
Submitted:
03 May 2024
Posted:
07 May 2024
You are already at the latest version
Abstract
Oxidative stress-mediated cell death is a major cause of dopaminergic neuronal loss in the substantia nigra (SN) of Parkinson’s disease patients. Ergothioneine (ET), a natural dietary compound, has been shown to have cytoprotective functions, but neuroprotective actions against PD have not been well established. 6-Hydroxydopamine (6-OHDA) is a widely used neurotoxin to simulate the degeneration of dopaminergic (DA) neurons in Parkinson's disease. In this study, we investigated the protective effect of ET on 6-OHDA treated iPSC-derived dopaminergic neurons (iDAs) and further confirmed the protective effects in 6-OHDA treated human neuroblastoma SH-SY5Y cells. In 6-OHDA treated cells, metabolic stress in the form of decreased mitochondrial membrane potential (ΔΨm), increased mitochondrial reactive oxygen species (mROS), reduced cellular ATP levels, and increased total protein carbonylation levels was observed. 6-OHDA treatment also significantly decreased tyrosine hydroxylase levels. These effects were significantly abrogated when ET was present. Verapamil hydrochloride (VHCL), a non-specific inhibitor of the ET transporter, OCTN1, abolished ET’s cytoprotective effects, indicative of an intracellular action. These results suggest that ET could be a potential therapeutic for Parkinson’s disease.
Keywords:
neurodegeneration
; ergothioneine
; mitochondrial dysfunction
; 6-OHDA
; Parkinson’s disease
1. Introduction
Parkinson's disease (PD) is one of the most prevalent neurological diseases globally (second after Alzheimer disease) [1], affecting approximately 2% to 3% of people aged 65 years and above [2]. There is increased oxidative damage in the affected brain region, involving iron-iron dependent generation of reactive oxygen species (ROS) [3,4,5,6,7,8]. Mitochondrial dysfunction is closely related to the pathophysiology of PD. Brain samples from PD patients showed evidence of mitochondrial damage [9,10,11] and mitochondrial complex I and II defects were detected in the substantia nigra pars compacta (SNc) of patients with PD [12,13,14,15]. Mitochondrial modulators with neuroprotective potential are currently being explored for treatment of PD. These include creatine (N-aminoiminomethyl-N-methylglycine) and coenzyme Q10 (CoQ10) [16], which have shown some neuroprotective effects in vitro and in vivo animal PD studies [17,18].
Ergothioneine (ET) is a naturally occuring amino thione synthesized by certain fungi and bacteria. It has gained much attention recently due to its cytoprotective, including neuroprotective, effects [19,20,21]. ET is an antioxidant that scavenges certain ROS [19,22,23] and chelates pro-oxidant divalent metal ions [5,19]. It can cross the blood-brain barrier (BBB) and accumulate in the brain following oral administration to mice [24], and is also found in human brain [19,22,25]. ET has shown protective effects against oxidative damage by oxysterols in human brain endothelial cells [26,27], in animal models of stroke [28], cardiovascular disease [29,30], inflammation [31,32], cognitive impairment and dementia in mice [33,34,35], and aging [36]. ET levels in the blood are lower in PD patients [37]. Dietary ET is taken up in the gut before entering cells via the organic cation transporter novel type-1 (OCTN1) [20,38,39]. The latter can be inhibited by verapamil hydrochloride, VHCL, a non-specific ion channel inhibitor [26]. Knockout mice that lacked OCTN1 are more susceptible to inflammation and oxidative damage [40].
Discovery of potential mitochondrial protectants that could cross the BBB could be highly valuable, given the role of mitochondria in PD as a source of oxidative stress [41]. Thus far, however, it is not known if ET could protect against mitochondrial damage in PD. Moreover, there is still a lack of comprehensive translational studies of neurodegenerative therapeutic strategies integrating cutting-edge models. Dopaminergic cultures produced from induced pluripotent stem cells (iPSCs) have been demonstrated to replicate the development of the midbrain, and Parkinsonian cellular networks can be formed from neurons that survive and integrate in vivo [42]. 6-hydroxydopamine (6-OHDA) has been used to induce oxidative stress-induced degeneration of dopaminergic neurons in cellular models of PD [43,44] and in vivo in rodents [45]. 6-OHDA enters dopaminergic neurons via dopamine transporters (DAT), leading to mitochondrial dysfunction and cell death [46,47]. In this study, we sought to determine whether ET could protect against 6-OHDA-induced cell death and mitochondrial damage in tyrosine hydroxylase positive (TH+) cells, in vitro. TH is a marker for dopamine-containing neurons [48]. Human-iPSC derived dopaminergic neurons and human-derived neuroblastoma SH-SY5Y cells were chosen as they are widely used as a cellular model for the investigation of neuronal differentiation and neuroprotection events including PD [42,49,50,51].
2. Materials and Methods
Chemicals
Ergothioneine (ET) was provided by Tetrahedron (Paris, France). 6-Hydroxydopamine hydrobromide (6-OHDA) and Verapamil hydrochloride (VHCL) were purchased from Sigma Aldrich (St. Louis, Missouri, USA).
Cell Culture
To make a 2mM stock solution, 1mg of 6-OHDA was dissolved in 2ml of 0.1% ascorbic acid solution in ice cold MiliQ water. The solution was filtered and can be stored at -20 °C for up to 1 week. SH-SY5Y cells were cultured in a 5% CO2 humidified incubator at 37 °C in high-glucose Dulbecco's modified eagle medium (DMEM-H) with 10% Fetal Bovine Serum (FBS) including glutamine and sodium pyruvate, and 1% Penicillin-Streptomycin. Cells were then cultured in either 6-well plates (qPCR/Immunofluorescence), 12-well plates (Flow cytometry/ATP assay) or 96-well plates (MTT assay) for 24 hours and at 80-90% confluency prior to treatments.
Pharmacological Patterning of Dopaminergic Neurons
The embryonic stem cells (ESCs) and healthy patient derived induced pluripotent stem cells used in this study are BJ, H9 and GM23720 from Corning (Corning, New York, USA). The induction of iDAs from stem cells was performed as described in [52]. In brief, stem cells were cultured on Matrigel-coated plates in StemMACS iPS-Brew XF (130104368) (Miltenyi Biotec., Bergisch Gladbach, Germany). On the day of differentiation (D0), stem cells were treated with Accutase (12679-54) (Nacalai Tesque, Kyoto, Japan) to achieve single cell suspension. Cells were then seeded at 70% confluency on dishes pre-coated with Matrigel and cultured in StemMACS iPS-Brew XF + 5 μM Y-27632 (72304) (STEMCELL Tech., Vancouver, Canada). On day 1 (D1), the medium was replaced with Neural Induction Medium (NIM) comprising the Neural Medium (NM) with SMAD (suppressor of mothers against decapentaplegic) inhibitors, + 0.5 μM LDN193189 (72147) (STEMCELL Tech.) + 10μM SB431542 (Miltenyi Biotec.), where NM is a mixture of 50% [v/v] DMEM: F12 + 50% [v/v] MACS Neuro Medium (130-093-570) (Miltenyi Biotec.) + GlutaMax (35050061) (Thermo Fisher Scientific, Massachusetts, United States) + NEAA (non-essential amino acids) (11140050) (Thermo Fisher Scientific) + N-2 (neuro-2) (17502048) (Thermo Fisher Scientific) + MACS NeuroBrew-21 (130-093-566) (Miltenyi Biotec.). On D3, SHH (sonic hedgehog) inducers, 100ng/ml SHH-C24II (130-095-723) (Miltenyi Biotec.), 100ng/ml FGF8b (fibroblast growth factor 8) (130-095-738) (Miltenyi Biotec.), 2μM Purmorphamine (130-104-465) (Miltenyi Biotec.), and GSK3β (glycogen synthase kinase 3 beta) inhibitor, 3μM CHIR99021 (130-104-172) (Miltenyi Biotec.) were supplemented to the medium, which was then changed every 2 - 3 days until D10. At this point, stem cells have differentiated into iDA progenitors. These progenitor cells were cultured in NM + 100ng/ml FGF8b + 20 ng/mL BDNF (bone-derived neurotrophic factor) (130-103-435) (Miltenyi Biotec.) + 0.2mM L-AA (L-ascorbic acid) (A5960) (Sigma) and the medium was changed every day. From D15, DA maturation medium, i.e., NM + 20 ng/mL GDNF (glia-derived neurotrophic factor) (130-108-986) (Miltenyi Biotec.) + 20 ng/mL BDNF + 0.2mM L-AA + 1ng/ml TGFβ3 (transforming growth factor beta 3) (243-B3-002/CF) (R&D Systems, Minneapolis, United States) + 0.5mM db-cAMP (dibutyryl cyclic adenosine monophosphate) (D0260) (Sigma) + 10μM DAPT (gamma secretase inhibitor) (2634) (Tocris Bio., Bristol, United Kingdom), was added and changed every other day until iDAs were ready on D40. Successful differentiation of iDAs was verified using gene expression assay (RT-qPCR) and protein expression assays (western blotting, immunofluorescence, and flow cytometry) for various stem cell, neuronal and dopaminergic markers.
Cellular ET Uptake and Liquid Chromatography Mass Spectrometry
ET was quantified as previously described [24]. Cells were washed thrice with ice cold PBS (Thermo Fisher Scientific) before addition of methanol spiked with internal standard (ISTD), ET-d9. Next, samples were centrifuged at 20,000g at 4 °C for 10 min and the supernatant was collected in glass vials and the contents evaporated at 37 °C under a N2 stream. Glass vials containing the sample residues were reconstituted in pure methanol and ET levels were analysed via liquid chromatography mass spectrometry (LC-MS/MS), using an Agilent 1290 UPLC system coupled with Agilent 6460 ESI mass spectrometer (Agilent Technologies, CA, USA). Samples were kept at 10 °C in the autosampler. 2 µl of the processed samples were injected into a Cogent Diamond-Hydride column (4 µm, 150 × 2.1 mm, 100 Å; MicroSolv Technology Corporation, NC, USA) maintained at 30 °C. Solvent A was acetonitrile in 0.1% formic acid, and Solvent B was 0.1% formic acid in ultrapure water. Chromatography was carried out at a flow rate of 0.5 ml/min using the following gradient: 1 min of 20% solvent B, followed by a 3 min gradient increase of solvent B to 40% to elute ET. The retention time for ET is 4.2 min.
Mass spectrometry was carried out using the positive ion, electrospray ionization mode, with multiple reaction monitoring (MRM) for quantification of specific target ions. Capillary voltage was set at 3200 V, and gas temperature was kept at 350 °C. Nitrogen sheath gas pressure for nebulizing sample was at 50 psi, and gas flow set at 12 L/min. Ultra-high purity nitrogen was used as collision gas. Precursor to product ion transitions and fragmentor voltages (V)/collision energies (eV) for each compound were as follows: ET; 230.1 → 186, 103 V/9 eV and ET-d9; 239.1 → 195.1, 98 V/9 eV.
MTT Assay
The MTT Assay Kit (Sigma) was used to detect the impact of 6-OHDA on cellular metabolic activity. 15μM 6-OHDA was found to induce 60-70% loss of cellular metabolic activity based on preliminary experiments. 1mg of MTT was added into 1mL of medium, which was incubated for 4h at 37 °C. The medium in the plate was discarded after 4 h, and 200 µL DMSO (dimethylsulphoxide) was added to each well before being shaken for one minute for dissolution. Absorbance at 570nm based on cellular dehydrogenase activity was measured in a microplate reader (Tecan, Switzerland). The different experimental conditions were normalised to control absorbance. Experiments were carried out three times.
Fluorescence Microscopy
Immunocytochemistry was employed to visualise OCTN1 expression, mitochondrial morphology, and mitochondrial ROS production. Cells were seeded onto 13mm coverslips in 6-well plates prior to respective treatment conditions. For live staining, cells were washed twice with PBS before incubation with either 2.5μM MitoSOX (InvitrogenTM, Waltham, Massachusetts, USA) or 50nM of MitoTrackerTM Green FM (Thermo Fisher Scientific) for 15min. Cells attached on the coverslips were washed thrice with PBS before removal. ProLong™ Gold Antifade Mountant with DAPI (Thermo Fisher Scientific) was added before visualisation with a fluorescence microscope. For fixed staining, cells were washed twice withPBS before fixing with 3.7% paraformaldehye (Thermo Fisher Scientific) for 20mins. Fixed cells were washed thrice with ultrapure water before permeabilisation with PBS containing 0.02% tween (Thermo Fisher Scientific) for 5min. Blocking was done in blocking buffer comprising of PBS, 0.02% tween and 1% bovine serum albumin (BSA) (Thermo Fisher Scientific) for 1h. After blocking, cells were incubated overnight (for at least 16 hours) at 4°C with primary antibody in blocking buffer. Mouse anti-neurofilament (#2F11) (Sigma), Mouse anti-NeuN (LV1825845) (Milipore, Massachusetts, United States), goat anti-beta tubulin III (MAB1195) (R & D Systems) and goat anti-OCTN1 (sc-19819) (Santa Cruz, California, United States) antibody were used at 1:200 dilution. Expression of OCTN1 was further confirmed using western blot after membrane extraction. APC -conjugated Tyrosine hydroxylase (130-120-352) (Miltenyi Biotec.) and FITC-conjugated Tyrosine hydroxylase (130-120-350) (Miltenyi Biotec.) were used at 1:50 dilution. Binding of primary antibodies was followed by incubation with either anti-mouse secondary AF488 (green) conjugated antibody (1: 1000) (A28175) (Thermo Fisher Scientific), anti-goat secondary APC conjugated antibody (1:1000) (A-865) (Thermo Fisher Scientific), anti-goat secondary AF647 (violet) conjugated antibody (1: 1000) (A-21447) (Thermo Fisher Scientific) in blocking buffer for 1h at room temperature. Cells were washed thrice with PBS before addition of ProLong™ Gold Antifade Mountant with DAPI (Thermo Fisher Scientific) and visualization with fluorescence microscope. Protein expression was visualized with Olympus FV3000 confocal microscope (Tokyo, Japan) and mROS production was visualized with Olympus DP70 fluorescence microscope (Tokyo, Japan).
Quantitative RT-PCR
TRIzol Reagent (InvitrogenTM) was used for RNA extraction as per the manufacturer’s instructions. cDNA was produced from reverse transcription of 1,000 ng of RNA (High-Capacity cDNA Reverse Transcription Kit; Applied Biosystems, Waltham, Massachusetts, USA). Reverse transcription was performed in a T-Personal Thermocycler (Biometra, Germany) with condition of 25°C for 10 min, 37°C for 30 min followed by 85°C for 5 min. qPCR was carried out to quantify OCT4 (octamer-binding transcription factor 4), NANOG (NANOG homeobox), MAP2 (microtubule associated protein 2), NeuN (neuronal nuclei), NeuroD1 (neurogenic differentiation factor 1), BDNF, FOXA2 (foxhead box protein A2) and DAT (dopamine transporter) mRNA expression, using SYBR Green Gene Expression Assay Probes (Applied Biosystems, USA) and SYBR Green Universal PCR Master Mix (Applied Biosystems, USA). GAPDH was used as the housekeeping gene for genes of interest. The RT-qPCR was performed in a 7500 Real-time PCR System (Applied Biosystems, USA) with conditions of 95°C for 10 min, followed by 95°C for 15s and 60°C for 1 min, for 40 cycles. Subsequently, the relative mRNA expression for the respective genes of interest was quantitated via the comparative CT (ΔΔCT) method. Primer sequences can be found in Supplementary Table S1.
Flow Cytometry
Flow cytometry measurements were performed using a CytoFlex LX flow cytometer (Beckman Coulter Life Sciences, Indianapolis, Indiana, USA), using 106 cells per sample for analysis with 10,000 events per sample recorded. The FL1 channel was used to quantify cell death (Propidium Iodide, Ex/Em = 535/615 nm), free intracellular calcium (Fluo-4, Ex/Em = 488/525 nm), mitochondrial membrane potential (Tetramethylrhodamine, methyl ester (TMRM), Ex/Em = 555/575nm), mitochondrial ROS (MitoSOX, Ex/Em = 510/580nm), and mitochondria (MitoTracker green Ex/Em= 490/526nm). iDAs were cultured on coverslips washed with PBS before being stained with either propidium iodide, MitoSOX or TMRM for 30 minutes. Cells were washed with PBS before 15 minutes fixation in 3.7% paraformaldehyde. Cells were permeabilized in perm buffer (Miltenyi Biotec.) before blocking and primary antibody incubation of tyrosine hydroxylase that was FITC conjugated (Miltenyi Biotec.). Cells were then washed with PBS before analysis using flow cytometry. For flow cytometry data acquisition, fluorescent signals were measured on a logarithmic scale of four decades of log. Raw data were processed using FlowJo version 10.5.3.
ATP Assay
A commercially available ATP determination kit (InvitrogenTM) was used to study ATP levels in cells. Trypsinised cells were collected and a Trypan blue assay was performed to ensure all samples contain equal number of cells (106 cells). Upon centrifugation and removal of supernatant, 1ml of boiling ultrapure water from an Arium pro® ultrapure system was added into the cell pellet and incubated in a water bath for 10min at 100°C [53]. Samples were then cooled on ice for 30s and supernatant utilised for ATP assay as per manufacturer instructions. Luminescence readings of the samples were performed using Synergy H1 Microplate Reader (BioTek, Winooski, Vermont, USA). For all experiments, ATP standard curves were run in the range of 0.2 to 1.4μM.
Western Blot
Cells were lysed with ProteoExtract® Native Membrane Protein Extraction Kit (Millipore) for OCTN1 expression, performed as per the manufacturer’s instructions. Supernatant was collected after centrifugation at 13,000 g at 4°C for 20 min. Protein concentration was determined using a bicinchoninic acid (BCA) protein assay kit (Sigma Aldrich). The samples were loaded and separated on precast SDS-polyacrylamide gels (Bio-Rad). Proteins were electro-transferred to a nitrocellulose membrane (Bio-Rad) in transfer buffer containing 48 mmol/l Tris-HCl, 39 mmol/l glycine, 0.037% SDS, and 20% methanol, at 4°C for 1 hour. Blocking was done in 2.5% non-fat milk for 1h at room temperature. Binding of OCTN1 primary goat anti-OCTN1 (sc-19819) (Santa Cruz, California, United States) antibody at 1:200 dilution, overnight at 4°C, was followed by incubation with secondary horseradish peroxidase-conjugated IgG in 2.5% non-fat milk for 1h at room temperature. Anti-mouse IgG HRP were obtained from Sigma Aldrich and dilution of 1:5000 was used. The blots were visualised with SuperSignal™ West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific) using iBright Imaging systems (InvitrogenTM). The blots were processed using open access software platform FIJI (ImageJ).
Protein Carbonylation Assay
Cells were lysed with Pierce® RIPA Lysis and Extraction buffer (Thermo Fisher Scientific) for protein carbonylation assay. Supernatant was collected after centrifugation at 13,000 g at 4°C for 20 min. Protein concentration was determined using a bicinchoninic acid (BCA) protein assay kit (Sigma Aldrich). OxyBlot Protein Oxidation Detection Kit (Chemicon/Millipore, USA) was utilised. The samples were derivatised and prepared as per manufacturer’s instructions prior to being loaded and separated on precast SDS-polyacrylamide gels (Bio-Rad). Proteins were electro-transferred to a nitrocellulose membrane (Bio-Rad) in transfer buffer containing 48 mmol/l Tris-HCl, 39 mmol/l glycine, 0.037% SDS, and 20% methanol, at 4°C for 1 hour. Blocking, incubation with 2,4-Dinitrophenylhydrazine (DNPH) antibody and secondary antibody respectively were also performed as specified by the manufacturer. Binding of primary antibodies was followed by incubation with secondary horseradish peroxidase (HRP) conjugated IgG in 2.5% non-fat milk for 1h at room temperature. Anti-mouse IgG HRP was obtained from Sigma Aldrich and dilution of 1:5000 was used. The blots were visualised with SuperSignal™ West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific) using iBright Imaging systems (InvitrogenTM). The blots were processed using open access software platform FIJI (ImageJ).
Tyrosine Hydroxylase Protein Assay
Flow cytometry was employed to assess tyrosine hydroxylase (TH) expression in SH-SY5Y cells. Cells were first detached, and cell pellets were washed with PBS. Cells were incubated for 30min with 3.7% paraformaldehyde before permeabilization step for another 30min. Blocking was done with 5% BSA before 30min incubation with FITC conjugated TH antibody and APC conjugated Tau antibody (130-119-363) (Miltenyi Biotec). Flow cytometry measurements were performed on Cytoflex LX flow cytometer (Beckman Coulter Life Sciences), using 1,000,000 cells per sample for analysis. 10,000 events per sample was recorded. Fluorescent signals were measured on a logarithmic scale of four decades of log. Raw data was processed using CytoFlex software. Raw data was processed using FlowJo version 10.5.3.
Dopamine Detection ELISA Kit
Dopamine levels were quantified using a commercially available dopamine detection kit (Novus Biologicals, Littleton, Colorado, USA). The colorimetric assay is a 90min, single-wash sandwich ELISA designed for quantitative measurement of dopamine. Conditioned media from treated cells were collected and centrifuged to remove cellular debris. Normalization of protein concentration was performed using a Pierce BCA protein assay kit. Next, 50ul of sample and 50ul of antibody cocktail was added to each well, followed by 1h incubation on a shaker at 400 rpm. After 1h, 3,3',5,5'-Tetramethylbenzidine (TMB) solution provided was added before the addition of stop solution, as per manufacturer’s instructions. Dopamine standards were prepared as well. Absorbance readings at 450nm of the samples were performed using Synergy H1 Microplate Reader (BioTek, USA). Standard curve for dopamine: For all experiments, dopamine standard curves were run in the range of 2.19 to 140 ng/ml.
Statistical Analysis
Data are shown as mean ± SD (standard deviation). All analysis was done in GraphPad prism 9.0 software. Comparisons were conducted by one-way ANOVAs, with Bonferroni correction for multiple comparison tests. P< 0.05 was regarded as statistically significant.
3. Results
3.1. Generation of hiPSC-Derived Day 40 Dopaminergic Neurons
We differentiated hiPSCs (BJ and GM23720) and hESC (H9) to investigate the protective effect of ET on 6-OHDA treated induced human dopaminergic neurons, a model highly relevant to human PD. We adopted a 40-day differentiation protocol, using various small molecules and growth factors (Figure 1A). Results show a significant decrease in the pluripotency markers: OCT4 and NANOG (Supp. Fig. 1A) but significant increases in neuronal markers: MAP2, NeuN, BDNF and NeuroD1 (Supp. Fig. 1B) and dopaminergic-specific markers: FOXA2 and DAT (Supp. Fig. 1C). Protein expression of neuronal and dopaminergic markers was also performed using confocal imaging which showed that our day 40 iDAs express neuronal proteins of NeuN and TUBB3, and dopaminergic neuron-specific marker TH (Figure 1B). Neuronal yield and dopaminergic yield were found to be approximately 80% (Figure 1C) and 40% (Figure 1D), respectively, which is consistent with other studies [54,55].
3.2. ET Protects iDAs Against 6-OHDA Induced Cell Death and Loss of Dopamine Secretion
MTT assay showed that 6-OHDA induced a 70%-80% loss of metabolic activity in iDAs (Figure 2A). 6-OHDA was also found to induce neuronal degeneration observed under the microscope (Figure 2B,C). Degenerating neurons appear fragmented with neurite degeneration, similar to other in vitro neuronal degeneration phenotypes [56]. 6-OHDA treatment led to a significant increase in non-viable cells, while co-treatment with ET was able to attenuate the cytotoxic effects of 6-OHDA (Figure 2D). Co-treatment with a non-specific inhibitor of OCTN1, verapamil hydrochloride (VHCL), on the other hand abrogated the protective effects of ET (Figure 2D). Results from the ATP assay also show that 6-OHDA treatment caused a decrease in intracellular ATP levels while co-treatment with ET was able to attenuate the decrease (Figure 2E). Protection against 6-OHDA toxicity by ET was dose dependent (Supplementary Figure S2A). 1mM of ET was used in subsequent experiments as it is similar to achievable cellular levels [57,58]. Co-treatment with VHCL abrogated the effects of ET (Figure 2E). Dopamine secretion assay show that 6-OHDA caused a significant reduction in the amount of dopamine secreted by day 40 iDAs, while co-treatment with ET was able to attenuate the decrease (Figure 2E). Co-treatment with VHCL abrogated the effects of ET (Figure 2E).
3.3. ET Also Protects Day 40 iDAs Against 6-OHDA-Induced Increase in mROS and Loss of MMP.
We investigated the effects of 6-OHDA on mitochondrial function, as measured by the TMRM (mitochondrial membrane potential - MMP) and MitoSOX (mitochondrial ROS) probes. Murphy et al. [59] discussed the pros and cons of this method and results must always be interpreted with caution, e.g., they can be affected by changes in mitochondrial size, shape and membrane potential. From the results, we found that 6-OHDA caused a significant decrease in MMP (Figure 3A,B) and an apparent significant increase in mROS levels (Figure 3C,D). The co-treatment with ET was able to attenuate the changes induced by 6-OHDA, while co-treatment with VHCL abrogated the effects of ET.
3.4. ET Uptake Protects TH+ SH-SY5Y Cells Against 6-OHDA Induced Cell Death, Metabolic Dysfunction, and Oxidative Stress
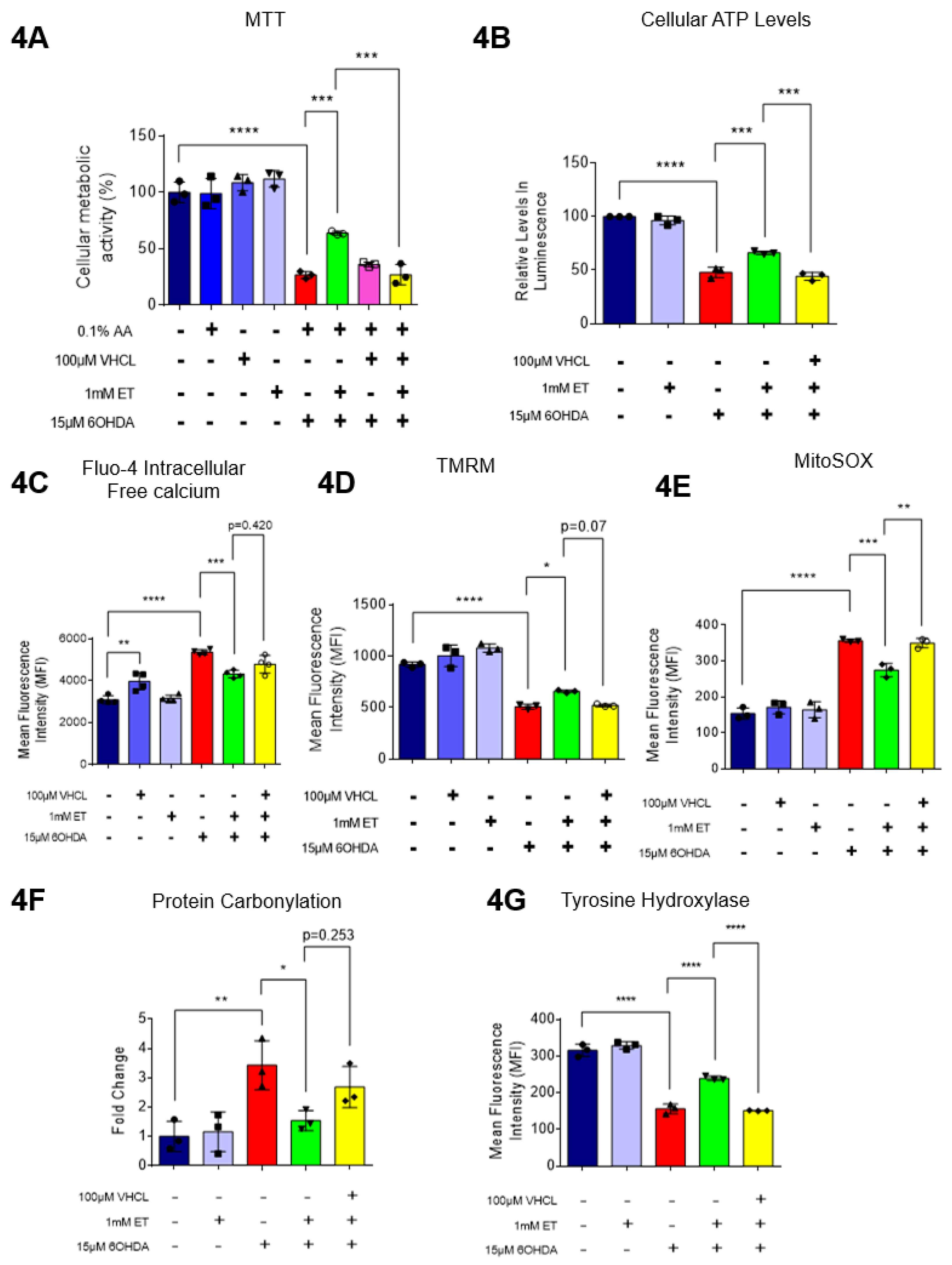
To investigate if ET can also protect other TH+ cells from the neurotoxicity of 6-OHDA, the SH-SY5Y human neuroblastoma cell line, a well-accepted model in PD studies[49], was also used. MTT assay on 6-OHDA-treated SH-SY5Y cells revealed a 60-70% loss of metabolic activity (Figure 4A). 6-OHDA was also found to significantly affect cellular metabolism as evidenced by reduction in intracellular ATP levels (Figure 4B), increased intracellular free calcium (Figure 4C), decreased mitochondrial membrane potential (MMP) (Figure 4D), and increased mitochondrial ROS (mROS) levels (Figure 4E), as measured by the MitoSOX probe. For all the above assays, co-treatment of SH-SY5Y cells with 1mM ET was able to attenuate the dysfunction caused by 6-OHDA. (Figure 4A-D). Since heightened levels of ROS can promote oxidative protein damage, as revealed by protein carbonylation formation [60], we performed a protein carbonylation assay by immunoblotting (Supp. Fig. 3C). Results show that 6-OHDA treatment led to a significant increase in oxidized proteins (approximately 3.5-fold) as compared to control but ET attenuated this increase (Figure 4F). Tyrosine hydroxylase (TH), a critical enzyme involved in the production of dopamine and implicated in PD, was also investigated. Using flow cytometry, 6-OHDA treatment was found to significantly reduce TH protein levels as compared to control, but ET was able to attenuate the decrease in TH levels (Figure 4G). In conjunction with the results from Figure 2F, which showed that 6-OHDA decreased dopamine secretion in day 40 iDAs, TH protein expression in SH-SY5Y cells was also decreased upon exposure to 6-OHDA. Tau protein, which is also implicated in PD [61], did not appear to have its levels affected by either 6-OHDA or ET treatment (Supp. Fig. 3D). Addition of VHCL tended to abrogate (note p values on the figure) the effects of ET, although the results were not always significant (p > 0.05) (Figure 4). These findings are consistent with our results involving D40 iDAs.
4. Discussion
In this study, we investigated the effect of ET on cell death caused by 6-OHDA in an iPSC model of PD. 6-OHDA induced oxidative stress occurs as early as 4h incubation but cell death occurs within 24h in rat [62], mouse [44], and human [63] dopaminergic cells. A concentration-dependent decrease in cellular metabolic activity with 60-70% loss of cells was observed at 15μM 6-OHDA concentration, with 24h incubation. This concentration of 6-OHDA was selected for further experiments while 1mM of ET was used on the basis of preliminary data [57,58]. Dose dependent ET protection against 6-OHDA is presented in Supplementary Figure 2A. Ergothioneine (ET) is a cytoprotective compound that accumulates at high levels in tissues during times of oxidative damage [22,64]. LC-MS result confirmed that verapamil hydrochloride (VHCL), a non-specific inhibitor of OCTN1, reduced intracellular ET levels (Supp. Fig. 2C-D), and decreased protective effect of ET. This demonstrated that any protective effect of ET observed in our experiments was dependent on cellular uptake of ET, and not through, perhaps, extracellular neutralization of 6-OHDA by direct reaction of ET with it or its oxidation products. Previous studies have found that OCTN1 is important for cellular uptake of ET in endothelial cells [65], and that OCTN1 levels could be elevated in response to tissue injury, e.g., in fatty liver [66] or kidney disease [67]. To demonstrate that OCTN1 is present in the cells, western blot (Supplementary Figure 2B) and fluorescence imaging (Supplementary Figure 2E,F) were carried out. Tyrosine hydroxylase (an enzyme involved in the production of dopamine precursor) was expressed in both D40 iDAs (Figure 1B) and SH-SY5Y cells (Supplementary Figure 2E). Results showed that ET could protect against 6-OHDA-induced degeneration of both human iDAs and SH-SY5Y cells. Coincubation of ET with a non-specific inhibitor of OCTN1, VHCL, abolished the protective effect of ET on cell viability.
Cotreatment with ET protects mature human induced dopaminergic neurons against 6-OHDA neurotoxicity, shown by MTT cell viability and propidium iodide cell death assay. iDA neurons also express OCTN1 and co-incubation of VHCL prevented ET uptake in these cells. Mitochondrial dysfunction was also ameliorated with ET co-incubation in 6-OHDA treated iDA neurons. Mitochondrial dysfunction is characterised by a decrease in ΔΨm, reduced ATP generation and increase in mitochondrial ROS (mROS) production [68]. 6-OHDA is thought to induce mitochondrial dysfunction in part through inhibition of complex I of the mitochondrial electron transport chain which contributes to a decreased ΔΨm, reduced ATP generation, increase in ROS production and apoptotic cell death [69]. Similar to other studies [70,71], we found that 6-OHDA induced mitochondrial dysfunction as evidenced by a reduction of ΔΨm, decreased cellular ATP levels and increase in mROS levels. ET ameliorated the effects of 6-OHDA with a smaller decrease in ΔΨm and cellular ATP levels, and lower increase in mROS levels. VHCL inhibited the protective effect of ET on mitochondrial dysfunction. Our results add to previous findings that ET has neuroprotective functions [26,72,73] which could potentially aid in the prevention and treatment of PD. We would also like to acknowledge the paper that was published by Yuzawa et al. (2024) [73], while we were preparing this manuscript. Yuzawa et al. reported the protective effect of ET against 6-OHDA neurotoxin in immortalized mouse hypothalamic cells (GT-17), a cell line perhaps less relevant to PD. Nevertheless, their data further illustrate the potential protective effect of ET against neurodegeneration [21].
The protective effect of ET against 6-OHDA neurotoxin was also observed in TH+ SHSY5Y cells. Besides loss of cell viability, treatment of SH-SY5Y cells with 15μM 6-OHDA resulted in a significant increase of intracellular free calcium levels. Increases in intracellular free calcium levels are known to induce mitochondrial oxidative stress-mediated apoptosis [74], and administration of nimodipine, an L-type calcium channel blocker, was shown to protect against MPTP-induced dopaminergic neuronal death in an animal model of PD [75]. ET reduced the 6-OHDA-induced increase in intracellular calcium levels. The reduced intracellular calcium levels could be due to an effect of ET as a cytoprotectant, or chelator of divalent metal ions including Ca2+ [76]. Our results demonstrate that ET could aid the maintenance of intracellular calcium homeostasis. Treatment of SH-SY5Y cells with 15μM 6-OHDA also resulted in a significant increase in total carbonylated proteins. Protein carbonylation is increased in cells that are undergoing oxidative stress [77]. Oxidized proteins contribute to increased ER stress and increased unfolded protein response (UPR) [78] and can result in cellular apoptosis [79,80]. Diseases such as PD have been linked to carbonylated protein accumulation [8,81]. 6-OHDA was found to induce an increase in carbonylated proteins in SH-SY5Y cells [8,82], PC12 cells [83] and rats [84]. Results indicate a protective effect of ET against protein carbonyl formation. Moreover, ET was able to ameliorate the 6-OHDA-induced decrease in tyrosine hydroxylase levels. Tyrosine hydroxylase (TH) the rate-limiting enzyme in dopamine production, and low dopamine levels play a key role in PD pathogenesis [91–93]. Similar to our study in SH-SY5Y cells, 6-OHDA treated rats were found to have reduced tyrosine hydroxylase enzyme [94,95]. The effect of ET in restoring TH levels could be clinically relevant since PD is caused by loss of dopamine-producing neurons.
In summary, findings demonstrate that ET can protect human dopaminergic neurons against 6-OHDA induced increase in cell death and metabolic dysfunction. Results also suggest that ET has disease modifying potential by increasing dopamine levels secreted by iDA neurons. Protective effects of ET were abrogated when cell cultures were cotreated with VHCL, a non-specific inhibitor of OCTN1, the ET transporter; suggesting that ET uptake into cells is necessary for protection against 6-OHDA. Results in SH-SY5Y cells also demonstrated protective effect of ET against 6-OHDA induced increase in intracellular free calcium, increase in total carbonylated proteins and reduction in tyrosine hydroxylase levels. In this paper, we have also shown the protective effect of ET against mitochondrial dysfunction. Uptake of ET into mitochondria is also unclear due to lack of substantial evidence with regards to localisation of OCTN1 on mitochondria [39,85,86]. Further studies also need to be carried out to validate the protective effect of ET in animal models of PD.
Supplementary Materials
The data are available upon request.
Author Contributions
D.M-K.L. contributed most of the lab work, experimental design, data collection, data analysis, and wrote the paper, I.C. and C.J-J.Y. contributed the experimental design, discussion and data analysis, L.C. contributed the lab work, Y-K.N. contributed the lab work and discussion, W-Y.O. and B.H. contributed the idea forming, discussion and helped write the paper. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by grants from the Ministry of Education (NUHSRO/2019/051/T1/Seed-Mar/04), National Medical Research Council of Singapore (HLCA21Jan-0019), Healthy Longevity Catalyst Award and Centennial Fund. Tetrahedron (14 avenue de l’Opera, Paris, France) provided Ergothioneine.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data are available upon request.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Ou, Z.; et al. Global Trends in the Incidence, Prevalence, and Years Lived With Disability of Parkinson's Disease in 204 Countries/Territories From 1990 to 2019. Frontiers in Public Health 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Poewe, W.; et al. Parkinson disease. Nat Rev Dis Primers 2017, 3, 17013. [Google Scholar] [CrossRef]
- Chinta, S.J.; Andersen, J.K. Redox imbalance in Parkinson's disease. Biochim Biophys Acta 2008, 1780, 1362–1367. [Google Scholar] [CrossRef]
- Jenner, P.; Olanow, C.W. Oxidative stress and the pathogenesis of Parkinson's disease. Neurology 1996, 47 (6_suppl_3), 161S–170S. [Google Scholar] [CrossRef] [PubMed]
- Dexter, D.T.; Jenner, P. Parkinson disease: From pathology to molecular disease mechanisms. Free Radic Biol Med 2013, 62, 132–144. [Google Scholar] [CrossRef] [PubMed]
- Seet, R.C.; et al. Oxidative damage in Parkinson disease: Measurement using accurate biomarkers. Free Radic Biol Med 2010, 48, 560–566. [Google Scholar] [CrossRef] [PubMed]
- Alam, Z.I.; et al. Oxidative DNA damage in the parkinsonian brain: An apparent selective increase in 8-hydroxyguanine levels in substantia nigra. J Neurochem 1997, 69, 1196–1203. [Google Scholar] [CrossRef]
- Alam, Z.I.; et al. A generalised increase in protein carbonyls in the brain in Parkinson's but not incidental Lewy body disease. J Neurochem 1997, 69, 1326–1329. [Google Scholar]
- Henchcliffe, C.; Beal, M.F. Mitochondrial biology and oxidative stress in Parkinson disease pathogenesis. Nat Clin Pract Neurol 2008, 4, 600–609. [Google Scholar] [CrossRef]
- Abou-Sleiman, P.M.; Muqit, M.M.K.; Wood, N.W. Expanding insights of mitochondrial dysfunction in Parkinson's disease. Nature Reviews Neuroscience 2006, 7, 207–219. [Google Scholar] [CrossRef]
- Bindoff, L.A.; et al. Mitochondrial function in Parkinson's disease. Lancet 1989, 2, 49. [Google Scholar] [CrossRef]
- Mythri, R.B.; et al. Mitochondrial complex I inhibition in Parkinson's disease: How can curcumin protect mitochondria? Antioxid Redox Signal 2007, 9, 399–408. [Google Scholar] [CrossRef]
- Schapira, A.H.; et al. Mitochondrial complex I deficiency in Parkinson's disease. Lancet 1989, 1, 1269. [Google Scholar] [CrossRef]
- Grünewald, A.; et al. Mitochondrial DNA Depletion in Respiratory Chain–Deficient Parkinson Disease Neurons. Annals of Neurology 2016, 79, 366–378. [Google Scholar] [CrossRef]
- González-Rodríguez, P.; et al. Disruption of mitochondrial complex I induces progressive parkinsonism. Nature 2021, 599, 650–656. [Google Scholar] [CrossRef]
- Thomas, B.; Beal, M.F. Mitochondrial therapies for Parkinson's disease. Movement Disorders, 2010, 25(S1), S155-S160.
- Matthews, R.T.; et al. Creatine and cyclocreatine attenuate MPTP neurotoxicity. Exp Neurol 1999, 157, 142–149. [Google Scholar] [CrossRef]
- Klivenyi, P.; et al. Additive neuroprotective effects of creatine and a cyclooxygenase 2 inhibitor against dopamine depletion in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mouse model of Parkinson's disease. J Mol Neurosci 2003, 21, 191–198. [Google Scholar] [CrossRef]
- Cheah, I.K.; Halliwell, B. Ergothioneine, recent developments. Redox Biol 2021, 42, 101868. [Google Scholar] [CrossRef]
- Halliwell, B.; Cheah, I. Ergothioneine, where are we now? FEBS Letters 2022, 596, 1227–1230. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Cheah, I. Are age-related neurodegenerative diseases caused by a lack of the diet-derived compound ergothioneine? Free Radic Biol Med 2024, 217, 60–67. [Google Scholar] [CrossRef]
- Halliwell, B.; Tang, R.M.Y.; Cheah, I.K. Diet-Derived Antioxidants: The Special Case of Ergothioneine. Annual Review of Food Science and Technology 2023, 14. [Google Scholar] [CrossRef] [PubMed]
- Hartman, P.E., [32] Ergothioneine as antioxidant, in Methods in Enzymology. 1990, Academic Press. p. 310-318.
- Tang, R.M.Y.; et al. Distribution and accumulation of dietary ergothioneine and its metabolites in mouse tissues. Sci Rep 2018, 8, 1601. [Google Scholar] [CrossRef] [PubMed]
- Novotny, B.C.; et al. Metabolomic and lipidomic signatures in autosomal dominant and late-onset Alzheimer's disease brains. Alzheimers Dement 2023, 19, 1785–1799. [Google Scholar] [CrossRef] [PubMed]
- Koh, S.S.; et al. Effect of Ergothioneine on 7-Ketocholesterol-Induced Endothelial Injury. NeuroMolecular Medicine 2021, 23, 184–198. [Google Scholar] [CrossRef] [PubMed]
- Leow, D.M.; et al. Protective Effect of Ergothioneine against 7-Ketocholesterol-Induced Mitochondrial Damage in hCMEC/D3 Human Brain Endothelial Cells. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef] [PubMed]
- Ong, W.Y.; et al. Protective Effect of Ergothioneine Against Stroke in Rodent Models. Neuromolecular Med 2023, 25, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Randhawa, P.K.; et al. Eugenol attenuates ischemia-mediated oxidative stress in cardiomyocytes via acetylation of histone at H3K27. Free Radic Biol Med 2023, 194, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.; et al. Ergothioneine is associated with reduced mortality and decreased risk of cardiovascular disease. Heart 2020, 106, 691–697. [Google Scholar] [CrossRef]
- Brancaccio, M.; et al. First evidence of dermo-protective activity of marine sulfur-containing histidine compounds. Free Radic Biol Med 2022, 192, 224–234. [Google Scholar] [CrossRef]
- Gao, Y.; et al. l-Ergothioneine Exhibits Protective Effects against Dextran Sulfate Sodium-Induced Colitis in Mice. ACS Omega 2022, 7, 21554–21565. [Google Scholar] [CrossRef]
- Roda, E.; et al. Cognitive Healthy Aging in Mice: Boosting Memory by an Ergothioneine-Rich Hericium erinaceus Primordium Extract. Biology 2023, 12. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.-C.; et al. Ergothioneine protects against neuronal injury induced by β-amyloid in mice. Food and Chemical Toxicology 2012, 50, 3902–3911. [Google Scholar] [CrossRef] [PubMed]
- Ishimoto, T.; et al. TrkB phosphorylation in serum extracellular vesicles correlates with cognitive function enhanced by ergothioneine in humans. npj Science of Food 2024, 8, 11. [Google Scholar] [CrossRef] [PubMed]
- Apparoo, Y.; et al. Ergothioneine and its prospects as an anti-ageing compound. Exp Gerontol 2022, 170, 111982. [Google Scholar] [CrossRef] [PubMed]
- Hatano, T.; et al. Identification of novel biomarkers for Parkinson's disease by metabolomic technologies. J Neurol Neurosurg Psychiatry 2016, 87, 295–301. [Google Scholar] [CrossRef]
- Gründemann, D.; et al. Discovery of the ergothioneine transporter. Proc Natl Acad Sci U S A 2005, 102, 5256–5261. [Google Scholar] [CrossRef] [PubMed]
- Gründemann, D.; Hartmann, L.; Flögel, S. The ergothioneine transporter (ETT): Substrates and locations, an inventory. FEBS Letters 2022, 596, 1252–1269. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; et al. Gene knockout and metabolome analysis of carnitine/organic cation transporter OCTN1. Pharm Res 2010, 27, 832–840. [Google Scholar] [CrossRef]
- Park, J.S.; Davis, R.L.; Sue, C.M. Mitochondrial Dysfunction in Parkinson's Disease: New Mechanistic Insights and Therapeutic Perspectives. Curr Neurol Neurosci Rep 2018, 18, 21. [Google Scholar] [CrossRef]
- Hiller, B.M.; et al. Optimizing maturity and dose of iPSC-derived dopamine progenitor cell therapy for Parkinson’s disease. npj Regenerative Medicine 2022, 7, 24. [Google Scholar] [CrossRef]
- Salari, S.; Bagheri, M. In vivo, in vitro and pharmacologic models of Parkinson's disease. Physiol Res 2019, 68, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; et al. The Parkinsonian mimetic, 6-OHDA, impairs axonal transport in dopaminergic axons. Molecular Neurodegeneration 2014, 9, 17. [Google Scholar] [CrossRef]
- Mendes-Pinheiro, B.; et al. Unilateral Intrastriatal 6-Hydroxydopamine Lesion in Mice: A Closer Look into Non-Motor Phenotype and Glial Response. International Journal of Molecular Sciences 2021, 22, 11530. [Google Scholar] [CrossRef] [PubMed]
- Blum, D.; et al. Molecular pathways involved in the neurotoxicity of 6-OHDA, dopamine and MPTP: Contribution to the apoptotic theory in Parkinson's disease. Prog Neurobiol 2001, 65, 135–172. [Google Scholar] [CrossRef] [PubMed]
- Latchoumycandane, C.; et al. Dopaminergic neurotoxicant 6-OHDA induces oxidative damage through proteolytic activation of PKCδ in cell culture and animal models of Parkinson's disease. Toxicol Appl Pharmacol 2011, 256, 314–323. [Google Scholar] [CrossRef]
- Weihe, E.; et al. Three types of tyrosine hydroxylase-positive CNS neurons distinguished by dopa decarboxylase and VMAT2 co-expression. Cell Mol Neurobiol, 2006; 26, 659–678. [Google Scholar]
- Xicoy, H.; Wieringa, B.; Martens, G.J.M. The SH-SY5Y cell line in Parkinson’s disease research: A systematic review. Molecular Neurodegeneration 2017, 12, 10. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.R., L.S. Hu, and G.Y. Li, SH-SY5Y human neuroblastoma cell line: In vitro cell model of dopaminergic neurons in Parkinson's disease. Chin Med J (Engl) 2010, 123, 1086–1092.
- Avazzadeh, S.; et al. Modelling Parkinson's Disease: iPSCs towards Better Understanding of Human Pathology. Brain Sci 2021, 11. [Google Scholar] [CrossRef]
- Mahajani, S.; et al. Homogenous generation of dopaminergic neurons from multiple hiPSC lines by transient expression of transcription factors. Cell Death & Disease 2019, 10, 898. [Google Scholar]
- Yang, N.-C., A Convenient One-Step Extraction of Cellular ATP Using Boiling Water for the Luciferin–Luciferase Assay of ATP. Analytical Biochemistry 2002, 306, 323–327. [CrossRef]
- Engel, M.; et al. Common pitfalls of stem cell differentiation: A guide to improving protocols for neurodegenerative disease models and research. Cellular and Molecular Life Sciences 2016, 73, 3693–3709. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; et al. Combined small-molecule inhibition accelerates the derivation of functional cortical neurons from human pluripotent stem cells. Nat Biotechnol 2017, 35, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Gaia, S.; et al. Mutant LRRK2 Toxicity in Neurons Depends on LRRK2 Levels and Synuclein But Not Kinase Activity or Inclusion Bodies. The Journal of Neuroscience 2014, 34, 418. [Google Scholar]
- Fu, T.-T.; L. Shen, Ergothioneine as a Natural Antioxidant Against Oxidative Stress-Related Diseases. Frontiers in Pharmacology 2022, 13.
- Nguyen, T.H., R. Nagasaka, and T. Ohshima, CHAPTER 12 - The Natural Antioxidant Ergothioneine: Resources, Chemical Characterization, and Applications, in Lipid Oxidation, A. Logan, U. Nienaber, and X. Pan, Editors. 2013, AOCS Press. p. 381-415.
- Murphy, M.P.; et al. Guidelines for measuring reactive oxygen species and oxidative damage in cells and in vivo. Nature Metabolism 2022, 4, 651–662. [Google Scholar] [CrossRef]
- Suzuki, Y.J., M. Carini, and D.A. Butterfield, Protein carbonylation. Antioxid Redox Signal 2010, 12, 323–325.
- Zhang, X.; et al. Tau Pathology in Parkinson's Disease. Front Neurol 2018, 9, 809. [Google Scholar] [CrossRef] [PubMed]
- Callizot, N.; et al. Necrosis, apoptosis, necroptosis, three modes of action of dopaminergic neuron neurotoxins. PLoS ONE 2019, 14, e0215277. [Google Scholar] [CrossRef]
- Wang, S.-F.; et al. Baicalein prevents 6-OHDA/ascorbic acid-induced calcium-dependent dopaminergic neuronal cell death. Scientific Reports 2017, 7, 8398. [Google Scholar] [CrossRef]
- Halliwell, B., I.K. Cheah, and C.L. Drum, Ergothioneine, an adaptive antioxidant for the protection of injured tissues? A hypothesis. Biochem Biophys Res Commun 2016, 470, 245–250. [CrossRef]
- Li; et al., Uptake and protective effects of ergothioneine in human endothelial cells. J Pharmacol Exp Ther 2014, 350, 691–700. [CrossRef] [PubMed]
- Cheah; et al., Liver ergothioneine accumulation in a guinea pig model of non-alcoholic fatty liver disease. A possible mechanism of defence? Free Radic Res 2016, 50, 14–25. [CrossRef] [PubMed]
- Shinozaki, Y.; et al. Impairment of the carnitine/organic cation transporter 1–ergothioneine axis is mediated by intestinal transporter dysfunction in chronic kidney disease. Kidney International 2017, 92, 1356–1369. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; et al. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen Res 2013, 8, 2003–2014. [Google Scholar] [PubMed]
- Blum, D.; et al. Molecular pathways involved in the neurotoxicity of 6-OHDA, dopamine and MPTP: Contribution to the apoptotic theory in Parkinson's disease. Progress in Neurobiology 2001, 65, 135–172. [Google Scholar] [CrossRef] [PubMed]
- Park, B.C.; et al. Protective effects of fustin, a flavonoid from Rhus verniciflua Stokes, on 6-hydroxydopamine-induced neuronal cell death. Experimental & Molecular Medicine 2007, 39, 316–326. [Google Scholar]
- Kwon, S.H.; et al. Suppression of 6-Hydroxydopamine-Induced Oxidative Stress by Hyperoside Via Activation of Nrf2/HO-1 Signaling in Dopaminergic Neurons. Int J Mol Sci 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Ong, W.Y.; et al. Protective Effect of Ergothioneine Against Stroke in Rodent Models. Neuromolecular Med, 2022.
- Yuzawa, S.; et al. Ergothioneine Prevents Neuronal Cell Death Caused by the Neurotoxin 6-Hydroxydopamine. Cells 2024, 13. [Google Scholar] [CrossRef] [PubMed]
- Brookes, P.S.; et al. Calcium, ATP, and ROS: A mitochondrial love-hate triangle. American Journal of Physiology-Cell Physiology 2004, 287, C817–C833. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; et al. Nimodipine, an L-type calcium channel blocker attenuates mitochondrial dysfunctions to protect against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinsonism in mice. Neurochem Int 2016, 99, 221–232. [Google Scholar] [CrossRef]
- Epand, R.M. The role of dietary ergothioneine in the development of diabetes mellitus. Med Hypotheses 1982, 9, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Fedorova, M., R.C. Bollineni, and R. Hoffmann, Protein carbonylation as a major hallmark of oxidative damage: Update of analytical strategies. Mass Spectrom Rev 2014, 33, 79–97. [CrossRef]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nature Reviews Molecular Cell Biology 2012, 13, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Estébanez, B.; et al. Endoplasmic Reticulum Unfolded Protein Response, Aging and Exercise: An Update. Front Physiol 2018, 9, 1744. [Google Scholar] [CrossRef]
- Holtz, W.A.; et al. Oxidative stress-triggered unfolded protein response is upstream of intrinsic cell death evoked by parkinsonian mimetics. Journal of Neurochemistry 2006, 99, 54–69. [Google Scholar] [CrossRef]
- Bizzozero, O.A., Protein Carbonylation in Neurodegenerative and Demyelinating CNS Diseases, in Handbook of Neurochemistry and Molecular Neurobiology: Brain and Spinal Cord Trauma, A. Lajtha, N. Banik, and S.K. Ray, Editors. 2009, Springer US: Boston, MA. p. 543-562.
- Di Rita, A.; et al. AMBRA1-Mediated Mitophagy Counteracts Oxidative Stress and Apoptosis Induced by Neurotoxicity in Human Neuroblastoma SH-SY5Y Cells. Frontiers in Cellular Neuroscience 2018, 12. [Google Scholar] [CrossRef]
- Elkon, H., E. Melamed, and D. Offen, Oxidative stress, induced by 6-hydroxydopamine, reduces proteasome activities in PC12 cells: Implications for the pathogenesis of Parkinson's disease. J Mol Neurosci 2004, 24, 387–400. [CrossRef]
- Smith, M.P.; W.A. Cass, Oxidative stress and dopamine depletion in an intrastriatal 6-hydroxydopamine model of Parkinson's disease. Neuroscience 2007, 144, 1057–1066. [CrossRef]
- Lamhonwah, A.M.; I. Tein, Novel localization of OCTN1, an organic cation/carnitine transporter, to mammalian mitochondria. Biochem Biophys Res Commun 2006, 345, 1315–1325. [CrossRef]
- Shitara, Y.; et al. Role of Organic Cation/Carnitine Transporter 1 in Uptake of Phenformin and Inhibitory Effect on Complex I Respiration in Mitochondria. Toxicological Sciences 2012, 132, 32–42. [Google Scholar] [CrossRef]
Figure 1.
Derivation of day 40 dopaminergic neurons and quality control analysis (A) Schematic representation of differentiation protocol to derive day 40 induced dopaminergic neurons from hiPSCs (BJ and GM23720) and hESC (H9). (B) Confocal imaging of neuronal and dopaminergic markers. DAPI (blue) stains the nucleus. (C) ImageJ analysis of confocal images of TH and NeuN to quantify the percentage of dopaminergic neurons present in the culture. (C–D) Data are represented as mean ± SD (n=3). Data was analyzed by unpaired student’s t-test. * p ≤ 0.05. ** p ≤ 0.01. *** p ≤ 0.001. **** p ≤ 0.0001.
Figure 1.
Derivation of day 40 dopaminergic neurons and quality control analysis (A) Schematic representation of differentiation protocol to derive day 40 induced dopaminergic neurons from hiPSCs (BJ and GM23720) and hESC (H9). (B) Confocal imaging of neuronal and dopaminergic markers. DAPI (blue) stains the nucleus. (C) ImageJ analysis of confocal images of TH and NeuN to quantify the percentage of dopaminergic neurons present in the culture. (C–D) Data are represented as mean ± SD (n=3). Data was analyzed by unpaired student’s t-test. * p ≤ 0.05. ** p ≤ 0.01. *** p ≤ 0.001. **** p ≤ 0.0001.
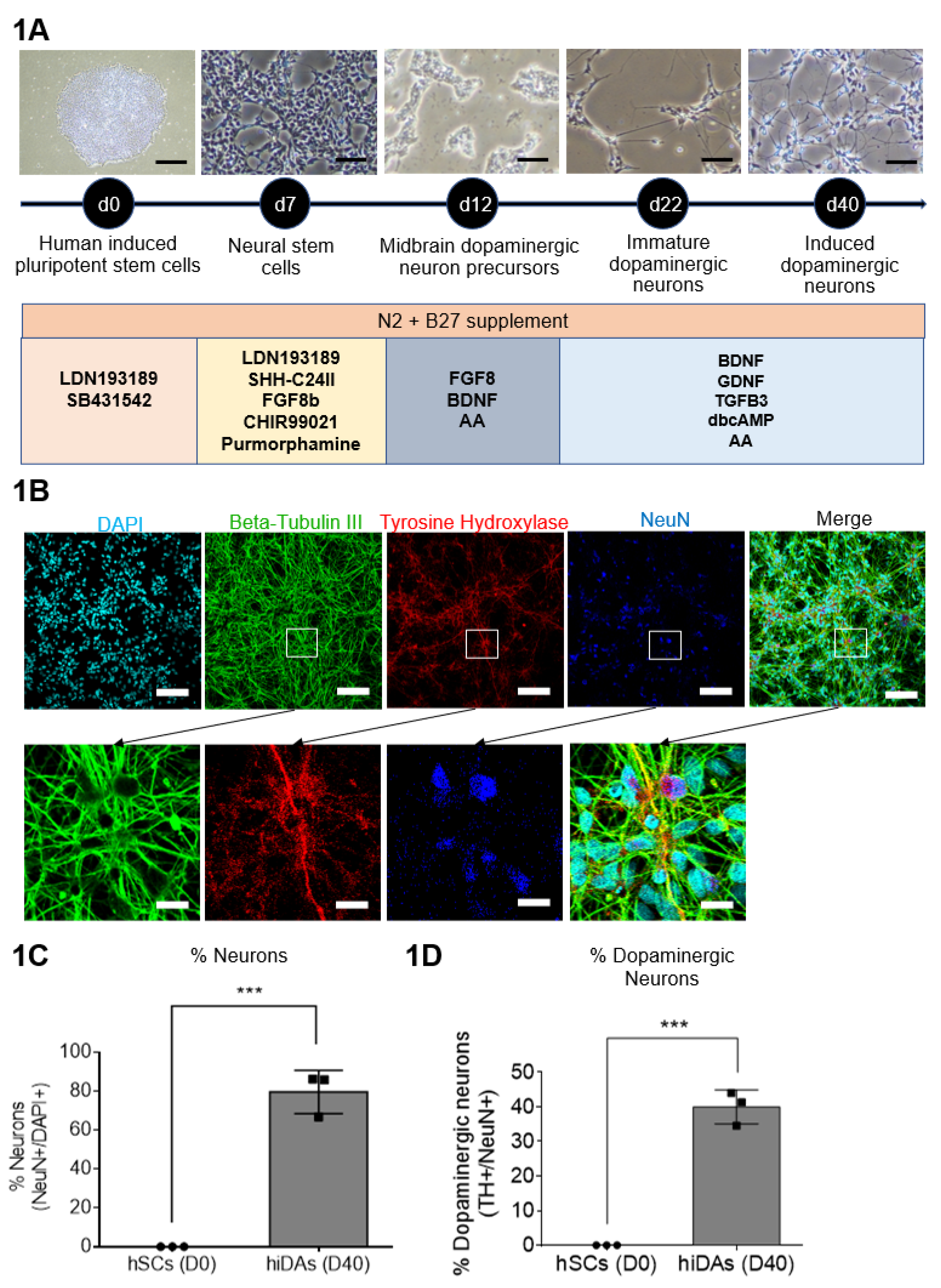
Figure 2.
ET intracellular uptake protects iDAs against 6-OHDA induced cell death, reduction in ATP levels and loss of dopamine secretion. (A) MTT assay showing quantitative changes in cellular metabolic activity in iDAs from the various treatment groups (B) Qualitative changes in iDAs from the various treatment groups using MTT assay, under microscope. (C) Hematoxylin and Eosin (H&E) staining to show changes of neuronal morphology from various treatment groups. (D) Propidium iodide assay using flow cytometry showing changes in quantity of non-viable TH+ cells. Higher MFI readings denote an increase in non-viable TH+ cells from the treatment group and vice versa. (E) ATP assay showing relative changes to intracellular ATP levels. Higher absorbance values denote greater quantities of ATP in the cell lysates of the treatment group. (F) Dopamine secretion assay showing the changes to the amounts of dopamine being secreted by the day 40 iDAs in each treatment group. Higher absorbance values denote greater quantities of dopamine. (A, D-F) Data are represented as mean ± SD (n=3). Data were analyzed by One-Way ANOVA with Bonferroni’s multiple comparison post-hoc test. * p ≤ 0.05. ** p ≤ 0.01. *** p ≤ 0.001. **** p ≤ 0.0001.
Figure 2.
ET intracellular uptake protects iDAs against 6-OHDA induced cell death, reduction in ATP levels and loss of dopamine secretion. (A) MTT assay showing quantitative changes in cellular metabolic activity in iDAs from the various treatment groups (B) Qualitative changes in iDAs from the various treatment groups using MTT assay, under microscope. (C) Hematoxylin and Eosin (H&E) staining to show changes of neuronal morphology from various treatment groups. (D) Propidium iodide assay using flow cytometry showing changes in quantity of non-viable TH+ cells. Higher MFI readings denote an increase in non-viable TH+ cells from the treatment group and vice versa. (E) ATP assay showing relative changes to intracellular ATP levels. Higher absorbance values denote greater quantities of ATP in the cell lysates of the treatment group. (F) Dopamine secretion assay showing the changes to the amounts of dopamine being secreted by the day 40 iDAs in each treatment group. Higher absorbance values denote greater quantities of dopamine. (A, D-F) Data are represented as mean ± SD (n=3). Data were analyzed by One-Way ANOVA with Bonferroni’s multiple comparison post-hoc test. * p ≤ 0.05. ** p ≤ 0.01. *** p ≤ 0.001. **** p ≤ 0.0001.
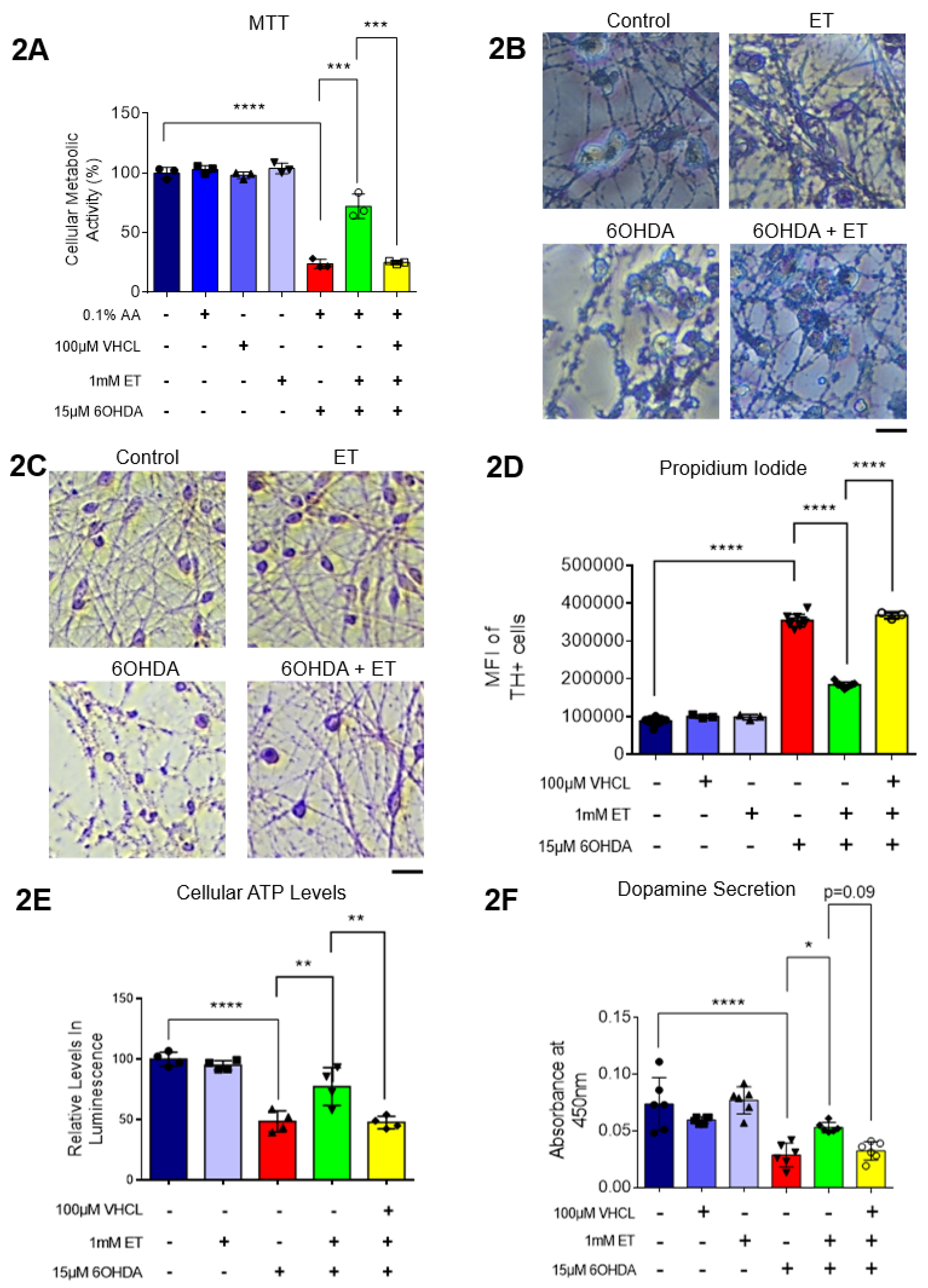
Figure 3.
TH+ Day 40 iDAs were analyzed for mROS and TMRM levels. (A) Scatter plot of flow cytometry data (TH-TMRM co-staining) of the day 40 iDAs from the different treatment groups. (B) Bar chart of flow cytometry data for TH and TMRM co-staining to show relative changes in the different treatment groups. (C) Scatter plot of flow cytometry data (TH-MitoSOX co-staining) of the day 40 iDAs from the different treatment groups. (D) Bar chart of flow cytometry data for TH and MitoSOX co-staining to show relative changes in the different treatment groups. (B,D) Data are represented as mean ± SD (n=3). Data were analyzed by One-Way ANOVA with Bonferroni’s multiple comparison post-hoc test. * p ≤ 0.05. ** p ≤ 0.01. *** p ≤ 0.001. **** p ≤ 0.0001.
Figure 3.
TH+ Day 40 iDAs were analyzed for mROS and TMRM levels. (A) Scatter plot of flow cytometry data (TH-TMRM co-staining) of the day 40 iDAs from the different treatment groups. (B) Bar chart of flow cytometry data for TH and TMRM co-staining to show relative changes in the different treatment groups. (C) Scatter plot of flow cytometry data (TH-MitoSOX co-staining) of the day 40 iDAs from the different treatment groups. (D) Bar chart of flow cytometry data for TH and MitoSOX co-staining to show relative changes in the different treatment groups. (B,D) Data are represented as mean ± SD (n=3). Data were analyzed by One-Way ANOVA with Bonferroni’s multiple comparison post-hoc test. * p ≤ 0.05. ** p ≤ 0.01. *** p ≤ 0.001. **** p ≤ 0.0001.
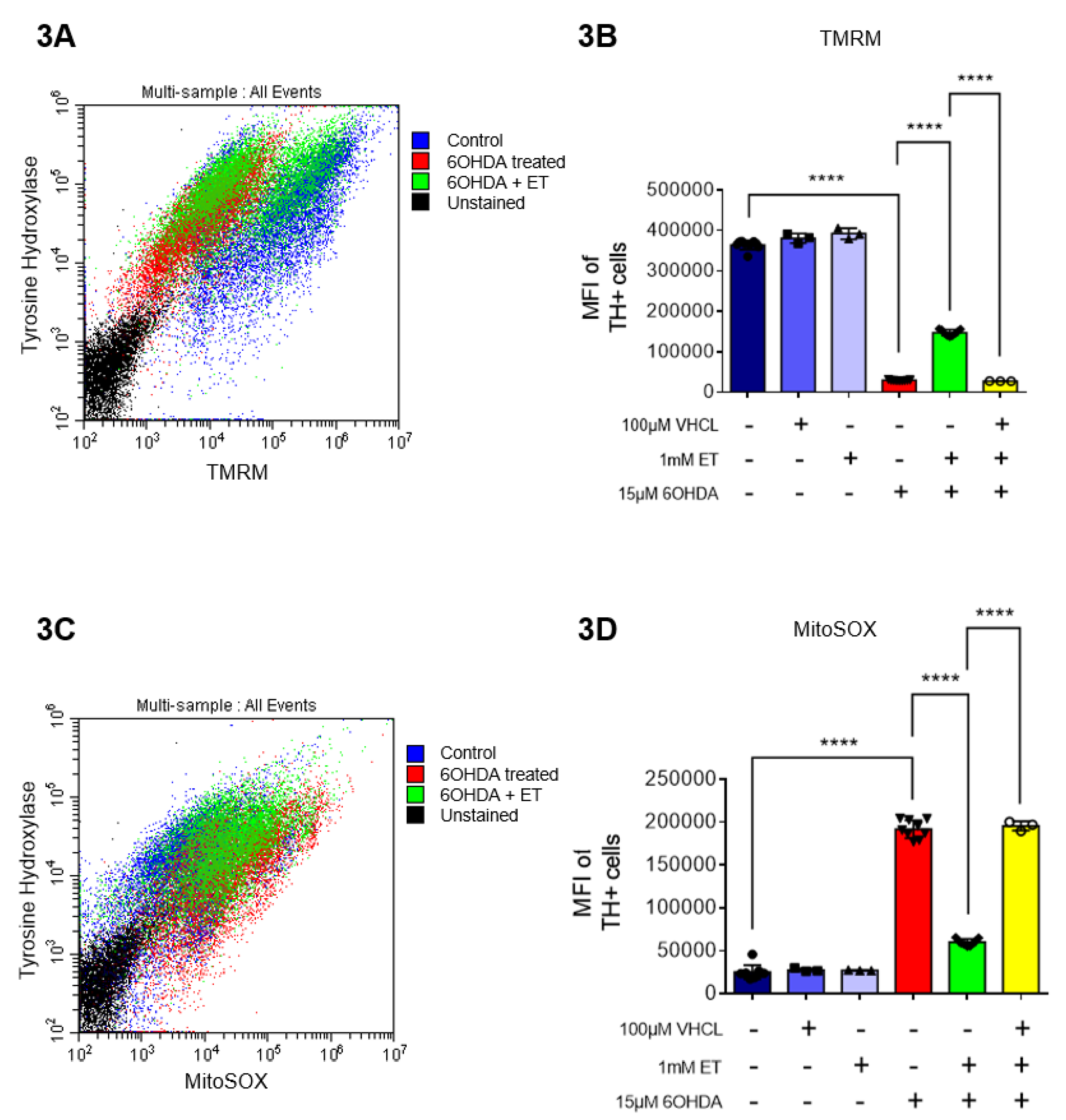
Figure 4.
Effect of intracellular ET on cellular metabolism and oxidative stress in SH-SY5Y cells. (A) MTT assay showing changes to cellular metabolic activity in the SH-SY5Y cells from the various treatment groups. (B) ATP assay showing relative levels in intracellular ATP as compared to the control group. (C) Fluo-4 AM assay measures intracellular free calcium levels. Data are represented as mean ± SD (n=4). (D) TMRM assay measures mitochondrial membrane potential (MMP). Data are represented as mean ± SD (n=3). (E) MitoSOX assay using flow cytometry showing changes in mROS levels. Data are represented as mean ± SD (n=3). (F) ImageJ analysis of immunoblot to quantify relative amounts of oxidized proteins between the treatment groups, as compared to control. (G) Flow cytometry analysis to quantify relative amounts of tyrosine hydroxylase (TH) enzyme expression as compared to control. Data were analyzed by One-Way ANOVA with Bonferroni’s multiple comparison post-hoc test. Unless stated in figures, * p ≤ 0.05. ** p ≤ 0.01. *** p ≤ 0.001. **** p ≤ 0.0001.
Figure 4.
Effect of intracellular ET on cellular metabolism and oxidative stress in SH-SY5Y cells. (A) MTT assay showing changes to cellular metabolic activity in the SH-SY5Y cells from the various treatment groups. (B) ATP assay showing relative levels in intracellular ATP as compared to the control group. (C) Fluo-4 AM assay measures intracellular free calcium levels. Data are represented as mean ± SD (n=4). (D) TMRM assay measures mitochondrial membrane potential (MMP). Data are represented as mean ± SD (n=3). (E) MitoSOX assay using flow cytometry showing changes in mROS levels. Data are represented as mean ± SD (n=3). (F) ImageJ analysis of immunoblot to quantify relative amounts of oxidized proteins between the treatment groups, as compared to control. (G) Flow cytometry analysis to quantify relative amounts of tyrosine hydroxylase (TH) enzyme expression as compared to control. Data were analyzed by One-Way ANOVA with Bonferroni’s multiple comparison post-hoc test. Unless stated in figures, * p ≤ 0.05. ** p ≤ 0.01. *** p ≤ 0.001. **** p ≤ 0.0001.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



