
Submitted:
02 May 2024
Posted:
06 May 2024
You are already at the latest version
Abstract
Cancer cells show altered antioxidant defense systems, dysregulated redox signaling, and increased generation of reactive oxygen species (ROS). Targeting cancer cells through ROS-mediated mechanisms has emerged as a significant therapeutic strategy due to its implications in cancer progression, survival, and resistance. Extensive research has focused on selective generation of H2O2 in cancer cells for selective cancer cell killing by employing various strategies such as metal-based prodrugs, photodynamic therapy, enzyme-based systems, nano-particle mediated approaches, chemical modulators, and combination therapies. Many of these H2O2-amplifying approaches have demonstrated promising anticancer effects and selectivity in preclinical investigations. They selectively induce cytotoxicity in cancer cells while sparing normal cells, sensitize resistant cells, and modulate the tumor microenvironment. However, challenges remain in achieving selectivity, addressing tumor heterogeneity, ensuring efficient delivery, and managing safety and toxicity. To address those issues, H2O2-generating agents have been combined with other treatments leading to optimized combination therapies. This review focuses on various chemical agents/approaches that kill cancer cells via H2O2-mediated mechanisms. Different categories of compounds that selectively generate H2O2 in cancer cells are summarized, their underlying mechanisms and function are elucidated, preclinical and clinical studies as well as recent advancements are discussed, and their prospects as targeted therapeutic agents and their therapeutic utility in combination with other treatments are explored. By understanding the potential of these compounds, researchers can pave the way for the development of effective and personalized cancer treatments.
Keywords:
Prooxidants
; Hydrogen peroxide generation
; reactive oxygen species
; Anticancer effects
; phenols and quinones
; vitamines
; metal peroxides
1. Introduction
Cancer is a complex and multifaceted disease that poses immense challenge for researchers around the globe to develop targeted therapies. Serious side-effects are common with current cancer therapies due to their lack of cellular specificity. Extensive efforts have been made to develop more selective therapeutics to specifically kill cancer cells, and a major focus is to exploit the inherent vulnerabilities among cancers cells in order to improve the selectivity of cancer treatment. One such approach involves manipulation of reactive oxygen species (ROS), due to the fact that cancer cells are known to exhibit increased intrinsic oxidative stress compared to normal cells.1-5 This intrinsic feature of cancer paves the way for the development of tumor-selective therapeutic strategies.3, 6-9 This review specifically focuses on strategies to selectively modulate the most stable ROS, hydrogen peroxide (H2O2) in cancer cells to achieve therapeutic effects.
Mildly increased levels of H2O2 are known to contribute to tumor growth by promoting the transformation, proliferation, and survival of cancerous cells, as well as angiogenesis and metastasis. High levels of H2O2 can cause damage to various biomolecules, such as DNA, proteins, and lipids, and thus are detrimental to cell survival and growth.5, 10 Although cancer cells produce large quantities of H2O2, their natural H2O2 levels are not sufficient to achieve noticeable therapeutic results. Amplification of H2O2 production, specifically within cancer cells, has emerged as a valuable approach for cancer treatment.7, 10 In healthy somatic cells, ROS generation and elimination are in a delicate equilibrium,, and thus a low level of basal ROS is achieved, ensuring redox homeostasis. This allows them to quickly adapt to any changes in oxidative stress. However, due to heightened metabolic irregularities and increased oncogenic signaling, cancer cells have high levels of H2O2 to begin with, which can trigger redox adaptation and lead to upregulation of antioxidant concentrations, such as glutathione (GSH) and thioredoxin. This further increases the redox capacity of the cancer cells, thus allowing them to maintain high levels of H2O2 that are very close to the cytotoxic threshold (Figure 1). Consequently, cancer cells are susceptible to prooxidants that generate H2O2 beyond the cytotoxic threshold, which leads to cell death.11 This phenomenon provides a biochemical foundation for developing therapeutics that selectively target cancer cells through H2O2-mediated mechanisms. Various strategies have been developed to boost H2O2 levels specifically within cancer cells to achieve selective therapeutic effects, such as treatment with glucose oxidase, lactate oxidase, peroxides, H2O2-releasing chemical agents, inhibitors of antioxidative enzymes, such as superoxide dismutase (SOD), glutathione peroxidase (GPx), and catalase that eliminate H2O2 in cells to maintain homeostasis. This review focuses on various chemical agents that kill cancer cells via selective H2O2 generation. This paper, which is not meant to be a thorough review, will discuss different categories of H2O2-producing agents and the mechanism underlying H2O2 production through a few selected examples.
2. Hydrogen Peroxide Generation and Its Reactivity with Biomolecules
H2O2 is naturally formed in living organisms and plays an important role as a redox metabolite that is involved in redox signaling, sensing, and regulation.7, 12 It functions as a messenger molecule that permeates through cells and tissues, triggering rapid cellular effects such as alteration in cell shape, cellular proliferation, and recruitment of immune responses. There are three major pathways for H2O2 formation in normal mammalian cells, all of which involve the reduction of O2 into superoxide anion (O2•−) (Figure 2).13 The first pathway involves the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase that catalyzes the conversion of oxygen (O2) into O2•−, which then dismutates to H2O2 by SOD. The second pathway pertains to the mitochondrial respiratory chain, particularly the cytochrome complex, in the production of superoxide radicals. The third mechanism involves the action of oxidases in specific cell types or subcellular organelles, such as xanthine oxidase, glucose oxidase, monoamine oxidase, or D-amino acid oxidase. Other cellular compartment that contributes to H2O2 production is the endoplasmic reticulum.14
In biological context, H2O2 causes damage to biomolecules via several biochemical reactions. It can oxidize thiol groups such as cysteine-containing proteins, forming sulfenic acids, which can further oxidize to form sulfinic and sulfonic acids, causing permanent protein inactivation (Figure 3).15 However, the chemical reactivity of H2O2 alone towards biomolecules remains low. In the presence of transition metal ions such as Fe2+, Cu+, Mn2+ etc., H2O2 is converted to highly reactive hydroxyl radical (•OH) via the Fenton-like reaction, which causes damage to both DNA and proteins. Hydroxyl radicals can cleave the sugar phosphate backbone and modify the nucleobases, such as Guanine and Thymine. Proteins are also very vulnerable to •OH attack, especially those with thiol groups like methionine. Hydroxyl radicals also interact with several other amino acids such as lysine, proline, arginine, and histidine, forming protein carbonyls and 2-oxo-histidines, compromising protein functionality. The collective impact of this oxidative damage results in heightened oxidative stress on several components of the cells, which is detrimental to the cell and often leads to cell death.16
3. Increasing H2O2 Level as an Anticancer Therapy
While excessive ROS production can stimulate cellular proliferation and genetic instability, it can also trigger apoptosis, suggesting that ROS-mediated mechanisms can be harnessed for cancer treatment.17 Many cancer therapies induce ROS (i.e. H2O2) production as a possible mechanism, such as chemotherapy,18 photodynamic therapy,19 radiotherapy,20 and enzyme-based therapies.17, 21 Various chemotherapeutic agents and radiotherapy directly generate ROS, causing apoptosis in cancer cells. Photodynamic therapy uses photons to activate photosensitizers to produce ROS causing cytotoxicity. However, there are still many challenges for these ROS-amplifying therapies including off-target effects, limited penetration, and safety concerns. Various strategies have been developed to improve cancer specificity and reduce systemic toxicity, such as the use of prooxidants to amplify ROS production selectively in cancer cells,22 selective delivery of ROS-producing agents to cancer cells via nanoparticles,23 gene therapy for encoding enzymes involved in ROS production to obtain targeted ROS production in cancer cells,24 and immunotherapy that harnesses the body’s defenses to recognize and attack cancer cells through ROS production.25 Collectively these approaches provide a wide range of applications that include customization, personalization, precision treatment with reduced toxicity, enhanced efficacy, and minimized resistance towards the ongoing battle against cancer.
The use of prooxidants in cancer therapies has been widespread and gained attention due to various advantages, such as selective cytotoxicity towards cancer cells, reduced off-target effects, and the potential to be used in combination with conventional chemotherapeutic agents to achieve synergistic anticancer effects.7, 22 Their distinct mechanism allows them to overcome drug resistance,3 and the understanding of ROS levels and the antioxidant capacity in individual cancer cells provides an avenue for researchers to tailor treatment strategies and make therapies more personalized and effective.26 The diverse selection of prooxidants, including natural and synthetic molecules, offer a wide range of options for researchers and clinicians. These compounds hold potential not only because of their direct killing of cancer, but also because of their ability to change H2O2 levels, thus allowing them to enable synergistic combination therapies with H2O2-activated prodrugs. Unraveling their potential and understanding their mechanisms of H2O2 generation will speed up the development of more effective and personalized cancer treatments. Based on the pathways for H2O2 production, the prooxidants fall into two major categories, either the category of directly producing H2O2 (direct approach) or the category of inhibiting the excessive antioxidative defense system within cancer cells) indirect approach). There are three major mechanisms for direct H2O2 production induced by prooxidants, including autooxidation, redox cycling, and metal ion interaction (Figure 4). Many prooxidants are directly involved in the electron transport chain for producing H2O2 in cancer cells, such as phenol and polyphenol analogues, quinone moieties, vitamin C, metal oxides, and many FDA-approved anticancer drugs. They can donate electron/hydrogen atoms for the reduction of molecular O2 to O2•─ and H2O2 either via autooxidation, redox cycling, or metal ion interaction.
3.1. Phenol and Polyphenol Analogues
Among various naturally occurring and synthesized compounds, structures with multiple phenolic groups facilitate H2O2 generation via either repeated steps of autooxidation in the presence of molecular oxygen (O2),27-28 or via redox cycling that involves NAD(P)H (Figure 4 and Figure 5). Autooxidation is often a slow reaction due to the lower redox potential of O2/ O2•− and produces H2O2 through dismutation. During the autooxidation process, phenols are oxidized to semiquinone that rapidly transforms into quinone, while O2 is reduced to superoxide radical (O2•−) that undergoes dismutation to generate H2O2 at the same time (Figure 5). The formed quinone then undergoes a subsequent reduction in the presence of NAD(P)H enzyme to regenerate polyphenols that spontaneously revert to their quinone form via a semiquinone intermediate. This continuous cycle between oxidation and reduction creates a self-perpetuating cycle known as redox cycling.27 Compounds that feature polyphenolic (hydroxyl) groups include a wide range of compounds such as flavonoids, hydroxytyrosol, propyl gallate, hydroxycinnamic acids, etc.
Flavonoids
Flavonoids are a diverse set of polyphenolic compounds found in plant-based foods and beverages. They have been extensively explored for their vast range of pharmacological properties such as antibacterial, antimutagenic, antiresorptive, antioxidative, and anticancer effects.29 A fundamental flavonoid structure includes two benzene rings (A and B) connected by a heterocyclic pyran ring (ring C) (Figure 6). There are five subclasses of flavonoids: Flavan-3-ols (such as catechins and gallocatechins), Flavones (such as apigenin, luteolin, and baicalein), Flavonols (such as kaempferol, quercetin, and myricetin), Flavanones (such as naringenin and carthamidin), and anthocyanins (such as delphinidine). These subclasses vary in their structural arrangements of hydroxyl and methoxy groups, and also in their ring conjugations (Figure 6). They have been found to act as prooxidants, and a wide variety of flavonoids (such as catechins,30-33 baicalein, quercetin, morin, myricetin,34-36 and wogonin37-39) have been reported to produce high level of H2O2.These flavonoids selectively kill malignant cells via H2O2 mediated processes and interactions with cellular functions that lead to apoptosis, such as enhanced hydroxyl radical formation via the Fenton reaction which causes DNA, protein, and cell membrane damage.30-32 The production of H2O2 was observed in flavonoid-treated media as well as in cell cultures. Their ability to generate ROS can be influenced by the presence and location of hydroxyl groups.40 In order to harness their therapeutic benefits while avoiding unintended harmful effects, it is essential to attain a better understanding of their prooxidant activities.
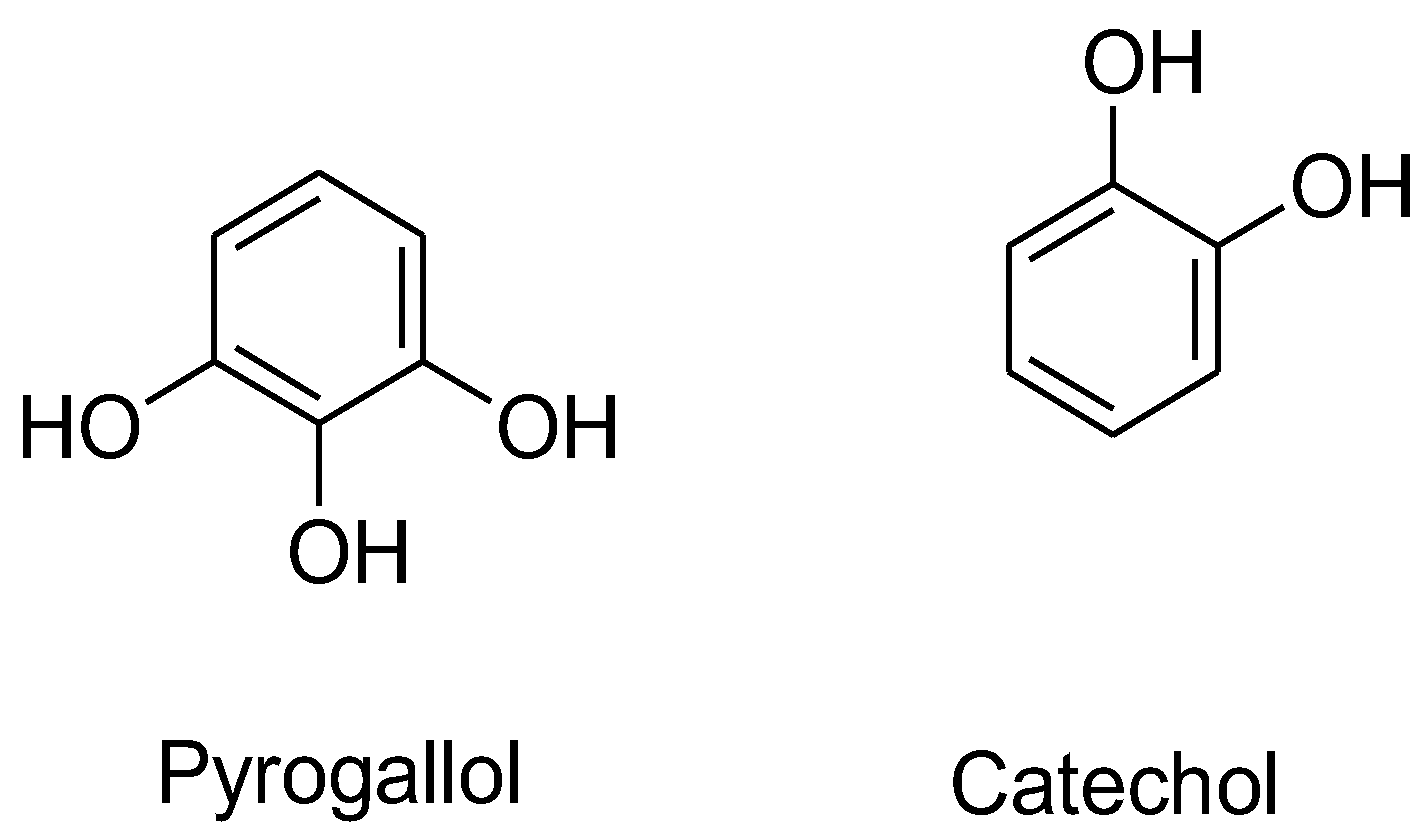
Among various flavonoids, catechins are a popular subclass and have been widely investigated for their prooxidative effects. Catechins belong to the category of flavanols, which have two isomeric forms, a positive (+) form and a negative (−) form (epicatechin). The (+)-catechins have antioxidative properties, whereas the (-)-epicatechins act as pro-oxidants inducing oxidative effects.41 The presence of phenolic hydroxyl groups in catechins makes them susceptible to repeated redox reactions where these groups donate electrons and form H2O2.30 The process becomes more feasible in ortho-dihydroxyl and ortho-trihydroxyl structures, where two or more adjacent hydroxyl groups facilitate electron transfer and enhance redox activity. The presence of oxygen in the tumor environment can cause auto-oxidation of these phenols into semiquinones in a process where oxygen gets reduced to O2•− and leads to H2O2 production.28, 42 It was observed that a pyrogallol-type structure in the B-ring (epigallocatechins, i.e. EGC and EGCG, R2 = OH) possesses H2O2-producing properties, which is responsible for its cytotoxic effect in Jurkat cells.32, 43 Hong et al. reported that a 50 μM dose of EGCG lead to generation of up to 25 μM of H2O2 in HT-29 human colon adenocarcinoma cells.44 Among other antioxidants, EGCG produced the highest concentration of H2O2 at neutral pH in human oral tumor cell lines.33 Nakagawa et al. suggested that a possible deprotonation or deprotonated form of EGCG in the pyrogallol moiety may contribute to H2O2 generation as the pKa for EGCG is 7.59-7.75. 32
Pyrogallol itself has been shown to effectively generate H2O2 and O2•− in various cell types, inducing O2•− mediated cell death (Figure 6).43 Its concentration and incubation time affect intracellular H2O2 levels, with 100 μM pyrogallol causing a rapid and acute increase in H2O2 levels. The presence of pyrogallol reduces intracellular GSH content in HeLa cells, and the addition of Tempol, SOD, and CAT rescues cells from pyrogallol-induced apoptosis by increasing intracellular GSH content. 45-46 Miura also reported that the flavonoids with a pyrogallol structure generated more H2O2 than flavonoids with a catechol structure.36 For example, myricetin and baicalein demonstrated higher H2O2-generating abilities than quercetin and (-)-epicatechin. The distinct placement of hydroxyl groups introduces a capability to interact with several cellular components such as DNA, enzymes, proteins, and many others, which make these compounds more diverse in the field of medicine.47
Figure 7.
The structures of Pyrogallol and Catechol.
Findings reported by Nakagawa et al. suggested that the cytotoxic effects of flavonoids are not only due to a higher prooxidant ability to generate H2O2, but also the cell’s ability to metabolize it.32 Additionally, the production of flavonoid phenoxy radicals during antioxidative reactions can also generate prooxidant effects. These highly reactive phenoxy radical species undergo oxidation to generate flavonoid quinones that can conjugate with nucleophiles such as GSH, cysteine, or nucleic acids.48 Phenoxy radicals of apigenin, naringenin, and naringin have been noted to rapidly oxidize NADH, leading to enhanced oxygen uptake and superoxide formation followed by H2O2 generation.49-50
Besides autooxidation, many phenol analogues can interact with metal ions. Their metal chelation activity is mainly associated with the presence of ortho-dihydroxy groups.51 These phenol compounds can get oxidized into semiquinones in the presence of metal ions via a one-electron transfer mechanism (Figure 5),52 and along with reduced metal ions, react with O2, generating O2•−, oxidized metal ions and quinones. Involvement of these metal ions in redox reactions facilitate their regeneration and allow repeated cycles of redox reactions. Consequently, O2•− accumulates and dismutates to produce H2O2.27 Figure 8 lists some examples of phenols that interact with metal ions to produce H2O2, such as caffeic acid, rosmarinic acid, hydroxytyrosol, and propyl gallate.
Hydroxycinnamic acid
Caffeic acid (CA) and rosmarinic acid (RA) are hydroxycinnamic acid (HCA) analogues containing phenol moiety, which are found in various dietary sources, including green tea, red wine, fruits, vegetables, coffee, as well as in medicinal plants such as rosemary and salvia. Their extensive range of properties encompass anti-cancer, antioxidant, anti-proliferative, and anti-inflammatory effects.53-54 CA showed pro-oxidant potential due to its ability to interact with metals like copper, inducing lipid peroxidation and causing DNA damage within tumor cells through either oxidation or covalent adduct formation.55-57 Zheng et al. proposed that the ortho-dihydroxyl groups can chelate Cu(II) to form a CA-Cu(II) complex A, therefore facilitating intramolecular electron transfer to generate a hydroxy phenoxy radical-Cu(I) complex C (Figure 9).55-56 During this process, CA undergoes deprotonation in response to copper, yielding a phenoxide anion that acts as a good ligand for metal ions due to the high electronic density of oxygen. Further deprotonation of C generated the semiquinone radical anion-Cu(I) complex D, which transfers an electron to O2 producing O2•− and the final products, H2O2 and ortho-quinone. H2O2 can be subsequently converted into •OH via a Fenton reaction, inducing DNA damage. Ortho-quinone can form covalent adducts with the DNA of cancer cells. Similarly, RA with two diphenolic rings induces O2•− and H2O2 formation in the presence of transition metals (i.e. iron) while producing the final product of o-quinones, which is corelated to the cytotoxicity of RA.58-60
Hydroxytyrosol
Hydroxytyrosol (HT), abundantly present in olives, has demonstrated anticancer properties in vitro.61-62 Sun et al. demonstrated that HT exhibited anti-proliferative and pro-apoptotic effects in cancer cells through H2O2 generation.62-63 The mechanism of prooxidant activity of HT involves O2 and transition metals. First, the phenol moiety undergoes oxidation by Cu(II) or Fe(II), forming semiquinones, which then react with O2 generating O2•− and finally producing H2O2.64 HT has been documented to generate H2O2 in colon cancer cells, ultimately leading to apoptotic cell death and mitochondrial dysfunction.62 Similarly, in prostate cancer PC3 cells, HT has been linked to superoxide and H2O2 generation, triggering apoptosis.65-66 Fabiani et al. have also reported that the chemo-preventive effects of HT rely on its prooxidant properties, hinging on its ability to generate H2O2 in the culture medium.67-68 Their work has reveals that various amounts of H2O2 accumulate in the culture medium, influenced by its components and the cell's ability to eliminate this peroxide. This clarifies the need for a wide range of HT concentrations to observe its chemo-preventive effects.
Propyl gallate
Propyl gallate (PG), chemically known as propyl-3,4,5-trihydroxybenzoate is widely present in processed food and cosmetics, hair products, and lubricants.69-72 This versatile compound boasts various biological properties, including potential anti-tumor effects. PG alone demonstrated antioxidative and cytoprotective properties against cellular damage, and gained a pro-oxidative property in combination with copper (II).73 It was reported that PG was one of the most active compounds capable of generating H2O2 in DMEM media, which contributes to the cytotoxic effects observed in vitro.74
Polyphenols are also widely used in many combination studies and possess a promising adjuvant property.38 Several chemotherapeutic drugs have been shown to have significantly increased efficacy when combined with polyphenols. This includes Cisplatin, 5-fluorouracil, celecoxib, doxorubicin, and tamoxifen. The synergistic effect between the two has been associated with drug resistance reduction, enhanced drug sensitivity, induction of cell cycle arrest, apoptosis, restricted angiogenesis, and anti-inflammatory and pro-oxidant effects.39,40
3.2. Compounds with Quinone Moieties
A wide range of quinone-containing compounds showed anticancer, antimicrobial and antiparasitic effects, such as naphthoquinones, aziridinylquinones, anthracyclines, indolequinones (i.e. mitomycins), aminoquinones (i.e. streptonigrins) and certain vitamins (Figure 10). H2O2 generation induced by these compounds is one of the possible mechanisms for their function. Quinones can induce H2O2 production in cells via autooxidation and redox cycling mechanisms. They undergo either one-electron reduction catalyzed by NAD(P)H-cytochrome P-450 reductase to form semiquinone radicals or two-electron reduction catalyzed by NAD(P)H:quinone oxidoreductase (NQO1 or NQO2) to generate hydroquinones (Figure 11). Semiquinone radicals and hydroquinones participate in redox cycling and undergo oxidation by O2 to regenerate quinones, while O2 is reduced to O2•− that dismutates to form H2O2.
Naphthoquinones. Many naturally occurring naphthoquinones and their derivatives showed cytotoxicity, which has been investigated for the development of anti-cancer drugs, such as menadione (2-methyl-1,4-naphthoquinone, also termed vitamin K3), plumbagin, and juglone (Figure 12). Their toxic effects on cells are mostly caused by ROS species including H2O2 generated through redox cycling.75-76 Criddle et al. has reported that menadione-induced ROS generation catalyzed by reductive enzymes, such as NADPH-cytochrome P-450 reductase, xanthine oxidase, and NQO1, promotes apoptosis of murine pancreatic acinar cells.75 Menadione-induced ROS generation is concentration-dependent and high concentrations trigger cell death.77 Clinical trials conducted on patients with prostate cancer showed that ascorbic acid-menadione produced an immediate drop in tumor cell numbers through a mechanism named autoschizis.78 It has been proposed that autoschizis induced by ascorbic acid-menadione was caused by oxidative radicals generated by H2O2 leading tocellular damage.
Hydroxyl naphthoquinone
Plumbagin and Juglone are hydroxyl naphthoquinone derivatives found in various plants, such as plants of the Plumbago genus and black walnuts. They possess a wide range of pharmacological properties such as antioxidant, anti-inflammatory, antifungal, antibacterial, antidiabetic, and anticancer effects. Their cytotoxicity is caused by two possible mechanisms: redox cycling and reaction with GSH, which both result in generation of the corresponding semiquinone radical and O2•−, leading to DNA damage and oxidative stress.79-82 For example, PL interferes with mitochondrial electron transport due to its structural similarity to ubiquinone (Coenzyme Q, CoQ), which lowers oxygen consumption while generating oxygen radicals. Inbaraj et al. indicated that exposure of plumbagin and juglone to HaCaT keratinocytes caused a concentration dependent reduction of cell viability, which was attributed to two primary mechanisms (Figure 13).83 First, plumbagin and juglone undergo one-electron reduction by enzyme NAD(P)H-cytochrome P-450 reductase or two-electron reduction by mitochondrial NADH-ubiquinone oxidoreductase, resulting in the formation of semiquinone radicals and hydroquinones.84 Under aerobic conditions, the semiquinone radicals or the hydroquinones formed participate in redox cycling and induce the reduction of O2 to generate O2•− and H2O2. Second, quinone functional groups can directly react with thiol groups in proteins and GSH, resulting in GSH depletion and cell death.83 It was found that a hydroxyl group at the C-5 position of naphthoquinones is correlated to heightened cytotoxicity due to improved redox cycling.85 Plumbagin and Juglone with a hydroxyl group at C-5 are much more reactive than lawsone and lapachol with an OH group in position C-2. Tautomerization of the C-2/C-3 enol structure of lawsone and lapachol will result in a saturated C-3 that prohibits Michael-type addition reactions in that position. This tautomerization also stabilizes the quinoid structure, which leads to a very low one-electron reduction potential, decreasing redox cycling.83
1,2-Naphthoquinone
β-Lapachone, a 1,2-naphthoquinone natural product isolated from the lapacho tree, is a potent anticancer drug that has been advanced into clinical trials based on its tumor-selective cytotoxic properties.86 Its antitumor mechanism is related to NQO1-mediated redox cycling. β-Lapachone and its derivatives undergo two-electron reduction catalyzed by NQO1 to form hydro-β-lapachone (β-lapachol) which is highly reactive and unstable (Figure 14) It then undergoes auto-oxidation within the cell in two steps, first generating β-lapachone-semiquinone that transforms into quinone-β-lapachone (Q). This redox cycle produces O2•− that dismutates into H2O2.87 Many studies indicated that β-lapachone enhances H2O2 generation in cancer cells. Chau et al. reported that human leukemia cells treated with β-lapachone had a substantial increase in intracellular H2O2, especially the ones with lower levels of GSH, including HL-60, U937, and Molt-4.88 The generated ROS have been linked to different pathways to apoptosis. It has been noted that the ROS generated by β-lapachone resulted in the oxidation of ubiquitin specific protease 2 (USP2) which is known to protect tumor cells from apoptosis by preventing protein degradation. This oxidation happens by transforming its thiol groups to cysteine sulfinic or sulfonic acids. The deactivation of USP2 by β-lapachone triggers proteasomal degradation pathways that contribute towards β-lapachone-induced anticancer effects.86
β-Lapachone consists of a benzene ring (A ring), an ortho-quinone ring (B ring), and a dihydropyran ring (C ring). Modifications have been made to A-, B-, and C-rings, resulting in a wide variety of promising derivatives with enhanced specificity and safety profiles.86 Various derivatives of β-lapachone have entered Phase I and Phase II clinical trials, either as a monotherapeutic agent or in combination with other cancer drugs. However, its rapid elimination and low bioavailability due to poor water solubility have posed challenges. Many derivatives such as ARQ 761, designed to overcome water solubility issues, also entered Phase I trials but exhibited only moderate potency.89 MB12066, a derivative with an undisclosed structure, activated mitochondrial metabolism through NQO1 and entered Phase I and Phase II trials, demonstrating good safety profiles. However, these trials were ultimately terminated.90 These trials aimed to explore the clinical potential of these β-lapachone derivatives in cancer treatment, highlighting the importance of addressing solubility and potency challenges. These structural properties and modifications hold considerable promise for a range of biomedical applications, including cancer therapy, treatment of Chagas disease and tuberculosis, development of antifungal and antibacterial agents, and antimalarial drugs, ultimately contributing to advances in these critical fields of medicine.86
Anthracyclines
Some naphthoquinone-containing compounds, such as anthracyclines, are FDA-approved anticancer agents. Anthracyclines are among the most effective anticancer drugs ever developed. Doxorubicin (DOX) and daunorubicin (DNR) were the first anthracyclines that were isolated from Streptomyces peucetius bacteria (Figure 15),91 and are commonly used for the treatment of both hematologic and solid tumors, such as breast cancer, childhood solid tumors, soft tissue sarcomas, aggressive lymphomas, and acute leukemias. Anthracyclines share a common structural framework characterized by a tetracyclic ring containing adjacent quinone-hydroquinone groups in rings B-C, a methoxy group at C-4 in ring D, and a short side chain at C-9 with a carbonyl at C-13. In addition, a sugar molecule called daunosamine is attached to the C-7 of ring A via a glycosidic bond.92-95 Inducing oxidative damage in tumor cells has been considered an important mechanism for the anticancer effect of anthracyclines.92 There are two mechanisms for superoxide production by anthracyclines (Figure 16). First, these drugs alter the properties of endogenous respiratory chain components, making them more susceptible to autooxidation by molecular oxygen.96 The quinone moiety in ring C undergoes one-electron reduction to form a semiquinone that quickly regenerates its parent quinone by reducing O2 to O2•− and H2O2. During this cycle, the glycosylic bond between ring A and daunosamine can also undergo reductive deglycosidation leading to the formation of 7-deoxyaglycone (Figure 16). 7-Deoxyaglycone has increased lipid solubility that allows for intercalation into biologic membranes and site-specific ROS production. One-electron redox cycling of DOX is also accompanied by a release of iron from intracellular stores which leads to the formation of drug-iron complexes that convert H2O2 into more potent hydroxyl radicals by a Fenton reaction. Second, due to their quinone nature, anthracyclines can function as artificial electron acceptors, withdrawing electrons from the respiratory chain. This action can lead to non-enzymatic oxidation of reduced anthracycline by O2, resulting in superoxide production. Superoxide produced in this process can contribute to the formation of H2O2 via dismutation.95
Efforts to enhance anthracycline drugs have led to the development of around 2000 analogs, but with only a few advancing to clinical use.92 Notable alternatives to DOX and DNR include epirubicin (EPI) and idarubicin (IDA) (Figure 15). EPI, derived from DOX, features an alteration in the hydroxyl group at C-4 in daunosamine, primarily affecting pharmacokinetics. Despite changes in volume of distribution and shorter half-life, EPI can be used at higher cumulative doses without increased cardiotoxicity. IDA, derived from DNR, exhibits activity against various cancers, attributed to increased lipophilicity and improved stabilization of the drug-topoisomerase II-DNA complex.97
Aziridinylquinones.
Aziridinylquinones, such as carbazilquinone, diaziquone (AZQ), BZQ, triaziquone, and apaziquone, have a unique structural composition with an aziridine ring attached to a quinone group (Figure 17). These compounds possess the ability to alkylate DNA and generate ROS, both of which contribute to their cytotoxic effects.98-99 Aziridinylquinones undergo enzymatic reduction within cells, leading to the transformation of the quinone into a hydroquinone variant, which results in an elevation of the pKa of the nitrogen atom within the aziridine ring (Figure 18). The increased pKa makes the aziridine nitrogen atom in the hydroquinone variant more easily protonated, forming a highly reactive aziridinium cation that is a powerful DNA alkylating agent.98 Meanwhile, molecular O2 reduces to H2O2 and other ROS in the redox cycling of semiquinone radicals formed via the reduction of aziridinylquinones catalyzed by enzymes like NADPH-cytochrome P-450 reductase. Under aerobic conditions, these radicals undergo redox cycling, generating O2•− and H2O2.99 Under hypoxic tumor conditions where the availability of O2 is limited, on the other hand, aziridinylquinones such as AZQ, undergo activation through a two-electron reduction mechanism facilitated by enzymes like DT-diaphorase (NQO1), forming semiquinone radical anions, which subsequently undergo redox cycling to produce cytotoxic H2O2. The ability of AZQ to exploit hypoxic environments enhances its cytotoxic effects in tumor cells.100
Indolequinones
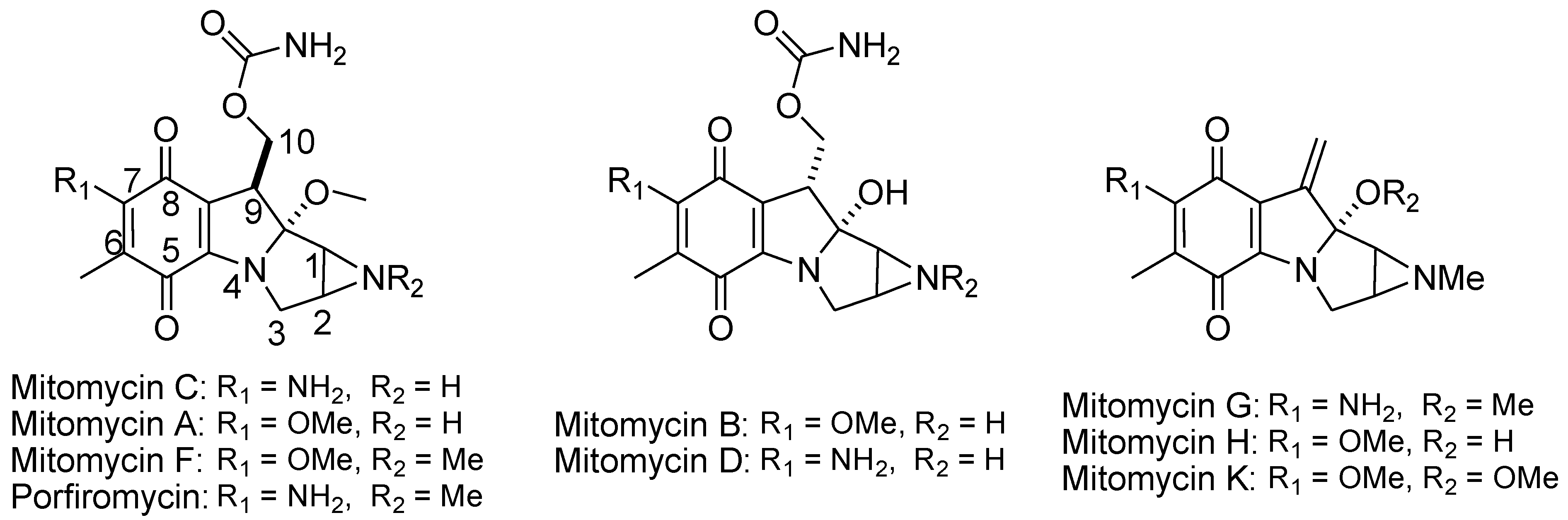
Many naturally occurring indolequinone analogues, such as mitomycins, showed potent anticancer properties.101 They were recognized as prodrugs which undergo bioreduction in vivo to form irreversible bis-alkylation of DNA. The reduction of mitomycin initiates a reduction-oxidation cycle, which generates H2O2 as a byproduct.102 Mitomycin C was the first discovered member of this class of compounds, and was isolated from the fermentation broth of Streptomyces caespetosius. It demonstrates the most potent anticancer efficacy, and has been used for the treatment of various tumors for decades.103 Several mitomycin analogues have been identified with various modifications on the aziridine ring or quinone ring substituents, each retaining the biological activity of mitomycin C. Hydrophilic compounds, such as mitomycin C and porfiromycin, demonstrate the most effective anticancer effects against L1210 leukemia. The presence of an aziridine ring is essential for antileukemia activity, while quinone reduction potential strongly influences antibacterial activity.104Among these mitomycin analogues, mitomycin C possesses unique chemical and physical properties, including good water solubility, low lipophilicity, and minimal binding to serum proteins, contributing to its potent anticancer properties. Correlations between partition coefficients and antitumor potency have been observed in some analogs, but correlations with quinone reduction potential or substituent size have been found to be insignificant in several studies.104-105
Figure 19.
Structures of mitomycins that contain a complex tetracyclic pyrrolo-indole core adorned with an aziridine ring, carbamoyl group, and bridged carbinolamine, rendering them moderately stable under certain conditions but highly reactive with reducing agents.
Figure 19.
Structures of mitomycins that contain a complex tetracyclic pyrrolo-indole core adorned with an aziridine ring, carbamoyl group, and bridged carbinolamine, rendering them moderately stable under certain conditions but highly reactive with reducing agents.
Aminoquinones.
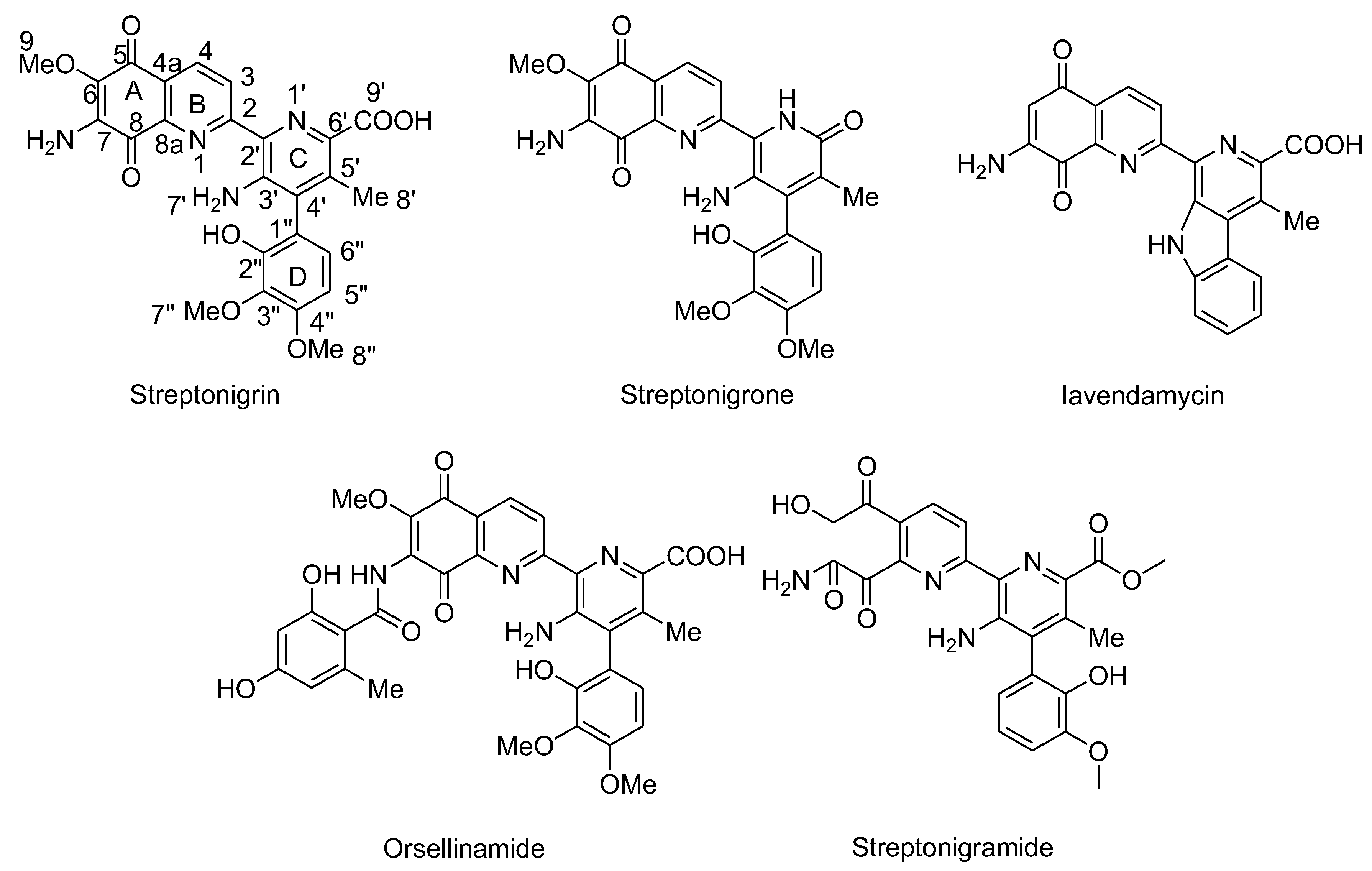
Streptonigrin and its derivatives contain aminoquinone moieties. They were isolated from Streptomyces flocculus and exhibit potent antitumor and antibiotic effects. Streptonigrin interacts with oxygen to generate superoxide radicals that undergo dismutation, producing H2O2 (Figure 11).106 The genotoxic effects of Streptonigrin are partly attributed to its ability to cleave DNA through a complex mechanism involving metal ions and autoxidation of its quinone moiety in the presence of NADH, leading to the production of oxygen-derived reactive species, including free radicals.106-109 Recent studies have explored the involvement of free radicals in SN-induced DNA and chromosome damage. Antioxidant enzymes such as SOD and catalase, when added, prevent SN-induced DNA and chromosome damage.107, 109 Conversely, the hydroxyl radical scavenger mannitol intensified DNA and chromosome damage induced by SN.109 However, when various antioxidants were encapsulated into liposomes and added to cell cultures, either alone or in combinations, a significant reduction in SN-induced chromosome aberrations and DNA damage was observed. This suggests that free radicals play a role in SN-induced genotoxicity and that this damage can be partially mitigated by incorporating antioxidants into cells.109 Streptonigrin was previously used as an anticancer drug but has been discontinued because of its toxic effects. Analogues of SN, such as streptonigrone, Lavendamycin, Orsellinamide, Streptonigramide have been designed and investigated, which did not lead to better anticancer activity and selectivity (Figure 20).
Figure 20.
Proposed mechanism of Mitomycins' activity via H2O2 production and DNA adducts formation upon bioreduction.
Figure 20.
Proposed mechanism of Mitomycins' activity via H2O2 production and DNA adducts formation upon bioreduction.
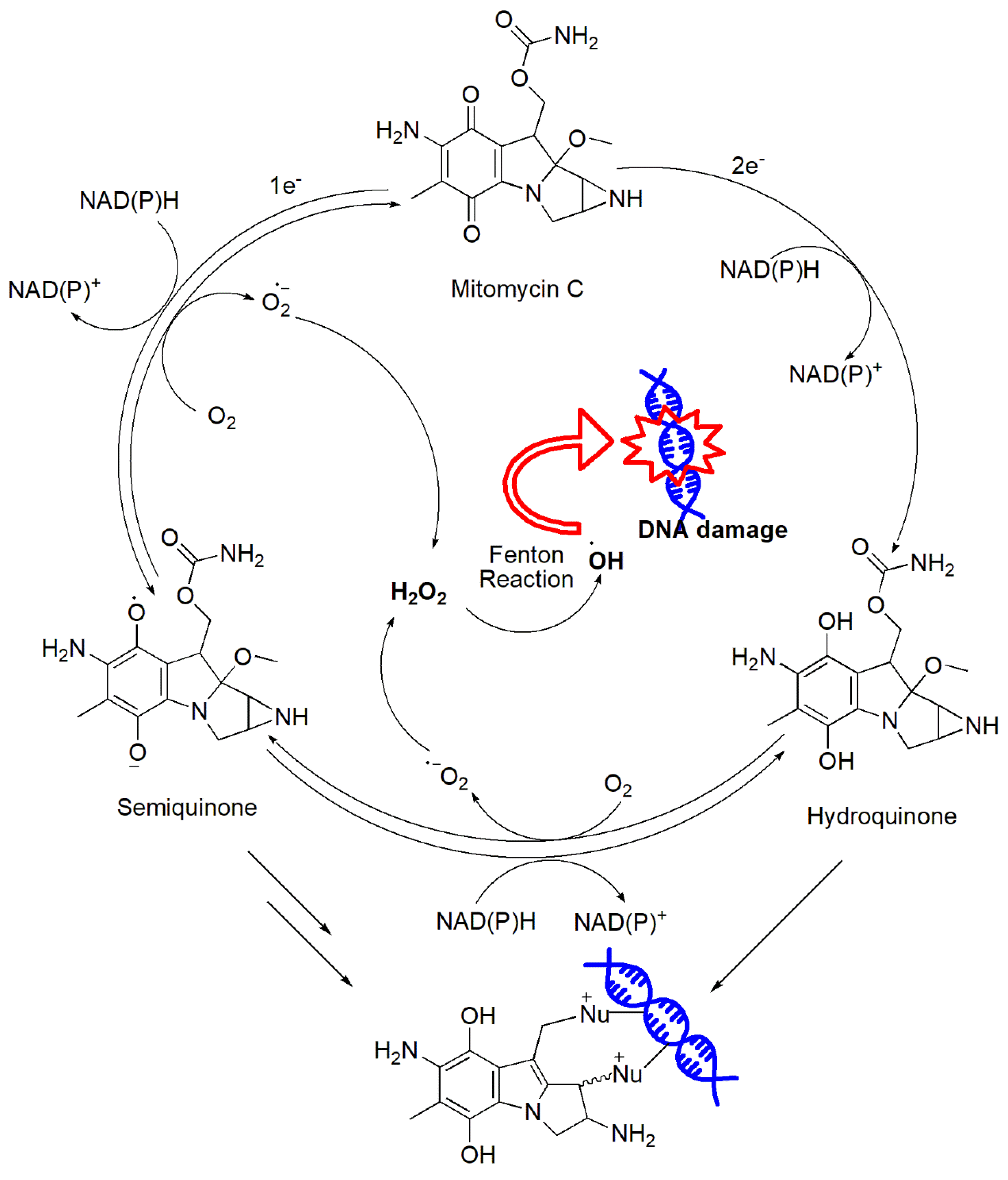
Figure 21.
Structures of Streptonigrin and its derivatives.
3.3. Vitamin C
Vitamin C (also known as ascorbic acid or ascorbate) is an essential vitamin in the body’s daily function. It allows for the biosynthesis of collagen and various neurotransmitters, is involved in protein metabolism, and strengthens the body’s immune system. In recent years, ascorbic acid has been shown to have selective anticancer properties at millimolar (mM) concentrations, with such an anticancer effect demonstrated both in vitro and in vivo.110-113 The main mechanism through which vitamin C kills tumor cells is by formation of H2O2.111, 114 At the beginning of this process, the ionized vitamin C is transformed into ascorbate radical by losing one electron (Figure 22). This electron then reduces a protein-centered metal, such as Fe3+ to Fe2+. The created Fe2+ then donates an electron to O2, forming O2●─ that is subsequently dismutated to form H2O2 and O2. The created H2O2 can cause damage to DNA, lipids, and proteins, inducing cancer cell death. Notably, these concentrations of Vitamin C are not enough to kill healthy, non-cancerous cells due to the high level of plasma catalase and/or GSH peroxidase that inhibit the redox reaction or destroy any formed H2O2 molecules, thus making cancer treatment via vitamin C even more appealing due to its selective nature.
Although early randomized clinical trials concluded that oral administration of high-dose vitC to patients with advanced cancers does not have any therapeutic benefits, parenteral (i.v. or i.p.) injection of vitC can result in concentrations as high as 70x (up to 20 mM) that of an oral dose in the plasma, a concentration high enough to damage cancer cells.115-117 Currently, there are more than a dozen completed and active clinical trials investigating the effectiveness of high-dose vitC to treat cancer (https://clinicaltrials.gov). Derivatives of vitamin C and combination therapy have also been developed to improve its efficacy. For example, hydrophobized palmitoyl ascorbate was used to construct polymeric micellar nanoparticles to further enhance anticancer efficacy and selectivity.118-119 Another possible avenue that could be taken with vitamin C cancer treatment is using it in conjunction with other therapeutics, such as vitamin K3, triethylenetetramine, or other H2O2-responsive chemotherapeutic drugs (i.e. camptothecin) to achieve synergistic anticancer effect while minimizing unwanted side effects.120-122
3.4. Metal, Metal Oxides, and Metal Peroxides
Metals play an essential role in biological systems and human health. Many enzymatic reactions require metals for their catalytic action.123 Essential metals such as calcium, sodium, potassium, magnesium, and transition metals iron, copper, and zinc are vital as well. Deficiency or excess of these metals can cause various diseases including cancer.124 Exposure to heavy metals like arsenic, cadmium, chromium, nickel, lead, and mercury, even at low levels, can be toxic and contribute to various cancers including skin and lung cancers. Although the molecular mechanism is not completely understood, their potential to generate ROS and alter cellular redox status is considered significant in metal-induced carcinogenesis.125 On the other hand, many metals, metal complexes, or metal peroxides have gained significant attention in cancer treatment, which has been highlighted in several reviews.126-128 Some metal oxides and peroxides are reported to enhance H2O2 production, which is one of the possible mechanisms for their anticancer efficacy and selectivity.128 This section does not intend to give a comprehensive review on metals, their oxides, and peroxides, but instead aims to discuss examples of metal oxides and peroxides that directly generate H2O2 in cancer cells and to highlight their role in cancer treatment.
There are several pathways for metal oxides or peroxides to induce H2O2 production, including inhibition ofantioxidant enzymes, photocatalysis, or via a chemical reaction with water. For example, trisenox, also known as As2O3, induces H2O2 production by inhibiting GPx and catalase.129-131 Titanium dioxide (TiO2) generates H2O2 primarily via photocatalysis.132-133 Many metal peroxides, such as MgO2, CaO2 can react with H2O to produce H2O2. Such a reaction is facilitated under acidic conditions (Figure 25)127-128.
Figure 25.
A reaction between metal peroxide and H2O to produce H2O2 using CaO2 as a representative example.
Figure 25.
A reaction between metal peroxide and H2O to produce H2O2 using CaO2 as a representative example.

Many metals and their oxide forms have limited therapeutic potential due to metal carcinogenesis. Modification of metal oxides with less toxicity, i.e. titanium dioxide (TiO2),132 zinc oxide (ZnO),134 etc. into nanoparticle (NP) forms allows for the targeting of cancer tissues more accurately due to their smaller size and greater bioavailability. For example, TiO2 NPs can be activated upon UV irradiation to produce various ROS (i.e. H2O2), leading to cytotoxicity. This process has been applied in photodynamic therapy to treat cancer.132 The popularity of ZnO has also risen due to being safe and efficient delivery,134-135 and being categorized as “generally recognized as safe” (GRAS) by the U.S. FDA ( 21CFR182.8991). Its functionality as an antibacterial and anticancer agent primarily relies on its ability to generate ROS.134, 136 A wide variety of ZnO NPs have been developed, which showed selective cytotoxicity towards cancer cells. The ZnO NPs undergo low-pH dependent dissolution into Zn2+ ions, which can disrupt cell membrane and mitochondrial functions, inducing ROS generation and leading to cancer cell death.
Metal peroxides (MO2) have also gained popularity recently due to their ability to react with water to form H2O2 which is facilitated under acidic conditions (Figure 25). Many metal peroxide NPs have been constructed to target unique tumor microenvironment, including hypoxia, low acidity, and high H2O2 and GSH levels. Such NPs include ones containing CaO2, MgO2, BaO2, ZnO2, or CuO2.128, 134, 136-137 For example, Zhang’s group constructed a CaO2-based nanocatalytic medicine, which simultaneously supplies O2 and H2O2 to achieve enhanced chemo/chemodynamic therapy.138 Tang and co-authors developed a biodegradable transferrin-modified MgO2 nanosheet that produced large quantities of H2O2 selectively in cancer cells in response to the acidic and low catalase activity of the tumor microenvironment.128 Chen’s group reported a method of fabricating CuO2 nanodots which are sensitive to the acidic environment of tumor cells, leading to simultaneous release of H2O2 and Cu2+.139 These tumor-targeting metal-peroxide NPs showed enhanced tumor growth inhibition and minimal side effects in vivo.
Among various metal peroxides, CaO2 shows the most promise due to its biocompatibility and potential for use in cancer treatments like calcium overload therapy and treatment of bone-related cancers. In catalytic medicine, H2O2 can be utilized to generate large amounts of hydroxyl radicals through a Fenton-like reaction. MO2 has been found to be effective in enhancing therapeutic effectiveness in procedures that involve O2, such as photodynamic therapy and radiotherapy. Thus, metal peroxide-based nanoparticles are emerging as a novel avenue for cancer treatment. They have been extensively investigated in the field of biomedical science for their H2O2 and O2 generation capabilities.
3.5. FDA-Approved Drugs
Many FDA-approved anticancer drugs not belonging to categories already touched on also undergo oxidation and induce O2 reduction to generate O2●─ and H2O2. These drugs include procarbazine, Paclitaxel, Motexafin Gadolinium, and others (Figure 23).
Procarbazine is a hydrazine derivative that is widely used for the treatment of various types of cancer, including Hodgkin's lymphoma, non-Hodgkin's lymphoma, and primary brain tumors.140 It was one of the first drugs to be developed that generates ROS, notably H2O2, in order to combat cancer cells.141 When exposed to oxygen, procarbazine undergoes oxidation and triggers the reduction of O2 to form H2O2, which is combined with iron (Fe2+) to produce OH•, causing damage to cellular components, such as DNA (Figure 24).141-142 Procarbazine can cross the blood-brain barrier, which has made it a valuable treatment for primary brain tumors.141-142 In clinical applications, procarbazine is mostly administered in combination with other drugs to treat Hodgkin's lymphoma, non-Hodgkin's lymphoma, and specific primary brain tumors. It has also shown remarkable efficacy in regimens like MOPP (Mechlorethamine, Vincristine, Procarbazine, and Prednisone) when used to treat Hodgkin's lymphoma.143-144
Paclitaxel (PTX), also known as taxol, was the first microtube stabilizing agent widely used in chemotherapy. Its primary mechanism of action is to bind and stabilize microtubules and inhibit cell division.145 Recently, it has been found that paclitaxel cytotoxicity is also correlated with ROS production.146 PTX has been shown to induce excessive production of O2•− and H2O2, leading to oxidative stress in various cancer cell types, such as lung and breast cancer cells. This process may involve activation by NADPH oxidase (Nox), which is found in cytosol and plays a role in generating O2•− from oxygen and NADPH.147 Paclitaxel-induced stabilization of microtubules within cells could potentially trigger the activation of Nox via a pathway that involves Rac GTPase, which is known to closely interact with microtubules. Therefore, paclitaxel's impact on microtubules might influence Rac GTPase activity, subsequently activating Nox and leading to O2•− production.148-149 Spitz et al. has demonstrated that combination of PTX with inhibitors of glucose and H2O2 metabolism greatly elevate H2O2 levels, which enhances killing of breast cancer cells.150 The clinical implications of these findings are profound. Combining inhibitors of glucose and H2O2 metabolism with PTX could represent a novel strategy to amplify oxidative stress selectively in cancer cells, making them more susceptible to cytotoxicity while minimizing harm to normal cells. This approach holds potential for improving the therapeutic efficacy of PTX, especially in breast cancer treatment.150
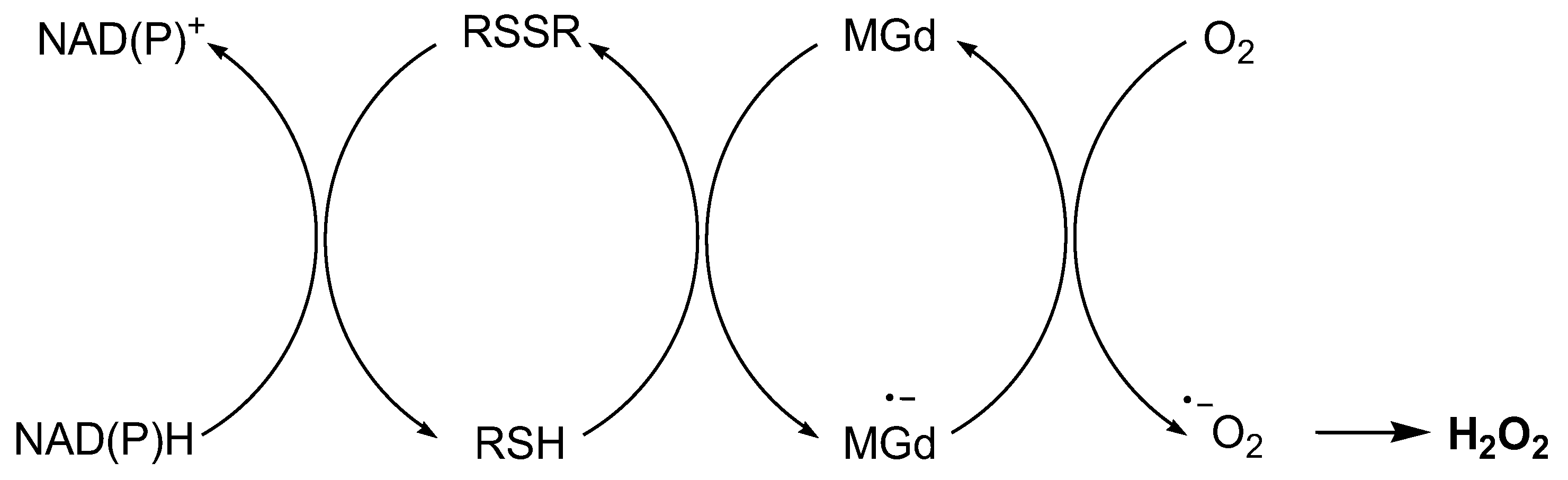
Motexafin gadolinium (MGd) is a gadolinium texaphyrin complex that has a strong affinity for electrons. MGd can accept electrons from various reducing metabolites, such as protein thiols, thioredoxin, nicotinamide adenine dinucleotide phosphate (NADPH), and GSH, and transfer them to oxygen, resulting in the production of O2•−.151 This electron transfer process interferes with ATP production and promotes apoptosis.152 MGd is specifically designed to localize within tumor cells, targeting cancerous tissue due to its affinity for the abnormal metabolic processes found in these cells. In contrast to normal cells, cancer cells predominantly employ anaerobic glycolysis for their energy production. This abnormal metabolism provides a crucial distinction for the drug's action. Once inside cancer cells, MGd initiates a unique mechanism where electrons are transferred from reducing metabolites within the cancer cells to O2, generating superoxide anions that are disproportionate to form oxygen and H2O2.151 Crucially, H2O2 and other ROS produced during this process are selectively trapped within tumor cells, resulting in damage to cellular DNA, proteins, and lipids inside cancer cells, eventually leading to apoptosis.151
Figure 25.
MGd induces H2O2 production via transferring electrons from biological reducing agents, such as protein thiols, thioredoxin, and glutathione, to form MGd radical anion that reduces O2 to O2•− and form the final product H2O2.
Figure 25.
MGd induces H2O2 production via transferring electrons from biological reducing agents, such as protein thiols, thioredoxin, and glutathione, to form MGd radical anion that reduces O2 to O2•− and form the final product H2O2.
Conclusion
Redox adaptations among cancer cells have been characterized by their heightened oxidative stress thresholds as well as their increased intracellular antioxidant defenses. To push cancer cells beyond their cytotoxic threshold necessitates approaches that disrupt these adaptations. One such approach is the manipulation of ROS, particularly H2O2. This can be achieved in a few ways, including inhibition of endogenous antioxidants, modulation of proteins responsible for maintaining redox homeostasis, and use of compounds that generate H2O2 selectively within cancer cells. This strategy to selectively enhance H2O2 levels in cancer cells has emerged as a potential therapeutic approach. Vulnerabilities within cancer cells make them susceptible towards H2O2-mediated toxicity or growth inhibition mechanisms. This review has discussed various chemicals agents capable of producing H2O2 in cancer cells and explained their mechanism of function. A wide range of compounds, including phenols and polyphenols, quinone-containing compounds, various vitamins (i.e. vitamin C), metal oxides and peroxides, and some FDA-approved chemotherapeutic drugs, etc. have shown significant potential in preclinical and clinical investigation to selectively induce H2O2-mediated cytotoxicity, sensitize resistance, and modulate tumor microenvironments. However, there are still challenges that must be overcome before H2O2-amplifying agents can be used for targeted cancer therapies, such as ROS heterogeneity, targeted drug delivery, difficulty to achieve optimal selectivity, safety, and toxicity concerns. Researchers have been addressing these challenges by combining H2O2-generating agents with other chemotherapeutic agents, nanoparticles, or other prooxidant-based therapies, for an effective and personalized cancer treatment. We recently observed that combination of H2O2-generating prooxidants with H2O2-activated prodrugs has potential to enhance anticancer efficacy and lower the required dosage, which in turn reduces the risk of toxicity towards healthy tissues.153 Understanding the functions and mechanisms of these H2O2-generating compounds facilitates the design and development of innovative approaches to combat cancer, which offer improved therapeutic outcomes.
References
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic Res 2010, 44, 479–496. [Google Scholar] [CrossRef]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov 2013, 12, 931–947. [Google Scholar] [CrossRef] [PubMed]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat Rev Drug Discov 2009, 8, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, V.; Hay, N. Molecular pathways: Reactive oxygen species homeostasis in cancer cells and implications for cancer therapy. Clin. Cancer Res. 2013, 19, 4309–4314. [Google Scholar] [CrossRef] [PubMed]
- Schumacker, P.T. Reactive oxygen species in cancer cells: Live by the sword, die by the sword. Cancer Cell 2006, 10, 175–176. [Google Scholar] [CrossRef] [PubMed]
- Hileman, E.O.; Liu, J.; Albitar, M.; Keating, M.J.; Huang, P. Intrinsic oxidative stress in cancer cells: A biochemical basis for therapeutic selectivity. Cancer Chemother. Pharmacol. 2004, 53, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Chu, Z.Y.J.; Zheng, W.; Sun, J.; Wang, W.; Qian, H. Recent Advances on Modulation of H2O2 in Tumor Microenvironment for Enhanced Cancer Therapeutic Efficacy. Coord. Chem. Rev. 2023, 481, 215049. [Google Scholar] [CrossRef]
- Dharmaraja, A.T. Role of Reactive Oxygen Species (ROS) in Therapeutics and Drug Resistance in Cancer and Bacteria. J Med Chem 2017, 60, 3221–3240. [Google Scholar] [CrossRef]
- Kim, S.J.; Kim, H.S.; Seo, Y.R. Understanding of ROS-Inducing Strategy in Anticancer Therapy. Oxid Med Cell Longev 2019, 2019, 5381692. [Google Scholar] [CrossRef]
- Lopez-Lazaro, M. Dual role of hydrogen peroxide in cancer: Possible relevance to cancer chemoprevention and therapy. Cancer Lett. 2007, 252, 1–8. [Google Scholar] [CrossRef]
- Panieri, E.; Santoro, M.M. ROS homeostasis and metabolism: A dangerous liason in cancer cells. Cell Death Dis. 2016, 7, e2253. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol 2017, 11, 613–619. [Google Scholar] [CrossRef]
- Celia María Curieses Andrés, J.M.P.d.l.L. , Celia Andrés Juan, Francisco J. Plou; Pérez-Lebeña, a. E., Chemistry of Hydrogen Peroxide Formation and Elimination in Mammalian Cells, and Its Role in Various Pathologies. Stresses 2022, 2, 256–274. [Google Scholar] [CrossRef]
- Nicco, C.; Laurent, A.; Chereau, C.; Weill, B.; Batteux, F. Differential modulation of normal and tumor cell proliferation by reactive oxygen species. Biomed. Pharmacother. 2005, 59, 169–174. [Google Scholar] [CrossRef]
- Dickinson, B.C.; Chang, C.J. Chemistry and biology of reactive oxygen species in signaling or stress responses. Nat Chem Biol 2011, 7, 504–511. [Google Scholar] [CrossRef]
- Imlay, J.A. The molecular mechanisms and physiological consequences of oxidative stress: Lessons from a model bacterium. Nat Rev Microbiol 2013, 11, 443–454. [Google Scholar] [CrossRef]
- Kong, Q.; Lillehei, K.O. Antioxidant inhibitors for cancer therapy. Med Hypotheses 1998, 51, 405–409. [Google Scholar] [CrossRef] [PubMed]
- Conklin, K.A. Chemotherapy-associated oxidative stress: Impact on chemotherapeutic effectiveness. Integr Cancer Ther 2004, 3, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Buytaert, E.; Dewaele, M.; Agostinis, P. Molecular effectors of multiple cell death pathways initiated by photodynamic therapy. Biochim. Biophys. Acta 2007, 1776, 86–107. [Google Scholar] [CrossRef]
- Kim, W.; Lee, S.; Seo, D.; Kim, D.; Kim, K.; Kim, E.; Kang, J.; Seong, K.M.; Youn, H.; Youn, B. Cellular Stress Responses in Radiotherapy. Cells, 2019; 8. [Google Scholar]
- Wang, C.Y. J.; Dong, C.; Shi, S. Glucose Oxidase-Related Cancer Therapies. Adv.Ther. 2020; 3, No.2000110. [Google Scholar]
- Firczuk, M.; Bajor, M.; Graczyk-Jarzynka, A.; Fidyt, K.; Goral, A.; Zagozdzon, R. Harnessing altered oxidative metabolism in cancer by augmented prooxidant therapy. Cancer Lett. 2020, 471, 1–11. [Google Scholar] [CrossRef]
- Foglietta, F.; Serpe, L.; Canaparo, R. ROS-generating nanoplatforms as selective and tunable therapeutic weapons against cancer. Discov Nano 2023, 18, 151. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Tian, X.; Ai, H.W. Molecular Tools to Generate Reactive Oxygen Species in Biological Systems. Bioconjug. Chem. 2019, 30, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Peng, L.; Zhou, L.; Huang, Z.; Zhou, C.; Huang, C. Oxidative Stress in Cancer Immunotherapy: Molecular Mechanisms and Potential Applications. Antioxidants (Basel), 2022; 11. [Google Scholar]
- Giansanti, M.; Karimi, T.; Faraoni, I.; Graziani, G. High-Dose Vitamin C: Preclinical Evidence for Tailoring Treatment in Cancer Patients. Cancers (Basel), 2021; 13. [Google Scholar]
- Akagawa, M.; Shigemitsu, T.; Suyama, K. Production of hydrogen peroxide by polyphenols and polyphenol-rich beverages under quasi-physiological conditions. Biosci. Biotechnol. Biochem. 2003, 67, 2632–2640. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, M.; Yamazaki, S.; Kano, K.; Ikeda, T. Kinetic analysis and mechanistic aspects of autoxidation of catechins. Biochim. Biophys. Acta, 2002; 1569, 35–44. [Google Scholar]
- Zhang, Z.; Shi, J.; Nice, E.C.; Huang, C.; Shi, Z. The Multifaceted Role of Flavonoids in Cancer Therapy: Leveraging Autophagy with a Double-Edged Sword. Antioxidants (Basel), 2021; 10. [Google Scholar]
- Oikawa, S.; Furukawaa, A.; Asada, H.; Hirakawa, K.; Kawanishi, S. Catechins induce oxidative damage to cellular and isolated DNA through the generation of reactive oxygen species. Free Radic Res 2003, 37, 881–890. [Google Scholar] [CrossRef]
- Furukawa, A.; Oikawa, S.; Murata, M.; Hiraku, Y.; Kawanishi, S. (-)-Epigallocatechin gallate causes oxidative damage to isolated and cellular DNA. Biochem. Pharmacol. 2003, 66, 1769–1778. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Hasumi, K.; Woo, J.T.; Nagai, K.; Wachi, M. Generation of hydrogen peroxide primarily contributes to the induction of Fe(II)-dependent apoptosis in Jurkat cells by (-)-epigallocatechin gallate. Carcinogenesis 2004, 25, 1567–1574. [Google Scholar] [CrossRef] [PubMed]
- Sakagami, H.; Arakawa, H.; Maeda, M.; Satoh, K.; Kadofuku, T.; Fukuchi, K.; Gomi, K. Production of hydrogen peroxide and methionine sulfoxide by epigallocatechin gallate and antioxidants. Anticancer Res. 2001, 21 (4A), 2633–2641. [Google Scholar]
- Yen, G.C.; Duh, P.D.; Tsai, H.L.; Huang, S.L. Pro-oxidative properties of flavonoids in human lymphocytes. Biosci. Biotechnol. Biochem. 2003, 67, 1215–1222. [Google Scholar] [CrossRef]
- Canada, A.T.; Giannella, E.; Nguyen, T.D.; Mason, R.P. The production of reactive oxygen species by dietary flavonols. Free Radic Biol Med 1990, 9, 441–449. [Google Scholar] [CrossRef]
- Miura, Y.H.; Tomita, I.; Watanabe, T.; Hirayama, T.; Fukui, S. Active oxygens generation by flavonoids. Biol. Pharm. Bull. 1998, 21, 93–96. [Google Scholar] [CrossRef]
- Baumann, S.; Fas, S.C.; Giaisi, M.; Muller, W.W.; Merling, A.; Gulow, K.; Edler, L.; Krammer, P.H.; Li-Weber, M. Wogonin preferentially kills malignant lymphocytes and suppresses T-cell tumor growth by inducing PLCgamma1- and Ca2+-dependent apoptosis. Blood 2008, 111, 2354–2363. [Google Scholar] [CrossRef]
- Wei, L.; Lu, N.; Dai, Q.; Rong, J.; Chen, Y.; Li, Z.; You, Q.; Guo, Q. Different apoptotic effects of wogonin via induction of H(2)O(2) generation and Ca(2+) overload in malignant hepatoma and normal hepatic cells. J Cell Biochem 2010, 111, 1629–1641. [Google Scholar] [CrossRef]
- Tsai, C.F.; Yeh, W.L.; Huang, S.M.; Tan, T.W.; Lu, D.Y. Wogonin induces reactive oxygen species production and cell apoptosis in human glioma cancer cells. Int J Mol Sci 2012, 13, 9877–9892. [Google Scholar] [CrossRef]
- Dzah, C.S.Z. H.; Gobe, V.; Asante-Donyinah, D.A.; Duan, Y. Anti- and pro-oxidant properties of polyphenols and their role in modulating glutathione synthesis, activity and cellular redox potential: Potential synergies for disease management. Adv. Red. Res. 2024, 11, 100099. [Google Scholar] [CrossRef]
- Braicu, C.; Ladomery, M.R.; Chedea, V.S.; Irimie, A.; Berindan-Neagoe, I. The relationship between the structure and biological actions of green tea catechins. Food Chem 2013, 141, 3282–3289. [Google Scholar] [CrossRef]
- Ouyang, J.; Zhu, K.; Liu, Z.; Huang, J. Prooxidant Effects of Epigallocatechin-3-Gallate in Health Benefits and Potential Adverse Effect. Oxid Med Cell Longev 2020, 2020, 9723686. [Google Scholar] [CrossRef]
- Saeki, K.; Hayakawa, S.; Isemura, M.; Miyase, T. Importance of a pyrogallol-type structure in catechin compounds for apoptosis-inducing activity. Phytochemistry 2000, 53, 391–394. [Google Scholar] [CrossRef]
- Hong, J.; Lu, H.; Meng, X.; Ryu, J.H.; Hara, Y.; Yang, C.S. Stability, cellular uptake, biotransformation, and efflux of tea polyphenol (-)-epigallocatechin-3-gallate in HT-29 human colon adenocarcinoma cells. Cancer Res. 2002, 62, 7241–7246. [Google Scholar]
- Park, W.H.; Han, Y.W.; Kim, S.H.; Kim, S.Z. A superoxide anion generator, pyrogallol induces apoptosis in As4.1 cells through the depletion of intracellular GSH content. Mutat Res, 2007; 619, 81–92. [Google Scholar]
- Han, Y.H.; Kim, S.Z.; Kim, S.H.; Park, W.H. Pyrogallol as a glutathione depletor induces apoptosis in HeLa cells. Int J Mol Med 2008, 21, 721–730. [Google Scholar]
- Liu, T.P. P.; Shi, H.; Feng, J.; Zhang, X. Assembled polyphenol-based systems as advanced therapeutics. J. Poly. Sci. 62(2).
- Awad, H.M.; Boersma, M.G.; Boeren, S.; van Bladeren, P.J.; Vervoort, J.; Rietjens, I.M. The regioselectivity of glutathione adduct formation with flavonoid quinone/quinone methides is pH-dependent. Chem. Res. Toxicol. 2002, 15, 343–351. [Google Scholar] [CrossRef]
- Galati, G.; Sabzevari, O.; Wilson, J.X.; O'Brien, P.J. Prooxidant activity and cellular effects of the phenoxyl radicals of dietary flavonoids and other polyphenolics. Toxicology 2002, 177, 91–104. [Google Scholar] [CrossRef]
- Chan, T.; Galati, G.; O'Brien, P.J. Oxygen activation during peroxidase catalysed metabolism of flavones or flavanones. Chem. Biol. Interact. 1999, 122, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Hider, R.C.; Liu, Z.D.; Khodr, H.H. Metal chelation of polyphenols. Methods Enzymol 2001, 335, 190–203. [Google Scholar]
- Pan, Y.Q. R.; Hou, M.; Xue, J.; Zhou, M.; Xu, L.; Zhang, Y. The interactions of polyphenols with Fe and their application in Fenton/Fenton-like reactions. Sep. Purif. Technol. 2022, 300, 121831. [Google Scholar] [CrossRef]
- Aruoma, O.I.; Spencer, J.P.; Rossi, R.; Aeschbach, R.; Khan, A.; Mahmood, N.; Munoz, A.; Murcia, A.; Butler, J.; Halliwell, B. An evaluation of the antioxidant and antiviral action of extracts of rosemary and Provencal herbs. Food Chem Toxicol 1996, 34, 449–456. [Google Scholar] [CrossRef] [PubMed]
- al-Sereiti, M.R.; Abu-Amer, K.M.; Sen, P. Pharmacology of rosemary (Rosmarinus officinalis Linn.) and its therapeutic potentials. Indian J Exp Biol 1999, 37, 124–130. [Google Scholar] [PubMed]
- Zheng, L.F.; Dai, F.; Zhou, B.; Yang, L.; Liu, Z.L. Prooxidant activity of hydroxycinnamic acids on DNA damage in the presence of Cu(II) ions: Mechanism and structure-activity relationship. Food Chem Toxicol 2008, 46, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Damasceno, S.S.; Dantas, B.B.; Ribeiro-Filho, J.; Antonio, M.A.D.; Galberto, M. d. C. J. Chemical Properties of Caffeic and Ferulic Acids in Biological System: Implications in Cancer Therapy. A Review. Curr Pharm Des 2017, 23, 3015–3023. [Google Scholar] [CrossRef]
- Li, Y.; Trush, M.A. Reactive oxygen-dependent DNA damage resulting from the oxidation of phenolic compounds by a copper-redox cycle mechanism. Cancer Res. 1994, 54 (7 Suppl), 1895s–1898s. [Google Scholar]
- Pirker, K.F.; Kay, C.W.; Stolze, K.; Tunega, D.; Reichenauer, T.G.; Goodman, B.A. Free radical generation in rosmarinic acid investigated by electron paramagnetic resonance spectroscopy. Free Radic Res 2009, 43, 47–57. [Google Scholar] [CrossRef]
- Murakami, K.; Haneda, M.; Qiao, S.; Naruse, M.; Yoshino, M. Prooxidant action of rosmarinic acid: Transition metal-dependent generation of reactive oxygen species. Toxicol In Vitro 2007, 21, 613–617. [Google Scholar] [CrossRef]
- Muñoz-Muñoz, J. l. G.-M. F.; Ros, E.; Tudela, J.; García-Canovas, F.; Rodriguez-Lopez, J.N. , Prooxidant and Antioxidant Activities of Rosmarinic Acid. Journal of Food Biochemistry,. 2013,, 37 (4).
- Fabiani, R.; De Bartolomeo, A.; Rosignoli, P.; Servili, M.; Montedoro, G.F.; Morozzi, G. Cancer chemoprevention by hydroxytyrosol isolated from virgin olive oil through G1 cell cycle arrest and apoptosis. Eur J Cancer Prev 2002, 11, 351–358. [Google Scholar] [CrossRef]
- Sun, L.; Luo, C.; Liu, J. Hydroxytyrosol induces apoptosis in human colon cancer cells through ROS generation. Food Funct 2014, 5, 1909–1914. [Google Scholar] [CrossRef] [PubMed]
- Long, L.H.; Hoi, A.; Halliwell, B. Instability of, and generation of hydrogen peroxide by, phenolic compounds in cell culture media. Arch. Biochem. Biophys. 2010, 501, 162–169. [Google Scholar] [CrossRef]
- Sakihama, Y.; Cohen, M.F.; Grace, S.C.; Yamasaki, H. Plant phenolic antioxidant and prooxidant activities: Phenolics-induced oxidative damage mediated by metals in plants. Toxicology 2002, 177, 67–80. [Google Scholar] [CrossRef] [PubMed]
- Guichard, C.; Pedruzzi, E.; Fay, M.; Marie, J.C.; Braut-Boucher, F.; Daniel, F.; Grodet, A.; Gougerot-Pocidalo, M.A.; Chastre, E.; Kotelevets, L.; Lizard, G.; Vandewalle, A.; Driss, F.; Ogier-Denis, E. Dihydroxyphenylethanol induces apoptosis by activating serine/threonine protein phosphatase PP2A and promotes the endoplasmic reticulum stress response in human colon carcinoma cells. Carcinogenesis 2006, 27, 1812–1827. [Google Scholar] [CrossRef]
- Luo, C.; Li, Y.; Wang, H.; Cui, Y.; Feng, Z.; Li, H.; Li, Y.; Wang, Y.; Wurtz, K.; Weber, P.; Long, J.; Liu, J. Hydroxytyrosol promotes superoxide production and defects in autophagy leading to anti-proliferation and apoptosis on human prostate cancer cells. Curr. Cancer Drug Targets 2013, 13, 625–639. [Google Scholar] [CrossRef]
- Fabiani, R.; Fuccelli, R.; Pieravanti, F.; De Bartolomeo, A.; Morozzi, G. Production of hydrogen peroxide is responsible for the induction of apoptosis by hydroxytyrosol on HL60 cells. Mol Nutr Food Res 2009, 53, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Fabiani, R.; Sepporta, M.V.; Rosignoli, P.; De Bartolomeo, A.; Crescimanno, M.; Morozzi, G. Anti-proliferative and pro-apoptotic activities of hydroxytyrosol on different tumour cells: The role of extracellular production of hydrogen peroxide. Eur J Nutr 2012, 51, 455–464. [Google Scholar] [CrossRef]
- Shingai, Y.; Fujimoto, A.; Nakamura, M.; Masuda, T. Structure and function of the oxidation products of polyphenols and identification of potent lipoxygenase inhibitors from Fe-catalyzed oxidation of resveratrol. J Agric Food Chem 2011, 59, 8180–8186. [Google Scholar] [CrossRef]
- Chen, C.H.; Lin, W.C.; Kuo, C.N.; Lu, F.J. Role of redox signaling regulation in propyl gallate-induced apoptosis of human leukemia cells. Food Chem Toxicol 2011, 49, 494–501. [Google Scholar] [CrossRef] [PubMed]
- You, B.R.; Park, W.H. The enhancement of propyl gallate-induced apoptosis in HeLa cells by a proteasome inhibitor MG132. Oncol Rep 2011, 25, 871–877. [Google Scholar]
- Han, Y.H.; Park, W.H. Propyl gallate inhibits the growth of HeLa cells via regulating intracellular GSH level. Food Chem Toxicol 2009, 47, 2531–2538. [Google Scholar] [CrossRef]
- Jacobi, H.; Eicke, B.; Witte, I. DNA strand break induction and enhanced cytotoxicity of propyl gallate in the presence of copper(II). Free Radic Biol Med 1998, 24, 972–978. [Google Scholar] [CrossRef] [PubMed]
- Grzesik, M.; Bartosz, G.; Stefaniuk, I.; Pichla, M.; Namiesnik, J.; Sadowska-Bartosz, I. Dietary antioxidants as a source of hydrogen peroxide. Food Chem 2019, 278, 692–699. [Google Scholar] [CrossRef]
- Criddle, D.N.; Gillies, S.; Baumgartner-Wilson, H.K.; Jaffar, M.; Chinje, E.C.; Passmore, S.; Chvanov, M.; Barrow, S.; Gerasimenko, O.V.; Tepikin, A.V.; Sutton, R.; Petersen, O.H. Menadione-induced reactive oxygen species generation via redox cycling promotes apoptosis of murine pancreatic acinar cells. J Biol Chem 2006, 281, 40485–40492. [Google Scholar] [CrossRef]
- Mishra, P.K.; Park, I.; Sharma, N.; Yoo, C.M.; Lee, H.Y.; Rhee, H.W. Enzymatic Recording of Local Hydrogen Peroxide Generation Using Genetically Encodable Enzyme. Anal. Chem. 2022, 94, 14869–14877. [Google Scholar] [CrossRef]
- Loor, G.; Kondapalli, J.; Schriewer, J.M.; Chandel, N.S.; Vanden Hoek, T.L.; Schumacker, P.T. Menadione triggers cell death through ROS-dependent mechanisms involving PARP activation without requiring apoptosis. Free Radic Biol Med 2010, 49, 1925–1936. [Google Scholar] [CrossRef] [PubMed]
- Lasalvia-Prisco, E.; Cucchi, S.; Vazquez, J.; Lasalvia-Galante, E.; Golomar, W.; Gordon, W. Serum markers variation consistent with autoschizis induced by ascorbic acid-menadione in patients with prostate cancer. Med Oncol 2003, 20, 45–52. [Google Scholar] [CrossRef]
- Checker, R.; Patwardhan, R.S.; Sharma, D.; Menon, J.; Thoh, M.; Sandur, S.K.; Sainis, K.B.; Poduval, T.B. Plumbagin, a vitamin K3 analogue, abrogates lipopolysaccharide-induced oxidative stress, inflammation and endotoxic shock via NF-kappaB suppression. Inflammation 2014, 37, 542–554. [Google Scholar] [CrossRef]
- Sand, J.M.; Bin Hafeez, B.; Jamal, M.S.; Witkowsky, O.; Siebers, E.M.; Fischer, J.; Verma, A.K. Plumbagin (5-hydroxy-2-methyl-1,4-naphthoquinone), isolated from Plumbago zeylanica, inhibits ultraviolet radiation-induced development of squamous cell carcinomas. Carcinogenesis 2012, 33, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Klaus, V.; Hartmann, T.; Gambini, J.; Graf, P.; Stahl, W.; Hartwig, A.; Klotz, L.O. 1,4-Naphthoquinones as inducers of oxidative damage and stress signaling in HaCaT human keratinocytes. Arch. Biochem. Biophys. 2010, 496, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Kapur, A.; Beres, T.; Rathi, K.; Nayak, A.P.; Czarnecki, A.; Felder, M.; Gillette, A.; Ericksen, S.S.; Sampene, E.; Skala, M.C.; Barroilhet, L.; Patankar, M.S. Oxidative stress via inhibition of the mitochondrial electron transport and Nrf-2-mediated anti-oxidative response regulate the cytotoxic activity of plumbagin. Sci Rep 2018, 8, 1073. [Google Scholar] [CrossRef] [PubMed]
- Inbaraj, J.J.; Chignell, C.F. Cytotoxic action of juglone and plumbagin: A mechanistic study using HaCaT keratinocytes. Chem. Res. Toxicol. 2004, 17, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Kappus, H. Overview of enzyme systems involved in bio-reduction of drugs and in redox cycling. Biochem. Pharmacol. 1986, 35, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Ollinger, K.; Brunmark, A. Effect of hydroxy substituent position on 1,4-naphthoquinone toxicity to rat hepatocytes. J Biol Chem 1991, 266, 21496–21503. [Google Scholar] [CrossRef] [PubMed]
- Gong, Q.; Hu, J.; Wang, P.; Li, X.; Zhang, X. A comprehensive review on beta-lapachone: Mechanisms, structural modifications, and therapeutic potentials. Eur J Med Chem 2021, 210, 112962. [Google Scholar] [CrossRef] [PubMed]
- Pink, J.J.; Planchon, S.M.; Tagliarino, C.; Varnes, M.E.; Siegel, D.; Boothman, D.A. NAD(P)H:Quinone oxidoreductase activity is the principal determinant of beta-lapachone cytotoxicity. J Biol Chem 2000, 275, 5416–5424. [Google Scholar] [CrossRef] [PubMed]
- Chau, Y.P.; Shiah, S.G.; Don, M.J.; Kuo, M.L. Involvement of hydrogen peroxide in topoisomerase inhibitor beta-lapachone-induced apoptosis and differentiation in human leukemia cells. Free Radic Biol Med 1998, 24, 660–670. [Google Scholar] [CrossRef]
- Gerber, D.E.; Beg, M.S.; Fattah, F.; Frankel, A.E.; Fatunde, O.; Arriaga, Y.; Dowell, J.E.; Bisen, A.; Leff, R.D.; Meek, C.C.; Putnam, W.C.; Kallem, R.R.; Subramaniyan, I.; Dong, Y.; Bolluyt, J.; Sarode, V.; Luo, X.; Xie, Y.; Schwartz, B.; Boothman, D.A. Phase 1 study of ARQ 761, a beta-lapachone analogue that promotes NQO1-mediated programmed cancer cell necrosis. Br. J. Cancer 2018, 119, 928–936. [Google Scholar] [CrossRef]
- Kim, S.; Lee, S.; Cho, J.Y.; Yoon, S.H.; Jang, I.J.; Yu, K.S. Pharmacokinetics and tolerability of MB12066, a beta-lapachone derivative targeting NAD(P)H: Quinone oxidoreductase 1: Two independent, double-blind, placebo-controlled, combined single and multiple ascending dose first-in-human clinical trials. Drug Des Devel Ther 2017, 11, 3187–3195. [Google Scholar] [CrossRef]
- Weiss, R.B. The anthracyclines: Will we ever find a better doxorubicin? Semin Oncol 1992, 19, 670–686. [Google Scholar] [PubMed]
- Minotti, G.; Menna, P.; Salvatorelli, E.; Cairo, G.; Gianni, L. Anthracyclines: Molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol Rev 2004, 56, 185–229. [Google Scholar] [CrossRef] [PubMed]
- Sinha, B.K.; Mimnaugh, E.G.; Rajagopalan, S.; Myers, C.E. Adriamycin activation and oxygen free radical formation in human breast tumor cells: Protective role of glutathione peroxidase in adriamycin resistance. Cancer Res. 1989, 49, 3844–3848. [Google Scholar] [PubMed]
- Ubezio, P.; Civoli, F. Flow cytometric detection of hydrogen peroxide production induced by doxorubicin in cancer cells. Free Radic Biol Med 1994, 16, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Wagner, B.A.; Evig, C.B.; Reszka, K.J.; Buettner, G.R.; Burns, C.P. Doxorubicin increases intracellular hydrogen peroxide in PC3 prostate cancer cells. Arch. Biochem. Biophys. 2005, 440, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Thayer, W.S. Adriamycin stimulated superoxide formation in submitochondrial particles. Chem. Biol. Interact. 1977, 19, 265–278. [Google Scholar] [CrossRef] [PubMed]
- Binaschi, M.; Farinosi, R.; Borgnetto, M.E.; Capranico, G. In vivo site specificity and human isoenzyme selectivity of two topoisomerase II-poisoning anthracyclines. Cancer Res. 2000, 60, 3770–3776. [Google Scholar] [PubMed]
- Skibo, E.B.; Xing, C. Chemistry and DNA alkylation reactions of aziridinyl quinones: Development of an efficient alkylating agent of the phosphate backbone. Biochemistry 1998, 37, 15199–15213. [Google Scholar] [CrossRef]
- Rashid, M.H.; Babu, D.; Siraki, A.G. Interactions of the antioxidant enzymes NAD(P)H: Quinone oxidoreductase 1 (NQO1) and NRH: Quinone oxidoreductase 2 (NQO2) with pharmacological agents, endogenous biochemicals and environmental contaminants. Chem. Biol. Interact. 2021, 345, 109574. [Google Scholar] [CrossRef]
- Avendaño, C.M.J.C. Chapter 11 - Drug Targeting in Anticancer. In Medicinal Chemistry of Anticancer Drugs; Avendaño, C., Menéndez, J.C., Eds.; Elsevier: Amsterdam, 2008; pp. 351–385. [Google Scholar]
- Webb, J.S.C. D.B.; Mowat, J.H.; Patrick, J.B.; Broschard, R.W.; Meyer, W.E.; Williams, R.P.; Wolf, C.F.; Fulmor, William. ; Pidacks, Charles.; Lancaster, J.E., The Structures of Mitomycins A, B and C and Porfiromycin--Part I. J. Am. Chem. Soc. 1962, 84, 3185–3187. [Google Scholar] [CrossRef]
- Tomasz, M. H2O2 generation during the redox cycle of mitomycin C and dna-bound mitomycin C. Chem. Biol. Interact. 1976, 13, 89–97. [Google Scholar] [CrossRef]
- Andrez, J.C. Mitomycins syntheses: A recent update. Beilstein J. Org. Chem. 2009, 5, 33. [Google Scholar] [CrossRef] [PubMed]
- Remers, W.A.; Schepman, C.S. Structure-activity relationships of the mitomycins and certain synthetic analogs. J Med Chem 1974, 17, 729–732. [Google Scholar] [CrossRef] [PubMed]
- Hornemann, U.H. M.J. , Stereochemical Relationship between Mitomycins A, B, and C. J. Org. Chem. 1985, 50, 1301–1302. [Google Scholar] [CrossRef]
- Mizuno, N.S. Effects of streptonigrin on nucleic acid metabolism of tissue culture cells. Biochim. Biophys. Acta 1965, 108, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Cone, R.; Hasan, S.K.; Lown, J.W.; Morgan, A.R. The mechanism of the degradation of DNA by streptonigrin. Can. J. Biochem. 1976, 54, 219–223. [Google Scholar] [CrossRef]
- Bolzan, A.D.; Bianchi, M.S. Genotoxicity of streptonigrin: A review. Mutat Res 2001, 488, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Testoni, M.I.; Bolzan, A.D.; Bianchi, M.S.; Bianchi, N.O. Effects of antioxidants on streptonigrin-induced DNA damage and clastogenesis in CHO cells. Mutat Res 1997, 373, 201–206. [Google Scholar] [CrossRef]
- Chen, Q.; Espey, M.G.; Krishna, M.C.; Mitchell, J.B.; Corpe, C.P.; Buettner, G.R.; Shacter, E.; Levine, M. Pharmacologic ascorbic acid concentrations selectively kill cancer cells: Action as a pro-drug to deliver hydrogen peroxide to tissues. Proc Natl Acad Sci U S A 2005, 102, 13604–13609. [Google Scholar] [CrossRef]
- Chen, Q.; Espey, M.G.; Sun, A.Y.; Lee, J.H.; Krishna, M.C.; Shacter, E.; Choyke, P.L.; Pooput, C.; Kirk, K.L.; Buettner, G.R.; Levine, M. Ascorbate in pharmacologic concentrations selectively generates ascorbate radical and hydrogen peroxide in extracellular fluid in vivo. Proc Natl Acad Sci U S A 2007, 104, 8749–8754. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Espey, M.G.; Sun, A.Y.; Pooput, C.; Kirk, K.L.; Krishna, M.C.; Khosh, D.B.; Drisko, J.; Levine, M. Pharmacologic doses of ascorbate act as a prooxidant and decrease growth of aggressive tumor xenografts in mice. Proc Natl Acad Sci U S A 2008, 105, 11105–11109. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, N.; Creagan, E.; Witzig, T.; Levine, M. Ascorbic Acid in Cancer Treatment: Let the Phoenix Fly. Cancer Cell 2018, 34, 700–706. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Martin, S.M.; Levine, M.; Wagner, B.A.; Buettner, G.R.; Wang, S.H.; Taghiyev, A.F.; Du, C.; Knudson, C.M.; Cullen, J.J. Mechanisms of ascorbate-induced cytotoxicity in pancreatic cancer. Clin. Cancer Res. 2010, 16, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Chapman, J.; Levine, M.; Polireddy, K.; Drisko, J.; Chen, Q. High-dose parenteral ascorbate enhanced chemosensitivity of ovarian cancer and reduced toxicity of chemotherapy. Sci Transl Med 2014, 6, 222ra18. [Google Scholar] [CrossRef] [PubMed]
- Nauman, G.; Gray, J.C.; Parkinson, R.; Levine, M.; Paller, C.J. Systematic Review of Intravenous Ascorbate in Cancer Clinical Trials. Antioxidants (Basel), 2018; 7. [Google Scholar]
- Padayatty, S.J.; Sun, H.; Wang, Y.; Riordan, H.D.; Hewitt, S.M.; Katz, A.; Wesley, R.A.; Levine, M. Vitamin C pharmacokinetics: Implications for oral and intravenous use. Ann. Intern. Med. 2004, 140, 533–537. [Google Scholar] [CrossRef] [PubMed]
- Sawant, R.R.; Vaze, O.; D'Souza, G.G.; Rockwell, K.; Torchilin, V.P. Palmitoyl ascorbate-loaded polymeric micelles: Cancer cell targeting and cytotoxicity. Pharm Res 2011, 28, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Sawant, R.R.; Vaze, O.S.; Wang, T.; D'Souza, G.G.; Rockwell, K.; Gada, K.; Khaw, B.A.; Torchilin, V.P. Palmitoyl ascorbate liposomes and free ascorbic acid: Comparison of anticancer therapeutic effects upon parenteral administration. Pharm Res 2012, 29, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, M.; Strafella, E.; Staffolani, S.; Santarelli, L.; Neuzil, J.; Guerrieri, R. alpha-Tocopheryl succinate promotes selective cell death induced by vitamin K3 in combination with ascorbate. Br. J. Cancer 2010, 102, 1224–1234. [Google Scholar] [CrossRef]
- Wang, L.; Luo, X.; Li, C.; Huang, Y.; Xu, P.; Lloyd-Davies, L.H.; Delplancke, T.; Peng, C.; Gao, R.; Qi, H.; Tong, C.; Baker, P. Triethylenetetramine Synergizes with Pharmacologic Ascorbic Acid in Hydrogen Peroxide Mediated Selective Toxicity to Breast Cancer Cell. Oxid Med Cell Longev 2017, 2017, 3481710. [Google Scholar] [CrossRef]
- Li, J.; Ke, W.; Wang, L.; Huang, M.; Yin, W.; Zhang, P.; Chen, Q.; Ge, Z. Self-sufficing H2O2-responsive nanocarriers through tumor-specific H2O2 production for synergistic oxidation-chemotherapy. J Control Release 2016, 225, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Orvig, C.; Abrams, M.J. Medicinal inorganic chemistry: Introduction. Chem. Rev. 1999, 99, 2201–2204. [Google Scholar] [CrossRef]
- Yaman, M.; Kaya, G.; Yekeler, H. Distribution of trace metal concentrations in paired cancerous and non-cancerous human stomach tissues. World J Gastroenterol 2007, 13, 612–618. [Google Scholar] [CrossRef]
- Lee, J.C.; Son, Y.O.; Pratheeshkumar, P.; Shi, X. Oxidative stress and metal carcinogenesis. Free Radic Biol Med 2012, 53, 742–757. [Google Scholar] [CrossRef]
- Frezza, M.; Hindo, S.; Chen, D.; Davenport, A.; Schmitt, S.; Tomco, D.; Dou, Q.P. Novel metals and metal complexes as platforms for cancer therapy. Curr Pharm Des 2010, 16, 1813–1825. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Fu, L.H.; Qi, C.; Lin, J.; Huang, P. Metal peroxides for cancer treatment. Bioact Mater 2021, 6, 2698–2710. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Li, Y.; Lv, G.; Bu, W. Redox dyshomeostasis strategy for tumor therapy based on nanomaterials chemistry. Chem. Sci. 2022, 13, 2202–2217. [Google Scholar] [CrossRef]
- Dugo, E.B.; Yedjou, C.G.; Stevens, J.J.; Tchounwou, P.B. Therapeutic Potential of Arsenic Trioxide (ATO) in Treatment of Hepatocellular Carcinoma: Role of Oxidative Stress in ATO-Induced Apoptosis. Ann Clin Pathol, 2017; 5. [Google Scholar]
- Chou, W.C.; Jie, C.; Kenedy, A.A.; Jones, R.J.; Trush, M.A.; Dang, C.V. Role of NADPH oxidase in arsenic-induced reactive oxygen species formation and cytotoxicity in myeloid leukemia cells. Proc Natl Acad Sci U S A 2004, 101, 4578–4583. [Google Scholar] [CrossRef]
- Jing, Y.; Dai, J.; Chalmers-Redman, R.M.; Tatton, W.G.; Waxman, S. Arsenic trioxide selectively induces acute promyelocytic leukemia cell apoptosis via a hydrogen peroxide-dependent pathway. Blood 1999, 94, 2102–2111. [Google Scholar] [CrossRef]
- Ziental, D.; Czarczynska-Goslinska, B.; Mlynarczyk, D.T.; Glowacka-Sobotta, A.; Stanisz, B.; Goslinski, T.; Sobotta, L. Titanium Dioxide Nanoparticles: Prospects and Applications in Medicine. Nanomaterials (Basel), 2020; 10. [Google Scholar]
- Kormann, C.; Bahnemann, D.W.; Hoffmann, M.R. Photocatalytic production of hydrogen peroxides and organic peroxides in aqueous suspensions of titanium dioxide, zinc oxide, and desert sand. Environ Sci Technol 1988, 22, 798–806. [Google Scholar] [CrossRef]
- Murali, M.; Kalegowda, N.; Gowtham, H.G.; Ansari, M.A.; Alomary, M.N.; Alghamdi, S.; Shilpa, N.; Singh, S.B.; Thriveni, M.C.; Aiyaz, M.; Angaswamy, N.; Lakshmidevi, N.; Adil, S.F.; Hatshan, M.R.; Amruthesh, K.N. Plant-Mediated Zinc Oxide Nanoparticles: Advances in the New Millennium towards Understanding Their Therapeutic Role in Biomedical Applications. Pharmaceutics, 2021; 13. [Google Scholar]
- Singh, T.A.; Das, J.; Sil, P.C. Zinc oxide nanoparticles: A comprehensive review on its synthesis, anticancer and drug delivery applications as well as health risks. Adv. Colloid Interface Sci. 2020, 286, 102317. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.S.; Wang, J.F.; Song, J.; Liu, Y.; Zhu, G.; Dai, Y.; Shen, Z.; Tian, R.; Song, J.; Wang, Z.; Tang, W.; Yu, G.; Zhou, Z.; Yang, Z.; Huang, T.; Niu, G.; Yang, H.H.; Chen, Z.Y.; Chen, X. Cooperation of endogenous and exogenous reactive oxygen species induced by zinc peroxide nanoparticles to enhance oxidative stress-based cancer therapy. Theranostics 2019, 9, 7200–7209. [Google Scholar] [CrossRef] [PubMed]
- Mbugua, S.N. Targeting Tumor Microenvironment by Metal Peroxide Nanoparticles in Cancer Therapy. Bioinorg. Chem. Appl. 2022, 2022, 5041399. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Jin, Y.; Ge, K.; Li, Z.; Liu, H.; Dai, X.; Zhang, Y.; Chen, S.; Liang, X.; Zhang, J. Self-Supply of O(2) and H(2)O(2) by a Nanocatalytic Medicine to Enhance Combined Chemo/Chemodynamic Therapy. Adv Sci (Weinh) 2019, 6, 1902137. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.-S.; Huang, T.; Song, J.; Ou, X.-Y.; Wang, Z.; Deng, H.; Tian, R.; Liu, Y.; Wang, J.-F.; Liu, Y.; Yu, G.; Zhou, Z.; Wang, S.; Niu, G.; Yang, H.-H.; Chen, X. Synthesis of Copper Peroxide Nanodots for H2O2 Self-Supplying Chemodynamic Therapy. Journal of the American Chemical Society 2019, 141, 9937–9945. [Google Scholar] [CrossRef] [PubMed]
- Mathe, G.; Schweisguth, O.; Schneider, M.; Amiel, J.L.; Berumen, L.; Brule, G.; Cattan, A.; Schwarzenberg, L. Methyl-Hydrazine in Treatment of Hodgkin's Disease and Various Forms of Haematosarcoma and Leukaemia. Lancet 1963, 2, 1077–1080. [Google Scholar] [CrossRef] [PubMed]
- Berneis, K.; Kofler, M.; Bollag, W.; Kaiser, A.; Langemann, A. The degradation of deoxyribonucleic acid by new tumour inhibiting compounds: The intermediate formation of hydrogen peroxide. Experientia 1963, 19, 132–133. [Google Scholar] [CrossRef]
- Berneis, K.; Kofler, M.; Bollag, W. The influence of chelating agents on the prooxidative effect of a hydrogen peroxide producing methylhydrazine compound. Experientia 1964, 20, 73–74. [Google Scholar] [CrossRef] [PubMed]
- Devita, V.T. Jr.; Serpick, A.A.; Carbone, P.P. Combination chemotherapy in the treatment of advanced Hodgkin's disease. Ann. Intern. Med. 1970, 73, 881–895. [Google Scholar] [CrossRef]
- DeVita, V.T. Jr.; Simon, R.M.; Hubbard, S.M.; Young, R.C.; Berard, C.W.; Moxley, J.H. 3rd; Frei, E. 3rd; Carbone, P.P.; Canellos, G.P. Curability of advanced Hodgkin's disease with chemotherapy. Long-term follow-up of MOPP-treated patients at the National Cancer Institute. Ann. Intern. Med. 1980, 92, 587–595. [Google Scholar] [CrossRef]
- Singh, S.P.; Gao, Y.; Singh, L.D.; Kunapuli, S.P.; Ravindra, R. Role of microtubules in glucose uptake by C6 glioma cells. Pharmacol Toxicol 1998, 83, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Alexandre, J.; Batteux, F.; Nicco, C.; Chereau, C.; Laurent, A.; Guillevin, L.; Weill, B.; Goldwasser, F. Accumulation of hydrogen peroxide is an early and crucial step for paclitaxel-induced cancer cell death both in vitro and in vivo. Int J Cancer 2006, 119, 41–48. [Google Scholar] [CrossRef]
- Babior, B.M. NADPH oxidase. Curr Opin Immunol 2004, 16, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Diebold, B.A.; Fowler, B.; Lu, J.; Dinauer, M.C.; Bokoch, G.M. Antagonistic cross-talk between Rac and Cdc42 GTPases regulates generation of reactive oxygen species. J Biol Chem 2004, 279, 28136–28142. [Google Scholar] [CrossRef] [PubMed]
- Zang, M.; Waelde, C.A.; Xiang, X.; Rana, A.; Wen, R.; Luo, Z. Microtubule integrity regulates Pak leading to Ras-independent activation of Raf-1. insights into mechanisms of Raf-1 activation. J Biol Chem 2001, 276, 25157–25165. [Google Scholar] [CrossRef] [PubMed]
- Hadzic, T.; Aykin-Burns, N.; Zhu, Y.; Coleman, M.C.; Leick, K.; Jacobson, G.M.; Spitz, D.R. Paclitaxel combined with inhibitors of glucose and hydroperoxide metabolism enhances breast cancer cell killing via H2O2-mediated oxidative stress. Free Radic Biol Med 2010, 48, 1024–1033. [Google Scholar] [CrossRef]
- Magda, D.; Lepp, C.; Gerasimchuk, N.; Lee, I.; Sessler, J.L.; Lin, A.; Biaglow, J.E.; Miller, R.A. Redox cycling by motexafin gadolinium enhances cellular response to ionizing radiation by forming reactive oxygen species. Int J Radiat Oncol Biol Phys 2001, 51, 1025–1036. [Google Scholar] [CrossRef]
- Xu, S.; Zakian, K.; Thaler, H.; Matei, C.; Alfieri, A.; Chen, Y.; Koutcher, J.A. Effects of Motexafin gadolinium on tumor metabolism and radiation sensitivity. Int J Radiat Oncol Biol Phys 2001, 49, 1381–1390. [Google Scholar] [CrossRef]
- Peng, X. Ali, T., Combination therapy for cancer using ROS-activated prodrugs and ROS-amplifying therapeutics. Patent Application 2024, US. provisional patent No. 63/480 347; (Atty. File No. 020871-0011-WO01),
Figure 1.
The redox imbalance of cancer cells and its therapeutic potential. The elevated levels of H2O2 in cancer cells trigger a redox adaptation response which further increase their antioxidant capacity to maintain H2O2 levels near the cytotoxic threshold and allow selectively targeting cancer cells by prooxidants that push H2O2 levels beyond the threshold and inducing cell death.
Figure 1.
The redox imbalance of cancer cells and its therapeutic potential. The elevated levels of H2O2 in cancer cells trigger a redox adaptation response which further increase their antioxidant capacity to maintain H2O2 levels near the cytotoxic threshold and allow selectively targeting cancer cells by prooxidants that push H2O2 levels beyond the threshold and inducing cell death.
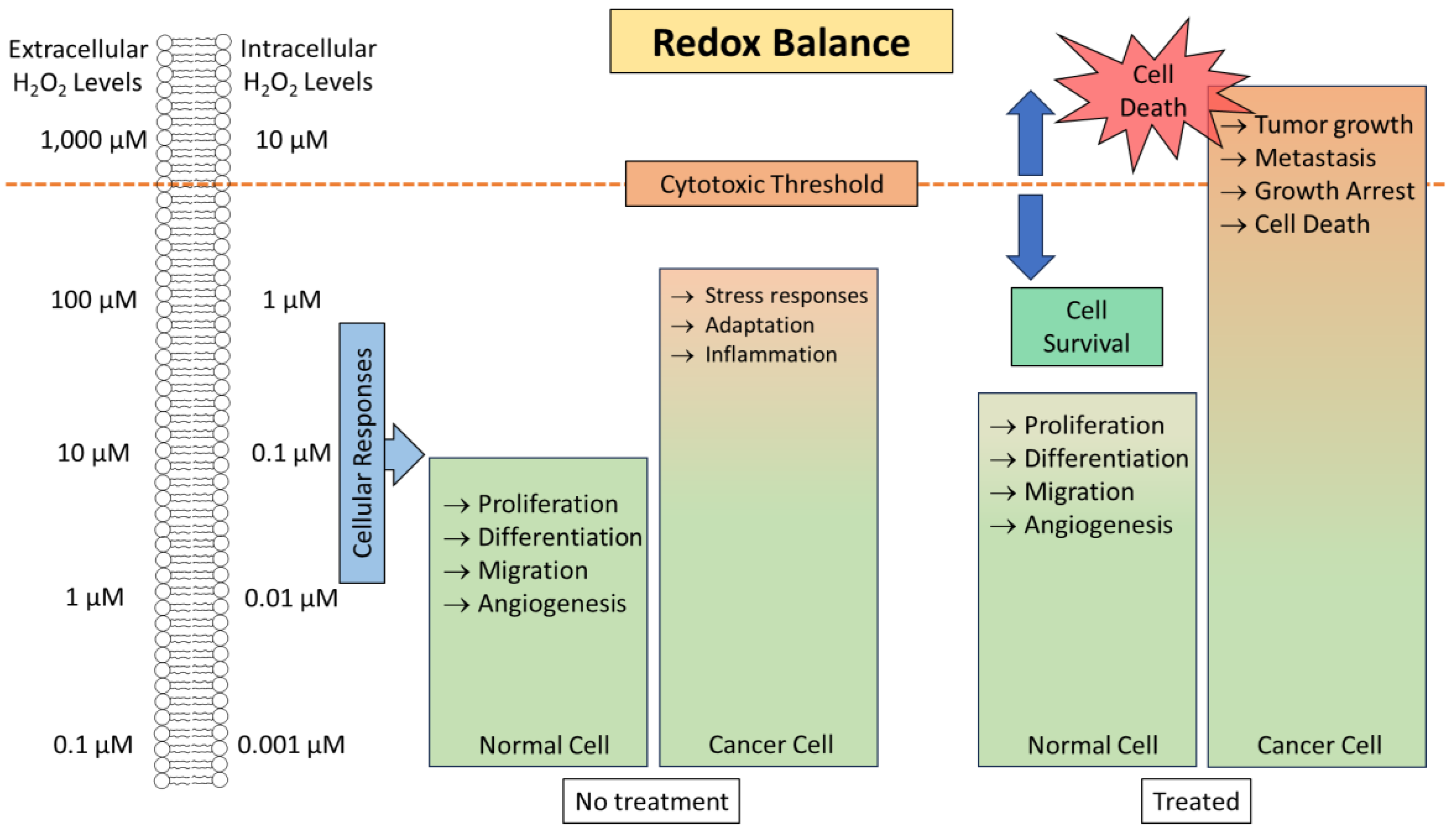
Figure 2.
Three major pathways for cellular H2O2 production: (1) NADPH catalyzed conversion of O2 to superoxide radical anion (O2•−), followed by transformation of O2•− to H2O2 via SOD; (2) Involvement of the mitochondrial respiratory chain, where cytochrome complex generates O2•−; (3) H2O2 production mediated by the oxidases and endoplasmic reticulum.
Figure 2.
Three major pathways for cellular H2O2 production: (1) NADPH catalyzed conversion of O2 to superoxide radical anion (O2•−), followed by transformation of O2•− to H2O2 via SOD; (2) Involvement of the mitochondrial respiratory chain, where cytochrome complex generates O2•−; (3) H2O2 production mediated by the oxidases and endoplasmic reticulum.
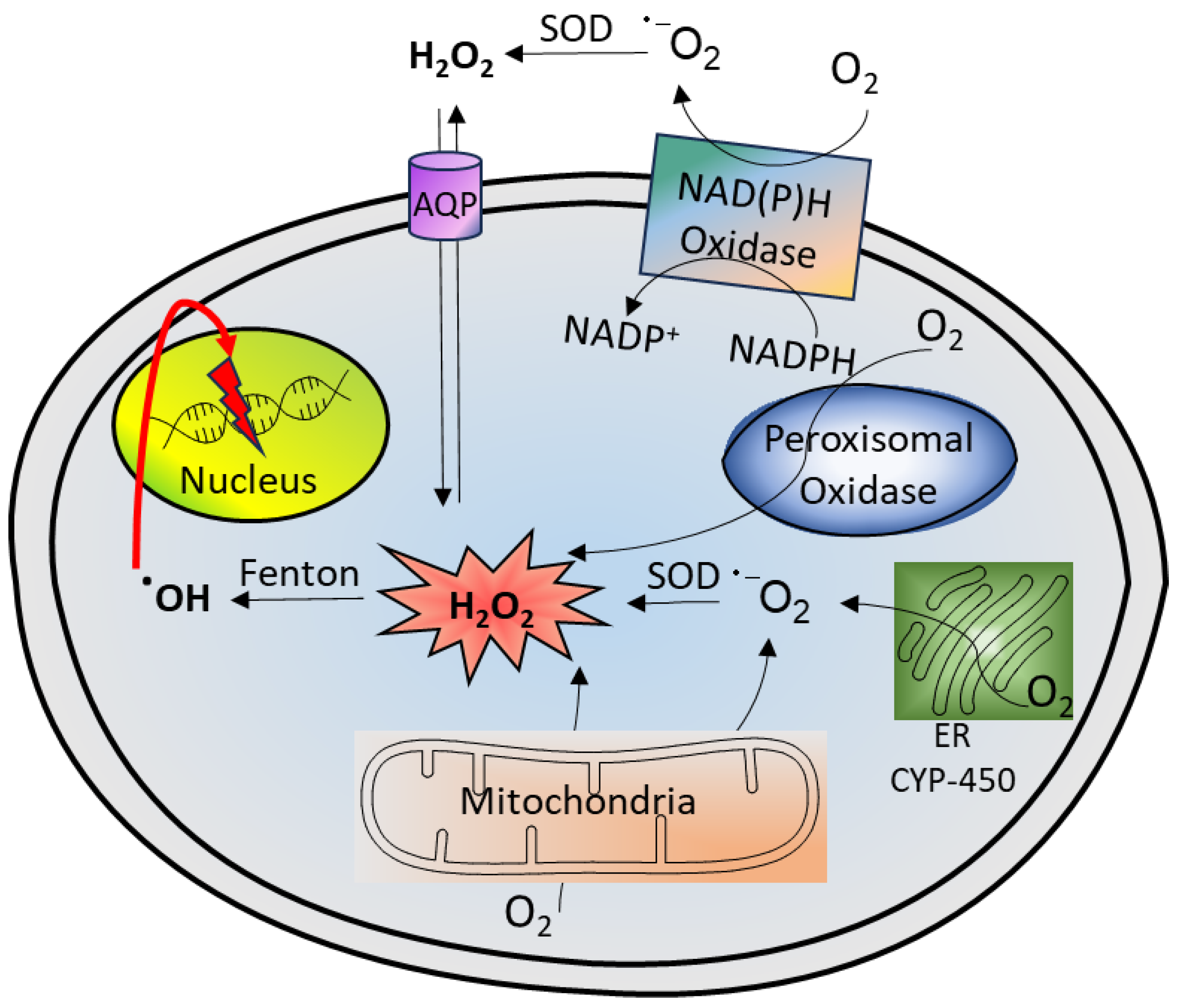
Figure 3.
Damaging effects of H2O2 to biomolecules via several biochemical reactions: A) direct oxidation of the thiol groups by H2O2; B) direct lipid oxidation by H2O2; C) conversion of H2O2 to highly reactive •OH via the Fenton-like reaction, which induces sugar phosphate backbone cleavage, nucleobase, or protein modification.
Figure 3.
Damaging effects of H2O2 to biomolecules via several biochemical reactions: A) direct oxidation of the thiol groups by H2O2; B) direct lipid oxidation by H2O2; C) conversion of H2O2 to highly reactive •OH via the Fenton-like reaction, which induces sugar phosphate backbone cleavage, nucleobase, or protein modification.
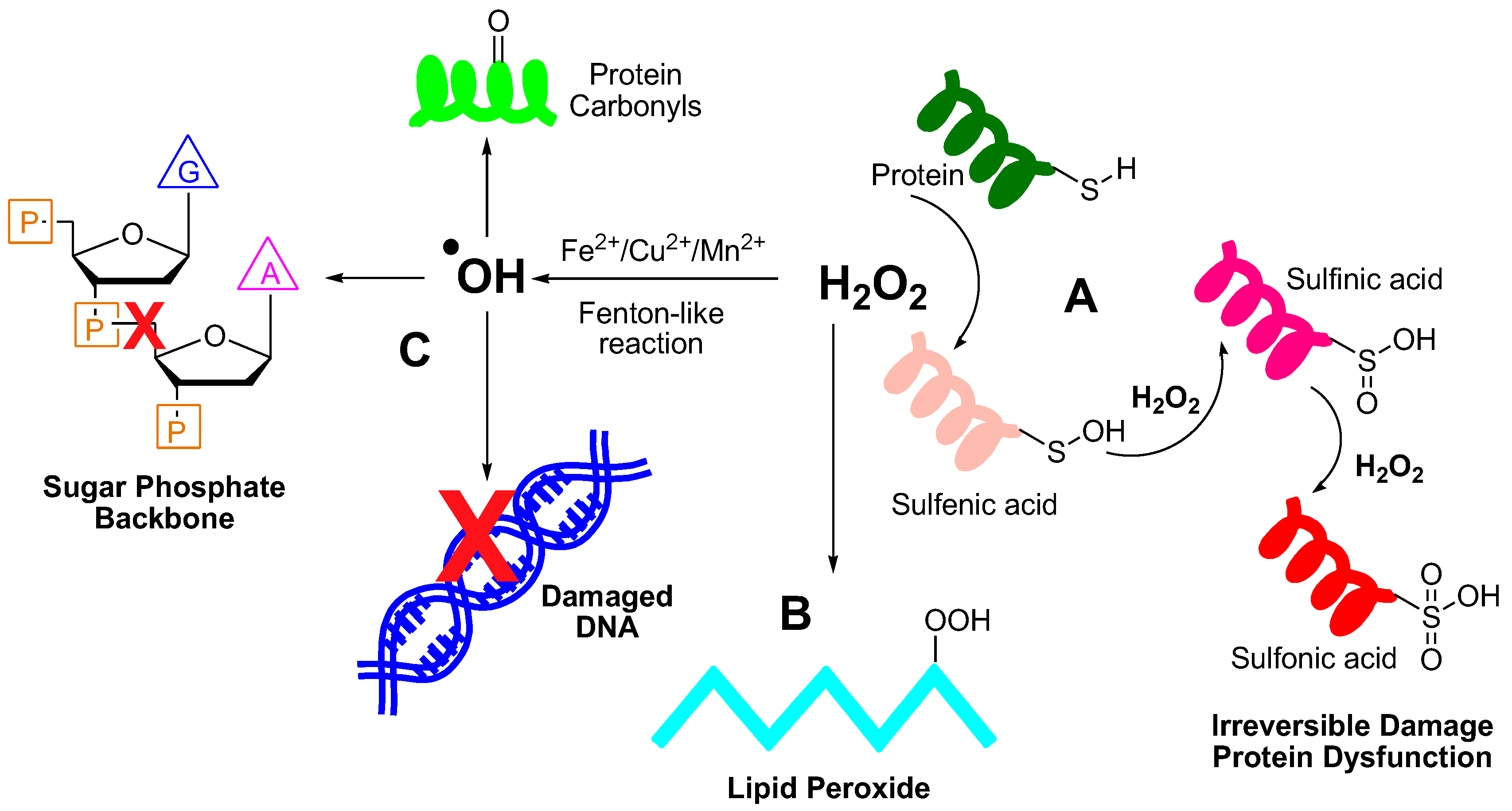
Figure 4.
Strategies for H2O2-induction in cancer cells: Direct approaches include autooxidation, redox cycling, and metal ion interactions whereas indirect approaches include inhibitors of antioxidants defense system such as SOD, GSH, and Catalase and inhibitors of cellular organelles responsible for Redox balance such as mitochondria and endoplasmic reticulum.
Figure 4.
Strategies for H2O2-induction in cancer cells: Direct approaches include autooxidation, redox cycling, and metal ion interactions whereas indirect approaches include inhibitors of antioxidants defense system such as SOD, GSH, and Catalase and inhibitors of cellular organelles responsible for Redox balance such as mitochondria and endoplasmic reticulum.
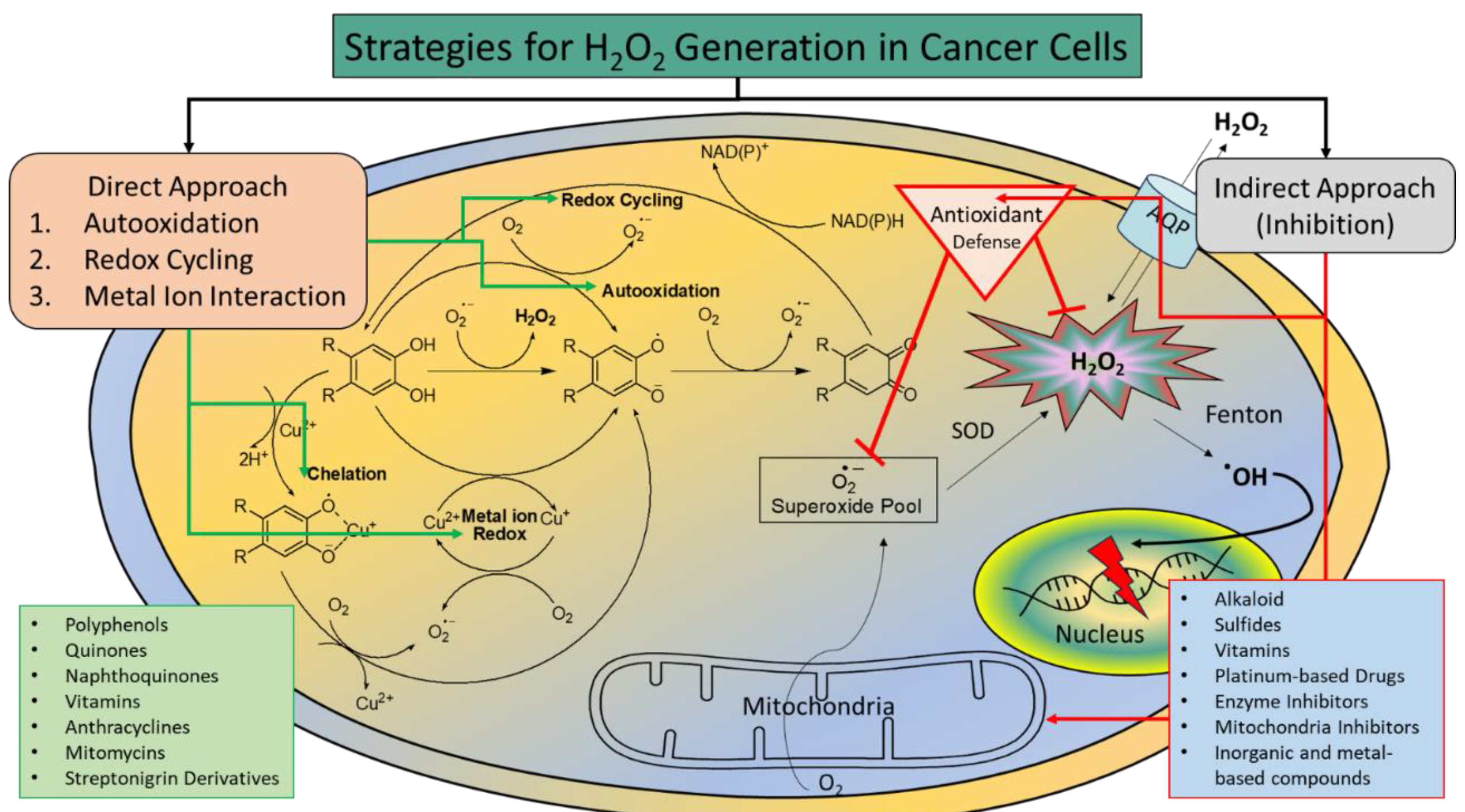
Figure 5.
Potential mechanisms for H2O2 generation induced by compounds containing phenolic groups.
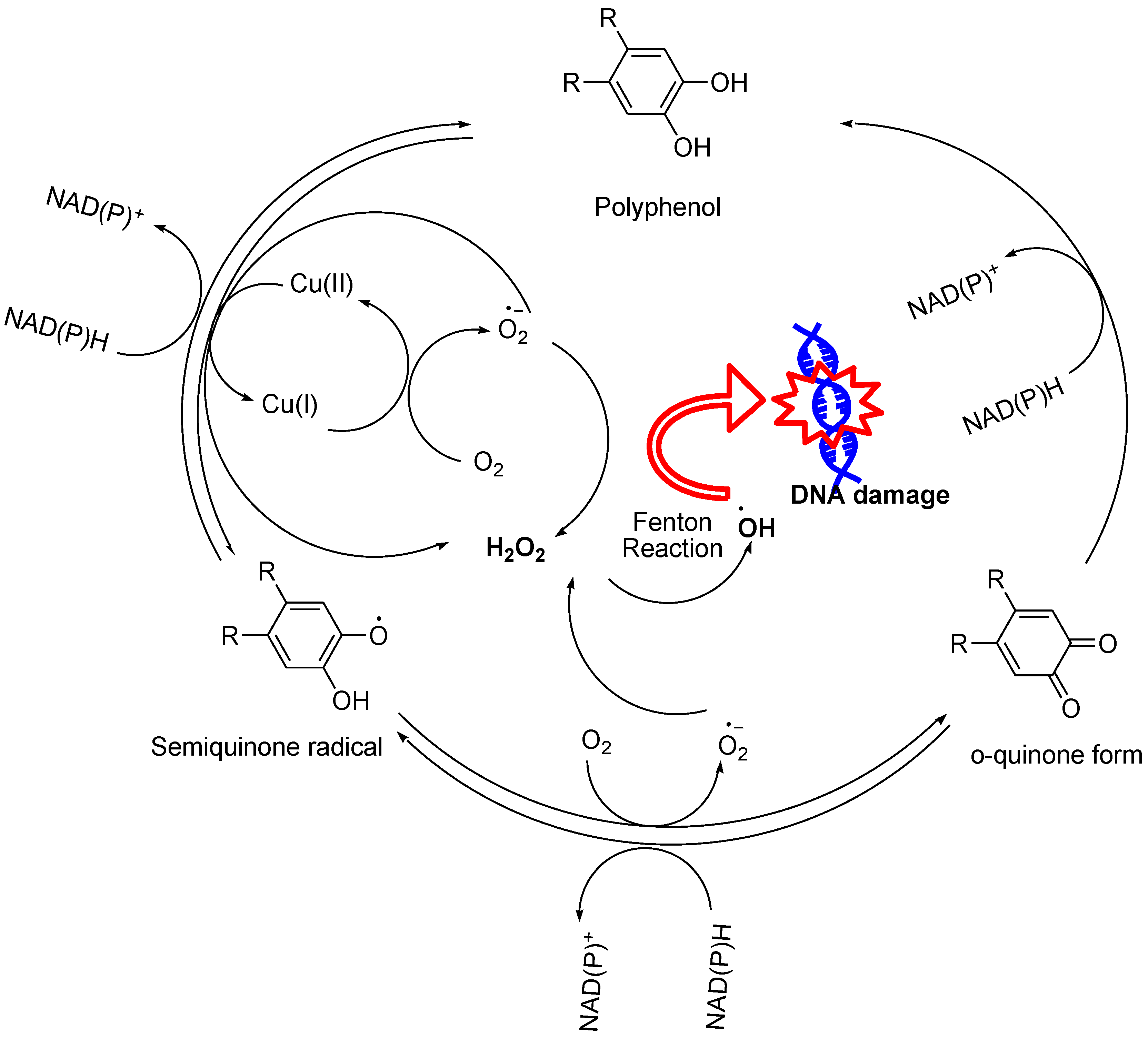
Figure 6.
Classification of Prooxidative Flavonoids.
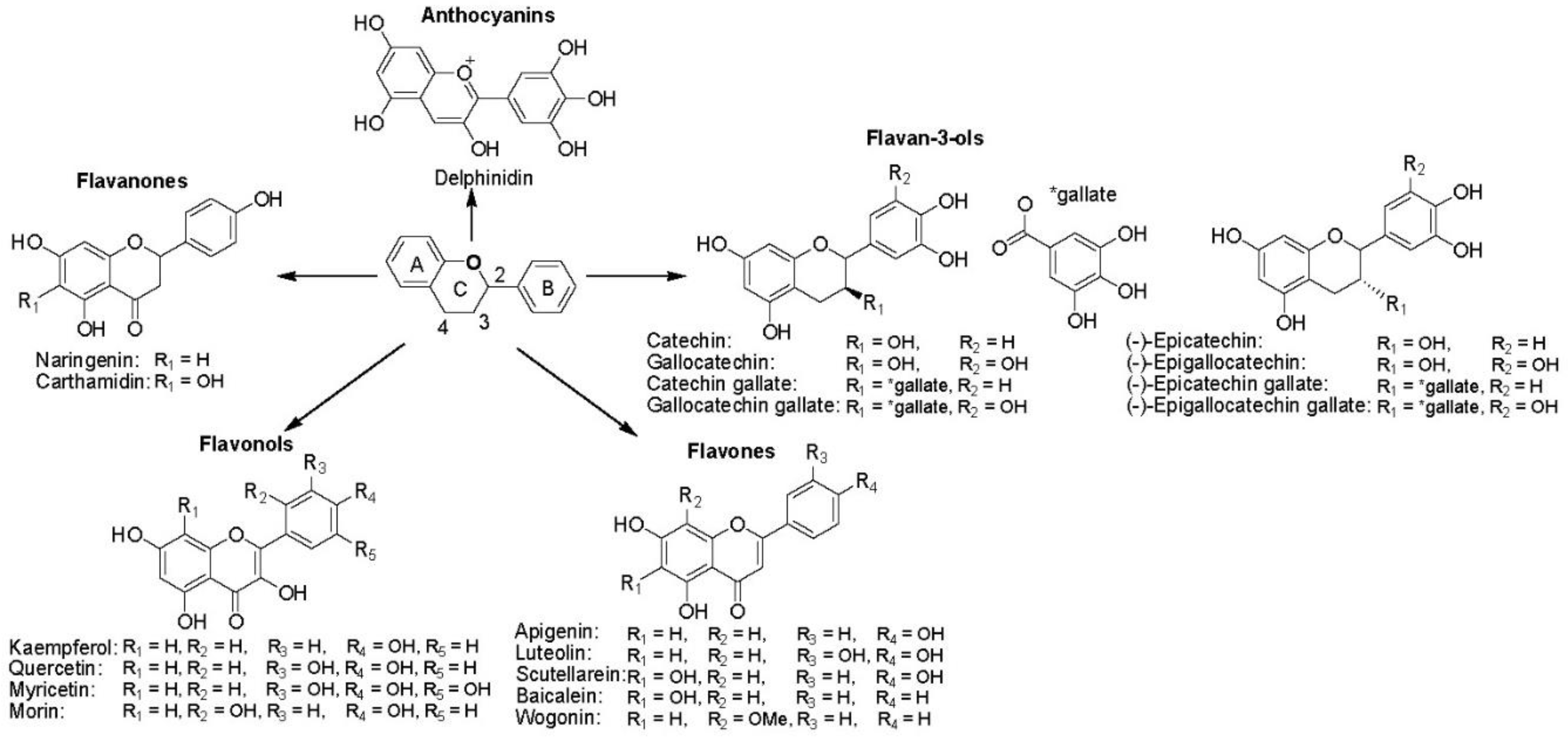
Figure 8.
Polyphenols reported for their metal ion interactions to generate H2O2.
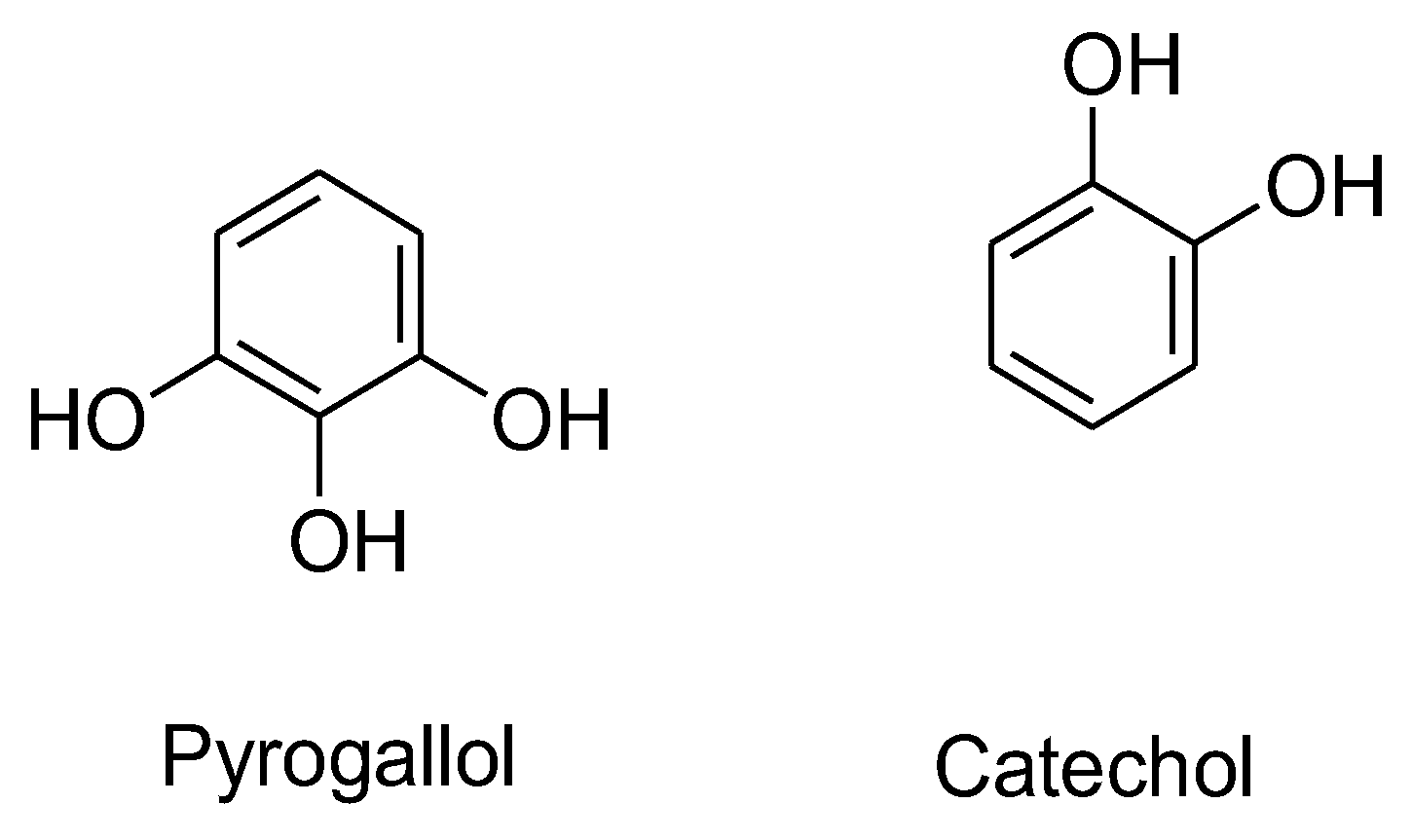
Figure 9.
Mechanism of metal mediated H2O2 generation by Caffeic acid (CA).
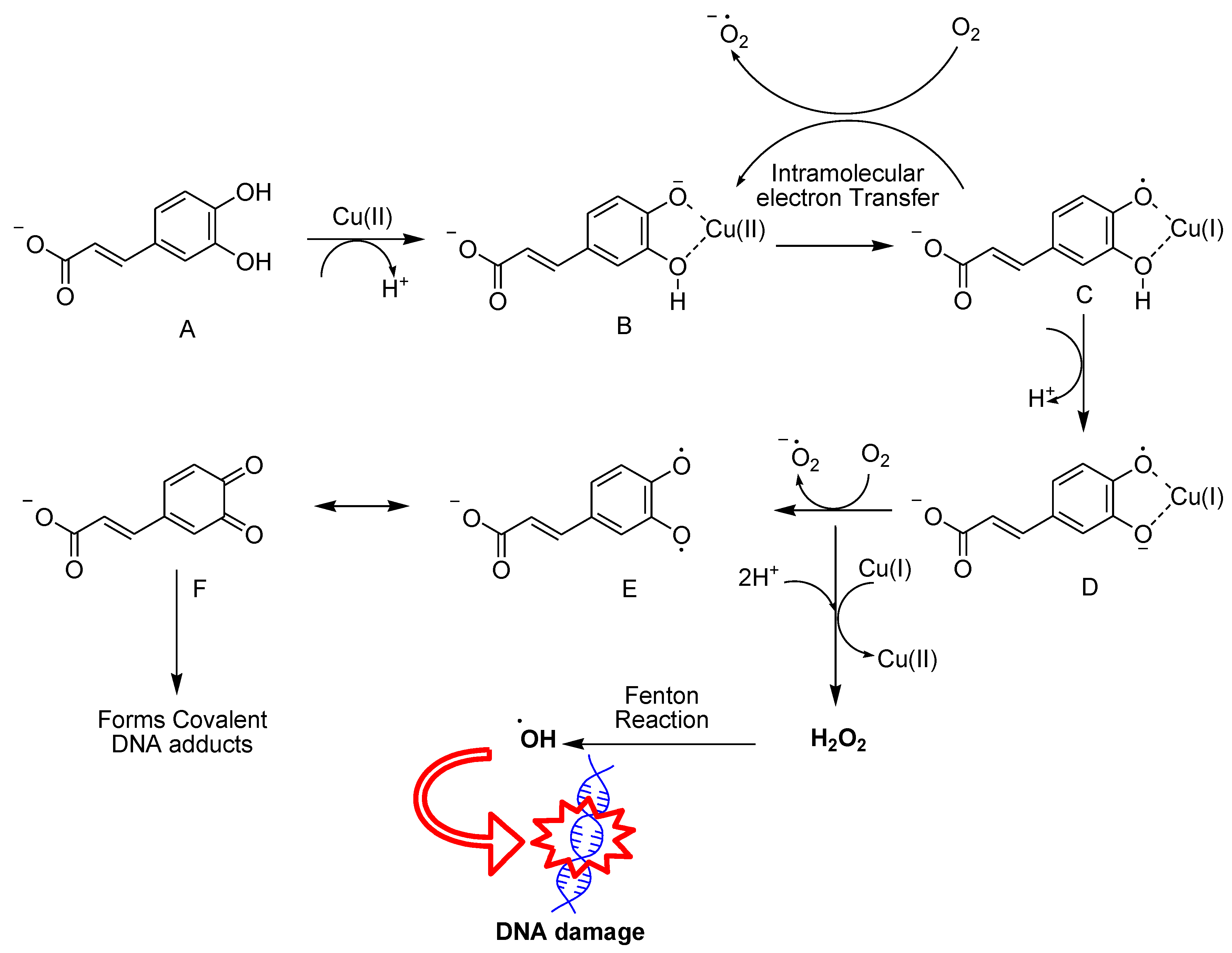
Figure 10.
Structures of compounds containing quinone moiety that can induce H2O2 production via autooxidation and redox cycling.
Figure 10.
Structures of compounds containing quinone moiety that can induce H2O2 production via autooxidation and redox cycling.
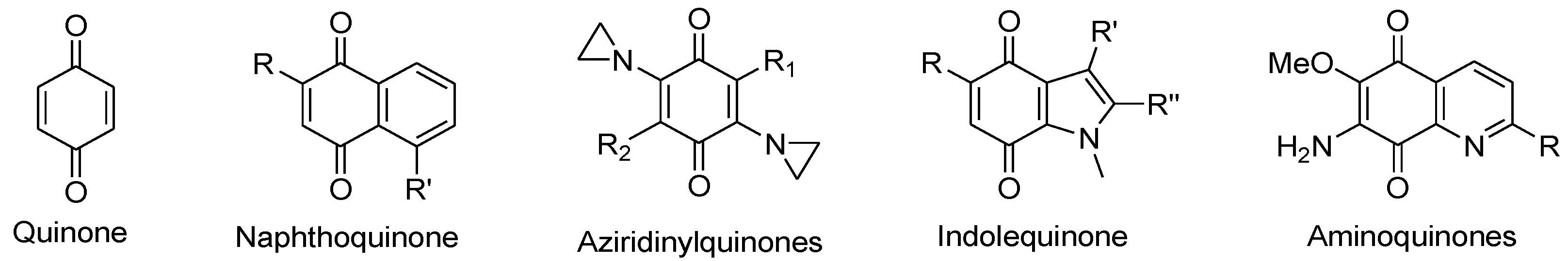
Figure 11.
Mechanism of H2O2 generation by compounds containing quinone moiety that can induce H2O2 production via autooxidation and redox cycling mechanism.
Figure 11.
Mechanism of H2O2 generation by compounds containing quinone moiety that can induce H2O2 production via autooxidation and redox cycling mechanism.
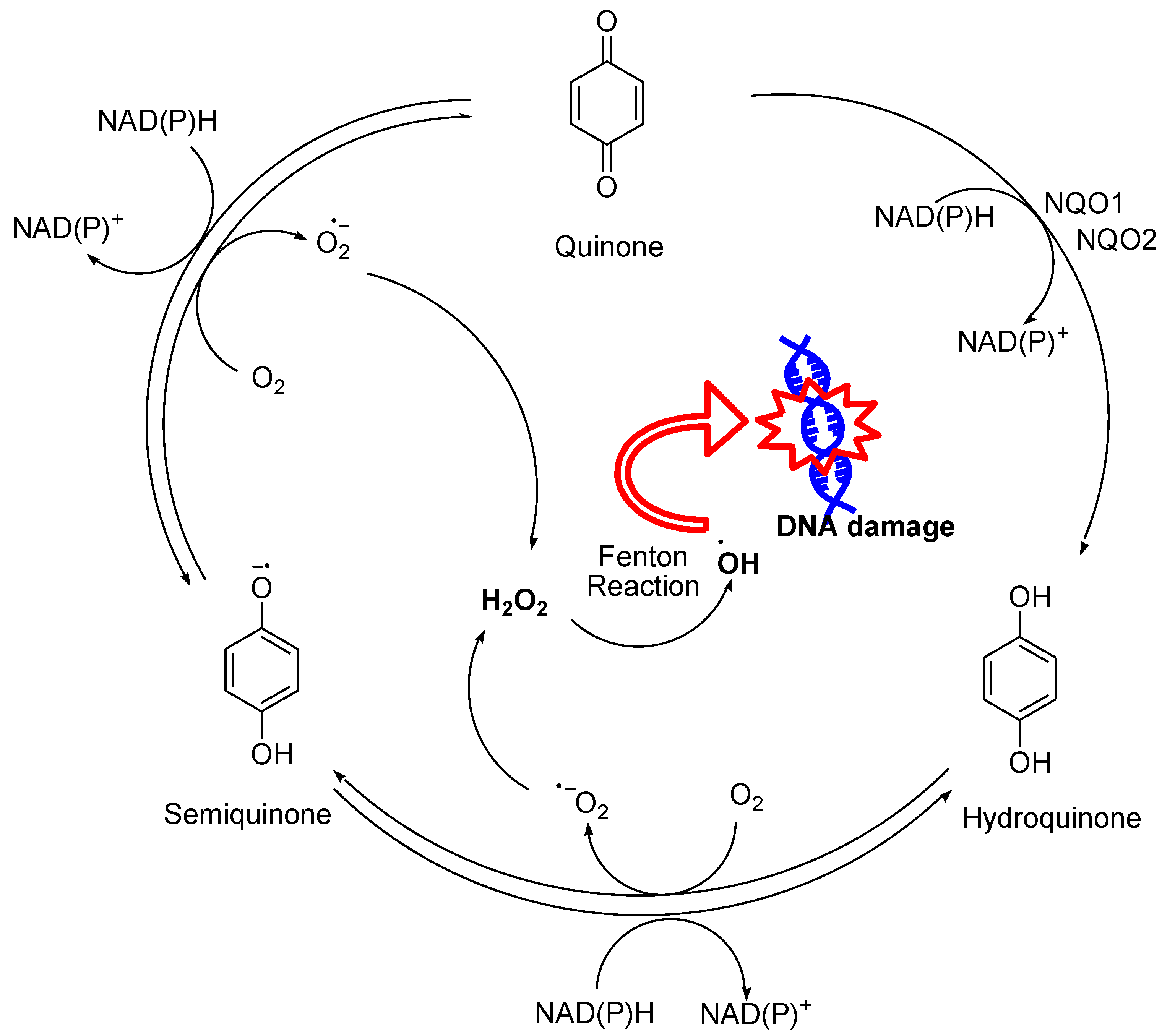
Figure 12.
Structures of compounds containing naphthoquinone moiety that can induce H2O2 production through redox cycling.
Figure 12.
Structures of compounds containing naphthoquinone moiety that can induce H2O2 production through redox cycling.
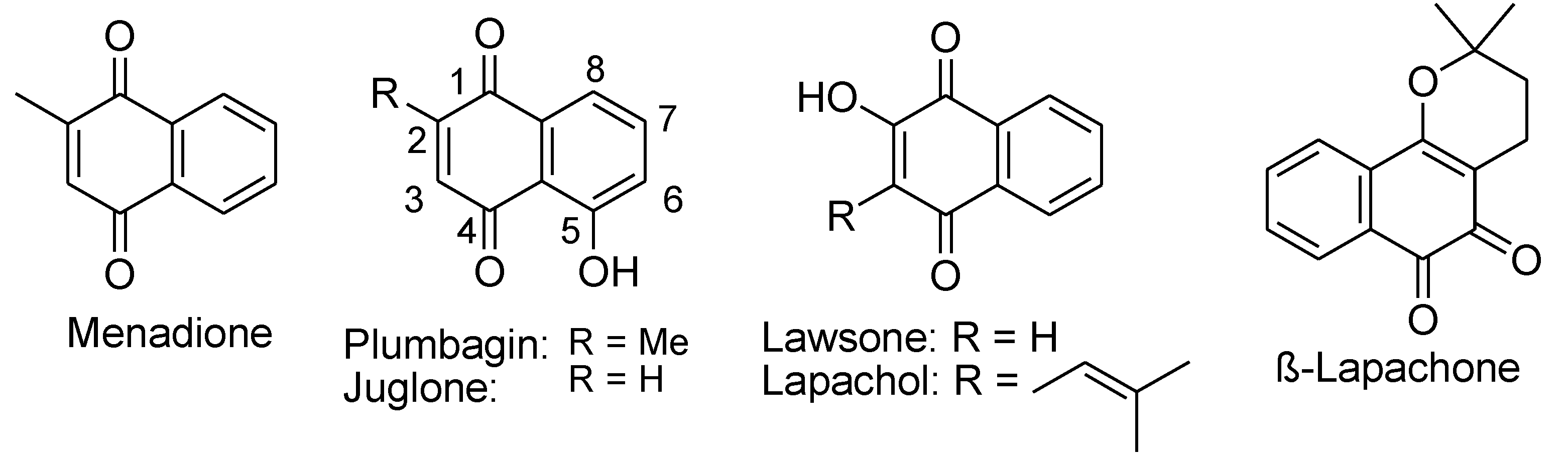
Figure 13.
Two primary mechanisms for the cytotoxic effects of PL: A) One-electron reduction of the quinone moiety generates semiquinone radicals that were engaged in redox cycling, generating both superoxide anions and H2O2 under aerobic conditions; B) Direct interaction of the quinone moiety with thiol groups present in proteins and GSH causes the depletion of the reducing biomolecules and ROS amplification.
Figure 13.
Two primary mechanisms for the cytotoxic effects of PL: A) One-electron reduction of the quinone moiety generates semiquinone radicals that were engaged in redox cycling, generating both superoxide anions and H2O2 under aerobic conditions; B) Direct interaction of the quinone moiety with thiol groups present in proteins and GSH causes the depletion of the reducing biomolecules and ROS amplification.
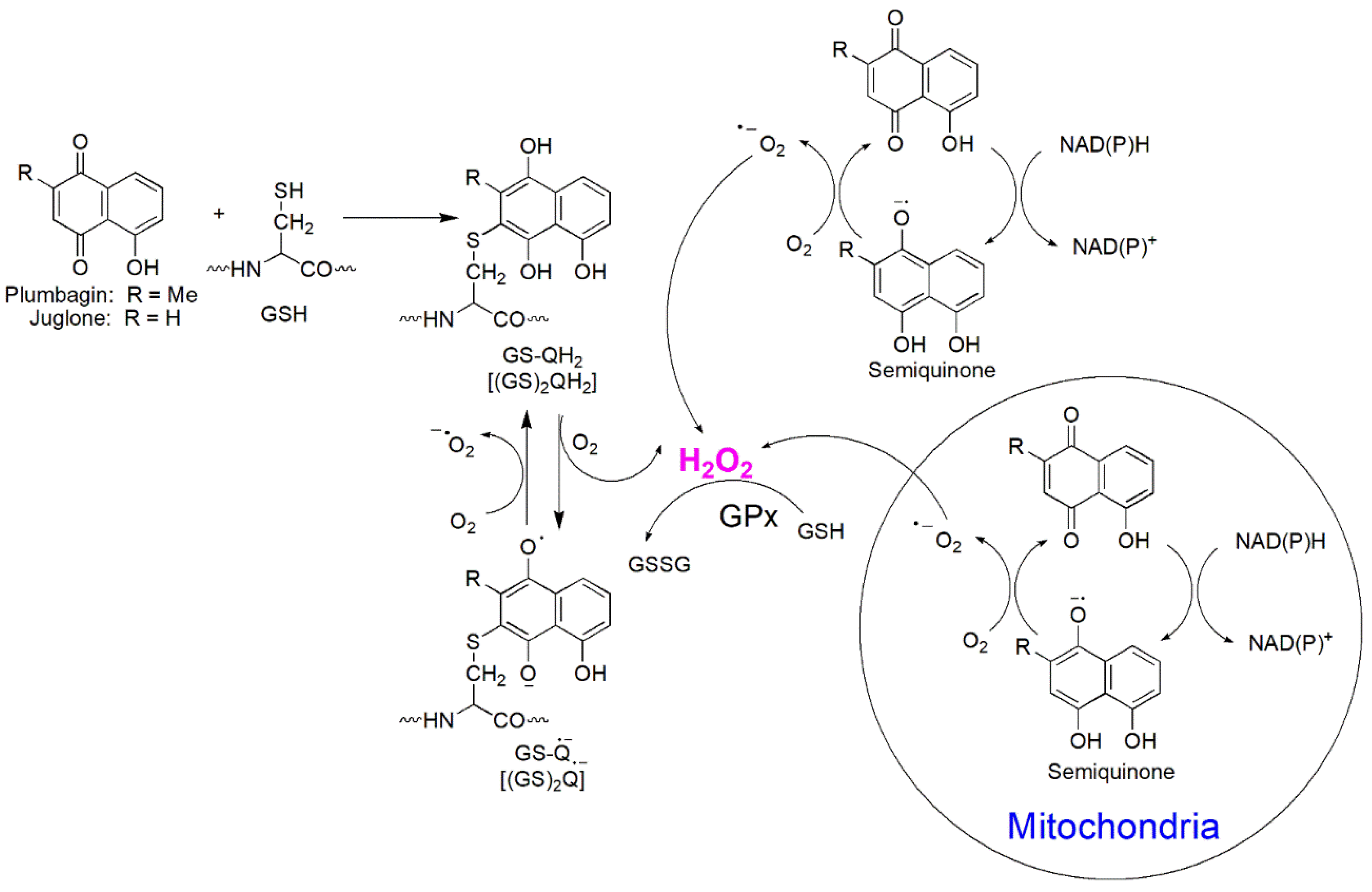
Figure 14.
Proposed mechanism detailing the conversion of β-lapachone into its semiquinone and hydroquinone forms through redox cycling, leading to the generation of H2O2.
Figure 14.
Proposed mechanism detailing the conversion of β-lapachone into its semiquinone and hydroquinone forms through redox cycling, leading to the generation of H2O2.
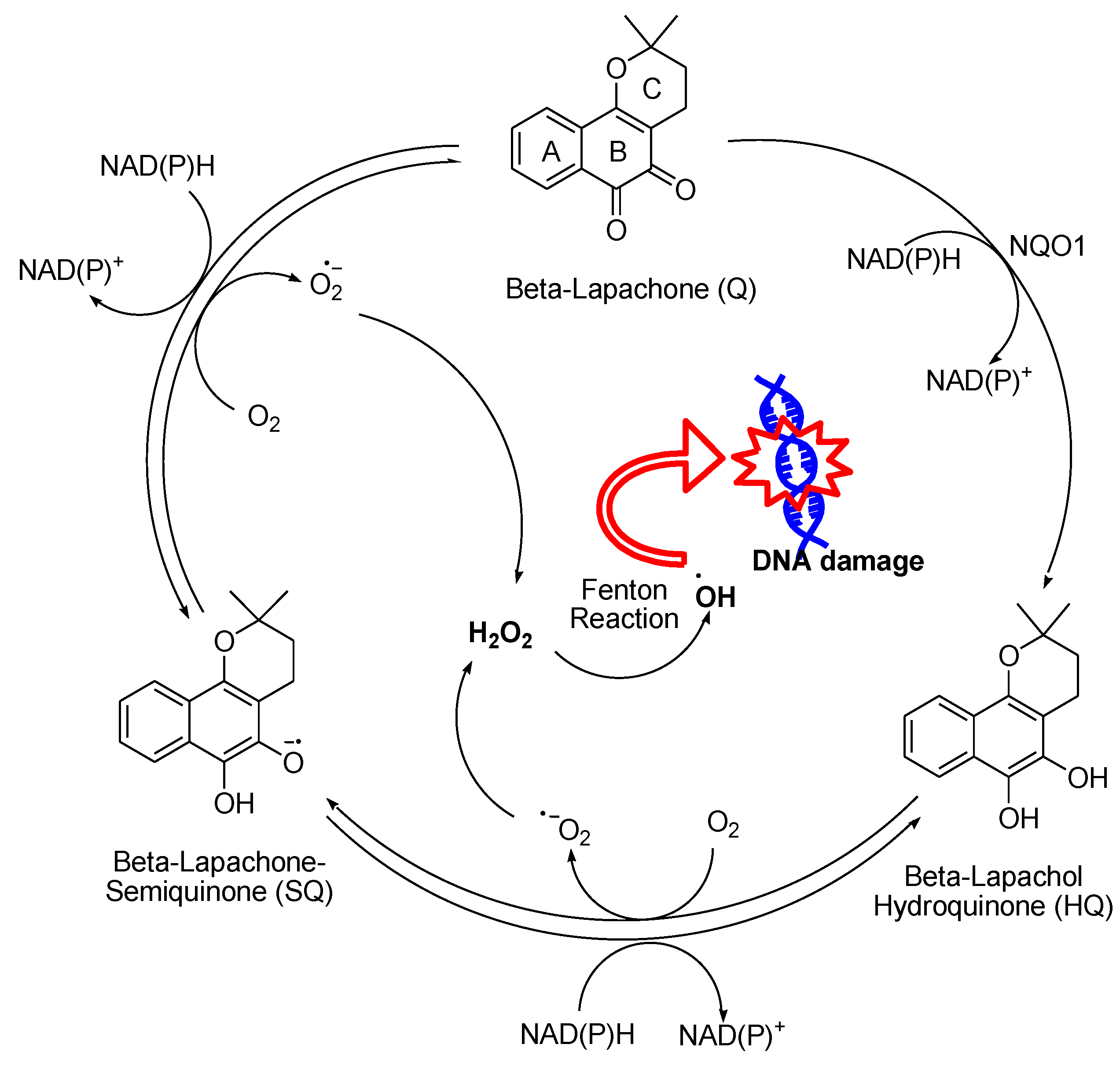
Figure 15.
Structures of anthracyclines commonly used for cancer treatment.
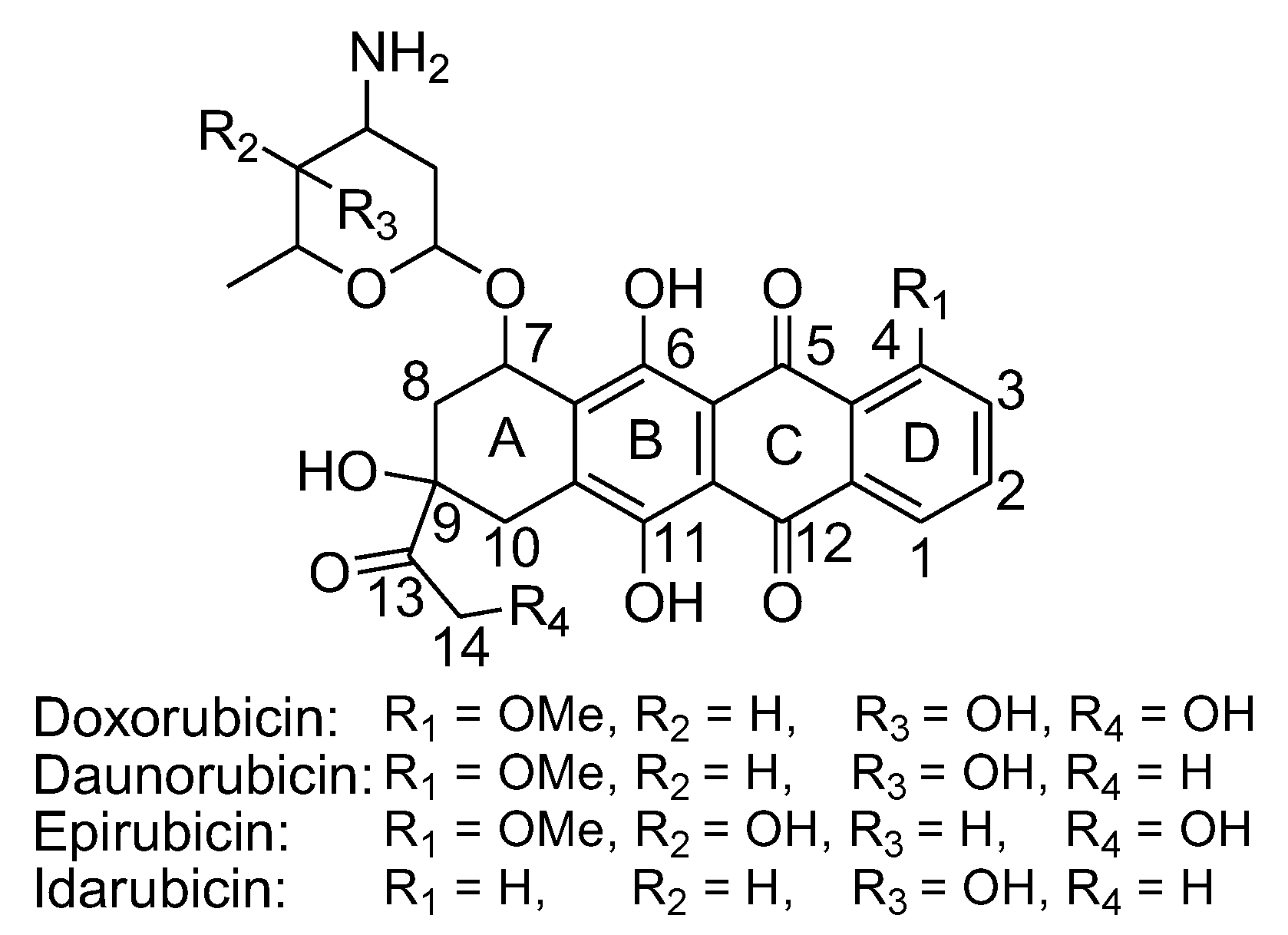
Figure 16.
Two plausible mechanisms for ROS production induced by anthracyclines: (I) one-electron reduction of ring C generates a semiquinone radical, which triggers redox cycling catalyzed by NAD(P)H-oxidoreductases generating O2•− and H2O2; (II) Reductive deglycosidation forms 7-deoxyaglycone with increased lipid solubility that intercalates into membranes producing site-specific ROS.
Figure 16.
Two plausible mechanisms for ROS production induced by anthracyclines: (I) one-electron reduction of ring C generates a semiquinone radical, which triggers redox cycling catalyzed by NAD(P)H-oxidoreductases generating O2•− and H2O2; (II) Reductive deglycosidation forms 7-deoxyaglycone with increased lipid solubility that intercalates into membranes producing site-specific ROS.
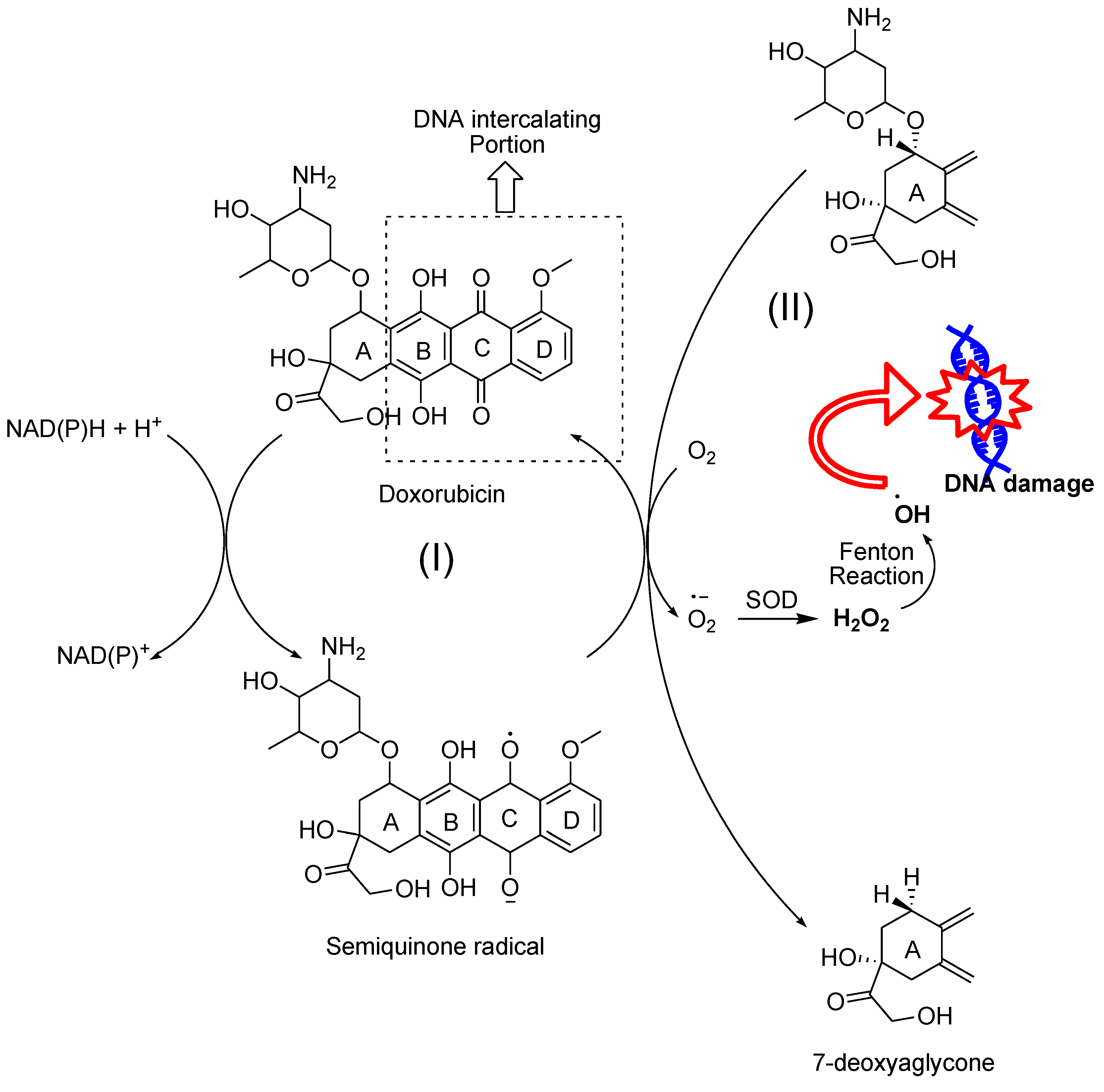
Figure 17.
Structures of aziridinylquinones with an aziridine ring attached to a quinone group.
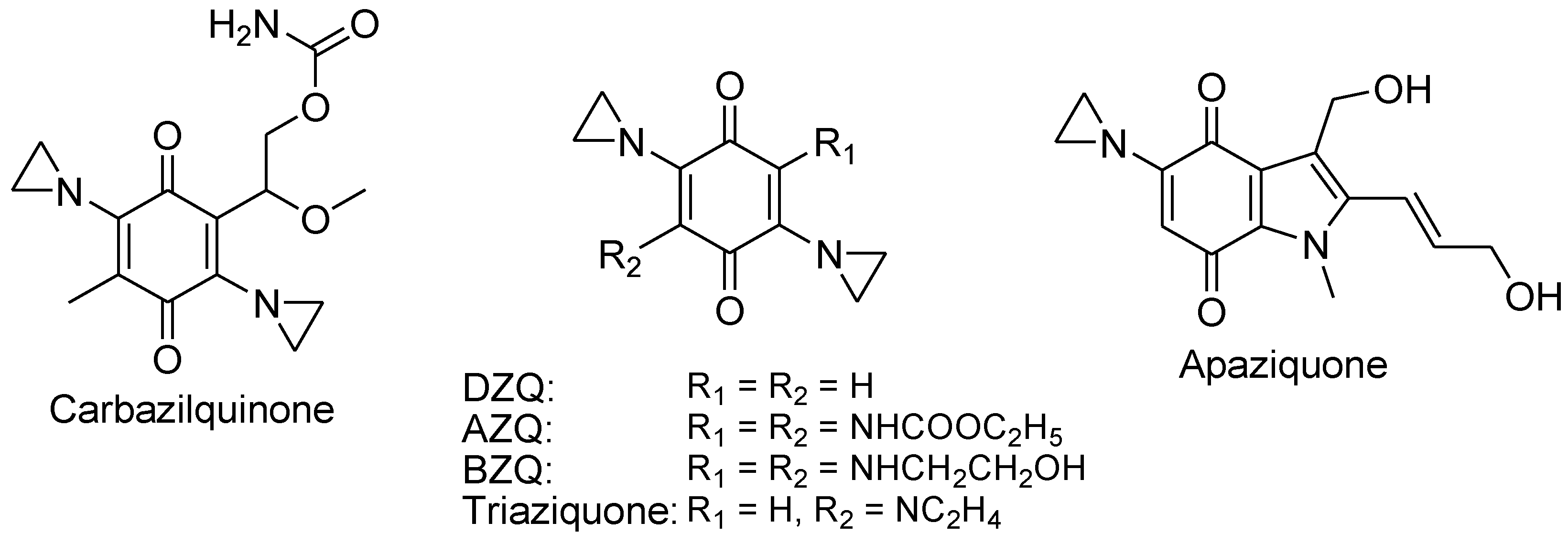
Figure 18.
Possible mechanisms for the cytotoxic effects of aziridinylquinones: (A) Enzyme-catalyzed reduction leads to hydroquinone formation and redox cycling of semiquinone radicals, generating superoxide radicals and H2O2 under aerobic conditions; (B) Aziridinylquinones possess dual abilities, causing DNA damage and cell death while generating ROS, including H2O2. AZQ relies on enzymatic reduction for antitumor activity, forming DNA-alkylating species.
Figure 18.
Possible mechanisms for the cytotoxic effects of aziridinylquinones: (A) Enzyme-catalyzed reduction leads to hydroquinone formation and redox cycling of semiquinone radicals, generating superoxide radicals and H2O2 under aerobic conditions; (B) Aziridinylquinones possess dual abilities, causing DNA damage and cell death while generating ROS, including H2O2. AZQ relies on enzymatic reduction for antitumor activity, forming DNA-alkylating species.
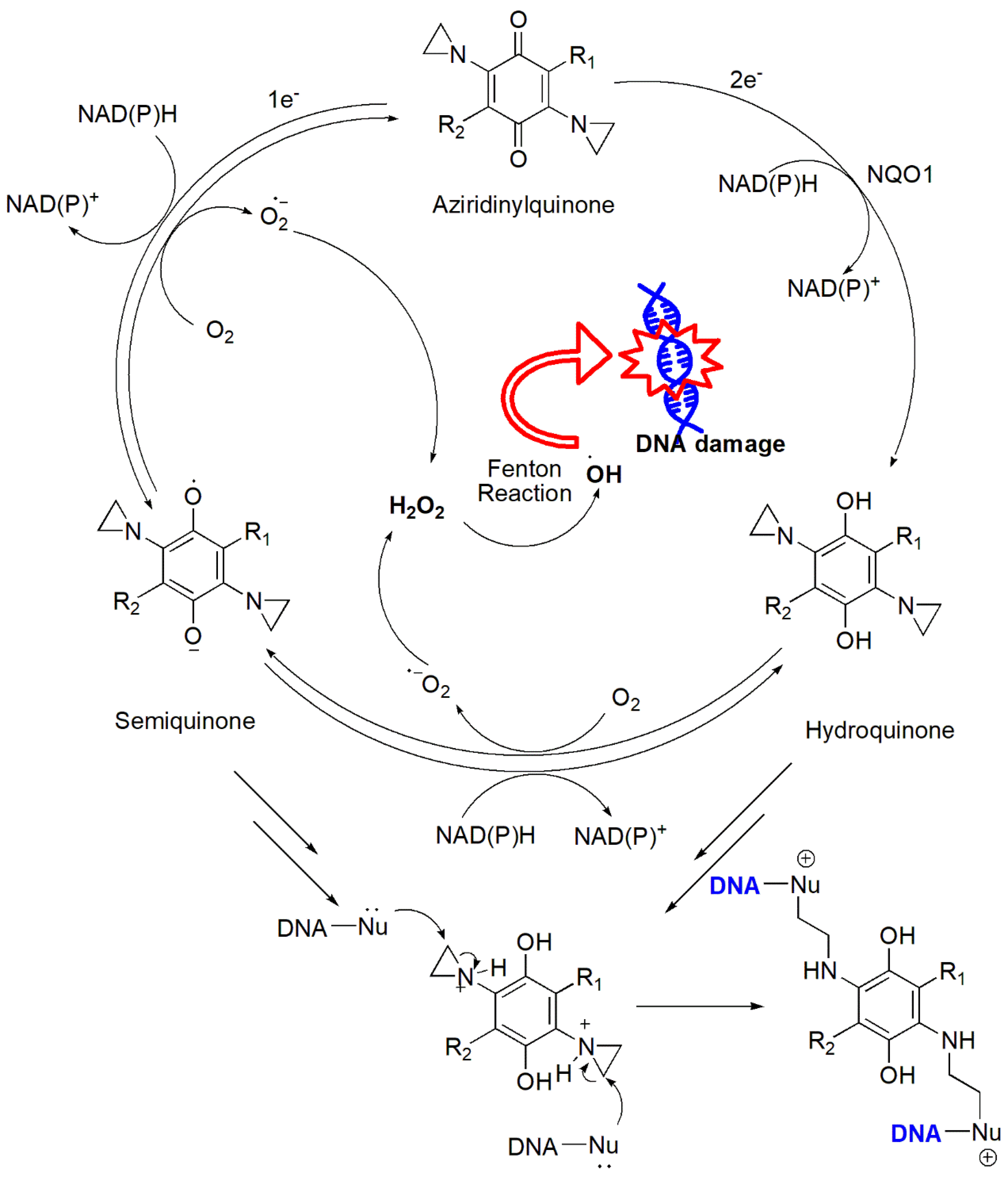
Figure 22.
The structure of ascorbic acid and derivatives and the proposed mechanism for the formation of H2O2 induced by ascorbic acid.
Figure 22.
The structure of ascorbic acid and derivatives and the proposed mechanism for the formation of H2O2 induced by ascorbic acid.
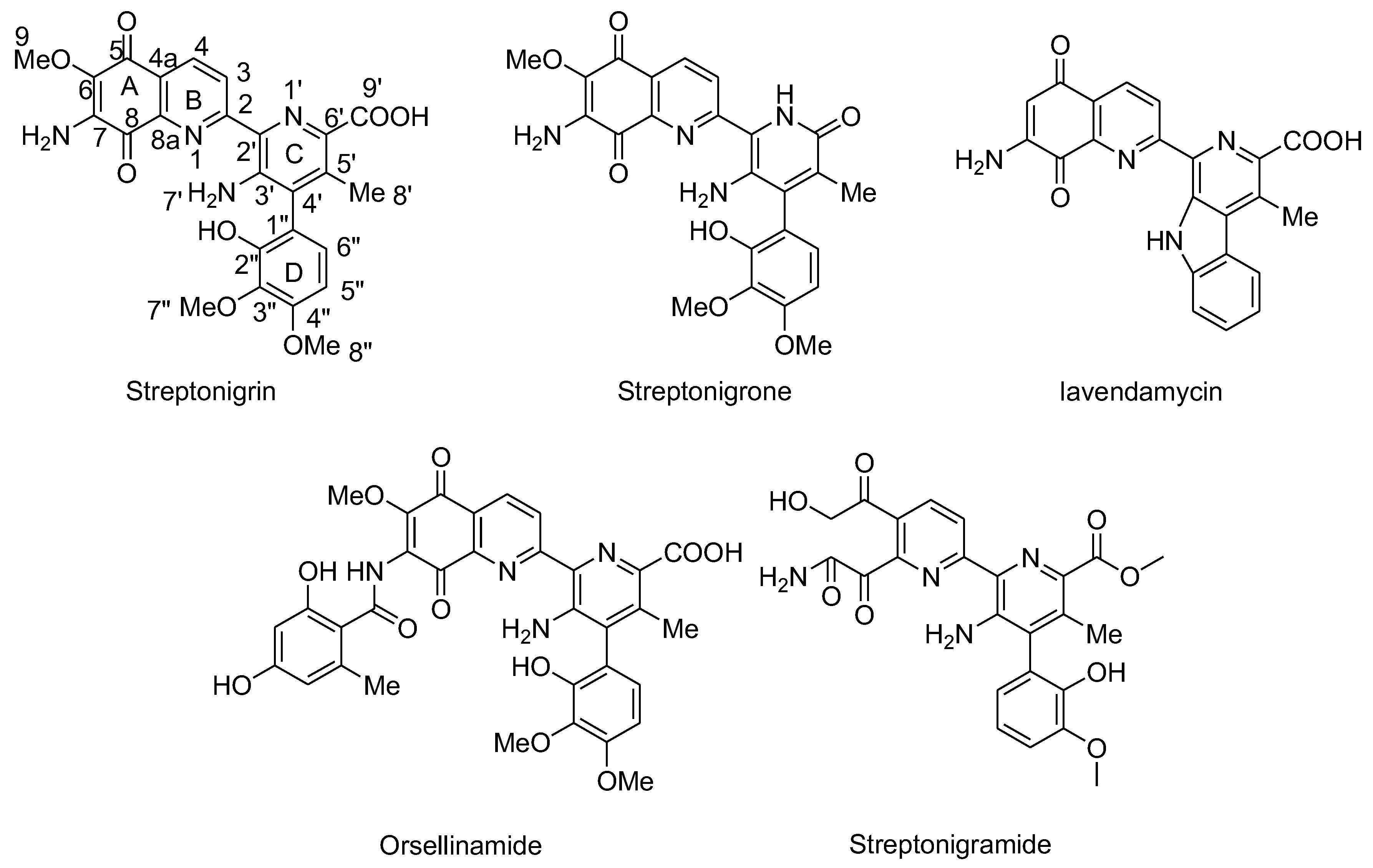
Figure 23.
FDA-approved drugs that generate H2O2.
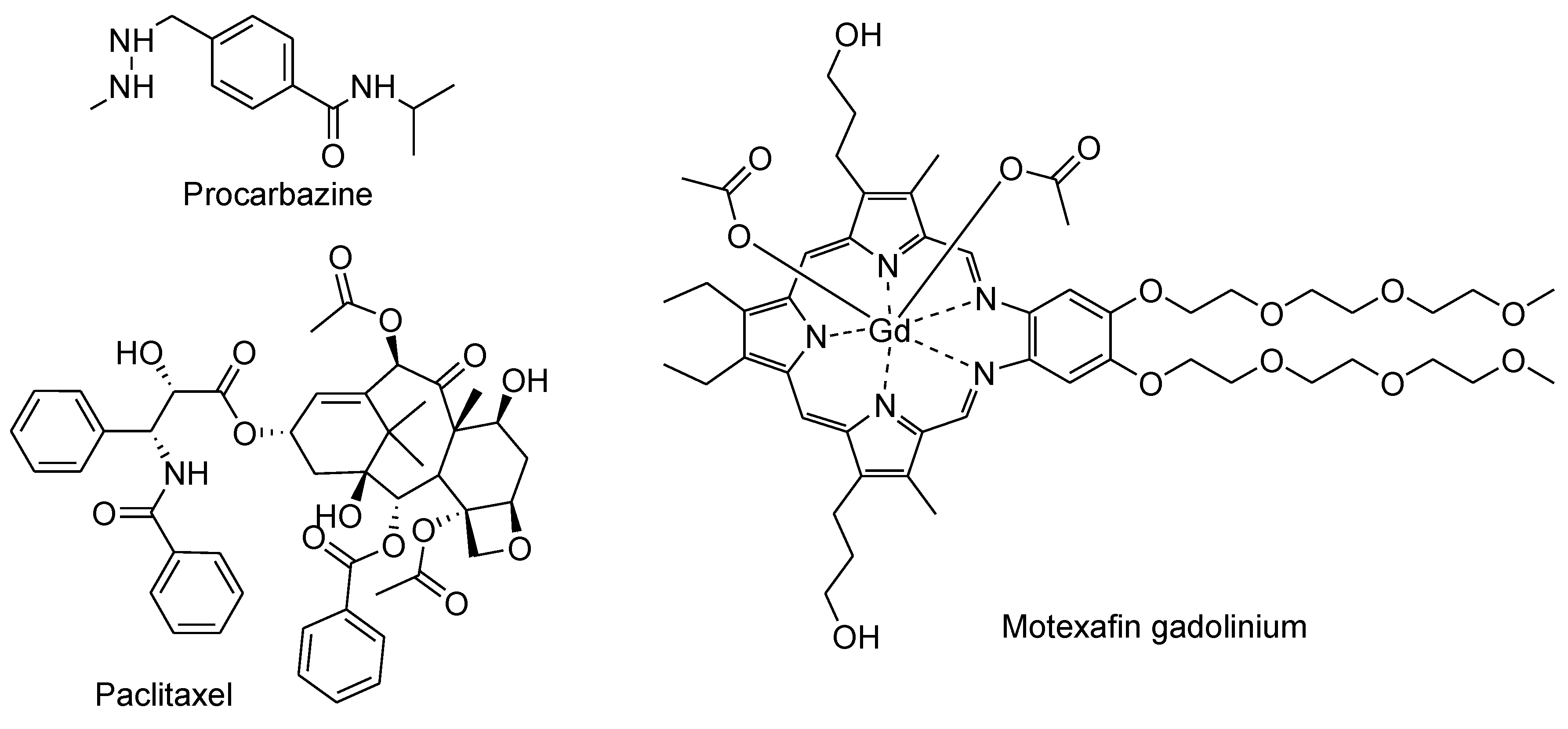
Figure 24.
Oxidation of procarbazine to azo derivative generates H2O2 that subsequently undergoes Fenton reaction to form OH• causing DNA damage.
Figure 24.
Oxidation of procarbazine to azo derivative generates H2O2 that subsequently undergoes Fenton reaction to form OH• causing DNA damage.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



