
Submitted:
02 May 2024
Posted:
06 May 2024
You are already at the latest version
Abstract
Evidence is emerging on the role of maternal diet, gut microbiota, and other lifestyle factors in establishing lifelong health and disease, which are determined by trans-generationally inherited epigenetic modifications. Understanding epigenetic mechanisms may help identify novel biomarkers for pregnancy-related exposure, burden, or disease risk. Such biomarkers are essential for developing tools for the early detection of risk factors and exposure levels. It is necessary to establish the exposure threshold to the fetus due to nutrient deficiencies or other environmental factors that can lead to clinically relevant epigenetic changes that modulate disease risks. This narrative review summarizes the latest updates on the roles of maternal nutrients (n-3 fatty acids, polyphenols, vitamins) and gut microbiota on the placental epigenome and its impacts on fetal brain development.
Keywords:
fetal development
; placental epigenome
; gut microbiota
; immune function
; brain development
; maternal diet
Introduction
Adequate maternal nutrition and optimal environmental factors support a healthy fetus and reduce the risks of adverse outcomes for the offspring and adult life [1]. Several environmental and maternal factors, including gut microbiota, regulate the developmental stages of the fetal-placental axis via epigenetic, endocrinal, and other pathways [2,3]. Maternal, placental, and fetal coordination and dynamic interaction maintain a healthy pregnancy. The placenta is the vital organ that affects these processes by supplying the fetus with oxygen, nutrients, hormones, and growth factors throughout pregnancy [4]. Therefore, optimizing maternal nutrition and limiting exposure to adverse environmental factors can ensure the healthy functioning of the fetoplacental interface [5]. Several fetal factors influence the delicate epigenetic balance in mammals, including maternal-fetal nutrient resources and fetal gut microbiota [6]. Maternal nutritional and environmental factors can alter the epigenetic state of the placental genome, which affects fetal growth and development. Epigenetics refers to the potent interplay between genetic and environmental components regulating gene expression via mechanisms involving alterations to the DNA coding sequence [7], resulting in all cells in an organism showing phenotypic plasticity without change in their genome. Studies aiming at maternal diet manipulation during pregnancy and examining fetal epigenetics provide the most convincing evidence that prenatal nutrition influences fetal methylation and gene expression patterns [8].
In addition to folic acid’s role in countering the development of neural tube defects, its deficiency first pointed to the fetal nutritional programming hypothesis through epigenetic programming. A diet deficient in methyl donor (restricted B12 and folate) during the critical period of the methylation programming phase, which spans from oocytes to embryonic development, resulted in excess adiposity, altered immune response, insulin resistance, and elevated blood pressure in male offspring [9]. Genome scanning has confirmed a widespread change that is connected with the effects of nutritional programming [10]. This nutritional epigenetic programming has also been observed with a prenatal n-3 fatty acid-restricted diet and its impact on the offspring’s phenotype [11]. Maternal n-3 fatty acid restriction typically altered placental epigenetic machinery by changing the methylation pattern [12] and affected gene expression of neurogenesis modulators in the young mice brains [13]. Maternal one-carbon metabolism is critical for maintaining fetal epigenetic stability and showed intergenerational effects. When pregnant mice were fed a diet low in betaine, choline, and methionine, the Cdkn3 gene, which encodes for kinase-associated phosphatase (Kap) in the fetal brain, was hypomethylated. Hypomethylation of Cdkn3 correlated with its increased expression, suppressing cell cycling and reducing neural progenitor cell proliferation in the fetal brain [14,15]. Again, maternal choline intake modified the epigenetic signature of the cortisol-regulating genes in the placenta by hyper-methylating the glucocorticoid receptor (NR3C1) and corticotrophin-releasing hormone (CRH) genes [16].
The DNA methylation landscape undergoes considerable changes throughout the pregnancy to assist placental function, fetal development, and maternal-fetal interactions. Soon after fertilization, maternal and paternal genomes exhibit demethylation patterns to erase parental distinct methylation patterns and prepare for early embryonic developmental reprogramming. After the formation of the blastocyst and its implantation in the uterine lining, DNA re-methylation occurs to build lineage-specific DNA methylation patterns [17,18]. DNA methylation levels change globally by connecting with the development of implantation and subsequent progress in fetal growth, and they also coordinate in a tissue-specific manner. The DNA methylation dynamics reveal that the proportion of methylome remodeled during developmental phases is more substantial than previously thought [19]. The tissues exhibit a specific DNA methylation signature as early as gestational day 9 of fetal development [20]. There are several windows of fetal development where dietary factors can affect the epigenome during gestation. For example, during embryogenesis, the epigenome is more vulnerable to environmental stressors due to the high DNA synthetic rate, and the typical DNA methylation patterns required for normal development and differentiation are recognized [21]. However, the extent to which the maternal intake of methyl groups through gestation can influence maternal DNA methylation is unclear. Dietary methyl sources can alter the epigenome entirely. The apparent mechanism is that the diet changes the ratio of S-adenosyl-L-methionine to S-adenosyl-L-homocysteine concentrations in tissues and directly alters the activity of DNA methyltransferase [22]. Food bioactive and phytochemicals also alter the epigenetic landscape of phenotypes. For example, sulforaphane, a cruciferous bioactive, downregulates DNA methyltransferase 1 (DNMT1) expression and induces cyclin D2 (CCND2) demethylation [23]. Genistein (a phytoestrogen in soybeans) also affected DNA methylation by inhibiting DNMT1 in vitro [24]. When genistein was fed to pregnant Avy methylation indicator mice during pregnancy, at levels comparable to humans consuming high-soy diets, they had offspring with shifted coat color (yellow to brown). This phenotypic change was significantly associated with increased methylation of six CpG sites at the inhibitor of the apoptosis protein upstream of the transcription start site of the Agouti gene [25,26]. Although the mechanism of epigenetic perturbation is unclear, interestingly, this outcome is similar to that observed when methyl donors were depleted using the same genetic model. This reflects the sensitivity of the Avy allele to dietary changes and that these are likely indirect effects of the two diets. The sex-specific effects of micronutrients, including folate, zinc, and vitamins A, B12, C, and D, on reducing methylation levels in DMRs in IGF2R girls and GTL2–2 in boys were observed [27]. Moreover, increased expression of several placental methyltransferases was observed [28]. Early-life exposures to dietary methyl group supplementation elevated the epigenetic variation over six generations in isogenic mice [29]. Thus, a substantial body of evidence from models and clinical studies reveals that methyl donors and cofactors in the diet can alter the DNA methylation of the fetus.
In addition to epigenetic methylation contributors, early-life metabolic adaptation of the fetus relies on maternal gut microbiota. The gut microbiota can influence the placental epigenome by producing bioactive metabolites such as short-chain fatty acids (SCFAs), trimethylamine (TMA), and trimethylamine N-oxide (TMAO), and modifications of the epigenome could be responsible for fetal programming [30]. Moreover, gut microbial metabolites can also affect pregnancy outcomes via diverse mechanisms [31]. The microbial capability to modify the host metabolic and immune functions is also evidenced by its interaction with the epigenome, suggesting that the maternal microbiome and its metabolites could be involved in fetal programming through epigenetic mechanisms. Maternal diet, BMI, metabolic disorders, ethnicity, and geographic and environmental factors can influence maternal-fetal microbiome composition. For example, during maternal obesity, a change in gut microbial diversity significantly associated with the fetal brain development [32]. Gut microbiota also regulates the onset of pre-eclampsia, a placental disease, by producing a different composition of gut metabolites such as SCFAs, TMAs, TMAOs, and others [33]. Maternal gut microbiota also affects offspring’s metabolism and immune system [34]. The potential correlation between the gut microbiota and the metabolic requirement of the developing brain has been studied [32,35,36]. A broad perspective on the human brain's early postnatal development and the involvement of the gut in crucial neurodevelopmental processes is also documented [32,36].
The thrifty phenotype hypothesis relies on early-life metabolic adaptation for the fetus's survival by choosing an appropriate growth trajectory in response to adverse dietary challenges and environmental exposure. In response to nutrient availability or scarcity, maternal metabolic control minimizes in-utero energy expenditure and maximizes the storage of reserve calories that the maternal microbiome might regulate [37]. The imprinted genes, such as IGF2, regulate the development of critical metabolic organs and their functions in controlling metabolic axes in postnatal life [38]. The imprinted genes expressed from one chromosome in a parent-of-origin manner depend on epigenetic modulation of expression control, making them sensitive to environmental changes in utero. Moreover, early fetal growth relies on the activation of imprinted genes since the latter is required to successfully develop both embryo and extraembryonic tissues. Therefore, it is possible that maternal nutrition and its gut microbiome, as metabolic modulators, closely interact with the fetal epigenome in determining the growth and development of the fetus. However, a little knowledge about host nutrition and microbiome on placental epigenomic programming that determines fetal development is available. This narrative review details the latest updates on maternal nutrients, gut microbiota, and their impacts on epigenetic controls of fetal growth, brain development, and functions.
Heritability of Diet-Induced Epigenetic Changes: Effects on Fetal Development
Although it is well accepted that diet-induced epigenetic changes can be mitotically heritable, the question of how changes induced in one generation can persist into the next remains a topic of debate. The multigenerational effects of epigenetic changes persist as many as three generations after dietary insult, in which case they are deemed “transgenerationally inherited” [39,40,41]. Evidence in mice for diet-induced multigenerational epigenetic changes was first shown at IAP-regulated metastable epialleles Avy and axin-fused. Many studies have reported epigenetic changes in the unexposed offspring of parental dietary challenges, sometimes with subsequent transmission to future generations. For example, a preconception paternal diet (low protein and high fat) in rodents altered energy metabolism in the unexposed offspring [42,43]. This demonstrates the transmission of the diet-induced incorporated epigenomic traits of the male germline or semen composition in a way that perturbed offspring metabolic programming. Multigeneration epigenetic inheritance occurs after maternal exposure to various diets (high fat, caloric restriction, vitamin D depletion) during gestation. Evidence of epigenetic inheritance in humans remains limited due to the challenges of time, cost, and participant retention rate required for longitudinal studies. More importantly, beyond these challenges, humans do not live in an uncontrolled environment and are exposed to various environmental factors throughout their lifespan, making it impossible to assess direct links between diet and epigenetic outcomes.
Different epigenetic mechanisms may explain in part by which dietary factors in critical developmental stage might affect the susceptibility to develop metabolic diseases in adulthood. Fetal epigenetic modifications to the maternal dietary intervention are substantiated by a large number pre-clinical trial (Table 1). MicroRNAs are versatile regulators of gene expression and play a significant role during tissue homeostasis and disease. Dietary factors have also been shown to modify microRNA expression. However, the role of microRNAs in fetal programming remains largely unstudied. Several in vivo studies analyzed the effect of maternal diet on the modulation of the microRNA expression in the offspring and their influence on the development of metabolic and cardiovascular disease in later life. Evidence suggests that nutritional status during pregnancy influences offspring susceptibility to developing cardiometabolic risk factors, partly through microRNA action. Thus, therapeutic modulation of microRNAs can open up new strategies to combat – later in life – the effects of nutritional insult during critical development points. Chen et al. studied the gene expression and DNA methylation in male and female placentas of mice exposed to either caloric restriction or ad libitum diets [44]. Caloric restriction resulted not only in several differentially expressed pathways associated with intrauterine growth restricted (IUGR) phenotypes but also in a significant decrease in the overall methylation, which was more pronounced in males [44]. Furthermore, several differentially methylated genes enriched for known imprinted genes were identified, suggesting that imprinted loci may be particularly susceptible to dietary changes. Maloyan et al. studied the sequence and profiled the cardiac miRNA expression in baboon fetuses born to high-fat diet (HFD)/high fructose-diet-fed mothers during the four months before pregnancy compared to fetal hearts from mothers eating a regular diet [45]. Furthermore, fetuses of HFD mothers showed a significant decrease in fetal weight and an increase in the weight of the brain and thymus. 55 miRNAs out of 80 differentially expressed were upregulated and 25 downregulated with maternal HFD/high fructose diet.
Maternal HFD/high fructose diet modulates the expression of several miRNAs that were changed in adult cardiac impairments such as miR-143, miR-499, and miR-21 [46] , heart failure such as miR-21 and miR-223 [47] , and myocardial infarction such as miR-30c, miR-451 and miR-139 [48]. In animal models, dams fed with HF/high fructose diet before and during pregnancy resulted in fetal cardiac fibrosis. They showed differential expression of miRNAs implicated in heart disease that may participate in the programming of heart development.Khorram et al. [49] studied the possible role of miRNA-29c in carotid artery remodeling of rats' offspring at week 3, month 3, and month 9 of age and whose mothers were undernourished (50%) in the latter half of gestation. Maternal undernutrition increased the protein expression of extracellular matrix in carotid arteries, including collagen 3A1 (Col3A1), collagen 4A5 (Col4A5), elastin (ELN) or matrix metalloproteinase 2 (MMP2). In this condition, miR-29c – which targets most of these genes – was repressed. Interestingly, some of these changes were blocked when metyrapone, a glucocorticoid inhibitor, was administered in drinking water from day 10 of gestation to term. The latter changes were accompanied by increased expression of miR-29c, suggesting a close relationship between maternal undernutrition, miR-29c, and carotid artery remodeling in offspring. Despite these studies, models using genetic manipulation of differentially expressed miRNAs are necessary to understand epigenetics’ role in fetal heart development due to maternal overnutrition.
Among different theories of fetal programming, Barker supports the thesis that maternal undernutrition during pregnancy may be the origin of several diseases in later life. In contrast, Plagemann et al. describe that gestational oversupply of nutrients may cause predispositions to developing disorders [50]. However, similar adult phenotypes may result in different programming pathways, such as intrauterine growth restriction (IUGR) with a low protein diet and maternal obesogenic environment with an HFD. While the mechanism for the low protein diet is most likely due to the mismatch between the intra-uterine and postnatal environment [51] , the HFD results in “malprogramming” of the hypothalamus and beta-cells [50]. However, the role of miRNAs in the modulation of fetal programming and the differentiation between these two theories is unclear. Intrauterine growth restriction is most likely associated with the induction of persistent changes in tissue structure and functionality.
In contrast, a maternal obesogenic environment is most likely associated with metabolic reprogramming of glucose and lipid metabolism and future risk of metabolic syndrome, fatty liver, and insulin resistance [52]. Despite several theories of fetal programming on epigenetic regulation , deciphering modulation of specific miRNAs roles in undernutrition or excess nutrients could eventually lead to working hypothesis of fetal programming. As many genes are differentially modulated in both scenarios in different tissues, but given a similar phenotype (for example, cardiometabolic disease), miRNAs – by regulating many transcripts – could eventually tell us if this still unexplored mechanism may be responsible for the observed effects.
Placental Interactions with Environments: Impacts on Fetoplacental Development
Obesity in pregnancy has various impacts on the structure and function of the human placenta. During early pregnancy, the human placenta responds to increased maternal insulin levels in obese women. The cellular signaling system likely plays a role in mediating these effects, influencing inflammation, metabolism, and oxidative stress. These alterations in placental function can independently and synergistically affect pregnancy outcomes, potentially interacting with other risk factors [53,54]. The placenta has intricate vascularization for fetal blood supply, necessitating considerable angiogenesis. Suboptimal angiogenesis causes aberrant placental size and vasculature. The placentas of obese women at term show increased total lipid content, infiltrating neutrophils, foam-loaded macrophages, and elevated levels of pro-inflammatory mediators [55,56]. Maternal obesity-induced metabolic alterations influence early placental growth, gene expression, and further changes in placental structure and function, clinically evident in late pregnancy [57]. Placental dysfunctions may negatively impact fetal growth [5,57].
Genetics, food, and lifestyle factors all contribute to suboptimal placental angiogenesis [58]. Dysregulated angiogenesis in the placenta may directly or indirectly influence pregnancies, such as pre-eclampsia, pre-term birth, gestational diabetes mellitus (GDM), and IUGR [5,59]. n-3 fatty acid deficit impaired placental transport of fatty acids in pre-eclampsia and GDM-associated fetuses [60,61]. Several medical disorders include abnormal placental vasculature, including pre-term birth, IUGR, and pre-eclampsia. Several angiogenic factors, including vascular endothelial growth factor A (VEGFA), angiopoietin-like 4 (ANGPTL4), fibroblast growth factor (FGF), placental growth factor (PlGF), and docosahexaenoic acid, 22:6 n-3 (DHA), assist optimal placentation [62,63,64]. High-fat diets and maternal obesity modify the metabolome and early modifications in the placental transcriptome, reducing placental vascularity [65]. A high-fat diet during pregnancy promotes ectopic lipid accumulation, which leads to lipotoxicity and persistent inflammation in the placenta [66].
Furthermore, the high-fat diet causes the placenta to modify its metabolic response and structural alteration (thickness) by influencing angiogenesis. In vivo data revealed lower placental labyrinth depth and higher insulin-like growth factor 2 (IGF2) expression and its receptor genes in the fetuses of high-fat diet dams [67]. Deficiency in n-3 polyunsaturated fatty acids (PUFA) is similar to the impaired placental phenotypes caused by a high-fat diet. The maternal n-3 PUFA insufficiency impaired the vascular development of decidua; the fetoplacental unit indicates that maternal fatty acid status affects placental vascularity [12]. Obese placentas accumulate lipids due to changes fatty acid transporter expression, lipoprotein lipase activity, and mitochondrial oxidative metabolism [4,68]. The genome-wide transcriptome, epigenome, and proteome revealed the impact of maternal obesity on placental lipid transport and metabolism [69,70]. The altered lipid transport and metabolism of the obese placenta, as reflected by changes in fatty acid transporter expression, had deleterious consequences on smooth placental functioning in transport and the metabolism of lipids throughout the fetoplacental unit [71,72]. The obese placenta had elevated total lipids, triglycerides, free fatty acids, and cholesterol. Obese placental phenotype supports increased lipid storage with impaired lipid transfer to the developing fetus, particularly long-chain polyunsaturated fatty acids (LCPUFAs), critical for fetal brain development [73]. Optimal LCPUFA supply is crucial for fetoplacental development, and any alterations observed in obesity can harm fetal brain development and performance [74,75]. During maternal obesity, pro-inflammatory M1 macrophages dominate over less pro-inflammatory M2 macrophages and thereby shift the trigger towards pro-inflammatory cascade. An obese state promotes low-grade chronic inflammation that may exacerbate immune functions in pathogenic pregnancies, such as pre-eclampsia, by lowering uterine natural killer (uNK) cell populations [76]. Furthermore, maternal obesity correlated with epigenetic dysregulations in leptin and adiponectin systems [77]. Thus, dysregulated endocrine controls in the placenta reduce the protective effects of these adipokines on placental development, showing the placenta's response to a hazardous maternal environment.
Maternal obesity and GDM both influence fatty acid transport throughout the placenta. Obese women with diabetes showed elevated levels of placental fatty acid binding protein 4 (FABP4) and endothelial lipase [78]. Obese placentas, on the other hand, had lower levels of FABP5 and decreased uptake of n-6 LCPUFAs [55,69] . Obese women's placentas expressed low and high levels of the fatty acid translocase CD36/FAT[78]. Obesity-related inflammation and metabolic dysfunction increase placental oxidative, endoplasmic reticulum stress, and downstream activation of the placental unfolded protein response, all of which have been linked to pregnancy complications such as fetal growth restriction, pre-eclampsia, and gestational diabetes. The placenta is exposed to high insulin during early pregnancy, which alters steroid hormones in mitochondria and impairs energy metabolism. Maternal lipid transport and metabolism influence fetal obesity through placental function. IUGR and GDM impair the placental transfer of maternal lipids. Inadequate placental LCPUFA transfers and fat-soluble vitamins may cause metabolic dysfunction and poor fetal growth. The interaction between placental ANGPTL4 and lipoprotein lipase causes fetal adiposity in GDM [79].
Placental Epigenome and Birth Outcomes
Nutrients modulate epigenetic enzymes such as DNMT, histone deacetylases (HDAC), or histone acetyltransferases (HAT) by changing the substrate accessibility required for these enzymatic reactions. HATs catalyze the transfer of an acetyl group from acetyl coenzyme A, while HDACs perform the antagonistic action of removing the acetyl group. This, in turn, changes the expression of essential genes that impact global health and longevity [80]. Mitsuya et al. studied patterns of placental DNA methylation and hydroxymethylation on a genomic scale and observed a partial but essential intersection among the genes that exhibited a rise in DNA methylation and a reciprocal reduction in DNA hydroxy methylation with increased maternal obesity, suggesting a possible decrease in the conversion efficiency of methylation to hydroxy methylation. Other studies have also provided evidence in support of a connection between metabolism and epigenetics. The dysregulation of adenosine monophosphate-activated protein kinase (AMPK) and the mammalian target of rapamycin (mTOR) homeostasis are linked to gestational obesity [81]. AMPK activation occurs in response to reduced energy levels suggested by high and low ATP concentrations.
Nutrients can reverse or change epigenetic phenomena such as DNA methylation, histone modifications, and miRNA expression, thereby modifying the expression of critical genes associated with physiologic and pathologic processes, including embryonic development (Figure 1). Folic acid is recognized as a one-carbon donor for DNA methylation and synthesis. DNA methylation of miR-203 and miR375 was higher when cells were cultured in a medium enriched with folic acid [83]. Expression of miR-615-5p, miR-3079-5p, miR-124, and miR-101b were downregulated, whereas miR-143 was upregulated in livers from offspring from high-choline diet-fed dams [84]. MiRNAs also regulate some genes of methyl metabolism; miR-125b targets DNMT3b in vascular smooth muscle [85], while miR-22 and miR-29b directly target rat Mthfr and Mat1 genes, respectively [86]. Several miRNAs are epigenetically regulated by DNA methylation, and these include miRs 375, 149, 27b, 196b, 203, 375, 129-2/-137/-935/-3663/-3665 and -428, 211, 153, 145a-5p [83,87,88,89] and many more. DNA methylation of miR 1451–5p inhibits its expression [89] , indicating feedback regulation by epigenetic control.
Despite the limited epigenomic studies integrating exposures and outcomes, omics approaches are emerging to investigate parturition and birth outcomes. Within the last five years, several studies have utilized placental epigenomics to gain biological insight into perturbations of the placenta related to birth outcomes, including preterm birth, intrauterine growth restriction, birth weight, or birth size. This large body of research associating birth outcomes with placental epigenomic measures attempted to develop new hypotheses for exposure-outcome pairings. For example, regarding birth weight, Tekola-Ayele et al. [90] evaluated DNA methylation in the NICHD-FGS singleton cohort (N = 301). To contextualize the relevance of their findings, pathway analysis was performed on the genes annotated to the top 100 CpG sites associated with birth weight [90]. This analysis identified multiple pathways such as RNA polymerase I complex assembly, CTLA4 signaling in cytotoxic T lymphocytes, and DNA methylation and transcriptional repression signaling [90]. Future research can leverage these findings to develop hypotheses surrounding exposure-birth weight associations by identifying exposures that have previously been associated with these same pathways based on placental epigenomics.Several studies use placental omics data to link prenatal exposures and accompanying measurements of birth or early childhood developmental outcomes within the population. Methodologies are used to connect exposure and outcome results included (a) statistical mediation analyses in a subset of genes, (b) analyses that identified commonly enriched placental gene networks between exposure and outcome using approaches like weighted gene co-expression network analyses (i.e., WGCNA), or (c) identification of gene or CpG overlaps that were independently associated with the exposure or outcome (d) co-methylation where local CG sites of one short chromosome region methylate or do not methylate together in a single sample and others [91]. A multi-omics analysis of the placenta measured the expression of mRNA, microRNA, and DNA methylation to identify signatures that may predict perinatal outcomes, including birth weight , placental weight, and placental inflammation [92]. The predictivity of top-ranking sites (257 microRNAs) was tested in an independent cohort (n = 36) by microarray, identifying 32 overlapping microRNAs [92]. Of these 32 microRNAs, six were associated with prenatal Cd exposure, implying the utility of these microRNAs as predictors of perinatal outcomes following prenatal exposures [92]. Nutrients can play an epigenetic modulatory role by inhibiting activities of the epigenetic enzymes such as DNMT, histone deacetylases (HDAC), or histone acetyltransferases (HAT), or by changing the accessibility of the substrate required for these enzymatic reactions [93]. HATs catalyze the transfer of an acetyl group from acetyl coenzyme A, while HDACs perform the antagonistic action of removing the acetyl group. This, in turn, changes the expression of essential genes that impact global health and longevity [80]. Mitsuya et al. studied patterns of placental DNA methylation and hydroxymethylation on a genomic scale and observed a partial but essential intersection among the genes that exhibited a rise in DNA methylation and a reciprocal reduction in DNA hydroxy methylation with increased maternal obesity, suggesting a possible decrease in the conversion efficiency of methylation to hydroxy methylation. Other studies have also provided evidence in support of a connection between metabolism and epigenetics. Epigenetic alterations via DNA methylation and hydroxymethylation affect the development of the fetal brain and are modulated by several factors associated with prenatal exposure (Figure 2)
There are several central windows of fetal development where dietary factors can affect the epigenome during gestation. For example, during embryogenesis, the epigenome is more vulnerable to environmental stressors due to the high DNA synthetic rate, and the DNA methylation patterns required for normal development and differentiation are recognized [21]. However, the extent to which the maternal intake of methyl groups through gestation can influence maternal DNA methylation is unclear.
The perception that pregnancy is associated with the suppression of the immune system has created a myth of pregnancy as a condition of immunological fragility and, hence, of enhanced sensitivity to infectious disorders. Given their high turnover rates, most immune cells are highly susceptible to environmental alteration and can adapt to various stressors. Myeloid-derived cells, particularly, can activate stress response pathways that cause plasticity in their transcriptional activity. During normal pregnancies, the human decidua contains many immune cells, including macrophages, natural killer (NK) cells, and regulatory T cells. Accordingly, immune cells infiltrate the decidua during the first trimester and accumulate around the invading trophoblast cells. Pregnancy hormones are crucial to maintaining pregnancies and can profoundly impact the immune response. A growing body of evidence has established that sex hormones can regulate the most important epigenetic alterations involving the modulation of miRNA expression, DNA methylation status, and chromatin rearrangement.
Maternal Dietary Fats on Placental Epigenome
The maternal nutrients including dietary fats can alter the epigenetic state of the placental genome. Maternal fats and ecological cues to fetoplacental pathology are being studied, with consequences for fetal development and adult life. The placenta is crucial in facilitating normal fetal development by serving as a primary barrier between the maternal environment and the fetus. It regulates key functions such as gas exchange, hormone production, and nutrient secretion. Additionally, in healthy pregnancies, the term placenta contains microbiota. There is speculation that the microbiota in the placenta might play a role in the development of the fetal immune system, particularly during the second trimester of pregnancy [94].
Maternal fatty acids such as n-3 and n-6 long-chain polyunsaturated fatty acids (LCPUFAs) status are potential predictors of fetoplacental growth and development [1,95,96,97,98]. Maternal docosahexaenoic acid, 22:6n-3 (DHA) deficiency could affect fetal neurodevelopment, changes in fetoplacental epigenetics, offspring growth, and lipogenic capacity [5]. Alterations in membrane phospholipid fatty acid composition can affect the function of the neurons by changing the membrane receptors, ion channels, enzymes, and fatty acid-derived second messengers. Mice studies showed that n-3 PUFA deficiency during fetal development impacted the offspring's adipose browning and postnatal musculoskeletal development [99]; however, no such information is available in humans. A high-fat diet during pregnancy can increase the risk of abnormal neurological behaviors later in offspring’s life [32,100]. Fat (adipose) tissue is also target for signals of nutrients, hormones, and epigenetics that regulate fetal growth and development [101]. Therefore, emerging data revealed whether disruptions in the maternal, nutritional, hormonal, epigenetic, and gut microbiota in obesity alter fetal growth and later-life adiposity [101]. DNA methylation was assessed at IGF2 promoter 3 (IGF2 P3), IGF2 differentially methylated region (DMR), and the H19 DMR using cord blood mononuclear cells of pregnant women supplemented without or with 400 mg DHA (DHA supplemented, n = 131; control group, n = 130) during gestation week 18–22 to parturition. DNA methylation levels at only one CpG in IGF2 P3 were significantly higher in the DHA group than control infants. No changes were observed at the nine other CpGs (including the H19 DMR) [102] . The same study assessed DNA methylation states in Th1, Th2, Th17, and regulatory T-relevant genes and LINE1 repetitive elements of cord blood mononuclear cells [103]. No significant difference in promoter methylation levels was shown between the supplemented and control groups for the genes analyzed. This study monitored DNA from cord blood, which might not reflect changes in the epigenome of target tissues that express IGF2. The observed differential methylation of a single CpG may not alter the gene expression, and no data were presented that DHA supplementation was associated with altered gene expression or altered growth of the children. DHA supplementation with 400 mg/day during pregnancy induced DNA methylation changes in inflammatory genes and LINE-1, especially in infants born to mothers who smoked during pregnancy [103].
One-Carbon Metabolism-Related Nutrients and Placental Epigenome
The importance of one-carbon metabolism in fetal programming was reported through evidence indicating that low maternal vitamin B12 levels correlate strongly with hyperhomocysteine, predicted higher offspring adiposity, and insulin resistance in humans [104]. Global methylation and imprinted gene methylation were also modified by one-carbon metabolism [27,105]. Among the nutrients involved in one-carbon metabolism, folic acid has been studied widely and is implemented universally for disease prevention. In particular, the methylation of imprinted genes like IGF2/H19 DMR was well studied with maternal folate supplementation [106]. For the effect of choline supplementation, a higher maternal choline intake (930 vs. 480 mg/day) increased placental CRH and GR methylation and decreased placental CRH transcript abundance, indicating that extra choline during pregnancy decreased placental expression of cortisol-regulating genes [28]. Impaired activity of one-carbon metabolism enzymes may lead to an aberrant DNA methylation pattern and increased plasma Hcy, a toxic derivative that leads to vascular lesions. In addition, it was reported that restriction of folate, methionine, and B vitamins during the periconceptional period resulted in altered DNA methylation, insulin resistance, and elevated blood pressure observed in adult male offspring , as well as gene expression and methylation changes involved in the renin-angiotensin system, mitochondrion metabolism, and phospholipid homeostasis [107]. Additionally, deficiencies in folate and vitamin B12 during gestation and lactation produced manifestations of fetal programming, with decreased birth weight, increased central fat mass, liver steatosis, and myocardium hypertrophy in pups from deficient rat mothers [108].
Choline may have essential roles in fetal brain development, as maternal choline deficiency during pregnancy has been shown to modify the epigenome of the fetal brain. For example, Vegfc and Angpt2 [109], which are involved in angiogenic signaling, and Cdkn3 [14], for cell proliferation, were hypomethylated in the fetal brain. Additionally, H3K9me2 and H3K27Me3 (repressed) levels were upregulated in the fetal liver and brain by maternal choline supplementation, whereas H3K4me2 (active) levels were the highest in choline-deficient rats. Gestational choline deficiency also altered imprinted gene like IGF2 methylation, and expression through effects on DNTM1 in the liver [110]. However, choline supply has also been shown to modify the DNA methylation and expression of genes involved in methionine and lipid metabolism in Wilson disease [111]. Therefore, the nutrients regulating maternal-fetal one-carbon metabolism may be essential in fetal programming and metabolic diseases.
Folic acid is recognized as a one-carbon donor for DNA methylation and synthesis. Additionally, folic acid is required to produce S-adenosyl methionine (SAM), a methyl donor in DNA methyltransferase-mediated DNA modification. It has been established that SAM is generated in the cytoplasm and drives epigenetic modifications [112]. Folic acid supplementation is related to global DNA methylation (hydroxyl), in which several studies have shown an inhibition in DNA methylation in response to folic acid consumption. In vivo, the delay in the DNA methylation response to folic acid intake can be partially associated with the prolonged revenue of folate stores all over the body. Pauwels et al. found that there may be a delay in the DNA methylation response in women who took a folic acid supplement before and during gestation. This would have occurred before gestation [113].
Vitamin D Levels and Fetoplacental Epigenome
Vitamin D is another micronutrient that alters epigenetic pathways. Low vitamin D concentrations can cause increased inflammation and anti-inflammatory effects due to changes in DNA methylation and histone modifications [114]. During pregnancy, the maternal metabolism undergoes multiple alterations of physiological processes to ensure the healthy development of the fetus. A close relationship between maternal and fetal vitamin D status during pregnancy underscores the importance of an optimal supply in this critical time. Maternal supply status could significantly affect the offspring's development and health in utero and later life [115]. Observational studies from all over the world show a link between low vitamin D levels and adverse pregnancy-related outcomes for mother and child [116]. Low vitamin D concentrations can increase inflammation due to changes in DNA methylation and histone modifications [114]. The complex system of vitamin D metabolism comprises many targets, including the vitamin D receptor (VDR) and enzymatic molecules whose function could be crucially changed by epigenetic modulation of the respective genes [117].
On the other hand, vitamin D status itself may induce epigenetic alterations in various genes, including genes of vitamin D metabolism. However, little is known about the interrelationship of vitamin D and epigenetic mechanisms. Vitamin D, as an essential micronutrient, may influence the genome’s methylation status, comparable to other nutritional components of the maternal diet [118]. The first indications of an association between maternal vitamin D status and epigenetic changes in the offspring come from animal experiments. A mouse model of maternal vitamin D depletion showed changes in DNA methylation in two generations of offspring, thus indicating transgenerational effects of the vitamin D status on the epigenome [119]. In a small human study, Jung et al. found significant differences in DNA methylation profiles in the cord blood of newborns with high versus low 25OHD levels [120]. However, a recent genome-wide study did not find association between maternal 25OHD concentrations at midterm and cord blood DNA methylation [121]. Another study examining the influence of midterm maternal 25OHD levels on DNA methylation levels of crucial genes linked with fetal growth did not associate either [122],
Epigenetic modifications also affect the expression of genes involved in vitamin D synthesis and degradation and, as such, directly impact vitamin D status. DNA methylation can influence how a particular gene is expressed. In general, hypermethylation in the promoter region of a gene is usually linked to gene silencing and decreased gene expression [123] . In contrast, promoter hypomethylation generally leads to increased gene expression [123]. Hypomethylation of hepatic CYP2R1, the gene encoding a crucial 25-hydroxylase for producing 25OHD, could account for an increased expression of the CYP2R1 enzyme and a subsequent increase in 25OHD levels [124]. On the other hand, hypermethylation and concomitant lower expression [125] levels in the CYP24A1 gene, which encodes an essential enzyme of vitamin D catabolism, would lead to an increase of 1,25(OH)2D.
Interestingly, placental CYP24A1 is specifically hypermethylated during pregnancy. A decreased expression of placental CYP24A1 could lead to a reduced local degradation of 1,25(OH)2D. Therefore, it might improve the fetus's 1,25(OH)2D availability at the maternal-fetal interface. A relationship between the maternal vitamin D status and the methylation of genes in the offspring must be further investigated.
Gut Microbiota and Placental Epigenome
The gut microbiota changes throughout pregnancy, crucial in fetal and infant growth and development. Changes in gut microbiota occur throughout life and are particularly pronounced throughout fetal development, infancy, childhood, and puberty when the microbiota gains sexually dimorphic traits, as well as with aging [126,127,128,129]. The maternal gut microbiota in the early postnatal phase influences the infant's nervous system activity. Recent breakthroughs in metagenomics have revealed that the placenta contains its varied microbiota, which has been examined and documented in healthy pregnancies (Pelzer et al., 2017). An altered placental microbiome (dysbiosis) can cause preterm labor, chorioamnionitis, premature membrane rupture (PROM), intrauterine growth restriction (IUGR), and even postpartum hemorrhage (PPH) [53]. The use of antibiotics to deplete the maternal microbiota during pregnancy led to a shortage of thalamocortical axons, hindered thalamic axon growth in the fetus, and caused changes in tactile sensitivity in adult offspring. Animal studies have illustrated that the maternal microbiota during pregnancy can influence fetal brain development and postnatal behavior [130,131,132]. Targeted restoration of the maternal gut microbiome prevented abnormalities in fetal thalamocortical axogenesis [132].
In a recent study involving 1064 pregnant mothers and 1074 children, a connection was found between the makeup of maternal fecal microbiota during pregnancy and internalizing behavior linked to anxiety in two-year-old children. Additionally, fecal samples from pregnant mothers whose children exhibited typical behavior indicated greater microbial alpha diversity and increased levels of butyrate-producing bacteria [133]. Animal and human research highlights the crucial role of maternal microbiota in promoting fetal neurodevelopment during pregnancy.
Microbial invasion (by non-commensal microorganisms) of the fetoplacental junction has been linked to maternal and newborn illness. The presence of several bacteria in the placenta and amniotic cavity is associated with miscarriage, chorioamnionitis, premature membrane rupture, preterm birth, and stillbirth [134,135]. Click or tap here to enter text.However, emerging information suggests that the same bacteria may be present in babies without associated problems. As a result, genetic and/or environmental processes may allow the progression of unfavorable perinatal outcomes caused by germs during a given gestational stage [136]. Placental and amniotic microbiota alterations have been linked to various pregnancy-related diseases, including bacterial vaginosis [136]. Analyses of placental tissue in pathological pregnancies reveal a prevalence of anaerobic germs over beneficial Lactobacillus.
Preterm birth cases show an increased presence of species such as Prevotella, Bacteroides, Peptostreptococcus, Gardnerella, Mobiluncus, and Mycoplasma [137,138,139]. Women with chorioamnionitis exhibit higher proportions of Streptococcus agalactiae, Fusobacterium nucleatum, and Ureaplasma parvum species [136]. Despite their typically low virulence outside the intrauterine environment, these microorganisms can impact placental vasculature when spread hematogenously, modifying endothelial permeability and allowing the entry of other pathogenic organisms, including Escherichia coli [53]. Oral bacteria like Fusobacterium or Capnocytophaga in placenta samples of women with preterm birth have been linked to periodontal disease that develops toward the end of pregnancy [140]. However, it's crucial to note that changes in flora are not solely associated with oral microorganisms. As pregnancy progresses, hormonal variations lead to shifts in gastrointestinal microbiota. Studies demonstrate significant alterations in the fecal flora composition from the first to the third trimester, including increased Proteobacteria and Actinobacteria and decreased Lactobacillus [53]. Microorganisms from the oral cavity and gastrointestinal tract, particularly Enterobacteriaceae, have been identified in the placental environment. This is linked to heightened immune tolerance during pregnancy, facilitating the movement of bacteria from the gastrointestinal system into the bloodstream. This establishes [136] a pathway through the bloodstream, connecting various organ systems to the uterine cavity. These changes, influenced by the evolving maternal immune environment, mainly manifest toward the end of pregnancy. The modified flora translocates into the bloodstream, reaching the amniotic and placental cavities, creating a proinflammatory environment.
Maternal Microbiome and Its Impact on Fetoplacental Growth and Development
The relationship between the maternal gut microbiota and the fetus is a topic of considerable discussion, primarily revolving around the concept of a "sterile womb." This idea proposes no direct contact between maternal gut bacteria and the fetus, suggesting that the fetus relies on metabolites derived from the gut microbiota. However, emerging evidence indicates the potential translocation of maternal gut microbiota to the uteroplacental unit [142]. Regardless of the microbiota in the uterus, maternal gut microbial metabolites play a vital role in supplying energy, nutrients, and essential vitamins like B complex, folate, choline, betaine, and SCFAs. Environmental variables, such as early life stress, can have long-lasting effects on the brain and behavior via the epigenome, which regulates gene expression [143,144]. The metabolic and immunological alterations observed in pregnant women are accompanied by changes in maternal gut microbiota, which begin during the second trimester and increase throughout the third trimester [36]. A shift towards low microbial alpha diversity marks these modifications (indicating reduced richness and abundance of taxa) and a heightened beta index (indicating increased variability in composition) [145]. This is linked to increased glycogen- and lactose-producing bacteria, decreased butyrate-producing bacteria, diminished diversity, and augmentation in Actinobacteria and Proteobacteria phyla.
Healthy human gut microbiota is dominated by phyla Firmicutes, Bacteroidetes, Actinobacteria, Proteobacteria, and Fusobacteria. The Firmicutes phylum comprises genera such as Lactobacillus, Bacillus, Clostridium, Bacillus, Enterococcus, and Ruminicoccus. Bacteroidetes consist of predominant genera such as Bacteroides and Prevotella. The Bifidobacterium genus mainly represents the less abundant Actinobacteria phylum [141]. Faecalibacterium, a butyrate-producing bacterium with anti-inflammatory properties and a member of the Firmicutes phylum, experiences a notable decrease in the third trimester of pregnancy. The third trimester shows elevated beta diversity, which correlates with weight gain, insulin insensitivity, and elevated fecal cytokines, indicating inflammation [140,146,147]. There is an increase in Firmicutes, related to the rise in the requirement for energy storage, and in Proteobacteria and Actinobacteria levels, which have a protective effect on both the mother and the fetus through pro-inflammatory processes [147]. These alterations in maternal gut flora match the fetus' metabolic demands, contribute to fetal body weight gain, and supply glucose, but they also cause maternal hyperglycemia [148]. During a typical pregnancy, the maternal gut's operational dynamics and bacterial makeup undergo transition in their diversity due to the inflammatory and immune adaptations essential for maintaining pregnancy. These changes are instigated by modified hormonal levels, particularly the pregnancy-specific hormone human chorionic gonadotropin (hCG).
Consequently, hCG plays a role in regulating the secretion of estrogen and progesterone, influencing the composition of the maternal gut microbiota. Elevated levels of progesterone extend gastrointestinal transit time, a pivotal factor in shaping the composition and functionality of the gut microbiota [149]. The alterations in the gut microbiota during a normal pregnancy are crucial for maintaining maternal well-being and fostering fetal development. Distinctive changes in the composition of the maternal gut microbiota are observed in complicated pregnancies, potentially linked to the increase in progesterone levels [150].
The microbial signatures have been associated with embryonic development, influencing the CNS and immune systems while balancing health and disease delicately. Microbial gut dysbiosis, characterized by an imbalance in microbiota homeostasis, has significant implications for overall health. In pregnancy, maternal gut dysbiosis denotes a disturbance in the adaptation of the microbiota to the specific conditions of pregnancy, posing risks to both the mother and the fetus. Various factors contribute to microbial gut dysbiosis in pregnant women, including maternal obesity, dietary patterns, stress, inflammation, infection, antibiotics, and antidepressants. Maternal obesity during pregnancy is linked to an elevation in Firmicutes, resulting in an increased ratio of gut Firmicutes to Bacteroidetes. This rise in Firmicutes may enhance calorie absorption, potentially contributing to weight gain and correlating with gut inflammation and increased intestinal permeability. Additionally, obesity during pregnancy can induce alterations in bacterial phyla compared to non-obese pregnancies [151].
Gut metabolites, derived from the fermentation of dietary fiber by microbiota, also include polyphenols that contribute to the nutritional programming of fetal growth and development through epigenetic mechanisms. These maternal metabolites traverse from the gut lumen to the bloodstream, gaining access to the fetus through the placenta and reaching fetal circulation, providing the necessary nutrients for fetal growth and development. Furthermore, these nutrients impact gene expression and contribute to fetal brain development. Some metabolites breach the fetal blood-brain barrier (BBB) and play a role in fetal CNS and immune system development by influencing dendritic T-cell activity and cytokine production [152]. Gut dysbiosis in preeclamptic patients had increased plasma levels of LPS and TMAO related to inflammation status [153].
Microbiota and Fetal Immune Development
The placental microbiome is distinct, resembles the mother‘s oral microbiota, and can negatively impact pregnancy outcomes [154]. Notably, the placental membranes act as a protective barrier in the fetal environment and possess bactericidal properties. This is attributed to cells like extravillous trophoblasts, natural killer cells, leukocytes, and macrophages. Despite bacteria potentially breaching the barrier through bacterial ligands, they became non-viable and fragmented upon passage. Additionally, there is a possibility that certain microorganisms may conceal themselves within placental trophoblasts [155]. During gestation in mice, fetal placental vascularization establishes contact with maternal circulation, enabling the transfer of metabolites, such as SCFAs, from maternal gut microbiota to the fetus. These metabolites contribute to the development of the BBB and innate immunity.
Preterm infants have been found to have a fetal inflammatory response syndrome (FIRS), which is characterized by higher fetal proinflammatory cytokines and hypoxic-ischemic events, as well as myelination failure [156] . Both rodent and human data suggest the role of maternal microbiota during pregnancy in the development of fetal innate and adaptive immunity [157]. Studies on germ-free pregnant mouse dams transiently colonized with E. coli revealed altered innate lymphoid cells in pups. Furthermore, mouse pups from dams treated with antibiotics during pregnancy showed impaired innate immunity at 14 days postpartum [36]. Furthermore, exposing nonobese diabetic mice to vancomycin during pregnancy enhanced offspring susceptibility to type 1 diabetes [158] . In mice, a limited amount of pre-B cells was discovered in bone marrow by gestation day 19, while T lymphocytes were detected at birth [159]. B- and T-cells emerge between 12 and 14 weeks of gestation in humans, steadily increasing until birth. Both innate and adaptive immunity are established by 16 weeks of gestation. The initial lymphoid tissues, including mesenteric lymph nodes and Peyer's patches, develop in the sterile environment of the fetus [160] . The immaturity of the immune system in early development aligns with the function of the underdeveloped intestinal barrier, leading to increased antigen passage across the intestine. Notably, in humans, mothers provide natural passive immunization during fetal life. Microbial antigens from maternal gut microbiota combine with maternal antibodies (antigen-IgG) and are transferred to the fetus through the placenta, triggering immune activation. Free dietary antigens may also traverse the placental barrier [161]. During the prenatal stage, intestinal digestion is minimal, and amniotic fluid components such as proteinase inhibitors influence the luminal environment and the production of antigenic compounds. It has been proposed that maternal microbiota metabolites are essential for immunological activities [162]. SCFAs produced by the maternal microbiota during pregnancy have been found to affect intestinal immunity, T-cell development, dendritic cell (DC) activity, and epithelial integrity [157]. Specific neuropeptides, vasoactive intestinal peptides, and norepinephrine play a role in modulating the functions of dendritic cells and cells in the intestinal wall and secondary lymphoid tissues, such as Peyer's patches. Furthermore, there is a suggestion that bacterial DNA transferred from the mother to the fetal gut stimulates mucosal immune development and influences the fetal immune system in preparation for the transfer of maternal-fetal microbiota postnatally [163] . In germ-free mice, the development of fetal thymic CD4+T cells and regulatory T cells (Treg cells) is compromised. Still, supplementation with the bacterial metabolite SCFA-acetate rescues this deficiency. Acetate induces the upregulation of the autoimmune regulator, contributing to Treg cell generation. In humans, low maternal serum acetate is associated with preeclampsia, a pregnancy-associated disorder affecting maternal cardiovascular and immune systems, leading to reduced Treg cells. Maternal immune changes in preeclampsia are reflected in the fetal immune system, suggesting a role for SCFAs produced by maternal microbiota in regulating maternal and fetal immune systems during pregnancy [158]. A Danish study found that antibiotic usage during pregnancy changed maternal microbiota and their metabolites and was associated with increased development of immunological diseases such as immune atopic dermatitis in newborns of mothers with atopy and increased risk of asthma in children 2-10 years old [164]. Maternal microbiome changes during pregnancy have been linked to type 1 diabetes and inflammatory bowel disease [165].
The enteric nervous system (ENS) undergoes development in the fetal window, originating from vagal and sacral neural crest cells. Maternal gut microbial metabolites, particularly SCFAs, regulate fetal ENS. SCFAs, transmitted from the maternal gut through the placenta to the fetus, serve as an energy source, regulate fetal gut epithelium, and influence the development of the fetal metabolic system, neural system, and immune responses [166,167]. Studies in germ-free mice demonstrate a significant reduction in the cross-placental transfer of microbial metabolites compared to specific pathogen-free mice, emphasizing the importance of maternal gut microbiota [168]. Maternal gut dysbiosis negatively impacts fetal intestine permeability and integrity [169]. Recent evidence indicates a direct influence of bacterial exposure on fetal gut colonization [170]. The placenta and amniotic fluid contain a distinct microbiota characterized by low richness, low diversity, and a prevalence of Proteobacteria. Similarities among the placenta, amniotic fluid, and infant meconium microbiota suggest cross-placental microbial transfer and prenatal seeding of the fetal gut. Meconium microbiota shows more resemblance to amniotic fluid than maternal fecal and vaginal microbiota, suggesting seeding from multiple maternal sites, with amniotic fluid contributing significantly [171] . Microbial contact can initiate healthy immune maturation during fetal life [170]. In both rodents and humans, the development of the digestive system aligns with the maturation of the immune system and the ENS. In mice, the intestinal epithelium undergoes the formation of villi and crypt structures, leading to restricted epithelial proliferation and the cytodifferentiation of villi into functional cell types in the small intestine. This period also witnesses the development of smooth muscle layers around the gut tube and the ENS. In mice, sensory and motor neurons project into the gut around embryonic day 14. Expression of brush border, crypt, and Paneth cells at low levels is observed around 14 postnatal days in mice, with complete intestinal morphogenesis occurring postnatally [172]. While many anatomical and cellular features develop in utero, the ENS becomes functional during the postnatal period in mice [173]. In humans, the development of the digestive tube, a precursor to the gut, begins in the third week of gestation, with crypt formation occurring around the 12th week and intestinal functions developing by the 24th week of gestation. This development coincides with the ENS, which is not fully mature at birth and continues to develop postnatally [174]. Enteroendocrine cells emerge as early as the 13th week of gestation. Key cells like brush border, crypt, and Paneth cells are expressed at low levels in the fetal intestine at 20 weeks of gestation, and their maturation occurs before birth. Tight junction proteins, including claudin, control the intestinal epithelial barrier, with claudin expression beginning as early as the 18th week of gestation. The integrity of this barrier is crucial for regulating transport across the lumen and excluding pathogens.
Consequently, intestinal morphogenesis is completed before birth in humans. Mucosal immunity starts developing in the human fetal intestine by 11–14 weeks gestation, with dendritic cells populating the developing intestine capable of responding to microbial stimuli and initiating T cell responses. By the 13th gestational week, memory T cells become abundant in the fetal intestine, indicating early immune priming. Specific immunomodulatory microbes with bacterial-like morphology have been identified in mid-gestation human meconium, accompanied by enriched taxa and T-cell patterns. Viable bacteria observed in the fetal intestine at mid-gestation can limit inflammatory potential by interacting with fetal immune cell populations. Fetal T cells demonstrate the ability to form memory in the intestine, suggesting that bacterial antigens contribute to T cell activation. These specific bacteria persist under nutrient-limiting conditions, utilize pregnancy hormones for growth, and can survive within phagocytes [175]. In humans, immunoglobulins cross the placenta during the fetal phase, producing a single layer between fetal and maternal circulation that allows for selective maternal-fetal macromolecular transfer. The transfer of IgG is modest throughout the first and second trimesters but increases during the third trimester [161]. Multiple research studies have identified bacterial DNA in the placenta and amniotic fluid. Investigations in mice reveal similarities between fetal intestine bacterial DNA, placental bacterial DNA, and maternal oral and vaginal DNA, with additional overlap observed in meconium [176]. Intriguingly, the human fetus starts producing meconium as early as 12 weeks of gestation, hinting at a potential exchange of microbiota between the fetal and maternal environments [177].
Maternal stress in the first week of pregnancy induces persistent changes in maternal gut microbiota, impacting the placental transfer of nutrients. Notably, male offspring, not females, exhibit significant neurodevelopmental alterations in the hypothalamic and limbic circuits, affecting stress responsivity [178]. These findings demonstrate that the makeup of maternal gut microbiota during pregnancy is an essential contributor to the metabolic programming of offspring [179]. Initially shielded by the placenta, the fetus gains an additional layer of protection through the maturation of the BBB during pregnancy. In rodents, the BBB starts forming around gestational days 13.5-15.5, characterized by a higher concentration of tight junction proteins and extracellular matrix components than the adult BBB. Notably, in germ-free mice, the BBB appears leaky around gestational day 16.5, emphasizing the crucial role of maternal microbiota in developing a functional BBB [180]. Some efflux transporters capable of excluding poisons are present in humans as early as eight weeks of gestation, and BBB components appear at 12 weeks. It is widely accepted that during gestation and in newborns, this barrier is immature or "leaky,"" making the developing brain more sensitive to medications or toxins entering the fetal circulation from the mother [152,181]. Elevated intestinal permeability during early pregnancy is believed to be linked to higher maternal levels of LPS and cytokines at the endometrial level. This phenomenon is thought to enhance the translocation of bacterial metabolites and bacteria from the intestinal lumen into the maternal circulation [145]. Bacteria can reach the placenta through the bloodstream, potentially facilitated by dendritic cells translocating from the gut epithelium to lymphoid organs. Maternal oral bacteria entering the bloodstream are also suggested as a source of fetal microbiota. This early prenatal microbiota may play a role in priming the immune system for the subsequent colonization of microbiota after birth. In the first year of life, the human intestine quickly becomes populated with microbiota, primarily strict anaerobes. By 2-5 years, the microbiota in infants becomes individually distinct from the composition and diversity seen in adults [182]. The first diffusion of maternal metabolites across the placenta promotes the fetal nervous system and HPA axis development. It is followed by gut bacteria penetrating, transferring the placenta, and transferring it into the fetal bloodstream. Thus, gut microbiota colonization could occur throughout fetal life [152,183]. Some bacteria may enter the fetal gut, but they are insufficient and unique enough to activate intestinal epithelial tolerance during the first interaction with microbiota after birth.
Gut Microbiota and Fetal Brain Development
The maternal gut microbiota during pregnancy is critical for developing the fetal central nervous system. Traditionally, the prevailing belief, based on the concept of a sterile womb, is that the offspring's GBA is established postnatally and regulated by the maternal gut microbiota transmitted during birth. However, there is a growing recognition of a limited number of distinctive bacteria found in fetuses, suggesting their potential role as transitional species facilitating the establishment of a complete microbiota after birth [157,184].
Factors such as maternal genetics, diet, health condition, stress, and medication can determine the microbiota-gut-fetal brain axis [185]. Animal and human studies have established a relationship between the maternal gut microbiota and fetal neurodevelopment. Rodent studies have indicated gender-dependent impaired prenatal brain development in germ-free mice [186]. The microbiota-deficient mice demonstrated altered gene expression are associated with neurotransmission, neuroplasticity, metabolism, and morphology in the hippocampus and thalamocortical neurodevelopment with sensorimotor behavior and pain perception after birth [132]. The development of the human brain and nervous system initiates at six weeks during the embryonic development and extends throughout pregnancy, continuing into puberty and beyond. Specific neurodevelopmental changes involving axonal growth, synapse formation, and dendritic and axonal arborization occur during gestation, culminating in establishing synaptic connections. Maternal gut microbiota regulates metabolites that reach the fetus through transplacental signaling, influencing fetal brain development [132].
Existing research extensively explores the relationship between gut microbiota and the gut-brain axis (GBA) postnatally. Still, there needs to be more understanding regarding this connection during the prenatal period [187]. While the fully functional bidirectional offspring GBA develops after birth, the rudimentary GBA is established in fetus under maternal gut microbial control.
Due to the absence of a functional GBA in fetal life, the maternal gut microbiota influences the rudimentary developed GBA in humans. Newborns are exposed to significant amounts of maternal vaginal, fecal, and skin microbiota during birth, with the colonization influenced by the delivery method (vaginal or C-section). Nursing further contributes to the transfer of maternal microbiota to the infant, with the composition influenced by the mother's health and gestational age. Gut closure, a developmental stage of intestinal maturation around six months, signifies establishing a functional GBA [188]. The functional GBA involves a complex bidirectional network encompassing the central nervous system (CNS), autonomic nervous system (ANS), ENS, vagus nerve (VN), neuroendocrine and neuroimmune systems, the HPA, and the gut/gut microbiota. The microbiota plays a crucial role in the GBA, contributing to nutrient bioconversion, detoxification, immune regulation, and protection against pathogens. Research indicates that maternal gut microbiota plays a critical role in fetal development by transferring metabolites and other factors to the fetus through the placenta [189].
Maternal gut microbiota plays a crucial role in supporting the healthy development of the fetal brain, influencing both its structural and functional connectivity and ultimately affecting cognitive performance and behavioral outcomes in offspring. Clinical studies indicate that disruptions in maternal gut microbiota, such as dysbiosis during pregnancy, can negatively impact fetal CNS's physiological and functional development. Instances of microbial depletion, caused by factors like infection or antibiotic treatment during pregnancy, have been associated with abnormal brain structure and function, contributing to maladaptive behaviors reminiscent of autism in offspring [131,132]. Depletion of maternal gut microbiota during pregnancy impacts gene expression in the developing fetal brain, particularly genes that regulate the development of new axons connecting the thalamus to the cortex, which is responsible for sensory processes. Gut microbiota generates neurotransmitters and neuromodulators, including serotonin, gamma-aminobutyric acid (GABA), and SCFAs. These bioactive substances are transported to the fetal brain through the placenta and blood-brain barrier. Maternal gut microbiota, particularly during pregnancy, produces SCFAs crucial for various differentiations facilitated by G protein-coupled receptors.
Additionally, maternal gut metabolites support fetal thalamocortical axogenesis. Notably, germ-free mice exhibited region-specific changes in neurotransmitter systems, with increased serotonin (5-HT) concentration in the hippocampus [186]. The placenta plays a crucial role in synthesizing 5-HT, influencing fetal CNS development by regulating cell proliferation, migration, and wiring during prenatal development. Tryptophan, the precursor to 5-HT, is derived from maternal gut metabolites. Placental 5-HT reaches the fetal forebrain during cortical neurogenesis and initial axon growth, with serotonergic neurons appearing in the fetal hindbrain around embryonic day 10.5 in mice [190]. Chronic mild stress during rat pregnancy increases free tryptophan concentration in both maternal blood and the fetal brain, leading to heightened anxiety in the offspring. In humans, placental synthesis of 5-HT occurs in the first and second trimesters of pregnancy [191]. Disruption of placental 5-HT signaling causes long-term behavioral problems, including anxiety after birth. The maternal gut microbiome also influences the formation of the fetal BBB. Animal studies have demonstrated that higher BBB permeability in germ-free mice is related to poorer brain innate immunity and reduced thalamocortical axon development [35]. The VN plays a significant role in innervating the intestine from the proximal duodenum to the distal descending colon, serving as a crucial bidirectional communication pathway between the gut and the brain. It conveys information about the gut's status, including chemical content, distension, and inflammation, to key brain regions such as the nucleus of the solitary tract (NTS), the paraventricular nucleus of the hypothalamus (PVN), and the arcuate nucleus. Metabolites produced by maternal gut microbiota, including SCFAs are transported through specific transporters across the gut epithelium, activating the VN.
Afferent fibers of the VN carry signals from the gut microbiota to the brain. In response to these signals, the brain sends feedback signals back to entero-epithelial cells through efferent fibers of the VN. This bidirectional communication system allows for the regulation of gut-brain interactions [192]. VN sensory and motor nuclei were discovered in E11-E14 mouse embryos [193]. In humans, morphological studies reveal a rapid developmental rise in the total number of myelinated vagal fibers from 24 weeks until adolescence, with the most significant increases reported from roughly 30-32 weeks of gestation to approximately six months after birth [194]. During the transition from the late second to the early third trimester, sympathetic activation increases, accompanied by increased parasympathetic regulation and baseline stability ability [174]. During the final trimester, myelination of vagal efferent fibers associated with heart activities begins[169].
The gut microbiota engages in bidirectional communication with the brain through the GBA, encompassing the CNS and ANS, sympathetic and parasympathetic components, including the VN and ENS, as well as the immune, endocrine (including the hypothalamic–pituitary–adrenal or HPA), and gut/gut microbiota systems (Figure 3). The reciprocal nature of the GBA network enables communication between the emotional and cognitive centers of the brain and the peripheral intestinal functions of the ENS [195]. The gut microbiota produces metabolites, hormones, and neurotransmitters, creating a connection between the gut and the brain.
Conversely, the brain influences intestinal activities, including the behavior of functional immune effector cells. It is improbable that the prenatal gut microbiota can independently generate metabolites and influence CNS development. It is reasonable to infer that maternal microbial metabolites and other factors might be involved in fetal development, including the formation of the GBA. This inference gains support from observations indicating that disruptions in maternal gut microbiota during pregnancy are linked to irregularities in the developing GBA, encompassing gastrointestinal, neuronal, immune, and hormonal components [196]. Maternal gut dysbiosis activates microglia, leading to systemic inflammation, resulting in neuroinflammation [197]. While eubiosis promotes myelination, dysbiosis-associated increase in proinflammatory cytokines can damage differentiating neurons and oligodendrocytes during active myelination, supporting the direct impact of maternal gut dysbiosis-induced inflammation on fetal neuroinflammation [187].
Maternal gut microbiota activates the fetal neuroendocrine HPA axis, producing cortisol and influencing the physiological stress response [198]. This activation occurs by releasing proinflammatory mediators, microbial antigens, and prostaglandins that can cross the BBB. Dysbiosis in maternal gut microbiota may result in constitutive hyperactivity of the HPA axis [199]. The fetal HPA axis is crucial for the differentiation and maturation of fetal lungs, liver, and CNS. In species giving birth to mature young (such as primates, sheep, and guinea pigs), a significant portion of neuroendocrine maturation occurs in utero [146]. In contrast, species giving birth to immature young (like rats, rabbits, and mice) experience neuroendocrine development postnatally.
Consequently, manipulations during the fetal or neonatal stages affect different phases of neuroendocrine development, depending on the species studied [200]. The human HPA axis becomes active at 11 weeks’ gestation, with hormonal activity detectable between 8 and 12 weeks. The glucocorticoid receptor (GR) mRNA is present in the adrenal gland by 8–10 weeks of life, with limited knowledge about later developmental changes in GR expression [201]. Corticotropin-releasing hormone (CRH) immunoactivity and bioactivity are evident in fetal hypothalamic tissue extracts by 12–13 week gestation, increasing with gestational age [202]. During pregnancy, the fetal hypothalamus and the placenta produce CRH, regulating HPA axis maturation and adrenocorticotrophin (ACTH) secretion. ACTH, in turn, coordinates fetal adrenocortical growth, angiogenesis, differentiation, and steroidogenesis. Cortisol plays a crucial role in maintaining intrauterine homeostasis and fetal tissue maturation, with de novo synthesis starting in humans after 28 weeks of gestation. In response to acute stress like arterial hypotension, the fetal hypothalamus releases CRH, stimulating the secretion of fetal ACTH, which enhances cortisol production [203]. Placental estrogens also impact the fetal HPA axis by converting active cortisol to inactive cortisone, thereby reducing fetal cortisol levels. Recent epidemiological evidence suggests that stress during the fetal period and exposure to corticosteroids can lead to lasting changes in neural pathways, potentially predisposing individuals to diseases later in life [204].
Conclusions
Epigenetic alterations related to maternal nutrition and environmental exposures may affect fetal growth. Nutritional epigenetics has been viewed as an attractive tool to prevent pediatric developmental diseases. In recent years, epigenetics roles have been linked to the pathogenesis of type 2 diabetes mellitus, obesity, inflammation, and neurocognitive disorders. Although the possibility of developing a treatment or discovering preventative measures for these non-communicable diseases is exciting, current knowledge in nutritional epigenetics is limited and warrants studies to expand the available tools like AI, resources such as next-generation RNA sequencing, and others.
Epigenetic regulation maintains gestational integrity and fetoplacental development. Unveiling epigenomic modification can expand understanding of the dynamic developmental processes to intervene for a better new life. Disseminating epigenetic regulation may allow clinicians to advise women at increased risk of adverse pregnancy outcomes and develop precise, personalized, risk-specific interventions. Several clinical trials have highlighted the effects of maternal nutrients on fetal epigenetic programming, which influences fetal growth, birth weight, and brain performance in the subsequent stages (Table 2). Previous studies identified epigenetic biological markers specific to the placenta in maternal blood, suggesting that epigenetic biological markers from the placenta may be released or leaked into the mother’s bloodstream during pregnancy. A better understanding of using nutrients or bioactive food components for maintaining health and preventing diseases through modifiable epigenetic mechanisms is required.
Epigenetic reprogramming is essential in embryonic and early postnatal development, and epigenetic deregulation is recognized in the etiology of several developmental disorders. The various in-utero exposures and maternal nutritional state may affect the epigenome differently. Although it is widely agreed that epigenomic dysregulation can program the later disease risk in offspring, effective intervention strategies aiming to modulate the epigenome and prevent the development of complex human diseases because of nutritional programming are still challenging. Early biological markers should be established during the prenatal period to detect systemic pathophysiological processes (nonspecific tissue) or local processes (specific tissue, such as the placenta).

Table 1.
Fetal epigenetic modifications to the maternal dietary intervention: evidence from pre-clinical trials.
Table 1.
Fetal epigenetic modifications to the maternal dietary intervention: evidence from pre-clinical trials.
| Aim | Study design | Primary outcome | Ref |
|---|---|---|---|
| To distinguish between direct transmission of epigenetic states and de novo epigenetic-induced marks in each generation in response to energy diets | Female Wistar rats, dietary energy increased by 25% at conception in F0 and maintained at this level up to generation F3; Epigenetic marks over four generations studied | -The transgenerational effects on offspring’s phenotype directly reflected with altered DNA methylation of specific genes -Induction of de novo epigenetic marks in each generation indicated heritability of epigenetic changes due to germline transmission |
[223] |
| Effect of dietary choline on fetal brain and memory function in deficient mice | Timed-pregnant C57 BL/6 mice, choline diet- 1.1 g/kg choline chloride, from embryonic day 12-17, brain analysis on day 17 | -Decreased DNA global methylation in the ventricular and subventricular zones of the choline-deficient fetal brain -Decreased DNAm of the gene (Cdkn3) expression of a cell cycle regulator indicated altered brain development |
[14] |
| Prenatal restricted diet and trans-generational epigenetic alterations in four successive generations in rats | Wistar rats, control- chow diet, restricted group- 50% of their daily intake, from the appearance of vaginal plug until parturition | -Prenatal dietary restriction affected expression of genes involved in epigenetic mechanisms in the liver across generations -Influenced the global histone H3 acetylation in fetal liver |
[224] |
| The n-3 PUFA deficiency at the time of pregnancy and lactation and epigenetic changes in the brain of the adult offspring in mice | C57BL/6J mice, n-3 PUFA deficient diet- sunflower oil with high LA and no n-3 PUFA, n-3 PUFA adequate diet- fish oil, flax seed and sunflower oil containing 0.47% DHA and 5.25% of LA | -Decreased BDNF expression in deficient diet associated with a hypermethylated DNA at CpG sites indicated a potential long-term impacts of brain development in offspring | [225] |
| Folic acid availability or deficiency on oocytes development and epigenetic effects in next generation progeny | Female BALB/c mice, folic acid deficient (7-fold) or supplement (10-fold) starting from 4 wks prior mating, throughout gestation and lactation | -F2 litters showed a higher resorption rate, reduced litter size, abnormal embryo outcomes -Both low and high folate prior to oocyte maturation compromised oocyte quality and affecting early development of next generation by altering DNA methylation patterns |
[226] |
| Maternal n-3 PUFA deficiency during pregnancy and changes in the epigenetic modulators of the placenta | Female Swiss albino mice, n-3 PUFA deficient diet - n-6/n-3 PUFA= 50/1, 0.13% energy from ALA; n-3 sufficient diet- n-6/ n-3 PUFA= 2/1; 2.26% energy from ALA; | -Global hypermethylation in F0 and F1 of n-3 deficient placenta -Increased expression of DNMT3A and DNMT3B in F0 and F1 n-3 deficient placenta |
[227] |
| Maternal micronutrients and omega-3 fatty acids on global methylation patters in the brain | Pregnant rats fed with folic acid (normal and excess) in presence or absence of Vit B12; Omega-3 supplemented in Vit B12 deficient rats | -Global hypomethylation in offspring brain at birth. -Despite postnatal control diet, cortex of offspring showed hypermethylation at adult age -Omega-3 fatty acids reversed methylation patterns |
[228] |
| Effects of dietary combinations of B12 and folic acid on genome imprinting modulators | 6-week-old C57BL/6 males mice fed with standard chow diet and females with different levels of folic acid and B12 for two generations | Non-coding RNA expression of IGF2R and KCNQ1OT1 were sensitive to the different dietary combination folic acid and B12 in the mice offspring indicated epigenetic programming | [229] |
| Maternal low protein diet (MLPD) and epigenetic alterations in gene expression of brain renin-angiotensin system (RAS) in mice | FVB/NJ mice, MLPD-50% protein depletion, from gD10.5 to 17.5 | -MLPD results hypomethylation of ACE-1 promoter regions in fetal brain -Changes in DNA methylation and miRNA expression modulate key regulators of hypertension in adults |
[230] |
| Effect of a maternal HFD on fetal gluconeogenic gene expression and regulation of histone modifications | Timed-pregnant obese resistant rats, control - 64%, 20%, 16% and HFD- 35%, 20% and 45% of carbohydrate, protein and fat respectively from embryonic day 2 to 20 |
In utero exposure to HFD programmed the gluconeogenic capacity of offspring through epigenetic modifications and predispose offspring with altered insulin sensitivity in adulthood | [231] |
| Maternal calorie restriction (CR) on DNA methylation and placental gene expression | C57/BL6 mice, control- chow diet, CR- 50% (w/w) diet of their daily intake from gD 10 to 19 | Altered DMRs in CR mice associated with IUGR phenotypes enriched with micro-RNA target genes linked with risk factors of cardiovascular and neurological diseases | [232] |
| Maternal HFD during pregnancy and its effects on regulation of gene functions |
Female C57BL6J, HFD-22.6% fat, 48.6% carbohydrate, 23% protein; Standard chow diet- 10% fat, 68.8% carbohydrate, 18% protein; prior to conception, during pregnancy and lactation | Altered hepatic expression of insulin like growth factor-2 and key microRNAs in the adult offspring | [233] |
HFD- High fat diet; LA- Linoleic acid; ALA-alpha-linolenic acid; PUFA- polyunsaturated fatty acids; BDNF- Brain-derived neurotrophic factor; DHA- Docosahexaenoic acid; DMR-differentially methylated regions; Cdkn3- cyclin dependent kinase inhibitor 3; ACE-1- Angiotensin-converting enzyme 1; IUGR- Intrauterine growth restriction; DNMT3A- DNA methyltransferase 3A; DNMT3B- DNA methyltransferase 3B; gD- gestational day;.
Table 2.
Role of maternal nutrients in fetal epigenetic programming: consolidated clinical trials.
| Aim | Subjects & design | Analysis & method | Primary outcome | Ref. |
|---|---|---|---|---|
| In utero DHA supplement and fetal epigenome | -Pregnant women in Australia -DHA (800 mg), EPA (100 mg) supplement per day from gD 21 until delivery |
Blood spots at birth (n=991) |
- 21 differentially methylated regions (DMRs) in genes associated with appetite regulation, neurodevelopment -DMRs was greater in males than females |
[205] |
| Association between maternal fatty acids (FA) and newborn DNA methylation (DNAm) |
-Pregnant women in the United States -Plasma phospholipid FAs at preconception 4mo before pregnancy (n = 346) and at 8wks (n = 374) of gestation |
Cord blood DNA from singletons | -Preconception marine PUFA levels correlated with increased DNAm of GRAMD2A and HTR1B genes involved in neurological functions -Preconception SFA levels correlated with increased DNAm at KIF25-AS1 and SLC39A14 genes involved in microtubule motor activity and neurodegeneration respectively -FAs levels at 8 weeks of gestation were unrelated to DNAm |
[206] |
| Dietary PUFA during pregnancy and methylation of imprinted genes | DHA (400mg/day, n=131) or placebo (n=130) from gD 18-22 until delivery | DNA methylation of IGF2 promoter 3 (P3), IGF2 DMRs, and H19 DMR in cord blood mononuclear cells | - DNAm of IGF2 P3 higher (p=0.04) in preterm infants of DHA group than placebo - IGF2 DMRs higher in DHA group than placebo infants of overweight mothers - H19 DMR lower in infants of normal weight mothers in DHA group |
[207] |
| Maternal micronutrient supplementation and DNAm in their children | Indian (n=698) and Gambian (n=293) pregnant women before and during pregnancy |
-Micronutrient rich snack periconceptionally (India) -Micronutrient tablet daily until confirmation of pregnancy (Gambia) |
-No changes in DNAm children (5-7 yrs) from the Indian cohort -In the Gambian cohort, Changes in DMRs involved in angiogenesis and cell growth; cadherin 18 (CDH18) and catenin alpha 2 (CTNNA2) implicated to the brain development |
[208] |
| Prenatal n-3 PUFA intake and DMRs in cord blood | Pregnant women, n=577, Italy | Pregnant women 45–64% of daily calories from carbohydrates, 20–35% of daily calories from fats and 60g/day of proteins and 3 fishes per week | -Reduced methylation of MSTN, ATP8B3, IFNA13 and GABBR2 genes in newborns from either low or high n-3 PUFA diet compared to medium n-3 PUFA diet -Over expression of these genes may lead to insulin resistance and adiposity |
[209] |
| Maternal dietary FAs quality on epigenetic aging and newborn cardiometabolic risk | Healthy mothers and children, n=224, Australia | Body fat, aortic intima-media thickness, heart rate variability and epigenetic age acceleration in newborn infants | - Omega-3 FAs showed a beneficial association with newborn epigenetic age acceleration -Maternal dietary FAs quality associated with epigenetic aging in newborns |
[210] |
| Vitamin D supplementation and epigenetic gestational age acceleration (GAA) in newborn | Pregnant women, n=92, multiethnic population, USA | - 4000 IU/ day Vit D3 or placebo plus prenatal 400 IU vitamin D3 during pregnancy | -GAA associated with higher birth weight -No association between vitamin D3 supplementation and GAA |
[212] |
| Folic acid beyond first trimester and DNAm of genes related to brain development and function | Pregnant women, n=86, Ireland | 400 µg folic acid/day, through the second and third trimesters compared with placebo | -Lower DNAm of LINE-1, IGF2, and BDNF -Continued folic acid supplementation in pregnancy results in significant changes in DNAm in cord blood of genes related to brain development |
[213] |
| Maternal choline intake on methylations of cortisol regulating genes in placenta and cord venous blood | Pregnant women, n=26, USA | Choline supplementation- 480 mg/day and 930 mg/day for 12 weeks from 26-29 gestational weeks until delivery | -Higher placental promoter methylation of cortisol regulating genes, CRH, NR3C1 -Lower cord blood promoter methylation of CRH, NR3C1 -maternal choline intake modulates the epigenetic state of genes that regulate fetal HPA axis |
[16] |
| Maternal dietary glycaemic index on cord blood DNAm | Mother offspring pairs, n=2003, U.K, Netherlands and Spain | Maternal dietary glycaemic index calculated by FFQ and correlated with cord blood DNAm | Maternal glycaemic index associated with the changes in DNAm of genes associated with neurodevelopment, lipid metabolism in overweight/obesity subjects | [214] |
| Parental dietary quality on offspring DNAm | Families, n=1124, Ireland | Maternal diet in first trimester and paternal diet were assessed by FFQ | -Maternal healthy eating index inversely associated with DNAm of PLEKHM1 gene (bone development) -Prenatal parental dietary quality might influence offspring DNAm during childhood |
[215] |
| Mediterranean diet during pregnancy and neonatal DNA methylation at birth | Mother-infant pairs, n= 390, USA | Overall dietary assessment by FFQ in periconception and once in each trimester | -Lower adherence to Mediterranean diet results in a greater odd of hypomethylation at the MEG3-IG DMR indicating dietary effects on genome imprinting | [216] |
| Mediterranean diet adherence during pregnancy and imprinted gene methylation of brain task | Mother/infant pairs, n=325, USA | Maternal periconceptional FFQ and correlated with child behavioural outcomes at 2 years of age | - Adherence to a Mediterranean diet in early pregnancy was associated with favourable neurobehavioral outcomes in early childhood -Sex-dependent methylation differences of MEG3, IGF2 DMRs. |
[217] |
| Dietary fat intake during pregnancy and imprinting gene methylations | Pregnant women, N=154, Eastern Massachusetts | Maternal fat intake during first and second trimester by FFQ | Maternal total fat and PUFA intake inversely correlated with IGF2-DMR and positively correlated with H19-DMR (negative regulation of body weight and cell proliferation) methylation | [219] |
| Effect of substituted low glycaemic index (GI) diet or GI diet on epigenetic profile of the offspring at 5 years of age | 5-year-old children from ROLO kids’ study, n=60-63, Australia, Ireland | High GI diet substituted with low GI from second trimester until delivery | No association between maternal factors due to substituted GI or low GI diet on DNAm status of the offspring at 5 years age | [220] [221] |
| Mediterranean diet adherence during pregnancy, fetal gut microbiota and offspring epigenetic regulation | Pregnant women, n=41, USA | Dietary patterns assessed by FFQ, Mediterranean diet adherence scores correlated with neonatal microbiome and fetal epigenetic programming | With adherence to Mediterranean diet results a greater abundance of Pasteurellaceae, Bacteroidaceae and other short-chain fatty acid-producing species and connected with fetal DMRs of in utero development | [222] |
DHA-Docosahexaenoic acid; EPA-Eicosapentaenoic acid; DMR-differentially methylated regions; DNAm-DNA methylation; GRAMD2A-GRAM domain containing 2A; HTR1B- 5-Hydroxytryptamine Receptor 1B; KIF25-AS1- Kinesin Family Member 25 Antisense RNA 1; SLC39A14- solute carrier family 39 member 14; IGF2- insulin like growth factor 2; H19-gene for a long noncoding RNA; MSTN-myostatin; ATP8B3-ATPase Phospholipid Transporting 8B3; IFNA13-Interferon Alpha 13; GABBR2-gamma-aminobutyric acid type B receptor subunit 2; FFQ-food frequency questionnaire; SFA-saturated fatty acid; PUFA- polyunsaturated fatty acids; LINE-1-long interspersed element 1; BDNF-Brain-derived neurotrophic factor; CRH- Cortisol regulating hormone; NR3C1- glucocorticoid receptor; HPA- hypothalamic-pituitary-adrenal axis; PLEKHM1-pleckstrin homology and RUN domain containing M1; MEG3- maternally expressed 3 gene; gD- gestational day;.
Author Contributions
AKDR conceptualized the idea and wrote the original draft. SB wrote and edited the manuscript. RM edited the manuscript. BN wrote parts of the manuscript.
Funding
No funding was available.
Conflicts of Interest
Authors declare no conflict of interest.
References
- Duttaroy, A.K. Docosahexaenoic acid supports feto-placental growth and protects cardiovascular and cognitive function: A mini review. European Journal of Lipid Science and Technology 2016, 118, 1439-1449. [CrossRef]
- Dimasuay, K.G.; Boeuf, P.; Powell, T.L.; Jansson, T. Placental Responses to Changes in the Maternal Environment Determine Fetal Growth. Front Physiol 2016, 7, 12. [CrossRef]
- Duttaroy, A.K. Influence of Maternal Diet and Environmental Factors on Fetal Development. Nutrients 2023, 15. [CrossRef]
- Duttaroy, A.K. Transport of fatty acids across the human placenta: A review. Prog Lipid Res 2009, 48, 52-61. [CrossRef]
- Basak, S.; Duttaroy, A.K. Maternal PUFAs, Placental Epigenetics, and Their Relevance to Fetal Growth and Brain Development. Reprod Sci 2023, 30, 408-427. [CrossRef]
- Kuzawa, C.W. Fetal origins of developmental plasticity: Are fetal cues reliable predictors of future nutritional environments? American Journal of Human Biology 2005, 17, 5-21.
- Bird, A. Perceptions of epigenetics. Nature 2007, 447, 396-398. [CrossRef]
- Chandak, G.R.; Silver, M.J.; Saffari, A.; Lillycrop, K.A.; Shrestha, S.; Sahariah, S.A.; Di Gravio, C.; Goldberg, G.; Tomar, A.S.; Betts, M.; et al. Protocol for the EMPHASIS study; epigenetic mechanisms linking maternal pre-conceptional nutrition and children’s health in India and Sub-Saharan Africa. BMC Nutrition 2017, 3, 81. [CrossRef]
- Yajnik, C.S.; Deshpande, S.S.; Jackson, A.A.; Refsum, H.; Rao, S.; Fisher, D.J.; Bhat, D.S.; Naik, S.S.; Coyaji, K.J.; Joglekar, C.V.; et al. Vitamin B12 and folate concentrations during pregnancy and insulin resistance in the offspring: The Pune Maternal Nutrition Study. Diabetologia 2008, 51, 29-38. [CrossRef]
- Burdge, G.C.; Hanson, M.A.; Slater-Jefferies, J.L.; Lillycrop, K.A. Epigenetic regulation of transcription: A mechanism for inducing variations in phenotype (fetal programming) by differences in nutrition during early life? British Journal of Nutrition 2007, 97, 1036-1046. [CrossRef]
- Burdge, G.C.; Lillycrop, K.A. Fatty acids and epigenetics. Curr Opin Clin Nutr Metab Care 2014, 17, 156-161. [CrossRef]
- Srinivas, V.; Molangiri, A.; Mallepogu, A.; Kona, S.R.; Ibrahim, A.; Duttaroy, A.K.; Basak, S. Maternal n-3 PUFA deficiency alters uterine artery remodeling and placental epigenome in the mice. J Nutr Biochem 2021, 96, 108784-108796. [CrossRef]
- Srinivas, V.; Varma, S.; Kona, S.R.; Ibrahim, A.; Duttaroy, A.K.; Basak, S. Dietary omega-3 fatty acid deficiency from pre-pregnancy to lactation affects expression of genes involved in hippocampal neurogenesis of the offspring. Prostaglandins, Leukotrienes and Essential Fatty Acids 2023, 191, 102566. [CrossRef]
- Niculescu, M.D.; Craciunescu, C.N.; Zeisel, S.H. Dietary choline deficiency alters global and gene-specific DNA methylation in the developing hippocampus of mouse fetal brains. FASEB journal : Official publication of the Federation of American Societies for Experimental Biology 2006, 20, 43-49. [CrossRef]
- Craciunescu, C.N.; Albright, C.D.; Mar, M.H.; Song, J.; Zeisel, S.H. Choline availability during embryonic development alters progenitor cell mitosis in developing mouse hippocampus. J Nutr 2003, 133, 3614-3618. [CrossRef]
- Jiang, X.; Yan, J.; West, A.A.; Perry, C.A.; Malysheva, O.V.; Devapatla, S.; Pressman, E.; Vermeylen, F.; Caudill, M.A. Maternal choline intake alters the epigenetic state of fetal cortisol-regulating genes in humans. The FASEB Journal 2012, 26, 3563-3574. [CrossRef]
- Smith, Z.D.; Chan, M.M.; Humm, K.C.; Karnik, R.; Mekhoubad, S.; Regev, A.; Eggan, K.; Meissner, A. DNA methylation dynamics of the human preimplantation embryo. Nature 2014, 511, 611-615. [CrossRef]
- Okae, H.; Chiba, H.; Hiura, H.; Hamada, H.; Sato, A.; Utsunomiya, T.; Kikuchi, H.; Yoshida, H.; Tanaka, A.; Suyama, M.; et al. Genome-wide analysis of DNA methylation dynamics during early human development. PLoS Genet 2014, 10, e1004868. [CrossRef]
- Slieker, R.C.; Bos, S.D.; Goeman, J.J.; Bovee, J.V.; Talens, R.P.; van der Breggen, R.; Suchiman, H.E.; Lameijer, E.W.; Putter, H.; van den Akker, E.B.; et al. Identification and systematic annotation of tissue-specific differentially methylated regions using the Illumina 450k array. Epigenetics Chromatin 2013, 6, 26. [CrossRef]
- Slieker, R.C.; Roost, M.S.; van Iperen, L.; Suchiman, H.E.; Tobi, E.W.; Carlotti, F.; de Koning, E.J.; Slagboom, P.E.; Heijmans, B.T.; Chuva de Sousa Lopes, S.M. DNA Methylation Landscapes of Human Fetal Development. PLoS genetics 2015, 11, e1005583. [CrossRef]
- Faulk, C.; Dolinoy, D.C. Timing is everything: The when and how of environmentally induced changes in the epigenome of animals. Epigenetics 2011, 6, 791-797. [CrossRef]
- Fukumoto, K.; Ito, K.; Saer, B.; Taylor, G.; Ye, S.; Yamano, M.; Toriba, Y.; Hayes, A.; Okamura, H.; Fustin, J.-M. Excess S-adenosylmethionine inhibits methylation via catabolism to adenine. Communications Biology 2022, 5, 313. [CrossRef]
- Inoue, S.; Honma, K.; Mochizuki, K.; Goda, T. Induction of histone H3K4 methylation at the promoter, enhancer, and transcribed regions of the Si and Sglt1 genes in rat jejunum in response to a high-starch/low-fat diet. Nutrition 2015, 31, 366-372. [CrossRef]
- Fang, M.; Chen, D.; Yang, C.S. Dietary polyphenols may affect DNA methylation. J Nutr 2007, 137, 223S-228S. [CrossRef]
- Dolinoy, D.C.; Weidman, J.R.; Waterland, R.A.; Jirtle, R.L. Maternal genistein alters coat color and protects Avy mouse offspring from obesity by modifying the fetal epigenome. Environ Health Perspect 2006, 114, 567-572. [CrossRef]
- Day, J.K.; Bauer, A.M.; DesBordes, C.; Zhuang, Y.; Kim, B.E.; Newton, L.G.; Nehra, V.; Forsee, K.M.; MacDonald, R.S.; Besch-Williford, C.; et al. Genistein alters methylation patterns in mice. J Nutr 2002, 132, 2419S-2423S. [CrossRef]
- Cooper, W.N.; Khulan, B.; Owens, S.; Elks, C.E.; Seidel, V.; Prentice, A.M.; Belteki, G.; Ong, K.K.; Affara, N.A.; Constancia, M.; et al. DNA methylation profiling at imprinted loci after periconceptional micronutrient supplementation in humans: Results of a pilot randomized controlled trial. FASEB J 2012, 26, 1782-1790. [CrossRef]
- Jiang, X.; Yan, J.; West, A.A.; Perry, C.A.; Malysheva, O.V.; Devapatla, S.; Pressman, E.; Vermeylen, F.; Caudill, M.A. Maternal choline intake alters the epigenetic state of fetal cortisol-regulating genes in humans. FASEB J 2012, 26, 3563-3574. [CrossRef]
- Li, C.C.; Cropley, J.E.; Cowley, M.J.; Preiss, T.; Martin, D.I.; Suter, C.M. A sustained dietary change increases epigenetic variation in isogenic mice. PLoS Genet 2011, 7, e1001380. [CrossRef]
- Li, Y. Epigenetic Mechanisms Link Maternal Diets and Gut Microbiome to Obesity in the Offspring. Front Genet 2018, 9, 342. [CrossRef]
- Ruiz-Trivino, J.; Alvarez, D.; Cadavid, J.A.; Alvarez, A.M. From gut to placenta: Understanding how the maternal microbiome models life-long conditions. Front Endocrinol (Lausanne) 2023, 14, 1304727. [CrossRef]
- Basak, S.; Das, R.K.; Banerjee, A.; Paul, S.; Pathak, S.; Duttaroy, A.K. Maternal Obesity and Gut Microbiota Are Associated with Fetal Brain Development. Nutrients 2022, 14. [CrossRef]
- Cui, J.; Wang, J.; Wang, Y. The role of short-chain fatty acids produced by gut microbiota in the regulation of pre-eclampsia onset. Front Cell Infect Microbiol 2023, 13, 1177768. [CrossRef]
- Nakajima, A.; Habu, S.; Kasai, M.; Okumura, K.; Ishikawa, D.; Shibuya, T.; Kobayashi, O.; Osada, T.; Ohkusa, T.; Watanabe, S.; et al. Impact of maternal dietary gut microbial metabolites on an offspring's systemic immune response in mouse models. Bioscience of microbiota, food and health 2020, 39, 33-38. [CrossRef]
- Braniste, V.; Al-Asmakh, M.; Kowal, C.; Anuar, F.; Abbaspour, A.; Toth, M.; Korecka, A.; Bakocevic, N.; Ng, L.G.; Kundu, P.; et al. The gut microbiota influences blood-brain barrier permeability in mice. Sci Transl Med 2014, 6, 263ra158. [CrossRef]
- Miko, E.; Csaszar, A.; Bodis, J.; Kovacs, K. The Maternal-Fetal Gut Microbiota Axis: Physiological Changes, Dietary Influence, and Modulation Possibilities. Life (Basel) 2022, 12. [CrossRef]
- Basak, S.; Das, R.K.; Banerjee, A.; Paul, S.; Pathak, S.; Duttaroy, A.K. Maternal Obesity and Gut Microbiota Are Associated with Fetal Brain Development. Nutrients 2022, 14, 4515.
- Millership, S.J.; Van de Pette, M.; Withers, D.J. Genomic imprinting and its effects on postnatal growth and adult metabolism. Cellular and molecular life sciences : CMLS 2019, 76, 4009-4021. [CrossRef]
- Anway, M.D.; Cupp, A.S.; Uzumcu, M.; Skinner, M.K. Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science 2005, 308, 1466-1469. [CrossRef]
- Cropley, J.E.; Suter, C.M.; Beckman, K.B.; Martin, D.I. Germ-line epigenetic modification of the murine A vy allele by nutritional supplementation. Proc Natl Acad Sci U S A 2006, 103, 17308-17312. [CrossRef]
- Rakyan, V.K.; Beck, S. Epigenetic variation and inheritance in mammals. Curr Opin Genet Dev 2006, 16, 573-577. [CrossRef]
- Ng, S.F.; Lin, R.C.; Laybutt, D.R.; Barres, R.; Owens, J.A.; Morris, M.J. Chronic high-fat diet in fathers programs beta-cell dysfunction in female rat offspring. Nature 2010, 467, 963-966. [CrossRef]
- Lesch, B.J.; Tothova, Z.; Morgan, E.A.; Liao, Z.; Bronson, R.T.; Ebert, B.L.; Page, D.C. Intergenerational epigenetic inheritance of cancer susceptibility in mammals. Elife 2019, 8. [CrossRef]
- Chen, P.Y.; Ganguly, A.; Rubbi, L.; Orozco, L.D.; Morselli, M.; Ashraf, D.; Jaroszewicz, A.; Feng, S.; Jacobsen, S.E.; Nakano, A.; et al. Intrauterine calorie restriction affects placental DNA methylation and gene expression. Physiol Genomics 2013, 45, 565-576. [CrossRef]
- Maloyan, A.; Muralimanoharan, S.; Huffman, S.; Cox, L.A.; Nathanielsz, P.W.; Myatt, L.; Nijland, M.J. Identification and comparative analyses of myocardial miRNAs involved in the fetal response to maternal obesity. Physiol Genomics 2013, 45, 889-900. [CrossRef]
- Wehbe, N.; Nasser, S.A.; Pintus, G.; Badran, A.; Eid, A.H.; Baydoun, E. MicroRNAs in Cardiac Hypertrophy. Int J Mol Sci 2019, 20. [CrossRef]
- Colpaert, R.M.W.; Calore, M. MicroRNAs in Cardiac Diseases. Cells 2019, 8. [CrossRef]
- Duisters, R.F.; Tijsen, A.J.; Schroen, B.; Leenders, J.J.; Lentink, V.; van der Made, I.; Herias, V.; van Leeuwen, R.E.; Schellings, M.W.; Barenbrug, P.; et al. miR-133 and miR-30 regulate connective tissue growth factor: Implications for a role of microRNAs in myocardial matrix remodeling. Circ Res 2009, 104, 170-178, 176p following 178. [CrossRef]
- Khorram, O.; Chuang, T.D.; Pearce, W.J. Long-term effects of maternal undernutrition on offspring carotid artery remodeling: Role of miR-29c. J Dev Orig Health Dis 2015, 6, 342-349. [CrossRef]
- Plagemann, A. 'Fetal programming' and 'functional teratogenesis': On epigenetic mechanisms and prevention of perinatally acquired lasting health risks. J Perinat Med 2004, 32, 297-305. [CrossRef]
- Godfrey, K.M.; Lillycrop, K.A.; Burdge, G.C.; Gluckman, P.D.; Hanson, M.A. Epigenetic mechanisms and the mismatch concept of the developmental origins of health and disease. Pediatr Res 2007, 61, 5R-10R. [CrossRef]
- Sookoian, S.; Gianotti, T.F.; Burgueno, A.L.; Pirola, C.J. Fetal metabolic programming and epigenetic modifications: A systems biology approach. Pediatr Res 2013, 73, 531-542. [CrossRef]
- Dunlop, A.L.; Mulle, J.G.; Ferranti, E.P.; Edwards, S.; Dunn, A.B.; Corwin, E.J. Maternal Microbiome and Pregnancy Outcomes That Impact Infant Health: A Review. Adv Neonatal Care 2015, 15, 377-385. [CrossRef]
- Enstad, S.; Cheema, S.; Thomas, R.; Fichorova, R.N.; Martin, C.R.; O'Tierney-Ginn, P.; Wagner, C.L.; Sen, S. The impact of maternal obesity and breast milk inflammation on developmental programming of infant growth. Eur J Clin Nutr 2021, 75, 180-188. [CrossRef]
- Calabuig-Navarro, V.; Puchowicz, M.; Glazebrook, P.; Haghiac, M.; Minium, J.; Catalano, P.; Hauguel deMouzon, S.; O'Tierney-Ginn, P. Effect of omega-3 supplementation on placental lipid metabolism in overweight and obese women. Am J Clin Nutr 2016, 103, 1064-1072. [CrossRef]
- Khanal, P.; Duttaroy, A.K. Prospect of potential intrauterine programming impacts associated with COVID-19. Front Public Health 2022, 10, 986162. [CrossRef]
- Lassance, L.; Haghiac, M.; Leahy, P.; Basu, S.; Minium, J.; Zhou, J.; Reider, M.; Catalano, P.M.; Hauguel-de Mouzon, S. Identification of early transcriptome signatures in placenta exposed to insulin and obesity. American journal of obstetrics and gynecology 2015, 212, 647 e641-611. [CrossRef]
- Apicella, C.; Ruano, C.S.M.; Mehats, C.; Miralles, F.; Vaiman, D. The Role of Epigenetics in Placental Development and the Etiology of Preeclampsia. Int J Mol Sci 2019, 20. [CrossRef]
- Troncoso, F.; Acurio, J.; Herlitz, K.; Aguayo, C.; Bertoglia, P.; Guzman-Gutierrez, E.; Loyola, M.; Gonzalez, M.; Rezgaoui, M.; Desoye, G.; et al. Gestational diabetes mellitus is associated with increased pro-migratory activation of vascular endothelial growth factor receptor 2 and reduced expression of vascular endothelial growth factor receptor 1. PLoS ONE 2017, 12, e0182509. [CrossRef]
- Devarshi, P.P.; Grant, R.W.; Ikonte, C.J.; Hazels Mitmesser, S. Maternal Omega-3 Nutrition, Placental Transfer and Fetal Brain Development in Gestational Diabetes and Preeclampsia. Nutrients 2019, 11. [CrossRef]
- Basak, S.; Vilasagaram, S.; Duttaroy, A.K. Maternal dietary deficiency of n-3 fatty acids affects metabolic and epigenetic phenotypes of the developing fetus. Prostaglandins Leukot Essent Fatty Acids 2020, 158, 102109-102120. [CrossRef]
- Johnsen, G.M.; Basak, S.; Weedon-Fekjaer, M.S.; Staff, A.C.; Duttaroy, A.K. Docosahexaenoic acid stimulates tube formation in first trimester trophoblast cells, HTR8/SVneo. Placenta 2011, 32, 626-632. [CrossRef]
- Duttaroy, A.K.; Basak, S. Maternal Fatty Acid Metabolism in Pregnancy and Its Consequences in the Feto-Placental Development. Front Physiol 2021, 12, 787848. [CrossRef]
- Basak, S.; Das, M.K.; Duttaroy, A.K. Fatty acid-induced angiogenesis in first trimester placental trophoblast cells: Possible roles of cellular fatty acid-binding proteins. Life sciences 2013, 93, 755-762. [CrossRef]
- Stuart, T.J.; O'Neill, K.; Condon, D.; Sasson, I.; Sen, P.; Xia, Y.; Simmons, R.A. Diet-induced obesity alters the maternal metabolome and early placenta transcriptome and decreases placenta vascularity in the mouse. Biol Reprod 2018, 98, 795-809. [CrossRef]
- Mohammed, S.; Qadri, S.S.Y.; Mir, I.A.; Kondapalli, N.B.; Basak, S.; Rajkumar, H. Fructooligosaccharide ameliorates high-fat induced intrauterine inflammation and improves lipid profile in the hamster offspring. J Nutr Biochem 2022, 101, 108925. [CrossRef]
- Song, L.; Sun, B.; Boersma, G.J.; Cordner, Z.A.; Yan, J.; Moran, T.H.; Tamashiro, K.L.K. Prenatal high-fat diet alters placental morphology, nutrient transporter expression, and mtorc1 signaling in rat. Obesity (Silver Spring) 2017, 25, 909-919. [CrossRef]
- Duttaroy, A.K.; Basak, S. Maternal Fatty Acid Metabolism in Pregnancy and Its Consequences in the Feto-Placental Development. Frontiers in Physiology 2022, 12. [CrossRef]
- Saben, J.; Lindsey, F.; Zhong, Y.; Thakali, K.; Badger, T.M.; Andres, A.; Gomez-Acevedo, H.; Shankar, K. Maternal obesity is associated with a lipotoxic placental environment. Placenta 2014, 35, 171-177. [CrossRef]
- Shrestha, D.; Workalemahu, T.; Tekola-Ayele, F. Maternal dyslipidemia during early pregnancy and epigenetic ageing of the placenta. Epigenetics 2019, 14, 1030-1039. [CrossRef]
- Dube, E.; Gravel, A.; Martin, C.; Desparois, G.; Moussa, I.; Ethier-Chiasson, M.; Forest, J.C.; Giguere, Y.; Masse, A.; Lafond, J. Modulation of fatty acid transport and metabolism by maternal obesity in the human full-term placenta. Biol Reprod 2012, 87, 14, 11-11. [CrossRef]
- Segura, M.T.; Demmelmair, H.; Krauss-Etschmann, S.; Nathan, P.; Dehmel, S.; Padilla, M.C.; Rueda, R.; Koletzko, B.; Campoy, C. Maternal BMI and gestational diabetes alter placental lipid transporters and fatty acid composition. Placenta 2017, 57, 144-151. [CrossRef]
- Dutta-Roy, A.K. Transport mechanisms for long-chain polyunsaturated fatty acids in the human placenta. Am J Clin Nutr 2000, 71, 315S-322S.
- Gazquez, A.; Prieto-Sanchez, M.T.; Blanco-Carnero, J.E.; Ruiz-Palacios, M.; Nieto, A.; van Harskamp, D.; Oosterink, J.E.; Schierbeek, H.; van Goudoever, J.B.; Demmelmair, H.; et al. Altered materno-fetal transfer of 13C-polyunsaturated fatty acids in obese pregnant women. Clin Nutr 2020, 39, 1101-1107. [CrossRef]
- Hirschmugl, B.; Desoye, G.; Catalano, P.; Klymiuk, I.; Scharnagl, H.; Payr, S.; Kitzinger, E.; Schliefsteiner, C.; Lang, U.; Wadsack, C.; et al. Maternal obesity modulates intracellular lipid turnover in the human term placenta. Int J Obes (Lond) 2017, 41, 317-323. [CrossRef]
- Laskewitz, A.; van Benthem, K.L.; Kieffer, T.E.C.; Faas, M.M.; Verkaik-Schakel, R.N.; Plosch, T.; Scherjon, S.A.; Prins, J.R. The influence of maternal obesity on macrophage subsets in the human decidua. Cell Immunol 2019, 336, 75-82. [CrossRef]
- Nogues, P.; Dos Santos, E.; Jammes, H.; Berveiller, P.; Arnould, L.; Vialard, F.; Dieudonne, M.N. Maternal obesity influences expression and DNA methylation of the adiponectin and leptin systems in human third-trimester placenta. Clin Epigenetics 2019, 11, 20. [CrossRef]
- Gauster, M.; Hiden, U.; van Poppel, M.; Frank, S.; Wadsack, C.; Hauguel-de Mouzon, S.; Desoye, G. Dysregulation of placental endothelial lipase in obese women with gestational diabetes mellitus. Diabetes 2011, 60, 2457-2464. [CrossRef]
- Herrera, E.; Ortega-Senovilla, H. Lipid metabolism during pregnancy and its implications for fetal growth. Curr Pharm Biotechnol 2014, 15, 24-31. [CrossRef]
- Davis, C.D.; Ross, S.A. Dietary components impact histone modifications and cancer risk. Nutr Rev 2007, 65, 88-94. [CrossRef]
- Jansson, N.; Rosario, F.J.; Gaccioli, F.; Lager, S.; Jones, H.N.; Roos, S.; Jansson, T.; Powell, T.L. Activation of placental mTOR signaling and amino acid transporters in obese women giving birth to large babies. J Clin Endocrinol Metab 2013, 98, 105-113. [CrossRef]
- Jacobs, D.I.; Hansen, J.; Fu, A.; Stevens, R.G.; Tjonneland, A.; Vogel, U.B.; Zheng, T.; Zhu, Y. Methylation alterations at imprinted genes detected among long-term shiftworkers. Environ Mol Mutagen 2013, 54, 141-146. [CrossRef]
- Hao, M.; Zhao, W.; Zhang, L.; Wang, H.; Yang, X. Low folate levels are associated with methylation-mediated transcriptional repression of miR-203 and miR-375 during cervical carcinogenesis. Oncol Lett 2016, 11, 3863-3869. [CrossRef]
- Zheng, J.; Zhang, Q.; Mul, J.D.; Yu, M.; Xu, J.; Qi, C.; Wang, T.; Xiao, X. Maternal high-calorie diet is associated with altered hepatic microRNA expression and impaired metabolic health in offspring at weaning age. Endocrine 2016, 54, 70-80. [CrossRef]
- Cao, C.; Zhang, H.; Zhao, L.; Zhou, L.; Zhang, M.; Xu, H.; Han, X.; Li, G.; Yang, X.; Jiang, Y. miR-125b targets DNMT3b and mediates p53 DNA methylation involving in the vascular smooth muscle cells proliferation induced by homocysteine. Exp Cell Res 2016, 347, 95-104. [CrossRef]
- Koturbash, I.; Melnyk, S.; James, S.J.; Beland, F.A.; Pogribny, I.P. Role of epigenetic and miR-22 and miR-29b alterations in the downregulation of Mat1a and Mthfr genes in early preneoplastic livers in rats induced by 2-acetylaminofluorene. Mol Carcinog 2013, 52, 318-327. [CrossRef]
- Li, X.; Wu, Y.; Liu, A.; Tang, X. MiR-27b is epigenetically downregulated in tamoxifen resistant breast cancer cells due to promoter methylation and regulates tamoxifen sensitivity by targeting HMGB3. Biochem Biophys Res Commun 2016, 477, 768-773. [CrossRef]
- Wilting, S.M.; Miok, V.; Jaspers, A.; Boon, D.; Sorgard, H.; Lando, M.; Snoek, B.C.; van Wieringen, W.N.; Meijer, C.J.; Lyng, H.; et al. Aberrant methylation-mediated silencing of microRNAs contributes to HPV-induced anchorage independence. Oncotarget 2016, 7, 43805-43819. [CrossRef]
- Du, J.; Cheng, X.; Shen, L.; Tan, Z.; Luo, J.; Wu, X.; Liu, C.; Yang, Q.; Jiang, Y.; Tang, G.; et al. Methylation of miR-145a-5p promoter mediates adipocytes differentiation. Biochem Biophys Res Commun 2016, 475, 140-148. [CrossRef]
- Tekola-Ayele, F.; Zeng, X.; Ouidir, M.; Workalemahu, T.; Zhang, C.; Delahaye, F.; Wapner, R. DNA methylation loci in placenta associated with birthweight and expression of genes relevant for early development and adult diseases. Clin Epigenetics 2020, 12, 78. [CrossRef]
- Sun, L.; Sun, S. Within-sample co-methylation patterns in normal tissues. BioData Mining 2019, 12, 9. [CrossRef]
- Clark, J.; Avula, V.; Ring, C.; Eaves, L.A.; Howard, T.; Santos, H.P.; Smeester, L.; Bangma, J.T.; O'Shea, T.M.; Fry, R.C.; et al. Comparing the Predictivity of Human Placental Gene, microRNA, and CpG Methylation Signatures in Relation to Perinatal Outcomes. Toxicol Sci 2021, 183, 269-284. [CrossRef]
- Agarwal, P.; Morriseau, T.S.; Kereliuk, S.M.; Doucette, C.A.; Wicklow, B.A.; Dolinsky, V.W. Maternal obesity, diabetes during pregnancy and epigenetic mechanisms that influence the developmental origins of cardiometabolic disease in the offspring. Crit Rev Clin Lab Sci 2018, 55, 71-101. [CrossRef]
- McGovern, N.; Shin, A.; Low, G.; Low, D.; Duan, K.; Yao, L.J.; Msallam, R.; Low, I.; Shadan, N.B.; Sumatoh, H.R.; et al. Human fetal dendritic cells promote prenatal T-cell immune suppression through arginase-2. Nature 2017, 546, 662-666. [CrossRef]
- Innis, S.M. Essential fatty acids in growth and development. Prog Lipid Res 1991, 30, 39-103. [CrossRef]
- Basak, S.; Mallick, R.; Banerjee, A.; Pathak, S.; Duttaroy, A.K. Maternal Supply of Both Arachidonic and Docosahexaenoic Acids Is Required for Optimal Neurodevelopment. Nutrients 2021, 13. [CrossRef]
- Basak, S.; Mallick, R.; Duttaroy, A.K. Maternal Docosahexaenoic Acid Status during Pregnancy and Its Impact on Infant Neurodevelopment. Nutrients 2020, 12. [CrossRef]
- Crabtree, J.T.; Gordon, M.J.; Campbell, F.M.; Dutta-Roy, A.K. Differential distribution and metabolism of arachidonic acid and docosahexaenoic acid by human placental choriocarcinoma (BeWo) cells. Mol. Cell. Biochem. 1998, 185, 191-198.
- Srinivas, V.; Molangiri, A.; Varma, S.; Mallepogu, A.; Kona, S.R.; Ibrahim, A.; Duttaroy, A.K.; Basak, S. Maternal omega-3 fatty acid deficiency affects fetal thermogenic development and postnatal musculoskeletal growth in mice. J Nutr Biochem 2023, 112, 109218. [CrossRef]
- DeCapo, M.; Thompson, J.R.; Dunn, G.; Sullivan, E.L. Perinatal Nutrition and Programmed Risk for Neuropsychiatric Disorders: A Focus on Animal Models. Biol Psychiatry 2019, 85, 122-134. [CrossRef]
- Moreno-Mendez, E.; Quintero-Fabian, S.; Fernandez-Mejia, C.; Lazo-de-la-Vega-Monroy, M.L. Early-life programming of adipose tissue. Nutr Res Rev 2020, 33, 244-259. [CrossRef]
- Lee, H.S.; Barraza-Villarreal, A.; Biessy, C.; Duarte-Salles, T.; Sly, P.D.; Ramakrishnan, U.; Rivera, J.; Herceg, Z.; Romieu, I. Dietary supplementation with polyunsaturated fatty acid during pregnancy modulates DNA methylation at IGF2/H19 imprinted genes and growth of infants. Physiol Genomics 2014, 46, 851-857. [CrossRef]
- Lee, H.S.; Barraza-Villarreal, A.; Hernandez-Vargas, H.; Sly, P.D.; Biessy, C.; Ramakrishnan, U.; Romieu, I.; Herceg, Z. Modulation of DNA methylation states and infant immune system by dietary supplementation with omega-3 PUFA during pregnancy in an intervention study. Am J Clin Nutr 2013, 98, 480-487. [CrossRef]
- Refsum, H.; Yajnik, C.S.; Gadkari, M.; Schneede, J.; Vollset, S.E.; Orning, L.; Guttormsen, A.B.; Joglekar, A.; Sayyad, M.G.; Ulvik, A.; et al. Hyperhomocysteinemia and elevated methylmalonic acid indicate a high prevalence of cobalamin deficiency in Asian Indians. Am J Clin Nutr 2001, 74, 233-241. [CrossRef]
- Fryer, A.A.; Nafee, T.M.; Ismail, K.M.; Carroll, W.D.; Emes, R.D.; Farrell, W.E. LINE-1 DNA methylation is inversely correlated with cord plasma homocysteine in man: A preliminary study. Epigenetics 2009, 4, 394-398. [CrossRef]
- Haggarty, P.; Hoad, G.; Campbell, D.M.; Horgan, G.W.; Piyathilake, C.; McNeill, G. Folate in pregnancy and imprinted gene and repeat element methylation in the offspring. Am J Clin Nutr 2013, 97, 94-99. [CrossRef]
- Chen, G.; Broseus, J.; Hergalant, S.; Donnart, A.; Chevalier, C.; Bolanos-Jimenez, F.; Gueant, J.L.; Houlgatte, R. Identification of master genes involved in liver key functions through transcriptomics and epigenomics of methyl donor deficiency in rat: Relevance to nonalcoholic liver disease. Mol Nutr Food Res 2015, 59, 293-302. [CrossRef]
- Gueant, J.L.; Namour, F.; Gueant-Rodriguez, R.M.; Daval, J.L. Folate and fetal programming: A play in epigenomics? Trends Endocrinol Metab 2013, 24, 279-289. [CrossRef]
- Mehedint, M.G.; Craciunescu, C.N.; Zeisel, S.H. Maternal dietary choline deficiency alters angiogenesis in fetal mouse hippocampus. Proc Natl Acad Sci U S A 2010, 107, 12834-12839. [CrossRef]
- Kovacheva, V.P.; Mellott, T.J.; Davison, J.M.; Wagner, N.; Lopez-Coviella, I.; Schnitzler, A.C.; Blusztajn, J.K. Gestational choline deficiency causes global and Igf2 gene DNA hypermethylation by up-regulation of Dnmt1 expression. J Biol Chem 2007, 282, 31777-31788. [CrossRef]
- Medici, V.; Shibata, N.M.; Kharbanda, K.K.; Islam, M.S.; Keen, C.L.; Kim, K.; Tillman, B.; French, S.W.; Halsted, C.H.; LaSalle, J.M. Maternal choline modifies fetal liver copper, gene expression, DNA methylation, and neonatal growth in the tx-j mouse model of Wilson disease. Epigenetics 2014, 9, 286-296. [CrossRef]
- Aon, M.A.; Cortassa, S.; Juhaszova, M.; Sollott, S.J. Mitochondrial health, the epigenome and healthspan. Clin Sci (Lond) 2016, 130, 1285-1305. [CrossRef]
- Pauwels, S.; Duca, R.C.; Devlieger, R.; Freson, K.; Straetmans, D.; Van Herck, E.; Huybrechts, I.; Koppen, G.; Godderis, L. Maternal Methyl-Group Donor Intake and Global DNA (Hydroxy)Methylation before and during Pregnancy. Nutrients 2016, 8. [CrossRef]
- Smolders, J.; Menheere, P.; Kessels, A.; Damoiseaux, J.; Hupperts, R. Association of vitamin D metabolite levels with relapse rate and disability in multiple sclerosis. Mult Scler 2008, 14, 1220-1224. [CrossRef]
- Hollis, B.W.; Wagner, C.L. Vitamin D supplementation during pregnancy: Improvements in birth outcomes and complications through direct genomic alteration. Mol Cell Endocrinol 2017, 453, 113-130. [CrossRef]
- Sablok, A.; Batra, A.; Thariani, K.; Batra, A.; Bharti, R.; Aggarwal, A.R.; Kabi, B.C.; Chellani, H. Supplementation of vitamin D in pregnancy and its correlation with feto-maternal outcome. Clin Endocrinol (Oxf) 2015, 83, 536-541. [CrossRef]
- Fetahu, I.S.; Hobaus, J.; Kallay, E. Vitamin D and the epigenome. Front Physiol 2014, 5, 164. [CrossRef]
- Dominguez-Salas, P.; Moore, S.E.; Baker, M.S.; Bergen, A.W.; Cox, S.E.; Dyer, R.A.; Fulford, A.J.; Guan, Y.; Laritsky, E.; Silver, M.J.; et al. Maternal nutrition at conception modulates DNA methylation of human metastable epialleles. Nat Commun 2014, 5, 3746. [CrossRef]
- Xue, J.; Schoenrock, S.A.; Valdar, W.; Tarantino, L.M.; Ideraabdullah, F.Y. Maternal vitamin D depletion alters DNA methylation at imprinted loci in multiple generations. Clin Epigenetics 2016, 8, 107. [CrossRef]
- Junge, K.M.; Bauer, T.; Geissler, S.; Hirche, F.; Thurmann, L.; Bauer, M.; Trump, S.; Bieg, M.; Weichenhan, D.; Gu, L.; et al. Increased vitamin D levels at birth and in early infancy increase offspring allergy risk-evidence for involvement of epigenetic mechanisms. J Allergy Clin Immunol 2016, 137, 610-613. [CrossRef]
- Suderman, M.; Stene, L.C.; Bohlin, J.; Page, C.M.; Holvik, K.; Parr, C.L.; Magnus, M.C.; Haberg, S.E.; Joubert, B.R.; Wu, M.C.; et al. 25-Hydroxyvitamin D in pregnancy and genome wide cord blood DNA methylation in two pregnancy cohorts (MoBa and ALSPAC). J Steroid Biochem Mol Biol 2016, 159, 102-109. [CrossRef]
- Benjamin Neelon, S.E.; White, A.J.; Vidal, A.C.; Schildkraut, J.M.; Murtha, A.P.; Murphy, S.K.; Kullman, S.W.; Hoyo, C. Maternal vitamin D, DNA methylation at imprint regulatory regions and offspring weight at birth, 1 year and 3 years. Int J Obes (Lond) 2018, 42, 587-593. [CrossRef]
- Reichetzeder, C.; Dwi Putra, S.E.; Li, J.; Hocher, B. Developmental Origins of Disease - Crisis Precipitates Change. Cell Physiol Biochem 2016, 39, 919-938. [CrossRef]
- Zhou, Y.; Zhao, L.J.; Xu, X.; Ye, A.; Travers-Gustafson, D.; Zhou, B.; Wang, H.W.; Zhang, W.; Lee Hamm, L.; Deng, H.W.; et al. DNA methylation levels of CYP2R1 and CYP24A1 predict vitamin D response variation. J Steroid Biochem Mol Biol 2014, 144 Pt A, 207-214. [CrossRef]
- Adams, J.S.; Hewison, M. Extrarenal expression of the 25-hydroxyvitamin D-1-hydroxylase. Arch Biochem Biophys 2012, 523, 95-102. [CrossRef]
- Fransen, F.; van Beek, A.A.; Borghuis, T.; Meijer, B.; Hugenholtz, F.; van der Gaast-de Jongh, C.; Savelkoul, H.F.; de Jonge, M.I.; Faas, M.M.; Boekschoten, M.V.; et al. The Impact of Gut Microbiota on Gender-Specific Differences in Immunity. Front Immunol 2017, 8, 754. [CrossRef]
- Korpela, K.; Kallio, S.; Salonen, A.; Hero, M.; Kukkonen, A.K.; Miettinen, P.J.; Savilahti, E.; Kohva, E.; Kariola, L.; Suutela, M.; et al. Gut microbiota develop towards an adult profile in a sex-specific manner during puberty. Sci Rep 2021, 11, 23297. [CrossRef]
- Wilmanski, T.; Diener, C.; Rappaport, N.; Patwardhan, S.; Wiedrick, J.; Lapidus, J.; Earls, J.C.; Zimmer, A.; Glusman, G.; Robinson, M.; et al. Gut microbiome pattern reflects healthy ageing and predicts survival in humans. Nat Metab 2021, 3, 274-286. [CrossRef]
- Yuan, X.; Chen, R.; Zhang, Y.; Lin, X.; Yang, X. Gut microbiota: Effect of pubertal status. BMC Microbiol 2020, 20, 334. [CrossRef]
- Buffington, S.A.; Di Prisco, G.V.; Auchtung, T.A.; Ajami, N.J.; Petrosino, J.F.; Costa-Mattioli, M. Microbial Reconstitution Reverses Maternal Diet-Induced Social and Synaptic Deficits in Offspring. Cell 2016, 165, 1762-1775. [CrossRef]
- Kim, S.; Kim, H.; Yim, Y.S.; Ha, S.; Atarashi, K.; Tan, T.G.; Longman, R.S.; Honda, K.; Littman, D.R.; Choi, G.B.; et al. Maternal gut bacteria promote neurodevelopmental abnormalities in mouse offspring. Nature 2017, 549, 528-532. [CrossRef]
- Vuong, H.E.; Pronovost, G.N.; Williams, D.W.; Coley, E.J.L.; Siegler, E.L.; Qiu, A.; Kazantsev, M.; Wilson, C.J.; Rendon, T.; Hsiao, E.Y. The maternal microbiome modulates fetal neurodevelopment in mice. Nature 2020, 586, 281-286. [CrossRef]
- Dawson, S.L.; O'Hely, M.; Jacka, F.N.; Ponsonby, A.L.; Symeonides, C.; Loughman, A.; Collier, F.; Moreno-Betancur, M.; Sly, P.; Burgner, D.; et al. Maternal prenatal gut microbiota composition predicts child behaviour. EBioMedicine 2021, 68, 103400. [CrossRef]
- Aagaard, K.; Ma, J.; Antony, K.M.; Ganu, R.; Petrosino, J.; Versalovic, J. The placenta harbors a unique microbiome. Sci Transl Med 2014, 6, 237ra265. [CrossRef]
- Solt, I. The human microbiome and the great obstetrical syndromes: A new frontier in maternal-fetal medicine. Best Pract Res Clin Obstet Gynaecol 2015, 29, 165-175. [CrossRef]
- Pelzer, E.; Gomez-Arango, L.F.; Barrett, H.L.; Nitert, M.D. Review: Maternal health and the placental microbiome. Placenta 2017, 54, 30-37. [CrossRef]
- DiGiulio, D.B.; Romero, R.; Amogan, H.P.; Kusanovic, J.P.; Bik, E.M.; Gotsch, F.; Kim, C.J.; Erez, O.; Edwin, S.; Relman, D.A. Microbial prevalence, diversity and abundance in amniotic fluid during preterm labor: A molecular and culture-based investigation. PLoS ONE 2008, 3, e3056. [CrossRef]
- DiGiulio, D.B.; Romero, R.; Kusanovic, J.P.; Gomez, R.; Kim, C.J.; Seok, K.S.; Gotsch, F.; Mazaki-Tovi, S.; Vaisbuch, E.; Sanders, K.; et al. Prevalence and diversity of microbes in the amniotic fluid, the fetal inflammatory response, and pregnancy outcome in women with preterm pre-labor rupture of membranes. Am J Reprod Immunol 2010, 64, 38-57. [CrossRef]
- Onderdonk, A.B.; Hecht, J.L.; McElrath, T.F.; Delaney, M.L.; Allred, E.N.; Leviton, A.; Investigators, E.S. Colonization of second-trimester placenta parenchyma. American journal of obstetrics and gynecology 2008, 199, 52 e51-52 e10. [CrossRef]
- Collado, M.C.; Isolauri, E.; Laitinen, K.; Salminen, S. Distinct composition of gut microbiota during pregnancy in overweight and normal-weight women. Am J Clin Nutr 2008, 88, 894-899. [CrossRef]
- Bhatia, P.; Chhabra, S. Physiological and anatomical changes of pregnancy: Implications for anaesthesia. Indian J Anaesth 2018, 62, 651-657. [CrossRef]
- Chen, J.J.; Zeng, B.H.; Li, W.W.; Zhou, C.J.; Fan, S.H.; Cheng, K.; Zeng, L.; Zheng, P.; Fang, L.; Wei, H.; et al. Effects of gut microbiota on the microRNA and mRNA expression in the hippocampus of mice. Behav Brain Res 2017, 322, 34-41. [CrossRef]
- Bale, T.L. Epigenetic and transgenerational reprogramming of brain development. Nat Rev Neurosci 2015, 16, 332-344. [CrossRef]
- Mallick, R.; Duttaroy, A.K. Epigenetic modification impacting brain functions: Effects of physical activity, micronutrients, caffeine, toxins, and addictive substances. Neurochem Int 2023, 171, 105627. [CrossRef]
- Di Simone, N.; Santamaria Ortiz, A.; Specchia, M.; Tersigni, C.; Villa, P.; Gasbarrini, A.; Scambia, G.; D'Ippolito, S. Recent Insights on the Maternal Microbiota: Impact on Pregnancy Outcomes. Front Immunol 2020, 11, 528202. [CrossRef]
- Koren, O.; Goodrich, J.K.; Cullender, T.C.; Spor, A.; Laitinen, K.; Backhed, H.K.; Gonzalez, A.; Werner, J.J.; Angenent, L.T.; Knight, R.; et al. Host remodeling of the gut microbiome and metabolic changes during pregnancy. Cell 2012, 150, 470-480. [CrossRef]
- Rinninella, E.; Raoul, P.; Cintoni, M.; Franceschi, F.; Miggiano, G.A.D.; Gasbarrini, A.; Mele, M.C. What is the Healthy Gut Microbiota Composition? A Changing Ecosystem across Age, Environment, Diet, and Diseases. Microorganisms 2019, 7. [CrossRef]
- Shin, N.R.; Whon, T.W.; Bae, J.W. Proteobacteria: Microbial signature of dysbiosis in gut microbiota. Trends Biotechnol 2015, 33, 496-503. [CrossRef]
- Prochazkova, N.; Falony, G.; Dragsted, L.O.; Licht, T.R.; Raes, J.; Roager, H.M. Advancing human gut microbiota research by considering gut transit time. Gut 2023, 72, 180-191. [CrossRef]
- Zakaria, Z.Z.; Al-Rumaihi, S.; Al-Absi, R.S.; Farah, H.; Elamin, M.; Nader, R.; Bouabidi, S.; Suleiman, S.E.; Nasr, S.; Al-Asmakh, M. Physiological Changes and Interactions Between Microbiome and the Host During Pregnancy. Front Cell Infect Microbiol 2022, 12, 824925. [CrossRef]
- Rubini, E.; Schenkelaars, N.; Rousian, M.; Sinclair, K.D.; Wekema, L.; Faas, M.M.; Steegers-Theunissen, R.P.M.; Schoenmakers, S. Maternal obesity during pregnancy leads to derangements in one-carbon metabolism and the gut microbiota: Implications for fetal development and offspring wellbeing. American journal of obstetrics and gynecology 2022, 227, 392-400. [CrossRef]
- Goasdoue, K.; Miller, S.M.; Colditz, P.B.; Bjorkman, S.T. Review: The blood-brain barrier; protecting the developing fetal brain. Placenta 2017, 54, 111-116. [CrossRef]
- Wang, J.; Gu, X.; Yang, J.; Wei, Y.; Zhao, Y. Gut Microbiota Dysbiosis and Increased Plasma LPS and TMAO Levels in Patients With Preeclampsia. Front Cell Infect Microbiol 2019, 9, 409. [CrossRef]
- Manrique-Corredor, E.J.; Orozco-Beltran, D.; Lopez-Pineda, A.; Quesada, J.A.; Gil-Guillen, V.F.; Carratala-Munuera, C. Maternal periodontitis and preterm birth: Systematic review and meta-analysis. Community Dent Oral Epidemiol 2019, 47, 243-251. [CrossRef]
- Zakis, D.R.; Paulissen, E.; Kornete, L.; Kaan, A.M.M.; Nicu, E.A.; Zaura, E. The evidence for placental microbiome and its composition in healthy pregnancies: A systematic review. J Reprod Immunol 2022, 149, 103455. [CrossRef]
- Mousa, W.K.; Chehadeh, F.; Husband, S. Microbial dysbiosis in the gut drives systemic autoimmune diseases. Front Immunol 2022, 13, 906258. [CrossRef]
- Nyangahu, D.D.; Jaspan, H.B. Influence of maternal microbiota during pregnancy on infant immunity. Clin Exp Immunol 2019, 198, 47-56. [CrossRef]
- Hu, Y.; Jin, P.; Peng, J.; Zhang, X.; Wong, F.S.; Wen, L. Different immunological responses to early-life antibiotic exposure affecting autoimmune diabetes development in NOD mice. J Autoimmun 2016, 72, 47-56. [CrossRef]
- Holladay, S.D.; Smialowicz, R.J. Development of the murine and human immune system: Differential effects of immunotoxicants depend on time of exposure. Environ Health Perspect 2000, 108 Suppl 3, 463-473. [CrossRef]
- Eberl, G.; Lochner, M. The development of intestinal lymphoid tissues at the interface of self and microbiota. Mucosal Immunol 2009, 2, 478-485. [CrossRef]
- Westrom, B.; Arevalo Sureda, E.; Pierzynowska, K.; Pierzynowski, S.G.; Perez-Cano, F.J. The Immature Gut Barrier and Its Importance in Establishing Immunity in Newborn Mammals. Front Immunol 2020, 11, 1153. [CrossRef]
- Holmes, E.; Li, J.V.; Marchesi, J.R.; Nicholson, J.K. Gut microbiota composition and activity in relation to host metabolic phenotype and disease risk. Cell Metab 2012, 16, 559-564. [CrossRef]
- McElroy, S.J.; Weitkamp, J.H. Innate Immunity in the Small Intestine of the Preterm Infant. Neoreviews 2011, 12, e517-e526. [CrossRef]
- Timm, S.; Schlunssen, V.; Olsen, J.; Ramlau-Hansen, C.H. Prenatal antibiotics and atopic dermatitis among 18-month-old children in the Danish National Birth Cohort. Clin Exp Allergy 2017, 47, 929-936. [CrossRef]
- Ortqvist, A.K.; Lundholm, C.; Halfvarson, J.; Ludvigsson, J.F.; Almqvist, C. Fetal and early life antibiotics exposure and very early onset inflammatory bowel disease: A population-based study. Gut 2019, 68, 218-225. [CrossRef]
- Joly, A.; Leulier, F.; De Vadder, F. Microbial Modulation of the Development and Physiology of the Enteric Nervous System. Trends Microbiol 2021, 29, 686-699. [CrossRef]
- Yang, L.L.; Millischer, V.; Rodin, S.; MacFabe, D.F.; Villaescusa, J.C.; Lavebratt, C. Enteric short-chain fatty acids promote proliferation of human neural progenitor cells. J Neurochem 2020, 154, 635-646. [CrossRef]
- Pessa-Morikawa, T.; Husso, A.; Karkkainen, O.; Koistinen, V.; Hanhineva, K.; Iivanainen, A.; Niku, M. Maternal microbiota-derived metabolic profile in fetal murine intestine, brain and placenta. BMC Microbiol 2022, 22, 46. [CrossRef]
- Garzoni, L.; Faure, C.; Frasch, M.G. Fetal cholinergic anti-inflammatory pathway and necrotizing enterocolitis: The brain-gut connection begins in utero. Front Integr Neurosci 2013, 7, 57. [CrossRef]
- Collado, M.C.; Rautava, S.; Aakko, J.; Isolauri, E.; Salminen, S. Human gut colonisation may be initiated in utero by distinct microbial communities in the placenta and amniotic fluid. Sci Rep 2016, 6, 23129. [CrossRef]
- He, Q.; Kwok, L.Y.; Xi, X.; Zhong, Z.; Ma, T.; Xu, H.; Meng, H.; Zhao, F.; Zhang, H. The meconium microbiota shares more features with the amniotic fluid microbiota than the maternal fecal and vaginal microbiota. Gut Microbes 2020, 12, 1794266. [CrossRef]
- Chin, A.M.; Hill, D.R.; Aurora, M.; Spence, J.R. Morphogenesis and maturation of the embryonic and postnatal intestine. Semin Cell Dev Biol 2017, 66, 81-93. [CrossRef]
- Toure, A.M.; Landry, M.; Souchkova, O.; Kembel, S.W.; Pilon, N. Gut microbiota-mediated Gene-Environment interaction in the TashT mouse model of Hirschsprung disease. Sci Rep 2019, 9, 492. [CrossRef]
- Schneider, U.; Bode, F.; Schmidt, A.; Nowack, S.; Rudolph, A.; Dolker, E.M.; Schlattmann, P.; Gotz, T.; Hoyer, D. Developmental milestones of the autonomic nervous system revealed via longitudinal monitoring of fetal heart rate variability. PLoS ONE 2018, 13, e0200799. [CrossRef]
- Rackaityte, E.; Halkias, J.; Fukui, E.M.; Mendoza, V.F.; Hayzelden, C.; Crawford, E.D.; Fujimura, K.E.; Burt, T.D.; Lynch, S.V. Viable bacterial colonization is highly limited in the human intestine in utero. Nat Med 2020, 26, 599-607. [CrossRef]
- Martinez, K.A., 2nd; Romano-Keeler, J.; Zackular, J.P.; Moore, D.J.; Brucker, R.M.; Hooper, C.; Meng, S.; Brown, N.; Mallal, S.; Reese, J.; et al. Bacterial DNA is present in the fetal intestine and overlaps with that in the placenta in mice. PLoS ONE 2018, 13, e0197439. [CrossRef]
- Ostrea, E.M., Jr.; Bielawski, D.M.; Posecion, N.C., Jr. Meconium analysis to detect fetal exposure to neurotoxicants. Arch Dis Child 2006, 91, 628-629. [CrossRef]
- Jasarevic, E.; Howard, C.D.; Misic, A.M.; Beiting, D.P.; Bale, T.L. Stress during pregnancy alters temporal and spatial dynamics of the maternal and offspring microbiome in a sex-specific manner. Sci Rep 2017, 7, 44182. [CrossRef]
- Kimura, I.; Miyamoto, J.; Ohue-Kitano, R.; Watanabe, K.; Yamada, T.; Onuki, M.; Aoki, R.; Isobe, Y.; Kashihara, D.; Inoue, D.; et al. Maternal gut microbiota in pregnancy influences offspring metabolic phenotype in mice. Science 2020, 367. [CrossRef]
- Segarra, M.; Aburto, M.R.; Acker-Palmer, A. Blood-Brain Barrier Dynamics to Maintain Brain Homeostasis. Trends Neurosci 2021, 44, 393-405. [CrossRef]
- Saunders, N.R.; Liddelow, S.A.; Dziegielewska, K.M. Barrier mechanisms in the developing brain. Front Pharmacol 2012, 3, 46. [CrossRef]
- Rodriguez, J.M.; Murphy, K.; Stanton, C.; Ross, R.P.; Kober, O.I.; Juge, N.; Avershina, E.; Rudi, K.; Narbad, A.; Jenmalm, M.C.; et al. The composition of the gut microbiota throughout life, with an emphasis on early life. Microb Ecol Health Dis 2015, 26, 26050. [CrossRef]
- Guzzardi, M.A.; Ait Ali, L.; D'Aurizio, R.; Rizzo, F.; Saggese, P.; Sanguinetti, E.; Weisz, A.; Pellegrini, M.; Iozzo, P. Fetal cardiac growth is associated with in utero gut colonization. Nutr Metab Cardiovasc Dis 2019, 29, 170-176. [CrossRef]
- Increases in platelet and red cell counts, blood viscosity, and arterial pressure during mild surface cooling. Br Med J (Clin Res Ed) 1985, 290, 74-75.
- Ratsika, A.; Codagnone, M.C.; O'Mahony, S.; Stanton, C.; Cryan, J.F. Priming for Life: Early Life Nutrition and the Microbiota-Gut-Brain Axis. Nutrients 2021, 13. [CrossRef]
- Clarke, G.; Grenham, S.; Scully, P.; Fitzgerald, P.; Moloney, R.D.; Shanahan, F.; Dinan, T.G.; Cryan, J.F. The microbiome-gut-brain axis during early life regulates the hippocampal serotonergic system in a sex-dependent manner. Mol Psychiatry 2013, 18, 666-673. [CrossRef]
- Jena, A.; Montoya, C.A.; Mullaney, J.A.; Dilger, R.N.; Young, W.; McNabb, W.C.; Roy, N.C. Gut-Brain Axis in the Early Postnatal Years of Life: A Developmental Perspective. Front Integr Neurosci 2020, 14, 44. [CrossRef]
- Mueller, N.T.; Bakacs, E.; Combellick, J.; Grigoryan, Z.; Dominguez-Bello, M.G. The infant microbiome development: Mom matters. Trends in molecular medicine 2015, 21, 109-117. [CrossRef]
- Sajdel-Sulkowska, E.M. The Impact of Maternal Gut Microbiota during Pregnancy on Fetal Gut-Brain Axis Development and Life-Long Health Outcomes. Microorganisms 2023, 11. [CrossRef]
- Bonnin, A.; Goeden, N.; Chen, K.; Wilson, M.L.; King, J.; Shih, J.C.; Blakely, R.D.; Deneris, E.S.; Levitt, P. A transient placental source of serotonin for the fetal forebrain. Nature 2011, 472, 347-350. [CrossRef]
- Bonnin, A.; Levitt, P. Fetal, maternal, and placental sources of serotonin and new implications for developmental programming of the brain. Neuroscience 2011, 197, 1-7. [CrossRef]
- Dicks, L.M.T. Gut Bacteria and Neurotransmitters. Microorganisms 2022, 10. [CrossRef]
- Momose-Sato, Y.; Sato, K. Development of synaptic networks in the mouse vagal pathway revealed by optical mapping with a voltage-sensitive dye. Eur J Neurosci 2016, 44, 1906-1918. [CrossRef]
- Sachis, P.N.; Armstrong, D.L.; Becker, L.E.; Bryan, A.C. Myelination of the human vagus nerve from 24 weeks postconceptional age to adolescence. J Neuropathol Exp Neurol 1982, 41, 466-472. [CrossRef]
- Carabotti, M.; Scirocco, A.; Maselli, M.A.; Severi, C. The gut-brain axis: Interactions between enteric microbiota, central and enteric nervous systems. Ann Gastroenterol 2015, 28, 203-209.
- Chaudhry, T.S.; Senapati, S.G.; Gadam, S.; Mannam, H.; Voruganti, H.V.; Abbasi, Z.; Abhinav, T.; Challa, A.B.; Pallipamu, N.; Bheemisetty, N.; et al. The Impact of Microbiota on the Gut-Brain Axis: Examining the Complex Interplay and Implications. Journal of clinical medicine 2023, 12. [CrossRef]
- Dash, S.; Syed, Y.A.; Khan, M.R. Understanding the Role of the Gut Microbiome in Brain Development and Its Association With Neurodevelopmental Psychiatric Disorders. Front Cell Dev Biol 2022, 10, 880544. [CrossRef]
- Rusch, J.A.; Layden, B.T.; Dugas, L.R. Signalling cognition: The gut microbiota and hypothalamic-pituitary-adrenal axis. Front Endocrinol (Lausanne) 2023, 14, 1130689. [CrossRef]
- Farzi, A.; Frohlich, E.E.; Holzer, P. Gut Microbiota and the Neuroendocrine System. Neurotherapeutics 2018, 15, 5-22. [CrossRef]
- Kapoor, A.; Dunn, E.; Kostaki, A.; Andrews, M.H.; Matthews, S.G. Fetal programming of hypothalamo-pituitary-adrenal function: Prenatal stress and glucocorticoids. J Physiol 2006, 572, 31-44. [CrossRef]
- Condon, J.; Gosden, C.; Gardener, D.; Nickson, P.; Hewison, M.; Howie, A.J.; Stewart, P.M. Expression of type 2 11beta-hydroxysteroid dehydrogenase and corticosteroid hormone receptors in early human fetal life. J Clin Endocrinol Metab 1998, 83, 4490-4497. [CrossRef]
- Howland, M.A.; Sandman, C.A.; Glynn, L.M. Developmental origins of the human hypothalamic-pituitary-adrenal axis. Expert Rev Endocrinol Metab 2017, 12, 321-339. [CrossRef]
- Sheng, J.A.; Bales, N.J.; Myers, S.A.; Bautista, A.I.; Roueinfar, M.; Hale, T.M.; Handa, R.J. The Hypothalamic-Pituitary-Adrenal Axis: Development, Programming Actions of Hormones, and Maternal-Fetal Interactions. Front Behav Neurosci 2020, 14, 601939. [CrossRef]
- Ng, P.C. The fetal and neonatal hypothalamic-pituitary-adrenal axis. Arch Dis Child Fetal Neonatal Ed 2000, 82, F250-254. [CrossRef]
- van Dijk, S.J.; Zhou, J.; Peters, T.J.; Buckley, M.; Sutcliffe, B.; Oytam, Y.; Gibson, R.A.; McPhee, A.; Yelland, L.N.; Makrides, M.; et al. Effect of prenatal DHA supplementation on the infant epigenome: Results from a randomized controlled trial. Clinical Epigenetics 2016, 8, 114. [CrossRef]
- Robinson, S.L.; Mumford, S.L.; Guan, W.; Zeng, X.; Kim, K.; Radoc, J.G.; Trinh, M.-H.; Flannagan, K.; Schisterman, E.F.; Yeung, E. Maternal fatty acid concentrations and newborn DNA methylation. The American Journal of Clinical Nutrition 2020, 111, 613-621. [CrossRef]
- Lee, H.-S.; Barraza-Villarreal, A.; Biessy, C.; Duarte-Salles, T.; Sly, P.D.; Ramakrishnan, U.; Rivera, J.; Herceg, Z.; Romieu, I. Dietary supplementation with polyunsaturated fatty acid during pregnancy modulates DNA methylation at IGF2/H19 imprinted genes and growth of infants. Physiological Genomics 2014, 46, 851-857. [CrossRef]
- Saffari, A.; Shrestha, S.; Issarapu, P.; Sajjadi, S.; Betts, M.; Sahariah, S.A.; Tomar, A.S.; James, P.; Dedaniya, A.; Yadav, D.K.; et al. Effect of maternal preconceptional and pregnancy micronutrient interventions on children’s DNA methylation: Findings from the EMPHASIS study. The American Journal of Clinical Nutrition 2020, 112, 1099-1113. [CrossRef]
- Bianchi, M.; Alisi, A.; Fabrizi, M.; Vallone, C.; Ravà, L.; Giannico, R.; Vernocchi, P.; Signore, F.; Manco, M. Maternal Intake of n-3 Polyunsaturated Fatty Acids During Pregnancy Is Associated With Differential Methylation Profiles in Cord Blood White Cells. Frontiers in Genetics 2019, 10. [CrossRef]
- Koemel, N.A.; Senior, A.M.; Dissanayake, H.U.; Ross, J.; McMullan, R.L.; Kong, Y.; Phang, M.; Hyett, J.; Raubenheimer, D.; Gordon, A.; et al. Maternal dietary fatty acid composition and newborn epigenetic aging—a geometric framework approach. The American Journal of Clinical Nutrition 2022, 115, 118-127. [CrossRef]
- Phang, M.; Ross, J.; Raythatha, J.H.; Dissanayake, H.U.; McMullan, R.L.; Kong, Y.; Hyett, J.; Gordon, A.; Molloy, P.; Skilton, M.R. Epigenetic aging in newborns: Role of maternal diet. The American Journal of Clinical Nutrition 2020, 111, 555-561. [CrossRef]
- Chen, L.; Wagner, C.L.; Dong, Y.; Wang, X.; Shary, J.R.; Huang, Y.; Hollis, B.W.; Zhu, H. Effects of Maternal Vitamin D3 Supplementation on Offspring Epigenetic Clock of Gestational Age at Birth: A Post-hoc Analysis of a Randomized Controlled Trial. Epigenetics 2020, 15, 830-840. [CrossRef]
- Caffrey, A.; Irwin, R.E.; McNulty, H.; Strain, J.J.; Lees-Murdock, D.J.; McNulty, B.A.; Ward, M.; Walsh, C.P.; Pentieva, K. Gene-specific DNA methylation in newborns in response to folic acid supplementation during the second and third trimesters of pregnancy: Epigenetic analysis from a randomized controlled trial. The American Journal of Clinical Nutrition 2018, 107, 566-575. [CrossRef]
- Küpers, L.K.; Fernández-Barrés, S.; Mancano, G.; Johnson, L.; Ott, R.; Vioque, J.; Colombo, M.; Landgraf, K.; Tobi, E.W.; Körner, A.; et al. Maternal Dietary Glycemic Index and Glycemic Load in Pregnancy and Offspring Cord Blood DNA Methylation. Diabetes Care 2022, 45, 1822-1832. [CrossRef]
- Lecorguillé, M.; Navarro, P.; Chen, L.-W.; Murrin, C.; Viljoen, K.; Mehegan, J.; Shivappa, N.; Hébert, J.R.; Kelleher, C.C.; Suderman, M.; et al. Maternal and Paternal Dietary Quality and Dietary Inflammation Associations with Offspring DNA Methylation and Epigenetic Biomarkers of Aging in the Lifeways Cross-Generation Study. The Journal of nutrition 2023, 153, 1075-1088. [CrossRef]
- Gonzalez-Nahm, S.; Mendez, M.; Robinson, W.; Murphy, S.K.; Hoyo, C.; Hogan, V.; Rowley, D. Low maternal adherence to a Mediterranean diet is associated with increase in methylation at the MEG3-IG differentially methylated region in female infants. Environmental Epigenetics 2017, 3. [CrossRef]
- House, J.S.; Mendez, M.; Maguire, R.L.; Gonzalez-Nahm, S.; Huang, Z.; Daniels, J.; Murphy, S.K.; Fuemmeler, B.F.; Wright, F.A.; Hoyo, C. Periconceptional Maternal Mediterranean Diet Is Associated With Favorable Offspring Behaviors and Altered CpG Methylation of Imprinted Genes. Frontiers in Cell and Developmental Biology 2018, 6. [CrossRef]
- Zhang, C.; Wu, J.; Ouidir, M.; Hinkle, S.; Tekola-Ayele, F. Abstract MP42: Adherence To Healthy Dietary Patterns In Pregnancy And Placental Dna Methylation- An Epigenome Wide Association Study. Circulation 2021, 143. [CrossRef]
- Chiu, Y.-H.; Fadadu, R.P.; Gaskins, A.J.; Rifas-Shiman, S.L.; Laue, H.E.; Moley, K.H.; Hivert, M.-F.; Baccarelli, A.; Oken, E.; Chavarro, J.E.; et al. Dietary fat intake during early pregnancy is associated with cord blood DNA methylation at IGF2 and H19 genes in newborns. Environmental and Molecular Mutagenesis 2021, 62, 388-398. [CrossRef]
- Geraghty, A.A.; Sexton-Oates, A.; O’Brien, E.C.; Saffery, R.; McAuliffe, F.M. Epigenetic Patterns in Five-Year-Old Children Exposed to a Low Glycemic Index Dietary Intervention during Pregnancy: Results from the ROLO Kids Study. Nutrients 2020, 12, 3602.
- Geraghty, A.A.; Sexton-Oates, A.; O’Brien, E.C.; Alberdi, G.; Fransquet, P.; Saffery, R.; McAuliffe, F.M. A Low Glycaemic Index Diet in Pregnancy Induces DNA Methylation Variation in Blood of Newborns: Results from the ROLO Randomised Controlled Trial. Nutrients 2018, 10, 455.
- Sasaki, T.; Kawamura, M.; Okuno, C.; Lau, K.; Riel, J.; Lee, M.-J.; Miller, C. Impact of Maternal Mediterranean-Type Diet Adherence on Microbiota Composition and Epigenetic Programming of Offspring. Nutrients 2024, 16, 47.
- Burdge, G.C.; Hoile, S.P.; Uller, T.; Thomas, N.A.; Gluckman, P.D.; Hanson, M.A.; Lillycrop, K.A. Progressive, Transgenerational Changes in Offspring Phenotype and Epigenotype following Nutritional Transition. PLoS ONE 2011, 6, e28282. [CrossRef]
- Nowacka-Woszuk, J.; Szczerbal, I.; Malinowska, A.M.; Chmurzynska, A. Transgenerational effects of prenatal restricted diet on gene expression and histone modifications in the rat. PLoS ONE 2018, 13, e0193464. [CrossRef]
- Fan, C.; Fu, H.; Dong, H.; Lu, Y.; Lu, Y.; Qi, K. Maternal n-3 polyunsaturated fatty acid deprivation during pregnancy and lactation affects neurogenesis and apoptosis in adult offspring: Associated with DNA methylation of brain-derived neurotrophic factor transcripts. Nutrition Research 2016, 36, 1013-1021. [CrossRef]
- Ly, L.; Chan, D.; Landry, M.; Angle, C.; Martel, J.; Trasler, J. Impact of mothers' early life exposure to low or high folate on progeny outcome and DNA methylation patterns. Environmental Epigenetics 2020, 6. [CrossRef]
- Srinivas, V.; Molangiri, A.; Mallepogu, A.; Kona, S.R.; Ibrahim, A.; Duttaroy, A.K.; Basak, S. Maternal n-3 PUFA deficiency alters uterine artery remodeling and placental epigenome in the mice. The Journal of nutritional biochemistry 2021, 96, 108784. [CrossRef]
- Sable, P.; Randhir, K.; Kale, A.; Chavan-Gautam, P.; Joshi, S. Maternal micronutrients and brain global methylation patterns in the offspring. Nutritional Neuroscience 2015, 18, 30-36. [CrossRef]
- Mahajan, A.; Sapehia, D.; Bagga, R.; Kaur, J. Different dietary combinations of folic acid and vitamin B12 in parental diet results in epigenetic reprogramming of IGF2R and KCNQ1OT1 in placenta and fetal tissues in mice. Molecular Reproduction and Development 2021, 88, 437-458. [CrossRef]
- Goyal, R.; Goyal, D.; Leitzke, A.; Gheorghe, C.P.; Longo, L.D. Brain Renin-Angiotensin System: Fetal Epigenetic Programming by Maternal Protein Restriction During Pregnancy. Reproductive Sciences 2010, 17, 227-238. [CrossRef]
- Strakovsky, R.S.; Zhang, X.; Zhou, D.; Pan, Y.-X. Gestational high fat diet programs hepatic phosphoenolpyruvate carboxykinase gene expression and histone modification in neonatal offspring rats. The Journal of physiology 2011, 589, 2707-2717. [CrossRef]
- Chen, P.-Y.; Ganguly, A.; Rubbi, L.; Orozco, L.D.; Morselli, M.; Ashraf, D.; Jaroszewicz, A.; Feng, S.; Jacobsen, S.E.; Nakano, A.; et al. Intrauterine calorie restriction affects placental DNA methylation and gene expression. Physiological Genomics 2013, 45, 565-576. [CrossRef]
- Zhang, J.; Zhang, F.; Didelot, X.; Bruce, K.D.; Cagampang, F.R.; Vatish, M.; Hanson, M.; Lehnert, H.; Ceriello, A.; Byrne, C.D. Maternal high fat diet during pregnancy and lactation alters hepatic expression of insulin like growth factor-2 and key microRNAs in the adult offspring. BMC genomics 2009, 10, 478. [CrossRef]
Figure 1.
Epigenetic methylation switch modulates gene transcription and expression of functional proteins involved in the growth, metabolism, and development of the fetus.
Figure 1.
Epigenetic methylation switch modulates gene transcription and expression of functional proteins involved in the growth, metabolism, and development of the fetus.
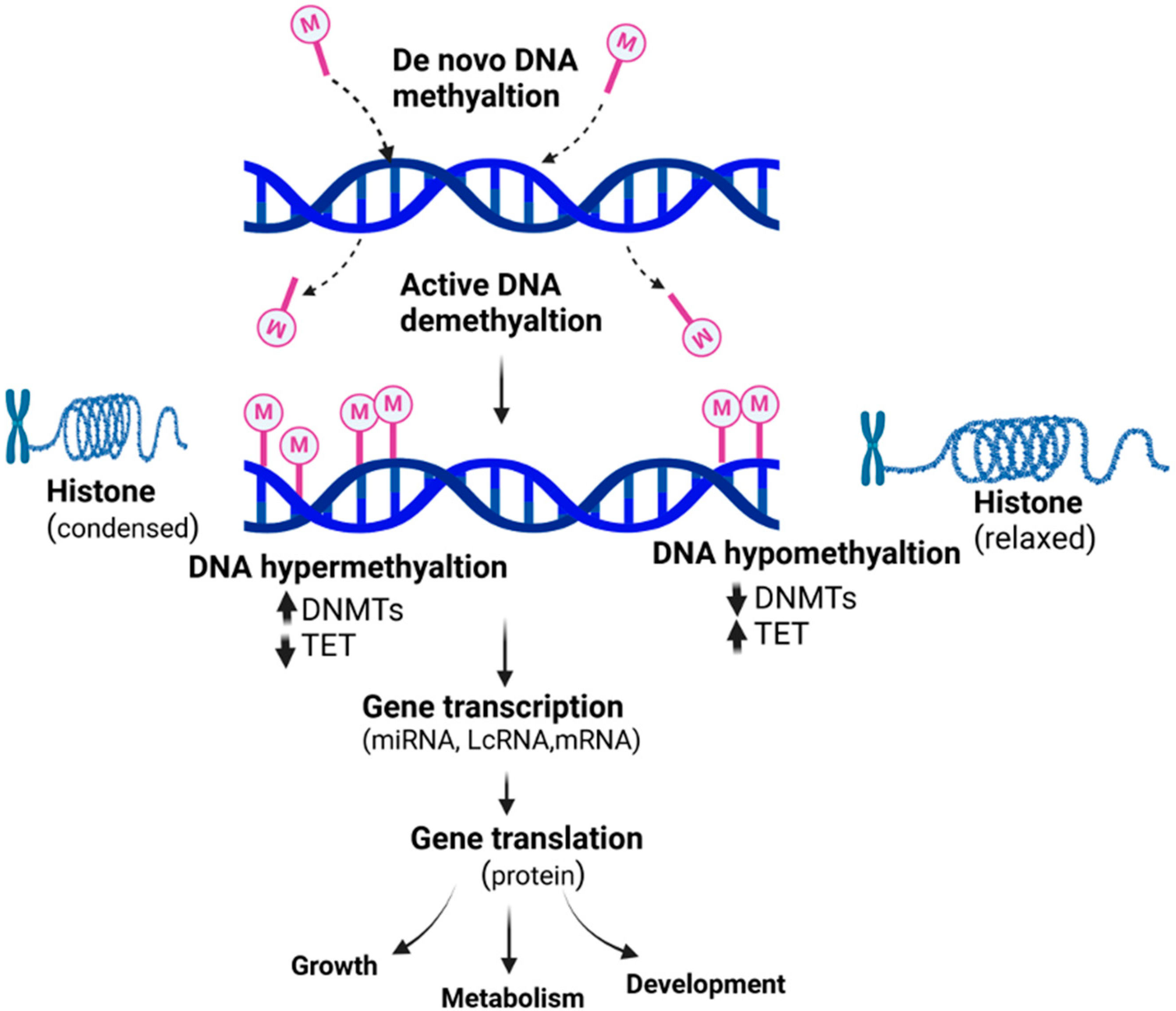
Figure 2.
In utero development of the fetal brain is also regulated by epigenetic alterations of DNA methylation and hydroxymethylation primed by several factors associated with prenatal exposure.
Figure 2.
In utero development of the fetal brain is also regulated by epigenetic alterations of DNA methylation and hydroxymethylation primed by several factors associated with prenatal exposure.
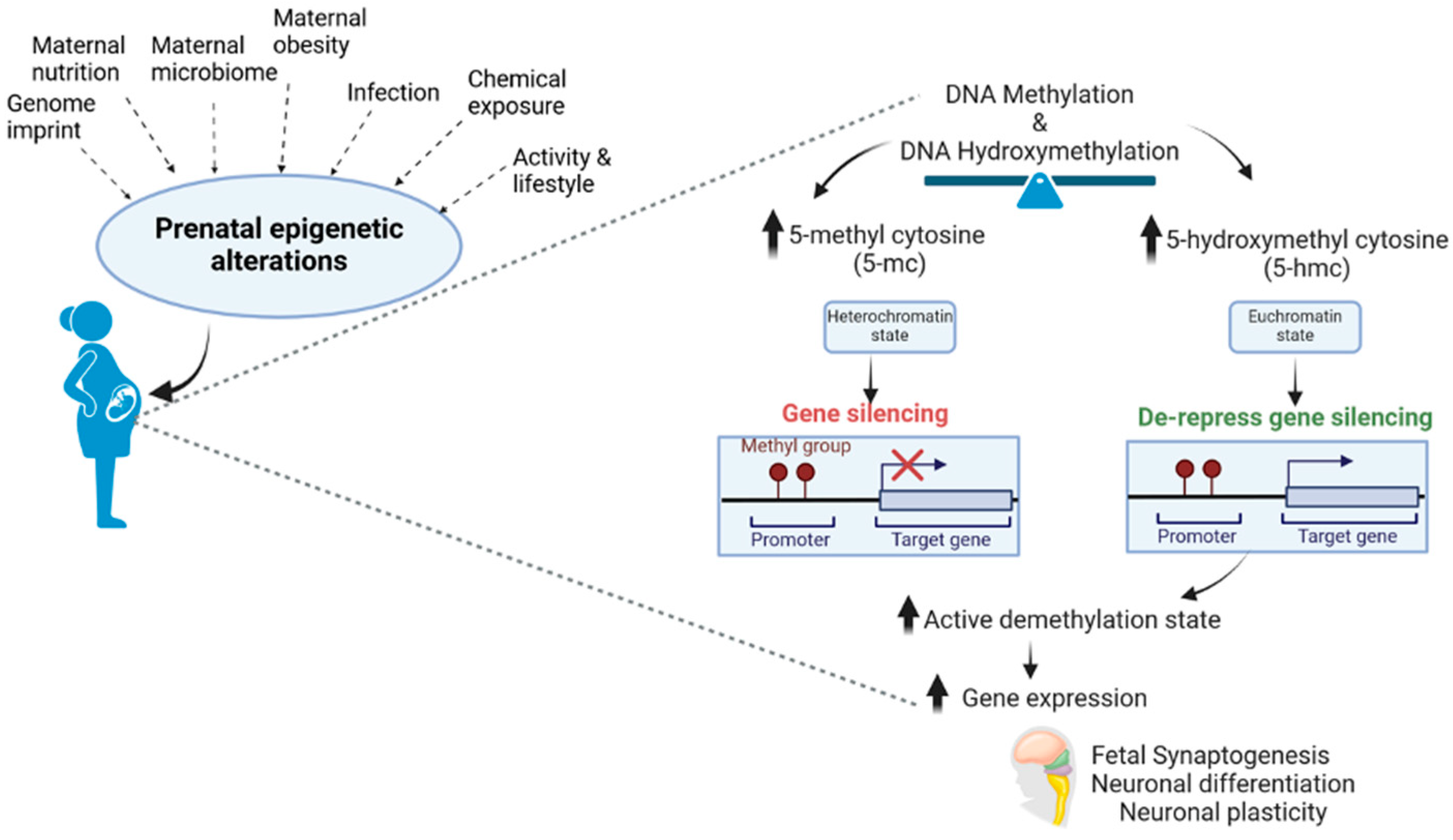
Figure 3.
Mechanistic overview of the gut microbial effects on fetal brain plasticity. Gut microflora produces several neuroactive metabolites such as GABA, tryptophan, and serotonin, potentially regulating neurotransmitter homeostasis in the fetal gut-brain axis. The gut microbiota also affects neuroinflammation and microglial maturation, and it functions in a mechanism involving SCFA action and/or modulation of neuroactive molecules. These metabolites, in turn, activate the expression of epigenetic marks, including mRNA, miRNA, hydroxymethylation, and histone remodeling in the brain cells. BDNF – Brain-derived neurotrophic factor; GABA-Gamma-aminobutyric acid; DNAm : DNA methylation.
Figure 3.
Mechanistic overview of the gut microbial effects on fetal brain plasticity. Gut microflora produces several neuroactive metabolites such as GABA, tryptophan, and serotonin, potentially regulating neurotransmitter homeostasis in the fetal gut-brain axis. The gut microbiota also affects neuroinflammation and microglial maturation, and it functions in a mechanism involving SCFA action and/or modulation of neuroactive molecules. These metabolites, in turn, activate the expression of epigenetic marks, including mRNA, miRNA, hydroxymethylation, and histone remodeling in the brain cells. BDNF – Brain-derived neurotrophic factor; GABA-Gamma-aminobutyric acid; DNAm : DNA methylation.
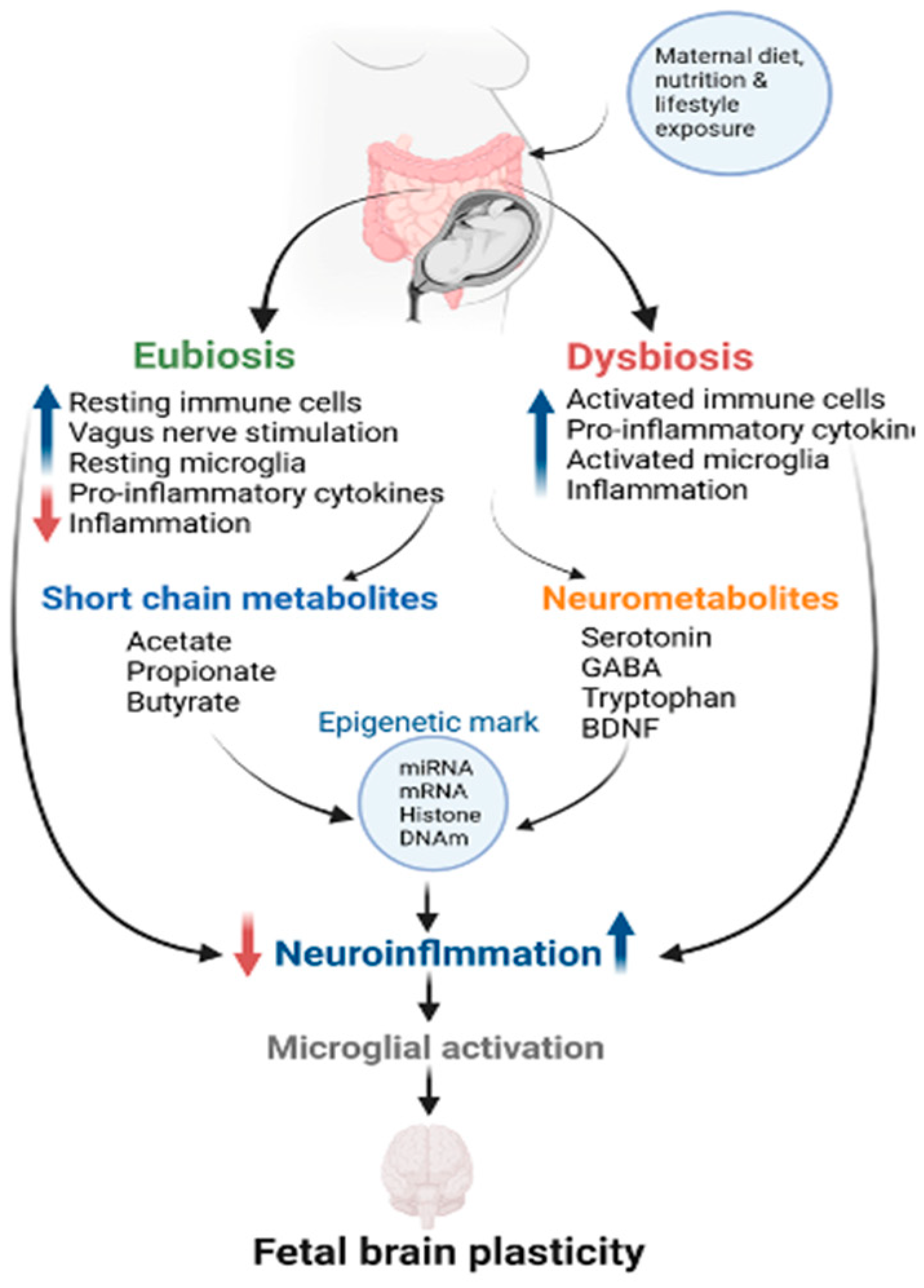
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



