
Submitted:
25 April 2024
Posted:
26 April 2024
You are already at the latest version
Abstract
The conformations of trifluoroacetyl triflate, CF3C(O)OSO2CF3, were investigated through vibrational experimental methods (gas-phase FTIR, liquid-phase Raman, and Ar-matrix FTIR spectroscopy) and Density Functional Theory (DFT) calculations. A potential energy surface was computed using the B3P86 /6-31+g(d) approximation as a function of the dihedral angles τ1 = CCOS and τ2 = COSC. The surface reveals three minima, which were further optimized using the B3LYP method with various basis sets (6-31++G(d), 6-311++G(d), tzvp and cc-pvtz). The global minimum corresponds to a syn-anti conformer (the CO double bound syn with respect the O−S single bond and the C−O single bond anti with respect to de S−C single bond). The other two minima represent enantiomeric syn-gauche forms. The Ar-matrix FTIR spectrum exhibited clear evidence of the presence of two conformers. Furthermore, the randomization process observed following broad-band UV-visible irradiation facilitated the identification of the IR absorption of each conformer. Based on the Ar-matrix FTIR experiments, the vapour phase of trifluoroacetyl triflate at room temperatures was composed of approximately 60-70% of the syn-anti conformer and 30-40% of the syn-gauche form.
Keywords:
conformations
; matrix-FTIR
; gas-phase FTIR
; Raman
; DFT
1. Introduction
Trifluoroacetyl triflate (CF3C(O)OSO2CF3, TFAT) is a convenient reagent for trifluoroacetylation reactions in organic synthesis [1]. Its efficacy in trifluoroacylation reaction with various nucleophilic molecules, including alcohols [2], amines [2], aromatic activated substrates [3], covalent fluorides [4] and halides [5], between others, has been reported. However, TFAT's high reactivity imposes limitations on the choice of solvents for these reactions, with benzene, saturated hydrocarbons, and common halogenates solvents being among the few compatible options [1]. Despite its widespread use as a reagent and its availability from commercial sources, the structural and vibrational properties of trifluoroacetyl triflate remain unexplored, to the best of our knowledge.
Our group has successfully prepared molecules with the general formula XC(O)OSO2CF3, where X = F [6] or Cl [7,8], by reacting halocarbonylsulphenyl chloride, XC(O)SCl, with silver triflate salt, AgSO3CF3. These molecules exhibit interesting conformational equilibrium. In the gas phase, FC(O)OSO2CF3 exists as a mixture of anti (the C−O single bond anti with respect to the S−C single bond) and gauche (the C−O doble bond gauche with respect to the S−C single bond) forms. In both conformers the C=O doble bond is synperiplanar with respect to the O-S single bond. Gas Electron Diffraction (GED) measurements revealed that the syn-anti rotamer predominates at ambient temperature, comprising 67(8)% of the gas-phase composition, whereas matrix-FTIR estimated its presence at 59(5)% [6]. Similar results were obtained for ClC(O)OSO2CF3, with the syn-anti conformer constituting 66(8)% according to matrix-FTIR measurements [7] and 67(11) % based on GED experiments [8]. Notably, FC(O)OSO2CF3 and ClC(O)OSO2CF3 are unique in exhibiting prevailing anti conformation around the S−O single bond, unlike other XOSO2CF3 species, which typically adopt a gauche conformation (see Ref. 6 and 8 and references cited therein). In this sense, a close and well-known example is the ClC(O)OSO2Cl molecule, where the gauche conformation predominates over the anti [9].
Matrix IR spectroscopy emerged as a valuable technique for investigated conformational equilibrium. Under such conditions, IR absorptions are narrowed compared to those observed in gas and condensed phases. This phenomenon primarily arises from the inhibition of rotation and intermolecular interactions in the isolated environment. Consequently, this method enables the discernment of bands with small wavenumber differences, particularly useful in distinguishing spectral features between conformers.
The experimental setup employed in matrix formation ensures the preservation of the conformational composition existing at the temperature of the mixture prior to deposition. However, irradiating a compound isolated within a matrix can induce interconversion between conformers, leading to a randomization process. This results in a final state where the proportion of each conformer approximates 50%. This phenomenon was observed in FC(O)SBr isolated in matrices [10]. It was explained by a transition to an excited state, wherein the minimum in the potential curve as a function of the torsion angle coincides with the maximum of the energy surface in the ground state. Consequently, this leads to a decay with equal probability to the two conformers. This process has been observed for several other molecules (see, for example, Ref. 11−13, and references cited therein). This behaviour constitutes a very important tool for a clear assignment of the IR absorptions to each conformer.
In this work, we explored two distinct routes for synthesizing trifluoroacetyl triflate. The first route involved the reaction of trifluoroacetyl chloride with silver triflate, while the second route by the reaction between trifluoroacetic and triflic acid. For the latter method, which has been reported in several works in the literature [2,14−16]. we sought to optimize the conditions to yield a highly pure sample. We conducted a comprehensive analysis using various spectroscopic techniques. Specifically, we measured the FTIR spectra of the vapor phase, the Raman spectrum of the liquid phase, and the FTIR spectra of the sample isolated in solid argon immediately after deposition and at different intervals following broad-band UV-visible irradiation. To provide theoretical insights, we employed Density Functional Theory (DFT) methods to calculate a potential energy surface. Subsequently, we optimized the minima over this surface. The assignment of vibrational spectra and the experimental estimation of the proportion of conformers in the vapour phase at ambient temperature were carried out based on the Ar-matrix FTIR spectra before and after irradiation, aided by predictions from theoretical calculations.
2. Materials and Methods
Chemicals. Trifluoroacetic acid (CF3C(O)OH, Carlo Erba), phosphorus pentachloride (PCl5, Sigma Aldrich), silver triflate (AgSO3CF3, Fluka), and triflic acid (HOSO2CF3, Sigma Aldrich) were utilised without further purification. Ar (AGA) was purified by passing it through two U-traps. The first trap contained thermally activated Molecular Sieve and the second was cooled to approximately –90 °C with an acetone-bath to retain potential traces of impurities, primarily H2O and CO2.
The FTIR spectra were acquired using a Nexus Nicolet instrument, equipped with either a cryogenic MCTB (for the range 4000–400 cm−1) or a DTGS detector (for the range 600–180 cm−1). For vapour-phase IR studies at ambient temperature, a 10 cm gas cell fitted with CsI windows was employed.
Raman spectra were obtained using a Horiba-Jobin-Yvon T64000 Raman spectrometer equipped with a confocal microscope and CCD detection system. Excitation was achieved using a 514.5 nm excitation wavelength from an Ar multiline laser. Wavenumbers were calibrated using the 459 cm−1 band of CCl4. The net liquid sample was contained within a sealed capillary for ambient temperature measurements.
A 1 L vacuum flask was filled with TFAT and Ar in a proportion 1:1000 using standard manometric techniques. The gas mixture was deposited onto a CsI window, cooled to approximately 15 K using a Displex closed-cycle refrigerator (SHI-APD Cryogenics, model DE-202) employing the pulse deposition technique. Matrix-isolated FTIR spectra were recorded in the 4000–400 cm−1 range using a Thermo Nicolet 6700 instrument equipped with a DTGS detector. The matrices were exposed to broad-band UV-visible radiation (200 ≤ λ ≤ 800 nm) emitted by a Spectra-Physics Hg–Xe arc lamp operating at 1000 W. To mitigate heating effects, the lamp output was restricted by a water filter to absorb infrared radiation.
All of the quantum chemical calculations were conducted using the Gaussian 03 program system [17]. A relaxed potential energy surface was computed using the B3P86 [18] functional in conjunction with the 6-31+G(d) basis set. Geometry optimizations of the energy minima employing the B3LYP [19,20] method and various basis set functions were sought using standard gradient techniques, allowing for simultaneous relaxation of all geometrical parameters. Subsequently these optimised structures were characterised by their vibrational properties. Natural Bond Orbital (NBO) analysis was performed for each structure and the orbital stabilization energies were calculated [21].
3. Results and Discussion
3.1. Synthesis of Trifluoroacetyl Triflate (TFAT)
Trifluoroacetyl triflate was synthetised via the reaction between trifluoroacetic acid and triflic acid, as illustrated in Scheme 1. Phosphorus pentoxide served as the dehydrating agent. These reactions were conducted at ambient temperature under an inert atmosphere, utilising either nitrogen or argon.
The formation reaction of TFAT by this route competes with the dehydration of trifluoroacetic and triflic acids caused by phosphorus pentoxide, according to the reactions described in Scheme 2. Taking into account that the spectroscopic studies carried out in this work, particularly matrix-isolation spectroscopy, require highly pure samples, various stoichiometric ratios of the reagents were explored. This optimization aimed to minimize secondary reactions and ensure optimal conditions for the desired reactions.
TFAT undergoes hydrolysis at room temperature, yielding trifluoroacetic acid and triflic acid [1], as well as the two anhydrides depicted in Scheme 2. Due to its hydrophilicity, purification must be conducted in a moisture-free environment. Initially, the reaction between trifluoroacetic acid and triflic acid was carried out with an equimolar ratio of both acids, following previously reported conditions [1,14]. Under these reaction conditions, a high proportion of the CF3C(O)OC(O)CF3 anhydride relative to TFAT was obtained. Subsequently, experiments were conducted using a 2:1 ratio between trifluoroacetic and triflic acids, respectively, as described in existing literature [2,15]. However, these tests yielded even poorer results than the previous ones.
To minimise the formation of the CF3C(O)OC(O)CF3 anhydride, the reaction was conducted with a 1:2 ratio between trifluoroacetic and triflic acids. This ratio was chosen deliberately to favour the formation of the CF3SO2OSO2CF3 anhydride over the CF3C(O)OC(O)CF3 anhydride, which exhibits lower volatility (approximately 8 mbar at ambient temperature) and facilitates its subsequent separation from TFAT. The synthesis of the TFAT used for the spectroscopic studies presented in this work was carried out under these conditions, using an Ar atmosphere to mitigate potential hydrolysis reactions.
Efforts to synthesise trichloroacetyl triflate by reacting silver triflate with trifluoroacetyl chloride, aiming to eliminate water presence, were unsuccessful. Initially, trifluoroacetic acid (CF3C(O)OH) was reacted with an approximately 50% excess of phosphorus pentachloride (PCl5) to yield trifluoroacetyl chloride (CF3C(O)Cl) [22]. The reaction was conducted in a flask, with CF3C(O)OH being added dropwise to PCl5 at room temperature (25 °C). The apparatus was connected to two traps cooled with refrigerated baths for subsequent distillation, and CF3C(O)Cl was isolated in the trap at −60°C. Subsequently, trifluoroacetyl chloride was reacted with ~50% excess of silver triflate at −20 °C. Before utilisation, the salt was dried in an oven at 120 °C. The progress of the reaction was monitored using FTIR and Raman spectroscopy. Unfortunately, this reaction failed to yield any favourable results, and after numerous attempts, we decided to abandon this route.
3.2. Quantum Chemical Calculations
To identify different conformational isomers, we computed a potential energy surface as a function of two dihedral angles τ1 = CC−OS and τ2 = CO−SC, as shown in Figure 1. Each torsional angle was varied from 0 to 360° in steps of 10°. The energy at each data point was determined using the B3P86/6-31+g(d) approximation, while all geometric parameters were simultaneously optimized, except for the torsion angles τ1 and τ2.
Figure 2 displays the calculated potential energy surface (left panel) and the contour map (right panel). The global minimum is evident, corresponding to τ1 = τ2 = 180°. Moreover, the surface reveals two additional local minima, where τ2 is approximately 180, while τ1 is around 70 and 290°, respectively. Subsequently, each of these minima underwent optimization using the B3LYP method with various basis functions, involving the simultaneous relaxation of all geometric parameters. The global energy minimum is denoted as the syn-anti conformer, characterised by the C=O double bound being syn with respect the O−S single bond and the C−O single bond anti with respect to de S−C single bond. The second is termed as the syn-gauche, where the C=O double bound is syn with respect the O−S single bond and the C−O single bond gauche with respect to de S−C single bond.
Figure 3 exhibits the molecular models of the optimised structures for both rotamers of trifluoroacetyl triflate, computed using the B3LYP/cc-pvtz approximation. The geometrical parameters are detailed in Table S1 of the Supplementary Material, while the Cartesian coordinates for the syn-anti and syn-gauche conformers are provided in Tables S2 and S3, respectively. In all instances, energy minima devoid of imaginary frequencies were achieved. Furthermore, Table S4 presents a comprehensive compilation of wavenumbers, IR and Raman intensities, along with tentative assignment for each conformer, calculated using the B3LYP/cc-pvtz approximation.
Table 1 presents the energy differences and Gibbs free energy differences between the syn-gauche and syn-anti conformers, calculated with B3LYP theoretical approximation with various basis sets. Additionally, the relative population of the rotamers at 25 °C was determined using the Boltzmann equation, considering a degeneracy of 2 for the syn-gauche form. The table reveals that, across all cases, the syn-anti form corresponds to the lowest energy conformer. However, the computed percentage at room temperature fluctuates depending on the basis set utilized, ranging from 96 to 69%. These findings will be discussed further along with the analysis of the experimental results.
To comprehensively understand the conformational preferences of TFAT, we conducted orbital interaction analyses using Natural Bond Orbital (NBO) formalism. Table 2 summarizes the key NBO stabilization energies computed, encompassing both hyperconjugative and anomeric effects. The hyperconjugative effect involves the interaction between a non-bonding orbital with π character, known as nπ(O), located on the oxygen atom (−O−), and the π antibonding orbital of the C=O double bond, referred to as π*(C=O). On the other hand, the syn conformation is stabilized by the anomeric effect corresponding to the lpσO → σ*C=O interaction. The syn-anti conformer is additionally stabilised by two lpσO → σ*S=O interactions, while for the syn-gauche form the lpσO → σ*S−C interaction emerges as the most significant factor contributing to its stabilization (see Table 2 for details). Figures S1 and S2 depicts a schematic representation of these orbital interactions. Upon analysis of the values presented in Table 2, it becomes evident that both hyperconjugative and anomeric effects play significant roles in stabilizing the syn-anti over the syn-gauche rotamer.
The vibrational spectra (IR and Raman) of each TFTA conformer were simulated using the B3LYP theoretical approximation in conjunction with various basis sets. The results will be discussed in the following section, alongside the experimental findings.
3.3. Vibrational Studies
As stated in the introduction, to the best of our knowledge, there have been no prior vibrational studies conducted on trifluoroacetyl triflate reported in the literature. In this study, we conducted measurements and analyses of the FTIR spectra in the gas phase, the Raman spectrum in the liquid phase, and the FTIR spectra of matrix-isolated CF3C(O)OSO2CF3. Our focus was particularly on identifying signals that could be attributed to different conformers. Figure 4 illustrates the experimental spectra, while Table 3 presents the experimental wavenumbers alongside the predicted values for the syn-anti and syn-gauche conformers, calculated using the B3LYP/cc-pvtz approximation, and a tentative assignment. For band assignment, we considered the following criteria: i) theoretical predictions, especially differences in wavenumbers for the vibrational modes of each conformer; ii) the behaviour of IR absorptions of TFAT isolated in an argon matrix following UV-visible irradiation; and iii) comparison with reported values for molecules similar to TFAT.
The expected differences between the IR spectra of both conformers were meticulously scrutinized based on the computationally simulated spectra. As shown in Table 3, certain vibrational modes are expected to exhibit nearly identical wavenumbers for both forms. Conversely, several vibrational modes display significant wavenumber disparities, enabling their distinct detection, particularly in the Ar-matrix FTIR spectra, which feature narrower bands compared to the gas and liquid phase spectra. Figure 5 illustrates the plot of the IR spectra simulated using the B3LYP/cc-pvtz approximation for the syn-anti and syn-gauche conformers, each multiplied by the relative abundance calculated using the same theoretical approximation, as well as the weighted sum of the individual spectra. This plot highlights selected regions where appreciable wavenumber differences were observed. These simulated spectra were then compared with the experimental spectra, with particular emphasis on the Ar-matrix FTIR spectrum, which served as a valuable tool for assignment purposes.
Another significant aspect aiding in the interpretation and assignment of the spectra was the analysis of the FTIR spectra following broad-band UV-visible irradiation of TFAT isolated in an Ar matrix. The matrix underwent exposure to UV-visible light for varying durations (15 and 30 s, and 1, 2, 4, 7, 12, 20, 30, 50 and 80 min). Changes occurring after each irradiation period were monitored using FTIR spectroscopy. During photolysis, it was observed that the IR absorptions corresponding to the syn-gauche conformer increased, while those of the syn-anti form decreased. The final two spectra, taken after 50 and 80 minutes of photolysis, exhibited no further changes, indicating the system had reached a stationary state. Figure 6 illustrates selected regions of the FTIR spectrum of the matrix taken immediately after deposition and after 12 and 50 min of photolysis. These spectral regions were chosen based on clear identification of both conformers, corresponding to the ν(C=O), ν(C−O) and δ(OCO) vibrational modes. For clarity, the spectra were normalized to the bands of the high-energy conformer. The pronounced increase in the relative intensities of the signals from the syn-gauche conformer compared to those of the syn-anti rotamer is evident in the Figure.
The abundances of the conformers were estimated from the IR matrix spectra using two different approaches. Firstly, it was assumed that in the last two spectra, taken after 50 and 80 min respectively, where a constant ratio of intensities of the bands assigned to the two conformers was achieved, the proportion of each conformer was approximately 50%, following a process known as conformational randomization. Experimental absorptivity coefficients were then calculated from the area ratio. Utilizing this relationship along with the intensity ratio in the spectrum acquired prior to irradiation, the percentage of each rotamer at room temperature was estimated. In a second method, absorptivity coefficients calculated using the B3LYP/cc-pvtz approximation were employed to correct the relationship of experimental intensities in the IR spectra of the matrix before irradiation. These procedures were performed for each of the normal modes depicted in Figure 6 (ν(C=O), ν(C−O) and δ(OCO)). Estimates between approximately 60-70% were obtained for the syn-anti form. The percentage determined for each normal mode using both described methods are presented in Table S5 of the supplementary material. Considering the various factors influencing these measurements, particularly the presence of matrix sites affecting the relative intensity of some bands, whose changes may also be influenced by irradiation, the dispersion of these values can be deemed acceptable. Moreover, they align well with the relative population predictions obtained using the B3LYP/cc-pvtz method.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Schematic representation of the hyperconjugative and anomeric interactions in the syn-anti conformer of trifluoroacetyl triflate calculated with the NBO formalism using the B3LYP/cc-pvtz approximation: (a) lpπ O → π* C=O; (b) lpσ O → σ* C=O; (c) lpσ O → σ* S=O; Figure S2: Schematic representation of the hyperconjugative and anomeric interactions in the syn-gauche conformer of trifluoroacetyl triflate calculated with the NBO formalism using the B3LYP/cc-pvtz approximation: (a) lpπ O → π* C=O; (b) lpσ O → σ* C=O; (c) lpσ O → σ* S−C; Table S1: Geometrical parameters (distances in Å and angles in degrees) for the syn-anti and syn-gauche conformers of CF3C(O)OSO2CF3 calculated with the B3LYP/cc-pvtz approximation; Table S2: Cartesian coordinates (in Å) of the syn-anti conformer of CF3C(O)OSO2CF3 calculated with the B3LYP/cc-pvtz approximation, Table S3: Cartesian coordinates (in Å) of the syn-gauche conformer of CF3C(O)OSO2CF3 calculated with the B3LYP/cc-pvtz approximation; Table S4: Wavenumbers, IR and Raman intensities, and tentative assignment, calculated with the B3LYP/cc-pvtz approximation for the syn-anti and syn-gauche conformers of CF3C(O)OSO2CF3; Table S5: Percentage of the syn-anti conformer of CF3C(O)OSO2CF3 estimated from the relative intensities of selected IR bands of the Ar-matrix FTIR spectrum of TFAT. I, using the relationship of the absorption coefficients of the involved bands when the concentrations of the two rotamers becomes equal after the randomization process (in this case, broad-band UV-visible irradiation of the matrix for 50 min); II) through the use of the absorption coefficients obtained from the B3LYP/cc-pvtz approximation.
Author Contributions
Conceptualization, R.M.R and C.O.D.V; formal analysis, A.S., MGP and R.M.R.; investigation, A.S. and M.G.P; writing—original draft preparation, R.M.R; writing—review and editing, R.M.R and C.O.D.V; funding acquisition, R.M.R and C.O.D.V. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), PIP 0352 and PUE-2017-22920170100053, Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT), PICT-2018-04355 and PICT-2020-03746, and Universidad Nacional de La Plata, UNLP-X822.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors on request.
Acknowledgments
We are grateful to Luciana Tamone from the Matrix Laboratory of CEQUINOR, as well as to Gustavo Pozzi and Gino Pietrodángelo from the Raman Laboratory of CEQUINOR for their assistance during the measurements.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- López, S.E.; Restrepo, J.; Salazar, J. Trifluoroacetylation in Organic Synthesis: Reagents, Developments and Applications in the Construction of Trifluoromethylated Compounds. Curr. Org. Synth. 2010, 7, 414-432. [CrossRef]
- Forbus, T.R.; Taylor, S.L.; Martin, J.C. Reactions of the Readily Accessible Electrophile, Trifluoroacetyl Triflate: A Very Reactive Agent for Trifluoroacetylations at Oxygen, Nitrogen, Carbon, or Halogen Centers. J. Org. Chem. 1987, 52 (19), 4156–4159. [CrossRef]
- Kiselyov, A.S.; Harvey, R.G. Acylation of Activated Aromatic Substrates under Mild Conditions with (RCO)2O/Me2S/BF3. Tetrahedron Lett. 1995, 23, 4005−4008. [CrossRef]
- Maas, G.; Stang, P.J. Dication disulfides by reaction of thioureas and related compounds with trifluoromethanesulfonic anhydride. The role of triflic anhydride as an oxidizing agent. J. Org. Chem. 1981, 46, 1606−1610.
- Michalak, R.S.; Martin, J.C. Persulfonium salts: the reaction of a difluoropersulfurane with Lewis acids. J. Am. Chem. Soc. 1980, 102, 5921−5923. [CrossRef]
- Della Védova, C.O.; Downs, A.J.; Novikov, V.P.; Oberhammer, H.; Parsons, S.; Romano, R. M.; Zawadski, A. Fluorocarbonyl Trifluoromethanesulfonate, FC(O)OSO2CF3: Structure and Conformational Properties in the Gaseous and Condensed Phases. Inorg. Chem. 2004, 43 (13), 4064–4071.
- Della Védova, C.O.; Downs, A.J.; Moschione, E.; Parsons, S.; Romano, R. M. Chlorocarbonyl Trifluoromethanesulfonate, ClC(O)OSO2CF3: Structure and Conformational Properties in the Gaseous and Condensed Phases. Inorg. Chem. 2004, 43 (25), 8143–8149.
- Trautner, F.; Della Védova, C.O.; Romano, R.M.; Oberhammer, H. Gas phase structure and conformational properties of chlorocarbonyl trifluoromethanesulfonate, ClC(O)OSO2CF3. J. Mol. Struct. 2006, 784 (1−3), 272–275. [CrossRef]
- Romano, R.M.; Moreno Betancourt, A.; Della Vedova, C.O.; Zeng, X.; Beckers, H.; Willner, H.; Schwabedissen, J.; Mitzel, N.W. Preparation and properties of chlorosulfuryl chloroformate, ClC(O)OSO2Cl. Inorg. Chem. 2018 57(23), 14834−14842. [CrossRef]
- Della Védova, C.O.; Mack, H.G. A matrix photochemistry study on (fluorocarbonyl) sulfenyl bromide: the precursor of sulfur bromide fluoride. Inorg. Chem. 1993, 32 (6), 948−950. [CrossRef]
- Tamone, L.M.; Picone, A.L.; Romano, R.M. New insights into the Ar-matrix-isolation FTIR spectroscopy and photochemistry of dichloroacetyl chloride, ClC(O)CHCl2: Influence of O2 and comparison with gas-phase photochemistry. J. Photochem. Photobiol. 2021, 6, 100019. [CrossRef]
- Bava, Y.B.; Cozzarín, M.V.; Della Védova, C.O.; Willner, H.; Romano, R.M. Preparation of FC(S)SF, FC(S) SeF and FC(Se)SeF through matrix photochemical reactions of F2 with CS2, SCSe, and CSe2. Phys. Chem. Chem. Phys 2021, 23 (37), 20892−20900.
- Romano, R.M.; Della Vedova, C.O.; Downs, A.J.; Greene, T.M. Matrix Photochemistry of syn-(Chlorocarbonyl) sulfenyl Bromide, syn-ClC(O) SBr: Precursor to the Novel Species anti-ClC(O)SBr, syn-BrC(O)SCl, and BrSCl. J. Am. Chem. Soc. 2001, 123 (24), 5794−5801.
- Forbus, T.R.; Martin, J.C. Trifluoroacetyl Triflate: An Easily Accessible, Highly Electrophilic Trifluoroacetylating Agent. J. Org. Chem. 1979, 44 (2), 313–314. [CrossRef]
- Taylor, S.L.; Forbus, T.R.; Martin, J.C. Trifloroacetyl Triflate: Acetic Acid, Trifluoro-, Anhydride with Trifluoromethanesulfonic Acid. Organic Syntheses; Kende, A.S., Freeman, J.P., Eds.; Wiley, 2003; 217.
- Taylor, S.L., Forbus, T.R. Jr, Martin, J.C. Trifluoroacetyl Triflate. Org. Synth. 1986, 64, 217.
- Gaussian 09, Revision A.02, Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; Li, X.; Caricato, M.; Marenich, A.; Bloino, J.; Janesko, B. G.; Gomperts, R.; Mennucci, B.; Hratchian, H.P.; Ortiz, J.V.; Izmaylov, A.F.; Sonnenberg, J.L.; Williams-Young, D.; Ding, F.; Lipparini, F.; Egidi, F.; Goings, J.; Peng, B.; Petrone, A.; Henderson, T.; Ranasinghe, D.; Zakrzewski, V. G.; Gao, J.; Rega, N.; Zheng, G.; Liang, W.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Throssell, K.; Montgomery, J.A. Jr., Peralta, J.E.; Ogliaro, F.; Bearpark, M.; Heyd, J.J.; Brothers, E.; Kudin, K.N.; Staroverov, V.N.; Keith, T.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J.C.;Iyengar, S.S.; Tomasi, J.; Cossi, M.; Millam, J.M.; Klene, M.; Adamo, C.; Cammi, R.; Ochterski, J.W.; Martin, R.L.; Morokuma, K.; Farkas, O.; Foresman, J.B.; Fox D.J. Gaussian, Inc., Wallingford CT, 2016.
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822−8824. [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785−789. [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098−3100. [CrossRef]
- Reed, A.E; Curtiss, L.A.; Weinhold, F. Chem. Rev. 1988, 88, 899−926.
- Della Védova, C.O.; Rubio, R.E. Photolysis of matrix-isolated perfluoroacetyl chloride, CF3C(O)Cl. J Mol. Struct. 1994, 321, 279−281. [CrossRef]
Scheme 1.
Synthesis of trifluoroacetyl triflate.
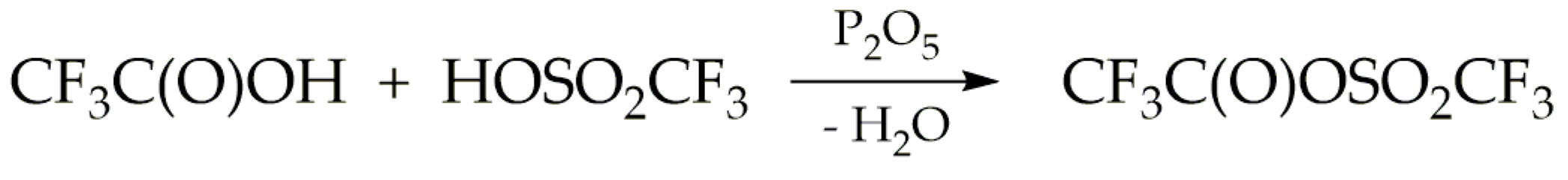
Scheme 2.
Secondary reactions in the synthesis of trifluoroacetyl triflate.
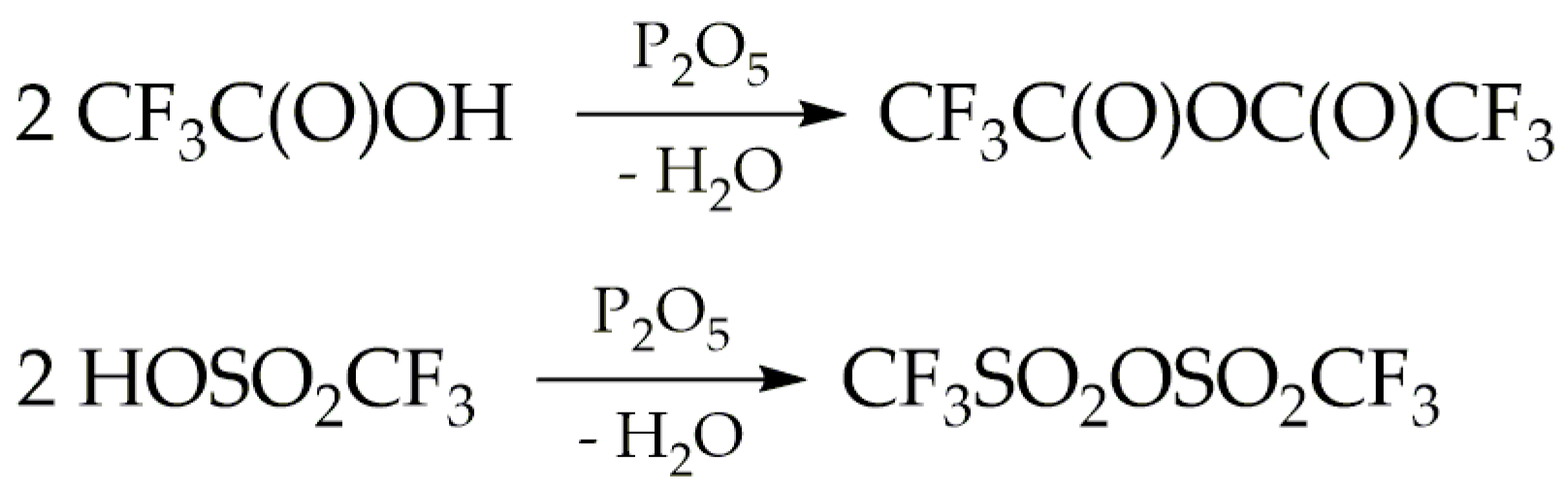
Figure 1.
Definition of the dihedral angles τ1 = CC−OS and τ2 = CO−SC of trifluoroacetyl triflate.
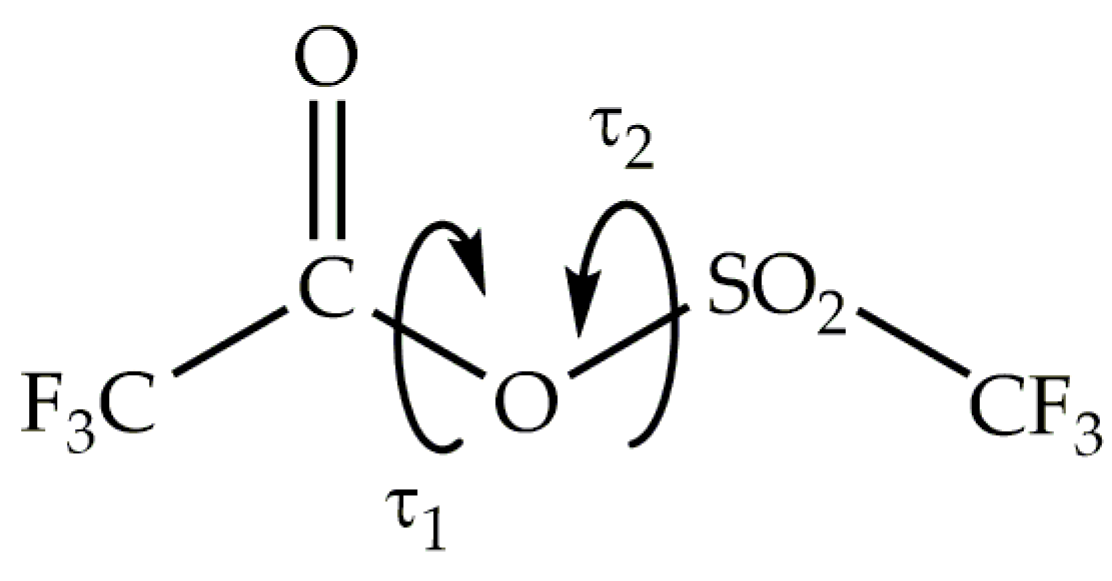
Figure 2.
(a) Potential energy surface and (b) contour map computed with the B3P86 /6-31+g(d) approximation as a function of the dihedral angles τ1 = CC−OS and τ2 = CO−SC of trifluoroacetyl triflate varied from 0 to 360° in steps of 10°.
Figure 2.
(a) Potential energy surface and (b) contour map computed with the B3P86 /6-31+g(d) approximation as a function of the dihedral angles τ1 = CC−OS and τ2 = CO−SC of trifluoroacetyl triflate varied from 0 to 360° in steps of 10°.
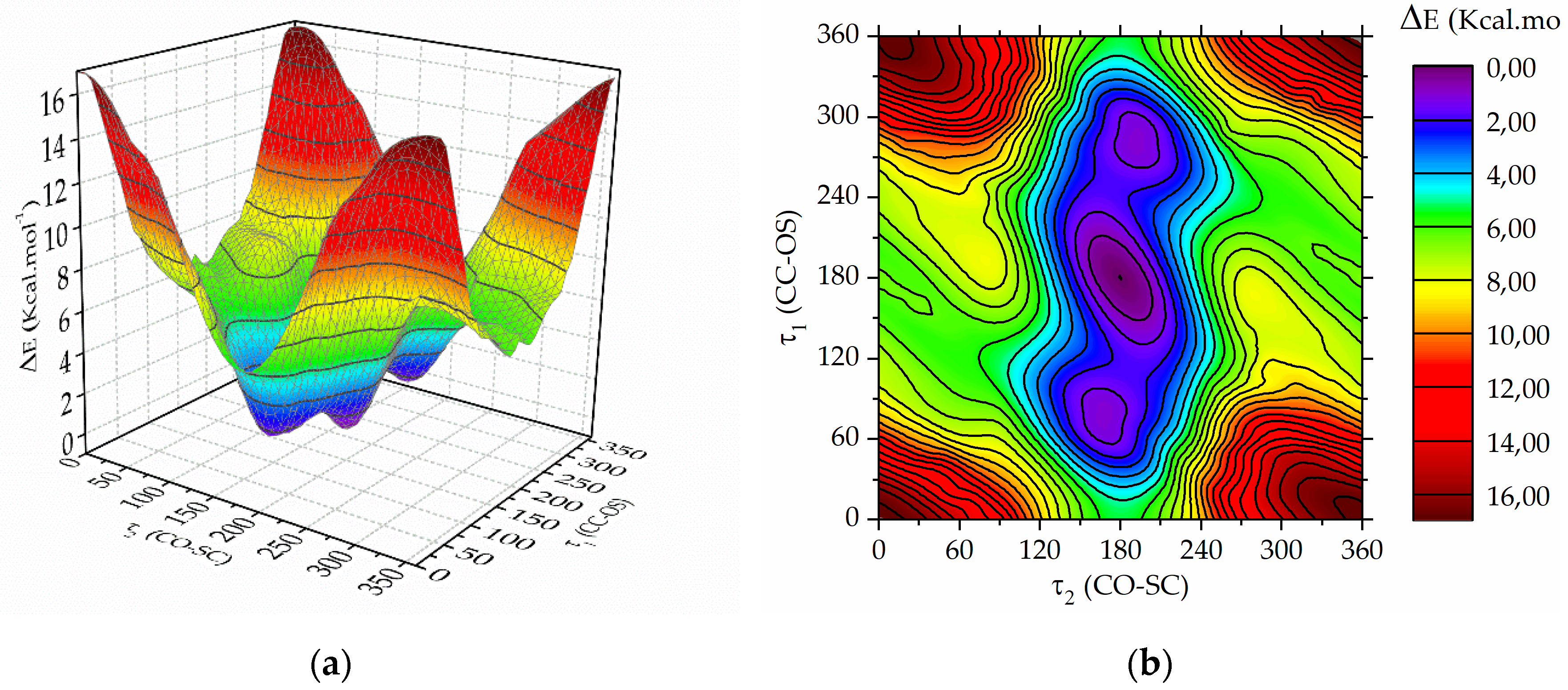
Figure 3.
Molecular models of the conformers of trifluoroacetyl triflate computed with the B3LYP/cc-pvtz approximation: (a) syn-anti (the C=O double bond syn with respect to the O−S single bond and the C−O single bond anti with respect to the S−C bond); (b) syn-gauche (the C=O double bond syn with respect to the O−S single bond and the C−O single bond gauche with respect to the S−C bond).
Figure 3.
Molecular models of the conformers of trifluoroacetyl triflate computed with the B3LYP/cc-pvtz approximation: (a) syn-anti (the C=O double bond syn with respect to the O−S single bond and the C−O single bond anti with respect to the S−C bond); (b) syn-gauche (the C=O double bond syn with respect to the O−S single bond and the C−O single bond gauche with respect to the S−C bond).
Figure 4.
Gas-phase FTIR spectrum (blue-trace, pressure 1.7 mbar, 0.5 cm−1 resolution, 64 scans), Ar-matrix FTIR spectrum (red-trace, 1:1000, 0.5 cm−1 resolution, 256 scans) and liquid-phase Raman spectrum (green-trace, λexc. 514.5 nm) of trifluoroacetyl triflate.
Figure 4.
Gas-phase FTIR spectrum (blue-trace, pressure 1.7 mbar, 0.5 cm−1 resolution, 64 scans), Ar-matrix FTIR spectrum (red-trace, 1:1000, 0.5 cm−1 resolution, 256 scans) and liquid-phase Raman spectrum (green-trace, λexc. 514.5 nm) of trifluoroacetyl triflate.
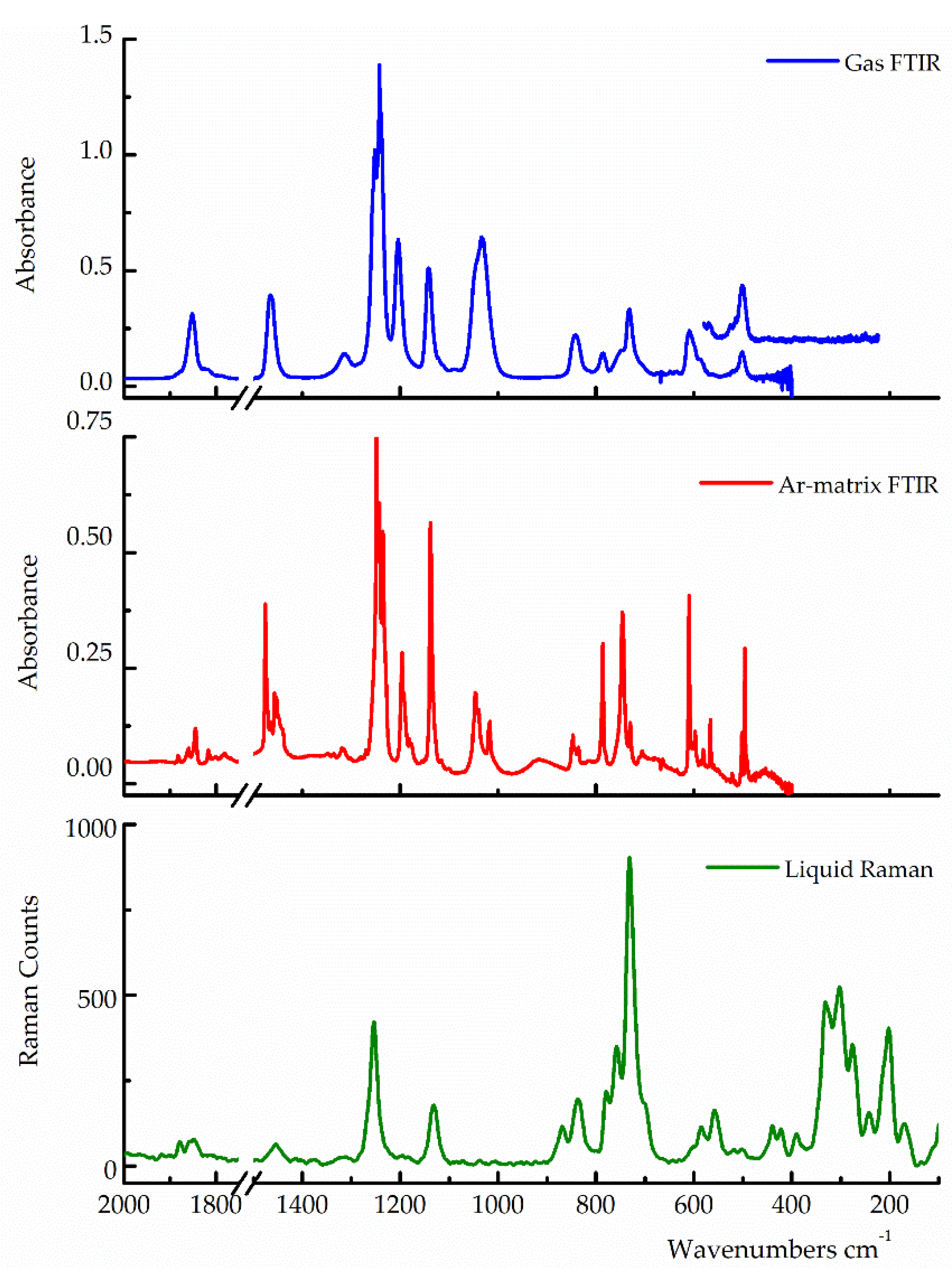
Figure 5.
Simulated IR spectra in selected regions for the conformers of TFAT, scaled by their relative abundances at 25 °C, calculated with the B3LYP/cc-pvtz approximation: syn-anti conformer, blue spectrum scaled by 0.69 factor; syn-gauche, green spectrum scaled by 0.31 factor; weighted sum of the spectra of the two conformers, red spectrum.
Figure 5.
Simulated IR spectra in selected regions for the conformers of TFAT, scaled by their relative abundances at 25 °C, calculated with the B3LYP/cc-pvtz approximation: syn-anti conformer, blue spectrum scaled by 0.69 factor; syn-gauche, green spectrum scaled by 0.31 factor; weighted sum of the spectra of the two conformers, red spectrum.
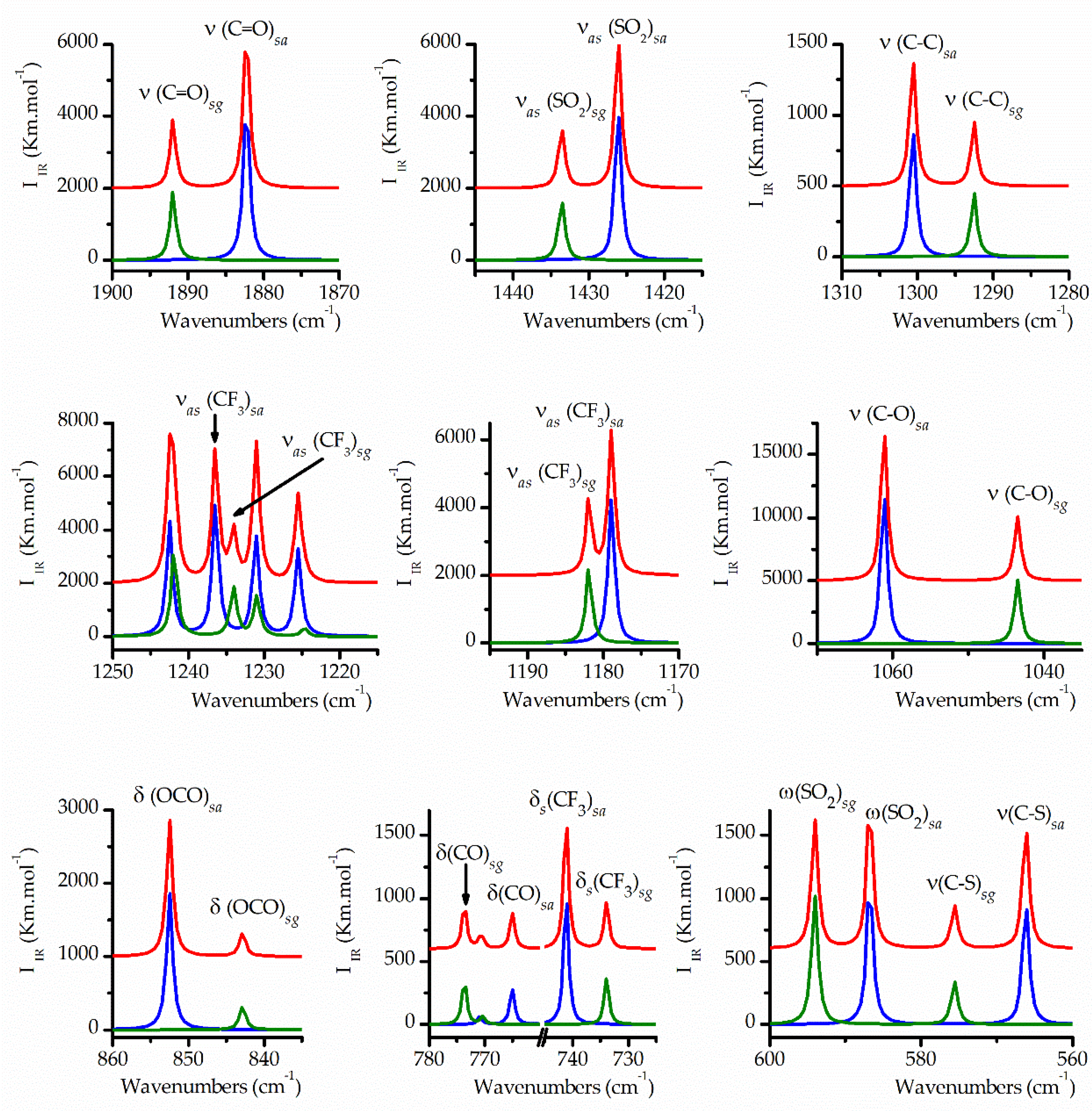
Figure 6.
Selected regions of FTIR spectra of an Ar-matrix containing TFAT in 1:1000 proportion: after deposition (black trace) and 12 (red trace) and 50 minutes (blue trace) photolysis with broad-band UV-visible light. The spectra are normalized to the intensities of the absorption assigned to the syn-anti conformer.
Figure 6.
Selected regions of FTIR spectra of an Ar-matrix containing TFAT in 1:1000 proportion: after deposition (black trace) and 12 (red trace) and 50 minutes (blue trace) photolysis with broad-band UV-visible light. The spectra are normalized to the intensities of the absorption assigned to the syn-anti conformer.
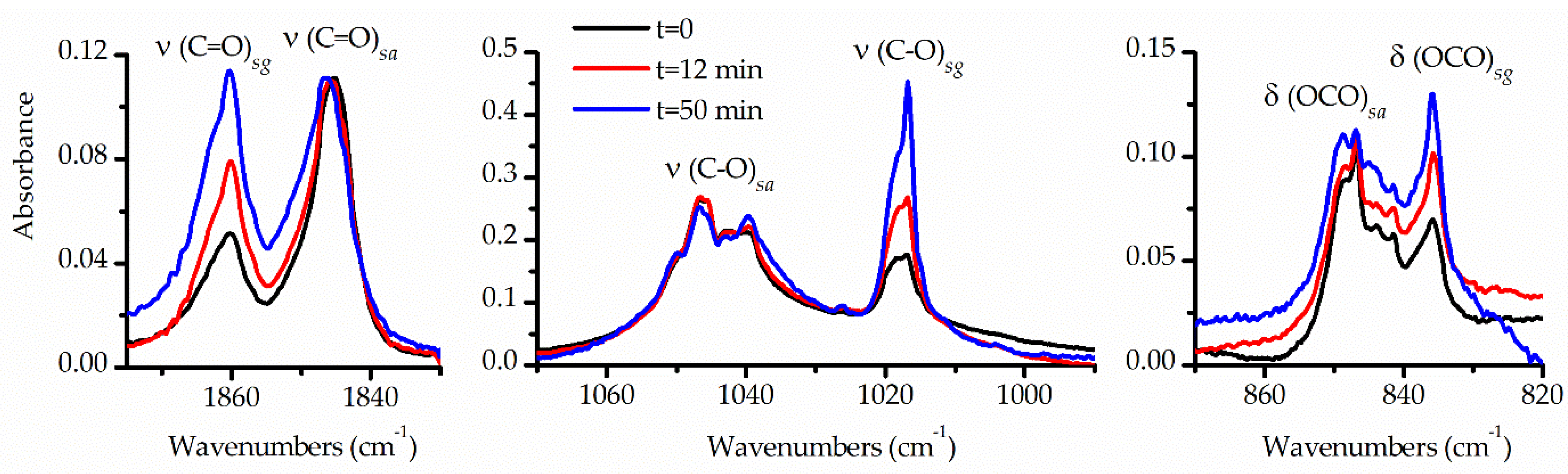

Table 1.
Energy differences and Gibbs free energy differences between syn-gauche and syn-anti conformers of trifluoroacetyl triflate and % of the syn-anti rotamer calculated with B3LYP theoretical approximation and different basis sets.
Table 1.
Energy differences and Gibbs free energy differences between syn-gauche and syn-anti conformers of trifluoroacetyl triflate and % of the syn-anti rotamer calculated with B3LYP theoretical approximation and different basis sets.
| Theoretical approximation | ΔE (Kcal.mol−1) 1 | ΔG° (Kcal.mol−1) 2 | % syn-anti 3 |
|---|---|---|---|
| B3LYP/6-31++G(d) | 1.26 | 2.22 | 96 |
| B3LYP/6-311++G(d) | 1.23 | 2.02 | 94 |
| B3LYP/tzvp | 1.11 | 1.34 | 83 |
| B3LYP/cc-pvtz | 0.87 | 0.88 | 69 |
1 ΔE = E(syn-gauche) − E(syn-anti); 1 ΔG° = G°(syn-gauche) − G°(syn-anti); 3 Calculated at ambient temperature (25 °C) and considering degeneracy 2 for the syn-gauche conformer.
Table 2.
Main orbital interaction energies (Kcal.mol−1) involved in the stabilization of the syn-anti and syn-gauche conformers of trifluoroacetyl triflate, calculated with the NBO formalism using the B3LYP/cc-pvtz approximation.
Table 2.
Main orbital interaction energies (Kcal.mol−1) involved in the stabilization of the syn-anti and syn-gauche conformers of trifluoroacetyl triflate, calculated with the NBO formalism using the B3LYP/cc-pvtz approximation.
| Orbital interaction 1 | syn-anti | syn-gauche |
|---|---|---|
| lpπO6 → π*C2=O8 | 39.20 | 35.65 |
| lpσO6 → σ*C2=O8 | 7.20 | 7.48 |
| lpσO6 → σ*S=O4 | 5.42 | − |
| lpσO6 → σ*S=O5 | 5.42 | − |
| lpσO6 → σ*S−C3 | − | 5.93 |
| Total anomeric effect | 18.04 | 13.41 |
| Total hyperconjugative effect | 39.20 | 35.65 |
| Total | 57.24 | 49.06 |
1 Atom numbering refers to Figure 3.
Table 3.
Experimental wavenumber (gas FTIR, Ar-matrix FTIR and liquid Raman) in cm−1 of trifluoroacetyl triflate, comparison with the calculated wavenumbers using the B3LYP/cc-pvtz approximation for the syn-anti and syn-gauche conformers, and tentative assignment.
Table 3.
Experimental wavenumber (gas FTIR, Ar-matrix FTIR and liquid Raman) in cm−1 of trifluoroacetyl triflate, comparison with the calculated wavenumbers using the B3LYP/cc-pvtz approximation for the syn-anti and syn-gauche conformers, and tentative assignment.
| Experimental | B3LYP/cc-pvtz | Tentative assignment | |||
|---|---|---|---|---|---|
| Gas-FTIR ν (cm−1) |
Ar-matrix FTIR ν (cm−1)a |
Liquid Raman ν (cm−1) | syn-anti | syn-gauche | |
| 1852 | 1860 | 1880 | 1892 | ν C=O syn-gauche | |
| 1845 | 1851 | 1882 | ν C=O syn-anti | ||
| 1465 | 1475 | 1455 | 1434 | νas SO2 syn-anti | |
| 1453 | 1426 | νas SO2 syn-gauche | |||
| 1314 | 1319 | 1317 | 1301 | ν C−CF3 syn-anti | |
| 1315 | 1293 | ν C−CF3 syn-gauche | |||
| 1252 | 1249 | 1242 | νas CF3 (−SO2) syn-anti | ||
| 1248 | 1241 | νas CF3 (−SO2) syn-gauche | |||
| 1241 | 1243 | 1266 | 1236 | νas CF3 (−C=O)syn-anti | |
| 1241 | 1234 | νas CF3 (−C=O) syn-gauche | |||
| 1234 sh | 1236 | 1231 | 1231 | νas CF3 (−SO2) | |
| 1228 | 1254 | 1225 | 1225 | νs SO2 syn-anti | |
| 1204 | 1200 | 1182 | νas CF3 (−C=O) syn-gauche | ||
| 1196 | 1179 | νas CF3 (−C=O) syn-anti | |||
| 1142 | 1136 | 1133 | 1106 | 1106 | νs CF3 (−SO2) |
| 1040 | 1047 | 1061 | ν C−O syn-anti | ||
| 1018 | 1043 | ν C−O syn-gauche | |||
| 842 | 847 | 840 | 852 | δ OCO syn-anti | |
| 836 | 843 | δ OCO syn-gauche | |||
| 788 | 788 | 780 | 774 | δo.o.p. (C=O) syn-gauche | |
| 786 | 765 | δo.o.p. (C=O) syn-anti | |||
| 779 sh | 780 | 770 | 770 | νs CF3 (−SO2) | |
| 751 | 746 | 759 | 741 | δs CF3 (−C=O)syn-anti | |
| 733 | 730 | 734 | δs CF3 (−C=O)syn-gauche | ||
| 704 sh | 705 | 733 | 686 | 686 | ν S−O |
| 611 | 611 | 594 | ω SO2 syn-gauche | ||
| 609 | 587 | ω SO2 syn-anti | |||
| 600 | 601 | 585 | 575 | ν C−Ssyn-gauche | |
| 598 | 566 | ν C−Ssyn-anti | |||
| 585 sh | 580 | 556 | 554 | δ CF3 (−SO2) | |
| 566 | 566 | 559 | 544 | 547 | δ CF3 (−SO2) |
| 522 | 521 | 519 | 517 | δ CF3 (−C=O) | |
| 498 | 496 | 489 | δ SO2 syn-anti | ||
| 492 | 487 | δ SO2 syn-gauche | |||
| 438 | 432 | 428 | δ O=S=O | ||
| 421 | 415 | 381 | δ C−C=O | ||
| 330 | 325 | 335 | δ F−C−C | ||
| 302 | 300 | 284 | δ F−C−C | ||
| 276 | 257 | 277 | δ F−C−S | ||
| 242 | 236 | 243 | ω CF3 (−C=O) | ||
| 202 | 186 | 200 | δ C−O−S | ||
| 170 | 159 | 149 | δ O−S−C | ||
a Only the most intense matrix sites are listed.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



