This study, to the best of our knowledge, is the first ever genetic comparative study of Taiyuan and Wuhan cities. This study will help better understand the virus, cope with the emergence, and spread of new variants at local level as well as at international level and inform the public health authorities to make better informed decisions in designing new viral vaccines and therapeutics.
Keywords:
SARS-CoV-2
; Phylogenetic tree
; Phylogenetic network
; Evolutionary dynamics
1. Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) belongs to the subgenus sarbecovirus under genus beta coronavirus. It originated from Wuhan city of China infecting humans in December 2020 [1,2]. It caused the disease coronavirus disease 2019 (COVID-19) which was declared a pandemic by the World Health Organization (WHO) in March 2020 [3]. It possibly originated from bats as confirmed by the phylogenetic analysis studies showing high similarity to the bat coronaviruses [4]. Globally, a total number of 773.8198 million people have been infected with cumulative deaths of 7.0105 million people with this virus so far as of December 31, 2023 (https://covid19.who.int/). In China, from January 3, 2020 to December 31, 2023, a total of 99.3 million people have been infected with cumulative deaths of 121,900, as reported to the WHO (https://covid19.who.int/region/wpro/country/cn).
SARS-CoV-2 is an enveloped, single-stranded, positive-sense RNA containing virus with a genome size of approximately 29.9kb which encodes four structural proteins (SPs) (spike-S, envelope-E, membrane-E and nucleocapsid-N) and 16 non-structural proteins (NSPs) (NSP 1-16) [5,6]. In addition, it also encodes several accessory proteins, ORF3a-b, ORF6, ORF7a-b, ORF8, ORF9b-c and ORF10 [6].
Due to high mutation rate because of lack of proofreading mechanism, the presence of more conducive environment and genetic recombination mechanisms, the SARS-CoV-2 has the advantage of evolving so rapidly so that resulting in the emergence of new variants and subtypes like Alpha, Beta, Gamma, Delta, Kappa, Omicron, and others. Within Omicron, there are many subtypes like Omicron BA.1, BA.2, BA.3, BA.4, BA.5, BQ.1.1, XBB.1.5, FY.3, EG.5.1, FU.1, HK.3, etc. [7,8,9,10,11]. The mutation rate of SARS-CoV-2 is ~1×10-3 nucleotide substitutions per year [12,13]. The presence of these mutations in the structural proteins especially the spike protein renders the anti-SARS-CoV-2 vaccines therapeutically ineffective [14].
All the information related to the full genome sequences of SARS-CoV-2 is available on the Global Initiative on Sharing All Influenza Data (GISAID) website which helps the researchers in the relevant fields to study on these emerging and rapidly evolving sequences to better understand the origin of the disease and the other aspects including the phylogenetics and phylodynamics of the virus [15]. In-depth genetic analysis of complete genomes of SARS-CoV-2 variants is crucial for understanding the evolutionary dynamics of the virus. Many researchers used different bioinformatics tools and techniques to study genetic characteristics of the SARS-CoV-2 variants from different locations. These types of analysis are very important to ultimately control the viral spread [16,17,18,19].
Hence, in this study, we characterized a total of 833 complete genomes of SARS-CoV-2 variants (including the wild-type or reference sequence) from two cities of China, that are Taiyuan and Wuhan, using lineage and subtyping analysis, phylogenetic tree, genetic similarity, and phylogenetic network analysis to help reveal a lot of useful information regarding the evolutionary dynamics of the viral variants, their genetic similarities/dissimilarities, and their phylogenetic relationships.
This comparative study, to the best of our knowledge, is the first study which genetically characterized the complete genomes of SARS-CoV-2 variants from Taiyuan and Wuhan cities of China. With the help of this study, researchers can gain valuable insights into the evolutionary and underlying adaptation mechanisms of SARS-CoV-2 at the local level. This will help the public health facilities to make better informed decisions for controlling the SARS-CoV-2 infection in particular and other viral infections in general at an early stage of the epidemic by designing new vaccines based on the genetic characteristics of the viral variants.
2. Materials and Methods
2.1. Sequences Acquisition
On December 7, 2023, we retrieved FASTA sequence congregations along with their rational meta data from GISAID EpiCoV server (www.epicov.org)[15] using the data filter as: virus name: hCoV-19, host: human, location: Asia/China/Shanxi/Taiyuan and Asia/China/Wuhan, complete, high coverage, clade: all. A total of 832 sequences were resulted of which 485 were from Wuhan and 347 from Taiyuan. The SARS-CoV-2 reference sequence (GenBank accession number: NC_045512.2) was accessed from NCBI database (www.ncbi.nlm.nih.gov).
2.2. Genome Annotation
SARS-CoV-2 Wuhan-Hu-1 (Accession No.: NC_045512) genome annotation was done by using Unipro UGENE v48.0 software (https://ugene.net/)[20] and the annotated genome was presented in both linear and circular forms.
Lineage and subtyping analysis
Clades and their Pango lineages of each of the sequence/isolate were determined using the Nextclade webserver (https://clades.nextstrain.org/) and Pangolin (Phylogenetic Assignment of Named Global Outbreak Lineages) Web (https://pangolin.cog-uk.io/). The Nextclade server uses, by default, Wuhan-Hu-1/2019 (MN908947) as the reference sequence. After the lineages and subtypes were determined by pangolin and nextstrain classification systems, the names of the sequences were modified as: Accession/EPI_ISL No.: Name of the virus/City-Year-Lineage name.
2.3. Sequence Alignment
All the sequences were aligned with a light-weight algorithm of Multiple Alignment using Fast Fourier Transform v7 Web server called the FFT-NS-fragment method using rapid calculation of full-length Multiple Sequence Alignment of closely related viral genomes, (https://mafft.cbrc.jp/alignment/software/closelyrelatedviralgenomes.html)[21]. The sequences were kept equal size with reference to the reference sequence. The multiple sequence alignment was trimmed manually by removing the ambiguous sequences and gaps using BioEdit v7.2.5 (https://bioedit.software.informer.com/)[22] based on the best of our knowledge keeping the alignment as intact as possible for the most accurate analysis.
2.4. Phylogenetic Tree Construction
The Maximum Likelihood (ML) rooted phylogenetic tree was constructed by using the IQ-Tree2 multicore v1.6.12 (http://www.iqtree.org/)[23] using the best-fit substitution model GTR+F+I+G4 (as determined by the ModelFinder tool built-in in IQ-Tree based on Bayesian Information Criterion-BIC) using 1000 bootstrap replicates and 1000 iterations [23]. The tree was rooted on the mid-point. The tree was visualized and edited by iTOL (Interactive Tree of Life) webserver (https://itol.embl.de/)[24] by showing the bootstrap values at each node/branch of the tree and coloring all the branches of a subtype with a different color.
2.5. Genetic Similarity Analysis
The genetic similarity analysis was performed using SimPlot v3.5.1 (https://mybiosoftware.com/simplot-3-5-1-sequence-similarity-plotting.html)[25]. Kimura 2-parameter distance model was used with 1000 bootstrap replicates, Ts/Tv ratio of 2 and neighbor-joining tree model. Window size and step size were set as 600 and 200 respectively to get the best-looking results. One sequence from each of the clades were included in the analysis. The Omicron XBB.2.3.2 from Taiyuan (name starting with 18495234) was used as query sequence.
2.6. Recombination Analysis
Recombination Detection Program (RDP) version (http://web.cbio.uct.ac.za/&hx0026;sim;darren/rdp.html) 5.29 [26] and Recombination Analysis Tool (RAT) (http://jic-bioinfo.bbsrc.ac.uk/bioinformatics-research/staff/graham_etherington/RAT.html) [27] were used to detect recombination in the multiple sequence aligned full-length genomes of SARS-CoV-2. For RDP, seven algorithms namely RDP, GENECONV, Bootscan, Chimaera, MaxChi, SiScan, and 3Seq were used separately (because of the large burden on the bioinformatics software and the computer being used for the analysis) to identify any recombination event (s), if any.
2.7. Phylogenetic Network Analysis
Phylogenetic network analysis is a powerful way to infer the genetic relationship among the analyzing sequences. Hence, the multiple aligned sequences (n= 834 including the reference SARS-CoV-2 and bat coronavirus sequences) were evaluated by constructing the phylogenetic network using Temporal Clustering of Sequences (TCS) method, which uses parsimony statistics to infer the network based on the number of mutations between sequences, implemented in the Population Analysis with Reticulate Trees (PopART) v1.7 software (http://popart.otago.ac.nz/)[28] where more than 5 percent sites containing undefined subtypes were masked.
3. Results
3.1. Sequence Acquisition
We analyzed a total of 834 SARS-CoV-2 full-length genome sequences including the wild-type reference genome and bat coronavirus genome (accessed from GISAID and NCBI database). The search filter location: Asia/China/Shanxi/Taiyuan resulted in a total number of 686 complete genomes of SARS-CoV-2 which was then narrowed down to 347 sequences by choosing the high coverage option. The search filter location: Asia/China/Hubei-Wuhan resulted in a total number of 485 complete sequences. Table 1 shows the details of the sequences used in this study.
3.2. Genome Annotation
Genome annotation of SARS-CoV-2 Wuhan-Hu-1 (Accession No.: NC_045512) was done by using Unipro UGENE v48.0 software and the annotated genome was presented in circular forms in Figure 1.
3.3. Subtype and Lineage Analysis
We determined and confirmed the subtype and lineage of the analyzed sequences through Nextclade and Pangolin classification, respectively. Due to the high burden of sequences, the sequences of Taiyuan and Wuhan were analyzed separately, and their results were then combined (Supplementary material No. 1).
The four most prevalent lineages of SARS-CoV-2 in Wuhan as determined by Pangolin lineage classification are the lineage B followed by the Omicron FY.3, lineage A and Omicron FL.2.3 whereas the four most prevalent clades in Wuhan as determined by Nextclade classification are the clades 19A followed by 22F (XBB), 23F (EG.5.1) and 23H (HK.3).
The four most prevalent lineages in Taiyuan as determined by the Pangolin classification are the Omicron lineages EG.5.1.1 followed by HK.3, FY.3 and XBB.1.16 whereas the four most prevalent clades in Taiyuan as determined by Nextclade classification are the clades 23F (EG.5.1) followed by 23H (HK.3), 22F (XBB) and 23D (XBB.1.9).
3.4. Phylogenetic Tree
To check and determine the genetic relationship between Taiyuan and Wuhan cities of China, we performed a phylogenetic analysis using complete genomes of SARS-CoV-2 from both cities. The phylogenetic tree showed some interesting relationships among the sequences. The tree shows that all the sequences of 2019 and 2020 (a total of 197 sequences all from Wuhan as none of the sequences was from Taiyuan) including the subtypes/clades 19A-B, 19B-A, were grouped together as one big clade which was then collapsed. Some other not so important sequences collapsed together to make the tree visibly better. The tree was divided into 14 clusters based on the topology and bootstrap values as can be seen in (Figure 2 and Supplementary Figure 1).
The Omicron BF.7.14, FR.1.1, GF.1 and XBB.1.16 from Taiyuan (names starting with 18495276, 18495161, 18495266 and 18495165) were shown to be more closely related to those from Wuhan (names starting with 17485739, 17672008, 17684335 and 17729945 respectively) but distantly related to those from Taiyuan itself (name starting with 18495416, 18495192, 18495282 and 18495390 respectively). The Omicron FU.2.1, FE.1.1, FY.3.1, FL.15, FL.2.4, EG.5.1, EG.5.1.4, HK.3, EG.5.1.1 from Taiyuan (names starting with 18495302, 18495297, 18495289, 18495442, 18495166, 18495417, 18495525, 18495522, 18495444) were closely related to those of the Wuhan (names starting with 17978548, 18146040, 17837024, 17672010, 17801857, 18376489, 18284743, 18438428, 18254244 respectively) with bootstrap values of 100%. The Omicron EG.5.1 from Taiyuan (name starting with 18495387, 18495518, 18495440) was closely related to those from Wuhan (name starting with 17988088, 18146042, 18401765) with bootstrap values of 76%, 58% and 38% respectively. The Omicron EG.5.1.1 from Wuhan (name starting with 18254243) was more closely related to that of the Taiyuan (name starting with 18495305) but distantly related to that of the Wuhan itself (name starting with 18105544). The Omicron EG.5.1.1 from Taiyuan (names starting with 18495407, 18495433, 18495438, 18495170, 18495338 and 18495286) were more closely related to those from Wuhan (names starting with 18146047, 18254254, 18146060, 18078299, 17978550 and 18254247) with 50%, 23%, 22%, 16% and 15% bootstraps respectively. The Omicron FL.2.3 from Taiyuan (names starting with 18495201, 18495361, 18495184) were more closely related to that from Wuhan (names starting with 17729943, 17672006, 17684329 respectively) but with relatively lower bootstrap values of 34%, 50%, 62% respectively. The Omicron FL.4 from Taiyuan (name starting with 18495316) was closely related to that of the Wuhan (name starting with 17794191) with bootstrap value of 98%. The Omicron XBB.1.9.1 from Taiyuan (name starting with 18495200) was closely related to that of the Wuhan (name starting with 17672025) but with only 25% bootstrap value and the Omicron XBB.1.9.1 from Taiyuan (name starting with 18495306) was closely related to the Omicron FL.18 from Wuhan (name starting with 17684330) but with only 18% bootstrap. The Omicron FY.3 from Taiyuan (name starting with 18495283) was closely related to the Omicron XBB.1.9.2 from Taiyuan (name starting with 18495281) with bootstrap 94%. The Omicron HK.3 from Wuhan and Taiyuan (names starting with 18146054 and 18495477 respectively) were closely related with the Omicron EG.5.1.1 from Wuhan and Taiyuan (names starting with 18105541 and 18495317 respectively) with 100% and 55% bootstraps respectively. The Omicron HK.3 from Taiyuan (names starting with 18495409, 18495504) were closely related with those from Wuhan (names starting with 18146053, 18535381 respectively) with bootstraps of 30% and 67%. The Omicron HK.2 from Taiyuan (name starting with 18495464) was closely related with those from Wuhan (name starting with 17978544) with bootstrap of 49%. The Omicron HK.1 from Taiyuan (name starting with 18495398) was closely related with the Omicron EG.5.1.1 from Wuhan (name starting with 18105543) with 99% bootstraps. One of the Omicron XBB.1.5 sequences from Wuhan (name starting with 17978557) showed more genetic similarity with the Omicron GR.1 from Wuhan (name starting with 17729936) than with the other Omicron XBB.1.5 from Wuhan (name starting with 17729949). The Omicron DY.2 from Taiyuan (name starting with 18495304) was shown to be more closely related to Omicron BA.5.2.48 from Taiyuan (name starting with 18495357) but distantly related to those from Taiyuan itself (names starting with 18495303 and 18495280).
3.5. Genetic Similarity Analysis
The genetic similarity plot showed that the SARS-CoV-2 clade 19A-B.4 from Wuhan (name starting with 412981) has the lowest genetic similarity (that is about 95.5%) between the nucleotide position 22000-24000; that is the spike protein nucleotide position compared to the query sequence of Omicron XBB.2.3.2 from Taiyuan (name starting with 18495234) followed by the Omicron FR.1.4 from Taiyuan (name starting with 18495199) (~97.2% similarity) and Omicron DY.3 (name starting with 17485740) with ~97.9% similarity. The rest of the variants showed equal to or more than 98% similarity with the query sequence of Omicron XBB.2.3.2 from Taiyuan (name starting with 18495234) as can be seen in Figure 3.
3.6. Recombination Analysis
Genetic recombination plays a crucial role in virus evolution resulting in the emergence of new variants of SARS-CoV-2. It helps to better understand the evolution and genetic diversity of the virus. In our study, we analyzed 832 complete genomes of SARS-CoV-2 from Wuhan and Taiyuan for any potential recombination event (s). A total of nine recombination events were detected by the RDP5 software as can be seen in Table 2 and supplementary material No.2.
Out of nine recombination events detected, only three (event Nos. 3, 5 and 7) were shown to be verified by four or more detection methods implemented in the RDP5 software but all these events were at the 3’ end of the genome.
The event 3 has been verified by five detection methods and showed that the minor parent was 19B-A of Wuhan (455406), and the major parent was unknown (but the software showed the most probable major parent i.e., Omicron-FL.13.1 of Wuhan (17672007)) resulting in the recombinant Omicron-XBB.1.16 of Wuhan (17672023). The event 5 has been verified by six detection methods and showed that the minor parent was 19A-B of Wuhan (454997), and the major parent was unknown (but the software showed the most probable major parent i.e., Omicron-FY.3 of Wuhan (17978543)) resulting in the recombinant Omicron-FY.3 of Wuhan (17672021). The event 7 has also been verified by five detection methods and showed that the minor parent was Omicron-DY.4 from Wuhan (18284748), and the major parent was unknown (but the software showed the most probable major parent i.e., Omicron-FD.2 of Wuhan (17684337)) resulting in the recombinant Omicron-FL.2.4 of Wuhan (17801857).
Interestingly, event 8 showed that the minor parent is Omicron FR.1-Taiyuan (18495199) and the major parent is Omicron EG.5.1.1-Taiyuan (18495364) resulting in the recombinant Omicron EG.5.1-Taiyuan (18495231). Also, only event 8 showed recombination at the spike protein nucleotide positions i.e., around 22,000 nucleotide positions in the alignment but this event has been verified by only one detection method.
3.7. Phylogenetic Network Analysis
Due to the presence of reticulate evolutionary phenomena, for example recombination, the evolution of many species cannot be inferred using phylogenetic trees. Hence, phylogenetic network analysis was performed to better understand the evolution of the SARS-CoV-2 variants between the Wuhan and Taiyuan cities of China.
A total of 834 (including reference and bat coronavirus sequences) multiple aligned complete genomes of SARS-CoV-2 were analyzed by constructing a phylogenetic network using Temporal Clustering of Sequences (TCS) method. The analysis showed that there are a total of 11466 segregating sites including 1000 parsimony-informative (PI) sites. The sequences of SARS-CoV-2 variants have been distributed based on the number of mutations. The details of the network are given in Table 3. The negative value (-2.33398) of the Tajima’s D statistic shows that the segregating and PI sites are statistically significant in the evolution of the viral target genomes.
The phylogenetic network of SARS-CoV-2 genomes probably originated from the bat-CoV with a result of more than 10,000 mutations and the network was divided into 14 clusters: cluster I-XIV which was consistent with the phylogenetic tree. Cluster-I contained all the sequences of 2019-2020 from Wuhan i.e., subtypes A, B and B.4 including the wild-type (reference) sequence. Cluster-II has probably been originated by 47 mutations from cluster-I and contained the sequences of Omicron DY.2/3, BF and, interestingly, the Omicron BA.5.2.48. Cluster-III contained Omicron FR with one Omicron BN sequence.
Cluster-IV contained the sequences of Omicron GF while cluster-V contained a few sequences of Omicron XBB and one of each of the Omicron FD and GR. Cluster-VI and VIII contained the sequences of Omicron XBB while cluster VII contained the sequences of Omicron GY. Cluster VIX contained the sequences of Omicron FU while cluster X contained the sequences of Omicron FE. Cluster XI contained the sequences of Omicron FY with one Omicron XBB.1.22.1 from Taiyuan (name starting with 18495245) while cluster XII contained the sequences of Omicron FL from both Taiyuan and Wuhan cities with one Omicron GR.1 from Wuhan (name starting with 17729936). Cluster XIII contained some of the sequences of Omicron XBB while cluster XIV contained the sequences of Omicron EG and HK with a sequence of Omicron XBB.1.9.2 from Taiyuan (name starting with 18495281). The phylogenetic network analysis results are consistent with the phylogenetic tree. The number of mutations between each genome/variant and clusters of the phylogenetic network can be seen in Figure 4.
4. Discussion
During the last two to three years, a lot of research has been done on the evolution of SARS-CoV-2 using different bioinformatics tools and techniques. Due to the mutations in SARS-CoV-2, multiple variants and lineages have been originated. Regular monitoring of these mutations and tracking of the travel history between and within a particular city or location is very important to unveil the true route of viral evolution and for better understanding of the comparative genetic characteristics between two locations or cities.
4.1. Subtype and Lineage Analysis
The subtyping and lineage analysis of our study shows that the most predominant lineage in both Wuhan and Taiyuan cities of China is Omicron and its sub-lineages. The emergence and prevalence of FY.3, FL.2.3, EG.5.1.1, HK.3, and XBB.1.16 Omicron sub-lineages is consistent with the global trend of SARS-CoV-2 (https://www.who.int/activities/tracking-SARS-CoV-2-variants) [29,30,31].
4.2. Phylogenetic Analysis
The phylogenetic analysis revealed that all the 197 sequences from 2019 and 2020, exclusively from Wuhan, China were grouped together in one clade. This grouping included subtypes/clades 19A-B and 19B-A both of which formed a significant clade that was collapsed to better visualize the tree. Also, some less important sequences were also collapsed to enhance the tree's visibility. These results align with a study on the transmission dynamics of SARS-CoV-2 in Wuhan, in which the Sun and colleagues highlighted the significance of lockdown and medical resources in controlling the spread of the virus using mathematical modelling [32]. In addition, another group of researchers studied the spatial distribution characteristics of SARS-CoV-2 in China using spatial analysis methods [33].
The close relatedness of SARS-CoV-2 variants of Taiyuan and Wuhan indicates a shared evolutionary history. It is crucial for tracking the transmission dynamics of the SARS-CoV-2 variants between Taiyuan and Wuhan. Our results provide valuable data for researchers and healthcare departments to carefully monitor the evolution of SARS-CoV-2 which can help in controlling the spread of SARS-CoV-2 in particular and other viruses in general [34,35].
Interestingly, some sequences of SARS-CoV-2 from Taiyuan showed a closer genetic similarity with those from Wuhan, the epicenter of the initial outbreak, rather than those of the other sequences of SARS-CoV-2 from Taiyuan. For example, the Omicron DY.2 from Taiyuan (name starting with 18495304) was shown to be more closely related to Omicron BA.5.2.48 but distantly related to those from Taiyuan itself. The reason might be that the Omicron BA.5.2.48 sequence has not been classified correctly by the Pangolin and Nextclade classifications. Also, the Omicron HK.1 from Taiyuan (name starting with 18495398) was shown to be closely related with the Omicron EG.5.1.1 from Wuhan (name starting with 18105543) with 99% bootstraps. The high bootstrap value suggests that both sequences might have shared a common epidemiological link or transmission route. It is probable that the individuals infected from these viral variants from Taiyuan and Wuhan were exposed to a same or similar infection source. In addition, it also suggests that the Omicron HK.1 from Taiyuan and Omicron EG5.1.1 from Wuhan have relatively low genetic divergence which might indicate a relatively recent introduction with limited genetic mutations.
4.3. Genetic Similarity Analysis
The genome of SARS-CoV-2 has a positive sense RNA of ~29.9kb in size (Wu et al 2020). The viral genomic RNA encodes nonstructural proteins (nsps) from two open reading frames (ORFs) called ORF1a and ORF1b. A large polyprotein is then produced from ORF1b, which is then cleaved to produce 15nsps. Shorter subgenomic RNAs (sgRNAs) are also produced which encode spike (S), envelope (E), membrane (M) and nucleocapside (N) structural proteins. The genome is flanked by 5’ and 3’ untranslated regions (UTRs) [1].
The clade 19A-B.4 from Wuhan (name starting with 412981) has the lowest genetic similarity compared to the Omicron XBB.2.3.2 from Taiyuan (name starting with 18495234) at the region of nucleotide position 22,000-24,000 which encodes the spike protein which means that the former is more genetically distinct from the latter. This suggests significant evolutionary divergence between the two. This difference might contribute to different phenotypic characteristics that warrant further research. The variants showing 98% or more genetic similarity might exhibit more similar genetic and biological characteristics.
4.4. Recombination Analysis
A lot of researchers have developed different methods to check recombination in SARS-CoV-2. Most of the time, recombination occurs in the spike gene and near its 5’ end [36,37]. The major and minor parents of a recombinant SARS-CoV-2 can both be from 5’-end as well as 3’-end of the viral genome [38]. Recombination can happen between different variants or subtypes. In our study, we analyzed the complete genomes of SARS-CoV-2 from Wuhan and Taiyuan cities of China for any potential recombination.
Interestingly, in event 8, the minor parent is Omicron FR.1-Taiyuan (18495199), and major parent is Omicron EG.5.1.1-Taiyuan (18495364) resulting in the recombinant Omicron EG.5.1-Taiyuan (18495231) but only one detection method detected this recombination event. The reasons might be the wrong assignment of the lineage names and might also be the convergent evolution by which the Omicron EG.5.1 may have independently acquired similar genetic changes as those found in the Omicron EG.5.1.1 rather than the parent-offspring relationship [39]. In addition, recombination event 8 was shown to occur at the spike protein at around 22,000 nucleotides positions which is particularly significant. The spike protein is the key target for therapeutics and vaccine designing and mutation/recombination in this region affects the virus-host interaction giving the virus immune escape benefit and can also affect the vaccine efficacy.
4.5. Phylogenetic Network Analysis
The evolution of genes is tree-like, but the evolution of species is no longer tree-like because of the phenomena of recombination, horizontal gene transfer and hybridization. In our phylogenetic network analysis, we used Temporal Clustering of Sequences (TCS) method which showed negative value of the Tajima’s D statistic which shows changes in the viral population, possibly influenced by genetic drift, natural selection and/or other mutations. This is consistent with the study of Al-Jawabreh and colleagues, but they constructed a phylogenetic network analysis using median-joining method [40]. In addition, the occurrence of a significant PI and segregating sites shows genetic and evolutionary dynamics within the genomes of SARS-CoV-2 between the two cities indicating the ongoing genetic diversity and adaptation of the virus.
Our phylogenetic network analysis showed fourteen (XIV) clusters of SARS-CoV-2 genomes based on their mutations which align with our phylogenetic tree. Al-Jawabreh and colleagues constructed median-joining phylogeographic network and reported three clusters of SARS-CoV-2 based on different geographical locations [40]. These analyses can help better understand the relatedness between different SARS-CoV-2 variants from Wuhan and Taiyuan which can help the public health authorities to effectively control the spread of the virus and mitigate its emerging mutants using targeted vaccine designing and other surveillance strategies (https://cov-lineages.org/) [41,42].
5. Conclusions
From our study, it is concluded that there is a continuous need to study the genetic characteristics of the SARS-CoV-2 variants in particular and other viruses in general as the rapidly mutating viral variants can render the modern-day vaccines and therapeutics ineffective. It has been reported in this comparative study that the SARS-CoV-2 has been evolved over time and spread from the epicenter, Wuhan city of China, to Taiyuan city and there are many important things determined for example the presence of multiple recombination events and clustering of viral variants from both cities as some viral variants from Taiyuan shared more similarity with those from Wuhan rather than with those from Taiyuan itself and the recombination of Omicron EG.5.1 from Taiyuan as a result of potential convergent evolution. This study will help the healthcare and scientific community to better understand the genetic characteristics, phylodynamics and evolutionary pathways of SARS-CoV-2 variants between local populations of China in particular and the whole world in general.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: Phylogenetic tree; Table S1: Pangolin classification; Table S2: Recombination.
Author Contributions
Conceptualization, B.H.; methodology, B.H.; software, B.H.; validation, B.H., W.C.; formal analysis, B.H.; investigation, C.W.; resources, C.W.; data curation, B.H.; writing—original draft preparation, B.H.; writing—review and editing, B.H., C.W.; visualization, B.H.; supervision, C.W.; project administration, B.H., C.W.; funding acquisition, C.W. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Shanxi Provincial Key R&D Project (202003D31005/GZ) and the Transformation of Scientific and Technological Achievements Programs of Higher Education Institutions in Shanxi (TSTAP).
Institutional Review Board Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors acknowledge the researchers of GISAID and NCBI for the genome sequences of SARS-CoV-2.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.J.N. A new coronavirus associated with human respiratory disease in China. 2020, 579, 265-269.
- Zhu, J.; Guo, J.; Xu, Y.; Chen, X.J.J.o.I. Viral dynamics of SARS-CoV-2 in saliva from infected patients. 2020, 81, e48-e50.
- Cucinotta, D.; Vanelli, M.J.A.b.m.A.p. WHO declares COVID-19 a pandemic. 2020, 91, 157.
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.J.T.l. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. 2020, 395, 565-574.
- Day, T.; Gandon, S.; Lion, S.; Otto, S.P.J.C.B. On the evolutionary epidemiology of SARS-CoV-2. 2020, 30, R849-R857.
- Tang, D.; Comish, P.; Kang, R.J.P.p. The hallmarks of COVID-19 disease. 2020, 16, e1008536.
- Gribble, J.; Stevens, L.J.; Agostini, M.L.; Anderson-Daniels, J.; Chappell, J.D.; Lu, X.; Pruijssers, A.J.; Routh, A.L.; Denison, M.R.J.P.p. The coronavirus proofreading exoribonuclease mediates extensive viral recombination. 2021, 17, e1009226.
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.J.C. Tracking changes in SARS-CoV-2 spike: evidence that D614G increases infectivity of the COVID-19 virus. 2020, 182, 812-827. e819.
- O’Toole, Á.; Scher, E.; Underwood, A.; Jackson, B.; Hill, V.; McCrone, J.T.; Colquhoun, R.; Ruis, C.; Abu-Dahab, K.; Taylor, B.J.V.e. Assignment of epidemiological lineages in an emerging pandemic using the pangolin tool. 2021, 7, veab064.
- Channabasappa, N.K.; Niranjan, A.K.; Emran, T.B.J.I.J.o.S. SARS-CoV-2 variant omicron XBB. 1.5: Challenges and prospects–correspondence. 2023, 109, 1054–1055. [Google Scholar]
- Haddad, D.; John, S.E.; Mohammad, A.; Hammad, M.M.; Hebbar, P.; Channanath, A.; Nizam, R.; Al-Qabandi, S.; Al Madhoun, A.; Alshukry, A.J.P.o. SARS-CoV-2: Possible recombination and emergence of potentially more virulent strains. 2021, 16, e0251368.
- Hussain, I.; Pervaiz, N.; Khan, A.; Saleem, S.; Shireen, H.; Wei, D.-Q.; Labrie, V.; Bao, Y.; Abbasi, A.A.J.G. ; Immunity. Evolutionary and structural analysis of SARS-CoV-2 specific evasion of host immunity. 2020, 21, 409–419. [Google Scholar]
- Duchene, S.; Featherstone, L.; Haritopoulou-Sinanidou, M.; Rambaut, A.; Lemey, P.; Baele, G.J.V.e. Temporal signal and the phylodynamic threshold of SARS-CoV-2. 2020, 6, veaa061.
- Peacock, T.P.; Goldhill, D.H.; Zhou, J.; Baillon, L.; Frise, R.; Swann, O.C.; Kugathasan, R.; Penn, R.; Brown, J.C.; Sanchez-David, R.Y.J.N.m. The furin cleavage site in the SARS-CoV-2 spike protein is required for transmission in ferrets. 2021, 6, 899-909.
- Shu, Y.; McCauley, J.J.E. GISAID: Global initiative on sharing all influenza data–from vision to reality. 2017, 22, 30494.
- Bhattacharjee, M.J.; Bhattacharya, A.; Kashyap, B.; Taw, M.J.; Li, W.-H.; Mukherjee, A.K.; Khan, M.R.J.V.J. Genome analysis of SARS-CoV-2 isolates from a population reveals the rapid selective sweep of a haplotype carrying many pre-existing and new mutations. 2023, 20, 201.
- Khailany, R.A.; Safdar, M.; Ozaslan, M.J.G.r. Genomic characterization of a novel SARS-CoV-2. 2020, 19, 100682.
- Boni, M.F.; Lemey, P.; Jiang, X.; Lam, T.T.-Y.; Perry, B.W.; Castoe, T.A.; Rambaut, A.; Robertson, D.L.J.N.m. Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. 2020, 5, 1408-1417.
- Rehman, S.U.; Shafique, L.; Ihsan, A.; Liu, Q.J.P. Evolutionary trajectory for the emergence of novel coronavirus SARS-CoV-2. 2020, 9, 240.
- Okonechnikov, K.; Golosova, O.; Fursov, M.; Bioinformatics, U.T.J. Unipro UGENE: a unified bioinformatics toolkit. 2012, 28, 1166-1167.
- Katoh, K.; Rozewicki, J.; Yamada, K.D.J.B.i.b. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. 2019, 20, 1160-1166.
- Hall, T.A. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. In Proceedings of the Nucleic acids symposium series; 1999; pp. 95–98. [Google Scholar]
- Trifinopoulos, J.; Nguyen, L.-T.; von Haeseler, A.; Minh, B.Q.J.N.a.r. W-IQ-TREE: a fast online phylogenetic tool for maximum likelihood analysis. 2016, 44, W232-W235.
- Letunic, I.; Bork, P.J.B. Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. 2007, 23, 127-128.
- Lole, K.S.; Bollinger, R.C.; Paranjape, R.S.; Gadkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C.J.J.o.v. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. 1999, 73, 152-160.
- Martin, D.P.; Varsani, A.; Roumagnac, P.; Botha, G.; Maslamoney, S.; Schwab, T.; Kelz, Z.; Kumar, V.; Murrell, B.J.V.E. RDP5: a computer program for analyzing recombination in, and removing signals of recombination from, nucleotide sequence datasets. 2021, 7, veaa087.
- Etherington, G.J.; Dicks, J.; Roberts, I.N.J.B. Recombination Analysis Tool (RAT): a program for the high-throughput detection of recombination. 2005, 21, 278-281.
- Leigh, J.W.; Bryant, D.J.M.i.e. ; evolution. POPART: full-feature software for haplotype network construction. 2015, 6, 1110–1116. [Google Scholar]
- VBM, V.B.M. SARS-CoV-2 Variant Classifications and Definitions.
- Li, J.; Lai, S.; Gao, G.F.; Shi, W.J.N. The emergence, genomic diversity and global spread of SARS-CoV-2. 2021, 600, 408-418.
- Fengjiao, S.; Xiaodong, L.; Jian, L.; Hui, L.J.M. Epidemiologic characteristics of SARS-CoV-2 in Wuhan, other regions of China, and globally based on data gathered from January 2020 to February 2021. 2022, 101, e30019.
- Sun, G.-Q.; Wang, S.-F.; Li, M.-T.; Li, L.; Zhang, J.; Zhang, W.; Jin, Z.; Feng, G.-L.J.N.D. Transmission dynamics of COVID-19 in Wuhan, China: effects of lockdown and medical resources. 2020, 101, 1981-1993.
- Ma, Q.; Gao, J.; Zhang, W.; Wang, L.; Li, M.; Shi, J.; Zhai, Y.; Sun, D.; Wang, L.; Chen, B.J.B.I.D. Spatio-temporal distribution characteristics of COVID-19 in China: a city-level modeling study. 2021, 21, 1-14.
- Cicaloni, V.; Costanti, F.; Pasqui, A.; Niccolai, N.; Bongini, P.J.F.i.G. A bioinformatics approach to investigate structural and non-structural proteins in human coronaviruses. 2022, 13, 891418.
- Ahmadi, E.; Zabihi, M.R.; Hosseinzadeh, R.; Mohamed Khosroshahi, L.; Noorbakhsh, F.J.P.O. SARS-CoV-2 spike protein displays sequence similarities with paramyxovirus surface proteins; a bioinformatics study. 2021, 16, e0260360.
- Turkahia, Y.; Thornlow, B.; Hinrichs, A.; McBroome, J.; Ayala, N.; Ye, C.; De Maio, N.; Haussler, D.; Lanfear, R.; Corbett-Detig, R.J.B. Pandemic-scale phylogenomics reveals elevated recombination rates in the SARS-CoV-2 spike region. 2021.
- Jackson, B.; Boni, M.F.; Bull, M.J.; Colleran, A.; Colquhoun, R.M.; Darby, A.C.; Haldenby, S.; Hill, V.; Lucaci, A.; McCrone, J.T.J.C. Generation and transmission of interlineage recombinants in the SARS-CoV-2 pandemic. 2021, 184, 5179-5188. e5178.
- Bolze, A.; Basler, T.; White, S.; Dei Rossi, A.; Wyman, D.; Dai, H.; Roychoudhury, P.; Greninger, A.L.; Hayashibara, K.; Beatty, M.J.M. Evidence for SARS-CoV-2 Delta and Omicron co-infections and recombination. 2022, 3, 848-859. e844.
- Focosi, D.; Maggi, F.J.V. Recombination in Coronaviruses, with a Focus on SARS-CoV-2. 2022, 14, 1239.
- Al-Jawabreh, A.; Ereqat, S.; Dumaidi, K.; Al-Jawabreh, H.; Nasereddin, A.J.B.R.N. Complete genome sequencing of SARS-CoV-2 strains: A pilot survey in Palestine reveals spike mutation H245N. 2021, 14, 466.
- Tang, X.; Ying, R.; Yao, X.; Li, G.; Wu, C.; Tang, Y.; Li, Z.; Kuang, B.; Wu, F.; Chi, C.J.S.b. Evolutionary analysis and lineage designation of SARS-CoV-2 genomes. 2021, 66, 2297-2311.
- Bai, Y.; Jiang, D.; Lon, J.R.; Chen, X.; Hu, M.; Lin, S.; Chen, Z.; Wang, X.; Meng, Y.; Du, H.J.I.J.o.I.D. Comprehensive evolution and molecular characteristics of a large number of SARS-CoV-2 genomes reveal its epidemic trends. 2020, 100, 164-173.
Figure 1.
Genome annotation of SARS-CoV-2 Wuhan-Hu-1. The figure shows the circular form of annotated genome of SARS-CoV-2 original Wuhan strain (Wuhan-Hu-1; accession number.: NC_045512). The complete genome is of 29903 nucleotide bases size. It shows different open reading frames (ORFs) along with other gene sequences. The figure was created with Unipro UGENE v48.0 software.
Figure 1.
Genome annotation of SARS-CoV-2 Wuhan-Hu-1. The figure shows the circular form of annotated genome of SARS-CoV-2 original Wuhan strain (Wuhan-Hu-1; accession number.: NC_045512). The complete genome is of 29903 nucleotide bases size. It shows different open reading frames (ORFs) along with other gene sequences. The figure was created with Unipro UGENE v48.0 software.
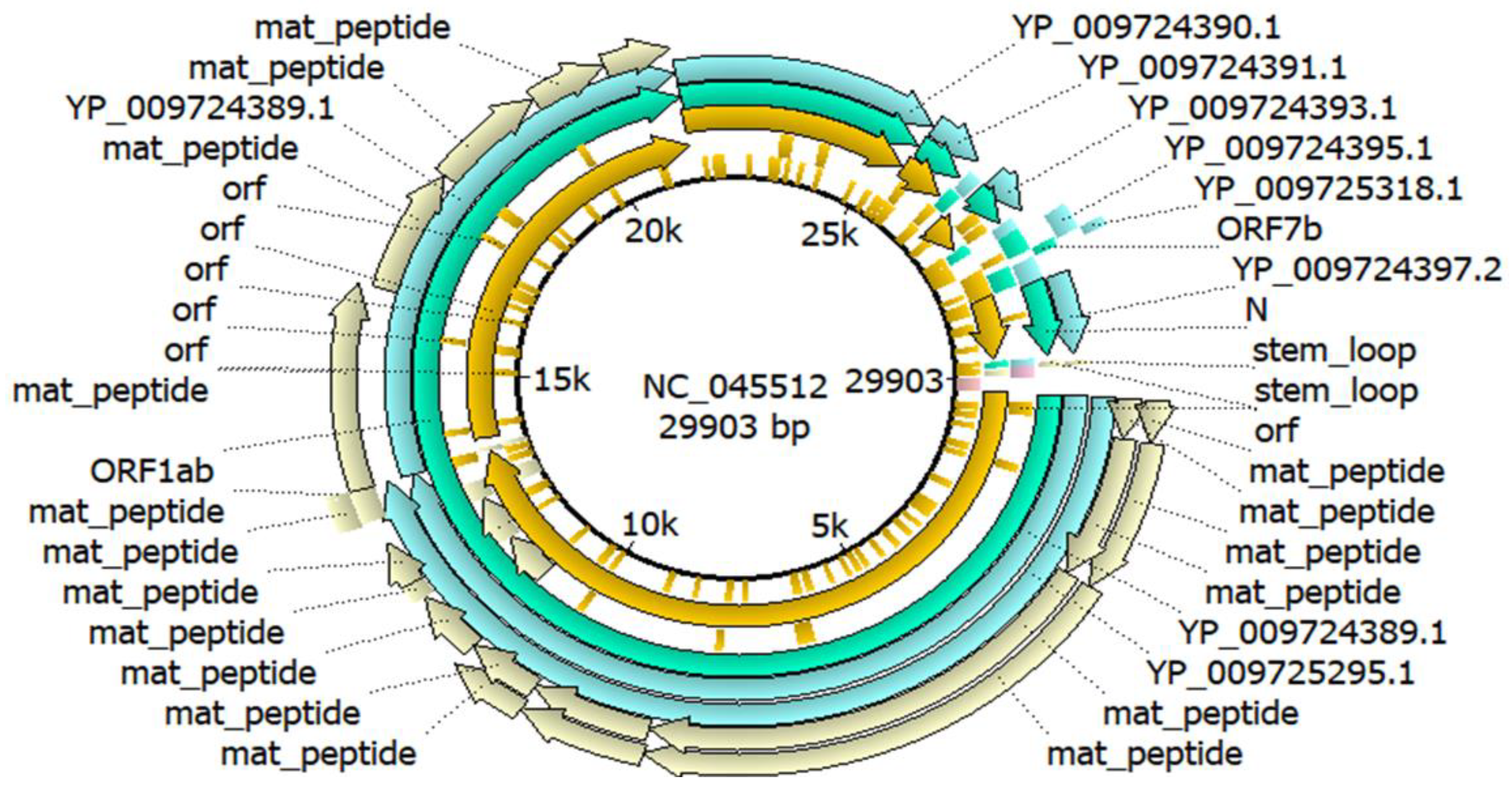
Figure 2.
Maximum Likelihood (ML) Phylogenetic tree. The figure shows the maximum likelihood (ML) phylogenetic tree constructed using IQ-Tree2 multicore software using the best-fit substitution model GTR+F+I+G4. The tree is rooted on mid-point and it shows 14 clusters containing different variants of SARS-CoV-2. Each cluster is given a different color. Some sequences have been collapsed to better visualize the tree. The tree was edited and visualized by iTOL webserver. Bootstrap values have been shown on each node.
Figure 2.
Maximum Likelihood (ML) Phylogenetic tree. The figure shows the maximum likelihood (ML) phylogenetic tree constructed using IQ-Tree2 multicore software using the best-fit substitution model GTR+F+I+G4. The tree is rooted on mid-point and it shows 14 clusters containing different variants of SARS-CoV-2. Each cluster is given a different color. Some sequences have been collapsed to better visualize the tree. The tree was edited and visualized by iTOL webserver. Bootstrap values have been shown on each node.
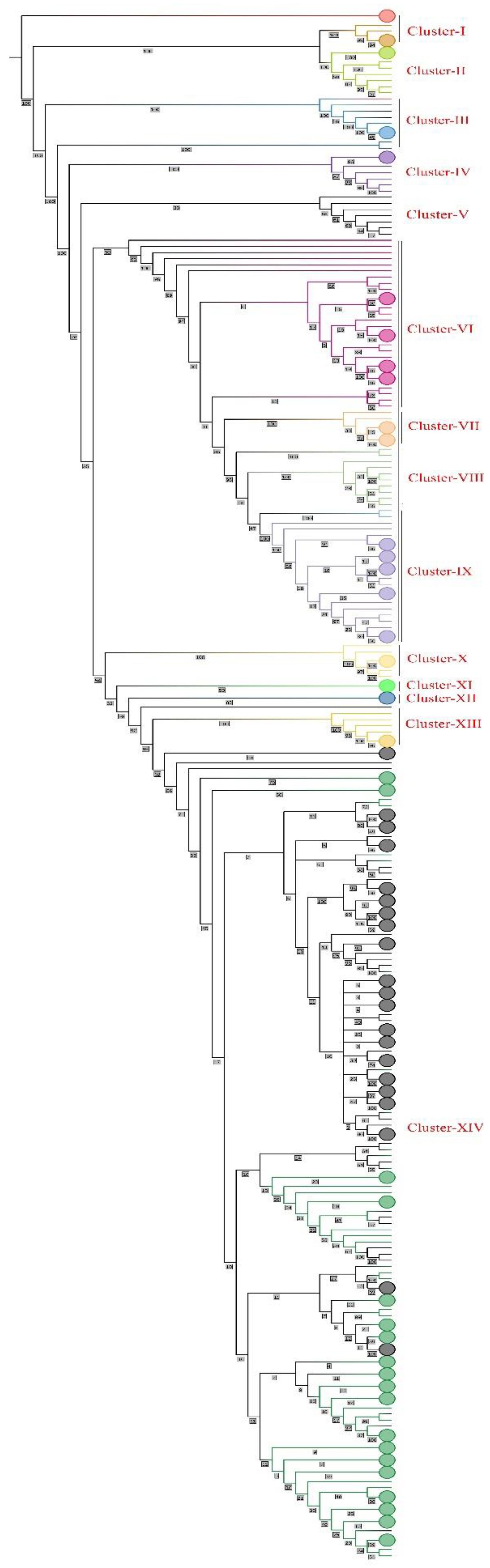
Figure 3.
Genetic similarity plot of representative SARS-CoV-2 variants. The figure shows the genetic similarity plots of the selected variants of SARS-CoV-2 in comparison with the query question of Omicron XBB.2.3.2 from Taiyuan (18495234). Genetic similarity (%) has been shown on Y-axis and nucleotide position has been shown on X-axis. Clade 19A-B.4 from Wuhan (412981) has the lowest genetic similarity of about 95.5% between the nucleotide position 22000-24000 which is the spike protein nucleotide position. Most of the variants show genetic similarity of 98% or more.
Figure 3.
Genetic similarity plot of representative SARS-CoV-2 variants. The figure shows the genetic similarity plots of the selected variants of SARS-CoV-2 in comparison with the query question of Omicron XBB.2.3.2 from Taiyuan (18495234). Genetic similarity (%) has been shown on Y-axis and nucleotide position has been shown on X-axis. Clade 19A-B.4 from Wuhan (412981) has the lowest genetic similarity of about 95.5% between the nucleotide position 22000-24000 which is the spike protein nucleotide position. Most of the variants show genetic similarity of 98% or more.
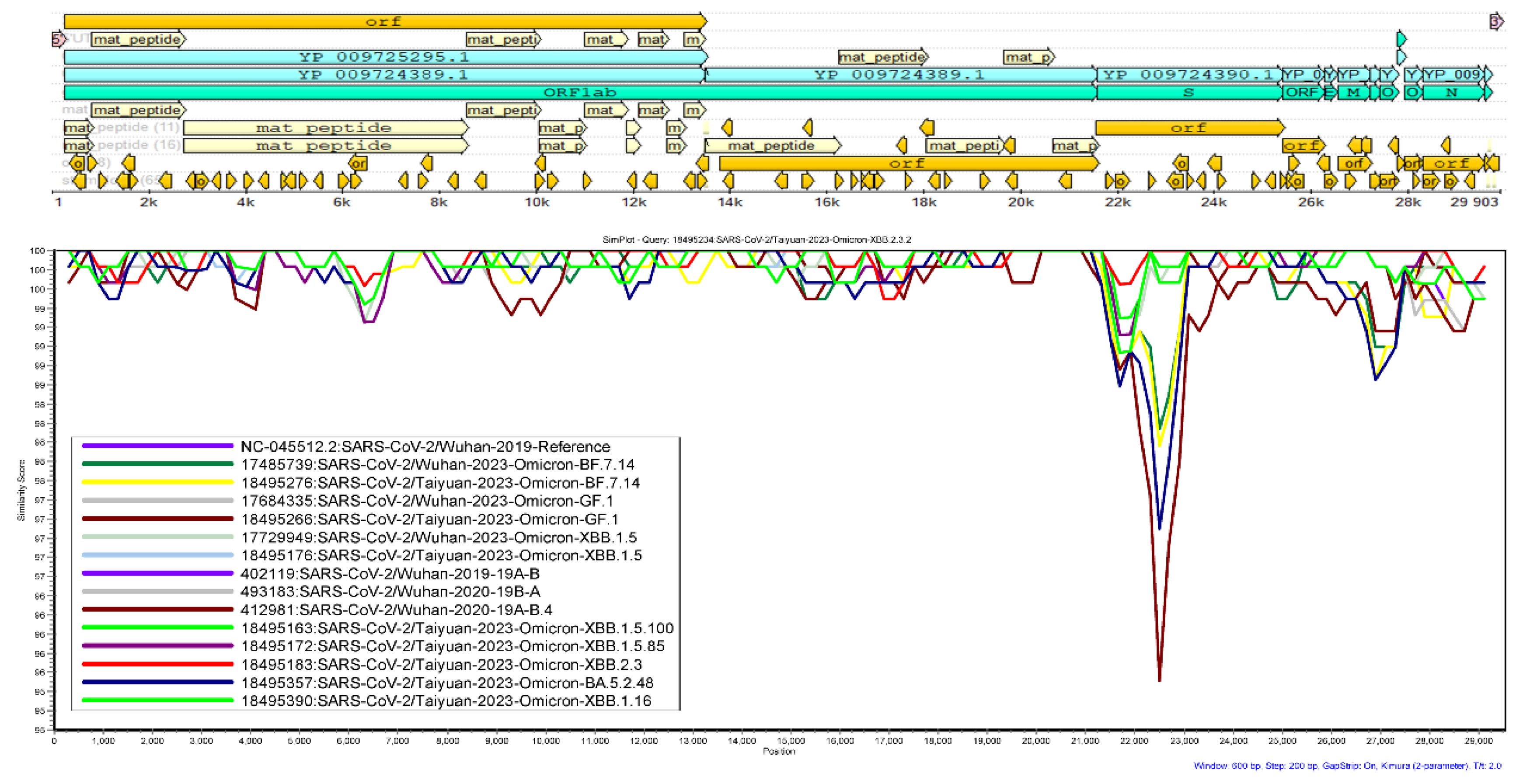
Figure 4.
Phylogenetic network of SARS-CoV-2 variants from Taiyuan and Wuhan. The figure shows the phylogenetic network of all the 833 complete genomes of SARS-CoV-2 from Taiyuan and Wuhan constructed by PopArt software. The network was constructed using TCS method and it shows that the SARS-CoV-2 genomes probably originated from the bat-CoV with a result of more than 10,000 mutations and the network was divided into 14 clusters in consistent with the phylogenetic tree. Numbers show the number of mutations, and each color represents a different main variant. Some interesting variants have been labeled on the network.
Figure 4.
Phylogenetic network of SARS-CoV-2 variants from Taiyuan and Wuhan. The figure shows the phylogenetic network of all the 833 complete genomes of SARS-CoV-2 from Taiyuan and Wuhan constructed by PopArt software. The network was constructed using TCS method and it shows that the SARS-CoV-2 genomes probably originated from the bat-CoV with a result of more than 10,000 mutations and the network was divided into 14 clusters in consistent with the phylogenetic tree. Numbers show the number of mutations, and each color represents a different main variant. Some interesting variants have been labeled on the network.
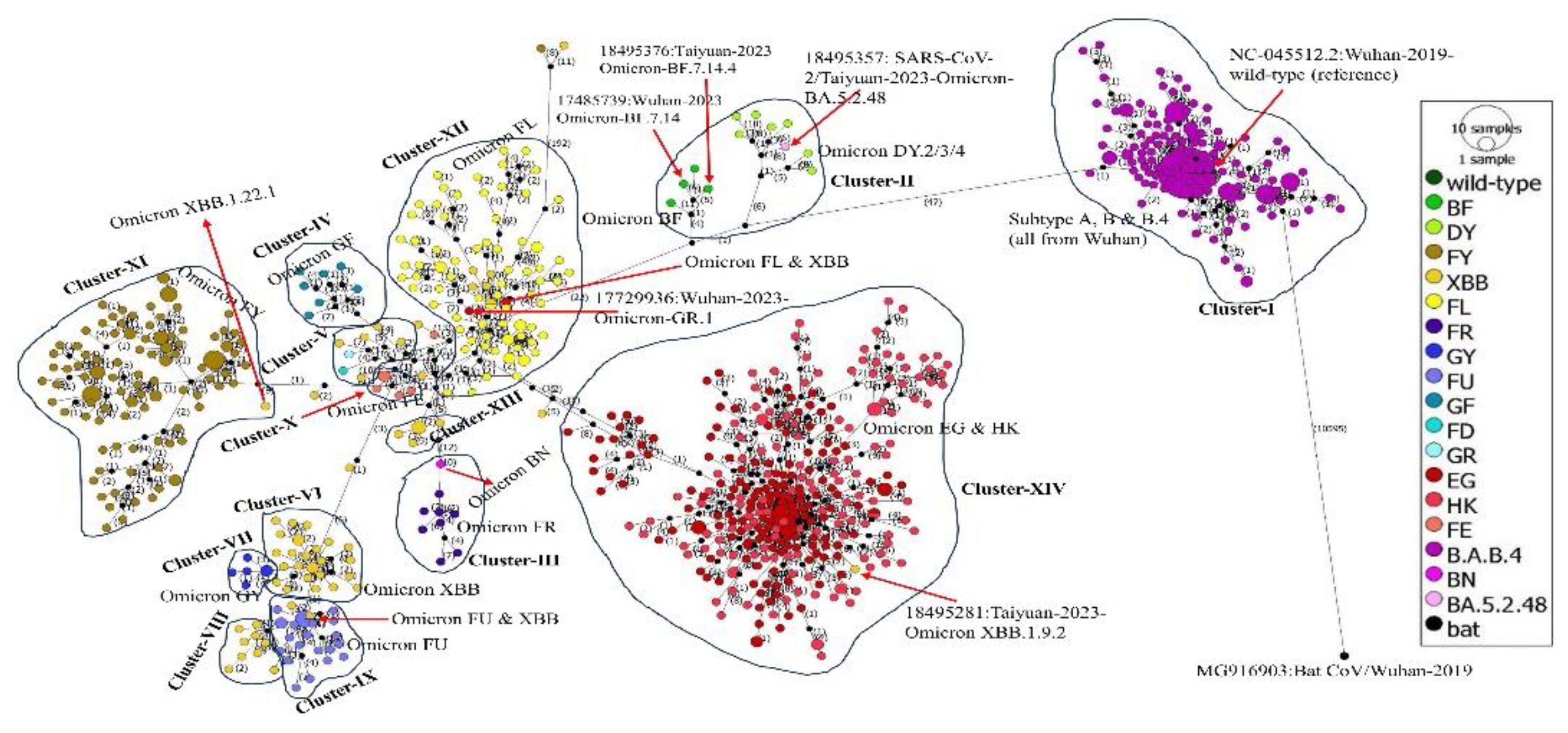

Table 1.
Sequences used in this study.
| SARS-CoV-2 variant | City name* | Total No.of sequences |
|---|---|---|
| BF.7.14 | T (2), W (1) | 3 |
| DY.3 | W (2) | 2 |
| FY.3 | T (28), W (69) | 97 |
| XBB.1.16.1 | T (16), W (11) | 27 |
| XBB.1.16.1.1 | T (7), W (3) | 10 |
| FL.2.3 | T (7), W (14) | 21 |
| FL.13.1 | W (1) | 1 |
| FR.1.1 | T (4), W (1) | 5 |
| GY.1 | T (3), W (3) | 6 |
| FL.15 | T (2), W (7) | 9 |
| FL.21.2 | W (1) | 1 |
| FU.1 | T (9), W (13) | 22 |
| XBB.1.9.1 | T (3), W (2) | 5 |
| XBB.1.16.18 | W (2) | 2 |
| FL.16 | T (3), W (4) | 7 |
| FL.18 | W (1) | 1 |
| FL.4 | T (9), W (4) | 13 |
| GF.1 | T (2), W (5) | 7 |
| FD.2 | W (1) | 1 |
| FL.2.4 | T (4), W (2) | 6 |
| GR.1 | W (1) | 1 |
| EG.5.1.1 | T (113), W (44) | 157 |
| XBB.1.5 | T (1), W (2) | 3 |
| HK.5 | W (1) | 1 |
| FL.2 | T (3), W (3) | 6 |
| FL.21 | W (1) | 1 |
| EG.5.1 | T (9), W (5) | 14 |
| FY.3.1 | T (12), W (9) | 21 |
| XBB.1.42.2 | T (4), W (1) | 5 |
| DY.4 | W (2) | 2 |
| HK.1 | T (1), W (1) | 2 |
| HK.2 | T (4), W (1) | 5 |
| FU.2.1 | T (1), W (1) | 2 |
| XBB.1.42 | W (1) | 1 |
| HK.3 | T (68), W (58) | 126 |
| FL.2.3.1 | W (2) | 2 |
| FL.13.2 | W (1) | 1 |
| FL.1.5.1 | W (1) | 1 |
| FE.1 | W (1) | 1 |
| FE.1.1 | T (3), W (1) | 4 |
| HK.4 | W (4) | 4 |
| EG.5.1.4 | T (2), W (1) | 3 |
| 19A-B | W (167) | 167 |
| 19B-B | W (6) | 6 |
| 19B-A | W (20) | 20 |
| 19A-B.4 | W (3) | 3 |
| XBB.1.5.100 | T (1) | 1 |
| FL.23.1 | T (3) | 3 |
| XBB.1.5.85 | T (1) | 1 |
| XBB.2.3 | T (1) | 1 |
| FR.1.4 | T (1) | 1 |
| XBB.1.17.1 | T (1) | 1 |
| FR.1 | T (1) | 1 |
| BN.1.3.5 | T (1) | 1 |
| FL.4.6 | T (1) | 1 |
| XBB.1.9.2 | T (2) | 2 |
| FL.5 | T (2) | 2 |
| XBB.2.3.2 | T (1) | 1 |
| XBB.1.22.1 | T (1) | 1 |
| FR.1.3 | T (1) | 1 |
| FL.24 | T (3) | 3 |
| DY.2 | T (3) | 3 |
| FE.1.1.3 | T (1) | 1 |
| BA.5.2.48 | T (1) | 1 |
| BF.7.14.4 | T (1) | 1 |
*T=Taiyuan city; W=Wuhan city.
Table 2.
Identification of potential recombination events in the complete genome of SARS-CoV-2 variants.
Table 2.
Identification of potential recombination events in the complete genome of SARS-CoV-2 variants.
| Event | Recombinant | Minor Parent | Major Parent | Detection*(RGBMCST) |
| 1 | 17672021:SARS-CoV-2/Wuhan-2023-Omicron-FY.3 | Unknown (455406:SARS-CoV-2/Wuhan-2020-19B-A) | 17684330:SARS-CoV-2/Wuhan-2023-Omicron-FL.18 | ------- |
| 2 | 17684330:SARS-CoV-2/Wuhan-2023-Omicron-FL.18 | 17729935:SARS-CoV-2/Wuhan-2023-Omicron-XBB.1.16 | 17672023:SARS-CoV-2/Wuhan-2023-Omicron-XBB.1.16 | ------- |
| 3 | 17672023:SARS-CoV-2/Wuhan-2023-Omicron-XBB.1.16 | 455406:SARS-CoV-2/Wuhan-2020-19B-A | Unknown (17672007:SARS-CoV-2/Wuhan-2023-Omicron-FL.13.1) | -+-++++ |
| 4 | 17684330:SARS-CoV-2/Wuhan-2023-Omicron-FL.18 | Unknown (455406:SARS-CoV-2/Wuhan-2020-19B-A) | 17672007:SARS-CoV-2/Wuhan-2023-Omicron-FL.13.1 | ------+ |
| 5 | 17672021:SARS-CoV-2/Wuhan-2023-Omicron-FY.3 | 454997:SARS-CoV-2/Wuhan-2020-19A-B | Unknown (17978543:SARS-CoV-2/Wuhan-2023-Omicron-FY.3) | -++++++ |
| 6 | 17801857:SARS-CoV-2/Wuhan-2023-Omicron-FL.2.4 | Unknown (17672023:SARS-CoV-2/Wuhan-2023-Omicron-XBB.1.16) | 17672007:SARS-CoV-2/Wuhan-2023-Omicron-FL.13.1 | -+----- |
| 7 | 17801857:SARS-CoV-2/Wuhan-2023-Omicron-FL.2.4 | 18284748:SARS-CoV-2/Wuhan-2023-Omicron-DY.4 | Unknown (17684337:SARS-CoV-2/Wuhan-2023-Omicron-FD.2) | -+-++++ |
| 8 | 18495231:SARS-CoV-2/Taiyuan-2023-Omicron-EG.5.1 | 18495199:SARS-CoV-2/Taiyuan-2023-Omicron-FR.1.4 | 18495364:SARS-CoV-2/Taiyuan-2023-Omicron-EG.5.1.1 | ------+ |
| 9 | 17801857:SARS-CoV-2/Wuhan-2023-Omicron-FL.2.4 | 455406:SARS-CoV-2/Wuhan-2020-19B-A | Unknown (17729949:SARS-CoV-2/Wuhan-2023-Omicron-XBB.1.5) | ------+ |
*Detection methods: R, RDP; G, GENECONV; B, BootScan; M, MaxChi; C, Chimaera; S, SiScan; T, 3Seq. ‘+’=verified; ‘-’=not verified.
Table 3.
Phylogenetic network analysis results.
| Sr. No. | Network method | Nucleotide diversity | Segregating sites | PI sites* | Tajima's D statistic |
| 1 | TCS network | pi= 0.0142049 | 11466 | 1000 | D = -2.33398; p (D > = -2.33398) = 0.999236 |
*PI sites=parsimony informative sites.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



