
Submitted:
24 April 2024
Posted:
25 April 2024
You are already at the latest version
Abstract
Ixodes scapularis is a blood-feeding obligate ectoparasite responsible for transmitting the Lyme disease (LD) agent, Borrelia burgdorferi. During the feeding process, I. scapularis injects B. burgdorferi into the host along with its saliva, facilitating the transmission and colonization of the LD agent. Tick calreticulin (CRT) is one of the earliest tick saliva proteins identified and is currently utilized as a biomarker for tick bites. Our recent findings revealed elevated levels of CRT in the saliva proteome of B. burgdorferi-infected I. scapularis nymphs compared to uninfected ticks. Differential precipitation of proteins (DiffPOP) and LC-MS/MS analyses were used to identify the interactions between Ixs (I. scapularis) CRT and human plasma proteins and further exploring its potential role in shielding B. burgdorferi from complement killing. We observed that although yeast expressed recombinant (r) IxsCRT binds to the C1 complex (C1q, C1r, and C1s), the activator of complement via the classical cascade, it did not inhibit deposition of the membrane attack complex (MAC) via the classical pathway. Intriguingly, rIxsCRT binds intermediate complement proteins (C3, C5, and C9) and reduces MAC deposition through the lectin pathway. Despite inhibition of MAC deposition in the lectin pathway, rIxsCRT did not protect a serum sensitive B. burgdorferi strain (B314/pBBE22Luc) from complement-induced killing. As B. burgdorferi establishes a local dermal infection before disseminating to secondary organs, it is noteworthy that rIxsCRT promotes the replication of B. burgdorferi in culture. We hypothesize that rIxsCRT may contribute to the transmission and/or host colonization of B. burgdorferi by acting as a decoy activator of complement and by fostering B. burgdorferi replication at the transmission site.
Keywords:
Ixodes scapularis
; complement cascade
; tick calreticulin
; Borrelia burgdorferi
1. Introduction
The black legged tick, Ixodes scapularis, is a 3-host tick blood feeding ectoparasite that completes its life cycle (larvae to nymph to adult) over 2 years. This tick is considered endemic to several parts of USA including Midwest, Northeast, West, Southeast, and Southern USA and its distribution is dependent on the various ecological factors such as climates, vegetation, landscape and availability of host [1,2]. I. scapularis, which transmits 7 of 16 human tick borne disease (TBD) agents in the USA [2] including causative agents of anaplasmosis (Anaplasma phagocytophilum), babesiosis (Babesia microti), ehrlichiosis (Ehrlichia chaffeensis), hard Tick Relapsing Fever (Borrelia miyamotoi), Powassan encephalitis virus [3,4,5], is famously known for transmission of causative agents of Lyme disease (LD): Borrelia burgdorferi [6] and the recently described B. mayonii [7,8,9]. LD is among the most important human TBD with ≈476,000 diagnosed cases nationally per year and can cost up to $1 billion without including suspected, undiagnosed and nonacute cases. The accurate economic cost of LD will be much higher if all parameters were included when generating estimates [10]. In the absence of effective vaccines against LD and other TBD agents, killing of ticks using acaricides, personal protections and avoidance of infected ticks are the only available options to prevent TBD. However, limitations associated with acaricide use such as tick resistance [11], toxicity [12], cost effectiveness [13], and environmental contamination have necessitated the development of alternative tick control methods. Immunization against tick feeding has emerged as the most promising alternative tick control method [14]. Identification and target validation of key tick saliva proteins that facilitate feeding and transmission of TBD agents by ticks is needed before tick antigen-based vaccines to prevent TBD could be developed. Toward this goal, our lab has identified and functionally characterized tick saliva proteins secreted by different stages of ticks and different phases of feeding [15,16,17]. Recently, we found I. scapularis tick calreticulin (IxsCRT) among proteins that were highly secreted by B. burgdorferi infected I. scapularis nymph [15]. The objective of this study was to characterize the role of IxsCRT as a tick saliva transmission factor of B. burgdorferi.
Tick CRT is among highly conserved tick saliva proteins which is found to be expressed universally in all developmental stages and tissues of ticks and has been reported to be secretory protein in saliva of multiple tick species including Amblyomma americanum, Rhipicephalus microplus and I. scapularis [18,19,20]. Except for erythrocytes, CRT has been reported in every cell of higher organisms. CRT shows multiple functions including calcium homeostasis, chaperon activity, lytic activity through T and natural killer (NK) cells, boosting the phagocytosis of apoptotic cells, suppressing tumor growth [21,22]. Similarly, tick CRT is also a calcium ion binding protein and it is believed that since calcium ion is an important factor for blood clotting, the tick might be secreting this protein to prevent blood clotting [23]. This concept was explored in another study which showed that Haemonchus contortus CRT prevented blood clotting by binding to Ca2+ and sequestering blood clotting factors, prothrombin factor X, and C-reactive protein [24,25].
Several studies reported elevated antibodies to tick CRT following tick bites and consequently, tick CRT serves as a biomarker for tick bites [18,26,27]. However, the role(s) of tick CRT in feeding and pathogen transmission is not fully known. Functional analyses of CRT in other parasites including Trypanosoma cruzi, Haemonchus contortus, Entamoeba histolytica, Trypanosoma carassii have suggested that parasite CRT blocks the complement cascade by binding with complement component, C1q [21,24,28,29]. In our lab, we previously reported that recombinant Amblyomma americanum tick CRT bound C1q, a member of the C1 complex, but did not inhibit deposition of terminal complex of complement cascade [30,31]. Similarly, in this study, we report that yeast expressed recombinant I. scapularis CRT (rIxsCRT) binds complement C1 complex proteins (C1q, C1r, and C1s). Although, the C1 complex activates the complement system via classical pathway, rIxsCRT binding of the C1 complex did not inhibit deposition of the membrane attack complex (MAC) in the classical pathway. Interestingly, rIxsCRT also bound intermediate complete system factors, (C3, C5 and C9) and significantly reduced MAC deposition via the lectin pathway. Importantly, while rIxsCRT did not rescue B. burgdorferi from complement killing, it promoted the growth of B. burgdorferi in culture. Our data suggests that B. burgdorferi stimulates the tick to secrete high amounts of CRT that could promote its growth at the tick feeding site.
2. Material and Methods
2.1. Expression and Affinity Purification of Recombinant I. Scapularis Calreticulin (rIxsCRT)
Expression of rIxsCRT was done using pPICZαA and Pichia pastoris yeast expression system as previously described [31]. Synthesis of the rIxsCRT expression plasmid was outsourced (Biomatik, Wilmington, DE, USA). The mature protein coding domain was based on I. scapularis CRT sequence in GenBank (accession # AY690335.1) with inclusion of a C-terminus hexa-histidine fusion tag and was cloned between EcoRI and NotI sites in pPICZαA plasmid. The rIxsCRT expression plasmid was used to transform P. pastoris (X-33) as described [32]. Transformed colonies were selected on Yeast Extract Peptone Dextrose Medium with Sorbitol (YPDS) agar plates with zeocin (100 μg/mL) incubated at 28°C. Positive transformants (confirmed by PCR check) were inoculated in Buffered Glycerol-complex Medium (BMGY) and grown overnight at 28°C with shaking (230–250 rpm). Subsequently the cells were used to inoculate Buffered Methanol-complex Medium (BMMY) to A600 of 1 after which protein expression was induced by adding methanol up to 1% final concentration every 24 h. Pilot expression showed that expression levels of rIxsCRT peaked at day 3. Thus, for large-scale 1 L cultures, we cultured yeast for up to three days. The pPICZαA plasmid secretes the recombinant protein into media, and thus, rIxsCRT was salted out using ammonium sulfate saturation as described [31]. Precipitated rIxsCRT was dialyzed against column binding buffer (1M NaCl, 0.4M Tris, 0.2M imidazole, pH7.4) and was affinity purified using HiTrap chelating HP column as per the manufacturer’s instructions (Cytiva, Marlborough, MA, USA). Expression and affinity purification of rIxsCRT was confirmed by SDS-PAGE and visualized by silver staining and western blotting analysis using antibodies against C-terminus hexa-histidine fusion tag (Thermo Fisher Scientific, Waltham, MA, USA). The affinity-purified rIxsCRT was dialyzed against 10 mM HEPES buffer pH 7.4, 1X PBS pH 7.4 or normal saline based on appropriate assays described below and stored in −80°C until used.
2.2. Differential Precipitation (DIffPOP) of rIxsCRT and Human Plasma Proteins
To identify plasma proteins that might interact with tick saliva CRT, protein-to-protein interaction using differential precipitation was conducted as published [33,34]. Briefly, the reaction volume of 150µl was prepared by preincubating rIxsCRT (10 µg) with 10% Normal Human Serum (NHS) for 90min at 37˚C. NHS only and rIxsCRT only were included as negative controls. After incubation, 100 µL of stabilizing buffer (Phosphoprotein Kit, Buffer A, Takara Bio Company, CA, USA) was added to the reaction. Subsequently, to fractionate, an escalating amount of precipitation buffer (90% methanol/1% acetic acid) starting at 3.75 µL (or 0.9% of reaction volume) in fraction 1 to 317 µL (or 79.25% of reaction) in fraction 10 was added to the reaction mixture (Supplemental Table S1) followed by vortexing to mix. The reaction was incubated at RT (room temperature) for 5 min and centrifuged at 13000xg for 10 min at 4˚C. The supernatant was carefully transferred to another tube without disturbing the pellet and transfer to another 1.5 mL centrifuge tube and repeated the precipitation process for 9 more times. The collected pellets (n=10) were washed with ice cold acetone, airdried, and dissolved in PBS and considered as fractions 1-10. The plasma proteins that interacted with rIxsCRT were expected to co-precipitate with rIxsCRT. Subsequently, each fraction was resolved on 10% SDS-PAGE gel, silver stained, and subjected to the standard western blot analysis using antibodies to the hexa-histidine tag to track rIxsCRT fractionation. To identify plasma proteins that interacted with rIxsCRT, fractions were subjected to LC-MS/MS analysis.
2.3. LC-MS/MS Analysis
The samples for LC-MS/MS analysis were prepared by in-solution protein digestion method. In this method, 10 µg of differentially fractionated proteins were acetone precipitated and reconstituted in 100 µL of digestion buffer (50 mM ammonium bicarbonate in water, pH 8.5) and reduced with DTT (2 mM). The solution was incubated at 60˚C for 30 min and then alkylated with 10 mM of iodoacetamide and incubated in dark for 30 min at 60˚C. Subsequently, proteins were digested overnight at 37˚C with trypsin at 50:1 ratio (protein-to-trypsin) and submitted for LC-MS/MS analysis. LC-MS/MS analysis was performed on the Thermo Scientific Orbitrap Fusion tribrid mass spectrometer equipped with a Dionex UltiMate 3000 reverse-phase nano-UHPLC system. 1 µl of each sample was injected onto and separated by a 150 × 0.075 mm C18 column (Phenomenex bioZen XB-C18, 2.6 µm particle size) at a flow rate of 0.400 µl/min. The total duration of the method was 60 minutes, with the gradient as follows: equilibration at 2% B (98% acetonitrile, 2% water, 0.1% formic acid), ramp to 45% B at 37 minutes, ramp to 90% B at 40 minutes and hold until 46 minutes, ramp down to 2% B at 47 minutes and hold at 2% B until the end of the run at 60 minutes.
Eluent was introduced into the Fusion mass spectrometer by nano-ESI at a static voltage of 2450 V, with a transfer capillary temperature of 275 °C. Mass spectrometry data were acquired in positive mode at a resolution of 120,000 (at m/z 200) in the m/z range 400-1600. The RF lens was set to 60%. Maximum injection time was 100 ms. Scans were acquired in top speed mode with a cycle duration set to 3 seconds. The intensity threshold for precursors of interest was 5.0e3. Charge states 1-6 were considered. Dynamic exclusion was set to 60 seconds with a mass tolerance of 10 ppm. MS/MS data were acquired by HCD at a fixed collision energy of 28% with a precursor ion isolation window of 1.6 m/z; fragments were detected in the ion trap at a rapid scan rate. Proteome Discoverer 2.4 software (Thermo Fisher Scientific, Waltham, MA, USA) was used to analyze the MS/MS spectra of peptide ions to search against the publicly available NCBI protein database (www.ncbi.nlm.nih.gov). Plasma protein and rIxsCRT interactions were confirmed if the normalized spectral abundance factor was significantly different between controls (plasma only) and treatment (plasma and rIxsCRT) treatments.
Subsequently, the pathways that were represented by interactions between rIxsCRT and plasma proteins were revealed on the Reactome database (http://reactome.org) [35]. Accession numbers of plasma protein interactors with rIxsCRT were loaded onto the Reactome database server and highly significant pathways were reported.
2.4. Pull-Down Assays to Validate Interactions between rIxsCRT and Human Complement Proteins
Prompted by DiffPOP and LC-MS/MS analyses interactions between complement proteins and rIxsCRT, Dynabeads™ His-Tag Isolation and Pulldown magnetic beads (Thermo Fisher Scientific, Waltham, MA, USA) was used to perform the pull-down assay to validate interactions. The experiment was performed according to manufacturer’s guidelines with minor modifications. Briefly, 50 µL (2 mg) of magnetic Dynabeads™ was transferred to 1.5 mL microcentrifuge tube and placed on the magnetic stand for 2 min and the supernatant was discarded. Purified rIxsCRT (40 µg) was added to the beads and briefly vortexed to mix. The mixture was incubated on a roller at RT for 2 h. The tube was placed on the magnet for 2 min and supernatant was discarded. To remove nonspecific interactions, the beads were washed 4 times with the washing buffer (100 mM Sodium Phosphate (pH 8), 600 mM NaCl, 0.02% Tween-20). The test proteins, human complement serum (HCS; Innovative Research, Inc, Novi, MI, USA) were diluted to 10% in the pull-down buffer (6.5 mM Sodium phosphate pH 7.4, 140 mM NaCl, 0.02% Tween-20) and were added to the bead-bait complex and incubated on a roller for 2 h at RT. Then, the mixture was placed on the magnetic stand for 2 min and the supernatant was discarded. The beads were washed 4 times with the wash buffer (100 mM sodium phosphate, pH 8.0, 600mM NaCl, 0.02% Tween-20) and test protein complex were eluted by incubating the beads with elusion buffer (300mM Imidazole, 50mM Sodium phosphate pH 8.0, 300mM NaCl, 0.01% Tween-20) for 5min at RT. Subsequently, eluted proteins were subjected to standard SDS-PAGE and western blot analysis using antibodies to specific complement proteins: C1q (Thermo Fisher Scientific, Waltham, MA, USA), C1r (Thermo Fisher Scientific, Waltham, MA, USA) and C1s (Sino Biological, Inc., Houston, TX, USA); Activated C3 Antibody (I3/15) (Santa Cruz Biotechnology, Inc. Dallas, TX, USA), inactive C3 (Complement Technology, Inc., Tyler, TX, USA), C5 (Complement Technology, Inc., Tyler, TX, USA), C9 (Complement Technology, Inc., Tyler, TX, USA), and the C5-9 complex or membrane attack complex: MAC (Santa Cruz Biotechnology, Inc. Dallas, TX, USA). For controls, rIxsCRT only and HCS only samples were processed.
2.5. ELISA Analysis to Validate rIxsCRT Binding of Complement Proteins
Preliminary differential precipitation and pulldown assays showed that rIxsCRT likely interacted with complement proteins. To further substantiate these interactions, conventional ELISA was used. High Binding plates (Thermo Fisher Scientific, Waltham, MA, USA) were coated with rIxsCRT (250 ng) in ELISA coating buffer (carbonate-bicarbonate buffer, pH 9.6) overnight at 4˚C. Subsequently, wells were washed 3 times with phosphate buffer saline with Tween 20 (PBST) and the non-specific binding sites were blocked using blocking buffer (5% skim milk dissolved in PBST) for 2 h at RT. Following washing, 10% normal human serum (NHS) was added to the wells and was incubated for 2 h at RT. The coated plate was washed with PBST and wells were incubated for 3 h with respective primary polyclonal antibodies (Anti-C1q, Anti-C1r, Anti-C1s) and anti-sera (Anti-C3, Anti-C5 and Anti-C9) to complement proteins, at 1:5000 dilution. Wells were washed 3 times with PBST and incubated with respective HRP conjugated secondary antibody for 1 h (Anti-Rabbit for C1q, C1r and C1s, and anti-Goat for C3, C5 and C9). Wells were again washed 3 times with PBST, and bound peroxidase activity was observed by adding 1-Step Ultra TMB-ELISA substrate (Thermo Fisher Scientific, Waltham, MA, USA) and reaction was stopped using 2M sulfuric acid. The intensity of color development was quantified by measuring intensity at 450nm using microplate reader (Biotek Instrument Inc., Winooski, VT, USA).
2.6. Effect of rIxsCRT on Complement Activation
The effects of rIxsCRT of complement activation was assayed using the Wieslab® Complement System Screen (Svar Life Science AB, Malmo, Sweden) according to the manufacturer’s recommendation and as previously published [34,36]. This kit evaluates effects on membrane attack complex (MAC) deposition via each of the three complement system activation pathways: Classical, Lectin, and Alternative. To test the effect of the rIxsCRT on the complement pathways, NHS (provided with the kit) was diluted according to the manufacturer’s instructions and incubated at RT for 15 min. Briefly, rIxsCRT (4 μM), was added to the NHS and incubated at 37˚C for 30min, before adding the samples to the wells of the Wieslab® plates. The 100 μL of rIxsCRT and NHS samples (provided in the kit) were then added to the wells of the Wieslab® plates along with positive control (human serum, provided with kit, negative controls (provided with kit) and blank (diluent only) in duplicates, and incubated at 37°C for 1 h. Finally, absorbance was read at 405 nm on a Biotek Synergy H1 microplate reader (Biotek Instrument Inc., Winooski, VT, USA). The MAC deposition rate was calculated as follows: (Sample-NC)/(PC-NC) x100 where NC is negative control and PC is positive control.
2.7. Complement Sensitivity Assay
Our preliminary findings shows that rIxsCRT dose dependently reduced deposition of the MAC by mannose-binding lectin pathway, so we were interested to study its effect to rescue complement sensitive B. burgdorferi spirochete. The complement sensitive B. burgdorferi (B314/pBBE22Luc) and resistant (B314/pCD100) strains [37] were a kind gift from Jon. T. Skare laboratory (Texas A&M University Health Science Center, College Station, TX, USA). Both strains were maintained in BSK-II media supplemented with 6% rabbit serum at 32°C, 1% CO2 for this assay. 15 µL of normal human serum (NHS) (Complement Technology, Inc., Tyler, TX, USA) was pre-incubated with serial dilutions of rIxsCRT (0.25, 0.5, 0.75, and 1µM) at 37°C for 30 min prior to addition of 85 µl of B. burgdorferi B314/pBBE22luc (106 cells/ml) and inoculated in a bio-shaker at 32°C with shaking at 100 rpm, PBS (Phosphate buffered saline) was used as blank. NHS with B. burgdorferi B314/pBBE22luc and B314/pPCD100 were included as negative and positive controls respectively. Survival of spirochetes was assessed at 1.5 h, 2 h, 2.5 h and 3 h post incubation. Spirochetes were counted from randomly chosen fields (10-15 fields) under a dark-field microscope. Spirochete viability was arbitrated based on cell mobility, membrane integrity, and cell lysis as described elsewhere [37]. Spirochete survival rates were calculated from 3 biological replicates. Heat-inactivated NHS (hiNHS) was used as the no complement-activity control.
2.8. Blood Recalcification Time Assay
To assess the effect of rIxsCRT on blood clotting, the blood recalcification time was calculated as described elsewhere [38,39]. In this assay, normal blood plasma, Pacific Hemostasis™ Coagulation Reference Plasma (Thermo Fisher Scientific, Waltham, MA, USA) was used to measure the recalcification time in the presence of rIxsCRT. In this assay, 50 µl of plasma was incubated with tris-buffer (20mM Tris-HCl, pH 7.5), and rIxsCRT (2.6 mM) at room temperature for 30 min. Simultaneously, 150mM CaCl2 was incubated at 37˚C for 30 min. After incubation, plasma with rIxsCRT (140 µL total volume) along with prewarmed CaCl2 (10µL) was placed in triplicate (for each sample) in microwell plate and the absorbance was measured at 650 nm wavelength for 30min at 55 seconds interval. Sample without rIxsCRT was used as positive control and tris-buffer only was used as blank.
2.9. Assessing the Effect of rIxsCRT on B. burgdorferi Growth in In-Vitro Culture
To gauge insight on effect of IxsCRT on B. burgdorferi, low passaged (<6 passages) B. burgdorferi sensu stricto B31 (strain MSK5) was co-cultured with empirically determined rIxsCRT (2.6 mM) final concentration in BSK-H medium (Sigma-Aldrich, MO, USA) supplemented with 6% rabbit serum. Spirochetes were monitored each day under dark field microscopy to observe any morphological changes. Using the Petroff-Hausser Hemacytometer (Hausser Scientific Co., PA, USA), the starting concentration of B. burgdorferi was adjusted to 105 cells/ml. From each 5ml culture tube, 5 μL of BSK-H medium was pipetted on a Petroff-Hausser Hemacytometer (Hausser Scientific Co., PA, USA) and the cells were counted in all cells. The number of spirochetes were determined by using following formula: Spirochetes/mL = Average of spirochetes counted in all wells x Dilution factor x 50000 (50,000 = 50 (cell depth is 1/50) X 1000 (1000 cubic mm = 1 milliliter).
To quantify B. burgdorferi 200 µL of cells were aliquoted from the culture on day 0, 2, 5 and 7. Genomic DNA (gDNA) for each aliquot were extracted by using the DNeasy Kit according to manufacturer`s instructions (Qiagens, USA). The DNA concentration was determined by Nanodrop (Thermo Fisher Scientific, Waltham, MA, USA). In triplicate, gDNA (500ng) was mixed with empirically determined 300 nM FlaB qPCR primers (sense TCTTTTCTCTGGTGAGGGAGCT, anti-sense TCCTTCCTGTTGAACACCCTCT) and the PowerUp SYBR® Green Supermix (Thermo Fisher Scientific, Waltham, MA, USA) in 20 µL reaction volume and subjected to cycling on iQ5 Multicolor Real-Time PCR Detection System (Bio-Rad). B. burgdorferi was quantified by comparative analysis using BioRad CFX Maestro 3.2v software which analyzes the Ct values between the standards (101, 102, 103, 104, 105, 106, 107, 108 cells/ml B. burgdorferi) and the test cultures (without treated and rIxsCRT treated B. burgdorferi). Borrelia count was calculated by plotting the standard curves of Ct values versus the log of copy numbers included in each PCR run [40].
2.10. Identifying the Reaction Intensity of rIxsCRT against B. burgdorferi Infected Rabbit IgG
To ascertain the reactivity of rIxsCRT towards B. burgdorferi-infected rabbit IgG, both ELISA and western blot assays were conducted. The ELISA and western blot were performed following the similar steps as mentioned above. For both the assays, 40µg/ml of pre-infested (PI), B. burgdorferi infected and uninfected rabbit IgG, was used as primary antibody and anti-Rabbit IgG-HRP conjugated (Southern Biotech, Birmingham, AL, USA) was used as secondary antibody (1:104 dilution). Primary antibody used in these assays was purified from the rabbit serum which was infested once with B. burgdorferi infected and uninfected I. scapularis. The IgG was purified using Protein A/G column (Cytiva, Marlborough, MA, USA) by following the company’s instructions.
2.11. Statistical Analysis
To evaluate statistical significance, the data was analyzed using GraphPad Prism 9 software (GraphPad Software Inc., La Jolla, CA, USA) which was represented as mean ± SEM (standard error of the mean) with statistical significance (P < 0.05) detected by using the non-parametric Student’s t-test and two-tailed ANOVA to 95% confidence interval.
3. Results
3.1. Differential Precipitation of Proteins (DiffPOP) and LC-MS/MS Analyses Reveal Multiple Interactions between rIxsCRT Human and Plasma Proteins
Recombinant (r) IxsCRT was successfully expressed in P. pastoris (Figure 1). Affinity purified rIxsCRT migrated at the expected molecular weight size (47.61 kDa) as shown by silver staining (Figure 1A) and western blotting analyses using antibodies to the histidine fusion tag (Figure 1B).
We previously reported that although A. americanum tick CRT bound the C1q complement protein, it did not inhibit activation of classical complement cascade [31]. To expand on these findings, we employed DiffPOP (Figure 2) assay [33]. In this assay, plasma proteins that interacted with rIxsCRT co-precipitated with rIxCRT and western blotting analysis using the antibody to the histidine tag identified co-precipitating fractions 1-8 (Figure 2C). These fractions were subjected to LC-MS/MS analysis and were divided into groups as, pooled lanes 1-5 (hereafter called group A), lane 6 (hereafter called group B), and pooled lanes 7 and 8 (hereafter called group C) to identify the plasma proteins that interacted with rIxsCRT.
Normalized abundance values were used to construct a volcano plot (Figure 3) as graphical representation of differential co-precipitation of human proteins without or with rIxsCRT. Analysis of abundance values identified 1074, 1617, and 1936 unique proteins that interacted with rIxsCRT in groups A-C respectively (Supplemental tables S2-S4). To gain further insights, plasma proteins that were detected at least 1.5 folds higher in rIxsCRT and plasma reactions compared to plasma only control were subjected to reactome analysis (Table 1 and Supplemental table S5).
Group A (pooled fractions 1-5) proteins mapped to nine pathways (Table 1) that included 3 complement function pathways, the initial triggering of complement cascade, regulation of complement cascade, and terminal pathway of complement (Table 1). Group A proteins were identified as defective ABCA12 (ATP-binding cassette sub-family A member 12) which causes ARCI4B (autosomal recessive congenital ichthyosis 4B) that is associated with reduced protease regulation and skin-barrier dysfunction [41,42,43,44], defective ABCC2 (ATP binding cassette subfamily 2) that causes DJS (Dubin–Johnson syndrome) associated with hyperbilirubinemia [45]. Other group A proteins mapped to pathways of defective factor XII and defective SERPING1 which cause hereditary angioedema [46], CD22 (receptor predominantly restricted to B cells) mediated BCR (B-cell receptors) regulation, and recycling of bile acids and salts [47].
Group B (fraction 6) proteins mapped to 12 pathways (Table 1) that included terminal pathway of complement, regulation of Complement cascade, and complement cascade. The other pathways associated with group B proteins include response to elevated platelet cytosolic Ca2+, regulation of Insulin-like Growth Factor (IGF) transport and uptake by Insulin-like Growth Factor Binding Proteins (IGFBPs), platelet degranulation, dissolution of Fibrin Clot, post-translational protein phosphorylation, semaphorin interactions, sema4D induced cell migration and growth-cone collapse, RHO GTPases activate CIT, and Common Pathway of Fibrin Clot Formation.
Finally, group C (pooled fraction 7 and 8) proteins mapped to 17 pathways (Table 1) associated with signaling, extracellular matrix (ECM), and cell adhesion functions. The signaling related pathways include the NGF (nerve growth factor) activated Trk (neurotrophin receptor), netrin-1 signaling which leads to release of intracellular calcium [48] and signaling by PDGF (platelet-derived growth factor) associated with cellular immune response [49]. The ECM related pathways include multiple collagen related functions including collagen chain trimerization, biosynthesis, formation, assembly, crosslinking, and degradation. The other ECM functions that mapped group C proteins include anchoring fibril formation, degradation of ECM, and laminin interactions.
3.2. rIxsCRT and Complement Protein Interactions Validated by Western Blotting and ELISA Analyses
Relative abundance analysis revealed that complement proteins co-precipitated at high levels in presence of rIxsCRT as revealed by normalized spectral abundance (Figure 4A,B). To validate these findings, all DiffPOP fractions (Figure 2) were subjected to western blotting analysis using antibodies to complement proteins including C1q, C1r, C1s, C3 (inactive and activated), C5, C9, and C5b-9 (or membrane attack complex). This analysis confirmed our differential precipitation and LC-MS/MS analyses data (Figure 3 and Figure 4) as indicated by high protein band intensity of complement proteins that co-precipitated with rIxsCRT compared to NHS only (red arrowhead marked in Figure 5A-L). Our data also show rIxsCRT bound all three subunits of the complement complex 1 (C1q, C1r, and C1s) along with the C3, C5, and C9 (Figure 5A–L).
To definitively ascertain the binding of complement proteins to rIxsCRT, we employed Dynabeads™ His-Tag magnetic beads for the pull-down of rIxsCRT and plasma protein complexes (Figure 6). Subsequently, these complexes underwent the standard western blotting analysis using antibodies similar to those utilized in Figure 5. Our rigorous analysis unequivocally validated the association of complement C1 complex proteins (C1q, C1r, and C1s), as well as intermediate complement proteins C3, C5, and C9, with rIxsCRT. Specifically, distinct signals were detected in the rIxsCRT and NHS pull-down samples, while being conspicuously absent in the NHS-only control (Figure 6). These findings provide robust evidence of the direct interaction between rIxsCRT and key components of the host complement system, further elucidating the molecular mechanisms underlying tick-host interactions.
To further validate differential fractionation and pull-down data (Figure 5 and Figure 6), conventional ELISA was used to confirm rIxsCRT binding of complement proteins. We coated ELISA plates with rIxsCRT and then incubated with NHS. Uncoated wells were used as negative control. As shown, we observed significantly high antibody binding in wells that were coated with rIxsCRT than uncoated control wells. Our data confirm that rIxsCRT bound the C1 complex proteins (C1q, C1r and C1s) and intermediate complement proteins, C3, and C5 but not activated C3, and the C5b-C9 (or membrane attack complex) (Figure 7).
3.4. rIxsCRT Moderately Inhibits Membrane Attack Complex (MAC) Deposition via Lectin Complement Cascade but Does Not Protect B. burgdorferi from Complement Killing
Prompted by findings that rIxsCRT bound complement proteins, we investigated the effect of this protein on activation of the complement system using the WIESLAB® complement system kit, which allows for independent assessment of the three complement activation pathways. Our data show that rIxsCRT inhibited deposition of the complement membrane attack complex (MAC) by 40% via the Mannan-Binding Lectin complement activation pathway but not via the classical and alternative complement activation pathways (Figure 8). We next evaluated if rIxsCRT could protect complement sensitive B. burgdorferi strain (B314/pBBE22luc) against killing by complement. Our data showed that pre-incubating serum with rIxsCRT did not protect complement sensitive strain from killing by complement (not shown).
Prompted by findings in our differential precipitation analysis that rIxsCRT interacted with blood clotting factor XII and fibrin clot formation pathways, we investigated the effect of this protein on blood clotting using the recalcification time assay. This analysis showed that rIxsCRT did not affect blood clotting (not shown).
3.5. rIxsCRT Promotes Borrelia burgdorferi Growth in Culture
Next, we have investigated the effects of rIxsCRT on growth of B. burgdorferi in culture. In this study we have observed that the B. burgdorferi (105 cells per mL) grown with rIxsCRT showed significant elevation in the growth rate of B. burgdorferi (Figure 9). The spirochetes were quantified by Petroff-Hausser Chamber and processed for genomic DNA extraction for FlaB gene quantification by qPCR. Both of these analyses showed that rIxsCRT promoted B. burgdorferi growth in culture by more than 10 folds at its log phase (Figure 9A,B).
3.6. B. burgdorferi Infected I. scapularis Feeding Elicit High IgG Level to rIxsCRT in Rabbits
In an earlier study, we identified rIxsCRT as one of the tick saliva proteins that were abundantly secreted by B. burgdorferi-infected I. scapularis nymphs [17]. Consistent with the dynamic secretion pattern of rIxsCRT protein, rabbits that were fed upon by B. burgdorferi-infected ticks exhibited significantly higher levels of IgG antibodies reacting to rIxsCRT compared to rabbits fed upon by uninfected ticks (Figure 10A-D).
4. Discussion
Calreticulin (CRT) is a well-known protein which was originally identified as calcium ion binding protein localized to the sarcoplasmic reticulum in rabbits [50]. It was originally believed to retain in endoplasmic reticulum and functions as molecular chaperon and Ca2+ homeostasis [51]. However, in ixodid ticks it was surprisingly found to lack endoplasmic reticulum (ER) retention signal (KDEL) [23] and it has been confirmed to be injected into the host during tick feeding [17] and it is currently used as the biomarker for tick bites [18]. The findings in this study elucidates the role of CRT in tick infestation dynamics. We demonstrate that rabbits infested only once against both uninfected or B. burgdorferi infected I. scapularis nymph feeding exhibited elevated antibody titers against recombinant I. scapularis calreticulin (rIxsCRT), affirming its potential as a reliable marker for tick bites. These findings not only underscore the multifaceted functions of CRT but also highlight its practical application in diagnosing tick infestations.
Studies in mammals have reported a plethora of CRT functions including calcium storage, cell proliferation, ion binding, apoptosis, and cell differentiation [51]. Likewise, other parasite CRT was linked to parasite evasion of the complement through sequestration of complement proteins and blocking the blood clotting system through binding of blood clotting factor Xa [52]. While further validation studies are needed, our differential precipitation and LC-MS/MS analyses data showing rIxsCRT interacted with multiple pathways indicate that tick CRT is likely involved with regulating several functions at the tick feeding site. Consistent with published studies that reported tick and other parasite CRT interactions with complement and blood clotting pathway proteins [53,54], rIxsCRT differentially co-precipitated with proteins associated these two pathways. Since the tick must evade both complement and blood clotting host defenses to successfully feed and transmit tick borne disease pathogens [55], the findings in this study suggest a role for IxsCRT in mediating tick evasion of host defense functions.
Evasion of the complement system by B. burgdorferi is critical in the pathogenesis of this spirochete [56], and thus we were curious to further explore interactions between rIxsCRT and complement proteins. Published studies have reported that tick CRT could bind to C1q, one of the 3 subunits (C1q, C1 and C1s) of the C1 complex[27,31]. This study has provided new insights into functions of tick CRT by showing that rIxsCRT binds to all three C1 complex complement proteins (C1q, C1r, and C1s) and to multiple intermediate complement proteins including C3, C5, C6, C7, C8, C9, complement factor I, and complement factor H. Further, our pull-down and ELISA analyses data demonstrates that although rIxsCRT directly binds C1 complex proteins, intermediate, and late complement proteins, it does not bind the activated C5b-9 or the membrane attack complex (MAC).
Complement cascades are activated via 3 different pathways, classical, alternative, and mannose-binding lectin (MBL). Activation of the classical complement pathway starts with C1q binding onto microbe surface of antigen which in turn activates 2 subunits of C1 complex (C1r and C1s) [28,57]. Likewise, the MBL pathway is activated by the binding of MBL and ficolins to microbial surface oligosaccharides on microbe surfaces [58]. Lastly, the alternative pathway is activated by spontaneous hydrolysis of the internal C3 thioester bond and binding of properdin to proteins, lipids, and carbohydrate on microbe surfaces [59]. The activation of the 3 pathways culminates in activation of the C3 convertase which leads to C5 convertase, which reacts with complement proteins C6, C7, C8, and C9 to form C5b-9 or MAC. It is notable that despite binding of the C1 complex proteins, C3, C5, and C9, rIxsCRT moderately blocked MAC deposition via the MBL pathway but not via the classical and alternative pathways. In our study we have noticed an increase in the MAC deposition in presence of rIxsCRT in classical and alternate pathway, which is consistent with our previous study that showed that increasing amounts of recombinant A. americanum tick CRT led to deposition of high amounts of MAC [31]. The fact that tick CRT binds complement proteins but moderately affects MAC deposition via the MBL pathway but not via the 2 other pathways is interesting and warrants additional investigations. The complement sensitive strain of B. burgdorferi (B314/pBBE22luc) has been utilized to study inhibitors of the complement system in B. burgdorferi pathogenesis [37,60]. Our findings that rIxsCRT did not rescue complement sensitive strain from complement killing may be explained by the fact that rIxsCRT had modest impact in the MBL pathway and no effect in classical and alternate pathway. We speculate that the protective effects of IxsCRT against complement killing of B. burgdorferi nullified by its lack of effect against classical and alternative activation pathways of complement despite reducing MAC deposition via the MBL pathway.
We have previously shown that B. burgdorferi infected I. scapularis nymphs highly secrete IxsCRT during the 48-72 feeding time points aligning with high B. burgdorferi transmission events after the tick has fed for more than 48 h [15,17]. This suggested that B. burgdorferi might directly interact with IxsCRT to promote its colonization of the host. It is interesting to note that supplementing the BSK-II media with rIxsCRT enhanced the growth pattern of B. burgdorferi under in vitro conditions. We speculate that exposure to IxsCRT triggers the rapid growth of B. burgdorferi at the site of its transmission and thus facilitating the establishment of local dermal infection that precedes colonization of secondary organs [61].
This study adds to our knowledge and raises questions warranting further investigations on the role of tick CRT in feeding and transmission of B. burgdorferi by ticks. It is particularly interesting that although IxsCRT bound the C1 complex complement proteins, it did not block deposition of the MAC via the classical pathway. We speculate that in the complex environment of host skin, the IxsCRT protein may serve as decoy that deviates the activation surface of complement from B. burgdorferi to IxsCRT. Our DiffPOP data revealed that, rIxsCRT interacted with extracellular matrix proteins such as collagen. It is possible that at the tick feeding site, native CRT bound to exposed extracellular matrix proteins could serve as the site of complement activation and in the process deviating complement attack of transmitted B. burgdorferi.
Supplementary Materials
The following supporting information can be downloaded at: www.mdpi.com/xxx/s1, Supplementary Table S1: Precipitation Buffer Volume for Differential Fractionation. This table provides details on the amount of precipitation buffer (in mL) added to the reaction to obtain the precipitate for the differential fractionation process. Supplementary Table S2: LC-MS/MS Analysis of Fractions 1-5. This table presents the results of LC-MS/MS analysis conducted on fractions 1-5 obtained from the differential precipitation assay of rIxsCRT with normal human plasma. The data is provided in Excel format. Supplementary Table S3: LC-MS/MS Analysis of Fraction 6. This table displays the results of LC-MS/MS analysis performed on fraction 6 obtained from the differential precipitation assay of rIxsCRT with normal human plasma. The data is presented in Excel format. Supplementary Table S4: Reactome Analysis of Complete LCMS/MS Data. This table presents the results of Reactome analysis conducted on the complete LCMS/MS dataset obtained from the differential precipitation assay of rIxsCRT with normal human plasma. The data is provided in Excel format.
Author Contributions
Conceived and designed the experiment: AM, MAA. Performed the experiment: MAA, TTN, THK. Analyzed the data: AM, MAA, TNN, THK, TKK. Wrote the paper: MAA, AM.
Funding
This research was supported by National Institutes of Health grants (AI093858, AI074789, AI138129, and AI119873) to AM.
Acknowledgments
The author would like to thank Drs. Jon T Skare and Alexandra D Powell-Pierce for providing the B. burgdorferi strains and sharing their complement sensitivity assay protocols. We are grateful to Dr. Klaudia Kocurek for helping us to acquire LC-MS/MS data with assistance of the Mass Spectrometry Facility at the Department of Chemistry.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Sosa, J.P.; Ferreira Caceres, M.M.; Agadi, K.; Pandav, K.; Mehendale, M.; Mehta, J.M.; Go, C.C.; Matos, W.F.; Guntipalli, P.; Belizaire, M.E. Diseases Transmitted by the Black-Legged Ticks in the United States: A Comprehensive Review of the Literature. Cureus 2021, 13, e17526. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.J.; Watkins, H.R.; Schwan, W.R. Ixodes scapularis: Vector to an Increasing Diversity of Human Pathogens in the Upper Midwest. WMJ 2020, 119, 16–21. [Google Scholar] [PubMed]
- Guzman, N.; Yarrarapu, S.N.S.; Beidas, S.O. Anaplasma Phagocytophilum. In StatPearls; Treasure Island (FL) ineligible companies. Disclosure: Siva Naga Yarrarapu declares no relevant financial relationships with ineligible companies. Disclosure: Sary Beidas declares no relevant financial relationships with ineligible companies, 2023. [Google Scholar]
- Krause, P.J. Human babesiosis. Int J Parasitol 2019, 49, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Piantadosi, A.; Solomon, I.H. Powassan Virus Encephalitis. Infect Dis Clin North Am 2022, 36, 671–688. [Google Scholar] [CrossRef] [PubMed]
- Wikel, S.K. Ticks and Tick-Borne Infections: Complex Ecology, Agents, and Host Interactions. Vet Sci 2018, 5. [Google Scholar] [CrossRef]
- Pritt, B.S.; Fernholz, E.C.; Replogle, A.J.; Kingry, L.C.; Sciotto, M.P.; Petersen, J.M. Borrelia mayonii - A cause of Lyme borreliosis that can be visualized by microscopy of thin blood films. Clin Microbiol Infect 2022, 28, 823–824. [Google Scholar] [CrossRef] [PubMed]
- Pritt, B.S.; Respicio-Kingry, L.B.; Sloan, L.M.; Schriefer, M.E.; Replogle, A.J.; Bjork, J.; Liu, G.; Kingry, L.C.; Mead, P.S.; Neitzel, D.F.; et al. Borrelia mayonii sp. nov., a member of the Borrelia burgdorferi sensu lato complex, detected in patients and ticks in the upper midwestern United States. Int J Syst Evol Microbiol 2016, 66, 4878–4880. [Google Scholar] [CrossRef]
- Stevenson, B. The Lyme disease spirochete, Borrelia burgdorferi, as a model vector-borne pathogen: insights on regulation of gene and protein expression. Curr Opin Microbiol 2023, 74, 102332. [Google Scholar] [CrossRef] [PubMed]
- Hook, S.A.; Jeon, S.; Niesobecki, S.A.; Hansen, A.P.; Meek, J.I.; Bjork, J.K.H.; Dorr, F.M.; Rutz, H.J.; Feldman, K.A.; White, J.L.; et al. Economic Burden of Reported Lyme Disease in High-Incidence Areas, United States, 2014-2016. Emerg Infect Dis 2022, 28, 1170–1179. [Google Scholar] [CrossRef]
- Obaid, M.K.; Islam, N.; Alouffi, A.; Khan, A.Z.; da Silva Vaz, I., Jr.; Tanaka, T.; Ali, A. Acaricides Resistance in Ticks: Selection, Diagnosis, Mechanisms, and Mitigation. Front Cell Infect Microbiol 2022, 12, 941831. [Google Scholar] [CrossRef]
- Nauen, R.; Stumpf, N.; Elbert, A.; Zebitz, C.P.; Kraus, W. Acaricide toxicity and resistance in larvae of different strains of Tetranychus urticae and Panonychus ulmi (Acari: Tetranychidae). Pest Manag Sci 2001, 57, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Paucar-Quishpe, V.; Perez-Otanez, X.; Rodriguez-Hidalgo, R.; Cepeda-Bastidas, D.; Perez-Escalante, C.; Grijalva-Olmedo, J.; Enriquez, S.; Arciniegas-Ortega, S.; Sandoval-Travez, L.; Benavides-Erazo, B.; et al. An economic evaluation of cattle tick acaricide-resistances and the financial losses in subtropical dairy farms of Ecuador: A farm system approach. PLoS One 2023, 18, e0287104. [Google Scholar] [CrossRef] [PubMed]
- Trager, W. Acquired Immunity to Ticks. The Journal of Parasitology 1939, 25, 57–81. [Google Scholar] [CrossRef]
- Kim, T.K.; Tirloni, L.; Bencosme-Cuevas, E.; Kim, T.H.; Diedrich, J.K.; Yates, J.R., 3rd; Mulenga, A. Borrelia burgdorferi infection modifies protein content in saliva of Ixodes scapularis nymphs. BMC Genomics 2021, 22, 152. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.K.; Tirloni, L.; Pinto, A.F.M.; Diedrich, J.K.; Moresco, J.J.; Yates, J.R., 3rd; da Silva Vaz, I., Jr.; Mulenga, A. Time-resolved proteomic profile of Amblyomma americanum tick saliva during feeding. PLoS Negl Trop Dis 2020, 14, e0007758. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.K.; Tirloni, L.; Pinto, A.F.; Moresco, J.; Yates, J.R., 3rd; da Silva Vaz, I., Jr.; Mulenga, A. Ixodes scapularis Tick Saliva Proteins Sequentially Secreted Every 24 h during Blood Feeding. PLoS Negl Trop Dis 2016, 10, e0004323. [Google Scholar] [CrossRef] [PubMed]
- Alarcon-Chaidez, F.; Ryan, R.; Wikel, S.; Dardick, K.; Lawler, C.; Foppa, I.M.; Tomas, P.; Cushman, A.; Hsieh, A.; Spielman, A.; et al. Confirmation of tick bite by detection of antibody to Ixodes calreticulin salivary protein. Clin Vaccine Immunol 2006, 13, 1217–1222. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, C.A.; Da Silva Vaz, I.; da Silva, S.S.; Haag, K.L.; Valenzuela, J.G.; Masuda, A. Cloning and partial characterization of a Boophilus microplus (Acari: Ixodidae) calreticulin. Exp Parasitol 2002, 101, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Jaworski, D.C. , Simmen, F.A., Lamoreaux, W., Coons, L.B., Muller, M.T., Needham, G.R. A secreted calreticulin protein in ixodid tick (Amblyomma americanum) saliva. Journal of Insect Physiology 1995, 41, 369–375. [Google Scholar] [CrossRef]
- Oladiran, A.; Belosevic, M. Trypanosoma carassii calreticulin binds host complement component C1q and inhibits classical complement pathway-mediated lysis. Dev Comp Immunol 2010, 34, 396–405. [Google Scholar] [CrossRef]
- Lu, Y.C.; Weng, W.C.; Lee, H. Functional roles of calreticulin in cancer biology. Biomed Res Int 2015, 2015, 526524. [Google Scholar] [CrossRef]
- Deborah, C. Jaworski, F.A.S., William Lamoreaux, Lewis B.Coons, Mark T.Muller, Glen R.Needham. A secreted calreticulin protein in ixodid tick (Amblyomma americanum) saliva. Journal of Insect Physiology 1995, 41, 369–375. [Google Scholar] [CrossRef]
- Suchitra, S.; Joshi, P. Characterization of Haemonchus contortus calreticulin suggests its role in feeding and immune evasion by the parasite. Biochim Biophys Acta 2005, 1722, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Suchitra, S.; Anbu, K.A.; Rathore, D.K.; Mahawar, M.; Singh, B.P.; Joshi, P. Haemonchus contortus calreticulin binds to C-reactive protein of its host, a novel survival strategy of the parasite. Parasite Immunol 2008, 30, 371–374. [Google Scholar] [CrossRef] [PubMed]
- Sanders, M.L.; Glass, G.E.; Nadelman, R.B.; Wormser, G.P.; Scott, A.L.; Raha, S.; Ritchie, B.C.; Jaworski, D.C.; Schwartz, B.S. Antibody levels to recombinant tick calreticulin increase in humans after exposure to Ixodes scapularis (Say) and are correlated with tick engorgement indices. Am J Epidemiol 1999, 149, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Sanders, M.L.; Jaworski, D.C.; Sanchez, J.L.; DeFraites, R.F.; Glass, G.E.; Scott, A.L.; Raha, S.; Ritchie, B.C.; Needham, G.R.; Schwartz, B.S. Antibody to a cDNA-derived calreticulin protein from Amblyomma americanum as a biomarker of tick exposure in humans. Am J Trop Med Hyg 1998, 59, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, V.; Valck, C.; Sanchez, G.; Gingras, A.; Tzima, S.; Molina, M.C.; Sim, R.; Schwaeble, W.; Ferreira, A. The classical activation pathway of the human complement system is specifically inhibited by calreticulin from Trypanosoma cruzi. J Immunol 2004, 172, 3042–3050. [Google Scholar] [CrossRef] [PubMed]
- Vaithilingam, A.; Teixeira, J.E.; Miller, P.J.; Heron, B.T.; Huston, C.D. Entamoeba histolytica cell surface calreticulin binds human c1q and functions in amebic phagocytosis of host cells. Infect Immun 2012, 80, 2008–2018. [Google Scholar] [CrossRef] [PubMed]
- Radulovic, Z.M.; Kim, T.K.; Porter, L.M.; Sze, S.H.; Lewis, L.; Mulenga, A. A 24-48 h fed Amblyomma americanum tick saliva immuno-proteome. BMC Genomics 2014, 15, 518. [Google Scholar] [CrossRef]
- Kim, T.K.; Ibelli, A.M.; Mulenga, A. Amblyomma americanum tick calreticulin binds C1q but does not inhibit activation of the classical complement cascade. Ticks Tick Borne Dis 2015, 6, 91–101. [Google Scholar] [CrossRef]
- Mulenga, A.; Kim, T.K.; Ibelli, A.M. Deorphanization and target validation of cross-tick species conserved novel Amblyomma americanum tick saliva protein. Int J Parasitol 2013, 43, 439–451. [Google Scholar] [CrossRef] [PubMed]
- Ryan, B.J.; Kinsella, G.K. Differential Precipitation and Solubilization of Proteins. Methods Mol Biol 2017, 1485, 191–208. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.; Kim, T.H.; Bencosme-Cuevas, E.; Berry, J.; Gaithuma, A.S.K.; Ansari, M.A.; Kim, T.K.; Tirloni, L.; Radulovic, Z.; Moresco, J.J.; et al. A tick saliva serpin, IxsS17 inhibits host innate immune system proteases and enhances host colonization by Lyme disease agent. PLoS Pathog 2024, 20, e1012032. [Google Scholar] [CrossRef] [PubMed]
- Croft, D.; O’Kelly, G.; Wu, G.; Haw, R.; Gillespie, M.; Matthews, L.; Caudy, M.; Garapati, P.; Gopinath, G.; Jassal, B.; et al. Reactome: a database of reactions, pathways and biological processes. Nucleic Acids Res 2011, 39, D691–697. [Google Scholar] [CrossRef] [PubMed]
- Bencosme-Cuevas, E.; Kim, T.K.; Nguyen, T.T.; Berry, J.; Li, J.; Adams, L.G.; Smith, L.A.; Batool, S.A.; Swale, D.R.; Kaufmann, S.H.E.; et al. Ixodes scapularis nymph saliva protein blocks host inflammation and complement-mediated killing of Lyme disease agent, Borrelia burgdorferi. Front Cell Infect Microbiol 2023, 13, 1253670. [Google Scholar] [CrossRef] [PubMed]
- Garcia, B.L.; Zhi, H.; Wager, B.; Hook, M.; Skare, J.T. Borrelia burgdorferi BBK32 Inhibits the Classical Pathway by Blocking Activation of the C1 Complement Complex. PLoS Pathog 2016, 12, e1005404. [Google Scholar] [CrossRef]
- Gulliani, G.L.; Hyun, B.H.; Litten, M.B. Blood recalcification time. A simple and reliable test to monitor heparin therapy. Am J Clin Pathol 1976, 65, 390–396. [Google Scholar] [CrossRef] [PubMed]
- Fanning, J.P.; Dubeau, A.M. The whole blood recalcification clotting time. A suggested simple and reliable method for monitoring heparin therapy. J Maine Med Assoc 1974, 65, 211–213. [Google Scholar] [PubMed]
- Pahl, A.; Kuhlbrandt, U.; Brune, K.; Rollinghoff, M.; Gessner, A. Quantitative detection of Borrelia burgdorferi by real-time PCR. J Clin Microbiol 1999, 37, 1958–1963. [Google Scholar] [CrossRef]
- Richard, G. Autosomal Recessive Congenital Ichthyosis. In GeneReviews((R)), Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; Seattle (WA), 1993.
- Akiyama, M. ABCA12 mutations and autosomal recessive congenital ichthyosis: a review of genotype/phenotype correlations and of pathogenetic concepts. Hum Mutat 2010, 31, 1090–1096. [Google Scholar] [CrossRef]
- Akiyama, M. The roles of ABCA12 in epidermal lipid barrier formation and keratinocyte differentiation. Biochim Biophys Acta 2014, 1841, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, M.; Sugiyama-Nakagiri, Y.; Sakai, K.; McMillan, J.R.; Goto, M.; Arita, K.; Tsuji-Abe, Y.; Tabata, N.; Matsuoka, K.; Sasaki, R.; et al. Mutations in lipid transporter ABCA12 in harlequin ichthyosis and functional recovery by corrective gene transfer. J Clin Invest 2005, 115, 1777–1784. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Li, Y.; Song, Y.; Zhou, D.; Jia, S.; Xu, A.; Zhang, W.; You, H.; Jia, J.; Huang, J.; et al. A recurrent ABCC2 p.G693R mutation resulting in loss of function of MRP2 and hyperbilirubinemia in Dubin-Johnson syndrome in China. Orphanet J Rare Dis 2020, 15, 74. [Google Scholar] [CrossRef] [PubMed]
- Deroux, A.; Boccon-Gibod, I.; Fain, O.; Pralong, P.; Ollivier, Y.; Pagnier, A.; Djenouhat, K.; Du-Thanh, A.; Gompel, A.; Faisant, C.; et al. Hereditary angioedema with normal C1 inhibitor and factor XII mutation: a series of 57 patients from the French National Center of Reference for Angioedema. Clin Exp Immunol 2016, 185, 332–337. [Google Scholar] [CrossRef]
- Clark, E.A.; Giltiay, N.V. CD22: A Regulator of Innate and Adaptive B Cell Responses and Autoimmunity. Front Immunol 2018, 9, 2235. [Google Scholar] [CrossRef] [PubMed]
- Hong, K.; Nishiyama, M.; Henley, J.; Tessier-Lavigne, M.; Poo, M. Calcium signalling in the guidance of nerve growth by netrin-1. Nature 2000, 403, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Z.; Wang, Q.; Li, W.; Huang, L.; Zhang, J.; Zhu, J.; Xie, B.; Wang, S.; Kuang, H.; Lin, X.; et al. Platelet-Derived Growth Factor-D Activates Complement System to Propagate Macrophage Polarization and Neovascularization. Front Cell Dev Biol 2021, 9, 686886. [Google Scholar] [CrossRef] [PubMed]
- Ostwald, T.J.; MacLennan, D.H. Isolation of a high affinity calcium-binding protein from sarcoplasmic reticulum. J Biol Chem 1974, 249, 974–979. [Google Scholar] [CrossRef] [PubMed]
- Michalak, M.; Corbett, E.F.; Mesaeli, N.; Nakamura, K.; Opas, M. Calreticulin: one protein, one gene, many functions. Biochem J 1999, 344 Pt 2, 281–292. [Google Scholar] [CrossRef]
- Kuwabara, K.; Pinsky, D.J.; Schmidt, A.M.; Benedict, C.; Brett, J.; Ogawa, S.; Broekman, M.J.; Marcus, A.J.; Sciacca, R.R.; Michalak, M.; et al. Calreticulin, an antithrombotic agent which binds to vitamin K-dependent coagulation factors, stimulates endothelial nitric oxide production, and limits thrombosis in canine coronary arteries. J Biol Chem 1995, 270, 8179–8187. [Google Scholar] [CrossRef]
- Ramirez-Toloza, G.; Aguilar-Guzman, L.; Valck, C.; Ferreira, V.P.; Ferreira, A. The Interactions of Parasite Calreticulin With Initial Complement Components: Consequences in Immunity and Virulence. Front Immunol 2020, 11, 1561. [Google Scholar] [CrossRef] [PubMed]
- Esperante, D.; Flisser, A.; Mendlovic, F. The many faces of parasite calreticulin. Front Immunol 2023, 14, 1101390. [Google Scholar] [CrossRef] [PubMed]
- Simo, L.; Kazimirova, M.; Richardson, J.; Bonnet, S.I. The Essential Role of Tick Salivary Glands and Saliva in Tick Feeding and Pathogen Transmission. Front Cell Infect Microbiol 2017, 7, 281. [Google Scholar] [CrossRef] [PubMed]
- Skare, J.T.; Garcia, B.L. Complement Evasion by Lyme Disease Spirochetes. Trends Microbiol 2020, 28, 889–899. [Google Scholar] [CrossRef] [PubMed]
- Dunkelberger, J.R.; Song, W.C. Complement and its role in innate and adaptive immune responses. Cell Res 2010, 20, 34–50. [Google Scholar] [CrossRef] [PubMed]
- Beltrame, M.H.; Catarino, S.J.; Goeldner, I.; Boldt, A.B.; de Messias-Reason, I.J. The lectin pathway of complement and rheumatic heart disease. Front Pediatr 2014, 2, 148. [Google Scholar] [CrossRef]
- Harboe, M.; Mollnes, T.E. The alternative complement pathway revisited. J Cell Mol Med 2008, 12, 1074–1084. [Google Scholar] [CrossRef] [PubMed]
- van Dam, A.P.; Oei, A.; Jaspars, R.; Fijen, C.; Wilske, B.; Spanjaard, L.; Dankert, J. Complement-mediated serum sensitivity among spirochetes that cause Lyme disease. Infect Immun 1997, 65, 1228–1236. [Google Scholar] [CrossRef]
- Hyde, J.A. Borrelia burgdorferi Keeps Moving and Carries on: A Review of Borrelial Dissemination and Invasion. Front Immunol 2017, 8, 114. [Google Scholar] [CrossRef]
Figure 1.
Expression and affinity purification of recombinant I. scapularis calreticulin. Recombinant I. scapularis calreticulin (rIxsCRT) with His-tag was expressed in Pichia pastoris over 3 days and affinity purified under native conditions. Affinity purification of rIxsCRT was validated by standard SDS-PAGE followed by silver staining (A) and western blotting using the antibody to the Histidine fusion tag (B). Lanes 1-4 represents the 50, 75, 100 and 200 imidazole concentration (mM) at which the rIxsCRT was eluted, dialyzed in PBS, concentrated, and were used for the assays.
Figure 1.
Expression and affinity purification of recombinant I. scapularis calreticulin. Recombinant I. scapularis calreticulin (rIxsCRT) with His-tag was expressed in Pichia pastoris over 3 days and affinity purified under native conditions. Affinity purification of rIxsCRT was validated by standard SDS-PAGE followed by silver staining (A) and western blotting using the antibody to the Histidine fusion tag (B). Lanes 1-4 represents the 50, 75, 100 and 200 imidazole concentration (mM) at which the rIxsCRT was eluted, dialyzed in PBS, concentrated, and were used for the assays.
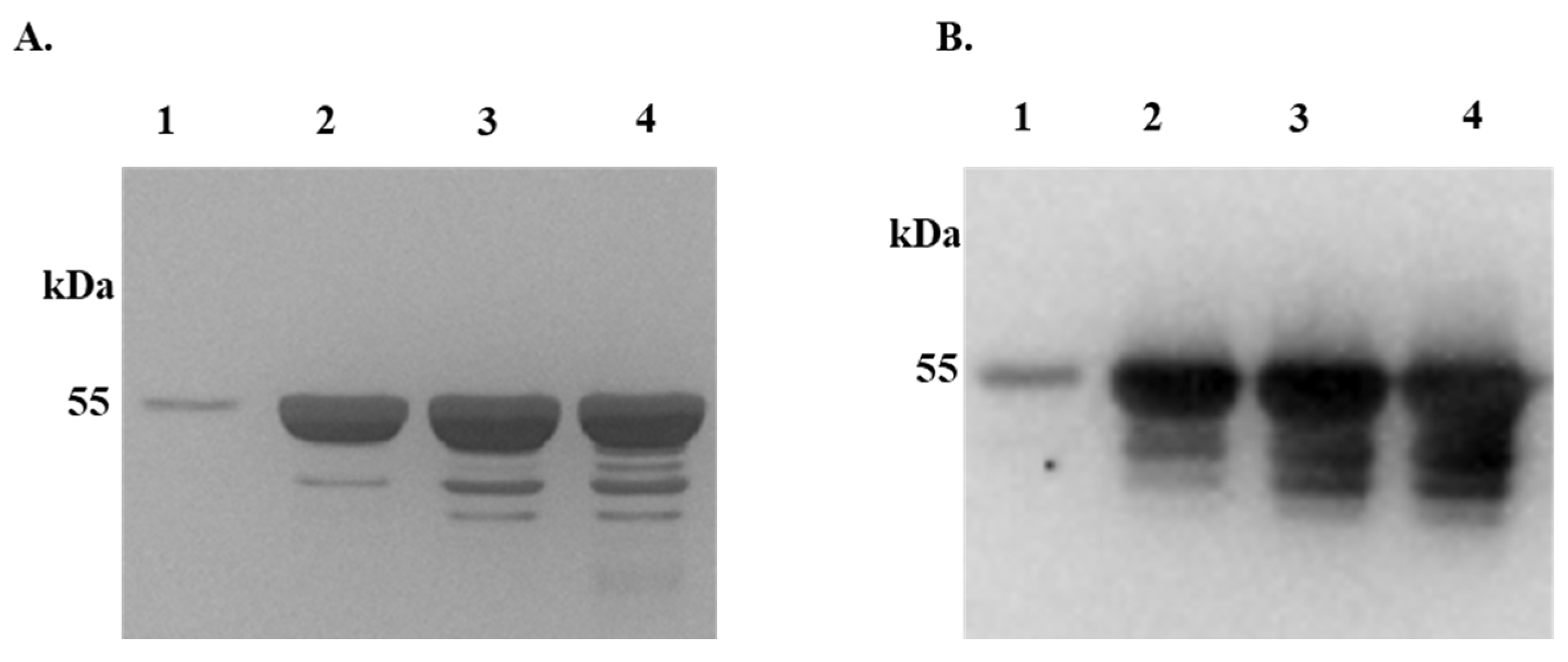
Figure 2.
Differential Precipitation of Proteins (DiffPOP) reveals interactions between rIxsCRT and complement proteins. Affinity-purified rIxsCRT (10 µg) was pre-incubated with 10% normal human serum (NHS) for 90 minutes at 37˚C, while NHS and rIxsCRT served as controls. The reactions were then stabilized and subjected to differential precipitation using escalating amount of precipitating buffer as described in the materials and methods (see supplemental Table S1). Different fractions obtained from the precipitation process were analyzed by standard SDS-PAGE and silver staining. Figure 2A shows the protein profile of NHS only, Figure 2B illustrates the profile of NHS + rIxsCRT mixture, and Figure 2C displays the profile of rIxsCRT only. Additionally, the NHS + rIxsCRT mixture was subjected to western blotting analysis using an antibody against the histidine fusion tag to track the precipitation of rIxsCRT (Figure 2D). The distinct protein profile between NHS and NHS + rIxsCRT, highlighted by the red arrowhead in Panels A and B, indicates potential interactions between rIxsCRT and specific proteins. The precipitation profile depicted in Figure 2D guided the selection of fractions for subsequent LCMS/MS analysis, shedding light on the specific complement proteins interacting with rIxsCRT. L: Ladder depicting molecular weight (kDa), Lanes 1-10 = DiffPOP fractions 1-10.
Figure 2.
Differential Precipitation of Proteins (DiffPOP) reveals interactions between rIxsCRT and complement proteins. Affinity-purified rIxsCRT (10 µg) was pre-incubated with 10% normal human serum (NHS) for 90 minutes at 37˚C, while NHS and rIxsCRT served as controls. The reactions were then stabilized and subjected to differential precipitation using escalating amount of precipitating buffer as described in the materials and methods (see supplemental Table S1). Different fractions obtained from the precipitation process were analyzed by standard SDS-PAGE and silver staining. Figure 2A shows the protein profile of NHS only, Figure 2B illustrates the profile of NHS + rIxsCRT mixture, and Figure 2C displays the profile of rIxsCRT only. Additionally, the NHS + rIxsCRT mixture was subjected to western blotting analysis using an antibody against the histidine fusion tag to track the precipitation of rIxsCRT (Figure 2D). The distinct protein profile between NHS and NHS + rIxsCRT, highlighted by the red arrowhead in Panels A and B, indicates potential interactions between rIxsCRT and specific proteins. The precipitation profile depicted in Figure 2D guided the selection of fractions for subsequent LCMS/MS analysis, shedding light on the specific complement proteins interacting with rIxsCRT. L: Ladder depicting molecular weight (kDa), Lanes 1-10 = DiffPOP fractions 1-10.
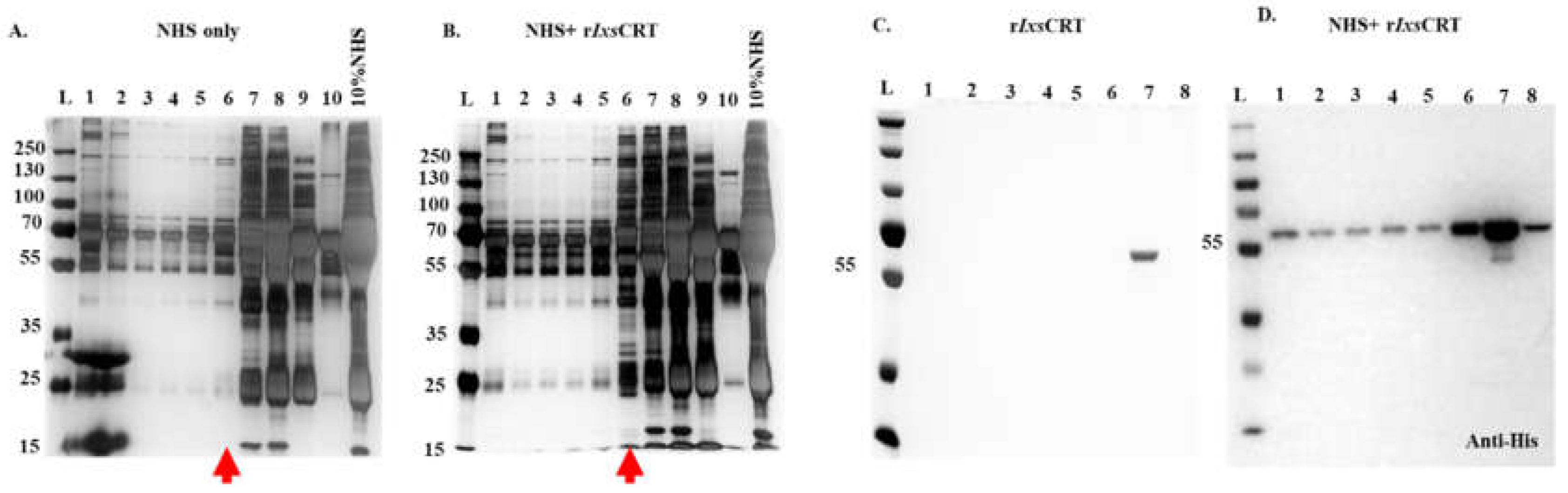
Figure 3.
Volcano plot analyses reveal human serum proteins that differentially co-precipitated with rIxsCRT. Normalized abundance values of three biological replicates were used in Proteome Discoverer™ Software (Thermo Fisher Scientific, Tx, USA) to generate the volcano plot. In volcano plot, the Y-axis shows -log10 p-value and X-axis shows magnitude of change (log2 fold change). Red dots represent proteins that co-precipitated in high amounts with rIxsCRT while green dots represent that were absent or co-precipitated in low amounts in presence of rIxsCRT. The adjusted p-value ≤ 0.05 and fold change log2 of more than 2 was used as cut off to select proteins that co-precipitated in high amounts with rIxsCRT.
Figure 3.
Volcano plot analyses reveal human serum proteins that differentially co-precipitated with rIxsCRT. Normalized abundance values of three biological replicates were used in Proteome Discoverer™ Software (Thermo Fisher Scientific, Tx, USA) to generate the volcano plot. In volcano plot, the Y-axis shows -log10 p-value and X-axis shows magnitude of change (log2 fold change). Red dots represent proteins that co-precipitated in high amounts with rIxsCRT while green dots represent that were absent or co-precipitated in low amounts in presence of rIxsCRT. The adjusted p-value ≤ 0.05 and fold change log2 of more than 2 was used as cut off to select proteins that co-precipitated in high amounts with rIxsCRT.
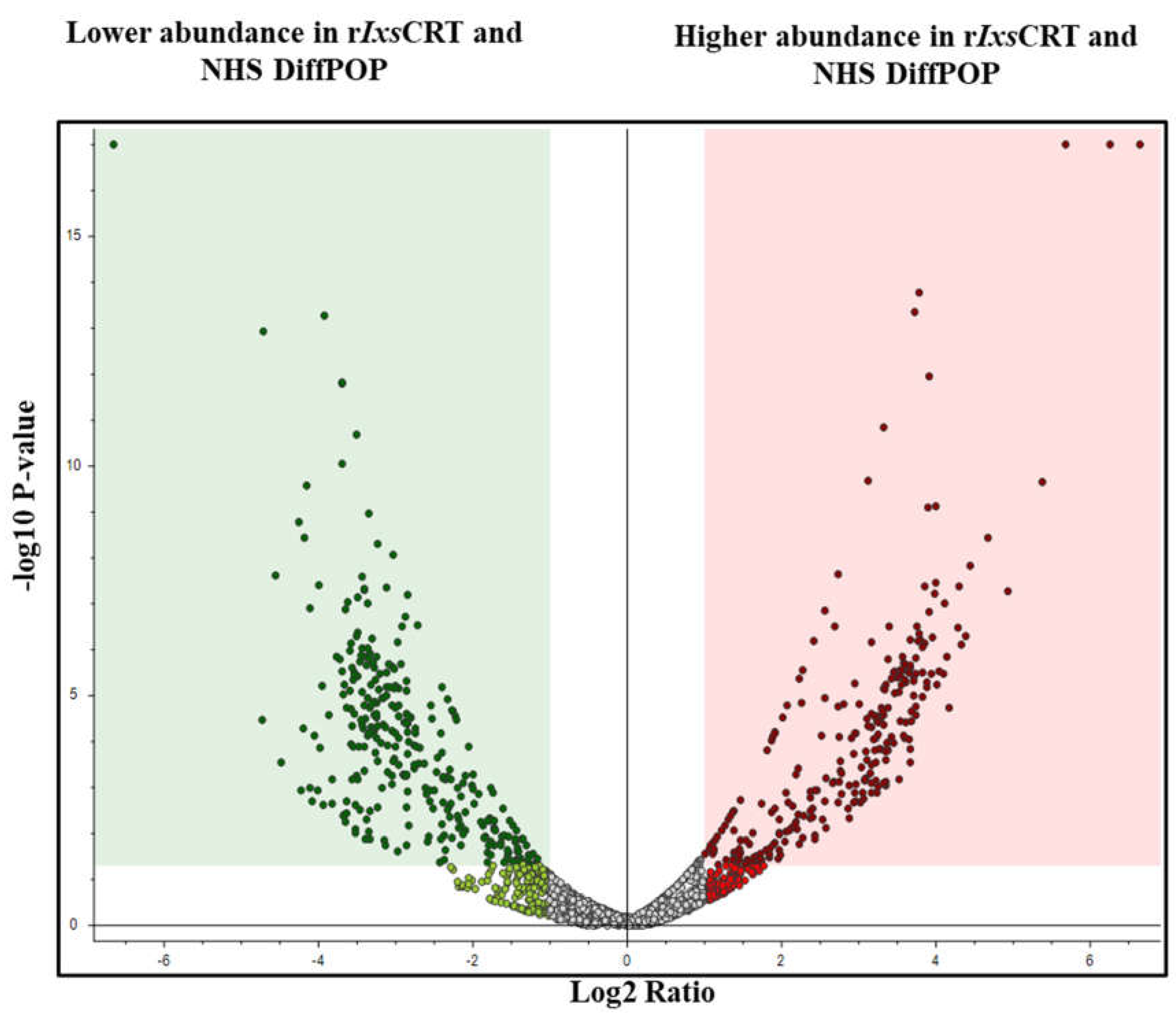
Figure 4.
Differential precipitation of complement proteins in the presence of rIxsCRT. Fractions obtained from the Differential Precipitation (DiffPOP) were pooled into group A (fractions 1-5), group B (Fraction 6), and group C (fractions 7 and 8). These fractions were subjected to Liquid Chromatography-Mass Spectrometry (LCMS/MS) analysis to investigate the abundance of complement proteins interacting with rIxsCRT. Figure 4A illustrates the abundance of complement proteins interacting with rIxsCRT in Group A, while Figure 4B depicts the corresponding interactions in Group B. The Y-axis in Figure 4A and 4B illustrates the protein abundance, indicating the levels of proteins co-precipitated with rIxsCRT (depicted by the red graph) in comparison to NHS only (represented by the blue graph).
Figure 4.
Differential precipitation of complement proteins in the presence of rIxsCRT. Fractions obtained from the Differential Precipitation (DiffPOP) were pooled into group A (fractions 1-5), group B (Fraction 6), and group C (fractions 7 and 8). These fractions were subjected to Liquid Chromatography-Mass Spectrometry (LCMS/MS) analysis to investigate the abundance of complement proteins interacting with rIxsCRT. Figure 4A illustrates the abundance of complement proteins interacting with rIxsCRT in Group A, while Figure 4B depicts the corresponding interactions in Group B. The Y-axis in Figure 4A and 4B illustrates the protein abundance, indicating the levels of proteins co-precipitated with rIxsCRT (depicted by the red graph) in comparison to NHS only (represented by the blue graph).
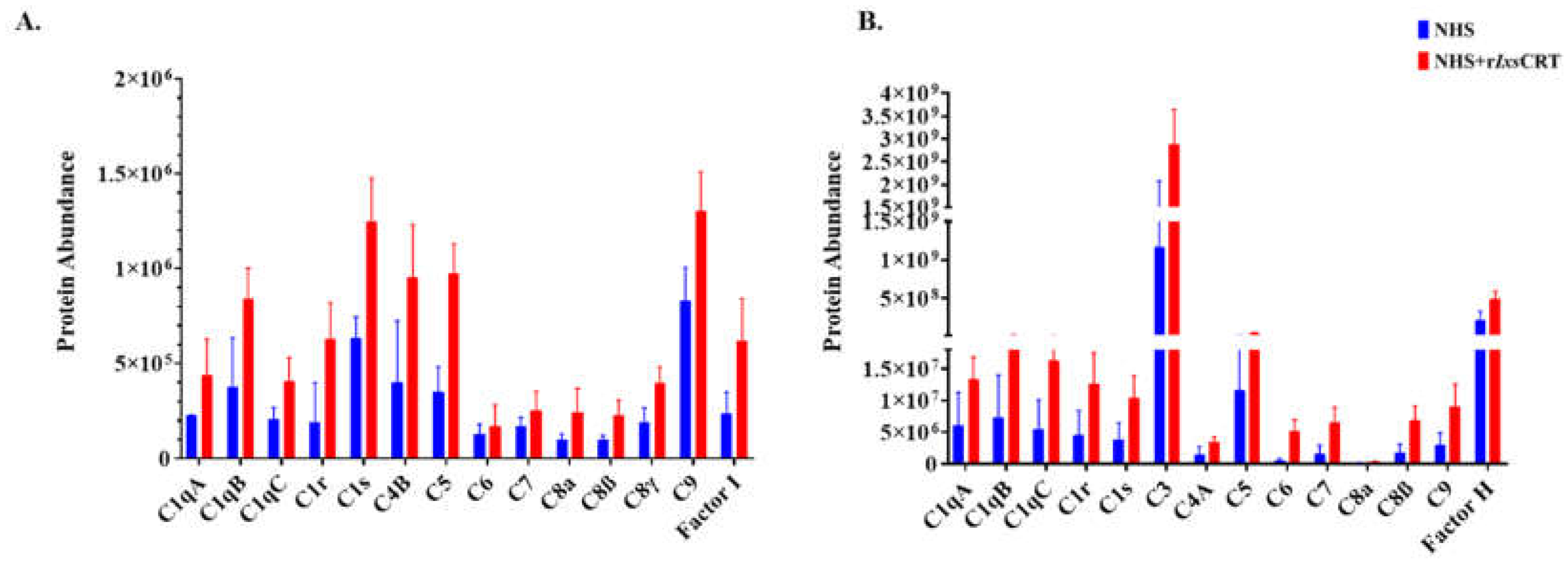
Figure 5.
Validation of complement protein and rIxsCRT interactions via western blotting analysis. To confirm the interactions observed in LCMS/MS analysis (referenced in Figure 3), DiffPOP fractions were subjected to standard western blotting using antibodies specific to complement proteins: C1q, C1r, C1s, C3, C5, and C9, as indicated. Unique binding patterns between NHS control and NHS + rIxsCRT are indicated by red arrowheads. Panels A, C, E, G, I, and K, depict immunoblots of NHS-only samples, while panels B, D, F, H, J, and L represent immunoblots of NHS + rIxsCRT samples.
Figure 5.
Validation of complement protein and rIxsCRT interactions via western blotting analysis. To confirm the interactions observed in LCMS/MS analysis (referenced in Figure 3), DiffPOP fractions were subjected to standard western blotting using antibodies specific to complement proteins: C1q, C1r, C1s, C3, C5, and C9, as indicated. Unique binding patterns between NHS control and NHS + rIxsCRT are indicated by red arrowheads. Panels A, C, E, G, I, and K, depict immunoblots of NHS-only samples, while panels B, D, F, H, J, and L represent immunoblots of NHS + rIxsCRT samples.
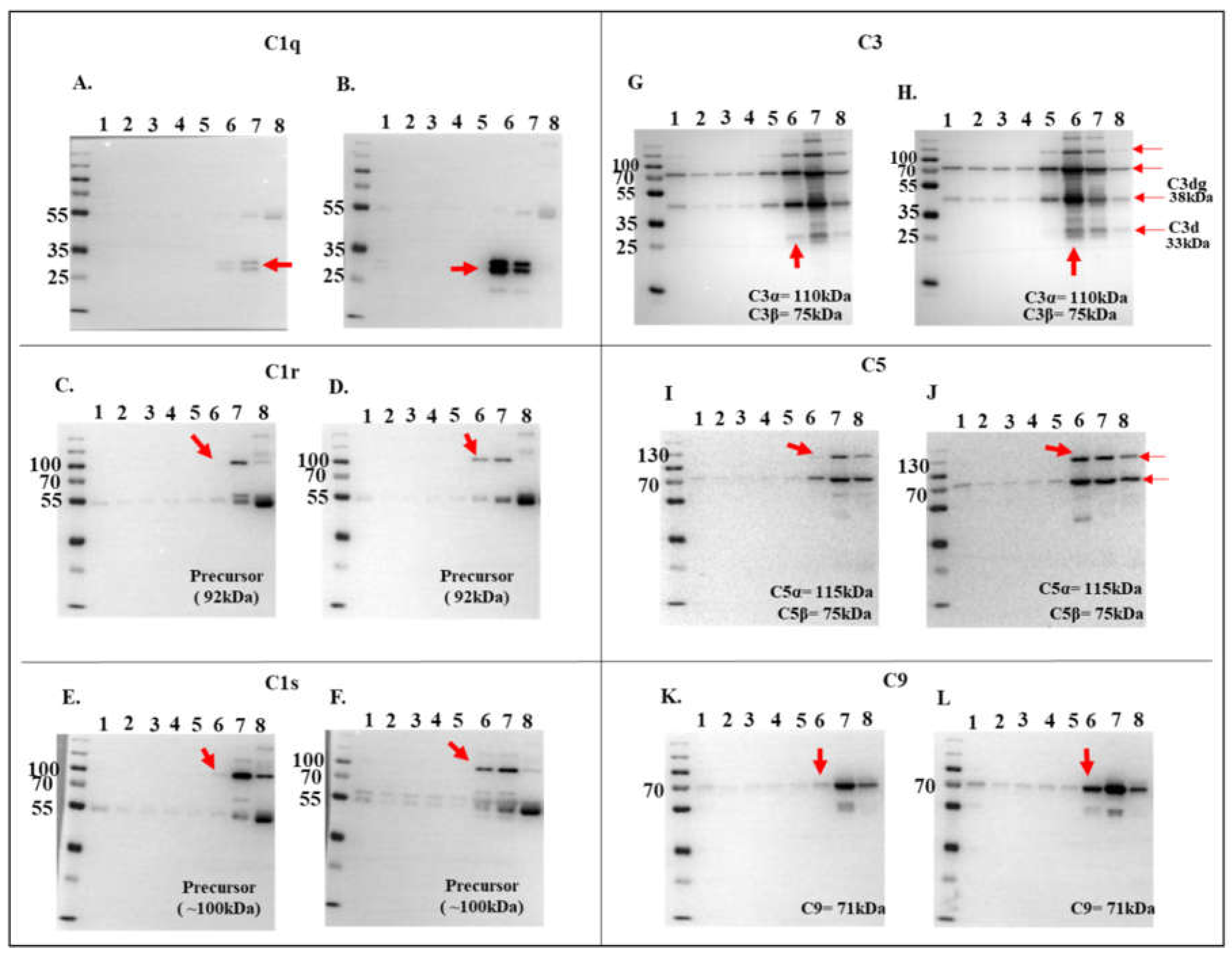
Figure 6.
Pull down assay validates rIxsCRT binding of complement proteins. Affinity purified rIxsCRT bound onto His specific magnetic beads (Dynabeads™) was used to pull down complement proteins from human complement serum (HCS). The beads were washed and the eluted protein complexes were subjected to western blotting analysis using antibodies to histidine tag (A) and complement proteins C1q (B), C1r (C), C1s (D), C3 (E), C5 (F), C9 (G). In all the panels (A-G), HCS is the human complement serum which was eluted from the empty beads i.e., without the bait protein (rIxsCRT) and HCS+rIxsCRT denotes the human complement proteins which were pulled down and eluted from the beads loaded with rIxsCRT. Red arrowheads denote the detected complement proteins at their expected molecular weight size (kDa).
Figure 6.
Pull down assay validates rIxsCRT binding of complement proteins. Affinity purified rIxsCRT bound onto His specific magnetic beads (Dynabeads™) was used to pull down complement proteins from human complement serum (HCS). The beads were washed and the eluted protein complexes were subjected to western blotting analysis using antibodies to histidine tag (A) and complement proteins C1q (B), C1r (C), C1s (D), C3 (E), C5 (F), C9 (G). In all the panels (A-G), HCS is the human complement serum which was eluted from the empty beads i.e., without the bait protein (rIxsCRT) and HCS+rIxsCRT denotes the human complement proteins which were pulled down and eluted from the beads loaded with rIxsCRT. Red arrowheads denote the detected complement proteins at their expected molecular weight size (kDa).
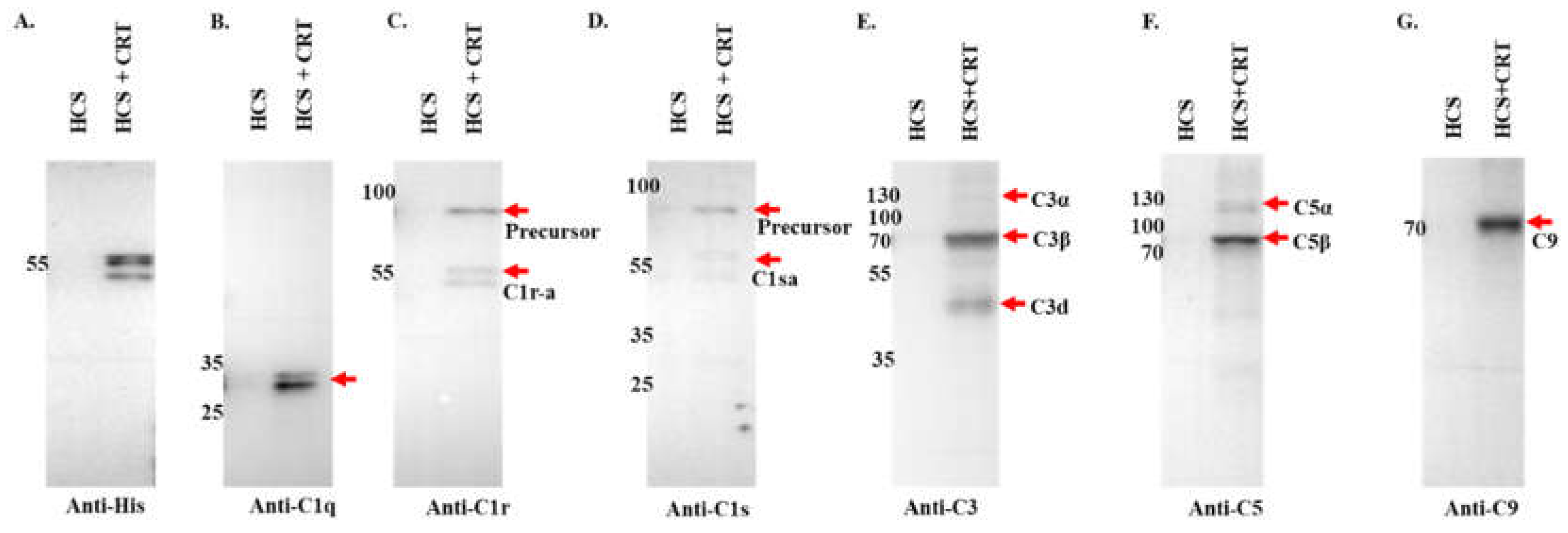
Figure 7.
ELISA analysis demonstrates rIxsCRT binding of complement proteins. High Binding ELISA plates coated with affinity purified rIxsCRT (250ng) was incubated with normal human serum (NHS) followed by antibodies to C1q (A), C1r (B), C1s (C), C3 (D), activated C3 (E), C5 (F), C9 (G), and C5b-9 or MAC (H) antibodies. Y-axis denotes the absorbance measured at A450nm which reflects to the intensity of the specific antibody binding to rIxsCRT. Non-coated well blocked with 1% BSA was incubated with NHS and used as negative control (Neg. Cont.). Data represents mean ± SEM of 3 biological replicate. For statistical analysis, t-test was performed on GraphPad Prism 9 and ** represents p < 0.01; *** represents p < 0.001, **** represents p < 0.0001, ns represents not significant.
Figure 7.
ELISA analysis demonstrates rIxsCRT binding of complement proteins. High Binding ELISA plates coated with affinity purified rIxsCRT (250ng) was incubated with normal human serum (NHS) followed by antibodies to C1q (A), C1r (B), C1s (C), C3 (D), activated C3 (E), C5 (F), C9 (G), and C5b-9 or MAC (H) antibodies. Y-axis denotes the absorbance measured at A450nm which reflects to the intensity of the specific antibody binding to rIxsCRT. Non-coated well blocked with 1% BSA was incubated with NHS and used as negative control (Neg. Cont.). Data represents mean ± SEM of 3 biological replicate. For statistical analysis, t-test was performed on GraphPad Prism 9 and ** represents p < 0.01; *** represents p < 0.001, **** represents p < 0.0001, ns represents not significant.
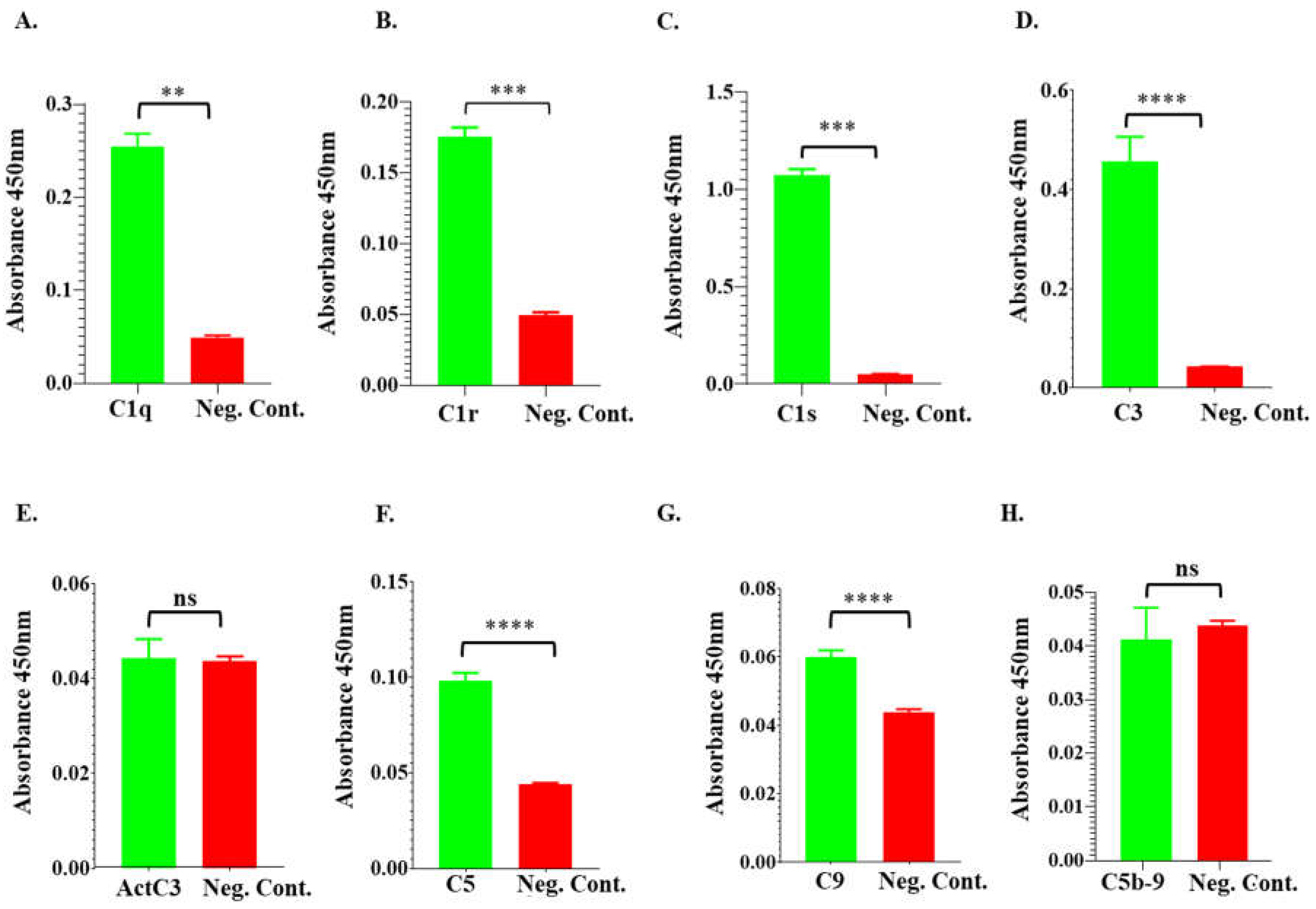
Figure 8.
rIxsCRT apparently enhances membrane attack complex (MAC) deposition in classical pathway and alternate pathway but inhibits MAC deposition in lectin pathway. Wieslab® Complement System Kit (Svar Life Science AB, Sweden) was used to detect the effects of rIxsCRT on MAC deposition via Classical pathway, Alternative pathways and Lectin pathway as described in materials and methods. In brief, rIxsCRT (4μM) was incubated with NHS (provided with the kit) at 37˚C for 30min and then added to wells pre-coated with the antibody to MAC. Diluent and kit provided reagent served as negative and positive control respectively. After washing, the conjugate and substrate were added according to the manufacturer instructions for each kit. NC is negative control and NHS is normal human serum used as positive control. % MAC deposition (Y-axis) was calculated as mentioned in methodology section and the MAC deposition in positive control was represented as 100% (denoted by black dotted line). Data is presented as deposited MAC ± SEM calculated from 3 biological replicates. Statistical significance was determined by student’s t-test in GraphPad Prism 9. * Represents p ≤ 0.05, ** represents p ≤ 0.01 and ns represents not significant.
Figure 8.
rIxsCRT apparently enhances membrane attack complex (MAC) deposition in classical pathway and alternate pathway but inhibits MAC deposition in lectin pathway. Wieslab® Complement System Kit (Svar Life Science AB, Sweden) was used to detect the effects of rIxsCRT on MAC deposition via Classical pathway, Alternative pathways and Lectin pathway as described in materials and methods. In brief, rIxsCRT (4μM) was incubated with NHS (provided with the kit) at 37˚C for 30min and then added to wells pre-coated with the antibody to MAC. Diluent and kit provided reagent served as negative and positive control respectively. After washing, the conjugate and substrate were added according to the manufacturer instructions for each kit. NC is negative control and NHS is normal human serum used as positive control. % MAC deposition (Y-axis) was calculated as mentioned in methodology section and the MAC deposition in positive control was represented as 100% (denoted by black dotted line). Data is presented as deposited MAC ± SEM calculated from 3 biological replicates. Statistical significance was determined by student’s t-test in GraphPad Prism 9. * Represents p ≤ 0.05, ** represents p ≤ 0.01 and ns represents not significant.
Figure 9.
rIxsCRT promotes the growth of B. burgdorferi in culture. B. burgdorferi cultured in presence of rIxsCRT (indicated concentration) were sampled at days 0, 2, 5 and 7. (A) In triplicate, cells were quantified by manual counts on Petroff-Hausser Chamber and quantified using the following formula: number of cells/mL = Average of cells counted in all chambers x dilution factor x 50,000. (B) B. burgdorferi was quantified by qPCR using the genomic DNA of the B. burgdorferi and fLaB primers. For ELISA, student’s t-test was used for statistical analysis on GraphPad Prism 9 and p value ≤ 0.05 (denoted by **) is considered as significant value for 3 biologicals replicates.
Figure 9.
rIxsCRT promotes the growth of B. burgdorferi in culture. B. burgdorferi cultured in presence of rIxsCRT (indicated concentration) were sampled at days 0, 2, 5 and 7. (A) In triplicate, cells were quantified by manual counts on Petroff-Hausser Chamber and quantified using the following formula: number of cells/mL = Average of cells counted in all chambers x dilution factor x 50,000. (B) B. burgdorferi was quantified by qPCR using the genomic DNA of the B. burgdorferi and fLaB primers. For ELISA, student’s t-test was used for statistical analysis on GraphPad Prism 9 and p value ≤ 0.05 (denoted by **) is considered as significant value for 3 biologicals replicates.
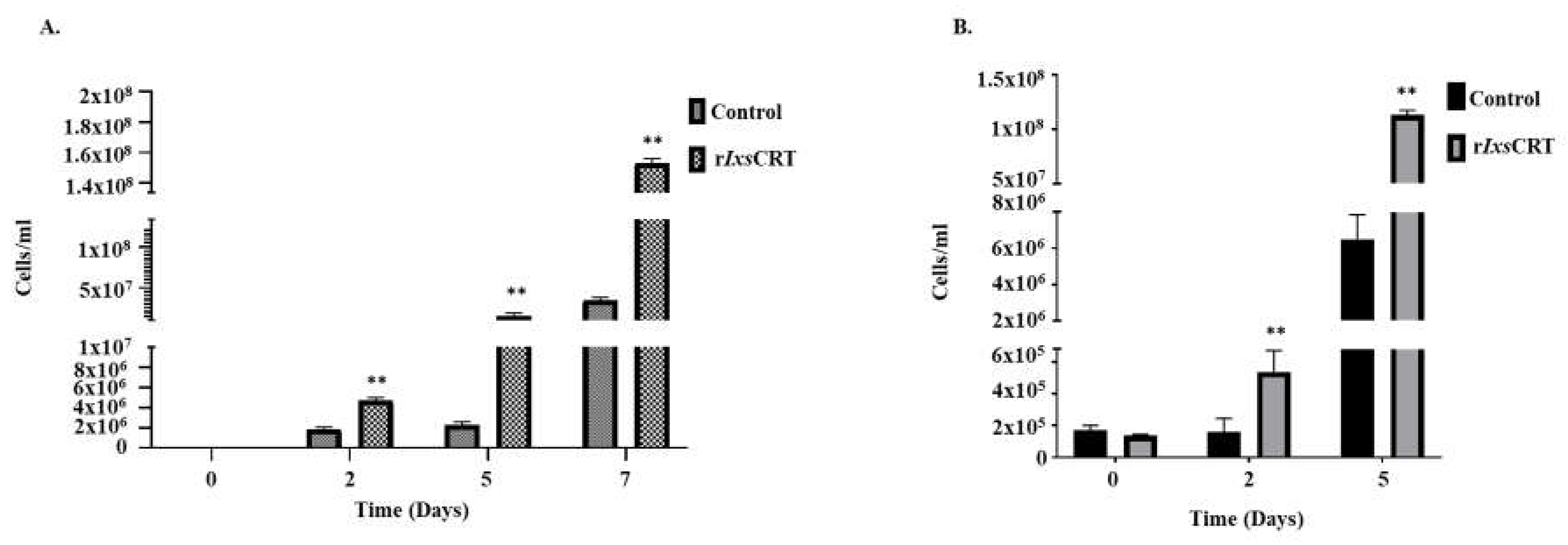
Figure 10.
One-time exposure of rabbits to B. burgdorferi infected I. scapularis nymphs triggers high IgG antibody levels to rIxsCRT. (A) Affinity purified rIxsCRT (250 ng) was subjected to standard ELISA using serially diluted purified IgG of rabbits that were fed on for a single time by uninfected (blue line graph) and B. burgdorferi infected (red line graph) I. scapularis nymph ticks. Black line graph represents pre-immune IgG binding that was used for negative control. (B-D) Various quantities of affinity purified rIxsCRT (100, 300, and 500 ng) was subjected to western blotting analysis using purified IgG from rabbits that were fed on for a single time by uninfected (B) and B. burgdorferi infected ticks (C) as well as the antibody to the histidine tag that served as positive control (D).
Figure 10.
One-time exposure of rabbits to B. burgdorferi infected I. scapularis nymphs triggers high IgG antibody levels to rIxsCRT. (A) Affinity purified rIxsCRT (250 ng) was subjected to standard ELISA using serially diluted purified IgG of rabbits that were fed on for a single time by uninfected (blue line graph) and B. burgdorferi infected (red line graph) I. scapularis nymph ticks. Black line graph represents pre-immune IgG binding that was used for negative control. (B-D) Various quantities of affinity purified rIxsCRT (100, 300, and 500 ng) was subjected to western blotting analysis using purified IgG from rabbits that were fed on for a single time by uninfected (B) and B. burgdorferi infected ticks (C) as well as the antibody to the histidine tag that served as positive control (D).
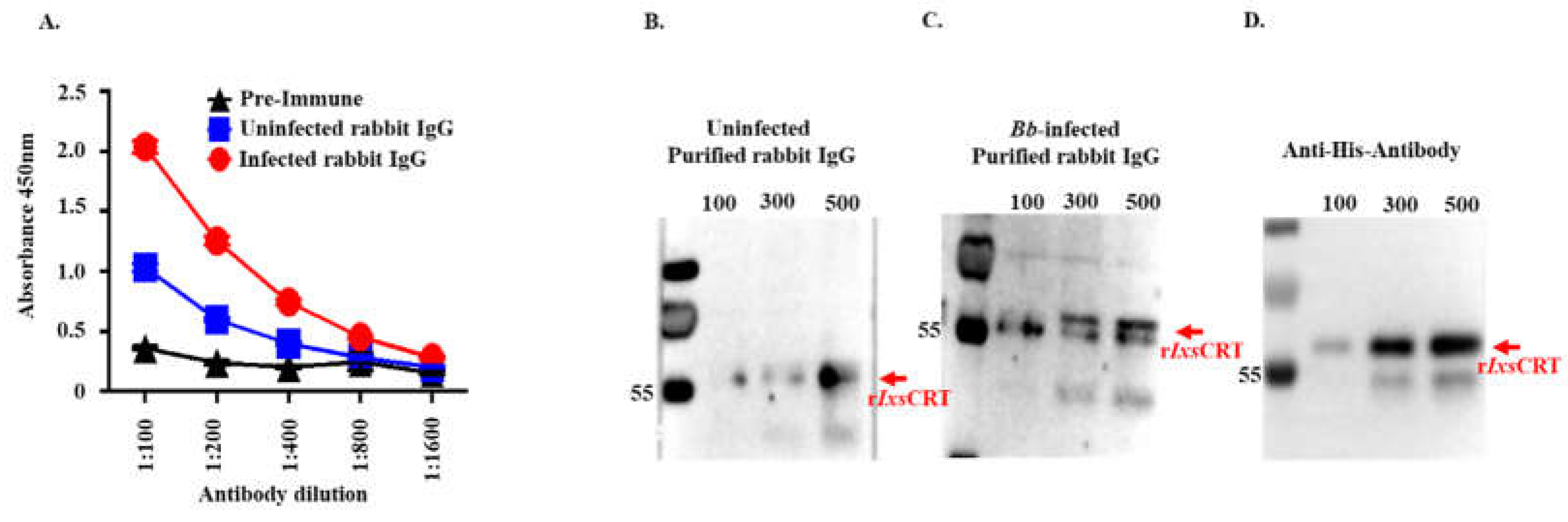

Table 1.
Reactome analysis depicting the pathways linked with proteins which are abundant ≥1.5-fold in NHS treated with CRT.
Table 1.
Reactome analysis depicting the pathways linked with proteins which are abundant ≥1.5-fold in NHS treated with CRT.
| Pathway name (reactome identifier) | Entities | Reactions | |||||
|---|---|---|---|---|---|---|---|
| found | ratio | p-value | FDR* | found | ratio | ||
| Fraction 1-5 | Terminal pathway of complement (R-HSA-166665.4) | 2 / 8 | 6.88e-04 | 0.009 | 0.578 | 4 / 5 | 3.49e-04 |
| Defective ABCA12 causes ARCI4B (R-HSA-5682294.3) | 1 / 1 | 8.60e-05 | 0.017 | 0.578 | 1 / 1 | 6.99e-05 | |
| Defective ABCC2 causes DJS (R-HSA-5679001.4) | 1 / 1 | 8.60e-05 | 0.017 | 0.578 | 1 / 1 | 6.99e-05 | |
| Regulation of Complement cascade (R-HSA-977606.6) | 6 / 135 | 0.012 | 0.03 | 0.578 | 20 / 42 | 0.003 | |
| CD22 mediated BCR regulation (R-HSA-5690714.3) | 4 / 70 | 0.006 | 0.033 | 0.578 | 3 / 4 | 2.80e-04 | |
| Recycling of bile acids and salts (R-HSA-159418.4) | 2 / 18 | 0.002 | 0.039 | 0.578 | 2 / 17 | 0.001 | |
| Complement cascade (R-HSA-166658.5) | 6 / 146 | 0.013 | 0.041 | 0.578 | 33 / 72 | 0.005 | |
| Defective factor XII causes hereditary angioedema (R-HSA-9657688.2) |
1 / 3 | 2.58e-04 | 0.05 | 0.578 | 2 / 2 |
1.40e-04 |
|
| Defective SERPING1 causes. hereditary angioedema (R-HSA-9657689.2) |
1 / 3 | 2.58e-04 | 0.05 | 0.578 | 1 / 3 | 2.10e-04 | |
| Fraction 6 | Terminal pathway of complement (R-HSA-166665.4) | 4 / 8 | 6.88e-04 | 9.00e-06 | 0.004 | 5 / 5 | 3.49e-04 |
| Regulation of Complement cascade (R-HSA-977606.6) | 9 / 135 | 0.012 | 3.14e-04 | 0.064 | 13 / 42 | 0.003 | |
| Complement cascade (R-HSA-166658.5) | 9 / 146 | 0.013 | 5.50e-04 | 0.075 | 22 / 72 | 0.005 | |
| Response to elevated platelet cytosolic Ca2+ (R-HSA-76005.3) | 7 / 133 | 0.011 | 0.005 | 0.429 | 2 / 14 | 9.79e-04 | |
| Regulation of Insulin-like Growth Factor (IGF) transport and uptake by Insulin-like Growth Factor Binding Proteins (IGFBPs) (R-HSA-381426.3) | 6 / 124 | 0.011 | 0.014 | 0.61 | 2 / 14 | 9.79e-04 | |
| Platelet degranulation (R-HSA-114608.4) | 7 / 128 | 0.011 | 0.004 | 0.429 | 2 / 11 | 7.69e-04 | |
| Dissolution of Fibrin Clot (R-HSA-75205.4) | 2 / 13 | 0.001 | 0.018 | 0.61 | 15 / 21 | 0.001 | |
| Post-translational protein phosphorylation (R-HSA-8957275.2) | 5 / 107 | 0.009 | 0.027 | 0.61 | 1 / 1 | 6.99e-05 | |
| Other semaphoring interactions (R-HSA-416700.2) | 2 / 19 | 0.002 | 0.036 | 0.61 | 3 / 9 | 6.29e-04 | |
| Sema4D induced cell migration and growth-cone collapse (R-HSA-416572.4) | 2 / 20 | 0.002 | 0.04 | 0.61 | 4 / 7 | 4.89e-04 | |
| RHO GTPases activate CIT (R-HSA-5625900.3) | 2 / 20 | 0.002 | 0.04 | 0.61 | 3 / 6 | 4.19e-04 | |
| Common Pathway of Fibrin Clot Formation (R-HSA-140875.5) | 2 / 22 | 0.002 | 0.047 | 0.61 | 6 / 29 | 0.002 | |
| Fraction 7-8 | Collagen chain trimerization (R-HSA-8948216.4) | 8 / 44 | 0.004 | 1.24e-04 | 0.091 | 7 / 28 | 0.002 |
| Collagen degradation (R-HSA-1442490.4) | 8 / 64 | 0.006 | 0.001 | 0.331 | 11 / 34 | 0.002 | |
| Anchoring fibril formation (R-HSA-2214320.4) | 4 / 15 | 0.001 | 0.002 | 0.331 | 4 / 4 | 2.80e-04 | |
| Collagen biosynthesis and modifying enzymes (R-HSA-1650814.5) | 8 / 67 | 0.006 | 0.002 | 0.331 | 30 / 51 | 0.004 | |
| Integrin cell surface interactions (R-HSA-216083.5) | 9 / 85 | 0.007 | 0.002 | 0.331 | 11 / 55 | 0.004 | |
| TRKA activation by NGF (R-HSA-187042.2) | 2 / 3 | 2.58e-04 | 0.005 | 0.551 | 4 / 4 | 2.80e-04 | |
| Collagen formation (R-HSA-1474290.4) | 8 / 90 | 0.008 | 0.011 | 0.763 | 39 / 77 | 0.005 | |
| NCAM1 interactions (R-HSA-419037.2) | 5 / 42 | 0.004 | 0.013 | 0.763 | 1 / 10 | 6.99e-04 | |
| ECM proteoglycans (R-HSA-3000178.5) | 7 / 76 | 0.007 | 0.014 | 0.763 | 7 / 23 | 0.002 | |
| Assembly of collagen fibrils and other multimeric structures (R-HSA-2022090.4) | 6 / 61 | 0.005 | 0.016 | 0.763 | 9 / 26 | 0.002 | |
| Laminin interactions (R-HSA-3000157.3) | 4 / 30 | 0.003 | 0.018 | 0.763 | 4 / 15 | 0.001 | |
| Degradation of the extracellular matrix (R-HSA-1474228.5) | 10 / 140 | 0.012 | 0.019 | 0.763 | 13 / 105 | 0.007 | |
| Crosslinking of collagen fibrils (R-HSA-2243919.4) | 3 / 18 | 0.002 | 0.022 | 0.763 | 1 / 13 | 9.09e-04 | |
| Activation of TRKA receptors (R-HSA-187015.3) | 2 / 7 | 6.02e-04 | 0.023 | 0.763 | 8 / 8 | 5.59e-04 | |
| Defective SLC2A9 causes hypouricemia renal 2 (RHUC2) (R-HSA-5619047.3) | 1 / 1 | 8.60e-05 | 0.032 | 0.763 | 1 / 1 | 6.99e-05 | |
| Role of second messengers in netrin- 1 signaling (R-HSA-418890.2) | 2 / 10 | 8.60e-04 | 0.043 | 0.763 | 1 / 4 | 2.80e-04 | |
| Signaling by PDGF (R-HSA-186797.5) | 5 / 60 | 0.005 | 0.049 | 0.763 | 1 / 31 | 0.002 | |
* FDR: False Discovery Rate.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



