
Submitted:
29 April 2024
Posted:
01 May 2024
You are already at the latest version
Abstract
Lymphocyte collection by apheresis for CAR-T production usually does not include mobilized blood using Granulocyte Colony Stimulating Factor (G-CSF) due to the widespread knowledge that it causes a decrease in the number and functionality of lymphocytes. However, it is used for stem cell transplant, which is a common treatment for hematological malignancies. The growing demand for CAR therapies (CAR-T and NK-CAR), both in research and clinic, makes necessary to evaluate whether mobilized PBSC products may be potential candidates for use in such therapies. This review collects recent work that experimentally verifies the role and functionality of T and NK lymphocytes, and the generation of CAR-T, from apheresis after G-CSF mobilization. As discussed, T cells do not vary significantly in their phenotype, the ratio of CD4+ and CD8+ remains constant and the different sub-populations remain stable. In addition, its expansion and proliferation rate are invariant regardless mobilization with G-CSF, as well as the secretion of proinflammatory cytokines and the cytotoxic ability. Therefore, cells mobilized before apheresis are postulated as a new alternative source of T cells for adoptive therapies that will serve to alleviate high demand, increase availability, and take advantage of the substantial number of existing cryopreserved products.
Keywords:
G-CSF
; Mobilization
; CAR-T
; CAR-NK
1. Introduction
Historically, treatment for hematologic cancers has primarily included chemotherapy, radiotherapy, and hematopoietic stem cell transplantation (HSCT) [1]. The latter is the one that remains a cornerstone in clinical practice for some hematological malignancies. However, with recent advances in both molecular and cellular knowledge and a greater understanding of tumor functionality, numerous alternatives have emerged within the field of targeted immunotherapy. In this area, some therapies are highlighted such as monoclonal and bispecific antibodies, conjugated antibodies, checkpoint inhibitors, and more recently adoptive cell therapy [2].
Adoptive cell therapy and concretely its development in the form of specific therapy has emerged as a solid alternative for patients with refractory or relapsed hematological cancers [3,4]. Among these, the application of chimeric antigen receptor (CAR) therapy in addressing hematologic malignancies has sparked considerable enthusiasm in recent years. Viewing it through the lens of transfusion medicine, the adoption of CAR-T therapies holds the potential to become a fundamental and enduring treatment not only for hematologic cancers but also for solid-organ malignancies. CAR-T has revolutionized the treatment of hematological malignancies and has achieved unprecedented responses in recent years, especially in B-cell acute lymphocytic leukemia (B-ALL), non-Hodgkin lymphoma (NHL) and multiple myeloma (MM). This presents a noteworthy prospect for the advancement and broadening of therapeutic options. However, these therapies are still far from being perfect and are not exempt from some negative effects and inconveniences that have been the subject of intense study [4]. To alleviate these disadvantages, different generations of CAR have emerged with specific molecular constructions that improve the functionality of the therapy. In addition, due to the unique characteristics of alloreactivity, recognition, persistence, and cytotoxicity presented by natural killer cells (NK), efforts are being focused on new CAR-NK therapies [5].
In this context, one of the main challenges that clinicians and researchers encounter daily is obtaining both T lymphocytes and NK cells, efficiently and on a large scale to be able to perform this therapy. This fact, together with the growing demand for CAR therapy, makes it necessary to find new ways to advance CAR immunotherapy. An important alternative advance would be the search for new sources of collection of these cells to generate CAR products. Typically, these cells are isolated from PBMCs, UCBs, or immortalized cell lines [6,7,8,9]. However, it is common to find cell products from donors or patients previously mobilized using Granulocyte Colony Stimulating Factor (G-CSF), since it is the usual procedure for HSCT treatment. Being PBMCs the preferred way to obtain them due to the ease of collection and the number of cells obtained, in this article we review the feasibility of using these mobilized G-CSF-mobilized PBMC to obtain CAR products, both for CAR-T and CAR-NK [10,11].
2. Adoptive Cell Therapy (ACT): The CAR Immunotherapy
Adoptive cell therapy (ACT) has emerged as a prominent form of anti-tumor immunotherapy, particularly for patients with refractory or recurrent hematologic malignancies (R/R). This personalized cancer treatment implies the direct administration of immune cells with anticancer properties. The process involves collecting autologous lymphocytes from the patient or allogeneic lymphocytes from healthy donors, usually from apheresis processes. Also, these cells can come from sources such as PBMCs, UCBs, or immortalized lines. Subsequently, these cells are infused in the patient, usually in cases of malignancy that have not responded to conventional treatments [3]. ACT can be classified into specific and non-specific cellular therapies. Non-specific cell therapy includes infusion of various immune cells, such as cytokine-induced killer cells (CIK), γ/δ T cells, and natural killer cells (NK). While the main goal is to improve overall immune function, these cells lack specificity against tumor targets, limiting their effectiveness [12,13,14,15].
In response to the limitations of non-specific cellular therapy, specific cellular therapy gained attention. Early studies recognized the drawbacks of non-specific approaches and proposed isolating specific cells, engineering genetically to express certain receptors against tumors, and expanding their numbers in vivo using cytokines and growth factors for further administration to the patient [4]. However, this therapy faces difficulties associated with obtaining, isolating, and expanding these cells. ACT therapies have used both natural cells derived from a host, and, more recently, cells genetically modified with antitumor T cell receptors (TCRs) or chimeric antigen receptors (CARs). This last option is the one that has gained the most weight in recent years [3,4]
The highest development in specific ACT research has been through the use of redirected T cells to recognize antitumor antigens. In the late 1980s, Dr. Zelig Eshhar's team developed the idea of redirecting T cells from the MD45 murine T-cell line to target antigens in a non-MHC-restricted manner of choice by inserting a newly constructed cell receptor [16]. From this publication, several research studies were developed to generate the prototypes of modern CAR [17]. Nowadays, CAR-T can be defined as T cells that have been genetically modified for the addition of artificial receptors that are capable of specifically recognizing antigens found in the membranes of cancer cells. This field of research continued for many years, and in 2009 it resulted in the first publication that detailed the manufacturing process for CAR-T obtained from PBMCs, specifically, non-mobilized frozen patients-derived apheresis products, to treat patients with R/R leukemia [18]. Several years later, in 2012, Davila et al published the first results of CAR-T cells applied to the treatment of Chronic Lymphocytic Leukemia (CLL) achieving complete remission in three patients [19].
Initially, first-generation CARs consisted simply of a TCR-like construct with a single CD3+ zeta signalling domain [16]. The low persistence in vivo resulted in the second generation of CAR with the incorporation of cytoplasmic domains into the construct, with costimulatory ability such as 4-1BB or CD28 to promote activation, proliferation, and persistence [20]. Subsequently, the research for CAR optimization led to the evolution towards the third generation CAR-T [21], thanks to the addition of two costimulatory molecules such as 4-1BB, CD28, CD27, ICOS, or OX40. A fourth generation called TRUCKS (T cell Redirected for antigen-unrestricted Cytokine-initiated Killing) [22] has recently been developed. In this four-generation CAR, the transgene encoding a second-generation CAR-T is completed with a gene encoding a cytokine such as IL-12 or IL-15 (Figure 1).
CAR-T therapy has been consolidating as a great promise in the treatment of hematologic malignancies since its commercial launch in 2017 with the FDA approval of Kymriah® (tisagenlecleucel) by Novartis [23] and Yescarta® (axicabtagene ciloleucel) by Gilead/KitePharma [24]. Both treatments are based on T cells genetically modified to express CARs targeting CD19 receptors and they have been indicated for R/R diffuse large B-cell lymphoma (DLBCL) and patients up to 25 years of age with R/R ALL. The beginning of commercial CAR-T therapy brought with it the necessity for safe protocols throughout its administration process. Both commercial therapies are based on autologous cells, so they contain the patient's T lymphocytes. The CAR-T cell manufacturing process begins with the collection of non-mobilized PBMCs from the patient [25], which is accomplished through a leukapheresis process. The collected apheresis products can be processed in several ways depending on the subsequent procedures. For example, in the case of the Novartis product, specific collected cell counts for CD3+ lymphocytes (≥1 × 109 CD3+ cells) are required. Once collected, the apheresis products are sent to a commercial facility where T cells are isolated, activated, genetically modified with a CAR-encoding vector, and expanded before cryopreservation, finally sent back for infusion into the patient [26].
CD19-targeting CAR T cell products currently approved by the FDA and EMA include axicabtagene ciloleucel [27], tisagenlecleucel [28], lisocabtagene maraleucel [29], and brexucabtagene autoleucel [30]. Among the CAR-T therapies approved, recent meta-analyses show long-term results of CAR-T treatment [31]. In the case of CAR T-cell therapy targeting CD19 in patients with B-cell lymphoma and CLL, 10 studies with more than 24 months of follow-up were evaluated, establishing overall response rates (ORR) of 44% to 91% and complete remission rate (CR) from 28% to 68%. In the case of therapy for B-ALL with CAR T-targeting CD19, up to 16 studies have been evaluated with a follow-up of more than one year indicating CR rates of more than 80% [32]. Regarding RRMM patients, there are two approved CAR-T products, idecabtagene vicleucel and ciltacabtagene autoleucel targeting B Cell Maturation Antigen (BCMA). There is less data from long-term studies with CAR-T targeting BCMA because these constructs are more recent. Specifically, 6 studies have been recorded with a follow-up of around 1 year, reporting ORR rates of 73% to 100% and CR rates of between 33% and 83% [31].
This enormous success reflected in 6 currently available CAR-T products on the market shows a growing increase in clinical trials launched aimed at the treatment of B malignancies, for instance, multiple myeloma, acute myeloid leukemia and solid tumors. In fact, 1381 CAR-T cell-based clinical trials were counted on ClinicalTrials.gov on April 8, 2024.
Despite the fact that CAR-T therapy has obtained successful results it still has several challenges that remain unresolved. Among them, there have been described some toxic effects on patients such as immune cell-associated neurological syndrome (ICANS) and cytokine release syndrome (CRS) [33]. Moreover, CAR-T therapy relies on autologous peripheral blood-derived and personally engineered cells which is both time-consuming and costly for patients [34]. All together with additional limitations that involve off-targeted toxicity and lack of effectiveness in solid tumors within the immunosuppressive microenvironment [35], it seems preceptive to optimize CAR products.
Recently, new CAR cell options have been developed, for instance, NK-CAR which could mitigate some CAR-T-related side effects. Similarly, to CAR-T cells, CAR-NK cells are NK cells genetically modified to express CARs that recognize a specific antigen uniquely expressed or overexpressed by tumor target cells. It is important to highlight that CAR-NK cells have NK cell characteristics which means these cells are not only capable of recognizing tumor antigens due to the CAR construction but also eliminating tumors due to its crucial role in immune surveillance by identifying and targeting cancerous or virally infected cells that down-regulate HLA class I molecules or express stress markers [5]. Furthermore, the NK cell mechanism of action relies on non-specific stimulatory and inhibitory signals thus it additionally could eliminate antigen-negative cancer cells. Besides that, allogenic NK-CAR does not exert GVHD [36], eliminating the necessity of developing different CAR products for each patient, hence could be a potential candidate for universal off-the-shelf therapy. All these advantages, added to nice preliminary results, are making CAR-NK therapy a quite promising field in clinical research [37]. There are many published studies regarding NK-CAR applied to preclinical and clinical research, most of them developed using the NK-92 cell line, NK derived from Umbilical Cordon Blood (UCB) or hematopoietic pluripotent stem cells (HPSC) [8,38]. Furthermore, there are other numerous CAR-NK clinical trials, concretely, 75 clinical trials were counted from ClinicalTrials.gov on 8 April 2024.
In this scenario, and with the growing demand for CAR therapy, it seems convenient to find new ways to progress in CAR immunotherapy, enhancing the clinical trial results. An important alternative approach to optimize this therapy would be the search for new sources of collecting cells to generate large amounts of CAR-T. The design of innovative studies aimed at developing ready-to-use CAR seems essential considering that a single treatment can lead to long-lasting results compared to other therapies, for example, monoclonal antibodies that require prolonged treatment at considerable cost.
3. New Cellular Sources for CAR Immunotherapy: G-CSF Mobilized PBSC Products
The development of CAR therapy has introduced a new field of therapeutic possibilities for patients with certain hematologic malignancies, however, the HSCT remains a cornerstone in clinical practice. This treatment is considered a preferred treatment for some high-risk and R/R hematological malignancies. Moreover, with the improved supportive care and increasing acceptance of haploidentical transplantations as an alternative treatment modality, more patients are opting for HSCT as a definite treatment for hematological malignancies. The database which includes all of the stem cell transplants conducted, includes information on more than 500,000 transplants. Over the last 5 years, 35,000 new patients have been treated annually. In Europe, more than 400,000 patients treated with HSCT have survived [39].
This therapy aims to infuse hematopoietic stem cells derived from the patient himself (autologous) or healthy donors (allogeneic). HSCs could be isolated from bone marrow (BM), peripheral blood (PB), or UCB [6,7]. The first successful human HSCT was a bone marrow transplant, performed in 1956 by Dr. E. Donnall Thomas and his research team [40]. The patient was a 5-year-old child with leukemia. The procedure involved transferring bone marrow from his healthy identical twin. This landmark achievement laid the foundation for modern-day bone marrow transplantation. In those past years, hematopoietic cells were obtained directly from large volumes of bone marrow, aspirated from the iliac crests and using general anesthesia. However, since 1994 and the initial demonstration that peripheral blood stem cells (PBSC) mobilized by cytokines (G-CSF first and more recently when needed plerixafor) could be used as well as BM, the proportion of PB transplants has increased to reach about 70–95% [41].
Over the past decades, apheresis has become a standard procedure in clinical hematology providing HSCs for hematopoietic reconstitution after myeloablative chemotherapy. For this reason and due to the reduced proportion of HSCs in peripheral blood (hovering at 0,04%) G-CSF is routinely utilized before apheresis to mobilize HSCs from BM to PB [39].
Granulocyte colony-stimulating factor (G-CSF) is a 19 kDa glycoprotein containing 175 amino acid residues that functions similarly to a hormone or cytokine [42]. Naturally, the pleiotropic cytokine is produced by activated monocytes, macrophages, endothelial cells, fibroblasts, astrocytes, osteoblasts and bone marrow cells. Overall, G-CSF binds to G-CSFR (receptor) in monocytes, neutrophils, HSPCs and endothelial cells, enhancing the bone marrow to produce progenitor cells and release them into the bloodstream. It is noticeable that this molecule not only exerts its function in hematopoietic stem cells but also in different progenitors such as myeloid, erythroid and megakaryocytic HSPCs [43,44].
Since G-CSF increases neutrophil mobilization and maturation, it was initially used in clinical practice to prevent and treat neutropenia [45]. However, nowadays its ability to mobilize HSC from the bone marrow [46] has positioned this molecule as the most commonly used factor for this purpose, being the gold standard and changing the paradigm of stem cell transplantation [47]. That great performance could be related to some advantages such as the increase in the number of different peripheral blood white cells such as lymphocytes, monocytes, and obviously HSC [48]; the reduced time for the restoration of neutrophils and platelets post-transplant [49], major safety, and importantly, according to clinical trials, normal donors prefer donation of HSPCs from blood, instead of donation from pelvic marrow, which is more invasive and dangerous [50].
Despite the fact that its mechanism of action has been quite studied, our knowledge is not complete. There is a hypothesis that envelopes several combinational actors in the mobilization process [51]. In this approach, the adhesion molecules in the BM niche gain special attention. In the normal state, hematopoietic stem cells (CD34+) are situated and retained in the bone marrow through multiple retention axes, such as SCF (Stem Cell Factor)/c-kit; VCAM-1 (Vascular Cell Adhesion molecule-1)/VLA-4; and SDF-1 (Stromal-Derived Factor-1)/Chemokine Receptor-4 (CXCR4) [52]. Studies have confirmed that these interactions are interrupted when G-CSF is used and this is sufficient for mobilization [53] (Figure 2).
In short, this hypothesis suggests that, on one hand, G-CSF prompts neutrophil activation, ending up in a degranulation, releasing some neutrophil’s protease enzymes such as neutrophil elastase, cathepsin G, dipeptidyl peptidase 1I and matrix metalloprotease-9 [54] that can ultimately accumulate in BM and lead to the degradation of cell adhesion-related molecules such as VCAM-1[55], SDF-1 and C-kit [53]. Moreover, G-CSF increases CD26, a serine exopeptidase on the surface of endothelial cells that causes internalization and degradation of VE-cadherin, opening endothelial boundaries. G-CSF also triggers erythroblasts to secrete fibroblast growth factor-23 (FGF-23), which counteracts the function of CXCR-4. On the other hand, following G-CSF administration it has been registered a reduce in the number of monocytes, macrophages and suppression of osteoblasts in bone marrow [56], which could enhance the mobilization process [57,58]. In addition to that, it seems that G-CSF induces sympathetic neurons to release noradrenaline which promotes a suppression of osteoblast [59], and macrophages [60] to release unknown factors that suppress SDF-1 expression on the surface of niche cells.
Overall, G-CSF affects various processes that in combination can disrupt the bone marrow microenvironment, leading to the mobilization of hematopoietic stem cells. These processes include reducing SDF-1 expression, opening endothelial barriers, and counteracting the function of CXCR4 due to the increase of erythroblasts-derived fibroblast growth factor-23 (FGF-23) [61].
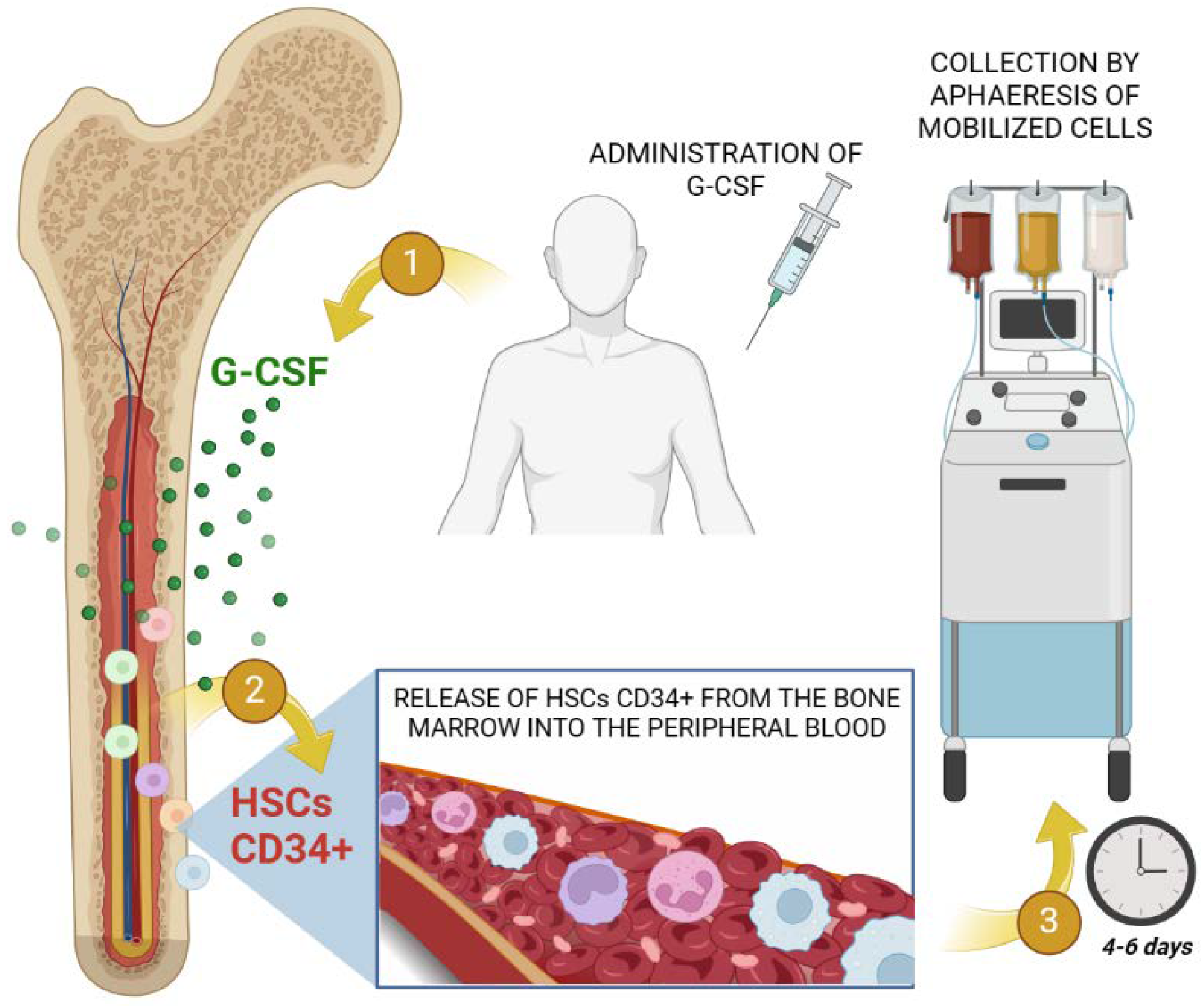
Mobilization of HSC from bone marrow via G-CSF is undoubtedly a crucial phase in both auto-HSCT and allo-HSCT. The process encompasses a G-CSF average dosage administration in patients of approximately 5-10 μg/kg for 5-7 days, obtaining around 2 x 106 CD34+ cells per kg weight. Leukapheresis is generally performed on day 5 (Figure 3). It is not compulsory to quantify the amount of CD34+ cells but nowadays CD34+ cell count in mobilized peripheral blood product is the most important parameter of graft quality, due to the fact that it is the only predictor of stable engraftment after auto-HSCT. Finally, HSC will be cryopreserved using dimethyl sulfoxide (DMSO) until infusion [39].
Therefore, it is fairly noticeable that nowadays, there is a large number of G-CSF mobilized and cryopreserved apheresis products both from donors and patients, stored in biobanks for HSCTs main purpose, which could be also used to obtain T lymphocytes and NK cells for subsequent generation of CAR products.
3.1. CAR-T Immunotherapy
Since CAR-T therapy has been proposed as an ultimate procedure after standard cancer treatment, it seems reasonable to think that T lymphocytes for CAR-T therapy could be isolated and expanded from the same cryopreserved product obtained for HSCT, in order to combine both treatments. The paradigmatic case of this potential use would be multiple myeloma or NHL, where many patients undergo CAR-T therapy after relapse following autologous transplantation [62]. In the case of LAL, successful cases have been described with the infusion of allogeneic CAR-T cells from the donor after early relapse following allogeneic HSCT [63,64,65]. On the other hand, the hematopoietic toxicities of autologous CAR-T cell therapies are a major concern, and there are many cases reported that have required the infusion of hematopoietic progenitors as rescue [66,67].
In all these circumstances it would be beneficial to have a single procedure for apheresis, but all of them require the administration of G-CSF as a mobilization agent, and this growth factor has not been considered for the manufacture of the CAR-T products among the different parameters that have been previously well determined [4].
Several studies on the functionality of T lymphocytes after mobilization provide reasons not to use this cell source for CAR-T cell production. In the late 1990s, a series of publications studied mobilized stem cell samples and concluded that G-CSF has a pleiotropic effect in different cell populations, including monocytes which inhibit T cell proliferation and function [48,68]. First, Young et al. informed the induction of immunosuppressive cells associated with myelopoiesis and stimulated by GM-CSF and IL-3 [69]. Subsequently, Ino K. et al analyzed the mobilized apheresis product of 21 patients with malignancies. They carried out different cellular inhibition assays and showed that the low-density fraction enriched with CD14+ significantly inhibited the functions of T cells and led to activation-induced apoptosis of these cells [70]. These findings suggest that the augmented monocyte fraction is responsible for the impaired function and inhibition of T-cells after mobilization
This could be an important reason to not utilize G-CSF mobilized PBMNC for immunotherapeutic purposes. However, one feasible way to mitigate this T-cell misfunction is to isolate and expand the T-cell CD3+ fraction specifically [71]. Nowadays there are a variety of kits widely used that are designed to enrich solely this CD3+ fraction [72,73]. Studies such as that by Ji et al. demonstrate that G-CSF priming did not change the total number of CD3+ cells in marrow grafts but decreased CD4+ cells and increased CD8+ cells, resulting in a significant reduction in CD4:CD8 ratio [74].
Besides the interactions with other non-lymphocytic cells, several publications have concluded that G-CSF promotes the generation of Treg phenotype in T cells [75], which produces IL-10 [76] and transforming growth factor-β promoting an increase in lymphocyte T helper Th2 differentiation while suppressing Th1 differentiation [77]. This information was also supported by other publications in which transcriptomic analysis of G-CSF mobilized peripheral blood from donors, revealed an upregulation of Th2 genes, Tregs; and a downregulation of Th17 and Th1 genes [78]. Overall, these findings suggest that G-CSF negatively affects antigen-specific T cells, and T-cell banking before mobilization might be the best option to optimize T-cell production [79] because the efficiency of CAR-T cells that are generated from T cells exposed to G-CSF could be reduced
In sharp contrast to all these results questioning the functionality of T lymphocytes in PBMNCs obtained after mobilization are the clinical results of thousands of allogeneic transplants of mobilized PB in which neither graft versus host disease (GVHD) nor graft-versus leukemia (GVL) effect are reduced by mobilization. Moreover, frozen T-lymphocytes from the original mobilized PBMC graft product are used clinically for immunotherapeutic purposes for the generation of functional virus-specific T cells with anti-viral and tumorigenic functions [80,81,82], and more routinely for donor lymphocyte infusions (DLI) as a treatment for recurrent malignant neoplasms. DLI is capable of eradicating minimal residual disease and rescuing hematological decline, being able to induce lasting remissions [83]. It mainly works well to treat mixed chimerism in which there is a persistent or increasing number of malignant host cells after allo-HSCT, which is a predictor of disease relapse. DLI [84] has the potential to improve the GVL effect, reducing the risk of relapse in patients with mixed chimerism. With the increased use of unrelated donors for hematopoietic cell transplantation, there is renewed interest in the use of large volumes of frozen mobilized apheresis products that could be the source for DLI as a complementary treatment to prevent or treat the appearance of mixed chimeras.
Considering the benefits previously mentioned and with the knowledge of the clinical success of T lymphocytes after mobilization [80,81,82] researchers have recently opened the debate about its utilization for purposes such as CAR-T cell production. In fact, it is not unreasonable to think that with the current sample processing and enrichment techniques, one could use a determined volume of the mobilized product to isolate solely the T-cell fraction without altering the functionality of these cells[72]. Some examples using a variety of cytokines and activation protocols for T-cells [85] can obtain different subpopulations. In addition, other techniques such as Cell Sorting could solve the problems resulting from the effect that other cells like monocytes have on the suppression of T lymphocytes by choosing the optimal CD3+ subpopulation [86].
Recently, innovative studies have analyzed the effect of the administration of G-CSF to obtain CAR-T cells such as the study conducted by Cummins et al [11]. They have extensively evaluated this effect through the analysis of cryopreserved apheresis samples derived from 4 healthy donors, both mobilized with G-CSFrh and unmobilized. The study included the cellular culture where T cells were activated ex vivo using CD3/CD28 beads, transduced with a lentiviral vector, and expanded in a specific cell culture media enriched with human serum and IL-7/IL-15. Subsequent analysis by flow cytometry, CyTOF, single-cell RNA sequencing (RNA-Seq), and metabolomics by mass spectrometry showed, in a non-significant manner, a higher expression of the CAR construct in non-mobilized cells. Remarkably, no significant differences were found in terms of the CD4/CD8 ratio obtained in both groups, nor differences resulted in terms of degranulation, cytokine production, and in vitro tumor cytotoxicity assays. In vivo analysis of xenografts in an acute myeloid leukemia mouse model showed no statistical differences in mouse body weight, toxicity, or survival in both analyzed groups. Finally, the RNAseq analysis showed a similar expression transcriptomic profile for both groups. The research indicates that the antitumor efficacy and in vivo toxicity of these products are comparable, with no significant differences observed in the product exposed to G-CSF, as determined through multi-omics analyses
In another recent comprehensive study, Canesin et al [87] determined the impact on apheresis product of mobilization or not with G-CSF and plerixafor from 30 healthy donors by assessing immune cell composition, T cell phenotype, and T cell functionality in controlling AML tumor growth following anti-CD33 CAR transduction. The resulting in vivo immunophenotypic analysis yields remarkably interesting results showing that mobilization decreases the overall percentage of CD3+ T cells but increases naive (CD45RA+/CCR7+) T cells and decreases the T cell population of effector memory (CD45RA-/CCR7-) and central memory (CD45RA-/CCR7+). In vitro functional cytotoxic assays demonstrated that mobilized-antiCD33-CAR T cells were as effective as non-mobilized-antiCD33-CAR T cells in killing CD33+ AML cells.
In the realm of multiple myeloma treatment, Battram et al [88] propose utilizing G-CSF-mobilized leukapheresis products to obtain CAR-T cells targeting BCMA. Their study reveals the minimal impact of G-CSF on T cell phenotype, both in vitro and in patients. Pre-treatment with G-CSF does not affect T cell survival or apoptosis during culturing, activation, transduction, and expansion. CAR T production is unaffected, with no impact on cell growth, differentiation, or anti-tumor killing capacity. Exhaustion markers like PD-1, LAG-3, TIM-3, and TIGIT show no significant increase with G-CSF, except for reduced TIM-3 expression in CD8+ cells, this being the only significant difference indicating less exhaustion.
In PBMCs obtained from MM patients through G-CSF mobilized and non-mobilized apheresis [88], no significant differences exist in the CD4:CD8 ratio or Treg population. G-CSF reduces TSCM cell frequency, specifically CD8+ TSCM cells, without affecting other memory and effector T cell populations. Although CAR transduction is statistically lower in mobilized cells, the high expansion rates and increased T-cell numbers compensate for this reduction. In vivo functionality of CAR-T cells, assessed in a mouse model with MM xenografts, shows similar disease development in CAR-T-treated mice from mobilized and non-mobilized cells. In summary, the study supports the feasibility of using CAR-T cells obtained from G-CSF-mobilized leukapheresis products for treating multiple myeloma. Despite a reduction in CAR transduction, the high cell expansion compensates for this effect, suggesting that these CAR-T cells could be an effective therapeutic option for multiple myeloma.
Another recent retrospective study by Künkele et al [10] has been carried out using cryopreserved PBMCs mobilized apheresis products derived from 8 patients diagnosed with neuroblastoma. Samples were obtained early in the treatment protocol. To activate and expand the T cells, a monocyte depletion of thawed PBSC units was required due to the outgrowth of monocytes. Furthermore, the CAR transduction efficiency in CD4+ and CD8+ mobilized products to generate the CAR-T cells product ranged from 65-75%. Besides that, flow cytometric analysis showed that cryopreserved G-CSF-stimulated apheresis products contain sufficient numbers of CD4+ and CD8+ T cell precursors with a naïve and central memory phenotype that showed increased replicating potential and high capacity to generate large numbers of effector T cells after tumor stimulation.
Lastly, other research works related to human γδ T Cells such as the study performed by Otto et al [89], have shown that the PBMCs G-CSF mobilized derived from healthy donors succeeded in retaining their cytotoxicity and in the production of a variety of cytokines. These cells produced considerable amounts of IL-6, IFNγ, TNFα, and GM-CSF, cytokines that are important in mediating cytotoxicity and supporting inflammation. Other cytokines released were IL-4, IL-5, IL-8, IL-13, G-CSF, MCP-1, and MIP-1β, relevant factors that help attract other effector cells. As for the cell phenotype, significant differences were observed in the phenotype of isolated γδ-T cells compared to non-mobilized γδ-peripheral donor T cells. There was increased expression of CD8, CD56, CD28, and CD11b/CD18 (MAC-1) in these isolated cells. This evidence suggests that these cells would have the ability to modulate the immune responses and play a significant role in adoptive immunotherapy in the same way the non-mobilized cells would do.
Overall, these experimental publications have some things in common. As Battram et al mentioned, the way they proceed is different from the late 90’s past experimental approaches due to the reduced exposure time of the G-CSF and the starting material for the ex vivo culture of the CAR-T. This source of starting material should be highly purified T cells and not the whole PBMC fraction [88]. This would lead to a major advantage thanks to the removal of T cell inhibitory cells such as monocytes CD14+. This particular principle is not against the Advanced Therapy Medicinal Products (ATMP) guidelines of CAR-T therapy. Guidance [90] of ATMPs indicates that the CAR T cell manufacturing process usually requires 12 days, and starts, in short, with the isolation of T cells from the leukapheresis product of a patient, followed by activation and genetic modification of the cells to express the respective CAR. Thus, given that these cells have to be isolated, cultured, usually sorted by cell phenotype and treated appropriately, it seems reasonable to assume that prior mobilization of G-CSF would not significantly affect the protocol, with these lymphocytes being fully functional as explained throughout this section.
Besides that, as we mentioned previously, G-CSF mobilization products may not be the first indicated option to obtain T-cell fraction, however, it can be seen that cryopreserved stored mobilized PBMCs could perform as well as non-mobilized do in terms of CAR-T production.
3.2. CAR-NK Immunotherapy
One of the main limitations in the development of CAR-NK is obtaining NK cells in massive quantities since these cells are found in a lower proportion (from 5% to 15% of blood circulating lymphocytes) than T or B lymphocytes under normal circumstances [38]. Source and clinical scale-up of NK cells with long-lasting cytotoxicity activity are the main challenges and the strategies differ depending on whether the sample is freshly isolated, activated, or in vitro expanded because of the variety of phenotypic and functional differences found. Nowadays, the principal source of obtaining NK-CAR for clinical trials is the NK92 [91,92] cell line due to the lack of variability that this cell line offers, together with its unlimited proliferation ability and modest cost. Nevertheless, there are some drawbacks including the lack of CD16 expression and higher tumorigenicity risk because is a tumoral cell line[93]. To circumvent these problems, it seems necessary to find new tools to broaden the source of these cells in order to obtain an “Off-The-Shelf” therapy. Other NK cell sources have been studied, for instance, isolated from PBMCs, UCBs, CD34+ hematopoietic progenitor cells [8] and induced Pluripotent Stem cells (iPSCs) [9].
One source being recently quite used is PBMC-derived CAR-NK. There are some fairly good advantages such as the uncomplicated way of collection and the lack of GVHD that allows being isolated from matched and mismatched HLA donors [94]. Besides that, these collected cells using isolation kits such as CD3+ depletion and CD56+ enrichment, are CD56dimCD16+ which is a more cytotoxic and mature phenotype than NK92 line [95]. Also, even though they have a reduced capacity for proliferation when compared to other phenotypes, with proper isolation and cell culture these cells can expand quite nicely. Another option is to obtain NK from UCB in the same manner although it can be seen reduced numbers of UCB NK due to the limited volume of UCB units [96].
Since PBMCs seem to be a good option to obtain NK cells for CAR manufacture, it is not far-fetched to think that, similarly to CAR-T, cryopreserved G-CSF mobilized apheresis products could be used to obtain these cells. Nevertheless, as with CAR-T, the apheresis product mobilized using G-CSF is not a common protocol to obtain the NK cell isolate, since the apheresis mobilization process does not aim to increase the number of lymphocytes or NK cells [97]. Some studies indicate that the mobilization process can negatively affect the functionality of NK cells [98], due to the over-proliferation of polymorphonuclear myeloid-derived suppressor cells (PMN-MDSCs) subpopulations [99], and the inhibition of IFN-Y secretion by them [100].
However, there is a debate well reviewed by Gazitt Y. [101] in which different publications related to cell populations from mobilized PBMCs such as CD34+ cells, T-cells, NK cells and dendritic cells are analyzed including his own experimental results. In this paper is highlighted that there are other studies that do not find differences between the mobilized and non-mobilized products in terms of NK, noticing a lack of consensus. In addition, a great heterogeneity is observed in terms of NK cell mobilization between patients, perceiving that those patients with relatively low NK cell or poor NK activity before mobilization had poor PBSC collection post-mobilization in terms of quantity and functionality. Thus, it is pointed out that these variables, together with the small number of patients included in some of the experimental publications analyzed, should be taken into consideration and the mobilization process itself could not be an issue
A recent publication by Xiong et al [98], showed significant differences between non-mobilized and mobilized-derived PBSC NK. In this case, mobilized NK fraction showed a CD56bright+ CD16- phenotype population suggesting that G-CSF favors the accumulation of less mature NK cell subsets. Besides that, they observed a decreased expansion rate in mobilized NK compared to non-mobilized NK. However, when IL-15 is added to the culture, it can be observed a progression and restoration in cytokine secretion profile in vitro, suggesting that these cells can experience a maturation. Notwithstanding the potential perception of this aspect as a drawback, expeditious dismissal of its optimization is unwarranted. It is imperative to recognize that presently, one of the preeminent sources of NK-CAR resides in NK-92, a cell line characterized by CD56+CD16- which exhibits low cytotoxicity and in vivo persistence. Besides that, as we mentioned before, the manufacture of CAR requires an optimal culture of the highly purified cells, thus it is reasonable to think that to design a CAR NK cell product, a lower maturation stage and an ex vivo culture modulation through cytokines should be a promising alternative source to certain NK-CAR phenotype [102,103].
Otherwise, as NK cells originate from CD34+ hematopoietic stem cells, one feasible way to obtain NK cells is to focus on the source of this population, which encompasses bone marrow or umbilical cord blood. As shown by Oberoi et al. [104] in their study, the effect of G-CSF can be used to obtain the mobilized CD34+ population also from peripheral blood in massive quantities and in a simple manner, to subsequently differentiate into NK cells. These cells are then expanded and differentiated into mature NK cells using a cocktail of cytokines in a culture system. There have been many advances in the way of culturing, expanding and differentiating functional NKs, for example, by co-culture with K562 feeder cells[105] that co-express the 4-1BB ligand and membrane-anchored IL-15 and IL-21, giving excellent results and solving the problem found in variability between donors. The resulting CD56+CD3− NK cells are mostly similar to PB NK cells, express NK cell-activating receptors, and exhibit potent cytotoxicity against leukemic cells in vitro and in vivo.
Some studies, such as the one published by Patel et al [97], have designed and evaluated a CAR-NK development protocol using NK derived from PBSC mobilized with G-CSF. The results derived from these studies achieved the differentiation of NK cells in the culture using cytokines such as IL-7 and IL-15 obtaining cell proliferation and expansion rates equivalent to those obtained from UCB, with a CD56+/CD16+/CD94+ phenotype showed by 10 to 40% of the CD56+ cells. They concluded that large-scale generation of CAR-NK products from PBSC source is feasible and compatible with good manufacturing practice (GMP) which is mandatory for ATMP products, and it is defined as potent products manufactured safely according to standardized methods under controlled, reproducible, and auditable conditions [39].
Finally, it is remarkable the success of Zhu et al [106] in the invariant natural killer (iNKT) cell generation, through TCR genetic engineering of peripheral CD34+ HSCs samples mobilized by G-CSF. iNKT are immune system cells with a strong potential to fight cancer. However, its clinical use has been limited due to its scarcity in cancer patients. In this study, they developed a proof of concept for cell therapy using HSC-iNKT, to provide sustained therapeutic levels of iNKT throughout treatment. Using a mouse model with human hematopoietic stem cell grafts and human T cell receptor genetic engineering for iNKT, these researchers demonstrated the efficient and long-term generation of HSC-iNKT cells in vivo.
These HSC-iNKT cells showed similarity to natural human iNKT cells and exhibited multiple mechanisms to attack tumor cells, effectively suppressing tumor growth in mouse models with human tumor xenografts. In addition, preclinical safety studies that were performed revealed no toxicity or tumor formation associated with HSC-iNKT cell therapy. Mobilized HSCs source presents a greater number of iNKT than non-mobilized samples, emphasizing its potential and safety in hematologic malignancy cell therapy.
Overall, similarly to CAR-T Immunotherapy, as CAR-NK manufacture is yet to be explored, it seems necessary to increase the experimental work related to NK cells and to exploit other potential sources available.
4. Conclusions
In the context of manufacturing CAR immunotherapeutic products, mobilization with G-CSF that is performed to increase the number of precursor hematopoietic cells in the peripheral blood before cell collection does not adversely affect the manufacture of CAR-engineered cells for several reasons.
Although G-CSF mobilization can increase the number of T cells or NK cells in the peripheral blood, it usually preserves the functionality and essential features of these cells. This includes the ability of these cells to be genetically modified and subsequently expanded in the CAR-T or CAR-NK manufacturing process. T cells and NK cells, mobilized with G-CSF are compatible with the collection, genetic modification, and expansion processes carried out in the laboratory to produce CAR-T or CAR-NK. Mobilization with G-CSF does not adversely affect the ability of these cells to be modified with CAR or their ability to proliferate in sufficient quantities.
Specific studies and analyses have evaluated the impact of G-CSF on these cells used in cell therapy, and the results have suggested that cells mobilized by G-CSF are suitable for CAR manufacturing, with comparable results in terms of efficiency and safety. It is important to note that although mobilization with G-CSF is well tolerated, there may be variations among patients, and it is essential to conduct specific evaluations in each case to ensure the efficacy and safety of CAR therapy.
This review highlights the benefits and the potential feasibility of creating and designing cellular immunotherapies that complement hematopoietic stem cell transplantation. The focus is on using the same mobilized collection for both purposes and finding a method to utilize previously cryopreserved mobilized units. This approach not only streamlines the process by using the same collection for multiple therapeutic purposes but also considers the potential advantages associated with the use of historically cryopreserved T cells, particularly in terms of reduced exposure to chemotherapy.
Author Contributions
MDC-L and CH: study conception and design; AB-R and AM-L: searching and selecting publications; MDC-L, AB-R and AM-L: writing original draft preparation; CH, AB-R and AM-L: writing, review and editing. MDC-L and CH: supervision. All authors contributed to the article and approved the submitted version.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Xu, Z.; Huang, X. Cellular Immunotherapy for Hematological Malignancy: Recent Progress and Future Perspectives. Cancer Biol Med 2021, 18. [CrossRef]
- Minda, A.G.; Awel, F.S.; Seifudin, K.A.; Gezahegne, M.K. Immunotherapy against Cancer: A Comprehensive Review. Journal of Cancer Research and Experimental Oncology 2016, 8. [CrossRef]
- Rohaan, M.W.; Wilgenhof, S.; Haanen, J.B.A.G. Adoptive Cellular Therapies: The Current Landscape. Virchows Archiv 2019, 474.
- Fesnak, A.; Lin, C.Y.; Siegel, D.L.; Maus, M. V. CAR-T Cell Therapies From the Transfusion Medicine Perspective. Transfus Med Rev 2016, 30.
- Li, Y.; Hermanson, D.L.; Moriarity, B.S.; Kaufman, D.S. Human IPSC-Derived Natural Killer Cells Engineered with Chimeric Antigen Receptors Enhance Anti-Tumor Activity. Cell Stem Cell 2018, 23. [CrossRef]
- Berger, M.; Barone, M.; Spadea, M.; Saglio, F.; Pessolano, R.; Fagioli, F. HSCT with Mismatched Unrelated Donors: Bone Marrow versus Peripheral Blood Stem Cells Sources in Pediatric Patients. Pediatr Transplant 2022, 26. [CrossRef]
- Grieco, D.; Lacetera, N.; Macis, M.; Di Martino, D. Motivating Cord Blood Donation with Information and Behavioral Nudges. Sci Rep 2018, 8. [CrossRef]
- Spanholtz, J.; Preijers, F.; Tordoir, M.; Trilsbeek, C.; Paardekooper, J.; de Witte, T.; Schaap, N.; Dolstra, H. Clinical-Grade Generation of Active NK Cells from Cord Blood Hematopoietic Progenitor Cells for Immunotherapy Using a Closed-System Culture Process. PLoS ONE 2011, 6. [CrossRef]
- Cany, J.; van der Waart, A.B.; Spanholtz, J.; Tordoir, M.; Jansen, J.H.; van der Voort, R.; Schaap, N.M.; Dolstra, H. Combined IL-15 and IL-12 Drives the Generation of CD34+-Derived Natural Killer Cells with Superior Maturation and Alloreactivity Potential Following Adoptive Transfer. Oncoimmunology 2015, 4. [CrossRef]
- Künkele, A.; Brown, C.; Beebe, A.; Mgebroff, S.; Johnson, A.J.; Taraseviciute, A.; Rolczynski, L.S.; Chang, C.A.; Finney, O.C.; Park, J.R.; et al. Manufacture of Chimeric Antigen Receptor T Cells from Mobilized Cyropreserved Peripheral Blood Stem Cell Units Depends on Monocyte Depletion. Biology of Blood and Marrow Transplantation 2019, 25. [CrossRef]
- Cummins, K.D.; Gupta, A.; Beyar-Katz, O.; Li, Y.; Chatterjee, P.; Kippner, L.; Shestova, O.; Eiva, M.A.; Salas-Mckee, J.; Yeago, C.; et al. G-CSF Mobilized Apheresis As an Alternative Source of CAR T-Cells. Blood 2022, 140. [CrossRef]
- Rettinger, E.; Kuçi, S.; Naumann, I.; Becker, P.; Kreyenberg, H.; Anzaghe, M.; Willasch, A.; Koehl, U.; Bug, G.; Ruthardt, M.; et al. The Cytotoxic Potential of Interleukin-15-Stimulated Cytokine-Induced Killer Cells against Leukemia Cells. Cytotherapy 2012, 14. [CrossRef]
- Rohaan, M.W.; Borch, T.H.; van den Berg, J.H.; Met, Ö.; Kessels, R.; Geukes Foppen, M.H.; Stoltenborg Granhøj, J.; Nuijen, B.; Nijenhuis, C.; Jedema, I.; et al. Tumor-Infiltrating Lymphocyte Therapy or Ipilimumab in Advanced Melanoma. New England Journal of Medicine 2022, 387. [CrossRef]
- Perko, R.; Kang, G.; Sunkara, A.; Leung, W.; Thomas, P.G.; Dallas, M.H. Gamma Delta T Cell Reconstitution Is Associated with Fewer Infections and Improved Event-Free Survival after Hematopoietic Stem Cell Transplantation for Pediatric Leukemia. Biology of Blood and Marrow Transplantation 2015, 21. [CrossRef]
- Douka, S.; Brandenburg, L.E.; Casadidio, C.; Walther, J.; Garcia, B.B.M.; Spanholtz, J.; Raimo, M.; Hennink, W.E.; Mastrobattista, E.; Caiazzo, M. Lipid Nanoparticle-Mediated Messenger RNA Delivery for Ex Vivo Engineering of Natural Killer Cells. Journal of Controlled Release 2023, 361. [CrossRef]
- Eshhar, Z.; Waks, T.; Gross, G.; Schindler, D.G. Specific Activation and Targeting of Cytotoxic Lymphocytes through Chimeric Single Chains Consisting of Antibody-Binding Domains and the γ or ζ Subunits of the Immunoglobulin and T-Cell Receptors. Proc Natl Acad Sci U S A 1993, 90. [CrossRef]
- Eshhar, Z. Tumor-Specific T-Bodies: Towards Clinical Application. Cancer Immunology Immunotherapy 1997, 45. [CrossRef]
- Hollyman, D.; Stefanski, J.; Przybylowski, M.; Bartido, S.; Borquez-Ojeda, O.; Taylor, C.; Yeh, R.; Capacio, V.; Olszewska, M.; Hosey, J.; et al. Manufacturing Validation of Biologically Functional T Cells Targeted to CD19 Antigen for Autologous Adoptive Cell Therapy. Journal of Immunotherapy 2009, 32. [CrossRef]
- Davila, M.L.; Brentjens, R.; Wang, X.; Rivière, I.; Sadelain, M. How Do CARs Work? Oncoimmunology 2012, 1. [CrossRef]
- Maher, J.; Brentjens, R.J.; Gunset, G.; Rivière, I.; Sadelain, M. Human T-Lymphocyte Cytotoxicity and Proliferation Directed by a Single Chimeric TCRζ/CD28 Receptor. Nat Biotechnol 2002, 20. [CrossRef]
- Carpenito, C.; Milone, M.C.; Hassan, R.; Simonet, J.C.; Lakhal, M.; Suhoski, M.M.; Varela-Rohena, A.; Haines, K.M.; Heitjan, D.F.; Albelda, S.M.; et al. Control of Large, Established Tumor Xenografts with Genetically Retargeted Human T Cells Containing CD28 and CD137 Domains. Proc Natl Acad Sci U S A 2009, 106. [CrossRef]
- Chmielewski, M.; Abken, H. TRUCKs: The Fourth Generation of CARs. Expert Opin Biol Ther 2015, 15.
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. New England Journal of Medicine 2018, 378. [CrossRef]
- Locke, F.L.; Ghobadi, A.; Jacobson, C.A.; Miklos, D.B.; Lekakis, L.J.; Oluwole, O.O.; Lin, Y.; Braunschweig, I.; Hill, B.T.; Timmerman, J.M.; et al. Long-Term Safety and Activity of Axicabtagene Ciloleucel in Refractory Large B-Cell Lymphoma (ZUMA-1): A Single-Arm, Multicentre, Phase 1–2 Trial. Lancet Oncol 2019, 20. [CrossRef]
- Qayed, M.; McGuirk, J.P.; Myers, G.D.; Parameswaran, V.; Waller, E.K.; Holman, P.; Rodrigues, M.; Clough, L.F.; Willert, J. Leukapheresis Guidance and Best Practices for Optimal Chimeric Antigen Receptor T-Cell Manufacturing. Cytotherapy 2022, 24.
- Wang, X.; Rivière, I. Clinical Manufacturing of CAR T Cells: Foundation of a Promising Therapy. Mol Ther Oncolytics 2016, 3.
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. New England Journal of Medicine 2017, 377. [CrossRef]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. New England Journal of Medicine 2019, 380. [CrossRef]
- Abramson, J.S.; Palomba, M.L.; Gordon, L.I.; Lunning, M.A.; Wang, M.; Arnason, J.; Mehta, A.; Purev, E.; Maloney, D.G.; Andreadis, C.; et al. Lisocabtagene Maraleucel for Patients with Relapsed or Refractory Large B-Cell Lymphomas (TRANSCEND NHL 001): A Multicentre Seamless Design Study. The Lancet 2020, 396. [CrossRef]
- Wang, M.; Munoz, J.; Goy, A.; Locke, F.L.; Jacobson, C.A.; Hill, B.T.; Timmerman, J.M.; Holmes, H.; Jaglowski, S.; Flinn, I.W.; et al. KTE-X19 CAR T-Cell Therapy in Relapsed or Refractory Mantle-Cell Lymphoma. New England Journal of Medicine 2020, 382. [CrossRef]
- Cappell, K.M.; Kochenderfer, J.N. Long-Term Outcomes Following CAR T Cell Therapy: What We Know so Far. Nat Rev Clin Oncol 2023, 20.
- Grover, P.; Veilleux, O.; Tian, L.; Sun, R.; Previtera, M.; Curran, E.; Muffly, L. Chimeric Antigen Receptor T-Cell Therapy in Adults with B-Cell Acute Lymphoblastic Leukemia. Blood Adv 2022, 6.
- Park, J.H.; Nath, K.; Devlin, S.M.; Sauter, C.S.; Palomba, M.L.; Shah, G.; Dahi, P.; Lin, R.J.; Scordo, M.; Perales, M.A.; et al. CD19 CAR T-Cell Therapy and Prophylactic Anakinra in Relapsed or Refractory Lymphoma: Phase 2 Trial Interim Results. Nat Med 2023, 29. [CrossRef]
- Cliff, E.R.S.; Kelkar, A.H.; Russler-Germain, D.A.; Tessema, F.A.; Raymakers, A.J.N.; Feldman, W.B.; Kesselheim, A.S. High Cost of Chimeric Antigen Receptor T-Cells: Challenges and Solutions. American Society of Clinical Oncology Educational Book 2023. [CrossRef]
- Beatty, G.L.; Haas, A.R.; Maus, M. V.; Torigian, D.A.; Soulen, M.C.; Plesa, G.; Chew, A.; Zhao, Y.; Levine, B.L.; Albelda, S.M.; et al. Mesothelin-Specific Chimeric Antigen Receptor MRNA-Engineered T Cells Induce Anti-Tumor Activity in Solid Malignancies. Cancer Immunol Res 2014, 2. [CrossRef]
- Rubnitz, J.E.; Inaba, H.; Ribeiro, R.C.; Pounds, S.; Rooney, B.; Bell, T.; Pui, C.H.; Leung, W. NKAML: A Pilot Study to Determine the Safety and Feasibility of Haploidentical Natural Killer Cell Transplantation in Childhood Acute Myeloid Leukemia. Journal of Clinical Oncology 2010, 28. [CrossRef]
- Myers, J.A.; Miller, J.S. Exploring the NK Cell Platform for Cancer Immunotherapy. Nat Rev Clin Oncol 2021, 18.
- Freud, A.G.; Mundy-Bosse, B.L.; Yu, J.; Caligiuri, M.A. The Broad Spectrum of Human Natural Killer Cell Diversity. Immunity 2017, 47.
- Carreras, E.; Dufour, C.; Mohty, M.; Kröger, N. The EBMT Handbook: Hematopoietic Stem Cell Transplantation and Cellular Therapies; 2018;
- Peter Gale, R. E Donnall Thomas (1920-2012). Leukemia 2013, 27. [CrossRef]
- Scheding, S.; Brugger, W.; Mertelsmann, R.; Kanz, L. Peripheral Blood Stem Cells: In Vivo Biology and Therapeutic Potential. Stem Cells 1994, 12. [CrossRef]
- Theyab, A.; Alsharif, K.F.; Alzahrani, K.J.; Oyouni, A.A.A.; Hawsawi, Y.M.R.; Algahtani, M.; Alghamdi, S.; Alshammary, A.F. New Insight into Strategies Used to Develop Long-Acting G-CSF Biologics for Neutropenia Therapy. Front Oncol 2023, 12.
- Demetri, G.D.; Griffin, J.D. Granulocyte Colony-Stimulating Factor and Its Receptor. Blood 1991, 78.
- Bussolino, F.; Ziche, M.; Ming Wang, J.; Alessi, D.; Morbidelli, L.; Cremona, O.; Bosia, A.; Marchisio, P.C.; Mantovani, A. In Vitro and in Vivo Activation of Endothelial Cells by Colony-Stimulating Factors. Journal of Clinical Investigation 1991, 87. [CrossRef]
- Gabrilove, J.L.; Jakubowski, A.; Scher, H.; Sternberg, C.; Wong, G.; Grous, J.; Yagoda, A.; Fain, K.; Moore, M.A.S.; Clarkson, B.; et al. Effect of Granulocyte Colony-Stimulating Factor on Neutropenia and Associated Morbidity Due to Chemotherapy for Transitional-Cell Carcinoma of the Urothelium. New England Journal of Medicine 1988, 318. [CrossRef]
- Deluca, E.; Sheridan, W.P.; Watson, D.; Szer, J.; Begley, C.G. Prior Chemotherapy Does Not Prevent Effective Mobilisation by G-Csf of Peripheral Blood Progenitor Cells. Br J Cancer 1992, 66. [CrossRef]
- Chang, H.H.; Liou, Y.S.; Sun, D.S. Hematopoietic Stem Cell Mobilization. Tzu Chi Med J 2022, 34.
- Reyes, E.; García-Castro, I.; Esquivel, F.; Hornedo, J.; Cortes-Funes, H.; Solovera, J.; Alvarez-Mon, M. Granulocyte Colony-Stimulating Factor (G-CSF) Transiently Suppresses Mitogen-Stimulated T-Cell Proliferative Response. Br J Cancer 1999, 80. [CrossRef]
- Russell, N.H.; Byrne, J.L. Allogeneic Transplantation Using Peripheral Blood Stem Cells. Best Pract Res Clin Haematol 2001, 14. [CrossRef]
- Favre, G.; Beksaç, M.; Bacigalupo, A.; Ruutu, T.; Nagler, A.; Gluckman, E.; Russell, N.; Apperley, J.; Szer, J.; Bradstock, K.; et al. Differences between Graft Product and Donor Side Effects Following Bone Marrow or Stem Cell Donation. Bone Marrow Transplant 2003, 32. [CrossRef]
- Bendall, L.J.; Bradstock, K.F. G-CSF: From Granulopoietic Stimulant to Bone Marrow Stem Cell Mobilizing Agent. Cytokine Growth Factor Rev 2014, 25.
- Fruehauf, S.; Tricot, G. Comparison of Unmobilized and Mobilized Graft Characteristics and the Implications of Cell Subsets on Autologous and Allogeneic Transplantation Outcomes. Biology of Blood and Marrow Transplantation 2010, 16.
- Lévesque, J.P.; Hendy, J.; Takamatsu, Y.; Simmons, P.J.; Bendall, L.J. Disruption of the CXCR4/CXCL12 Chemotactic Interaction during Hematopoietic Stem Cell Mobilization Induced by Gcsf or Cyclophosphamide. Journal of Clinical Investigation 2003, 111. [CrossRef]
- Heissig, B.; Hattori, K.; Dias, S.; Friedrich, M.; Ferris, B.; Hackett, N.R.; Crystal, R.G.; Besmer, P.; Lyden, D.; Moore, M.A.S.; et al. Recruitment of Stem and Progenitor Cells from the Bone Marrow Niche Requires MMP-9 Mediated Release of Kit-Ligand. Cell 2002, 109. [CrossRef]
- Lévesque, J.P.; Takamatsu, Y.; Nilsson, S.K.; Haylock, D.N.; Simmons, P.J. Vascular Cell Adhesion Molecule-1 (CD106) Is Cleaved by Neutrophil Proteases in the Bone Marrow Following Hematopoietic Progenitor Cell Mobilization by Granulocyte Colony-Stimulating Factor. Blood 2001, 98. [CrossRef]
- Semerad, C.L.; Christopher, M.J.; Liu, F.; Short, B.; Simmons, P.J.; Winkler, I.; Levesque, J.P.; Chappel, J.; Ross, F.P.; Link, D.C. G-CSF Potently Inhibits Osteoblast Activity and CXCL12 MRNA Expression in the Bone Marrow. Blood 2005, 106. [CrossRef]
- Miyamoto, K.; Yoshida, S.; Kawasumi, M.; Hashimoto, K.; Kimura, T.; Sato, Y.; Kobayashi, T.; Miyauchi, Y.; Hoshi, H.; Iwasaki, R.; et al. Osteoclasts Are Dispensable for Hematopoietic Stem Cell Maintenance and Mobilization. Journal of Experimental Medicine 2011, 208. [CrossRef]
- Winkler, I.G.; Sims, N.A.; Pettit, A.R.; Barbier, V.; Nowlan, B.; Helwani, F.; Poulton, I.J.; Van Rooijen, N.; Alexander, K.A.; Raggatt, L.J.; et al. Bone Marrow Macrophages Maintain Hematopoietic Stem Cell (HSC) Niches and Their Depletion Mobilizes HSCs. Blood 2010, 116. [CrossRef]
- Asada, N.; Katayama, Y.; Sato, M.; Minagawa, K.; Wakahashi, K.; Kawano, H.; Kawano, Y.; Sada, A.; Ikeda, K.; Matsui, T.; et al. Matrix-Embedded Osteocytes Regulate Mobilization of Hematopoietic Stem/Progenitor Cells. Cell Stem Cell 2013, 12. [CrossRef]
- Chow, A.; Lucas, D.; Hidalgo, A.; Méndez-Ferrer, S.; Hashimoto, D.; Scheiermann, C.; Battista, M.; Leboeuf, M.; Prophete, C.; Van Rooijen, N.; et al. Bone Marrow CD169+ Macrophages Promote the Retention of Hematopoietic Stem and Progenitor Cells in the Mesenchymal Stem Cell Niche. Journal of Experimental Medicine 2011, 208. [CrossRef]
- Ishii, S.; Suzuki, T.; Wakahashi, K.; Asada, N.; Kawano, Y.; Kawano, H.; Sada, A.; Minagawa, K.; Nakamura, Y.; Mizuno, S.; et al. FGF-23 from Erythroblasts Promotes Hematopoietic Progenitor Mobilization. Blood 2021, 137. [CrossRef]
- Shi, X.; Yan, L.; Shang, J.; Kang, L.; Yan, Z.; Jin, S.; Zhu, M.; Chang, H.; Gong, F.; Zhou, J.; et al. Anti-CD19 and Anti-BCMA CAR T Cell Therapy Followed by Lenalidomide Maintenance after Autologous Stem-Cell Transplantation for High-Risk Newly Diagnosed Multiple Myeloma. Am J Hematol 2022, 97. [CrossRef]
- Cao, X.Y.; Zhang, J.P.; Zhao, Y.L.; Xiong, M.; Zhou, J.R.; Lu, Y.; Sun, R.J.; Wei, Z.J.; Liu, D.Y.; Zhang, X.; et al. Analysis Benefits of a Second Allo-HSCT after CAR-T Cell Therapy in Patients with Relapsed/Refractory B-Cell Acute Lymphoblastic Leukemia Who Relapsed after Transplant. Front Immunol 2023, 14. [CrossRef]
- Zurko, J.; Ramdial, J.; Shadman, M.; Ahmed, S.; Szabo, A.; Iovino, L.; Tomas, A.A.; Sauter, C.; Perales, M.A.; Shah, N.N.; et al. Allogeneic Transplant Following CAR T-Cell Therapy for Large B-Cell Lymphoma. Haematologica 2023, 108. [CrossRef]
- Liu, W.; Li, C.; Cao, Y.; Wang, N.; Huang, L.; Shang, Z.; Wang, J.; Huang, L.; Xu, J.; Xiao, M.; et al. Sequential CAR T-Cell Therapy After Autologous Stem Cell Transplantation for the Treatment of Relapsed/Refractory Intravascular Large B-Cell Lymphoma With Central Nervous System Involvement: A Case Report. Front Oncol 2022, 12. [CrossRef]
- Reinhardt, B.; Lee, P.; Sasine, J.P. Chimeric Antigen Receptor T-Cell Therapy and Hematopoiesis. Cells 2023, 12.
- Rejeski, K.; Burchert, A.; Iacoboni, G.; Sesques, P.; Fransecky, L.; Bücklein, V.; Trenker, C.; Hernani, R.; Naumann, R.; Schäfer, J.; et al. Safety and Feasibility of Stem Cell Boost as a Salvage Therapy for Severe Hematotoxicity after CD19 CAR T-Cell Therapy. Blood Adv 2022, 6.
- Ageitos, A.G.; Varney, M.L.; Bierman, P.J.; Vose, J.M.; Warkentin, P.I.; Talmadge, J.E. Comparison of Monocyte-Dependent T Cell Inhibitory Activity in GM-CSF vs G-CSF Mobilized PSC Products. Bone Marrow Transplant 1999, 23. [CrossRef]
- Young, M.R.I.; Young, M.E.; Wright, M.A. Stimulation of Immune-Suppressive Bone Marrow Cells by Colony-Stimulating Factors. Exp Hematol 1990, 18.
- Ino, K.; Singh, R.K.; Talmadge, J.E. Monocytes from Mobilized Stem Cells Inhibit C Cell Function. J Leukoc Biol 1997, 61. [CrossRef]
- Samuel, E.R.; Newton, K.; MacKinnon, S.; Lowdell, M.W. Successful Isolation and Expansion of CMV-Reactive T Cells from G-CSF Mobilized Donors That Retain a Strong Cytotoxic Effector Function. Br J Haematol 2013, 160. [CrossRef]
- Weiss, R.; Gerdes, W.; Berthold, R.; Sack, U.; Koehl, U.; Hauschildt, S.; Grahnert, A. Comparison of Three CD3-Specific Separation Methods Leading to Labeled and Label-Free T Cells. Cells 2021, 10. [CrossRef]
- Tristán-Manzano, M.; Maldonado-Pérez, N.; Justicia-Lirio, P.; Muñoz, P.; Cortijo-Gutiérrez, M.; Pavlovic, K.; Jiménez-Moreno, R.; Nogueras, S.; Carmona, M.D.; Sánchez-Hernández, S.; et al. Physiological Lentiviral Vectors for the Generation of Improved CAR-T Cells. Mol Ther Oncolytics 2022, 25. [CrossRef]
- Ji, S.Q.; Chen, H.R.; Wang, H.X.; Yan, H.M.; Pan, S.P.; Xun, C.Q. Comparison of Outcome of Allogeneic Bone Marrow Transplantation with and without Granulocyte Colony-Stimulating Factor (Lenograstim) Donor-Marrow Priming in Patients with Chronic Myelogenous Leukemia. Biology of Blood and Marrow Transplantation 2002, 8. [CrossRef]
- Rutella, S.; Bonanno, G.; Pierelli, L.; Mariotti, A.; Capoluongo, E.; Contemi, A.M.; Ameglio, F.; Curti, A.; de Ritis, D.G.; Voso, M.T.; et al. Granulocyte Colony-Stimulating Factor Promotes the Generation of Regulatory DC through Induction of IL-10 and IFN-α. Eur J Immunol 2004, 34. [CrossRef]
- Morris, E.S.; MacDonald, K.P.A.; Rowe, V.; Johnson, D.H.; Banovic, T.; Clouston, A.D.; Hill, G.R. Donor Treatment with Pegylated G-CSF Augments the Generation of IL-10-Producing Regulatory T Cells and Promotes Transplantation Tolerance. Blood 2004, 103. [CrossRef]
- Rutella, S.; Zavala, F.; Danese, S.; Kared, H.; Leone, G. Granulocyte Colony-Stimulating Factor: A Novel Mediator of T Cell Tolerance. The Journal of Immunology 2005, 175. [CrossRef]
- Toh, H.C.; Sun, L.; Soe, Y.; Wu, Y.; Phoon, Y.P.; Chia, W.K.; Wu, J.; Wong, K.Y.; Tan, P. G-CSF Induces a Potentially Tolerant Gene and Immunophenotype Profile in T Cells in Vivo. Clinical Immunology 2009, 132. [CrossRef]
- Bunse, C.E.; Borchers, S.; Varanasi, P.R.; Tischer, S.; Figueiredo, C.; Immenschuh, S.; Kalinke, U.; Köhl, U.; Goudeva, L.; Maecker-Kolhoff, B.; et al. Impaired Functionality of Antiviral T Cells in G-CSF Mobilized Stem Cell Donors: Implications for the Selection of CTL Donor. PLoS ONE 2013, 8. [CrossRef]
- Beloki, L.; Ramírez, N.; Olavarría, E.; Samuel, E.R.; Lowdell, M.W. Manufacturing of Highly Functional and Specific T Cells for Adoptive Immunotherapy against Virus from Granulocyte Colony-Stimulating Factor-Mobilized Donors. Cytotherapy 2014, 16. [CrossRef]
- Clancy, L.E.; Blyth, E.; Simms, R.M.; Micklethwaite, K.P.; Ma, C.K.K.; Burgess, J.S.; Antonenas, V.; Shaw, P.J.; Gottlieb, D.J. Cytomegalovirus-Specific Cytotoxic t Lymphocytes Can Be Efficiently Expanded from Granulocyte Colony-Stimulating Factor-Mobilized Hemopoietic Progenitor Cell Products Ex Vivo and Safely Transferred to Stem Cell Transplantation Recipients to Facilitate Immune Reconstitution. Biology of Blood and Marrow Transplantation 2013, 19. [CrossRef]
- Samuel, E.R.; Beloki, L.; Newton, K.; Mackinnon, S.; Lowdell, M.W. Isolation of Highly Suppressive CD25 +FoxP3+ T Regulatory Cells from G-CSF-Mobilized Donors with Retention of Cytotoxic Anti-Viral CTLs: Application for Multi-Functional Immunotherapy Post Stem Cell Transplantation. PLoS ONE 2014, 9. [CrossRef]
- Ye, Y.; Yang, L.; Yuan, X.; Huang, H.; Luo, Y. Optimization of Donor Lymphocyte Infusion for AML Relapse After Allo-HCT in the Era of New Drugs and Cell Engineering. Front Oncol 2022, 11.
- Caldemeyer, L.E.; Akard, L.P.; Edwards, J.R.; Tandra, A.; Wagenknecht, D.R.; Dugan, M.J. Donor Lymphocyte Infusions Used to Treat Mixed-Chimeric and High-Risk Patient Populations in the Relapsed and Nonrelapsed Settings after Allogeneic Transplantation for Hematologic Malignancies Are Associated with High Five-Year Survival If Persistent Full Donor Chimerism Is Obtained or Maintained. Biology of Blood and Marrow Transplantation 2017, 23. [CrossRef]
- Sudarsanam, H.; Buhmann, R.; Henschler, R. Influence of Culture Conditions on Ex Vivo Expansion of T Lymphocytes and Their Function for Therapy: Current Insights and Open Questions. Front Bioeng Biotechnol 2022, 10.
- Klebanoff, C.A.; Gattinoni, L.; Restifo, N.P. Sorting through Subsets: Which T-Cell Populations Mediate Highly Effective Adoptive Immunotherapy? Journal of Immunotherapy 2012, 35. [CrossRef]
- Canesin, G.; Hoyt, H.; Williams, R.; Silva, M.; Chng, M.; Cummins, C.; Ung, M.; Qiu, H.; Shin, J.; Hu, J.; et al. G-CSF/Plerixafor Dual-Mobilized Donor Derived CD33CAR T-Cells As Potent and Effective AML Therapy in Pre-Clinical Models. Blood 2021, 138. [CrossRef]
- Battram, A.M.; Oliver-Caldés, A.; Suárez-Lledó, M.; Lozano, M.; Bosch i Crespo, M.; Martínez-Cibrián, N.; Cid, J.; Moreno, D.F.; Rodríguez-Lobato, L.G.; Urbano-Ispizua, A.; et al. T Cells Isolated from G-CSF-Treated Multiple Myeloma Patients Are Suitable for the Generation of BCMA-Directed CAR-T Cells. Mol Ther Methods Clin Dev 2022, 26. [CrossRef]
- Otto, M.; Barfield, R.C.; Iyengar, R.; Gatewood, J.; Müller, I.; Holladay, M.S.; Houston, J.; Leung, W.; Handgretinger, R. Human Γδ T Cells from G-CSF-Mobilized Donors Retain Strong Tumoricidal Activity and Produce Immunomodulatory Cytokines after Clinical-Scale Isolation. Journal of Immunotherapy 2005, 28. [CrossRef]
- Holzinger, A.; Abken, H. Treatment with Living Drugs: Pharmaceutical Aspects of CAR T Cells. Pharmacology 2022.
- Klingemann, H. The NK-92 Cell Line—30 Years Later: Its Impact on Natural Killer Cell Research and Treatment of Cancer. Cytotherapy 2023, 25.
- Bachiller, M.; Battram, A.M.; Perez-Amill, L.; Martín-Antonio, B. Natural Killer Cells in Immunotherapy: Are We Nearly There? Cancers (Basel) 2020, 12.
- Cheng, Z.F.; Li, H.K.; Yang, H.P.; Lee, C.Y.; Tang, S.W.; Lin, Y.L.; Hsiao, S.C. A Novel Endogenous CD16-Expressing Natural Killer Cell for Cancer Immunotherapy. Biochem Biophys Rep 2021, 26. [CrossRef]
- Ruggeri, L.; Capanni, M.; Urbani, E.; Perruccio, K.; Shlomchik, W.D.; Tosti, A.; Posati, S.; Rogaia, D.; Frassoni, F.; Aversa, F.; et al. Effectiveness of Donor Natural Killer Cell Aloreactivity in Mismatched Hematopoietic Transplants. Science (1979) 2002, 295. [CrossRef]
- Gong, J.H.; Maki, G.; Klingemann, H.G. Characterization of a Human Cell Line (NK-92) with Phenotypical and Functional Characteristics of Activated Natural Killer Cells. Leukemia 1994, 8.
- Sarvaria, A.; Jawdat, D.; Madrigal, J.A.; Saudemont, A. Umbilical Cord Blood Natural Killer Cells, Their Characteristics, and Potential Clinical Applications. Front Immunol 2017, 8.
- Patel, A.; Truscott, L.C.; De Oliveira, S.N. Development of Optimized Protocol for Generation of NK Cells Expressing Chimeric Antigen Receptors from Hematopoietic Stem Cells for Cancer Immunotherapy. Blood 2015, 126. [CrossRef]
- Xiong, Y.; Mouginot, M.; Reppel, L.; Qian, C.; Stoltz, J. francois; Bensoussan, D.; Decot, V. Modification of NK Cell Subset Repartition and Functions in Granulocyte Colony-Stimulating Factor-Mobilized Leukapheresis after Expansion with IL-15. Immunol Res 2017, 65. [CrossRef]
- Pelosi, A.; Besi, F.; Tumino, N.; Merli, P.; Quatrini, L.; Li Pira, G.; Algeri, M.; Moretta, L.; Vacca, P. NK Cells and PMN-MDSCs in the Graft From G-CSF Mobilized Haploidentical Donors Display Distinct Gene Expression Profiles From Those of the Non-Mobilized Counterpart. Front Immunol 2021, 12. [CrossRef]
- Zhao, X.; Peng, T.; Cao, X.; Hou, Y.; Li, R.; Han, T.; Fan, Z.; Zhao, M.; Chang, Y.; Chen, H.; et al. In Vivo G-CSF Treatment Activates the GR-SOCS1 Axis to Suppress IFN-γ Secretion by Natural Killer Cells. Cell Rep 2022, 40. [CrossRef]
- Gazitt, Y. Immunologic Profiles of Effector Cells and Peripheral Blood Stem Cells Mobilized with Different Hematopoietic Growth Factors. Stem Cells 2000, 18. [CrossRef]
- Romee, R.; Rosario, M.; Berrien-Elliott, M.M.; Wagner, J.A.; Jewell, B.A.; Schappe, T.; Leong, J.W.; Abdel-Latif, S.; Schneider, S.E.; Willey, S.; et al. Cytokine-Induced Memory-like Natural Killer Cells Exhibit Enhanced Responses against Myeloid Leukemia. Sci Transl Med 2016, 8. [CrossRef]
- Dubois, S.P.; Miljkovic, M.D.; Fleisher, T.A.; Pittaluga, S.; Hsu-Albert, J.; Bryant, B.R.; Petrus, M.N.; Perera, L.P.; Müller, J.R.; Shih, J.H.; et al. Short-Course IL-15 given as a Continuous Infusion Led to a Massive Expansion of Effective NK Cells: Implications for Combination Therapy with Antitumor Antibodies. J Immunother Cancer 2021, 9. [CrossRef]
- Oberoi, P.; Kamenjarin, K.; Ossa, J.F.V.; Uherek, B.; Bönig, H.; Wels, W.S. Directed Differentiation of Mobilized Hematopoietic Stem and Progenitor Cells into Functional NK Cells with Enhanced Antitumor Activity. Cells 2020, 9. [CrossRef]
- Ojo, E.O.; Sharma, A.A.; Liu, R.; Moreton, S.; Checkley-Luttge, M.A.; Gupta, K.; Lee, G.; Lee, D.A.; Otegbeye, F.; Sekaly, R.P.; et al. Membrane Bound IL-21 Based NK Cell Feeder Cells Drive Robust Expansion and Metabolic Activation of NK Cells. Sci Rep 2019, 9. [CrossRef]
- Zhu, Y.; Smith, D.J.; Zhou, Y.; Li, Y.R.; Yu, J.; Lee, D.; Wang, Y.C.; Di Biase, S.; Wang, X.; Hardoy, C.; et al. Development of Hematopoietic Stem Cell-Engineered Invariant Natural Killer T Cell Therapy for Cancer. Cell Stem Cell 2019, 25. [CrossRef]
Figure 1.
Generations of CAR cells: First-generation CAR cells, equipped with an extracellular antigen-recognizing domain combined with intracellular CD3z accounting for signal transduction. Second-generation CAR cells, equipped with an extracellular antigen-recognizing domain combined with two intracellular domains: CD3z and an additional costimulatory domain like CD28 or 4-1BB. Third-generation CAR cells, equipped with an extracellular antigen-recognizing domain combined with three intracellular domains: CD3z and two additional costimulatory domains. Fourth-generation CAR cells, a diversified group of CAR constructs like cytokine-expressing CAR cells. Created with BioRender.com.
Figure 1.
Generations of CAR cells: First-generation CAR cells, equipped with an extracellular antigen-recognizing domain combined with intracellular CD3z accounting for signal transduction. Second-generation CAR cells, equipped with an extracellular antigen-recognizing domain combined with two intracellular domains: CD3z and an additional costimulatory domain like CD28 or 4-1BB. Third-generation CAR cells, equipped with an extracellular antigen-recognizing domain combined with three intracellular domains: CD3z and two additional costimulatory domains. Fourth-generation CAR cells, a diversified group of CAR constructs like cytokine-expressing CAR cells. Created with BioRender.com.
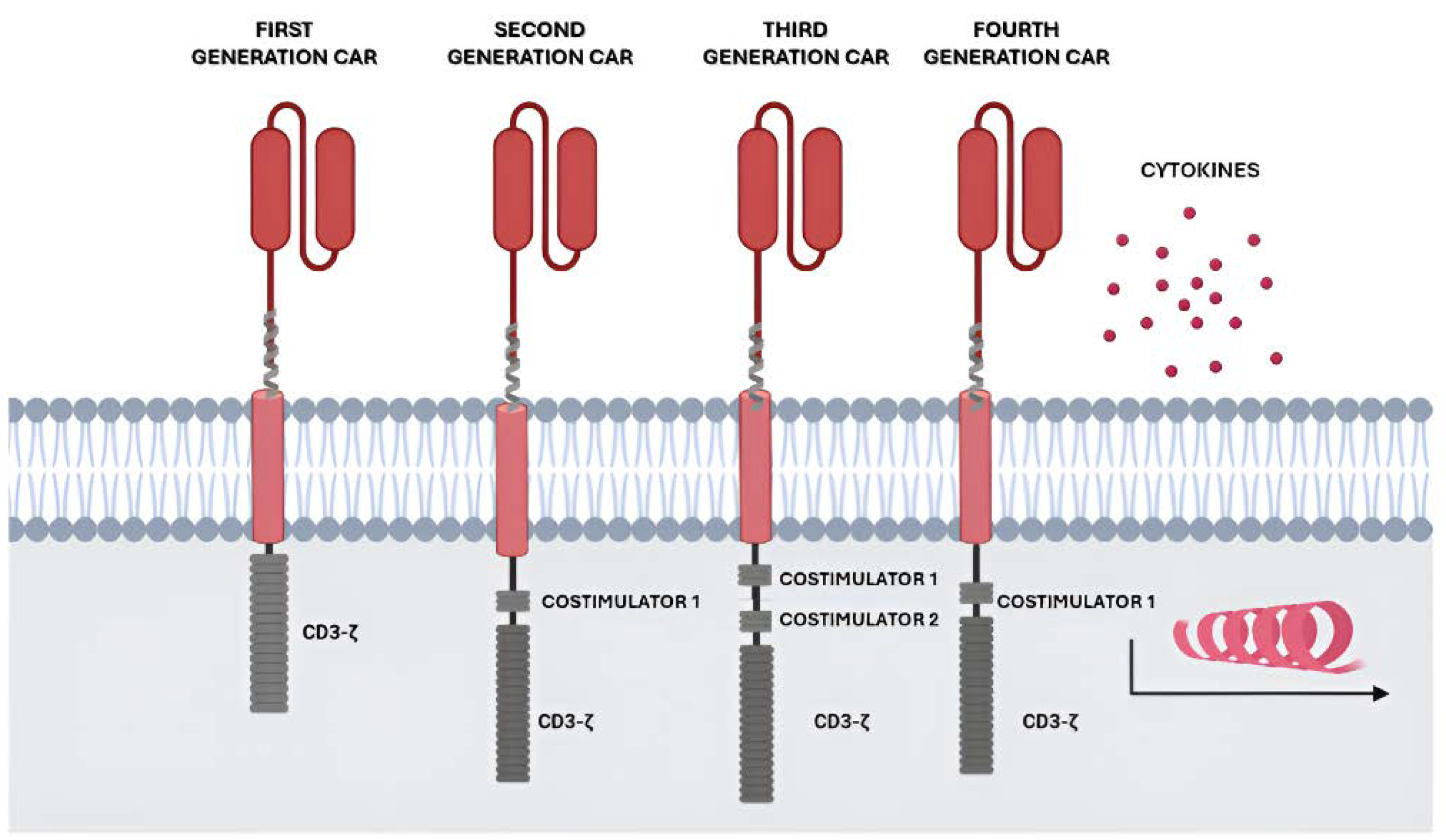
Figure 2.
On the left side, cells have not been exposed to G-CSF, therefore, osteoblasts and endothelial cells show on their surface a high expression of cell adhesion-related molecules, such as SDF-1, VCAM- 1 or kitL. On the right side, cells have been exposed to G-CSF, a molecule recognized by the G-CSFR receptor. This causes neutrophils to release protease enzymes that accumulate in the BM and degrade molecules related to cell adhesion. Furthermore, both the secretion of adrenaline by sympathetic neurons and the secretion of certain factors by macrophages promote a reduction in SDF-1 expression, which leads to disjunction and mobilization of HSPC cells from the bone marrow. Created with BioRender.com.
Figure 2.
On the left side, cells have not been exposed to G-CSF, therefore, osteoblasts and endothelial cells show on their surface a high expression of cell adhesion-related molecules, such as SDF-1, VCAM- 1 or kitL. On the right side, cells have been exposed to G-CSF, a molecule recognized by the G-CSFR receptor. This causes neutrophils to release protease enzymes that accumulate in the BM and degrade molecules related to cell adhesion. Furthermore, both the secretion of adrenaline by sympathetic neurons and the secretion of certain factors by macrophages promote a reduction in SDF-1 expression, which leads to disjunction and mobilization of HSPC cells from the bone marrow. Created with BioRender.com.
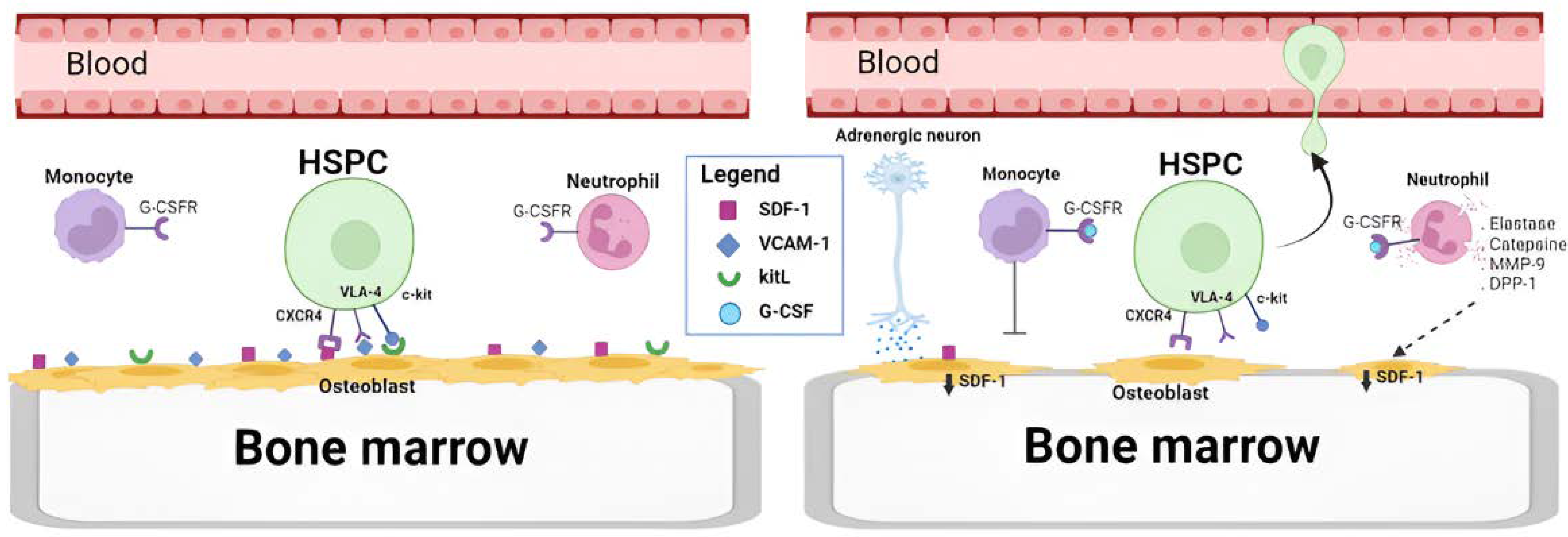
Figure 3.
Illustration of the clinical aspects of the apheresis process for the collection of HSCs through the therapeutic administration of G-CSF, showing the process of mobilization of CD34+ cells from the bone marrow to the peripheral blood for subsequent collection. Created with BioRender.com.
Figure 3.
Illustration of the clinical aspects of the apheresis process for the collection of HSCs through the therapeutic administration of G-CSF, showing the process of mobilization of CD34+ cells from the bone marrow to the peripheral blood for subsequent collection. Created with BioRender.com.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



