Submitted:
25 April 2024
Posted:
26 April 2024
You are already at the latest version
Abstract
LONP1 is the principal AAA+ unfoldase and bulk protease in the mitochondrial matrix, so its deletion causes embryonic lethality. The AAA+ unfoldase CLPX and the peptidase CLPP also act in the matrix, conspicuously during stress periods, but their substrates are poorly defined. Mammalian CLPP deletion triggers infertility, deafness, growth retardation, and cGAS-STING activated cytosolic innate immunity. CLPX mutations impair heme biosynthesis and heavy metal homeostasis. CLPP and CLPX are conserved from bacteria to human, despite their secondary role for proteolysis. Based on recent proteomic-metabolomic evidence from knockout mice and patient cells, we propose that CLPP acts on phase-separated ribonucleoprotein granules, and CLPX on multi-enzyme condensates, near the inner mitochondrial membrane, as first-aid system. Trimming within assemblies, CLPP rescues stalled processes in mitoribosomes, in mitochondrial RNA granules and nucleoids, and in D-foci-mediated degradation of toxic double-stranded mtRNA / mtDNA. Unfolding multi-enzyme condensates, CLPX maximizes PLP-dependent delta-transamination, and rescues malformed nascent peptides. Overall, their actions occur in granules with multivalent or hydrophobic interactions, separated from the aqueous phase. Thus, the role of CLPXP in the matrix is compartment-selective, like peptidases MPP at precursor import pores, m-AAA and i-AAA at either IMM face, PARL within the IMM, and OMA1/HTRA2 in the intermembrane space.
Keywords:
Perrault syndrome type 3 (PRLTS3)
; iron toxicity
; pyridoxal-5'-phosphate
; VWA8
; GFM1
; PNPT1
; RNA-G4
; ISC
; ALAS
; OAT
1. CLPP is a Key Modifier of Growth and Lifespan, But Its Substrates Remain Unclear
LONP1 is the principal proteolysis factor of the mitochondrial matrix, combining an AAA+ ATPase unfoldase domain with a serine peptidase domain within the same protein sequence. LONP1 homo-multimerizes in ring- or barrel-shape to maximize its efficiency, and plays the crucial role for the turnover of respiratory chain complexes and most other proteins in this compartment [1,2,3]. According to studies in bacteria, CLPXP is perceived as similar but stress-responsive proteolysis machine, also in the mitochondrial matrix. However, CLPX as AAA+ ATPase unfoldase component and CLPP as serine peptidase component are separate proteins. To obtain proteolytic capacity, via assembly in a barrel-like conformation in analogy to LONP1, they can hetero-multimerize. Nonetheless, in proteolysis they play a secondary role, becoming prominent only after cellular stress [4]. Both systems have been conserved from bacteria to human, so each of them has to play very relevant roles in the mitochondrial matrix. Indeed, the loss of LONP1 in homozygous state causes lethality already at early embryonic development [1]. In contrast, the loss of CLPP was observed to extend lifespan in the eukaryotic fungus Podospora anserina and CLPP is constitutively absent from the yeast Saccharomyces cerevisiae [5,6]. This emphasizes a dramatic difference in functional impact for these two systems, at odds with the concept that both act similarly in proteolytic degradation.
Genetic analyses of human diseases recently showed mild LONP1 mutations to be responsible for CODAS syndrome, where craniofacial dysmorphia, cataracts, ptosis, median nasal groove, delayed tooth eruption, delayed epiphyseal ossification, metaphyseal hip dysplasia, vertebral coronal clefts, short stature, psychomotor and developmental delay, with hearing loss are diagnostic hallmarks [7,8]. In contrast, CLPP mutations cause Perrault syndrome type 3 (PRLTS3) [9,10,11,12,13,14,15]. Perrault syndrome was clinically and genetically defined, as the combination of early female infertility due to primary ovarian failure, with subsequent onset of progressive sensorineural hearing impairment, and autosomal recessive inheritance. Later it was observed that not only deafness, but also sensory neuropathy, ataxia and brain white matter changes can appear as neurodegenerative features [16,17,18,19,20]. Judging by human genetics, the functions of LONP1 and CLPP therefore appear to target different pathways and their dysfunctions have widely different consequences.
Genetic causes of PRTLS are almost exclusively caused by mtDNA / mtRNA or mitoribosome machinery errors [13,21]. The role of mtDNA for Perrault syndrome pathogenesis is substantiated by causal mutations in the mitochondrial DNA/RNA helicase TWNK/PEO1, and in the mitochondrial transcription factor TFAM [22,23]. The role of mtRNA processing is corroborated by causal mutations in the mitochondrial rRNA chaperone ERAL1 and the mitochondrial RNase P component PRORP [16,24,25]. The role of mt-tRNA processing and mitoribosomal translation is evident from causal mutations in the mitochondrial tRNA-aminoacid ligases HARS2 and LARS2, as well as the mitoribosome-associated factor RMND1 [13,26,27,28,29,30,31,32,33,34,35]. Detailed correlation of mutant mitochondrial factors with the consequent phenotypes (Figure 4 in [36]) indicates that primary infertility is mostly due to abnormal mtDNA or mt-tRNA processing, whereas hearing impairment is frequently due to abnormal mtRNA processing or mitoribosomal translation. Thus, CLPP appears to selectively modulate mitochondrial RNA processing and translation.
Again in complete phenotypic contrast, human CLPX mutations were observed to cause erythropoietic protoporphyria 2 (leading to acute skin photosensitivity, mild microcytic anemia, and rarely, severe liver disease) [37,38]. The credibility of these findings is enhanced by observations from yeast to human that CLPX, independent from CLPP, activates heme biosynthesis [5,37,39].
The recent generation of several independent Clpp-knockout (KO) mouse lines, by Gene-Trap random insertion [40] and by targeted conditional technology [41], provided authentic genetic animal models of PRLTS3 with the characteristic phenotypes, and allowed the elucidation of the underlying molecular and functional deficits. Independently with perfect agreement, biochemical analyses of each research team showed CLPP homo-multimer rings to exist normally without CLPX association in blue-native electrophoresis where endogenous protein complexes are resolved according to their interaction stability and molecular weight (Figure 2 in [42], Supplementary Figure EV3B in [43]). Thus, in unstressed mammalian cells, CLPP rings do not associate with to the energy-providing AAA+ ATPase CLPX, and cannot perform the degradation of protein substrates that would require ATP hydrolysis [4]. Therefore, CLPP functions would normally be limited to act as peptidase like chymotrypsin [44], trimming proteins or multi-protein assemblies, rather than completely eliminating them. This concept is in agreement with a recent review where the role of CLPX and CLPP was seen in the fine-tuning of mitochondrial matrix multi-protein assemblies rather than in proteolysis [44]. CLPX as monomer or homo-multimer ring would then employ its energy from ATP hydrolysis to unfold proteins or protein complexes without subsequent destruction. Of course, CLPX and CLPP may join forces under conditions of cell stress, e.g. when mitoribosomal translation is stalled and a misfolded nascent polypeptide has to be degraded, or after cell damage to disaggregate and cleave toxic oligomers of ribonucleoproteins. An illustrated synopsis of this emerging scenario is provided in Figure 1, and the detailed evidence is compiled in the subsequent text paragraphs with citations of recent literature.
To elucidate the exact roles of CLPP serves an urgent unmet medical need, given that CLPP-activity modulation by drugs is consistently observed as very efficacious to counteract solid cancers [45,46,47]. Indeed, the effect of CLPP on cell growth is strong even under physiological conditions: its dysfunction leads to short stature of patients [15]. In the PRLTS3 mouse models, a reduction of weight up to 50% was observed, together with an underlying similar decrease of the nitrogen-storing amino acid L-arginine, which gets consumed in the maximized biosynthesis of heme instead of fueling growth [48]. In a preliminary meta-analysis of CLPP substrate trapping experiments and of CLPP-null proteome profiling in many organisms from bacteria to man, an enrichment of ribonucleoproteins was observed, with the unfoldase CLPX, the mitoribosomal translation factor GFM1 and the RNA degradation factor PNPT1 emerging as most consistent proteins that interact with CLPP and show accumulation upon its loss [36].
2. Novel Evidence on CLPP and CLPX Functions from Clpp-KO Mice and PRLTS3 Patients
2.1. Prominent Impact of CLPP Absence and CLPX Excess on Mitochondrial Nucleoids
Upon the first generation of two independent Clpp-KO mouse embryonic stem cells by inactivating gene-trap insertions in intron 1 and 2 at the Texas Institute of Genomic Medicine (TIGM), the derived mice were shown to serve as authentic model of Perrault syndrome [40]. These homozygous Clpp-KO mice exhibited complete infertility even at earliest age, an average weight reduction to 70% and length reduction to 90% at ages from 12 weeks, impaired locomotor activity by the age of 6 months, sensorineural hearing impairment by ages from 12 to 18 months, and a relative resistance to microbial infections [40]. In contrast to other forms of Perrault syndrome with exclusively female infertility due to primary ovarian insufficiency, CLPP absence according to mouse data causes also male infertility due to azoospermia after diplotene arrest [49]. Further analyses of the lean phenotype of several Clpp-KO mouse lines showed a protection from diet-induced obesity and from insulin resistance, but also a deficit to adapt body thermogenesis [50,51].
The molecular analyses of tissues showed the absence of CLPP to cause a >3-fold accumulation for the unfoldase CLPX, together with increased amounts of the mitochondrial protein chaperone mtHSP75 (but not HSPD1) [40,52]. This corresponds partially to previous observations in C. elegans studies of the unfolded protein response of mitochondria (UPRmt), where hsp-6 and hsp-60 were induced [53]. Beyond the expected impairment of proteostasis, careful quantification of mtDNA with qPCR in testis, ovary, heart, brain, liver and blood demonstrated a 1.5- to 4-fold increase of mtDNA copy number [53]. This observation was not reproduced in mice with conditionally targeted Clpp deletion when full-length mtDNA from heart muscle was assessed by Southern blotting [43], but was confirmed by an independent team in white adipose tissue from the Gene-Trap Clpp-KO mice with qPCR [51], and also in CLPP-mutant patient skin fibroblasts by qPCR [54]. Furthermore, the patient fibroblast analysis by microscopy demonstrated an enlargement of the nucleoid area with an apparent elevation of mtDNA signals [54]. It is important to note that the increased mtDNA copy number is not accompanied by an elevated abundance of TFAM as its primary binding partner. Instead, proteome profiling of targeted Clpp-KO mouse heart tissue, Gene-Trap Clpp-KO mouse embryonic fibroblasts, and patient fibroblasts, documented prominent accumulation of the nucleoid factor POLDIP2 [52,54,55], a protein known to associate with mtSSB [55] and with CLPX, which maximizes activity as well as stability of CLPXP [56]. Mechanistic analyses of the drug ZG36, which acts as CLPP agonist, showed a converse impact with reduction of mtDNA to half [57]. In addition, in Clpp-KO testis at early stages of spermatogenesis, a consistent accumulation was observed for the Twinky isoform of the mtDNA helicase TWNK/PEO1, which differs from the Twinkle isoform by absent binding to the D-loop [58]. This finding appears particularly relevant given that TWNK mutations can cause Perrault syndrome [23,59,60,61,62,63,64,65,66].
These observations are compatible with the concept that CLPP absence results in an increased dosage of mtDNA fragments rather than full-length copies, in a manner that their assembly with associated proteins is impaired, and the generation of the polycistronic transcript may be affected.
2.2. Prominent Impact of CLPP Absence and CLPX Excess on Mitoribosomes, Mostly on the tRNA-/mRNA-Associated and rRNA-Containing mtSSU
Further evidence that CLPP and CLPX target granular components of the mitochondrial matrix was reported for mitoribosomes, initially in the targeted Clpp-KO mouse [43] and then in the Gene-Trap Clpp-KO mouse as well [42].
Regarding ribosomal RNA, the 12S rRNA and the MRPS15-MRPS35 protein components of the mitoribosomal 28S small subunit (mtSSU) showed much more elevated abundance than the 16S rRNA and the MRPL12-MRPL37 protein component of the large subunit of mitoribosomes (mtLSU) in Clpp-KO mice [41]. Further analyses of the co-migration of mitochondrial proteins in blue-native gel electrophoresis to define interaction in assembled complexes confirmed a general accumulation of all components of the mtSSU in Clpp-KO testis, brain and heart, in the absence of CLPX co-migration [42].
Regarding the translation-associated enzymes, the elongation factor GFM1 (also known as EFG1, ortholog of bacterial fusA-encoded EF-G, see [67]), exhibited elevated protein abundance in Clpp-KO mice as well, together with abnormal sedimentation in sucrose gradients [41]. Indeed, the co-accumulation of CLPX together with its interactor GFM1 was subsequently confirmed in proteome profiling studies of mouse brain, MEFs, and patient fibroblasts [54]. This is in agreement with the notion that CLPX acts not only in heme biosynthesis, but is also able to target the GFM1-associated L7/L12 stalk and central protuberance of mitoribosomes to act in the translation elongation / recycling apparatus [68,69,70,71,72].
These findings identify the molecular details that underlie previous observations that CLPXP is necessary to rescue stalled translation complexes by unfolding the mitoribosome, so that translation elongation via addition of a CAT-tail to the nascent misfolded polypeptide can occur. CLPXP then eliminates this aberrant translation product before its aggregation tendency has toxic effects [73,74,75,76].
2.3. Prominent Impact of CLPP Absence and CLPX Excess on Mitochondrial RNA Processing Granules
A third line of evidence on the role of CLPP for mitochondrial matrix granules concerns the RNA processing compartment. It was observed that mtSSU rRNA accumulates in the Clpp-KO mice [41]. ERAL1 serves as mitochondrial rRNA chaperone while the 12S rRNA associates with ribonucleoproteins to form the mtSSU. Indeed, ERAL1 exhibited not only elevated protein abundance in Clpp-KO heart mitochondria, but also abnormal sedimentation in sucrose gradients [41]. Again, ERAL1 accumulation appears particularly relevant, given that ERAL1 mutations can trigger Perrault syndrome [25,77,78,79].
Clpp-KO triggered accumulation was also documented for a few mitochondrial tRNAs [41]. In particular, the tRNAs for valine (Val) and phenylalanine (Phe) exhibited higher aminoacylation in Clpp-KO heart [41]. Therefore, it is interesting that a very selective protein accumulation exists for mtLSU components like MRPL18 and MRPL38, which assemble with tRNA-Val/Phe in the central protuberance of mitoribosomes, and that this CLPP-null effect on central protuberance subunits of the mtLSU is conserved across eukaryotes until the ascomycete fungus Podospora anserina [42,48]. This selective impact on the mtLSU may also be relevant for mtDNA and the cytosolic stress response: MRPL38 influences the maintenance of the mitochondrial nucleoid at least in yeast [80]. Furthermore, there is a cytosolic isoform of MRPL18, which modulates ribosomal translation of molecular chaperones after cell stress [81] and can thus influence UPR outside of mitochondria.
A key role of tRNA-associated pathology in CLPP-dependent pathogenesis is evident also from human genetics data. Mutations in the mitochondrial tRNA-aminotransferases for histidine and leucine, HARS2 and LARS2, cause the typical features of Perrault syndrome [21,30,82,83,84,85,86], while mutations in the mitochondrially encoded tRNA sequences trigger different mostly neurodegenerative phenotypes that include progressive deafness [87]. Mirroring a joint pathogenetic pathway for different variants of Perrault syndrome, Clpp-KO testes from early stages of spermatogenesis contain elevated amounts of HARS2 [42]. It is curious to note furthermore that the deleterious effects of mutations in DARS2 (mitochondrial tRNA-aspartate aminotransferase) in mouse can be partially rescued by CLPP absence [41,88], so in conditions of enhanced mitochondrial RNA processing and translation blockade, it can be advantageous to have a CLPP loss-of-function that reduces UPRmt and prolongs lifespan.
The folding of mitochondrial tRNAs and rRNAs represents another pathway that is affected both by the impact of CLXP on heme and by the impact of CLPP on ribonucleoprotein condensates. Bacterial tRNA and rRNA contain guanine-rich sequences that can adopt quadruplex structures [89]. Also for mammalian cytosolic ribosomes, the importance of such rRNA quadruplexes for the mature conformation was already documented [90]. When guanine-rich sequences adopt quadruplex conformation (G-tetrads) with four RNA or DNA strands (RNA-G4 or DNA-G4), their structure can be stabilized by association with quadrangular porphyrin and heme molecules [90,91,92,93]. This interaction may activate peroxidase- or oxidase-mimicking features in this DNAzyme/RNAzyme complex [92,93], may modify the compaction and processing of DNA/RNA [94], and is crucial in ribosomes for optimal translation efficiency [95]. The high abundance of such rRNA-G4 structures even limits the bioavailability of heme in cells [90]. This pathway seems to be altered in PRLTS3, in view of the selective accumulation of RNA granule factor GRSF1 (G-Rich Sequence Factor 1) in Clpp-KO tissues [41,54]. Within mitochondria, GRSF1 is responsible for non-coding RNA in G4 conformation [96,97]. GRSF1 interacts with RNase P to influence the cleavage of polycistronic transcripts [98] and its dysfunction leads to RNA processing defects, accumulation of mtRNA breakdown products as well as increased levels of dsRNA and RNA:DNA hybrids [99]. These problems lead to the formation of distinct mitochondrial dsRNA foci [100]. In addition, GRSF1 dysfunction triggers abnormal loading of mRNAs and lncRNAs on the mitochondrial ribosome, and impaired ribosome assembly [101]. GRSF1 also influences the degradation of mtRNA in the degradosome in cooperation with PNPT1 [96,102]. Overall, it is not surprising that GRSF1 is also involved in iron toxicity like CLPX [103], and in lean body phenotypes like CLPP [104,105]. GRSF1 was observed in protein-protein-interactions with CLPX [36].
RNA-G4 structures also control the activity of the mitochondrial GTPase NOA1 (also known as C4ORF14) for mitoribosomal assembly [106,107,108]. NOA1 was also identified as a CLPXP target protein [109].
The joint roles of absent CLPP and excess CLPX during the assembly of mitoribosomes are further supported by the selective accumulation of VWA8 in Clpp-KO tissues [42]. The mitochondrial matrix protein VWA8 [110] contains a domain related to porphyrin chelatases [42], so it might interact with heme or its precursors. VWA8 also contains an AAA+ unfoldase domain, whose protein targets are undefined in mammals. Its yeast ortholog midasin (also known as Rea1) was clearly shown to be responsible for the maturation of the mitoribosomal LSU [111,112,113].
With excess heme being released from mitochondria in PRLTS3, abnormal G-tetrad processing might also occur in the nucleus, where homologous recombination is known to depend on DNA-G4 structures [114]. Thus, the complete infertility of PRLTS3 patients with abortion of nuclear meiosis-I after diplotene arrest [49] might partially be a consequence of defective G4 conformations.
Furthermore, the mitochondrial RNA granule factor LRPPRC undergoes selective accumulation in Clpp-KO tissues [41,54]. LRPPRC is known to modulate the poly(A) tail of mRNAs in mitochondria [115,116,117,118,119], and its dysfunction influences the efficiency of the RNA degradosome together with the accumulation of toxic dsRNA [120].
Jointly all this evidence indicates that the processing of polycistronic RNA, which is transcribed from mtDNA and then cleaved to tRNAs, rRNAs, other non-coding RNAs, and mRNAs, is selectively altered by CLPP absence. CLPP could trim components that are stuck within the RNA-protein complexes. CLPX clearly has a function in the disassembly of stalled translation complexes, and might play a role for the G4 conformation of rRNA that is important for the assembly of mitoribosomes.
2.4. Prominent Impact of CLPP Absence and CLPX Excess on Mitochondrial D-foci where RNA Degradation, Extrusion and Innate Immunity Activation are Decided
A fourth indication regarding the role of CLPP for mitochondrial matrix granules concerns the RNA degradosome in the so-called D-foci [121,122,123]. Its main component, the ribonuclease PNPT1 (orthologous to the bacterial polynucleotide phosphorylase/polyadenylase pnp, or PNPase), is associated with CLPP and dysregulated upon CLPP deletion, with consistency from E. coli to mouse [36]. Together with the RNA helicase SUPV3L1 (best known as SUV3, see [124]) and the RNA-G4-quadruplex modulating factor GRSF1, PNPT1 eliminates dsRNA, acting as 3’-5’ exonuclease [102,122,125,126,127,128,129], and even its bacterial ortholog pnp is responsible for antiviral immunity [130]. The matrix degradosome in D-foci appears to act not only on the abundant mtRNA, since PNPase and SUV3 show preference for mtDNA [131,132,133,134,135]. In analogy to mutations in CLPP, also mutations in PNPT1 are the cause of progressive deafness and of a sensory neuropathy with ataxia [136,137,138,139,140,141]. PNPT1 dysfunction causes the accumulation of toxic dsRNAs and their extrusion from mitochondria into the cytosol, where antiviral innate immunity responses get activated [142]. Similarly, homozygous absence of CLPP or heterozygous absence of mtDNA-binding TFAM in the mitochondrial matrix trigger cytosolic antiviral innate immunity responses, like the induction of AAA+ unfoldase RNF213. This unfoldase is also activated by toxic dsRNA mimics such as poly(I:C) administration [143]. In Clpp-KO mouse brain and MEFs, the selective activation of various cytosolic sensors for toxic DNA and for toxic RNA was documented [144]. The problems in packaging of mtDNA and in degrading / extruding toxic nucleic acids from mitochondria in Clpp-KO cells were shown to activate antiviral cytosolic responses via the cGAS / STING pathway [145].
Overall, the resulting steady-state activation of type I interferon signaling explains the marked resistance of CLPP-null mice to bacterial and RNA/DNA virus infections [40,145].
As preliminary conclusion, the above four paragraphs represent solid evidence that CLPP has a selective impact on matrix granules with RNA as component, which mediates liquid-liquid phase separation (LLPS) around these condensates.
2.5. Prominent Impact of CLPP Absence via CLPX Accumulation on Heme Biosynthesis and Incorporation into the Complex-IV of the Respiratory Chain
The absence of CLPP causes a several-fold accumulation of CLPX, as explained above. CLPX has an important role for the heme metabolism multi-enzyme complex, which is associated with the IMM [146], and serves to separate ferrous iron and reduced porphyrin intermediates from unwanted reactions in the matrix [147,148]. This multi-enzyme chain was previously shown to serve as metabolon, by definition held together by non-covalent interactions, as condensate with minimal hydration, to allow substrate channeling and maximal productivity [149,150,151]. The complex contains ALAS as the first enzyme of heme biosynthesis, whose product delta-aminolevulinic acid (deltaALA) is exported from mitochondria into the cytosol. It also contains CPOX-PPOX-FECH on different IMM surfaces as the three terminal enzymes of the biosynthesis chain, whose product heme gets incorporated into complexes II, III and IV of the respiratory chain within the IMM [147,152]. ALAS is furthermore associated with SUCLA2 in differentiating erythroid cells [147]. This IMM-associated multi-enzyme complex also serves as bridge [153,154] between at least three transmembrane proteins. Firstly, MFRN1 (also known as SLC25A37 or mitoferrin-1), which imports iron into the mitochondrial matrix [155], secondly ABCB10, which exports biliverdin to the cytosol [156], and thirdly ABCB7, which exports glutathione-coordinated iron-sulfur clusters to the cytosol [157] are connected to the IMM-associated heme biosynthesis complex according to several consistent reports. There is still debate [147] whether the tight association of IMM transmembrane proteins with this metabolon goes beyond the biliverdin / zinc-mesoporphyrin transporter ABCB10 [158,159,160] to include also the TMEM14C protoporphyrin-IX transporter [161,162,163,164], the protoporphyrin-IX transport modulator ANT2 [165] and the glutathione/succinate transport modulator OGC [166,167]. Enzyme complexes with similar isolation of reaction intermediates from the surrounding matrix have been observed also e.g. during L-arginine metabolism and bacterial cobalamin metabolism [168,169]. Heme and porphyrins are compounds that need a hydrophobic environment [170,171,172,173]. According to recent human genetics findings, mutations in FECH, ALAS2 and CLPX [174] underlie most cases of the disorder erythropoietic protoporphyria, while mutations in ALAS2 and in the mitochondrial glycine transporter SLC25A38 are the most frequent causes of congenital sideroblastic anemia [175].
CLPX was shown to unfold ALAS so that its cofactor pyridoxal-5’-phosphate (PLP) can bind and activate it, to consume succinate-CoA and glycine for production of the heme precursor delta-aminolevulinic acid (deltaALA) with optimal efficiency [5,37,38,176,177,178]. CLPXP was claimed to be responsible for ALAS degradation [179]. In the Clpp-KO mouse, the consequent elevation of CLPX abundance will unfold also OAT (ornithine delta-aminotransferase) so that PLP binds it and triggers the consumption of L-arginine and L-ornithine via a delta-transaminase and delta-aminomutase reaction to produce GSA as precursor of heme. In parallel, a recruitment of L-glutamate occurs into maximized deltaALA generation via accumulation of the enzyme ALDH18A1 (also known as delta-1-pyrroline-5-carboxylate synthase) [48,52]. Thus, ALAS and OAT are both acquiring the ability to perform transaminations at the delta-carbon position, when CLPX unfolds them and enables them to bind PLP as cofactor [180]. At the same time, iron accumulates in Clpp-KO tissue, together with the heavy metals molybdenum, cobalt, and manganese [42]. Thus, heavy metal toxicity and ferroptosis [181,182] may also be part of PRLTS3 pathogenesis mechanisms. The accumulation of the metal- and heme-binding protein COX15 and the preferential affection of respiratory complex-IV in the IMM of Clpp-KO mice can thus be explained as consequence of iron/heme dysregulation [42]. Indeed, the expression dysregulation of the heme-binding, mtDNA-encoded Cox1 membrane subunit in complex-IV had stood out across Clpp-KO testis, heart, liver and brain as a main molecular underpinning of respiratory dysfunction [40]. While heme is a protoporphyrin-IX that was chelated with Fe2+, plant chlorophyll is a protoporphyrin chelated with Mg2+, so both heme and chlorophyll biosynthesis depend on ALAS control by PLP and CLPX. Indeed, the regulation of heme/chlorophyll metabolism by CLPX is conserved from bacteria across phylogenesis to plants [183,184,185,186,187].
Unsurprisingly, the accumulation of CLPX in Clpp-KO tissues not only modulates the binding of PLP to target enzymes, but also leads to increased amounts of the PLP storage/transport protein PLPBP in some cell types [42].
Altogether, CLPX appears to fine-tune the biosynthesis and maturation of porphyrins and heme, with great consequences for iron and heavy metal utilization as well as respiratory competence, by continuous modulation of the IMM-associated multi-enzyme complex, which channels hydrophobic reaction intermediates and isolates them from the aqueous phase.
2.6. Prominent Impact of CLPP Absence on the Fe-S Cluster Containing Peripheral Arm of Respiratory Complex-I
The fine-tuning of multi-protein assemblies rather than proteolytic degradation seems to characterize also the selective role of CLPP absence and CLPX accumulation for the respiratory complex-I N-module [188]. Complex-I consists of a membrane arm embedded in the lipid bilayer of the IMM and a peripheral arm with the N / Q modules that protrudes into the aqueous phase of the matrix. The two modules serve to surround, isolate and channel electrons to a tunnel within the IMM, and to protect the many embedded iron-sulfur (Fe-S) clusters from oxidation [189,190,191,192,193]. The absence of CLPP results in a mild reduction of complex-I dependent state 3 respiration only in mouse heart mitochondria but not other tissues, so the mutation-triggered functional deficit is subtle [40,43]. As molecular underpinning, it was clearly shown that the turnover of the core subunits NDUFV1-NDUFV2-NDUFS2 in the NADH-oxidizing N-module of complex-I has a selective dependence on CLPP, in an ongoing exchange process where oxidatively damaged, inactive N-modules get substituted on the tip of the complex-I peripheral arm [194]. In addition, the selective accumulation SFXN4 in Clpp-KO tissue, as a component of the complex-I assembly machinery that controls metal association, indicates that the biogenesis of complex-I may be altered [42]. It has to be mentioned, however, that none of the established complex-I subunits accumulate in Clpp-KO mouse tissues, and that proteome profiling in the CLPP-null fungal eukaryote P. anserina did reveal some accumulated complex-I subunits but not for any specific module [48]. Thus, we assume that complex-I assembly is not a primary and conserved target of CLPP. Our team has observed upregulations of most factors in the iron-sulfur cluster (ISC) biogenesis pathway, most strongly for 4Fe–4S cluster generating enzymes, in the brain of Clpp-KO mice, but of course, this effect may simply represent a molecular adaptation to maintain sufficient ISC production despite the maximized iron utilization for heme biosynthesis.
Clearly, the functions of CLPP and CLPX seem to consist in the rapid refolding/trimming or substitution of a selected subunit within a complex that keeps working, but not the complete disassembly and disposal of entire respiratory complexes or supercomplexes.
3. Phase-Separated Condensates in Mitochondria and the Cytosol
Research over the past four years showed nucleoid components, the RNA processing granules and the RNA degradasome of bacteria and mitochondria to assemble in phase-separation [100,195,196,197,198,199]. The original concept of LLPS over twenty years ago [200,201] was derived from lipid droplets where components can move freely within a round compartment that excludes the aqueous phase. In the meantime, it has become clear that such condensates need not be liquid but can also assume a gelatinous or even solidified state, particularly in a disease context [202]. Although the mobility of individual components within the condensate may be high, they would certainly move along given structures within the phase-separated condensate, e.g. in the case of nucleolar ribosome biogenesis, or mtDNA transcription, or the processing of polycistronic mtRNA. Thus, these condensates may be defined by the multivalent interaction forces that keep long and flexible molecules such as lipids or RNA together [203,204]. They are also defined by the vulnerable reaction intermediates that need protection from the aqueous or membrane phases, such as cleaved unfolded RNA without modifications, or unchelated porphyrins that are unassembled with proteins, or pre-fibril oligomers with a propensity to disrupt membranes [205,206,207]. Regarding its multivalent interaction forces and its long flexible structure, RNA was the prime example to understand LLPS, based on the phase contrast during microscopic visualization of the nucleolus. Therefore, other RNA-containing granules in the nucleus and cytosol (e.g. paraspeckles, Cajal bodies, U bodies, PcG bodies, Balbiani bodies, stress granules, P-bodies, germ granules and RNA transport granules) constituted an early focus of LLPS research (Figure 1 in [202]). It was shown that also the ribonucleoproteins contribute to phase separation, with some binding domains deciding the specificity of interactions (known as “stickers”), while the intrinsically disordered regions (IDR) often intervening have solvation properties that influence density transition (known as “spacers”) [203]. Indeed, it was proposed that cells use RNAs and IDR-proteins to separate multi-enzyme complexes such as glycolysis in granular compartments that have a different phase than the surrounding cytosol [208]. Other mitochondrial metabolons such as the TCA cycle, heme biosynthesis, the urea cycle, the respiratory chain, and the breakdown of branched amino acids, also require efficient channeling of reaction intermediates, which are usually achieved by tight subunit docking and by hydrophobic interactions [209]. Thus, these metabolons might also be separated from the aqueous phase by the multivalent forces of associated non-coding RNA.
Thus, the recently defined targets of CLPP and CLPX are all condensates where phase separation or multi-enzyme assembly protect unstable reaction intermediates from the aqueous phase, and which need frequently a rapid repair of individual subunits while these assemblies keep fulfilling their function. It seems plausible that CLPP and CLPX have the ability to access these condensates and to provide the necessary first aid.
4. Proposal
Thus, it may be impossible to define consistent protein targets of CLPX and CLPP across phylogenesis, given that each organism is adapted to a different environment, has specific metabolic needs, and has to protect other reaction intermediates inside phase-separated multivalent or hydrophobic condensates from the aqueous phase. The polypeptide sequences of degrons recognized by CLPX in E. coli might differ from such sequences in mice, and the cleavage pattern of CLPP, while it fine-tunes multi-enzyme complexes, may vary according to steric constraints. There could be no protein that is exclusively the substrate of CLPXP-mediated proteolysis, all matrix proteins might be degraded finally by LONP1. This is exemplified by CLPX, which is certainly the prominent example of a protein whose abundance depends on CLPP in all organisms studied [36], and yet its proteolytic destruction was observed to be executed by LONP1 [210]. Instead of eliminating specific proteins in the mitochondrial matrix, CLPX and CLPP seem to have unique access properties to granular compartments where they can rescue a suboptimal or stalled process, either by unfolding a protein or by the excision of a misassembled component, so that the multi-enzyme complex can improve its performance (see Figure 1). Overall, it is necessary to validate in other organisms if the Clpp-KO mouse evidence holds true - that the specific roles of CLPX and CLPP are defined by the granular compartments they are monitoring. This would be analogous to most other peptidases in mitochondria, where MPP cleaves all precursor proteins at the import pore, m-AAA and i-AAA are responsible for protein quality control at either IMM face, PARL cleaves proteins within the IMM, and OMA1/HTRA2 perform surveillance in the intermembrane space [211,212,213,214].
Author Contributions
Conceptualization, G.A.; writing—original draft preparation, G.A.; writing—review and editing, J.K., S.G., and G.A.; visualization, J.K. and G.A.; project administration, G.A.; funding acquisition, G.A.
Funding
This project was supported by funds from the Klinikum Goethe Universität Frankfurt/Main.
Acknowledgments
We thank Hildegard König for help with lab and office infrastructure.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Abbreviations
| 4Fe–4S | iron sulfur clusters consisting of two interleaved 4Fe- and 4S-tetrahedra |
| AAA+ | ATPases Associated with diverse cellular Activities, and other ring-shaped P-loop NTPases |
| ABCB7 | ATP Binding Cassette Subfamily B Member 7 |
| ABCB10 | ATP Binding Cassette Subfamily B Member 10 |
| ALAS | delta-Amino-Levulinic Acid Synthase |
| ALAS2 | delta-Amino-Levulinic Acid Synthase 2, erythroid-specific |
| ALDH18A1 | Aldehyde Dehydrogenase 18 family member A1, = P5CS, delta-1-Pyrroline-5-Carboxylate Synthase |
| ANT2 | Adenine Nucleotide Translocator 2, = SLC25A25 |
| ATP | Adenosine Tri-Phosphate |
| ATPase | Adenosine Tri-Phosphatase |
| CAT-tail | C-terminal alanine and threonine tail |
| cGAS | cyclic GMP-AMP Synthase |
| CLPP | Caseinolytic Mitochondrial Matrix Peptidase Proteolytic Subunit |
| CLPX | Caseinolytic Mitochondrial Matrix Peptidase Chaperone Subunit X |
| CODAS | multiple anomalies syndrome with Cerebral, Ocular, Dental, Auricular and Skeletal anomalies |
| Cox1 | mitochondrially encoded Cytochrome C Oxidase I, mRNA |
| COX15 | Cytochrome C Oxidase assembly homolog COX15 |
| CPOX | Copro-Porphyrinogen OXidase |
| D-foci | degradosome granules in mitochondrial matrix |
| D-loop | displacement loop within the mtDNA |
| DARS2 | Aspartyl-tRNA Synthetase 2, mitochondrial |
| deltaALA | delta-aminolevulinic acid |
| DNA | Deoxyribo-Nucleic Acid |
| DNAzyme | catalytically active DNA sequences |
| dsRNA | double-stranded RNA |
| EF-G | Elongation Factor G |
| EFG1 | G Elongation Factor, mitochondrial 1 |
| ERAL1 | Era (E. coli) -Like 12S mitochondrial rRNA chaperone 1 |
| Fe2+ | divalent cation of iron, ferrous iron |
| FECH | Ferrochelatase |
| Fe-S | iron-sulfur |
| G4 | Guanine-quadruplex where RNA or DNA acquires four-stranded conformation |
| GFM1 | G elongation Factor Mitochondrial 1 |
| GRSF1 | G-rich RNA Sequence binding Factor 1 |
| GSA | Glutamate-5-Semi-Aldehyde |
| GTP | Guanosine-5'-triphosphate |
| HARS2 | Histidyl-tRNA Synthetase 2, mitochondrial |
| hsp-6 | heat shock protein family B (small) member 6 |
| hsp-60 | heat shock protein family D (hsp60) member 1, = human HSPD1 |
| HSPD1 | Heat Shock Protein family D (hsp60) member 1, = human HSP60, chaperonin |
| HTRA2 | High Temperature Requirement A serine peptidase 2 |
| i-AAA | ATP-dependent zinc metalloprotease YME1 (S. cerevisiae) -Like 1 |
| IDR | Intrinsically Disordered Region |
| IMM | Inner Membrane of Mitochondria |
| ISC | Iron-Sulfur-Cluster |
| KO | Knock-Out |
| LARS2 | Leucyl-tRNA Synthetase 2, mitochondrial |
| lncRNA | long non-coding RNA |
| LLPS | liquid-liquid phase separation |
| LONP1 | Lon Peptidase 1, Mitochondrial |
| LRPPRC | Leucine Rich Pentatrico-Peptide Repeat Containing |
| m-AAA | AFG3-like matrix AAA peptidase subunit 2 and SPG7 matrix AAA peptidase subunit paraplegin |
| MEFs | mouse embryonic fibroblasts |
| MFRN1 | mitoferrin 1, = SLC25A37, Solute Carrier Family 25 Member 37 |
| Mg2+ | divalent cation of magnesium |
| MPP | mitochondrial processing peptidase |
| MRPL12 | Mitochondrial Ribosomal Protein L12 |
| MRPL18 | Mitochondrial Ribosomal Protein L18 |
| MRPL37 | Mitochondrial Ribosomal Protein L37 |
| MRPL38 | Mitochondrial Ribosomal Protein L38 |
| MRPS15 | Mitochondrial Ribosomal Protein S15 |
| MRPS35 | Mitochondrial Ribosomal Protein S35 |
| mtDNA | mitochondrial DNA |
| mtHSP75 | mitochondrial heat shock protein 75, = human TRAP1 |
| mtLSU | mitoribosomal 39S large subunit |
| mtRNA | mitochondrial RNA |
| mtSSB | mitochondrial single-stranded DNA binding protein |
| mtSSU | mitoribosomal 28S small subunit |
| mt-tRNA | mitochondrial transfer RNA |
| NADH | Nicotinamide Adenine Dinucleotide, reduced form |
| NDUFS2 | NADH:Ubiquinone Oxidoreductase Core Subunit S2 |
| NDUFV1 | NADH:Ubiquinone Oxidoreductase Core Subunit V1 |
| NDUFV2 | NADH:Ubiquinone Oxidoreductase Core Subunit V2 |
| NOA1 | Nitric Oxide Associated 1 |
| NTPase | Nucleoside-Tri-Phosphatase |
| OAT | Ornithine delta-Amino-Transferase |
| OGC | 2-Oxoglutarate/Malate Carrier protein, mitochondrial |
| OMA1 | Overlapping with the M-AAA protease 1 homolog, zinc metallopeptidase |
| OMM | Outer Membrane of Mitochondria |
| PARL | Presenilin Associated Rhomboid Like |
| PcG bodies | polycomb bodies |
| PEO1 | Progressive External Ophthalmoplegia 1 protein = TWNK |
| Phe | phenylalanine |
| PLP | Pyridoxal-5’-Phosphate |
| PLPBP | PLP-binding protein |
| PNPase | Polyribo-Nucleotide Phosphorylase / Nucleotidyl-Transferase 1 = PNPT1 in human |
| PNPT1 | Polyribo-Nucleotide Phosphorylase / Nucleotidyl-Transferase 1 = PNPase |
| POLDIP2 | DNA Polymerase Delta Interacting Protein 2 |
| poly(A) tail | poly(adenine) tail of messenger RNAs |
| poly(I:C) | poly(inosinic : cytidylic) acid |
| PPOX | Proto-Porphyrinogen OXidase |
| PRLTS3 | Perrault Syndrome type 3 |
| PRORP | Protein Only RNase P catalytic subunit |
| qPCR | quantitative Polymerase Chain Reaction |
| RMND1 | Required for Meiotic Nuclear Division 1 homolog |
| RNA | Ribo-Nucleic Acid |
| RNA-G4 | RNA, guanine-rich, in quadruplex conformation |
| RNase | ribonuclease |
| RNAzyme | catalytically active RNA sequences |
| RNF213 | Ring Finger protein 213 |
| rRNA | ribosomal RNA |
| SFXN4 | Sideroflexin 4 |
| SLC25A37 | Solute Carrier family 25 member 37, Mitoferrin 1 |
| SLC25A38 | Solute Carrier Family 25 Member 38, mitochondrial glycine transporter |
| STING | STimulator of INterferon response cGAMP interactor 1 |
| SUCLA2 | Succinate-CoA Ligase ADP-forming subunit beta |
| SUPV3L1 | = SUV3, Suv3-like RNA helicase |
| TCA | Tri-Carboxylic Acid cycle, = Krebs cycle |
| TFAM | Transcription Factor A, Mitochondrial |
| TIGM | Texas Institute of Genomic Medicine |
| TMEM14C | Transmembrane Protein 14C |
| tRNA | transfer RNA |
| TWNK | Twinkle mtDNA helicase, = PEO1 |
| UPR | unfolded protein response |
| UPRmt | unfolded protein response in mitochondria |
| Val | valine |
| VWA8 | von Willebrand Factor A domain containing 8 |
References
- Key, J.; Kohli, A.; Barcena, C.; Lopez-Otin, C.; Heidler, J.; Wittig, I.; Auburger, G. Global Proteome of LonP1(+/-) Mouse Embryonal Fibroblasts Reveals Impact on Respiratory Chain, but No Interdependence between Eral1 and Mitoribosomes. Int J Mol Sci 2019, 20. [CrossRef]
- Bezawork-Geleta, A.; Brodie, E.J.; Dougan, D.A.; Truscott, K.N. LON is the master protease that protects against protein aggregation in human mitochondria through direct degradation of misfolded proteins. Sci Rep 2015, 5, 17397. [CrossRef]
- Stahlberg, H.; Kutejova, E.; Suda, K.; Wolpensinger, B.; Lustig, A.; Schatz, G.; Engel, A.; Suzuki, C.K. Mitochondrial Lon of Saccharomyces cerevisiae is a ring-shaped protease with seven flexible subunits. Proc Natl Acad Sci U S A 1999, 96, 6787-6790. [CrossRef]
- Baker, T.A.; Sauer, R.T. ClpXP, an ATP-powered unfolding and protein-degradation machine. Biochim Biophys Acta 2012, 1823, 15-28. [CrossRef]
- Kardon, J.R.; Yien, Y.Y.; Huston, N.C.; Branco, D.S.; Hildick-Smith, G.J.; Rhee, K.Y.; Paw, B.H.; Baker, T.A. Mitochondrial ClpX Activates a Key Enzyme for Heme Biosynthesis and Erythropoiesis. Cell 2015, 161, 858-867. [CrossRef]
- Fischer, F.; Weil, A.; Hamann, A.; Osiewacz, H.D. Human CLPP reverts the longevity phenotype of a fungal ClpP deletion strain. Nat Commun 2013, 4, 1397. [CrossRef]
- Dikoglu, E.; Alfaiz, A.; Gorna, M.; Bertola, D.; Chae, J.H.; Cho, T.J.; Derbent, M.; Alanay, Y.; Guran, T.; Kim, O.H.; et al. Mutations in LONP1, a mitochondrial matrix protease, cause CODAS syndrome. Am J Med Genet A 2015, 167, 1501-1509. [CrossRef]
- Strauss, K.A.; Jinks, R.N.; Puffenberger, E.G.; Venkatesh, S.; Singh, K.; Cheng, I.; Mikita, N.; Thilagavathi, J.; Lee, J.; Sarafianos, S.; et al. CODAS syndrome is associated with mutations of LONP1, encoding mitochondrial AAA+ Lon protease. Am J Hum Genet 2015, 96, 121-135. [CrossRef]
- Faridi, R.; Stratton, P.; Salmeri, N.; Morell, R.J.; Khan, A.A.; Usmani, M.A.; Newman, W.G.; Riazuddin, S.; Friedman, T.B. Homozygous novel truncating variant of CLPP associated with severe Perrault syndrome. Clin Genet 2024, 105, 584-586. [CrossRef]
- Brodie, E.J.; Zhan, H.; Saiyed, T.; Truscott, K.N.; Dougan, D.A. Perrault syndrome type 3 caused by diverse molecular defects in CLPP. Sci Rep 2018, 8, 12862. [CrossRef]
- Theunissen, T.E.; Szklarczyk, R.; Gerards, M.; Hellebrekers, D.M.; Mulder-Den Hartog, E.N.; Vanoevelen, J.; Kamps, R.; de Koning, B.; Rutledge, S.L.; Schmitt-Mechelke, T.; et al. Specific MRI Abnormalities Reveal Severe Perrault Syndrome due to CLPP Defects. Front Neurol 2016, 7, 203. [CrossRef]
- Dursun, F.; Mohamoud, H.S.; Karim, N.; Naeem, M.; Jelani, M.; Kirmizibekmez, H. A Novel Missense Mutation in the CLPP Gene Causing Perrault Syndrome Type 3 in a Turkish Family. J Clin Res Pediatr Endocrinol 2016, 8, 472-477. [CrossRef]
- Demain, L.A.; Urquhart, J.E.; O'Sullivan, J.; Williams, S.G.; Bhaskar, S.S.; Jenkinson, E.M.; Lourenco, C.M.; Heiberg, A.; Pearce, S.H.; Shalev, S.A.; et al. Expanding the genotypic spectrum of Perrault syndrome. Clin Genet 2017, 91, 302-312. [CrossRef]
- Ahmed, S.; Jelani, M.; Alrayes, N.; Mohamoud, H.S.; Almramhi, M.M.; Anshasi, W.; Ahmed, N.A.; Wang, J.; Nasir, J.; Al-Aama, J.Y. Exome analysis identified a novel missense mutation in the CLPP gene in a consanguineous Saudi family expanding the clinical spectrum of Perrault Syndrome type-3. J Neurol Sci 2015, 353, 149-154. [CrossRef]
- Jenkinson, E.M.; Rehman, A.U.; Walsh, T.; Clayton-Smith, J.; Lee, K.; Morell, R.J.; Drummond, M.C.; Khan, S.N.; Naeem, M.A.; Rauf, B.; et al. Perrault syndrome is caused by recessive mutations in CLPP, encoding a mitochondrial ATP-dependent chambered protease. Am J Hum Genet 2013, 92, 605-613. [CrossRef]
- Hochberg, I.; Demain, L.A.M.; Richer, J.; Thompson, K.; Urquhart, J.E.; Rea, A.; Pagarkar, W.; Rodriguez-Palmero, A.; Schluter, A.; Verdura, E.; et al. Bi-allelic variants in the mitochondrial RNase P subunit PRORP cause mitochondrial tRNA processing defects and pleiotropic multisystem presentations. Am J Hum Genet 2021, 108, 2195-2204. [CrossRef]
- Newman, W.G.; Friedman, T.B.; Conway, G.S.; Demain, L.A.M. Perrault Syndrome. In GeneReviews((R)), Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; Seattle (WA), 1993.
- Kobe, C.; Kracht, L.W.; Timmermann, L.; Bachmann, J.; Schmidt, M.C. Perrault Syndrome with progressive nervous system involvement. Clin Nucl Med 2008, 33, 922-924. [CrossRef]
- Gottschalk, M.E.; Coker, S.B.; Fox, L.A. Neurologic anomalies of Perrault syndrome. Am J Med Genet 1996, 65, 274-276. [CrossRef]
- Linssen, W.H.; Van den Bent, M.J.; Brunner, H.G.; Poels, P.J. Deafness, sensory neuropathy, and ovarian dysgenesis: a new syndrome or a broader spectrum of Perrault syndrome? Am J Med Genet 1994, 51, 81-82. [CrossRef]
- Faridi, R.; Rea, A.; Fenollar-Ferrer, C.; O'Keefe, R.T.; Gu, S.; Munir, Z.; Khan, A.A.; Riazuddin, S.; Hoa, M.; Naz, S.; et al. New insights into Perrault syndrome, a clinically and genetically heterogeneous disorder. Hum Genet 2022, 141, 805-819. [CrossRef]
- Tucker, E.J.; Rius, R.; Jaillard, S.; Bell, K.; Lamont, P.J.; Travessa, A.; Dupont, J.; Sampaio, L.; Dulon, J.; Vuillaumier-Barrot, S.; et al. Genomic sequencing highlights the diverse molecular causes of Perrault syndrome: a peroxisomal disorder (PEX6), metabolic disorders (CLPP, GGPS1), and mtDNA maintenance/translation disorders (LARS2, TFAM). Hum Genet 2020, 139, 1325-1343. [CrossRef]
- Gotta, F.; Lamp, M.; Geroldi, A.; Trevisan, L.; Origone, P.; Fugazza, G.; Fabbri, S.; Nesti, C.; Rubegni, A.; Morani, F.; et al. A novel mutation of Twinkle in Perrault syndrome: A not rare diagnosis? Ann Hum Genet 2020, 84, 417-422. [CrossRef]
- Smith, T.B.; Rea, A.; Thomas, H.B.; Thompson, K.; Olahova, M.; Maroofian, R.; Zamani, M.; He, L.; Sadeghian, S.; Galehdari, H.; et al. Novel homozygous variants in PRORP expand the genotypic spectrum of combined oxidative phosphorylation deficiency 54. Eur J Hum Genet 2023, 31, 1190-1194. [CrossRef]
- Chatzispyrou, I.A.; Alders, M.; Guerrero-Castillo, S.; Zapata Perez, R.; Haagmans, M.A.; Mouchiroud, L.; Koster, J.; Ofman, R.; Baas, F.; Waterham, H.R.; et al. A homozygous missense mutation in ERAL1, encoding a mitochondrial rRNA chaperone, causes Perrault syndrome. Hum Mol Genet 2017, 26, 2541-2550. [CrossRef]
- Rioux, A.V.; Bergeron, N.A.; Riopel, J.; Marcoux, N.; Theriault, C.; Gould, P.V.; Garneau, A.P.; Isenring, P. The ever wider clinical spectrum of RMND1-related disorders and limitedness of phenotype-based classifications. J Mol Med (Berl) 2023, 101, 1229-1236. [CrossRef]
- Neyroud, A.S.; Rudinger-Thirion, J.; Frugier, M.; Riley, L.G.; Bidet, M.; Akloul, L.; Simpson, A.; Gilot, D.; Christodoulou, J.; Ravel, C.; et al. LARS2 variants can present as premature ovarian insufficiency in the absence of overt hearing loss. Eur J Hum Genet 2023, 31, 453-460. [CrossRef]
- Ozieblo, D.; Pazik, J.; Stepniak, I.; Skarzynski, H.; Oldak, M. Two Novel Pathogenic Variants Confirm RMND1 Causative Role in Perrault Syndrome with Renal Involvement. Genes (Basel) 2020, 11. [CrossRef]
- Riley, L.G.; Rudinger-Thirion, J.; Frugier, M.; Wilson, M.; Luig, M.; Alahakoon, T.I.; Nixon, C.Y.; Kirk, E.P.; Roscioli, T.; Lunke, S.; et al. The expanding LARS2 phenotypic spectrum: HLASA, Perrault syndrome with leukodystrophy, and mitochondrial myopathy. Hum Mutat 2020, 41, 1425-1434. [CrossRef]
- Demain, L.A.M.; Gerkes, E.H.; Smith, R.J.H.; Molina-Ramirez, L.P.; O'Keefe, R.T.; Newman, W.G. A recurrent missense variant in HARS2 results in variable sensorineural hearing loss in three unrelated families. J Hum Genet 2020, 65, 305-311. [CrossRef]
- Karstensen, H.G.; Rendtorff, N.D.; Hindbaek, L.S.; Colombo, R.; Stein, A.; Birkebaek, N.H.; Hartmann-Petersen, R.; Lindorff-Larsen, K.; Hojland, A.T.; Petersen, M.B.; et al. Novel HARS2 missense variants identified in individuals with sensorineural hearing impairment and Perrault syndrome. Eur J Med Genet 2020, 63, 103733. [CrossRef]
- Demain, L.A.M.; Antunes, D.; O'Sullivan, J.; Bhaskhar, S.S.; O'Keefe, R.T.; Newman, W.G. A known pathogenic variant in the essential mitochondrial translation gene RMND1 causes a Perrault-like syndrome with renal defects. Clin Genet 2018, 94, 276-277. [CrossRef]
- Kosaki, R.; Horikawa, R.; Fujii, E.; Kosaki, K. Biallelic mutations in LARS2 can cause Perrault syndrome type 2 with neurologic symptoms. Am J Med Genet A 2018, 176, 404-408. [CrossRef]
- Pierce, S.B.; Gersak, K.; Michaelson-Cohen, R.; Walsh, T.; Lee, M.K.; Malach, D.; Klevit, R.E.; King, M.C.; Levy-Lahad, E. Mutations in LARS2, encoding mitochondrial leucyl-tRNA synthetase, lead to premature ovarian failure and hearing loss in Perrault syndrome. Am J Hum Genet 2013, 92, 614-620. [CrossRef]
- Pierce, S.B.; Chisholm, K.M.; Lynch, E.D.; Lee, M.K.; Walsh, T.; Opitz, J.M.; Li, W.; Klevit, R.E.; King, M.C. Mutations in mitochondrial histidyl tRNA synthetase HARS2 cause ovarian dysgenesis and sensorineural hearing loss of Perrault syndrome. Proc Natl Acad Sci U S A 2011, 108, 6543-6548. [CrossRef]
- Auburger, G.; Key, J.; Gispert, S. The Bacterial ClpXP-ClpB Family Is Enriched with RNA-Binding Protein Complexes. Cells 2022, 11. [CrossRef]
- Ducamp, S.; Luscieti, S.; Ferrer-Cortes, X.; Nicolas, G.; Manceau, H.; Peoc'h, K.; Yien, Y.Y.; Kannengiesser, C.; Gouya, L.; Puy, H.; et al. A mutation in the iron-responsive element of ALAS2 is a modifier of disease severity in a patient suffering from CLPX associated erythropoietic protoporphyria. Haematologica 2021, 106, 2030-2033. [CrossRef]
- Yien, Y.Y.; Ducamp, S.; van der Vorm, L.N.; Kardon, J.R.; Manceau, H.; Kannengiesser, C.; Bergonia, H.A.; Kafina, M.D.; Karim, Z.; Gouya, L.; et al. Mutation in human CLPX elevates levels of delta-aminolevulinate synthase and protoporphyrin IX to promote erythropoietic protoporphyria. Proc Natl Acad Sci U S A 2017, 114, E8045-E8052. [CrossRef]
- van der Vorm, L.N.; Paw, B.H. Studying disorders of vertebrate iron and heme metabolism using zebrafish. Methods Cell Biol 2017, 138, 193-220. [CrossRef]
- Gispert, S.; Parganlija, D.; Klinkenberg, M.; Drose, S.; Wittig, I.; Mittelbronn, M.; Grzmil, P.; Koob, S.; Hamann, A.; Walter, M.; et al. Loss of mitochondrial peptidase Clpp leads to infertility, hearing loss plus growth retardation via accumulation of CLPX, mtDNA and inflammatory factors. Hum Mol Genet 2013, 22, 4871-4887. [CrossRef]
- Seiferling, D.; Szczepanowska, K.; Becker, C.; Senft, K.; Hermans, S.; Maiti, P.; Konig, T.; Kukat, A.; Trifunovic, A. Loss of CLPP alleviates mitochondrial cardiomyopathy without affecting the mammalian UPRmt. EMBO Rep 2016, 17, 953-964. [CrossRef]
- Key, J.; Gispert, S.; Koepf, G.; Steinhoff-Wagner, J.; Reichlmeir, M.; Auburger, G. Translation Fidelity and Respiration Deficits in CLPP-Deficient Tissues: Mechanistic Insights from Mitochondrial Complexome Profiling. Int J Mol Sci 2023, 24. [CrossRef]
- Szczepanowska, K.; Maiti, P.; Kukat, A.; Hofsetz, E.; Nolte, H.; Senft, K.; Becker, C.; Ruzzenente, B.; Hornig-Do, H.T.; Wibom, R.; et al. CLPP coordinates mitoribosomal assembly through the regulation of ERAL1 levels. EMBO J 2016, 35, 2566-2583. [CrossRef]
- Arribas, J.; Castano, J.G. A comparative study of the chymotrypsin-like activity of the rat liver multicatalytic proteinase and the ClpP from Escherichia coli. J Biol Chem 1993, 268, 21165-21171.
- Mabanglo, M.F.; Wong, K.S.; Barghash, M.M.; Leung, E.; Chuang, S.H.W.; Ardalan, A.; Majaesic, E.M.; Wong, C.J.; Zhang, S.; Lang, H.; et al. Potent ClpP agonists with anticancer properties bind with improved structural complementarity and alter the mitochondrial N-terminome. Structure 2023, 31, 185-200 e110. [CrossRef]
- Prabhu, V.V.; Morrow, S.; Rahman Kawakibi, A.; Zhou, L.; Ralff, M.; Ray, J.; Jhaveri, A.; Ferrarini, I.; Lee, Y.; Parker, C.; et al. ONC201 and imipridones: Anti-cancer compounds with clinical efficacy. Neoplasia 2020, 22, 725-744. [CrossRef]
- Wong, K.S.; Houry, W.A. Chemical Modulation of Human Mitochondrial ClpP: Potential Application in Cancer Therapeutics. ACS Chem Biol 2019, 14, 2349-2360. [CrossRef]
- Key, J.; Gispert, S.; Kandi, A.R.; Heinz, D.; Hamann, A.; Osiewacz, H.D.; Meierhofer, D.; Auburger, G. CLPP-Null Eukaryotes with Excess Heme Biosynthesis Show Reduced L-arginine Levels, Probably via CLPX-Mediated OAT Activation. Biomolecules 2024, 14. [CrossRef]
- Key, J.; Gispert, S.; Koornneef, L.; Sleddens-Linkels, E.; Kohli, A.; Torres-Odio, S.; Koepf, G.; Amr, S.; Reichlmeir, M.; Harter, P.N.; et al. CLPP Depletion Causes Diplotene Arrest; Underlying Testis Mitochondrial Dysfunction Occurs with Accumulation of Perrault Proteins ERAL1, PEO1, and HARS2. Cells 2022, 12. [CrossRef]
- Becker, C.; Kukat, A.; Szczepanowska, K.; Hermans, S.; Senft, K.; Brandscheid, C.P.; Maiti, P.; Trifunovic, A. CLPP deficiency protects against metabolic syndrome but hinders adaptive thermogenesis. EMBO Rep 2018, 19. [CrossRef]
- Bhaskaran, S.; Pharaoh, G.; Ranjit, R.; Murphy, A.; Matsuzaki, S.; Nair, B.C.; Forbes, B.; Gispert, S.; Auburger, G.; Humphries, K.M.; et al. Loss of mitochondrial protease ClpP protects mice from diet-induced obesity and insulin resistance. EMBO Rep 2018, 19. [CrossRef]
- Hofsetz, E.; Demir, F.; Szczepanowska, K.; Kukat, A.; Kizhakkedathu, J.N.; Trifunovic, A.; Huesgen, P.F. The Mouse Heart Mitochondria N Terminome Provides Insights into ClpXP-Mediated Proteolysis. Mol Cell Proteomics 2020, 19, 1330-1345. [CrossRef]
- Benedetti, C.; Haynes, C.M.; Yang, Y.; Harding, H.P.; Ron, D. Ubiquitin-like protein 5 positively regulates chaperone gene expression in the mitochondrial unfolded protein response. Genetics 2006, 174, 229-239. [CrossRef]
- Key, J.; Torres-Odio, S.; Bach, N.C.; Gispert, S.; Koepf, G.; Reichlmeir, M.; West, A.P.; Prokisch, H.; Freisinger, P.; Newman, W.G.; et al. Inactivity of Peptidase ClpP Causes Primary Accumulation of Mitochondrial Disaggregase ClpX with Its Interacting Nucleoid Proteins, and of mtDNA. Cells 2021, 10. [CrossRef]
- Cheng, X.; Kanki, T.; Fukuoh, A.; Ohgaki, K.; Takeya, R.; Aoki, Y.; Hamasaki, N.; Kang, D. PDIP38 associates with proteins constituting the mitochondrial DNA nucleoid. J Biochem 2005, 138, 673-678. [CrossRef]
- Strack, P.R.; Brodie, E.J.; Zhan, H.; Schuenemann, V.J.; Valente, L.J.; Saiyed, T.; Lowth, B.R.; Angley, L.M.; Perugini, M.A.; Zeth, K.; et al. Polymerase delta-interacting protein 38 (PDIP38) modulates the stability and activity of the mitochondrial AAA+ protease CLPXP. Commun Biol 2020, 3, 646. [CrossRef]
- Zhang, R.; Wang, P.; Wei, B.; Chen, L.; Song, X.; Pan, Y.; Li, J.; Gan, J.; Zhang, T.; Yang, C.G. Assessment of the structure-activity relationship and antileukemic activity of diacylpyramide compounds as human ClpP agonists. Eur J Med Chem 2023, 258, 115577. [CrossRef]
- Nikali, K.; Suomalainen, A.; Saharinen, J.; Kuokkanen, M.; Spelbrink, J.N.; Lonnqvist, T.; Peltonen, L. Infantile onset spinocerebellar ataxia is caused by recessive mutations in mitochondrial proteins Twinkle and Twinky. Hum Mol Genet 2005, 14, 2981-2990. [CrossRef]
- Munson, H.E.; De Simone, L.; Schwaede, A.; Bhatia, A.; Mithal, D.S.; Young, N.; Kuntz, N.; Rao, V.K. Axonal polyneuropathy and ataxia in children: consider Perrault Syndrome, a case report. BMC Med Genomics 2023, 16, 278. [CrossRef]
- Wei, L.; Hou, L.; Ying, Y.Q.; Luo, X.P. A Novel Missense Mutation in TWNK Gene Causing Perrault Syndrome Type 5 in a Chinese Family and Review of the Literature. Pharmgenomics Pers Med 2022, 15, 1-8. [CrossRef]
- Chen, Z.; Tang, S.; Li, H.; Xu, X.; Lyu, J. [Analysis of TWNK variant in a family affected with Perrault syndrome]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2020, 37, 739-742. [CrossRef]
- Kume, K.; Morino, H.; Miyamoto, R.; Matsuda, Y.; Ohsawa, R.; Kanaya, Y.; Tada, Y.; Kurashige, T.; Kawakami, H. Middle-age-onset cerebellar ataxia caused by a homozygous TWNK variant: a case report. BMC Med Genet 2020, 21, 68. [CrossRef]
- Fekete, B.; Pentelenyi, K.; Rudas, G.; Gal, A.; Grosz, Z.; Illes, A.; Idris, J.; Csukly, G.; Domonkos, A.; Molnar, M.J. Broadening the phenotype of the TWNK gene associated Perrault syndrome. BMC Med Genet 2019, 20, 198. [CrossRef]
- Dominguez-Ruiz, M.; Garcia-Martinez, A.; Corral-Juan, M.; Perez-Alvarez, A.I.; Plasencia, A.M.; Villamar, M.; Moreno-Pelayo, M.A.; Matilla-Duenas, A.; Menendez-Gonzalez, M.; Del Castillo, I. Perrault syndrome with neurological features in a compound heterozygote for two TWNK mutations: overlap of TWNK-related recessive disorders. J Transl Med 2019, 17, 290. [CrossRef]
- Oldak, M.; Ozieblo, D.; Pollak, A.; Stepniak, I.; Lazniewski, M.; Lechowicz, U.; Kochanek, K.; Furmanek, M.; Tacikowska, G.; Plewczynski, D.; et al. Novel neuro-audiological findings and further evidence for TWNK involvement in Perrault syndrome. J Transl Med 2017, 15, 25. [CrossRef]
- Morino, H.; Pierce, S.B.; Matsuda, Y.; Walsh, T.; Ohsawa, R.; Newby, M.; Hiraki-Kamon, K.; Kuramochi, M.; Lee, M.K.; Klevit, R.E.; et al. Mutations in Twinkle primase-helicase cause Perrault syndrome with neurologic features. Neurology 2014, 83, 2054-2061. [CrossRef]
- Yamamoto, H.; Qin, Y.; Achenbach, J.; Li, C.; Kijek, J.; Spahn, C.M.; Nierhaus, K.H. EF-G and EF4: translocation and back-translocation on the bacterial ribosome. Nat Rev Microbiol 2014, 12, 89-100. [CrossRef]
- Carbone, C.E.; Loveland, A.B.; Gamper, H.B., Jr.; Hou, Y.M.; Demo, G.; Korostelev, A.A. Time-resolved cryo-EM visualizes ribosomal translocation with EF-G and GTP. Nat Commun 2021, 12, 7236. [CrossRef]
- Koripella, R.K.; Sharma, M.R.; Bhargava, K.; Datta, P.P.; Kaushal, P.S.; Keshavan, P.; Spremulli, L.L.; Banavali, N.K.; Agrawal, R.K. Structures of the human mitochondrial ribosome bound to EF-G1 reveal distinct features of mitochondrial translation elongation. Nat Commun 2020, 11, 3830. [CrossRef]
- Kummer, E.; Ban, N. Structural insights into mammalian mitochondrial translation elongation catalyzed by mtEFG1. EMBO J 2020, 39, e104820. [CrossRef]
- Chen, Y.; Feng, S.; Kumar, V.; Ero, R.; Gao, Y.G. Structure of EF-G-ribosome complex in a pretranslocation state. Nat Struct Mol Biol 2013, 20, 1077-1084. [CrossRef]
- Moraleda, J.M.; Gonzalez, R.; Alegre, A.; Anta, J.P.; San Miguel, J.F. Bone marrow necrosis and treatment with interferon. J Clin Pathol 1986, 39, 1045. [CrossRef]
- Sitron, C.S.; Park, J.H.; Giafaglione, J.M.; Brandman, O. Aggregation of CAT tails blocks their degradation and causes proteotoxicity in S. cerevisiae. PLoS One 2020, 15, e0227841. [CrossRef]
- Wu, Z.; Tantray, I.; Lim, J.; Chen, S.; Li, Y.; Davis, Z.; Sitron, C.; Dong, J.; Gispert, S.; Auburger, G.; et al. MISTERMINATE Mechanistically Links Mitochondrial Dysfunction with Proteostasis Failure. Mol Cell 2019, 75, 835-848 e838. [CrossRef]
- Lytvynenko, I.; Paternoga, H.; Thrun, A.; Balke, A.; Muller, T.A.; Chiang, C.H.; Nagler, K.; Tsaprailis, G.; Anders, S.; Bischofs, I.; et al. Alanine Tails Signal Proteolysis in Bacterial Ribosome-Associated Quality Control. Cell 2019, 178, 76-90 e22. [CrossRef]
- Gottesman, S.; Roche, E.; Zhou, Y.; Sauer, R.T. The ClpXP and ClpAP proteases degrade proteins with carboxy-terminal peptide tails added by the SsrA-tagging system. Genes Dev 1998, 12, 1338-1347. [CrossRef]
- Gruffaz, C.; Smirnov, A. GTPase Era at the heart of ribosome assembly. Front Mol Biosci 2023, 10, 1263433. [CrossRef]
- Dennerlein, S.; Rozanska, A.; Wydro, M.; Chrzanowska-Lightowlers, Z.M.; Lightowlers, R.N. Human ERAL1 is a mitochondrial RNA chaperone involved in the assembly of the 28S small mitochondrial ribosomal subunit. Biochem J 2010, 430, 551-558. [CrossRef]
- Uchiumi, T.; Ohgaki, K.; Yagi, M.; Aoki, Y.; Sakai, A.; Matsumoto, S.; Kang, D. ERAL1 is associated with mitochondrial ribosome and elimination of ERAL1 leads to mitochondrial dysfunction and growth retardation. Nucleic Acids Res 2010, 38, 5554-5568. [CrossRef]
- Accardi, R.; Oxelmark, E.; Jauniaux, N.; de Pinto, V.; Marchini, A.; Tommasino, M. High levels of the mitochondrial large ribosomal subunit protein 40 prevent loss of mitochondrial DNA in null mmf1 Saccharomyces cerevisiae cells. Yeast 2004, 21, 539-548. [CrossRef]
- Zhang, X.; Gao, X.; Coots, R.A.; Conn, C.S.; Liu, B.; Qian, S.B. Translational control of the cytosolic stress response by mitochondrial ribosomal protein L18. Nat Struct Mol Biol 2015, 22, 404-410. [CrossRef]
- Xu, P.; Wang, L.; Peng, H.; Liu, H.; Liu, H.; Yuan, Q.; Lin, Y.; Xu, J.; Pang, X.; Wu, H.; et al. Disruption of Hars2 in Cochlear Hair Cells Causes Progressive Mitochondrial Dysfunction and Hearing Loss in Mice. Front Cell Neurosci 2021, 15, 804345. [CrossRef]
- Gong, S.; Wang, X.; Meng, F.; Cui, L.; Yi, Q.; Zhao, Q.; Cang, X.; Cai, Z.; Mo, J.Q.; Liang, Y.; et al. Overexpression of mitochondrial histidyl-tRNA synthetase restores mitochondrial dysfunction caused by a deafness-associated tRNA(His) mutation. J Biol Chem 2020, 295, 940-954. [CrossRef]
- van der Knaap, M.S.; Bugiani, M.; Mendes, M.I.; Riley, L.G.; Smith, D.E.C.; Rudinger-Thirion, J.; Frugier, M.; Breur, M.; Crawford, J.; van Gaalen, J.; et al. Biallelic variants in LARS2 and KARS cause deafness and (ovario)leukodystrophy. Neurology 2019, 92, e1225-e1237. [CrossRef]
- Perli, E.; Fiorillo, A.; Giordano, C.; Pisano, A.; Montanari, A.; Grazioli, P.; Campese, A.F.; Di Micco, P.; Tuppen, H.A.; Genovese, I.; et al. Short peptides from leucyl-tRNA synthetase rescue disease-causing mitochondrial tRNA point mutations. Hum Mol Genet 2016, 25, 903-915. [CrossRef]
- Hornig-Do, H.T.; Montanari, A.; Rozanska, A.; Tuppen, H.A.; Almalki, A.A.; Abg-Kamaludin, D.P.; Frontali, L.; Francisci, S.; Lightowlers, R.N.; Chrzanowska-Lightowlers, Z.M. Human mitochondrial leucyl tRNA synthetase can suppress non cognate pathogenic mt-tRNA mutations. EMBO Mol Med 2014, 6, 183-193. [CrossRef]
- Levinger, L.; Morl, M.; Florentz, C. Mitochondrial tRNA 3' end metabolism and human disease. Nucleic Acids Res 2004, 32, 5430-5441. [CrossRef]
- Rumyantseva, A.; Popovic, M.; Trifunovic, A. CLPP deficiency ameliorates neurodegeneration caused by impaired mitochondrial protein synthesis. Brain 2022, 145, 92-104. [CrossRef]
- Lyu, B.; Song, Q. The intricate relationship of G-Quadruplexes and bacterial pathogenicity islands. Elife 2024, 12. [CrossRef]
- Mestre-Fos, S.; Ito, C.; Moore, C.M.; Reddi, A.R.; Williams, L.D. Human ribosomal G-quadruplexes regulate heme bioavailability. J Biol Chem 2020, 295, 14855-14865. [CrossRef]
- Chang, T.; Liu, X.; Cheng, X.; Qi, C.; Mei, H.; Shangguan, D. Selective isolation of G-quadruplexes by affinity chromatography. J Chromatogr A 2012, 1246, 62-68. [CrossRef]
- Li, W.; Li, Y.; Liu, Z.; Lin, B.; Yi, H.; Xu, F.; Nie, Z.; Yao, S. Insight into G-quadruplex-hemin DNAzyme/RNAzyme: adjacent adenine as the intramolecular species for remarkable enhancement of enzymatic activity. Nucleic Acids Res 2016, 44, 7373-7384. [CrossRef]
- Grigg, J.C.; Shumayrikh, N.; Sen, D. G-quadruplex structures formed by expanded hexanucleotide repeat RNA and DNA from the neurodegenerative disease-linked C9orf72 gene efficiently sequester and activate heme. PLoS One 2014, 9, e106449. [CrossRef]
- Li, C.; Yin, Z.; Xiao, R.; Huang, B.; Cui, Y.; Wang, H.; Xiang, Y.; Wang, L.; Lei, L.; Ye, J.; et al. G-quadruplexes sense natural porphyrin metabolites for regulation of gene transcription and chromatin landscapes. Genome Biol 2022, 23, 259. [CrossRef]
- Varshney, D.; Cuesta, S.M.; Herdy, B.; Abdullah, U.B.; Tannahill, D.; Balasubramanian, S. RNA G-quadruplex structures control ribosomal protein production. Sci Rep 2021, 11, 22735. [CrossRef]
- Pietras, Z.; Wojcik, M.A.; Borowski, L.S.; Szewczyk, M.; Kulinski, T.M.; Cysewski, D.; Stepien, P.P.; Dziembowski, A.; Szczesny, R.J. Dedicated surveillance mechanism controls G-quadruplex forming non-coding RNAs in human mitochondria. Nat Commun 2018, 9, 2558. [CrossRef]
- Noh, J.H.; Kim, K.M.; Abdelmohsen, K.; Yoon, J.H.; Panda, A.C.; Munk, R.; Kim, J.; Curtis, J.; Moad, C.A.; Wohler, C.M.; et al. HuR and GRSF1 modulate the nuclear export and mitochondrial localization of the lncRNA RMRP. Genes Dev 2016, 30, 1224-1239. [CrossRef]
- Jourdain, A.A.; Koppen, M.; Wydro, M.; Rodley, C.D.; Lightowlers, R.N.; Chrzanowska-Lightowlers, Z.M.; Martinou, J.C. GRSF1 regulates RNA processing in mitochondrial RNA granules. Cell Metab 2013, 17, 399-410. [CrossRef]
- Hensen, F.; Potter, A.; van Esveld, S.L.; Tarres-Sole, A.; Chakraborty, A.; Sola, M.; Spelbrink, J.N. Mitochondrial RNA granules are critically dependent on mtDNA replication factors Twinkle and mtSSB. Nucleic Acids Res 2019, 47, 3680-3698. [CrossRef]
- Xavier, V.J.; Martinou, J.C. RNA Granules in the Mitochondria and Their Organization under Mitochondrial Stresses. Int J Mol Sci 2021, 22. [CrossRef]
- Antonicka, H.; Sasarman, F.; Nishimura, T.; Paupe, V.; Shoubridge, E.A. The mitochondrial RNA-binding protein GRSF1 localizes to RNA granules and is required for posttranscriptional mitochondrial gene expression. Cell Metab 2013, 17, 386-398. [CrossRef]
- Pietras, Z.; Wojcik, M.A.; Borowski, L.S.; Szewczyk, M.; Kulinski, T.M.; Cysewski, D.; Stepien, P.P.; Dziembowski, A.; Szczesny, R.J. Controlling the mitochondrial antisense - role of the SUV3-PNPase complex and its co-factor GRSF1 in mitochondrial RNA surveillance. Mol Cell Oncol 2018, 5, e1516452. [CrossRef]
- Wang, J.; Lu, J.; Zhu, Y.; Huang, Q.; Gu, Q.; Tian, S.; Ge, J.; Lin, X.; Sha, W. Guanine-rich RNA sequence binding factor 1 regulates neuronal ferroptosis after spinal cord injury in rats via the GPX4 signaling pathway. Brain Res 2023, 1818, 148497. [CrossRef]
- Dumoulin, B.; Heydeck, D.; Jahn, D.; Lasse, M.; Sofi, S.; Ufer, C.; Kuhn, H. Male guanine-rich RNA sequence binding factor 1 knockout mice (Grsf1(-/-)) gain less body weight during adolescence and adulthood. Cell Biosci 2022, 12, 199. [CrossRef]
- Noh, J.H.; Kim, K.M.; Pandey, P.R.; Noren Hooten, N.; Munk, R.; Kundu, G.; De, S.; Martindale, J.L.; Yang, X.; Evans, M.K.; et al. Loss of RNA-binding protein GRSF1 activates mTOR to elicit a proinflammatory transcriptional program. Nucleic Acids Res 2019, 47, 2472-2486. [CrossRef]
- Al-Furoukh, N.; Goffart, S.; Szibor, M.; Wanrooij, S.; Braun, T. Binding to G-quadruplex RNA activates the mitochondrial GTPase NOA1. Biochim Biophys Acta 2013, 1833, 2933-2942. [CrossRef]
- He, J.; Cooper, H.M.; Reyes, A.; Di Re, M.; Kazak, L.; Wood, S.R.; Mao, C.C.; Fearnley, I.M.; Walker, J.E.; Holt, I.J. Human C4orf14 interacts with the mitochondrial nucleoid and is involved in the biogenesis of the small mitochondrial ribosomal subunit. Nucleic Acids Res 2012, 40, 6097-6108. [CrossRef]
- Kolanczyk, M.; Pech, M.; Zemojtel, T.; Yamamoto, H.; Mikula, I.; Calvaruso, M.A.; van den Brand, M.; Richter, R.; Fischer, B.; Ritz, A.; et al. NOA1 is an essential GTPase required for mitochondrial protein synthesis. Mol Biol Cell 2011, 22, 1-11. [CrossRef]
- Al-Furoukh, N.; Kardon, J.R.; Kruger, M.; Szibor, M.; Baker, T.A.; Braun, T. NOA1, a novel ClpXP substrate, takes an unexpected nuclear detour prior to mitochondrial import. PLoS One 2014, 9, e103141. [CrossRef]
- Luo, M.; Ma, W.; Sand, Z.; Finlayson, J.; Wang, T.; Brinton, R.D.; Willis, W.T.; Mandarino, L.J. Von Willebrand factor A domain-containing protein 8 (VWA8) localizes to the matrix side of the inner mitochondrial membrane. Biochem Biophys Res Commun 2020, 521, 158-163. [CrossRef]
- Chen, Z.; Suzuki, H.; Kobayashi, Y.; Wang, A.C.; DiMaio, F.; Kawashima, S.A.; Walz, T.; Kapoor, T.M. Structural Insights into Mdn1, an Essential AAA Protein Required for Ribosome Biogenesis. Cell 2018, 175, 822-834 e818. [CrossRef]
- Kawashima, S.A.; Chen, Z.; Aoi, Y.; Patgiri, A.; Kobayashi, Y.; Nurse, P.; Kapoor, T.M. Potent, Reversible, and Specific Chemical Inhibitors of Eukaryotic Ribosome Biogenesis. Cell 2016, 167, 512-524 e514. [CrossRef]
- Prattes, M.; Lo, Y.H.; Bergler, H.; Stanley, R.E. Shaping the Nascent Ribosome: AAA-ATPases in Eukaryotic Ribosome Biogenesis. Biomolecules 2019, 9. [CrossRef]
- Linke, R.; Limmer, M.; Juranek, S.A.; Heine, A.; Paeschke, K. The Relevance of G-Quadruplexes for DNA Repair. Int J Mol Sci 2021, 22. [CrossRef]
- McShane, E.; Couvillion, M.; Ietswaart, R.; Prakash, G.; Smalec, B.M.; Soto, I.; Baxter-Koenigs, A.R.; Choquet, K.; Churchman, L.S. A kinetic dichotomy between mitochondrial and nuclear gene expression processes. Mol Cell 2024. [CrossRef]
- Honarmand, S.; Shoubridge, E.A. Poly (A) tail length of human mitochondrial mRNAs is tissue-specific and a mutation in LRPPRC results in transcript-specific patterns of deadenylation. Mol Genet Metab Rep 2020, 25, 100687. [CrossRef]
- Wilson, W.C.; Hornig-Do, H.T.; Bruni, F.; Chang, J.H.; Jourdain, A.A.; Martinou, J.C.; Falkenberg, M.; Spahr, H.; Larsson, N.G.; Lewis, R.J.; et al. A human mitochondrial poly(A) polymerase mutation reveals the complexities of post-transcriptional mitochondrial gene expression. Hum Mol Genet 2014, 23, 6345-6355. [CrossRef]
- Chujo, T.; Ohira, T.; Sakaguchi, Y.; Goshima, N.; Nomura, N.; Nagao, A.; Suzuki, T. LRPPRC/SLIRP suppresses PNPase-mediated mRNA decay and promotes polyadenylation in human mitochondria. Nucleic Acids Res 2012, 40, 8033-8047. [CrossRef]
- Ruzzenente, B.; Metodiev, M.D.; Wredenberg, A.; Bratic, A.; Park, C.B.; Camara, Y.; Milenkovic, D.; Zickermann, V.; Wibom, R.; Hultenby, K.; et al. LRPPRC is necessary for polyadenylation and coordination of translation of mitochondrial mRNAs. EMBO J 2012, 31, 443-456. [CrossRef]
- Pajak, A.; Laine, I.; Clemente, P.; El-Fissi, N.; Schober, F.A.; Maffezzini, C.; Calvo-Garrido, J.; Wibom, R.; Filograna, R.; Dhir, A.; et al. Defects of mitochondrial RNA turnover lead to the accumulation of double-stranded RNA in vivo. PLoS Genet 2019, 15, e1008240. [CrossRef]
- Szczesny, R.J.; Wojcik, M.A.; Borowski, L.S.; Szewczyk, M.J.; Skrok, M.M.; Golik, P.; Stepien, P.P. Yeast and human mitochondrial helicases. Biochim Biophys Acta 2013, 1829, 842-853. [CrossRef]
- Borowski, L.S.; Dziembowski, A.; Hejnowicz, M.S.; Stepien, P.P.; Szczesny, R.J. Human mitochondrial RNA decay mediated by PNPase-hSuv3 complex takes place in distinct foci. Nucleic Acids Res 2013, 41, 1223-1240. [CrossRef]
- Szczesny, R.J.; Borowski, L.S.; Malecki, M.; Wojcik, M.A.; Stepien, P.P.; Golik, P. RNA degradation in yeast and human mitochondria. Biochim Biophys Acta 2012, 1819, 1027-1034. [CrossRef]
- Clemente, P.; Pajak, A.; Laine, I.; Wibom, R.; Wedell, A.; Freyer, C.; Wredenberg, A. SUV3 helicase is required for correct processing of mitochondrial transcripts. Nucleic Acids Res 2015, 43, 7398-7413. [CrossRef]
- Jain, M.; Golzarroshan, B.; Lin, C.L.; Agrawal, S.; Tang, W.H.; Wu, C.J.; Yuan, H.S. Dimeric assembly of human Suv3 helicase promotes its RNA unwinding function in mitochondrial RNA degradosome for RNA decay. Protein Sci 2022, 31, e4312. [CrossRef]
- Toompuu, M.; Tuomela, T.; Laine, P.; Paulin, L.; Dufour, E.; Jacobs, H.T. Polyadenylation and degradation of structurally abnormal mitochondrial tRNAs in human cells. Nucleic Acids Res 2018, 46, 5209-5226. [CrossRef]
- Szczesny, R.J.; Borowski, L.S.; Brzezniak, L.K.; Dmochowska, A.; Gewartowski, K.; Bartnik, E.; Stepien, P.P. Human mitochondrial RNA turnover caught in flagranti: involvement of hSuv3p helicase in RNA surveillance. Nucleic Acids Res 2010, 38, 279-298. [CrossRef]
- Wang, D.D.; Shu, Z.; Lieser, S.A.; Chen, P.L.; Lee, W.H. Human mitochondrial SUV3 and polynucleotide phosphorylase form a 330-kDa heteropentamer to cooperatively degrade double-stranded RNA with a 3'-to-5' directionality. J Biol Chem 2009, 284, 20812-20821. [CrossRef]
- Sarkar, D.; Fisher, P.B. Human polynucleotide phosphorylase (hPNPase old-35): an RNA degradation enzyme with pleiotrophic biological effects. Cell Cycle 2006, 5, 1080-1084. [CrossRef]
- Piazza, F.; Zappone, M.; Sana, M.; Briani, F.; Deho, G. Polynucleotide phosphorylase of Escherichia coli is required for the establishment of bacteriophage P4 immunity. J Bacteriol 1996, 178, 5513-5521. [CrossRef]
- Chen, P.L. SUV3 Helicase and Mitochondrial Homeostasis. Int J Mol Sci 2023, 24. [CrossRef]
- Silva, S.; Camino, L.P.; Aguilera, A. Human mitochondrial degradosome prevents harmful mitochondrial R loops and mitochondrial genome instability. Proc Natl Acad Sci U S A 2018, 115, 11024-11029. [CrossRef]
- Carzaniga, T.; Sbarufatti, G.; Briani, F.; Deho, G. Polynucleotide phosphorylase is implicated in homologous recombination and DNA repair in Escherichia coli. BMC Microbiol 2017, 17, 81. [CrossRef]
- Tuteja, N.; Tarique, M.; Tuteja, R. Rice SUV3 is a bidirectional helicase that binds both DNA and RNA. BMC Plant Biol 2014, 14, 283. [CrossRef]
- Minczuk, M.; Piwowarski, J.; Papworth, M.A.; Awiszus, K.; Schalinski, S.; Dziembowski, A.; Dmochowska, A.; Bartnik, E.; Tokatlidis, K.; Stepien, P.P.; et al. Localisation of the human hSuv3p helicase in the mitochondrial matrix and its preferential unwinding of dsDNA. Nucleic Acids Res 2002, 30, 5074-5086. [CrossRef]
- Barbier, M.; Bahlo, M.; Pennisi, A.; Jacoupy, M.; Tankard, R.M.; Ewenczyk, C.; Davies, K.C.; Lino-Coulon, P.; Colace, C.; Rafehi, H.; et al. Heterozygous PNPT1 Variants Cause Spinocerebellar Ataxia Type 25. Ann Neurol 2022, 92, 122-137. [CrossRef]
- Eaton, A.; Bernier, F.P.; Goedhart, C.; Caluseriu, O.; Lamont, R.E.; Boycott, K.M.; Parboosingh, J.S.; Innes, A.M.; Care4Rare Canada, C. Is PNPT1-related hearing loss ever non-syndromic? Whole exome sequencing of adult siblings expands the natural history of PNPT1-related disorders. Am J Med Genet A 2018, 176, 2487-2493. [CrossRef]
- Matilainen, S.; Carroll, C.J.; Richter, U.; Euro, L.; Pohjanpelto, M.; Paetau, A.; Isohanni, P.; Suomalainen, A. Defective mitochondrial RNA processing due to PNPT1 variants causes Leigh syndrome. Hum Mol Genet 2017, 26, 3352-3361. [CrossRef]
- Sato, R.; Arai-Ichinoi, N.; Kikuchi, A.; Matsuhashi, T.; Numata-Uematsu, Y.; Uematsu, M.; Fujii, Y.; Murayama, K.; Ohtake, A.; Abe, T.; et al. Novel biallelic mutations in the PNPT1 gene encoding a mitochondrial-RNA-import protein PNPase cause delayed myelination. Clin Genet 2018, 93, 242-247. [CrossRef]
- Slavotinek, A.M.; Garcia, S.T.; Chandratillake, G.; Bardakjian, T.; Ullah, E.; Wu, D.; Umeda, K.; Lao, R.; Tang, P.L.; Wan, E.; et al. Exome sequencing in 32 patients with anophthalmia/microphthalmia and developmental eye defects. Clin Genet 2015, 88, 468-473. [CrossRef]
- von Ameln, S.; Wang, G.; Boulouiz, R.; Rutherford, M.A.; Smith, G.M.; Li, Y.; Pogoda, H.M.; Nurnberg, G.; Stiller, B.; Volk, A.E.; et al. A mutation in PNPT1, encoding mitochondrial-RNA-import protein PNPase, causes hereditary hearing loss. Am J Hum Genet 2012, 91, 919-927. [CrossRef]
- Dhir, A.; Dhir, S.; Borowski, L.S.; Jimenez, L.; Teitell, M.; Rotig, A.; Crow, Y.J.; Rice, G.I.; Duffy, D.; Tamby, C.; et al. Mitochondrial double-stranded RNA triggers antiviral signalling in humans. Nature 2018, 560, 238-242. [CrossRef]
- Key, J.; Maletzko, A.; Kohli, A.; Gispert, S.; Torres-Odio, S.; Wittig, I.; Heidler, J.; Barcena, C.; Lopez-Otin, C.; Lei, Y.; et al. Loss of mitochondrial ClpP, Lonp1, and Tfam triggers transcriptional induction of Rnf213, a susceptibility factor for moyamoya disease. Neurogenetics 2020, 21, 187-203. [CrossRef]
- Maletzko, A.; Key, J.; Wittig, I.; Gispert, S.; Koepf, G.; Canet-Pons, J.; Torres-Odio, S.; West, A.P.; Auburger, G. Increased presence of nuclear DNAJA3 and upregulation of cytosolic STAT1 and of nucleic acid sensors trigger innate immunity in the ClpP-null mouse. Neurogenetics 2021, 22, 297-312. [CrossRef]
- Torres-Odio, S.; Lei, Y.; Gispert, S.; Maletzko, A.; Key, J.; Menissy, S.S.; Wittig, I.; Auburger, G.; West, A.P. Loss of Mitochondrial Protease CLPP Activates Type I IFN Responses through the Mitochondrial DNA-cGAS-STING Signaling Axis. J Immunol 2021, 206, 1890-1900. [CrossRef]
- McKay, R.; Druyan, R.; Getz, G.S.; Rabinowitz, M. Intramitochondrial localization of delta-aminolaevulate synthetase and ferrochelatase in rat liver. Biochem J 1969, 114, 455-461. [CrossRef]
- Medlock, A.E.; Shiferaw, M.T.; Marcero, J.R.; Vashisht, A.A.; Wohlschlegel, J.A.; Phillips, J.D.; Dailey, H.A. Identification of the Mitochondrial Heme Metabolism Complex. PLoS One 2015, 10, e0135896. [CrossRef]
- Ferreira, G.C.; Andrew, T.L.; Karr, S.W.; Dailey, H.A. Organization of the terminal two enzymes of the heme biosynthetic pathway. Orientation of protoporphyrinogen oxidase and evidence for a membrane complex. J Biol Chem 1988, 263, 3835-3839.
- Lim, S.; Clark, D.S. Phase-separated biomolecular condensates for biocatalysis. Trends Biotechnol 2024, 42, 496-509. [CrossRef]
- Dahmani, I.; Qin, K.; Zhang, Y.; Fernie, A.R. The formation and function of plant metabolons. Plant J 2023, 114, 1080-1092. [CrossRef]
- Kastritis, P.L.; Gavin, A.C. Enzymatic complexes across scales. Essays Biochem 2018, 62, 501-514. [CrossRef]
- Kim, H.J.; Khalimonchuk, O.; Smith, P.M.; Winge, D.R. Structure, function, and assembly of heme centers in mitochondrial respiratory complexes. Biochim Biophys Acta 2012, 1823, 1604-1616. [CrossRef]
- Maio, N.; Kim, K.S.; Holmes-Hampton, G.; Singh, A.; Rouault, T.A. Dimeric ferrochelatase bridges ABCB7 and ABCB10 homodimers in an architecturally defined molecular complex required for heme biosynthesis. Haematologica 2019, 104, 1756-1767. [CrossRef]
- Chen, W.; Dailey, H.A.; Paw, B.H. Ferrochelatase forms an oligomeric complex with mitoferrin-1 and Abcb10 for erythroid heme biosynthesis. Blood 2010, 116, 628-630. [CrossRef]
- Richardson, D.R.; Lane, D.J.; Becker, E.M.; Huang, M.L.; Whitnall, M.; Suryo Rahmanto, Y.; Sheftel, A.D.; Ponka, P. Mitochondrial iron trafficking and the integration of iron metabolism between the mitochondrion and cytosol. Proc Natl Acad Sci U S A 2010, 107, 10775-10782. [CrossRef]
- Shum, M.; Shintre, C.A.; Althoff, T.; Gutierrez, V.; Segawa, M.; Saxberg, A.D.; Martinez, M.; Adamson, R.; Young, M.R.; Faust, B.; et al. ABCB10 exports mitochondrial biliverdin, driving metabolic maladaptation in obesity. Sci Transl Med 2021, 13. [CrossRef]
- Pearson, S.A.; Cowan, J.A. Evolution of the human mitochondrial ABCB7 [2Fe-2S](GS)(4) cluster exporter and the molecular mechanism of an E433K disease-causing mutation. Arch Biochem Biophys 2021, 697, 108661. [CrossRef]
- Martinez, M.; Fendley, G.A.; Saxberg, A.D.; Zoghbi, M.E. Stimulation of the human mitochondrial transporter ABCB10 by zinc-mesoporphrin. PLoS One 2020, 15, e0238754. [CrossRef]
- Yamamoto, M.; Arimura, H.; Fukushige, T.; Minami, K.; Nishizawa, Y.; Tanimoto, A.; Kanekura, T.; Nakagawa, M.; Akiyama, S.; Furukawa, T. Abcb10 role in heme biosynthesis in vivo: Abcb10 knockout in mice causes anemia with protoporphyrin IX and iron accumulation. Mol Cell Biol 2014, 34, 1077-1084. [CrossRef]
- Chen, W.; Paradkar, P.N.; Li, L.; Pierce, E.L.; Langer, N.B.; Takahashi-Makise, N.; Hyde, B.B.; Shirihai, O.S.; Ward, D.M.; Kaplan, J.; et al. Abcb10 physically interacts with mitoferrin-1 (Slc25a37) to enhance its stability and function in the erythroid mitochondria. Proc Natl Acad Sci U S A 2009, 106, 16263-16268. [CrossRef]
- Clough, C.A.; Pangallo, J.; Sarchi, M.; Ilagan, J.O.; North, K.; Bergantinos, R.; Stolla, M.C.; Naru, J.; Nugent, P.; Kim, E.; et al. Coordinated missplicing of TMEM14C and ABCB7 causes ring sideroblast formation in SF3B1-mutant myelodysplastic syndrome. Blood 2022, 139, 2038-2049. [CrossRef]
- Harding, C.R.; Sidik, S.M.; Petrova, B.; Gnadig, N.F.; Okombo, J.; Herneisen, A.L.; Ward, K.E.; Markus, B.M.; Boydston, E.A.; Fidock, D.A.; et al. Genetic screens reveal a central role for heme metabolism in artemisinin susceptibility. Nat Commun 2020, 11, 4813. [CrossRef]
- Yien, Y.Y.; Robledo, R.F.; Schultz, I.J.; Takahashi-Makise, N.; Gwynn, B.; Bauer, D.E.; Dass, A.; Yi, G.; Li, L.; Hildick-Smith, G.J.; et al. TMEM14C is required for erythroid mitochondrial heme metabolism. J Clin Invest 2014, 124, 4294-4304. [CrossRef]
- Nilsson, R.; Schultz, I.J.; Pierce, E.L.; Soltis, K.A.; Naranuntarat, A.; Ward, D.M.; Baughman, J.M.; Paradkar, P.N.; Kingsley, P.D.; Culotta, V.C.; et al. Discovery of genes essential for heme biosynthesis through large-scale gene expression analysis. Cell Metab 2009, 10, 119-130. [CrossRef]
- Azuma, M.; Kabe, Y.; Kuramori, C.; Kondo, M.; Yamaguchi, Y.; Handa, H. Adenine nucleotide translocator transports haem precursors into mitochondria. PLoS One 2008, 3, e3070. [CrossRef]
- Kabe, Y.; Ohmori, M.; Shinouchi, K.; Tsuboi, Y.; Hirao, S.; Azuma, M.; Watanabe, H.; Okura, I.; Handa, H. Porphyrin accumulation in mitochondria is mediated by 2-oxoglutarate carrier. J Biol Chem 2006, 281, 31729-31735. [CrossRef]
- Lash, L.H. Mitochondrial glutathione transport: physiological, pathological and toxicological implications. Chem Biol Interact 2006, 163, 54-67. [CrossRef]
- Deery, E.; Schroeder, S.; Lawrence, A.D.; Taylor, S.L.; Seyedarabi, A.; Waterman, J.; Wilson, K.S.; Brown, D.; Geeves, M.A.; Howard, M.J.; et al. An enzyme-trap approach allows isolation of intermediates in cobalamin biosynthesis. Nat Chem Biol 2012, 8, 933-940. [CrossRef]
- Cheung, C.W.; Cohen, N.S.; Raijman, L. Channeling of urea cycle intermediates in situ in permeabilized hepatocytes. J Biol Chem 1989, 264, 4038-4044.
- Chen, C.; Hamza, I. Notes from the Underground: Heme Homeostasis in C. elegans. Biomolecules 2023, 13. [CrossRef]
- Chambers, I.G.; Willoughby, M.M.; Hamza, I.; Reddi, A.R. One ring to bring them all and in the darkness bind them: The trafficking of heme without deliverers. Biochim Biophys Acta Mol Cell Res 2021, 1868, 118881. [CrossRef]
- Sachar, M.; Anderson, K.E.; Ma, X. Protoporphyrin IX: the Good, the Bad, and the Ugly. J Pharmacol Exp Ther 2016, 356, 267-275. [CrossRef]
- Liou, Y.F.; Charoenkwan, P.; Srinivasulu, Y.; Vasylenko, T.; Lai, S.C.; Lee, H.C.; Chen, Y.H.; Huang, H.L.; Ho, S.Y. SCMHBP: prediction and analysis of heme binding proteins using propensity scores of dipeptides. BMC Bioinformatics 2014, 15 Suppl 16, S4. [CrossRef]
- Whitman, J.C.; Paw, B.H.; Chung, J. The role of ClpX in erythropoietic protoporphyria. Hematol Transfus Cell Ther 2018, 40, 182-188. [CrossRef]
- Fouquet, C.; Le Rouzic, M.A.; Leblanc, T.; Fouyssac, F.; Leverger, G.; Hessissen, L.; Marlin, S.; Bourrat, E.; Fahd, M.; Raffoux, E.; et al. Genotype/phenotype correlations of childhood-onset congenital sideroblastic anaemia in a European cohort. Br J Haematol 2019, 187, 530-542. [CrossRef]
- Kardon, J.R.; Moroco, J.A.; Engen, J.R.; Baker, T.A. Mitochondrial ClpX activates an essential biosynthetic enzyme through partial unfolding. Elife 2020, 9. [CrossRef]
- Brown, B.L.; Kardon, J.R.; Sauer, R.T.; Baker, T.A. Structure of the Mitochondrial Aminolevulinic Acid Synthase, a Key Heme Biosynthetic Enzyme. Structure 2018, 26, 580-589 e584. [CrossRef]
- Huang, L. An experimental study of the principle of electronic root canal measurement. J Endod 1987, 13, 60-64. [CrossRef]
- Kubota, Y.; Nomura, K.; Katoh, Y.; Yamashita, R.; Kaneko, K.; Furuyama, K. Novel Mechanisms for Heme-dependent Degradation of ALAS1 Protein as a Component of Negative Feedback Regulation of Heme Biosynthesis. J Biol Chem 2016, 291, 20516-20529. [CrossRef]
- Schiroli, D.; Peracchi, A. A subfamily of PLP-dependent enzymes specialized in handling terminal amines. Biochim Biophys Acta 2015, 1854, 1200-1211. [CrossRef]
- Tarnacka, B.; Jopowicz, A.; Maslinska, M. Copper, Iron, and Manganese Toxicity in Neuropsychiatric Conditions. Int J Mol Sci 2021, 22. [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol 2021, 22, 266-282. [CrossRef]
- Laut, C.L.; Leasure, C.S.; Pi, H.; Carlin, S.M.; Chu, M.L.; Hillebrand, G.H.; Lin, H.K.; Yi, X.I.; Stauff, D.L.; Skaar, E.P. DnaJ and ClpX Are Required for HitRS and HssRS Two-Component System Signaling in Bacillus anthracis. Infect Immun 2022, 90, e0056021. [CrossRef]
- Farrand, A.J.; Friedman, D.B.; Reniere, M.L.; Ingmer, H.; Frees, D.; Skaar, E.P. Proteomic analyses of iron-responsive, Clp-dependent changes in Staphylococcus aureus. Pathog Dis 2015, 73. [CrossRef]
- Farrand, A.J.; Reniere, M.L.; Ingmer, H.; Frees, D.; Skaar, E.P. Regulation of host hemoglobin binding by the Staphylococcus aureus Clp proteolytic system. J Bacteriol 2013, 195, 5041-5050. [CrossRef]
- Stanne, T.M.; Sjogren, L.L.; Koussevitzky, S.; Clarke, A.K. Identification of new protein substrates for the chloroplast ATP-dependent Clp protease supports its constitutive role in Arabidopsis. Biochem J 2009, 417, 257-268. [CrossRef]
- Wang, L.; Elliott, M.; Elliott, T. Conditional stability of the HemA protein (glutamyl-tRNA reductase) regulates heme biosynthesis in Salmonella typhimurium. J Bacteriol 1999, 181, 1211-1219. [CrossRef]
- Szczepanowska, K.; Trifunovic, A. Tune instead of destroy: How proteolysis keeps OXPHOS in shape. Biochim Biophys Acta Bioenerg 2021, 1862, 148365. [CrossRef]
- Vercellino, I.; Sazanov, L.A. The assembly, regulation and function of the mitochondrial respiratory chain. Nat Rev Mol Cell Biol 2022, 23, 141-161. [CrossRef]
- Giachin, G.; Bouverot, R.; Acajjaoui, S.; Pantalone, S.; Soler-Lopez, M. Dynamics of Human Mitochondrial Complex I Assembly: Implications for Neurodegenerative Diseases. Front Mol Biosci 2016, 3, 43. [CrossRef]
- Berrisford, J.M.; Baradaran, R.; Sazanov, L.A. Structure of bacterial respiratory complex I. Biochim Biophys Acta 2016, 1857, 892-901. [CrossRef]
- Friedrich, T. On the mechanism of respiratory complex I. J Bioenerg Biomembr 2014, 46, 255-268. [CrossRef]
- Clason, T.; Ruiz, T.; Schagger, H.; Peng, G.; Zickermann, V.; Brandt, U.; Michel, H.; Radermacher, M. The structure of eukaryotic and prokaryotic complex I. J Struct Biol 2010, 169, 81-88. [CrossRef]
- Szczepanowska, K.; Senft, K.; Heidler, J.; Herholz, M.; Kukat, A.; Hohne, M.N.; Hofsetz, E.; Becker, C.; Kaspar, S.; Giese, H.; et al. A salvage pathway maintains highly functional respiratory complex I. Nat Commun 2020, 11, 1643. [CrossRef]
- Feric, M.; Demarest, T.G.; Tian, J.; Croteau, D.L.; Bohr, V.A.; Misteli, T. Self-assembly of multi-component mitochondrial nucleoids via phase separation. EMBO J 2021, 40, e107165. [CrossRef]
- Rey, T.; Zaganelli, S.; Cuillery, E.; Vartholomaiou, E.; Croisier, M.; Martinou, J.C.; Manley, S. Mitochondrial RNA granules are fluid condensates positioned by membrane dynamics. Nat Cell Biol 2020, 22, 1180-1186. [CrossRef]
- Guilhas, B.; Walter, J.C.; Rech, J.; David, G.; Walliser, N.O.; Palmeri, J.; Mathieu-Demaziere, C.; Parmeggiani, A.; Bouet, J.Y.; Le Gall, A.; et al. ATP-Driven Separation of Liquid Phase Condensates in Bacteria. Mol Cell 2020, 79, 293-303 e294. [CrossRef]
- Ladouceur, A.M.; Parmar, B.S.; Biedzinski, S.; Wall, J.; Tope, S.G.; Cohn, D.; Kim, A.; Soubry, N.; Reyes-Lamothe, R.; Weber, S.C. Clusters of bacterial RNA polymerase are biomolecular condensates that assemble through liquid-liquid phase separation. Proc Natl Acad Sci U S A 2020, 117, 18540-18549. [CrossRef]
- Muthunayake, N.S.; Tomares, D.T.; Childers, W.S.; Schrader, J.M. Phase-separated bacterial ribonucleoprotein bodies organize mRNA decay. Wiley Interdiscip Rev RNA 2020, 11, e1599. [CrossRef]
- Kato, M.; Han, T.W.; Xie, S.; Shi, K.; Du, X.; Wu, L.C.; Mirzaei, H.; Goldsmith, E.J.; Longgood, J.; Pei, J.; et al. Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell 2012, 149, 753-767. [CrossRef]
- Annunziata, O.; Asherie, N.; Lomakin, A.; Pande, J.; Ogun, O.; Benedek, G.B. Effect of polyethylene glycol on the liquid-liquid phase transition in aqueous protein solutions. Proc Natl Acad Sci U S A 2002, 99, 14165-14170. [CrossRef]
- Zhang, L.; Wang, S.; Wang, W.; Shi, J.; Stovall, D.B.; Li, D.; Sui, G. Phase-Separated Subcellular Compartmentation and Related Human Diseases. Int J Mol Sci 2022, 23. [CrossRef]
- Peran, I.; Mittag, T. Molecular structure in biomolecular condensates. Curr Opin Struct Biol 2020, 60, 17-26. [CrossRef]
- Alberti, S.; Dormann, D. Liquid-Liquid Phase Separation in Disease. Annu Rev Genet 2019, 53, 171-194. [CrossRef]
- Dutta, A.; Sepehri, A.; Lazaridis, T. Putative Pore Structures of Amyloid beta 25-35 in Lipid Bilayers. Biochemistry 2023, 62, 2549-2558. [CrossRef]
- Vendruscolo, M.; Fuxreiter, M. Sequence Determinants of the Aggregation of Proteins Within Condensates Generated by Liquid-liquid Phase Separation. J Mol Biol 2022, 434, 167201. [CrossRef]
- Stockl, M.T.; Zijlstra, N.; Subramaniam, V. alpha-Synuclein oligomers: an amyloid pore? Insights into mechanisms of alpha-synuclein oligomer-lipid interactions. Mol Neurobiol 2013, 47, 613-621. [CrossRef]
- Fuller, G.G.; Kim, J.K. Compartmentalization and metabolic regulation of glycolysis. J Cell Sci 2021, 134. [CrossRef]
- Nesterov, S.V.; Ilyinsky, N.S.; Plokhikh, K.S.; Manuylov, V.D.; Chesnokov, Y.M.; Vasilov, R.G.; Kuznetsova, I.M.; Turoverov, K.K.; Gordeliy, V.I.; Fonin, A.V.; et al. Order wrapped in chaos: On the roles of intrinsically disordered proteins and RNAs in the arrangement of the mitochondrial enzymatic machines. Int J Biol Macromol 2024, 267, 131455. [CrossRef]
- Zurita Rendon, O.; Shoubridge, E.A. LONP1 Is Required for Maturation of a Subset of Mitochondrial Proteins, and Its Loss Elicits an Integrated Stress Response. Mol Cell Biol 2018, 38. [CrossRef]
- Szczepanowska, K.; Trifunovic, A. Mitochondrial matrix proteases: quality control and beyond. FEBS J 2022, 289, 7128-7146. [CrossRef]
- Feng, Y.; Nouri, K.; Schimmer, A.D. Mitochondrial ATP-Dependent Proteases-Biological Function and Potential Anti-Cancer Targets. Cancers (Basel) 2021, 13. [CrossRef]
- Quiros, P.M.; Langer, T.; Lopez-Otin, C. New roles for mitochondrial proteases in health, ageing and disease. Nat Rev Mol Cell Biol 2015, 16, 345-359. [CrossRef]
- Bohovych, I.; Chan, S.S.; Khalimonchuk, O. Mitochondrial protein quality control: the mechanisms guarding mitochondrial health. Antioxid Redox Signal 2015, 22, 977-994. [CrossRef]
Figure 1.
CLPX and CLPP perform first aid in matrix granules where reaction intermediates are separated from the aqueous phase. Depiction of a mitochondrial matrix compartment between two cristae, where the LONP1 homomultimer is responsible for bulk proteolysis, while the homohexameric ring of AAA+ unfoldase CLPX and the homoheptameric ring of peptidase CLPP perform first aid in IMM-associated granular condensates, in cooperation with AAA+ unfoldase VWA8. CLPX maximizes flux within the heme-biosynthesis multi-enzyme metabolon, unfolding ALAS and OAT so that they can bind their cofactor PLP, to perform transaminations at delta-carbon positions. CLPX also associates with the translation elongation factor GFM1 when a nascent peptide is misfolded. Indirectly via heme or directly via GRSF1, CLPX may also influence mtRNA-G4 processing. Heme availability has impact also on the respiratory chain, and many processes outside mitochondria. Rather than performing proteolytic degradation, CLPP without assistance by the ATPase CLPX can only trim short polypeptides from proteins and assemblies, like chymotrypsin. CLPP apparently has access to diverse phase-separated, ribonucleoprotein-containing condensates in the matrix, where transcription, processing, translation, degradation and extrusion of mtRNA are decided. The illustration presents the respiratory chain at each crista with its complexes I-V and in association with iron (red dots) sulfur (yellow dots) clusters as well as the heme quadrangular molecule. The mitoribosomal large subunit (green globe) and small subunit (light blue) is shown with the sites for aminoacyl binding (A), peptidyl extension (P), and exit to tunnel (E), where the GTPase GFM1 determines elongation. The various terms and protein symbols are defined in the Abbreviation List below.
Figure 1.
CLPX and CLPP perform first aid in matrix granules where reaction intermediates are separated from the aqueous phase. Depiction of a mitochondrial matrix compartment between two cristae, where the LONP1 homomultimer is responsible for bulk proteolysis, while the homohexameric ring of AAA+ unfoldase CLPX and the homoheptameric ring of peptidase CLPP perform first aid in IMM-associated granular condensates, in cooperation with AAA+ unfoldase VWA8. CLPX maximizes flux within the heme-biosynthesis multi-enzyme metabolon, unfolding ALAS and OAT so that they can bind their cofactor PLP, to perform transaminations at delta-carbon positions. CLPX also associates with the translation elongation factor GFM1 when a nascent peptide is misfolded. Indirectly via heme or directly via GRSF1, CLPX may also influence mtRNA-G4 processing. Heme availability has impact also on the respiratory chain, and many processes outside mitochondria. Rather than performing proteolytic degradation, CLPP without assistance by the ATPase CLPX can only trim short polypeptides from proteins and assemblies, like chymotrypsin. CLPP apparently has access to diverse phase-separated, ribonucleoprotein-containing condensates in the matrix, where transcription, processing, translation, degradation and extrusion of mtRNA are decided. The illustration presents the respiratory chain at each crista with its complexes I-V and in association with iron (red dots) sulfur (yellow dots) clusters as well as the heme quadrangular molecule. The mitoribosomal large subunit (green globe) and small subunit (light blue) is shown with the sites for aminoacyl binding (A), peptidyl extension (P), and exit to tunnel (E), where the GTPase GFM1 determines elongation. The various terms and protein symbols are defined in the Abbreviation List below.
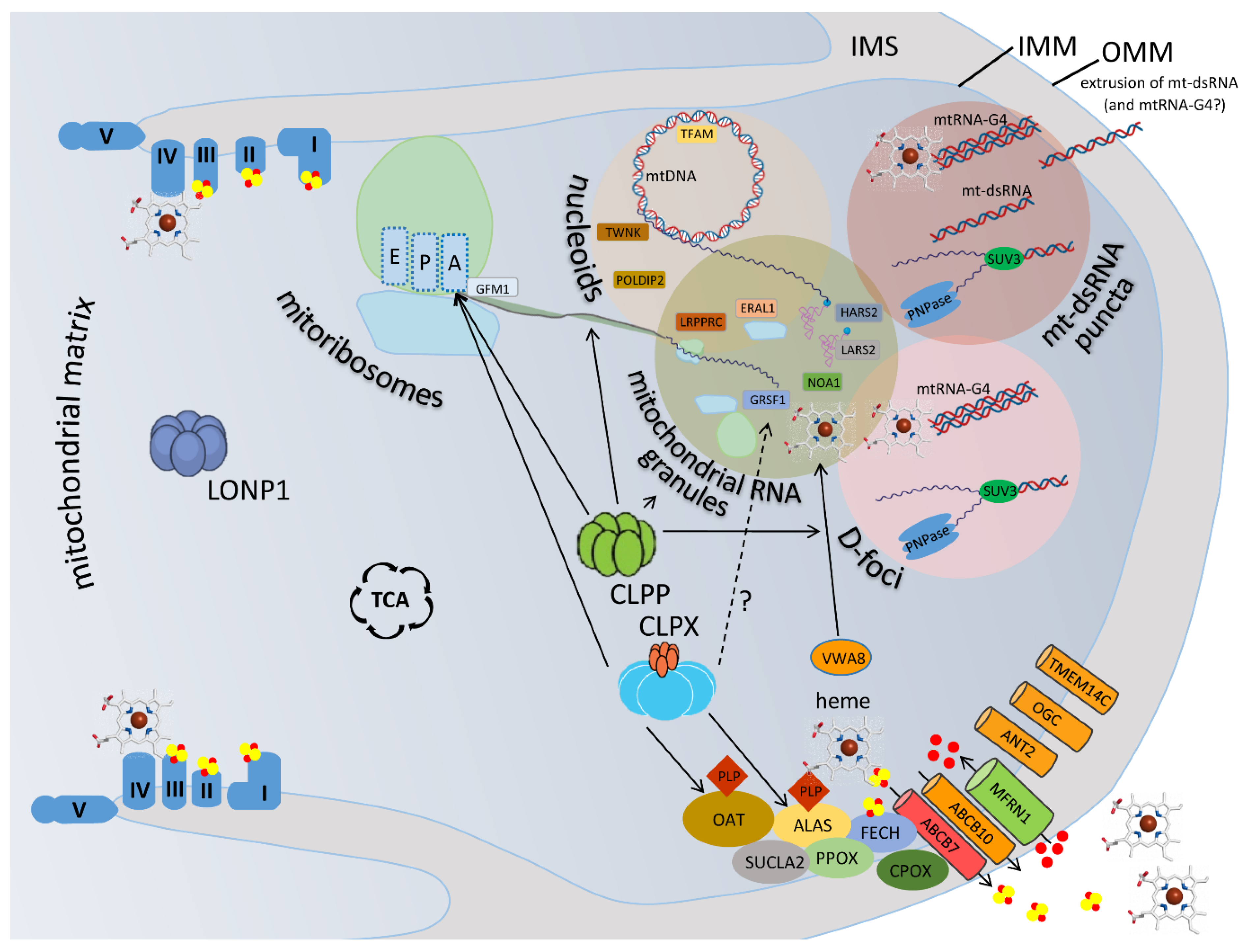
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



