
Submitted:
23 April 2024
Posted:
24 April 2024
You are already at the latest version
Abstract
Recent studies, typically using patient cerebrospinal fluid (CSF), have suggested that different autoantibodies (Aabs) acting on their respective receptors, may underlie neuropsychiatric disorders. The NR1 subunit of the N-methyl-D-aspartate receptor (NMDAR) has been identified as a target of anti-NMDAR Aabs in a number of central nervous system (CNS) diseases including encephalitis and autoimmune epilepsy. However, the role or the nature of Aabs responsible for effects on neuronal excitability and synaptic plasticity is yet to be established fully. Patient-derived anti-NMDAR Aabs have been shown to bind to specific regions within the NR1 subunit; here, peptide immunisation was used to generate Aabs against selected specific NR1 extracellular sequences. ‘Protein A’ purification was used to obtain total IgG and further peptide purification used to obtain a greater percentage of NMDAR-target specific IgG Aabs. Binding and specificity of these anti-NMDAR Aabs was determined using a range of methodologies including enzyme-linked immunosorbent assay, immunocytochemistry and immunoblotting. Functional effects were determined using different in vitro electrophysiology techniques: two-electrode voltage-clamp in Xenopus oocytes and measures of long-term potentiation (LTP) in ex vivo hippocampal brain slices using multi-electrode arrays. We show that anti-NMDAR Aabs generated from peptide immunisation had specificity for NR1 immunisation peptides as well as target-specific binding to the native protein. Anti-NMDAR Aabs had no clear effect on isolated NMDARs in an oocyte expression system. However, we show that peptide-purified anti-NMDAR Aabs prevented the induction of LTP at Schaffer collateral-CA1 synapses in ex vivo brain slices. Our data demonstrate that anti-NMDAR Aabs cause synaptic NMDAR hypofunction at a network level. This work provides a solid basis to address outstanding questions regarding anti-NMDAR Aabs mechanisms of action and, potentially, development of therapies against CNS diseases.
Keywords:
NMDA receptor autoantibodies
; hippocampal neuron
; long-term potentiation
; multi-electrode arrays
1. Introduction
In the CNS, NMDARs are one of the main ionotropic glutamate receptors, alongside α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and kainate receptors, and have critical functions in synaptic plasticity and learning and memory [1]. NMDARs are typically heterotetramers comprised of 2 GluN1 subunits present in all NMDARs (encoded by GRIN1, although with a number of splice variants) and 2 GluN2 subunits, with 4 different sub-type (GluN2A-D, encoded by GRIN2A-D respectively) that exhibit different spatio-temporal expression and functional properties. A third subunit type, GluN3, has 2 subtypes (GluN3A and GluN3B, encoded by GRIN3A and GRIN3B respectively), which also exhibit different spatiotemporal expression and functional properties (reviewed in [2]). Autoantibodies (Aabs) produced by the body against self-neurotransmitter receptors including NMDARs have been implicated in CNS diseases. Anti-NMDAR Aabs of the IgG class directed against the NR1 subunit have been found in in CSF of patients with anti-NMDAR encephalitis (ANRE), a condition characterised by seizures, psychosis and cognitive deficits [3], and in patients with forms of autoimmune epilepsy [4,5]. Such evidence sparked interest in investigating the functionality of anti-NMDAR Aabs [6]. For example, an early study showed that post-mortem hippocampus from ANRE patients expressed significantly less NMDARs than age-matched controls [7]. The consensus view is that anti-NMDAR Aabs in patient brains can induce pathological changes leading to symptoms; it has been shown that in vivo infusion of ANRE patient CSF, containing anti-NMDAR Aabs, resulted in increased chemical-induced epileptic seizures, behavioural changes, and memory impairment in rodents [8,9,10]. Electroencephalogram recordings of mice infused intraventricularly with ANRE patient CSF showed a higher frequency of seizures [11]. At a cellular level, exposure of ANRE patient CSF to primary hippocampal neurons in vitro resulted in a reduction in NMDAR currents [7,12]; monoclonal antibodies cloned from an ANRE patient caused similar effects [13,14]. It was also reported that Aabs have the ability to activate the complement ‘membrane attack complex’ in NMDAR-expressing cells, proposed to be responsible for some symptoms observed in ANRE patients[15]. In order to investigate the mechanisms underlying potential pathophysiology, purified anti-NMDAR Aabs were produced. Peptide-purified anti-NMDAR Aabs had no clear effects on NMDAR gating in an in vitro oocyte expression system, but showed an inhibitory effect on long-term synaptic plasticity in ex vivo hippocampal brain slices. Increasing our mechanistic knowledge of how NMDAR Aabs cause effects in CNS may lead to better understanding/treatment of diseases such as ANRE and autoimmune epilepsies.
2. Results
2.1. Generation and Characterisation of NMDAR Aabs
Five immunisation peptides were generated based on NMDAR NR1 subunit sequences targeted by Aabs identified from patient CSF or sera (Figure 1A). Following immunisation with all five peptides, resulting sera were Protein A-, and then peptide-, purified (see Methods). Terminal serum titres were measured by ELISA following Protein A and peptide purification (Figure 1B). A comparative ELISA demonstrated increased anti-NMDAR Aab binding to the amino terminal domain (ATD) protein following peptide purification (Figure 1C). Thus, peptide-purified material required 0.51 µg/ml to elicit a 50% maximal concentration response, whereas the Protein A-purified material required 2.36 µg/ml to elicit a 50% maximal response, indicating an increase in binding to ATD protein due to a greater percentage of NMDAR-target specific IgG Aab. The specificity of peptide-purified anti-NMDAR Aabs for the ATD of the NR1 subunit was determined by western blot, with a strong band at the predicted ATD size of 60 kDa pre- and post-peptide purification, similar to a commercial anti-NR1 antibody that recognises the ATD (rNMDAR, Synaptic Systems, Cat no. 114 103, Figure 1D). Conversely, no band was observed with a commercial anti-NR1 antibody that does not target the ATD (mNMDAR, Synaptic Systems, Cat no. 114011) or negative control antibodies (rIgG, mIgG2b, secondary only or an unrelated primary, ENT1, Figure 1D).
HEK cells were transfected with a vector encoding the NR1 subunit. Immunocytochemistry performed using peptide-purified anti-NMDAR Aabs and a commercial mouse monoclonal anti-NR1 antibody (mNMDAR) showed positively co-labelled NR1-transfected HEK cells (Figure 2A). A commercial polyclonal anti-NR1 antibody (rNMDAR) showed similar co-labelling (Figure 2B). No positively-labelled cells were observed with class-specific negative control rIgG and secondary-only controls in NR1-transfected HEK cells or with NR1 Aabs or commercial anti-NR1 antibodies in empty vector-transfected HEK cells (Supplemental Figure S1). To confirm the presence of anti-NMDAR Aabs in neurons, immunocytochemistry was also performed in primary cortical neurons, where anti-NMDAR Aabs labelling colocalized with the neuronal marker βIII tubulin but not with the astrocyte marker GFAP (Figure 2C). Overall, further peptide purification resulted in more concentrated NMDAR NR1 Aabs, which showed specific labelling in both NR1-transfected HEK cells and in primary neurons.
2.2. Lack of Functional Effects of NMDAR Aabs in NR1/NR2A Expressing Xenopus Oocytes
An NMDAR expressing NR1/NR2A subunits Xenopus oocyte model was used to investigate effects of anti-NMADR Aabs on NMDAR currents [16,17]. NMDAR negative allosteric modulator (TCN-201) was used as a positive control to show inhibition of the currents (Figure 3). TCN-201 (0.01 – 3.0 µM) reduced NMDAR currents in oocytes (n = 6-7) in a concentration-dependent manner (Figure 3B,C) with a maximal inhibition of >90% at 3 µM. The half-maximal inhibitory concentration IC50 value for TCN-201 was 0.35 µM. In the vehicle-incubated oocytes, NMDA currents did not decrease with time, and even slightly increased AUC with time (Figure 3A,C). Peptide-purified anti-NMDAR Aabs or rIgG controls were then assessed in oocytes at 1:300 dilution (4 µg/ml, determined by ELISA, see Figure 1). Figure 4 shows results for peptide-purified anti-NMDAR Aabs (Figure 4B) and control peptide-purified IgG (Figure 4A). There was no significant difference between NMDA currents induced by glutamate application in oocytes incubated with IgG or anti-NMDAR Aabs (Figure 4C). Of further note, Protein A-purified anti-NMDAR were similarly without effects vs control Protein A-purified IgG on NMDA currents (Supplemental Figure S2). Therefore, anti-NMDAR Aabs had no effect on NMDAR currents in oocyte experiments.
2.3. Schaffer Collateral-CA1 LTP Is Inhibited by Anti-NMDAR Aabs
We next investigated the effects of anti-NMDAR Aabs on NMDAR-dependent synaptic plasticity using MEA electrophysiology on ex vivo mouse hippocampal brain slices to record Schaffer collateral-CA1 LTP. A high-frequency stimulation (HFS) protocol was used to induce LTP, identified by a potentiation in evoked field excitatory postsynaptic potential (fEPSP) slope for at least 1 h post-HFS in vehicle condition with data normalised to pre-LTP baselines (Figure 5A). Vehicle-treated slices generated a consistent HFS-induced potentiation at 1 h: 153.1 ± 31.6% (n=8), which was significantly inhibited by the non-competitive NMDAR antagonist, APV (50 µM, 109.2 ± 22.7% (n=6), p < 0.01 compared to vehicle) (Figure 5A,B). HFS-induced potentiation was significantly reduced in slices pre-incubated for 1 h with anti-NMDAR Aabs ((1:1000) 119.5 ± 13.8% (n=7), p < 0.05) compared to vehicle (Figure 5A,B). This level of inhibition was similar to that seen with APV. These data demonstrate a significant effect of the peptide-purified anti-NMDAR Aabs on NMDAR function in an ex vivo brain slice model. Of further note, Protein A-purified anti-NMDAR Aabs had no effect on HFS-induced potentiation compared to vehicle controls (Supplemental Figure S3A,B). Using the same protocol, we also tested two commercial anti-NR1 antibodies (rNMDAR and mNMDAR) and their class-specific negative controls (rIgG and mIgG2b, respectively), all at 1:1000 dilution. In all cases, no significant difference was observed in HFS-induced potentiation compared to vehicle controls (Supplemental Figure S3C,D). In summary, peptide-purified anti-NMDAR Aabs were able to functionally inhibit LTP to a similar extent to APV, but equivalent effects were not observed with the two commercial anti-NR1 antibodies tested.
3. Discussion
Here, we show that anti-NMDAR Aabs generated through peptide immunisation bind to native NMDARs and inhibit LTP in mouse hippocampal brain slices, similar to those effects reported in studies using CSF or, in some studies, using monoclonal antibodies from ANRE patients. We were unable to demonstrate clear effects of our anti-NMDAR Aabs on NMDAR in a validated oocyte expression system.
3.1. Generation of Anti-NMDA Aabs as an Experimental Tool
The NMDAR NR1 subunit is obligatory for a fully functioning receptor [18]. Our strategy was to use immunizing peptides based on five different human NR1 subunit extracellular loop regions predicted to be of high immunogenicity to generate anti-NMDAR Aabs able to bind to the NR1 subunit in its natural conformation. One of our peptides (cyclo-GIYNGTHVIPN) contained amino acids N368/G369; these residues, near the hinge region within the ATD, were shown to be crucial for human Aab recognition and binding to NMDARs from ANRE patients CSF [19] and to NR1 monoclonal antibodies [13].
The majority of studies investigating effects of anti-NMDAR Aabs use material derived from patient CSF. Whilst these studies are of clear value, Aabs present in these mixed component preparations represent heterogenous families, targeted against different epitopes, including to alternative receptor subtypes that may not contribute to functional effects of interest; overall, this makes the definitive assignment of function uncertain. In the present study, we use a strategy that involves a peptide purification step additional to a Protein A purification methodology. We demonstrate that additional purification produces anti-NMDAR Aab with improved target binding compared to Protein A purification, indicating a greater percentage of NMDAR-target specific IgG Aab. Of note here, is that Protein A-purified only anti-NMDAR Aabs had a statistically insignificantly effect on HFS-induced potentiation, whilst additional peptide purification produced anti-NMDAR Aabs that functionally inhibited LTP. Together, these data argue that the purification state of Aabs investigated here is of fundamental importance and that the peptide-purified anti-NMDAR Aabs represent useful experimental tools.
3.2. Functional Effects in Oocytes
Overall, our results indicate that anti-NMDAR Aabs had no significant effect on NMDAR current in NR1/NR2A-expressing oocytes following 60 min incubation. Although there was some variability during the incubation, the same variability was seen in rIgG control and in anti-NMDA Aabs treated oocytes. Of further interest was that the commercial NMDAR anti-NR1 antibody, mNMDAR (which successfully labelled NR1-transfected HEK cells, see Figure 1), was also without effect in the oocyte system (Brice Mullier, data not shown). These data confirm the lack of effect of anti-NMDAR antibodies on NMDA receptor function in oocytes. A previous study in oocytes has reported that in the presence of sera from both ill and healthy patients, or a commercial anti-NR1 antibody (mNMDAR, as used here), NMDA currents remained stable; whilst in the presence of sera from seronegative patients, NMDA current amplitude increased, leading authors to conclude that seropositive sera lowered NMDAR currents compared to seronegative controls [16]. A recent study generated expression-purified IgGs from a range of monoclonal antibodies produced in mice immunized with intact NMDAR NR1/NR2 protein [20]. One such species (termed IgG2) produced a rapid block of NMDAR currents, both in oocytes and in a mammalian HEK cell expression system. These anti-NMDAR Aab species were proposed to function via allosteric downregulation of NMDAR function due to stabilization of an inactive NMDAR confirmation, as supported by X-ray crystallography and cryo-EM structural evidence [20]. Of further interest was that another species (termed IgG5) had a stimulatory effect on NMDAR current, whilst other IgGs apparently had no effect. These data point to differential functional effects between heterogenous Aab components.
The lack of functional effect by anti-NMDAR Aabs here could be explained by a number of alternative factors. Anti-NMDAR Aabs may interact with signalling or scaffolding proteins, which are absent in the current oocyte system, but present in native neurons. A candidate protein here is the ephrin-B2 receptor, which stabilises and clusters NMDAR at synaptic surfaces [21]. In cultured neurons, anti-NMDAR Aabs were shown to disrupt NR1 subunit interaction with ephrin-B2 receptors and prevented receptor internalization [22]. Moreover, co-administration of ephrin B2 ligand with CSF maintained NMDAR cell-surface levels and prevented pathogenic behavioural effects [23]. Therefore, oocytes may not have the mechanistic capabilities to reproduce the protein complexes required for such modulatory process [24].
Our data in oocytes show that anti-NMDAR Aab lack any clear ionotropic NMDAR effects, such as channel block or negative allosteric modulation even after 60 min incubation. Longer term incubations (>24 hours) typically suppress excitatory postsynaptic currents (EPSCs) [7,25,26] and whole-cell NMDAR current [27]. However, acute application of different IgG species raised against NMDARs leads to differential effects, ranging from no effect, blockade, or potentiation of NMDAR current. Acute application of patient CSF combined with an NMDAR agonist increased mean open time of recombinant NMDARs in single channel recordings [19]. It was also reported that 30 min application of patient CSF had no effect on NMDAR-mediated mEPSCs in hippocampal neurons; by contrast, receptor internalization was seen after 12 h [12]. This study concluded that patient CSF effects occurred independently of NMDAR antagonism and are due to longer term receptor internalization. Related to this, it was reported that monoclonal antibodies from ANRE patients lacked effects on miniature spontaneous calcium transients in hippocampal neurons in response to 10 min exposure [28]; by contrast, similar monoclonal antibodies could bind NMDAR in hippocampal neurons and be internalized over a 45 min timeframe [14]. Thus, a hypothesis arises whereby heterogenous anti-NMDAR Aabs may engage different modes of actions; namely, an ionotropic effect on NMDAR channels and/or receptor internalization; moreover, such effects may be time-dependent for selected species.
3.3. Anti-NMDAR Aabs Suppress LTP
Anti-NMDA Aabs produced by peptide purification had a significant inhibitory effect on HFS-induced potentiation in MEA-LTP experiments. Effects of anti-NMDAR Aabs were of similar magnitude to the NMDAR antagonist APV. Our data in oocytes, which used a similar 60 min exposure, indicate that effects of anti-NMDAR Aabs do not mirror those of receptor antagonists (see also [12]). Our data are in general agreement with previous studies whereby exposure to patient CSF-containing anti-NMDAR Aabs for 1-8 days prior to testing caused a consistent, significant inhibition of LTP across several different pathways within the hippocampus [25,29,30]. Blome et al. (2018) further reported substantial variation among CSF samples [29]; these observations support the presence of different epitopes among patient-derived antibodies. Together, these studies are proposed to reflect a reduction in synaptic density of NMDAR due to receptor internalization. Such a mechanism was originally proposed by Hughes et al. (2010), whereby treatment with patients Aabs bound to and cross-linked NMDARs, leading to a selective decrease in synaptic NMDAR currents [7]. Studies using advanced single particle tracking to investigate receptor internalization demonstrated differential effects of patient CSF, whereby synaptic GluN2A (NR1) was removed from the surface, but extrasynaptic GluN2B (NR2) was mainly cross-linked by Aabs [22]. By contrast to the majority of studies above, an ‘acute’ 5 min pre-incubation was reported to cause a significant inhibition of LTP, suggesting direct receptor block rather than an internalization process [31]. Mikasova et al. (2012) investigated a time course of action, reporting that patient CSF caused a significant reduction in NMDAR cell surface localization after 2 h, which became greater after 20 h incubation [22]. As discussed above, such studies are consistent with some species of anti-NMDAR Aabs being able to mediate an acute and/or longer-term action on NMDARs.
Neither of the two commercial antibodies or class specific negative controls had effects on LTP here, the latter arguing against any non-NMDAR-specific IgG effects. Würdemann et al. (2016) reported that patient CSF-induced reductions in LTP were similarly shown by a commercial antibody raised against cytoplasmic C terminal amino acid (aa) 909-938 [25], also used by Hughes et al. (2010) [7]; however, it is unclear how this internal region can be accessed in these experiments. Würdemann et al. (2016) further reported that commercial NR1 or NR2 antibodies reduced LTP [25]); however, these were directed against different sequences to the ones targeted by the commercial antibodies used here. Overall, it was of interest that, despite immunocytochemical labelling of NMDARs, neither of the commercial antibodies had functional effects in ex vivo slices, nor were any functional effects seen for mNMDAR in oocyte experiments. These commercial antibodies are directed against single target epitope sequences within the ATD of the NR1 subunit (mNMDAR: aa660-811; rNMDAR: aa35-53); these sequences differ to those on which we based our five peptides used to generate anti-NMDAR Aabs. These data further suggest that, in mechanistic studies, it is important to target residues of functional importance. The data may also reflect that a strategy involving use of multiple immunizing peptides and/or cyclo-peptides to generate Aabs is advantageous to detect specific functional effects.
3.4. Clinical Relevance
It can be hypothesised that our anti-NMDAR Aabs caused NMDAR hypofunction, potentially via a mechanism involving receptor internalization, rather than a direct block of NMDAR channel gating. The consensus view is that anti-NMDAR Aabs present in patient CSF is deleterious and pathogenic in different diseases including, but not limited to, ANRE and autoimmune epilepsies [32,33,34]. It is also broadly recognised that NMDAR hypofunction causes the seizures associated with such diseases, due to disruption of neuronal circuitry and changes in excitatory /inhibitory balance [26,35]. The mainstay of treatment options for disorders such as ANRE are plasma exchange, intravenous immunoglobulins and steroids, with second-line agents including immunosuppressants such as rituximab [36,37]. There is also good pharmacological evidence that positive allosteric modulators may be useful to improve the receptor hypofunction caused by anti-NMDAR Aabs [27,38,39]. Recent advances in engineering immunological elements may also bear fruit in new therapies. Thus, anti-NMDAR Aab-mediated NMDAR internalization can be prevented by a fusion construct comprising the IgG Fc region and NR1 and NR2 subunit ATDs [40]. It has also been reported that the neonatal Fc receptor (FcRn) can prevent patient sera-induced inhibition of LTP [41]. As discussed above, patient CSF and/or sera may contain many different component Aabs to different receptors and NMDAR Aabs may exhibit heterogenous effects, some of which are paradoxical to those of NMDAR antibodies designed as therapeutic agents [42]. Therefore, it is important to develop appropriate tools, such as the peptide-purified anti-NMDAR Aabs investigated here, with which to investigate NMDAR physiology and potential pathophysiology, in order to best develop new therapies strategies to combat devastating NMDAR-associated diseases.
4. Materials and Methods
4.1. Antibody Design, Production and Purification
Five peptides were designed by targeting amino acid (aa) sequences in the extracellular loops of the human GluN1 subunit (GenBank accession Q05586): (1) aa125-135, MSIYSDKSIHL; (2) aa249-266, cyclo-VGEREISGNALRYAPDGI; (3) aa272-281, INGKNESAHI; (4) aa353-363, cyclo-IMNLQNRKLVQ; and (5) aa365-375, cyclo-GIYNGTHVIPN (see Figure 1A). Design and generation of peptides was performed by UCB and Peptide Synthetics, UK. All peptides were generated and modified with N-terminal acetylation and C-terminal amidation to help prevent degradation by exopeptidases. Peptides 2, 4 and 5 were cyclised via a thioester to aid in mimicking the natural 3D structure of their respective epitopes [43], therefore increasing the chances of generating Aabs that bind to the GluN1 subunit in its natural conformation.
Antibodies were raised using the 5 peptides, as described previously [44]. Briefly, peptides were conjugated to either keyhole limpet hemocyanin emulsified in an equal volume of Complete Freund’s Adjuvant, bovine serum albumin or ovalbumin (Peptide Synthetics, UK). A female New Zealand White rabbit (> 2 kg) was immunized sub-cutaneously with the 5 peptides (500 µg of peptide per immunization) with booster injections at 14-day intervals. The rabbit was sacrificed 14 days after the final immunisation by Schedule 1 methods in accordance with the UK Animals (Scientific Procedures) Act 1986 and the terminal serum collected.
Protein A-purified Aabs were prepared as described previously [44]. and further purified through high-capacity streptavidin agarose resin (Pierce; ThermoFisher Scientific, UK). Biotin-bound peptides (peptides 1-5) used for immunisation (133 μM each) were first mixed with 5 ml streptavidin agarose resin. This was then combined with Protein A-purified Aabs (overnight at 4°C), applied to columns, washed with PBS, and peptide-specific antibody eluted with 0.1 M sodium citrate (pH 3.2). Pooled fractions were pH neutralised with Tris-HCl (pH 8.5). Fractions were concentrated through buffer exchange with PBS using 10 kDa MWCO. Total peptide-specific IgG concentration was determined using absorbance at 280 nm, SDS-PAGE and ELISA, as described previously [44].
4.2. Expression of NMDARs in HEK Cells
Human embryonic kidney 293 (HEK) cells were grown in DMEM + 10% FBS media. Cells were transfected with plasmids encoding the GluN1 (NR1) subunit (pcDNA3.1(+) NR1-4a_HS) and GluN2b (NR2B, pcDNA3.1(-) NR2B_HS) respectively; control cells were transfected with an empty plasmid vector; pcDNA3.1(+). Transfections used a 2:1 ratio polyethylenimine (PEI):DNA inOptiMem (Gibco; ThermoFisher Scientific, UK) for 6 h before sub-culture onto poly-D-lysine-coated coverslips (PDL; 20 μg/ml; Sigma Aldrich) at 2.5x105 cells/well for 24 h. Cells were then fixed with 4% PFA for 10 min (Sigma Aldrich) before processing for immunocytochemistry.
4.3. Animals
The housing and use of animals in all experiments were carried out in accordance with UK Home Office regulations under the Animals (Scientific Procedures) Act, 1986. Mice were housed at 21°C in a 12-h light/dark cycle with food and water available ad libitum according to ARRIVE guidelines [45].
Preparation of acute mouse hippocampal slices
400 μm transverse hippocampal slices were prepared from male 4-6 week-old C57BL6/J mice. Mice were terminally anaesthetised with 4% isoflurane and immediately underwent cervical dislocation and decapitation. Slices were produced using high-sucrose artificial cerebrospinal fluid (aCSF) cutting solution, comprising (in mM): sucrose (75), NaCl (87), NaHCO3 (25), KCl (2.5), NaH2PO4 (1.25), CaCl2 (0.5), MgCl2 (7), glucose (25), pH 7.4, all Fisher Scientific) on a VT1200S vibrotome (Leica) and transferred to carboxygenated aCSF, comprising (in mM): NaCl (126), glucose (10), MgCl2 (2), KCl (2.5), NaH2PO4 (1.25), NaHCO3 (26), CaCl2 (0.5), pH 7.4 at 37°C for 30 min. Slices were equilibrated at room temperature for at least 1 h before use.
Embryonic day18 (E18) mouse primary neuronal cell culture
Cortical cultures were prepared as described previously [44]. Briefly, primary neuronal cultures were prepared from male and female E18 C57BL/6 mice; after removal of meninges, dissected cortices were chemically dissociated using papain and DNase (Sigma Aldrich, UK) and diluted in culture medium (Neurobasal medium with 1% B27, 2 mM GlutaMax, 2.5% FBS and 100 U/mL/100 µg/mL penicillin/streptomycin; all Gibco/Life Technologies). Cells were pelleted and resuspended in culture medium before plating at 2x105 cells/well on laminin-coated coverslips (Sigma) in 24-well plates. 50 % culture medium change was done every 2-3 days. Cells were washed with PBS and fixed with 4% PFA for 10 min (Sigma Aldrich) before processing for immunocytochemistry.
4.4. Immunocytochemistry and Immunoblotting
Primary and secondary antibodies used were as follows: rabbit anti-NMDAR (rNMDA: 1:100, polyclonal raised against residues 35–53 of human GluN1, Synaptic Systems, Cat No. 114 103,); mouse anti-NMDAR (mNMDA: 1:100, monoclonal raised against residues 660-811 of human GluN1, Synaptic Systems, Cat No. 114 011); rabbit anti-IgG (rIgG, 1:100, 011-000-003, Jackson ImmunoResearch); mouse anti-IgG2 (mIgG2b, 1:100, 70-4732, BioLegend); mouse anti-βIII-tubulin (1:500, 801201, BioLegend); mouse anti-glial fibrillary acidic protein (GFAP, 1:400, MAB3402, Millipore); goat anti-rabbit or anti-mouse Alexa Fluor 488/594/647 (all at 1:1000, Life Technologies). For transfected HEK cells, cells were washed with blocking buffer (1x PBS, 0.1% Triton X-100, 1% normal goat serum) on a shaker, transferred to a humidified chamber and incubated with primary antibodies in blocking buffer) overnight at 4°C. Coverslips were then washed with blocking buffer incubated with Alexa Fluor-coupled secondary antibodies in blocking buffer for 2 h at room temperature. Following incubation, cells were washed 3x with blocking buffer and subsequently 3x PBS (all 5 min each). Nuclei were counterstained using 4’,6-diamidino-2-phenylindole (DAPI; 1:10,000, Thermo Fisher Scientific, UK) and coverslips were mounted in ProLong Gold anti-fade mounting medium (Thermo Fisher Scientific, UK). Cells were visualised with an AxioImager A1 microscope (Zeiss) and images acquired using Axiovision 4.6.3 imaging software (Zeiss).
Immunocytochemistry on primary neurons
Immunocytochemistry was performed as described in [44]. Briefly, cortical neurons were incubated for 2 h at room temperature with primary antibodies (anti-NMDAR Aabs, rIgG, mIgG2b, anti-βIII tubulin or anti-GFAP) in blocking buffer (PBS + 10% normal goat serum). Cells were permeabilied using 0.1% Triton X/PBS. Specific Alexa Fluor-coupled secondary antibodies were added (in blocking buffer) and incubated for 30 min at room temperature. Cells were visualised with an AxioImager A1 microscope and Axiovision 4.6.3 imaging software (Zeiss).
Immunoblot of brain lysates
SDS-PAGE and Western blotting was performed as described previously [44]. Briefly, protein lysates were prepared from mouse whole brain tissue (C57BL6/J, male 4-6 weeks) using lysis buffer of NaCl (150 mM), Triton-X-100 1% (v/v) glycerol 10% (v/v), HEPES 30 mM, and SigmaFAST protease inhibitors (1 tablet/50 mL). Protein concentration was determined using a bicinchoninic acid protein assay kit (ThermoFisher Scientific, UK). SDS-PAGE gels were prepared with 10% separating gel and 3% stacking gels. Western blotting was used to assess the specificity of NMDAR Aabs against protein lysate.
4.5. Xenopus Oocyte Expression System
GRIN1/GRIN2A constructs encoding GluN1 and GluN2A were generated and linearized by Genscript (Piscataway, NJ) using restriction sites BamHI and PmeI, respectively. Plasmids were transcribed using mScript (Cellscript, Madison, WI). Transcripts were capped, polyA tailed and purified, and the size was confirmed by gel electrophoresis. mRNAs for oocyte injections were prepared by RD-Biotech (Besançon, France). GRIN1/GRIN2A human mRNAs (0.15 ng/oocyte, 3.75 ng/µl solution, total volume 40 nl ratio 1:5) were injected into Xenopus oocytes (stage V-VI) previously dissected and de-folliculated (provided by EcoCyte Bioscience, Castrop-Rauxel, Germany) via an automated micro-injector (Robocyte, MCS, Reutlingen, Germany) using a glass micropipette (5.5 μm diameter, MCS) in RNAse free water (Ambion, Thermo Fisher Scientific, Waltham, MA). Oocytes were left to express receptors for 72 h at 17°C in a Barth’s solution, comprising (in mM): NaCl (88), KCl (1), NaHCO3 (2.4), Ca(NO3)2 (0.33), CaCl2 (0.41), MgSO4 (0.82), Tris-HCl (5), supplemented with penicillin/streptomycin (100 IU/mL, pH 7.4), with solution changed daily.
Two Electrode voltage clamp (TEVC) electrophysiology in Xenopus oocytes
TEVC recordings were performed using an automated platform (HiClamp®, MCS). Electrodes (0.1-1 MΩ resistance) were filled with potassium chloride (KCl, 1.5 M) and potassium acetate (Kac, 1.5 M). Oocytes were impaled and voltage-clamped at a holding potential of -80 mV. Oocytes were then rinsed and recordings for glutamate response were performed in modified frog buffer (MFB), comprising (in mM): NaCl (88), KCl (2.5), CaCl2 (1.8), HEPES (5); pH 7.85 at 19 °C as described by [16] and by [17]. Oocytes were pre-incubated for 3 min in control buffer (MFB) and currents were induced by 10 s applications of 1 or 10 μM glutamate/10 μM glycine (glu/gly) every 3 min for 12 min to generate stable NMDAR-mediated currents, then incubated in test substance and challenged by 10 s applications of glu/gly every 15 min (total exposure to antibodies = 60 min). In different oocytes, the NMDAR negative allosteric modulator TCN-201 (Sigma-Aldrich, 0.01-3 µM) was used as a positive control to show inhibition of currents. Current responses were measured by the area under the curve (AUC) and normalized to AUC from control responses (Steps 3 and 4).
Data analysis oocytes
Currents were analysed off-line using the HiClamp Software (DataMining and DataMerger). Glu/gly current inhibition by the drug or the Aab was calculated based on the ratio AUC(glu+drug)/AUC(glu) or AUC(glu+Aab)/AUC(glu). To control for potential sources of variation, current AUC in the presence of the compound were normalised to the mean response of glu/gly current AUC values without the compound. Dose response curves were obtained by plotting average relative current AUC (AUC/AUC0) as function of compound concentration. Concentrations were log transformed before analysis to generate a data set amenable to non-linear regression and to parametric statistical analysis. Dose response curves were analyzed by GraphPad software version 9 for Windows (San Diego CA) and pIC50 values were calculated by non-linear regression analysis using a sigmoidal dose response equation equivalent to log(dose) vs. response (variable slope) Sigmoidal, 4PL, [Y=Bottom+(Top-Bottom)/(1+10^((LogEC50-X)*Hillslope)], X=log of dose; Y=response. No weighting of the points was applied.
4.6. Multi-Electrode Array (MEA) Recordings
Evoked electrical activity across hippocampal slices was monitored and recorded using titanium nitrate MEAs (MCS). Slices were positioned on MEAs in aCSF using a Leitz Diavert microscope using a slice harp (Harvard Apparatus), such that the Schaffer collateral and CA1 areas were covering the electrodes. Slices were continually perfused with carboxygenated aCSF (~3 mL/min) and maintained at 32°C. Recordings were made via head stage (MEA1060-Inv-BC, MCS, Germany), whereby, bi-phasic voltage pulses were applied to one electrode (STG2008 stimulator, MCS; 100μs biphasic pulses, ± 0.5-2.0 V, every 30 s) to evoke field excitatory postsynaptic potentials (fEPSPs) [46]. Signals were amplified by a 60-channel head-stage amplifier (MEA60 System, MCS), and simultaneously sampled at 10 kHz per channel and amplified at 1200x gain. Data acquisition to a computer was carried out using the software MC_Rack which monitored and recorded data for later offline analysis.
Schaffer collateral-CA1 LTP was induced using high frequency stimulation (HFS; 100Hz). Paired pulse fEPSPs were evoked every 30 s both pre- and post-LTP induction, being recorded for 30 min pre-LTP induction to establish a steady baseline and for 60 min post-LTP induction. HFS potentiation was measured as changes to fEPSP slope as a measure of excitatory drive. The fEPSPs could be isolated into separate glutamatergic components: AMPAR/kainate receptors or NMDARs were blocked by addition of 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX 5 μM; Abcam) or DL-2-amino-5- phosphonopentanoic acid (APV 50 μM; Abcam), respectively, to the perfusing aCSF. Slices were pre-incubated for 1 h in aCSF containing anti-NMDAR Aabs or IgG controls (both 1:1000 dilution) with continuous carboxygenation. Slices were also incubated with either mNMDAR or rNMDAR or mIgG2b/rIgG antibodies as positive and negative controls, respectively, (1:1000 dilution). One slice per animal was used per condition.
Statistical Analysis
All data are presented as mean ± standard deviation (SD) with number of independent experiments (n) detailed in text and analysed using GraphPad Prism 7.00 (GraphPad Software, Inc.). All data sets were tested for normality using D’Agostino Pearson tests; on this basis, parametric one- or two-way ANOVAs were performed. Throughout, data were considered significant at p < 0.05.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, J.-P.S., R.M., B.M., V.M.A., C.W., A.B., G.J.S.; methodology, J.-P.S., R.M., T.S.B., B.M., V.M.A., C.D., A.B., G.J.S.; formal analysis, C.D., B.M., V.M.A., A.B., G.J.S.; investigation, B.M., V.M.A., C.D., J.-P.S., R.M.; writing-original draft preparation, C.D., A.B., G.J.S.; writing-review and editing, C.D., J.-P.S., R.M., B.M., V.M.A., C.W., A.B., G.J.S.; supervision, J.-P.S., B.M., V.M.A., C.W., A.B., G.J.S.; project administration, C.W., A.B., G.J.S.; funding acquisition, C.W., A.B., G.J.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by a University of Reading Strategic Studentship award and a UCB Pharma Case award to A.B. and G.J.S. G.J.S. is a Section Editor for Pharmaceuticals and has APC waiver.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within the article and raw data presented in this study are available upon request.
Acknowledgments
We thank University of Reading and UCB Pharma for funding to CD. We acknowledge the support of Daniel Lightwood, Head of Antibody Discovery, UCB.
Conflicts of Interest
B.M., V.M.A., C.W., J.-P.S., R.M. are employees of UCB and may have access to shares/stock options.
References
- Dupuis, J.P.; Nicole, O.; Groc, L. NMDA receptor functions in health and disease: Old actor, new dimensions. Neuron 2023, 111, 2312–2328. [Google Scholar] [CrossRef] [PubMed]
- Paoletti, P.; Bellone, C.; Zhou, Q. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci 2013, 14, 383–400. [Google Scholar] [CrossRef] [PubMed]
- Dalmau, J.; Tuzun, E.; Wu, H.Y.; Masjuan, J.; Rossi, J.E.; Voloschin, A.; Baehring, J.M.; Shimazaki, H.; Koide, R.; King, D.; et al. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol 2007, 61, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Bien, C.G.; Holtkamp, M. "Autoimmune Epilepsy": Encephalitis With autoantibodies for epileptologists. Epilepsy Curr 2017, 17, 134–141. [Google Scholar] [CrossRef]
- Geis, C.; Planaguma, J.; Carreno, M.; Graus, F.; Dalmau, J. Autoimmune seizures and epilepsy. J Clin Invest 2019, 129, 926–940. [Google Scholar] [CrossRef]
- Bauer, J.; Becker, A.J.; Elyaman, W.; Peltola, J.; Ruegg, S.; Titulaer, M.J.; Varley, J.A.; Beghi, E. Innate and adaptive immunity in human epilepsies. Epilepsia 2017, 58 Suppl 3, 57–68. [Google Scholar] [CrossRef]
- Hughes, E.G.; Peng, X.; Gleichman, A.J.; Lai, M.; Zhou, L.; Tsou, R.; Parsons, T.D.; Lynch, D.R.; Dalmau, J.; Balice-Gordon, R.J. Cellular and synaptic mechanisms of anti-NMDA receptor encephalitis. J. Neurosci. 2010, 30, 5866–5875. [Google Scholar] [CrossRef]
- Manto, M.; Dalmau, J.; Didelot, A.; Rogemond, V.; Honnorat, J. In vivo effects of antibodies from patients with anti-NMDA receptor encephalitis: further evidence of synaptic glutamatergic dysfunction. Orphanet J. Rare Dis. 2010, 5, 31. [Google Scholar] [CrossRef]
- Planaguma, J.; Leypoldt, F.; Mannara, F.; Gutierrez-cuesta, J.; Martin-Garcia, E.; Aguilar, E.; Titulaer, M.J.; Petit-Pedrol, M.; Jain, A.; Balice-Gordon, R.; et al. Human N-methyl D-aspartate receptor antibodies alter memory and behaviour in mice. Brain 2015, 138, 94–109. [Google Scholar] [CrossRef]
- Wright, S.; Hashemi, K.; Stasiak, L.; Bartram, J.; Lang, B.; Vincent, A.; Upton, A.L. Epileptogenic effects of NMDAR antibodies in a passive transfer mouse model. Brain 2015, 138, 3159–3167. [Google Scholar] [CrossRef]
- Taraschenko, O.; Fox, H.S.; Pittock, S.J.; Zekeridou, A.; Gafurova, M.; Eldridge, E.; Liu, J.; Dravid, S.M.; Dingledine, R. A mouse model of seizures in anti-N-methyl-d-aspartate receptor encephalitis. Epilepsia 2019, 60, 452–463. [Google Scholar] [CrossRef]
- Moscato, E.H.; Peng, X.; Jain, A.; Parsons, T.D.; Dalmau, J.; Balice-Gordon, R.J. Acute mechanisms underlying antibody effects in anti-N-methyl-D-aspartate receptor encephalitis. Ann. Neurol. 2014, 76, 108–119. [Google Scholar] [CrossRef]
- Kreye, J.; Wenke, N.K.; Chayka, M.; Leubner, J.; Murugan, R.; Maier, N.; Jurek, B.; Ly, L.T.; Brandl, D.; Rost, B.R.; et al. Human cerebrospinal fluid monoclonal N-methyl-D-aspartate receptor autoantibodies are sufficient for encephalitis pathogenesis. Brain 2016, 139, 2641–2652. [Google Scholar] [CrossRef]
- Sharma, R.; Al-Saleem, F. H.; Panzer, J.; Lee, J.; Puligedda, R.D.; Felicori, L.F.; Kattala, C.D.; Rattelle, A.J.; Ippolito, G.; Cox, R.H.; et al. Monoclonal antibodies from a patient with anti-NMDA receptor encephalitis. Ann. Clin. Transl. Neurol. 2018, 5, 935–951. [Google Scholar] [CrossRef]
- Irani, S.R.; Bera, K.; Waters, P.; Zuliani, L.; Maxwell, S.; Zandi, M.S.; Friese, M.A.; Galea, I.; Kullmann, D.M.; Beeson, D.; et al. N-methyl-D-aspartate antibody encephalitis: temporal progression of clinical and paraclinical observations in a predominantly non-paraneoplastic disorder of both sexes. Brain 2010, 133, 1655–1667. [Google Scholar] [CrossRef]
- Castillo-Gomez, E.; Oliveira, B.; Tapken, D.; Bertrand, S.; Klein-Schmidt, C.; Pan, H.; Zafeiriou, P.; Steiner, J.; Jurek, B.; Trippe, R.; et al. All naturally occurring autoantibodies against the NMDA receptor subunit NR1 have pathogenic potential irrespective of epitope and immunoglobulin class. Mol. Psychiatry 2017, 22, 1776–1784. [Google Scholar] [CrossRef]
- Mullier, B.; Wolff, C.; Sands, Z.A.; Ghisdal, P.; Muglia, P.; Kaminski, R.M.; Andre, V.M. GRIN2B gain of function mutations are sensitive to radiprodil, a negative allosteric modulator of GluN2B-containing NMDA receptors. Neuropharmacol. 2017, 123, 322–331. [Google Scholar] [CrossRef]
- Paoletti, P.; Neyton, J. NMDA receptor subunits: function and pharmacology. Curr. Opin. Pharmacol. 2007, 7, 39–47. [Google Scholar] [CrossRef]
- Gleichman, A.J.; Spruce, L.A.; Dalmau, J.; Seeholzer, S.H.; Lynch, D.R. Anti-NMDA receptor encephalitis antibody binding is dependent on amino acid identity of a small region within the GluN1 amino terminal domain. J. Neurosci. 2012, 32, 11082–11094. [Google Scholar] [CrossRef]
- Tajima, N.; Simorowski, N.; Yovanno, R.A.; Regan, M.C.; Michalski, K.; Gomez, R.; Lau, A.Y.; Furukawa, H. Development and characterization of functional antibodies targeting NMDA receptors. Nat. Commun. 2022, 13, 923. [Google Scholar] [CrossRef]
- Washburn, H.R.; Xia, N.L.; Zhou, W.; Mao, Y.T.; Dalva, M.B. Positive surface charge of GluN1 N-terminus mediates the direct interaction with EphB2 and NMDAR mobility. Nat. Commun. 2020, 11, 570. [Google Scholar] [CrossRef]
- Mikasova, L.; De Rossi, P.; Bouchet, D.; Georges, F.; Rogemond, V.; Didelot, A.; Meissirel, C.; Honnorat, J.; Groc, L. Disrupted surface cross-talk between NMDA and Ephrin-B2 receptors in anti-NMDA encephalitis. Brain 2012, 135, 1606–1621. [Google Scholar] [CrossRef] [PubMed]
- Planaguma, J.; Haselmann, H.; Mannara, F.; Petit-Pedrol, M.; Grunewald, B.; Aguilar, E.; Ropke, L.; Martin-Garcia, E.; Titulaer, M.J.; Jercog, P.; et al. Ephrin-B2 prevents N-methyl-D-aspartate receptor antibody effects on memory and neuroplasticity. Ann. Neurol. 2016, 80, 388–400. [Google Scholar] [CrossRef]
- Goldin, A.L. Expression of ion channels in Xenopus oocytes. In Expression and analysis of recombinant ion channels; Clare, J.J., Trezise, D.J., Eds.; Wiley VCH Verlag GmbH & Co. KgaA: Weinheim, 2006; pp. 1–25. [Google Scholar]
- Würdemann, T.; Kersten, M.; Tokay, T.; Guli, X.; Kober, M.; Rohde, M.; Porath, K.; Sellmann, T.; Bien, C.G.; Kohling, R.; et al. Stereotactic injection of cerebrospinal fluid from anti-NMDA receptor encephalitis into rat dentate gyrus impairs NMDA receptor function. Brain Res. 2016, 1633, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Wright, S.K.; Rosch, R.E.; Wilson, M.A.; Upadhya, M.A.; Dhangar, D.R.; Clarke-Bland, C.; Wahid, T.T.; Barman, S.; Goebels, N.; Kreye, J.; et al. Multimodal electrophysiological analyses reveal that reduced synaptic excitatory neurotransmission underlies seizures in a model of NMDAR antibody-mediated encephalitis. Commun. Biol. 2021, 4, 1106. [Google Scholar] [CrossRef]
- Warikoo, N.; Brunwasser, S.J.; Benz, A.; Shu, H.J.; Paul, S.M.; Lewis, M.; Doherty, J.; Quirk, M.; Piccio, L.; Zorumski, C.F.; et al. Positive allosteric modulation as a potential therapeutic strategy in anti-NMDA receptor encephalitis. J. Neurosci. 2018, 38, 3218–3229. [Google Scholar] [CrossRef]
- Dean, C.A.; Metzbower, S.R.; Dessain, S.K.; Blanpied, T.A.; Benavides, D.R. Regulation of NMDA receptor signaling at single synapses by human anti-NMDA receptor antibodies. Front Mol. Neurosci. 2022, 15, 940005. [Google Scholar] [CrossRef]
- Blome, R.; Bach, W.; Guli, X.; Porath, K.; Sellmann, T.; Bien, C.G.; Kohling, R.; Kirschstein, T. Differentially altered NMDAR dependent and independent long-term potentiation in the CA3 Subfield in a model of anti-NMDAR encephalitis. Front. Synaptic Neurosci. 2018, 10, 26. [Google Scholar] [CrossRef] [PubMed]
- Kersten, M.; Rabbe, T.; Blome, R.; Porath, K.; Sellmann, T.; Bien, C.G.; Kohling, R.; Kirschstein, T. Novel object recognition in rats with NMDAR dysfunction in CA1 after stereotactic injection of anti-NMDAR encephalitis cerebrospinal fluid. Front. Neurol. 2019, 10, 586. [Google Scholar] [CrossRef]
- Zhang, Q.; Tanaka, K.; Sun, P.; Nakata, M.; Yamamoto, R.; Sakimura, K.; Matsui, M.; Kato, N. Suppression of synaptic plasticity by cerebrospinal fluid from anti-NMDA receptor encephalitis patients. Neurobiol. Dis. 2012, 45, 610–615. [Google Scholar] [CrossRef]
- Masdeu, J.C.; Dalmau, J.; Berman, K.F. NMDA receptor internalization by autoantibodies: a reversible mechanism underlying psychosis? Trends Neurosci. 2016, 39, 300–310. [Google Scholar] [CrossRef]
- Hunter, D.; Jamet, Z.; Groc, L. Autoimmunity and NMDA receptor in brain disorders: Where do we stand? Neurobiol. Dis. 2021, 147, 105161. [Google Scholar] [CrossRef]
- Olivero, G.; Roggeri, A.; Pittaluga, A. Anti-NMDA and anti-AMPA receptor antibodies in central disorders: preclinical approaches to assess their pathological role and translatability to clinic. Int. J. Mol. Sci. 2023, 24, 14905. [Google Scholar] [CrossRef] [PubMed]
- Ceanga, M.; Rahmati, V.; Haselmann, H.; Schmidl, L.; Hunter, D.; Brauer, A.K.; Liebscher, S.; Kreye, J.; Pruss, H.; Groc, L.; et al. Human NMDAR autoantibodies disrupt excitatory-inhibitory balance, leading to hippocampal network hypersynchrony. Cell Rep. 2023, 42, 113166. [Google Scholar] [CrossRef] [PubMed]
- Flammer, J.; Neziraj, T.; Ruegg, S.; Probstel, A.K. Immune mechanisms in epileptogenesis: update on diagnosis and treatment of autoimmune epilepsy syndromes. Drugs 2023, 83, 135–158. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.; Wang, C. Anti-NMDA receptor autoimmune encephalitis: diagnosis and management strategies. Int. J. Gen. Med. 2023, 16, 7–21. [Google Scholar] [CrossRef]
- Mannara, F.; Radosevic, M.; Planaguma, J.; Soto, D.; Aguilar, E.; Garcia-Serra, A.; Maudes, E.; Pedreno, M.; Paul, S.; Doherty, J.; et al. Allosteric modulation of NMDA receptors prevents the antibody effects of patients with anti-NMDAR encephalitis. Brain 2020, 143, 2709–2720. [Google Scholar] [CrossRef]
- Radosevic, M.; Planaguma, J.; Mannara, F.; Mellado, A.; Aguilar, E.; Sabater, L.; Landa, J.; Garcia-Serra, A.; Maudes, E.; Gasull, X.; et al. Allosteric modulation of NMDARs reverses patients' autoantibody effects in mice. Neurol. Neuroimmunol. Neuroinflamm. 2022, 9, e1122. [Google Scholar] [CrossRef]
- Steinke, S.; Kirmann, T.; Loi, E.A.; Nerlich, J.; Weichard, I.; Kuhn, P.; Bullmann, T.; Ritzau-Jost, A.; Rizalar, F.S.; Pruss, H.; et al. NMDA-receptor-Fc-fusion constructs neutralize anti-NMDA receptor antibodies. Brain 2023, 146, 1812–1820. [Google Scholar] [CrossRef]
- García-Serra, A.; Radosevic, M.; Ríos, J.; Aguilar, E.; Maudes, E.; Landa, J.; Sabater, L.; Martinez-Hernandez, E.; Planagumà, J.; Dalmau, J. Blocking placental class G immunoglobulin transfer prevents NMDA receptor antibody effects in newborn mice. Neurol. Neuroimmunol. Neuroinflamm. 2021, 8, e1061. [Google Scholar] [CrossRef]
- Young, D. The NMDA receptor antibody paradox: A possible approach to developing immunotherapies targeting the NMDA receptor. Front. Neurol. 2020, 11, 635. [Google Scholar] [CrossRef] [PubMed]
- Skwarczynski, M.; Toth, I. Peptide-based synthetic vaccines. Chem. Sci. 2016, 7, 842–854. [Google Scholar] [CrossRef] [PubMed]
- Day, C.; Silva, J.P.; Munro, R.; Baker, T.S.; Wolff, C.; Bithell, A.; Stephens, G.J. Anti-AMPA receptor autoantibodies reduce excitatory currents in rat hippocampal neurons. Pharmaceuticals 2023, 16, 77. [Google Scholar] [CrossRef] [PubMed]
- Percie du Sert, N.; Hurst, V.; Ahluwalia, A.; Alam, S.; Avey, M.T.; Baker, M.; Browne, W.J.; Clark, A.; Cuthill, I.C.; Dirnagl, U.; et al. The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. PLoS Biol. 2020, 40, e3000410. [Google Scholar]
- Hill, A.J.; Jones, N.A.; Williams, C.M.; Stephens, G.J.; Whalley, B.J. Development of multi-electrode array screening for anticonvulsants in acute rat brain slices. J. Neurosci. Methods 2010, 185, 246–56. [Google Scholar] [CrossRef]
Figure 1.
Purification of NMDAR Aabs. (A) NMDAR peptide sequences used for immunisation. All peptides were located within the amino terminal domain (ATD) of NR1 subunit and generated with the addition of a C residue and an Ac residue on either end. Peptides 2, 4 and 5 were cyclised via a thioester to help better represent the true epitope. (B) ELISA for Protein A-purified (upper panel) and peptide-purified (lower panel) anti-NMDAR Aabs; in particular, peptides 2, 4 and 5 exhibited robust binding following further peptide purification. (C) ELISA for peptide-purified vs Protein A-purified Aabs. Dotted lines represent 50% reduction in signal values. Each n=3 technical replicates per concentration. D) Human NR1 subunit ATD probed with anti-NMDAR Aabs (upper panel) and controls rIgG, mIgG2b, secondary only and equilibrative Nucleoside Transporter 1 (ENT1). Blots incubated with anti-NMDAR Aabs detected a strong band at 60 kDa, the expected size of the ATD. Representative blot selected from n=3 technical replicates.
Figure 1.
Purification of NMDAR Aabs. (A) NMDAR peptide sequences used for immunisation. All peptides were located within the amino terminal domain (ATD) of NR1 subunit and generated with the addition of a C residue and an Ac residue on either end. Peptides 2, 4 and 5 were cyclised via a thioester to help better represent the true epitope. (B) ELISA for Protein A-purified (upper panel) and peptide-purified (lower panel) anti-NMDAR Aabs; in particular, peptides 2, 4 and 5 exhibited robust binding following further peptide purification. (C) ELISA for peptide-purified vs Protein A-purified Aabs. Dotted lines represent 50% reduction in signal values. Each n=3 technical replicates per concentration. D) Human NR1 subunit ATD probed with anti-NMDAR Aabs (upper panel) and controls rIgG, mIgG2b, secondary only and equilibrative Nucleoside Transporter 1 (ENT1). Blots incubated with anti-NMDAR Aabs detected a strong band at 60 kDa, the expected size of the ATD. Representative blot selected from n=3 technical replicates.
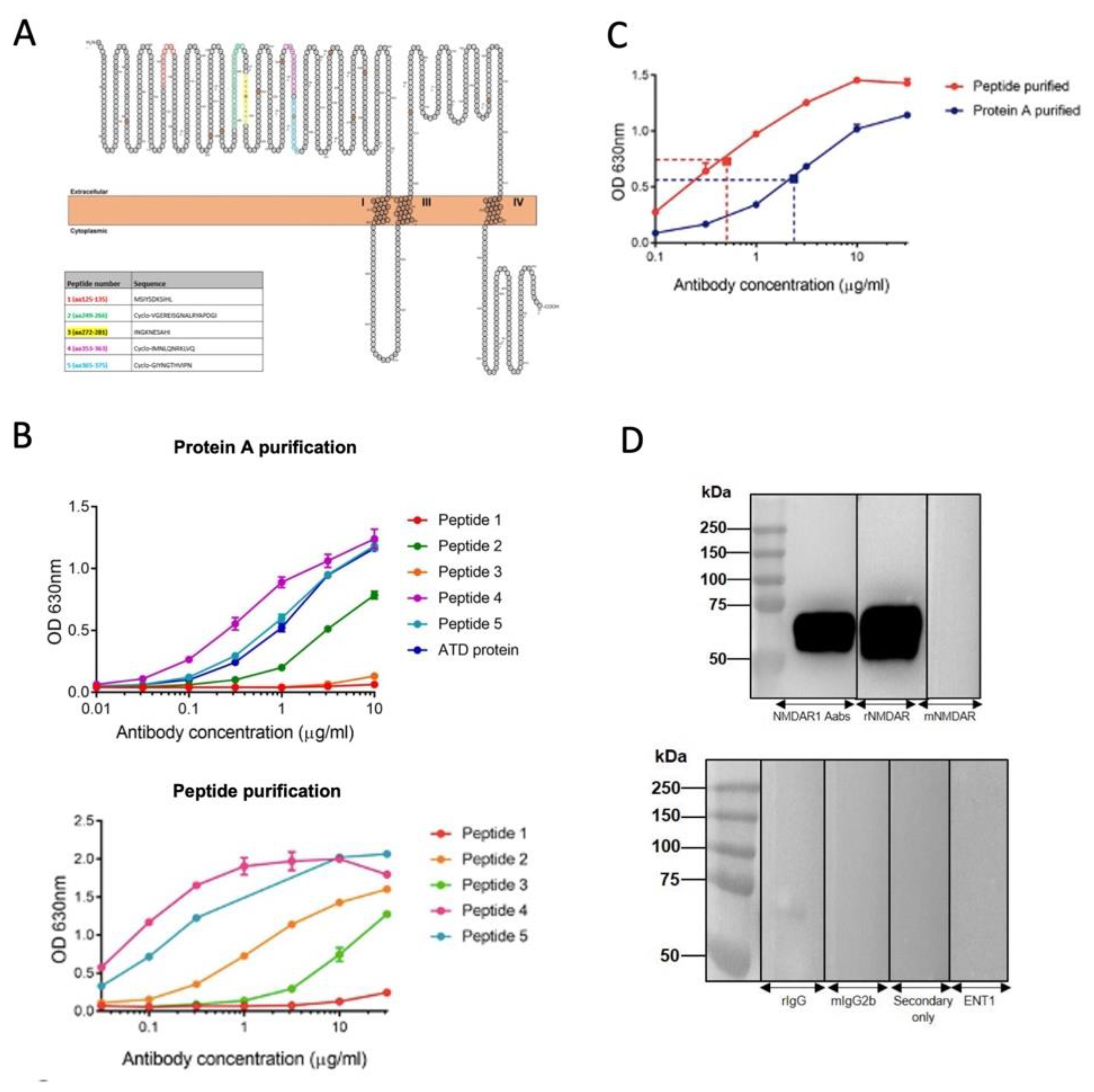
Figure 2.
Anti-NMDAR Aabs show selective immunocytochemical staining NMDAR NR1-transfected HEK cells were stained with peptide-purified anti-NMDAR Aabs (1:100), plus one of two commercial anti-NR1 antibodies: mouse anti-NR1 (mNMDAR, 1:100), rabbit anti-NR1 (rNMDAR, 1:100) and a nuclear stain (DAPI, 1:10,000, blue). (Ai-Aiv) Cells transfected with NR1 were detected by anti-NMDAR1 Aabs (Aii), which were co-labelled by the commercial antibody mNMDAR (Aiii), (as shown by white arrows). (Bi-Biv) Both commercial anti-NR1 antibodies rNMDAR (Bii) and mNMDAR (Biii) co-labelled the same NR1-transfected cells (as shown by white arrows). Representative images selected from n= 3 biological replicates. (Ci-Cv) Primary cortical neuronal cells (DIV14) were stained with anti-NMDAR Aabs (1:100, green, Cii); co-stained with the neuronal marker βIII tubulin (1:500, red, Ciii); the astrocytic marker GFAP (1:400, Civ) and DAPI (1:10,000, blue, Cv). Representative images selected from n= 3 biological replicates. Scale bar = 20 µm throughout.
Figure 2.
Anti-NMDAR Aabs show selective immunocytochemical staining NMDAR NR1-transfected HEK cells were stained with peptide-purified anti-NMDAR Aabs (1:100), plus one of two commercial anti-NR1 antibodies: mouse anti-NR1 (mNMDAR, 1:100), rabbit anti-NR1 (rNMDAR, 1:100) and a nuclear stain (DAPI, 1:10,000, blue). (Ai-Aiv) Cells transfected with NR1 were detected by anti-NMDAR1 Aabs (Aii), which were co-labelled by the commercial antibody mNMDAR (Aiii), (as shown by white arrows). (Bi-Biv) Both commercial anti-NR1 antibodies rNMDAR (Bii) and mNMDAR (Biii) co-labelled the same NR1-transfected cells (as shown by white arrows). Representative images selected from n= 3 biological replicates. (Ci-Cv) Primary cortical neuronal cells (DIV14) were stained with anti-NMDAR Aabs (1:100, green, Cii); co-stained with the neuronal marker βIII tubulin (1:500, red, Ciii); the astrocytic marker GFAP (1:400, Civ) and DAPI (1:10,000, blue, Cv). Representative images selected from n= 3 biological replicates. Scale bar = 20 µm throughout.
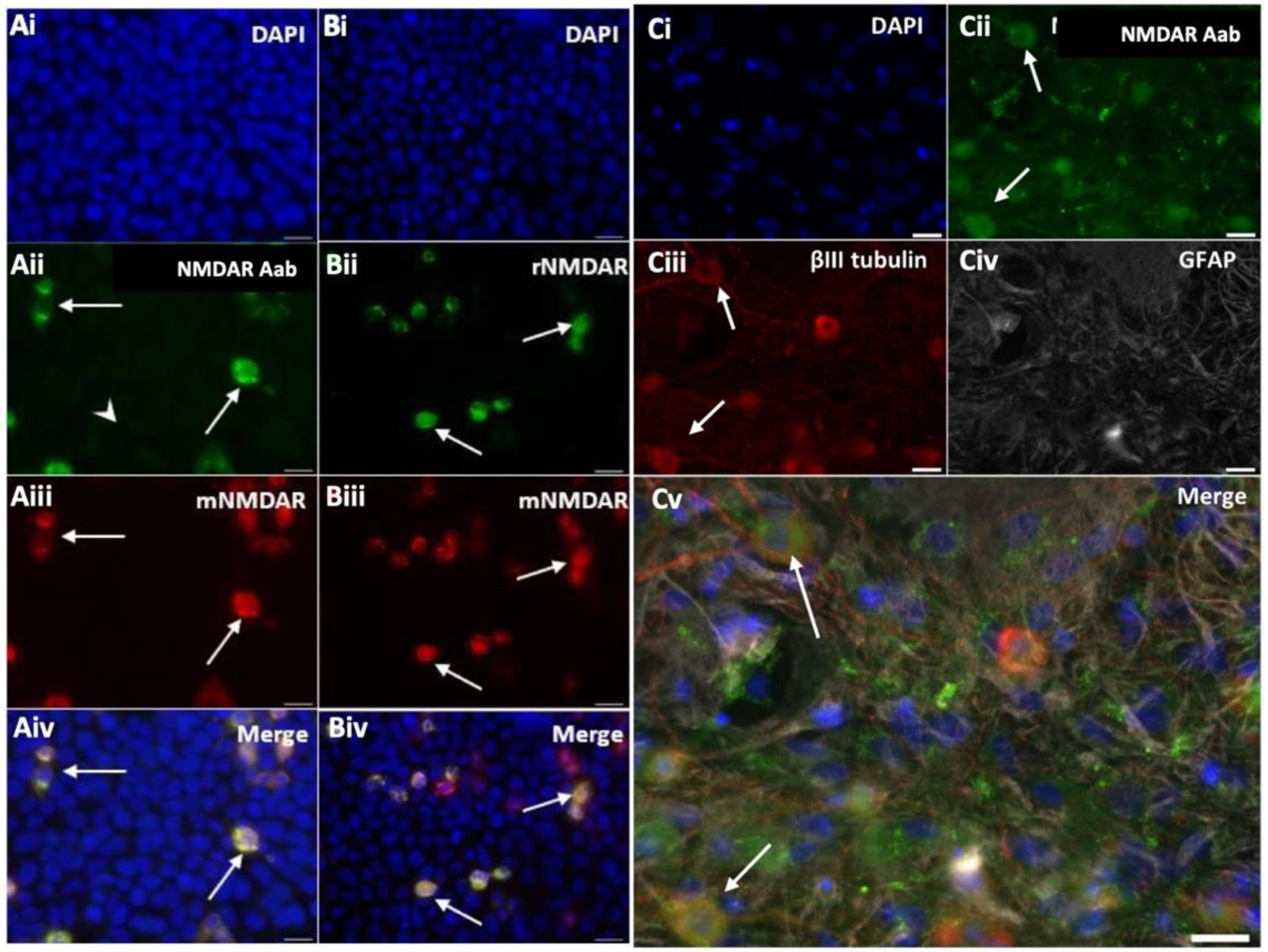
Figure 3.
NMDAR negative modulator significantly reduces NMDAR currents in Xenopus oocytes. (A) Traces of NMDAR-evoked responses over time: 1 µM glutamate/10 µM glycine-induced NMDA currents were elicited every 3 min. The 2 last baseline traces of 4 traces are depicted and used to normalise to control before switching to vehicle (control). (B) Traces of NMDAR-evoked responses before and after addition of increasing concentrations of TCN-201. (C) Graph shows a significant reduction in normalised glutamate-evoked (AUC) response with increasing concentration of TCN-201 vs vehicle control. A two-way ANOVA revealed both a significant effect of drug vs vehicle (*= p < 0.0001, n = 6-7 per group) and drug concentration used (*= p < 0.0001, n = 6-7 per group) respectively). Data are represented as mean ± SD. Each oocyte was used for baseline and treatment throughout the 30 min experiment.
Figure 3.
NMDAR negative modulator significantly reduces NMDAR currents in Xenopus oocytes. (A) Traces of NMDAR-evoked responses over time: 1 µM glutamate/10 µM glycine-induced NMDA currents were elicited every 3 min. The 2 last baseline traces of 4 traces are depicted and used to normalise to control before switching to vehicle (control). (B) Traces of NMDAR-evoked responses before and after addition of increasing concentrations of TCN-201. (C) Graph shows a significant reduction in normalised glutamate-evoked (AUC) response with increasing concentration of TCN-201 vs vehicle control. A two-way ANOVA revealed both a significant effect of drug vs vehicle (*= p < 0.0001, n = 6-7 per group) and drug concentration used (*= p < 0.0001, n = 6-7 per group) respectively). Data are represented as mean ± SD. Each oocyte was used for baseline and treatment throughout the 30 min experiment.
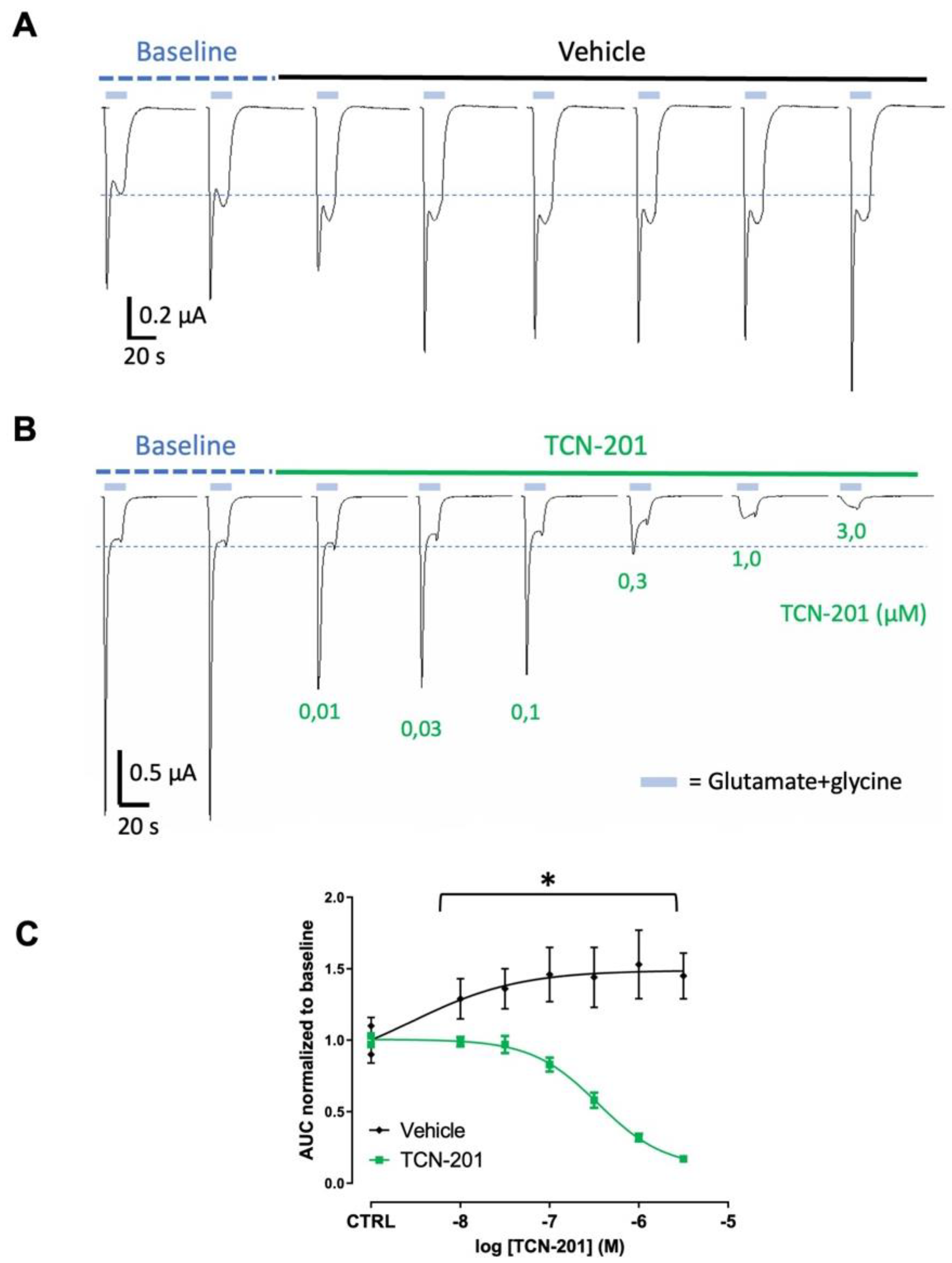
Figure 4.
Anti-NMDAR Aabs have no effect on NMDA current in Xenopus oocytes. (A) Traces of NMDAR-evoked responses over time before and after incubation with control peptide-purified IgG: 10 µM glutamate/10 µM glycine-induced NMDA currents were elicited every 15 min in the presence of IgG (1:300 dilution) applied for up to 60 min (B) Traces of NMDAR-evoked responses over time before and after incubation of peptide-purified anti-NMDA Aabs: 10 µM glutamate/10 µM glycine-induced NMDA currents were elicited every 15 min in the presence of anti-NMDAR Aabs (1:300 dilution) applied for up to 60 min. (C) Graph shows effect of peptide-purified anti-NMDAR Aab and rIgG glutamate-evoked (AUC) responses normalised to baseline (mean of applications 1 and 2 shown in A,B). There was no significant change in AUC when compared to baseline in anti-NMDAR Aabs or IgG incubated oocytes. All data are represented as mean ± SD. Each oocyte was used for baseline and treatment throughout the 60 min experiment.
Figure 4.
Anti-NMDAR Aabs have no effect on NMDA current in Xenopus oocytes. (A) Traces of NMDAR-evoked responses over time before and after incubation with control peptide-purified IgG: 10 µM glutamate/10 µM glycine-induced NMDA currents were elicited every 15 min in the presence of IgG (1:300 dilution) applied for up to 60 min (B) Traces of NMDAR-evoked responses over time before and after incubation of peptide-purified anti-NMDA Aabs: 10 µM glutamate/10 µM glycine-induced NMDA currents were elicited every 15 min in the presence of anti-NMDAR Aabs (1:300 dilution) applied for up to 60 min. (C) Graph shows effect of peptide-purified anti-NMDAR Aab and rIgG glutamate-evoked (AUC) responses normalised to baseline (mean of applications 1 and 2 shown in A,B). There was no significant change in AUC when compared to baseline in anti-NMDAR Aabs or IgG incubated oocytes. All data are represented as mean ± SD. Each oocyte was used for baseline and treatment throughout the 60 min experiment.

Figure 5.
Anti-NMDR Aabs inhibit HFS-induced LTP in hippocampal brain slices. (A) Normalised mean traces of HFS-induced LTP for vehicle, peptide-purified anti-NMDAR Aab (1:1000, 1 h pre-incubation) or 50 μM APV treated slices. HFS elicited a potentiation of ~150% in vehicle conditions which was maintained for at least 1 h. The addition of APV dramatically inhibited HFS-induced LTP. Similarly, anti-NMDAR Aabs inhibited HFS-induced LTP. Inset shows representative traces of paired pulse recordings post-HFS (in vehicle). The addition of the AMPAR blocker CNQX (5µM) caused a rapid reduction in HFS-induced LTP. Subsequent addition of the general voltage-gated sodium channel blocker, tetrodotoxin (TTX, 1 µM), abolished any remaining signal. (B) HFS-induced LTP (measured as mean fEPSP slope increase over baseline) revealed a significant reduction in APV-treated (n=6) slices vs vehicle (n=8), with anti-NMDAR Aabs-treated slices (n=7) demonstrating a similar significant reduction vs vehicle. Data represented as mean ± SD; one-way ANOVA with Dunnett’s multiple comparisons *= p < 0.05, ** = p < 0.01.
Figure 5.
Anti-NMDR Aabs inhibit HFS-induced LTP in hippocampal brain slices. (A) Normalised mean traces of HFS-induced LTP for vehicle, peptide-purified anti-NMDAR Aab (1:1000, 1 h pre-incubation) or 50 μM APV treated slices. HFS elicited a potentiation of ~150% in vehicle conditions which was maintained for at least 1 h. The addition of APV dramatically inhibited HFS-induced LTP. Similarly, anti-NMDAR Aabs inhibited HFS-induced LTP. Inset shows representative traces of paired pulse recordings post-HFS (in vehicle). The addition of the AMPAR blocker CNQX (5µM) caused a rapid reduction in HFS-induced LTP. Subsequent addition of the general voltage-gated sodium channel blocker, tetrodotoxin (TTX, 1 µM), abolished any remaining signal. (B) HFS-induced LTP (measured as mean fEPSP slope increase over baseline) revealed a significant reduction in APV-treated (n=6) slices vs vehicle (n=8), with anti-NMDAR Aabs-treated slices (n=7) demonstrating a similar significant reduction vs vehicle. Data represented as mean ± SD; one-way ANOVA with Dunnett’s multiple comparisons *= p < 0.05, ** = p < 0.01.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



