Submitted:
06 May 2024
Posted:
06 May 2024
Read the latest preprint version here
Abstract
Cuproptosis, a copper-dependent cellular death mechanism, is typically overlooked in glioblastoma diagnosis. Lipoylation is needed for cuproptosis, and FDX1 controls this process. Cuproptosis in prognostic models may drive cancer treatment. This study evaluated cuproptosis and glioblastoma cell proliferation. The Cancer Genome Atlas provided annotated clinical, genetic mutation, and RNA sequencing data for the TCGA_GBM and TCGA_LGG cohorts. Patients with gliomas were randomly allocated to the validation or training cohort. Least absolute shrinkage and selection operator (LASSO) and multivariate Cox regression models were utilized to evaluate which model best predicted prognosis in the training cohort. The models' independent predictive power was evaluated throughout the cohort. A previous study revealed 19 genes related to ferroptosis. LncRNAs associated with cuproptosis were identified by coexpression analysis. Cox discovered 17 cuproptosis-associated lncRNAs and established a predictive model. The median risk score classified patients as high- or low-risk. K‒M survival analysis demonstrated significant differences in overall survival among risk categories. Principal component analysis (PCA) and receiver operating characteristic (ROC) curve analysis were used to test the model’s predictive power. Univariate and multivariate Cox regression analyses revealed risk score-related independent prognostic factors. Multivariate Cox regression analysis was used to construct a nomogram for marker-based prognosis. The risk category affected the tumour mutation rate. High-risk glioma patients benefit from immunotherapy. Glioma drug sensitivity was also substantially linked with the risk score. The expression of 17 cuproptosis-related long noncoding RNAs (lncRNAs) may assist in the stratification of glioma patient prognosis, molecular characteristics, cell cycle gene regulation pathways, the TME, and clinicopathological aspects. Our clinical sample and database analysis revealed that cuproptosis influences glioma prognosis and may guide therapy.
Keywords:
cuproptosis
; LncRNAs
; prognosis
; prediction
; Tumour Microenvironment
; glioma
1. Introduction
Cuproptosis is an emerging cell death pathway, and further exploration of its regulatory mechanism in the context of glioblastoma (GBM) is needed [1] [1]. Our objective was to examine cuproptosis-associated genes (CRGs) and develop a novel prognostic model for GBM.
Gliomas are the most common solid tumours of the central nervous system (CNS) [2,3]. Despite the implementation of extensive treatment regimens and recent advancements in therapeutic approaches, glioma treatment remains a significant challenge in the field of medicine[4,5]. Glioma includes a variety of subtypes, including glioblastomas, ependymomas, medulloblastomas, astrocytomas, oligodendrogliomas, and glioblastoma multiforme [6]. The therapeutic approach for glioblastoma has undergone minimal alterations over the past decade [7], resulting in a persistently unfavourable prognosis characterized by a median survival of approximately 15 months [8]. An extensive multimodal therapeutic approach encompassing chemotherapy, radiation, and surgical resection (where appropriate) is typically applied, but this treatment regimen significantly decreases patients’ quality of life [9].
The primary approach for managing glioblastoma (GBM) comprises a multimodal treatment strategy that includes chemotherapy, surgery, and radiation therapy [10]. Therefore, it is crucial to identify dependable prognostic biomarkers to support the advancement of gene-targeted therapeutics for glioma [11]. Additionally, the development of novel pharmaceutical compounds is of critical importance. At present, chemotherapeutic agents designed to treat glioma are predominantly administered after the completion of clinical trials involving other malignant tumour types. Nonetheless, further investigation is warranted to ascertain specific therapeutic interventions that efficiently target glioma cells. The CRISPR/Cas9 approach is employed to target and suppress the expression of specific genes in embryos, hence establishing the most effective screening technique for clarifying pathogenesis mechanisms. Drug screening can also be conducted using this method. However, the inherent diversity of glioblastoma limits the effectiveness of this strategy in tackling this particular problem. Similarly, the injection of glioma stem cells (GSCs) into animal embryos for the purpose of creating glioma models does not adequately address the issue of heterogeneity. Consequently, the use of organoids or high-throughput screening might be considered supplementary alternatives.
The primary aim of this study was to examine the association between CRGs and commonly prescribed medications utilized in the treatment of individuals with glioblastoma. Drug sensitivity in individuals diagnosed with glioma is often assessed based on treatment response and the half-maximum inhibitory concentration (IC50) value. One approach for determining the IC50 value of a chemotherapy treatment involves utilizing the "prophytic" R package in conjunction with the necessary dependencies, such as "car, ridge preprocessCore, genefilter, and sva". IC50 values were determined for commonly used anticancer medicines in both high-risk and low-risk populations. Drug testing for glioblastoma multiforme (GBM) is currently a labour-intensive process, which limits its application.
We examined whether lncRNAs associated with cuproptosis are predictive of the prognosis of glioma patients. These findings could contribute to the understanding of the involvement of cuproptosis in the initiation and progression of glioma.
2. Results
2.1. Data Processing
According to Figure 1, the search and screening methods produced the following results. A total of 16,876 long noncoding RNAs (lncRNAs) were identified, and 19 genes involved in cuproptosis were associated with these lncRNAs. By using the 'GENCODE' database, protein-coding genes were removed from the TCGA-GBM and TCGA-LGG datasets, allowing for the identification of 16876 long noncoding RNAs (lncRNAs). Based on a comprehensive analysis of previous studies, 19 genes were identified as being associated with cuproptosis. Pearson correlation analysis revealed a correlation between long noncoding RNAs (lncRNAs) and cuproptosis. Figure 1B illustrates the relationships between long noncoding RNAs (lncRNAs) and cuproptosis-associated genes.
2.2. Prognostic Markers for Mutations Associated with Noncoding RNAs Are Constructed and Validated
As shown in Figure 2A, 17 lncRNAs associated with cuproptosis were studied by univariate Cox analysis. We further screened 17 lncRNAs via LASSO Cox regression. Cross-validation results were used to determine the evolutionary trajectory of regression coefficients for long noncoding RNAs (Figure 2B,C). On the basis of survival, 19 patients were assigned to the high CRLPM score group. The Cox regression model was designed to predict the overall survival (OS) of glioma patients. Afterwards, a heatmap was constructed to show the relationships between genes associated with cuproptosis and the CRLPM score (Figure 1B). Survival analysis was conducted on the patients in the training set based on their median risk scores, which were divided into high-risk and low-risk groups. The OS of those at high risk and those at low risk differed significantly (p < 0.05; Figure 2D). According to Figure 2E, patients had a low survival rate and a high risk score distribution. As shown in Figure 2F, 17 genes were differentially expressed between the high- and low-risk groups. There was a clear inverse relationship between the lncRNA-based risk score and both survival time and mortality rate. Both the validation cohort and the overall cohort showed significant differences in OS between the high-risk and low-risk groups (p < 0.05; Figure 2G-I, Figure 3A-C). Principal component analysis (PCA) was used to analyse the expression patterns of the identified ferroptosis-related lncRNAs and glioma model lncRNAs. LncRNAs appeared to have greater efficacy in separating patients into high-risk and low-risk cohorts than other factors in the glioma model. Our recommended model is relatively effective in distinguishing between risk groups on the basis of their characteristics. Using PCA, we assessed risk scores in terms of total gene expression profiles (Figure 3D), ferroptosis genes (Figure 3E), cuproptosis-associated lncRNAs (Figure 3F), and risk signatures (Figure 3G).
2.3. The Cuproptosis-Related lncRNA Prognostic Model Is Independent of Overall Survival
Univariate and multivariate analyses of the Cox regression model were conducted. Univariate Cox analysis revealed statistically significant differences among age, grade, and risk score (Figure 4A). Multivariate Cox regression analysis revealed that grade, age, and risk score were prognostic variables (Figure 4B). As a result of a review of the entire cohort, progression-free survival (PFS) was also substantially lower for the high-risk subgroup than for the low-risk subgroup (based on the total number of participants) (p < 0.05, Figure 4C). According to the ROC curve, the copper-related lncRNAs were accurate and useful for predicting OS, with AUCs of 0.904 after 1 year, 0.941 after 2 years, and 0.883 after 3 years. (Figure 4D). Furthermore, the CRLPM was assessed using the area under the curve (AUC) and receiver operating characteristic (ROC) curves. The R packages “time ROC” and “survival ROC” were used to calculate the statistical data. According to the C-index and ROC curves, the prognostic model performed better than other clinical factors, including age, sex, and tumour grade (Figure 4E,F). A nomogram is a tool for quantitatively analysing glioma patients' clinical outcomes. Therefore, we developed a nomogram based on numerous clinicopathological variables to predict patient prognosis (Figure 5A). Calibration plots were used to validate the prognostic value of the signature. The predicted values matched the actual values well. (Figure 5B).
2.4. Correlation Analysis between CRLs and Clinicopathological Features in Glioma
Subsequently, an investigation into the relationship between CRLPM and a variety of clinical parameters was carried out to assess the therapeutic efficacy of CRLPM. There were statistically significant differences between stage II and stage III-IV patients in terms of risk scores and clinical stages. (Figure 5C,D)
2.5. An Analysis of Gene Sets and Pathways that Enrich Pathways
Furthermore, both high- and low-risk CRLs were subjected to enrichment analyses via KEGG and GO enrichment analyses. In this bar plot, we show the top 30 enrichment terms based on the GO classification. A gene associated with biological processes is one that binds to structural components of the extracellular matrix, antigens, glycosaminoglycans, growth factors, or heparin. Components that are part of cells. In the GO cellular component category, genes were mainly associated with the collagen-containing extracellular matrix, the external surface of the plasma membrane, the lumen of the endoplasmic reticulum, the lumen of vesicles, the chromosome, and the centromeric region. In the GO category, genes are categorized primarily as cellular components. Figure 6A shows numerous structures, including encapsulant structure, extracellular matrix, and ossification. We generated bar plots based on the GO analysis of the 30 most important classification enrichment terms. GO genes in the category of biological processes focused mainly on extracellular matrix organization, extracellular structure organization, external encapsulating structure organization, external encapsulating structure organization and somatic adaptive immune response. In the cellular component category, genes in the GO category were mainly enriched in collagen-containing extracellular matrix, endoplasmic reticulum lumen, the external side of the plasma membrane, the basement membrane, and the collagen trimer. In the category of molecular function, genes in the GO category were enriched mainly in extracellular matrix structural constituents, antigen binding, collagen binding, and growth factor binding (Figure 6B). The results of the GO enrichment analysis are displayed in a circle diagram. In the category of biological processes, genes in the GO category were mainly focused on leukocyte-mediated immunity, extracellular matrix organization, extracellular structure organization, and external encapsulating structure organization. In the cellular component category, genes in the GO category were mainly enriched in the collagen-containing extracellular matrix, the external side of the plasma membrane, the endoplasmic reticulum lumen, the vesicle lumen, the chromosome, and the centromeric region. In the category of molecular function, genes in the GO category were mainly enriched in extracellular matrix structural constituents, antigen binding, glycosaminoglycan binding, growth factor binding, and heparin binding (Figure 6C). The genes in the KEGG category were enriched mainly in ECM−receptor interaction, haematopoietic cell lineage, phagosome, focal adhesion, cell cycle, complement and coagulation cascades, proteoglycans in cancer, Staphylococcus aureus infection, and allograft rejection, as shown in the KEGG bar plot (Figure 6D). The genes in the KEGG category were enriched mainly in human papillomavirus infection, cytokine−cytokine receptor interaction, focal adhesion, proteoglycans in cancer, human T−cell leukaemia virus 1 infection, phagosome, Epstein−Barr virus infection, cell cycle, transcriptional misregulation in cancer, tuberculosis, and ECM−receptor interaction, as shown in the KEGG bubble plot. Circle chart and bar plot for GO and KEGG analyses of biological process, cellular component, and molecular function (Figure 6F,G).
2.6. Estimated Infiltration of Immune Cells in Tumours and Immunotherapy Response
Figure 7A shows the heatmap of the immune response based on the ssGSEA algorithm. Correlation analysis of ssGSEA-inferred immune cell populations and related functions based on TCGA-GBM-LGG data revealed significant differences in the enrichment of the Type_II_IFN_Reponse, Cytolytic_activity, Inflammation-promoting, APC_co_inhibition, and T_cell_co-inhibition gene sets. These results suggest that the CRLPM score is associated with GBM immune cell infiltration. The TIDE algorithm was used to predict the effect of immunotherapy in patients. Figure 7B shows no significant difference in TIDE scores between the high- and low-risk groups. This further proved that there was no significant difference in immune escape potential or immunotherapy benefit between the high- and low-risk groups.
2.7. TMB Analysis Based on the Cuproptosis-Related Prognostic lncRNA Signature in Glioma
Mutations relevant to glioma were downloaded from the TCGA database; TMB levels for the two distinct groups were estimated. The mutation load of the high-risk group in GBM was greater than that of the low-risk group, as shown in Figure 7C. On the basis of the median cut-off point, we classified patients into "high TMB" and "low TMB" groups and conducted a survival analysis. According to the results, patients with high TMB had a better prognosis than those with low TMB. (Figure 7D). Through combined survival analysis, we were able to obtain the combined survival curve of the tumour mutation load and risk score. According to the results, the TMB and risk score were significantly related to OS for patients with gliomas (Figure 7E). CRLPM mutation patterns in the high-risk and low-risk groups were compared. According to the waterfall map, the 15 genes associated with higher mutation frequencies were located in the high-risk and low-risk groups. According to the results, the high-risk group had more mutations (Figure 7F,G), and IDH1, TP53, ATRX, PTEN, TTN, EGFR, CIC, MUC16 and PIK3CA were the 10 genes with the most frequent changes. IDH1, TP53 and ATRX mutations were more frequent in the low-risk group.
2.8. Drug Sensitivity Analysis of Glioma Patients
The correlation between IC50 values and the risk scores of drugs used in GBM therapy was examined to determine whether CRLPM could be used in individualized therapy for GBM. A comparison of commonly used cancer drugs in different risk categories was conducted to assess their sensitivity to risk scores. We found drugs that were strongly associated with risk scores. Figure 8 shows the comparison between the high-risk and low-risk groups in terms of drug sensitivity. The high-risk and low-risk groups showed significant differences in the sensitivity to anticancer drugs. Moreover, 26 drugs in the high-risk group had lower IC50 values, further indicating that this group is more susceptible to drug treatment. In addition, the risk scores were negatively correlated with the IC50 values. Further study of the correlation between risk scores and sensitivity to these drugs revealed that a higher risk score corresponded to a lower IC50 value. The results indicate that patients in the high-risk group are more sensitive to the drugs and that they will benefit more from the potential drugs. In addition, the risk score and drug sensitivity were negatively correlated. To conclude, these results can be used as a basis for future research aimed at evaluating potential pharmaceutical products that can be tested for the purpose of managing gliomas. Using the risk score to assess the effectiveness of immunotherapy for treating glioma may provide a more thorough assessment of its effectiveness than a previous study.
3. Discussion
Copper-rich environments are essential for the survival of living organisms. It plays a crucial role in mitochondrial respiration, iron absorption, and ti-oxidation, among other biological processes. [12,13,14,15]. Several studies have revealed that copper ions have a dual nature, acting as both vital enzyme cofactors and potential harmful agents [16]. In particular, elevated copper concentrations can bind lipoic acid-related enzymes connected to the tricarboxylic acid cycle, resulting in cellular death. Tsvetkov et al. (2022) introduced the term "cuproptosis" to describe a distinctive type of cell death that differs from previously identified mechanisms [17]. A wide range of lncRNAs are found in diverse bodily fluids; they play important roles in a wide range of pathophysiological processes, including screening for human diseases [18].
Approximately 308,000 cases of brain cancer are diagnosed worldwide every year, 80% of which are gliomas. [18,19,20]. The extreme prevalence of malignant brain lesions in the central nervous system indicates that they are extremely prevalent [21]. Most malignancies affecting the central nervous system are rapidly expanding, highly fatal malignancies [22]. Approximately 80% of gliomas are thought to arise from nonneuronal cells, particularly astrocytes, a subtype of glial cell that provides support and protection to neurons. [23,24]. This study focused on malignant tumours of the central nervous system to advance research, treatment, and scientific assessment. It is imperative to establish a diagnosis and initiate therapeutic interventions promptly to increase the overall survival rate of patients. In addition, more effective therapeutic interventions need to be explored in the modern medical field, and the underlying mechanisms contributing to tumour proliferation need to be elucidated.
The presence of long noncoding RNAs in central nervous system tissues is considered a marker of tumours in the central nervous system [25]. In particular, copper ion carrier small molecule anticancer drugs have been used to investigate the prognosis of patients with lung adenocarcinoma. An increasing amount of attention has been given to lncRNAs over the last few years. Studies have shown that long noncoding RNAs, such as linc01140, which interferes with osteosarcoma proliferation and invasion when targeted to miR-139-5p/HOXA9, play crucial roles in tumour progression. [26]. Numerous malignant tumours, including gastric cancer, cholecystitis, hepatocellular carcinoma, and breast cancer, exhibit aberrant expression of the HXA-AS2 gene [27,28]. Nine favourable factors were identified among the 17 lncRNAs studied in this study (AC00783.1, AC00795.2, AC010273.3, AC010319.4, AC021739.2, AC087501.4, AC104623.1, DLGAP1-AS2, PITPNA-AS1, and SOX21-AS1), as were 11 unfavourable factors (AL139243.1, AL35578.1, AL513534.2, AL592295.6, AL691432.4, LINC01574, and TMEM72-AS1). Previous studies have suggested an association between LINC01574 and poor prognosis in gliomas [29].
Current research has identified 17 lncRNAs associated with cuproptosis prognosis; patients with gliomas were selected for prognostic analysis based on these lncRNAs. A total of 19 transcripts related to cuproptosis were obtained. A combination of LASSO regression and Cox regression analyses was used to identify lncRNAs associated with prognosis in patients with cuprotosis. We also examined the associations of CRLPM with common clinical variables, upstream regulatory mechanisms, immune cell infiltration, and immunotherapy response in glioma patients.
K‒M analysis demonstrated that the OS for those at high risk was inferior to that for those at low risk. Next, the accuracy of the risk model was validated. As shown by the ROC curves, the CRLPM correctly predicted survival at one, three, and five years; all the AUC values exceeded 0.65. Furthermore, PCA distinguished the high-risk group from the low-risk group. Based on glioma patients' distant metastasis, lymph node metastasis, and prognosis, a novel nomogram was developed for predicting PFS.
Additionally, functional enrichment analysis provided valuable insight into the potential biological mechanisms underlying the prognostic effects of CRLPM. An examination of the primary signalling pathways of eight long noncoding RNAs (lncRNAs) was conducted in this study. According to gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses, the differentially expressed CRLs were significantly enriched in genes involved in cholesterol metabolism. Numerous signalling pathways, including the complement cascade and coagulation cascade, PPAR and Wnt signalling pathways, PI3K-Akt signalling pathways, and ECM-receptor interactions, as well as neuroactive ligand‒receptor interactions, are associated with salivary secretion.
In glioma patients at high risk, the TMB was significantly greater than that in patients at low risk, indicating a superior effect of immunotherapy. Mutations in IDH1 were most frequent among patients with gliomas among the initial 15 genes. Additionally, drug sensitivity analyses were performed based on the CRLPM to guide clinical treatment. For all 36 drugs assessed, significant differences were found between high-risk and low-risk patients in terms of the IC50.
The limitations of our work include the lack of in vitro tissue studies. Second, novel long noncoding RNAs of clinical significance in gliomas require further exploration to identify their molecular mechanisms. The third difference is that this study separated the training set from the test set equally. Through reduced sample differences between the two groups, statistical consistency of clinical measures can be achieved, but parameter estimation can be compromised.
A further limitation is that the analysis of DEGs associated with cuproptosis in glioma patients could not be performed. A larger sample size and a more comprehensive clinical prognostic dataset are recommended for future research, as determined by the TCGA dataset. Additionally, while our predictions were validated using data from public databases, additional biological evidence is required in addition to the statistical evidence we have presented. However, further research is needed to determine the precise mechanism underlying the observed correlations.
Additionally, functional enrichment analysis revealed significant insights into the postulated molecular mechanism that regulates the implicated CRLPM. Cuproptosis signalling pathways were studied for eight long noncoding RNAs (lncRNAs) linked to this process. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses revealed that the CRLPM gene was the most enriched gene.
Cuproptosis is a promising approach for cancer treatment that may play a significant role in future therapeutic interventions. [25,30]. Alternatively, several long noncoding RNAs (lncRNAs) can affect the response to treatment and progression of cancer through a variety of biological processes. Cuproptosis and long noncoding RNAs (lncRNAs) are still poorly understood. Using cuproptosis as a model, this study provides new insights into the development and formation of gliomas. A major objective of this study was to examine cuproptosis indicators that may be employed for assessing glioma prognosis, thus improving its management and treatment. Validation of the findings included the whole dataset as well as the TCGA validation set. However, additional patients should be included in the model to increase its reliability. Preclinical studies are therefore essential for further validation. Furthermore, we will need to perform in vitro and in vivo experiments to validate our findings.
4. Materials and Methods
Patient Data Sources
The RNA sequencing data and clinical information for glioma patients were acquired from the TCGA, which may be accessed at https://portal.gdc.cancer.gov/. The gene expression profiles and clinical data for glioma patients in the TCGA-GBM-LGG dataset were augmented with additional follow-up information, including age, sex, survival status, and overall survival. Data processing and analysis were performed using R (version 4.1.2) and Perl (Strawberry version) software. The lncRNA annotations were acquired from the GENCODE website, accessible at http://www.gencodegenes.org/. In addition, genes associated with cuproptosis were identified in accordance with a prior investigation [17]. The downloaded data were processed in accordance with TCGA regulations.
Selection of Cuproptosis-Related Genes
Tsvetkov et al. published their research paper titled "c" in 2010 [17]. Copper toxicity is associated with distinct mechanisms compared to apoptosis, ferroptosis, pyroptosis, and necrosis. Cuproptosis, an unidentified mechanism of cell death, was identified by researchers as the expression of ten regulatory genes, namely, LIPT1, LIPT2, LIAS, DLD, DBT, GCSH, DLAT, PDHB, ATP7B, MTF1 and PDHA1, and eight negative regulatory genes, namely, SLC31A1, NFE2L2, GLS, FDX1, DLST, CDKN2A, ATP7A, and NLRP3.
Nomogram and Calibration
The independent prognostic function of the risk model was examined using univariate and multivariate Cox regression. The survival, regplot, and rms R packages were used to generate a nomogram based on the outcomes of univariate and multivariate Cox regressions. Using the rms package, line graphs of 1-, 3-, and 5-year OS rates for glioma patients were constructed based on risk scores and clinical characteristics. The Hosmer–Lemeshow test was used to construct a calibration curve, which confirmed the effectiveness of the developed nomogram models for making predictions.
Univariate Cox Survival Analysis
Using the coxph function from the R package, we performed a univariate Cox survival analysis on the genes associated with cuproptosis. In addition, we performed further analysis of numerous cuproptosis-associated genes that were deemed significant for the prognosis of glioma patients, with a significance threshold of 0.05. Univariate and multivariate Cox regressions were used to determine whether the risk score model could independently predict patient prognosis.
Functional Enrichment Analysis
Principal component analysis (PCA) was employed to classify the expression profiles of compression-associated long noncoding RNAs (lncRNAs) in glioma specimens, aiming to visually depict the spatial arrangement of samples with diverse risk levels. The correlations between the variables of the samples were visualized using 3D scatter plots. A genetic analysis of differentially expressed genes (DEGs) was performed using the glm method of the "edgeR" R package. With each log fold change (log2FC) set to 1, the false discovery rate (FDR) was set at 0.05. Furthermore, gene ontology (GO) analysis was performed to determine the distinct genes among the low- and high-risk groups to investigate essential DEGs in three categories: biological process (BP), cell component (CC), and molecular function (MF). Differential Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways related to cellular components, biological processes, and molecular functions were also screened, with an FDR of 0.05.
Investigation of the Correlation between Clinical Stage and the Prognostic Risk Score
To determine whether the risk score can be used as an independent predictor of prognosis, both univariate and multivariate models were implemented using Cox regression analysis. Using the R programming language, histograms were generated to predict glioma patient survival at 1, 3, and 5 years in the TCGA cohort. Risk scores and clinicopathologic factors were used to generate histograms.
The Development of a Prognostic Model and Histopathological Analysis of Cuproptosis in Gliomas
The R programming language's "caret" function was used to randomize the samples into training and validation groups. Univariate Cox proportional hazard regression analysis was performed on each long noncoding RNA associated with cuproptosis using the survival R package. To prevent overfitting, we used the “glmnet” package in R, which uses least absolute shrinkage and selection operator-penalized Cox regression. We determined the optimal and minimum penalty criteria through ten cross-validation runs before performing a series of stepwise Cox regression analyses to generate a cuproptosis-related lncRNA prognostic model (CRLPM). A prognostic model based on the LASSO method was used to determine the risk associated with an individual patient.
Model Validation
Patients in the validation and training cohorts were categorized into high-risk and low-risk groups based on their median risk scores and corresponding coefficients. K‒M analysis was used for both the training and validation sets to determine the prognostic performance of the risk score model. Furthermore, the CRLPM was assessed using the area under the curve (AUC) and receiver operating characteristic (ROC) curves. R packages for time ROC and survival ROC were used to calculate the statistical measures. Principal component analysis (PCA) was also used by the researchers to evaluate the risk models. With the help of the R platform's scatterplot3D tool, the results of the PCA were visualized. Survivminer is a package built in the R programming language that provides incremental tools. C-indices were used to assess the accuracy of risk models based on "survival," "rms," "dplyr," and "pec.". Validating the model on full cohorts was primarily intended to authenticate and verify its accuracy and reliability.
TIDE
To assess the infiltration and function of tumour-infiltrating immune cells, the single-sample gene set enrichment analysis (ssGSEA) algorithm function package in the R software genome variation analysis package was used to explore the relationship between the CRLPM risk score and immune cell infiltration. Based on the simulated tumour immune escape mechanism (http://tide.dfci.harvard.edu), a heatmap was created; next, the tumour immune dysfunction and exclusion (TIDE) algorithm was utilized to predict the immunotherapy response. [31]. In general, we examined the impact of immunotherapy in high- and low-risk populations using the TIDE algorithm.
Estimation of the Tumour Mutational Burden
In malignant tumours, the tumour mutational burden (TMB) is a measurement of how many mutations are present. We used the R package "maftools" in this study to analyse mutation data from The Cancer Genome Atlas (TCGA). An analysis of the relationship between the TMB and risk score in patients with gliomas is shown in the cascade diagram.
Pathway Enrichment Analysis and Gene Set Enrichment Analysis
Using the R package "limma," we identified differentially expressed genes (DEGs) between high- and low-risk patients. Considering the log2-fold change, DEGs must have a false discovery rate less than 0.05. The KEGG and Gene Ontology databases were used for analysis of molecular functions and critical signalling pathways. ClusterProfiler, org.Hs.egdb, and enrichplot were used for this analysis.
Drug Sensitivity
A measure of drug efficacy or treatment effectiveness is the half-maximal inhibitory concentration (IC50). In this study, we examined the correlation between the CRG risk score and the sensitivity of gliomas to commonly prescribed medications. Several R packages, including "pRRophetic" and its dependent programs, including "car, ridge preprocessCore, genefilter, and sva", were used to evaluate drug sensitivity in individuals with gliomas. Using the Wilcoxon signed rank test, we calculated the difference between the high-risk and low-risk groups in terms of the IC50 values for common antineoplastic drugs.
Statistical Analysis
For analysis of the bioinformatics data, we used R version 4.1.3. To determine whether the groups differed significantly, we used a Wilcoxon rank-sum test. The data were analysed using the Perl programming language (version). To analyse the differences between two or more groups, Student's t test and one-way ANOVA were used. Using Kaplan‒Meier analysis, we assessed and compared the overall survival rates of two distinct patient cohorts. Cox regression analysis, LASSO analysis, and univariate analysis were used to assess the prognostic value of the signature. To assess the correlation of gene expression, Pearson correlation analysis was used. The dependability and sensitivity of the prognostic signature were studied using receiver operating characteristic (ROC) curves and areas under the curve (AUCs). Statistical significance was determined by a p value of 0.05.
5. Conclusions
Globally, gliomas rank among the most prevalent malignant tumours, and their inherent heterogeneity complicates their pathophysiology. In this way, many biomarkers could be integrated into a unified model, and their predictive precision could be evaluated, thus enhancing treatment regimen customization and effectiveness, immunological significance, and the ability to assess the responsiveness to specific drugs.
We systematically analysed the prognostic value of cuproptosis-related lncRNAs in gliomas. Several lncRNAs associated with cuproptosis have demonstrated high levels of accuracy in predicting patient outcomes during glioma treatment. Furthermore, 17 cuproptosis-related lncRNAs were found to be reliable for predicting the clinical outcome of patients with colon adenocarcinoma. Further investigation of the interactions between cuproptosis-related lncRNAs and glioma is warranted based on our findings. A further benefit of this study is that three specific lncRNAs associated with cuproptosis were identified, which could serve as potential targets for the treatment of gliomas.
As a result, we demonstrated that a novel therapeutic approach can be used to tailor treatment for glioma patients and predict the response to immunotherapy. A total of eight long noncoding RNAs (lncRNAs) implicated in cuproptosis have been proposed as potential therapeutic targets for gliomas.
Author Contributions
Jia and Qian contributed to the idea and design of the research. Xin and Shang were tasked with coordinating the database. Tang, Kang and Li were tasked with performing the statistical analysis. Shi and Wu were the original authors of the text. Wu and Fan made contributions to the text. Every contributor meticulously edited the document, scrutinized it, and gave their endorsement to the final version that was presented.
Funding
The authors declare that no funds, grants, or other support was received during the preparation of this manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study can be found in the TCGA database (https://portal.gdc.cancer.gov, accessed on 16 November 2023).
Conflicts of Interest
The authors confirm that there were no financial or commercial interactions that may be seen as a potential conflict of interest throughout the research.
References
- Chang, B.; Hu, Z.; Chen, L.; Jin, Z.; Yang, Y. Development and validation of cuproptosis-related genes in synovitis during osteoarthritis progress. Front. Immunol. 2023, 14, 1090596. [Google Scholar] [CrossRef]
- Sturm, D.; Pfister, S.M.; Jones, D.T. Pediatric Gliomas: Current Concepts on Diagnosis, Biology, and Clinical Management. J. Clin. Oncol. 2017, 35, 2370–2377. [Google Scholar] [CrossRef] [PubMed]
- Leng, Y.; Wang, X.; Liao, W.; Cao, Y. Radiomics in gliomas: A promising assistance for glioma clinical research. . 2018, 43, 354–359. [Google Scholar] [PubMed]
- Okolie, O.; Bago, J.R.; Schmid, R.S.; Irvin, D.M.; Bash, R.E.; Miller, C.R.; Hingtgen, S.D. Reactive astrocytes potentiate tumor aggressiveness in a murine glioma resection and recurrence model. Neuro-Oncology 2016, 18, 1622–1633. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Wei, Y.; Wang, X.; Zhang, Z.; Yin, J.; Li, W.; Chen, L.; Lyu, X.; Shi, Z.; Yan, W.; et al. DNA-methylation-mediated activating of lncRNA SNHG12 promotes temozolomide resistance in glioblastoma. Mol. Cancer 2020, 19, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.L.; DeAngelis, L.M. Reappraising the 2016 WHO classification for diffuse glioma. Neuro-Oncology 2017, 19, 609–610. [Google Scholar] [CrossRef]
- Stupp, R.; Brada, M.J.; van den Bent, M.; Tonn, J.-C.; Pentheroudakis, G.; on behalf of the ESMO Guidelines Working Group. High-grade glioma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2014, 25, iii93–iii101. [Google Scholar] [CrossRef] [PubMed]
- Weingart, J.; Grossman, S.A.; Carson, K.A.; Fisher, J.D.; Delaney, S.M.; Rosenblum, M.L.; Olivi, A.; Judy, K.; Tatter, S.B.; Dolan, M.E. Phase I Trial of Polifeprosan 20 With Carmustine Implant Plus Continuous Infusion of Intravenous O6-Benzylguanine in Adults With Recurrent Malignant Glioma: New Approaches to Brain Tumor Therapy CNS Consortium Trial. J. Clin. Oncol. 2007, 25, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Hu, B.; Pan, K.; Chang, J.; Zhao, X.; Chen, L.; Lin, H.; Wang, J.; Zhou, G.; Xu, W.; et al. SYVN1-MTR4-MAT2A Signaling Axis Regulates Methionine Metabolism in Glioma Cells. Front. Cell Dev. Biol. 2021, 9. [Google Scholar] [CrossRef]
- Kim, H.S.; Seo, M.; Park, T.-E.; Lee, D.Y. A novel therapeutic strategy of multimodal nanoconjugates for state-of-the-art brain tumor phototherapy. J. Nanobiotechnology 2022, 20, 1–27. [Google Scholar] [CrossRef]
- Wang, Z.-Q.; Zhang, M.-Y.; Deng, M.-L.; Weng, N.-Q.; Wang, H.-Y.; Wu, S.-X. Low serum level of miR-485-3p predicts poor survival in patients with glioblastoma. PLOS ONE 2017, 12, e0184969. [Google Scholar] [CrossRef]
- Schlecht, U.; Suresh, S.; Xu, W.; Aparicio, A.M.; Chu, A.; Proctor, M.J.; Davis, R.W.; Scharfe, C.; Onge, R.P.S. A functional screen for copper homeostasis genes identifies a pharmacologically tractable cellular system. BMC Genom. 2014, 15, 263–263. [Google Scholar] [CrossRef]
- Chun, H.; Sharma, A.K.; Lee, J.; Chan, J.; Jia, S.; Kim, B.-E. The Intestinal Copper Exporter CUA-1 Is Required for Systemic Copper Homeostasis in Caenorhabditis elegans. J. Biol. Chem. 2017, 292, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Mhaske, A.; Dileep, K.; Kumar, M.; Poojary, M.; Pandhare, K.; Zhang, K.Y.; Scaria, V.; Binukumar, B. ATP7A Clinical Genetics Resource – A comprehensive clinically annotated database and resource for genetic variants in ATP7A gene. Comput. Struct. Biotechnol. J. 2020, 18, 2347–2356. [Google Scholar] [CrossRef] [PubMed]
- Chi, H.; Peng, G.; Wang, R.; Yang, F.; Xie, X.; Zhang, J.; Xu, K.; Gu, T.; Yang, X.; Tian, G. Cuprotosis Programmed-Cell-Death-Related lncRNA Signature Predicts Prognosis and Immune Landscape in PAAD Patients. Cells 2022, 11, 3436. [Google Scholar] [CrossRef]
- Dang, J.; Wang, T.; Wen, J.; Liang, H. An Important Role of the Type VI Secretion System of Pseudomonas aeruginosa Regulated by Dnr in Response to Anaerobic Environments. Microbiol. Spectr. 2022, 10, e0153322. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkov, P.; Coy, S.; Petrova, B.; Dreishpoon, M.; Verma, A.; Abdusamad, M.; Rossen, J.; Joesch-Cohen, L.; Humeidi, R.; Spangler, R.D.; et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science 2022, 375, 1254–1261. [Google Scholar] [CrossRef]
- Zhang, L.; Fang, W.; Zhang, K.; Jiang, W.; Chen, M.; Liao, W.; Pan, W. Long noncoding RNA expression profile from cryptococcal meningitis patients identifies DPY19L1p1 as a new disease marker. CNS Neurosci. Ther. 2019, 25, 772–782. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R. L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. , Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, X.; Zhu, X.-L.; Wang, Z.-Z.; Bai, H.; Zhang, J.-J.; Hao, C.-Y.; Duan, H.-B. A Novel Immune-Related Prognostic Biomarker and Target Associated With Malignant Progression of Glioma. Front. Oncol. 2021, 11. [Google Scholar] [CrossRef]
- Miranda-Gonçalves, V.; Gonçalves, C.S.; Granja, S.; de Castro, J.V.; Reis, R.M.; Costa, B.M.; Baltazar, F. MCT1 Is a New Prognostic Biomarker and Its Therapeutic Inhibition Boosts Response to Temozolomide in Human Glioblastoma. Cancers 2021, 13, 3468. [Google Scholar] [CrossRef] [PubMed]
- Watson, J.R.; Martirosyan, N.; Lemole, G.M.; Trouard, T.P.; Romanowski, M. Intraoperative brain tumor resection with indocyanine green using augmented microscopy. J. Biomed. Opt. 2018, 23, 090501–4. [Google Scholar] [CrossRef]
- Zou, H.; Feng, R.; Huang, Y.; Tripodi, J.; Najfeld, V.; Tsankova, N.M.; Jahanshahi, M.; Olson, L.E.; Soriano, P.; Friedel, R.H. Double minute amplification of mutant PDGF receptor α in a mouse glioma model. Sci. Rep. 2015, 5, 8468–8468. [Google Scholar] [CrossRef]
- Gao, Y.; Wu, Y.; Zhang, N.; Yuan, H.; Wang, F.; Xu, H.; Yu, J.; Ma, J.; Hou, S.; Cao, X. IDH1 gene mutation activates Smad signaling molecules to regulate the expression levels of cell cycle and biological rhythm genes in human glioma U87-MG cells. Mol. Med. Rep. 2021, 23, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Yang, Y.; Gao, Y.; He, J. Cuproptosis: mechanisms and links with cancers. Mol. Cancer 2023, 22, 1–30. [Google Scholar] [CrossRef]
- Zhang, S.; Chen, R. LINC01140 regulates osteosarcoma proliferation and invasion by targeting the miR-139-5p/HOXA9 axis. Biochem. Biophys. Rep. 2022, 31, 101301. [Google Scholar] [CrossRef]
- Tong, G.; Wu, X.; Cheng, B.; Li, L.; Li, X.; Li, Z.; Nong, Q.; Chen, X.; Liu, Y.; Wang, S. Knockdown of HOXA-AS2 suppresses proliferation and induces apoptosis in colorectal cancer. . 2017, 9, 4545–4552. [Google Scholar]
- Soghala, S.; Harsiny, K.; Momeni, P.; Hatami, M.; Oskooei, V.K.; Hussen, B.M.; Taheri, M.; Ghafouri-Fard, S. Down-regulation of LINC-ROR, HOXA-AS2 and MEG3 in gastric cancer. Heliyon 2022, 8, e11155. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, Y.; Gao, Y.; Wang, L.; Shi, H.; Xuan, C. , H19- and hsa-miR-338-3p-mediated NRP1 expression is an independent predictor of poor prognosis in glioblastoma. PLoS One 2021, 16, e0260103. [Google Scholar] [CrossRef]
- Zhang, L.; Di, L.; Liu, J.; Lei, X.; Gu, M.; Zhang, W.; Wang, Y. The LncRNA signature associated with cuproptosis as a novel biomarker of prognosis in immunotherapy and drug screening for clear cell renal cell carcinoma. Front. Genet. 2023, 14, 1039813. [Google Scholar] [CrossRef]
- Wu, Z.; Xu, J.; Tang, R.; Wang, W.; Zhang, B.; Yu, X.; Liu, J.; Shi, S. The Role of PDGFRA in Predicting Oncological and Immune Characteristics in Pancreatic Ductal Adenocarcinoma. J. Oncol. 2022, 2022, 1–16. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Sankey diagram and heatmap. (A) According to the Sankey diagram, 1177 cuproptosis-related long noncoding RNAs (lncRNAs) coexpressed with 19 cuproptosis-related genes are shown. (B) The correlation between 19 genes associated with cuproptosis and 17 long noncoding RNAs associated with prognosis. *p < 0.05, **p < 0.01, and ***p < 0.001.
Figure 1.
Sankey diagram and heatmap. (A) According to the Sankey diagram, 1177 cuproptosis-related long noncoding RNAs (lncRNAs) coexpressed with 19 cuproptosis-related genes are shown. (B) The correlation between 19 genes associated with cuproptosis and 17 long noncoding RNAs associated with prognosis. *p < 0.05, **p < 0.01, and ***p < 0.001.
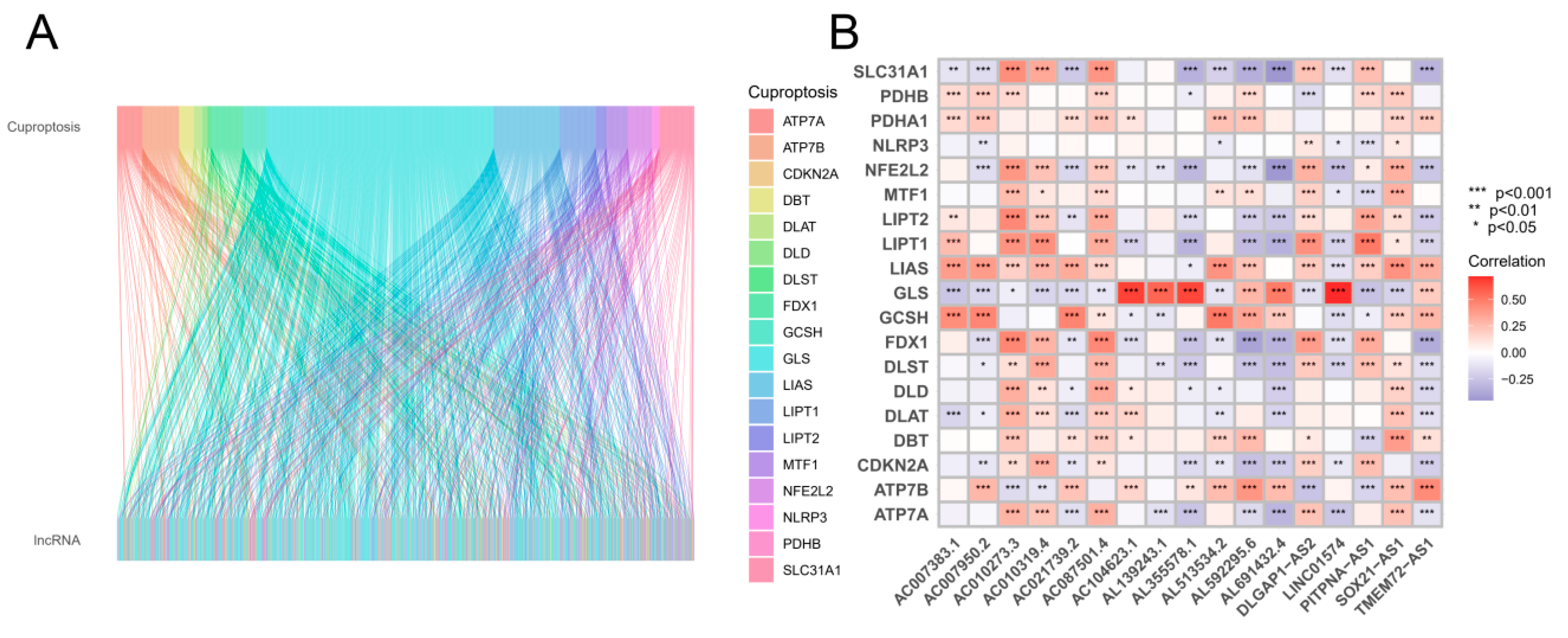
Figure 2.
The development of a prognostic cuprotosis-associated long noncoding RNA (lncRNA) risk model for gliomas. (A) Univariate Cox regression analysis of lncRNAs associated with cuproptosis. (B–C) For the construction of prognostic models, LASSO Cox regression analysis was used. (D) Survival curves for the high-risk and low-risk groups based on Kaplan‒Meier curves. (E) The distribution of risk scores and survival status among patients with GBM. (F) Prognostic markers and overall survival heatmap. (G) Kaplan‒Meier survival curves from the validation cohort. (H) Risk score distribution and survival status of the validation cohort. (I) Validation cohort heatmap showing prognostic markers and overall survival.
Figure 2.
The development of a prognostic cuprotosis-associated long noncoding RNA (lncRNA) risk model for gliomas. (A) Univariate Cox regression analysis of lncRNAs associated with cuproptosis. (B–C) For the construction of prognostic models, LASSO Cox regression analysis was used. (D) Survival curves for the high-risk and low-risk groups based on Kaplan‒Meier curves. (E) The distribution of risk scores and survival status among patients with GBM. (F) Prognostic markers and overall survival heatmap. (G) Kaplan‒Meier survival curves from the validation cohort. (H) Risk score distribution and survival status of the validation cohort. (I) Validation cohort heatmap showing prognostic markers and overall survival.
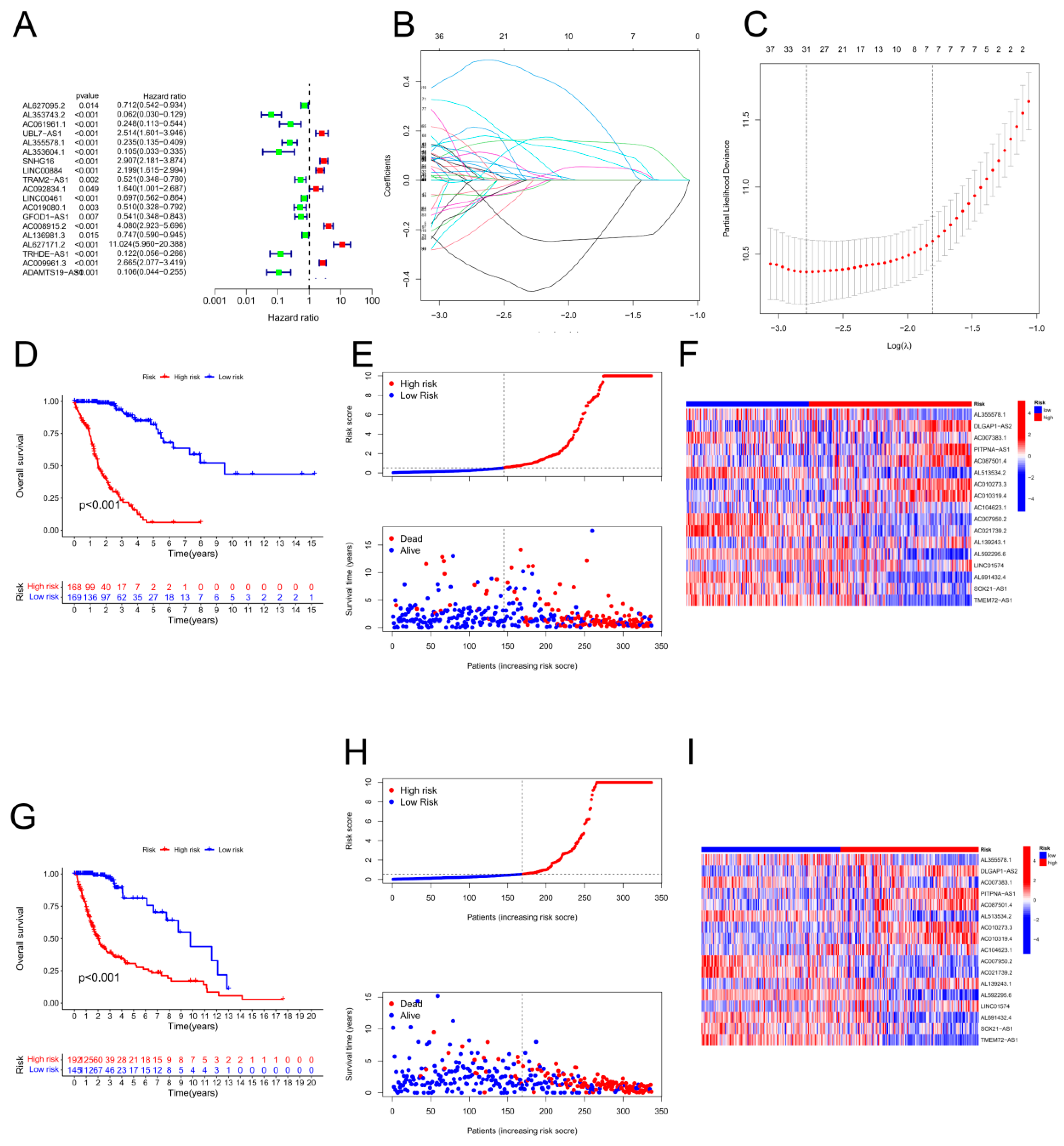
Figure 3.
Validation of the risk model within the entire cohort and principal component analysis. (A) Kaplan–Meier curves for survival analysis in the entire cohort. (B) Survival status of the entire cohort based on risk score distribution. (C) Overall survival and prognostic markers for the entire cohort in a heatmap. PCA was performed by using the following genes as a proxy for risk: (D) all genes, (E) cuproptosis-related genes, (F) cuproptosis-related long noncoding RNAs (lncRNAs), and (G) cuproptosis-related lncRNAs.
Figure 3.
Validation of the risk model within the entire cohort and principal component analysis. (A) Kaplan–Meier curves for survival analysis in the entire cohort. (B) Survival status of the entire cohort based on risk score distribution. (C) Overall survival and prognostic markers for the entire cohort in a heatmap. PCA was performed by using the following genes as a proxy for risk: (D) all genes, (E) cuproptosis-related genes, (F) cuproptosis-related long noncoding RNAs (lncRNAs), and (G) cuproptosis-related lncRNAs.
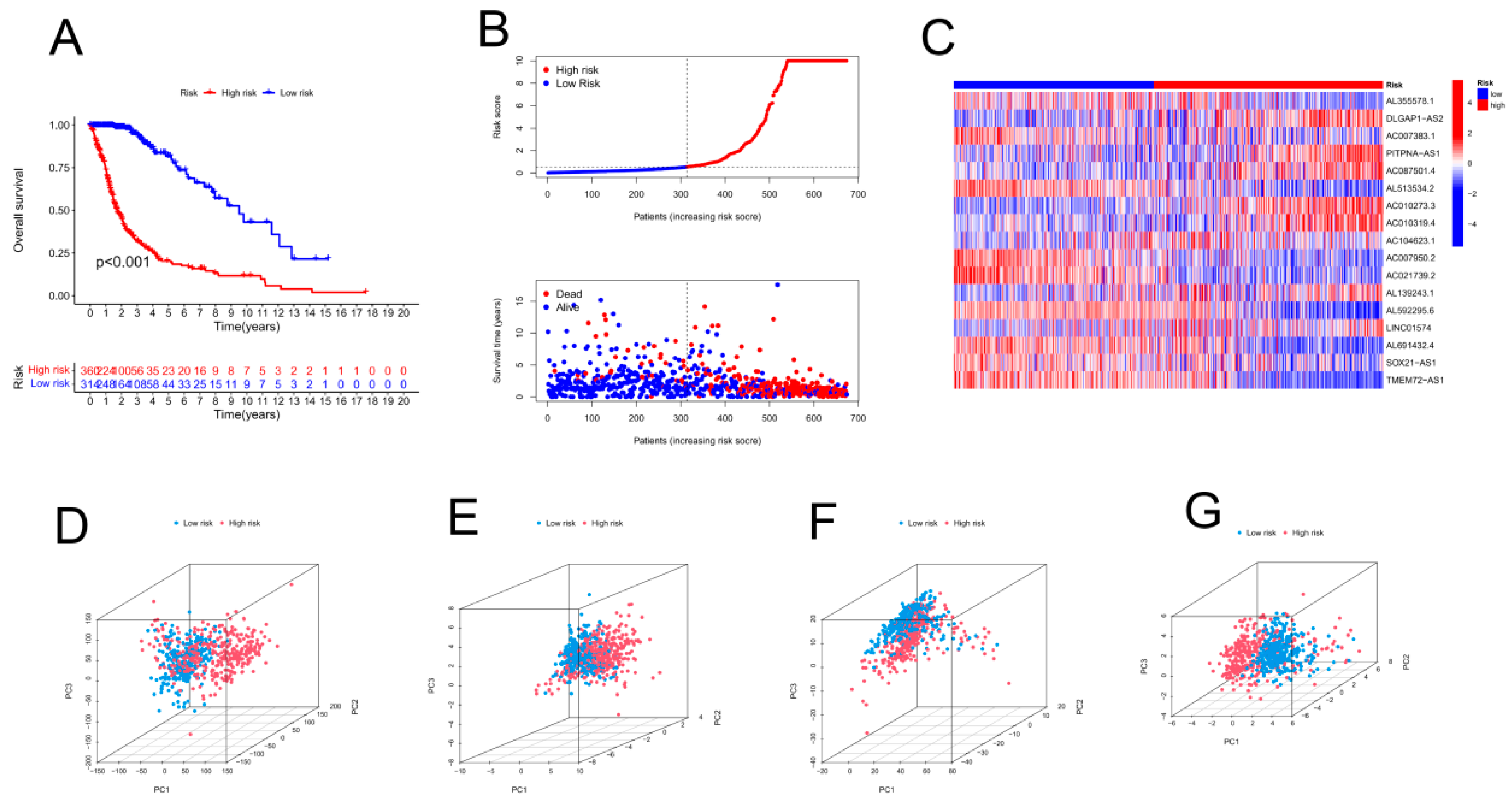
Figure 4.
Prognostic analysis of overall survival for gliomas (GBMs). (A) Univariate Cox analysis was conducted. Age, stage, and risk score were statistically significant. (B) Statistical significance was found for age, stage, and risk score in a multivariate Cox analysis. (C) PFS curves based on the Kaplan‒Meier method. (D) On the basis of the TimeROC curve, 1-, 3-, and 5-year OS was predicted for GBM patients. (E) ROC curve analysis demonstrated that the risk model was more accurate than other clinical parameters in predicting patient outcome. (F) According to the C-index, the risk model was more accurate than other clinical parameters for predicting mortality.
Figure 4.
Prognostic analysis of overall survival for gliomas (GBMs). (A) Univariate Cox analysis was conducted. Age, stage, and risk score were statistically significant. (B) Statistical significance was found for age, stage, and risk score in a multivariate Cox analysis. (C) PFS curves based on the Kaplan‒Meier method. (D) On the basis of the TimeROC curve, 1-, 3-, and 5-year OS was predicted for GBM patients. (E) ROC curve analysis demonstrated that the risk model was more accurate than other clinical parameters in predicting patient outcome. (F) According to the C-index, the risk model was more accurate than other clinical parameters for predicting mortality.
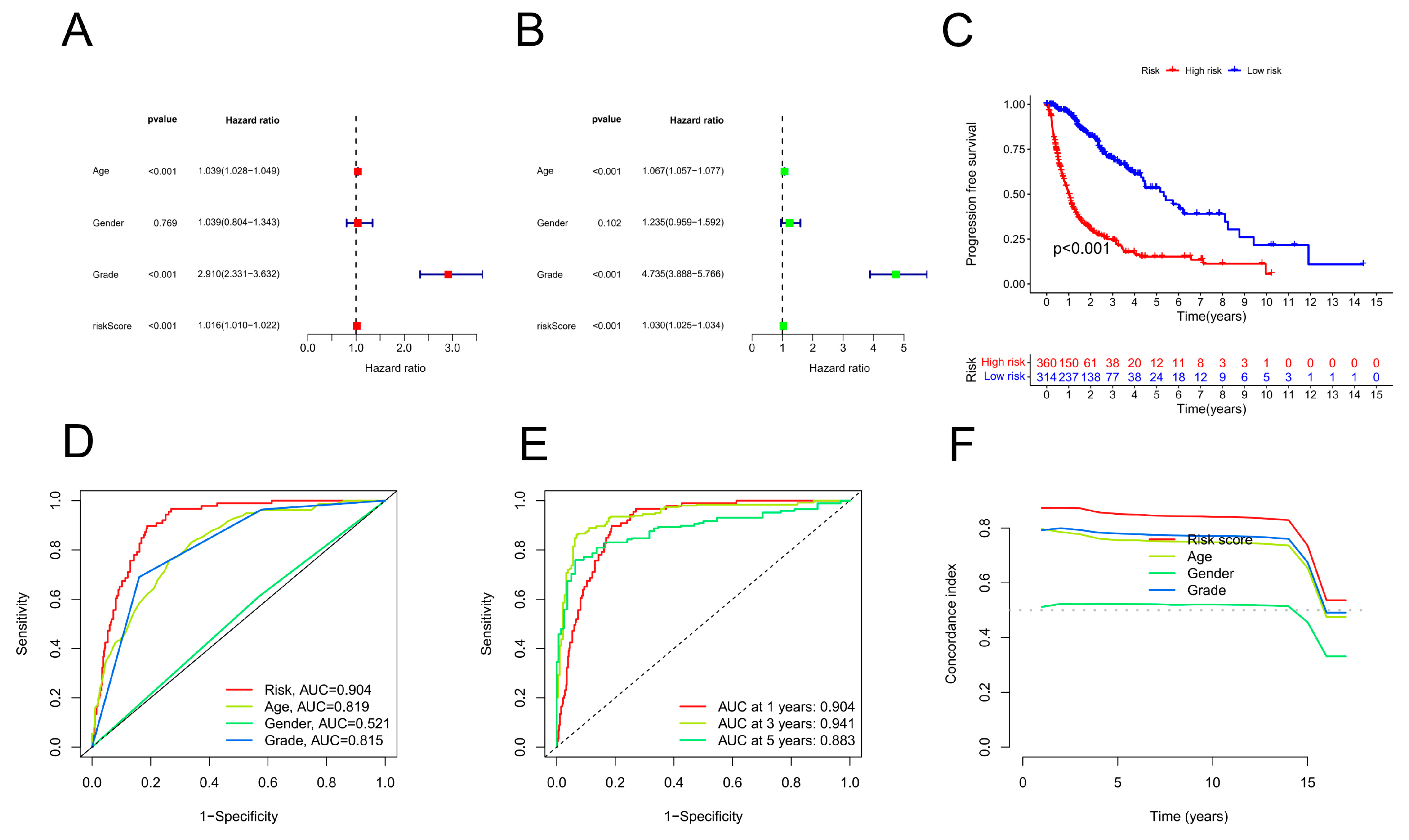
Figure 5.
Evaluation of the CRLPM-based nomogram. (A) CRLPM was used to construct the nomogram for predicting prognosis. (B) Predictions of survival over 1, 3, and 5 years are based on calibration curves. (C) Patients with stage II cancer and Kaplan–Meier curves. (D) Kaplan‒Meier curves for patients with stage III through IV disease.
Figure 5.
Evaluation of the CRLPM-based nomogram. (A) CRLPM was used to construct the nomogram for predicting prognosis. (B) Predictions of survival over 1, 3, and 5 years are based on calibration curves. (C) Patients with stage II cancer and Kaplan–Meier curves. (D) Kaplan‒Meier curves for patients with stage III through IV disease.
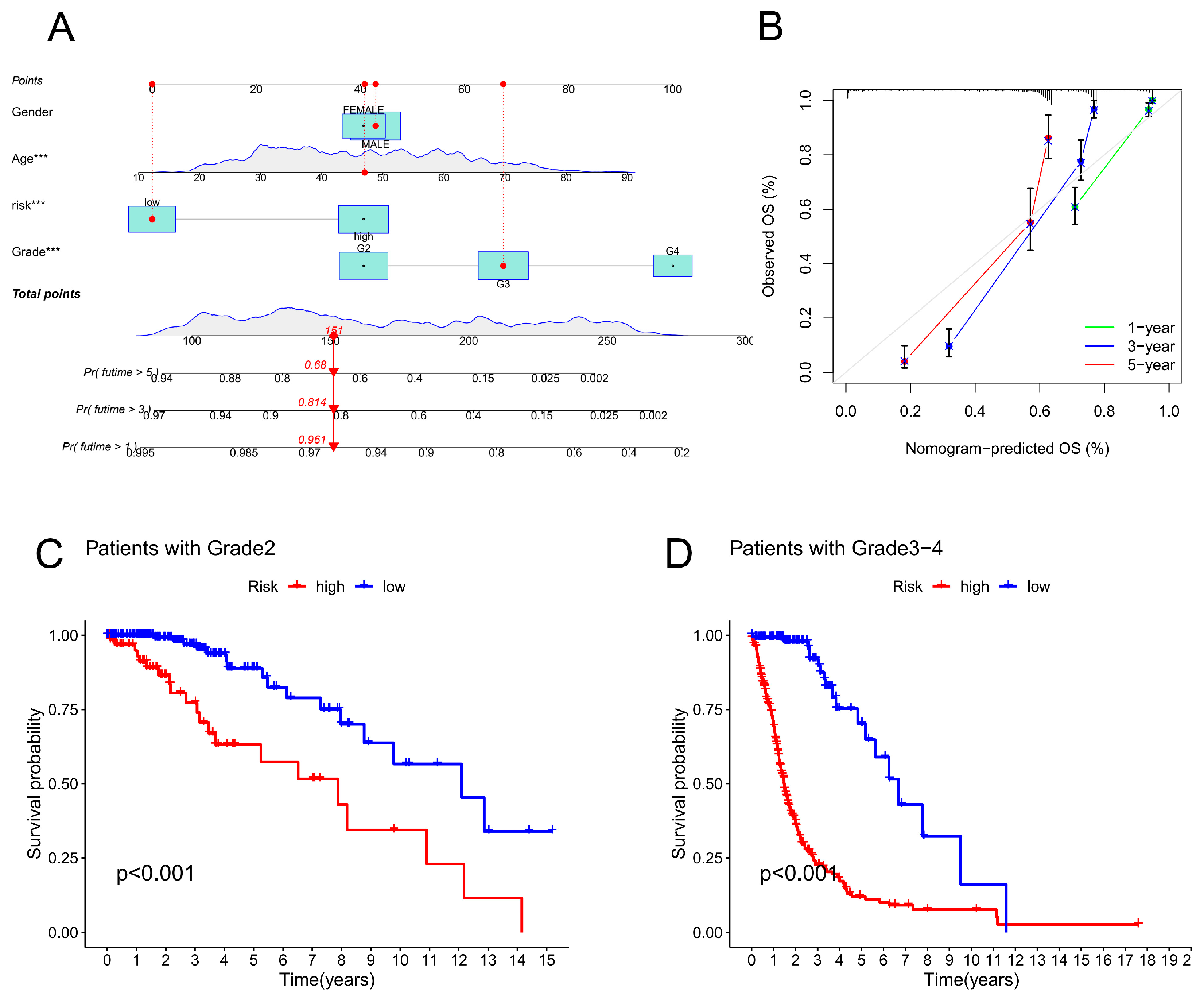
Figure 6.
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses. (A) A bar plot showing the top 30 enriched GO terms by category is shown below. (B) Bar plot of the top 30 enriched GO terms by categorys. (C) Bubble plot of the top 30 GO enrichment terms. (D) Bar plot of the top 30 enriched KEGG terms. (E) Bubble chart of the top 30 enriched KEGG terms. (F) Circle diagram of KEGG enrichment analysis of biological processes, cellular components, and molecular functions. (G) Circle diagram of GO enrichment analysis of biological processes, cellular components, and molecular functions.
Figure 6.
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses. (A) A bar plot showing the top 30 enriched GO terms by category is shown below. (B) Bar plot of the top 30 enriched GO terms by categorys. (C) Bubble plot of the top 30 GO enrichment terms. (D) Bar plot of the top 30 enriched KEGG terms. (E) Bubble chart of the top 30 enriched KEGG terms. (F) Circle diagram of KEGG enrichment analysis of biological processes, cellular components, and molecular functions. (G) Circle diagram of GO enrichment analysis of biological processes, cellular components, and molecular functions.
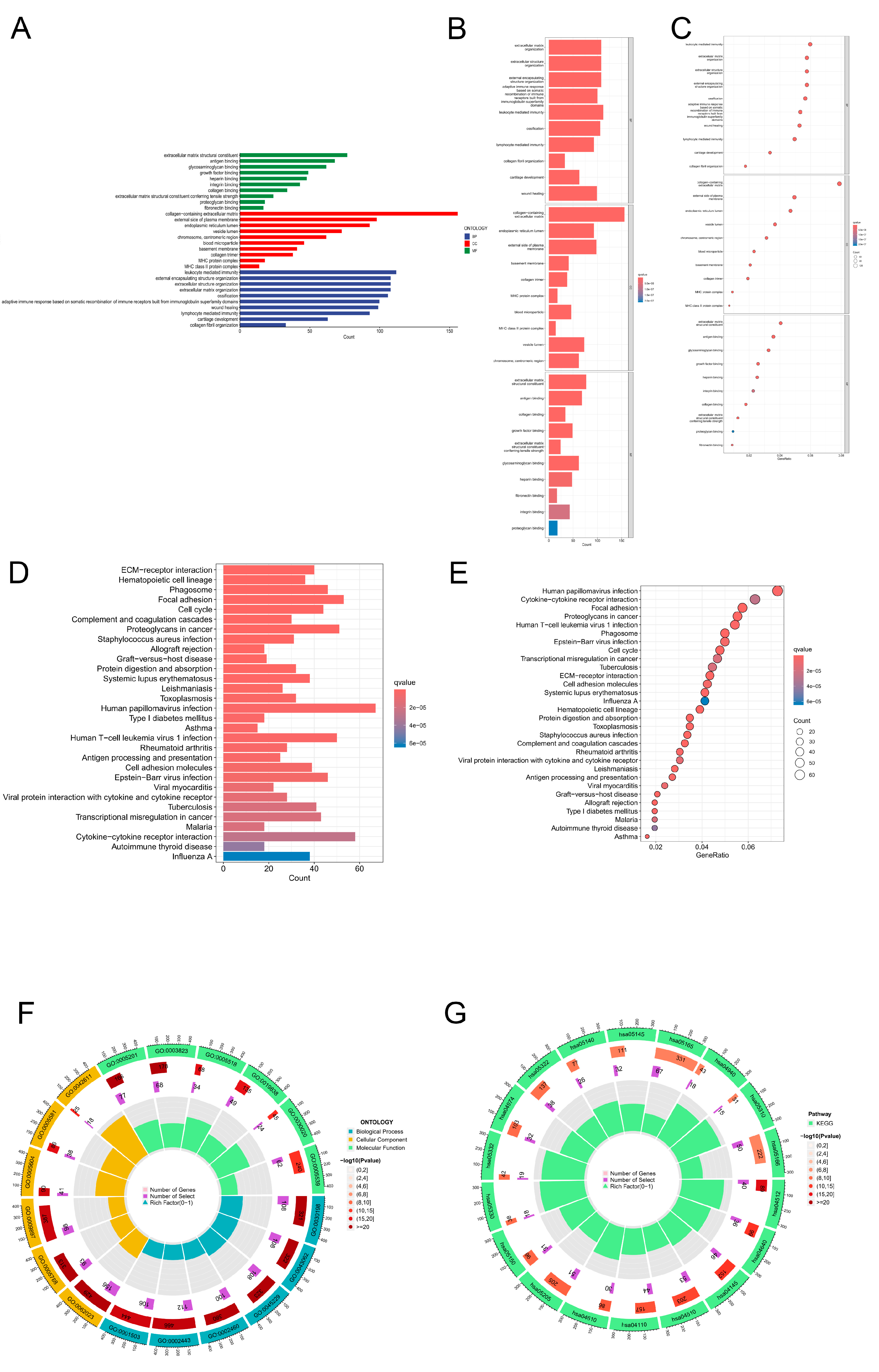
Figure 7.
The immunological landscape of glioblastoma (GBM) patients and their cancer mutation burden (TMB). (A) An analysis of single-sample gene set enrichment algorithms selected from the highest-risk and lowest-risk groups in GBM was conducted to create a heatmap showing tumour-infiltrating lymphocytes. *p < 0.05, **p < 0.01, and ***p < 0.001. (B) TIDE prediction scores for the high-risk vs. low-risk groups. (C) TMB differences between the high- and low-risk groups in GBM patients. (D) Survival analysis of the high- and low-TMB groups. (E) Combined TMB risk and survival curve of patients with GBM. (F) The top 15 mutated genes in the high-risk group of GBM patients are plotted in a waterfall graph. (G) The top 15 mutated genes in patients at low risk of GBM are shown in a waterfall plot.
Figure 7.
The immunological landscape of glioblastoma (GBM) patients and their cancer mutation burden (TMB). (A) An analysis of single-sample gene set enrichment algorithms selected from the highest-risk and lowest-risk groups in GBM was conducted to create a heatmap showing tumour-infiltrating lymphocytes. *p < 0.05, **p < 0.01, and ***p < 0.001. (B) TIDE prediction scores for the high-risk vs. low-risk groups. (C) TMB differences between the high- and low-risk groups in GBM patients. (D) Survival analysis of the high- and low-TMB groups. (E) Combined TMB risk and survival curve of patients with GBM. (F) The top 15 mutated genes in the high-risk group of GBM patients are plotted in a waterfall graph. (G) The top 15 mutated genes in patients at low risk of GBM are shown in a waterfall plot.
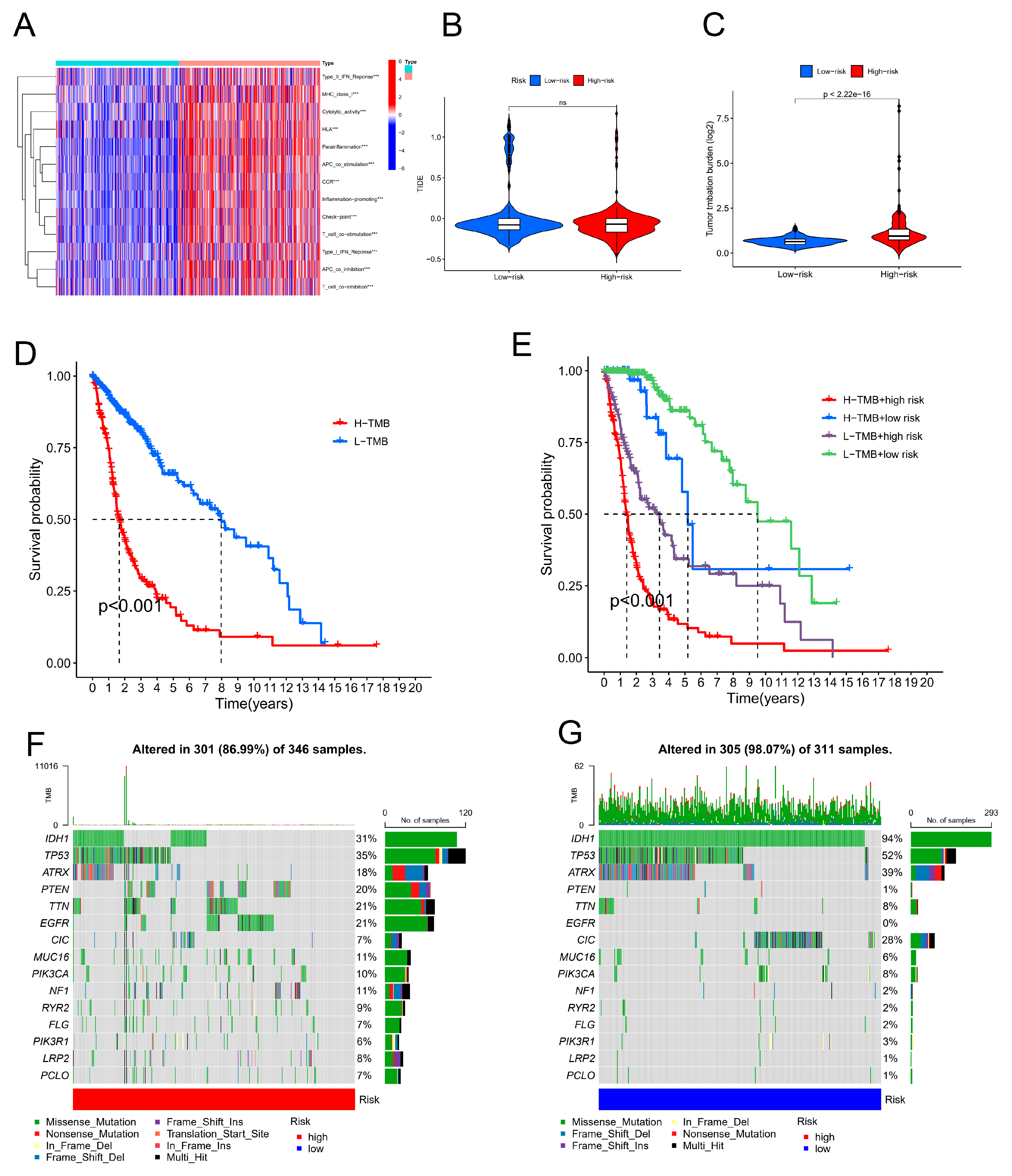
Figure 8.
Drug sensitivity (IC50) correlated with high- and low-risk patients with glioma.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



