Submitted:
26 April 2024
Posted:
28 April 2024
You are already at the latest version
Abstract
Viral diseases pose a significant threat to tomato crops (Solanum lycopersicum L.), one of the world's most economically important vegetable crops. The limited genetic diversity of cultivated tomatoes contributes to their high susceptibility to viral infections. To address this challenge, tomato breeding programs must harness the genetic resources found in native populations and wild relatives. Breeding efforts may aim to develop broad-spectrum resistance against the virome. To identify the viruses naturally infecting 19 advanced lines, derived from native tomatoes, high-throughput sequencing (HTS) of small RNAs and validation by PCR and RT-PCR were used. Single and mixed infections with Tomato mosaic virus (ToMV), Tomato golden mosaic virus (ToGMoV), and Pepper huasteco yellow vein virus (PHYVV) were detected. The complete consensus genomes of three variants of Mexican ToMV isolates were reconstructed, potentially forming a new ToMV clade with a distinct 3' UTR. The absence of reported mutations associated with resistance-breaking to ToMV suggests that the Tm-1, Tm-2, and Tm-22 genes could theoretically be used to confer resistance. However, the high mutation rates and a 63 nucleotide insertion in the 3' UTR of Mexican ToMV isolates suggest the need to evaluate known resistance genes against these viral variants. The precise identification and characterization of viruses that naturally infect tomato germplasm could be used as a strategy for selecting the appropriate disease management and guiding breeding efforts.
Keywords:
sRNA-seq
; virome
; Solanum lycopersicum
; native germplasm
1. Introduction
Tomato (Solanum lycopersicum L.) is one of the world's most economically valuable vegetable crops and holds cultural and economic importance for Mexico. Viral diseases are among the most damaging threats to tomato crops, causing a significant decrease in yield and quality [1]. The yield reduction induced by viruses can range between 42.1%–95.5% [2]. However, economic and productive losses vary greatly and often depend on the viral species and the growing region [3].
The management of viral diseases in agriculture relies on two main strategies: immunization to get resistant plants and prophylactic measures to restrain virus spread [4]. Among immunization strategies, virus resistance breeding is one of the most promising methods for controlling viral diseases [2]. Identifying both pathogen and host genes influencing disease outcomes is crucial for plant breeding strategies.
Additionally, understanding the inheritance pattern of the plant's resistance trait, along with the presence of pathogen strains or physiological races, is essential for effective breeding [5]. Therefore, the virus disease management strategy must be developed specifically for each viral species, strain, host, and environment. A successful strategy heavily relies on a fast and accurate identification of the causal agent, as well as knowledge of virus vectors, transmission biology, and ecology [4].
The high number of tomato-infecting viral species and frequent mixed infections with different species or variants of the same species [6,7,8] challenge standard diagnostic tests. Molecular and serological tests require prior knowledge of the target virus and are often unsuitable for detecting unexpected or unknown viruses. Furthermore, their high specificity can limit the detection of diverse isolates, variants, or strains, potentially leading to false negative results [9]. High-throughput sequencing (HTS) offers a powerful solution, identifying plant viruses regardless of their genome nature or structure [10]. In theory, HTS can detect all viruses present in a single assay, providing an exhaustive view of a plant´s viral phytosanitary status. Moreover, the sequence information obtained can be used for a variety of purposes, including elucidating virus population structure, ecology, and evolution, as well as differentiating variants with varying contributions to disease etiology. Additionally, HTS data can be shared and re-analyzed by multiple users as databases expand [11].
The cultivated tomato has low genetic diversity due to intensive selection and genetic bottlenecks that emerged during its evolution and domestication [12]. This could explain its high susceptibility to viruses, with over 300 documented, many causing disease symptoms and yield losses [8]. To address this, tomato breeding programs require a broader genetic base, readily found in native populations [13,14] and wild relatives [15].
Mexico, a center of tomato genetic diversity, holds native germplasm potentially harboring various degrees of tolerance to biotic and abiotic stresses, along with unique desirable organoleptic characteristics [16]. In a variety trials conducted in 2021 and 2022 at the Instituto Nacional de Investigaciones Forestales, Agrícolas y Pecuarias, virus-like symptoms were observed in 19 advanced lines derived from native tomatoes collected in different locations of Southeastern Mexico. The symptoms, observed over two growing cycles, suggest potential seed-borne viral infection and susceptibility in these advanced lines. Seed-borne viruses pose a particular threat as they can spread widely through seed exchange and establish infected plants within plantations, acting as a continuous source of inoculum [17]. To diagnose the specific viruses, recommend appropriate management strategies, and guide breeding efforts, the virome of these 19 tomato lines was analyzed using High-throughput sequencing of small RNA (sRNA) from leaf tissue composite samples. The analysis was performed in conjunction with PCR and RT-PCR validation of the viruses identified in each sample.
2. Materials and Methods
2.1. Plant Material Sampling
In February 2023, young leaves exhibiting virus-like symptoms were collected from 36 plants of 19 advanced lines of tomato (Solanum lycopersicum L.) at a greenhouse evaluation facility at the “Instituto Nacional de Investigaciones Forestales, Agrícolas y Pecuarias” in Celaya, Guanajuato, México (20°34’42.06’’N 100°49’12.37’’W). These advanced lines were developed from native tomatoes collected across six states in Southeast Mexico (Table 1). The observed symptoms included foliar necrosis, local necrotic lesions, yellow mottling, green vein banding, blistering, upward and downward curling, leaf malformation, yellowing, and interveinal chlorosis (Supplementary Figure S1).
For high-throughput sequencing (HTS) identification of viruses, foliar tissues were immediately frozen in liquid nitrogen and stored at –72 °C until further processing. To validate the HTS-identified viruses using PCR or RT-PCR, separate foliar tissues were dried and preserved in silica gel with a moisture indicator, as described in Chiquito-Almanza et al. [18].
2.2. Library Construction and sRNA Sequencing
HTS of sRNA and assembly was employed to identify viral species [19]. Total RNA was extracted from approximately 0.4 g of leaf tissue per sample using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) and quantified using a NanoDrop 8000™ analyzer (Thermo Fisher Scientific, Waltham, MA, USA). Agarose gel electrophoresis was used to assess RNA quality. Three composite samples (JM1, JM2, and JM3) were created by pooling equimolar mixtures of total RNA from 15, 10, and 11 plants, respectively (composition details in Table 1). Approximately 3 μg of RNA from each composite sample was shipped to Macrogen Inc. (Seoul, Korea) for sRNA library construction using the TruSeq® Small RNA Library Preparation Kit (Illumina, San Diego, CA, USA). The libraries were then sequenced on an Illumina HiSeq 2500 (rapid) system in single-end, 50-bp mode.
2.3. Small RNA Bioinformatic Analysis
Bioinformatic analysis and virus identification were performed according to Chiquito-Almanza et al. [20], following the procedure outlined by Zheng et al. [21]. Briefly, the quality of raw sRNA reads was assessed using the sRNA_clean.pl script (https://github.com/kentnf/VirusDetect/tree/master/tools/sRNA_clean, accessed on 01 September 2023). This script trimmed 3’ adaptor sequences, removed low-quality reads and reads without adaptor sequences, and eliminated reads shorter than 15 nucleotides (nt) after trimming. The average number of sRNA mapped and unmapped to the host was obtained by aligning the trimmed sRNA sequences (15–40 nt) to the tomato genome (NCBI: GCA_000499845.1) using the Bowtie v1.1.1 program [22].
De novo assembly of reads into contigs and subsequent virus identification were conducted using the online VirusDetect pipeline with default parameters (coverage = 0.1 and depth = 5) (http://virusdetect.feilab.net/cgi-bin/virusdetect/index.cgi, accessed on 08 March 2024). The tomato reference genome and plant virus reference database group 248 U100 available on the VirusDetect platform were used for host sRNA subtraction and guided assemblies through sRNA sequence alignment.
To determine consensus sequences, the longest contigs from each library were mapped to the corresponding virus reference genome for each virus identified. For Tomato mosaic virus (ToMV), the 3' end was additionally amplified by RT-PCR and Sanger sequencing. The RT-PCR was performed on each of the 36 samples using the specific direct primer ToMV_GSPD1 (5'-GGTTGCAATTCGGTCTGCTAT-3') designed from the assembled contigs and the reverse primer ToMV_3R from the study by [Lyu et al., 2023], with a 16 (T>A) base substitution (3'-TTATATATGGGCCCCTACCG-5'). These primers flank a 124 nt region of the coat protein (CP) end and the complete 3' untranslated region (UTR) of ToMV. The consensus and the contig sequences of each virus were aligned against their respective taxa using the NCBI database with BLASTn [23]. Consensus viral genomes were reconstructed and annotated using SnapGene V4.1.9. Software (GSL Biotech LLC, Chicago, IL, USA). Protein molecular weights were predicted using the Compute pI/Mw tool (https://web.expasy.org/compute_pi/).
2.4. Recombination and Phylogenetic Analysis
The phylogenetic analysis was performed using the complete genome sequences of the isolates of ToMV available in the NCBI GenBank (Supplementary Table S1). The MEGA11 software package [24] was utilized for nucleotide sequence alignment, selection of the best-fit substitution model, construction of the phylogenetic tree, and mutation rate. Nucleotide sequences were aligned using the ClustalW algorithm with default settings. The number of mutations and mutation rates were determined by removing alignment sites containing gaps. The maximum likelihood method with 1000 bootstrap replicates was employed for phylogenetic tree construction. A tobacco mosaic virus (TMV) isolate (NC_001367) was used as an outgroup. Nodes with bootstrap values less than 70% were collapsed. Assessment of potential recombination events among ToMV isolates was carried out using the RDP v.4.101 program [25] on the same sequence set used for phylogenetic analysis. A putative recombination event was considered significant if supported by at least four of the seven implemented methods, with an associated p-value < 1 × 10-6.
2.5. PCR and RT-PCR Validation of HTS
To validate the presence of the viruses identified by HTS, total nucleic acids (DNA and RNA) were extracted from each of the 36 leaf tissue samples preserved in silica gel. The extraction method employed the cetyl trimethyl ammonium bromide (CTAB) protocol described by Abarshi et al. [26], with the modifications outlined in Chiquito-Almanza et al. [18]. The integrity of total nucleic acids was assessed using 1% agarose gel electrophoresis. Quality and quantity were determined by spectrophotometry, measuring absorbance at 260 nm and 280 nm wavelengths using a NanoDrop 8000™ instrument (Thermo Fisher Scientific, Waltham, MA, USA). For all samples, reverse transcription (RT) was performed using 1 µg of total nucleic acids and a SuperScript III First-Strand synthesis kit (Invitrogen, Carlsbad, CA, USA) in a 20 µL reaction volume. The RT products were stored at -20 °C and subsequently used as templates for the detection of both DNA and RNA viruses by PCR and RT-PCR, respectively. Specific primers reported by Yan et al. [27] were used for RT-PCR detection of Tomato mosaic virus (ToMV). Detection of Pepper huasteco yellow vein virus (PHYVV), and Tomato golden mottle virus (ToGMoV) was achieved using PCR with primers described by Nakhla et al. [28].
3. Results
3.1. HTS Data
Three sRNA libraries, designated JM1Lc, JM2Lc, and JM3Lc, were constructed from total RNA composite samples JM1, JM2, and JM3 (Table 1). These sRNA libraries were subjected to HTS, generating between 72.0 and 72.4 million raw reads per library. Following adapter trimming and removal of low-quality reads, 58.9, 57.4, and 58.5 million clean reads within the 15-40 nucleotide range were obtained from the JM1Lc, JM2Lc, and JM3Lc libraries, respectively. Between 90.6 to 92.7% of these reads mapped to the host tomato genome, while 4.3, 5.4, and 4.8 million reads were identified as non-host.
3.2. Virome Characterization of Tomato Advanced Lines
After the VirusDetect process, between 3-11 contigs exhibiting nucleotide identity to DNA-A and DNA-B components of ToGMoV were assembled from the three sRNA libraries, and 10 and 2 contigs with nt identity to DNA-A and DNA-B components of PHYVV were assembled from the JM3Lc library. Except for the PHYVV DNA-A component (96.8% coverage, 93.3% nt identity) and the ToGMoV DNA-B component (90.9-94.5% coverage, 90.0-90.7% nt identity), the remaining contigs with identity to begomoviruses nearly covered the complete viral genomes with 96.8-98.4% nt identity compared to the respective reference viral genome sequences (Table 2). Furthermore, one contig that nearly covered the complete genome reference sequence with >99% nt identity to ToMV was assembled from each library (Table 2). No unknown viruses were identified.
Consensus sequences exhibiting the typical genome features described for DNA-A and DNA-B components of PHYVV and ToGMoV were generated by mapping the contigs from each library against their respective reference genomes (Supplementary Figures S2 and S3). Three nearly complete consensus sequences of the ToGMoV DNA-A component comprising 2599, 2583, and 2608 nt with 99% nt identity among them were reconstructed, exhibiting the highest nucleotide identity (97.5-98.4%) with the Rioverde SLP2 DNA-A isolate (EF501976.1) from Mexico. Similarly, the 2546 nt PHYVV DNA-A consensus sequence shared 93.3% nt identity with the DNA-A component of the PEL95W2007 isolate (LN848879.1), also originating from Mexico.
The presence of PHYVV, ToGMoV, and ToMV in all the samples was validated by PCR and RT-PCR. PHYVV was identified only in the sample JCM07-5 in co-infection with ToMV. ToGMoV was identified in 13 samples in co-infection with ToMV, and ToMV was identified in all samples in single and mixed infection (Table 1).
3.3. Analysis of ToMV Reconstructed Sequences
To perform the recombination, mutation rates, and phylogenetic analysis, and identify mutations reported as associated with resistance-breaking, the complete consensus genome sequences of ToMV were determined. For this purpose, the assembled contigs from composite samples JM1, JM2, and JM3 were used as a reference, and the complete 3' UTR sequence (264 nt) was obtained by RT-PCR and Sanger sequencing using cDNA synthesized from each of the 36 samples (Table 1).
The contigs assembled from composite samples JM1 and JM2 exhibited 100% nucleotide identity, while the contig from sample JM3 showed 99.9% identity to JM1 and JM2. These contigs, ranging from 6,290 to 6,300 nt in length, spanned the complete 5' UTR (72-73 nt), four open reading frames (ORFs), as well as a 107-118 nt fragment of the 3' UTR (Supplementary Figure S4). No differences were detected across the nucleotide sequences of the 3' UTR of the 36 samples analyzed. As a result, the full-length consensus genomes, designated INIFAP JM1, INIFAP JM2, and INIFAP JM3, were reconstructed and deposited in the NCBI GenBank database with the accession numbers PP481218–PP481220.
While INIFAP JM1 and INIFAP JM2 have identical 6,447 nt sequences, INIFAP JM3 has 6,448 nt, with a cytosine insertion at the beginning of the 5' UTR and silent mutations at positions 3114 (G>A) and 3117 (A>G) compared to INIFAP JM1 and INIFAP JM2. The ORF analysis revealed the presence of four ORFs encoding the putative viral replicase (126.3 kDa), the RNA-dependent RNA polymerase (RdRp) (183.5 kDa), the movement protein (MP) (29.2 kDa), and the CP (17.7 kDa).
The 3’ UTR region of INIFAP JM1-3 differed from most 3’ UTR sequences of ToMV isolates available in the NCBI GenBank database. The alignment of this region with the 3' UTR of 52 ToMV isolates revealed a 63 nt insertion present solely in the 3' UTR of INIFAP JM1-3, and the isolate SRVP22_05 (OQ722333).
The highest nucleotide identity of the complete consensus genome of INIFAP JM1-3 ranges from 99.34 to 99.37% with the isolate SRVP22_05 (OQ722333) from Russia. None of the previously reported mutations of ToMV associated with resistance-breaking were identified in the reconstructed consensus sequences.
3.4. Recombination, Mutation Rates, and Phylogenetic Analysis
The Tamura-Nei model [29] with gamma-distributed rates and invariant sites was determined to be the best-fitting nucleotide substitution model for phylogenetic analysis. Phylogenetic analysis revealed that the INIFAP JM1-3 isolates clustered in a compact group outside the principal clade containing 47 of the ToMV strains from China, Japan, Australia, Germany, Russia, South Korea, and other countries (Clade I). The other 8 isolates formed separate branches outside this principal clade, including the Mexican isolates (Figure 1).
Four putative recombination events, supported by at least four of the seven different methods employed, were detected in the ToMV isolates NVWA36783860, Tai'an 2, and N5 from China, and SRVP22_05 from Russia (Table 3). Notably, the third recombination event, identified at positions 6126-6442 nt within the 3' UTR of the SRVP22_05 isolate, was also present in the isolates 99-1 from USA, Tianjin from China, and the consensus sequences of Mexican isolates INIFAP JM1-3. Analysis indicated that the major parent for this recombination event was the isolate AH4 from Egypt, while the minor parent remains unknown (Table 3).
The mutation rates of the INIFAP JM1-3 isolates were compared to the reference Queensland isolate from Australia (NC_002692), and the SRVP22_05 isolate. The ORFs of all isolates had the same nucleotide length, but length variations were observed across the 5' and 3’ UTRs. The mutations in the ORFs range between 2-47 nt and 0.42-1.01 amino acids (aa), with mutation rates between 0-2% and 0-0.76%, respectively (Table 4). In the 5’ UTR, between 0-2 nt were mutated, with a mutation rate between 0.0-2.74%, while in the 3’ UTR, between 11 to 16 nt were mutated, with a mutation rate of 4.8-6.06% (Table 4).
4. Discussion
Early and precise detection of viruses, along with cultural practices and immunization, is a crucial component of plant virus disease control strategies [4]. The HTS technology has emerged as a valuable tool that facilitates the identification of phytopathogenic organisms and enhances disease management approaches. HTS enables the detection of novel and emerging pathogens, facilitates the tracking of disease outbreaks, and contributes to the development of disease-resistant cultivars [30,31].
4.1. Selecting the Virus Identification Strategy
In theory, HTS can detect all DNA and RNA viruses present in a sample, and be implemented using individual, bulked, or mixed samples [19,32,33], with reported repeatability and reproducibility of up to 100% [34,35,36]. However, major challenges in using HTS for virus detection still arise from the lack of established methodological standards, especially regarding bioinformatic tools, input material, nucleic acid preparation, and the overall cost of the analysis [37].
In the current study, sRNA high-throughput sequencing was utilized to identify the viruses naturally infecting advanced lines of native tomatoes. To minimize library construction costs, three composite samples were created by pooling multiple tomato plants and lines (Table 1). Virus identification was performed using the online VirusDetect software and validation was realized by PCR and RT-PCR. This strategy was chosen based on our bioinformatic skills, economic capacity, and infrastructure.
The sRNA sequencing strategy is used to detect virus-derived small interfering RNAs (siRNA) generated as part of the plant's gene silencing-based immune response [19]. With this approach, viroids, RNA viruses, DNA viruses, and even persistent viruses can be identified [19,38,39,40,41].
Compared to HTS of total RNA for virus diagnosis, sRNA sequencing yields a very high number of reads and depth of coverage [35,42]. The relative enrichment of viral signatures in sRNA data is likely responsible for the enhanced ability to detect viruses and viroids with relatively low expression levels, which can be challenging using total RNA approaches [43]. Furthermore, the purification of sRNA is simpler compared to the time-consuming and experimentally challenging methods for other types of viral molecules such as double-stranded RNA or virion purification, which require sophisticated and expensive laboratory equipment such as ultracentrifugation [37,41]. Although the best approach to capture the entire viral diversity is perhaps a combination of all techniques, this strategy is not cost-feasible for routine application in a breeding program.
We selected VirusDetect software because it is an automated bioinformatic service for plant virus discovery with a public web interface dedicated to the analysis of sRNA data [21]. It is recommended for virologists with low to moderate bioinformatic skills and has proven useful for detecting known viruses and discovering new ones [20,37,44,45,46], including persistent viruses [20,41]. VirusDetect has achieved 100% sensitivity between sequencing depths of 2.5M and 250K, without false positives [47]. In our analysis, between 4.3M to 5.4M non-host reads were obtained from the libraries comprising 10 to 15 samples. This number of reads corresponds to an average of 286K to 540K reads per sample. Therefore, our sequencing depth falls within the highly sensitive range of VirusDetect.
4.2. Virus Identification and Implications of Co-Infections
With the implemented strategy, the near-complete consensus nucleotide sequences of the PHYVV, ToGMoV, and ToMV genomes were reconstructed. These consensus sequences represent an overall view of the viruses present in the samples. The validation by PCR/RT-PCR (Table 1) not only confirmed the identity of the viruses but also enabled the determination of the presence of single and mixed infections.
The complex interplay between viruses during viral co-infections can have significant implications for disease development, symptom expression, and virus epidemiology [48]. These viral co-infections can result in diverse interactions between the coexisting viruses, which can lead to at least four different types of interactions: neutralism, synergism, antagonism, and synergism/antagonism [49,50].
Studies on tomato co-infections with other viral species have revealed the complexity of the interactions. For example, the co-infection of the crinivirus Tomato chlorosis virus (ToCV) with the begomovirus Tomato yellow leaf curl virus (TYLCV) or with the tospovirus Tomato spotted wilt virus (TSWV) induced disease synergism. The co-infection of ToCV with TYLCV resulted in increased disease severity, reduced plant growth, and higher viral accumulation [51], while the co-infection of ToCV with TSWV led to rapid death of susceptible cultivars and the breaking of TSWV resistance in resistant plants when pre-infected with ToCV [52]. On the other hand, the co-infection with the begomoviruses Tomato yellow spot virus (ToYSV) and Tomato rugose mosaic virus (ToRMV) reduced the viral titers of both viruses, but the symptoms were more severe compared to single infections [53].
Understanding the nature of these virus-virus interactions is an important consideration in the management of plant viral diseases, as the outcomes can vary widely depending on the specific species or strain of viruses involved, the environmental conditions, and the cultivar. The implications of the presence of mixed infections, such as the co-infection of ToMV with ToGMoV or with PHYVV, and how this may affect the dynamics and impact of these viral diseases, need to be further investigated.
4.3. Genomic Characterization and Phylogenetic Relationships of ToMV Isolates
The integration of the phylogenetic, recombination, and mutation data provides a more thorough picture of the evolutionary dynamics of ToMV, which may have implications for predicting the emergence of new viral variants and designing durable resistance strategies in tomato cultivation.
The reconstructed sequences INIFAP JM1-3 isolates share the highest nucleotide identity with the sequence of the SRVP22_05 isolate obtained from a wastewater sample [54]. The SRVP22_05 isolate was classified within the order Martellivirales, but no specific species was assigned. BLASTn analysis of its sequence indicated that it shares >99% identity with various ToMV isolates. Its detection in wastewater aligns with the known ability of ToMV to survive in aqueous solutions under different pH and temperature conditions [55,56]. This evidence strongly suggests that SRVP22_05 is a ToMV isolate.
While the SRVP22_05 isolate sequence was reported as a complete genome, the 5' and 3' end sequences are likely partial. Alignment of this sequence with 52 complete ToMV genomes shows 23 fewer nucleotides at the beginning of the 5' UTR, and 35 nt fewer, upstream of the 63 nt insertion, in the 3' UTR. Because the SRVP22_05 sequence was obtained through metagenome-assembled genome analysis and not confirmed by Sanger sequencing [54], it is possible that the 63 nt insertion in the 3' UTR is an assembly artifact. However, the sequences of the 3' UTR of the INIFAP JM1-3 isolates were obtained through Sanger sequencing and were identical across all analyzed samples. The unique 63 nt insertion could be a signature feature of this ToMV lineage and may have implications for its molecular epidemiology and evolution.
Interestingly, Russia is the fourth most important importer of Mexican tomatoes, which could explain the similarity of the isolates in such geographically separated locations. This characteristic could be used for the design of diagnostic assays or to further the understanding of ToMV biology.
A high number of mutations were detected in the ORFs encoding the putative viral replicase (126.3 kDa) and the RdRp (183.5 kDa) of the INIFAP JM1-3 isolates, the majority of which being silent mutations (Table 4). Although none of the previously reported mutations associated with ToMV resistance-breaking [57,58,59,60] were identified, the high rate of mutations identified in the 3' UTR region (Table 4) suggests a potential genomic divergence and could have important implications for pathogenesis.
The function of the ToMV 3' UTR is poorly studied, but some evidence suggests this region is important for pathogenesis [61]. In the related tobamovirus TMV, the 3' UTR contains a complex secondary structure consisting of three consecutive pseudoknots immediately downstream of the CP ORF stop codon, followed by a 3'-terminal transfer RNA (tRNA)-like structure with two additional pseudoknots [62,63]. The upstream pseudoknot domain in TMV has a crucial role in viral replication and translation. Mutational analysis shows this domain could interact with eEF1A/GTP with high affinity [64], and modifications in the TMV 3' UTR can detrimentally affect infectivity [65,66], virus accumulation, and symptom expression [67]. Given the functional importance of the 3' UTR in the closely related TMV, the ToMV 3' UTR likely has similar critical roles.
Furthermore, the detection of recombination events, particularly the one shared between the SRVP22_05 isolate and the Mexican INIFAP JM1-3 isolates, highlights the importance of considering recombination as a mechanism contributing to the genetic variation of ToMV. Interestingly, the third recombination event, with an unknown minor parent (Table 3), coincides with the position of the 63 nt insertion found in the 3' UTR of the INIFAP JM1-3 sequences. This suggests that recombination may have contributed to the acquisition of this unique genomic feature, which could have implications for the biology or evolution of these Mexican ToMV isolates. Further characterization of the potential functional significance of this 3' UTR insertion and mutations would be an important area for future research.
The phylogenetic analysis provides insights into the global diversity of ToMV strains. The majority of the strains cluster within a primary clade, which is consistent with the findings reported by Lyu et al. [68]. Notably, the isolates INIFAP JM1-3 form a distinct group. This information in conjunction with the high sequence identity (99.9-100%) between the Mexican isolates suggests a low degree of genetic diversity among these isolates, and a potential regional differentiation of the INIFAP JM1-3 isolates, which could be influenced by geographical isolation, host-pathogen co-evolution, or other factors.
The tomato lines analyzed in this study were developed from native germplasm collected in different states of southeastern Mexico and correspond to accessions with distinct fruit shapes. Due to the ability of ToMV to be seed-transmitted, it is necessary to determine whether the ToMV variants identified originate from any of the collection sites or were acquired at the site where the genetic improvement is being carried out. Further investigations into the evolutionary dynamics and the factors driving the diversification of ToMV in Mexico would be valuable.
4.4. Disease Management and Resistance Breeding Implications of the Viruses Identified
Crop breeding efforts may aim to develop resistance against the virome rather than targeting only one or two specific viruses. This approach becomes feasible when the viromes present in different agroecological zones have been thoroughly characterized, methodologies are available to expose breeding materials to the relevant viromes during the breeding process [30], and resistance genes are available. The implications for disease management and resistance breeding of the viral species identified in this studio are discussed below.
4.4.1. Begomoviruses ToGMoV and PHYVV
ToGMoV and PHYVV are two circular single-stranded DNA begomoviruses with bipartite genomes comprised of DNA-A and DNA-B components [69,70]. PHYVV is an endemic and widely distributed species in Mexico that infect pepper, Physalis philadelphica, and tomato [69,71], while ToGMoV was first reported infecting S. Lycopersicum and S. rostratum in San Luis Potosi State in the north-central region of Mexico in 2007 [70]. Both viruses have also been identified infecting cucumber plants in Colima, Mexico [72].
Strains of PHYVV compromise the tolerance or resistance of tomato to Tomato yellow leaf curl virus (TYLCV), the most damaging and widely distributed monopartite begomovirus [73], and although resistance to mixed infections of PHYVV and Pepper golden mosaic virus (PepGMV) has been identified in wild pepper accessions [74], to date, no resistance genes to ToGMoV or PHYVV have been reported in tomato cultivars. Neither of these viruses is seed-transmitted; their transmission occurs in a circulative persistent manner by the vector Bemisia tabaci [75]. In general, insect-transmitted viruses are geographically restricted. Therefore, as with other begomoviruses, their control relies mainly on implementing good management practices and strict sanitary measures to prevent the presence of the vector [76].
4.4.2. ToMV
ToMV is a positive-sense single-stranded RNA tobamovirus [77]. It infects all parts of the tomato plant at any growth stage and can produce a wide range of symptoms such as leaf discoloration and deformation, fruit malformation and size reduction, internal necrotic alterations, and flower abortion [78,79]. This virus is seed-transmitted in susceptible plants and represents a major problem in greenhouse production because it is highly stable, contagious, and cosmopolitan; can survive in the soil for several years and remain infectious in nutrient solutions for at least 6 months regardless of temperature and storage conditions [55,56]. Additionally, it can be easily transferred from contaminated workers' hands, tools, and clothing through simple contact between an infected plant and a healthy one during cultivation practices [80,81]. Therefore, prevention relies on implementing strict sanitary measures through the use of certified pathogen-free propagation material, disinfection of tomato seeds by thermotherapy or chemical treatment [82], sterilizing cutting tools during cultural and manipulation operations, and large crop rotations in soils free of vegetation residues and weeds that can be hosts of the virus [83]. However, genetic resistance is the most suitable option to control this virus.
At present, the three predominant strategies employed for antiviral resistance breeding in tomato crops comprise marker-assisted selection (MAS) for the development of hybrid cultivars, the generation of transgenic lines, and the application of the CRISPR/Cas system for targeted gene editing. Combining conventional hybrid breeding techniques with MAS for introgressing known ToMV resistance genes has proven to be effective, economical, and sustainable. This integrated strategy circumvents current controversies and regulations restricting the use of transgenic crops in many countries [84,85] while expediting the release of improved virus-resistant tomato cultivars.
Three major resistance genes to ToMV: Tm-1, Tm-2, and Tm-22 have been characterized. These genes were introgressed into tomato cultivars from the wild tomato species Solanum hirsutum and S. peruvianum [79,86]. Among these, Tm-22 confers resistance to all the most common ToMV strains (0, 1, and 2), except for the 22 strain (not very common in crops) [83,84]; thus, Tm-22 gene is widely deployed in most breeding programs [87].
Nearly all commercial tomato varieties exhibit high resistance to ToMV, so this viral species is not regarded as one of the economically important viruses affecting tomato crops in Mexico [88]. However, the detection of ToMV in all evaluated native tomato lines (Table 1) indicates their susceptibility to this viral pathogen. This finding aligns with the unimproved genetic background of these materials, which have not been subjected to breeding efforts targeting ToMV resistance. To harness the genetic diversity present in these native resources, a strategic breeding approach is required for the development of resistant cultivars.
The Tm-2 and Tm-22 genes induce a defense reaction in tomato plants by recognizing the movement protein of ToMV [89]. However, this resistance can be overcome by even a few amino acid substitutions in the viral genome. Although no mutations associated with resistance-breaking were identified in the consensus genome sequence of the INIFAP JM1-3 isolates, the high rate of mutations and the 63 nt insertion identified in the 3' UTR region requires evaluating whether these ToMV variants possess the ability to overcome the resistance conferred by the Tm-1, Tm-2, and Tm-22 genes.
5. Conclusions
The High-throughput sequencing of sRNA using composite samples, and the validation by PCR or RT-PCR of individual samples, allowed us to determine that the advanced tomato lines were infected with either single or mixed infections of ToGMoV, PHYVV, and ToMV. Furthermore, a potential new clade of ToMV with a distinct 3' UTR sequence was discovered. Because all the evaluated lines were susceptible to ToMV, it is necessary to implement proper virus management strategies and initiate the identification of resistance sources to develop ToMV-resistant tomato cultivars.
The high rate of mutations and the presence of a 63 nt insertion in the 3' UTR region of the Mexican ToMV isolates necessitates evaluating whether the known resistance genes are effective in conferring protection or resistance against these viral variants.
The precise identification of the viral species that naturally infect tomato germplasm could be used as a strategy for selecting appropriate disease management approaches and guiding resistance breeding efforts.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Symptoms observed in selected native tomato lines; Figure S2: Schematic representation of the coverage of assembled contigs across the viral reference genome and the reconstructed genome sequences of PHYVV DNA-A and DNA-B components; Figure S3: Schematic representation of the coverage of assembled contigs across the viral reference genome and the reconstructed genome sequences of ToGMoV DNA-A and DNA-B components; Figure S4: Schematic representation of the coverage of assembled contigs across the viral reference genome and the reconstructed genome sequences of ToMV; Table S1: Details of the 52 complete genomes of ToMV isolates used in this study.
Author Contributions
Conceptualization, JLAL and ECA; methodology, ECA; validation, ECA; formal analysis, JLAL, and ECA; investigation, ECA; resources, EGP, SVR, JKC, and JLAL; data curation, JLAL, and ECA; writing—original draft preparation, JLAL; review and editing, JLAL, ECA; supervision, JLAL; project administration, JLAL and EGP; funding acquisition, JLAL and EGP. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by INIFAP (SIGI number: 16592536649).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Genomic sequences of viruses described in this study are available at the GenBank database under accession numbers PP481218–PP481220.
Acknowledgments
We express our sincere gratitude to the anonymous reviewers for their valuable feedback that significantly improved this manuscript. We also thank the project administrators for their crucial support throughout this investigation.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Ong, S.N.; Taheri, S.; Othman, R.Y.; Teo, C.H. Viral disease of tomato crops (Solanum lycopersicum L.): an overview. J. Plant Dis. Prot. 2020, 127, 725–739. [Google Scholar] [CrossRef]
- Anikina, I.; Kamarova, A.; Issayeva, K.; Issakhanova, S.; Mustafayeva, N.; Insebayeva, M.; Mukhamedzhanova, A.; Khan, S.M.; Ahmad, Z.; Lho, L.H.; Han, H.; Raposo, A. Plant protection from virus: a review of different approaches. Front. Plant Sci. 2023, 14, 1163270. [Google Scholar] [CrossRef] [PubMed]
- Selda-Rivarez, M.P.; Vučurović, A.; Mehle, N.; Ravnikar, M.; Kutnjak, D. Global advances in tomato virome research: Current status and the impact of high-throughput sequencing. Front. Microbiol. 2021, 12, 671925. [Google Scholar]
- Rubio, L.; Galipienso, L.; Ferriol, I. Detection of plant viruses and disease management: Relevance of genetic diversity and evolution. Front. Plant Sci. 2020, 11, 1092. [Google Scholar] [CrossRef] [PubMed]
- Nelson, R.; Wiesner-Hanks, T.; Wisser, R.; Balint-Kurti, P. Navigating complexity to breed disease-resistant crops. Nat. Rev. Genet. 2018, 19, 21–33. [Google Scholar] [CrossRef]
- Xu, C.; Sun, X.; Taylor, A.; Jiao, C.; Xu, Y.; Cai, X.; Wang, X.; Ge, C.; Pan, G.; Wang, Q.; Zhangjun, F.; Wang, Q. Diversity, distribution, and evolution of tomato viruses in China uncovered by small RNA sequencing. J. Virol. 2017, 91, e00173-17. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Jo, Y.; Cho, W.K.; Yu, J.; Tran, P.T.; Salaipeth, L.; Kwak, H.R.; Choi, H.S.; Kim, K.H. Identification of viruses and viroids infecting tomato and pepper plants in Vietnam by metatranscriptomics. Int. J. Mol. Sci. 2020, 21, 7565. [Google Scholar] [CrossRef] [PubMed]
- Rivarez, M.P.S.; Vucurovic, A.; Mehle, N.; Ravnikar, M.; Kutnjak, D. Global advances in tomato virome research: Current status and the impact of High-Throughput Sequencing. Front. Microbiol. 2021, 12, 671925. [Google Scholar] [CrossRef] [PubMed]
- Mehetre, G.T.; Leo, V.V.; Singh, G.; Sorokan, A.; Maksimov, I.; Yadav, M.K.; Upadhyaya, K.; Hashem, A.; Alsaleh, A.N.; Dawoud, T.M.; Almaary, K.S.; Singh, B.P. Current developments and challenges in plant viral diagnostics: A systematic review. Viruses. 2021, 13, 412. [Google Scholar] [CrossRef]
- Roossinck, M.J. Metagenomics of plant and fungal viruses reveals an abundance of persistent lifestyles. Front. Microbiol. 2015, 5, 767. [Google Scholar] [CrossRef]
- Maree, H.J.; Fox, A.; Al Rwahnih, M.; Boonham, N.; Candresse, T. Application of HTS for routine plant virus diagnostics: State of the art and challenges. Front. Plant Sci. 2018, 9, 1082. [Google Scholar] [CrossRef] [PubMed]
- Blanca, J.; Montero-Pau, J.; Sauvage, C.; Bauchet, G.; Illa, E.; Díez, M.J.; Francis, D.; Causse, M.; van der Knaap, E.; Cañizares, J. Genomic variation in tomato, from wild ancestors to contemporary breeding accessions. BMC Genom. 2015, 16, 257. [Google Scholar] [CrossRef] [PubMed]
- Maldonado-Peralta, R.; Ramírez-Vallejo, P.; González Hernández, V.A.; Castillo-González, F. ; Sandoval-Villa, M, Livera-Muñoz, M. ; Cruz-Huerta, N. Agronomic wealth in Mexican collections of native tomatoes (Solanum lycopersicon L.). Agroproductividad. 2016, 9, 68–75. [Google Scholar]
- Martínez-Vázquez, E.A.; Hernández-Bautista, A.; Lobato-Ortiza, R.; García-Zavala, J.J.; Reyes-López, D. Exploring the breeding potential of Mexican tomato landraces. Sci. Hortic-Amsterdam 2017, 220, 317–325. [Google Scholar] [CrossRef]
- Marín-Montes, I.M.; Lobato-Ortiz, R.; Carrillo-Castañeda, G.; Rodríguez-Pérez, J.E.; García-Zavala, J.J.; Hernández-Rodríguez, M.; Velasco-García, A.M. Parámetros genéticos de una cruza interespecífica de S. lycopersicum L. y S. habrochaites Knapp & Spooner. Rev. Chapingo Ser. Hortic. 2020, 26, 111–123. [Google Scholar]
- Tavitas, F.L.; Hernández, A.L. Banco de germoplasma de jitomate (Lycopersicon sp. L) (Solanum lycopersicon L.) del Campo Experimental Zacatepec. Folleto técnico núm. 104, 1st ed.; INIFAP: Zacatepec, Morelos, México, 2017; pp. 1–30. [Google Scholar]
- Sastry, K.S. Seed-Borne Plant Virus Diseases, 1st ed.; Springer: New Delhi, India, 2013; pp. 1–145. [Google Scholar]
- Chiquito-Almanza, E.; Acosta-Gallegos, J.A.; García-Álvarez, N.C.; Garrido-Ramírez, E.R.; Montero-Tavera, V.; Guevara-Olvera, L.; Anaya-López, J.L. Simultaneous detection of both RNA and DNA viruses infecting dry bean and occurrence of mixed infections by BGYMV, BCMV and BCMNV in the Central-west region of Mexico. Viruses. 2017, 9, 63. [Google Scholar] [CrossRef] [PubMed]
- Kreuze, J.F.; Perez, A.; Untiveros, M.; Quispe, D.; Fuentes, S.; Barker, I. , Simon, R. Complete viral genome sequence and discovery of novel viruses by deep sequencing of small RNAs: a generic method for diagnosis, discovery and sequencing of viruses. Virology. 2009, 388, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Chiquito-Almanza, E.; Caballero-Pérez, J.; Acosta-Gallegos, J.A.; Montero-Tavera, V.; Mariscal-Amaro, L.A.; Anaya-López, J.L. Diversity and distribution of viruses infecting wild and domesticated Phaseolus spp. in the Mesoamerican Center of domestication. Viruses. 2021, 13, 1153. [Google Scholar] [CrossRef]
- Zheng, Y.; Gao, S.; Padmanabhan, C.; Li, R.; Galvez, M.; Gutierrez, D.; Fuentes, S.; Ling, K.S.; Kreuze, J.; Fei, Z. VirusDetect: An automated pipeline for efficient virus discovery using deep sequencing of small RNAs. Virology. 2017, 500, 130–138. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [PubMed]
- Abarshi, M.M.; Mohammed, I.U.; Wasswa, P.; Hillocks, R.J.; Holt, J.; Legg, J.P.; Seal, S.E.; Maruthi, M.N. Optimization of diagnostic RT-PCR protocols and sampling procedures for the reliable and cost-effective detection of Cassava brown streak virus. J. Virol. Methods. 2010, 163, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.Y.; Zhao, M.S.; Ma, H.Y.; Liu, L.Z.; Yang, G.L.; Geng, C.; Tian, Y.P.; Li, X.D. Biological and molecular characterization of tomato brown rugose fruit virus and development of quadruplex RT-PCR detection. J. Integr. Agr. 2021, 20, 1871–1879. [Google Scholar] [CrossRef]
- Nakhla, M.K.; Sorensen, A.; Maxwell, D.; Mejía, L.; Ramírez, P.; Karkashian, J.P. Molecular characterization of tomato-infecting begomoviruses in Central America and development of DNA-based detection methods. Acta Hortic. 2005, 695, 277–288. [Google Scholar] [CrossRef]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar]
- Alcalá Briseño, R.I.; Batuman, O.; Brawner, J.; Cuellar, W.J.; Delaquis, E.; Etherton, B.A.; Kreuze, J.F.; Navarrete, I.; Ogero, K.; Plex-Súla, A.I.; Yilmaz, S.; Garret, K.A. Translating virome analyses to support biosecurity, on-farm management, and crop breeding. Front. Plant Sci. 2023, 14, 1056603. [Google Scholar] [CrossRef] [PubMed]
- Nizamani, M.M.; Zhang, Q.; Muhae-Ud-Din, G.; Wang, Y. High-throughput sequencing in plant disease management: a comprehensive review of benefits, challenges, and future perspectives. Phytopathol. Res. 2023, 5, 1–17. [Google Scholar] [CrossRef]
- Alcalá-Briseño, R.I.; Casarrubias-Castillo, K.; López-Ley, D.; Garrett, K.A.; Silva-Rosales, L. Network analysis of the papaya orchard virome from two agroecological regions of Chiapas, Mexico. mSystems. 2020, 5, e00423-19. [Google Scholar] [CrossRef]
- Moubset, O.; Francois, S.; Maclot, F.; Palanga, E.; Julian, C.; Claude, L.; et al. Virion-associated nucleic acid-based metagenomics: A decade of advances in molecular characterization of plant viruses. Phytopathology. 2022, 112, 2253–2272. [Google Scholar] [CrossRef] [PubMed]
- Bester, R.; Cook, G.; Breytenbach, J.H.J.; Steyn, C.; De Bruyn, R.; Maree, H.J. Towards the validation of high-throughput sequencing (HTS) for routine plant virus diagnostics: Measurement of variation linked to HTS detection of citrus viruses and viroids. Virol. J. 2021, 18, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Di Gaspero, G.; Radovic, S.; De Luca, E.; Spadotto, A.; Magris, G.; Falginella, L.; Cattonaro, F.; Marroni, F. Evaluation of sensitivity and specificity in RNA-Seq-based detection of grapevine viral pathogens. J. Virol. Methods. 2022, 300, 114383. [Google Scholar] [CrossRef] [PubMed]
- Rong, W.; Rollin, J.; Hanafi, M.; Roux, N.; Massart, S. Validation of high-throughput sequencing as virus indexing test for Musa germplasm: Performance criteria evaluation and contamination monitoring using an alien control. PhytoFrontiers. 2023, 3, 91–102. [Google Scholar] [CrossRef]
- Kutnjak, D.; Tamisier, L.; Adams, I.; Boonham, N.; Candresse, T.; Chiumenti, M.; et al. A primer on the analysis of high-throughput sequencing data for detection of plant viruses. Microorganisms. 2021, 9, 841. [Google Scholar] [CrossRef] [PubMed]
- Seguin, J.; Rajeswaran, R.; Malpica-López, N.; Martin, R.R.; Kasschau, K.; Dolja, V.V.; Otten, P.; Farinelli, L.; Pooggin, M.M. De novo reconstruction of consensus master genomes of plant RNA and DNA viruses from siRNAs. PLoS ONE. 2014, 9, e88513. [Google Scholar] [CrossRef]
- Kutnjak, D.; Rupar, M.; Gutierrez-Aguirre, I.; Curk, T.; Kreuze, J.F.; Ravnikar, M. Deep sequencing of virus-derived small interfering RNAs and RNA from viral particles shows highly similar mutational landscapes of a plant virus population. J. Virol. 2015, 89, 4760–4769. [Google Scholar] [CrossRef] [PubMed]
- Pecman, A.; Kutnjak, D.; Gutiérrez-Aguirre, I.; Adams, I.; Fox, A.; Boonham, N.; Ravnikar, M. Next generation sequencing for detection and discovery of plant viruses and viroids: Comparison of two approaches. Front. Microbiol. 2017, 8, 1998. [Google Scholar] [CrossRef] [PubMed]
- Galipienso, L.; Elvira-González, L.; Velasco, L.; Herrera-Vásquez, J.Á.; Rubio, L. Detection of persistent viruses by high-throughput sequencing in tomato and pepper from Panama: Phylogenetic and evolutionary studies. Plants. 2021, 10, 2295. [Google Scholar] [CrossRef]
- Botella, L.; Jung, T. Multiple viral infections detected in Phytophthora condilina by total and small RNA sequencing. Viruses. 2021, 13, 620. [Google Scholar] [CrossRef]
- Gauthier, M.E.A.; Lelwala, R.V.; Elliott, C.E.; Windell, C.; Fiorito, S.; Dinsdale, A.; Whattam, M.; Pattemore, J.; Barrero, R.A. Side-by-side comparison of post-entry quarantine and high throughput sequencing methods for virus and viroid diagnosis. Biology. 2022, 11, 263. [Google Scholar] [CrossRef] [PubMed]
- Villamor, D.E.V.; Ho, T.; Al Rwahnih, M.; Martin, R.R.; Tzanetakis, I.E. High throughput sequencing for plant virus detection and discovery. Phytopathology. 2019, 109, 716–725. [Google Scholar] [CrossRef] [PubMed]
- Chiquito-Almanza, E.; Zamora-Aboytes, J.M.; Medina, H.R.; Acosta-Gallegos, J.A.; Anaya-López, J.L. Complete genome sequence of a novel comovirus infecting common bean. Arch. Virol. 2020, 165, 1505–1509. [Google Scholar] [CrossRef] [PubMed]
- Chiquito-Almanza, E.; Acosta-Gallegos, J.A.; Anaya-López, J.L. Complete genome sequence of a novel polerovirus infecting chickpea (Cicer arietinum L.). Arch. Virol. 2022, 167, 2783–2788. [Google Scholar] [CrossRef] [PubMed]
- Massart, S.; Chiumenti, M.; De Jonghe, K.; Glover, R.; Haegeman, A.; Koloniuk, I.; et al. Virus detection by high-throughput sequencing of small RNAs: Large-scale performance testing of sequence analysis strategies. Phytopathology. 2019, 109, 488–497. [Google Scholar] [CrossRef] [PubMed]
- Singhal, P.; Nabi, S.U.; Yadav, M.K.; Dubey, A. Mixed infection of plant viruses: diagnostics, interactions and impact on host. J. Plant Dis. Protect. 2021, 128, 353–368. [Google Scholar] [CrossRef]
- Moreno, A.B.; López-Moya, J.J. When viruses play team sports: Mixed infections in plants. J. Phytopathol. 2020, 2020. 110, 29–48. [Google Scholar] [CrossRef]
- Mascia, T.; Gallitelli, D. Synergies and antagonisms in virus interactions. Plant Sci. 2016, 252, 176–192. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, J.C.; Ding, T.B.; Chu, D. Synergistic effects of a Tomato chlorosis virus and Tomato yellow leaf curl virus mixed infection on host tomato plants and the whitefly vector. Front. Plant Sci. 2021, 12, 672400. [Google Scholar] [CrossRef]
- García-Cano, E.; Resende, R.O.; Fernández-Muñoz, R.; Moriones, E. Synergistic interaction between Tomato chlorosis virus and Tomato spotted wilt virus results in breakdown of resistance in tomato. Phytopathology. 2006, 96, 1263–1269. [Google Scholar] [CrossRef]
- Alvez-Junior, M.; Alfenas-Zerbini, P.; Andrade, E.C.; Esposito, D.A.; Silva, F.N.; da Cruz, A.C.F.; Ventrella, M.C.; Otoni, W.C.; Zerbini, F.M. Synergism and negative interference during co-infection of tomato and Nicotiana benthamiana with two bipartite begomoviruses. Virology. 2009, 387, 257–266. [Google Scholar] [CrossRef]
- Potapov, S.; Gorshkova, A.; Krasnopeev, A.; Podlesnaya, G.; Tikhonova, I.; Suslova, M.; Kwon, D.; Patrushev, M.; Drucker, V.; Belykh, O. RNA-Seq virus fraction in lake Baikal and treated wastewaters. Int. J. Mol. Sci. 2023, 24, 12049. [Google Scholar] [CrossRef] [PubMed]
- Broadbent, L. Epidemiology and control of tomato mosaic virus. Annu. Rev. Phytopathol. 1976, 14, 75–96. [Google Scholar] [CrossRef]
- Pares, R.D.; Gunn, L.V.; Cresswell, G.C. Tomato mosaic virus infection in a recirculating nutrient solution. J. Phytopathol. 1992, 135, 192–198. [Google Scholar] [CrossRef]
- Hamamoto, H.; Watanabe, Y.; Kamada, H.; Okada, Y. Amino acid changes in the putative replicase of tomato mosaic tobamovirus that overcome resistance in Tm-1 tomato. J. Gen. Virol. 1997, 78, 461–464. [Google Scholar] [CrossRef] [PubMed]
- Strasser, M.; Pfitzner, A.J.P. The double-resistance-breaking Tomato mosaic virus strain ToMV1-2 contains two independent single resistance-breaking domains. Arch. Virol. 2007, 152, 903–914. [Google Scholar] [CrossRef] [PubMed]
- Weber, H.; Schultze, S.; Pfitzner, A.J. Two amino acid substitutions in the tomato mosaic virus 30-kilodalton movement protein confer the ability to overcome the Tm-22 resistance gene in the tomato. J. Virol. 1993, 67, 6432–6438. [Google Scholar] [CrossRef]
- Kuroiwa, M.; Handa, S.; Gyoutoku, Y.; Moriyama, M.; Neriya, Y.; Nishigawa, H.; Natsuaki, T. Characterization of a ToMV isolate overcoming Tm-22 resistance gene in tomato. Virus Genes. 2022, 58, 478–482. [Google Scholar] [CrossRef] [PubMed]
- Xue, C.Y.; Tao, X.R.; Zhou, X.P. Coat protein gene and 3’ noncoding region of Tobacco mosaic virus and Tomato mosaic virus are associated with viral pathogenesis in Nicotiana tabacum. Prog. Nat. Sci. 2002, 12, 679–683. [Google Scholar]
- van Belkum, A.; Abrahams, J.P.; Pleij, C.W.; Bosch, L. Five pseudoknots are present at the 204 nucleotides long 3′ noncoding region of tobacco mosaic virus RNA. Nucleic Acids Res. 1985, 13, 7673–7686. [Google Scholar] [CrossRef]
- Osman, T.A.M.; Buck, K.W. Identification of a region of the tobacco mosaic virus 126- and 183-kilodalton replication proteins which binds specifically to the viral 3′ -terminal tRNA-like structure. J. Virol. 2003, 77, 8669–8675. [Google Scholar] [CrossRef] [PubMed]
- Zeenko, V.V.; Ryabova, L.A.; Spirin, A.S.; Rothnie, H.M.; Hess, D.; Browning, K.S.; Hohn, T. ; Eukaryotic elongation factor 1A interacts with the upstream pseudoknot domain in the 3′ untranslated region of tobacco mosaic virus RNA. J. Virol. 2002, 76, 5678–5691. [Google Scholar] [CrossRef]
- Takamatsu, N.; Watanabe, Y.; Meshi, T.; Okada, Y. Mutational analysis of the pseudoknot region in the 3′ noncoding region of tobacco mosaic virus RNA. J. Virol. 1990, 64, 3686–3693. [Google Scholar] [CrossRef] [PubMed]
- Takamatsu, N.; Watanabe, Y.; Iwasaki, T.; Shiba, T.; Meshi, T.; Okada, Y. Deletion analysis of the 5′ untranslated leader sequence of tobacco mosaic virus RNA. J. Virol. 1991, 65, 1619–1622. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Kierzek, E.; Chen, G.; Zhou, Y.J.; Wong, S.M. TMV mutants with poly (A) tracts of different lengths demonstrate structural variations in 3′ UTR affecting viral RNAs accumulation and symptom expression. Sci. Rep-UK. 2015, 5, 18412. [Google Scholar] [CrossRef] [PubMed]
- Lyu, J.; Yang, Y.; Sun, X.; Jiang, S.; Hong, H.; Zhu, X.; Liu, Y. Genetic variability and molecular evolution of Tomato Mosaic Virus populations in three Northern China provinces. Viruses. 2023, 15, 1617. [Google Scholar] [CrossRef]
- Torres-Pacheco, I.; Garzón-Tiznado, J.A.; Brown, J.K.; Becerra-Flora, A.; Rivera-Bustamante, R.F. Detection and distribution of geminiviruses in Mexico and Southern United States. Phytopathology. 1996, 86, 1186–1192. [Google Scholar] [CrossRef]
- Mauricio-Castillo, J.A.; Argüello-Astorga, G.R.; Ambriz-Granados, S.; Alpuche-Solís, A.G.; Monreal-Vargas, C.T. First report of Tomato Golden Mottle Virus on Lycopersicon esculentum and Solanum rostratum in Mexico. Plant Dis. 2007, 91, 1513. [Google Scholar] [CrossRef]
- Idris, A.M.; Lee, S.H.; Brown, J.K. First report of Chino del tomate and Pepper hausteco geminiviruses in greenhouse-grown tomato in Sonora, Mexico. Plant Dis. 1999, 83, 396–396. [Google Scholar] [CrossRef]
- Sanchez-Chavez, S.; Regla-Marquez, C.F.; Cardenas-Conejo, Z.E.; Garcia-Rodriguez, D.A.; Centeno-Leija, S.; Serrano-Posada, H.; Liñan-Rico, A.; Partida-Palacios, B.L.; Cardenas-Conejo, Y. First report of begomoviruses infecting Cucumis sativus L. in North America and identification of a proposed new begomovirus species. PeerJ. 2020, 8, e9245. [Google Scholar] [CrossRef]
- Moreno-Félix, M.L.; Rodríguez-Negrete, E.A.; Meléndrez-Bojórquez, N.; Camacho-Beltrán, E.; Leyva-López, N.E.; Méndez-Lozano, J. A new isolate of Pepper huasteco yellow vein virus (PHYVV) breaks geminivirus tolerance in tomato (Solanum lycopersicum) commercial lines. Acta Hortic. 2018, 1207, 35–44. [Google Scholar] [CrossRef]
- Anaya-López, J.L.; Torres-Pacheco, I.; González-Chavira, M.; Garzon-Tiznado, J.A.; Pons-Hernandez, J.L.; Guevara-González, R.G.; Muñoz-Sánchez, C.I.; Guevara-Olvera, L.; Rivera-Bustamante, R.F.; Hernández-Verdugo, S. Resistance to geminivirus mixed infections in Mexican wild peppers. HortScience. 2003, 38, 251–255. [Google Scholar] [CrossRef]
- Barboza, N.; Hernández, E.; Inoue-Nagata, A.K.; Moriones, E.; Hilje, L. Achievements in the epidemiology of begomoviruses and their vector Bemisia tabaci in Costa Rica. Rev. Biol. Trop. 2019, 67, 419–453. [Google Scholar]
- Kumar, S.; Srivastava, A.; Kumari, A.; Raj, R.; Jaidi, M.; Raj, S.K. Begomovirus disease management measures, now and then. In Begomoviruses: Occurrence and management in Asia and Africa, 1st ed.; Saxena, S., Tiwari, A.K., Eds.; Eds.; Springer: Singapore, 2017; Volume 1, pp. 71–92. [Google Scholar]
- Lefkowitz, E.J.; Dempsey, D.M.; Hendrickson, R.C.; Orton, R.J.; Siddell, S.G.; Smith, D.B. Virus taxonomy: the database of the International Committee on Taxonomy of Viruses (ICTV). Nucleic Acids Res. 2018, 46, D708–D717. [Google Scholar] [CrossRef]
- Park, W.M.; Lee, G.P.; Ryu, K.H.; Park, K.W. Transmission of tobacco mosaic virus in recirculating hydroponic system. Sci. Hortic-Amsterdam. 1999, 79, 217–226. [Google Scholar] [CrossRef]
- Panthee, D.R.; Brown, A.F.; Yousef, G.G.; Ibrahem, R.; Anderson, C. Novel molecular marker associated with Tm2a gene conferring resistance to tomato mosaic virus in tomato. Plant. Breeding. 2013, 132, 413–416. [Google Scholar] [CrossRef]
- Ullah, N.; Ali, A.S.A.D.; Ahmad, M.; Fahim, M.; Din, N.; Ahmad, F. Evaluation of tomato genotypes against tomato mosaic virus (ToMV) and its effect on yield contributing parameters. Pak. J. Bot. 2017, 49, 1585–1592. [Google Scholar]
- Ullah, N.; Akhtar, K.P.; Saleem, M.Y.; Habib, M. Characterization of tomato mosaic virus and search for its resistance in Solanum species. Eur. J. Plant Pathol. 2019, 155, 1195–1209. [Google Scholar] [CrossRef]
- Dombrovsky, A.; Smith, E. Seed transmission of Tobamoviruses: Aspects of global disease distribution. Adv. Seed Biol. 2017, 12, 233–260. [Google Scholar]
- Panno, S.; Davino, S.; Caruso, A.G.; Bertacca, S.; Crnogorac, A.; Mandić, A.; Noris, E.; Matić, S. A review of the most common and economically important diseases that undermine the cultivation of tomato crop in the Mediterranean basin. Agronomy. 2021, 11, 2188. [Google Scholar] [CrossRef]
- Shi, A.; Vierling, R.; Grazzini, R.; Chen, P.; Caton, H.; Panthee, D. Molecular markers for Tm-2 alleles of Tomato mosaic virus resistance in tomato. Am. J. Plant Sci. 2011, 2, 180–189. [Google Scholar] [CrossRef]
- Shahriari, Z.; Su, X.; Zheng, K.; Zhang, Z. Advances and prospects of virus-resistant breeding in tomatoes. Int. J. Mol. Sci. 2023, 24, 15448. [Google Scholar] [CrossRef] [PubMed]
- Young, N.D.; Zamir, D.; Ganal, M.W.; Tanksley, S.D. Use of isogenic lines and simultaneous probing to identify DNA markers tightly linked to the Tm-2a gene in tomato. Genetics. 1988, 120, 579–585. [Google Scholar] [CrossRef] [PubMed]
- Rezk, A.; Abhary, M.; Akhkha, A. Tomato (Solanum lycopersicum L.) breeding strategies for biotic and abiotic stresses. In Advances in Plant Breeding Strategies: Vegetable Crops, 1st ed.; Al-Khayri, J.M., Jain, S.M., Johnson, D.V., Eds.; Springer Nature: Switzerland, 2021; Volume 9, pp. 363–405. [Google Scholar]
- García-Estrada, R.S.; Díaz-Lara, A.; Aguilar-Molina, V.H.; Tovar-Pedraza, J.M. Viruses of economic impact on tomato crops in Mexico: From diagnosis to management-A review. Viruses. 2022, 14, 1251. [Google Scholar] [CrossRef]
- Weber, H.; Ohnesorge, S.; Silber, M.V.; Pfitzner, A.J.P. The Tomato mosaic virus 30 kDa movement protein interacts differentially with the resistance genes Tm-2 and Tm-2 2. Arch. Virol. 2004, 2004. 149, 1499–1514. [Google Scholar] [CrossRef]
Figure 1.
Phylogenetic relationships of tomato mosaic virus Mexican isolates INIFAP JM1-3 with global isolates. The phylogenetic tree was constructed using the complete genome sequences of 57 Tomato mosaic virus isolates, including the Mexican isolates INIFAP JM1-3 shown in green color. The tree was inferred using the Maximum-Likelihood method (TN93 + G + I model) with 1,000 bootstrap replicates. A Tobacco mosaic virus isolate was included as an outgroup. Nodes with bootstrap support less than 70% are collapsed.
Figure 1.
Phylogenetic relationships of tomato mosaic virus Mexican isolates INIFAP JM1-3 with global isolates. The phylogenetic tree was constructed using the complete genome sequences of 57 Tomato mosaic virus isolates, including the Mexican isolates INIFAP JM1-3 shown in green color. The tree was inferred using the Maximum-Likelihood method (TN93 + G + I model) with 1,000 bootstrap replicates. A Tobacco mosaic virus isolate was included as an outgroup. Nodes with bootstrap support less than 70% are collapsed.
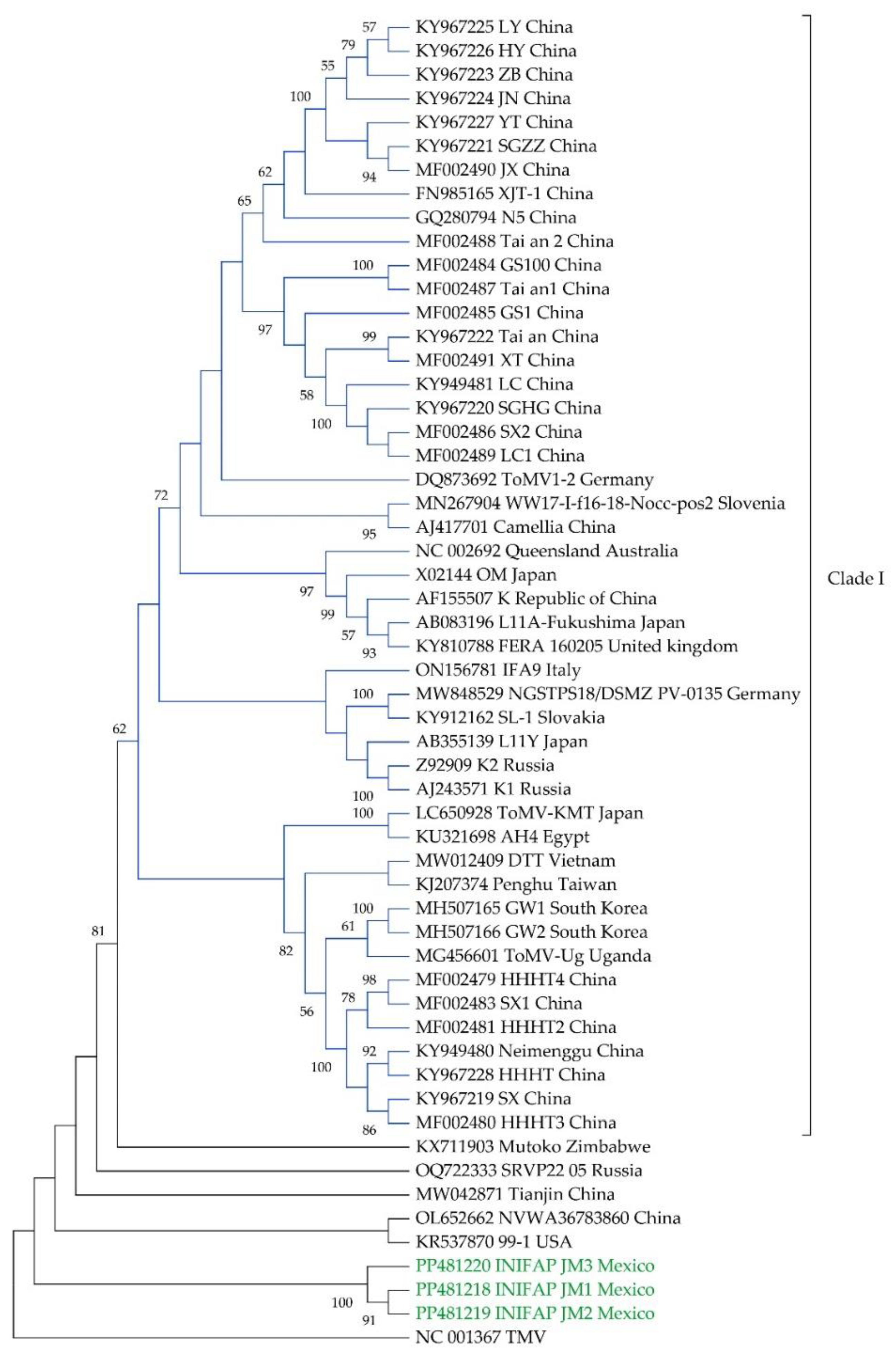

Table 1.
sRNA composite libraries from advanced tomato lines of native tomatoes from southeastern Mexico and virus validation by PCR/RT-PCR.
Table 1.
sRNA composite libraries from advanced tomato lines of native tomatoes from southeastern Mexico and virus validation by PCR/RT-PCR.
| Sample/ Library a |
Tomato lines | Fruit shape | Municipality | State | Samples | PCR or RT-PCR confirmation | ||
|---|---|---|---|---|---|---|---|---|
| ToGMoV | PHYVV | ToMV | ||||||
| JM1/JM1Lc | JCM02 | Riñón | Huachinango | Puebla | JCM02-1 | + | - | + |
| JCM02-7 | - | - | + | |||||
| JCM05 | Cherry | Xoxocotla | Morelos | JCM05-1 | - | - | + | |
| JCM05-2 | - | - | + | |||||
| JCM10 | Riñón | Huachinango | Puebla | JCM10-2 | - | - | + | |
| JCM10-7 | + | - | + | |||||
| JCM11 | Riñón | Zitlala | Puebla | JCM11-2 | + | - | + | |
| JCM11-4 | + | - | + | |||||
| JCM14 | Chino criollo | Altepexi | Puebla | JCM14-1 | - | - | + | |
| JCM15 | JCM15-2 | - | - | + | ||||
| JCM16 | Chino criollo | Zinacatepec | Puebla | JCM16-4 | - | - | + | |
| JCM16-6 | - | - | + | |||||
| JCM17 | Chino criollo | Miahuatlán | Puebla | JCM17-3 | - | - | + | |
| JCM18 | Chino criollo | Tehuacán | Puebla | JCM18-4 | + | - | + | |
| JCM19 | Chino criollo | Zinacatepec | Puebla | JCM19-1 | - | - | + | |
| JM2/JM2Lc | JCM03 | Riñón | Tlacolula | Oaxaca | JCM03-1 | - | - | + |
| JCM03-5 | - | - | + | |||||
| JCM03-11 | - | - | + | |||||
| JCM04 | Riñón | Poza Rica | Veracruz | JCM04-2 | - | - | + | |
| JCM04-9 | - | - | + | |||||
| JCM04-13 | + | - | + | |||||
| JCM06 | Cherry | Tlacolula | Oaxaca | JCM06-1 | + | - | + | |
| JCM09 | Medio saladette | Tlacolula | Oaxaca | JCM09-1 | + | - | + | |
| JCM09-8 | + | - | + | |||||
| JCM12 | Riñón | Zozocolco | Veracruz | JCM12-2 | + | - | + | |
| JM3/JM3Lc | JCM01 | Riñón | Dzitbalché | Campeche | JCM01-6 | - | - | + |
| JCM01-7 | - | - | + | |||||
| JCM01-9 | - | - | + | |||||
| JCM01-11 | - | - | + | |||||
| JCM07 | Riñón | Teapa | Tabasco | JCM07-1 | - | - | + | |
| JCM07-5 | - | + | + | |||||
| JCM07-11 | - | - | + | |||||
| JCM08 | Medio riñón | Dzitbalché | Campeche | JCM08-2 | + | - | + | |
| JCM08-3 | + | - | + | |||||
| JCM08-4 | + | - | + | |||||
| JCM20 | Riñón | Dzitbalché | Campeche | JCM20-2 | - | - | + | |
a: Composite sample and library names. +: Detected. -: Not detected.
Table 2.
Identity of viral contigs assembled via high-throughput sequencing of sRNA from composite samples JM1, JM2, and JM3.
Table 2.
Identity of viral contigs assembled via high-throughput sequencing of sRNA from composite samples JM1, JM2, and JM3.
| Virus species | Reference accession a | Genome length (pb) |
Contig coverage (%) | Number of contigs | Sequence identity (%) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| JM1 | JM2 | JM3 | JM1 | JM2 | JM3 | JM1 | JM2 | JM3 | |||
| ToGMoV DNA-A b | EF501976 | 2615 | 99.4 | 98.8 | 99.7 | 3 | 5 | 3 | 98.1 | 97.5 | 98.4 |
| ToGMoV DNA-B b | DQ406674 | 2558 | 90.9 | 94.5 | 91.9 | 7 | 11 | 7 | 90.7 | 90.2 | 90.0 |
| PHYVV DNA-A b | LN848879 | 2630 | - | - | 96.8 | - | - | 10 | - | - | 93.3 |
| PHYVV DNA-B b | LN848915 | 2595 | - | - | 99.3 | - | - | 2 | - | - | 96.8 |
| ToMV c | KR537870 | 6383 | 100 | 100 | 99 | 1 | 1 | 1 | 99.4 | 99.5 | 99.3 |
a: GenBank accession number of the viral genome reference sequence used for identification. b: Genus Begomovirus. c: Genus Tobamovirus. -: Not detected.
Table 3.
Recombination events in complete nucleotide sequences of ToMV isolates detected by RDP4.
| Event | Position | Recombinant | Major parent | Minor Parent | Detection methods a |
|---|---|---|---|---|---|
| 1 | 6214-6442 | NVWA36783860 (OL652662) | 99-1 (KR537870) | Camellia (AJ417701) | R, G, B, M, C, S, 3S |
| 2 | 4045-6442 | Tai'an 2 (MF002488) |
Unknown | GS100 (MF002484) | G, M, C, S, 3S |
| 3 b | 6126-6442 | SRVP22_05 (OQ722333) | AH4 (KU321698) | Unknown | R, G, B, M, C, 3S |
| 4 | 3138-5570 | N5 (GQ280794) |
XJT-1 (FN985165) | (AF155507) c | M, C, S, 3S |
a R: RDP, G: GENECONV, B: BootScan, M: MaxChi, C: Chimera, S: SiScan, 3S: 3Seq. b Event found also in isolates INIFAP JM1 (PP481218), INIFAP JM2 (PP481219), INIFAP JM3 (PP481220), 99-1 (KR537870), and Tianjin (MW042871). c Attenuated mutant strain.
Table 4.
Mutation rate of the isolates INIFAP JM1, JM2 and JM3.
| Genomic Region (Number of nt/aa) | Isolates | Nucleotide (nt) | Amino Acid (aa) | ||
|---|---|---|---|---|---|
| Mutations | Mutation Rate (%) | Mutations | Mutation Rate (%) | ||
| 5' UTR (49-73 nt) a | INIFAP JM1 and JM2 | 1 | 1.39 | - | - |
| INIFAP JM3 | 2 | 2.74 | - | - | |
| SRVP22_05 | 0 | 0.00 | - | - | |
| 126 KDa (3351 nt/1116 aa) | INIFAP JM1 and JM2 | 34 | 1.01 | 1 | 0.09 |
| INIFAP JM3 | 32 | 0.95 | 1 | 0.09 | |
| SRVP22_05 | 19 | 0.57 | 0 | 0.00 | |
| 183 KDa (4851 nt/1616 aa) | INIFAP JM1 and JM2 | 47 | 0.97 | 1 | 0.06 |
| INIFAP JM3 | 45 | 0.93 | 1 | 0.06 | |
| SRVP22_05 | 23 | 0.47 | 0 | 0.00 | |
| MP (795 nt/264 aa) | INIFAP JM1 and JM2 | 8 | 1.01 | 2 | 0.76 |
| INIFAP JM3 | 8 | 1.01 | 2 | 0.76 | |
| SRVP22_05 | 4 | 0.50 | 2 | 0.76 | |
| CP (480 nt/159 aa) | INIFAP JM1 and JM2 | 3 | 0.63 | 0 | 0.00 |
| INIFAP JM3 | 3 | 0.63 | 0 | 0.00 | |
| SRVP22_05 | 2 | 0.42 | 0 | 0.00 | |
| 3' UTR (201-264 nt) b | INIFAP JM1 and JM2 | 16 | 6.06 | - | - |
| INIFAP JM3 | 16 | 6.06 | - | - | |
| SRVP22_05 | 11 | 4.80 | - | - | |
The number of mutations and mutation rates were determined by removing alignment sites containing gaps. a The 5' UTR nucleotide lengths of the Queensland, SRVP22_05, INIFAP JM1, JM2, and JM3 isolates are 71, 49, 72, 72, and 73 nucleotides, respectively. b The 3' UTR nucleotide lengths of the Queensland, SRVP22_05, INIFAP JM1, JM2, and JM3 isolates are 201, 229, 264, 264, and 264 nucleotides, respectively. GenBank accession numbers: Queensland (NC_002692); INIFAP JM1 (PP481218); INIFAP JM2 (PP481219); INIFAP JM3 (PP481220); SRVP22_05 (OQ722333).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



