Submitted:
02 May 2024
Posted:
07 May 2024
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
Proteases represent common targets in combating infectious diseases including COVID-19. The 3-chymotrypsin-like protease (3CLpro) is a validated molecular target for COVID-19 and it is key for developing potent and selective inhibitors for inhibiting viral replication of SARS-CoV-2. In this review, we discuss structural relationships and diverse subsites of 3CLpro, shedding light on the pivotal role of dimerization and active site architecture in substrate recognition and catalysis. Our analysis of bioinformatics and other published studies unveil a proposal of a novel catalytic mechanism for the SARS-CoV-2 polyprotein cleavage by 3CLpro, centering on the triad mechanism involving His41-Cys145-Asp187 and its indispensable role in viral replication. Recognizing Asp187 as a crucial component in the catalytic process underscores its significance as a fundamental pharmacophoric element in drug design. Next, we provide an overview of both covalent and non-covalent inhibitors, elucidating advancements in drug development observed in preclinical and clinical trials. By highlighting various chemical classes and their pharmacokinetic profiles, our review aims to guide future research directions toward the development of highly selective inhibitors, underscore the significance of 3CLpro as a validated therapeutic target, and propel the progression of drug candidates through preclinical and clinical phases.
Keywords:
SARS-CoV-2
; 3CLpro
; Novel Mechanism of Catalysis
; FDA-approved drugs
; Triad
Introduction
Coronaviruses (CoVs) are single-stranded positive-sense ribonucleic acid (RNA) viruses, being divided into four subgroups Alphacoronavirus, Betacoronavirus, Gammacoronavirus, and Deltacoronavirus [1] (Figura S1). They have a single large genome (27-32 kilobases) directly translated by host cells [2]. The hosts of CoVs are vertebrates that range from human beings to birds, generally causing respiratory and gastrointestinal tract disorders [3]. To date, seven coronaviruses (CoVs) have emerged to infect humans: HCoV-NL63, HCoV-229E, HCoV-HKU1, HCoV-OC43, Middle East respiratory syndrome coronavirus (MERS-CoV), severe acute respiratory syndrome coronavirus (SARS-CoV), and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Among these, HCoV-NL63, HCoV-229E, HCoV-HKU1, and HCoV-OC43 typically cause seasonal flu with mild-to-moderate respiratory symptoms. MERS-CoV and SARS caused epidemics, while SARS-CoV-2 caused the 2019 pandemic and is now considered a seasonal respiratory disease. . According to the latest release from the International Committee on Taxonomy of Viruses (https://talk.ictvonline.org/), SARS-CoV-2 and SARS-CoV are classified under the genus of β-CoVs, subgenus of Sarbecovirus and specie of Betacoronavirus pandemicum.
In late 2019, a novel severe pneumonia was reported in Wuhan, Hubei province, China [4]. The World Health Organization (WHO) named this disease “coronavirus disease 2019’, or COVID-19, referring to the first cases of this disease reported in 2019 [5]. March 2024, SARS-CoV-2 caused a total of 7,033,430 deaths and infected more than 774 million individuals. Since the virus's emergence, until March 2024, more than 3,9 thousand different genomes have been recorded [6]. The virus accesses the host cell through the Spike protein, which binds to the human ACE2 receptor [5,7,8,9]. The virus may infect the human cells bys two primary entry pathways: (1) Fusion of the virus membrane with the cell membrane, facilitating the release of its genetic material into the cytoplasm of the host cell or/and (2) Internalization of the virus via endocytosis. Following these entry mechanisms, the virus proceeds to initiate its replicative process, whereby its genome is released into the cytoplasm of the host cell. Upon entry into the host cell, the viral genome is released and acts as a template for the synthesis of viral proteins and genome replication. The ORFa and ORF1ab genes are then translated into two important polyproteins, respectively, pp1a and pp1ab [10,11,12]. The production of these polyproteins involves overlapping regions and is initiated by a ribosomal frameshift within this region. Collectively, the genome of SARS-CoV-2 encodes four structural proteins, sixteen non-structural proteins (nsps), and nine accessory proteins [13]. Two proteases—papain-like and 3-chymotrypsin-like (PL and 3CLpro, respectively)—cleave the polyprotein [13,14]. The papain-like protease cleaves three sites of the polyproteins, while the protease (Mpro or 3CLpro) cleaves thirteen sites [15], making this last protease be called the main protease [10].
Given the vital role of these proteases in viral replication, especially during the pandemic caused by SARS-CoV-2, they have emerged as promising targets for the development of broad-spectrum anti-SARS-CoV-2 agents [16]. The absence of human homologs of 3CLpro enables the development of specific inhibitors with minimal side effects on human proteases, potentially reducing the adverse effects associated with these inhibitors [17]. The 3CLpro has sparked significant interest in both academic field and pharmaceutical industry as a molecular target to inhibit viral replication and pathogenesis in various coronaviruses [18]. Several drug discovery strategies have been employed to find 3CLpro inhibitors, including drug repurposing, high-throughput virtual screening (HTVS), high-throughput screening (HTS), and structure- and ligand-based drug designs (SBDD and LBBD, respectively) [19,20,21,22,23,24,25]. Additionally, the strategy of exploring natural products as 3CLpro inhibitors has been one of the most important approaches for developing novel anti-SARS-CoV-2 agents [26,27]. These efforts have led to chemically diverse synthetic compounds and natural products emerging as effective inhibitors of SARS-CoV-2 3CLpro [28,29].
Herein, we reviewed the main protease of SARS-CoV-2 as a pivotal and promising therapeutic target for the treatment of COVID-19. Among the methodologies discussed, we propose that the cleavage of polyproteins pp1a and pp1ab is facilitated by the triad H41-Cys145-Asp187, based on bioinformatics analyses coupled with computational and experimental data from the literature. This hypothesis assumes critical significance, as Asp187 emerges as a pharmacophoric determinant justifying consideration in the optimization of bioactive compounds targeting the virus's principal enzyme. Next, we discuss the compounds evaluated both in vitro and in vivo, alongside those advancing through clinical drug development phases. From a pharmacokinetic standpoint, our effort is to correlate biological attributes with the physicochemical features of the drug-like compound, with the aiming goal of discerning requisite patterns favoring the effective attenuation of viral pathogenesis. Taken together, our study brings important insights in optimizing lead compounds in the pre-clinical stage of development.
3-chymotrypsin-Like Protease: A Validated Molecular Target
A crucial strategy in rational drug design is to target molecular entities that lack homologs in humans. The absence of homolog proteins of 3CLpro in humans is critical because inhibitors must be designed to specifically interact with the viral protease, thereby reducing the probability of affecting human proteases. Homologs are similar proteins found in different species, and the presence of human homologs of 3CLpro could increase the risk of adverse effects during administration in humans. By employing the Basic Local Alignment Search Tool (BLASTP) and Position-Specific Iterated BLAST (PSIBLAST) tools on the National Center for Biotechnology Information (NCBI, https://www.ncbi.nlm.nih.gov/), considering the sequence of 3CLpro from SARS-CoV-2 (PDB ID 7WOF) (Table S1), we examined the sequence of 3CLpro from SARS-CoV-2 and restricted the search only to Homo sapiens (taxid:9606). Although we did not find human proteins with sequence similarity to 3CLpro of SARS-CoV-2, it is possible that some human proteins share structural similarities. In this regard, we also searched the Dali server [30,31] using the structure of the 3CLpro A chain (PDB ID 7EN8) [32] as a query to search for distant homologs in humans. The results of the search for structural homologues of 3CLpro in human proteins, obtained from the Dali server, are available in Table S2. We found serine protease Fam111a, prothrombin, serine protease Hepsin, mannan-binding lectin serine protease, complement C3b beta chain, enteropeptidase, protein Z-dependent protease inhibitor, human complement factor I, coagulation factor XI and plasminogen, in which root-mean-square deviation (RMSD) ranged from 2.7 to 3.5 Å, similarity of amino acid sequences between 8 and 16%, and Z-score between 9.1 and 11.5 (Table S2). The presence of structural similarities does not necessarily imply adverse effects in individuals, demanding experimental studies for investigating inhibitor selectivity. These results help for developing more selective inhibitors, reducing associated adverse effects and enhancing safety and efficacy in COVID-19 treatment.
In addition to identifying possible homologs of 3CLpro in humans, it is crucial to verify the specificity of this molecular target, ensuring its essentiality in viral replication. One approach to demonstrate this is the inhibition of 3CLpro in several strains of coronavirus. By validating this essentiality, it is possible to extrapolate its importance to all viral lineages, consolidating it as a fundamental molecular target for drug development. Yang and co-authors were pioneers in identifying molecular targets for the development of broad-spectrum inhibitors against coronavirus diseases [33]. They carried out an initial screening of targets, excluding structural proteins due to notable variations between different strains of coronavirus [33]. After this selection, they focused on RNA-dependent RNA polymerase (RdRp), RNA helicase, and Mpro as possible targets among the non-structural proteins [33]. Despite the differences in the primary sequence of Mpro between coronaviruses, three-dimensional analysis revealed a high similarity, especially in the active site [33]. Based on that, Yang and co-authors developed irreversible inhibitors, demonstrating efficacy against several Mpros of different coronaviruses, including SARS-CoV, MERS-CoV, and others [33], showing that 3CLpro is a key protease for replication of the coronavirus.
Structure and Function of Mpro: A Novel Proposal for the Mechanism of Polyprotein Processing
MproSARS-CoV-2 is a cysteine protease [34] sharing 93.2% sequence similarity with SARS-CoV. It demonstrates comparable catalytic behavior [18]. Structurally, SARS-CoV-2 Mpro exists as a dimer, with a dissociation constant (KD) in the micromolar range [34]. Its three-dimensional structure includes domains I and II (residues 10–99 and 100–182, respectively), resembling pancreatic trypsin, featuring six barrel structures arranged into antiparallel β-strands (Figure 1a-b) [35]. Conversely, domain III (residues 198–303) comprises five α-helices governing 3CLpro dimerization [36]. Mutation within domain III leads to a monomeric Mpro, and therefore, resulting in catalytic activity loss [37,38,39]. The enzyme's active site is localized between domains I and II, in which it has specific subsites (S4, S3, S2, S1, and S1') (Figure 1c-d) [40,41,42]. Notably, catalytic histidine, H41 residue, occupies the catalytic S1' subsite, while residues Cys145, His172, Glu166, His163, His164, and Phe140 occupy the S1 subsite. Hydrophobic residues such as Met49, Tyr54, Met165, Pro168, and Val186 belong to the S2 subsite, engaging with the substrate's hydrophobic segment at the P2 position [35]. Subsites S3 and S4, consisting of residues Gln189, Gly251, Gln192, and Ala191, remain more solvent-exposed. The limited catalytic activity of monomeric Mpro underscores the importance of dimerization in substrate recognition, especially at the S1 subsite. Consequently, several inhibitors of 3CLpro have been developed to target protein dimerization.
The structure and function of 3CLpro are crucial for understanding the processing of viral polyproteins [46]. Since the active site of SARS-CoV-2 3CLpro is broad and contains subsites with different physicochemical features, the substrate binds in a manner that occupies the entire active site [47]. Positions P1, P2, and P1ʹ on the substrate play a crucial role in enzymatic specificity, while positions P4, P3, and P3ʹ expand the interaction area and stabilize the substrate [48]. Zhao and colleagues demonstrated the sequence alignment of residues surrounding eleven cleavage sites, revealing a specific pattern within the polyprotein for substrate recognition by the enzyme. This pattern includes P4 (A/V/P/T), P3 (T/K/R/V/M), P2 (L, highly conserved/F/V), P1 (conserved for Q), and P1’ (S, reasonably frequent/A/N) [49]. Therefore, these data demonstrate that, for certain positions, 3CLpro keeps specificity for this pattern.
The currently accepted catalytic mechanism involves the His41/Cys145 dyad in polyprotein cleavage [50]. Typically, position P1 is occupied by glutamine [48,49], which is an almost universal requirement. The catalytic dyad is activated by water, maintained by residue Asp187 [51]. Once activated, the catalytic cysteine acts as a nucleophile in the proteolytic process, with its activation occurring through deprotonation of the thiol group by His41 [52]. The resulting thiolate then undergoes nucleophilic attack on the substrate's glutamine, resulting in amide bond cleavage. Subsequently, the catalytic histidine restores its deprotonated form, allowing the thioester to be attacked, with water acting as a nucleophile and releasing the hydrolyzed C-terminal, restoring the catalytic dyad [51].
It is known that chymotrypsin, a homolog of 3CLpro, has a catalytic mechanism containing a catalytic triad [53]. As described earlier, SARS-CoV-2 3CLpro has a mechanism containing a dyad. We searched in published articles site-directed mutagenesis studies, computational and biochemical studies that demonstrate the possibility of a catalytic triad mechanism participating in the catalysis process. Our previous molecular dynamics study showed that Asp187 played an important role in interacting with the catalytic histidine in different 3CLpro/substrate complexes [36], presenting extremely significant hydrogen bonds (hydrogen bonding occupancy above 50%) [36]. Our previous study in identification of novel 3CLpro inhibitors, we observed that the contribution for binding affinity of the Asp187 was comparable to Glu166, suggesting that both are critical for investigation of novel inhibitors [22]. Other computational simulation works have corroborated the hypothesis that Asp187 is crucial in interacting with the catalytic histidine [54].
In the initial analysis, we included in this review bioinformatics studies to verify the conservation of Asp187 in different families of coronaviruses. To analyze the conserved residues of the Mpro protein, we conducted a search using PSI-BLAST (Position-Specific Iterated BLAST) with a single iteration, using the SARS-CoV-2 3CLpro sequence (UniProt accession code P0DTD1, residues 3264 to 3569 and structure solved with PDBID 7WOF)[43]. We adjusted the search parameters to a maximum of 2 million sequences, resulting in a total of 1,228,540 hits after applying an e-value filter of 10-3. We then grouped these sequences using the MMseqs2 (Many-against-Many sequence searching) program [55], using the region identified as a hit by PSI-BLAST and setting 80% coverage and 0% identity to eliminate redundancy [56]. Subsequently, we used the hmmscan module of the HMMER 3.0 package and the Pfam HMM model (protein family databases) for Mpro to extract the region of interest from the polyprotein [57,58]. We also applied a filter to remove duplicated organisms and filtered sequences with lengths less than 274 residues, combined with the removal of sequences with incorrect annotations or truncations. This process resulted in 18 sequences. We performed a multiple sequence alignment (MSA) using fast-multiple sequence alignment (FAMSA) [59] to produce a preliminary phylogenetic tree using IQ-TREE [60] and refined the MSA removing outliers sequences detected. This MSA was then represented by a sequence logo (Figure 2), produced using the WebLogo 3 online server (https://weblogo.threeplusone.com/) [61,62]. Our bioinformatics analysis shows that among the different members of the Coronavidae family, Asp187 is highly conserved, indicating its importance for the different 3CLpro, including MERS-CoV, SARS-CoV, and SARS-CoV-2 (Figure 2).
A quantum mechanics-molecular mechanics (QM-MM) study seeking to understand the mechanism of action of the PF-0721332 inhibitor on SARS-CoV-2 3CLpro described that Asp187 was close to the His41 residue and could form a catalytic triad, Cys145–His41–Asp187, which would facilitate the deprotonation of Cys145 [54]. In this regard, Asp187 was included in the QM region and it was verified that in this covalent inhibition mechanism between Mpro and PF-0721332 there was a strong hydrogen bond (distance of 1.64 Å between His41 and Asp187) [54]. These results show that the catalytic triad Cys145–His41–Asp187 plays an important role in the covalent inhibition of SARS-CoV-2 Mpro, allowing the deprotonation of Cys145 and thus facilitating subsequent reaction. This finding is consistent with a previous study that demonstrated that Asp187 favors the transfer of protons from Cys145 to His41 [54].
Additionally, Adem and co-authors performed a series of experiments using site-directed mutations in the main residues of the different subsites of 3CLpro, comparing the proteolytic activity of the mutants relative to 3CLproWT. The alanine substitution in D187 (3CLproD187A) abolished 3CLpro activity [63]. Furthermore, the D187E and D187N substitutions resulted in 3CLpro inactivation [63]. Taken together, all results combinated with our bioinformatics data suggest that the catalytic mechanism of 3CLpro is formed by a His41-Cys145-Asp187 triad. Indeed, the proposal of the catalytic triad participating in reactions is classic in several catalytic reactions of various enzyme classes [64,65,66,67,68]. The proposed mechanism in this review follows two phases, acylation followed by deacylation, where the roles of these residues would be analogous to the catalytic triad of 3CLpro.
I. Acylation step. At the active site of the main protease, Cys145 acts as a nucleophile. Initially, His41 acts as a base, taking a proton from Cys145, activating it as a nucleophile. Then, the nucleophilic Cys145 attacks the carbon of the carbonyl group of the main chain of the substrate forming a tetrahedral intermediate. This leads to cleavage of the peptide bond, releasing a substrate fragment with a alcohol group and forming an acyl bond between Cys145 and the other fragment of the substrate (Figure 3).
II. Deacylation step. In this step, a water molecule acts as a nucleophile by a hydrolysis reaction. His41 now acts as an acid taking a proton from the water molecule activating the water to perform the nucleophilic attack on the acyl group in Cys145 forming a tetrahedral intermediate. This results in cleavage of the acyl bond to form a carboxylic acid and the regeneration of the active form of the main protease. The second fragment of the substrate is released, completing the reaction.
This catalytic mechanism, adapted for the main protease of SARS-CoV-2 and other coronavirus lineages, is crucial for the processing of viral polyproteins. As a result, we have suggested that Asp187 as a pharmacophore in the design of 3CLpro inhibitors, where inhibitors that interact with this residue destabilize the hydrogen bond interaction between the residue pair His41/Asp187.
Pre-Clinical and Clinical Trials of Reversible and Irreversible 3CLpro Inhibitors Against SARS-CoV-2
The compounds tested on the Mpro originated from various sources such as peptides, non-peptides, chemicals, flavonoid derivatives, among others [48,69,70,71]. Most are competitive inhibitors focused on allosteric inhibition [72]. The global emergence of COVID-19 led to the fast repurposing of protease inhibitors previously developed for other viral indications [73]. Although HIV protease inhibitors did not provide any benefit for COVID-19 treatment [74], it was the first-generation peptidomimetic protease inhibitors that were initially tested against the main enzyme of SARS-CoV-2. These molecules have a Gln substitution at the P1 position but do not exhibit significant inhibitory activity against SARS-CoV-2's 3CLpro (for example, rupintrivir see reference [75]). Second-generation Mpro inhibitors showed good in vitro and in vivo activity in animal models against MERS and in vitro activity against SARS-CoV-2 [76]. Although the SARS-CoV-2 primarily infects the respiratory tract (pharynx, trachea, lungs) in the early stages of infection, a broader organotropism has been reported in more severe and advanced cases of the disease [77].
Remdesivir, the nucleotide analog, presented good results in the pre-clinical phase, showing IC50 and EC50 equal to 43 μM and 10 nM, respectively (Figura 4). This chemical structure is an U.S. FDA-approved drug and its administration is intravenous in hospitalized patients [78]. This type of administration is undesirable for drugs due to their unfavorable pharmacokinetic properties such as low oral bioavailability. CDI-45205 (structure unavailable) also is administered by intravenous or inhalation [78,79]. Other 3CLpro inhibitors have also been administered intravenously or intraperitoneally to increase oral bioavailability. PF-00835231 and GC376 inhibitors presented IC50 values of 4 and 13 μM and EC50 values of 15 and 1,600 nM, respectively (Figure 4). Both presented low oral bioavailability in rats (1.4% and 3%, respectively) [80]. Remdesivir has one hydrogen bond donor and nine hydrogen bond acceptors, with a molecular weight of 602.61 g/mol, exceeding the limit of 500 Da established by Lipinski's rules for orally desirable drugs [81]. This high molecular weight indicates a possible difficulty in fully crossing the cell membrane and reaching the target enzyme, which may contribute to its low bioavailability. PF-00835231 and GC376 are examples that have a polar surface area (PSA) of 150 and 182 Ų, both greater than 140 Ų. In general, molecules with values above 140 Ų are considered difficult to permeate through membranes [82], which may result in low bioavailability in the circulatory system. Conversely, pharmacokinetic optimizations of PF-00835231 led to the PF-07304814[83] (Figure 5 and Table 1).
In general, second-generation inhibitors aim to enhance pharmacokinetic properties, particularly by increasing oral bioavailability [79]. For instance, peptidomimetic inhibitors targeting the human rhinovirus 3-chymotrypsin protease have demonstrated oral bioavailability exceeding 20% in rodent and other non-clinical models, suggesting their potential suitability for oral administration [79]. Pfizer has initiated a Phase I clinical trial (NCT04756531) with the second-generation oral 3CLpro inhibitor PF-07321332 (Nirmatrelvir) (Figure 6 and Table 1), which has been specifically optimized for oral administration [84]. This inhibitor features a nitrile head and has undergone optimization due to reduction of hydrogen bond donors and the incorporation of a trifluoroacetyl group, which presented a Ki value of 7 nM for Mpro and presented a high antiviral activity in Vero E6 cells infected with SARS-CoV-2, with EC50 value of 880 nM with good selectivity and safety in vivo. The National Medical Products Administration (NMPA) approved the SIM0417 SARS-CoV-2 Mpro inhibitor for the treatment of adults with mild to moderate COVID-19 symptoms on January 28, 2023. However, the structure of SIM0417 has not been disclosed. Similar to Nirmatrelvir, SIM0417 requires low-dose ritonavir administration, which slows down first-pass metabolism in vivo and enhances efficacy in inhibiting the virus life cycle.
Nirmatrelvir was developed by Pfizer in 2002 for the treatment of SARS-CoV; this pharmaceutical company backed studies in 2021 to evaluate its efficacy against SARS-CoV-2 [85,86]. The drug exhibits in vitro and in vivo antiviral activity, and currently, the study is in Phase II clinical trials to assess its safety, tolerability, and pharmacokinetics (Table 1). However, Mpro SARS-CoV-2 variants containing mutations in the E166N/V, M165T, G143S, Q189E, A173V, H172F/Q/Y, or Q192S/T/V residues rendering virus resistant to Nirmatrelvir treatment [87]. Recently approved by the FDA, on December 22, 2021, Paxlovid is a novel oral administration that combines a 3CLpro inhibitor (Nirmatrelvir) with Ritonavir or Paxlovid (Pfizer PF-07321332/ritonavir) to treat patients with moderate or severe COVID-19. Nirmatrelvir inhibited 3CLpro from several types of human coronaviruses, besides possessing antiviral activity in Vero E6 cells with an EC50 value of 74.5 nM. The clinical trial of Phase II/III with Nirmatrelvir and ritonavir (ID NCT04960202) in non-hospitalized COVID-19 adults reduced the risk of hospitalization or death by 89% [88]. In the Ômicron BA.2 subvariant, Paxlovid treatment decreased COVID-19 progression and mortality due to viral load reduction [89].
Boceprevir is used as a protease inhibitor for the hepatitis C virus (HCV) [90] by containing an α-ketoamide that forms a covalent bond with the serine active site in the 3CLpro protein. It proved to be an effective drug in virtual screening and in vitro assays. Boceprevir, in vitro showed effective inhibition, with an IC50 of 4 μM. However, in vivo, EC50 was more than 20 μM, leading it to be halted in the preclinical phase, requiring further in vitro and in vivo studies. The result may be explained because Boceprevir may violate Lipinski's rules due to its molecular weight (519.7 Da). Furthermore, we observed that PSA is more than 140 Ų, which makes it difficult to cross through cell membranes, exceeding the limits established for these parameters. Comparing Nirmatrelvir with Boceprevir, both present the same chemical scaffold (Figure 6). However, Boceprevir has the chemical groups cyclobutane, isopropyl urea, and oxopropyl amine group in the same position that pyrrolidinone, trifluoromethyl amide, and nitrile group in Nirmatrelvir, respectively. Consequently, these groups may significantly influence the electronic density of the reactive group in these molecules and their pharmacokinetic properties.
Lopinavir and Ritonavir are HIV protease inhibitors used in AIDS treatment (Figure 7) [91,92]. Both were employed in treating SARS-CoV-2 infections due to their ability to reduce SARS-CoV 3CLpro activity in vitro. Despite in vitro studies showing a decrease in viral replication, in vivo studies in animal models demonstrated that the drug combination was ineffective in the SARS-CoV-2 treatment [93]. In 2021, the World Health Organization (WHO) did not recommend the use of these medications for COVID-19 treatment. Despite their withdrawal from clinical trials, Ritonavir has found new utility when combined with Nirmatrelvir, currently undergoing phase II/III clinical trials (Table 1). Ritonavir is used as a combined strategy with Nirmatrelvir because it improves pharmacokinetic properties by inactivating CYP3A4 [94]. Nirmatrelvir's rapid metabolism may interfere with the treatment of COVID-19. Ritonavir acts by decreasing the metabolism of Nirmatrelvir, decreasing the expression of CYP3A4 and consequently improving the efficacy of Nirmatrelvir [95]. Furthermore, Ritonavir has a high binding affinity to plasma proteins, suggesting that the increased action of Paxlovid may also be related to the efficacy of this drug [96]. When combined, Ritonavir may occupy the binding sites of these proteins in blood plasma, increasing the free concentration of Nirmatrelvir to have its antiviral action [96].
The α-ketoamides were envisioned as broad-spectrum inhibitors of the main proteases of beta coronaviruses and alpha coronaviruses, as well as enterovirus 3C proteases [84,97]. Compound 11r showed promise, with an EC50 of 400 pM against MERS-CoV in Huh7 cells and EC50 against SARS-CoV and a variety of enteroviruses in various cell lines [44]. Building on these results, Zhang and co-authors modified compound 11r by replacing the amide at the P2-P3 position with a pyridone ring, aiming to enhance selectivity for the SARS-CoV-2 protease [44] (Figure 8). Another modification was made to improve one of the pharmacokinetic properties, plasma solubility, and reduce plasma protein binding [44]. The authors replaced the tert-butyloxycarbonyl protecting group or tert-butoxycarbonyl protecting group (Boc) group with a slightly less hydrophobic group, resulting in compound 13a (Figure 8) [44]. The α-ketoamides are a class of reversible inhibitors [98]. 3Clpro cys145 undergoes a nucleophilic attack on the α-keto group of the α-ketoamide, forming a thiohemiacetal in a reversible reaction [98]. Compound 13b was able to inhibit the recombinant purified SARS-CoV-2 Mpro with IC50 = 670 μM. In vitro assays showed that Compound 13b was effective against SARS-CoV and MERS-CoV, as well as inhibiting SARS-CoV-2 RNA replication in Calu-3 cells [44]. To enhance pharmacodynamic properties, the authors substituted the cyclohexyl group at the P2 position with a cyclopropyl in Compound 13b. The absence of the Boc group in this class of inhibitors is necessary for crossing the cell membrane but leads to higher plasma protein binding, a disadvantage in pharmacokinetic properties [44]. Compound 11r inhibited effectively MERS-CoV and SARS-CoV, leading to modifications as present in compound 13a to enhance selectivity for SARS-CoV-2 protease [44]. Compound 13b inhibited SARS-CoV-2 Mpro and showed effectiveness against SARS-CoV and MERS-CoV in vitro. Pharmacokinetic studies revealed adequate metabolic stability for compound 13a, with primarily tissue distribution and excretion via urine [44]. Compound 13b exhibited similar properties and pulmonary tropism, suggesting potential efficacy in the treatment of COVID-19 [44]. Inhalation administration was well tolerated, indicating the possibility of direct lung delivery [44]. Therefore, alpha-ketoamides represent a promising class of inhibitors for viral diseases, with compounds 13a and 13b showing potential in both in vitro studies and animal models.
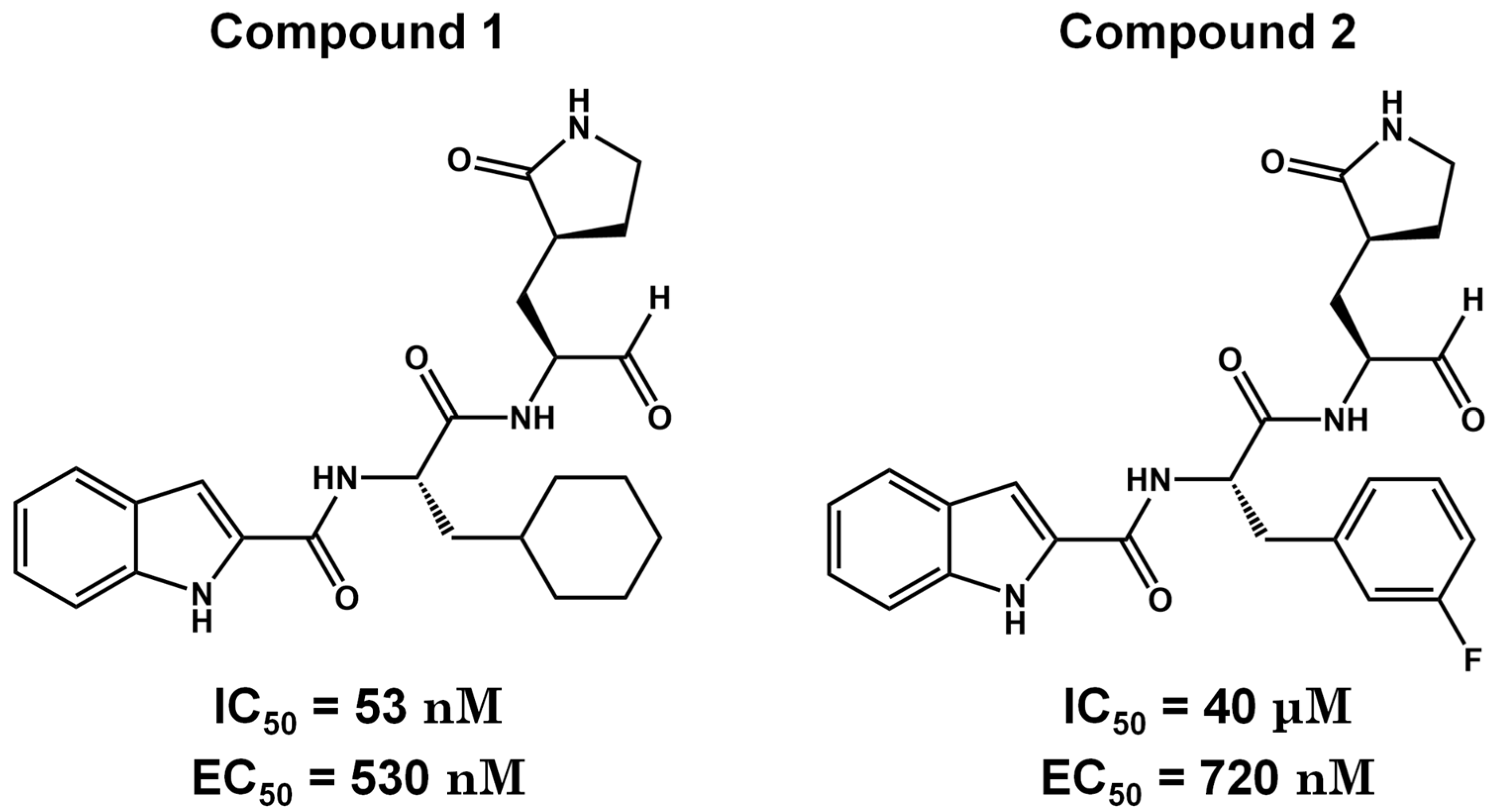
Carbonic groups such as aldehydes and ketones stand out as promising targets for the development of novel covalent inhibitors of SARS-CoV-2 Mpro [99,100,101]. Generally, the formation of the covalent bond depends on the electrophilic nature of the carbonyl carbon, susceptible to nucleophilic attack by the catalytic cysteine, resulting in the formation of a reversible thiohemiketal adduct [102]. The structural similarity between this adduct and the intermediate formed by the natural substrate during the enzymatic catalytic cycle ensures high stability of the inhibitor-protein complex and prolonged residence time [102]. Examples of these covalent inhibitors, such as compounds 1 and 2 (Figure 9), have been derived as peptidomimetics and have shown excellent inhibitory activity against SARS-CoV-2 Mpro (IC50 = 53 nM and 40 nM, respectively) and antiviral activity against SARS-CoV-2 (EC50 = 530 and 720 nM, respectively). Moreover, these compounds have demonstrated low cytotoxicity and favorable pharmacokinetic and toxicological properties in vivo. These data suggest that compounds 1 and 2 are promising candidates to advance to clinical trials phases in the treatment for COVID-19.

Table 1.
Drugs in preclinical and clinical trials. The table provides an overview of drugs in preclinical and clinical trials. Each row represents a specific drug under investigation, with information about the drug's name, the pharmaceutical company responsible for its development, the route of administration, the current state of drug development, the inhibitory concentration (IC50) in nanomolar for drugs in preclinical trials, the effective concentration (EC50) in micromolar for drugs in clinical trials, and the unique identifier (ID) of the registered clinical trial. These data offer a comprehensive perspective on drugs in the research and development phase for potential use in treating COVID-19 infectious diseases.
Table 1.
Drugs in preclinical and clinical trials. The table provides an overview of drugs in preclinical and clinical trials. Each row represents a specific drug under investigation, with information about the drug's name, the pharmaceutical company responsible for its development, the route of administration, the current state of drug development, the inhibitory concentration (IC50) in nanomolar for drugs in preclinical trials, the effective concentration (EC50) in micromolar for drugs in clinical trials, and the unique identifier (ID) of the registered clinical trial. These data offer a comprehensive perspective on drugs in the research and development phase for potential use in treating COVID-19 infectious diseases.
| Drug | Company | Delivery | State | IC50 (nM) | EC50 (μM) | ID (clinical trials) |
|---|---|---|---|---|---|---|
| Nirmatrelvir (PF-07321332) | Pfizer | Oral | Phase II | 10-100 | 0.07-0.3 | NCT05011513 |
| Lopinavir and Ritonavir | Abbvie | Oral | Discontinued | 12000.0# | 26.6* | NCT04381936 |
| Paxlovid™ (Nirmatrelvir and Ritonavir) | Pfizer | Oral | Phase II/III | 3.1 | 0.075 | NCT04960202 258 |
| Boceprevir | Merck | Oral | Preclinical | 1.6 | 2-10& | ND |
| GC376 | Kansas State University | Oral | Preclinical | 26.4 | 2.6 ± 0.2**/ 1.1 ± 0.2*** | ND |
| Lufotrelvir (PF-07304814) | Pfizer | IV | Phase I | 0.27 | 760 | NCT05050682 |
| Xiannuoxin™:Simnotrelvir (SIM0417) and Ritonavir (SSD8432) | Simcere | Oral | Phase II/III | 9 | 34 | NCT05373433 [103] |
# IC50 determined for Lopinavir; *Vero E6 and A549, overexpressing ACE2/TMPRSS2 cells; & Assays done in HEK293T/17 cells; **Assays performed in Vero cells; ***Assays performed in Calo-2 cells; % assays done in iPS-AT2 cells. IV = intravenous administration.
Conclusion
The 3CLpro protease plays a crucial role in viral replication, characterized by its high degree of protein conservation among members of the Coronaviridae family. Its specificity for viral proteins renders it a promising target for drug development aimed at inhibiting viral replication. However, the urgent need for anti-SARS-CoV-2 drugs during the pandemic prompted many countries to approve various 3CLpro inhibitors, some of which were originally developed as antivirals for other diseases [104]. Unfortunately, since 2020, many of these approved drugs have been discontinued due to their ineffectiveness in combating the virus in vivo, as well as their inability to reduce hospitalizations and due to their high toxicity [105]. Additionally, the emergence of SARS-CoV-2 variants has rendered some of these drugs ineffective against the new strains [106]. Hence, the search for novel molecules capable of inhibiting virus replication is paramount, serving as a preventive measure against future coronavirus-induced pandemics. Another significant contribution in this review is the hypothesis regarding Asp187 as a potential pharmacophoric component in the design of inhibitors. We have evidence to suggest that it is part of a catalytic triad involved in processing the virus polyprotein. Including Asp187 as a pharmacophoric element could be crucial in enhancing the activity of bioactive compounds undergoing molecular optimization. We believe that if the catalytic aspartate interacts with a specific inhibitor, it will destabilize the triad, thus impeding proton transfer in the acylation and deacylation stages, which are fundamental for catalysis.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, V.M.d.F.A., C.R.G., and A.S.d.S.; methodology, V.M.d.F.A., E.P.S., and A.S.d.S.; validation, A.S.d.S.; formal analysis, V.M.d.F.A., R.F.d.S., C.R.G., and A.S.d.S.; investigation, V.M.d.F.A., C.R.G., E.P.S, D.G.S.M, and A.S.d.S.; resources, R.F.d.S. and C.R.G.; data curation, V.M.d.F.A., E.P.S, and A.S.d.S.; writing—original draft, V.M.d.F.A., C.R.G., and A.S.d.S.; writing—review and editing, V.M.d.F.A., R.F.d.S., C.R.G., E.P.S, D.G.S.M, and A.S.d.S.; visualization, V.M.d.F.A, E.P.S.; supervision, C.R.G. and A.S.d.S.; project administration, C.R.G. and A.S.d.S.; funding acquisition, R.F.d.S and C.R.G. All authors have read and agreed to the published version of the manuscript.
Acknowledgment
The authors acknowledge the National Council for Scientific and Technological Development (CNPq), the Coordination for the Improvement of Higher Education Personnel (CAPES, grant 88887.374931/2019-00, Coordenação de Aperfeiçoamento de Pessoal de Nível Superior—Finance Code 01), and the São Paulo Research Foundation (FAPESP, grants 2019/00195-2, 2020/04680-0, 2016/09047-8, 2022/08730-7), Rede Virus MCTI (grant FINEP 0459/20), Brazil, for financial support. The authors also acknowledge Geoambiente Sensoriamento Remoto LTDA for their generous support and for providing access to the Google Cloud services supported by grant FAPESP 2021/00070-5.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kar, S.; Ganguly, M.; Sen, S. Lifting Scheme-Based Wavelet Transform Method for Improved Genomic Classification and Sequence Analysis of Coronavirus. Innovation and Emerging Technologies 2023. [Google Scholar] [CrossRef]
- Corman, J.F.D.; M. , V.; Drosten, C. Ecology, Evolution and Classification of Bat Coronaviruses in the Aftermath of SARS. Antiviral Res. 2014, 101, 45–56. [Google Scholar]
- Hassan, S.A.; Sheikh, F.N.; Jamal, S.; Ezeh, J.K.; Akhtar, A. Coronavirus (COVID-19): A Review of Clinical Features, Diagnosis, and Treatment. Cureus 2020, 12, e7355. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Wei, L.; Niu, P. The Novel Coronavirus Outbreak in Wuhan, China. Global Health Research and Policy 2020, 5, 1–3. [Google Scholar] [CrossRef] [PubMed]
- de Souza, A.S.; de Freitas Amorim, V.M.; Guardia, G.D.A.; Dos Santos, F.F.; Ulrich, H.; Galante, P.A.F.; de Souza, R.F.; Guzzo, C.R. Severe Acute Respiratory Syndrome Coronavirus 2 Variants of Concern: A Perspective for Emerging More Transmissible and Vaccine-Resistant Strains. Viruses 2022, 14. [Google Scholar] [CrossRef] [PubMed]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-Time Tracking of Pathogen Evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- de Souza, A.S.; Amorim, V.M. de F.; de Souza, R.F.; Guzzo, C.R. Molecular Dynamics Simulations of the Spike Trimeric Ectodomain of the SARS-CoV-2 Omicron Variant: Structural Relationships with Infectivity, Evasion to Immune System and Transmissibility. J. Biomol. Struct. Dyn. 2022, 1–18.
- de Souza, A.S.; de Souza, R.F.; Guzzo, C.R. Cooperative and Structural Relationships of the Trimeric Spike with Infectivity and Antibody Escape of the Strains Delta (B.1.617.2) and Omicron (BA.2, BA.5, and BQ.1). J. Comput. Aided Mol. Des. 2023, 37, 585–606. [Google Scholar] [CrossRef] [PubMed]
- Silva de Souza, A.; Rivera, J.D.; Almeida, V.M.; Ge, P.; de Souza, R.F.; Farah, C.S.; Ulrich, H.; Marana, S.R.; Salinas, R.K.; Guzzo, C.R. Molecular Dynamics Reveals Complex Compensatory Effects of Ionic Strength on the Severe Acute Respiratory Syndrome Coronavirus 2 Spike/Human Angiotensin-Converting Enzyme 2 Interaction. J. Phys. Chem. Lett. 2020, 11, 10446–10453. [Google Scholar] [CrossRef]
- V’kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus Biology and Replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2020, 19, 155–170. [Google Scholar] [CrossRef]
- Perlman, S.; Netland, J. Coronaviruses Post-SARS: Update on Replication and Pathogenesis. Nat. Rev. Microbiol. 2009, 7, 439–450. [Google Scholar] [CrossRef]
- Finkel, Y.; Mizrahi, O.; Nachshon, A.; Weingarten-Gabbay, S.; Morgenstern, D.; Yahalom-Ronen, Y.; Tamir, H.; Achdout, H.; Stein, D.; Israeli, O.; et al. The Coding Capacity of SARS-CoV-2. Nature 2020, 589, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Nyodu, R.; Maurya, V.K.; Saxena, S.K. Morphology, Genome Organization, Replication, and Pathogenesis of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2). Coronavirus Disease 2019 (COVID-19), 2020; 23–31. [Google Scholar]
- Cannalire, R.; Cerchia, C.; Beccari, A.R.; Di Leva, F.S.; Summa, V. Targeting SARS-CoV-2 Proteases and Polymerase for COVID-19 Treatment: State of the Art and Future Opportunities. J. Med. Chem. 2020. [Google Scholar] [CrossRef]
- Romano, M.; Ruggiero, A.; Squeglia, F.; Maga, G.; Berisio, R. A Structural View of SARS-CoV-2 RNA Replication Machinery: RNA Synthesis, Proofreading and Final Capping. Cells 2020, 9, 1267. [Google Scholar] [CrossRef]
- Zhang, Y.; Tang, L.V. Overview of Targets and Potential Drugs of SARS-CoV-2 According to the Viral Replication. J. Proteome Res. 2020. [Google Scholar] [CrossRef]
- Kuzikov, M.; Costanzi, E.; Reinshagen, J.; Esposito, F.; Vangeel, L.; Wolf, M.; Ellinger, B.; Claussen, C.; Geisslinger, G.; Corona, A.; et al. Identification of Inhibitors of SARS-CoV-2 3CL-Pro Enzymatic Activity Using a Small Molecule in Vitro Repurposing Screen. ACS Pharmacol Transl Sci 2021, 4, 1096–1110. [Google Scholar] [CrossRef]
- Ullrich, S.; Nitsche, C. The SARS-CoV-2 Main Protease as Drug Target. Bioorg. Med. Chem. Lett. 2020, 30, 127377. [Google Scholar] [CrossRef] [PubMed]
- Zhai, T.; Zhang, F.; Haider, S.; Kraut, D.; Huang, Z. An Integrated Computational and Experimental Approach to Identifying Inhibitors for SARS-CoV-2 3CL Protease. Front Mol Biosci 2021, 8, 661424. [Google Scholar] [CrossRef] [PubMed]
- Glaab, E.; Manoharan, G.B.; Abankwa, D. Pharmacophore Model for SARS-CoV-2 3CLpro Small-Molecule Inhibitors and in Vitro Experimental Validation of Computationally Screened Inhibitors. J. Chem. Inf. Model. 2021. [Google Scholar] [CrossRef]
- Kuang, Y.; Ma, X.; Shen, W.; Rao, Q.; Yang, S. Discovery of 3CLpro Inhibitor of SARS-CoV-2 Main Protease. Future Sci OA 2023, 9, FSO853. [Google Scholar] [CrossRef]
- de Souza, A.S.; de Souza, R.F.; Guzzo, C.R. Quantitative Structure-Activity Relationships, Molecular Docking and Molecular Dynamics Simulations Reveal Drug Repurposing Candidates as Potent SARS-CoV-2 Main Protease Inhibitors. J. Biomol. Struct. Dyn. 2021, 1–18. [Google Scholar] [CrossRef]
- Schake, P.; Dishnica, K.; Kaiser, F.; Leberecht, C.; Haupt, V.J.; Schroeder, M. An Interaction-Based Drug Discovery Screen Explains Known SARS-CoV-2 Inhibitors and Predicts New Compound Scaffolds. Sci. Rep. 2023, 13, 9204. [Google Scholar] [CrossRef] [PubMed]
- Mody, V.; Ho, J.; Wills, S.; Mawri, A.; Lawson, L.; Ebert, M.C.C.J.C.; Fortin, G.M.; Rayalam, S.; Taval, S. Identification of 3-Chymotrypsin like Protease (3CLPro) Inhibitors as Potential Anti-SARS-CoV-2 Agents. Commun Biol 2021, 4, 93. [Google Scholar] [CrossRef] [PubMed]
- Luttens, A.; Gullberg, H.; Abdurakhmanov, E.; Vo, D.D.; Akaberi, D.; Talibov, V.O.; Nekhotiaeva, N.; Vangeel, L.; De Jonghe, S.; Jochmans, D.; et al. Ultralarge Virtual Screening Identifies SARS-CoV-2 Main Protease Inhibitors with Broad-Spectrum Activity against Coronaviruses. J. Am. Chem. Soc. 2022, 144, 2905–2920. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.C.; Freitas, H.F.; Campos, J.M.; Kimani, N.M.; Silva, C.H.T.P.; Borges, R.S.; Pita, S.S.R.; Santos, C.B.R. Natural Products-Based Drug Design against SARS-CoV-2 Mpro 3CLpro. Int. J. Mol. Sci. 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Pokharkar, O.; Zyryanov, G.V.; Tsurkan, M.V. Natural Products from Marine Actinomycete Genus Salinispora Might Inhibit 3CLpro and PLpro Proteins of SARS-CoV-2: An in Silico Evidence. Microbiol. Res. 2023, 14, 1907–1941. [Google Scholar] [CrossRef]
- Singh, R.; Gautam, A.; Chandel, S.; Ghosh, A.; Dey, D.; Roy, S.; Ravichandiran, V.; Ghosh, D. Protease Inhibitory Effect of Natural Polyphenolic Compounds on SARS-CoV-2: An In Silico Study. Molecules 2020, 25, 4604. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Zhang, B.; Jiang, X.-M.; Su, H.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.; Peng, J.; Liu, F.; et al. Structure-Based Design of Antiviral Drug Candidates Targeting the SARS-CoV-2 Main Protease. Science 2020. [Google Scholar] [CrossRef] [PubMed]
- Holm, L. Dali Server: Structural Unification of Protein Families. Nucleic Acids Res. 2022, 50, W210–W215. [Google Scholar] [CrossRef] [PubMed]
- Holm, L.; Laiho, A.; Törönen, P.; Salgado, M. DALI Shines a Light on Remote Homologs: One Hundred Discoveries. Protein Sci. 2023, 32, e4519. [Google Scholar] [CrossRef]
- Hou, N.; Shuai, L.; Zhang, L.; Xie, X.; Tang, K.; Zhu, Y.; Yu, Y.; Zhang, W.; Tan, Q.; Zhong, G.; et al. Development of Highly Potent Noncovalent Inhibitors of SARS-CoV-2 3CLpro. ACS Cent Sci 2023, 9, 217–227. [Google Scholar] [CrossRef]
- Yang, H.; Xie, W.; Xue, X.; Yang, K.; Ma, J.; Liang, W.; Zhao, Q.; Zhou, Z.; Pei, D.; Ziebuhr, J.; et al. Design of Wide-Spectrum Inhibitors Targeting Coronavirus Main Proteases. PLoS Biol. 2005, 3, e324. [Google Scholar]
- Suárez, D.; Díaz, N. SARS-CoV-2 Main Protease: A Molecular Dynamics Study. J. Chem. Inf. Model. 2020. [Google Scholar] [CrossRef]
- La Monica, G.; Bono, A.; Lauria, A.; Martorana, A. Targeting SARS-CoV-2 Main Protease for Treatment of COVID-19: Covalent Inhibitors Structure–Activity Relationship Insights and Evolution Perspectives. J. Med. Chem. 2022. [Google Scholar] [CrossRef]
- de Freitas Amorim, V.M.; de Souza, R.F.; Guzzo, C.R.; de Souza, A.S. Molecular Dynamics Simulations Suggest SARS-CoV-2 3CLpro Mutations in Beta and Omicron Variants Do Not Alter Binding Affinities for Cleavage Sites of Non-Structural Proteins. COVID 2023, 3, 622–636. [Google Scholar] [CrossRef]
- Cheng, S.-C.; Chang, G.-G.; Chou, C.-Y. Mutation of Glu-166 Blocks the Substrate-Induced Dimerization of SARS Coronavirus Main Protease. Biophys. J. 2010, 98, 1327–1336. [Google Scholar] [CrossRef]
- Zhong, N.; Zhang, S.; Zou, P.; Chen, J.; Kang, X.; Li, Z.; Liang, C.; Jin, C.; Xia, B. Without Its N-Finger, the Main Protease of Severe Acute Respiratory Syndrome Coronavirus Can Form a Novel Dimer through Its C-Terminal Domain. J. Virol. 2008, 82, 4227–4234. [Google Scholar] [CrossRef]
- Chang, H.-P.; Chou, C.-Y.; Chang, G.-G. Reversible Unfolding of the Severe Acute Respiratory Syndrome Coronavirus Main Protease in Guanidinium Chloride. Biophys. J. 2007, 92, 1374–1383. [Google Scholar] [CrossRef]
- Świderek, K.; Moliner, V. Revealing the Molecular Mechanisms of Proteolysis of SARS-CoV-2 Mpro by QM/MM Computational Methods. Chem. Sci. 2020, 11, 10626–10630. [Google Scholar] [CrossRef]
- Duan, Y.; Wang, H.; Yuan, Z.; Yang, H. Structural Biology of SARS-CoV-2 Mpro and Drug Discovery. Curr. Opin. Struct. Biol. 2023, 82, 102667. [Google Scholar] [CrossRef]
- Tasci, H.S.; Akkus, E.; Yildiz, M.; Kocak, A. Computational Analysis of Substrate Recognition of Sars-Cov-2 Mpro Main Protease. Comput. Biol. Chem. 2023, 107, 107960. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, B.; Ma, S.; Wang, H.; Shang, L.; Zhu, C.; Ye, S. Discovery of SARS-CoV-2 3CLPro Peptidomimetic Inhibitors through the Catalytic Dyad Histidine-Specific Protein–Ligand Interactions. Int. J. Mol. Sci. 2022, 23, 2392. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal Structure of SARS-CoV-2 Main Protease Provides a Basis for Design of Improved α-Ketoamide Inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef]
- Tumskiy, R.S.; Tumskaia, A.V.; Klochkova, I.N.; Richardson, R.J. SARS-CoV-2 Proteases Mpro and PLpro: Design of Inhibitors with Predicted High Potency and Low Mammalian Toxicity Using Artificial Neural Networks, Ligand-Protein Docking, Molecular Dynamics Simulations, and ADMET Calculations. Comput. Biol. Med. 2023, 153, 106449. [Google Scholar] [CrossRef]
- Miczi, M.; Golda, M.; Kunkli, B.; Nagy, T.; Tőzsér, J.; Mótyán, J.A. Identification of Host Cellular Protein Substrates of SARS-COV-2 Main Protease. Int. J. Mol. Sci. 2020, 21, 9523. [Google Scholar] [CrossRef]
- Rut, W.; Groborz, K.; Zhang, L.; Sun, X.; Zmudzinski, M.; Pawlik, B.; Młynarski, W.; Hilgenfeld, R.; Drag, M. Substrate Specificity Profiling of SARS-CoV-2 Main Protease Enables Design of Activity-Based Probes for Patient-Sample Imaging. bioRxiv, 9819; 2020.03.07.981928. [Google Scholar]
- Henry Chan, H.T.; Moesser, M.A.; Walters, R.K.; Malla, T.R.; Twidale, R.M.; John, T.; Deeks, H.M.; Johnston-Wood, T.; Mikhailov, V.; Sessions, R.B.; et al. Discovery of SARS-CoV-2 Mpro Peptide Inhibitors from Modelling Substrate and Ligand Binding. Chem. Sci. 2021, 12, 13686–13703. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhu, Y.; Liu, X.; Jin, Z.; Duan, Y.; Zhang, Q.; Wu, C.; Feng, L.; Du, X.; Zhao, J.; et al. Structural Basis for Replicase Polyprotein Cleavage and Substrate Specificity of Main Protease from SARS-CoV-2. Proc. Natl. Acad. Sci. U. S. A. 2022, 119, e2117142119. [Google Scholar] [CrossRef]
- Paasche, A.; Zipper, A.; Schäfer, S.; Ziebuhr, J.; Schirmeister, T.; Engels, B. Evidence for Substrate Binding-Induced Zwitterion Formation in the Catalytic Cys-His Dyad of the SARS-CoV Main Protease. 2014. [Google Scholar] [CrossRef]
- Ramos-Guzmán, C.A.; Javier Ruiz-Pernía, J.; Tuñón, I. Unraveling the SARS-CoV-2 Main Protease Mechanism Using Multiscale Methods. ACS Catal. 2020. [Google Scholar] [CrossRef]
- Świderek, K.; Moliner, V. Revealing the Molecular Mechanisms of Proteolysis of SARS-CoV-2 Mpro by QM/MM Computational Methods. Chem. Sci. 2020, 11, 10626–10630. [Google Scholar] [CrossRef]
- Hudáky, P.; Perczel, A. A Self-Stabilized Model of the Chymotrypsin Catalytic Pocket. The Energy Profile of the Overall Catalytic Cycle. Proteins: Struct. Funct. Bioinf. 2006, 62, 749–759. [Google Scholar] [CrossRef]
- Ngo, S.T.; Nguyen, T.H.; Tung, N.T.; Mai, B.K. Insights into the Binding and Covalent Inhibition Mechanism of PF-07321332 to SARS-CoV-2 Mpro. RSC Adv. 2022, 12, 3729–3737. [Google Scholar] [CrossRef]
- Hauser, M.; Steinegger, M.; Söding, J. MMseqs Software Suite for Fast and Deep Clustering and Searching of Large Protein Sequence Sets. Bioinformatics 2016, 32, 1323–1330. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A New Generation of Protein Database Search Programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- HMMER HMMER Available online:. Available online: http://hmmer.org/ (accessed on 27 April 2024).
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The Protein Families Database in 2021. Nucleic Acids Res. 2020, 49, D412–D419. [Google Scholar] [CrossRef]
- Deorowicz, S.; Debudaj-Grabysz, A.; Gudyś, A. FAMSA: Fast and Accurate Multiple Sequence Alignment of Huge Protein Families. Sci. Rep. 2016, 6, 1–13. [Google Scholar]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Schneider, T.D.; Stephens, R.M. Sequence Logos: A New Way to Display Consensus Sequences. Nucleic Acids Res. 1990, 18, 6097–6100. [Google Scholar] [CrossRef]
- Crooks, G.E.; Hon, G.; Chandonia, J.-M.; Brenner, S.E. WebLogo: A Sequence Logo Generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef]
- Al Adem, K.; Ferreira, J.C.; Fadl, S.; Mustafa, M.; Rabeh, W.M. Key Allosteric and Active Site Residues of SARS-CoV-2 3CLpro Are Promising Drug Targets. Biochem. J 2023. [Google Scholar] [CrossRef]
- Chen, Y.; Wei, W.; Zhou, Y.; Xie, D. The Role of Hydrogen Bond in Catalytic Triad of Serine Proteases †. Chin. J. Chem. Phys. 2021, 34, 797–804. [Google Scholar] [CrossRef]
- Vinces, T.C.; de Souza, A.S.; Carvalho, C.F.; Visnardi, A.B.; Teixeira, R.D.; Llontop, E.E.; Bismara, B.A.P.; Vicente, E.J.; Pereira, J.O.; de Souza, R.F.; et al. Monomeric Esterase: Insights into Cooperative Behavior, Hysteresis/Allokairy. Biochemistry 2024. [Google Scholar] [CrossRef]
- Brinen, L.S.; Hansell, E.; Cheng, J.; Roush, W.R.; McKerrow, J.H.; Fletterick, R.J. A Target within the Target: Probing Cruzain’s P1' Site to Define Structural Determinants for the Chagas' Disease Protease. Structure 2000, 8, 831–840. [Google Scholar] [CrossRef]
- de Souza, A.S.; de Oliveira, M.T.; Andricopulo, A.D. Development of a Pharmacophore for Cruzain Using Oxadiazoles as Virtual Molecular Probes: Quantitative Structure-Activity Relationship Studies. J. Comput. Aided Mol. Des. 2017, 31, 801–816. [Google Scholar] [CrossRef]
- Blankenship, E.; Vukoti, K.; Miyagi, M.; Lodowski, D.T. Conformational Flexibility in the Catalytic Triad Revealed by the High-Resolution Crystal Structure of Streptomyces Erythraeus Trypsin in an Unliganded State. Acta Crystallogr. D Biol. Crystallogr. 2014, 70, 833–840. [Google Scholar] [CrossRef] [PubMed]
- Aljuhani, A.; Ahmed, H.E.A.; Ihmaid, S.K.; Omar, A.M.; Althagfan, S.S.; Alahmadi, Y.M.; Ahmad, I.; Patel, H.; Ahmed, S.; Almikhlafi, M.A.; et al. In Vitro and Computational Investigations of Novel Synthetic Carboxamide-Linked Pyridopyrrolopyrimidines with Potent Activity as SARS-CoV-2-MPro Inhibitors. RSC Adv. 2022, 12, 26895–26907. [Google Scholar] [CrossRef]
- da Silva, F.M.A.; da Silva, K.P.A.; de Oliveira, L.P.M.; Costa, E.V.; Koolen, H.H.F.; Pinheiro, M.L.B.; de Souza, A.Q.L.; de Souza, A.D.L. Flavonoid Glycosides and Their Putative Human Metabolites as Potential Inhibitors of the SARS-CoV-2 Main Protease (Mpro) and RNA-Dependent RNA Polymerase (RdRp). Mem. Inst. Oswaldo Cruz 2020, 115, e200207. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Lin, X.; Xing, N.; Zhang, Z.; Zhang, H.; Wu, H.; Xue, W. Structure-Based Discovery of Novel Nonpeptide Inhibitors Targeting SARS-CoV-2 Mpro. J. Chem. Inf. Model. 2021. [Google Scholar] [CrossRef] [PubMed]
- Fischer, C.; Feys, J.R. SARS-CoV-2 Mpro Inhibitors: Achieved Diversity, Developing Resistance and Future Strategies. Future Pharmacology 2023, 3, 80–107. [Google Scholar] [CrossRef]
- Pandey, A.; Nikam, A.N.; Shreya, A.B.; Mutalik, S.P.; Gopalan, D.; Kulkarni, S.; Padya, B.S.; Fernandes, G.; Mutalik, S.; Prassl, R. Potential Therapeutic Targets for Combating SARS-CoV-2: Drug Repurposing, Clinical Trials and Recent Advancements. Life Sci. 2020, 256, 117883. [Google Scholar] [CrossRef]
- Cao, B.; Wang, Y.; Wen, D.; Liu, W.; Wang, J.; Fan, G.; Ruan, L.; Song, B.; Cai, Y.; Wei, M.; et al. A Trial of Lopinavir-Ritonavir in Adults Hospitalized with Severe Covid-19. N. Engl. J. Med. 2020, 382, 1787–1799. [Google Scholar] [CrossRef]
- Liu, C.; Boland, S.; Scholle, M.D.; Bardiot, D.; Marchand, A.; Chaltin, P.; Blatt, L.M.; Beigelman, L.; Symons, J.A.; Raboisson, P.; et al. Dual Inhibition of SARS-CoV-2 and Human Rhinovirus with Protease Inhibitors in Clinical Development. Antiviral Res. 2021, 187, 105020. [Google Scholar] [CrossRef]
- Pharma, C. Cocrystal Pharma Selects Lead Compound for Further Development Against Coronaviruses . Available online: https://www.cocrystalpharma.com/news/press-releases/detail/105/cocrystal-pharma-selects-lead-compound-for-further (accessed on 28 April 2024).
- Nakayama, T.; Lee, I.T.; Jiang, S.; Matter, M.S.; Yan, C.H.; Overdevest, J.B.; Wu, C.T.; Goltsev, Y.; Shih, L.C.; Liao, C.K.; et al. Determinants of SARS-CoV-2 Entry and Replication in Airway Mucosal Tissue and Susceptibility in Smokers. Cell Reports Medicine 2021, 2, 100421. [Google Scholar] [CrossRef] [PubMed]
- Kalligeros, M.; Tashima, K.T.; Mylona, E.K.; Rybak, N.; Flanigan, T.P.; Farmakiotis, D.; Beckwith, C.G.; Sanchez, M.; Neill, M.; Johnson, J.E.; et al. Remdesivir Use Compared With Supportive Care in Hospitalized Patients With Severe COVID-19: A Single-Center Experience. Open Forum Infect Dis 2020, 7, ofaa319. [Google Scholar] [CrossRef]
- Vandyck, K.D.J. Considerations for the Discovery and Development of 3-Chymotrypsin-like Cysteine Protease Inhibitors Targeting SARS-CoV-2 Infection. Curr. Opin. Virol. 2021, 49, 36–40. [Google Scholar] [CrossRef]
- Boras, B.; Jones, R.M.; Anson, B.J.; Arenson, D.; Aschenbrenner, L.; Bakowski, M.A.; Beutler, N.; Binder, J.; Chen, E.; Eng, H.; et al. Discovery of a Novel Inhibitor of Coronavirus 3CL Protease for the Potential Treatment of COVID-19. bioRxiv 2021. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Pajouhesh, H.; Lenz, G.R. Medicinal Chemical Properties of Successful Central Nervous System Drugs. NeuroRx 2005, 2, 541–553. [Google Scholar] [CrossRef]
- Clinical Trials ID NCT04535167 History of Changes for Study: NCT04535167. Available online: https://classic.clinicaltrials.gov/ct2/history/NCT04535167?V_9=View (accessed on 28 April 2024).
- Owen, D.R.; Allerton, C.M.N.; Anderson, A.S.; Aschenbrenner, L.; Avery, M.; Berritt, S.; Boras, B.; Cardin, R.D.; Carlo, A.; Coffman, K.J.; et al. An Oral SARS-CoV-2 Mpro Inhibitor Clinical Candidate for the Treatment of COVID-19. Science 2021. [Google Scholar] [CrossRef]
- Abdelnabi, R.; Foo, C.S.; Jochmans, D.; Vangeel, L.; De Jonghe, S.; Augustijns, P.; Mols, R.; Weynand, B.; Wattanakul, T.; Hoglund, R.M.; et al. The Oral Protease Inhibitor (PF-07321332) Protects Syrian Hamsters against Infection with SARS-CoV-2 Variants of Concern. Nat. Commun. 2022, 13, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Lin, C.; Zhou, X.; Zhong, F.; Zeng, P.; Yang, Y.; Zhang, Y.; Yu, B.; Fan, X.; McCormick, P.J.; et al. Structural Basis of the Main Proteases of Coronavirus Bound to Drug Candidate PF-07321332. J. Virol. 2022. [Google Scholar] [CrossRef]
- Noske, G.D.; de Souza Silva, E.; de Godoy, M.O.; Dolci, I.; Fernandes, R.S.; Guido, R.V.C.; Sjö, P.; Oliva, G.; As. , G. Structural Basis of Nirmatrelvir and Ensitrelvir Activity against Naturally Occurring Polymorphisms of the SARS-CoV-2 Main Protease. J. Biol. Chem. 2023, 299, 103004. [Google Scholar] [CrossRef]
- Reina, J.; Iglesias, C. [Nirmatrelvir plus ritonavir (Paxlovid) a potent SARS-CoV-2 3CLpro protease inhibitor combination]. Rev. Esp. Quimioter. 2022, 35, 236–240. [Google Scholar] [CrossRef]
- Perelson, A.S.; Ribeiro, R.M.; Phan, T. An Explanation for SARS-CoV-2 Rebound after Paxlovid Treatment. medRxiv, 2023; 2023.05.30.23290747. [Google Scholar]
- Khalilieh, S.; Feng, H.-P.; Hulskotte, E.G.J.; Wenning, L.A.; Butterton, J.R. Clinical Pharmacology Profile of Boceprevir, a Hepatitis C Virus NS3 Protease Inhibitor: Focus on Drug-Drug Interactions. Clin. Pharmacokinet. 2015, 54, 599–614. [Google Scholar] [CrossRef]
- Nutho, B.; Mahalapbutr, P.; Hengphasatporn, K.; Pattaranggoon, N.C.; Simanon, N.; Shigeta, Y.; Hannongbua, S.; Rungrotmongkol, T. Why Are Lopinavir and Ritonavir Effective against the Newly Emerged Coronavirus 2019? Atomistic Insights into the Inhibitory Mechanisms. Biochemistry 2020. [Google Scholar] [CrossRef] [PubMed]
- Cvetkovic, R.S.; Goa, K.L. Lopinavir/Ritonavir. Drugs 2012, 63, 769–802. [Google Scholar] [CrossRef] [PubMed]
- Schoergenhofer, C.; Jilma, B.; Stimpfl, T.; Karolyi, M.; Zoufaly, A. Pharmacokinetics of Lopinavir and Ritonavir in Patients Hospitalized With Coronavirus Disease 2019 (COVID-19). Ann. Intern. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Eng, H.; Dantonio, A.L.; Kadar, E.P.; Obach, R.S.; Di, L.; Lin, J.; Patel, N.C.; Boras, B.; Walker, G.S.; Novak, J.J.; et al. Disposition of Nirmatrelvir, an Orally Bioavailable Inhibitor of SARS-CoV-2 3C-Like Protease, across Animals and Humans. Drug Metab. Dispos. 2022, 50, 576–590. [Google Scholar] [CrossRef] [PubMed]
- Denissen, J.F.; Grabowski, B.A.; Johnson, M.K.; Buko, A.M.; Kempf, D.J.; Thomas, S.B.; Surber, B.W. Metabolism and Disposition of the HIV-1 Protease Inhibitor Ritonavir (ABT-538) in Rats, Dogs, and Humans. Drug Metab. Dispos. 1997, 25, 489–501. [Google Scholar]
- Chen, W.; Liang, B.; Wu, X.; Li, L.; Wang, C.; Xing, D. Advances and Challenges in Using Nirmatrelvir and Its Derivatives against SARS-CoV-2 Infection. J Pharm Anal 2023, 13, 255–261. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Kusov, Y.; Nian, Y.; Ma, Q.; Wang, J.; von Brunn, A.; Leyssen, P.; Lanko, K.; Neyts, J.; et al. α-Ketoamides as Broad-Spectrum Inhibitors of Coronavirus and Enterovirus Replication: Structure-Based Design, Synthesis, and Activity Assessment. J. Med. Chem. 2020, 63, 4562–4578. [Google Scholar] [CrossRef]
- Stein, M.L.; Cui, H.; Beck, P.; Dubiella, C.; Voss, C.; Krüger, A.; Schmidt, B.; Groll, M. Systematic Comparison of Peptidic Proteasome Inhibitors Highlights the α-Ketoamide Electrophile as an Auspicious Reversible Lead Motif. Angew. Chem. Int. Ed Engl. 2014, 53, 1679–1683. [Google Scholar] [CrossRef]
- Ramos-Guzmán, C.A.; Javier Ruiz-Pernía, J.; Tuñón, I. Inhibition Mechanism of SARS-CoV-2 Main Protease with Ketone-Based Inhibitors Unveiled by Multiscale Simulations: Insights for Improved Designs**. Angew. Chem. Int. Ed. 2021, 60, 25933–25941. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Jochmans, D.; Xie, H.; Yang, H.; Li, J.; Su, H.; Chang, D.; Wang, J.; Peng, J.; Zhu, L.; et al. Design, Synthesis, and Biological Evaluation of Peptidomimetic Aldehydes as Broad-Spectrum Inhibitors against Enterovirus and SARS-CoV-2. J. Med. Chem. 2021. [Google Scholar] [CrossRef]
- Ma, Y.; Yang, K.S.; Geng, Z.Z.; Alugubelli, Y.R.; Shaabani, N.; Vatansever, E.C.; Ma, X.R.; Cho, C.C.; Khatua, K.; Xiao, J.; et al. A Multi-Pronged Evaluation of Aldehyde-Based Tripeptidyl Main Protease Inhibitors as SARS-CoV-2 Antivirals. Eur. J. Med. Chem. 2022, 240, 114570. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Chenna, B.C.; Yang, K.S.; Cole, T.R.; Goodall, Z.T.; Giardini, M.; Moghadamchargari, Z.; Hernandez, E.A.; Gomez, J.; Calvet, C.M.; et al. Self-Masked Aldehyde Inhibitors: A Novel Strategy for Inhibiting Cysteine Proteases. J. Med. Chem. 2021, 64, 11267–11287. [Google Scholar] [CrossRef]
- Jiang, X.; Su, H.; Shang, W.; Zhou, F.; Zhang, Y.; Zhao, W.; Zhang, Q.; Xie, H.; Jiang, L.; Nie, T.; et al. Structure-Based Development and Preclinical Evaluation of the SARS-CoV-2 3C-like Protease Inhibitor Simnotrelvir. Nat. Commun. 2023, 14, 6463. [Google Scholar] [CrossRef]
- Kandwal, S.; Fayne, D. Genetic Conservation across SARS-CoV-2 Non-Structural Proteins – Insights into Possible Targets for Treatment of Future Viral Outbreaks. Virology 2023, 581, 97–115. [Google Scholar] [CrossRef]
- Ray, A.K.; Sen Gupta, P.S.; Panda, S.K.; Biswal, S.; Bhattacharya, U.; Rana, M.K. Repurposing of FDA-Approved Drugs as Potential Inhibitors of the SARS-CoV-2 Main Protease: Molecular Insights into Improved Therapeutic Discovery. Comput. Biol. Med. 2022, 142, 105183. [Google Scholar] [CrossRef]
- Hu, Y.; Lewandowski, E.M.; Tan, H.; Zhang, X.; Morgan, R.T.; Zhang, X.; Jacobs, L.M.C.; Butler, S.G.; Gongora, M.V.; Choy, J.; et al. Naturally Occurring Mutations of SARS-CoV-2 Main Protease Confer Drug Resistance to Nirmatrelvir. ACS Central Science 2023. [Google Scholar] [CrossRef]
Figure 1.
3CLpro Structure and Function. a) The three-dimensional structure of 3CLpro colored based on chain (PDB ID 7WOF) [43] b) three distinct domains I, II, and III. Domains I (residues 10-99) and II (residues 100-182) unfold with six antiparallel β-strands, forming a stage for the catalytic site [44,45]. Meanwhile, Domain III (residues 198-303) unveils a captivating structure featuring a globular cluster of five α-helices, steering 3CLpro dimerization regulation through a vital salt bridge. c) The substrate binding site of 3CLpro is a composition of four subsites—S1, S1', S2, and S3/S4— d) following the Schecter-Berger nomenclature for proteases. 3CLpro's active site features S1, S1', S2, and S2 subsites, with Pis and Pi's denoting substrate positions. The S1-S1' interface marks the cleavage site, initiating numbering. X-ray studies confirm four subsites: S1', S1, S2, and S3/S4 [41,42]. The S1 subsite (F140, C145, H163, E166) ensures structural stability and substrate recognition, featuring key π-π interactions. S1' (T25, T26, H41, L27, N142, G143) is exposed to the aqueous environment, accommodating smaller side chains of the substrate. S2 (M49, Y54, P52, H164, M165, D187, R188, Q189) is highly hydrophobic, and S3/S4 (M165, E166, L167, P168, F185, T190, A191) completes the hydrophobic subsite composition [41,42].
Figure 1.
3CLpro Structure and Function. a) The three-dimensional structure of 3CLpro colored based on chain (PDB ID 7WOF) [43] b) three distinct domains I, II, and III. Domains I (residues 10-99) and II (residues 100-182) unfold with six antiparallel β-strands, forming a stage for the catalytic site [44,45]. Meanwhile, Domain III (residues 198-303) unveils a captivating structure featuring a globular cluster of five α-helices, steering 3CLpro dimerization regulation through a vital salt bridge. c) The substrate binding site of 3CLpro is a composition of four subsites—S1, S1', S2, and S3/S4— d) following the Schecter-Berger nomenclature for proteases. 3CLpro's active site features S1, S1', S2, and S2 subsites, with Pis and Pi's denoting substrate positions. The S1-S1' interface marks the cleavage site, initiating numbering. X-ray studies confirm four subsites: S1', S1, S2, and S3/S4 [41,42]. The S1 subsite (F140, C145, H163, E166) ensures structural stability and substrate recognition, featuring key π-π interactions. S1' (T25, T26, H41, L27, N142, G143) is exposed to the aqueous environment, accommodating smaller side chains of the substrate. S2 (M49, Y54, P52, H164, M165, D187, R188, Q189) is highly hydrophobic, and S3/S4 (M165, E166, L167, P168, F185, T190, A191) completes the hydrophobic subsite composition [41,42].
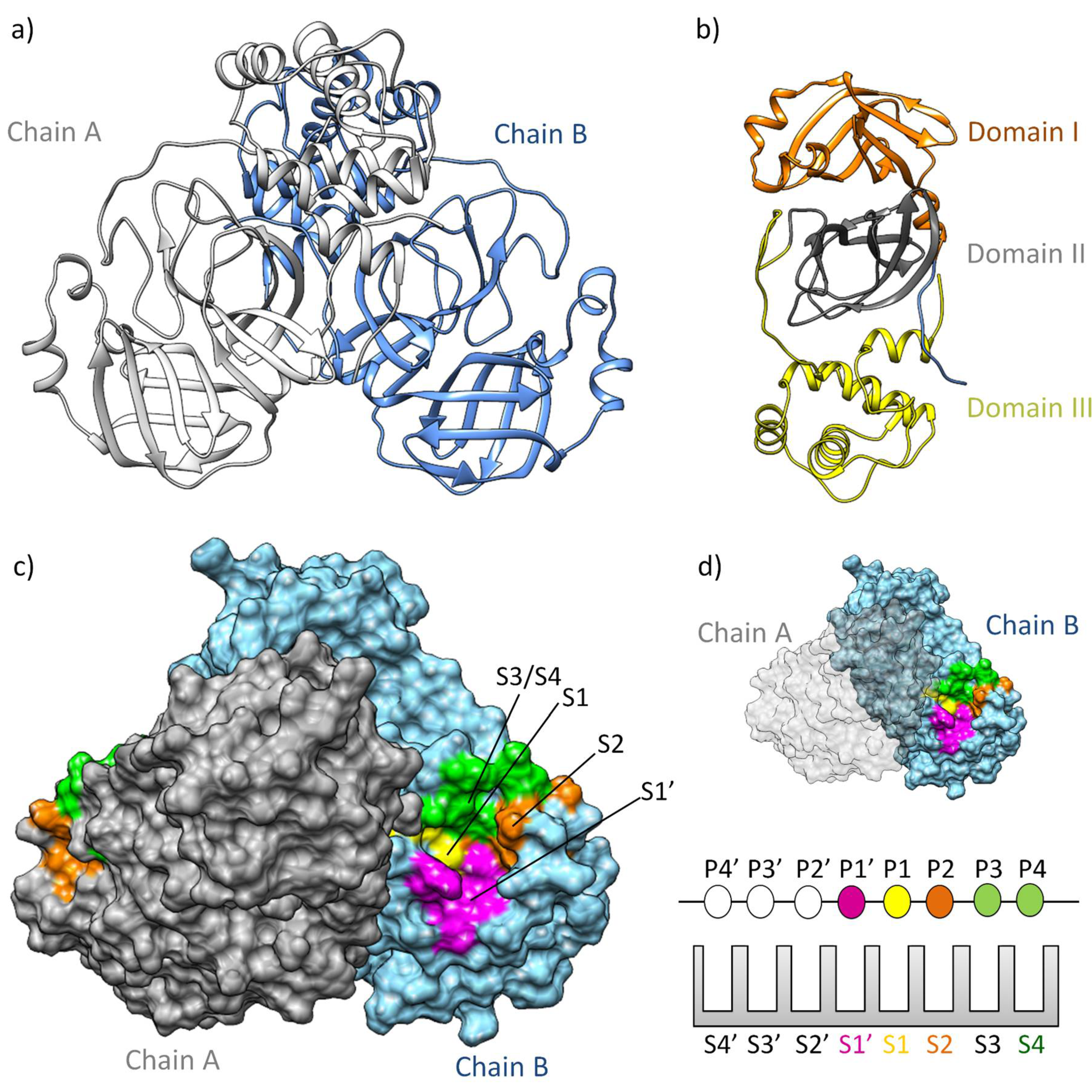
Figure 2.
Amino acid residue conservation profile of SARS-CoV-2 3CLpro and its relationships with the novel mechanism of catalysis. a) WebLogo representation displaying the conserved residues among different species of coronaviruses. b) Domains I, II, and III are denoted as DI, DII, and DIII. c) Between domains I and II lies the active site, where the proposed catalytic triad H41, C145, and D187 is located. The red arrow indicates the position of each conserved amino acid, and indeed, there is a pattern maintained by the catalytic residues. By aligning with the sequences ACA52156.1, ALJ99894.1, AWR88311.1, AXF38647.1, QSE37596.1, WCZ55891.1, YP_008439216.1, YP_009915659.1, YP_009924387.1, 7WOF_A, and 8E7C_A. We detected a significant conservation of amino acid residues, notably H41, C145, and D187 with probability equal to 1 (Figure S2).
Figure 2.
Amino acid residue conservation profile of SARS-CoV-2 3CLpro and its relationships with the novel mechanism of catalysis. a) WebLogo representation displaying the conserved residues among different species of coronaviruses. b) Domains I, II, and III are denoted as DI, DII, and DIII. c) Between domains I and II lies the active site, where the proposed catalytic triad H41, C145, and D187 is located. The red arrow indicates the position of each conserved amino acid, and indeed, there is a pattern maintained by the catalytic residues. By aligning with the sequences ACA52156.1, ALJ99894.1, AWR88311.1, AXF38647.1, QSE37596.1, WCZ55891.1, YP_008439216.1, YP_009915659.1, YP_009924387.1, 7WOF_A, and 8E7C_A. We detected a significant conservation of amino acid residues, notably H41, C145, and D187 with probability equal to 1 (Figure S2).
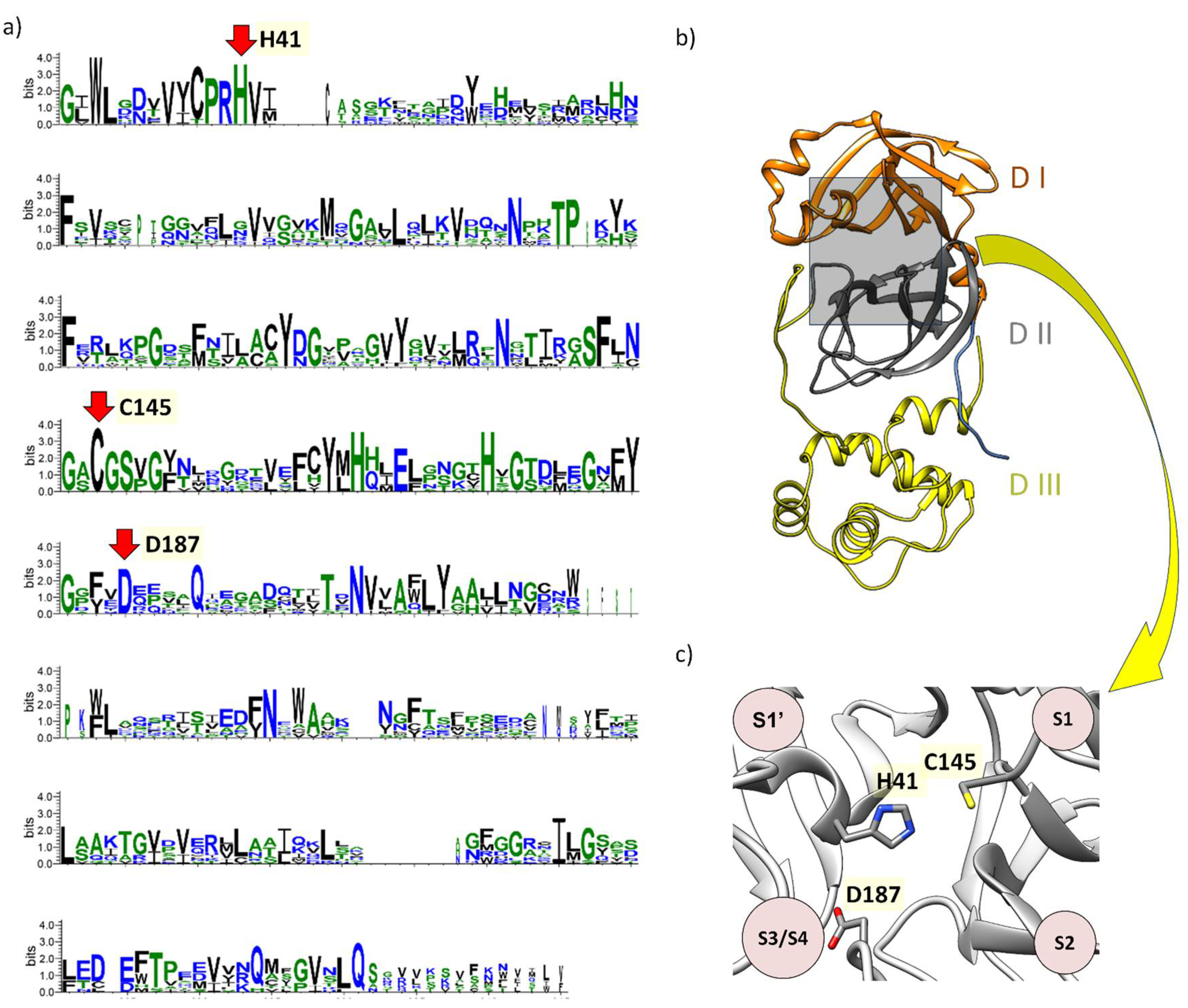
Figure 3.
Proposal for the mechanism of polyprotein processing. The proposed mechanism outlined in this review delineates two distinct phases: acylation (step I) followed by deacylation (step II), wherein the roles of specific residues mirror those of the catalytic triad observed in 3CLpro. The acylation step, the active site of the main protease, Cys145 functions as a nucleophile. Initially, His41 serves as a base, facilitating the donation of a proton to Cys145, thereby activating it as a nucleophile. Next, the nucleophilic Cys145 initiates an attack on the carbon atom of the carbonyl group within the substrate's amino acid residue, forming a tetrahedral intermediate. This enzymatic action leads to the cleavage of the peptide bond, resulting in the release of a substrate fragment with a alcohol group and the formation of an acyl bond between Cys145 and the residual fragment of the substrate. Deacylation step is the subsequent phase, where a water molecule assumes the role of a nucleophile and His41 functions as an acid, facilitating the donation of a proton from the water molecule to the His41, which in turn instigates an attack on the carbon atom of the acyl group bound to Cys145. Consequently, the acyl bond is cleaved, and the active form of the main protease is regenerated. Simultaneously, the second fragment with a carboxylic group of the substrate is released, thereby completing the enzymatic reaction.
Figure 3.
Proposal for the mechanism of polyprotein processing. The proposed mechanism outlined in this review delineates two distinct phases: acylation (step I) followed by deacylation (step II), wherein the roles of specific residues mirror those of the catalytic triad observed in 3CLpro. The acylation step, the active site of the main protease, Cys145 functions as a nucleophile. Initially, His41 serves as a base, facilitating the donation of a proton to Cys145, thereby activating it as a nucleophile. Next, the nucleophilic Cys145 initiates an attack on the carbon atom of the carbonyl group within the substrate's amino acid residue, forming a tetrahedral intermediate. This enzymatic action leads to the cleavage of the peptide bond, resulting in the release of a substrate fragment with a alcohol group and the formation of an acyl bond between Cys145 and the residual fragment of the substrate. Deacylation step is the subsequent phase, where a water molecule assumes the role of a nucleophile and His41 functions as an acid, facilitating the donation of a proton from the water molecule to the His41, which in turn instigates an attack on the carbon atom of the acyl group bound to Cys145. Consequently, the acyl bond is cleaved, and the active form of the main protease is regenerated. Simultaneously, the second fragment with a carboxylic group of the substrate is released, thereby completing the enzymatic reaction.
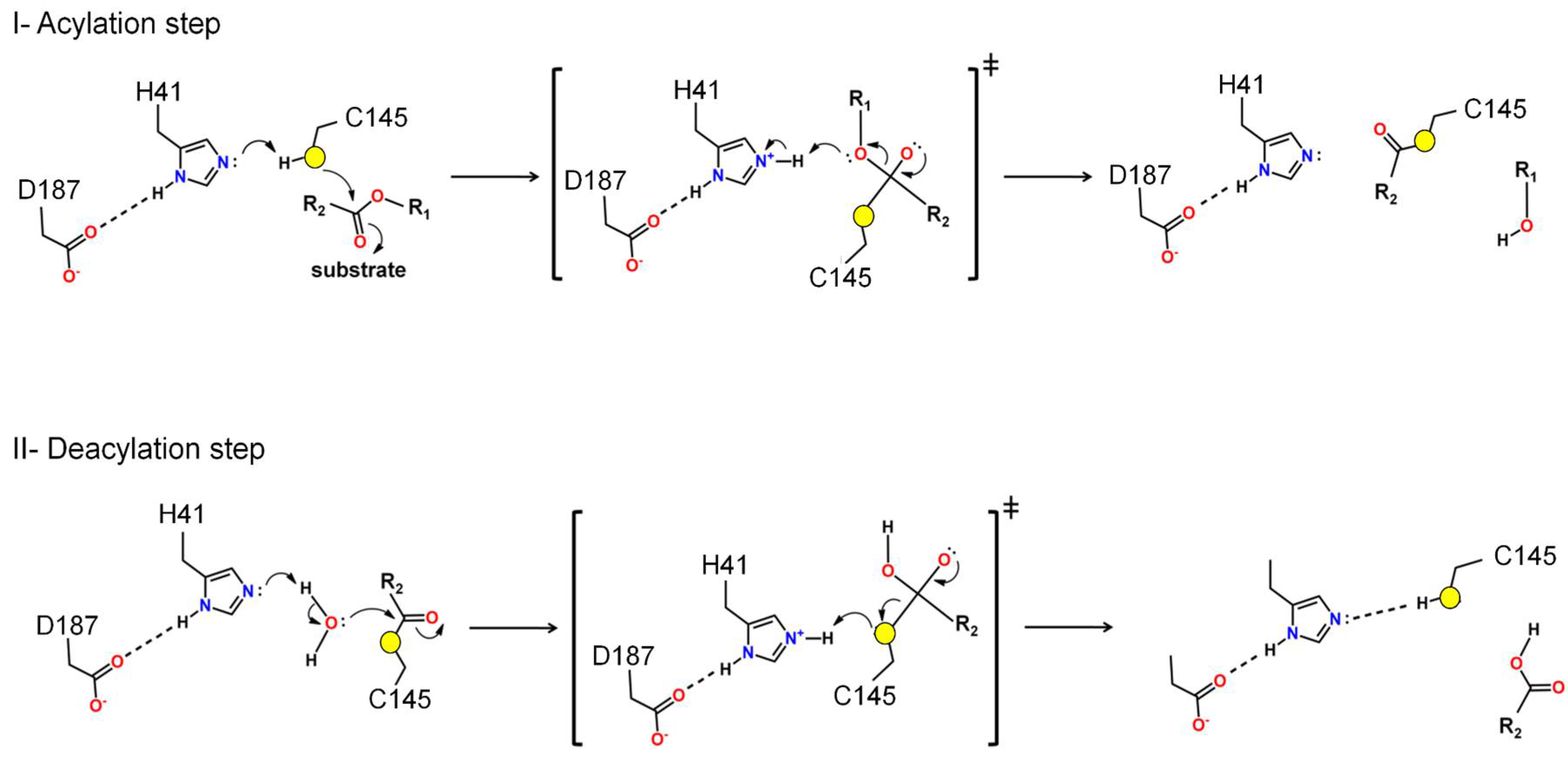
Figure 4.
Potent 3CLpro competitive inhibitors. Biological activities (IC50 and EC50) are shown for remdesivir, GC376, PF-00835231, and PF-07304814.
Figure 4.
Potent 3CLpro competitive inhibitors. Biological activities (IC50 and EC50) are shown for remdesivir, GC376, PF-00835231, and PF-07304814.
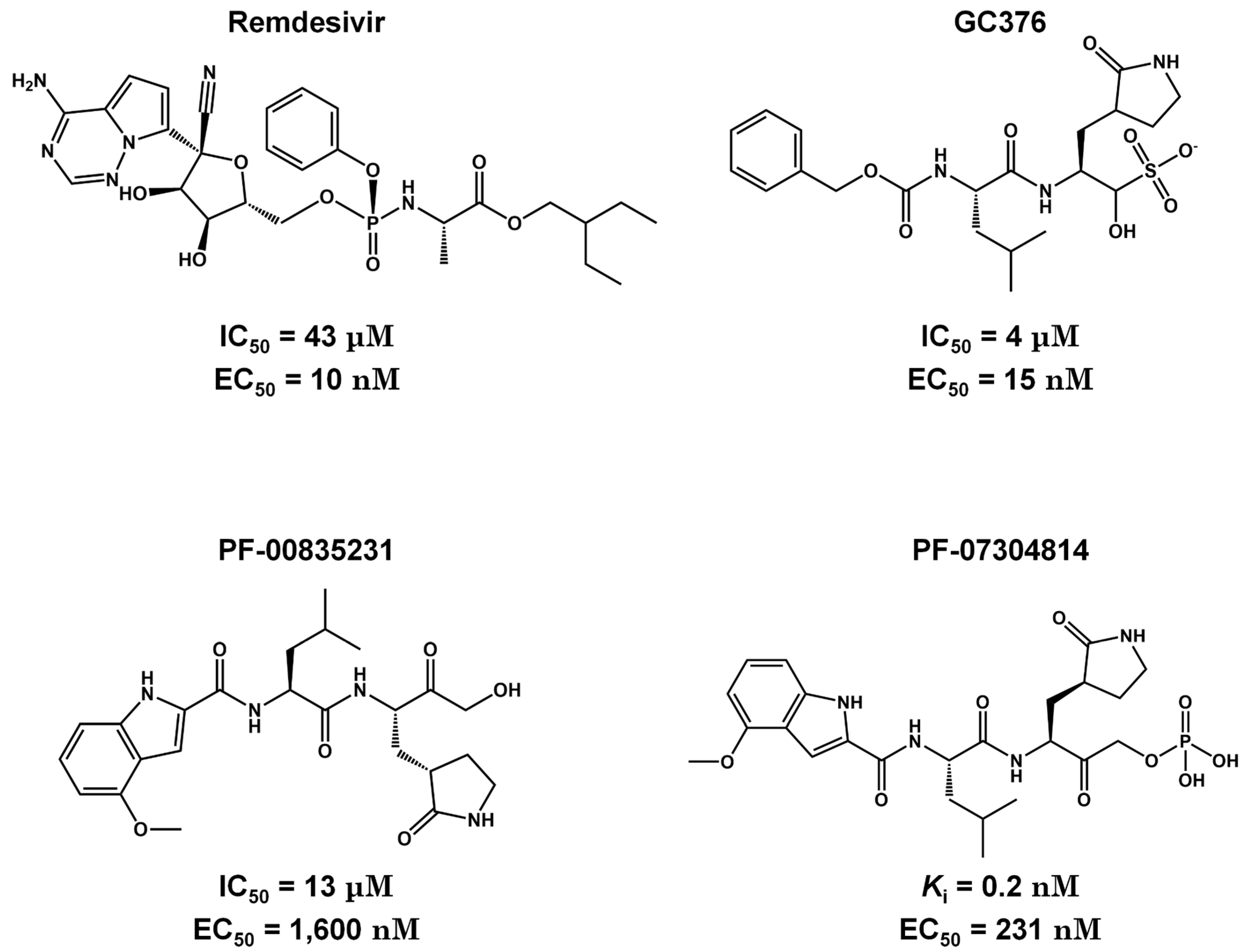
Figure 5.
Pharmacokinetic optimization for PF-00835231. Biological activities (IC50 or Ki and EC50) are shown for PF-00835231 and PF–7304814.
Figure 5.
Pharmacokinetic optimization for PF-00835231. Biological activities (IC50 or Ki and EC50) are shown for PF-00835231 and PF–7304814.
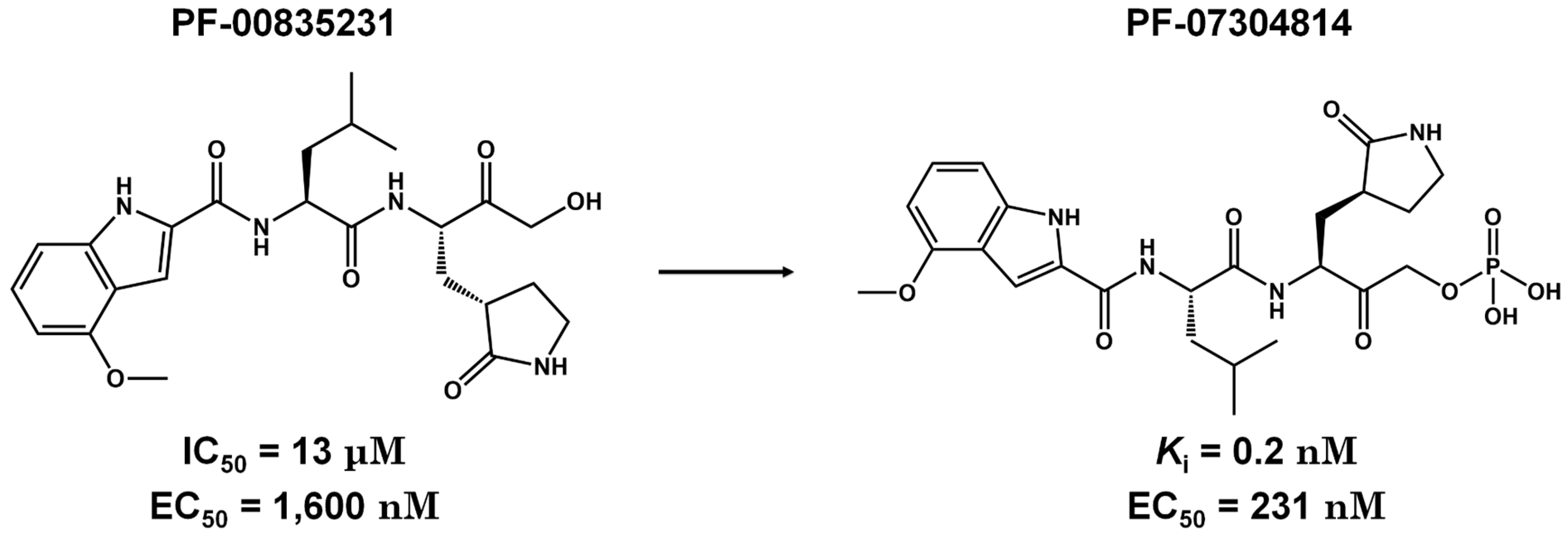
Figure 6.
Potent 3CLpro inhibitors. Biological activities (IC50 or Ki and EC50) are shown for PF-07321332 and Boceprevir.
Figure 6.
Potent 3CLpro inhibitors. Biological activities (IC50 or Ki and EC50) are shown for PF-07321332 and Boceprevir.
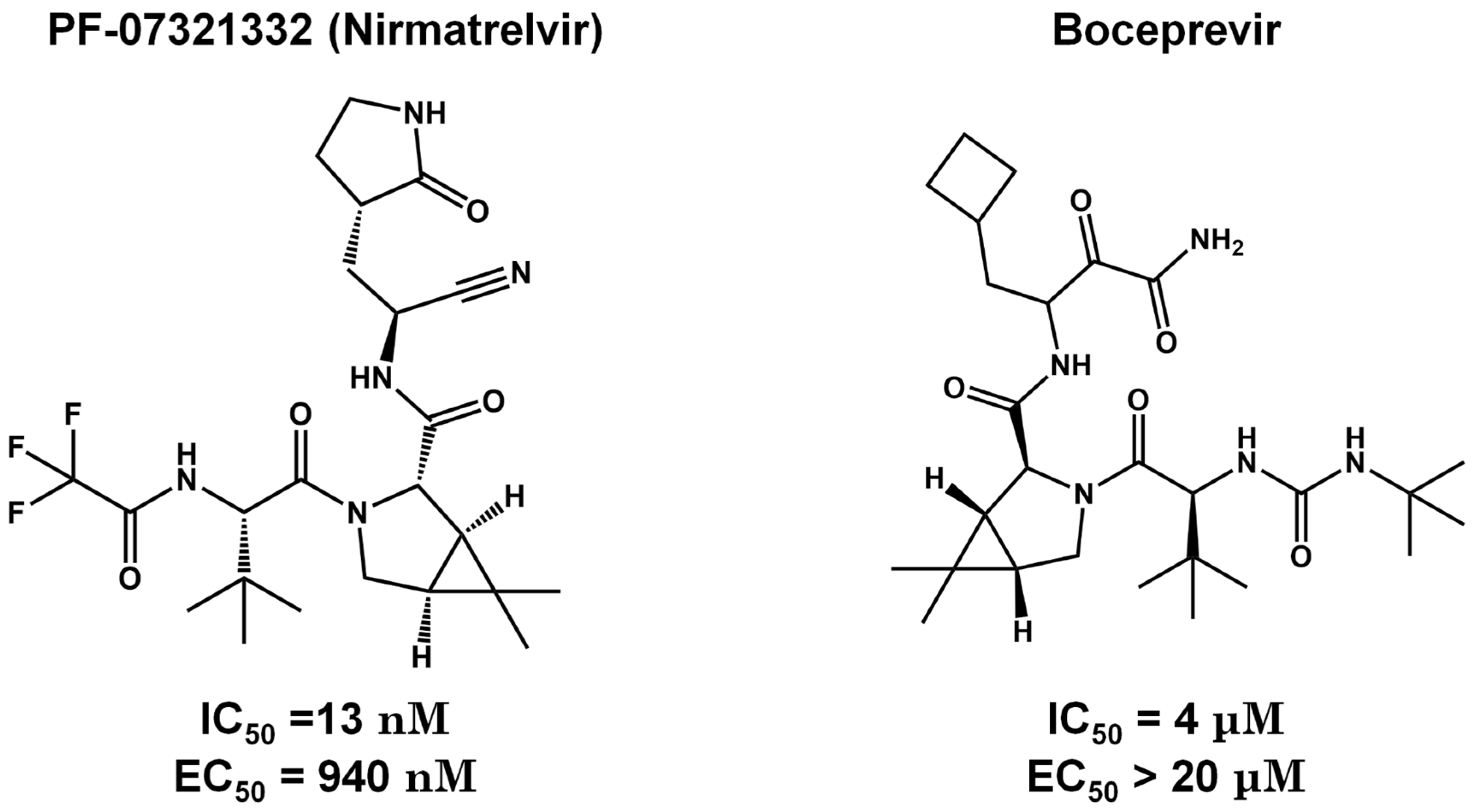
Figure 7.
Discontinued inhibitors. Both Lopinavir and Ritonavir are examples that did not advance through clinical trials. While Lopinavir and Ritonavir were discontinued from clinical trials, Ritonavir is now being utilized in combination with Nirmatrelvir in a phase II/III clinical trial.
Figure 7.
Discontinued inhibitors. Both Lopinavir and Ritonavir are examples that did not advance through clinical trials. While Lopinavir and Ritonavir were discontinued from clinical trials, Ritonavir is now being utilized in combination with Nirmatrelvir in a phase II/III clinical trial.
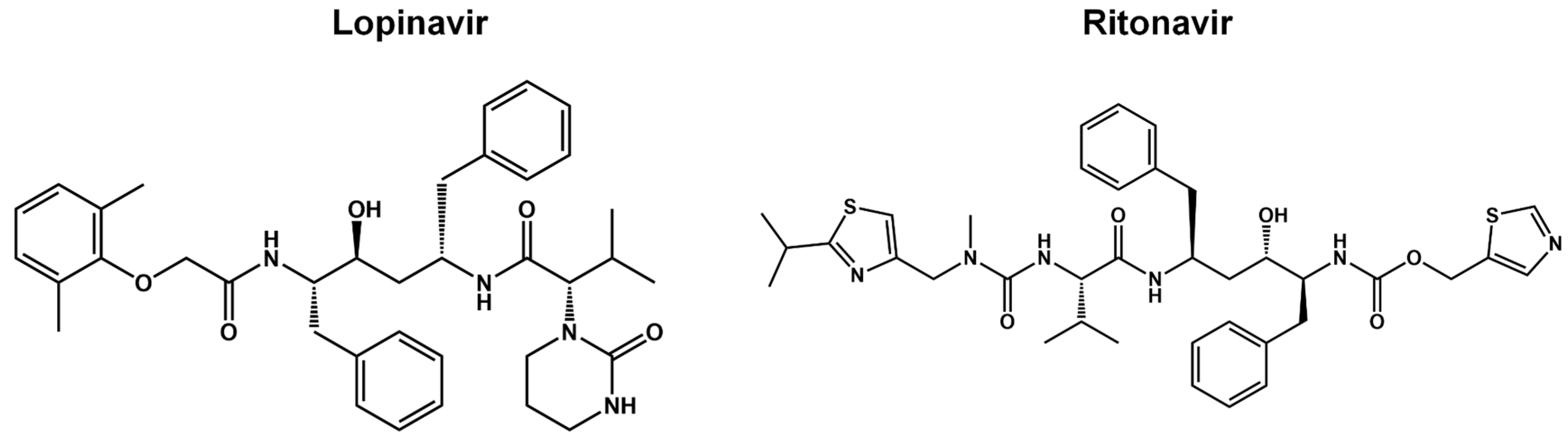
Figure 8.
Pharmacokinetic optimization for α-ketoamide derivatives. Biological activities (IC50 and EC50) are shown for compounds 11r, 13a, 13b, and 14b. NA = without inhibitory activity.
Figure 8.
Pharmacokinetic optimization for α-ketoamide derivatives. Biological activities (IC50 and EC50) are shown for compounds 11r, 13a, 13b, and 14b. NA = without inhibitory activity.
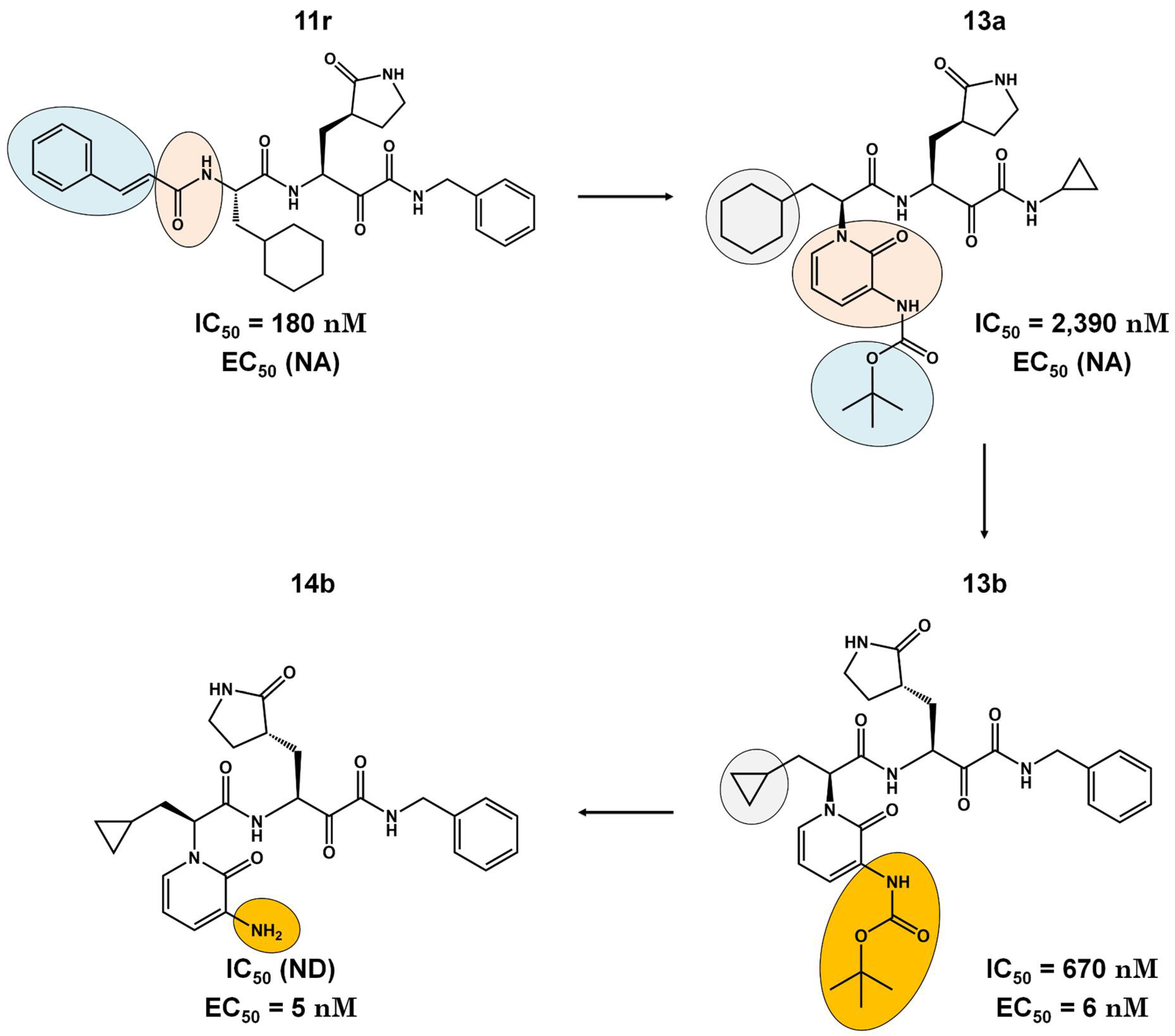
Figure 9.
Pharmacokinetic optimization for aldehyde derivatives of covalent inhibitors. Biological activities (IC50 and EC50) are shown for compounds 1 and 2.
Figure 9.
Pharmacokinetic optimization for aldehyde derivatives of covalent inhibitors. Biological activities (IC50 and EC50) are shown for compounds 1 and 2.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



