
Submitted:
22 April 2024
Posted:
23 April 2024
You are already at the latest version
Abstract
Natural killer (NK) cells play a crucial role in innate immunity, particularly in combating infections and tumors. However, in hematological cancers, NK cells often exhibit impaired functions. Therefore, it is very important to activate its endosomal toll-like receptors (TLRs) as a potential strategy to restore its antitumor activity. We stimulate NK cells from the peripheral blood mononuclear cells from children with acute lymphoblastic leukemia and NK cells isolated, the NK cells were stimulated with specific TLR ligands (Poly I:C, Imiquimod, R848, and ODN2006) and evaluated changes in IFN-γ, CD107a, NKG2D, NKp44 expression, Granzyme B secretion, cytokine/chemokine release, and cytotoxic activity. Results revealed that Poly I:C and Imiquimod enhanced activation of both immunoregulatory and cytotoxic NK cells, increasing IFN-γ, CD107a, NKG2D, and NKp44 expression. R848 activated immunoregulatory NK cells, while ODN2006 boosted CD107a, NKp44, NKG2D, and IFN-γ secretion in cytotoxic NK cells. R848 also increased secretion of various cytokines/chemokines. Importantly, R848 and ODN 2006 significantly improved cytotoxicity against leukemic cells. Overall, TLR stimulation enhances NK cell activation, suggesting TLR8 (R848) and TLR9 (ODN 2006) ligands as promising candidates for antitumor immunotherapy.
Keywords:
Natural-killer-cells
; Toll-like receptors
; cytotoxicity
; acute lymphoblastic leukemia
; IFN-γ
; NKG2D
; NKp44 7
1. Introduction
Natural Killer (NK) cells represent a highly specialized subpopulation of lymphoid cells (5-15%), integral to the innate immune system [1]. These cells provide early immunoregulatory cytokines and tumor cell lysis without previous stimulation, crucial for immunological surveillance and tumor suppression. The surface antigens CD56 and CD16 antigens allow the characterization of NK cells and categorize them as regulatory and cytotoxic, depending on their expression levels [2].
Approximately 90% of human NK cells in peripheral blood are cytotoxic, with low expression of CD56 (CD56dim/-) and high levels of the Fcγ receptor III (FcγRIII, CD16bright), while 10% are immunoregulatory, which are CD56brightCD16dim or CD16-. Of these two subpopulations, CD56dimCD16bright cells have greater cytotoxic activity, while CD56brightCD16dim favor the synthesis of inflammatory cytokines [2,3].
NK cells express numerous receptors on their membrane, which mediate different signals for the activation of their immune activity against infections and tumor cells. Since NK cells do not have clonal receptors, their repertoire is conformed of germline receptors, which have traditionally been classified as activating receptors (trigger the function of the NK cells) and inhibitory receptors (those that regulate the signal delivered by activating receptors and prevent NK cell activation) [2]. In addition, NK cells express pattern recognition receptors (PRRs), such as Toll-like receptors, through which they recognize pathogen-associated molecular patterns (PAMPs) as well as damage-associated molecular patterns (DAMPs) [4].
Acute lymphoblastic leukemia (ALL) constitutes 80% of all leukemias in children [5]. Even though the survival of patients with ALL has increased remarkably, 20% of patients are still resistant to treatment [6]. NK cells play a central role in identifying and eradicating leukemia cells [7,8] however, studies have reported that peripheral blood NK cells exhibit compromised cytotoxicity towards autologous blasts [9,10] with altered cell surface expression of inhibitory receptors (NKp46 and NKG2A) compared to age-matched healthy control subjects [11,12]. Therefore, the discovery that TLRs are also expressed in NK cells has increased the interest in their immune response against tumor cells.
TLRs constitute a family of transmembrane proteins, that recognize specific molecular patterns from microorganisms, as well as endogenous ligands that produce inflammatory responses [13]. Of the 11 TLRs recognized in humans, 10 are expressed as proteins, with TLRs 1, 2, 4, 5, 6, and 10 expressed on the cell membrane and recognizing mainly microbial components such as LPS, proteins, and lipoproteins [14], while TLRs 3, 7, 8, and 9 are mainly expressed in endosomes where they recognize nucleic acid components such as double-stranded RNA (dsRNA), single-stranded RNA (ssRNA), and unmethylated CpG DNA motifs [15,16,17]. TLR11 is considered a pseudogene [18].The binding of TLRs to their corresponding ligand triggers a signaling pathway leading to the activation of the immune response for the elimination of pathogens [19,20,21].
It has been described that NK cells express endosomal TLRs [22]; however, even though there is evidence of the direct activation of NK cell functions through these TLRs, this activation was evaluated in healthy donors NK cells and also discovered that can only occur by the complex interaction with other cells of the immune system and the microenvironment [23,24,25,26,27]. Although different preclinical studies have shown effective activation of donor NK cells with TLR ligands to induce blast lysis [28,29,30], the effects of this activation in the context of hematological disorders, such as ALL, remains unknown [31]. Since NK cells are important for effective tumor suppression demonstrating that the activation of this cells from hematological oncological disorders are possible, would provide a new panorama for the development of antitumoral therapies.
Previously in our work group, Sánchez et al. demonstrated that the NK cells from ALL pediatric patients (bone marrow and peripheral blood), express all the endosomal TLRs (TLR3, TLR7, TLR8 and TLR9) [22] therefore, the aim of this study was to evaluate the response of the different subpopulations of human NK cells from pediatric ALL patients to stimulation through their different endosomal TLRs using synthetic TLR agonists and compare this response with those from unstimulated human NK cells from pediatric ALL patients in order to evaluate the feasibility of inducing NK cell activation as a first approach as a possible adjuvant in antitumor immunotherapy.
2. Results
2.1. Characteristics of the Patients
Peripheral blood NK cells from patients recently diagnosed with ALL without treatment, transfusion, and/or infection were analyzed. There were 14 boys and 10 girls with ages between 1 and 17 years old. Ten patients were used for each determination, depending on the number of cells that could be purified from each patient. The characteristics of the study group are summarized in Table 1.
2.2. Identification of the Different NK Cell Subpopulations in Patients with ALL and IFN-γ, CD107a, NKp44, and NKG2D expression.
The different NK cell subpopulations of all samples were characterized in both whole PBMCs and enriched NK cells. After enrichment, the samples displayed more than 96% NK cells. NK cells were characterized by selecting cells with CD56/CD16 expression from the previously gated CD3-CD19- subpopulation. Within the NK cell populations of all samples, immunoregulatory cells (CD56+, CD16-) and cytotoxic cells (CD56+, CD16+ and CD56-, CD16+) were identified. For each NK cells subpopulation, the mean fluorescence intensities and percentages of alive positive cells were determined for key markers such as: CD107a, IFN-γ, NKG2D, and NKp44 (Figure 1, Figure 2 and Figure S1 and S2).
2.3. Poly I:C and R848 Increase the Activity of NK Subpopulations within the PBMC.
To assess the impact of TLR ligands on the function of NK cell subpopulations, both mononuclear cells and freshly enriched NK cells were cultured for 24 hours with R848 (TLR7/8 ligand), Poly I:C (TLR3 ligand), Imiquimod (TLR7 ligand), or ODN2006 (TLR9 ligand) and were analyzed for IFN-γ production and CD107a expression.
When the regulatory activity of the NK subpopulations after stimulation within the PBMC fraction was evaluated, none of the treatments modified the production of intracellular IFN-γ among the three subpopulations, (Figure 3A). However, the proportions of IFN-γ+ cells significantly increased after the stimulation with Poly I:C(p=0.0187) and R848 (p= 0.0288) in the immunoregulatory subpopulation CD56+CD16-(Figure 3B).
Subsequently, the degranulation of NK cells was evaluated by the expression of CD107a, a degranulation marker. We observed a significant increase in the CD107a expression on CD56+CD16- (p=0.0075) after stimulation with Poly I:C (p=0.0075) and on CD56+CD16+ after Poly I:C (p=0.0075), and ODN2006 (p=0.0075) stimulation (Figure 3C). When analyzing the proportions of cells expressing CD107a, we found that Poly I:C significantly increased the proportion of CD56+CD16- and in the CD56+CD16+ subpopulation (p=0.0036 and p=0.0012 respectively). Additionaly, R848 increased the proportion of the immunoregulatory subpopulation (CD56+CD16-) (P=0.0288) (Figure 3D).
2.4. Imiquimod and ODN2006 Increase the Function of Enriched NK Cells Subpopulations
To assess the response of the TLR activation on the NK cells, we stimulated enriched NK cells (purity 96.3%) with Poly I:C, Imiquimod, R848, and ODNThe mean fluorescence intensities and the percentages of cells expressing the different markers, CD107a, IFN-γ, NKG2D, and NKp44, were determined for each subpopulation and secretion of Granzyme B and IFN-γ was evaluated in the supernatants (Figure 4).
The immunoregulatory activity was evaluated measuring intracellular production of IFN-γ. In this case, only the cytotoxic subpopulation (CD56-CD16+) significantly increased the expression of IFN-γ (Poly I:C p<0.005, Imiquimod, R848 and ODN, p< 0.05) with the four treatments (Figure 4A). When the proportions of IFN-γ+ cells were analyzed, we observed that the three subpopulations remained without significant change in proportions after the four treatments (Figure 4B).
Additionally, to further evaluate the increase in the immunoregulatory function of NK cells, we measured the concentration of IFN-γ (pg/mL) in the supernatant of enriched NK cells cultured with the four different ligands (Figure 4C). We found that ODN significantly increased the production of IFN-γ (p=0.0352) compared to untreated cells.
When we assessed the degranulation of the NK cells through measuring the expression of CD107a, only the immunoregulatory (CD56+CD16-) subpopulation increased significantly the CD107a expression when stimulated with Imiquimod (p=0.0449) as compared with the non-treated cells (Figure 4D). Imiquimod also significantly increased the proportion of cells expressing this marker (p=0.0162) in the immunoregulatory subpopulation (CD56+CD16-) (Figure 4E).
To further evaluate degranulation, we evaluated the concentration of granzyme B in the supernatant of enriched NK cells cultured with the four different ligands (Figure 4F). We observed that ODN increased significantly the secretion of Granzyme B (p=0.0075).
In addition, to determine the increase of the immunoregulatory function, we measured the concentration of different proinflammatory cytokines and chemokines in the supernatant of the PBMC cultured with the four different ligands (Figure 5). We found that R848 induced a significant increase in secretion of IFN-γ (5A), IL-2 (5B), IL-1β (5C), TNF-α (5D), IL-6 (5E), IL-8/CXCL-8 (5F), and MCP-1/CCL-2 (5G), after 24 h of stimulation with this ligand compared with the non-treated cells.
2.5. Endosomal TLRs Increase the Activation of NK Subpopulations within the PBMC.
To determine if the activation of NK cells from leukemia patients is dependent on their NK receptors (NCR), we evaluated the expression of NKG2D and NKp44 receptors in the different NK subpopulations by culturing PBMC for 24 h with Poly I:C, Imiquimod, R848 or ODN2006 (Figure 6).
Imiquimod (TLR7) and ODN (TLR9) significantly increased NKG2D expression in the cytotoxic CD56+CD16+ (p=0.0356 and p=0.0233 respectively) subpopulation (Figure 6A). When we analyzed the proportions of NKG2D positive cells, we found that none of the treatments significantly increased the percentage of NKG2D positive cells in any of the subpopulations (Figure 6B).
When the expression of NKp44 on the NK subpopulations was assessed, Poly I:C and R848 significantly increased this marker expression on CD56+CD16- subpopulation (p=0.0436 and p=0.0436 respectively) (Figure 6C). Regarding the proportions of NKp44+ cells, R848 significantly increased the percentage of CD56+CD16- expressing NKp44 (p=0.0016); CD56-CD16+ expressing NKp44 significantly increased when they were stimulated with Poly I:C (p=0.0075) and ODN (p=0.0094), (Figure 6D).
2.6. Endosomal TLRs Ligands Increase the Activation of Enriched NK Cell Subpopulations
We analyzed the expression of NKG2D and NKp44 receptors in the three different NK cell subpopulations after culturing enriched NK cells for 24 h with Poly I:C, Imiquimod, R848, or ODN2006 (Figure 7). We observed that Imiquimod and R848 significantly increased NKG2D expression in the immunoregulatory subpopulation (CD56+CD16-) (p=0.0162 and p=0.0094 respectively), while poly I:C significantly increased the NKG2D expression in the cytotoxic subpopulation CD56+CD16+ (p= 0.0352) compared with the non-treated cells. (Figure 7A). When analyzing the proportions of NKG2D positive cells, we found that none of the treatments significantly modified it in any of the subpopulations (Figure 7B).
When we analyzed the expression of NKp44, we observed that only R848 significantly increased the expression of NKp44 on the CD56+CD16+ subpopulation (p=0.0274) (Figure 7C). In the evaluation of the proportions of NKp44+ cells, we observed that ODN significantly increased the percentage of NKp44 positive cells for the cytotoxic subpopulation CD56-CD16+ (p=0.0162) (Figure 7D).
2.7. R848 Increase HLA-I Expression in Immature B Cells of Patients with ALL
We determined if the stimulation with synthetic TLR ligands affects the density of HLA-I in the lymphoblastic cells in the patient’s blood. We observed that the expression of HLA-I on mature B cells (CD19+) (Figure. 8A) and B cell precursors (CD19+CD10+) (Figurw. 8B) significantly increased when PBMC were stimulated with R848 compared to untreated PMBC (p=0.0001, p=0.0288 respectively). In the CD10+ cells, the expression levels of HLA-I significantly increased when they were stimulated with imiquimod (p=0.0436) and ODN (p=0.0436) (Figure 8C) compared to untreated PMBC. Meanwhile the proportion of CD19+ (Figure 8D) and CD19+CD10+ (Figure 8E) cells that expressed MHC-I did not change, the proportion of CD10+ cells that expressed HLA-I significantly increased only with R848 (p=0.0021) compared to untreated PBMC (Figure 8F).
2.8. TLR8 and TLR9 Ligands Increase the Cytotoxic Activity of NK Cells
To evaluate if the functional and phenotypic changes of the NK cells previously observed affectd their antitumoral activity against leukemic cells, two cytotoxicity assays were performed (Figure 9).
We measured the viability of both NK cells and leukemic RS4; 11 cells in co-culture using Zombie NIR (viability marker) to determine if the blast lysis capacity was increased in the NK cells treated with the four TLRs ligands. We observed that with all the treatments the median of the proportion of dead NK cell decreased (Poly I:C=21.80, Imiquimod=22.90, R848=28.80, and ODN=26.90) when compared with the non-treated cells (M=31.20) (Figure 9A). Interestingly, we observed that R848 and ODN2006 significantly increased the cytotoxicity against the leukemic RS4 cells (p=0.0162 and p=0.0071 respectively) (Figure 9B).
To further confirm the cytotoxicity of the NK cells isolated from the ALL patients towards the RS4; 11, we measured LDH released in the supernatant from the RS4 co-culture with stimulated NK cells. We calculated the % of cytotoxicity of the target cells. Interestingly, R848 and ODN2006 significantly increased the % of cytotoxicity against the leukemic RS4; 11 cells (p=0.0325 and p=0.0186 respectively) (Figure 9C). These results in accordance with the Zombie NIR assay suggest that stimulation with R848 and ODN may increase the cytotoxicity of the NK cells from ALL patients against the leukemic RS4; 11 cells.
3. Discussion
This study aimed to evaluate the response of NK cell subpopulations from pediatric patients with ALL to endosomal TLR and compare them with those from the unstimulated NK cells with the objective of identifying the most suitable molecule(s) with potential as adjuvants in combination with cancer immunotherapy. We show that the agonists poly I:C, imiquimod, R848, and ODN2006 increase the function of NK cells, with R848 and ODN increasing the cytotoxicity. In addition, our study showed that the most sensitive NK subpopulation was the CD56+CD16-, which is considered mainly immunoregulatory, suggesting that this subpopulation may be also contributing to blast lysis.
Existing studies have shown that only NK cells derived from healthy donors or solid tumors can be directly activated by TLR agonist stimuli, increasing their immunoregulatory and cytotoxic activities [32,33], leaving uncertainties about the response patterns in hematological malignancies. This is the first study to assess the response of different NK subpopulations from ALL patients when are stimulated with TLR´s ligands, offering insight into the functional characteristics of NK cells in this context.
In cancer situations, including ALL, exposure to tumor cells and the tumor microenvironment components hinders NK cell functions, rendering them dysfunctional [9,34]. Hence we assessed the behavior of NK cells following stimulation in both PBMCs and in isolation in order to discern whether the NK cells from our patients exhibited a consistent response to stimuli both within their natural microenvironment and when isolated.
The first result of our study is that Poly I:C (TLR3) and R848 (TLR8) gives the strongest signal upregulating NK-cell functions within the PBMCs. We found that the IFN-γ positive cells of CD56+CD16- increased by both R848 and Poly I:C in NK cells. Meanwhile, we found that the four ligands upregulated the IFN-γ production of CD56-CD16+ subpopulation on isolated NK cells. These results are in agreement with the evidence that TLR agonists can increase production of IFN-γ by NK cells [35]
Other characteristic of functional NK cells is the release of the cytotoxic granules this degranulation is measured by the expression CD107a. In our study, we observed that Poly I:C, ODN, and R848 increased NK cells CD107 expression within the PBMCs, and the percent of CD56+CD16- and CD56+CD16+ subpopulations. On the other hand, the results on isolated NK cells showed that Imiquimod (TLR7) modified the expression and proportion of cells expressing CD107a for CD56+CD16- [36,37]
The difference between the PBMC and isolated NK cells on the TLRs increasing their functions suggest that the activation of the PBMC by the TLR ligands may have an adding effect on the NK cells.
To further evaluate the degranulation on the isolated NK cells, we evaluated the concentration of Granzyme B on the supernatant, for this enzyme is one contained in the cytotoxic granules of NK cells. [38] We observed that ODN was the one which stimulated the secretion of Granzyme B from these cells, this in odds with our finding regarding the expression of CD107a which increased with Imiquimod therefore, this shoud be further studied to fully understand the phenomenon.
It has been suggested that TLR7/8 activation promotes tumors by inducing robust pro-inflammatory cytokine secretion and activating NK at the tumor site [39]. IL-1β and IL-2 have been suggested as a ptent co-stimulus for IFN-γ production by the immunoregulatory NK cells [40,41]. TNF-α, IFN-γ, and CXCL8 are produced by NK cell and also enhance their activation and recruitment [42,43]. Concordantly, we observed that R848 increased these proinflammatory cytokines on the supernatant of PBMCs. These results suggest that monocytes, T cells, and dendritic cells are being stimulated and subsequently may promote the activation of NK cells which could explain the increase in IFN-γ and CD107a observed on the CD56+CD16- subpopulation.
Regarding the density of activating receptors in NK cells from ALL patients our findings that Imiquimod and ODN increased the expression of the NKG2D in the CD56+CD16+ subpopulation of the PBMCs suggests that this ligands may help the NK cells to undergo a polarization towards an activated state while on the isolated NK cells Imiquimod and R848 increased the expression of NKG2D on the CD56+CD16- subpopulation and Poly I:C on the CD56+CD16+ subpopulation. Our results agree with what Girart et al. stated about the increase of NKG2D expression after stimulation through TLR7 [44].
The other receptor, NKp44 is associated with an activated state of the NK cells [45]. Our results show that Poly I:C, and the R848 significantly increased the expression of this receptor and the proportion of the CD56+CD16- subpopulation while Poly I:C and ODN2006 increased the proportion of NKp44 expressing cells in the CD56-CD16+ cytotoxic population of the PBMCs. Regarding the isolated NK cells, R848 increased the proportion of CD56+CD16+ cells expressing NKp44, and ODN increased the expression of NKp44 on the CD56-CD16+ subpopulation. Taking these results together, they suggest that the Poly I:C, R848 and OD2006 ligands (TLR3, TLR8, and TLR9 respectively) promote an activated status on the NK cells.
The different response to TLR of the NK cells embedded in PBMC versus the isolated NK cells in our study, confirm the microenvironment cells influence on NK responses in addition to the TLR stimulation [34] and should be further evaluated. However, in both aspects CD56+CD16- was the most sensible to the stimulation, suggesting that this subpopulation could be less impaired and its potential for a therapeutic approach should be further analyze.
Some HLA-I molecules may play a role as ligands for activating NK receptors, such as NKG2D [44]. The stimulation with the TLR8 ligand (R848) significantly increased the expression of HLA-I on the CD19+ B lymphocytes and CD19+CD10 blast subpopulation, while Imiquimod (TLR7) and ODN (TLR9) increased it on all CD10+ cells. This results are relevant since knowing the nature of these HLA-I (inhibitory o activating ligands) may help to understand the answer that NK cells will elicit.
Finally, the measurement of the cytotoxic activity of NK cells through the release of LDH and by the assessment of cell death with Zombie NIR show that R848 and ODN were able to increase the cytotoxic activity of NK cells from patients with ALL against the RS4 cell line (leukemic blasts). These results aligned with those observed regarding Granzyme B, which can suggest that this is the mechanism unfolding the blast lysis, however this must be further evaluated to confirm. In addition, our results agree with Veziani et al. [41] who demonstrated that specific TLR8 agonists increased cytokine production and cytotoxic activity.
In our work, it was evident that when the isolated CD56-CD16+ were stimulated, the increase in degranulation was not present with none of the TLR ligands, this in concordance with the literature that this subpopulation is “dysfunctional” due to the reduced cytotoxic potential, notably against tumor target cells [46]. Recently, this population has been described in Kenyan children with Burkitt lymphoma [47] and in non-virally Acute Lymphoblastic Leukemia patients [48] with adverse clinical outcome. A proteomic analysis showed that CD56-CD16+ shared some similarities with CD56dimCD16+ NK cells [49], supporting the classification of this subset as NK cells. Furthermore, there has been suggested that this phenotype with a CD56 down regulated emerge in the presence of an immunosuppressive milieu [50], even when the mechanism by which this happens remains unknow. This may explain the presence of this subpopulation in our patients with ALL and the impaired (lack of) function even after the stimulation. In our study we reported CD56-CD16+ as NK cells when isolated since we used negative selection, assuring the elimination of non-NK cells (CD14+, CD11c) and in de PBMCs due to the expression of NKG2D in 90% of this subpopulation.
Our results support the hypothesis that the activation with TLR ligand may restore the functionality and promote the expression of activation receptors of NK cell from patients with ALL. Various clinical trials with these studied ligands in solid tumors lay the groundwork for a potential translation to clinical applications in hematological malignancies. Poly I:C has been studied in combination with anti-PD1 on solid tumors (NCT03721679) [51] and in Subjects with Unresectable Hepatocellular Carcinoma (NCT03732547) [52]. Some Imiquimod trials evaluated effectiveness in cutaneous metastases of melanoma (NCT01264731) [53] and cervical high-grade squamous intraepithelial neoplasia (NCT05405270) [54]. One clinical trial for R848 evaluated the effect on T-cells in in cutaneous T-cell lymphoma (NCT01676831) [55] and as an adjuvant with vaccine therapy for melanoma (NCT00470379) [56]. ODN has been studied as Injection Boosters against solid tumors (NCT04952272) [57].
We acknowledge the limitations of our study confined to in vitro scenarios. Nonetheless, the significance of this information in literature serves as a crucial precedent. It indicates that NK cells, known to exhibit compromised functionality in hematologic neoplasms, possess the capacity to restore and enhance their cytotoxic and immunoregulatory anti-tumor functions through their endosomal TLR receptor stimulation. This sets the stage for a promising avenue of research, suggesting that these molecules warrant further exploration with an immunotherapeutic approach against hematologic neoplasms.
4. Materials and Methods
4.1. Patients
Twenty-four pediatric patients newly diagnosed between March 2022 and July 2023 with ALL, who had not received any prior treatment or transfusions and who had no infectious complications were included. Peripheral blood samples were taken with the prior informed consent of the patients' parents or guardians, in accordance with institutional protocols.
This study was approved by the Research, Ethics, and Biosafety Committees of the Hospital Infantil de México Federico Gómez, following the international guidelines for biomedical research in humans (approval no. CIOMS-WHO 1993).
4.2. Obtaining Mononuclear Cells, Isolating NK Cells, Culture and Activation with TLR Ligands
Mononuclear cells were isolated from peripheral blood by density gradient centrifugation with Lymphoprep™ (Axis-Shield, Boston, USA), according to the manufacturers protocol. To assess the direct activation of NK cells through TLRs, they were enriched from peripheral blood mononuclear cells using an EasySep™ Human NK Cell Isolation Kit (STEMCELL Technologies Inc. Vancouver, Canada), according to the manufacturer's protocol.
The obtained mononuclear cells (1 x106) were cultured in 1 mL of RPMI medium supplemented with 2% fetal bovine serum (FBS), in 48-well plates, and the enriched NK cells (1 x105) were cultured in 0.5 mL of RPMI medium supplemented with 2% FBS. The TLR ligands used, and their doses were: poly:IC 1 μg/mL (Sigma-Aldrich, USA), Imiquimod 0.1 μg/mL (Enzo life science, Plymouth, PA, EU), R-848 5 μg/mL (Enzo life science, Plymouth, PA, EU), and ODN 2006 1 ng/mL (Invivo, San Diego, CA, USA) (all concentrations were previously determined). Stimulation was left to proceed at 37ºC in a 5% CO2 atmosphere for 24 h.
4.3. Characterization of NK Cells and Determination of Activation
To characterize NK cells, they were stained for 20 min at 4°C, with 20 μL of Staning mix containing staining buffer (phosphate-buffered saline (PBS) with 1% serum albumin) and the following antibodies antihuman: anti-CD3-pacific blue, (0.2 μg per 106 cells) anti-CD56-APC, (0.5 μg per 106 cells) anti-CD19-PECy7 (0.5 μg per 106 cells ) (All of them from BD Biosciences, San Jose, CA, USA), and anti-CD16-PerCPcy5.5 (2.5 μL per 106 cells) (BioLegend, San Diego, CA, USA). To determine the activation of NK cells, the change in the expression of CD107a and intracellular IFN-γ was used, as well as the expression of receptors NKG2D and NKp44; the expression of HLA class I was also determined. The cells were stained with anti-CD107a-FITC, (2.5 μL per 106 cells) anti-HLA ABC-PE, (5 μL per 106 cells) anti-NKG2D-APCCY7 (2.5 μL per 106 cells) (BioLegend, San Diego, CA, USA), and anti-NKp44-VioBrightFITC (1 μg per 106 cells (Merck Millipore)). For intracellular staining, monensin (2 μM) (Biolegend, San Diego, CA, USA) was added to the culture during the last 4 hours of treatment. To detect intracellular cytokine production, cells were fixed, permeabilized with CytoFix/Cytoperm (BD bioscience) following the manufacturer’s protocol and stained with anti-IFN-γ-PECy7 (2.5 μL per 106 cells (Abcam, Cambridge United Kingdom) according to manufacturer's instructions. The maximum of staning mix volume used for tube of treatment were 20 μL the staining buffer volume varied depending of the tubes to stain
Reading was performed on the CytoFLEX flow cytometer (Beckman Coulter) and analysis was performed with the FlowJo software V.10
4.4. Granzyme B Detection
The concentration of granzyme B in the supernatants of cells cultured and treated with TLR ligands was determined using the commercial Human Granzyme B DuoSet ELISA kit (R&D Systems, Minneapolis USA), following the manufacturer's instructions. The optical density of each well was measured at 450 and 570 nm in a microplate reader (Agilent BioTek™ Epoch, Santa Clara, USA) with a margin of no more than 30 minutes.
4.5. Cytokines Determination
The concentration of cytokines (IFN-γ, IL-2, IL-1β, TNF-α, and IL-6) and chemokines (IL-8 and MCP-1) in the supernatants of cells cultured and treated with TLR ligands was determined using the commercial kit Human Cytokine/Chemokine Magnetic Bead Panel-Immunology Multiplex Assay (Millipore, Massachusetts, USA), following the manufacturer's instructions. The optical density of each well was measured with the MagPix plate reader (Luminex, Austin, USA).
4.6. Interferon Gamma Determination
IFN-γ was determined in supernatants of enriched NK cells. After 24 h of stimulation, of the enriched NK cells, with the TLRs ligands, the supernatant was collected and stored at –20°C in aliquots until analysis. The sample was thawed only once to avoid degradation. IFN-γ was determined with the commercial ELISA IFN-γ OptEIA kit (BD Biosciences, San Diego, CA, USA), according to the manufacturer´s instructions. The optical density of each well was measured at 450 nm in a microplate reader (Agilent BioTek™ Epoch, Santa Clara, USA) with a margin of no more than 30 minutes.
4.7. Cytotoxicity Assays
4.7.1. Zombie NIR Assay
The increase in cytotoxic activity of NK cells stimulated with the specific TLR ligands was measured by using a viability marker (Zombie NIR, Biolegend, San Diego, USA). Briefly: enriched NK cells from patients with leukemia were co-cultured with the RS4;11 CRL-1873 cell line from ATCC in a 1:2 ratio in RPMI medium with 2% FBS; then, they were stimulated or not with each ligand of TLR3, TLR7, TLR8, and TLR9 and incubated for 24 h at 37°C with 5% COAfter incubation, cells were harvested and stained with 100 μL of Zombie NIR (1:1000 dilution with PBS) to evaluate viability in a CytoFLEX flow cytometer (Beckman Coulter). Analysis was performed with the FlowJo software version V.10.
4.7.2. LDH Release Assay
This assay was also used to measure the cytotoxicity of the effector cells (enriched NK cells from ALL patients) against the target cells (RS4;11 CRL-1873 cell line from ATCC). We used the commercial kit CytoTox 96 non-radioactive assay (Promega, Wisconsin, EU) to measure the concentration of LDH released by the dead target cells after the co-culture with the NK cells, following the manufacturer’s instructions. Briefly: stimulated enriched NK cells were co-cultured with the RS4 cell line in RPMI medium with 2% FBS, for 24 h at 37°C with 5% COAfter incubation, they were centrifuged and the supernatant was stored at -20 until used. The supernatant of co-cultures with each treatment and the supernatant of NK cells and RS4 cell lines alone were placed by triplicate in a 96-well plate. The substrate provided by the kit was added to each well and the plate was incubated for 30 min at 37ºC protected from light. Finally, stop solution was added and reading was carried out at 490 nm in a microplate reader (Agilent BioTek™ Epoch, Santa Clara, USA) with a margin of no more than 30 minutes. To determine the cytotoxic activity of the NK cells against target RS4; 11 cells, we used the following formula.
4.8. Statistical Analysis
The median frequencies of the different subpopulations of peripheral blood NK cells and enriched NK cells from patients with ALL from each treatment were compared with the non-treated cells using a Friedman test. GraphPad Prism version 8 (GraphPad Software Inc., Boston, USA) was used for the statistical analysis.
5. Conclusions
It is possible to directly activate the NK cells from ALL by endosomal TLRs this activation promotes a different behavior from the different NK cells subpopulations than when activated within the PBMCs. The activation via endosomal TLRs increase the functional activity of the NK cells from ALL patients. Furthermore, direct activation of the NK cells via TLR8 and TLR9 with R848 and ODN respectively increased the anti-tumoral activity of the NK cells, since they increased the blast lysis. These findings provide a foundation for further exploration of TLR ligands as potential immunotherapeutic agents to enhance NK cell-mediated anti-tumor responses in ALL.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Viability Cytometry; Figure S2: MFI of Activation markers of the NK cells within PBMCs after endosomal TLR stimulation.
Author Contributions
For research articles with several authors, a short paragraph specifying their individual contributions must be provided. The following statements should be used “Conceptualization, C.MB. and J.GZ.; methodology, J.GZ.; software, J.GZ.; validation, C.MB., J.GZ. and E.PF.; formal analysis, J.GZ.; investigation, J.GZ.; resources, J.GZ.; data curation, J.GZ and E-PF.; writing—original draft preparation, J.GZ.; writing—review and editing, C.MB., J.GZ., V.OL., A.MS., E.JH. and E.O.; visualization, J.GZ.; supervision, C.MB., E.O., E.PF., V.OL., A.MS. and E.JH.; project administration, C.MB.; funding acquisition, C.MB. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Hospital Infantil de México Federico Gómez, grant number “HIM/2019/035” and “The APC was funded by Hospital Infantil de Mexico Federico Gómez”.ing.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Research, Research Ethics and Biosafety Committees of the Children's Hospital of Mexico Federico Gómez (protocol no. HIM/2019/035: 2019-11-07).
Informed Consent Statement
Written informed consent from parents or legal representative of all our participants was obtained. Also Informed Assent from participants older than 8 years, was obtained for this study.
Data Availability Statement
The datasets during the current study are available from the corresponding author upon reasonable request.
Acknowledgments
Janet Gallardo-Zapata is a doctoral student from the Programa de Doctorado en Ciencias Biomédicas, Universidad Nacional Autónoma de México (UNAM) and has received CONAHCYT fellowship 669082.
Conflicts of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Maldonado-Bernal C, Sánchez-Herrera D. Toll-Like Receptors and Natural Killer Cells. Toll-like Receptors, IntechOpen; 2020. [CrossRef]
- Cooper MA, Fehniger TA, Caligiuri MA. The biology of human natural killer-cell subsets. Trends Immunol 2001;22:633–40. [CrossRef]
- Huntington ND, Vosshenrich CAJ, Di Santo JP. Developmental pathways that generate natural-killer-cell diversity in mice and humans. Nat Rev Immunol 2007;7:703–14. [CrossRef]
- Tang D, Kang R, Coyne CB, Zeh HJ, Lotze MT. PAMPs and DAMPs: Signal 0s that spur autophagy and immunity. Immunol Rev 2012;249:158–75. [CrossRef]
- Jiménez-Morales S, Hidalgo-Miranda A, Ramírez-Bello J. Acute lymphoblastic leukemia: A genomic perspective. Bol Med Hosp Infant Mex 2017;74:13–26. [CrossRef]
- Ceppi F, Cazzaniga G, Colombini A, Biondi A, Conter V. Risk factors for relapse in childhood acute lymphoblastic leukemia: Prediction and prevention. Expert Rev Hematol 2015;8:57–70. [CrossRef]
- Greaves M. A causal mechanism for childhood acute lymphoblastic leukaemia. Nat Rev Cancer 2018;18:471–84. [CrossRef]
- Chiossone L, Dumas P-Y, Vienne M, Vivier E. Natural killer cells and other innate lymphoid cells in cancer. Nat Rev Immunol 2018;18:671–88. [CrossRef]
- Valenzuela-Vazquez L, Núñez-Enríquez JC, Sánchez-Herrera J, Jiménez-Hernández E, Martín-Trejo JA, Espinoza-Hernández LE; et al. Functional characterization of NK cells in Mexican pediatric patients with acute lymphoblastic leukemia: Report from the Mexican Interinstitutional Group for the Identification of the Causes of Childhood Leukemia. PLoS ONE 2020;15:1–15. [CrossRef]
- Guillerey C, Huntington ND, Smyth MJ. Targeting natural killer cells in cancer immunotherapy. Nat Immunol 2016;17:1025–36. [CrossRef]
- Rouce RH, Shaim H, Sekine T, Weber G, Ballard B, Ku S; et al. The TGF-β/SMAD pathway is an important mechanism for NK cell immune evasion in childhood B-acute lymphoblastic leukemia. Leukemia 2016;30:800–11. [CrossRef]
- Valenzuela-Vázquez L, Nuñez-Enriquez JC, Sánchez-Herrera J, Medina-Sanson A, Pérez-Saldivar ML, Jiménez-Hernández E; et al. NK cells with decreased expression of multiple activating receptors is a dominant phenotype in pediatric patients with acute lymphoblastic leukemia. Front Oncol 2022;12. [CrossRef]
- Kawasaki T, Kawai T. Toll-Like Receptor Signaling Pathways. Front Immunol 2014;0:461. [CrossRef]
- Guo Q, Zhang C. Critical role of Toll-like receptor signaling in NK cell activation. Chinese Science Bulletin 2012;57:3192–202. [CrossRef]
- Vacchelli E, Eggermont A, Sautè;s-Fridman C, Galon J, Zitvogel L, Kroemer G; et al. Trial watch toll-like receptor agonists for cancer therapy. Oncoimmunology 2013;2:1–14. [CrossRef]
- Barbalat R, Ewald SE, Mouchess ML, Barton GM. Nucleic Acid Recognition by the Innate Immune System. Annu Rev Immunol 2011;29:185–214. [CrossRef]
- Blasius AL, Beutler B. Intracellular Toll-like Receptors. Immunity 2010;32:305–15. [CrossRef]
- Roach JC, Glusman G, Rowen L, Kaur A, Purcell MK, Smith KD; et al. The evolution of vertebrate Toll-like receptors. Proceedings of the National Academy of Sciences 2005;102:9577–82. [CrossRef]
- Takeda K, Akira S. TLR signaling pathways. Semin Immunol 2004;16:3–9. [CrossRef]
- Akira S, Takeda K, Kaisho T. Toll-like receptors: Critical proteins linking innate and acquired immunity. Nature Immunology 2001 2:8 2001;2:675–80. [CrossRef]
- Rakoff-Nahoum S, Medzhitov R. Toll-like receptors and cancer. Nat Rev Cancer 2009;9:57–63. [CrossRef]
- Sánchez-Herrera D, Contreras-Ramos A, Jiménez-Hernández E, Medina-Sansón A, Giono-Cerezo S, Maldonado-Bernal C. NK Cell Subpopulation Is Altered and the Expression of TLR1 and TLR9 Is Decreased in Patients with Acute Lymphoblastic Leukemia. J Oncol 2021;2021:1–12. [CrossRef]
- Della Chiesa M, De Maria A, Muccio L, Bozzano F, Sivori S, Moretta L. Human NK Cells and Herpesviruses: Mechanisms of Recognition, Response and Adaptation. Front Microbiol 2019;10. [CrossRef]
- Cooper MA, Fehniger TA, Fuchs A, Colonna M, Caligiuri MA. NK cell and DC interactions. Trends Immunol 2004;25:47–52. [CrossRef]
- Guillerey C, Chow MT, Miles K, Olver S, Sceneay J, Takeda K; et al. Toll-like receptor 3 regulates NK cell responses to cytokines and controls experimental metastasis. Oncoimmunology 2015;4:1–11. [CrossRef]
- Gruijs M, Ganzevles SH, Stigter-van Walsum M, van der Mast R, van Ostaijen-ten Dam MM, Tuk CW; et al. NK Cell-Dependent Antibody-Mediated Immunotherapy Is Improved In Vitro and In Vivo When Combined with Agonists for Toll-like Receptor 2 in Head and Neck Cancer Models. Int J Mol Sci 2021;22:11057. [CrossRef]
- Li T, Yang Y, Song H, Li H, Cui A, Liu Y; et al. Activated NK cells kill hepatic stellate cells via p38/PI3K signaling in a TRAIL-involved degranulation manner. J Leukoc Biol 2019;105:695–704. [CrossRef]
- Samudio I, Rezvani K, Shaim H, Hofs E, Ngom M, Bu L; et al. UV-inactivated HSV-1 potently activates NK cell killing of leukemic cells. Blood, The Journal of the American Society of Hematology 2016;127:2575–86. [CrossRef]
- Gorski KS, Waller EL, Bjornton-Severson J, Hanten JA, Riter CL, Kieper WC; et al. Distinct indirect pathways govern human NK-cell activation by TLR-7 and TLR-8 agonists. Int Immunol 2006;18:1115–26. [CrossRef]
- Lauzon NM, Mian F, MacKenzie R, Ashkar AA. The direct effects of Toll-like receptor ligands on human NK cell cytokine production and cytotoxicity. Cell Immunol 2006;241:102–12. [CrossRef]
- Ronsley R, Kariminia A, Ng B, Mostafavi S, Reid G, Subrt P; et al. The TLR9 agonist (GNKG168) induces a unique immune activation pattern in vivo in children with minimal residual disease positive acute leukemia: Results of the TACL T2009-008 phase I study. Pediatr Hematol Oncol 2019;36:468–81. [CrossRef]
- Sivori S, Falco M, Della Chiesa M, Carlomagno S, Vitale M, Moretta L; et al. CpG and double-stranded RNA trigger human NK cells by toll-like receptors: Induction of cytokine release and cytotoxicity against tumors dendritic cells. Proc Natl Acad Sci U S A 2004;101:10116–21. [CrossRef]
- Khanna V, Kim H, Zhang W, Larson P, Shah M, Griffith TS; et al. Novel TLR 7/8 agonists for improving NK cell mediated antibody-dependent cellular cytotoxicity (ADCC). Sci Rep 2021;11:3346. [CrossRef]
- Portale F, Di Mitri D. NK Cells in Cancer: Mechanisms of Dysfunction and Therapeutic Potential. Int J Mol Sci 2023;24:9521. [CrossRef]
- Veneziani I, Alicata C, Moretta L, Maggi E. The Latest Approach of Immunotherapy with Endosomal TLR Agonists Improving NK Cell Function: An Overview. Biomedicines 2022;11:64. [CrossRef]
- Cooper MA, Fehniger TA, Turner SC, Chen KS, Ghaheri BA, Carson WE; et al. Human natural killer cells: A unique innate immunoregulatory role for the CD56bright subset. Blood 2000;96:3146–51. [CrossRef]
- Poli A, Michel T, Thérésine M, Andrès E, Hentges F, Zimmer J. CD56bright natural killer (NK) cells: An important NK cell subset. Immunology 2009;126:458–65. [CrossRef]
- Prager I, Liesche C, van Ooijen H, Urlaub D, Verron Q, Sandström N; et al. NK cells switch from granzyme B to death receptor–mediated cytotoxicity during serial killing. Journal of Experimental Medicine 2019;216:2113–27. [CrossRef]
- Khanna V, Kim H, Zhang W, Larson P, Shah M, Griffith TS; et al. Novel TLR 7/8 agonists for improving NK cell mediated antibody-dependent cellular cytotoxicity (ADCC). Sci Rep 2021;11:3346. [CrossRef]
- Cooper MA, Fehniger TA, Turner SC, Chen KS, Ghaheri BA, Ghayur T; et al. Human natural killer cells: A unique innate immunoregulatory role for the CD56bright subset. Blood 2001;97:3146–51. [CrossRef]
- Veneziani I, Alicata C, Pelosi A, Landolina N, Ricci B, D’Oria V; et al. Toll-like receptor 8 agonists improve NK-cell function primarily targeting CD56brightCD16- subset. J Immunother Cancer 2022;10. [CrossRef]
- Wang R, Jaw JJ, Stutzman NC, Zou Z, Sun PD. Natural killer cell-produced IFN-γ and TNF-α induce target cell cytolysis through up-regulation of ICAM-J Leukoc Biol 2012;91:299. [CrossRef]
- Montaldo E, Vitale C, Cottalasso F, Conte R, Glatzer T, Ambrosini P; et al. Human NK cells at early stages of differentiation produce CXCL8 and express CD161 molecule that functions as an activating receptor. Blood 2012;119:3987–96. [CrossRef]
- Girart M V, Fuertes MB, Domaica CI, Rossi LE, Zwirner NW. Engagement of TLR3, TLR7, and NKG2D regulate IFN-γ secretion but not NKG2D-mediated cytotoxicity by human NK cells stimulated with suboptimal doses of IL-The Journal of Immunology 2007;179:3472–9. [CrossRef]
- Kasahara Y, Shin C, Kubo N, Mihara K, Iwabuchi H, Takachi T; et al. Development and characterisation of NKp44-based chimeric antigen receptors that confer T cells with NK cell-like specificity. Clin Transl Immunology 2020;9. [CrossRef]
- Müller-Durovic B, Grählert J, Devine OP, Akbar AN, Hess C. CD56-negative NK cells with impaired effector function expand in CMV and EBV co-infected healthy donors with age. Aging 2019;11:724–40. [CrossRef]
- Forconi CS, Cosgrove CP, Saikumar-Lakshmi P, Nixon CE, Foley J, Ong’echa JM; et al. Poorly cytotoxic terminally differentiated CD56negCD16pos NK cells accumulate in Kenyan children with Burkitt lymphomas. Blood Adv 2018;2:1101–14. [CrossRef]
- Chretien A-S, Devillier R, Granjeaud S, Cordier C, Demerle C, Salem N; et al. High-dimensional mass cytometry analysis of NK cell alterations in AML identifies a subgroup with adverse clinical outcome. Proceedings of the National Academy of Sciences 2021;118. [CrossRef]
- Voigt J, Malone DFG, Dias J, Leeansyah E, Björkström NK, Ljunggren H; et al. Proteome analysis of human CD56negNK cells reveals a homogeneous phenotype surprisingly similar to CD56dimNK cells. Eur J Immunol 2018;48:1456–69. [CrossRef]
- Reiners KS, Topolar D, Henke A, Simhadri VR, Kessler J, Sauer M; et al. Soluble ligands for NK cell receptors promote evasion of chronic lymphocytic leukemia cells from NK cell anti-tumor activity. Blood 2013;121:3658–65. [CrossRef]
- Clinicaltrials.gov. Poly-ICLC (Hiltonol) and Anti-PD1 or Anti-PD-L1 (NCT03721679). Bethesda (MD): National Library of Medicine (US) 2000 Feb 29 2018. https://clinicaltrials.gov/study/NCT03721679?cond=Cancer&term=Polyinosinic-polycytidylic%20acid&rank=1 (accessed January 15, 2024).
- Clinicaltrials.gov. Study of PolyIC and PD-1 mAb in Subjects With Unresectable Hepatocellular Carcinoma (NCT03732547). Bethesda (MD): National Library of Medicine (US) 2000 Feb 29 2018. https://clinicaltrials.gov/study/NCT03732547?cond=Cancer&term=poly-IC&intr=Therapy&page=2&rank=11 (accessed January 15, 2024).
- ClinicalTrials.gov. Combination Therapy of Topical Imiquimod Plus Multipeptide Vaccination for Cutaneous Metastases of Melanoma (MEL53) (NCT01264731). Bethesda (MD): National Library of Medicine (US) 2000 Feb 29 2011. https://clinicaltrials.gov/study/NCT01264731?cond=Cancer&term=imiquimod&intr=Therapy&page=3&rank=25 (accessed January 13, 2024).
- Clinicaltrials.Gov. Predicting Response In Cervical Intraepithelial Neoplasia to Topical Imiquimod Treatment (PRedICT-TOPIC) (NCT05405270). Bethesda (MD): National Library of Medicine (US) 2000 Feb 29 2022. https://clinicaltrials.gov/study/NCT05405270?cond=Cancer&term=imiquimod&intr=Therapy&page=1&rank=6 (accessed January 13, 2024).
- Rook AH, Gelfand JM, Wysocka M, Troxel AB, Benoit B, Surber C; et al. Topical resiquimod can induce disease regression and enhance T-cell effector functions in cutaneous T-cell lymphoma. Blood 2015;126:1452–61. [CrossRef]
- clinicaltrials.gov. Vaccine Therapy and Resiquimod in Treating Patients With Stage II, Stage III, or Stage IV Melanoma That Has Been Completely Removed by Surgery (NCT00470379). Bethesda (MD): National Library of Medicine (US) 2000 Feb 29 2006. https://clinicaltrials.gov/study/NCT00470379?cond=Cancer&term=R848&intr=Therapy&page=1&rank=2 (accessed January 15, 2024).
- Clinicaltrials.gov. Intratumor CpG-ODN Injection Boosters Immune Killing Against in Situ Tumor Antigen Release for Advanced Solid Tumors (NCT04952272). Bethesda (MD): National Library of Medicine (US) 2000 Feb 29 2021. https://clinicaltrials.gov/study/NCT04952272?cond=Cancer&term=ODN%20&intr=Therapy&rank=1 (accessed January 15, 2024).
Figure 1.
PBMC Flow cytometry analysis strategy. We obtained the total NK cells (CD3-, CD19-) from the lymphocyte’s population identified by granularity-size and from this population we obtained the NK cells subpopulations depending on their expression of CD56 and CDFinally, from each subpopulation, we obtained the expression of the markers to be analyzed.
Figure 1.
PBMC Flow cytometry analysis strategy. We obtained the total NK cells (CD3-, CD19-) from the lymphocyte’s population identified by granularity-size and from this population we obtained the NK cells subpopulations depending on their expression of CD56 and CDFinally, from each subpopulation, we obtained the expression of the markers to be analyzed.

Figure 2.
Enriched NK cells Flow cytometry analysis strategy. We obtained the total NK cells (CD3-, CD19-) from the lymphocytes population identified by granularity-size and from this population we obtained the NK cell subpopulations depending on their expression of CD56 and CDFinally, from each subpopulation, we obtained the expression of the markers to be analyzed.
Figure 2.
Enriched NK cells Flow cytometry analysis strategy. We obtained the total NK cells (CD3-, CD19-) from the lymphocytes population identified by granularity-size and from this population we obtained the NK cell subpopulations depending on their expression of CD56 and CDFinally, from each subpopulation, we obtained the expression of the markers to be analyzed.
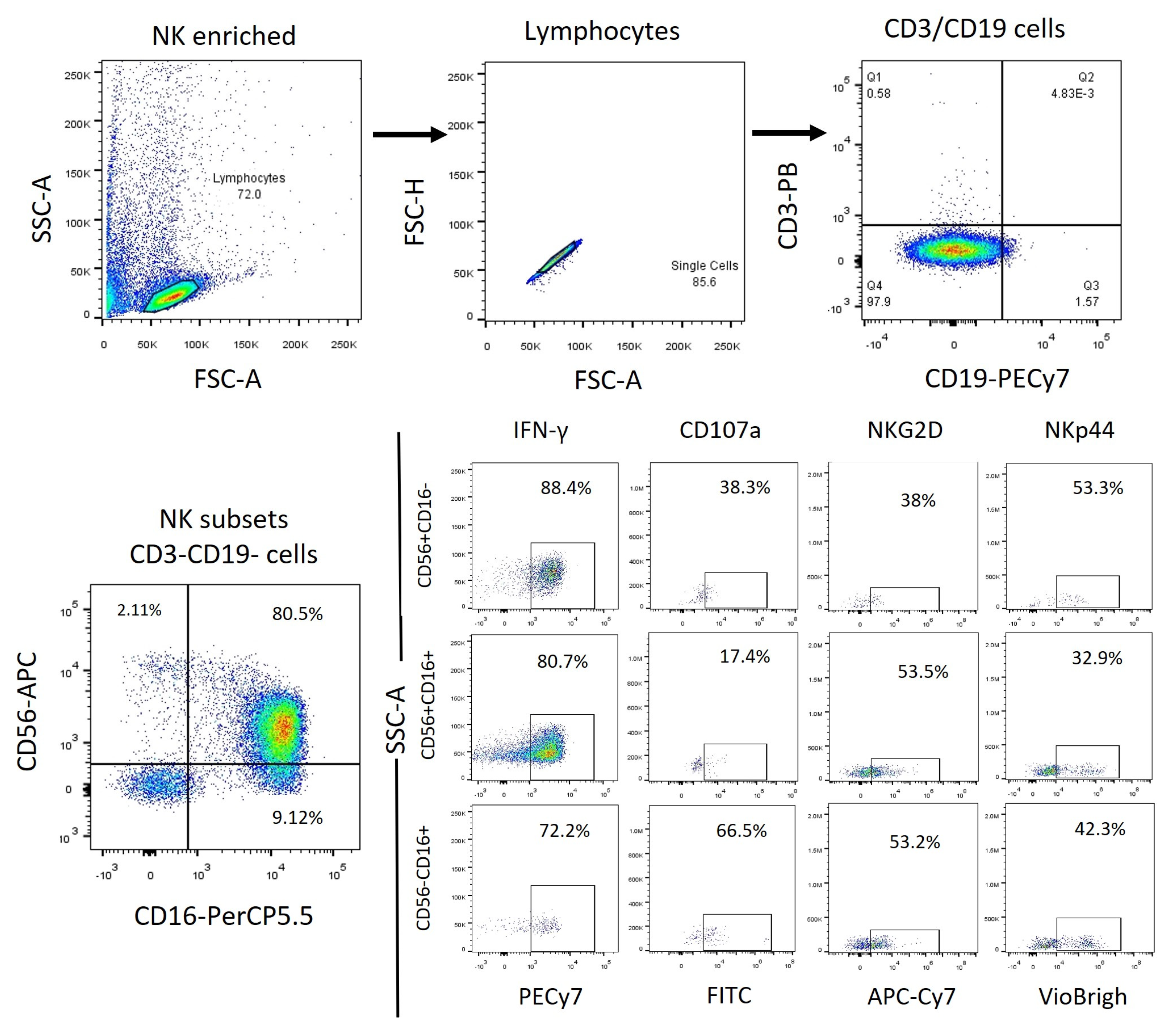
Figure 3.
Functional Activity of the NK cells within PBMCs after endosomal TLR stimulation. A The MFI of intracellular IFN-γ was determined in the three subpopulations for each treatment and compared to the MFI of non-treated cells B The median of the percentage of the IFN-γ expressing cells was determined and compared with the percentage of non-treated cells expressing IFN-γ. C The median of the Median Fluorescence Intensity (MFI) of CD107a was determined for each treatment in the three subpopulations and compared to the MFI of non-treated cells (control). D The median of the percentage of the CD107a expressing cells was determined for each treatment in the three subpopulations and compared with the percentage of non-treated cells expressing CD107a. The box and whisker plot was employed to visualize the distribution of the data, highlighting the central tendency, spread and the outlier’s values within the dataset. The Friedman test was used for the comparison. *p<0.05, **p<0.005.
Figure 3.
Functional Activity of the NK cells within PBMCs after endosomal TLR stimulation. A The MFI of intracellular IFN-γ was determined in the three subpopulations for each treatment and compared to the MFI of non-treated cells B The median of the percentage of the IFN-γ expressing cells was determined and compared with the percentage of non-treated cells expressing IFN-γ. C The median of the Median Fluorescence Intensity (MFI) of CD107a was determined for each treatment in the three subpopulations and compared to the MFI of non-treated cells (control). D The median of the percentage of the CD107a expressing cells was determined for each treatment in the three subpopulations and compared with the percentage of non-treated cells expressing CD107a. The box and whisker plot was employed to visualize the distribution of the data, highlighting the central tendency, spread and the outlier’s values within the dataset. The Friedman test was used for the comparison. *p<0.05, **p<0.005.
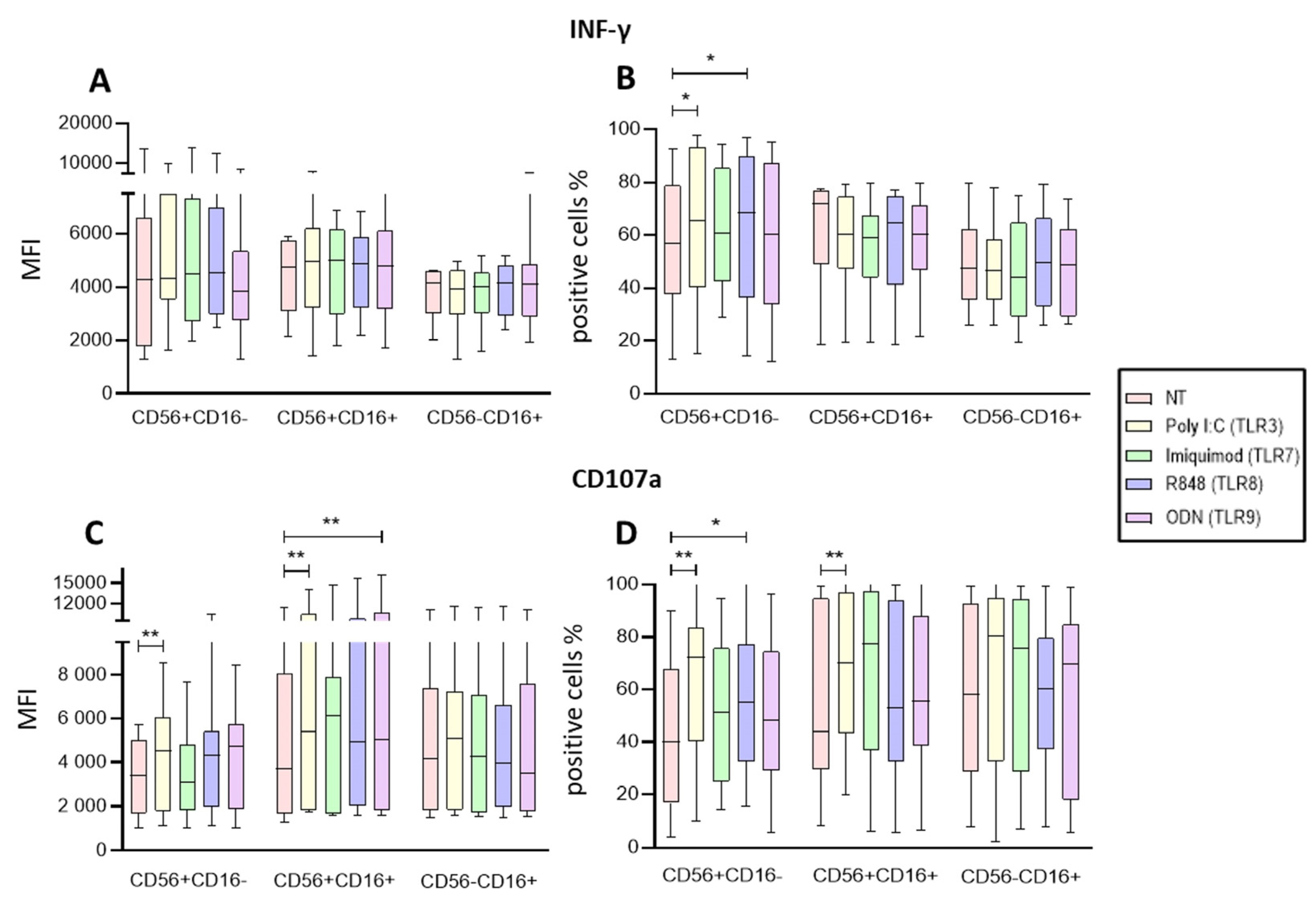
Figure 4.
Functional Activity of the enriched NK cells after endosomal TLR stimulation. A The MFI of IFN-γ was determined for each treatment in the three subpopulations and compared to the non-treated MFI. B The median of the percentage of the IFN-γ expressing cells was determined and compared with the percentage of non-treated cells expressing IFN-γ. C Concentration of IFN-γ in supernatants of enriched NK cells after endosomal TLR stimulation D The median of the Median Fluorescence Intensity (MFI) of CD107a was determined for each treatment in the three subpopulations and compared to the MFI of non-treated cells (control). E The median of the percentage of the CD107a expressing cells was determined and compared with the percentage of non-treated cells expressing CD107a. F The concentration of Granzyme B in supernatants of enriched NK cells after endosomal TLR stimulation. The violin plot was employed similar to the box-and-whiskers plot, to visualize the full distribution of the data, providing shape and density within the dataset. The Friedman test was used for the comparison. *p<0.05, **p<0.005, ***p<0.0005.
Figure 4.
Functional Activity of the enriched NK cells after endosomal TLR stimulation. A The MFI of IFN-γ was determined for each treatment in the three subpopulations and compared to the non-treated MFI. B The median of the percentage of the IFN-γ expressing cells was determined and compared with the percentage of non-treated cells expressing IFN-γ. C Concentration of IFN-γ in supernatants of enriched NK cells after endosomal TLR stimulation D The median of the Median Fluorescence Intensity (MFI) of CD107a was determined for each treatment in the three subpopulations and compared to the MFI of non-treated cells (control). E The median of the percentage of the CD107a expressing cells was determined and compared with the percentage of non-treated cells expressing CD107a. F The concentration of Granzyme B in supernatants of enriched NK cells after endosomal TLR stimulation. The violin plot was employed similar to the box-and-whiskers plot, to visualize the full distribution of the data, providing shape and density within the dataset. The Friedman test was used for the comparison. *p<0.05, **p<0.005, ***p<0.0005.
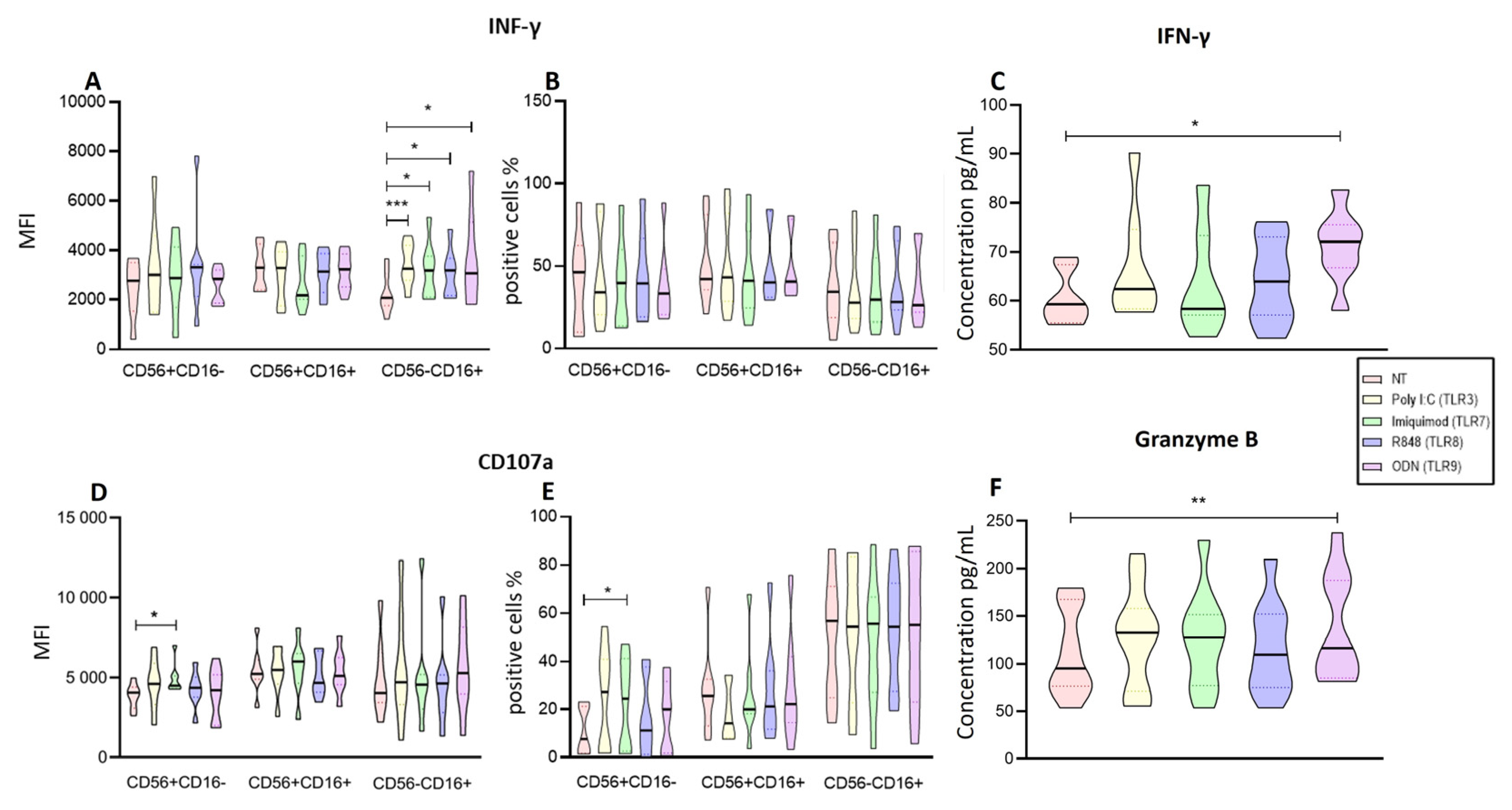
Figure 5.
C IL-2, D TNF-α, E IL-6, F IL-8, and G MCP-1 was determined for each treatment and compared to the non-treated cells. The Friedman test was used for the comparison. *p<0.05, **p<0.005.
Figure 5.
C IL-2, D TNF-α, E IL-6, F IL-8, and G MCP-1 was determined for each treatment and compared to the non-treated cells. The Friedman test was used for the comparison. *p<0.05, **p<0.005.
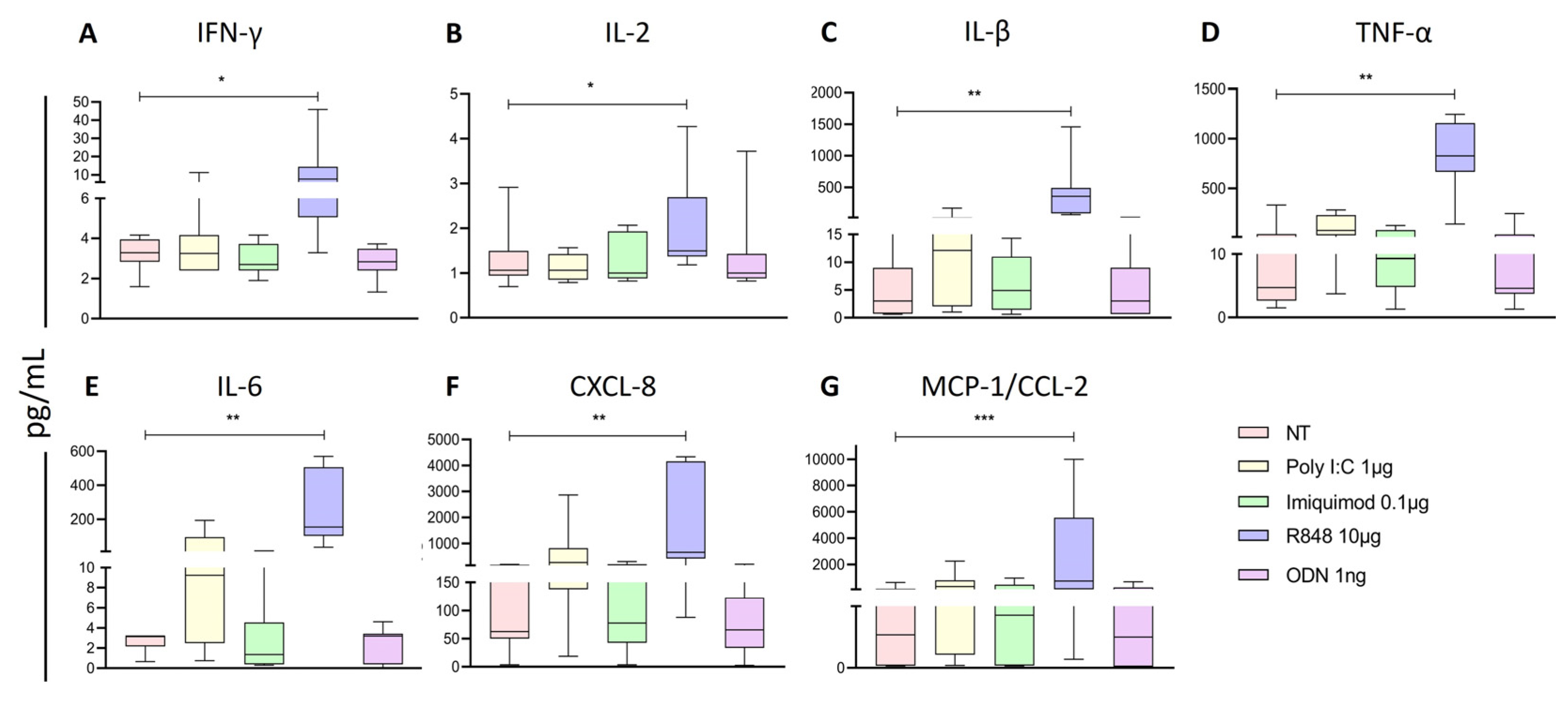
Figure 6.
Analysis of Activation markers of the different subpopulations of NK cells from PBMCs after endosomal TLR stimulation. A The MFI of NKG2D expression was determined for each treatment in the three subpopulations and compared to the MFI of non-treated cells. B The median of the percentage of NKG2D expressing cells after treatment with the TLRs ligands was determined and compared with the percentage of non-treated cells expressing it. C The MFI of NKp44 expression was determined for each treatment in the three subpopulations and compared to the MFI of non-treated cells. D The median of the percentage of NKp44 expressing cells after treatment with the TLR ligands was determined and compared with the percentage of non-treated cells expressing it. The box and whisker plot was employed to visualize the distribution of the data, highlighting the central tendency, spread and the outlier’s values within the dataset. The Friedman test was used for the comparison. *p<0.05, **p<0.005.
Figure 6.
Analysis of Activation markers of the different subpopulations of NK cells from PBMCs after endosomal TLR stimulation. A The MFI of NKG2D expression was determined for each treatment in the three subpopulations and compared to the MFI of non-treated cells. B The median of the percentage of NKG2D expressing cells after treatment with the TLRs ligands was determined and compared with the percentage of non-treated cells expressing it. C The MFI of NKp44 expression was determined for each treatment in the three subpopulations and compared to the MFI of non-treated cells. D The median of the percentage of NKp44 expressing cells after treatment with the TLR ligands was determined and compared with the percentage of non-treated cells expressing it. The box and whisker plot was employed to visualize the distribution of the data, highlighting the central tendency, spread and the outlier’s values within the dataset. The Friedman test was used for the comparison. *p<0.05, **p<0.005.
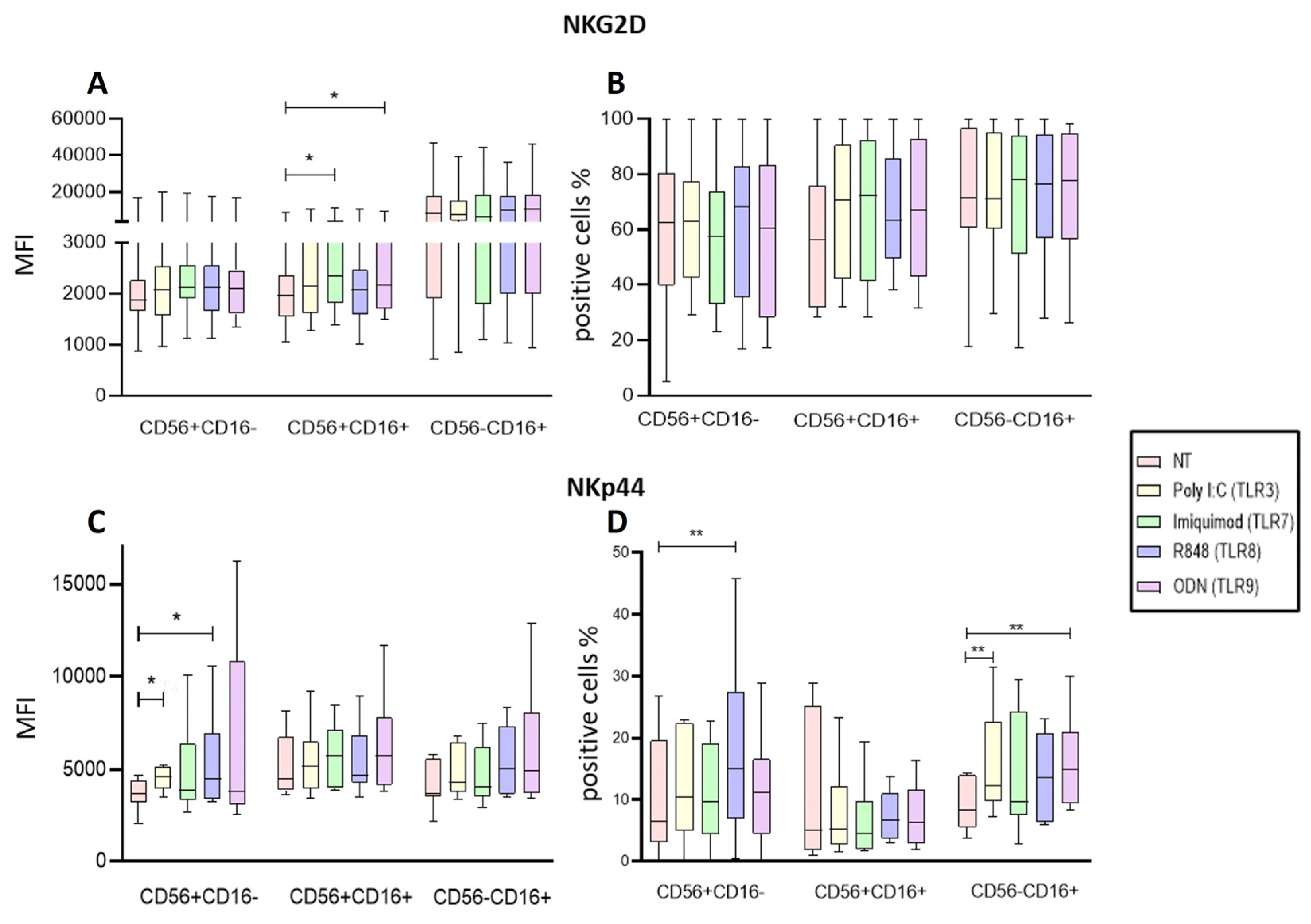
Figure 7.
Analysis of Activation markers of the different subpopulations of enriched NK cells after endosomal TLR stimulation. A The MFI of NKG2D expression was determined for each treatment in the three subpopulations and compared to the MFI of non-treated cells. B The median of the percentage of NKG2D expressing cells after treatment with the TLR ligands was determined and compared with the percentage of non-treated cells expressing it. C The MFI of NKp44 expression was determined for each treatment in the three subpopulations and compared to the MFI of non-treated cells. D The median of the percentage of NKp44 expressing cells after treatment with the TLR ligands was determined and compared with the percentage of non-treated cells expressing it. The violin plot was employed similar to the box-and-whiskers plot, to visualize the full distribution of the data, providing shape and density within the dataset. The Friedman test was used for the comparison. *p<0.05, **p<0.005.
Figure 7.
Analysis of Activation markers of the different subpopulations of enriched NK cells after endosomal TLR stimulation. A The MFI of NKG2D expression was determined for each treatment in the three subpopulations and compared to the MFI of non-treated cells. B The median of the percentage of NKG2D expressing cells after treatment with the TLR ligands was determined and compared with the percentage of non-treated cells expressing it. C The MFI of NKp44 expression was determined for each treatment in the three subpopulations and compared to the MFI of non-treated cells. D The median of the percentage of NKp44 expressing cells after treatment with the TLR ligands was determined and compared with the percentage of non-treated cells expressing it. The violin plot was employed similar to the box-and-whiskers plot, to visualize the full distribution of the data, providing shape and density within the dataset. The Friedman test was used for the comparison. *p<0.05, **p<0.005.
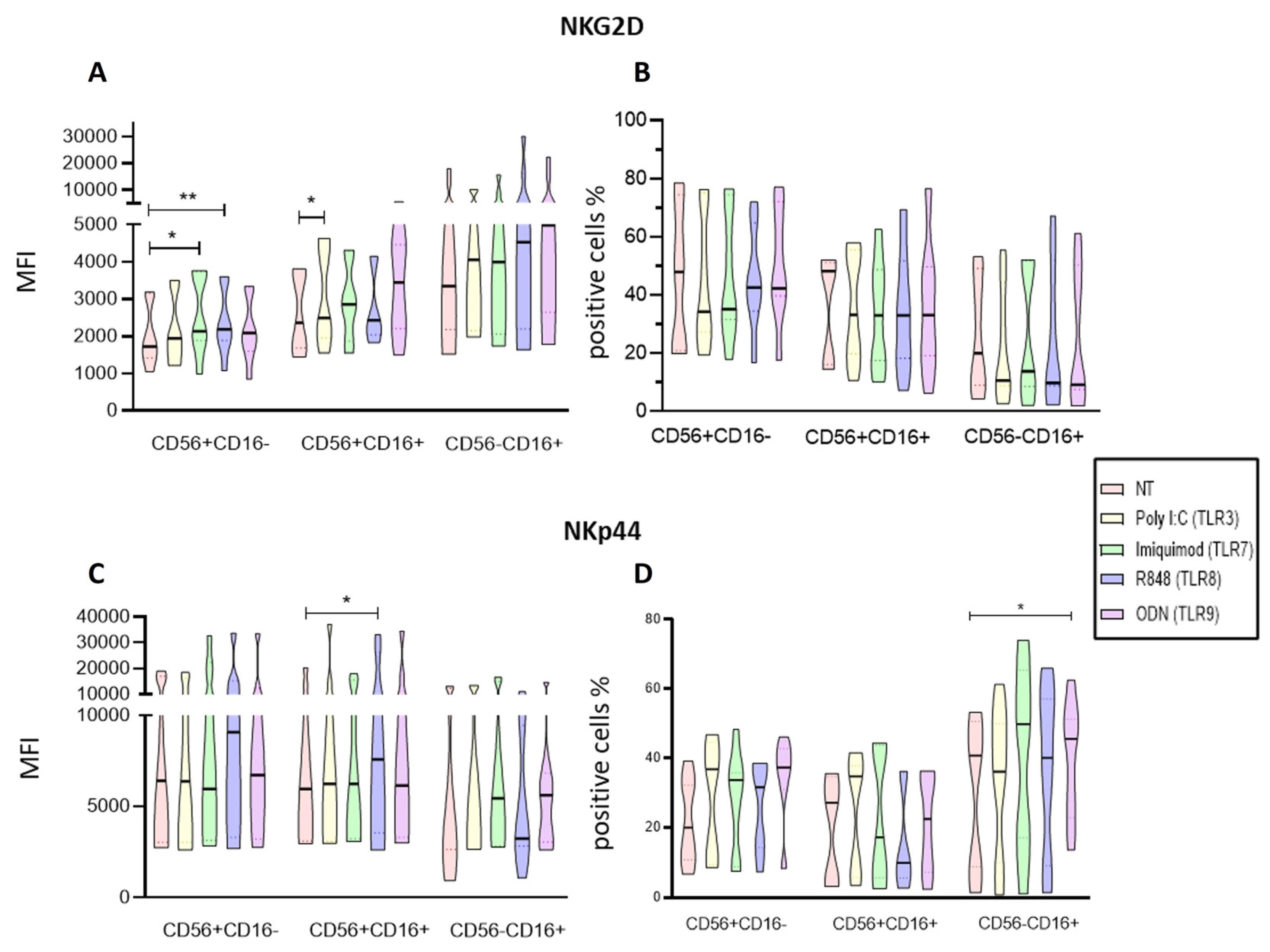
Figure 8.
represents the mature B cells, B CD10+ represents the marker of lymphoid progenitor cells, and C CD10+ CD19+ represents the pre-B cells from the patients with ALL. The median of MFI and the median of the percentage of HLA-I was determined for each treatment and compared to the non-treated cells. The box and whisker plot was employed to visualize the distribution of the data, highlighting the central tendency, spread and the outlier’s values within the dataset. The Friedman test was used for the comparison. *p<0.05, **p<0.005 ***p<0.0005.
Figure 8.
represents the mature B cells, B CD10+ represents the marker of lymphoid progenitor cells, and C CD10+ CD19+ represents the pre-B cells from the patients with ALL. The median of MFI and the median of the percentage of HLA-I was determined for each treatment and compared to the non-treated cells. The box and whisker plot was employed to visualize the distribution of the data, highlighting the central tendency, spread and the outlier’s values within the dataset. The Friedman test was used for the comparison. *p<0.05, **p<0.005 ***p<0.0005.
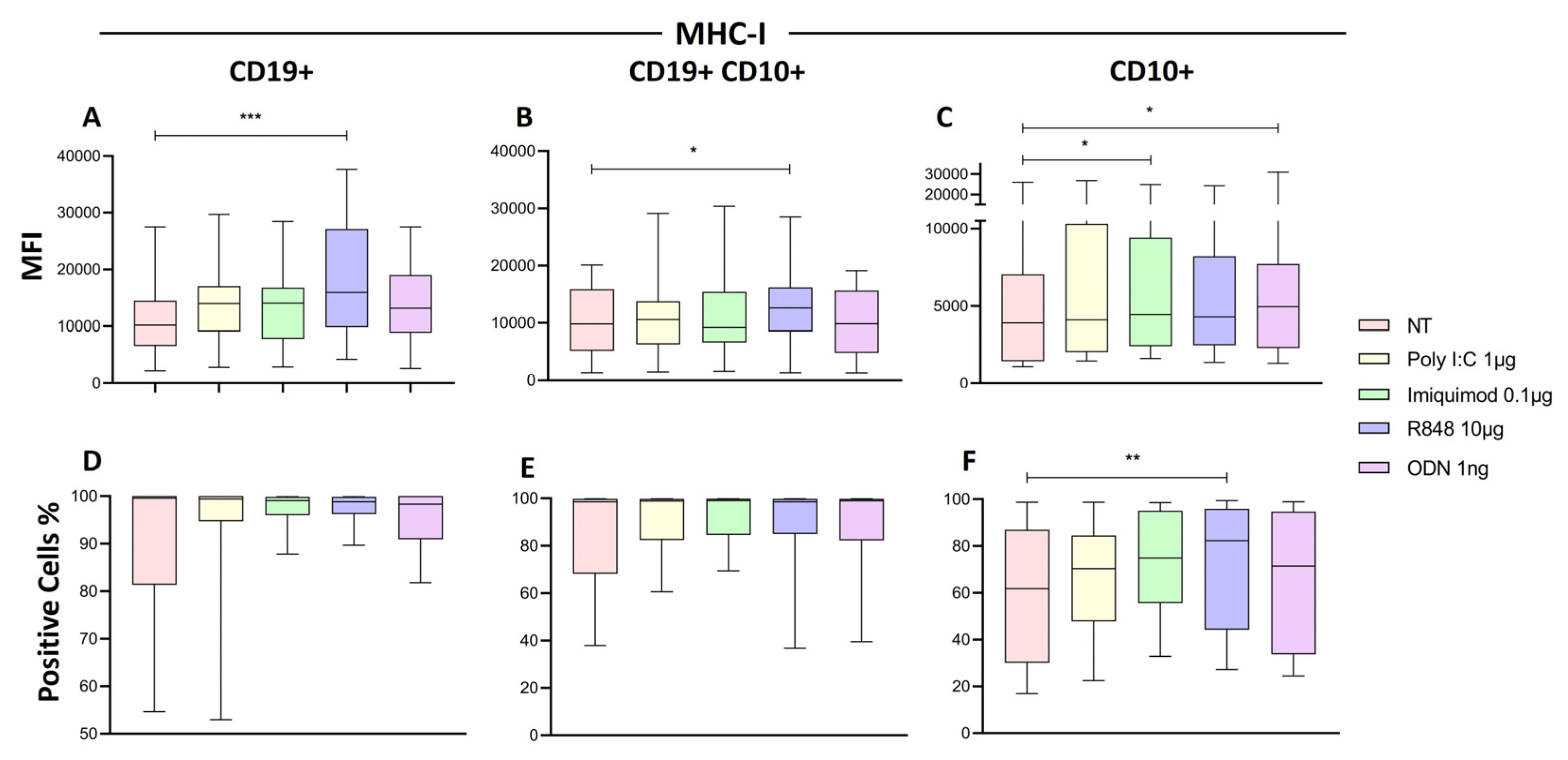
Figure 9.
Cytotoxic activity of NK cells. A The median of the percentage of NK cells death was determined for each treatment and compared with the percentage of non-treated cells death. B The median of the percentage of RS4 death was determined for each treatment and compared with the percentage of non-treated cells death. C Cell-mediated cytotoxic activity, the median of the percentage of LDH released into the supernatant of cocultures of RS4 cells with NK cells. The box and whisker plot was employed to visualize the distribution of the data, highlighting the central tendency, spread and the outlier’s values within the dataset. The Friedman test was used for the comparison. *p<0.05, **p<0.005 ***p<0.0005.
Figure 9.
Cytotoxic activity of NK cells. A The median of the percentage of NK cells death was determined for each treatment and compared with the percentage of non-treated cells death. B The median of the percentage of RS4 death was determined for each treatment and compared with the percentage of non-treated cells death. C Cell-mediated cytotoxic activity, the median of the percentage of LDH released into the supernatant of cocultures of RS4 cells with NK cells. The box and whisker plot was employed to visualize the distribution of the data, highlighting the central tendency, spread and the outlier’s values within the dataset. The Friedman test was used for the comparison. *p<0.05, **p<0.005 ***p<0.0005.
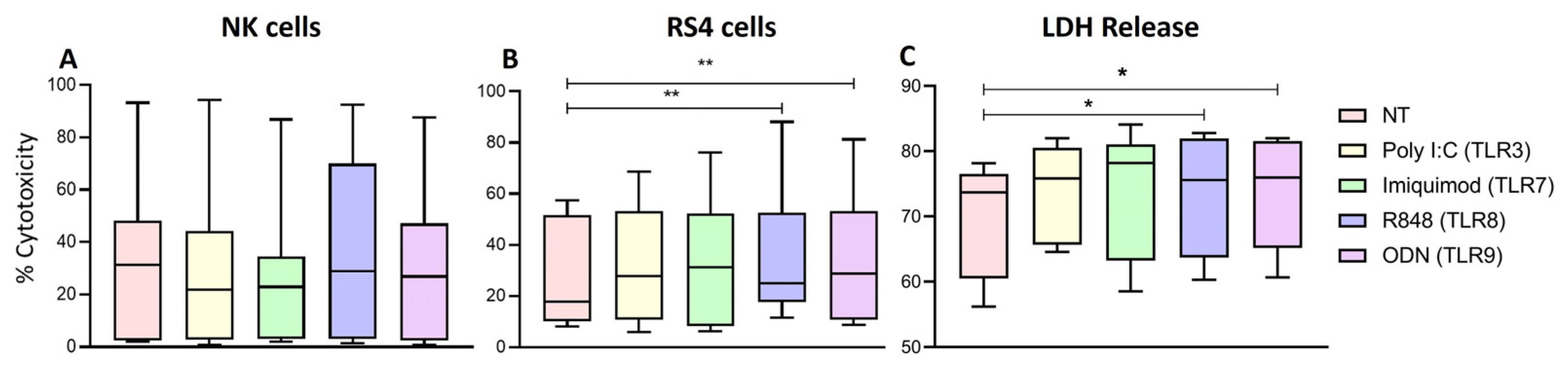

Table 1.
Demographic characteristic of the study group.
| Patients with acute lymphoblastic leukemia | |
|---|---|
| Characteristics | Peripheral blood |
| N° of cases | 24 |
| Sex (M:F) | 14:10 |
| Age media (range) | 7 (1-17) years |
| Immunophenotype | |
| Pre-B | 22 |
| not defined | 2 |
| % NK Cells (range) | 1.87 (0.4-5.97) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



