
Submitted:
02 May 2024
Posted:
03 May 2024
You are already at the latest version
Abstract
In this study, we demonstrate the direct preparation of dihalo-γ-lactams featuring two distinct halogens from dichloroamides using a novel Atom Exchange Radical Cyclization (AERC) procedure. This method integrates the established Atom Transfer Radical Cyclization with halogen exchange in solution. The technique operates under mild conditions and requires little amounts of metallic copper, serving as both a supplemental activator and reducing agent.
Keywords:
atom transfer radical cyclization
; transition metal catalysis
; γ-lactams
; halocompounds
1. Introduction
Exploiting the reactivity of open-shell organic compounds, radical synthetic methodologies often require low energy input. Atom transfer radical techniques provide the additional benefit of high atom economy, achieved through the termination of radical intermediates by active species, typically a halogen atom, that results in the conservation of the initially present functionality. These properties can be exploited to achieve high efficiency and sustainability in the synthesis of new molecular or polymeric targets. Furthermore, the use of catalytic amount of a transition metal complex, often allows optimal control over the generation and shut-off of the reactive radical species, which translates into high product selectivities.
Among the various atom transfer radical techniques, Atom Transfer Radical Polymerization (ATRP) stand out as the most widely used today. ATRP enables the construction of polymeric materials with precise control, i.e., narrow polydispersity, and halogen-capped chain ends. In addition to ATRP, applications for the synthesis of low molecular weight targets are also well developed. These include addition (Atom Transfer Radical Addition, ATRA) and/or cyclization (Atom Transfer Radical Cyclization, ATRC) processes, providing an environmental-friendly route to industrially appealing building blocks or pharmaceuticals.
A classic example is the transition metal-catalyzed ATRC of N-allyl-2,2-dichloroamides, which enables access to functionalized specialty chemicals [1]. The process starts with the reversible interaction between the substrate (e.g., 1 of Figure 1) and the metal catalyst, composed of a transition metal salt (e.g., CuCl) and a nitrogen polydentate ligand (L, e.g., tris(2-pyridilmethyl)amine, TPMA).
Through a monoelectronic halogen transfer, the active catalyst Cu(I)Cl·L is oxidized to Cu(II)Cl2·L, generating the radical 1’. This species can be considered electrophilic, since the radical-bearing carbon is surrounded by electron-withdrawing groups (EWGs). Featuring a low-lying SOMO, its interaction with the electron-rich alkenyl moiety is anticipated to be rapid, particularly when an appropriate auxiliary group on the nitrogen atom (e.g., Ph) induces a favorable conformational push from trans-1’ to cis-1’ [2], leading to the cyclic radical 1”. The absence of EWGs near the radical center in 1” renders it nucleophilic, thus slowing down its intermolecular addition to other substrate molecules. This allows the use of less solvent while maintaining a high productivity and a high selectivity towards the cyclic product, even when working at molar concentrations. Typically, 1” exhibits high affinity for the oxidized form of the metal catalyst, which acts as a persistent radical [3]. This interaction not only leads to the conversion of 1” into the product 2, but let the regeneration of the metal catalyst in its active form. This irreversible regeneration step suggests that the radical cycle may be sustained by a very low amount of catalyst but, in practice, inevitable termination events lead to the progressive build-up of the oxidized form of the catalyst. Thus, to achieve complete conversion, higher amounts of metal catalysts are employed, typically around 10 mol%.
To further improve reaction control and sustainability, a proper reducing agent (“red.” in Figure 1) can be added to reactivate the spent catalyst. Among the various alternatives, the “activators regenerated by electron transfer” (ARGET) process, which employs non-radical reducing agents, is well developed and can work with very low metal loads [4]. Thus, the ARGET-ATRC of easily assembled dichloroamides offers an effective and sustainable route to dichloro-γ-lactams. These compounds represent advanced intermediates found within various valuable targets, such as anti-hypertensives [5], psychotropic agents [6], proteolysis inhibitors [7], and antimuscarinic agents [8].
The numerous functional groups within of dichloro-γ-lactams backbone readily explain their extensive synthetic flexibility. An example of this is the well-known elimination/substitution sequence (solid arrow in the upper Figure 2), which provides access to maleimides or their related hydrolysis products, i.e., maleic anhydrides [9]. The notable acidity of the hydrogen in position 4 of these structures (A, Figure 2) make them highly susceptible to first undergo elimination of hydrogen halide, a feature that was exploited for the preparation of unsaturated pyrrolidin-2-ones (B) [10].
In addition to the described transformations, the selective substitution of the exocyclic halogen atom of A hold significant potential. Specifically, the use of oxygen nucleophiles shows promise for generating higher oxidation state structures, including paraconic acids, aza-paraconic acids, and their unsaturated counterparts (dashed arrows in the lower Figure 2), which represent valuable bioactive targets.
In an attempt to modify 2 (Figure 1), our initial approach was to evaluate several oxidative substitution protocols suitable for neutral or low-basicity environments, aiming to circumvent the described 3,4 elimination. Unfortunately, potentially suitable methods such as the Kornblum [11], H2O2/EtOH –based [12,13], H2O2/AlCl3 –based [14], or trimethylamine N-oxide-based systems [15], all failed to yield a clean exocyclic substitution in our experiments.
The replacement of the exocyclic chlorine atom with a more efficient leaving group such as iodine or the more atom-economical bromine was anticipated to yield improved outcomes. The attainment of a dihalo-γ-pyrrolidinone with a more activated halide on the exocyclic side can be achieved through various synthetic avenues: (i) an ATRC of a chloro-bromoamide, (ii) a selective halogen exchange process on the exocyclic chlorine atom of 2 [16], or (iii) a proper trapping of the cyclic radical 1” (Figure 1). The first approach is hindered by the non-trivial preparation of the dihaloamide [10], while the second option may warrant further investigation. However, among the three methods, the last one shows the highest potential in terms of sustainability, due to its ability to exploit the intrinsic reactivity of 1”. An already known approach involves the addition of a Kharasch radical trap [17], but its application is hampered by the use of toxic tetrahalomethanes.
Therefore, the aim of this study is to evaluate the feasibility of leveraging the innate affinity of 1” towards the metal complex catalyst, along with the option to modify the composition of the latter through some inorganic halides added in solution, accordingly to some recent studies related to ATRP [18]. This should enable an overall Atom Exchange Radical Cyclization (AERC) process.
2. Materials and Methods
2.1. General
Reagents and solvents were standard grade commercial products and were used without further purification. For the catalytic systems we used: CuCl2 from Fisher Scientific (≥ 97%), TPMA (98%) and ascorbic acid (> 99.5%) from Merck, Na2CO3 from Carlo Erba (> 99.5%). The starting amides 1 and 3 were obtained through standard aminodechlorination of acyl chlorides with allyl amines (all prepared by N-alkylation of amines, adapting the Shipman’s method [19]). 2,2-dichloropropanoyl chloride was prepared from commercial propanoyl chloride, as described in ref. [20]. Dichlorolactam 2 [4] and trichlorolactam 5 [21] are known compounds. Silica Gel 60 from Merck (40–63 µm) was employed for flash chromatography purifications.
Elemental Analysis (EA) were performed with a Thermo Scientific FLASH 2000 organic elemental analyzer. GC-MS spectra were acquired with a ‘HP G1800C GCD System Series II’ (Agilent Technologies Italy, Cernusco sul Naviglio (MI), Italy). IR spectra were acquired with a FTIR-4700LE spectrometer (Jasco Europe, Cremella (LC), Italy).
1H NMR and 13C NMR spectra were recorded on Bruker Avance 600 spectrometer (Billerica, MA, USA). All the mono- and bidimensional experiments were performed with standard pulses programs. 1H NMR and 13C NMR signals attribution was based on 1H,1H-DQF-COSY, 1H,13C-EditedHSQC, and 1H,13C-HMBC experiments. The structural assignment of compounds 3 and 6 were determined by homonuclear Nuclear Overhauser Enhancement NMR correlation techniques. Carbon signals were detectable and assigned only to major products.
2.1. ARGET-ATRC towards 2
In an oven-dried Schlenk tube (previously rinsed with a 10% aqueous NH4OH and water in sequence) were weighed ascorbic acid (0.2 mmol, 35.2 mg), Na2CO3 (0.4 mmol, 42.4 mg) and amide 1 (8 mmol, 2.065 g). After three cycles of vacuum/argon (around 10 min, each) ethyl acetate (AcOEt, 3 mL) and – once the substrate was fully solubilized – a 0.04 mol/L solution of CuCl2/TPMA in absolute ethanol (EtOH, 1 mL) were added under argon. The Schlenk tube was heated at 35 °C and stirred at 700 rpm for 8 h. The reaction was then quenched with water (8 mL), acidified with aqueous HCl 10% w/v and extracted with CH2Cl2 (3 × 6 mL). The combined organic layers were concentrated to dryness at the rotary evaporator, and the crude product was purified by flash chromatography on silica gel, eluting with a petroleum ether (PE bp 40–60 °C)/diethyl ether (Et2O) gradient (from 100/0 to 0/100). This gave the pyrrolidinone 2 as a colourless oil; yield: 1.941 g (94%), as an inseparable mixture of cis/trans diastereomers (87:13).
2.2. SARA-AERC towards 3 or 6
Preparation of 3 - In an oven-dried Schlenk tube (previously rinsed with a 10% aqueous NH4OH and water in sequence) were weighed Na2CO3 (0.2 mmol, 21 mg), NaBr (4 mmol, 0.412 g), amide 1 (4 mmol, 1.032 g), and metallic copper wire (length 40 mm, diameter 1 mm, 99.9 %). After three cycles of vacuum/argon (around 10 min, each) AcOEt (2 mL), EtOH (96 vol%, 1 mL) and TPMA (1 mL of 0.04 mol/L solution in absolute ethanol) were added under argon. The Schlenk tube was heated at 37 °C and stirred at 700 rpm for 16 h. The reaction was then quenched with water (6 mL), acidified with aqueous HCl (10% w/v, 1 mL) and extracted with CH2Cl2 (3 × 5 mL). The combined organic layers were concentrated to dryness at the rotary evaporator, giving the crude product 3 as a brownish solid (95 %, 3.8 mmol, 1.150 g), that resulted contaminated by a small amount (~5% GC) of ATRC dichlorolactam 2, and a small amount (~5% GC) of isomeric dibromolactam 3b (see Figure 3 and Figure S1). A purer version of 3 was obtained through (unoptimized) recrystallization from a mixture of CH2Cl2 : toluene : Et2O in 65: 15: 20 ratio, giving a beige solid in 82 % yield, as an inseparable mixture of cis/trans diastereomers (97:3).
Compound 3: (cis-isomer, 97 %): 1H NMR (600 MHz, CDCl3): δ = 1.86 (s, 3 H, CH3), 2.75 (m, 1 H, CH), 3.65 (pq, J = 9.4 Hz, 2 H, CHHBr, CHHNCO), 3.71 (pdd, J = 4.74, 10.4 Hz, 1 H, CHHBr), 4.02 (pdd, J = 6.9, 9.9 Hz, 1 H, CHHNCO), 7.21 (pt, J = 7.4 Hz, 1 H, p-ArH), 7.40 (pt, J = 7.6 Hz, 2 H, m-ArH), 7.65 (pd, J = 8.1 Hz, 2H, o-ArH) ppm. 13C NMR (150 MHz, CDCl3): δ = 25.0 (CH3), 29.2 (CH2Br), 47.3 (CH), 50.6 (CH2NCO), 120.3 (o-ArC), 125.7 (p-ArC), 129.2 (m-ArC), 138.7 (N-ArC), 170.0 (NCO) ppm. (trans-isomer, 3 %): 1H NMR (600 MHz, CDCl3): δ = 1.72 (s, 3 H, CH3), 3.15 (m, 1 H, CH), 3.36 (t, J = 10.4 Hz, 1 H, CHHBr), 3.69 (m, 1 H, CHHNCO), 4.04 (m, 1 H, CHHBr), 4.18 (pdd, J = 6.8, 10.1 Hz, 1 H, CHHNCO), 7.21 (pt, J = 7.4 Hz, 1 H, p-ArH), 7.40 (pt, J = 7.6 Hz, 2 H, m-ArH), 7.65 (pd, J = 8.1 Hz, 2H, o-ArH) ppm.
EA found: C 47.7 H 4.2 N 4.8; calcd for (C12H13NOClBr): C 47.63 H 4.33 N 4.63
Preparation of 6 - In an oven-dried Schlenk tube (previously rinsed with a 10% aqueous NH4OH and water in sequence) were weighed Na2CO3 (0.2 mmol, 21 mg), NaBr (4 mmol, 0.412 g), amide 4 (4 mmol, 1.312 g), and a piece of copper wire (length 40 mm, 1 mm diameter, 99.9 %). After three cycles of vacuum/argon (around 10 min, each) AcOEt (3 mL), EtOH (96 vol%, 1.5 mL) and TPMA (1 mL of 0.04 mol/L solution in absolute ethanol) were added under argon. The Schlenk tube was heated at 37 °C and stirred at 700 rpm for 16 h. The reaction was then quenched with water (6 mL), acidified with aqueous HCl (10% w/v, 1 mL) and extracted with CH2Cl2 (3 × 5 mL). The combined organic layers were concentrated to dryness at the rotary evaporator, giving the crude product 6 as a brownish solid (96 %, 3.84 mmol, 1.260 g), that resulted contaminated by a small amount (~ 9% GC) of trichlorolactam 5 (see also Figure S2). A purer version of 6 was obtained through (unoptimized) recrystallization from a mixture of CH2Cl2 : Petroleum Ether in 1 : 2 ratio, giving a yellowish solid in 78 % yield.
Compound 6: 1H NMR (600 MHz, CDCl3): δ = 3.06 (q, J = 9.3 Hz, 1 H, CHHNCO), 3.10 (m, 1 H, CH), 3.43–3.53 (m, 2 H, CHHNCO, CHHBr), 3.78 (dd, J = 3.9, 10.4 Hz, 1 H, CHHBr), 4.43 (d, J = 14.6 Hz, 1 H, CHHPh), 4.65 (d, J = 14.6 Hz, 1 H, CHHPh), 7.24 (pd, J = 7.5 Hz, 2 H, o-ArH), 7.31–7.39 (m, 3 H, m,p-ArH) ppm. 13C NMR (150 MHz, CDCl3): δ = 27.8 (CH2Br), 48.0 (CH2Ph), 48.3 (CH2NCO), 51.9 (CH), 84.5 (CCl2), 128.4 (o-ArC), 128.5 (p-ArC), 129.2 (m-ArC), 166.3 (NCO) ppm.
EA found: C 42.9 H 3.7 N 4.2; calcd for (C12H12NOCl2Br): C 42.76 H 3.59 N 4.16
3. Results and Discussion
The ratio between bromide and chloride concentrations in solution was expected to reflect the prevalent form of the radical shutting-off metal complex, as a result of fast halide exchange processes [18]. A higher concentration of Cu(II)Br·L was potentially associated with a dominant AERC, whereas an abundance of Cu(II)Cl·L was expected to favour predominant ATRC. The development of the AERC should, therefore, be based on a cyclization process requiring a minimal starting amount of metal chlorides.
The ARGET-ATRC of dichloroamide 1 (Figure 1) [4], that can work with only 0.01 mol of CuCl2 for each mol of substrate (see Materials and Methods section), was thus considered a suitable starting point. The inclusion of increasing amounts of NaBr within the reaction mixture, was then expected to bring the formation of increasing amounts of brominated compounds, as depicted in Figure 3. The desired product (3) may arise from the entrapment of the transient radical 1” by Cu(II)Br·L., i.e., the thought AERC process. Conversely, the same metal complex may promote the formation of the constitutional isomeric lactam 3a via radical substitution at the 3 position of the ATRC product 2 [4]. The formation of dibromide 3b from the AERC product 3 may also follow the same substitution mechanism. However, the formation of 3a and 3b is less likely due to low operating temperature (37 °C) and the higher C-Cl bond dissociation energy (83 kcal/mol) compared to that of the C-Br bond (77 kcal/mol) [22].
When a moderate amount of NaBr (0.25 mol for each mol of 1) was included into the described ARGET-ATRC reaction mixture, a new product was formed, together with the standard product 2. This new compound featured higher retention in GC-MS (r.t. ~ 13.95 min) compared to 2 (GC r.t. ~ 12.90 min) and is characterized by a MS pattern compatible with a bromo-chloro lactam. Further characterization through multinuclear NMR spectroscopy, after careful chromatographic fractionation, confirmed the identity of compound 3. The limited operativity of the radical substitution mechanisms affording 3a or 3b, in the AERC reaction conditions, was then ascertained.
A confirmation of that was obtained by heating a purified sample of compound 2 at 50°C for 24 hours in the presence of 10 mol% of Cu(II)Br2·TPMA metal complex. Under these conditions, 2 converted to another compound, characterized by a molecular ion very near to that of lactam 3 but different retention time (~ 13.85 min) and mass fragmentation (Figure 4). It was thus assigned to 3a.
As the operativity of the AERC mechanism was demonstrated, further experimentation was directed towards increasing the production of 3. Therefore, a set of ATRC of 1 were set up, incorporating increased amounts of NaBr. Unfortunately, the GC yield of 3 did not correlate linearly with the amount of added bromide. This unexpected behavior was attributed to the chemical complexity of the system. Indeed, the ARGET-ATRC involves a multitude of chemical species, such as Na2CO3, ascorbic acid, and the metal complex, whose mutual interactions and relative solubilities can be altered by the added salts.
To simplify the composition of the reaction mixture, a “Supplemental Activator Reducing Agent” (SARA) process was considered. In the SARA radical mechanism, copper comproportionation is assumed to dominate over disproportionation [23], making it possible to utilize Cu0 as both a reducing agent and a metal source for the catalytic complex. This eliminates the need for the addition of the copper salt and the reductant. Furthermore, provided that the metal possesses adequate surface area, the reaction rate can be controlled by adjusting the amount of nitrogen ligand (e.g., TPMA), which determines the concentration of the formed catalytic complex. As a result, in the SARA process, some species required in the ARGET process (CuCl2 and ascorbic acid) can be replaced by a piece of copper wire. A small amount of carbonate (~ 5 mol%) was also maintained [24], due to the ability of the EtOH/Na2CO3 couple to function as a reducing agent [25].
The SARA ATRC was successfully applied to 1, resulting in a clean and complete conversion into the expected dichloro-γ-lactam 2 within 16 h at 37 °C (entry 1, Table 1). Subsequently, one mole of bromide salt for each mole of substrate was added, so to achieve partial AERC. A marked effect of the metal cation used, whether lithium (entry 2), sodium (entry 3), or potassium (entry 4), on the formation of different proportions of bromolactam 3 and dichlorolactam 2 was observed. Specifically, sodium bromide/carbonate (entry 3) exhibited the best selectivity towards 3, while the potassium salts demonstrated a propensity towards 2.
Using the sodium salts as the reference, the solvent polarity (i.e., EtOAc/EtOH ratio) was explored. Lower polarities (entry 5) worsened the selectivity towards 3, while a slight improvement was observed when EtOH was increased to 50 vol% (entry 6) or higher (entry 7). Several other interventions aimed at improving selectivity were investigated. These include: (i) increasing the amount of bromide (1.5 moles NaBr/moles of 1), (ii) adding an anion transporter (5 mol% of Bu4N+Br-/Et4N+Br-), or (iii) adding of a cation-catching species (5 mol% of 15-crown-5 ether). Unfortunately, none of these interventions yielded improved results obtained in entry 6.
The greater selectivity observed at higher EtOH concentrations suggests that salt solubility might play a significant role in the outcome of the reaction. Accordingly, the addition of a minor amount of a polar co-solvent was evaluated, as shown in Table 2. Addition of MeCN (entry 1) resulted in a significant improvement in selectivity towards 3, but also in a reduced reaction rate, highlighted by incomplete conversion. Instead, DMSO (entries 2, 3, 4) [23] brought to high selectivities towards 3, together with quantitative conversions of 1. It is worth noting that the best result was attained including the lowest amount of DMSO (entry 2).
Lastly, the addition of water in small amount (entries 5 and 6) led to almost complete conversion of 1 into 3. Such an addition can be conveniently implemented by using 96 vol% ethanol instead of absolute ethanol.
Another parameter investigated, aimed at assessing possible increments of process productivity, was the solvent volume. We found that halving the volume of solvents has no impact on yield and selectivity, offering the opportunity to use reactors of half the volume. However, further reductions in volume worsened the results. The final procedure (see Materials and Methods) gave raw 3 in 95 % isolated yield. A small amount (~ 5 mol%) of dibromolactam 3b was also present as a contaminant (see Figure S1), likely deriving from 3 through a CuBr-promoted radical substitution (Figure 3).
Having demonstrated the facile generation of bromochloro-γ-lactams with precise halogen regioselectivity from dichloroamide 1, the generality of the method was further investigated by considering another substrate with marked structural variation. Trichloroacetamides are well-known reactive species in atom transfer radical processes, owing to the ease of forming of radical intermediates through halogen extraction [26]. However, in the absence of an effective catalytic cycle, the same radical intermediates are involved in uncontrolled processes, eroding the selectivity and synthetic appeal of the method. Hydrogen transfer from the solvent to radical species may also occur, giving de-halogenated byproducts. Moreover, the higher reaction rate should favor the formation of the “standard” ATRC product (5, Figure 5) instead of the AERC one (6). The technique was thus put to the test with trichloro compound 4, prompting a re-evaluation of the operating conditions.
Following the same approach used earlier, several cyclization experiments were conducted on 4, as shown in Table 3.
Proceeding in a similar manner, various solvent proportions were tested (entries 1- 4), revealing that a EtOAc:EtOH ratio of 1:1 (entry 3) works best, similarly to what observed on 1. Keeping this solvent system, its total volume was varied (entries 5 and 6), along with the inclusion of small amounts of water. These investigations resulted in high selectivities towards 6, especially when 1.5 mL of solvent mixture, and when 15 µL of water (added through the use of a portion of 96 vol% ethanol) were employed, for each mmol of substrate (entry 5). In these conditions (see also Materials and Methods) raw 6 was obtained in 96 % isolated yield. A minor amount (~ 15 mol%) of trichlorolactam 5 was also present as a contaminant (see Figure S2), coming from the ATRC of substrate 4 (Figure 5).
The obtained bromochloro-γ-lactams (3 and 6) can be further purified by crystallization (see Materials and Methods).
4. Conclusions
Through the AERC methodology developed herein, we have demonstrated the facile preparation of dihalo-γ-lactams containing two distinct halogens from dichloroamides. This method, which integrates the established SARA ATRC with halogen exchange in solution, operates under mild conditions and requires low amounts of metallic copper, serving as both a supplemental activator and reducing agent. Starting from readily available substrates, the obtained efficiencies and selectivities observed in the studied cases suggest significant potential for the sustainable preparation of novel valuable halogenated synthetic targets.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: 1H and 13C NMR of 3 (raw reaction mixture, after extraction), featuring small amounts of byproducts: Br,Br isomer (6%) and standard ATRC product 2 (6%); Figure S2: Table S1: title; Video S1: title.
Author Contributions
For research articles with several authors, a short paragraph specifying their individual contributions must be provided. The following statements should be used “Conceptualization, X.X. and Y.Y.; methodology, X.X.; software, X.X.; validation, X.X., Y.Y. and Z.Z.; formal analysis, X.X.; investigation, X.X.; resources, X.X.; data curation, X.X.; writing—original draft preparation, X.X.; writing—review and editing, X.X.; visualization, X.X.; supervision, X.X.; project administration, X.X.; funding acquisition, Y.Y. All authors have read and agreed to the published version of the manuscript.” Please turn to the CRediT taxonomy for the term explanation. Authorship must be limited to those who have contributed substantially to the work reported.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Clark, A.J. Copper Catalyzed Atom Transfer Radical Cyclization Reactions. Eur J Org Chem 2016, 2016, 2231–2243. [Google Scholar] [CrossRef]
- Clark, A.J.; Cornia, A.; Felluga, F.; Gennaro, A.; Ghelfi, F.; Isse, A.A.; Menziani, M.C.; Muniz-Miranda, F.; Roncaglia, F.; Spinelli, D. Arylsulfonyl Groups: The Best Cyclization Auxiliaries for the Preparation of ATRC Γ-Lactams Can Be Acidolytically Removed. Eur J Org Chem 2014, 2014, 6734–6745. [Google Scholar] [CrossRef]
- Fischer, H. The Persistent Radical Effect: A Principle for Selective Radical Reactions and Living Radical Polymerizations. Chem Rev 2001, 101, 3581–3610. [Google Scholar] [CrossRef] [PubMed]
- Bellesia, F.; Clark, A.J.; Felluga, F.; Gennaro, A.; Isse, A.A.; Roncaglia, F.; Ghelfi, F. Efficient and Green Route to Γ-Lactams by Copper-Catalysed Reversed Atom Transfer Radical Cyclisation of A-Polychloro- N -allylamides, Using a Low Load of Metal (0.5 Mol%). Adv Synth Catal 2013, 355, 1649–1660. [Google Scholar] [CrossRef]
- Bergmann, R.; Gericke, R. Synthesis and Antihypertensive Activity of 4-(1,2-Dihydro-2-Oxo-1-Pyridyl)-2H-1-Benzopyrans and Related Compounds, New Potassium Channel Activators. J Med Chem 1990, 33, 492–504. [Google Scholar] [CrossRef] [PubMed]
- Moody, C.M.; Young, D.W. Stereospecific Synthesis of 4-Alkylglutamates and 4-Alkylprolines. Tetrahedron Lett 1994, 35, 7277–7280. [Google Scholar] [CrossRef]
- Corey, E.J.; Li, W.-D.Z. Total Synthesis and Biological Activity of Lactacystin, Omuralide and Analogs. Chem Pharm Bull (Tokyo) 1999, 47, 1–10. [Google Scholar] [CrossRef]
- Nilsson, B.M.; Hacksell, U. Base-Catalyzed Cyclization of N -propargylamides to Oxazoles. J Heterocycl Chem 1989, 26, 269–275. [Google Scholar] [CrossRef]
- Ghelfi, F.; Pattarozzi, M.; Roncaglia, F.; Parsons, A.; Felluga, F.; Pagnoni, U.; Valentin, E.; Mucci, A.; Bellesia, F. Preparation of the Maleic Anhydride Nucleus from Dichloro γ-Lactams: Focus on the Role of the N-Substituent in the Functional Rearrangement and in the Hydrolytic Steps. Synthesis (Stuttg) 2008, 2008, 3131–3141. [Google Scholar] [CrossRef]
- Slough, G.A. (Ph3P)3RuCl2 Catalyzed Equilibration and Elimination of α-Chloro-N-Tosyl-2-Pyrrolidinones: A Unique Route to Unsaturated 2-Pyrrolidinones. Tetrahedron Lett 1993, 34, 6825–6828. [Google Scholar] [CrossRef]
- Kornblum, N.; Jones, W.J.; Anderson, G.J. A New and Selective Method of Oxidation. The Conversion of Alkyl Halides and Alkyl Tosylates to Aldehydes. J Am Chem Soc 1959, 81, 4113–4114. [Google Scholar] [CrossRef]
- Tang, J.; Zhu, J.; Shen, Z.; Zhang, Y. Efficient and Convenient Oxidation of Organic Halides to Carbonyl Compounds by H2O2 in Ethanol. Tetrahedron Lett 2007, 48, 1919–1921. [Google Scholar] [CrossRef]
- Patil, R.D.; Adimurthy, S. Direct and Selective Conversion of Benzyl Bromides to Benzaldehydes with Aqueous H 2 O 2 Without Catalyst. Synth Commun 2011, 41, 2712–2718. [Google Scholar] [CrossRef]
- Lei, Z.; Wang, R. Oxidation of Alcohols Using H2O2 as Oxidant Catalyzed by AlCl3. Catal Commun 2008, 9, 740–742. [Google Scholar] [CrossRef]
- Zheng, P.; Yan, L.; Ji, X.; Duan, X. A Green Procedure for the Oxidation of Benzyl Halides to Aromatic Aldehydes or Ketones in Aqueous Media. Synth Commun 2010, 41, 16–19. [Google Scholar] [CrossRef]
- Smith, M.B. March’s Advanced Organic Chemistry: Reactions, Mechanisms, and Structure. Ed. 8th; John Wiley & Sons.; 2020; Section 10.45; pp. 534.
- Boivin, J.; Yousfi, M.; Zard, S.Z. A Versatile Radical Based Synthesis of γ—Lactams Using Nickel Powder / Acetic Acid. Tetrahedron Lett 1994, 35, 5629–5632. [Google Scholar] [CrossRef]
- Peng, C.-H.; Kong, J.; Seeliger, F.; Matyjaszewski, K. Mechanism of Halogen Exchange in ATRP. Macromolecules 2011, 44, 7546–7557. [Google Scholar] [CrossRef]
- Ince, J.; Ross, T.M.; Shipman, M.; Slawin, A.M.Z.; Ennis, D.S. Observations on the Synthesis of Functionalised Methyleneaziridines. Tetrahedron 1996, 52, 7037–7044. [Google Scholar] [CrossRef]
- Bellesia, F.; D’Anna, F.; Felluga, F.; Frenna, V.; Ghelfi, F.; Parsons, A.; Reverberi, F.; Spinelli, D. Breakthrough in the α-Perchlorination of Acyl Chlorides. Synthesis (Stuttg) 2012, 2012, 605–609. [Google Scholar] [CrossRef]
- Benedetti, M.; Forti, L.; Ghelfi, F.; Pagnoni, U.M.; Ronzoni, R. Halogen Atom Transfer Radical Cyclization of N-Allyl-N-Benzyl-2,2-Dihaloamides to 2-Pyrrolidinones, Promoted by Fe0-FeCl3 or CuCl-TMEDA. Tetrahedron 1997, 53, 14031–14042. [Google Scholar] [CrossRef]
- McGivern, W.S.; Derecskei-Kovacs, A.; North, S.W.; Francisco, J.S. Computationally Efficient Methodology to Calculate C−H and C−X (X = F, Cl, and Br) Bond Dissociation Energies in Haloalkanes. J Phys Chem A 2000, 104, 436–442. [Google Scholar] [CrossRef]
- Peng, C.-H.; Zhong, M.; Wang, Y.; Kwak, Y.; Zhang, Y.; Zhu, W.; Tonge, M.; Buback, J.; Park, S.; Krys, P.; et al. Reversible-Deactivation Radical Polymerization in the Presence of Metallic Copper. Activation of Alkyl Halides by Cu 0. Macromolecules 2013, 46, 3803–3815. [Google Scholar] [CrossRef]
- Clark, A.J.; Duckmanton, J.N.; Felluga, F.; Gennaro, A.; Ghelfi, F.; Hardiman, J.R.D.; Isse, A.A.; Manferdini, C.; Spinelli, D. Cu 0 -Promoted Cyclisation of Unsaturated A-Halogeno Amides To Give Β- and Γ-Lactams. Eur J Org Chem 2016, 2016, 2479–2491. [Google Scholar] [CrossRef]
- Braidi, N.; Buffagni, M.; Ghelfi, F.; Parenti, F.; Gennaro, A.; Isse, A.A.; Bedogni, E.; Bonifaci, L.; Cavalca, G.; Ferrando, A.; et al. ARGET ATRP of Styrene in EtOAc/EtOH Using Only Na 2 CO 3 to Promote the Copper Catalyst Regeneration. J Macromol Sci A 2021, 58, 376–386. [Google Scholar] [CrossRef]
- Pattarozzi, M.; Roncaglia, F.; Giangiordano, V.; Davoli, P.; Prati, F.; Ghelfi, F. ‘Ligand-Free-Like’ CuCl-Catalyzed Atom Transfer Radical Cyclization of N-Substituted N-Allyl Polychloroamides to γ-Lactams. Synthesis (Stuttg) 2010, 2010, e2–e2. [Google Scholar] [CrossRef]
Figure 1.
Mechanism for the transition metal catalyzed ATRC of 1.
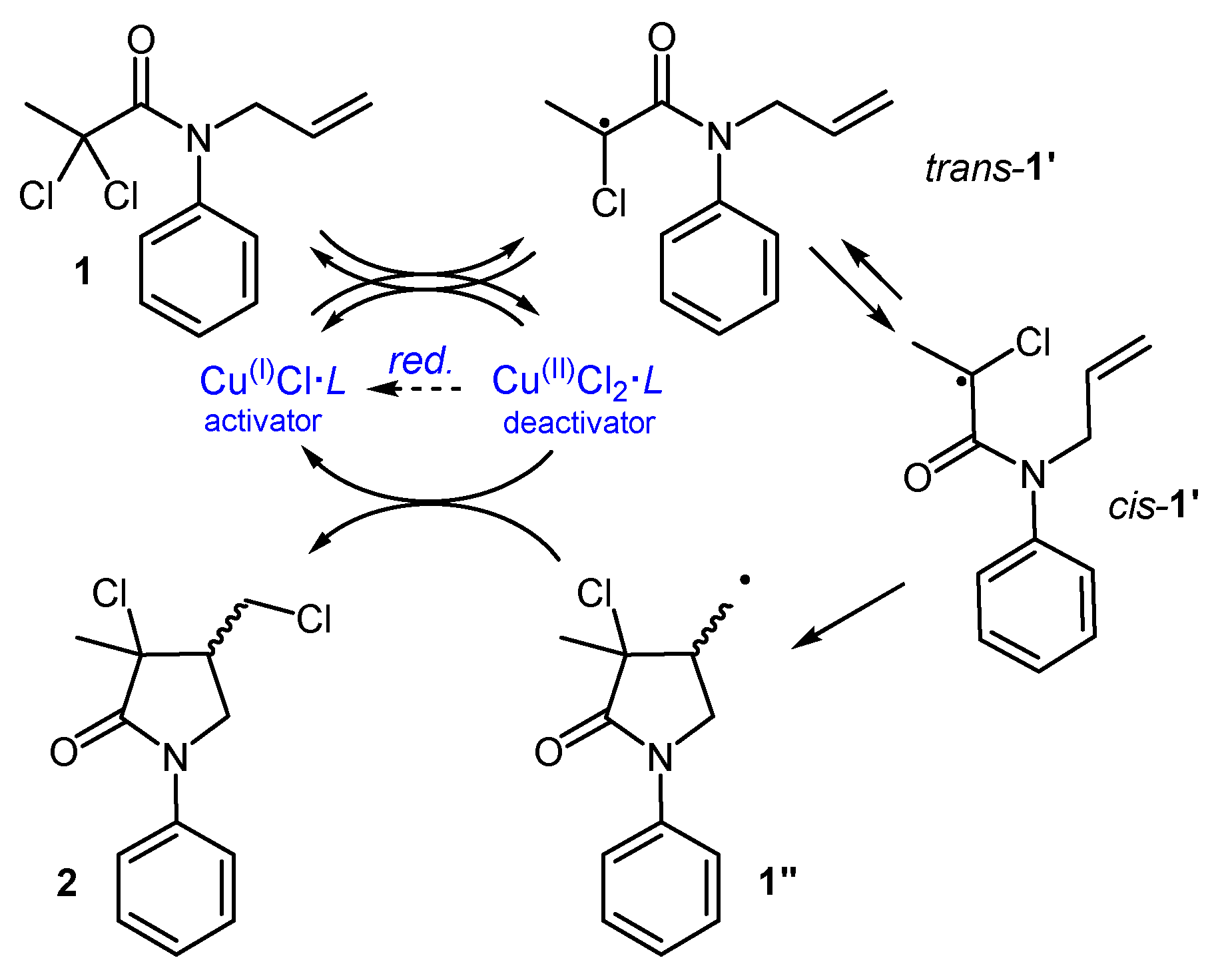
Figure 2.
Some synthetic applications of dichloro-γ-lactams.
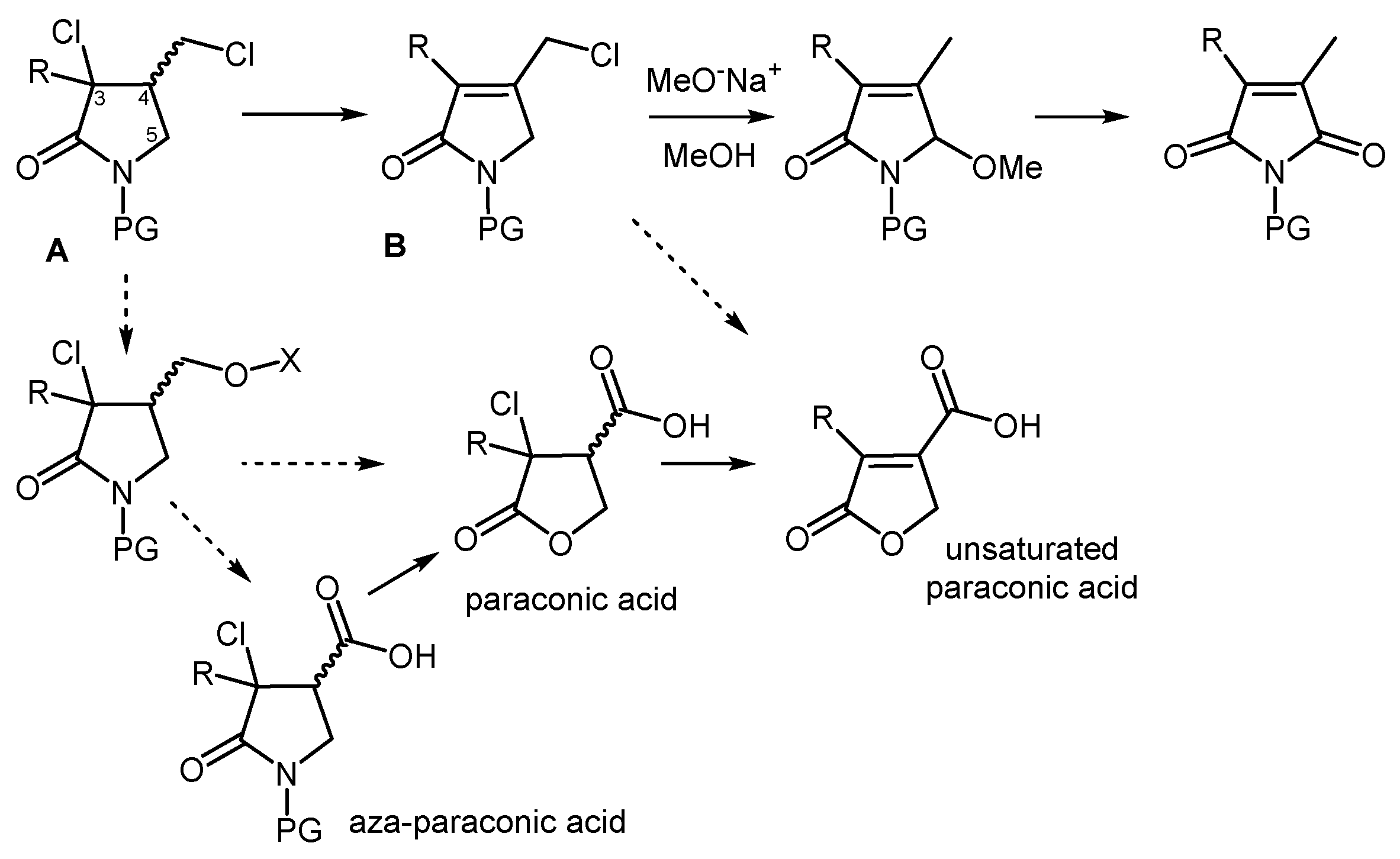
Figure 3.
Competition between ATRC and AERC of 1 and possible substitution on the products.
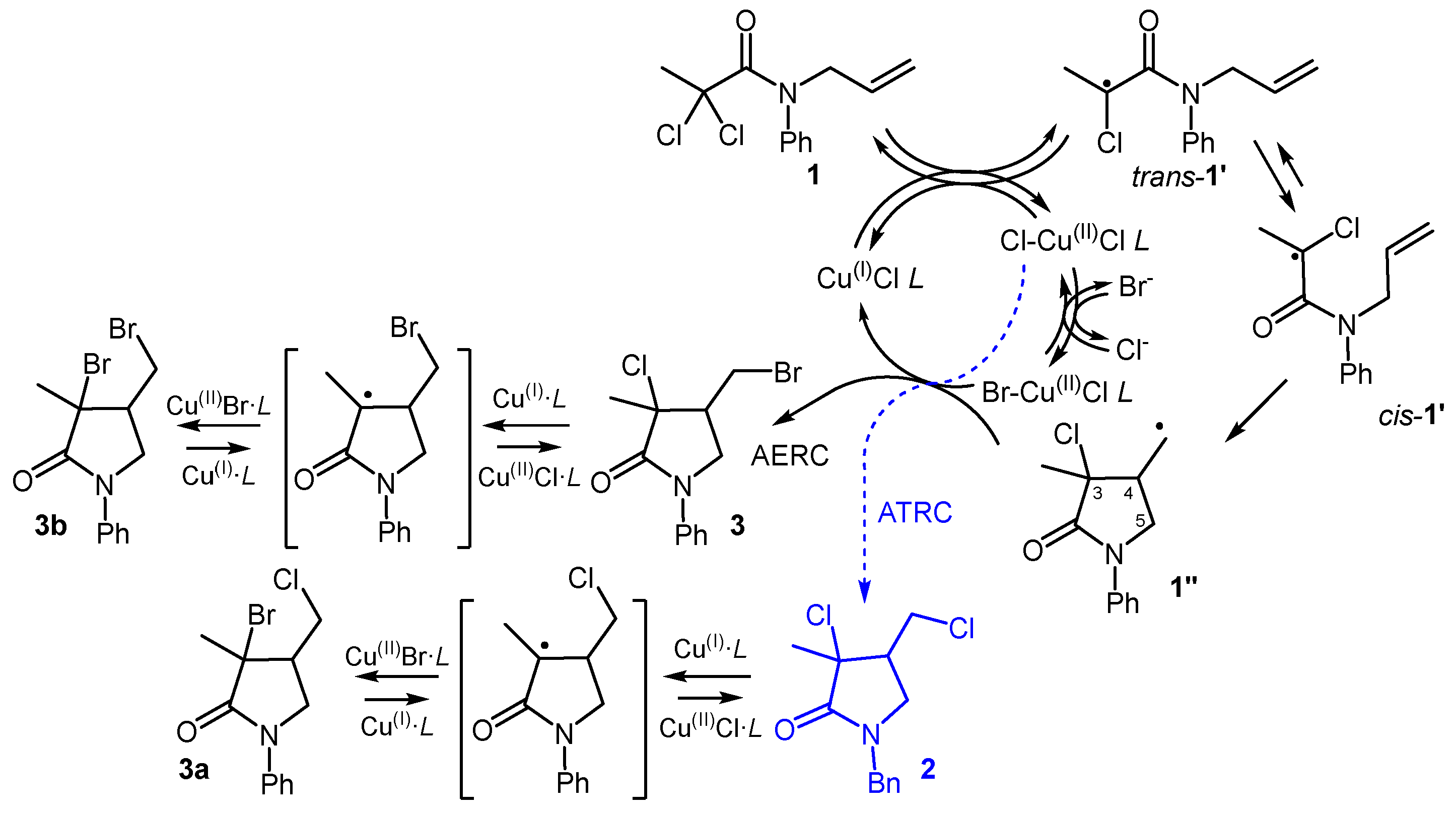
Figure 4.
MS characterization of 3 and 3a.
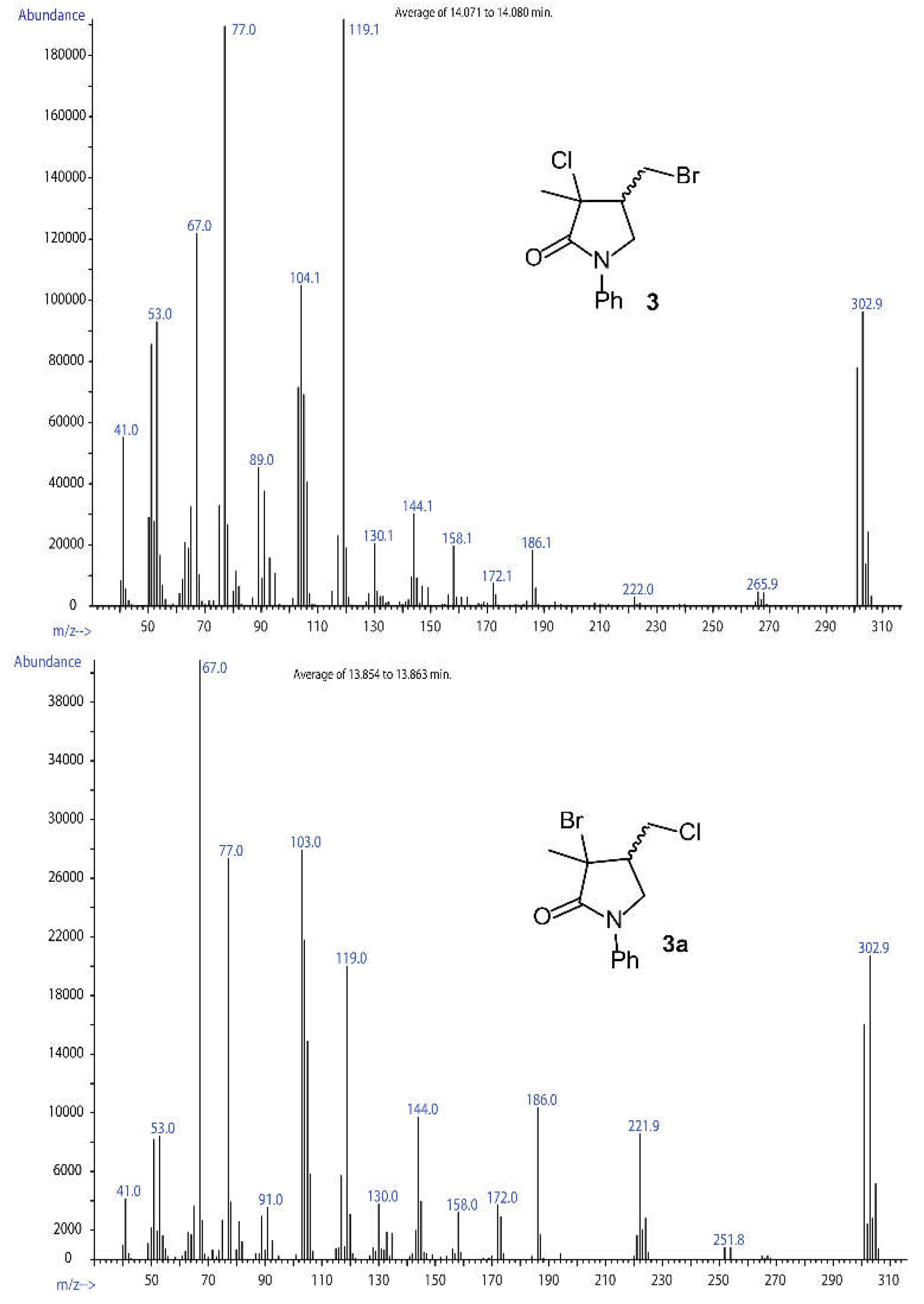
Figure 5.
AERC of trichloroacetamide 4.
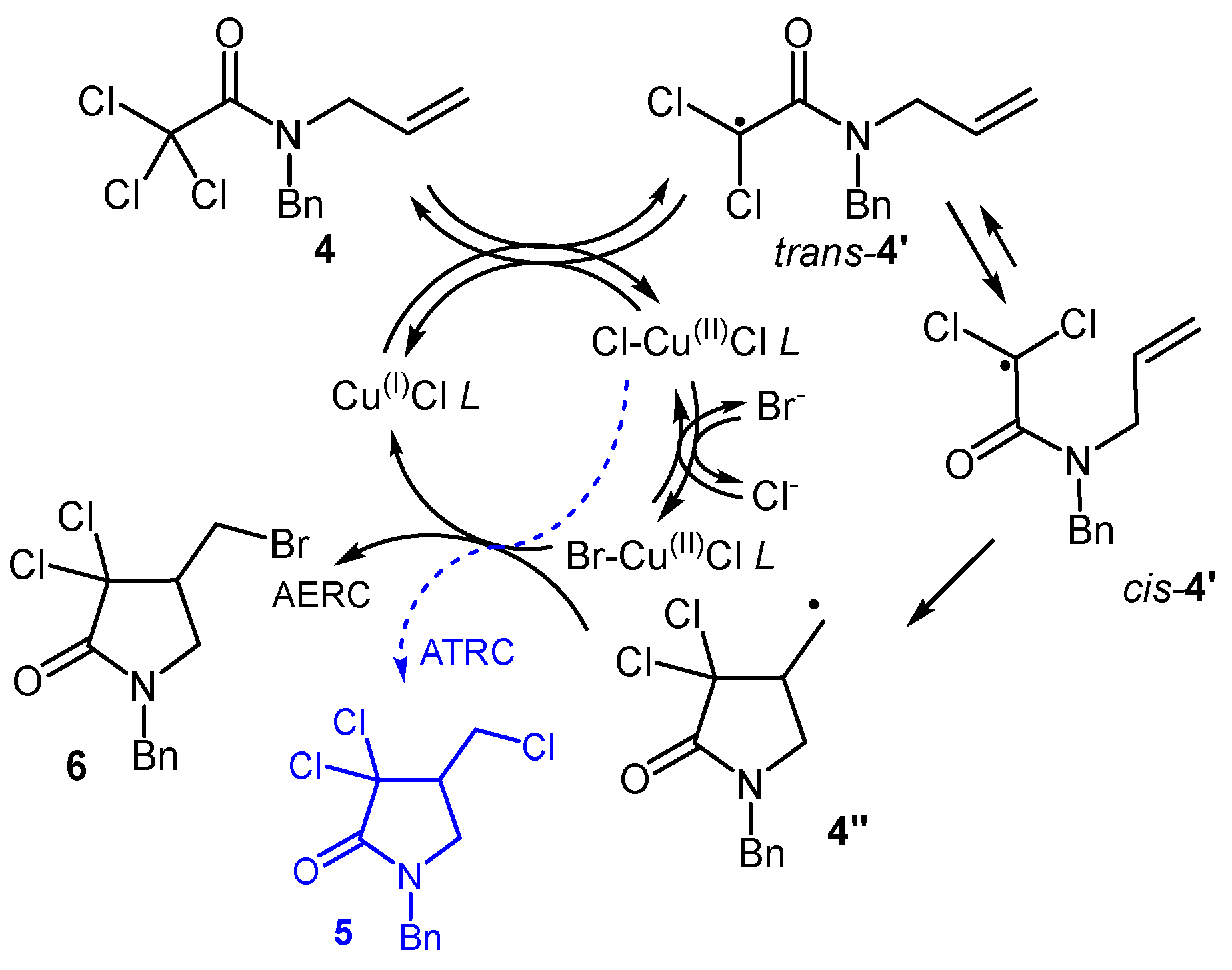

Table 1.
SARA ATRC / AERC of 1.1.
| entry | EtOAc (mL) | EtOH (mL) |
Bromide | Carbonate | yield of 2 (%) | yield of 3 (%) |
|---|---|---|---|---|---|---|
| 1 | 1.5 | 0.5 | - | Na2CO3 | 100 | - |
| 2 | 1.5 | 0.5 | LiBr | Li2CO3 | 30 | 57 |
| 3 | 1.5 | 0.5 | NaBr | Na2CO3 | 13 | 71 |
| 4 | 1.5 | 0.5 | KBr | K2CO3 | 54 | 30 |
| 5 | 1.7 | 0.3 | NaBr | Na2CO3 | 35 | 32 |
| 6 | 1.0 | 1.0 | NaBr | Na2CO3 | 15 | 75 |
| 7 | 0.5 | 1.0 | NaBr | Na2CO3 | 15 | 75 |
1 1 (1.00 mmol), carbonate (0.05 mmol), bromide (1.00 mmol), Cu(0) (wire, 1 mm diam. and 20 mm long), solvent (2.0 mL, see Table), TPMA (0.01 mmol), 16 h, 37 °C. GC yields are reported.
Table 2.
Solvent changes in the AERC of 1.
| entry | Cosolvent (mL) |
Conversion of 1 (%) | yield of 2 (%) | yield of 3 (%) |
|---|---|---|---|---|
| 1 | MeCN (0.20) | 98 | 7 | 76 |
| 2 | DMSO (0.10) | 100 | 3 | 87 |
| 3 | DMSO (0.20) | 100 | 2 | 83 |
| 4 | DMSO (0.40) | 100 | 1 | 82 |
| 5 | H2O (0.02) | 100 | 2 | 97 |
| 6 | H2O (0.05) | 100 | 3 | 95 |
1 1 (1 mmol), Na2CO3 (0.05 mmol), NaBr (1.00 mmol), Cu(0) (wire, 1 mm diam. and 20 mm long), solvent (2.0 mL, composed of EtOAc : EtOH (1:1) and cosolvent), TPMA (0.01 mmol), 16 h, 37 °C. GC conversions and yields are reported.
Table 3.
SARA ATRC / AERC of 4.
| entry | EtOAc (mL) | EtOH (mL) |
yield of 5 (%) | yield of 6 (%) |
|---|---|---|---|---|
| 1 | 1.75 | 0.25 | 76 | 20 |
| 2 | 1.50 | 0.50 | 13 | 81 |
| 3 | 1.00 | 1.00 | 12 | 82 |
| 4 | 0.50 | 1.50 | 22 | 73 |
| 5 | 0.75 | 0.752 | 5 | 96 |
| 6 | 0.50 | 0.502 | 13 | 86 |
1 4 (1.00 mmol), Na2CO3 (0.05 mmol), NaBr (1.00 mmol), Cu(0) (wire, 1 mm diam. and 20 mm long), solvent (see Table), TPMA (0.01 mmol), 16 h, 37 °C. GC yields are reported. 2 absolute EtOH : 96 vol% EtOH 1 : 1.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



