
Submitted:
30 April 2024
Posted:
01 May 2024
You are already at the latest version
Abstract
A 23-month-old neutered male dog of unknown ancestry presented with a history of progressive neurological signs that included anxiety, cognitive impairment, tremors, seizure activity, ataxia, and pronounced visual impairment. The clinical signs were accompanied by global brain atrophy. Due to progression in the severity of disease signs, the dog was euthanized at 26 months of age. Examination of tissues collected at necropsy revealed dramatic intracellular accumulations of autofluorescent inclusions in the brain, retina, and cardiac muscle. The inclusions were immunopositive for subunit c of mitochondrial ATP synthase, and their ultrastructural appearances were similar to those of lysosomal storage bodies that accumulate in some of the neuronal ceroid lipofuscinosis (NCL) diseases. The also dog exhibited widespread neuroinflammation. Based on these findings, the dog was deemed likely to have suffered from a form of NCL. A whole genome sequence analysis of the proband’s DNA revealed a homozygous C to T substitution that altered the intron 3 – exon 4 splice site of CLN6. Other mutations in CLN6 cause NCL diseases in humans and animals including dogs. CLN6 protein was undetectable with immunolabeling in the tissues of the proband. Based on the clinical history, fluorescence and electron-microscopy, immunohistochemistry, and molecular genetic findings, the disorder in this dog is classified as an NCL resulting from the absence of CLN6 protein. Screening the dog’s genome for a panel of breed-specific polymorphisms indicated that its ancestry included numerous breeds, with no single breed predominating. This suggests that the CLN6 disease variant is likely to be present in other mixed breed dogs and at least some of the ancestral breeds.
Keywords:
neurodegeneration
; lysosomal storage
; mitochondrial subunit c protein
; lipofusciin
; fluorescence
; whole genome sequencing
; electron microscopy
1. Introduction
Among the most common hereditary neurodegenerative diseases in dogs are the neuronal ceroid lipofuscinoses (NCLs) [1]. Mutations in at least 13 genes have been associated with different forms of NCL in human subjects [2,3]. Progressive neurodegenerative disorders in dogs have been associated with mutations in canine orthologs of the majority of these genes [1]. The NCLs are lysosomal storage disorders that have been distinguished from other lysosomal storage diseases by the combination of phenotypic features consisting of predominantly neurological signs that progress over time, atrophy of the central nervous system, and a characteristic autofluorescence of the lysosomal storage bodies [4]. In addition to the 13 genes that have been associated with NCLs by consensus among many researchers, other hereditary disorders in dogs with NCL-like phenotypes have been described that result from mutations in genes that have not been associated with NCL in human subjects. Among these genes are ARSG and CNP [5–7]. For dogs that exhibit the characteristic signs of NCL, whole genome sequence (WGS) analysis can be used to determine whether the disorder is associated with a mutation in any of these genes. In this study, a dog that had exhibited progressive neurological signs suggestive of NCL was euthanized due to disease progression, the tissues were examined for NCL-like pathology, and a WGS analysis was performed to determine whether the disorder was associated with a mutation in any of the genes previously associated with NCL.
2. Materials and Methods
A 23-month-old neutered male dog of unknown ancestry (Figure 1) presented after a 4 month history of progressive neurological signs that included anxiety, cognitive impairment, tremors, seizure activity, ataxia, incoordination, and pronounced visual impairment. Marked diffuse brain atrophy was documented with magnetic resonance imaging (Figure 2). The dog was euthanized at approximately 26 months of age due to the progression of the neurological signs. Following euthanasia, the eyes, brain and heart ventricular wall were collected and preserved with aldehyde fixatives for light and electron microscopy, as described previously [8]. “Immuno Fix” consisting of 3.5% paraformaldehyde, 0.05% glutaraldehyde, 120 mM sodium cacodylate, and 1 mM calcium chloride, pH 7.4 was used for the light microscopy and immunohistochemistry samples. “EM Fix” consisting of 2% glutaraldehyde, 1.12% paraformaldehyde, and 120 mM sodium cacodylate, pH 7.4 was used for electron microscopy samples. Unstained cryostat sections of retina, cerebral cortex, cerebellar cortex, and cardiac muscle fixed for light microscopy were examined for lipofuscin-like autofluorescence [7]. Slices of the same tissues fixed in “Immuno Fix” were embedded in paraffin. Sections of the paraffin-embedded samples were immunostained using Abcam (Cambridge, UK) anti-mitochondrial ATP synthase subunit c primary antibody (cat. no. ab180149, dilution 1:100), Agilent Dako (Agilent Technologies, Santa Clara, CA, USA) anti-GFAP primary antibody (cat. no. Z0334, dilution 1:200), Fujifilm Wako (Fujifilm North America, Lousiville, KY, USA), anti-Iba1 primary antibody (cat. no. 019-19741, dilution 1:100), and Abcam anti-CLN6 antibody (cat. no. ab272678, dilution 1:50). Antigen retrieval and immunostaining was performed as described previously [7,9]. Paraffin sections of Immuno-fixed brain samples from a 12-month-old Shiba Inu that was euthanized due to seizures were also immunolabeled with the anti-CLN6 antibody. Pieces of the same tissues fixed in “EM Fix” were post-fixed with osmium tetroxide and embedded in epoxy resin. Thin sections of the latter samples were examined with transmission electron microscopy using a JEOL JEM-1400 microscope equipped with a Gatan digital camera.
Genomic DNA was prepared from EDTA-anticoagulated blood of the proband (Katz et al., 2005) and was submitted to the University of Missouri Genomics Technology Core Facility for library preparation and 2 × 150 bp paired-end sequencing on their Illumina NovaSeq 6000 sequencer. Alignment of the sequence reads to the current canine reference genome assembly (Dog10K_Boxer_Tasha) and variant calling was performed initially using OVarFlow workflow software [11]. In addition, a previously described data-processing pipeline was used to align the sequence reads to the same genome assembly and to analyze them with Ensembl annotation in conjunction with reads from 334 other whole genome sequences generated by the University of Missouri Canine Genetics Laboratory and deposited in the NCBI Sequence Read Archive (SRA) [7]. The SRA BioSample identifier for the proband is SAMN39309507. The SRA BioSample identifiers of the remaining whole genome sequences used in this analysis were reported previously [6]. The amino acid positions for canine CLN6 were numbered according to XP_038298694.1 (NCBI).
An allelic discrimination assay was used to genotype individual dogs for a candidate variant at position 32,185,406 on chromosome 30. For this assay, the sequences of the PCR primers were 5′- AAGGTGATGATGCTGACGTAGATC -3′ and 5′- GCCACGGCCTCCCT -3′. The competing probes sequences were 5′-VIC- CTGACCTCAGCTCATC -NFQ-3′ (reference allele) and 5′-FAM- CTGACCTCAACTCATC -NFQ-3′ (variant allele). This assay was used to genotype 13 mixed breed dogs with neurological disorders of unknown etiology represented in our DNA archive.
A DNA sample from the proband was submitted to Wisdom Health (Portland, OR) for genotyping with the Wisdom Panel Premium. This panel includes genotyping for variants that indicate a dog’s breed composition. It also includes genotyping for over 235 mutations that have been shown to underlie hereditary disorders in dogs. Among the disease variants in the test panel are NCL-causing mutations in PPT1, ATP13A2, MFSD8, and CLN8.
3. Results
3.1. Microscopic Findings
The proband exhibited pronounced accumulations of autofluorescent inclusions in the cerebellum, cerebral cortex, retina, and cardiac muscle (Figure 3). These inclusions displayed yellow to orange emissions when illuminated with blue light. Cells containing large aggregates of these inclusions were present throughout the cerebellar and cerebral cortexes. In the retina, large aggregates of the autofluorescent inclusions were present only in the ganglion cells, while individual autofluorescent granules were scattered among cells in the other layers of the retina. In the cardiac muscle groups of autofluorescent inclusions were clustered in linear arrays within the muscle fibers.
Within cells of the cerebellar cortex, some of the disease-related intracellular inclusion bodies appeared to be autophagolysosomes that contained structures with morphological appearances suggesting that they were derived from mitochondria, as well as a heterogenous mixture of membrane-like components (Figure 4A). The majority of the inclusion bodies in the cerebellar cortex consisted of tightly packed membranous components (Figure 4B). In the cerebral cortex, the contents of the storage bodies consisted primarily of stacks of membrane-like components in random orientations (Figure 5). In the retinal ganglion cells tightly packed clusters of storage bodies contained membrane-like components that were mostly arranged in fingerprint-like patterns (Figure 6). In the cardiac muscle¸ the storage bodies were clustered among groups of mitochondria that flanked many of the muscle fiber cell nuclei. The contents of the storage bodies consisted of tightly packed vesicular structures of similar size and stacks of membrane-like structures (Figure 7, Figure 8 and Figure 9).
The subunit c protein of mitochondrial ATP-synthase is a major component of the lysosomal storage bodies that accumulate in many forms of NCL, including the form that results from CLN6 mutations [12]. Sections of brain, retina, and cardiac muscle of the proband showed substantial immunostaining of intracellular inclusions with an anti-subunit c antibody (Figure 10 and Figure 11). The localization of the immunostaining was the same as localization of the autofluorescent inclusions in these tissues.
Neuroinflammation is often characterized by glial activation. Activated astrocytes, detected with GFAP immunolabeling, were abundant throughout the cerebral cortex and cerebellum of the proband (Figure 12). The proband also exhibited abundant activated microglia, detected with Iba1 immunolabeling, in the cerebral cortex and cerebellum (Figure 13).
3.2. Molecular Genetic Findings
The proband was adopted from a rescue service and no pedigree information was available. Wisdom Panel genotyping was performed to characterize the breed background of the proband. Based on the dog’s pattern of DNA sequence variants, its ancestry was found to include over 25 breeds, with no one breed accounting for more than 10% of the genome. Thus, it was unlikely that the proband’s disorder resulted from known canine NCL mutations that are almost all either breed-specific or are shared by a few similar breeds. The genotyping analysis indicated that the degree of heterozygosity in the proband was 40%, which is typical for mixed breed dogs and lower than most purebred dogs.
To elucidate the potential molecular genetic cause of the disease, DNA from the proband to was used to generate a 41.2-fold average-coverage whole-genome sequence. This sequence contained 24,818 called variants relative to the canine reference sequence that were predicted to alter the primary structure of the encoded gene products. The proband was homozygous for 5,811 of these variants. The proband’s homozygous variants were sorted according to the allele frequency among all 335 canine whole genome sequences included in the analysis. Within this cohort, variants in 12 genes were uniquely homozygous in the proband (Supplemental File 1). Among these was a C to T substitution at position 32,185,406 on chromosome 30, which was predicted to disrupt the intron 3-exon 4 splice site of CLN6 (Figure 14). The validity of this variant call was confirmed by an Integrative-Genomics-Viewer-assisted inspection of aligned reads from the proband’s whole genome sequence to the Tasha reference sequence surrounding position 32,185,406 on chromosome 30 (Figure 15) and by Sanger sequencing of the region surrounding the variant position. The disease phenotype was consistent with phenotypes previously associated with other CLN6 variants [13,14], whereas a similar disease phenotype has not been associated with mutations in any of the other 11 genes that were uniquely homozygous in the proband (Supplemental File 1). The proband did not have potentially deleterious sequence variants in any of the other known NCL genes.
An allelic discrimination assay that distinguishes the reference and variant alleles at position 30: 32,185,406 was performed on 13 mixed breed dogs with a variety of neurological disorders of unknown etiology. All 13 dogs were homozygous for the reference allele. This indicates that the CLN6 variant is not a common cause of neurological disease in mixed breed dogs.
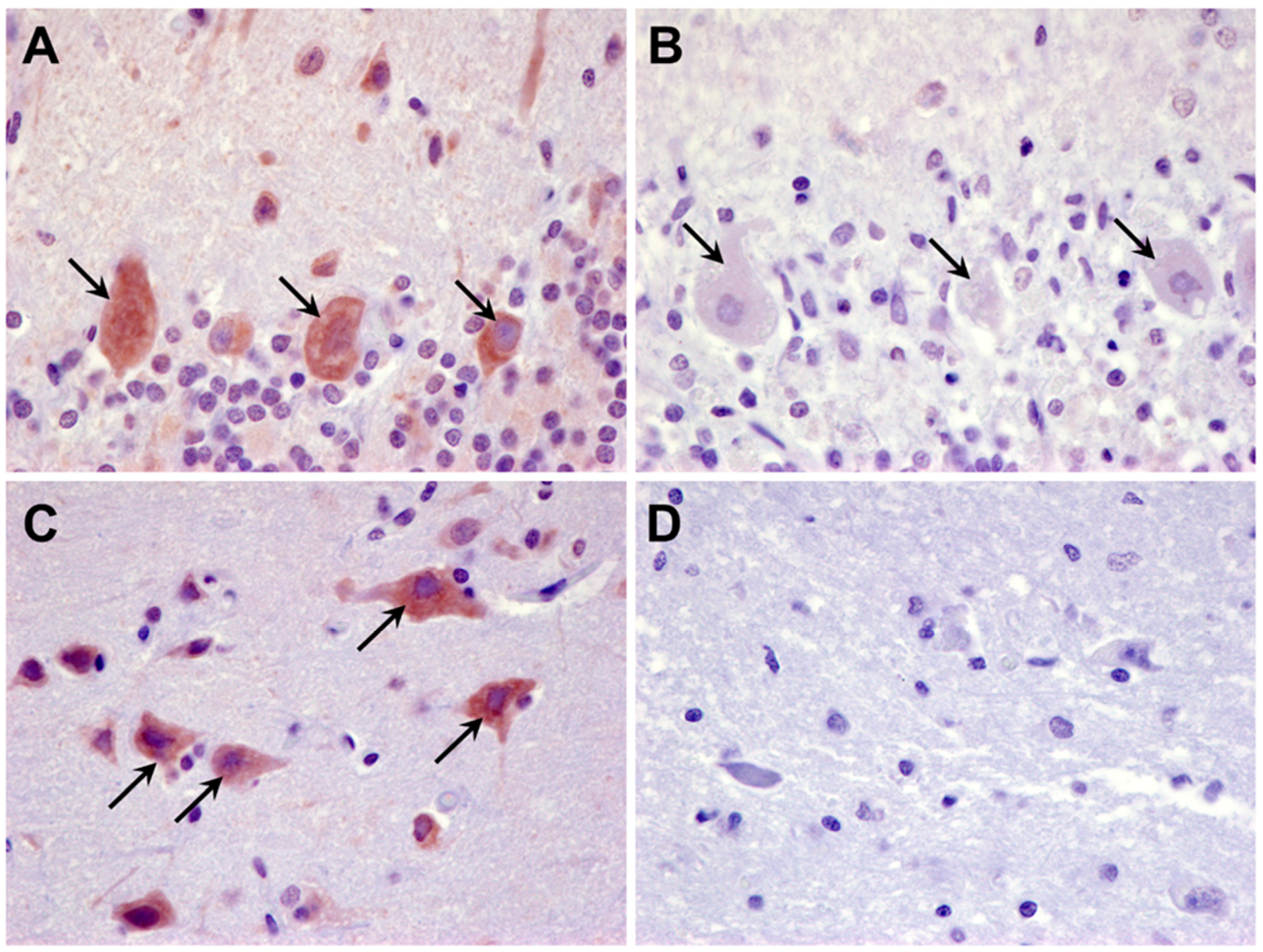
The CLN6 sequence variant is predicted to result in skipping of exon 4 during pre-mRNA processing and thus a lack of full-length CLN6 protein. To determine whether any CLN6 protein was made, tissue sections from the proband were immunolabeled with an antibody directed against amino acid residues 282 to 311 of the protein that are encoded by exon 7 of CLN6. Cells in the cerebellum and cerebral cortex of an unaffected Shiba Inu dog exhibited pronounced CLN6 immunolabeling that was not observed in these tissues from the proband (Figure 16).
4. Discussion
The disorder evaluated in this study included all of the features that have been used to classify diseases as NCLs: progressive neurological and behavioral signs, brain atrophy, and intracellular accumulation of storage bodies that exhibit lipofuscin-like autofluorescence. The ultrastructural appearances of the disease-related storage bodies were similar to those of other NCLs, as was the presence of mitochondrial ATP synthase subunit c protein in the storage bodies. Based on the ultrastructural features of the storage body contents, they appear to have derived, at least in part, from mitochondrial membranes. However, there were tissue-specific differences in the ultrastructure of the storage body contents. This may reflect differences in the molecular composition of the storage body precursors in different tissues, including tissue-specific differences in mitochondrial molecular composition [15–17]. Like other NCLs and similar neurodegenerative disorders, the proband exhibited significant neuroinflammation. Based on current consensus of most investigators in the field, human disorders classified as NCLs result from mutations in PPT1, TPP1, CLN3, DNAJC5, CLN5, CLN6, MFSD8, CLN8, CTSD, GRN, ATP13A2, CTSF, and KCTD7 [4,18]. All of these are autosomal recessive traits except the autosomal dominant disorder resulting from DNAJC5 mutations [19]. Canine NCLs have been associated with mutations in PPT1, TPP1, CLN5, CLN6, MFSD8, CLN8, CTSD, and ATP13A2 [10,13,20–35]. In addition, NCL-like disorders in dogs have been associated with mutations in ARSG, and CNP [5–7]. Among these genes, the only potential deleterious variant in the whole genome sequence of the proband was a homozygous C to T substitution at the intron 3-exon 4 splice site of CLN6. This mutation is predicted to result in the absence of full length CLN6 protein, and the protein was not detected with immunohistochemistry in the proband’s tissues.
Numerous pathogenic mutations in CLN6 have been reported in human subjects suffering from NCL [14,36–44]. Among these are splice site variants in introns 2 and 4 and missense variants in all 7 exons [36,37,45]. A missense mutation in CLN6 exon 7 was also associated with NCL in an Australian Shepherd dog [13]. Thus, it is not surprising that the splice site mutation in the proband resulted in NCL disease.
Of the canine NCL disease variants, all but one occur only within a single breed or in a few similar breeds in which interbreeding is likely to have occurred. The only previous exception is a mutation in CLN5, which has been associated with NCL in Border Collies, Australian Cattle Dogs, a German Shepherd-Australian Cattle Dog mix, and a mixed breed dog of unknown ancestry [26]. In general, one would expect homozygosity for disease alleles to be less common in mixed breed dogs than in purebred dogs as the latter tend to be much more highly inbred. An exception would be if two closely related mixed breed dogs were mated. The proband in this study does not appear to be such an exception. The Wisdom Panel genotyping analysis indicated that the proband had 40% heterozygosity, which is typical for mixed breed dogs and lower than most purebred dogs. The degree of heterozygosity would be significantly lower if the parents of the proband were closely related. Thus, it appears that the CLN6 variant in the proband could be widespread, although probably uncommon, in the mixed breed dog population and may be present in one or more of the ancestral breeds. Of the ancestral breeds identified in the proband’s Wisdom Panel, no cases of NCL have been reported that have not been associated with other previously identified NCL mutations. Should mixed breed dogs or dogs of any breed present with NCL-like signs similar to those of the proband, it would be reasonable to test for the CLN6 variant identified in this study. In a small sample of 13 mixed breed dogs with neurological disorders of unknown etiology, none had the CLN6 splice site variant. However, screening of mixed-breed dogs with NCL-like signs is warranted.
The mechanisms by which lack of functional CLN6 protein causes lysosomal storage body accumulation and neurodegeneration are not well understood. CLN6 is a membrane protein that has been localized to the endoplasmic reticulum [4,46–48]. Evidence suggests that among the functions of CLN6 may be regulation of sphingolipid and glycerophospholipid metabolism [49], mediation of delivery of proteins to lysosomes [50,51], prevention of protein aggregation [52–54], mediation of protein secretion from cells [55], and regulation of the lipid composition of lysosomes [56]. Impairment of any of these functions could result in a general impairment of lysosomal function and the accumulation of substrates of lysosomal degradative enzymes. However, this would not explain the specific accumulation of ATP synthase subunit c protein that occurs in CLN6 disease and some of the other NCL disorders. The ultrastructural findings in this study in conjunction with previous research suggest that most components of autophagocytosed mitochondria other than the subunit c protein are degraded normally in CLN6-deficient cells [57–60]. The subunit c protein is very hydrophobic and prone to aggregation, properties that may explain its resistance to degradation and specific accumulation in lysosomes. Evidence suggests that CLN6 may be required for normal turnover of the subunit c protein within mitochondria, and that the accumulation of the protein within lysosomes is secondary to impaired degradation of subunit c within the mitochondria. A selective impairment of mitochondrial subunit c degradation in CLN6 disease is supported by the fact that in the cardiac muscle of the proband, storage material accumulated specifically in the mitochondria-rich perinuclear regions of the muscle fibers. In ovine CLN6 disease it was found that excess subunit c protein selectively accumulates within the mitochondrial inner membrane, indicating that the disease results in impaired turnover of this protein within the mitochondria [61]. This suggests that in addition to its putative other roles in cellular metabolism, the CLN6 protein may be important for maintaining normal proteolytic degradation of subunit c within mitochondria and preventing secondary accumulation in lysosomal storage bodies [62,63]. Impaired turnover of subunit c within mitochondria may in turn result in impairment of mitochondrial function that could play an important role in CLN6 disease pathogenesis [64–66]. For example, lack of normal CLN6 results in increased levels of the mitochondrial antioxidant enzyme manganese-dependent superoxide dismutase, suggesting increased production of reactive oxygen species in mitochondria that can cause cell damage and death [67]. The evidence that lack of normal CLN6 protein results in impaired subunit c protein turnover within mitochondria warrants research into whether, in addition to its localization in the endoplasmic reticulum, CLN6 protein is also present in mitochondria, at least in brain, retina, and cardiac muscle.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Supplementary File 1 consists of a list of protein coding sequence variants that were uniquely homozygous in the proband relative to a cohort of 334 dogs with other disorders.
Author Contributions
Conceptualization, T.M. and M.L.K.; methodology, T.M., G.B., S.C., and M.L.K.; software, G.B.; validation, T.M. and M.L.K.; formal analysis, T.M., G.B., and M.L.K..; investigation, T.M. and M.L.K.; resources, G.B. and M.L.K. data curation, T.M.; writing—original draft preparation, T.M. and M.L.K.; writing—review and editing, T.M., G.B., S.C., and M.L.K..; supervision, M.L.K.; project administration, M.L.K.; funding acquisition, T.M., G.B. and M.L.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by American Kennel Club Canine Health Foundation Grant No. 02604, by U.S. National Institutes of Health grants S10 OD032246 and EY031674, and by the Orthopedic Foundation for Animals. The bioinformatic analyses used Bridges-2 at Pittsburgh Supercomputing Center through allocation mcb160068p from the Advanced Cyberinfrastructure Coordination Ecosystem: Services & Support (ACCESS) program, which is supported by National Science Foundation grants #2138259, #2138286, #2138307, #2137603, and #2138296, and the computing resources of the University of Missouri Bioinformatics and Analytics Core Facility.
Data Availability Statement
DNA sequence data for the proband has been archived and deposited in the NCBI Sequence Read Archive as BioSample SAMN39309507.
Acknowledgements
Our thanks to Robert Schnabel for assistance with whole genome sequence analyses. Dr. Gary S. Johnson, who passed away in January of 2024, played an important role in most aspects of this study.
Conflicts of Interest
The Canine Genetics Laboratory at the University of Missouri provides fee-for-service genetic testing for dogs. The authors disclose no other conflict of interest.
References
- Katz, M.L.; Rustad, E.; Robinson, G.O.; Whiting, R.E.H.; Student, J.T.; Coates, J.R.; Narfstrom, K. Canine Neuronal Ceroid Lipofuscinoses: Promising Models for Preclinical Testing of Therapeutic Interventions. Neurobiol Dis 2017, 108. [Google Scholar] [CrossRef] [PubMed]
- Butz, E.S.; Chandrachud, U.; Mole, S.E.; Cotman, S.L. Moving towards a New Era of Genomics in the Neuronal Ceroid Lipofuscinoses. Biochim Biophys Acta Mol Basis Dis 2020, 1866, 165571. [Google Scholar] [CrossRef]
- Beck-Wodl, S.; Harzer, K.; Sturm, M.; Buchert, R.; Ries, O.; Mennel, H.-D.; Latta, E.; Pagenstecher, A.; Keber, U. Homozygous TBC1 Domain-Containing Kinase (TBCK) Mutation Causes a Novel Lysosomal Storage Disease - a New Type of Neuronal Ceroid Lipofuscinosis (CLN15)? Acta Neuropathol Commun 2018, 6, 145. [Google Scholar] [CrossRef]
- Mole, S.E.; Williams, R.E.; Goebel, H.H. The Neuronal Ceroid Lipofuscinoses (Batten Disease), 2nd ed.; Mole, S.E., Willimas, R.E., Goebel, H.H., Eds.; Oxford University Press: Oxford, 2011. [Google Scholar]
- Abitbol, M.; Thibaud, J.-L.; Olby, N.J.; Hitte, C.; Puech, J.-P.; Maurer, M.; Pilot-Storck, F.; Hedan, B.; Dreano, S.; Brahimi, S.; et al. A Canine Arylsulfatase G (ARSG) Mutation Leading to a Sulfatase Deficiency Is Associated with Neuronal Ceroid Lipofuscinosis. Proc Natl Acad Sci U S A 2010, 107, 14775–14780, PT - Journal Article, Research Support, Non-U.S. Gov’t. [Google Scholar] [CrossRef]
- Keller, S.H.; Johnson, G.S.; Bullock, G.; Mhlanga-Mutangadura, T.; Schwartz, M.; Pattridge, S.G.; Guo, J.; Kortz, G.D.; Katz, M.L. Homozygous CNP Mutation and Neurodegeneration in Weimaraners: Myelin Abnormalities and Accumulation of Lipofuscin-like Inclusions. Genes (Basel) 2024, 15. [Google Scholar] [CrossRef] [PubMed]
- Bullock, G.; Johnson, G.S.; Mhlanga-Mutangadura, T.; Petesch, S.C.; Thompson, S.; Goebbels, S.; Katz, M.L. Lysosomal Storage Disease Associated with a CNP Sequence Variant in Dalmatian Dogs. Gene 2022, 830, 146513. [Google Scholar] [CrossRef] [PubMed]
- Bullock, G.; Johnson, G.S.; Pattridge, S.G.; Mhlanga-Matangadura, T.; Guo, T.; Cook, J.; Campbell, R.S.; Vite, C.H.; Katz, M.L. A Homozygous MAN2B1 Missense Mutation in a Doberman Pinscher Dog with Neurodegeneration, Cytoplasmic Vacuoles, Autofluorescent Storage Granules and an α-Mannosidase Deficiency. Genes (Basel) 2023. In Press. [Google Scholar] [CrossRef] [PubMed]
- Morgan, B.R.; Coates, J.R.; Johnson, G.C.; Shelton, G.D.; Katz, M.L. Characterization of Thoracic Motor and Sensory Neurons and Spinal Nerve Roots in Canine Degenerative Myelopathy, a Potential Disease Model of Amyotrophic Lateral Sclerosis. J Neurosci Res 2014, 92. [Google Scholar] [CrossRef] [PubMed]
- Katz, M.L.; Khan, S.; Awano, T.; Shahid, S.A.; Siakotos, A.N.; Johnson, G.S. A Mutation in the CLN8 Gene in English Setter Dogs with Neuronal Ceroid-Lipofuscinosis. Biochem Biophys Res Commun 2005, 327. [Google Scholar] [CrossRef] [PubMed]
- Bathke, J.; Luhken, G. OVarFlow: A Resource Optimized GATK 4 Based Open Source Variant Calling WorkFlow. BMC Bioinformatics 2021, 22, 402. [Google Scholar] [CrossRef] [PubMed]
- Palmer, D.N.; Barry, L.A.; Tyynela, J.; Cooper, J.D. NCL Disease Mechanisms. Biochim Biophys Acta 2013, 1832, 1882–1893. [Google Scholar] [CrossRef] [PubMed]
- Katz, M.L.; Farias, F.H.; Sanders, D.N.; Zeng, R.; Khan, S.; Johnson, G.S.; O’Brien, D.P. A Missense Mutation in Canine CLN6 in an Australian Shepherd with Neuronal Ceroid Lipofuscinosis. J Biomed Biotechnol 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Rus, C.-M.; Weissensteiner, T.; Pereira, C.; Susnea, I.; Danquah, B.D.; Morales Torres, G.; Rocha, M.E.; Cozma, C.; Saravanakumar, D.; Mannepalli, S.; et al. Clinical and Genetic Characterization of a Cohort of 97 CLN6 Patients Tested at a Single Center. Orphanet J Rare Dis 2022, 17, 179. [Google Scholar] [CrossRef] [PubMed]
- Hulbert, A.J.; Turner, N.; Hinde, J.; Else, P.; Guderley, H. How Might You Compare Mitochondria from Different Tissues and Different Species? J Comp Physiol B 2006, 176, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Sadeesh, E.M.; Singla, N.; Lahamge, M.S.; Kumari, S.; Ampadi, A.N.; Anuj, M. Tissue Heterogeneity of Mitochondrial Activity, Biogenesis and Mitochondrial Protein Gene Expression in Buffalo. Mol Biol Rep 2023, 50, 5255–5266. [Google Scholar] [CrossRef] [PubMed]
- Hansen, F.M.; Kremer, L.S.; Karayel, O.; Bludau, I.; Larsson, N.-G.; Kuhl, I.; Mann, M. Mitochondrial Phosphoproteomes Are Functionally Specialized across Tissues. Life Sci Alliance 2024, 7. [Google Scholar] [CrossRef] [PubMed]
- Kaminiow, K.; Kozak, S.; Paprocka, J. Recent Insight into the Genetic Basis, Clinical Features, and Diagnostic Methods for Neuronal Ceroid Lipofuscinosis. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Naseri, N.; Sharma, M.; Velinov, M. Autosomal Dominant Neuronal Ceroid Lipofuscinosis: Clinical Features and Molecular Basis. Clin Genet 2021, 99, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Melville, S.A.; Wilson, C.L.; Chiang, C.S.; Studdert, V.P.; Lingaas, F.; Wilton, A.N. A Mutation in Canine CLN5 Causes Neuronal Ceroid Lipofuscinosis in Border Collie Dogs. Genomics 2005, 86, 287–294. [Google Scholar] [CrossRef]
- Hirz, M.; Drogemuller, M.; Schanzer, A.; Jagannathan, V.; Dietschi, E.; Goebel, H.H.; Hecht, W.; Laubner, S.; Schmidt, M.J.; Steffen, F.; et al. Neuronal Ceroid Lipofuscinosis (NCL) Is Caused by the Entire Deletion of CLN8 in the Alpenlandische Dachsbracke Dog. Mol Genet Metab 2017, 120, 269–277. [Google Scholar] [CrossRef]
- Lingaas, F.; Guttersrud, O.-A.; Arnet, E.; Espenes, A. Neuronal Ceroid Lipofuscinosis in Salukis Is Caused by a Single Base Pair Insertion in CLN8. Anim Genet 2018, 49, 52–58. [Google Scholar] [CrossRef]
- Kolicheski, A.; Barnes Heller, H.L.; Arnold, S.; Schnabel, R.D.; Taylor, J.F.; Knox, C.A.; Mhlanga-Mutangadura, T.; O’Brien, D.P.; Johnson, G.S.; Dreyfus, J.; et al. Homozygous PPT1 Splice Donor Mutation in a Cane Corso Dog With Neuronal Ceroid Lipofuscinosis. J Vet Intern Med 2017, 31. [Google Scholar] [CrossRef]
- Sanders, D.N.; Farias, F.H.; Johnson, G.S.; Chiang, V.; Cook, J.R.; O’Brien, D.P.; Hofmann, S.L.; Lu, J.-Y.; Katz, M.L. A Mutation in Canine PPT1 Causes Early Onset Neuronal Ceroid Lipofuscinosis in a Dachshund. Mol Genet Metab 2010, 100. [Google Scholar] [CrossRef] [PubMed]
- Awano, T.; Katz, M.L.; O’Brien, D.P.; Sohar, I.; Lobel, P.; Coates, J.R.; Khan, S.; Johnson, G.C.; Giger, U.; Johnson, G.S. A Frame Shift Mutation in Canine TPP1 (the Ortholog of Human CLN2) in a Juvenile Dachshund with Neuronal Ceroid Lipofuscinosis. Mol Genet Metab 2006, 89. [Google Scholar] [CrossRef] [PubMed]
- Villani, N.A.; Bullock, G.; Michaels, J.R.; Yamato, O.; O’Brien, D.P.; Mhlanga-Mutangadura, T.; Johnson, G.S.; Katz, M.L. A Mixed Breed Dog with Neuronal Ceroid Lipofuscinosis Is Homozygous for a CLN5 Nonsense Mutation Previously Identified in Border Collies and Australian Cattle Dogs. Mol Genet Metab 2019, 127, 107–115, PT - Case Reports, Journal Article, Research Support, N.I.H., Extramural, Research Support, Non-U.S. Gov’t. [Google Scholar] [CrossRef]
- Schmutz, I.; Jagannathan, V.; Bartenschlager, F.; Stein, V.M.; Gruber, A.D.; Leeb, T.; Katz, M.L. ATP13A2 Missense Variant in Australian Cattle Dogs with Late Onset Neuronal Ceroid Lipofuscinosis. Mol Genet Metab PT - Case Reports, Journal Article, Research Support, N.I.H., Extramural, Research Support, Non-U.S. Gov’t. 2019, 127, 95–106. [Google Scholar] [CrossRef]
- Kolicheski, A.; Johnson, G.S.; O’Brien, D.P.; Mhlanga-Mutangadura, T.; Gilliam, D.; Guo, J.; Anderson-Sieg, T.D.; Schnabel, R.D.; Taylor, J.F.; Lebowitz, A.; et al. Australian Cattle Dogs with Neuronal Ceroid Lipofuscinosis Are Homozygous for a CLN5 Nonsense Mutation Previously Identified in Border Collies. J Vet Intern Med 2016. [Google Scholar] [CrossRef]
- Gilliam, D.; Kolicheski, A.; Johnson, G.S.; Mhlanga-Mutangadura, T.; Taylor, J.F.; Schnabel, R.D.; Katz, M.L. Golden Retriever Dogs with Neuronal Ceroid Lipofuscinosis Have a Two-Base-Pair Deletion and Frameshift in CLN5. Mol Genet Metab 2015, 115. [Google Scholar] [CrossRef]
- Guo, J.; Johnson, G.S.; Brown, H.A.; Provencher, M.L.; da Costa, R.C.; Mhlanga-Mutangadura, T.; Taylor, J.F.; Schnabel, R.D.; O’Brien, D.P.; Katz, M.L. A CLN8 Nonsense Mutation in the Whole Genome Sequence of a Mixed Breed Dog with Neuronal Ceroid Lipofuscinosis and Australian Shepherd Ancestry. Mol Genet Metab 2014, 112. [Google Scholar] [CrossRef] [PubMed]
- Ashwini, A.; D’Angelo, A.; Yamato, O.; Giordano, C.; Cagnotti, G.; Harcourt-Brown, T.; Mhlanga-Mutangadura, T.; Guo, J.; Johnson, G.S.; Katz, M.L. Neuronal Ceroid Lipofuscinosis Associated with an MFSD8 Mutation in Chihuahuas. Mol Genet Metab 2016, 118. [Google Scholar] [CrossRef]
- Guo, J.; O’Brien, D.P.; Mhlanga-Mutangadura, T.; Olby, N.J.; Taylor, J.F.; Schnabel, R.D.; Katz, M.L.; Johnson, G.S. A Rare Homozygous MFSD8 Single-Base-Pair Deletion and Frameshift in the Whole Genome Sequence of a Chinese Crested Dog with Neuronal Ceroid Lipofuscinosis. BMC Vet Res 2015, 10, 960. [Google Scholar] [CrossRef]
- Guo, J.; Johnson, G.S.; Cook, J.; Harris, O.K.; Mhlanga-Mutangadura, T.; Schnabel, R.D.; Jensen, C.A.; Katz, M.L. Neuronal Ceroid Lipofuscinosis in a German Shorthaired Pointer Associated with a Previously Reported CLN8 Nonsense Variant. Mol Genet Metab Rep 2019, 21, 100521. [Google Scholar] [CrossRef]
- Awano, T.; Katz, M.L.; O’Brien, D.P.; Taylor, J.F.; Evans, J.; Khan, S.; Sohar, I.; Lobel, P.; Johnson, G.S. A Mutation in the Cathepsin D Gene (CTSD) in American Bulldogs with Neuronal Ceroid Lipofuscinosis. Mol Genet Metab 2006, 87. [Google Scholar] [CrossRef] [PubMed]
- Farias, F.H.G.; Zeng, R.; Johnson, G.S.; Wininger, F.A.; Taylor, J.F.; Schnabel, R.D.; McKay, S.D.; Sanders, D.N.; Lohi, H.; Seppälä, E.H.; et al. A Truncating Mutation in ATP13A2 Is Responsible for Adult-Onset Neuronal Ceroid Lipofuscinosis in Tibetan Terriers. Neurobiol Dis 2011, 42. [Google Scholar] [CrossRef]
- Sharp, J.D.; Wheeler, R.B.; Parker, K.A.; Gardiner, R.M.; Williams, R.E.; Mole, S.E. Spectrum of CLN6 Mutations in Variant Late Infantile Neuronal Ceroid Lipofuscinosis. Hum Mutat 2003, 22, 35–42. [Google Scholar] [CrossRef]
- Teixeira, C.A.; Espinola, J.; Huo, L.; Kohlschutter, J.; Persaud Sawin, D.-A.; Minassian, B.; Bessa, C.J.P.; Guimaraes, A.; Stephan, D.A.; Sa Miranda, M.C.; et al. Novel Mutations in the CLN6 Gene Causing a Variant Late Infantile Neuronal Ceroid Lipofuscinosis. Hum Mutat 2003, 21, 502–508. [Google Scholar] [CrossRef]
- Bouhouche, A.; Regragui, W.; El Fahime, E.; Bouslam, N.; Tazi-Ahnini, R.; Melloul, M.; Benomar, A.; Yahyaoui, M. CLN6 p.I154del Mutation Causing Late Infantile Neuronal Ceroid Lipofuscinosis in a Large Consanguineous Moroccan Family. Indian J Pediatr 2013, 80, 694–696. [Google Scholar] [CrossRef]
- Chin, J.J.; Behnam, B.; Davids, M.; Sharma, P.; Zein, W.M.; Wang, C.; Chepa-Lotrea, X.; Gallantine, W.B.; Toro, C.; Adams, D.R.; et al. Novel Mutations in CLN6 Cause Late-Infantile Neuronal Ceroid Lipofuscinosis without Visual Impairment in Two Unrelated Patients. Mol Genet Metab 2019, 126, 188–195. [Google Scholar] [CrossRef]
- Onodera, M.; Tsujimoto, S.; Doi, S.; Yamashita, A.; Yamazaki, T.; Makifuchi, T.; Inazu, T. P. Asn77Lys Homozygous CLN6 Mutation in Two Unrelated Japanese Patients with Kufs Disease, an Adult Onset Neuronal Ceroid Lipofuscinosis. Clin Chim Acta 2021, 523, 191–195. [Google Scholar] [CrossRef]
- Sun, G.; Yao, F.; Tian, Z.; Ma, T.; Yang, Z. A First CLN6 Variant Case of Late Infantile Neuronal Ceroid Lipofuscinosis Caused by a Homozygous Mutation in a Boy from China: A Case Report. BMC Med Genet 2018, 19, 177. [Google Scholar] [CrossRef]
- Sato, R.; Inui, T.; Endo, W.; Okubo, Y.; Takezawa, Y.; Anzai, M.; Morita, H.; Saitsu, H.; Matsumoto, N.; Haginoya, K. First Japanese Variant of Late Infantile Neuronal Ceroid Lipofuscinosis Caused by Novel CLN6 Mutations. Brain Dev 2016, 38, 852–856. [Google Scholar] [CrossRef]
- Matsumoto, A.; Nagashima, M.; Iwama, K.; Mizuguchi, T.; Makino, S.; Ikeda, T.; Muramatsu, K.; Matsumoto, N.; Yamagata, T.; Osaka, H. Rapid Progression of a Walking Disability in a 5-Year-Old Boy with a CLN6 Mutation. Brain Dev 2019, 41, 726–730. [Google Scholar] [CrossRef]
- Panjeshahi, S.; Karimzadeh, P.; Movafagh, A.; Ahmadabadi, F.; Rahimian, E.; Alijanpour, S.; Miryounesi, M. Clinical and Genetic Characterization of Neuronal Ceroid Lipofuscinoses (NCLs) in 29 Iranian Patients: Identification of 11 Novel Mutations. Hum Genet 2023, 142, 1001–1016. [Google Scholar] [CrossRef]
- Kousi, M.; Lehesjoki, A.-E.; Mole, S.E. Update of the Mutation Spectrum and Clinical Correlations of over 360 Mutations in Eight Genes That Underlie the Neuronal Ceroid Lipofuscinoses. Hum Mutat 2012, 33, 42–63. [Google Scholar] [CrossRef]
- Wheeler, R.B.; Sharp, J.D.; Schultz, R.A.; Joslin, J.M.; Williams, R.E.; Mole, S.E. The Gene Mutated in Variant Late-Infantile Neuronal Ceroid Lipofuscinosis (CLN6) and in Nclf Mutant Mice Encodes a Novel Predicted Transmembrane Protein. Am J Hum Genet 2002, 70, 537–542. [Google Scholar] [CrossRef]
- Heine, C.; Quitsch, A.; Storch, S.; Martin, Y.; Lonka, L.; Lehesjoki, A.-E.; Mole, S.E.; Braulke, T. Topology and Endoplasmic Reticulum Retention Signals of the Lysosomal Storage Disease-Related Membrane Protein CLN6. Mol Membr Biol 2007, 24, 74–87. [Google Scholar] [CrossRef]
- Mole, S.E.; Michaux, G.; Codlin, S.; Wheeler, R.B.; Sharp, J.D.; Cutler, D.F. CLN6, Which Is Associated with a Lysosomal Storage Disease, Is an Endoplasmic Reticulum Protein. Exp Cell Res 2004, 298, 399–406. [Google Scholar] [CrossRef]
- Rus, C.-M.; Polla, D.L.; Di Bucchianico, S.; Fischer, S.; Hartkamp, J.; Hartmann, G.; Alpagu, Y.; Cozma, C.; Zimmermann, R.; Bauer, P. Neuronal Progenitor Cells-Based Metabolomics Study Reveals Dysregulated Lipid Metabolism and Identifies Putative Biomarkers for CLN6 Disease. Sci Rep 2023, 13, 18550. [Google Scholar] [CrossRef]
- Tuermer, A.; Mausbach, S.; Kaade, E.; Damme, M.; Sylvester, M.; Gieselmann, V.; Thelen, M. CLN6 Deficiency Causes Selective Changes in the Lysosomal Protein Composition. Proteomics 2021, 21, e2100043. [Google Scholar] [CrossRef]
- Bajaj, L.; Sharma, J.; di Ronza, A.; Zhang, P.; Eblimit, A.; Pal, R.; Roman, D.; Collette, J.R.; Booth, C.; Chang, K.T.; et al. A CLN6-CLN8 Complex Recruits Lysosomal Enzymes at the ER for Golgi Transfer. J Clin Invest 2020, 130, 4118–4132. [Google Scholar] [CrossRef]
- Yamashita, A.; Hiraki, Y.; Yamazaki, T. Identification of CLN6 as a Molecular Entity of Endoplasmic Reticulum-Driven Anti-Aggregate Activity. Biochem Biophys Res Commun 2017, 487, 917–922. [Google Scholar] [CrossRef]
- Yamashita, A.; Shiro, Y.; Hiraki, Y.; Yujiri, T.; Yamazaki, T. Implications of Graded Reductions in CLN6’s Anti-Aggregate Activity for the Development of the Neuronal Ceroid Lipofuscinoses. Biochem Biophys Res Commun 2020, 525, 883–888. [Google Scholar] [CrossRef]
- Shiro, Y.; Yamashita, A.; Watanabe, K.; Yamazaki, T. CLN6’s Luminal Tail-Mediated Functional Interference between CLN6 Mutants as a Novel Pathomechanism for the Neuronal Ceroid Lipofuscinoses. Biomed Res 2021, 42, 129–138. [Google Scholar] [CrossRef]
- Best, H.L.; Clare, A.J.; McDonald, K.O.; Wicky, H.E.; Hughes, S.M. An Altered Secretome Is an Early Marker of the Pathogenesis of CLN6 Batten Disease. J Neurochem 2021, 157, 764–780. [Google Scholar] [CrossRef]
- Teixeira, C.A.F.; Lin, S.; Mangas, M.; Quinta, R.; Bessa, C.J.P.; Ferreira, C.; Sa Miranda, M.C.; Boustany, R.-M.N.; Ribeiro, M.G. Gene Expression Profiling in VLINCL CLN6-Deficient Fibroblasts: Insights into Pathobiology. Biochim Biophys Acta 2006, 1762, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Palmer, D.N.; Fearnley, I.M.; Walker, J.E.; Hall, N.A.; Lake, B.D.; Wolfe, L.S.; Haltia, M.; Martinus, R.D.; Jolly, R.D. Mitochondrial ATP Synthase Subunit c Storage in the Ceroid-Lipofuscinoses (Batten Disease). Am J Med Genet 1992, 42, 561–567. [Google Scholar] [CrossRef]
- Martinus, R.D.; Harper, P.A.; Jolly, R.D.; Bayliss, S.L.; Midwinter, G.G.; Shaw, G.J.; Palmer, D.N. Bovine Ceroid-Lipofuscinosis (Batten’s Disease): The Major Component Stored Is the DCCD-Reactive Proteolipid, Subunit C, of Mitochondrial ATP Synthase. Vet Res Commun 1991, 15, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Palmer, D.N.; Fearnley, I.M.; Medd, S.M.; Walker, J.E.; Martinus, R.D.; Bayliss, S.L.; Hall, N.A.; Lake, B.D.; Wolfe, L.S.; Jolly, R.D. Lysosomal Storage of the DCCD Reactive Proteolipid Subunit of Mitochondrial ATP Synthase in Human and Ovine Ceroid Lipofuscinoses. Adv Exp Med Biol 1989, 266, 211–223. [Google Scholar] [CrossRef]
- Fearnley, I.M.; Walker, J.E.; Martinus, R.D.; Jolly, R.D.; Kirkland, K.B.; Shaw, G.J.; Palmer, D.N. The Sequence of the Major Protein Stored in Ovine Ceroid Lipofuscinosis Is Identical with That of the Dicyclohexylcarbodiimide-Reactive Proteolipid of Mitochondrial ATP Synthase. Biochem J 1990, 268, 751–758. [Google Scholar] [CrossRef]
- Hughes, S.M.; Moroni-Rawson, P.; Jolly, R.D.; Jordan, T.W. Submitochondrial Distribution and Delayed Proteolysis of Subunit c of the H+-Transporting ATP-Synthase in Ovine Ceroid-Lipofuscinosis. Electrophoresis 2001, 22, 1785–1794. [Google Scholar] [CrossRef]
- Kominami, E.; Ezaki, J.; Wolfe, L.S. New Insight into Lysosomal Protein Storage Disease: Delayed Catabolism of ATP Synthase Subunit c in Batten Disease. Neurochem Res 1995, 20, 1305–1309. [Google Scholar] [CrossRef]
- Tanner, A.J.; Dice, J.F. Batten Disease and Mitochondrial Pathways of Proteolysis. Biochem Mol Med 1996, 57, 1–9. [Google Scholar] [CrossRef]
- Das, A.M.; Jolly, R.D.; Kohlschutter, A. Anomalies of Mitochondrial ATP Synthase Regulation in Four Different Types of Neuronal Ceroid Lipofuscinosis. Mol Genet Metab 1999, 66, 349–355. [Google Scholar] [CrossRef]
- Pezzini, F.; Gismondi, F.; Tessa, A.; Tonin, P.; Carrozzo, R.; Mole, S.E.; Santorelli, F.M.; Simonati, A. Involvement of the Mitochondrial Compartment in Human NCL Fibroblasts. Biochem Biophys Res Commun 2011, 416, 159–164. [Google Scholar] [CrossRef]
- Cao, Y.; Staropoli, J.F.; Biswas, S.; Espinola, J.A.; MacDonald, M.E.; Lee, J.-M.; Cotman, S.L. Distinct Early Molecular Responses to Mutations Causing VLINCL and JNCL Presage ATP Synthase Subunit C Accumulation in Cerebellar Cells. PLoS One 2011, 6, e17118. [Google Scholar] [CrossRef] [PubMed]
- Heine, C.; Tyynela, J.; Cooper, J.D.; Palmer, D.N.; Elleder, M.; Kohlschutter, A.; Braulke, T. Enhanced Expression of Manganese-Dependent Superoxide Dismutase in Human and Sheep CLN6 Tissues. Biochem J 2003, 376, 369–376. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Photograph of the proband about the time of onset of neurological disease signs.

Figure 2.
Sagittal (A) and axial (B) T2-weighted MR images of the proband’s brain. The ventricular system (V) was dramatically enlarged, and there was a pronounced shrinkage of the parenchyma of both the cerebellum (green arrow in A) and the cerebral cortex (red arrow in B).
Figure 2.
Sagittal (A) and axial (B) T2-weighted MR images of the proband’s brain. The ventricular system (V) was dramatically enlarged, and there was a pronounced shrinkage of the parenchyma of both the cerebellum (green arrow in A) and the cerebral cortex (red arrow in B).
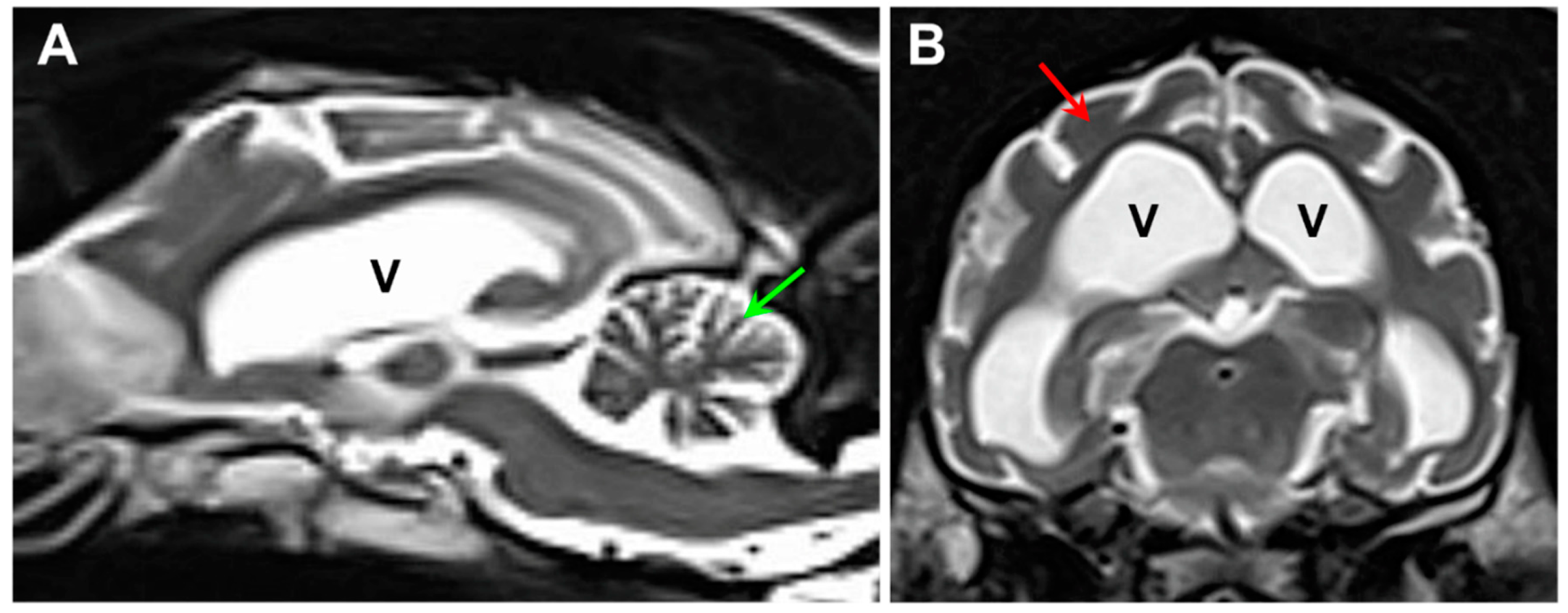
Figure 3.
Fluorescence micrographs of unstained cryostat sections of cerebellar cortex, cerebral cortex, retina, and cardiac muscle. In the cerebellum, aggregates of autofluorscent inclusions were present in cells of the molecular (m), Purkinje (p), and granule cell (g) layer, with particularly large granules in the Purkinje cells (white arrows). In the retina, accumulations of these inclusions were most prominent in the ganglion cells (blue arrows), with small individual autofluorescent granules in other layers of the retina (red arrows). Retinal layers shown include the ganglion cell layer (gcl), inner plexiform layer (ipl), and inner nuclear layer (inl).
Figure 3.
Fluorescence micrographs of unstained cryostat sections of cerebellar cortex, cerebral cortex, retina, and cardiac muscle. In the cerebellum, aggregates of autofluorscent inclusions were present in cells of the molecular (m), Purkinje (p), and granule cell (g) layer, with particularly large granules in the Purkinje cells (white arrows). In the retina, accumulations of these inclusions were most prominent in the ganglion cells (blue arrows), with small individual autofluorescent granules in other layers of the retina (red arrows). Retinal layers shown include the ganglion cell layer (gcl), inner plexiform layer (ipl), and inner nuclear layer (inl).
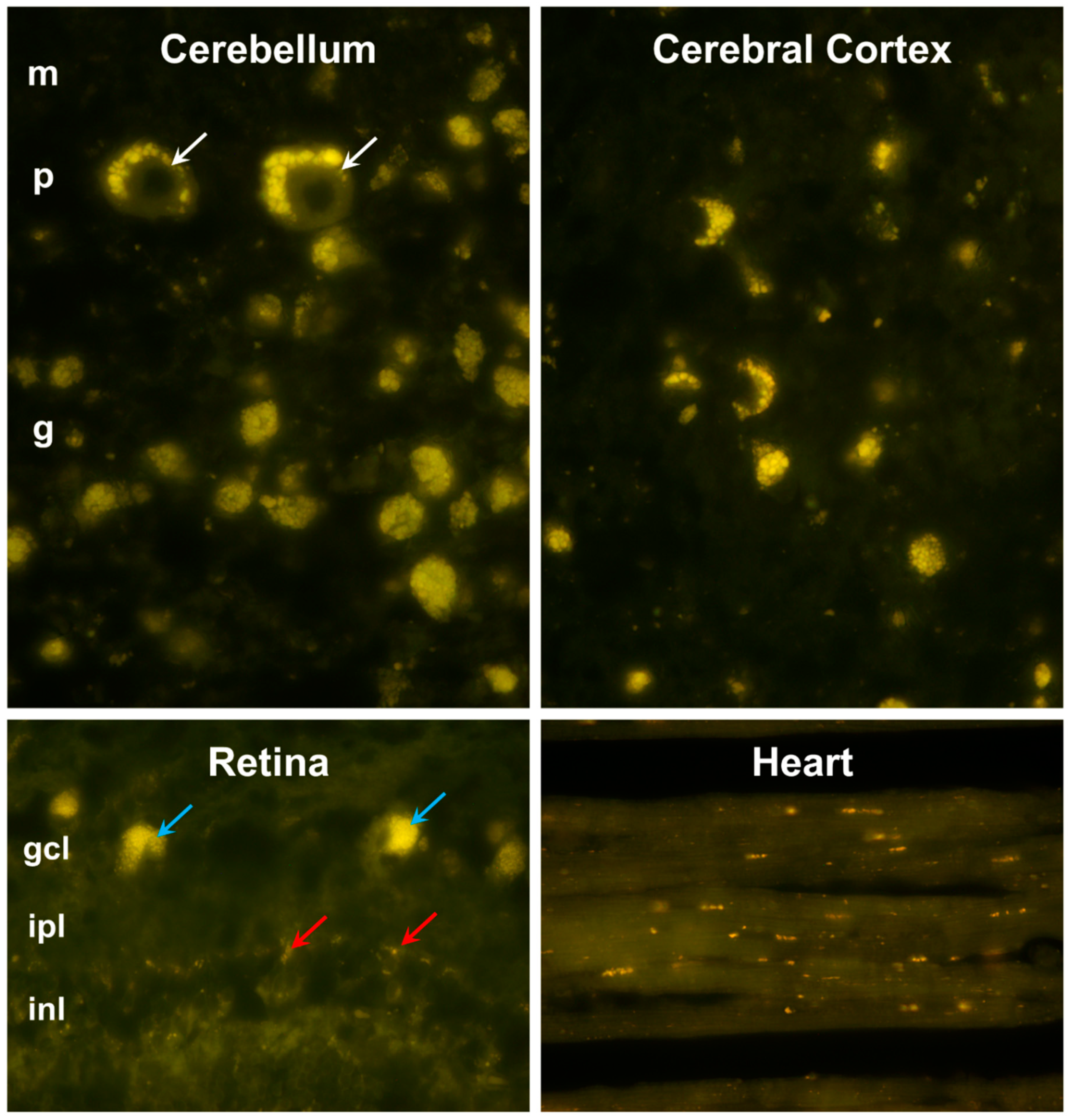
Figure 4.
Electron micrographs of representative storage bodies in cells of the cerebellar cortex. The storage body in (A) contains a number of inclusions that appear to be derived from mitochondria (m). In (B) the organization of the membrane-like contents of the storage body is more heterogeneous.
Figure 4.
Electron micrographs of representative storage bodies in cells of the cerebellar cortex. The storage body in (A) contains a number of inclusions that appear to be derived from mitochondria (m). In (B) the organization of the membrane-like contents of the storage body is more heterogeneous.
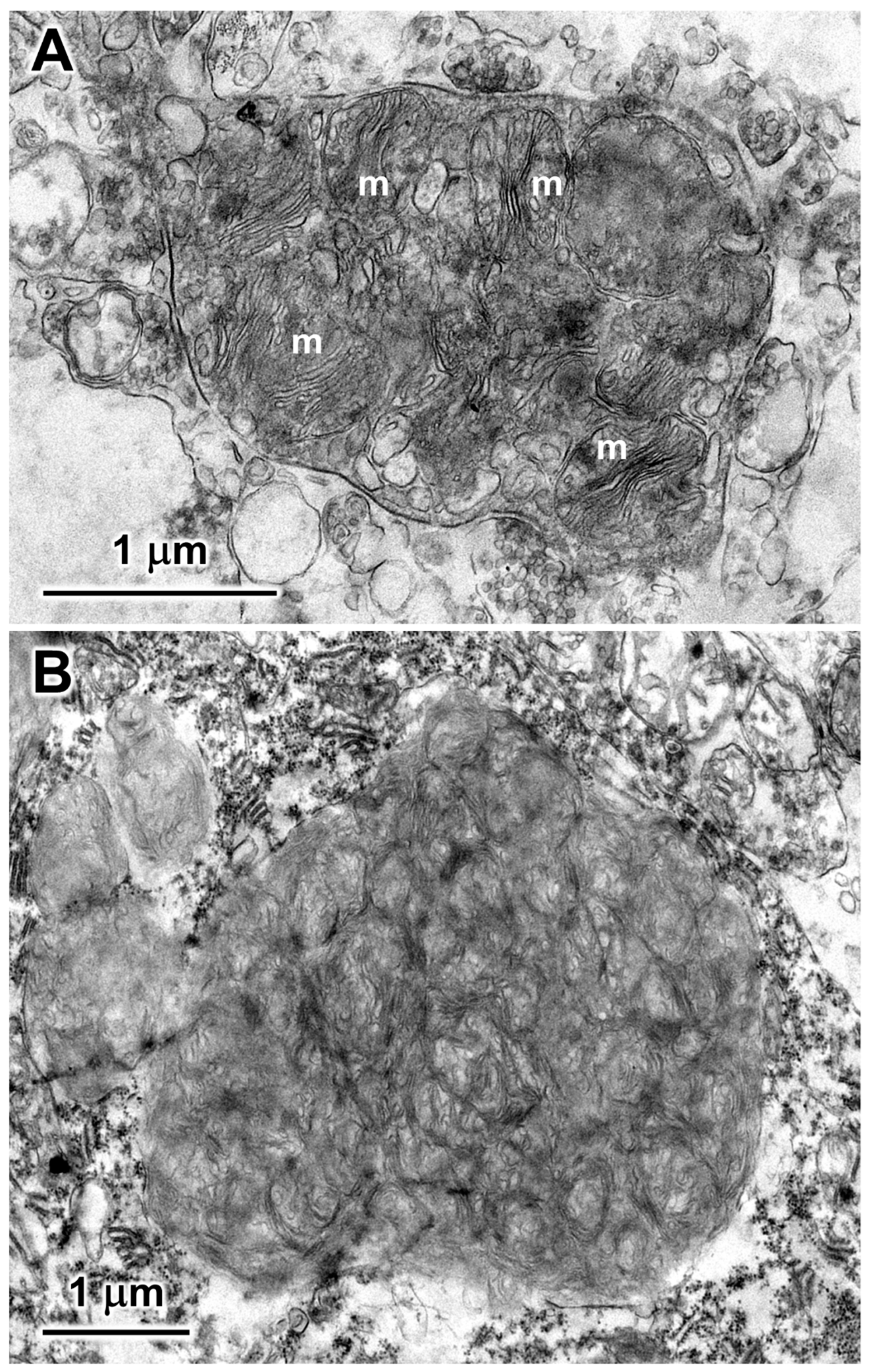
Figure 5.
Electron micrograph of a disease-related storage body in a cerebral cortical neuron of the proband.
Figure 5.
Electron micrograph of a disease-related storage body in a cerebral cortical neuron of the proband.
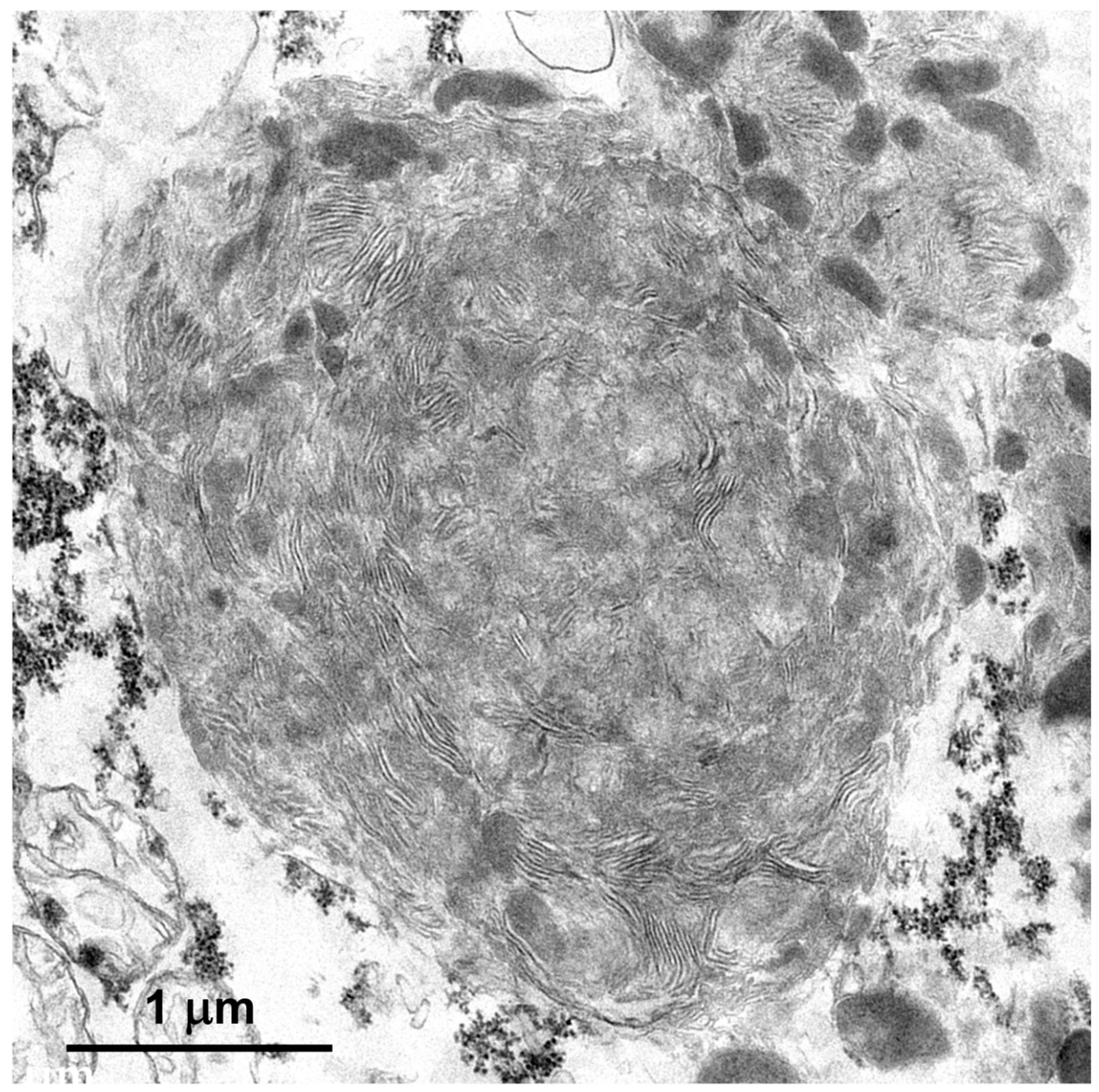
Figure 6.
Electron micrograph of a cluster of disease-related storage bodies in a retinal ganglion cell of the proband.
Figure 6.
Electron micrograph of a cluster of disease-related storage bodies in a retinal ganglion cell of the proband.
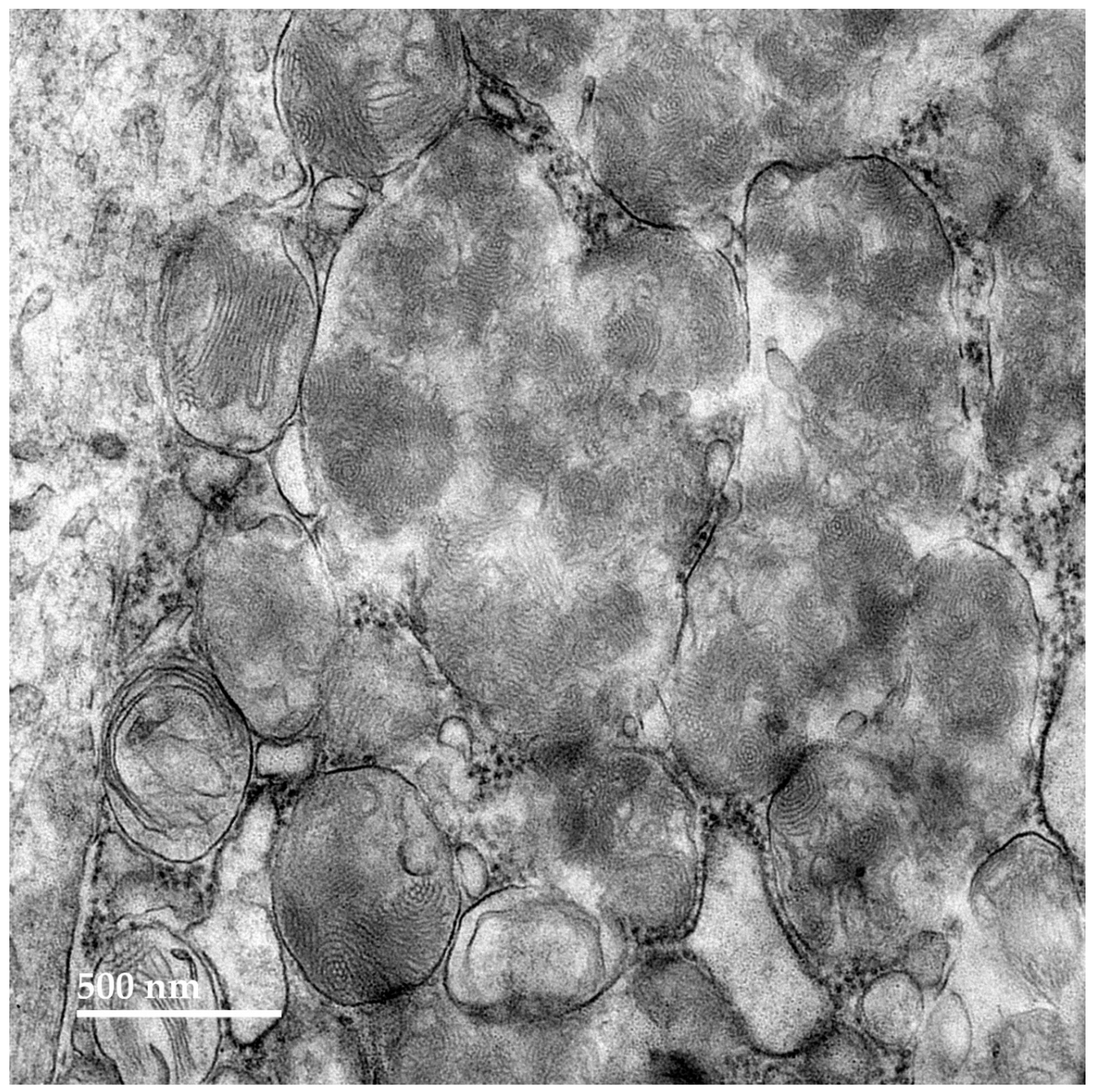
Figure 7.
Electron micrograph of disease-related storage body (SB) in cardiac muscle of the proband.
Figure 7.
Electron micrograph of disease-related storage body (SB) in cardiac muscle of the proband.
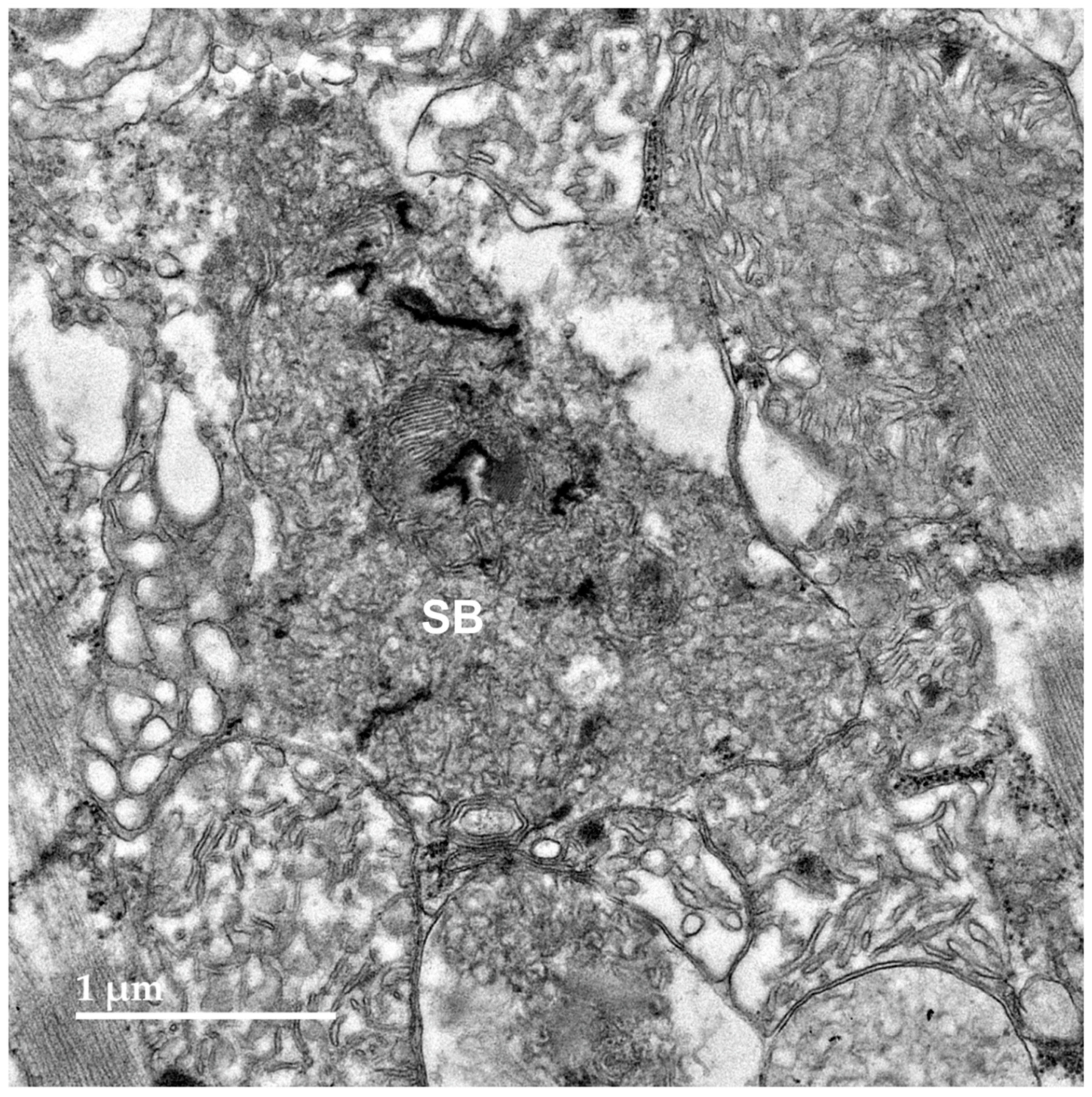
Figure 8.
Electron micrograph of a cluster of disease-related storage bodies (SB) in cardiac muscle of the proband containing primarily vesicular structures.
Figure 8.
Electron micrograph of a cluster of disease-related storage bodies (SB) in cardiac muscle of the proband containing primarily vesicular structures.
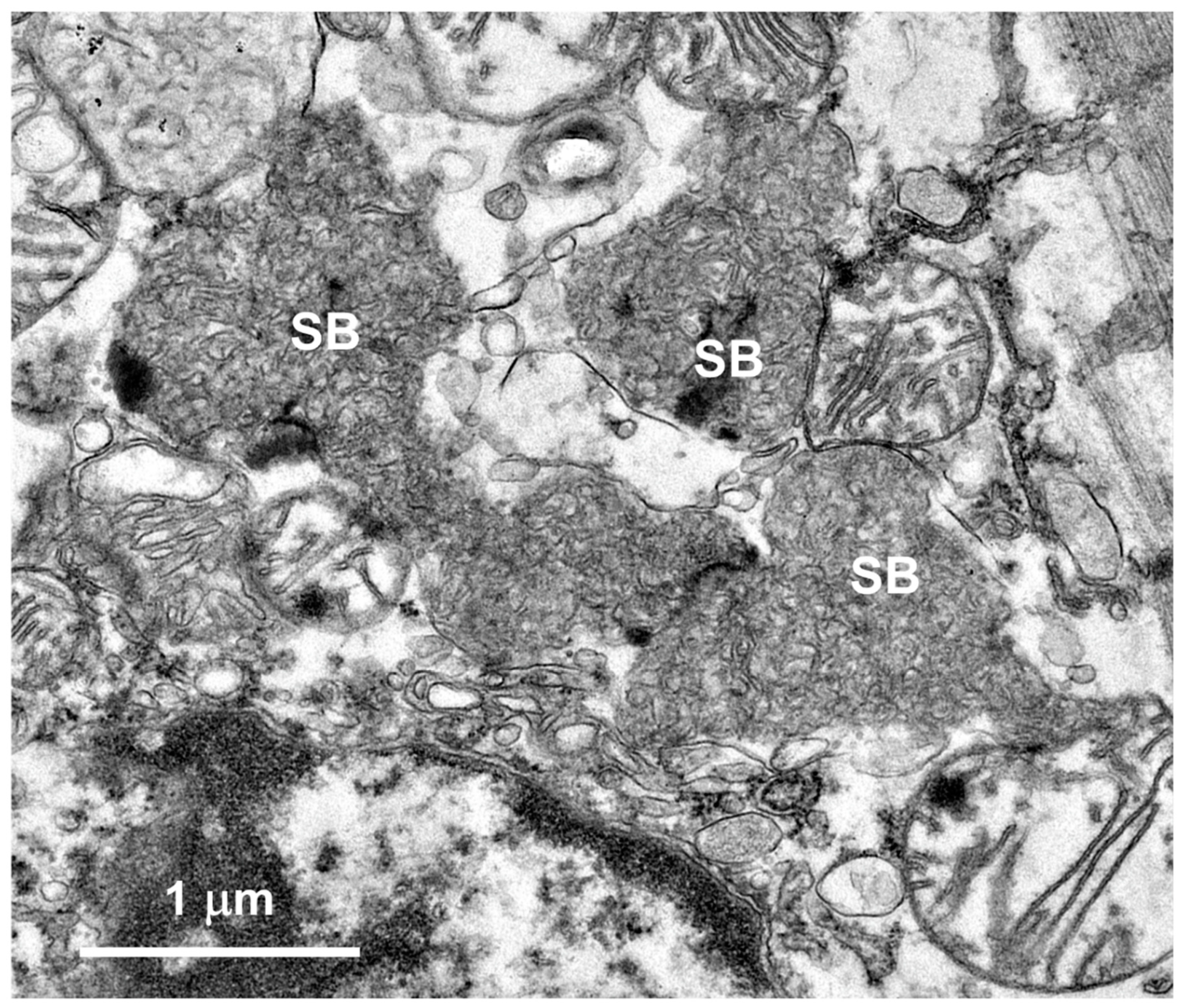
Figure 9.
Electron micrograph of disease-related storage body (SB) in cardiac muscle of the proband containing stacks of membrane-like components.
Figure 9.
Electron micrograph of disease-related storage body (SB) in cardiac muscle of the proband containing stacks of membrane-like components.
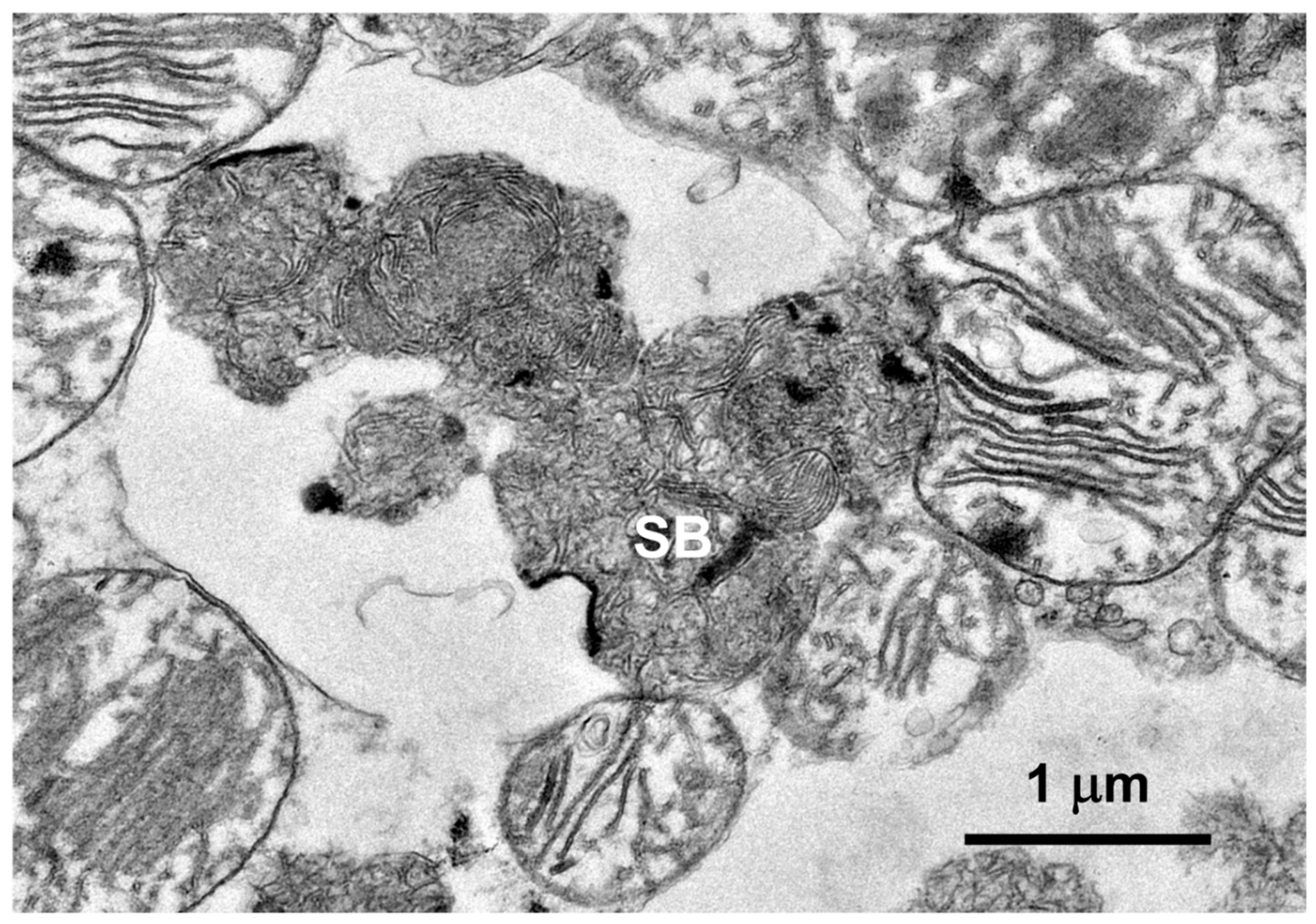
Figure 10.
Paraffin sections of cerebral cortex gray matter (A) and cerebellar cortex (B) of the proband immunostained with an anti-subunit c antibody. Dark brown immunostain (arrows) was present in the numerous cells of both brain regions, including Purkinje cells (p) of the cerebellum.
Figure 10.
Paraffin sections of cerebral cortex gray matter (A) and cerebellar cortex (B) of the proband immunostained with an anti-subunit c antibody. Dark brown immunostain (arrows) was present in the numerous cells of both brain regions, including Purkinje cells (p) of the cerebellum.
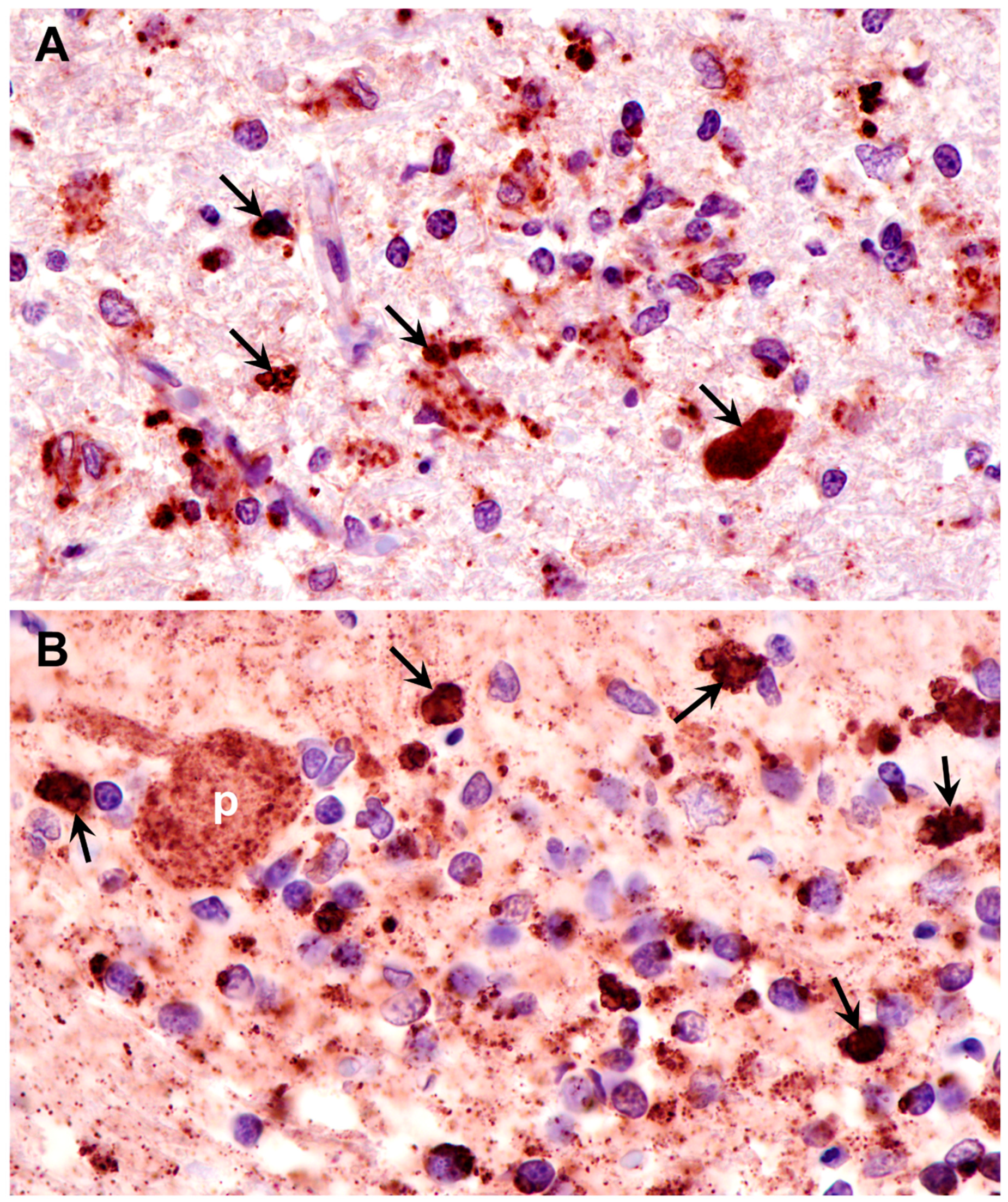
Figure 11.
Paraffin sections of retina (A) and cardiac muscle (B) of the proband immunostained with an anti-subunit c antibody. Dark brown subunit c immunostain (arrows and arrowheads) was present in retinal ganglion cells (g), scattered throughout the rest of the retina (arrowheads in A), and in rows of punctate inclusions flanking nuclei of cardiac muscle fibers (arrows in B).
Figure 11.
Paraffin sections of retina (A) and cardiac muscle (B) of the proband immunostained with an anti-subunit c antibody. Dark brown subunit c immunostain (arrows and arrowheads) was present in retinal ganglion cells (g), scattered throughout the rest of the retina (arrowheads in A), and in rows of punctate inclusions flanking nuclei of cardiac muscle fibers (arrows in B).
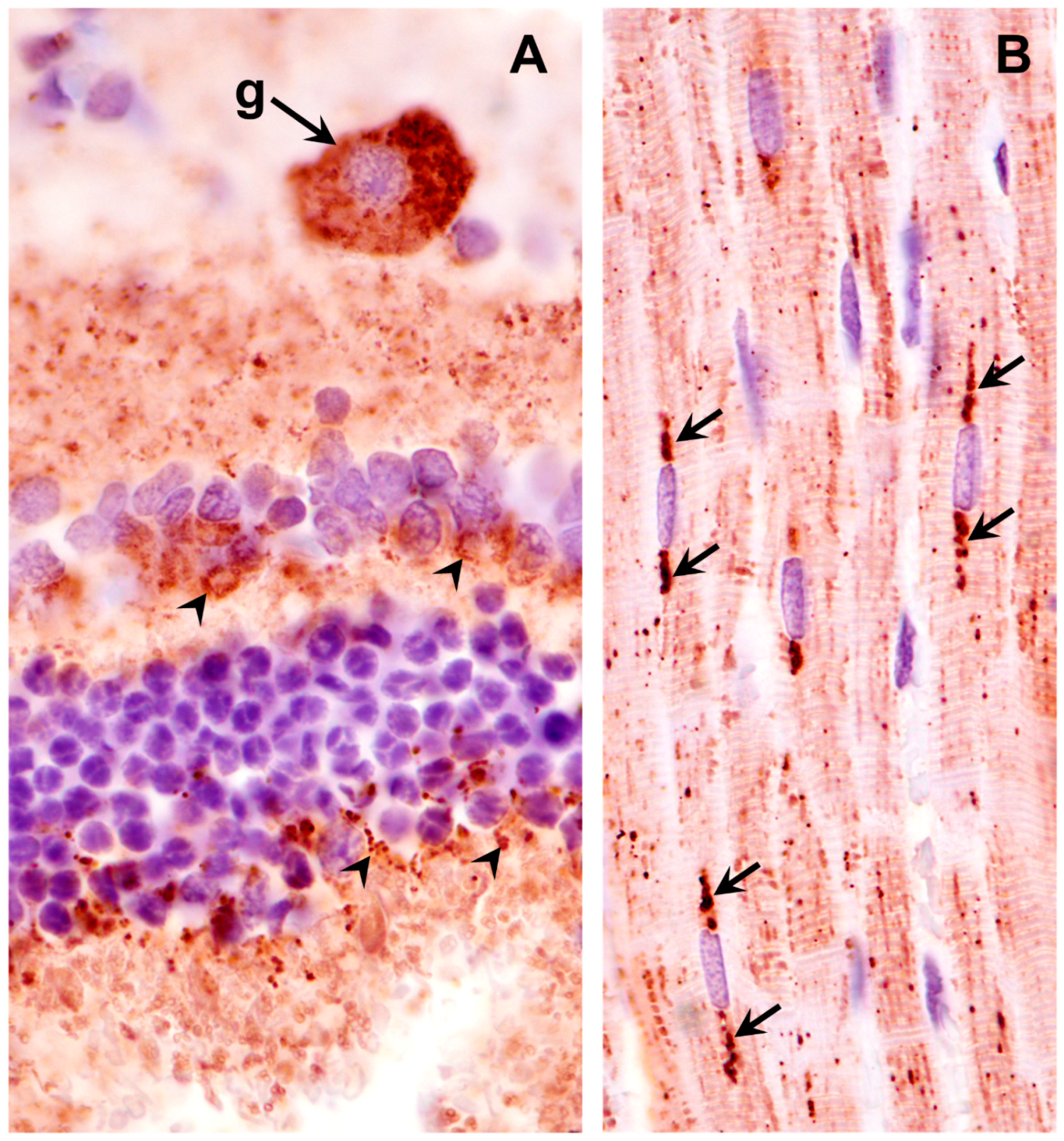
Figure 12.
Paraffin sections of cerebral cortex gray matter (A) and cerebellar cortex (B and C) immunostained for GFAP localization (dark brown stain). Intensely-stained cells (arrows) are activated astrocytes. Processes of the activated astrocytes extended throughout the molecular (m), Purkinje cell (p), and granular (g) layers of the cerebellar cortex (B). Granular layer of the cerebellar cortex is shown in (C).
Figure 12.
Paraffin sections of cerebral cortex gray matter (A) and cerebellar cortex (B and C) immunostained for GFAP localization (dark brown stain). Intensely-stained cells (arrows) are activated astrocytes. Processes of the activated astrocytes extended throughout the molecular (m), Purkinje cell (p), and granular (g) layers of the cerebellar cortex (B). Granular layer of the cerebellar cortex is shown in (C).
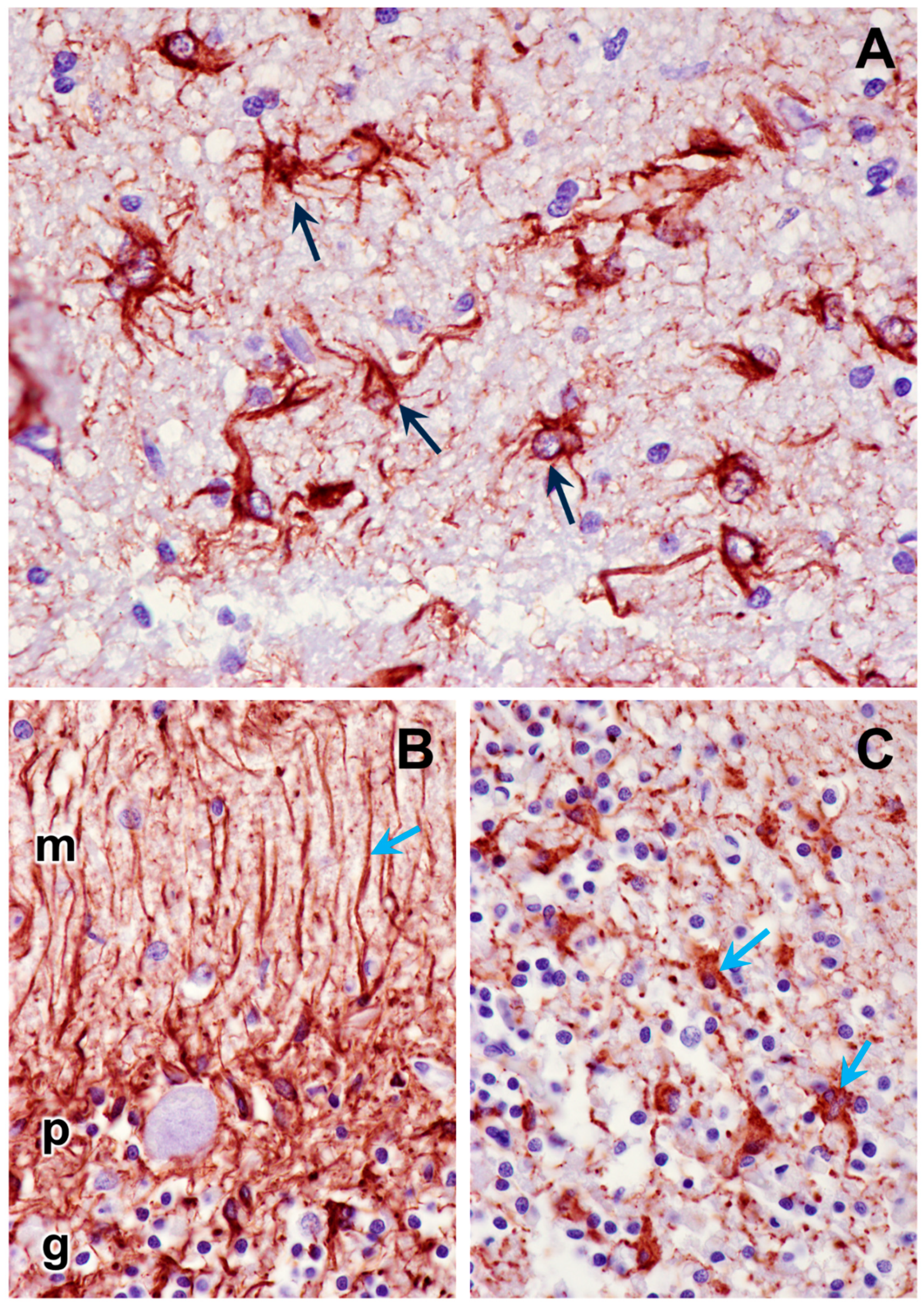
Figure 13.
Paraffin sections of cerebral cortex gray matter (A) and cerebellar cortex (B) immunostained for Iba1 localization (dark brown stain). Intensely-stained cells (arrows) are activated microglia. Activated microglia were present within the molecular (m), Purkinje cell (p), and granular (g) layers of the cerebellar cortex (B).
Figure 13.
Paraffin sections of cerebral cortex gray matter (A) and cerebellar cortex (B) immunostained for Iba1 localization (dark brown stain). Intensely-stained cells (arrows) are activated microglia. Activated microglia were present within the molecular (m), Purkinje cell (p), and granular (g) layers of the cerebellar cortex (B).
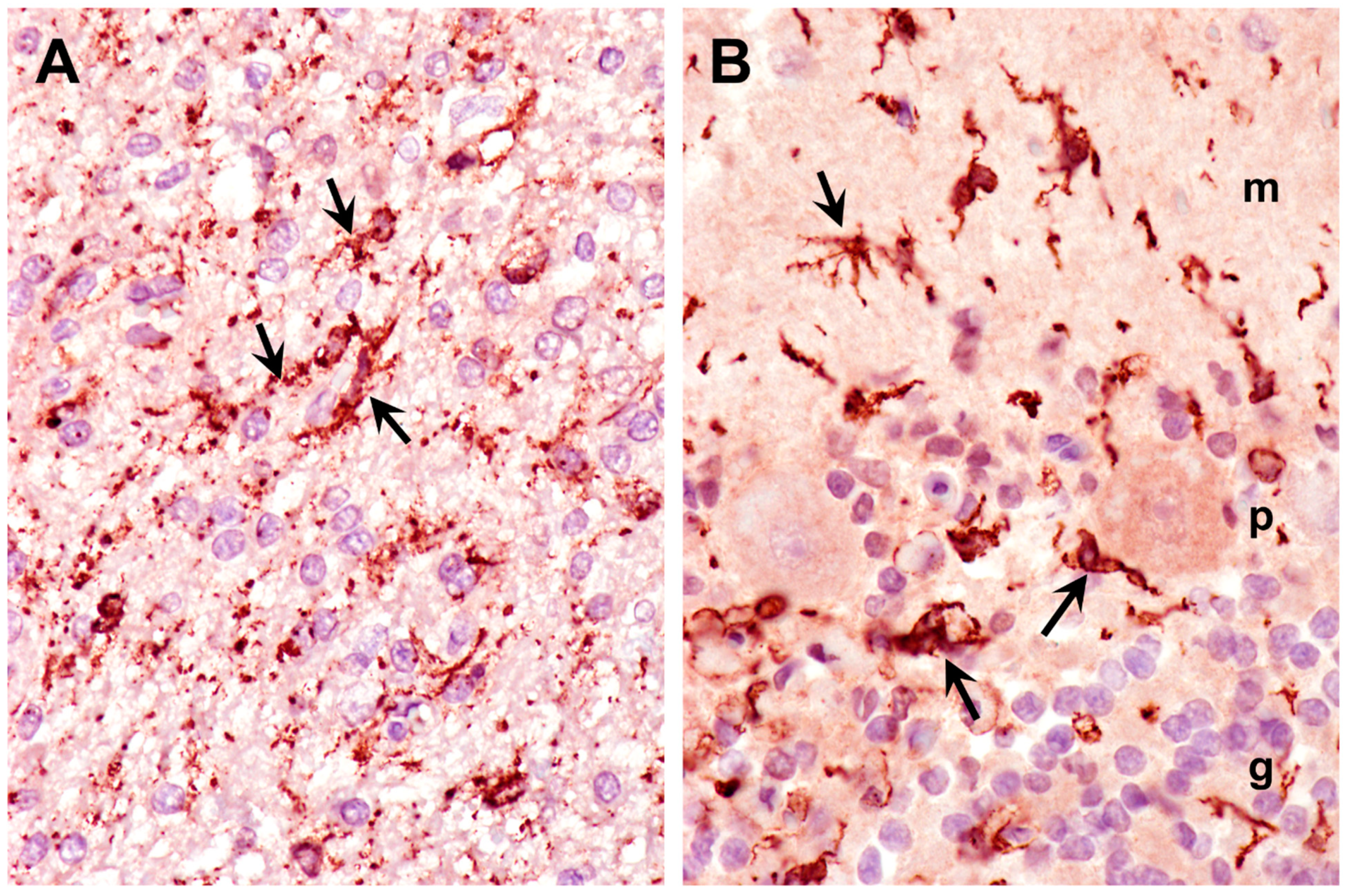
Figure 14.
Predicted partial CLN6 pre-mRNA sequences from the reference canine genome and the genome sequence of the proband showing the location of the G>A substitution at the 3’ end of intron 3.
Figure 14.
Predicted partial CLN6 pre-mRNA sequences from the reference canine genome and the genome sequence of the proband showing the location of the G>A substitution at the 3’ end of intron 3.

Figure 15.
Reads of the proband’s whole genome sequence aligned to the reference sequence in the vicinity of position 32,185,406 on chromosome 30 as viewed with the Integrative Genomics Viewer. The variant T is highlighted in red.
Figure 15.
Reads of the proband’s whole genome sequence aligned to the reference sequence in the vicinity of position 32,185,406 on chromosome 30 as viewed with the Integrative Genomics Viewer. The variant T is highlighted in red.

Figure 16.
Paraffin sections of the cerebellar cortex (A) and cerebral cortex (C) of an unaffected Shiba Inu dog, and the cerebellar cortex (B) and cerebral cortex (D) of the proband immunolabeled for localization of CLN6 protein (dark brown stain). In the cerebellar cortex of the of the unaffected dog, the Purkinje cells exhibited pronounced immunolabeling (arrows in A), that was not observed in the Purkinje cells of the proband (arrows in B). Likewise, cells throughout the cerebral cortex gray matter of the unaffected dog exhibited substantial immunolabeling (arrows in C) that was not observed in the cerebral cortex gray matter of the proband (D).
Figure 16.
Paraffin sections of the cerebellar cortex (A) and cerebral cortex (C) of an unaffected Shiba Inu dog, and the cerebellar cortex (B) and cerebral cortex (D) of the proband immunolabeled for localization of CLN6 protein (dark brown stain). In the cerebellar cortex of the of the unaffected dog, the Purkinje cells exhibited pronounced immunolabeling (arrows in A), that was not observed in the Purkinje cells of the proband (arrows in B). Likewise, cells throughout the cerebral cortex gray matter of the unaffected dog exhibited substantial immunolabeling (arrows in C) that was not observed in the cerebral cortex gray matter of the proband (D).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



