
Submitted:
26 April 2024
Posted:
29 April 2024
You are already at the latest version
Abstract
Background: Across the world, antibiotic resistance is a critical challenge. To address such a problem, it has to be done to investigate new antibiotic sources. Aim of the study: Study of the antibacterial activity of Corallopyronin A in preclinical animal testing and randomized human clinical trials stages 1/2, as well as investigating the purification of Corallopyronin A from different soil conditions in Egypt. Type of the study: Screening experimental study. Methodology: Egypt's various soil conditions were tested to develop bacterial isolates that produced the antibiotic compound Corallopyronin A. Reversed phase HPLC was used to purify Myxopyronin A. The broth microdilution method and the paper disc diffusion assay determined the test antibiotic's minimum inhibitory concentration( MIC) and in vitro antibacterial activity. Moreover, in vivo antibacterial spectrum, adverse medication responses, and pharmacokinetics were found in phases 1/2 of randomized clinical studies involving humans and animal models. Results: Corallopyronin A was generated from the culture supernatant of the soil bacterial isolate Corallococcus coralloides M2, cultivated on a Casein yeast peptone( CYP) plate. The test antibiotic prevented the growth of several Gram-ve bacteria, including Escherichia coli, at MICs higher than 100 mcg/ ml; while blocking the development of many Gram +ve bacteria, with MICs ranging from 3 to 15 mcg/ ml. However, eukaryotic cells—such as those found in fungi and humans—were unaffected. A bactericidal consequence of the test antibiotic was detected in the inhibition of prokaryotic DNA-dependent RNA polymerase( RNLP). Conclusion: The synthesis of the bactericidal antibiotic Corallopyronin A from Corallococcus coralloides M2 isolated from different soil settings in Egypt intrigued the current investigation.
Keywords:
Corallopyronin A
; infection
; antibiotic
; resistance
; myxobacteria
Introduction
For an antibiotic to be useful as a medicine, it must exhibit selective toxicity [1]. The activities of bacteria must be significantly suppressed compared to those of human cells [2]. The search for novel antibiotic sources is essential because antibiotic resistance is a serious, global issue that has to be addressed [3]. Cell walls, ribosomes, cell membranes, and nucleic acids are the four main targets of antibacterial drugs [4]. These drugs have no impact on human cells since they don't have a cell wall and have unique ribosomes, nucleic acid enzymes, and sterols in their membranes [5]. drugs known to be bactericidal are effective against bacteria [6]. Conversely, bacteriostatic drugs prevent the growth of germs [7]. The pathogen is eliminated by the patient's phagocytes while using bacteriostatic drugs [8].
Patients with low neutrophil counts should be treated with bacteriocidal medicines [9]. One class of alpha-pyrone antibiotics is Corallopyronins [10]. Pyranones or pyrones are a class of heterocyclic chemical compounds [11]. They have an unsaturated six-membered ring with one oxygen atom and a ketone functional group [12]. The two isomers are called 4-pyrone and 2-pyrone [13]. Corallopyronins may be able to assist in addressing the growing problem of drug resistance in tuberculosis as they do not cross-resistance with any other medication [14]. Therapy for Methicillin-resistant Staphylococcus aureus( MRSA) may also be beneficial [15]. The destination of the contemporary study was to assess how a novel antibiotic called Corallopyronin A was synthesized in various soil conditions in Egypt. Antibacterial activity was also investigated in phase 1/2 randomized human clinical investigations.
Patients and Methods
Ethical statement:
Prioritized in the current study were all pertinent institutional, national, and/or international standards concerning the use and care of humans and animals. All study protocols involving humans and animals were authorized by the Ethical Committee for Human and Animal Handling at Cairo University( ECAHCU), located at the Faculty of Pharmacy, Cairo University, Egypt, by the Weatherall Report's recommendations (approval number T-9-4-2022). At all costs, the study's human and animal subjects' numbers and suffering were kept to a minimum. Phase 1/2 registration number for the randomized human clinical trials was NCT00000714/ 2022.
Type of the study:
Screening experimental study.
Place and date of the study:
The study was completed at Cairo University's pharmacy faculty in Egypt between April 2022 and February 2024.
Source of animal models:
The department of pharmacology and toxicology at Cairo University's college of pharmacy provided animal models, which were deemed acceptable.
Inclusion criteria for animal models:
Animal models of adult male, obese rabbits weighing approximately 2 kg are available for Inoculation against several bacterial diseases. Before the trial, the rabbits were allowed to acclimatize for one week. At 50% ± 5% humidity, a 12-hour light-dark cycle, and a regulated temperature of 25± 2°C. Fresh grass was given to the bunnies to eat.
Exclusion criteria for animal models:
Young and female rabbits; Non-obese rabbits weighing less than 2 kg.
Collection of 100 soil samples:
The samples were randomly selected grassland soils that were taken from various soil settings in Egypt at a depth of 30 cm. Before being processed, samples were kept at 4 ℃ in sterile containers. Each soil sample was weighed out at one gramme, and each 250 ml Erlenmeyer flask had 99 ml of sterile distilled water. The flasks were shaken at 400 rpm for five minutes using a gyrator shaker. Following dilutions from 10-1 to 10-6 in sterile distilled water, the soil suspensions were plated on selective Casein yeast peptone agar medium (bought from Sigma-Aldrich, USA).
50 cc of nutrient broth liquid at PH 7 was added to 250 ml Erlenmeyer flasks to create the inoculum for the bacterial isolate under investigation. The medium was autoclaved and then infected with a loopful of culture from a nutritional agar slant that had been left overnight. The inoculum was the inoculated flasks, which were shaken for a whole day at 150 rpm.
Instruments

Table 1.
List of instruments:.
| Instrument | Model and manufacturer |
| Autoclaves | Tomy, japan |
| Aerobic incubator | Sanyo, Japan |
| Digital balance | Mettler Toledo, Switzerland |
| Oven | Binder, Germany |
| Deep freezer -70 0C | Artiko |
| Refrigerator 5 | whirpool |
| PH meter electrode | Mettler-toledo,UK |
| Deep freezer -20 0C | whirlpool |
| Gyrator shaker | Corning gyratory shaker, Japan |
| 190-1100nm Ultraviolet visible spectrophotometer | UV1600PC, China |
| Light(optical) microscope | Amscope 120X-1200X, China |
Material:
The suppliers of all chemical and biological materials were the Egyptian companies Alnasr Chemical Company and Algomhuria Pharmaceutical Company. Analytical grade chemical reagents were utilised in all cases.
Isolation of Corallococcus coralloides M2 producing Corallopyronin antibiotics:
The selective isolation of species of Corallococcus coralloides M2 from different soil samples was directly achieved using dilution plating. The technique comprised the suppression of competing bacteria exploiting antibiotics such as 10 mcg/ ml Vancomycin and/ or 10 mcg/ ml Chloramphenicol combined with wet heat treatment of soils and air drying. Fungi were eliminated via supplementing the plating medium with 2 mcg/ ml Terbinafine HCl. Swarming of Corallococcus coralloides M2 colonies was controlled with Casein Yeast Peptone( CYP) plates incubated at 30℃ and PH 7.2 for 5 days. The constitution of CYP plate included 0.4 % Peptone from Casein, tryptically digested, 0.3 % CaCl2.2H2O, 0.1 % MgSO4.7H2O, PH 7.2. The potent bacterial isolate producing Myxopyronin was performed utilizing 16 S rRNA sequencing technique. The predominant bacterial isolate with high antibacterial activity was identified using 16S rRNA sequencing and other biochemical tests. Nucleic acid was extracted from a swab by bead-beating in a buffered solution containing Phenol, Chloroform and Isoamyl alcohol. Variable region of 16S rRNA gene was then amplified from the resulting nucleic acid using PCR. The genomic DNA was extracted from 120 hours cultured cells using a DNA purification kit[ PurreLinkTM Genomic DNA Mini Kit with Catalog number: K182002 was purchased from Invitrogen, USA] according to the protocol provided by the manufacturer of DNA purification kit. The 16S rRNA gene was amplified by PCR[ PCR SuperMix kit was purchased from Invitrogen,USA] using forward[ 5-AGAGTTTGATCCTGGCTCAG-3-] and reverse[ 5-GGTTACCTTGTTACGACTT-3-] primers. PCR amplicons from up to hundreds of samples were then combined and sequenced on a single run. The resulting sequences were matched to a reference database to determine relative bacterial abundances. Polymerase Chain Reaction ( PCR) was a powerful method for amplifying particular segments of DNA. PCR used the enzyme PlatinumTM Taq DNA polymerase with catalog number 10966018[ purchased from Invitrogen, USA] that directed the synthesis of DNA from deoxynucleotide substrates on a single-stranded DNA template. DNA polymerase added nucleotides to the 3` end of a custom-designed oligonucleotide when it was annealed to a longer template DNA. Thus, if a synthetic oligonucleotide was annealed to a single-stranded template that contained a region complementary to the oligonucleotide, DNA polymerase could use the oligonucleotide as a primer and elongate its 3` end to generate an extended region of double stranded DNA. Denaturation was the initial PCR cycle stage The DNA template was heated to 94° C. This broke down the weak hydrogen bonds that held DNA strands together in a helix, allowing the strands to separate creating single stranded DNA. Annealing was the second PCR cycle. The mixture was cooled to anywhere from 50-70° C. This allowed the primers to bind (anneal) to their complementary sequence in the template DNA. Extension was the final step of PCR cycle. The reaction was ; then heated up to 72° C, the optimal temperature for DNA polymerase to act. DNA polymerase extended the primers, adding nucleotides onto the primer in a sequential manner, using the target DNA as a template. With one cycle, a single segment of double-stranded DNA template was amplified into two separate pieces of double-stranded DNA. These two pieces were then available for amplification in the next cycle. As the cycles were repeated, more and more copies were generated and the number of copies of the template was increased exponentially. The amplified PCR product was sequenced using a genetic analyzer 3130XL[ purchased from Applied biosystems, USA]. DNA sequence homology search analysis of the predominant bacterial isolate was achieved using Blastn algorithm at NCBI website. Fruiting bodies were examined using a Stereomicroscope( dissecting microscope) MSC-ST45T( purchased from Infetik, China). Wet mounts from crushed fruiting bodies were prepared. The refaractility, shape and the size of Myxospores were determined victimizing phase contrast microscopy. On the other hand the plates were exposed to 360 nm wavelength ultraviolet light to assess the fruiting bodies fluoresced [16].
Identification Myxopyronin A producing bacterial isolates:
Gram stain:
It classified bacteria into two categories based on the makeup of their cell walls. The bacterial cells became purple after being treated with a solution of crystal violet and subsequently iodine on a microscope slide. When colored cells were treated with a solvent such as alcohol or acetone, gram-positive organisms kept the stain whereas gram-negative organisms lost the stain and turned colorless. With the addition of the counter-stain safranin, the clear, gram-negative bacteria became pink [17].
Spore shape:
This was discovered using the spore staining method. To get rid of any fingerprints, the slide was wiped with alcohol and a Kim-wipe. On the bottom of the slide, a Sharpie was used to create two circles. Each circle was filled with two tiny droplets of water using an inoculation loop. A very small amount of germs was taken out of the culture tube using an aseptic method. The water droplet on the slide had microorganisms on it. The slide was thoroughly dried by air. Bypassing the slide through the flame three to four times with the smear side up, the slide was heat-fixed. It took a while for the slide to completely cool. A piece of paper towel placed inside the slide's border was used to hide the streaks. A beaker of heating water was situated over the slide. The slide was allowed to steam for three to five minutes; while the paper towel was covered with a malachite green liquid. Removed and thrown away was the discolored paper towel. To get rid of any stray paper towel bits, the slide was gently cleaned with water. The counter-stain was safranin for 1 minute. Before putting the slide on the microscope's stage and seeing it via the oil immersion lens, the slide's bottom was dried [18].
Spore site:
During the Gram stain test, the spore location was established [19].
Cell shape:
During the Gram stain test, the cell shape was assessed [20].
Blood haemolysis:
On blood agar media, the test antibiotic capacity to haemolyze the blood was tested [21].
Motility test:
It discriminated between motile bacteria and non-motile bacteria.
A sterile needle was used to penetrate the medium to within 1 cm of the tube's bottom to select a well-isolated colony and test for motility. The needle was certainly retained in the same position as it was inserted and removed from the medium. It took 18 hours of incubation at 35°C, or until noticeable growth appeared [22].
Nitrate reduction test:
0.5 ml of nitrate broth was added in a clean test tube, was autoclaved for 15 minutes at 15 lbs pressure and 121°C, and was let to cool to room temperature. The tube was inoculated with a heavy inoculum of fresh bacterial culture and was incubated at 35°C for 2 hours. 2 drops of reagent A and 2 drops of reagent B were added and mixed well. The development of red color within 2 minutes was observed for. If no red color was developed, a small amount of zinc dust was added and observed for the development of the red color within 5 minutes [23].
Methyl red test:
In the Methyl Red test, an infected tube of MR broth was used before adding the methyl red PH indicator. The buffers in the medium were overcome by the acids when an organism used the mixed acid fermentation pathway and produced stable acidic end products, resulting in an acidic environment [24].
Catalase test:
A little inoculum of a specific bacterial strain was introduced to a 3% hydrogen peroxide solution to see if it might produce catalase. It was observed for the rapid emergence of oxygen bubbles [25].
Oxidase test:
The 1% Kovács oxidase reagent was applied to a tiny piece of filter paper, which was then allowed to air dry. A well-isolated colony was taken from a fresh (18 to 24-hour culture) bacterial plate using a sterile loop, and it was then rubbed onto prepared filter paper. Color alterations were noticed [26].
Citrate utilization:
Five milliliters of a Simmon Koser's citrate medium were taken after it had been autoclaved at 15 pounds for 15 minutes. To create a clear slant and butt, the test tube containing melted citrate medium was slanted. Using sterilized wire and labeled tubes, the specified samples of microbe were injected on the media's incline. For 24 hours, the tubes were incubated at 37°C. The medium's color shift was watched for [27].
Starch hydrolysis:
For 48 hours at 37°C, the bacterium plates were injected. After incubation, a dropper was used to saturate the surface of the plates with an iodine solution for 30 seconds. Iodine that was in excess was afterward poured out. The area surrounding the bacterial growth line was looked at [28].
Tween 80 hydrolysis:
1% Tween 80 was used to create agar media. The supplied microorganism was added to the Tween 80 agar plates by utilizing an inoculating loop to create a single center streak in the plate. The plates were incubated for 24 hours at 37 °C. HgCl2 solution was poured over the plates. After a short while, the plates were examined. Positive test result; distinct halo-zone surrounding the injected region showed Tween 80 hydrolysis [29].
Growth at 10-45 0C:
On nutrient agar media, growth was observed to be possible at 45°C [30].
Indol test:
The test tube containing the microorganism for inoculation received 5 drops of the Kovács reagent directly. Within seconds after introducing the reagent to the media, the reagent layer formed a pink to red colour (cherry-red ring), which was a sign of a positive indol test [31].
Tolerance salinity test:
Its capacity to develop on nutrient agar while being responsive to 5% and 7 % NaCl was examined [32].
Voges-Proskauer(VP) test:
For the test, Voges-Proskauer broth, a glucose-phosphate broth loaded with microorganisms, was added to alpha-naphthol and potassium hydroxide. A successful outcome was indicated by a cherry red tint, whereas an unfortunate outcome was indicated by a yellow-brown color [33].
Casein hydrolysis test:
For testing the casein hydrolyzing activity of the test antibiotic, a single line streak of the given culture was made in the center of the skim milk agar plate under aseptic conditions and plate was incubated at 37°C in an incubator for 24-48 h [34].
Saccharide fermentation tests:
Glucose fermentation test:
The fermentation reactions of glucose were investigated using glucose purple broth. Peptone and the PH indicator bromcresol purple made up the purple broth. A 1% concentration of glucose was added. Isolated colonies from a 24-hour pure culture of microorganisms were added to the glucose purple broth as an inoculant. Parallel to the inoculation of the glucose-based medium, a control tube of purple broth base was used. The inoculated medium was incubated aerobically for 3 days at a temperature of 35–37 °C. The medium began to become yellow, which was a sign of a successful outcome. A poor carbohydrate fermentation response was indicated by the lack of yellow color development [35].
Fructose fermentation test:
A pure culture's inoculum was aseptically transferred to a sterile tube of phenol red fructose broth. The infected tube was incubated for 18–24 hours at 35–37 °C. A color shift from red to yellow, signifying an acidic PH alteration, was a sign of a favorable response [36].
Maltose fermentation test:
A pure culture inoculum was aseptically transferred to a sterile tube containing phenol red maltose broth. The infected tube was incubated for 18–24 hours at 35–37 °C. A color shift from red to yellow, signifying an acidic PH alteration, was a sign of a favorable response [37].
Sucrose fermentation test:
A pure culture's inoculum was aseptically transferred to a sterile tube containing phenol red sucrose broth. For 24 hours, the infected tube was incubated at 35–37 0C. A colour shift from red to yellow, signifying an acidic PH alteration, was a sign of a favourable response [38].
Purification of Corallopyronin A antibiotic:
This was achieved through reversed phase chromatography technique.
The aeration rate was 0.142 V/ V. min. The stirring rate was 500 rpm. PO2 was about 90 % of saturation; but decreased to about 20 % after 18 hours). The fermentation was stopped after 40 hours via centrifugation at 500 rpm in a gyrator shaker. The supernatants were collected; then tested for antimicrobial sensitivity using broth dilution technique to detect MICs and agar paper diffusion discs technique. The test antibiotic was extracted from the 2 liters of culture broth with 2/ 10 volume ethyl acetate. The ethyl acetate was then removed under the reduced pressure at 40 °C. Afterwards, the residue was dissolved in 398 ml of methanol-water( 90: 10) and chromatographed on reversed phase HPLC. Methanol was the mobile phase. The eluent was 70 part methanol: 16 part water: 4 part acetic acid with flow rate 300 ml/ min. Detection of the antibiotic components was achieved exploiting refractive index. The main peak with retention time 5 minutes contained the biological antibiotic activity which was determined via agar diffusion assay using paper discs and Staphylococcus aureus as an indicator organism. On the other hand, the main peak was subjected to neutralization via NaHCO3. Corallopyronin A was extracted using 10 % V/ V Methylene chloride. After the evaporation of the solvent, about 90 % of the antibiotic substance purified was Corallopyronin A. It was noticed that the retention rime of Corallopyronin A was 9 minutes. Molecular formula of the purified Corallopyronin A was detected through mass spectrometer( Quadrupole mass spectrometer, Advion, USA) [39]. It was detected also, that 10% of Corallopyronin mixture extract were 7% Corallopyronin B and 3% Corallopyronin C.
Procedure of Broth dilution assay for determination of MICs of Corallopyronin A:
A specific broth was added to several microtiter plates during the testing process based on the requirements of the target bacterium. The test microorganisms and antibiotics were then introduced to the plate in varying amounts. After that, the plate was put into a non-CO2 incubator and left there for sixteen to twenty hours at 37 degrees Celsius. The plate was taken out and examined for bacterial growth after the specified amount of time had passed. Bacterial growth was detected in the cloudiness of the broth. The lowest concentration of antibiotics that prevented bacterial growth, or Minimum Inhibitory Concentration( MIC), was used to describe the outcomes of the broth microdilution method [40].
Agar diffusion assay with paper discs procedure for the determination of Corallopyronin A antimicrobial activity:
The agar diffusion technique( ADM) was used to classify the disc diffusion method( DDM) because the test microorganism-seeded agar media allowed the test antibiotic extract to disperse from its reservoir. A filter paper disc put on an agar surface served as the reservoir most of the time. After the filter paper disc was incubated, an inhibitory zone formed around the tested extract chemicals that were microbiologically active [41]. The test extract's antibacterial potency was accurately reflected by the inhibition zone's diameter [42]. Both broth and selection or enrichment growing media were used to isolate the test microorganisms (Table 2).
Estimation of Corallopyronin A effect on bacterial RNA synthesis:
The concentration of RNA isolated with RNeasy Kits( purchased from QIAGEN, USA) was determined by measuring the absorbance at 260 nm in a spectrophotometer. An absorbance of 1 unit at 260 nm corresponds to 40 µg of RNA per ml( A260 = 1 = 40 µg/ ml) [42].
Estimation of Corallopyronin A effect on bacterial protein synthesis:
Absorbance was measured at 205 nm to calculate the protein concentration by comparison with a standard curve. A( 205) method could be used to quantify total protein in crude lysates and purified or partially purified protein. The UV spectrophotometer was set to read at 205 nm allowing 15 min for the instrument to equilibrate. The absorbance reading was set to zero with a solution of the buffer and all components except the protein present. The protein solution was placed in the 1 ml cuvette and the absorbance was determined. The dilution and readings of samples were performed in duplicate.The matched cuvettes for samples and controls were utilized during the test procedure. The extinction coefficient of the protein was known, the following equation was employed. Absorbance = Extinction coefficient × concentration of protein × path length( 1 cm) to determine the concentration of the protein [43].
Estimation of pharmacodynamic and pharmacokinetic effects of Corallopyronin A during experimental animal testing in preclinical clinical trials:
In the present study, the pharmacokinetics and the pharmacodynamics of Corallopyronin A were evaluated after dosing in male rabbit animal models weighing about 2 kg. Furthermore, compound concentrations were determined in target compartments, such as lung, kidney and thigh tissue, using LC-MS/ MS. Based on the pharmacokinetic results, the pharmacodynamic profile of Corallopyronin A was assessed victimizing the standard neutropenic thigh and lung infection models [44].
Estimation of pharmacodynamic and pharmacokinetic effects of Corallopyronin A in randomized human clinical trials phases 1/2:
This study was conducted in 150 human volunteer subjects to show the bioavailability, pharmacokinetics and the pharmacodynamics of the test antibiotic. The study was designed as randomized, single-dose, 2-treatment, 2-period crossover trial with a washout period of 1 week. Blood samples were collected at 0( baseline), 10, 20, and 40 minutes and at 1, 1.5, 2, 3, 4, 6, 9, 12, and 24 hours postdose. Plasma concentrations of the 4 drugs were measured by using a rapid chromatography-tandem mass spectrometry method. Pharmacokinetic parameters were calculated by using noncompartmental methods. Bioequivalence was determined if the 90 % CIs of the log-transformed test/ reference ratios AUC( 0-24), AUC( 0-∞), and Cmax were within the predetermined range of 80% to 125%. Tolerability was assessed by using clinical parameters and subject reports Pharmacodynamic effects were evaluated through the determination of MICs via agar diffusion assay and broth dilution technique During randomized human clinical trials phases 1/2 all utilized infectious bacterial cell counts were estimated spectrophotometrically [45].
Estimation of of phototoxicity, mutagenicity and carcinogenicity of the test antibiotic:
The phototoxicity was determined via 3T3 neutral red uptake phototoxicity technique [46]. On the other hand, mutagenicity and carcinogenicity of the test antibiotic were assessed using Ames test [47].
The determination of toxokinetic and toxodynamic effects:
Up and down method for acute toxicity detection of Corallopyronin A was utilized for this purpose.[48]
The determination of maximum bactericidal activity of Corallopyronin A:
A pure culture of a specified microorganism was grown overnight, then diluted in growth-supporting broth( typically Mueller Hinton Broth) to a concentration between 1 x 10^5 and 1 x 10^6 cfu/ ml. A stock dilution of the antimicrobial test substance was made at approximately 100 times the expected MIC. Further 1:1 dilutions were made in test tubes. All dilutions of the test antibiotic were inoculated with equal volumes of the specified microorganism. A positive and negative control tube was included for every test microorganism to demonstrate adequate microbial growth over the course of the incubation period and media sterility, respectively. An aliquot of the positive control was plated and used to establish a baseline concentration of the microorganism used.The tubes were then incubated at the appropriate temperature and duration. Turbidity indicated growth of the microorganism and the MIC was the lowest concentration where no growth was visually observed. To determine the MBC, the dilution representing the MIC and at least two of the more concentrated test product dilutions were plated and enumerated to determine viable CFU/ ml. The MBC was the lowest concentration that demonstrated a pre-determined reduction (such as 99.9%) in CFU/ ml when compared to the MIC dilution [49].
Determination of plasma protein binding capacity of Corallopyronin A:
Victimizing of an ultrafiltration technique, the protein binding( PB) extent and changeability of the test antibiotic medicates were settled when given simultaneously to 30 patients inoculated with infectious pneumococci inside hospitals in Egypt. Clinical samples used were routinely received by microbiological laboratory inside the faculty of Pharmacy, Cairo University, Egypt. Plasma proteins were likewise plumbed. A protein-free medium was used to determine the nonspecific binding. Plasma samples from 30 patients were enclosed, of which plasma proteins were deliberated for 24 patients.
Determination of liver, kidney and heart function tests:
These functional tests were performed to assess the vitality of liver, kidney and heart during the randomized human clinical trials phases 1/2. On the other hand, Urine, stool analyses were achieved in addition to estimation of complete blood counts to all experimental subjects which received graded doses of Corallopyronin A.
Formulation of Corallopyronin A( COR A):
A liquid solution( COR A > 30 mg ml−1) comprising polyethylenglycol-15-hydroxystearate( 35%), propylene glycol( 15%), and phosphate buffered saline pH 7.3( 75%), as excipients, was prepared for IV and SC administration. PEG 400( 50%) and phosphate buffered saline PH 7.3( 60%) were added to a liquid formulation that included COR A for human effectuality attempts administered by oral and SC methods. For toxicity tests, a liquid COR A formulation based on PEG 200 that permitted an oral dosage of 1500 mg kg−1( 150 mg ml−1) was created. Each formulation exhibited adequate COR A in-use stability.
Figure 1.
It demonstrates the structure of Corallopyronin A extracted from bacterial isolates Corallococcus coralloides M2 collected from different soil environments in Egypt . Molecular formula of the purified test antibiotic was noticed to be C30H41NO7 determined through mass spectrometer.
Figure 1.
It demonstrates the structure of Corallopyronin A extracted from bacterial isolates Corallococcus coralloides M2 collected from different soil environments in Egypt . Molecular formula of the purified test antibiotic was noticed to be C30H41NO7 determined through mass spectrometer.
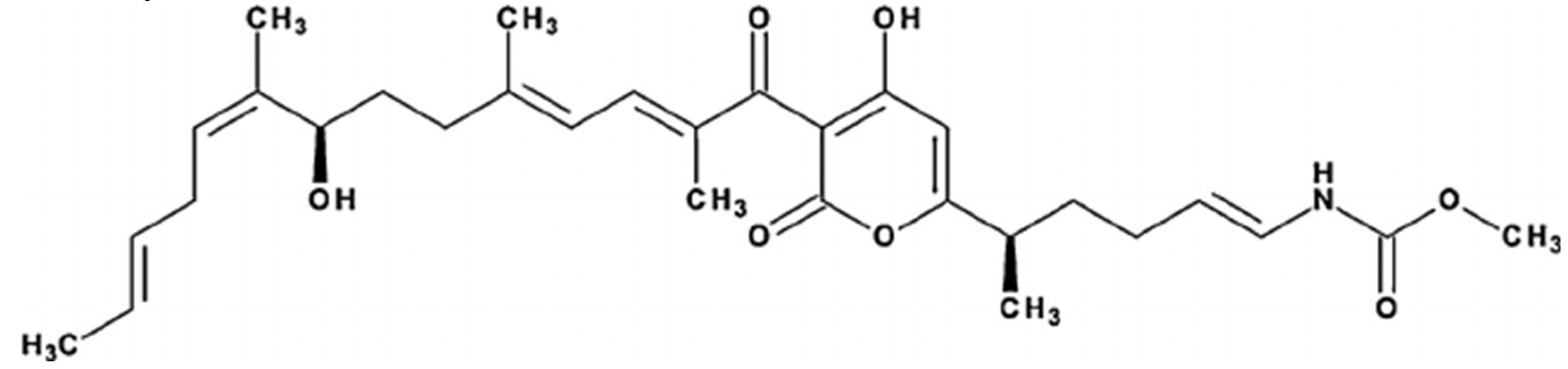
Figure 2.
It represents docking of Corallopyronin A ligand on Bacterial RNA polymerase. Corallopyronin A showed high affinity and inhibitory effect towards the switch region of RNA Polymerase.
Figure 2.
It represents docking of Corallopyronin A ligand on Bacterial RNA polymerase. Corallopyronin A showed high affinity and inhibitory effect towards the switch region of RNA Polymerase.
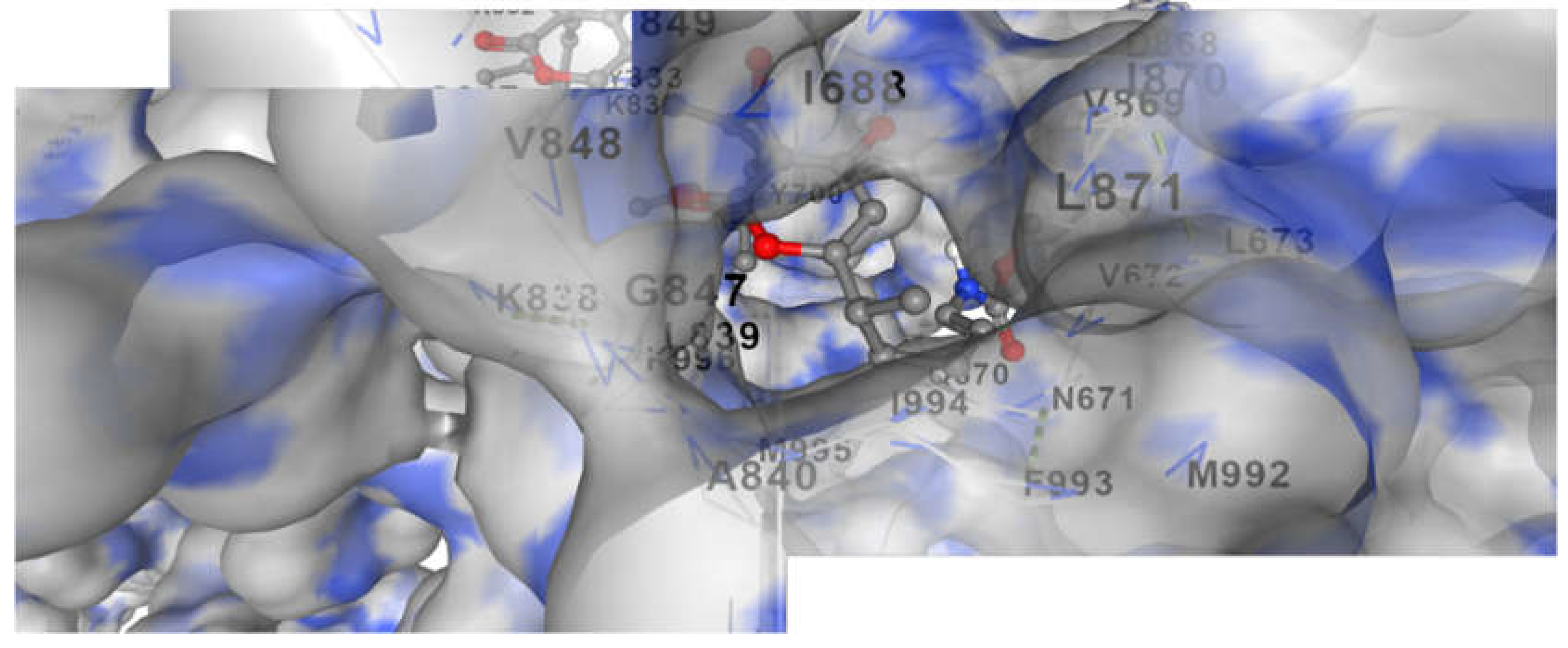
Figure 3.
It demonstrates 3D structure of bacterial prokaryotic RNA polymerase.
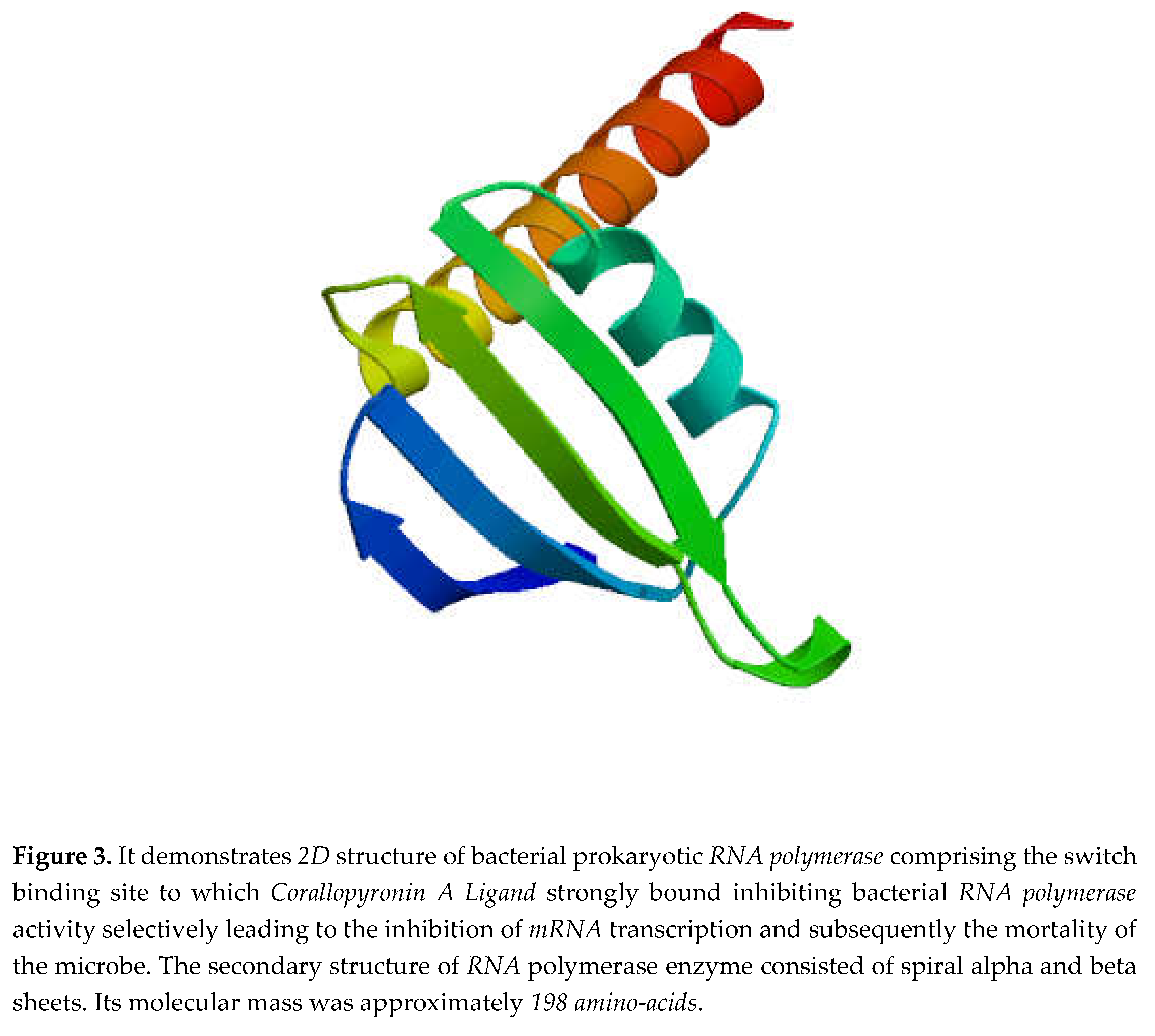
Figure 4.
It shows the impact of various concentrations of Soluble starch on the production of Corallopyronin A.
Figure 4.
It shows the impact of various concentrations of Soluble starch on the production of Corallopyronin A.
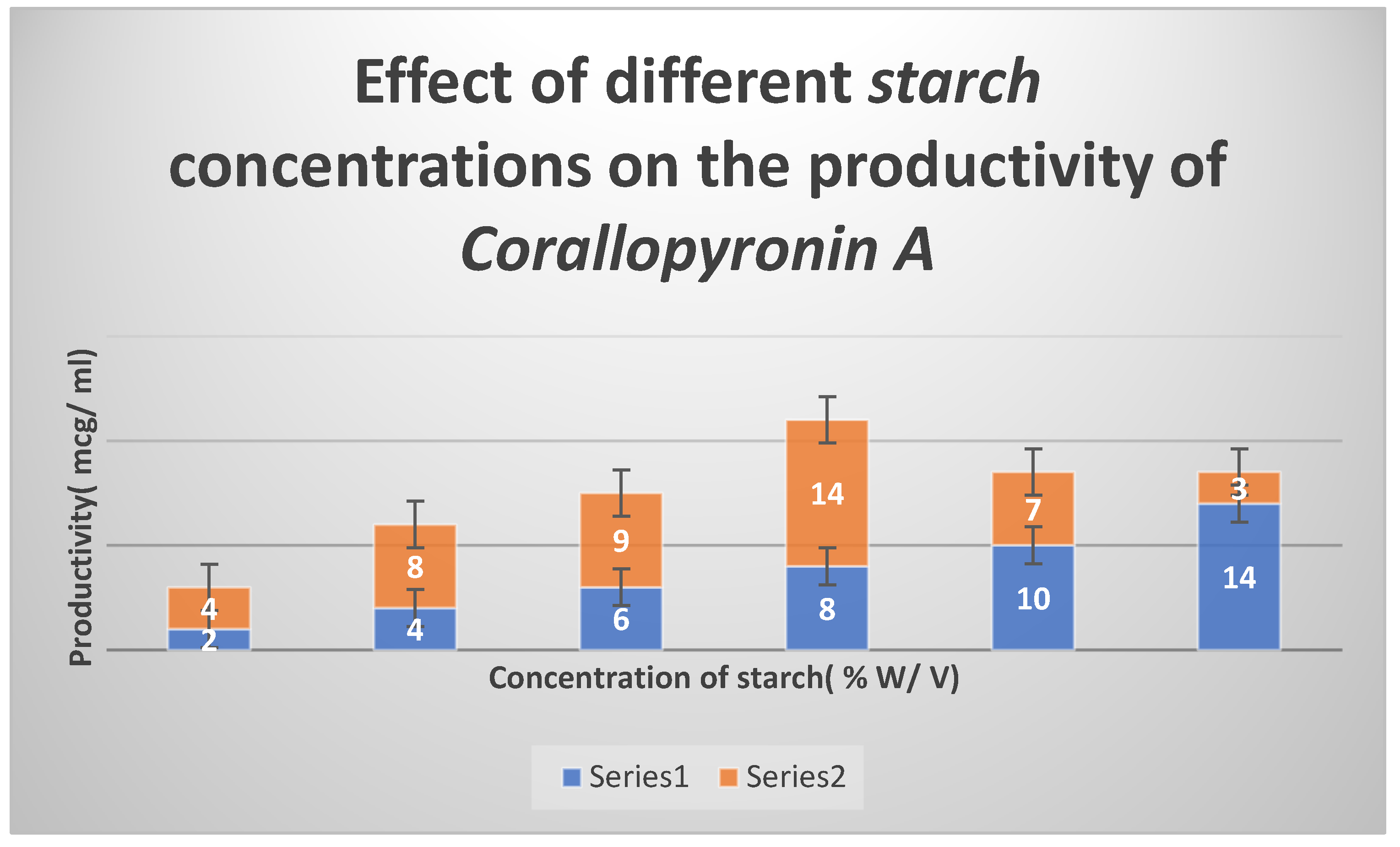
Figure 5.
It shows the effects of different Peptone concentrations as nitrogen growth factor on the productivity of Corallopyronin A.
Figure 5.
It shows the effects of different Peptone concentrations as nitrogen growth factor on the productivity of Corallopyronin A.
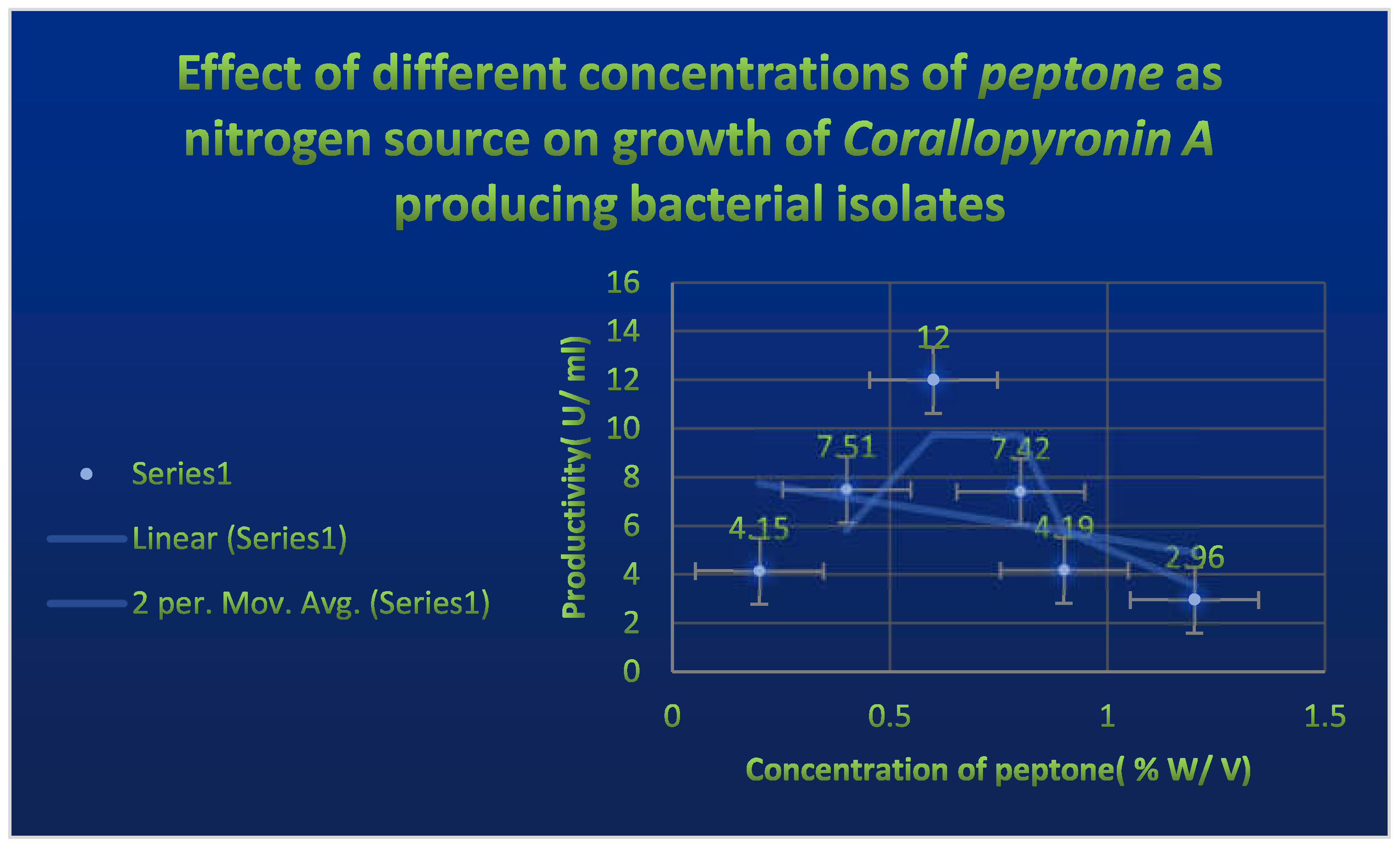
Figure 6.
It refers to the estimation of effect of Corallopyronin A on microbial mRNA productivity. mRNA synthesis was detected to be diminished proportionately up on employment of exploding doses of Myxopyronin A antibiotic.
Figure 6.
It refers to the estimation of effect of Corallopyronin A on microbial mRNA productivity. mRNA synthesis was detected to be diminished proportionately up on employment of exploding doses of Myxopyronin A antibiotic.
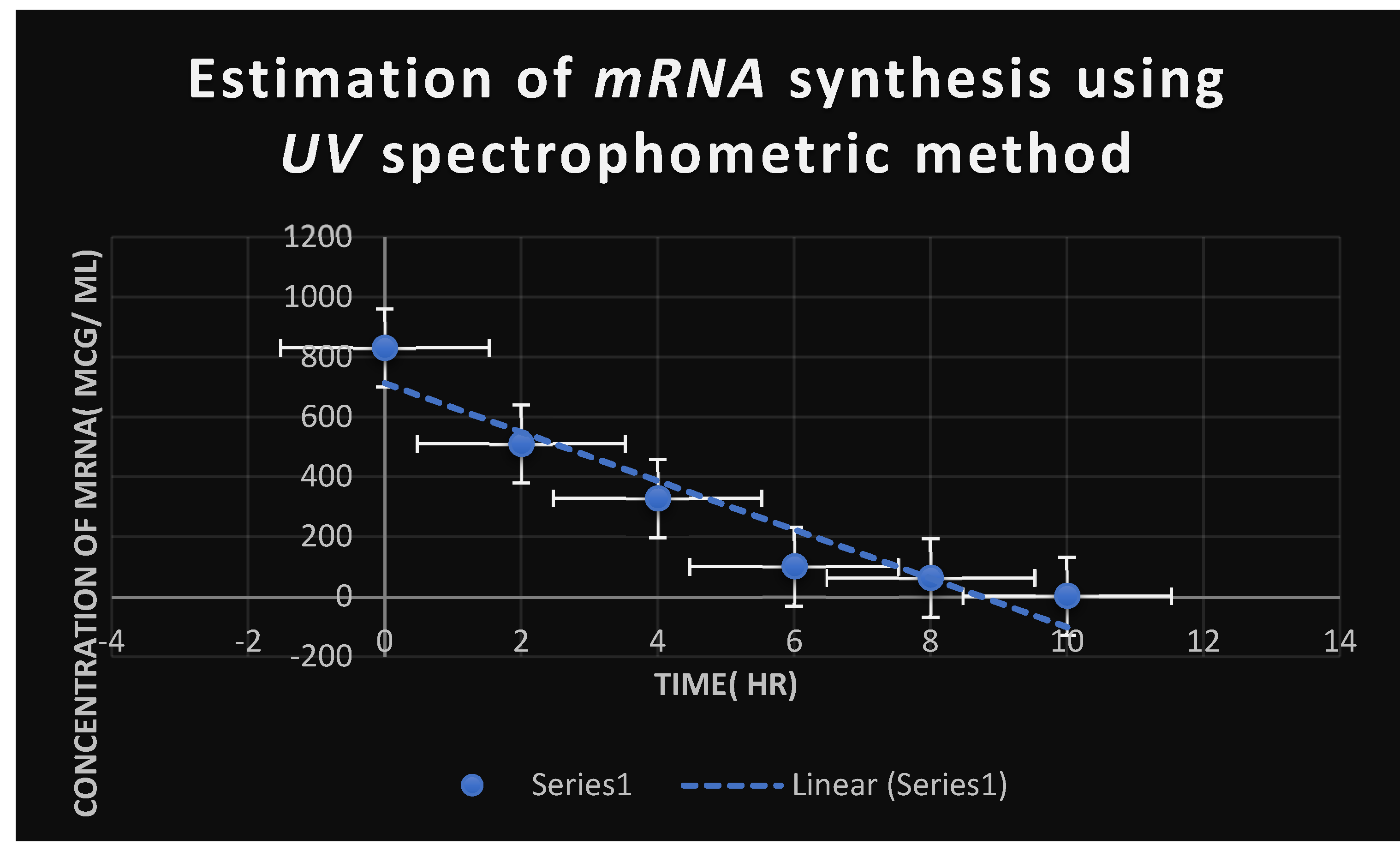
Figure 7.
It demonstrates the influence of Corallopyronin A on protein synthesis using UV spectrophotometer absorption at 205 nm. Protein synthesis was noticed to be decreased dramatically up on utilization of increasing doses of Myxopyronin A antibiotic.
Figure 7.
It demonstrates the influence of Corallopyronin A on protein synthesis using UV spectrophotometer absorption at 205 nm. Protein synthesis was noticed to be decreased dramatically up on utilization of increasing doses of Myxopyronin A antibiotic.
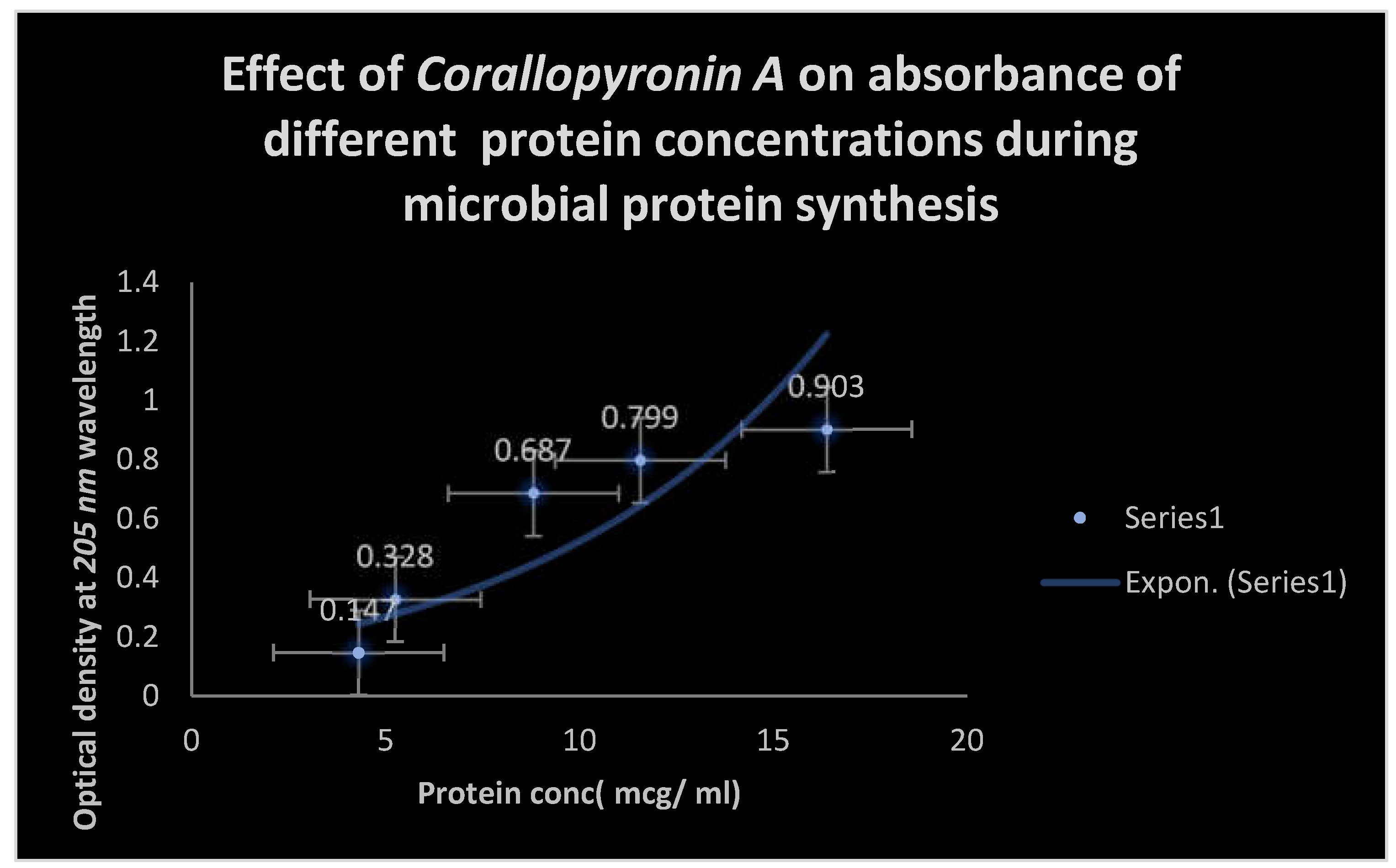
Figure 8.
It shows AUC of Corallopyronin A following SC administration in randomized human clinical trials stages 1/2. Efficacious dose ranged from 4-5 mg/ kg of body weight. Onset of action was observed following closely 20 minutes. It followed first order of elimination kinetics.
Figure 8.
It shows AUC of Corallopyronin A following SC administration in randomized human clinical trials stages 1/2. Efficacious dose ranged from 4-5 mg/ kg of body weight. Onset of action was observed following closely 20 minutes. It followed first order of elimination kinetics.
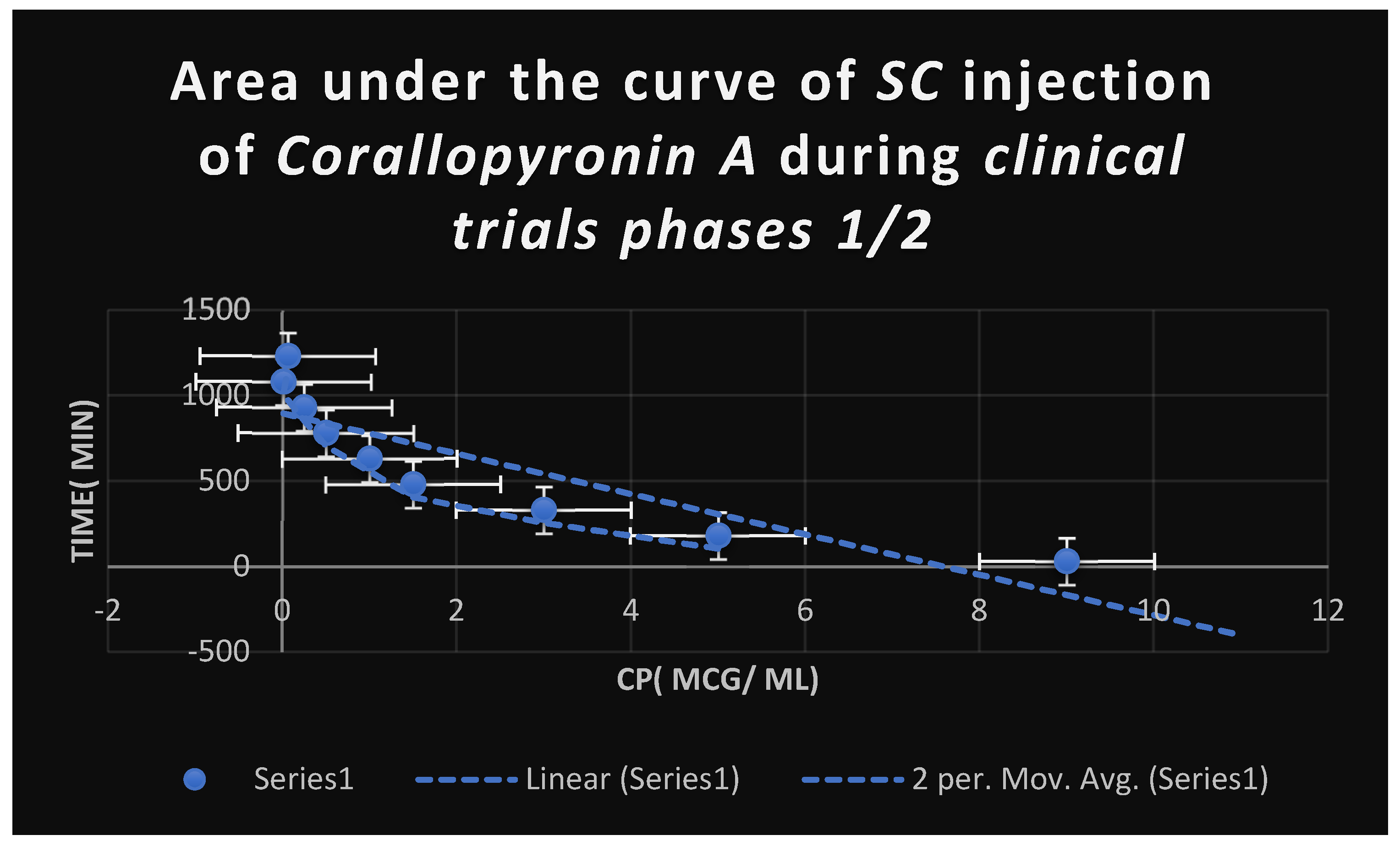
Figure 9.
Area under the curve( AUC) following oral administration of Corallopyronin A during clinical trials phases 1/2. Efficacious dose ranged from 9-10 mg/ kg of body weight. Onset of action was observed following nearly 25 minutes. It followed first order of elimination kinetics.
Figure 9.
Area under the curve( AUC) following oral administration of Corallopyronin A during clinical trials phases 1/2. Efficacious dose ranged from 9-10 mg/ kg of body weight. Onset of action was observed following nearly 25 minutes. It followed first order of elimination kinetics.
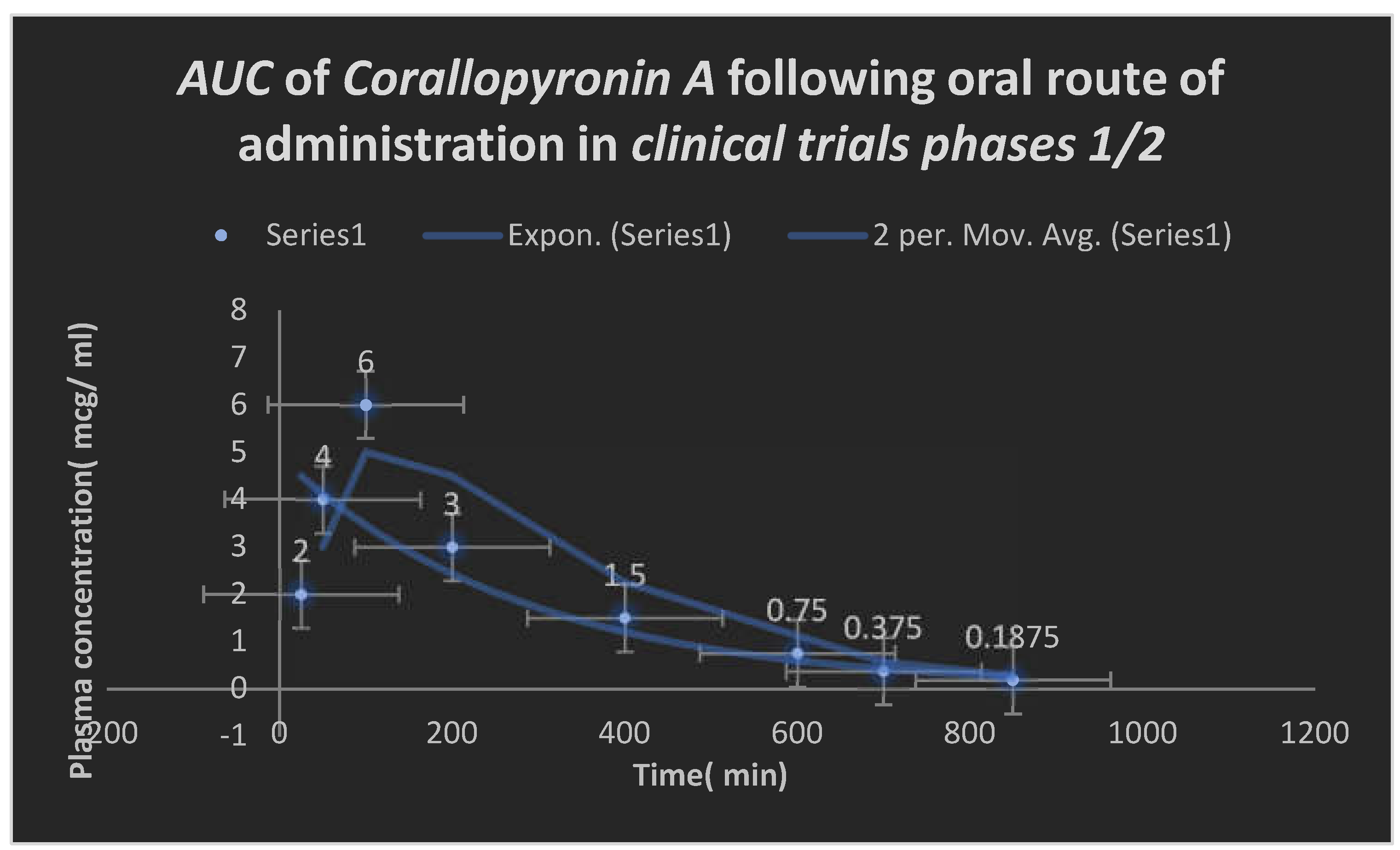
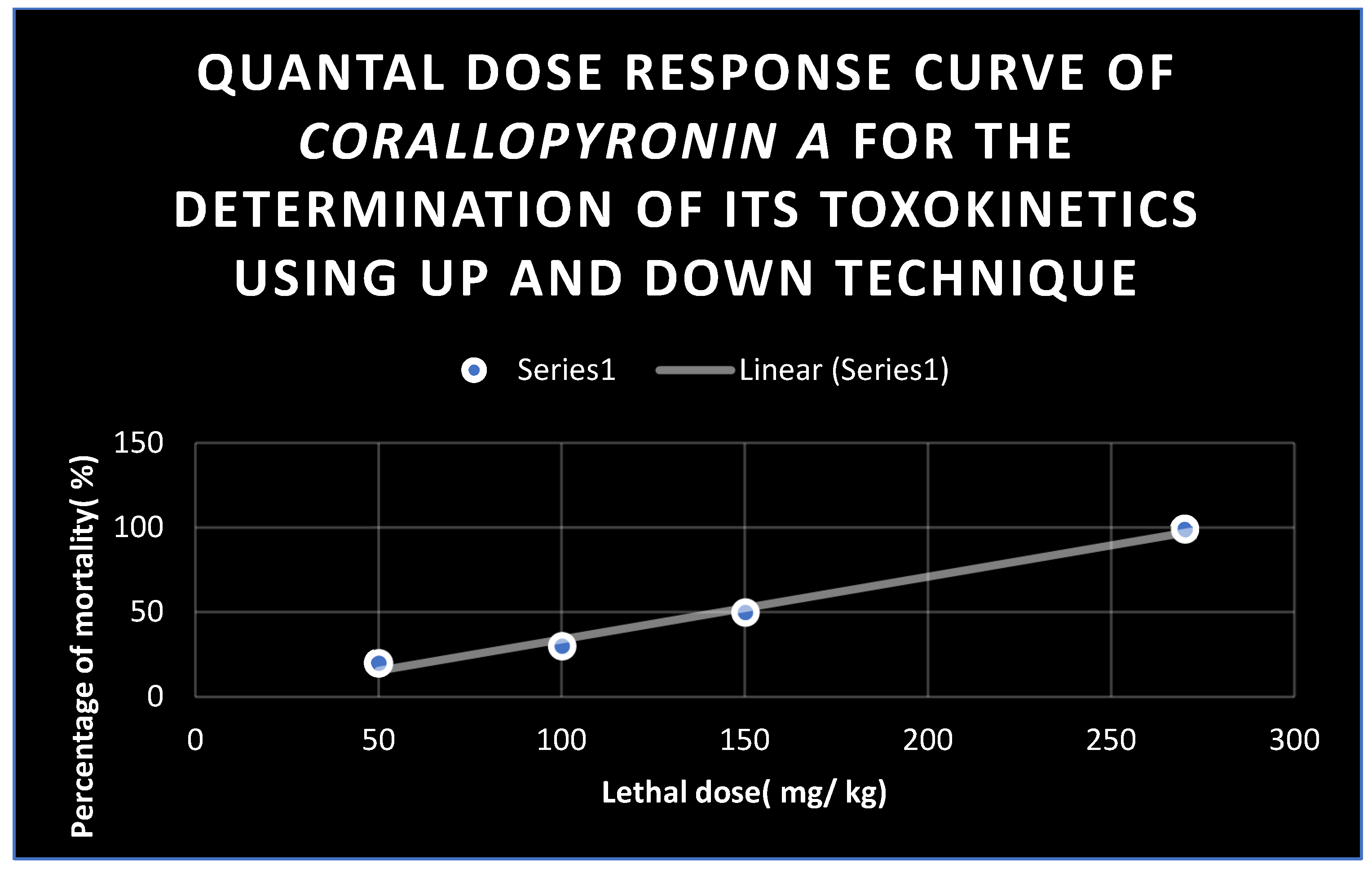
Figure 10.
Quantal dose response curve for the determination of toxokinetics of Corallopyronin A. LD50 % was found to be 150 mg / kg; while LD99 % was nearly 270 mg/ kg.
Figure 10.
Quantal dose response curve for the determination of toxokinetics of Corallopyronin A. LD50 % was found to be 150 mg / kg; while LD99 % was nearly 270 mg/ kg.
Statistical Analysis
All cultures were conducted in triplets. Their presentation was by means and standard deviation. One way analysis of variance( p value≤ 0.05) was used as means for performing statistical analysis and also, statistical analysis was based on excel-spreadsheet-software. The F statistical analysis test was utilized during the present study.
Figure 11.
It demonstrates the major Gram negative bacterial isolates producing Corallopyronin A antibiotic using Stereomicroscope.
Figure 11.
It demonstrates the major Gram negative bacterial isolates producing Corallopyronin A antibiotic using Stereomicroscope.
Results
From the culture supernatant of the Myxobacterium Corallococcus coralloides M2 which was the predominant soil bacterial isolate( 30 isolates, table 3) grown on Casein yeast peptone( CYP) plate, Myxopyronin A was was produced. The test antibiotic blocked the growth of many Gram +ve bacteria with MICs less than 100 mcg/ ml; whereas it inhibited the growth of few Gram -ve bacteria such as Escherichia coli at MICs greater than 100 mcg/ ml. On the other hand, Eukaryotic cells such as fungal and human cells were not affected. Prokaryotic DNA-dependant-RNA polymerase( RNLP) was noticed to be inhibited via the test antibiotic suggesting its bactericidal action. Cmax was 10 mcg/ ml at Tmax 2.5 hours when 500 mg dose was orally administered in randomized human clinical trials phases 1/2; as well as T1/2 reached 2.33 hours following first order kinetics of elimination. Duration of its action was nearly 8 hours after oral administration. Rare toxicity was detected during preclinical and randomized human clinical trials phases 1/2 in the form of mild diarrhea and cholestatic jaundice in less than 5 % of experimental candidates. Corallopyronin A was the predominating compound after reverse phase HPLC technique was utilized for the refinement and the purification of of the test antibiotics( Table 4). The phototoxicity was determined via 3T3 neutral red uptake phototoxicity technique which showed no phototoxicity. On the other hand, mutagenicity and carcinogenicity of the test antibiotic were assessed using Ames test which demonstrated no carcinogenicity and genotoxicity at all. Figure 11 demonstrates the major Gram negative bacterial isolates producing Corallopyronin A antibiotic using Stereomicroscope.
Table 3.
It shows the distribution of Corallopyronin A producing bacterial isolates:.
| No of +ve bacterial isolates producing Corallopyronin A | No of -ve bacterial isolates producing Corallopyronin A |
|---|---|
| 30 | 20 |
Table 4.
It demonstrates the degree of purity of test antibiotics following the purification via reversed phase HPLC technique:.
Table 4.
It demonstrates the degree of purity of test antibiotics following the purification via reversed phase HPLC technique:.
| Test antibiotic | Degree of purity( %) |
|---|---|
| Corallopyronin A | 90 |
| Corallopyronin B | 10 |
Table 5.
It demonstrates 16 S rRNA detection of Corallopyronin A producing isolates using BLASTn software:.
Table 5.
It demonstrates 16 S rRNA detection of Corallopyronin A producing isolates using BLASTn software:.
| Description | Query Cover | E value | Per. ident | Acc. Len |
| Corallococcus coralloides strain M2 23S ribosomal RNA gene, complete sequence | 100% | 0 | 100 | 2976 |
| Corallococcus coralloides DSM 2259, complete genome | 100% | 0 | 100 | 10080619 |
| Corallococcus sp. NCRR chromosome, complete genome | 100% | 0 | 99.05 | 9787125 |
| Corallococcus coralloides strain B035 chromosome, complete genome | 100% | 0 | 98.57 | 9587888 |
| Corallococcus sp. EGB chromosome, complete genome | 100% | 0 | 96.83 | 9431171 |
| Myxococcus fulvus 124B02, complete genome | 100% | 0 | 92.08 | 11048835 |
| Myxococcus sp. MH1 DNA, complete genome | 100% | 0 | 91.92 | 10778154 |
| Myxococcus sp. SDU36 chromosome, complete genome | 100% | 0 | 91.63 | 9016985 |
| Myxococcus xanthus strain GH3.5.6c2 chromosome, complete genome | 100% | 0 | 91.28 | 9321034 |
| Vulgatibacter incomptus strain DSM 27710, complete genome | 99% | 2.00E-153 | 82.99 | 4350553 |
| Anaeromyxobacter sp. Fw109-5, complete genome | 99% | 1.00E-125 | 80.43 | 5277990 |
| Uncultured bacterium clone F5K2Q4C04IF4QS 23S ribosomal RNA gene, partial sequence | 77% | 3.00E-122 | 83.53 | 492 |
| Uncultured bacterium clone F5K2Q4C04I5GUV 23S ribosomal RNA gene, partial sequence | 77% | 6.00E-114 | 82.73 | 491 |
Table 6.
It shows the estimation of zones of inhibition and minimum inhibitory concentrations of Corallopyronin A via Agar diffusion assay using paper discs:.
Table 6.
It shows the estimation of zones of inhibition and minimum inhibitory concentrations of Corallopyronin A via Agar diffusion assay using paper discs:.
| Test organism 1 | MIC( µg/ ml) | Diameter of inhibition zone( mm) |
|---|---|---|
| Bacillus subtilis | 6 | 12 |
| Staphylococcus aureus | 8 | 18 |
| Streptococcus pneumonae | 12 | 9 |
| Escherichia coli | 128 | 11 |
| Pseudomonas aeruginosa | 130 | 0 |
| Candida albicans | 115 | 0 |
| Sacchromyces cerevisiae | 110 | 0 |
| Salmonella typhimurium | 121 | 17 |
| Bacillus cereus | 15 | 10 |
| Micrococcus luteus | 21 | 13 |
| Serratia Marcescens | 137 | 9 |
| Mucor hiemalis | 0 | 19 |
| Shigella dysentery | 103 | 7 |
| Proteus mirabilis | 120 | 6 |
| Rickettsiae prowazaki | 119 | 9 |
| Chlamydiae pneumonae | 135 | 11 |
| Legionella pneumophilla | 140 | 8 |
The initial density of each organism during Agar diffusion assay for the determination of minimum inhibitory concentrations and zones of inhibition of gtowth was nearly 10^5/ ml.
Table 7.
It demonstrates MICs of Corallopyronin A on different microorganisms using broth microdilution technique:.
Table 7.
It demonstrates MICs of Corallopyronin A on different microorganisms using broth microdilution technique:.
| Pathogenic m.o | MIC( µg/ ml) |
|---|---|
| Bacillus subtilis | 9 |
| Bacillus cereus | 8 |
| Staphylococcus aureus | 10 |
| Pneumococci | 14 |
| E.coli | 120 |
| Pseudomonas aeruginosa | 134 |
| Candida albicans | 0 |
| Sacchromyces cerevisiae | 0 |
| Salmonella typhimurium | 112 |
| Haemophilus influenza | 0 |
| Gonococci | 116 |
| meningococci | 126 |
| Serratia Marcescens | 117 |
| Mucor hiemalis | 0 |
| Shigella dysenteriae | 108 |
| Micrococcus luteus | 0 |
| Proteus mirabilis | 0 |
| Rickettsiae prowazaki | 120 |
| Chlamydiae pneumonae | 128 |
| Legionella pneumophilla | 131 |
Table 8.
It demonstrates Minimum bactericidal concentrations( MBCs) of Corallopyronin A on different microorganisms using Broth microdilution technique:.
Table 8.
It demonstrates Minimum bactericidal concentrations( MBCs) of Corallopyronin A on different microorganisms using Broth microdilution technique:.
| Pathogenic m.o | MBC( µg/ ml) |
|---|---|
| Bacillus subtilis | 25 |
| Bacillus cereus | 22 |
| Staphylococcus aureus | 40 |
| Pneumococci | 45 |
| E.coli | 340 |
| Pseudomonas aeruginosa | 400 |
| Candida albicans | 0 |
| Sacchromyces cerevisiae | 0 |
| Salmonella typhimurium | 318 |
| Haemophilus influenza | 0 |
| Gonococci | 380 |
| meningococci | 385 |
| Serratia Marcescens | 319 |
| Mucor hiemalis | 0 |
| Shigella dysenteriae | 298 |
| Micrococcus luteus | 0 |
| Proteus mirabilis | 0 |
| Rickettsiae prowazaki | 370 |
| Chlamydiae pneumonae | 412 |
| Legionella pneumophilla | 266 |
Table 9.
It shows the estimation of mRNA quantity via UV spectrophotometer at 260 nm after addition of Corallopyronin A:.
Table 9.
It shows the estimation of mRNA quantity via UV spectrophotometer at 260 nm after addition of Corallopyronin A:.
| mRNA concentration( ng/ ml) | Absorbance( optical density) at 260 nm |
|---|---|
| 710 | 0.831 |
| 605 | 0.715 |
| 241 | 0.461 |
| 25 | 0.129 |
Table 10.
It shows the effect of Corallopyronin A on the microbial protein synthesis using UV spectrophotometer at 205 nm:.
Table 10.
It shows the effect of Corallopyronin A on the microbial protein synthesis using UV spectrophotometer at 205 nm:.
| Bacterial protein concentration( mcg/ ml) | Time( hr) |
|---|---|
| 80.3 | 2 |
| 31.06 | 4 |
| 17.51 | 7 |
| 2.18 | 9 |
| 0.39 | 11 |
Table 11.
The resolution of biochemical reactions:.
| Test | Result |
|---|---|
| Gram stain | -ve rods |
| Cell shape | Elongated bacilli with tapered ends |
| Spore shape | Ellipsoidal |
| Spore site | Central |
| Motility | + via gliding |
| Catalase | + |
| Oxidase | - |
| Blood haemolysis | - |
| Indol | - |
| Methyl red | - |
| Nitrate reduction test | + |
| Vogues proscauer | - |
| Citrate utilization | - |
| Starch hydrolysis | + |
| Casein hydrolysis | + |
| Growth at 45 ℃ | Bacterial isolates did not grow at 45 ℃; but were grown at 10-37 ℃ |
| Tween 80 | + |
| Tolerance salinity | |
| 5% NaCl | - |
| 7% NaCl | - |
| Saccharide fermentation | |
| Glucose | - |
| Fructose | - |
| Maltose | - |
| Sucrose | - |
Protein synthesis and mRNA synthesis were decreased significantly with increasing the dose Myxopyronin A, as demonstrated in tables 10 and 9 respectively. Docking studies using MCULE and SWISS DOCK softwares demonstrated that the mechanism of action of the test antibiotic was probably due to the inhibition of RNA Polymerase through binding with its switch region. High ∆G of the test antibiotic was observed to be approximately 17 J/ mole as determined via SWISS model software. On the other hand, low Kd of the test antibiotic towards the switch region was found to be approximately 250 nM using SWISS MODEL software. The biochemical profile and the morphology of the potent bacterial isolates producing the test antibiotic in the present study was summarized in table 11. The morphology and biochemical reactions indicated that the predominant bacterial isolates secreting the extracellular test antibiotic were Corallococcus coralloides M2. A total of 150 human subjects( mean SD age, 27.3[ 9.8] years were enrolled and completed the study. For the test antibiotic 90% CIs for the long transformed ratios of Cmax, AUC( 0-24) and AUC( 0-∞) were 90.2 to 95.3, 89.1 to 96.1 and 89.6 to 97.5 respectively. The mean PB was observed for Corallopyronin A which approximated 91%. The major protein binding for Corallopyronin A and Rifampicin was detected to be Albumin. The unbound fraction was found to be responsible for the therapeutic activity.
Discuss
Globally, exploding morbidity and mortality due to antibiotic-resistant micro-organism infections was observed. Hence, amended hindrance and touchstone of infectious diseases, as well as appropriate use of approved antibacterial drugs were essential. The in vitro and in vivo antimicrobial activity of Corallopyronin A, a novel antibiotic was evaluated in the present study. It demonstrated excellent bactericidal activity against a broad spectrum of G +ve bacteria with MICs did not exceed 20 mcg/ ml. On the other hand It showed broad bactericidal activities against G -ve bacteria with minimal inhibitory concentrations were greater than 100 mcg/ ml. Its mechanism of action was realized during the investigation of RNA synthesis to be via the inhibition of prokaryotic DNA-dependant-RNA polymerase; whereas no inhibitory impact was observed for Eukaryotic one. Docking studies through SWISS DOCK software confirmed this as well. The antibiotic activities Corallopyronin A and B were isolated from the culture supernatant of 29 bacterial isolates of Myxobacterium Corallococcus coralloides M2 detected molecularly using 16 S rRNA technique( table 3). The antibiotic activity did not inhibit the growth or kill eukaryotic cells such as human and fungal cells reflecting selectivity towards the inhibition of the growth of prokaryotic bacterial cells. This selectivity effect minimized the adverse effects noticed during the present study. Docking studies via SWISS DOCK software revealed that desmethylation of either Myxopyronin A or B enhanced its biological activity. Purification was performed through reversed phase HPLC. Myxopyronin A was the main refined antibiotic. Its purity degree reached approximately 80 %; while, the remaining purified antibiotic was detected to be Myxopyronin B. The antibacterial activity was assessed via the determination of MICs of the test antibiotics using the agar diffusion technique utilizing paper discs 5 mm in diameter and the broth dilution assay. The initial density of each test microorganism was about 105/ ml of the culture suspension. The MICs of test antibiotic against G +ve bacteria ranged from 6 to 20 mcg/ ml; Whereas MICs reached above 100 mcg/ ml against some selected G -ve bacteria. On the other hand no effect was detected against the growth of fungi and yeasts. ( Irschik H et al., 1983) stated that myxovalargin A was a novel peptide antibiotic isolated from the culture supernatant of the myxobacterium Myxococcus fulvus strain Mx f65. It was active against Gram-positive bacteria( MIC 0.3 approximately 5 micrograms/ ml), at higher concentrations also against Gram-negative ones( MIC 6 approximately 100 micrograms/ ml), and not at all against yeasts and molds. Its mechanism of action involved the inhibition of the bacterial protein synthesis [50]. According to( Glaus F et al., 2018) Ripostatin, a novel antibiotic, isolated from the culture supernatant of Myxobacterium, Sorangium cellulosum strain So ce377. On the other hand it interfered of the bacterial RNA synthesis [51]. On the other hand, Corallopyronin A was found to be structurally related to α-pyrone antibiotics from myxobacteria. Its ability to inhibit RNA polymerase was through interaction with the switch region of RNA polymerase; while Rifampicin inhibited the same enzyme through different region [52]. Corallopyronin A showed no phototoxicity and mutagenicity in rabbit animal models during the preclinical trials stage, in the present study. Rare adverse effects including cholestatic jaundice were reported in less than 5 % of the experimental subjects received the test antibiotics during randomized human clinical trials phases 1/2. The biological half life of Corallopyronin A reached approximately 2.33 hours. 0.6 % peptone and 8 % soluble starch were detected to be the optimal nitrogen and carbon growth factors for bacterial isolates producing the test antibiotics, respectively( figures 4 and 5). High ∆G of the test antibiotic was observed to be approximately 20 J/ mole as determined via SWISS MODEL software reflecting high catalytic activity of the test antibiotic towards the switch region. On the other hand, low Kd of the test antibiotic towards the switch region was found to be approximately -480 nM using SWISS MODEL software indicating high affinity and binding capacity. Bioavailability studies were performed using HPLC during randomized human clinical trials phases 1/2 revealed that Corallopyronin A reached nearly 91% oral bioavailability, 93% IM bioavailability and 100% IV bioavailability. Metabolic studies using HPLC revealed that the test antibiotic showed no in vivo induction of hepatic metabolizing Cytochrome P450 enzymatic system; while Rifampicin induced CYP3A4 hepatic metabolizing enzyme potently. Up and down procedure intended for the evaluation of acute toxicity profile of the test antibiotic showed that LD50% was about 150 mg/ kg body weight; while LD99% reached 270 mg/ kg. On the other hand, therapeutic margin of the test antibiotic ranged from 8 mcg/ ml to 103 mcg/ ml. Corallopyronin A producing bacterial isolates were gram negative, spore forming obligate aerobes and chemoorganotrophic. They were elongated rods with tapered ends. No flagella were present; but the cells moved via gliding. They fermented Tween 80, starch and casein. On the other hand they were positive for catalase while negative for oxidase tests. They reduced nitrates
And were able to grow at 10-37 ℃. A total of 150 human subjects( mean SD age, 27.3[ 8.6]. years were enrolled and completed the study. For the test antibiotic 90% CIs for the long transformed ratios of Cmax, AUC( 0-24) and AUC( 0-∞) were 90.2 to 95.3, 89.1 to 96.1 and 89.6 to 97.5 respectively. The point estimates for Cmax in the present study were outside the limit for bio-equivalence for Rifampicin standard drug. The mean PB was observed for Corallopyronin A which approximated 91% while that of rifampicin reached 88% [53]. It was noticed that plasma protein binding was proportionally increased with increasing the doses of the test antibiotic. The plasma protein binding participated in extending the Corallopyronin A duration of action. The major protein binding for Corallopyronin A and Rifampicin was noticed to be Albumin. The unbound fraction was detected to be responsible for the therapeutic activity.
Conclusion
Antibiotic resistance is a global challenge that the current study shows promise in solving. According to the findings of the current study, Corallopyronin A, which was isolated from the bacterial isolates Corallococcus coralloides M2 that were collected from various soil environments in Egypt, exhibited significant antibiotic activity both in vitro and in vivo against a moderate range of pathogenic bacteria, particularly G+ve varieties. Future research is recommended to investigate pharmacological interactions of the synergism type between Corallopyronin A and different antibiotic classes.
References
- Dalhoff, A. Selective toxicity of antibacterial agents-still a valid concept or do we miss chances and ignore risks? Infection. 2021 Feb;49(1):29-56. [CrossRef] [PubMed]
- Hutchings MI, Truman AW, Wilkinson B. Antibiotics: past, present and future. Curr Opin Microbiol. 2019 Oct;51:72-80. [CrossRef] [PubMed]
- Wencewicz, TA. Crossroads of Antibiotic Resistance and Biosynthesis. J Mol Biol. 2019 Aug 23;431(18):3370-3399. [CrossRef] [PubMed]
- Lepe JA, Martínez-Martínez L. Resistance mechanisms in Gram-negative bacteria. Med Intensiva (Engl Ed). 2022 Jul;46(7):392-402. [CrossRef] [PubMed]
- Vila J, Marco F. Lectura interpretada del antibiograma de bacilos gramnegativos no fermentadores [Interpretive reading of the non-fermenting gram-negative bacilli antibiogram]. Enferm Infecc Microbiol Clin. 2010 Dec;28(10):726-36. Spanish. [CrossRef] [PubMed]
- Mushtaq S, Vickers A, Woodford N, Livermore DM. WCK 4234, a novel diazabicyclooctane potentiating carbapenems against Enterobacteriaceae, Pseudomonas and Acinetobacter with class A, C and D β-lactamases. J Antimicrob Chemother. 2017 Jun 1;72(6):1688-1695. [CrossRef] [PubMed]
- Irwin SV, Fisher P, Graham E, Malek A, Robidoux A. Sulfites inhibit the growth of four species of beneficial gut bacteria at concentrations regarded as safe for food. PLoS One. 2017 Oct 18;12(10):e0186629. [CrossRef] [PubMed]
- Jeong S, Lee Y, Yun CH, Park OJ, Han SH. Propionate, together with triple antibiotics, inhibits the growth of Enterococci. J Microbiol. 2019 Nov;57(11):1019-1024. [CrossRef] [PubMed]
- Kohanski MA, Dwyer DJ, Hayete B, Lawrence CA, Collins JJ. A common mechanism of cellular death induced by bactericidal antibiotics. Cell. 2007 Sep 7;130(5):797-810. [CrossRef] [PubMed]
- Brauer M, Herrmann J, Zühlke D, Müller R, Riedel K, Sievers S. Myxopyronin B inhibits growth of a Fidaxomicin-resistant Clostridioides difficile isolate and interferes with toxin synthesis. Gut Pathog. 2022 Jan 6;14(1):4. [CrossRef] [PubMed]
- Doundoulakis T, Xiang AX, Lira R, Agrios KA, Webber SE, Sisson W, Aust RM, Shah AM, Showalter RE, Appleman JR, Simonsen KB. Myxopyronin B analogs as inhibitors of RNA polymerase, synthesis and biological evaluation. Bioorg Med Chem Lett. 2004 Nov 15;14(22):5667-72. [CrossRef] [PubMed]
- Lira R, Xiang AX, Doundoulakis T, Biller WT, Agrios KA, Simonsen KB, Webber SE, Sisson W, Aust RM, Shah AM, Showalter RE, Banh VN, Steffy KR, Appleman JR. Syntheses of novel myxopyronin B analogs as potential inhibitors of bacterial RNA polymerase. Bioorg Med Chem Lett. 2007 Dec 15;17(24):6797-800. [CrossRef] [PubMed]
- Moy TI, Daniel A, Hardy C, Jackson A, Rehrauer O, Hwang YS, Zou D, Nguyen K, Silverman JA, Li Q, Murphy C. Evaluating the activity of the RNA polymerase inhibitor myxopyronin B against Staphylococcus aureus. FEMS Microbiol Lett. 2011 Jun;319(2):176-9. [CrossRef] [PubMed]
- Srivastava A, Talaue M, Liu S, Degen D, Ebright RY, Sineva E, Chakraborty A, Druzhinin SY, Chatterjee S, Mukhopadhyay J, Ebright YW, Zozula A, Shen J, Sengupta S, Niedfeldt RR, Xin C, Kaneko T, Irschik H, Jansen R, Donadio S, Connell N, Ebright RH. New target for inhibition of bacterial RNA polymerase: 'switch region'. Curr Opin Microbiol. 2011 Oct;14(5):532-43. [CrossRef] [PubMed]
- Mosaei H, Harbottle J. Mechanisms of antibiotics inhibiting bacterial RNA polymerase. Biochem Soc Trans. 2019 Feb 28;47(1):339-350. [CrossRef] [PubMed]
- Sucipto H, Sahner JH, Prusov E, Wenzel SC, Hartmann RW, Koehnke J, Müller R. In vitro reconstitution of α-pyrone ring formation in myxopyronin biosynthesis. Chem Sci. 2015 Aug 1;6(8):5076-5085. [CrossRef] [PubMed]
- O'Toole, GA. Classic Spotlight: How the Gram Stain Works. J Bacteriol. 2016 Nov 4;198(23):3128. [CrossRef] [PubMed]
- Luhur J, Chan H, Kachappilly B, Mohamed A, Morlot C, Awad M, Lyras D, Taib N, Gribaldo S, Rudner DZ, Rodrigues CDA. A dynamic, ring-forming MucB / RseB-like protein influences spore shape in Bacillus subtilis. PLoS Genet. 2020 Dec 14;16(12):e1009246. [CrossRef] [PubMed]
- Qin Y, Faheem A, Hu Y. A spore-based portable kit for on-site detection of fluoride ions. J Hazard Mater. 2021 Oct 5;419:126467. [CrossRef] [PubMed]
- Cabeen MT, Jacobs-Wagner C. Bacterial cell shape. Nat Rev Microbiol. 2005 Aug;3(8):601-10. [CrossRef] [PubMed]
- Wang Q, Xiao L, He Q, Liu S, Zhang J, Li Y, Zhang Z, Nie F, Guo Y, Zhang L. Comparison of haemolytic activity of tentacle-only extract from jellyfish Cyanea capillata in diluted whole blood and erythrocyte suspension: diluted whole blood is a valid test system for haemolysis study. Exp Toxicol Pathol. 2012 Nov;64(7-8):831-5. [CrossRef] [PubMed]
- Dubay MM, Acres J, Riekeles M, Nadeau JL. Recent advances in experimental design and data analysis to characterize prokaryotic motility. J Microbiol Methods. 2023 Jan;204:106658. [CrossRef] [PubMed]
- Wang C, Zhang Y, Luo H, Zhang H, Li W, Zhang WX, Yang J. Iron-Based Nanocatalysts for Electrochemical Nitrate Reduction. Small Methods. 2022 Oct;6(10):e2200790. [CrossRef] [PubMed]
- Hu CY, Cheng HY, Yao XM, Li LZ, Liu HW, Guo WQ, Yan LS, Fu JL. Biodegradation and decolourization of methyl red by Aspergillus versicolor LH1. Prep Biochem Biotechnol. 2021;51(7):642-649. [CrossRef] [PubMed]
- Xu D, Wu L, Yao H, Zhao L. Catalase-Like Nanozymes: Classification, Catalytic Mechanisms, and Their Applications. Small. 2022 Sep;18(37):e2203400. [CrossRef] [PubMed]
- Pawlik A, Stefanek S, Janusz G. Properties, Physiological Functions and Involvement of Basidiomycetous Alcohol Oxidase in Wood Degradation. Int J Mol Sci. 2022 Nov 9;23(22):13808. [CrossRef] [PubMed]
- Cordaro JT, Sellers W. Blood coagulation test for citrate utilization. Appl Microbiol. 1968 Jan;16(1):168-9. [CrossRef] [PubMed]
- Krajang M, Malairuang K, Sukna J, Rattanapradit K, Chamsart S. Single-step ethanol production from raw cassava starch using a combination of raw starch hydrolysis and fermentation, scale-up from 5-L laboratory and 200-L pilot plant to 3000-L industrial fermenters. Biotechnol Biofuels. 2021 Mar 16;14(1):68. [CrossRef] [PubMed]
- Kerwin, BA. Polysorbates 20 and 80 used in the formulation of protein biotherapeutics: structure and degradation pathways. J Pharm Sci. 2008 Aug;97(8):2924-35. [CrossRef] [PubMed]
- Trueba FJ, Neijssel OM, Woldringh CL. Generality of the growth kinetics of the average individual cell in different bacterial populations. J Bacteriol. 1982 Jun;150(3):1048-55. [CrossRef] [PubMed]
- McCrea KW, Xie J, LaCross N, Patel M, Mukundan D, Murphy TF, Marrs CF, Gilsdorf JR. Relationships of nontypeable Haemophilus influenzae strains to hemolytic and nonhemolytic Haemophilus haemolyticus strains. J Clin Microbiol. 2008 Feb;46(2):406-16. [CrossRef] [PubMed]
- Jogawat A, Vadassery J, Verma N, Oelmüller R, Dua M, Nevo E, Johri AK. PiHOG1, a stress regulator MAP kinase from the root endophyte fungus Piriformospora indica, confers salinity stress tolerance in rice plants. Sci Rep. 2016 Nov 16;6:36765. [CrossRef] [PubMed]
- Barry AL, Feeney KL. Two quick methods for Voges-Proskauer test. Appl Microbiol. 1967 Sep;15(5):1138-41. [CrossRef] [PubMed]
- Wang J, Su Y, Jia F, Jin H. Characterization of casein hydrolysates derived from enzymatic hydrolysis. Chem Cent J. 2013 Apr 4;7(1):62. [CrossRef] [PubMed]
- de Bie TH, Witkamp RF, Balvers MG, Jongsma MA. Effects of γ-aminobutyric acid supplementation on glucose control in adults with prediabetes: A double-blind, randomized, placebo-controlled trial. Am J Clin Nutr. 2023 Sep;118(3):708-719. [CrossRef] [PubMed]
- Endoh R, Horiyama M, Ohkuma M. D-Fructose Assimilation and Fermentation by Yeasts Belonging to Saccharomycetes: Rediscovery of Universal Phenotypes and Elucidation of Fructophilic Behaviors in Ambrosiozyma platypodis and Cyberlindnera americana. Microorganisms. 2021 Apr 5;9(4):758. [CrossRef] [PubMed]
- Lu Z, Guo W, Liu C. Isolation, identification and characterization of novel Bacillus subtilis. J Vet Med Sci. 2018 Mar 24;80(3):427-433. [CrossRef] [PubMed]
- Zhao Y, Meng K, Fu J, Xu S, Cai G, Meng G, Nielsen J, Liu Z, Zhang Y. Protein engineering of invertase for enhancing yeast dough fermentation under high-sucrose conditions. Folia Microbiol (Praha). 2023 Apr;68(2):207-217. [CrossRef] [PubMed]
- Irschik H, Gerth K, Höfle G, Kohl W, Reichenbach H. The myxopyronins, new inhibitors of bacterial RNA synthesis from Myxococcus fulvus (Myxobacterales). J Antibiot (Tokyo). 1983 Dec;36(12):1651-8. [CrossRef] [PubMed]
- Wiegand I, Hilpert K, Hancock RE. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat Protoc. 2008;3(2):163-75. [CrossRef] [PubMed]
- Balouiri M, Sadiki M, Ibnsouda SK. Methods for in vitro evaluating antimicrobial activity: A review. J Pharm Anal. 2016 Apr;6(2):71-79. [CrossRef] [PubMed]
- Dell'Anno A, Fabiano M, Duineveld GCA, Kok A, Danovaro R. Nucleic acid (DNA, RNA) quantification and RNA/DNA ratio determination in marine sediments: comparison of spectrophotometric, fluorometric, and HighPerformance liquid chromatography methods and estimation of detrital DNA. Appl Environ Microbiol. 1998 Sep;64(9):3238-45. [CrossRef] [PubMed]
- Simonian, MH. Spectrophotometric determination of protein concentration. Curr Protoc Cell Biol. 2002 Aug;Appendix 3:Appendix 3B. [CrossRef] [PubMed]
- Rox K, Becker T, Schiefer A, Grosse M, Ehrens A, Jansen R, Aden T, Kehraus S, König GM, Krome AK, Hübner MP, Wagner KG, Stadler M, Pfarr K, Hoerauf A. Pharmacokinetics and Pharmacodynamics (PK/PD) of Corallopyronin A against Methicillin-Resistant Staphylococcus aureus. Pharmaceutics. 2022 Dec 30;15(1):131. [CrossRef] [PubMed]
- Xu J, Jin H, Zhu H, Zheng M, Wang B, Liu C, Chen M, Zhou L, Zhao W, Fu L, Lu Y. Oral bioavailability of rifampicin, isoniazid, ethambutol, and pyrazinamide in a 4-drug fixed-dose combination compared with the separate formulations in healthy Chinese male volunteers. Clin Ther. 2013 Feb;35(2):161-8. [CrossRef] [PubMed]
- Utku Türk EG, Jannuzzi AT, Alpertunga B. Determination of the Phototoxicity Potential of Commercially Available Tattoo Inks Using the 3T3-neutral Red Uptake Phototoxicity Test. Turk J Pharm Sci. 2022 Feb 28;19(1):70-75. [CrossRef] [PubMed]
- Thomas DN, Wills JW, Tracey H, Baldwin SJ, Burman M, Williams AN, Harte DSG, Buckley RA, Lynch AM. Ames Test study designs for nitrosamine mutagenicity testing: qualitative and quantitative analysis of key assay parameters. Mutagenesis. 2023 Dec 19:gead033. [CrossRef] [PubMed]
- Zhang, YY., Huang, YF., Liang, J. et al. Improved up-and-down procedure for acute toxicity measurement with reliable LD50 verified by typical toxic alkaloids and modified Karber method. BMC Pharmacol Toxicol 23, 3 (2022). [CrossRef]
- Heuser E, Becker K, Idelevich EA. Bactericidal Activity of Sodium Bituminosulfonate against Staphylococcus aureus. Antibiotics (Basel). 2022 Jul 5;11(7):896. [CrossRef] [PubMed]
- Irschik H, Gerth K, Kemmer T, Steinmetz H, Reichenbach H. The myxovalargins, new peptide antibiotics from Myxococcus fulvus (Myxobacterales). I. Cultivation, isolation, and some chemical and biological properties. J Antibiot (Tokyo). 1983 Jan;36(1):6-12. [CrossRef] [PubMed]
- Glaus F, Dedić D, Tare P, Nagaraja V, Rodrigues L, Aínsa JA, Kunze J, Schneider G, Hartkoorn RC, Cole ST, Altmann KH. Total Synthesis of Ripostatin B and Structure-Activity Relationship Studies on Ripostatin Analogs. J Org Chem. 2018 Jul 6;83(13):7150-7172. [CrossRef] [PubMed]
- Dennison TJ, Smith JC, Badhan RKS, Mohammed AR. Formulation and Bioequivalence Testing of Fixed-Dose Combination Orally Disintegrating Tablets for the Treatment of Tuberculosis in the Paediatric Population. J Pharm Sci. 2020 Oct;109(10):3105-3113. [CrossRef] [PubMed]
- Alghamdi WA, Al-Shaer MH, Peloquin CA. Protein Binding of First-Line Antituberculosis Drugs. Antimicrob Agents Chemother. 2018 Jun 26;62(7):e00641-18. [CrossRef] [PubMed]
Table 2.
It demonstrates different isolation media for different pathogenic m.os. utilized in the Broth microdilution test and agar diffusion assay using paper discs:.
Table 2.
It demonstrates different isolation media for different pathogenic m.os. utilized in the Broth microdilution test and agar diffusion assay using paper discs:.
| Pathogenic m.o | No of strains | Isolation media |
|---|---|---|
| Bacillus subtilis | 5 | Mannitol egg yolk polymixin agar( MEYP) |
| Bacillus cereus | 7 | Polymixin egg yolk mannitol bromothymol blue agar( PEMBA) |
| Staphylococcus aureus | 6 | Salt mannitol agar( SMA) |
| Pneumococci | 13 | Todd Hewitt broth with yeast extract |
| coli | 17 | Sorbitol- Macconkey agar |
| Pseudomonas aeruginosa | 10 | Pseudomonas isolation agar( PSA) |
| Candida albicans | 1 | Potato dextrose agar( PDA) |
| Sacchromyces cerevisiae | 5 | Sabourad dextrose agar( SDA) |
| Salmonella typhimurium | 4 | Bismuth sulfite agar( BSA) |
| Haemophilus influenza | 3 | Enriched chocolate agar |
| Gonococci | 4 | Thayer martin medium |
| meningococci | 6 | Mueller Hinton agar |
| Serratia Marcescens | 4 | Caprylate thallous agar medium |
| Mucor hiemalis | 1 | Potato dextrose broth |
| Shigella dysenteriae | 8 | Hekteon enteric agar |
| Micrococcus luteus | 1 | Tryptic soy agar |
| Proteus mirabilis | 1 | Blood agar |
| Chlamydiae pneumonae | 1 | Chlamydiae pneumonae Monkey cell culture |
| Rickettsiae typhi | 1 | Chicken embryos culture |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



