
Submitted:
26 April 2024
Posted:
27 April 2024
You are already at the latest version
Abstract
Pulmonary arteriovenous malformations (PAVMs) are vascular anomalies resulting in abnormal connection between pulmonary arteries and veins. In 80% of cases, PAVMs are present from birth, but clinical manifestations are rarely seen in childhood. These congenital malformations are typically associated with Hereditary Hemorrhagic Telangiectasia (HHT), a rare disease that affects 1 in 5,000/8,000 individuals. HHT disease is frequently caused by mutations in genes involved in the TGF-β pathway. However, approximately 15% of patients do not have a genetic diagnosis and, among the genetically diagnosed, more than 33% do not meet the Curaçao Criteria. This makes clinical diagnosis even more challenging in the pediatric age group. Here, we introduce an 8 years old patient bearing a severe phenotype of multiple diffuse PAVMs caused by an unknown mutation which ended in lung transplantation. Phenotypically, the case of study follows a molecular pattern HHT-like. Therefore, molecular biology and cellular functional analysis has been performed in primary endothelial cells (ECs) isolated from the explanted lung. The findings revealed a loss of functionality in lung endothelial tissue and a stimulation of endothelial to mesenchymal transition. Understanding the molecular basis of this transition could potentially offer new therapeutic strategies to delay lung transplantation in severe cases.
Keywords:
Vascular rare disease
; Pulmonary arteriovenous malformations (PAVMs)
; Endothelial to Mesenchymal Transition (EndMT)
; angiogenesis
; TGF-β
1. Introduction
Pulmonary arteriovenous malformations (PAVMs) structurally are the consequence of an incorrect connection between pulmonary veins and arteries, leading to dilated anomalous vessels and the disappearance of the capillary network. This generates a right-to-left shunt, which increases pressure in the pulmonary circulatory system, as well as impairs regular gas exchange.
PAVMs are typically asymptomatic and undiagnosed. Approximately 80% of PAVMs are congenital in origin, but they are not commonly diagnosed in children because clinical manifestations are rarely seen before adulthood. However, patients who remain undiagnosed may develop serious complications later in life. The severity of the clinical symptoms can vary from dyspnea to cyanosis, heart palpitations and chest pain [1,2].
The majority of patients with congenital PAVMs have underlying Rendu-Osler-Weber syndrome, also known as Hereditary Hemorrhagic Telangiectasia (HHT). HHT is a rare multisystemic vascular disease with an autosomal dominant pattern of inheritance and a prevalence of 1 in 5,000/8000 in the general population, which increases in certain regions due to the founder effect and geographical isolation [3,4]. Patients with HHT often present mucocutaneous telangiectasias, the rupture of which causes recurrent and spontaneous nose hemorrhages (epistaxis), which is usually the most frequent and the first symptom to appear in HHT. Bleeding usually increases throughout the patient's life, and in some of the most severe cases can even lead to anemia and iron deficiency. Another clinical manifestation is the presence of arteriovenous malformations (AVMs) in internal organs, such as the lungs and brain, more frequent in HHT1 type, while in HHT2 is more frequent in liver. Clinical diagnosis of HHT is possible following Curaçao criteria, which are epistasis, telangiectasia, arteriovenous malformation in internal organs and a familiar pattern of autosomal dominant inheritance [5]. Nevertheless, the Curaçao criteria are not accurate for infants, due to the late development of certain symptoms and to the intra-familial phenotypic variability.
In addition to a symptomatic perspective, HHT can be diagnosed genetically. All mutations leading to HHT are found in genes belonging to the BMP9/TGF-β signaling pathway. Mutations in ENG (HHT1) and ALK1/ACRVRL1(HHT2) genes are responsible for 90% of HHT cases [6,7]. Less common mutations, such as those found in MADH4/SMAD4 and GDF2 genes, have been described in less than 2% of the HHT population and result in HHT-like syndromes. Mutations in MADH4/SMAD4 cause juvenile polyposis/HHT overlap syndrome (JPHT) that generates additionally to HHT symptoms, colon polyps and thoracic aneurysms [8]. In the case of GDF2 mutations generate an HHT-like syndrome classified as HHT5 [9]. Recent research suggests that mutations in EPHB4 or RASA1 may be responsible for the previously known as HHT3 and HHT4 types, as there is no evidence for the existence of two independent genes linked to chromosomes 5 and 7 [10]. EPHB4 and RASA1 variants cause capillary malformations-arteriovenous malformation (CM)/AVM syndrome. This syndrome is characterized by the appearance of multiple small, red, randomly distributed spots with white halo known as capillary malformations. It can be difficult to distinguish CM/AVM from HHT [11].
BMPR2 gene is also involved in the BMP9/TGF-β signaling pathway. Its expression is particularly high in pulmonary vascular endothelium. Pathological changes in this gene can lead to pulmonary arterial hypertension (PAH) [12].
However, 15% of clinically diagnosed with HHT lack a genetic diagnosis. Furthermore, we can add the difficult diagnosis in pediatric cases and the existence of sporadic (non-inherited) cases of HHT. In this context, we have studied a clinical case with multiple diffuse pulmonary fistulas in pediatric age who underwent a bilateral lung transplantation. Previous genetic analysis did not show any genetic alterations associated with HHT or any of the other related syndromes above mentioned. Moreover, the patient does not have any family history of HHT. Given the severity of the clinical manifestations and their similarity to HHT, the aim of the present study is to establish the molecular parameters and differential gene expression profile of this symptomatology, aiming to a similarity to HHT.
Results
The Index case of this study is a 8-years-old child with clinical diagnosis of probable HHT without a familiar history of HHT. The case shows symptoms related to this syndrome since he was 3 years old, including strong cyanosis, hypoxemia (abnormal low oxygen in blood), epistaxis and multiple small diffuse pulmonary arteriovenous fistulas in both lungs. This phenotype was becoming more severe and non-compatible with life, due to the low oxygen saturation. In 2020, he was put on top of an emergency list for pediatric lung transplantation. In spite of all the genetic analysis carried out in the known genes related to HHT and HHT-like syndromes, the genetic origin of the disease remains unknown.
2.1. Visual Analysis of Primary Endothelial Cell Culture
Figure 1 displays photographs of two cultures of human pulmonary microvasculature: the Index case and the HMVEC_L as control of lung microvasculature. As a routinary way to confirm the identity of our established endothelial cell culture, we performed CD31/PECAM-1 and vWF staining (data not shown). The cultures appear similar at first, but over time, the cells from the case study changed in size and morphology. In the primary culture of the patient, two cell populations can be identified. One population maintains an endothelial appearance, while the other acquires a mesenchymal-like morphology and a clear increase in size is apparent. Ultimately, mesenchymal-like morphology became the dominant population in the culture.
Overall, the appearance of this mesenchymal-like cell population over time suggests the presence of an endothelial-mesenchymal transition (EndMT). Due to the prematurity and severity of the symptoms in this child, as well as the observed phenotypic in vitro changes in cells, we investigated the possibility of molecular-level alterations. As a result, experiments were designed to test the EndMT hypothesis, by analyzing the expression and functional profile of the ECs from the explanted lung versus the control HMVEC_L.
2.2. mRNA Expression Profile
In the present case, in which there was a vascular involvement, we decided to focus our gene expression analysis on endothelial and mesenchymal expression genes. The amount of RNA was normalized to the amount of 18S RNA as a housekeeping gene. Figure 2 provides additional details about the mRNA profile.
The gene expression was compared between primary cell culture of ECs from a healthy donor and the Index case. RT-qPCR analysis showed that all of the endothelial markers were downregulated in the case under study (Figure 2A). Although these results are to be expected in HHT, as seen in the literature [13], the expression levels were extremely low. In HHT patients, endoglin haploinsufficiency leads to increased COX2 expression and decreased NO synthesis, which is a compensatory mechanism [14] However, in this case, the compensatory system is not fulfilled (Figure 2B). On the other hand, the Index case expressed high mRNA levels of mesenchymal markers, such as MMP3, FN1 and Vimentin (Figure 2C). We did not find significant differences in SNAI1, whose functions are associated with development processes, neural differentiations and epithelium-mesenchymal transition [15]. The replacement of usual epithelial markers with mesenchymal markers, such as Vimentin and Fibronectin, determines these changes [15,16]. Figure 2A and 2C confirm a decrease in endothelial markers and an increase in mesenchymal markers, supporting our initial idea that an EndMT is occurring. EndMT happens during the embryonic development, but this process has been also previously described in adults [17,18].
Also, we examined the expression of SMAD genes since they play a significant role in the TGF-β pathway affected by the HHT syndrome. A downregulation of SMAD genes is observed (Figure 2D).
2.3. Protein Expression
To corroborate the previous expression profile comparing HMVEC_L against the Index case we performed protein analysis, by immunofluorescent assay and/or western blot.
An immunofluorescent assay was conducted to corroborate the gene expression levels of endothelial cell-specific genes (VE-Cadherin 2, PECAM-1) and a specific marker of dedifferentiated cells (N-Cadherin) at the cellular level (Figure 3A). The images confirm the presence of VE-Cadherin 2 on the cell surface of HMVEC_L control culture cells, observing strong accumulations of this protein observed in the areas of cell-cell contacts, forming adherent junctions. In contrast, VE-Cadherin 2 is less prominent in the patient's cells, and there is no accumulation of it on the cell surface. This suggests that cell-cell contacts have been lost. Since VE-Cadherin 2 plays a crucial role in maintaining endothelial integrity and leukocyte extravasation, a decrease in VE-Cadherin 2 would result in reduced cell adhesion and less cohesion among cells. To confirm endothelial identity of the cell culture CD31/PECAM-1 labelling was used as marker. This protein is almost exclusively expressed in ECs. The patient cells exhibited significantly lower levels of PECAM-1 compared to the control cells, which supports the results obtained from qPCR and confirms our initial hypothesis.
Furthermore, an examination of F-Actin in both cultures was conducted to determine if the mesenchymal shaped cells showed a different cytoskeleton distribution than healthy ECs, which might facilitate proper migration and contraction. Figure 3B illustrates significant disparities in the quantity and configuration/organization of actin filaments. When comparing both conditions, it is evident that the labelling of the control cells enables a precise delimitation of the cell shape, which is not the case in the patient samples. In the latter, the labelling accumulates in cytoplasmic fibers, thicker and more abundant than in the control. Additionally, a significant variability of cell sizes within the same culture was observed during the experiment (see Figure 3B). The larger, apparently mesenchymal cells showed a highly ordered network of filaments. The phase contrast allowed for the identification of smaller cells as ECs undergoing apoptosis (Figure 3C).
To analyze TGF-β and PI3K/MAPK pathways, we performed Western blot assays. The TGF-β pathway is closely related to the HHT syndrome and plays a crucial role in a wide range of cellular functions. The signaling status of the TGF-β pathway was analyzed through SMAD proteins (SMAD1, SMAD3 and SMAD4). We examined the levels of each of the SMAD proteins and their response to TGF-β treatment (Figure 4A). Figure 4A shows that the Index case has a complete inhibition of this pathway, and TGF-β stimulation has no effect.
Additionally, we aimed to investigate the status of the PI3K/MAPK pathway and its activation (phosphorylated proteins). Figure 3B illustrates that all proteins from the Index case were overexpressed when compared with control healthy HMVEC-L cells, indicating a higher activation of both pathways through AKT/pAKT and ERK/pERK. Additionally, we aimed to examine the protein levels of the mesenchymal marker Vimentin. As previously observed in the RT-qPCR, the western blot assay revealed a higher quantity of Vimentin in the Index case, while it was undetectable in the control (Figure 4B).
2.4. Functional Analysis
Due to the low expression levels of certain genes involved in processes such as angiogenesis (ANGPT2), migration, cell adhesion (PECAM-1 and VE-Cadherin 2) and genes involved in proper blood vessel physiology (NOS-3 and COX2), we performed functional assays of wound healing and tube formation (angiogenesis) to test how compromised patient´s cells were compared to healthy control cells.
Figure 5A shows the wound healing assay. These assays attempted to mimic a physiological condition of in vitro tissue damage and test cell function by assessing cell migration and cohesion. In both cases, the advancing front moved. However, it is evident that HMVEC_L control cells exhibited a more organized and uniform movement, while the cells of the patient under study displayed less connection with each one moving independently. It is obvious that control cells showed a greater healing capacity, as they were capable of closing almost completely the discontinuity (wound) after 6 hours. In contrast, the cells in the case under study moved towards the wound in an attempt to close it, but after 24 hours, gaps between the cells can be observed and the closure showed incomplete. Therefore, control cells have a greater capacity for repairing and forming cell cohesion, while the patient cultures experience delays and incomplete closure.
In terms of functional capacity for tubulogenesis, Figure 5B illustrates that healthy HMVEC_L cells were able to generate completely closed and defined tubes of various sizes within 3 hours on Matrigel®. After 6 hours, the tubes formed were reinforced by several layers of cells. However, cells from the Index case were unable to build defined tubes, and a large proportion of the cells were not able to adhere to Matrigel® after 6 hours of plating.
3. Discussion
Angiogenesis processes play a dual role: they are essential for the formation of new vessels from pre-existing ones but, when impaired, are also involved in pathogenic processes. ECs undergo morphological changes and are induced to migrate during angiogenesis, enabling them to migrate and remodel the ECM. This behavior may resemble that of mesenchymal cells. However, it is not typical for ECs to detach from their neighbor cells, indicating that angiogenesis may involve a partial transition to mesenchymal cells, EndMT, followed by vessel stabilization [19,20]. EndMT has also been observed in processes of cardiac fibrosis and pulmonary hypertension [17,21].
This paper presents the first documented case of endothelial to mesenchymal transition in a sample from a pediatric patient, subjected to lung transplantation. Although this transition has been observed in adults [17,22], this may be the only documented case in children so far. Our hypothesis is that damaged pulmonary ECs eventually transition into mesenchymal cells as a survival mechanism when they can no longer fulfil their endothelial function. In spite of the unknown origin of this damage in, which is currently under investigation, we have studied the outcomes. The consequences are the loss of endothelial integrity, which could explain the severity of the patient's symptoms, due to a collapse of the lung vasculature and therefore the lung function. The results of the in vitro experiments described in this work support this hypothesis.
The primary lung microvascular culture from the Index case showed the progressive emergence with passages of a second population with a mesenchymal-like morphology (Figure 1). To test this assumption, we compared the expression profile of this cell culture with a primary control culture (Figure 2). The analyzed genes are involved in various physiological processes, including angiogenesis, cell adhesion, migration, transmigration, TGF-β signaling, cell cycle control, cytoskeleton, and vascular physiology. A decrease in the expression of vascular genes was observed, as has been described in HHT patients14. However, characteristic genes of mesenchymal cells involved in transition processes, such as MMP3, FN1, Vimentin and SNAI1 were found significantly increased in their expression, in support of our EndMT hypothesis [20,23].
During EndMT, ECs detach and migrate, potentially invading the underlying tissue. As part of this process, the mesenchymal phenotype overlaps with the endothelial phenotype. This is characterized by the acquisition of mesenchymal markers, such as N-Cadherin, and the complementary loss of endothelial markers, such as CD31/PECAM-1 and VE-Cadherin 2. Additionally, ECs lose their cell-cell junctions and acquire characteristics adapted to migration and invasion [24].
To verify these changes, the expression and subcellular localization of these proteins was examined using laser confocal microscopy (Figure 3B). Our findings confirmed that the cells in the case study had lost the cell-cell connections typical of endothelial tissue and were expressing mesenchymal markers (N-Cadherin). Significant oscillations in VE-Cadherin 2 levels can increase N-Cadherin expression in the endothelium. N-Cadherin promotes cell migration and contributes to vascular elongation, while VE-Cadherin 2 induces vascular stabilization by limiting growth factor receptor signaling [25].
Several studies have shown that the TGF-β pathway promotes EndMT [22,26]. TGF-β can also activate SMAD-independent pathways such as MAPK/ERK/JNK, all of which are involved in EndMT [27]. This is similar to the situation we observe in the Index patient, where a total inhibition of the SMAD protein-dependent TGF- β pathway is found [28]. On the other hand, both in HHT patients, and in our case an increase in basal and functional protein levels of the PI3K/MAPK pathways is found [29,30]. These results are supporting the hypothesis of an EndMT present in the case under investigation in this work.
Recent studies suggest that vascular lesions in HHT may be originated by a second mutation event in ENG or ALK1, in angiogenic processes [31]. Mouse models of HHT1 and HHT2 provide strong evidence supporting a two-hit mechanism as the origin of AVM. This mechanism requires complete endothelial loss of ENG or ALK1 protein for the formation of robust vascular lesions. The current mouse heterozygous model, with only one alteration, is not effective in replicating HHT symptoms [32]. Singh et al. (2020) [33] noted that AVMs are primarily of venous origin, where cells express higher levels of endoglin. ENG restricts endothelial proliferation, and its absence in localized regions increases the frequency of AVMs [33]. Endoglin is also involved in cell intercalation processes, necessary for the extension and formation of new blood vessels while maintaining cell-cell contact. Therefore, its absence may be a factor for the development of these vascular lesions [34].
HHT patients exhibit at least a 50% reduction in ENG or ALK1 functional protein levels, due to their heterozygous condition for mutations in ENG (HHT1) or ALK1 (HHT2). These patients experience increased and anomalous angiogenic processes, where there is no proper control on the generation and stabilization of blood vessels. The reported finding suggests that low ENG levels adversely affect both cell migration and tubule formation [35,36,37]. Previous studies in human brain AVMs have found evidence that while SMAD-mediated TGF-β signaling is reduced in AVMs, non-canonical TGF-β signaling may contribute to the development of AVMs through the PI3K/MAPK pathway [28,38]. In HHT, due to mutations in components of the TGF-β pathway, a decrease in the canonical TGF-β pathway and an increase in the PI3K pathway is found, facilitating a partial endothelial-mesenchymal transition. In this scenario, some cells may undergo a partial EndMT transition in the AVM. However, cells are able to revert at least to some extent the EndMT transition, and HHT cells still maintain cell-cell junctions to an intermediate degree between healthy cells and the damaged cells observed in the present case [13].
Moreover, low levels of physiological oxygen saturation as those exhibited by the patient under study, 70-80%, are sensed by cells, as hypoxia, inducing an increase in VEGF levels, and leading to a continuous pro-angiogenic state. Additionally, we observed a very strong decrease, far below 50% in the expression of both ENG and ALK1, close to the conditional knock-out mouse models of Eng and Acvrl1/Alk1 published in the literature [34]. The study found a phenotype with greater aggressiveness in the knockout mouse of Acvrl1/Alk1, resulting in a complete inhibition of the TGF-β pathway, as seen in our Index case (Figure 4A) [32]. Additionally, decreased levels of ENG create an adequate environment for the generation of multiple AVMs, in our case, pulmonary. A decrease in the levels of ALK1 and ENG impairs TGF-β signaling, in addiction to an hypoxic environment leads to an EndMT and finally to the generation of AVM. In the Index case, the ECs undergo the EndMT, becoming individual cells without a proper alignment with other cells, apparently unable to reverse to endothelial cell phenotype. This results in a permanent EndMT that completely compromises the integrity and function of the vasculature. Furthermore, the Figure 3C demonstrate a significant variation in cell size, with some endothelial-like cells undergoing apoptotic processes. This suggests that ECs that are incapable of initiating an EndMT transition process may ultimately die (Figure 5B and 5C). We identified similarities in gene expression patterns and, in the signaling related to TGF-β and PI3K/MAPK pathways between HHT patients and our case study [13,29,30]. However, this case presents an extreme profile with low expression of endothelial markers and increased expression of mesenchymal cell-specific markers. This may explain a fatal compromise of vascular integrity and function.
The mechanism explained above is depicted in Figure 6 where three angiogenic situations are shown. The normal angiogenesis with transitory and reversible EndMT (Figure 6) is undergone by healthy ECs. The transition with only partial reversion would occur in an HHT patient. Finally, the EndMT suffered by the cells presented in this case under study would lead to a loss of endothelial function in the vasculature. Although, the origin of the anomalous transition occurred in this pediatric patient remains unknown, it is likely that this could have occurred somatically, restricted mainly to the lung, without involvement of the germline.
4. Materials and Methods
4.1. Human Samples and Ethics
The experiments included in this work were approved by the Ethical Committee of CSIC with the reference Number 075/2017. All donors were informed of the experimental procedures, and their informed consent was obtained. The tutor of the patient studied in this manuscript gave his informed consent to participate in the study.
Cells derived from patient’s explanted lung were isolated and cultured.
4.2. Cell Culture
Primary cultures of lung microvasculature cells from the patient under study were isolated following an established procedure from one of the explanted lungs after surgery performed for replacement of both lungs at the Hospital de La Fe, Valencia (Spain) [39]. A human lung microvasculature endothelial primary cell culture (HMVEC_L) was used as control (Innoprot, Derio, Spain). Both patient’s and HMVEC_L cultures required precoated collagen plates (Sigma-Aldrich, St. Louis, Missouri, USA) for attachment and were grown in EGM-2/EBM-2 medium (PromoCell, Heidelberg, Germany) supplemented with 20% FBS (Gibco, New York, USA), 2 mM L-glutamine, and 100 U/ml penicillin/streptomycin (Gibco). Experiments were carried out at cell passages lower than 7, and each parallel experiment between control HMVEC_L and patient-derived lung ECs was performed at similar passages. For passaging, cells were detached from the plates with 0.05% Trypsin and 0.02% EDTA solution (Gibco). HMVEC_L and patient-derived lung endothelial cell cultures were treated with two different concentrations of TGF-β (R&D Systems, Miami, Florida, USA) to stimulate the TGF-β pathways, either preferentially mediated by ALK5 (treatment with 10 ng/ml o/n), or preferentially by ALK1 (1 ng/ml for 3 h). The treatment was conducted in EGM-2/EBM-2 medium (PromoCell) with 2% FBS (Gibco), and a control was maintained without any treatment (EGM-2/EBM-2 medium with 2% FBS).
All cell cultures were grown in conditions of 37 °C and 5% C02 and humidity. Cell cultures brightfield images were taken using a Zeiss Axiovert 135 microscope.
4.3. Gene Expression Analysis at the RNA Level
4.3.1. Real-Time Quantitative PCR
Total RNA was extracted from Index case and control primary cultures using the NucleoSpin RNA kit (Macherey-Nagel, GmbH&Co., Düren, Germany). 500 ng of total RNA from each sample was retrotranscribed into cDNA in a final volume of 20 μl with a commercial High-Capacity cDNA Reverse Transcription kit (Thermo Fisher Scientific, Waltham, USA). This kit uses hexamer random sequencing primers (random primers). The iQ5 system (BioRad) was used to perform real-time PCR using FastStart Essential DNA Green Master (Roche, Basel, Switzerland) as reaction mix. The primers used in q-PCR (Sigma-Aldrich) are shown in the Table 1. Each gene was analyzed in triplicate for each of the samples used.
4.4. Analysis of Gene Expression at the Protein Level
4.4.1. Immunofluorescence Assay
Immunofluorescence analyses were conducted to test some of the Real-Time Quantitative PCR results and to assess the expression of endothelial cell-specific markers, VE-Cadherin 2 and PECAM-1, as well as the mesenchymal cell marker N-Cadherin. Additionally, we aimed to analyze differences in the cytoskeleton's F-Actin between control and patient cells.
Cells from both cultures, HMVEC_L and Index case, were plated on pre-coated Collagen sterile glass coverslips (12 mm diameter, VWR international, Radnor, USA). The control culture HMVEC_L was seeded with 2 x 104 cells/well, while the patient culture was seeded with 4 x 104 cells/well, due to the special characteristics of cells, as explained in results.
On the following day, the cells were washed with cold 1X HBSS (Gibco) and fixed with 3.5% paraformaldehyde in 1X PBS for 20 minutes at 4°C. In the case of F-Actin, we permeabilized with 100 μg/ml L-α-Lysophosphatidylcholine (LPC) (Sigma-Aldrich). The cells were then washed again with 1X HBSS and non-specific unions were blocked with 1% PBS 1X/BSA for 1 hour at 4°C. Mouse anti-human antibodies were used to detect VE-Cadherin 2 (1:200 in PBS 1x BSA 4%) (Abcam, Cambridge, United Kingdom), N-Cadherin, and PECAM-1 (1:50 in PBS 1x/BSA 4%) (Santa Cruz Biotechnology, Dallas, Texas, USA) at 4°C for 40 minutes. Then, cells were washed with 1X PBS and then incubated for 30 minutes at 4°C with secondary antibodies such as AlexaFluor 488 for VE-Cadherin 2 and N-Cadherin (1:250) and AlexaFluor 647 (BioLegend, San Diego, California, USA) or AlexaFluor 568 (Molecular Probes, Eugene, OR, USA), for PECAM-1and F-Actin, respectively.
Finally, ProLong mounting medium with DAPI (Invitrogen, Thermo Fisher Scientific, Waltham, USA) was used to mount the cells on glass coverslips. Immunofluorescence images were acquired using a SP5 fluorescence confocal microscope (DMI6000 CS Leica Microsystems, Wetzlar, Germany) at 63X magnification.
4.4.2. Western Blot
Cell lysates were obtained from primary cultures using TNE buffer (50 mM Tris, 150 mM NaCl, 1 mM EDTA and 0.5% Triton X100) and kept on ice for 30 minutes. Specific protease and proteasome inhibitors (Roche) were added, along with lactocystin (Sigma-Aldrich). The lysates were centrifuged at 14,000×g for 5 minutes. Similar amounts of proteins from the lysates were boiled in SDS sample buffer and analyzed by 4-20% SDS-PAGE gel under reducing conditions (BioRad). The proteins present in the gels were transferred to nitrocellulose membranes (Amersham, Little Chalfont, UK) through electrotransfer o/n. To prevent nonspecific binding, membrane was blocked with 5% BSA-TBS-0.05% Tween for 1 hour, followed by immunodetection with anti-SMAD1/4 (Rabbit mAb; Cell Signaling, Danvers, Massachusetts, USA), anti-SMAD3 (Rabbit mAb; Cell Signaling), anti-β Actin (Mouse mAb; Sigma-Aldrich), anti-AKT (Rabbit mAb; Cell Signaling), anti-pAKT (Rabbit mAb; Cell Signaling), anti-ERK (Mouse mAb; Cell Signaling), anti-pERK (Rabbit mAb; Cell Signaling), anti-Vimentin (Mouse mAb, Proteintech, Rosemont, USA) or anti-γ Tubulin (Mouse mAb; Sigma-Aldrich) antibodies. The primary antibody was incubated at 4°C o/n. Then, samples were washed and incubated with its corresponding secondary antibody from Dako (Glostrup, Denmark) at RT for 1 hour. The antibodies were used at the dilution recommended by the manufacturer, and the membranes were revealed using chemiluminescence (SuperSignal West Pico chemiluminescent substrate, Thermo Scientific). FIJI-Image J 1.53q software tool was used to quantify chemiluminescence.
4.5. Functional Analysis
4.5.1. Wound Healing
Wound formation assays are generated from scraping confluent cultures of HMVEC_L and the Index case. Both conditions were seeded the day before at a concentration of 8 x 104 cells/well in a 24-well plate. Cells were washed with HBSS (Gibco) and cultured in EBM-2/EGM-2 medium (PromoCell). Assays were monitored and photographed for 6 h with intervals of 90 min in those wells with similarly sized wounds.
4.5.2. Tubulogenesis: Endothelial Cell Tube Formation Assay
For the angiogenesis or tube formation assay, 8 x 104 cells/well were seeded in 24-well plates. The wells used were previously coated with Matrigel® (Gibco) at a 1:1 dilution in RPMI medium (Gibco) without any supplement. Cultures were monitored and photographed from 90 min after seeding until 6 h, at this time the formed tubes started to retract and finally collapse.
4.6. Statistics
Data shown are means ± SD. Those differences shown were analyzed using Student's t-test. Thus, those P-values < 0.05 were considered statistically significant and therefore, in each figure significance has been marked with asterisks. Values associated with asterisks are * p< 0.05; **p < 0.01; *** p< 0.001 and **** p< 0.0001.
Author Contributions
Conceptualization, V.A., A.M.C. and L.M.B.; methodology, L.R.-P., L.L.-H; software, V.A., L.L-H and .; validation, , V.A. and L.M.B.; formal analysis, V.A., and L.M.B.; investigation, L.L.-H., L.R.-P., V.A..; resources, L.L.-H and L.M.B; writing—original draft preparation, L.L.-H and L.M.B.; writing—review and editing, V.A., A.C.M., and L.M.B.; funding acquisition, L.M.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by MICINN (Ministry of Science and Innovation of Spain), grant number PID2020-115371RB-I00. L.R.-P. was the recipient of a contract from an internal project of CSIC (PIE201820E073). L.L.-H is contracted by CIBER of rare diseases (ISCIII, Spain).
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki and approved by the Institutional Review Board (Human Ethics Committee) of CSIC (National Research Council of Spain) for studies involving humans (protocol code 228/2020).
Informed Consent Statement
Written informed consent was obtained from the patient(s) to publish this paper.
Data Availability Statement
Data supporting the reported results can be found in our laboratory records. DNA, RNA, cell culture and tissue samples are part of the collection associated with the research of the group.
Acknowledgments
We acknowledge to the patients and Spanish HHT Association.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Majumdar S., McWilliams J.P. Approach to Pulmonary Arteriovenous Malformations: A Comprehensive Update. J Clin Med 2020, 9:1927. [CrossRef]
- Martinez-Pitre PJ, Khan YS. Pulmonary Arteriovenous Malformation (AVMs), 1st ed.; StatPearls: Treasure Island (FL), U.S.A., 2023. PMID: 32644715.
- Jessurun G.A., Kamphuis D.J., van der Zande F.H., Nossent J.C. Cerebral arteriovenous malformations in The Netherlands Antilles. High prevalence of hereditary hemorrhagic telangiectasia-related single and multiple cerebral arteriovenous malformations. Clin Neurol Neurosurg 1993, 95: 193-198. [CrossRef]
- Kjeldsen A.D., Vase P., Green A. Hereditary haemorrhagic telangiectasia: a population-based study of prevalence and mortality in Danish patients. J Intern Med 1999; 245:31-39. [CrossRef]
- Shovlin C.L., Guttmacher A.E., Buscarini E., et al. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Am J Med Genet 2000; 91:66-67. [CrossRef]
- Johnson D.W., Berg J.N., Baldwin M.A., et al. Mutations in the activin receptor–like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat Genet 1996; 13:189-195. [CrossRef]
- McAllister K.A., Grogg K.M., Johnson D.W., et al. Endoglin, a TGF-β binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat Genet 1994; 8:345-351. [CrossRef]
- Gallione C.J., Repetto G.M., Legius E., et al. A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4). Lancet 2004; 363:852-859. [CrossRef]
- Wooderchak-Donahue W.L., McDonald J., O’Fallon B., et al. BMP9 mutations cause a vascular-anomaly syndrome with phenotypic overlap with hereditary hemorrhagic telangiectasia. Am J Hum Genet 2013; 93:530-537. [CrossRef]
- Shovlin C.L., Almaghlouth F.I., Alsafi A., et al. Updates on diagnostic criteria for hereditary haemorrhagic telangiectasia in the light of whole genome sequencing of “gene-negative” individuals recruited to the 100 000 Genomes Project. J Med Genet 2024; 61:182-185. [CrossRef]
- Bayrak-Toydemir P., Stevenson D.A. Capillary Malformation Syndrome, 1st ed.; GeneReviews®: Seattle (WA): University of Washington, U.S.A., 2011. PMID: 21348050.
- Tatius B., Wasityastuti W., Astarini F.D., Nugrahaningsih D.A.A. Significance of BMPR2 mutations in pulmonary arterial hypertension. Respir Investig 2021; 59:397-407. [CrossRef]
- Fernandez-Lopez A., Garrido-Martin E.M., Sanz-Rodriguez F., et al. Gene expression fingerprinting for human hereditary hemorrhagic telangiectasia. Hum Mol Genet 2007; 16:1515-1533. [CrossRef]
- Jerkic M., Rivas-Elena J.V., Santibanez J.F., et al. Endoglin regulates cyclooxygenase-2 expression and activity. Circ Res 2006; 99:248-256. [CrossRef]
- Nieto M.A. The snail superfamily of zinc-finger transcription factors. Nat Rev Mol Cell Biol 2002; 3:155-166. [CrossRef]
- Kaufhold S., Bonavida B. Central role of Snail1 in the regulation of EMT and resistance in cancer: a target for therapeutic intervention. J Exp Clin Cancer Res 2014; 33:62. [CrossRef]
- Zeisberg E.M., Tarnavski O., Zeisberg M., et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med 2007; 13:952-961. [CrossRef]
- Platel V., Faure S., Corre I., Clere N. Endothelial-to-Mesenchymal Transition (EndoMT): Roles in Tumorigenesis, Metastatic Extravasation and Therapy Resistance. J Oncol 2019; 2019:8361945. [CrossRef]
- Welch-Reardon K.M., Wu N., Hughes C.C.W. A role for partial endothelial-mesenchymal transitions in angiogenesis? Arterioscler Thromb Vasc Biol 2015; 35:303-308. [CrossRef]
- Sun J.X., Chang T.F., Li M.H., et al. SNAI1, an endothelial–mesenchymal transition transcription factor, promotes the early phase of ocular neovascularization. Angiogenesis 2018; 21:635-652. [CrossRef]
- Gorelova A., Berman M., Ghouleh I.A. Endothelial-to-Mesenchymal Transition in Pulmonary Arterial Hypertension. Antioxid Redox Signal 2021; 34:891-914. [CrossRef]
- Ranchoux B., Antigny F., Rucker-Martin C., et al. Endothelial-to-Mesenchymal Transition in Pulmonary Hypertension. Circulation 2015; 131:1006-1018. [CrossRef]
- Lamouille S., Xu J., Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol 2014; 15:178-196. [CrossRef]
- van Meeteren L.A., ten Dijke P. Regulation of endothelial cell plasticity by TGF-β. Cell Tissue Res 2012; 347:177-186. [CrossRef]
- Giannotta M., Trani M., Dejana E. VE-Cadherin and Endothelial Adherens Junctions: Active Guardians of Vascular Integrity. Dev Cell 2013; 26:441-454. [CrossRef]
- Shu D.Y., Butcher E., Saint-Geniez M. EMT and EndMT: Emerging Roles in Age-Related Macular Degeneration. Int J Mol Sci 2020;21(12):4271. [CrossRef]
- Ma J., Sanchez-Duffhues G., Goumans M.J., ten Dijke P. TGF-β-Induced Endothelial to Mesenchymal Transition in Disease and Tissue Engineering. Front Cell Dev Biol 2020; 8:260. [CrossRef]
- Shoemaker L.D., McCormick A.K., Allen B.M., Chang S.D. Evidence for endothelial-to-mesenchymal transition in human brain arteriovenous malformations. Clin Transl Med 2020;10 (2):e99. [CrossRef]
- Alsina-Sanchís E., García-Ibáñez Y., Figueiredo A.M., et al. ALK1 Loss Results in Vascular Hyperplasia in Mice and Humans Through PI3K Activation. Arterioscler Thromb Vasc Biol 2018; 38:1216-1229. [CrossRef]
- Iriarte A., Figueras A., Cerdà P., et al. PI3K (Phosphatidylinositol 3-Kinase) Activation and Endothelial Cell Proliferation in Patients with Hemorrhagic Hereditary Telangiectasia Type 1. Cells 2019; 8:971. [CrossRef]
- Snellings D.A., Gallione C.J., Clark D.S., Vozoris N.T., Faughnan M.E., Marchuk D.A. Somatic Mutations in Vascular Malformations of Hereditary Hemorrhagic Telangiectasia Result in Bi-allelic Loss of ENG or ACVRL1. Am J Hum Genet 2019; 105:894-906. [CrossRef]
- Tual-Chalot S., Oh S.P., Arthur H.M. Mouse models of hereditary hemorrhagic telangiectasia: recent advances and future challenges. Front Genet 2015; 6:25. [CrossRef]
- Singh E., Redgrave R.E., Phillips H.M., Arthur H.M. Arterial endoglin does not protect against arteriovenous malformations. Angiogenesis 2020; 23:559-566. [CrossRef]
- Mahmoud M., Allinson K.R., Zhai Z., et al. Pathogenesis of Arteriovenous Malformations in the Absence of Endoglin. Circ Res 2010; 106:1425-1433. [CrossRef]
- Fernandez-L A., Sanz-Rodriguez F., Zarrabeitia R., et al. Blood outgrowth endothelial cells from Hereditary Haemorrhagic Telangiectasia patients reveal abnormalities compatible with vascular lesions. Cardiovasc Res 2005; 68:235-248. [CrossRef]
- Fernandez-L A., Sanz-Rodriguez F., Zarrabeitia R., et al. Mutation study of Spanish patients with hereditary hemorrhagic telangiectasia and expression analysis of Endoglin and ALK1. Hum Mutat 2006; 27:295-295. [CrossRef]
- Fernández-L A., Sanz-Rodriguez F., Blanco F.J., Bernabéu C., Botella L.M. Hereditary hemorrhagic telangiectasia, a vascular dysplasia affecting the TGF-beta signaling pathway. Clin Med Res 2006; 4:66-78. [CrossRef]
- Hauer A.J., Kleinloog R., Giuliani F., et al. RNA-Sequencing Highlights Inflammation and Impaired Integrity of the Vascular Wall in Brain Arteriovenous Malformations. Stroke 2020; 51:268-274. [CrossRef]
- Albiñana V., Villar Gómez de las Heras K., Serrano-Heras G., et al. Propranolol reduces viability and induces apoptosis in hemangioblastoma cells from von Hippel-Lindau patients. Orphanet J Rare Dis 2015; 10:118. [CrossRef]
Figure 1.
Primary cultures of lung endothelial cells are shown from a control condition (HMVEC_L) and the patient under study (ICase) along different cell passages. The white arrows indicate the appearance of a second, larger cell population which becomes dominant in the cellular pass 5.
Figure 1.
Primary cultures of lung endothelial cells are shown from a control condition (HMVEC_L) and the patient under study (ICase) along different cell passages. The white arrows indicate the appearance of a second, larger cell population which becomes dominant in the cellular pass 5.
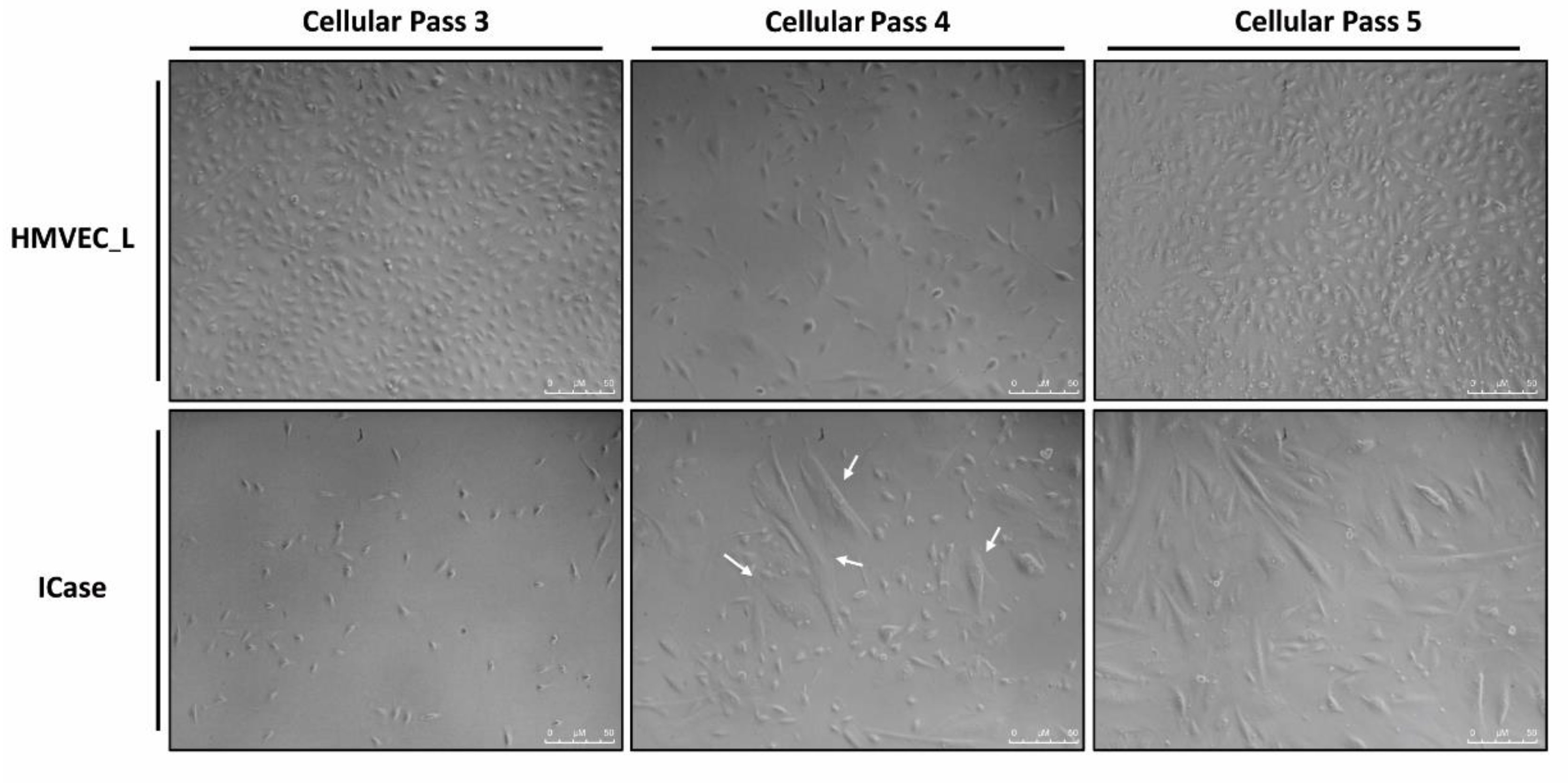
Figure 2.
RT-qPCR results for the control (dark grey) and the Index case (light grey). BioRad's iQ5 Optical System software was used to obtain these values, with the 18S gene used as a housekeeping gene to normalize the results. The β-Actin levels were altered and therefore could not be used as a housekeeping gene (see Figure 2A). Differential expression of genes involved in endothelial functions (Figure 2A and 2B), mesenchymal identity, or involved in mesenchymal transition (Figure 2C), as well as genes of the canonical TGF-β signaling pathway were observed. Error bars denote ± SEM. Student’s t-test. Statistical differences were marked with asterisks: *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001.
Figure 2.
RT-qPCR results for the control (dark grey) and the Index case (light grey). BioRad's iQ5 Optical System software was used to obtain these values, with the 18S gene used as a housekeeping gene to normalize the results. The β-Actin levels were altered and therefore could not be used as a housekeeping gene (see Figure 2A). Differential expression of genes involved in endothelial functions (Figure 2A and 2B), mesenchymal identity, or involved in mesenchymal transition (Figure 2C), as well as genes of the canonical TGF-β signaling pathway were observed. Error bars denote ± SEM. Student’s t-test. Statistical differences were marked with asterisks: *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001.
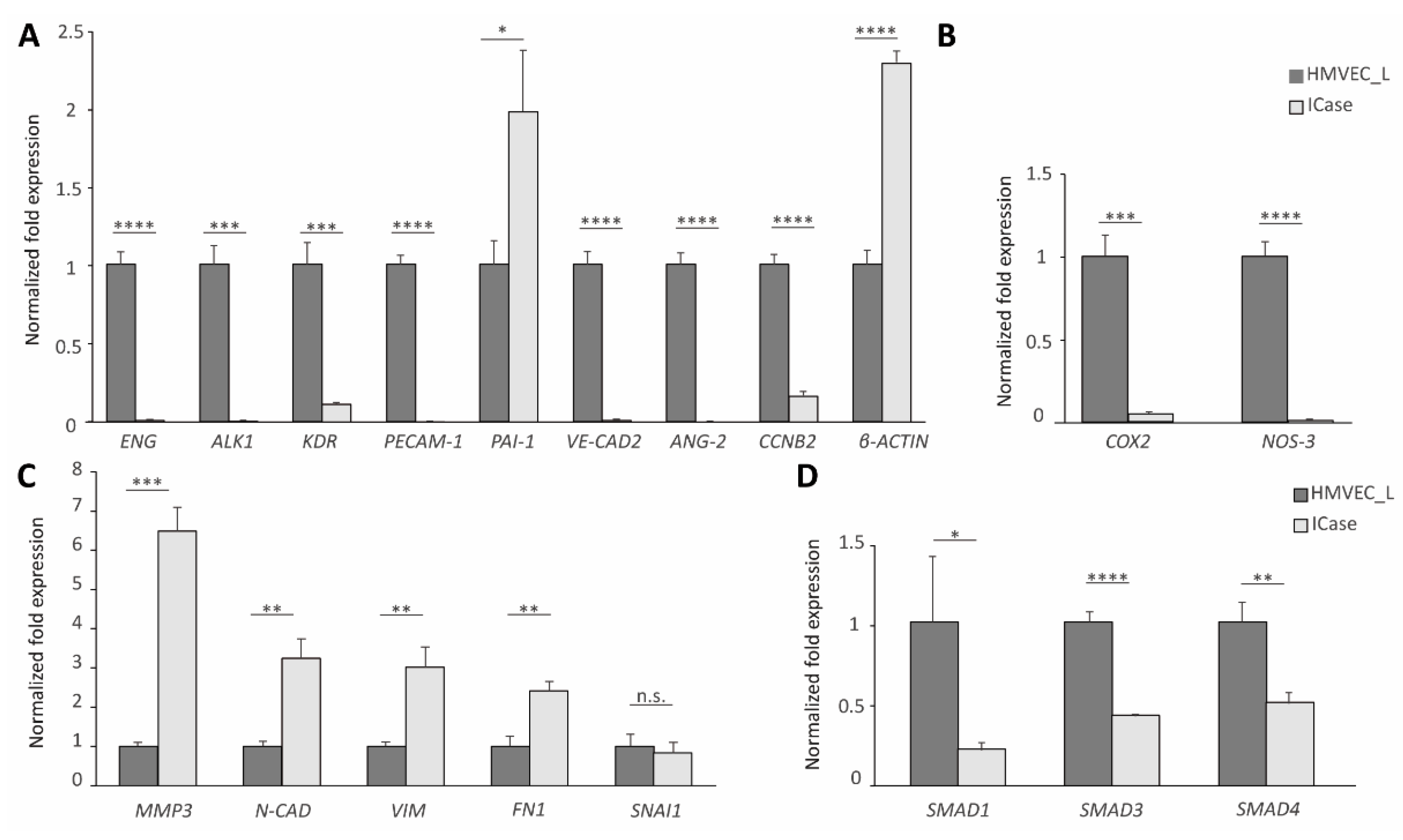
Figure 3.
Immunofluorescence, contrast phase, and light microscopy representative images. The confocal staining of HMVEC_L and ICase displays the distribution of the endothelial markers VE-Cadherin 2 (Alexa 488/green) and PECAM-1 (Alexa 647/red), as well as the mesenchymal marker N-Cadherin (Alexa 488/green) (Figure 3A). In Figure 3A, the PECAM-1 staining in the upper left corner captures the moment when filopodia are generated between two cells. Additionally, we used a cytoskeleton marker, F-Actin (Alexa 568/red) (Figure 3B). Nuclei are stain in blue (DAPI). Figure 3C displays two fields of the cell culture in phase contrast for the case study (ICase).
Figure 3.
Immunofluorescence, contrast phase, and light microscopy representative images. The confocal staining of HMVEC_L and ICase displays the distribution of the endothelial markers VE-Cadherin 2 (Alexa 488/green) and PECAM-1 (Alexa 647/red), as well as the mesenchymal marker N-Cadherin (Alexa 488/green) (Figure 3A). In Figure 3A, the PECAM-1 staining in the upper left corner captures the moment when filopodia are generated between two cells. Additionally, we used a cytoskeleton marker, F-Actin (Alexa 568/red) (Figure 3B). Nuclei are stain in blue (DAPI). Figure 3C displays two fields of the cell culture in phase contrast for the case study (ICase).
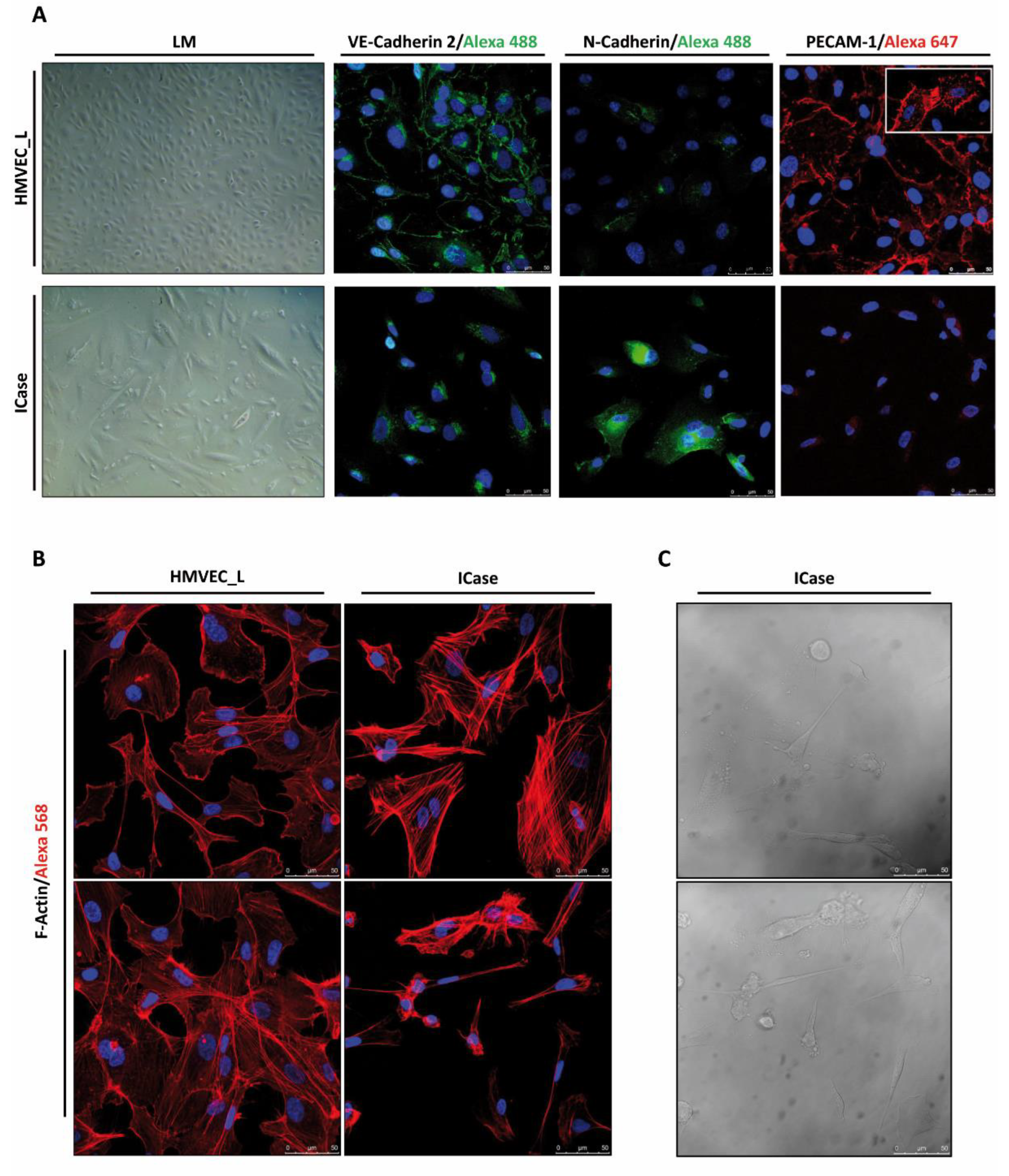
Figure 4.
The Western-blot analysis of SMAD proteins involved in signaling through the canonical TGF-β pathway was conducted in control (HMVEC_L) and case study (ICase) cultures, normalized against β-Actin, and treated with TGF-β (+) or not (-) (Figure 4A). Figure 4B displays the activation status of the PI3K/MAPK pathway, as well as the levels of the mesenchymal marker Vimentin in both conditions (control and ICase). Additionally, γ tubulin was used as a loading control for all membranes in Figure 4B. Quantifications of the band intensity is shown with the respective load normalized ratios.
Figure 4.
The Western-blot analysis of SMAD proteins involved in signaling through the canonical TGF-β pathway was conducted in control (HMVEC_L) and case study (ICase) cultures, normalized against β-Actin, and treated with TGF-β (+) or not (-) (Figure 4A). Figure 4B displays the activation status of the PI3K/MAPK pathway, as well as the levels of the mesenchymal marker Vimentin in both conditions (control and ICase). Additionally, γ tubulin was used as a loading control for all membranes in Figure 4B. Quantifications of the band intensity is shown with the respective load normalized ratios.
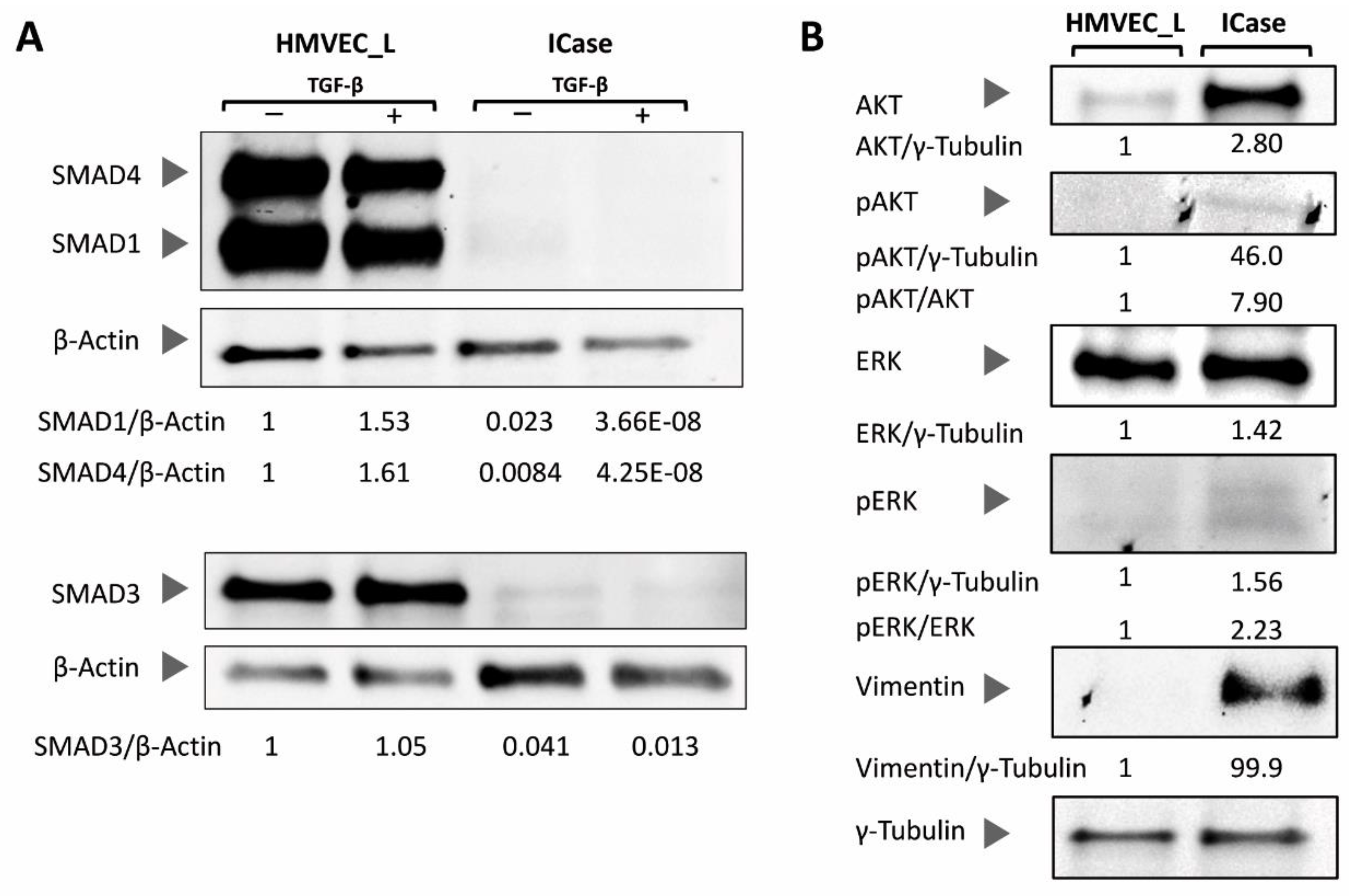
Figure 5.
Functional assays in HMVEC_L from a healthy donor (control) and the Index patient (ICase). Wound healing (Figure 5A). A wound was creating by a disruption of the confluent monolayer in both cultures. We monitored cell migration through photos taken at different times. Significant differences in the ability to close the wound were observed between the two cultures (Figure 5C). Tube formation process is abnormal or non-existent in ICase as compared to control culture (Figure 5B). Error bars denote ± SEM. Student’s t-test. Statistical differences were marked with asterisks: *p<0.05; **p<0.01.
Figure 5.
Functional assays in HMVEC_L from a healthy donor (control) and the Index patient (ICase). Wound healing (Figure 5A). A wound was creating by a disruption of the confluent monolayer in both cultures. We monitored cell migration through photos taken at different times. Significant differences in the ability to close the wound were observed between the two cultures (Figure 5C). Tube formation process is abnormal or non-existent in ICase as compared to control culture (Figure 5B). Error bars denote ± SEM. Student’s t-test. Statistical differences were marked with asterisks: *p<0.05; **p<0.01.
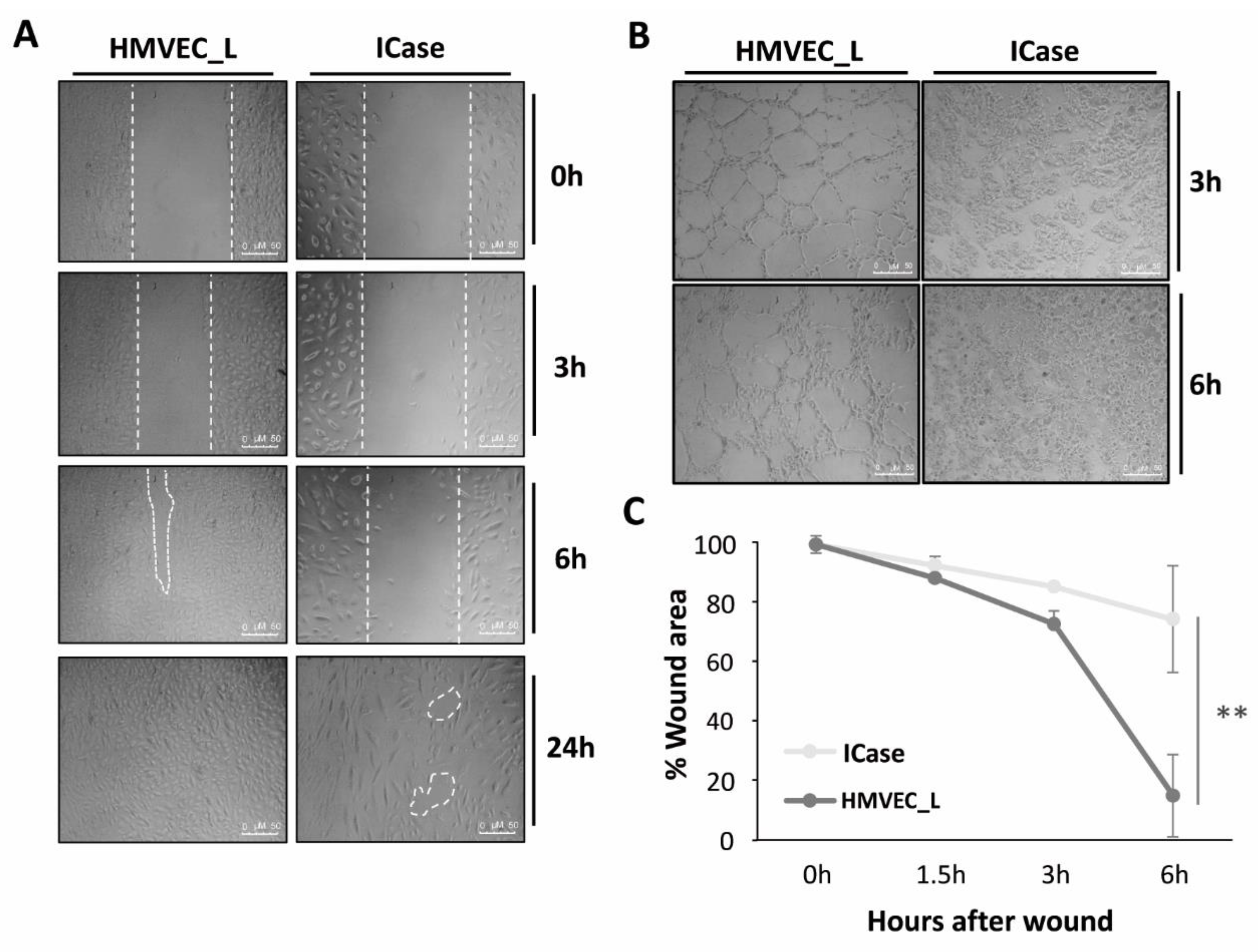
Figure 6.
Hypothetical graphic summary of the abnormal angiogenesis in PAVMs, where we observe three states from the normal to the most severe one.
Figure 6.
Hypothetical graphic summary of the abnormal angiogenesis in PAVMs, where we observe three states from the normal to the most severe one.
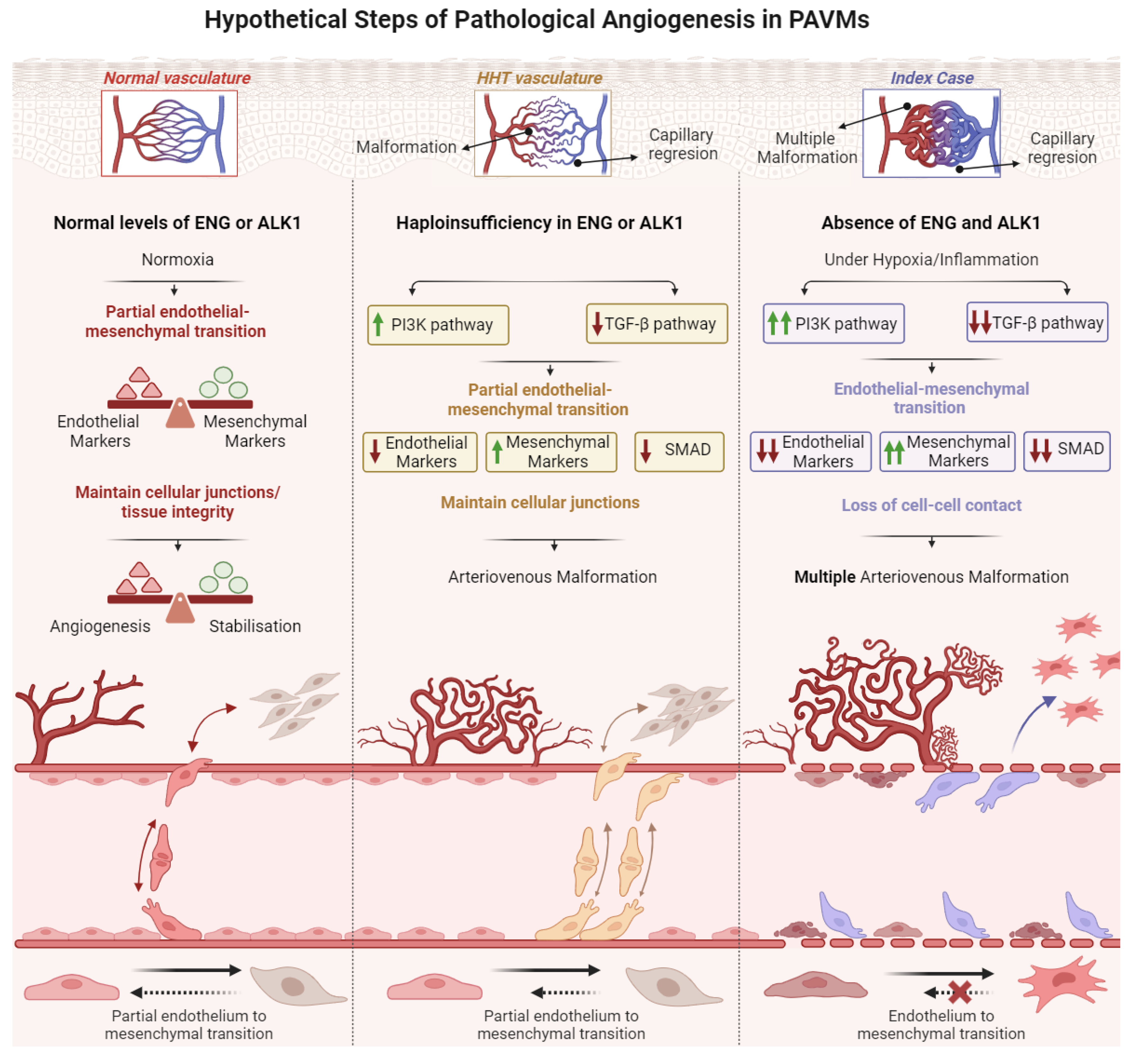

Table 1.
Primers used for q-PCR assays.
| Genes | Forward Sequences (5´-3´) | Reverse Sequences (5´-3´) |
|---|---|---|
| 18S | CTCAACACGGGAAACCTCAC | CGCTCCACCAACTAAGAACG |
| ACVRL1/ALK1 | ATCTGAGCAGGGCGACAC | ACTCCCTGTGGTGCAGTCA |
| ANG-2 | TGCAAATGTTCACAAATGCTAA | AAGTTGGAAGGACCACATGC |
| β-ACTIN | AGCCTCGCCTTTGCCGA | CTGGTGCCTGGGGCG |
| CCNB2 | TGGAAAAGTTGGCTCCAAAG | CTTCCTTCATGGAGACATCCTC |
| COX2 | TCACGCATCAGTTTTTCAAGA | TCACCGTAAATATGATTTAAGTCCAC |
| ENG | AGCCACATCGCTCAGACAC | GCCAATACGACCAAATCC |
| FN1 | TCCATTACCAAGACACACACACT | GGGAGAATAAGCTGTACCATCG |
| KDR | GAGTGAGGAAGGAGGACGAAGG | CCGTAGGATGATGACAAGAAGTAGC |
| MMP3 | CACTCACAGACCTGACTCGGTT | AAGCAGGATCACAGTTGGCTGG |
| N-CADHERIN | CCTCCAGAGTTTACTGCCATGAC | GTAGGATCTCCGCCACTGATTC |
| NOS-3 | GACCCTCACCGCTACAACAT | CCGGGTATCCAGGTCCAT |
| PAI-1 | TCCAGCAGCTGAATTCCTG | GCTGGAGACATCTGCATCCT |
| PECAM-1 | AGAAAACCACTGCAGAGTACCAG | GGCCTCTTTCTTGTCCAGTGT |
| SMAD1 | TTGATGTGCTTTGTGTGCCC | CCACAGCAAAATTCCGCCAG |
| SMAD3 | CAGGAGGAGAAGTGGTGCGA | TCCAGTGACCTGGGGATGGTAA |
| SMAD4 | CTACCAGCACTGCCAACTTTCC | CCTGATGCTATCTGCAACAGTCC |
| SNAI1 | TGCCCTCAAGATGCACATCCGA | GGGACAGGAGAAGGGCTTCTC |
| VE-CADHERIN 2 | GGAGGAGCTCACTGTGGATT | CTGATGCAGCAAGGACAGC |
| VIM | GAGAACTTTGCCGTTGAAGC | GCTTCCTGTAGGTGGCAATC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



