
Submitted:
05 May 2024
Posted:
06 May 2024
You are already at the latest version
Abstract
Alzheimer’s disease (AD), the most common cause of dementia, is characterized by disruptions in memory, cognition, and personality, significantly impacting morbidity and mortality rates among older adults. However, the exact pathophysiological mechanism of AD remains unknown, and effective treatment options for AD are still lacking. Human induced pluripotent stem cells (iPSC) are emerging as promising platforms for disease research, offering the ability to model genetic mutations associated with various conditions. Patient-derived iPSCs are useful for modeling neurodegenerative and neurodevelopmental disorders. In this study, we generated AD iPSCs from peripheral blood mononuclear cells obtained from a 65-year-old patient with AD carrying the E682K mutation in the amyloid precursor protein (APP) gene. Cerebral organoids derived from AD iPSCs recapitulated the AD phenotype, exhibiting significantly increased levels of tau protein. Our analysis revealed that an iPSC disease model of AD is a valuable assessment tool for pathophysiological research and drug screening.
Keywords:
Alzheimer’s disease
; amyloid precursor protein
; cerebral organoid
; disease modeling
; drug screening
; induced pluripotent stem cells
; organoids
; tau pathology
1. Introduction
The brain comprises a complex architecture that regulates the functions of several organs. It presides over essential cognitive processes, such as thought, decision-making, memory, emotional responses, movement, speech, respiratory control, temperature control, and the regulation of organ function. A wide range of diseases and disorders affect the brain, including brain tumors, neurodegenerative diseases, neurodevelopmental disorders, encephalitis, stroke, and traumatic brain injury. Alzheimer’s disease (AD) is an irreversible chronic disease that affects the brain and slowly destroys memory, cognition, and personality. The hallmark of AD is the accumulation of amyloid beta (Aβ) and tau proteins [1]. Familial AD, an autosomal dominant inherited form of AD, is caused by gene mutations in amyloid precursor protein (APP), presenilin 1, and presenilin 2. Approximately 70% of the risk for developing AD can be attributed to genetic factors [2]. In particular, APP gene mutations influence the Aβ levels and disease risk.
Globally, more than 55 million people have dementia, which is the leading cause of physical and cognitive disabilities [3]. The incidence, prevalence, and mortality rates have increased dramatically [4]. Currently, no cure, medications, treatments, or other therapies are available for AD. Challenges in distinguishing between human brain physiology and disease model systems have hindered both therapeutic development and the understanding of disease pathology [5]. Furthermore, biochemical and physiological differences exist between human and animal models.
One promising strategy for overcoming the scarcity of biomaterials in diseases is to generate induced pluripotent stem cells (iPSCs) [6]. Human iPSCs (hiPSCs) are obtained by reprogramming somatic cells (e.g., cord blood mononuclear cells, peripheral blood mononuclear cells [PBMC], fibroblasts, keratinocytes, and hair follicle cells) using lentiviruses, Sendai viruses, or episomal vectors. The hiPSCs are pluripotent and can differentiate into three germ layers in vitro. Patient-derived iPSCs are valuable tools for modeling brain diseases and can differentiate into disease-related cells [7,8]. Furthermore, iPSC-based systems and genome-editing tools will be critical for understanding the roles of numerous newly discovered genes and mutations that influence disease risk.
Many protocols and methods have been developed to differentiate iPSC-derived neural models [9]. Two-dimensional (2D) methods are relatively simple and require a short differentiation period. Three-dimensional (3D) methods, such as spheroids and organoids, offer a more functionally complex system that mimics the developmental process. Cerebral organoids (COs) derived from iPSCs are self-assembling 3D cellular aggregates that mimic the human brain. CO models have emerged as a new approach for investigating human brain diseases in vitro, overcoming the conventional limitations of in vitro human cell–based systems and animal models [10]. CO models facilitate studying pathological mechanisms and screening for potential drugs. Many hiPSC-derived brain organoid models have been developed to study brain diseases [11]. However, 3D methods require prolonged culture periods, ranging from 80 days to 1 year. Such extended culture times are constrained by both time and cost considerations. In addition, prolonged culture durations often lead to necrosis within the organoids [12].
This study generated an iPSC line from PBMCs obtained from patients with AD carrying the E682K mutation in the APP gene. Furthermore, we describe a shortened differentiation protocol for COs facilitating efficient disease modeling of AD.
2. Materials and Methods
2.1. Reprogramming of PBMCs Derived from Patients with AD and Maintenance of iPSCs
The hiPSCs were generated from the PBMCs of patients with AD using the CytoTune-iPS 2.0 Sendai reprogramming kit (Thermo Fisher Scientific; Waltham, MA, USA). The generated iPSCs were maintained under feeder-free conditions on iMatrix-511–coated (Nippi; Tokyo, Japan) dishes containing StemFit Basic 03 medium (Ajinomoto; Tokyo, Japan) with 100 ng/mL bFGF (Sigma-Aldrich; St Louis, MO, USA).
2.2. Alkaline Phosphatase Staining and Immunofluorescence Staining of AD iPSCs
Alkaline phosphatase staining was performed according to the manufacturer’s instructions. Briefly, iPSCs were fixed with 4% paraformaldehyde (Wako) for 3 min and incubated with the staining solution for 15 min in the dark. Stained iPSC colonies were examined using bright-field microscopy.
For immunofluorescence staining, the iPSCs were fixed with 4% paraformaldehyde (Wako) for 20 min and permeabilized with 0.2% Triton X-100 (Sigma-Aldrich) for 10 min. The cells were blocked for 30 min in 1% bovine serum albumin (BSA, Sigma-Aldrich) and incubated with primary antibodies. The cells were treated with the secondary antibodies for 1 h; counterstaining was performed with 4′,6-diamidino-2-phenylindole (DAPI). The images were analyzed using fluorescence microscopy (Olympus JP/IX83; Tokyo, Japan).
2.3. Real-Time Polymerase Chain Reaction (RT-PCR) Analysis of AD iPSC
Total RNA was isolated from AD iPSCs using the Maxwell RSC simplyRNA Cell Kit (Promega; Madison, WI, USA) and transcribed to cDNA using EcoDry Premix (Clontech; Kusatsu, Shiga, Japan). RT-PCR was performed using TaqMan gene expression Master Mix (Applied Biosystems; Waltham, MA, USA). The TaqMan probe IDs are listed in Table 1. Gene expression levels were normalized to GAPDH levels for standardization.
2.4. Mycoplasma Test of AD iPSC
Cell supernatants were analyzed using the e-Myco™ VALID Mycoplasma PCR Detection Kit (25239; Boca Scientific, Dedham, MA, USA) according to the manufacturer’s instructions. The amplified products were analyzed via gel electrophoresis.
2.5. Karyotyping of AD iPSC
Karyotypes were determined using standard cytogenetic procedures using the GTG-band method. The cells were treated with colcemid for 45 min, incubated in a hypotonic solution, and fixed with a methanol–acetic acid solution (3:1). After Giemsa trypsin banding, karyotypes were analyzed according to the International System for Human Cytogenetic Nomenclature using the standard G-banding method.
2.6. In Vitro Three Germ Layer Differentiation of AD iPSC
For three germ layer differentiation in vitro, hiPSCs were induced to form embryoid bodies (EBs) using Dulbecco’s Modified Eagle Medium F-12 with 20% knockout serum replacement, 1% nonessential amino acids, and 0.1 mM β-mercaptoethanol for 14 days. Differentiated EBs were analyzed via RT-qPCR using the TaqMan gene Expression Master Mix. The TaqMan probes are listed in Table 1. Gene expression levels were normalized to the GAPDH levels for standardization.
2.7. Differentiation of hiPSC into Cerebral Organoids
CO differentiation was performed using the STEMdiff Cerebral Organoid Kit (Stem Cell Technologies; Vancouver, Canada) with some modifications. For EB formation, an AggreWell plate was pretreated with an anti-adherence rinsing solution and centrifuged at 1,300 × g for 5 min. Each well was rinsed with warm EB-formation media, and warm media was added to each well. The hiPSCs were detached using Accutase, resuspended in EB formation medium (9×103 cells/EB) with 5 mM Y-27632, and transferred to an AggreWell plate. The AggreWell plates were incubated at 37°C. On days 2 and 4, 1 mL of medium was removed, and 1 mL of EB formation medium was added to each well. On day 5, the EBs were transferred to a six-well ultra-low attachment plate with induction medium and incubated at 37°C for 48 h. On day 7, EBs were transferred to an embedding surface (Parafilm) using a wide-bore 200-µL pipette tip, and the excess medium was removed from the EB. We added 15 µL of Matrigel onto each EB, repositioned the EB to the center of the Matrigel, and incubated the culture at 37°C for 30 min. Embedded EBs were transferred to ultra-low–attachment plates containing induction medium and incubated at 37°C for 3 days. On days 10–40, the organoid medium was replaced with maturation medium, and the cultures were incubated on an orbital shaker at 37°C.
2.8. Immunofluorescence
For cryosectioning, COs were fixed with 4% paraformaldehyde overnight and incubated with 0.1% Tween 20. Fixed COs were treated with 15% and 30% sucrose for cryoprotection. COs were embedded in optimal cutting temperature compound and sectioned into 10-µm slices using a microtome. The slides were dried for 30 min at room temperature and washed with 1× phosphate-buffered saline (PBS). The slides were blocked with 5% BSA in PBS with 0.5% Triton X-100 and incubated overnight with primary antibodies against TUJ1 (18207; Abcam, Cambridge, UK), MAP2 (13-1500; Invitrogen, Waltham, MA, USA), and Tau (13-6400; Invitrogen). Then, the cells were incubated with Alexa 488/594-conjugated anti-mouse or anti-rabbit secondary antibodies. Counterstaining was performed with DAPI. The stained slides were observed via confocal laser-scanning microscopy (FV3000-OSP; Olympus).
2.9. Whole Staining of Cerebral Organoids
For complete staining of the organoids, the medium and fixed organoids were removed using 4% paraformaldehyde. Fixed organoids were washed with 1× PBS to release the organoid from the Matrigel dome and permeabilized overnight at 4°C with blocking buffer (5% BSA with 0.5% Triton X-100 in 1× PBS). The organoids were incubated overnight with primary antibodies against TUJ1 (18207; Abcam), MAP2 (13-1500; Invitrogen), and Tau (13-6400; Invitrogen). Then, the cells were incubated overnight with Alexa488/594-conjugated anti-mouse or anti-rabbit secondary antibodies. Counterstaining was performed for 15 min using DAPI. Stained organoids were observed using confocal laser-scanning microscopy.
2.10. Enzyme-Linked Immunosorbent Assay (ELISA)
A human Tau ELISA kit (Invitrogen) was used to analyze Tau in the culture supernatant, according to the manufacturer’s instructions. The diluted supernatant was added to each well of the antibody-coated capture plate and incubated for 2 h at room temperature. The detection antibodies were added to each well, incubated for 1 h, and treated with an anti-rabbit IgG horseradish peroxidase solution for 30 min. A stabilized chromogen was added to each well and incubated in the dark for 30 min. The stop solution was added, and the absorbance was detected at 450 nm.
3. Results
3.1. Reprogramming of PBMCs and Characterization of iPSCs Derived from Patients with AD
For modeling AD, we generated an iPSC line from PBMCs of patients with AD (65, XY) with the E682K mutation in the APP gene (21q21.3). AD iPSCs were generated from patients with AD using a reprogramming vector containing the Yamanaka factors. The AD iPSCs exhibited a morphology similar to that of human embryonic stem cells and expressed alkaline phosphatase (Figure 1a). Alkaline phosphatase staining revealed that the AD iPSCs were maintained in an undifferentiated state. AD iPSCs also expressed the pluripotency markers OCT4, SSEA4, TRA-1-60, and TRA-1-81 (Figure 1b). The expression of pluripotency markers (OCT4, NANOG, SOX2, GABRB3, and GDF3) was confirmed in AD iPSCs, and the expression levels were compared with those in the H9 human embryonic stem cell line (Figure 1c). The mycoplasma test was negative (Figure 1d). Long-term iPSC culture involves the accumulation of mutations and genomic integration [13]. Karyotyping was performed using the GTG-band method, and the AD iPSCs were confirmed to have a normal karyotype (Figure 1e). To confirm the iPSC differentiation potency, we tested EB formation and determined the expression of ectoderm (PAX6), mesoderm (ITGA8), and endoderm (AFP) genes in AD iPSC-derived EBs (Figure 1f,g). AD iPSCs were successfully generated from the PBMCs of patients with AD, and pluripotency was confirmed.
3.2. Differentiation of Cerebral Organoids Using iPSCs Derived from Patients with AD
AD is characterized by a severe loss of neurons in the cortex and certain subcortical regions, followed by cortical dysfunction [14]. For disease modeling, we differentiated COs. To differentiate iPSC-derived COs, we induced iPSCs to form EBs in AggreWell plates. A schematic representation of the CO differentiation protocol is shown in Figure 2a. EBs were transferred to ultra-low–attachment plates on day 5 and embedded in Matrigel on day 7. The embedded EBs were maintained in an orbital shaker. The embedded EBs expanded into neuroepithelial cells with the budding of the organoid surface on day 10. On day 20, neural rosettes formed in the organoids. On days 30–40, the morphology of the organoids exhibited alterations, presenting a dense and dark core with cortical layering (Figure 2b). The size of the organoids did not differ from that of the healthy controls (HCs) and patients with AD (Figure 2c). For the histological analysis of the COs, we performed immunofluorescence staining. On day 40, the expression of the CO markers TUJ1 and MAP2 increased in HC and AD iPSC-derived COs (Figure 2d). For a more comprehensive analysis, we performed whole organoid staining. TUJ1 and MAP2 expression generally increased in the CO group (Figure 2e). These results confirm that HC and AD iPSC-derived COs recapitulate the cerebral cortex.
3.3. In Vitro Disease Modeling of AD iPSC-Derived Cerebral Organoids
Increased tau expression is a critical pathological feature in patients with AD; therefore, we analyzed the tau protein levels using cryosection slides (Figure 3a). Tau expression began to increase on day 21 and tended to increase three-fold on day 40 in AD iPSC-derived COs compared to HC iPSC-derived COs (Figure 3b). Furthermore, we analyzed whole organoids and found that the tau expression increased in AD iPSC-derived COs (Figure 3c). Fluorescence intensity analysis showed that the expression of tau increased by >6-fold in AD iPSC-derived COs (Figure 3d). Analysis of tau phosphorylation is essential for understanding its pathology and physiology. AD iPSC-derived COs showed greater phosphorylated tau levels than HC iPSC-derived COs in the culture supernatant (Figure 3e). These organoids exhibited increased expression of tau and secretion of phosphorylated tau, confirming the mimicking of the AD pathological phenotype in AD iPSC-derived COs.
4. Discussion
AD is an irreversible and progressive neurodegenerative disease characterized by a severe neuronal loss in the cerebral cortex and the accumulation of Aβ and phosphorylated tau proteins [1]. Aβ pathology might involve upstream pathophysiological signal and transfer as an activator of downstream signaling pathways, leading to tau phosphorylation, misfolding, and accumulation [15-17]. However, the pathology of tau hyperphosphorylation is unclear, and the exact pathological relationship between tau and Aβ remains unknown. Aβ are fragments of the transmembrane APP, while tau is a brain-specific microtubule-associated protein.
Tauopathy of phosphorylation and aggregation is the primary cause of neurodegeneration in AD, leading to neuronal dysfunction and death [18]. The tau pathology can also be found in the brain of patients with very mild dementia without Aβ pathology [19]. Moreover, the approaches to removing Aβ or reducing Aβ production are inadequate for clinical therapy. Thus, interest in tau-based therapies is emerging [20,21]. Experimental models characterized by human tau pathology are necessary to develop tau-based therapies. However, traditional animal models (e.g., transgenic mice) often fail to recapitulate clinical efficacy due to inherent differences in nervous system complexity, physiology, and drug metabolism between humans and animals [22,23]. The breakthrough was the proposal of patient-derived iPSCs as a promising strategy for disease modeling. Applying iPSC-derived disease models in drug development can account for differences between animal models and humans.
The hiPSCs can differentiate into the three germ layers and reveal a patient’s genetic background. High-quality undifferentiated patient-derived hiPSCs are essential for disease modeling. However, it is crucial to verify the identity of cell lines, confirming their undifferentiated state, pluripotency, absence of mycoplasma contamination, karyotype integrity, and capability for differentiation into the three germ layers [24,25]. Poor-quality iPSCs lead to low differentiation efficiency and faulty analysis. In the Korea National Stem Cell Bank, we confirmed the iPSC quality using their identity, sterility, consistency, stability, and safety to maintain high-quality cell lines.
Various iPSC-derived disease models have been developed for pathological research and drug screening. IPSC-derived neuronal disease modeling has traditionally been conducted in a 2D monolayer culture as a simple and low-cost method. However, 2D models are insufficient for mimicking the complex physiology of the human brain. The limitations of 2D models have been overcome by the development of 3D models (e.g., spheroids and organoids). The 3D models more closely recapitulate in vivo environments, cell-to-cell interactions, and neural networks [26] and show an increased expression of neuronal and maturation markers compared to their 2D monolayer counterparts. Furthermore, the 3D models effectively replicate tau pathology. Thus, 3D models are a more valuable tool for mimicking neurodegenerative diseases.
Organoids are self-assembling 3D structures that mimic the functions of human organs. They can be used as mini-organs in disease modeling, drug screening, and personalized medicine. COs recapitulate human brain development and simulate brain function. COs have overcome the differences between human and animal models and have emerged as new strategies for treating neurodegenerative diseases and neurodevelopmental disorders. The functionality of the organoids was evaluated through immunofluorescence and immunohistochemical analyses. Traditional cryosections and paraffin sections provide only partial analysis and incompletely reveal the 3D structure; therefore, we performed a whole analysis of the COs. Whole staining analysis is useful for high-definition structural morphology and localization of overall expression phenotypes. Moreover, this approach eliminates the need for serial sectioning steps, thereby reducing structural damage and saving time compared to cryosectioning or paraffin-sectioning methods.
Organoids require long-term culture for disease modeling; however, long-term culturing for at least 80 days to 1 year is time-consuming and expensive. In addition, long-term cultures are limited by the lack of nutrients and oxygen in the organoid core. This environment can lead to necrosis within the organoid due to hypoxic conditions. Therefore, improved methods are necessary for efficient disease modeling.
This study describes a rapid differentiation method for Cos, facilitating efficient modeling of AD. Notably, the expression of neuronal markers was evident by day 14 of differentiation, with a significant increase in tau expression observed in AD iPSC-derived COs, sustained until day 100. Our method can be used for modeling human tau-overexpressing diseases, particularly aiding in tau-targeted drug screening. Our method provides a shortened differentiation protocol for COs and efficient AD modeling, enabling effective screening of pharmaceutical compounds and drug development endeavors.
5. Conclusions
We generated iPSC lines from patients with AD harboring the E682K mutation in the APP gene. These iPSC lines were then differentiated into COs to model AD pathology. Our findings demonstrate that AD iPSC-derived COs effectively differentiated within a short time and recapitulated key features of the AD phenotype. Consequently, the AD iPSC-derived CO model holds immediate promise for disease modeling and may increase drug screening feasibility.
Author Contributions
Conceptualization, B.Y.K.; Methodology, Y.K. and B.Y.; Validation, Y.K and B.Y.; Formal analysis, Y.K. and B.Y.K.; Investigation, B.S.Y. and B.Y.K.; Resources, B.S.Y. and B.Y.K.; Data curation, Y. K. and B.Y.; Writing—original draft preparation, Y.K.; Writing—review and editing, B.Y.K.; Visualization, Y. K. and B.Y.; Supervision, B.Y.K.; Project administration, B.Y.K.; Funding acquisition, B.Y.K. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by an intramural research grant from the Korea National Institute of Health (grant number 2023-NI-009).
Institutional Review Board Statement
The study was conducted in accordance with the Korea National Institute of Health Institutional Review Board (KDCA-2023-05-05) and Severance Children’s Hospital Institutional Review Board (4-2016-1158).
Informed Consent Statement
Informed consent was obtained from all participants involved in the study. Written informed consent was obtained from the patient for the publication of this paper.
Data Availability Statement
All datasets of this article are included within the article.
Acknowledgments
We thank Hyun Jung Choi (Division of Biomedical Research Institute, Korea National Institute of Health) for assistance with confocal microscopy analysis. We would like to thank Editage (www.editage.co.kr) for English language editing.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Hardy, J.; Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer's disease. Trends Pharmacol Sci 1991, 12, 383–388. [CrossRef]
- Ballard, C.; Gauthier, S.; Corbett, A.; Brayne, C.; Aarsland, D.; Jones, E. Alzheimer's disease. Lancet 2011, 377, 1019–1031. [CrossRef]
- Feigin, V.L.; Vos, T.; Nichols, E.; Owolabi, M.O.; Carroll, W.M.; Dichgans, M.; Deuschl, G.; Parmar, P.; Brainin, M.; Murray, C. The global burden of neurological disorders: translating evidence into policy. Lancet Neurol 2020, 19, 255–265. [CrossRef]
- Li, X.; Feng, X.; Sun, X.; Hou, N.; Han, F.; Liu, Y. Global, regional, and national burden of Alzheimer's disease and other dementias, 1990-2019. Front Aging Neurosci 2022, 14, 937486. [CrossRef]
- Vadodaria, K.C.; Jones, J.R.; Linker, S.; Gage, F.H. Modeling Brain Disorders Using Induced Pluripotent Stem Cells. Cold Spring Harb Perspect Biol 2020, 12, a035659. [CrossRef]
- Shi, Y.; Inoue, H.; Wu, J.C.; Yamanaka, S. Induced pluripotent stem cell technology: a decade of progress. Nat Rev Drug Discov 2017, 16, 115–130. [CrossRef]
- Karagiannis, P.; Takahashi, K.; Saito, M.; Yoshida, Y.; Okita, K.; Watanabe, A.; Inoue, H.; Yamashita, J.K.; Todani, M.; Nakagawa, M.; et al. Induced pluripotent stem cells and their use in human models of disease and development. Physiol Rev 2019, 99, 79-114. [CrossRef]
- Inoue, H.; Yamanaka, S. The use of induced pluripotent stem cells in drug development. Clin Pharmacol Ther 2011, 89, 655–661. [CrossRef]
- Mayhew, C.N.; Singhania, R. A review of protocols for brain organoids and applications for disease modeling. STAR Protoc 2023, 4, 101860. [CrossRef]
- Barak, M.; Fedorova, V.; Pospisilova, V.; Raska, J.; Vochyanova, S.; Sedmik, J.; Hribkova, H.; Klimova, H.; Vanova, T.; Bohaciakova, D. Human iPSC-derived neural models for studying alzheimer's disease: from neural stem cells to cerebral organoids. Stem Cell Rev Rep 2022, 18, 792–820.. [CrossRef]
- Benito-Kwiecinski, S.; Lancaster, M.A. Brain organoids: Human neurodevelopment in a dish. Cold Spring Harb Perspect Biol 2020, 12, a035709. [CrossRef]
- Andrews, M.G.; Kriegstein, A.R. Challenges of organoid research. Annu Rev Neurosci 2022, 45, 23–39. [CrossRef]
- Peterson, S.E.; Westra, J.W.; Rehen, S.K.; Young, H.; Bushman, D.M.; Paczkowski, C.M.; Yung, Y.C.; Lynch, C.L.; Tran, H.T.; Nickey, K.S.; et al. Normal human pluripotent stem cell lines exhibit pervasive mosaic aneuploidy. PLoS One 2011, 6, e23018. [CrossRef]
- Vanova, T.; Sedmik, J.; Raska, J.; Amruz Cerna, K.; Taus, P.; Pospisilova, V.; Nezvedova, M.; Fedorova, V.; Kadakova, S.; Klimova, H.; et al. Cerebral organoids derived from patients with Alzheimer's disease with PSEN1/2 mutations have defective tissue patterning and altered development. Cell Rep 2023, 42, 113310. [CrossRef]
- He, Z.; Guo, J.L.; McBride, J.D.; Narasimhan, S.; Kim, H.; Changolkar, L.; Zhang, B.; Gathagan, R.J.; Yue, C.; Dengler, C.; et al. Amyloid-beta plaques enhance Alzheimer's brain tau-seeded pathologies by facilitating neuritic plaque tau aggregation. Nat Med 2018, 24, 29–38. [CrossRef]
- Wang, L.; Benzinger, T.L.; Su, Y.; Christensen, J.; Friedrichsen, K.; Aldea, P.; McConathy, J.; Cairns, N.J.; Fagan, A.M.; Morris, J.C.; et al. Evaluation of Tau imaging in staging alzheimer disease and revealing interactions between beta-amyloid and tauopathy. JAMA Neurol 2016, 73, 1070–1077. [CrossRef]
- Lewis, J.; Dickson, D.W.; Lin, W.L.; Chisholm, L.; Corral, A.; Jones, G.; Yen, S.H.; Sahara, N.; Skipper, L.; Yager, D.; et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science 2001, 293, 1487–1491. [CrossRef]
- Mashal, Y.; Abdelhady, H.; Iyer, A.K. Comparison of Tau and Amyloid-beta Targeted Immunotherapy Nanoparticles for Alzheimer's Disease. Biomolecules 2022, 12, 1001. [CrossRef]
- de Paula, V.J.R.; Guimaraes, F.M.; Diniz, B.S.; Forlenza, O.V. Neurobiological pathways to Alzheimer's disease: Amyloid-beta, TAU protein or both? Dement Neuropsychol 2009, 3, 188–194. [CrossRef]
- Congdon, E.E.; Ji, C.; Tetlow, A.M.; Jiang, Y.; Sigurdsson, E.M. Tau-targeting therapies for Alzheimer disease: current status and future directions. Nat Rev Neurol 2023, 19, 715–736. [CrossRef]
- Congdon, E.E.; Sigurdsson, E.M. Tau-targeting therapies for Alzheimer disease. Nat Rev Neurol 2018, 14, 399–415. [CrossRef]
- Haston, K.M.; Finkbeiner, S. Clinical Trials in a Dish: The potential of pluripotent stem cells to develop therapies for neurodegenerative diseases. Annu Rev Pharmacol Toxicol 2016, 56, 489–510. [CrossRef]
- Pasteuning-Vuhman, S.; de Jongh, R.; Timmers, A.; Pasterkamp, R.J. Towards advanced iPSC-based drug development for neurodegenerative disease. Trends Mol Med 2021, 27, 263–279. [CrossRef]
- Sullivan, S.; Stacey, G.N.; Akazawa, C.; Aoyama, N.; Baptista, R.; Bedford, P.; Bennaceur Griscelli, A.; Chandra, A.; Elwood, N.; Girard, M.; et al. Quality control guidelines for clinical-grade human induced pluripotent stem cell lines. Regen Med 2018, 13, 859–866. [CrossRef]
- Kim, J.H.; Jo, H.Y.; Ha, H.Y.; Kim, Y.O. Korea National Stem Cell Bank. Stem Cell Res 2021, 53, 102270. [CrossRef]
- D'Avanzo, C.; Aronson, J.; Kim, Y.H.; Choi, S.H.; Tanzi, R.E.; Kim, D.Y. Alzheimer's in 3D culture: challenges and perspectives. Bioessays 2015, 37, 1139–1148. [CrossRef]
Figure 1.
Characterization of induced pluripotent stem cells (iPSCs) derived from a patient with Alzheimer’s disease (AD). (a) Morphology and alkaline phosphatase staining of AD iPSC (scale bars: 100 µm). (b) Immunofluorescence analysis and DAPI staining of pluripotency markers OCT4, SSEA4, TRA-1-60, and TRA-1-81 (scale bars: 100 µm. (c) Real-time polymerase chain reaction (RT-PCR) analysis of the pluripotency marker genes OCT4, NANOG, SOX2, GABRB3, and GDF3. (d) Mycoplasma test of AD iPSC. (e) Karyotyping of AD iPSC. (f) Morphology of AD iPSC-derived EBs (scale bars: 100 µm). (g) RT-PCR analysis of three germ layer differentiation markers: PAX6 (ectoderm), ITGA8 (mesoderm), and AFP (endoderm). All graphs show the mean and standard error of the mean.
Figure 1.
Characterization of induced pluripotent stem cells (iPSCs) derived from a patient with Alzheimer’s disease (AD). (a) Morphology and alkaline phosphatase staining of AD iPSC (scale bars: 100 µm). (b) Immunofluorescence analysis and DAPI staining of pluripotency markers OCT4, SSEA4, TRA-1-60, and TRA-1-81 (scale bars: 100 µm. (c) Real-time polymerase chain reaction (RT-PCR) analysis of the pluripotency marker genes OCT4, NANOG, SOX2, GABRB3, and GDF3. (d) Mycoplasma test of AD iPSC. (e) Karyotyping of AD iPSC. (f) Morphology of AD iPSC-derived EBs (scale bars: 100 µm). (g) RT-PCR analysis of three germ layer differentiation markers: PAX6 (ectoderm), ITGA8 (mesoderm), and AFP (endoderm). All graphs show the mean and standard error of the mean.
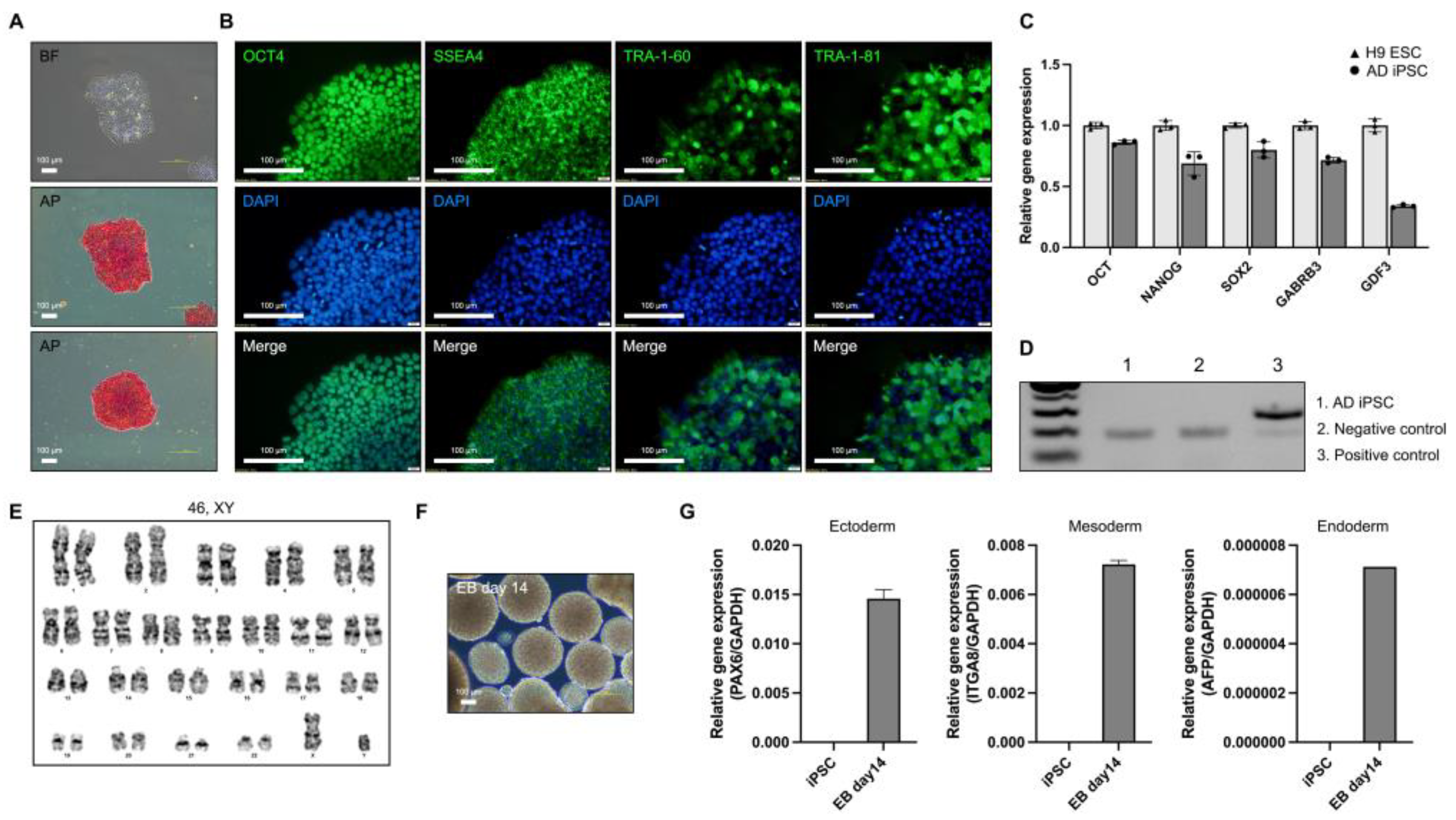
Figure 2.
Differentiation of iPSCs into cerebral organoids (COs) and their characterization (a) Scheme of the CO differentiation process. (b) Morphology of the iPSC-derived COs (scale bars: 100 µm). (c) Quantification of the organoid size using the ImageJ program. All graphs show the mean and standard error of the mean. (d) Immunofluorescence analysis of TUJ1 and MAP2 with DAPI staining on a cryosection slide (scale bars: 200 µm). (e) Whole staining analysis of TUJ1 and MAP2 in COs (scale bars: 50 µm).
Figure 2.
Differentiation of iPSCs into cerebral organoids (COs) and their characterization (a) Scheme of the CO differentiation process. (b) Morphology of the iPSC-derived COs (scale bars: 100 µm). (c) Quantification of the organoid size using the ImageJ program. All graphs show the mean and standard error of the mean. (d) Immunofluorescence analysis of TUJ1 and MAP2 with DAPI staining on a cryosection slide (scale bars: 200 µm). (e) Whole staining analysis of TUJ1 and MAP2 in COs (scale bars: 50 µm).
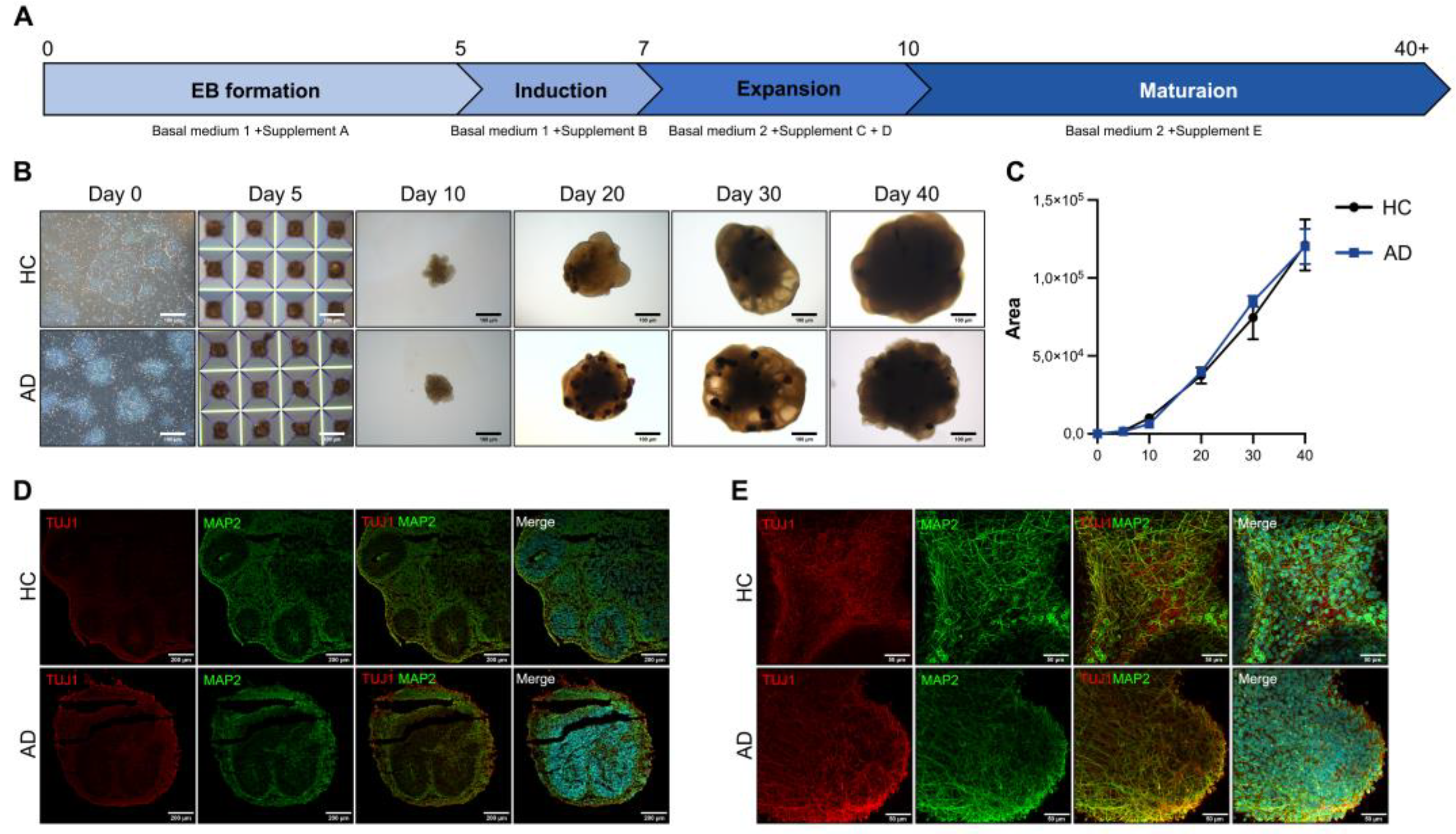
Figure 3.
In vitro modeling of Alzheimer’s disease (AD) using iPSCs derived from patients with AD. (a) Immunofluorescence staining of cryosectioned tissue. (b) Quantification of the fluorescence intensity of tau using cellSens imaging software (version 4.1, Olympus). (c) Whole staining analysis of cerebral organoids (COs) (scale bars: 50 µm). (d) Quantification of fluorescence intensity of tau in whole organoids. (e) Phosphorylation analysis of the tau protein in the culture supernatant. All graphs show the mean and standard error of the mean.
Figure 3.
In vitro modeling of Alzheimer’s disease (AD) using iPSCs derived from patients with AD. (a) Immunofluorescence staining of cryosectioned tissue. (b) Quantification of the fluorescence intensity of tau using cellSens imaging software (version 4.1, Olympus). (c) Whole staining analysis of cerebral organoids (COs) (scale bars: 50 µm). (d) Quantification of fluorescence intensity of tau in whole organoids. (e) Phosphorylation analysis of the tau protein in the culture supernatant. All graphs show the mean and standard error of the mean.
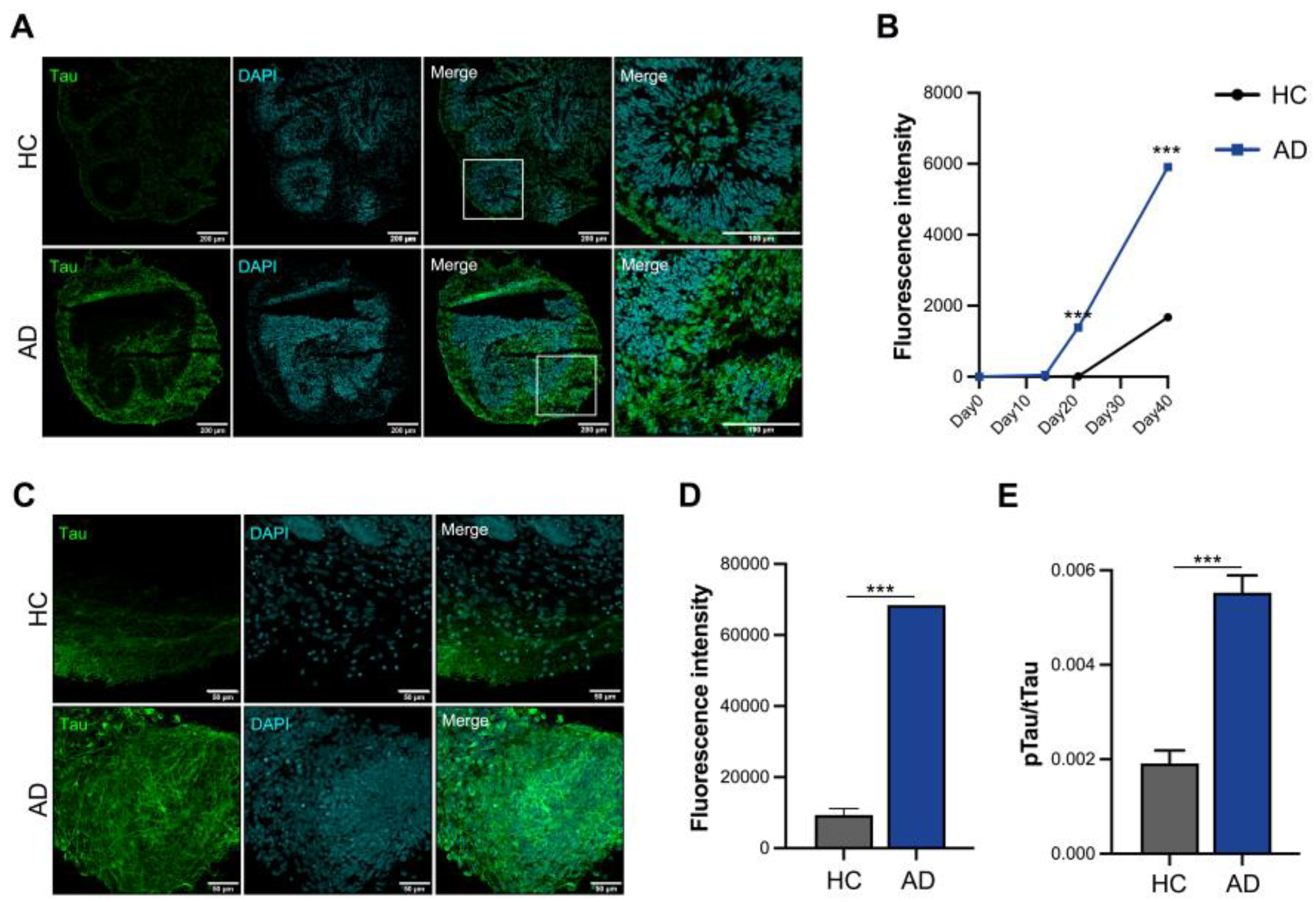

Table 1.
TaqMan® probe ID list.
| Analysis | Target | Amplicon Length | Assay ID |
|---|---|---|---|
| Pluripotency markers | OCT4 | 65bp | TaqMan Probe ID Hs00742896-s1 |
| NANOG | 109bp | TaqMan Probe ID Hs02387400-g1 | |
| SOX2 | 121bp | TaqMan Probe ID Hs00602736-s1 | |
| GABRB3 | 63bp | TaqMan Probe ID Hs00241459-m1 | |
| GDF3 | 65bp | TaqMan Probe ID Hs00220998-m1 | |
| House-keeping gene | GAPDH | 122bp | TaqMan Probe ID Hs999999905-m1 |
| Three germ layer differentiation markers | PAX6 | 76bp | TaqMan Probe ID Hs00240871-m1 |
| ITGA8 | 89bp | TaqMan Probe ID Hs00233321-m1 | |
| AFP | 82bp | TaqMan Probe ID Hs00173490-m1 | |
| House-keeping gene | GAPDH | 58bp | TaqMan Probe ID Hs03929097-G1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



