
Submitted:
07 May 2024
Posted:
07 May 2024
You are already at the latest version
Abstract
Epidemiological studies point cholesterol as a possible key factor for both prostate cancer incidence and progression. So, it could represent a targetable metabolite as the most aggressive tumors also appear to be sensitive to therapies designed to decrease hypercholesterolemia, such as statins. However, whether and how cholesterol, through its dietary uptake and its metabolism, could be important for early tumorigenesis, remains unknown. Oncogene clonal induction in Drosophila melanogaster accessory gland allows to reproduce tumorigenesis from initiation to early progression, where tumour cells undergo basal extrusion to form extra-epithelial tumors. Here we show that these tumors accumulate lipids, and especially esterified cholesterol, as in human late carcinogenesis. Interestingly, high cholesterol diet displays limited effect on accessory gland tumorigenesis. On the contrary, cell-specific downregulation of cholesterol uptake, intracellular transport or metabolic response, impairs the formation of such tumors. Furthermore, in this context, high cholesterol diet suppresses this impairment. Taken together, these results reveal that during early tumorigenesis, tumour cells strongly increase their uptake and use of dietary cholesterol to specifically promote the step of basal extrusion. Interestingly, expression data from primary prostate cancer tissues indicate an early signature of redirection from cholesterol de novo synthesis to uptake. Altogether, these results suggest by which mechanism reduction of dietary cholesterol could lower the risk and slow down the progression of prostate cancer.
Keywords:
prostate cancer
; cholesterol
; early tumorigenesis
; basal extrusion
1. Introduction
In the last decades, prostate cancer (PCa) incidence has reached the first rank of men cancers. Within the various factors involved, dietary fat and especially cholesterol has been pointed out as a potential modulator of prostate cancer risk and development [1,2].
In men, the association between cholesterol levels and aggressiveness has largely been reported [3,4,5,6,7] along with the correlation between cholesterol-lowering drugs, i.e. statins, and a decreased prostate cancer risk [7,8,9,10,11]. However, conflicting studies and meta-analyses have indicated that there is no clear association between plasma levels of total or fractions of cholesterol, and PCa, [5,12,13,14]. In the same way, the protective role of statins have recently been debated in regard to cancer progression [15,16], questioning the reality of the connection between plasma cholesterol levels and PCa. Nonetheless, cholesterol metabolism is indeed specifically altered in prostate tumors [17] and, in many preclinical models, cholesterol has been linked to prostate cancer progression notably via its role as lipid raft component promoting akt and erk signal transduction [18,19,20,21]. There, activation of cholesterol master regulators Liver X Receptors (LXRs) depletes cholesterol from lipid rafts and subsequently induces cancer cell apoptosis [22]. Furthermore, if high-cholesterol diet does not alter murine prostate architecture by itself, the knockout of LXRs in this context induces prostatic intra-epithelial neoplasia [23], suggesting that loss of cholesterol homeostasis could also pave the way for prostate cancer development in case of excessive cholesterol uptake.
We have previously developed an in vivo model of epithelial tumorigenesis in the accessory gland of drosophila melanogaster covering the steps from initiation to early progression characterized by tumour formation [24]. Indeed, drosophila represents a strong model for decrypting epithelial cancer-related mechanisms, for example for lung or colon [25,26], and accessory gland itself represents a functional equivalent of a prostatic acinus [27,28,29,30,31]. Furthermore, there is a strong conservation of cholesterol role and metabolism in this insect [32,33,34]. Also, in mammals, cholesterol is supplied through diet and de novo synthesis [35], possibly masking the direct impact of dietary cholesterol on tumorigenesis. Crucially, the cholesterol auxotrophy of the fruit fly makes it only dependent of dietary intake of this metabolite [36]. Together, this renders the drosophila accessory gland a particularly relevant model to assess whether and how cholesterol could play on the early steps of epithelial tumorigenesis.
Herein, we show that, in the accessory gland, oncogene-induced tumours accumulate cholesterol into lipid droplets as in human late carcinogenesis [37]. In this condition, forcing further the activation of cholesterol metabolism through genetic or dietetic approaches has no effect. On the contrary, tumour cell-specific downregulation of cholesterol metabolism decreases the critical step of basal extrusion, limiting the formation of tumours, in a cholesterol dose-dependent manner. Finally, using a cohort of adenocarcinoma, we show that a profound deregulation of cholesterol metabolism occurs already in primary prostate cancer, suggesting that in human also, this deregulation could be important for the early steps of tumorigenesis.
2. Materials and Methods
2.1. Fly Stocks and Experimental Crosses
y,w,HS:flp122/+;Act:FRTstopFRTGal4, UAS:GFP/CyO flies allowed conditional clonal expression of GFP. Combination with UAS:EgfrλTop (#59843) allowed conditional clonal co-expression of GFP and EgfrλTop (EGFRλ flies) . GFP-Egfrλ flies were then crossed with following stocks to realize experiments: UAS-GFP.nls (#4775, control condition, designed in figures as EGFRλ), UAS-LRP1 RNAi (#31151), UAS-LpR2 RNAi (#54461), UAS-Npc1a RNAi (#37504), UAS-Npc1b RNAi (#38296), UAS-CG8112 RNAi (#63035) and UAS-miR DHR96 (#27992, designed as UAS-DHR96 RNAi in figures) from the Bloomington Stock Center.
2.2. Conditional Expression Induction
Briefly, flippase (flp)/FRT system was activated by a 12 min heat-shock induction at 37°C during the pupal stage, to create an average of 4–6 clones per accessory gland (≈1% of total number of epithelial cells). Flies were then kept at 27°C until the end of pupal stage. Males were collected at emergence from pupae 3 to 3.5 days after heat shock and kept for another 2.5 days at 27°C before dissection.
2.3. High Cholesterol Diet
After the cross, flies were either raised on standard-diet or high-cholesterol-diet (HCD) complemented with 0.2% cholesterol (#3045, Sigma-Aldrich). These diet conditions were also conserved after males’ collection at the emergence from pupae.
2.4. Immunohistochemistry and Imaging
Accessory glands were dissected in PBS, fixed for 10 min in 4% formaldehyde, washed and permeabilized for 15 min in PBS containing 0.2% Triton (PBS-T). Glands were blocked for 1 hour with 0.5% of BSA in PBS-T then incubated overnight at 4°C with primary antibodies diluted in the same blocking solution. After three washes in PBS-T, glands were incubated in secondary antibody diluted 1:1000 in blocking solution for 1 h at room temperature with DAPI (DiAminidoPhenylIndol, D8417, Sigma) 1:1000 (DNA staining) and/or Alexa633-phalloidin (A22284, Life Technology) 1:5000 (to reveal F-actin). Glands were then washed three times with PBS and subsequently mounted in Vectashield (#-1000, Vector Laboratories). Imaging was realised on Leica SP8 confocal microscope, and image stacks were processed either in ImageJ or Imaris softwares.
List of antibodies: Mouse Coracle (1:400, #C566.9 DSHB), NileRed (1ng/mL, Sigma-Aldrich, Saint-Louis, USA), Bodipy (2ng/mL, #D3835 Invitrogen), secondary antibodies coupled to different fluorophores 488 (1:1000, A11055 Invitrogen), Cy3 or Cy5 (1:1000, 711-165-152, 715-165-151,715-175-150, Jackson Immunology).
2.5. Clones, Cells and Nuclei Size
Clones/tumours, cells and nuclei volumes were determined from 3D reconstruction and automatic quantification in Imaris software. For each clone/tumor, average cell size was determined by ratio between clone size and number of nuclei in the considered clone.
2.6. Invasive Tumour Frequency
Tumour frequency was determined as the percentage of flies that displayed at least one tumour on their accessory glands at dissection.
2.7. Statistical Analyses
All experiments were repeated independently a minimum of three times (N: number of independent experiments) on numerous glands (n: number of pairs of glands or number of imaged and quantified glands for tumour size and nuclei number quantification). Statistical analyses were performed using GraphPad Prism 6: for tumour volume, nuclei number and droplets volume, quantification were compared by Krustal-Wallis test as % of glands with extra-glandular tumours were compared by Chi2 test.
3. Results
3.1. Accessory Gland Tumors Accumulate Cholesterol into Lipid Droplets
Accessory gland represents a perfectly defined epithelial compartment comparable to a single prostatic acinus. It is composed of a monolayer of secretory epithelial cells surrounded by a basement membrane in which a well-organized network of muscle fibers is completely enclosed (revealed by phalloidin staining in Figure 1A,D, yellow). Here, in order to mimic tumour initiation, clonal expression of constitutively active version of EGFR (EGFRλ condition) coupled to GFP was realized in approximatively 1% of the accessory gland cells. As previously described [24], this expression leads to the formation of two kinds of GFP-positive tumour cells (Figure 1). First, epithelial clones composed of slightly hypertrophic cells whose cytoskeleton are disorganized (Figure 1. white arrowheads) and second, GFP-positive tumours that grow outside the epithelial compartment consequently of a phenomenon of basal extrusion (Figure 1. empty arrowheads). These tumors, which represent a state of early progression, are composed of cells that bare hallmarks of cancer, such as hypertrophy, hyperplasia, the loss of epithelial markers, and tumours are furthermore associated to neotracheogenesis (a neoangiogensis equivalent) [24]. In this context, we wondered whether this parallel could be extended to abnormal lipid storage. Indeed, we observe by classical Nile Red staining the accumulation of intracellular neutral lipid droplets specifically in tumour cells (Figure 1A,B: magenta; D: Nile Red staining only, grey). To further evaluate the content of these droplets, we used Bodipy, a marker of esterified cholesterol that proves the presence of stored cholesterol into these droplets (Figure 1E,F: magenta; H: Bodipy staining only, grey). So, we concluded that, as in human, formation of tumoursduring progression is accompanied by lipid, and especially esterified cholesterol, accumulation into the tumour cells.
3.2. Cholesterol Metabolism Downregulation Impairs EGFRλ Induced Cholesterol Storage
In order to test whether this excess of cholesterol is important for tumour formation, we then decided to downregulate expression of genes involved in cholesterol uptake (LRP1, LpR2) (Figure 2E-H and I-L, respectively) [38], intracellular trafficking (Npc1a) (Figure 2M-P) [39,40] and storage (Figure 2Q-T) (CG8112 – ortholog of SOAT1/ACAT1) [41] in a clone-specific manner. RNAi against Npc1b (Figure 2A-D), involved in cholesterol import was used as a negative control as its expression is described as restricted to the gut [42]. In all the conditions, except Npc1b RNAi, we observed a strong decrease in lipid droplets volume and number (Figure 2), indicating that tumor-specific cholesterol storage is indeed impaired in the tumours.
We so concluded that each of these actors (except Npc1b) is necessary for cholesterol accumulation in drosophila accessory gland tumours, showing that all of them represent targetable proteins for regulation of cholesterol abnormal accumulation.
3.3. Hyperactivation of Cell Autonomous Cholesterol Metabolism Specifically Drives Basal Extrusion
We then assessed the percentage of glands baring tumours in the previously described conditions. For all the genotypes, except Npc1b RNAi negative control, we observed a significant decrease in tumour frequency (Figure 3A), showing that not only uptake (LRP1, LpR2), but also intracellular metabolism (Npc1a, CG8112) of cholesterol is necessary for tumour formation itself. This indicates that, in case of cell transformation by oncogene expression, a common cause of initiation, cholesterol metabolism is implicated in early tumorigenesis.
In addition, in order to see if cholesterol could be involved in early progression as well, we also characterized tumour volume (Figure 3B), tumour cell number (Figure 3C) and cell volume (Figure 3D) for all considered genotypes. Interestingly, all these parameters remain unaffected, and this phenotype is not altered throughout time (Figure 1SD).
Altogether, these results demonstrate that the observed deregulation of cholesterol metabolism in tumour cells does not impact tumour growth itself, but is necessary for tumour formation. So, we conclude that this apparent hyperactivation of cholesterol metabolism is specifically important for the early step of basal extrusion, when tumour cells actively leave the accessory gland to form tumours outside the epithelial compartment.
3.4. Further Cholesterol Metabolism Deregulation Has No Effect on Early Tumorigenesis
As tumours accumulate high levels of cholesterol, we then wondered whether it was possible to push further this phenotype, and whether this could impact tumour characteristics or formation. In order to do so, we used two different strategies alone or combined. First, we mimicked the so-called western diet, with flies fed either with a standard diet or a diet supplemented with 0.2% of cholesterol (High Cholesterol Diet condition or HCD). Second, in order to maximize tumour cell specific deregulation of cholesterol metabolism, we downregulated the fly cholesterol sensor DHR96 expression [43,44].
Compared to tumour control condition (Nile red staining in Figure 4A-D and quantification in Figure 5A), high cholesteroldiet could slightly increase the accumulation of lipids into the tumours, denoting a limited capacity to exacerbate the abnormal lipid storage induced by the oncogene expression in the tumour cells. Interestingly, neither tumour characteristics (Figure 5B,C) nor basal extrusion (Figure 5D) were significantly affected by high cholesterol diet. We concluded that oncogene transformation is sufficient to obtain an independency to diet for promoting tumour formation. For genetic deregulation of cholesterol metabolism by co-expression of DHR96 RNAi, a limited increase of lipid storage was observed (Figure .4I-L and quantification in Figure 5A), but once more no effect on either tumour characteristics or tumour formation was observed (Figure 5B-D). We concluded that oncogene-driven deregulation of cholesterol metabolism may not exert a maximal effect in term of lipid accumulation, but is definitely on top for tumour promotion. Finally, double deregulation by downregulation of DHR96 in a high cholesterol diet does not result in a higher accumulation of lipid droplets (Figure 4M-P and Figure 5A) and does not affect the studied parameters of tumorigenesis (Figure 5B-D) compared to EGFRl conditions (see also Figure 1. SD).
Overall, these results show that oncogene transformation exerts a profound deregulation of cholesterol homeostasis which in turn promotes basal extrusion, and that further deregulation through diet or targeting cholesterol regulators in the tumour cells cannot potentiate the initial effect due to the oncogene expression.
3.5. High Cholesterol Diet Counteracts Effect of Cholesterol Metabolism Downregulation in Egfrλ Induced Tumorigenesis
As downregulation of cholesterol metabolism reduces basal extrusion, we then wondered if in this case, high cholesterol diet could now impact cholesterol storage and tumour formation. Interestingly, increasing dietary cholesterol does restore Nile red staining in all genotypes (compare Figure 6A-P to Figure 2E-T). Strikingly, it furthermore completely counteracts the reduction in tumour formation that was observed in all the conditions where cholesterol metabolism is genetically impaired (Figure 7A-E). This indicates that dietary intake of cholesterol can finally play a major role in tumour formation in a context of decreased cholesterol access or metabolism by the tumour cells.
3.6. Genes Coding for Cholesterol Metabolism Are Deregulated in Primary Prostate Cancer
In order to understand when cholesterol metabolism deregulation could be important during prostate carcinogenesis, we then determined from published data the expression of genes associated to this pathway in normal, primary or metastatic samples [45]. First, master regulators of cell cholesterol homeostasis are already deregulated in primary cancer, with a decreased NR1H2, expression, coding for LXRb which is associated to the management of excess of cholesterol (Figure 8A). On the contrary, SREBF1, which codes for SREBP1 whose role is to increase cholesterol cell levels, remains unchanged even though it tends to be upregulated (Figure 8B). This pleads for an increased uptake and retention of cholesterol. Second, genes involved in cholesterol uptake such as SCARB1, coding for the receptor for high density lipoprotein cholesterol(HDL), or LDLR, are either maintained or upregulated in primary cancer (Figure 8C,D). On the contrary, genes implicated in cholesterol de novo synthesis such as HMGCr (Figure 8E), HMGCS1 (Figure 8F), FDFT1 (squalene synthase, Figure 8G) and SQLE (Figure 8H) are either maintained or downregulated. This pleads for a role of dietary uptake rather than de novosynthesis in the building of higher levels of cholesterol into the tumour cells. Third, SOAT1, coding for the enzyme that catalyzes cholesterol esterification, is specifically overexpressed in primary tumours compared to either normal and metastatic samples (Figure 8I). This pleads for an early role for cholesterol accumulation on tumorigenesis.
So, as cholesterol homeostasis is so clearly disturbed in primary cancer already, it could indeed promote the early steps of prostate tumorigenesis in addition to its role in later phases of progression.
Overall, we conclude that targeting cholesterol metabolism in early phases of tumorigenesis could reduce tumour promotion but that this effect is dependent of the level of cholesterol intake.
4. Discussion
Dietary habits have been pointed out as a risk factor in plethora of cancers, and especially western diet characterized by a high content of lipids and sugar and a low level of vegetables. Prostate cancer follows the same trend with some data associating cholesterol blood levels to cancer incidence and aggressiveness [1,2,46]. Experiments with high cholesterol diet have been performed in murine models harboring human prostate cell lines xenografts, indicating that late evolution of cancer to castration resistance, possibly through higher androgen anabolism, correlates with high plasma cholesterol [47,48]. Also, cholesterol anabolism appears increased in CRPC through SQLE overexpression, with an impact on lymph node invasion in a xenografted mouse model [49]. However, whether cholesterol metabolism could also be implicated in early tumorigenesis remains unknown. High plasma cholesterol levels have been linked to a risk to develop high grade cancer [5,12], but the role of hypercholesterolemia on cancer incidence, if appearing in some clinical studies, is still not characterized by a meta-analysis approach [13]. Furthermore, relative part of diet versus intra-tumoral production is not resolved. In this context, we have decided to use a drosophila model of early tumorigenesis, in the accessory gland, a functional and structural equivalent of a prostatic acinus previously developed in the team [24]. As insects are auxotroph for cholesterol, by modifying the food content, we can directly evaluate the impact of normal and high diet cholesterol on tumour incidence.
In this context, we have first shown that tumours formed after basal extrusion in the gland accumulate lipids, and especially esterified cholesterol into droplets, a phenomenon to compare with the human pathology [37]. Neither this phenotype nor tumour incidence, volume and cell number are affected by high cholesterol diet. This shows that initiation, here by oncogene expression, is by itself sufficient to render the tumour cells independent of normal to high cholesterol diets. Indeed, lipid accumulation into droplets in tumour cells has never been associated to diet, but rather linked to genetic alterations appearing frequently in the PI3K/Akt pathway during the late stages of cancer progression [37]. However, this pathway is itself implicated in basal extrusion, and could be recruited well before being affected by a genetic alteration by autocrine activation [24] to sustain cholesterol accumulation and promote the early steps of cancer development. Overall, this could explain why, despite clear evidence of the role of western diet on cancer incidence [1,46], role of available cholesterol itself is still not forcefully established through its plasma levels [13].
We also decreased cholesterol metabolism specifically in tumour cells from initiation, by co-expression of RNAi targeting cholesterol import, transport or storage along with the oncogene expression. There, a decrease in lipid accumulation was obtained, and if the average number of tumours was significantly decreased in these conditions, their size and cell composition remained mostly unaffected. This shows that the growth of the tumour itself does not depend much on hyperactivated cholesterol metabolism, but that, on the contrary, formation of tumours outside the accessory gland does depend on it. This itself relies on the capacity for tumour cells to migrate through basement membrane of the epithelial compartment, a phenomenon called epithelial basal extrusion. Basal extrusion is thought to be a funding event in adenocarcinoma formation [50,51], i.e. in a majority of cancers. However, despite its central role in carcinogenesis, this elusive event is largely understudied, due to the paucity of specific tools dedicated to its analysis, and logically to the absence of close-to-normal human samples where this event could be in progress. Overall, the molecular events that are known to be necessary for basal extrusion concern mostly the Ras/MAPK pathway [24,52,53,54], along with PI3K/Akt pathway co-activation [24]. Furthermore, it has also been shown that T-box transcription factors are necessary for basal extrusion [55], as well as the sphingosine 1-phosphate (S1P) pathway that could play a role as well in cell survival as in cell morphology [52,56]. Here, we so add one component on the control of basal extrusion: hyperactivation of cholesterol metabolism. Then, basal extrusion appears more and more as a highly complex and highly controlled step of epithelial tumorigenesis. On the one hand, it provides a view on the richness of pathway deregulation appearing early in the carcinogenesis, and, on the other hand it indicates potential targetable metabolites to limit cancer progression.
Concerning the source of cholesterol in the tumour cells, by the use of an auxotroph model, we showed here that there is no need of intracellular cholesterol anabolism in order to acquire cholesterol accumulation in the tumour cells (Figure 1). This correlates with the expression of genes in cancer patients, where cholesterol uptake genes, as SCARB1 are overexpressed [17] and, in contrast, genes implicated in cholesterol anabolism are downregulated (Figure 8). If tumour cells are more dependent on dietary cholesterol rather than cell production, it could explain why western diet is a good indicator of prostate cancer development [38,39], and why statins, which block de novo synthesis of cholesterol, still have an uncertain role on prostate cancer incidence [13].
5. Conclusions
Altogether, this study highlights the pro-tumoral role of dietary cholesterol and of its metabolism in situ in a model of prostate cancer, and especially points out at its role in the critical step of basal extrusion leading to the formation of tumours (Figure 9). Considering the strong deregulation of cholesterol metabolism in primary adenocarcinoma, these findings could be indicative of the modus operandum by which high cholesterol metabolism could promote prostate carcinogenesis in human.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: cholesterol dietary intake and/or cholesterol metabolism has no effect on tumour phenotype itself.
Author Contributions
Conceptualization, M.V., J.M.L., S.B., L.M. and C.J; methodology, M.V., L.M., C.J.; validation, M.V., E.B. and C.J.; formal analysis, M.V., E.B. and C.J.; investigation, M.V., E.B. and C.J.; resources, M.V., E.B. A.T., A.K. and C.J.; writing—original draft preparation, M.V and C.J.; writing—review and editing, J.M.L, S.B, L.M.; visualization, M.V. and C.J.; supervision, C.J.; project administration, C.J.; funding acquisition, C.J. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by La Ligue contre le cancer (C.J.), La Fondation ARC (M.V.) and French Center for the 3Rs (FC3R, Re-innov project, C.J.).
Institutional Review Board Statement
Not applicable.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors on request.
Acknowledgments
The authors thank N. Anglaret, L. Babkina, A. de Haze, C. Gaudichet and C. Tamisier for technical help; P. Pouchin for support with imaging treatment; S. de Joussineau for scientific illustration; Bloomington Drosophila Stock Center (BDSC) for providing fly stocks, and Drosophila Studies Hybridoma Bank (DSHB) for providing antibodies. CLermont Imagerie Confocale (CLIC) facility for support with imaging.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Shimizu, H.; Ross, R.K.; Bernstein, L.; Yatani, R.; Henderson, B.E.; Mack, T.M. Cancers of the Prostate and Breast among Japanese and White Immigrants in Los Angeles County. Br. J. Cancer 1991, 63, 963–966. [Google Scholar] [CrossRef] [PubMed]
- Cook, L.S.; Goldoft, M.; Schwartz, S.M.; Weiss, N.S. Incidence of Adenocarcinoma of the Prostate in Asian Immigrants to the United States and Their Descendants. J. Urol. 1999, 161, 152–155. [Google Scholar] [CrossRef] [PubMed]
- Platz, E.A.; Clinton, S.K.; Giovannucci, E. Association between Plasma Cholesterol and Prostate Cancer in the PSA Era. Int. J. cancer 2008, 123, 1693–1698. [Google Scholar] [CrossRef] [PubMed]
- Platz, E.A.; Till, C.; Goodman, P.J.; Parnes, H.L.; Figg, W.D.; Albanes, D.; Neuhouser, M.L.; Klein, E.A.; Thompson, I.M.J.; Kristal, A.R. Men with Low Serum Cholesterol Have a Lower Risk of High-Grade Prostate Cancer in the Placebo Arm of the Prostate Cancer Prevention Trial. Cancer Epidemiol. biomarkers Prev. a Publ. Am. Assoc. Cancer Res. cosponsored by Am. Soc. Prev. Oncol. 2009, 18, 2807–2813. [Google Scholar] [CrossRef] [PubMed]
- Mondul, A.M.; Clipp, S.L.; Helzlsouer, K.J.; Platz, E.A. Association between Plasma Total Cholesterol Concentration and Incident Prostate Cancer in the CLUE II Cohort. Cancer Causes Control 2010, 21, 61–68. [Google Scholar] [CrossRef]
- Kitahara, C.M.; Berrington de González, A.; Freedman, N.D.; Huxley, R.; Mok, Y.; Jee, S.H.; Samet, J.M. Total Cholesterol and Cancer Risk in a Large Prospective Study in Korea. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2011, 29, 1592–1598. [Google Scholar] [CrossRef]
- Farwell, W.R.; D’Avolio, L.W.; Scranton, R.E.; Lawler, E. V; Gaziano, J.M. Statins and Prostate Cancer Diagnosis and Grade in a Veterans Population. J. Natl. Cancer Inst. 2011, 103, 885–892. [Google Scholar] [CrossRef] [PubMed]
- Ukomadu, C.; Dutta, A. Inhibition of Cdk2 Activating Phosphorylation by Mevastatin. J. Biol. Chem. 2003, 278, 4840–4846. [Google Scholar] [CrossRef]
- Platz, E.A.; Leitzmann, M.F.; Visvanathan, K.; Rimm, E.B.; Stampfer, M.J.; Willett, W.C.; Giovannucci, E. Statin Drugs and Risk of Advanced Prostate Cancer. J. Natl. Cancer Inst. 2006, 98, 1819–1825. [Google Scholar] [CrossRef]
- Flick, E.D.; Habel, L.A.; Chan, K.A.; Van Den Eeden, S.K.; Quinn, V.P.; Haque, R.; Orav, E.J.; Seeger, J.D.; Sadler, M.C.; Quesenberry, C.P.J.; et al. Statin Use and Risk of Prostate Cancer in the California Men’s Health Study Cohort. Cancer Epidemiol. biomarkers Prev. a Publ. Am. Assoc. Cancer Res. cosponsored by Am. Soc. Prev. Oncol. 2007, 16, 2218–2225. [Google Scholar] [CrossRef]
- Shannon, J.; Tewoderos, S.; Garzotto, M.; Beer, T.M.; Derenick, R.; Palma, A.; Farris, P.E. Statins and Prostate Cancer Risk: A Case-Control Study. Am. J. Epidemiol. 2005, 162, 318–325. [Google Scholar] [CrossRef]
- Shafique, K.; McLoone, P.; Qureshi, K.; Leung, H.; Hart, C.; Morrison, D.S. Cholesterol and the Risk of Grade-Specific Prostate Cancer Incidence: Evidence from Two Large Prospective Cohort Studies with up to 37 Years’ Follow Up. BMC Cancer 2012, 12, 25. [Google Scholar] [CrossRef] [PubMed]
- YuPeng, L.; YuXue, Z.; PengFei, L.; Cheng, C.; YaShuang, Z.; DaPeng, L.; Chen, D. Cholesterol Levels in Blood and the Risk of Prostate Cancer: A Meta-Analysis of 14 Prospective Studies. Cancer Epidemiol. biomarkers Prev. a Publ. Am. Assoc. Cancer Res. cosponsored by Am. Soc. Prev. Oncol. 2015, 24, 1086–1093. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Shui, I.M.; Keum, N.; Shen, X.; Wu, K.; Clinton, S.K.; Cao, Y.; Song, M.; Zhang, X.; Platz, E.A.; et al. Plasma Total Cholesterol Concentration and Risk of Higher-Grade Prostate Cancer: A Nested Case-Control Study and a Dose-Response Meta-Analysis. Int. J. cancer 2023, 153, 1337–1346. [Google Scholar] [CrossRef]
- Meijer, D.; van Moorselaar, R.J.A.; Vis, A.N.; Bijnsdorp, I. V Prostate Cancer Development Is Not Affected by Statin Use in Patients with Elevated PSA Levels. Cancers (Basel). 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Caro-Maldonado, A.; Camacho, L.; Zabala-Letona, A.; Torrano, V.; Fernández-Ruiz, S.; Zamacola-Bascaran, K.; Arreal, L.; Valcárcel-Jiménez, L.; Martín-Martín, N.; Flores, J.M.; et al. Low-Dose Statin Treatment Increases Prostate Cancer Aggressiveness. Oncotarget 2018, 9, 1494–1504. [Google Scholar] [CrossRef] [PubMed]
- Celhay, O.; Bousset, L.; Guy, L.; Kemeny, J.-L.; Leoni, V.; Caccia, C.; Trousson, A.; Damon-Soubeyrant, C.; De Haze, A.; Sabourin, L.; et al. Individual Comparison of Cholesterol Metabolism in Normal and Tumour Areas in Radical Prostatectomy Specimens from Patients with Prostate Cancer: Results of the CHOMECAP Study. Eur. Urol. Oncol. 2019, 2, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, L.; Lin, J.; Lu, M.L.; Solomon, K.R.; Freeman, M.R. Cholesterol-Rich Lipid Rafts Mediate Akt-Regulated Survival in Prostate Cancer Cells. Cancer Res. 2002, 62, 2227–2231. [Google Scholar] [PubMed]
- Patra, S.K. Dissecting Lipid Raft Facilitated Cell Signaling Pathways in Cancer. Biochim. Biophys. Acta 2008, 1785, 182–206. [Google Scholar] [CrossRef]
- Llaverias, G.; Danilo, C.; Wang, Y.; Witkiewicz, A.K.; Daumer, K.; Lisanti, M.P.; Frank, P.G. A Western-Type Diet Accelerates Tumor Progression in an Autochthonous Mouse Model of Prostate Cancer. Am. J. Pathol. 2010, 177, 3180–3191. [Google Scholar] [CrossRef]
- Oh, H.Y.; Lee, E.J.; Yoon, S.; Chung, B.H.; Cho, K.S.; Hong, S.J. Cholesterol Level of Lipid Raft Microdomains Regulates Apoptotic Cell Death in Prostate Cancer Cells through EGFR-Mediated Akt and ERK Signal Transduction. Prostate 2007, 67, 1061–1069. [Google Scholar] [CrossRef]
- Pommier, A.J.C.; Alves, G.; Viennois, E.; Bernard, S.; Communal, Y.; Sion, B.; Marceau, G.; Damon, C.; Mouzat, K.; Caira, F.; et al. Liver X Receptor Activation Downregulates AKT Survival Signaling in Lipid Rafts and Induces Apoptosis of Prostate Cancer Cells. Oncogene 2010, 29, 2712–2723. [Google Scholar] [CrossRef] [PubMed]
- Pommier, A.J.C.; Dufour, J.; Alves, G.; Viennois, E.; De Boussac, H.; Trousson, A.; Volle, D.H.; Caira, F.; Val, P.; Arnaud, P.; et al. Liver x Receptors Protect from Development of Prostatic Intra-Epithelial Neoplasia in Mice. PLoS Genet. 2013, 9, e1003483. [Google Scholar] [CrossRef] [PubMed]
- Rambur, A.; Lours-Calet, C.; Beaudoin, C.; Buñay, J.; Vialat, M.; Mirouse, V.; Trousson, A.; Renaud, Y.; Lobaccaro, J.-M.A.; Baron, S.; et al. Sequential Ras/MAPK and PI3K/AKT/MTOR Pathways Recruitment Drives Basal Extrusion in the Prostate-like Gland of Drosophila. Nat. Commun. 2020, 11, 2300. [Google Scholar] [CrossRef]
- Levine, B.D.; Cagan, R.L. Drosophila Lung Cancer Models Identify Trametinib plus Statin as Candidate Therapeutic. Cell Rep. 2016, 14, 1477–1487. [Google Scholar] [CrossRef] [PubMed]
- Bangi, E.; Murgia, C.; Teague, A.G.S.; Sansom, O.J.; Cagan, R.L. Functional Exploration of Colorectal Cancer Genomes Using Drosophila. Nat. Commun. 2016, 7, 13615. [Google Scholar] [CrossRef]
- Kalb, J.M.; DiBenedetto, A.J.; Wolfner, M.F. Probing the Function of Drosophila Melanogaster Accessory Glands by Directed Cell Ablation. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 8093–8097. [Google Scholar] [CrossRef]
- Wolfner, M.F. Tokens of Love: Functions and Regulation of Drosophila Male Accessory Gland Products. Insect Biochem. Mol. Biol. 1997, 27, 179–192. [Google Scholar] [CrossRef]
- Rylett, C.M.; Walker, M.J.; Howell, G.J.; Shirras, A.D.; Isaac, R.E. Male Accessory Glands of Drosophila Melanogaster Make a Secreted Angiotensin I-Converting Enzyme (ANCE), Suggesting a Role for the Peptide-Processing Enzyme in Seminal Fluid. J. Exp. Biol. 2007, 210, 3601–3606. [Google Scholar] [CrossRef]
- Rewitz, K.F.; O’Connor, M.B.; Gilbert, L.I. Molecular Evolution of the Insect Halloween Family of Cytochrome P450s: Phylogeny, Gene Organization and Functional Conservation. Insect Biochem. Mol. Biol. 2007, 37, 741–753. [Google Scholar] [CrossRef]
- Sharma, V.; Pandey, A.K.; Kumar, A.; Misra, S.; Gupta, H.P.K.; Gupta, S.; Singh, A.; Buehner, N.A.; Ravi Ram, K. Functional Male Accessory Glands and Fertility in Drosophila Require Novel Ecdysone Receptor. PLoS Genet. 2017, 13, e1006788. [Google Scholar] [CrossRef]
- Danielsen, E.T.; Moeller, M.E.; Yamanaka, N.; Ou, Q.; Laursen, J.M.; Soenderholm, C.; Zhuo, R.; Phelps, B.; Tang, K.; Zeng, J.; et al. A Drosophila Genome-Wide Screen Identifies Regulators of Steroid Hormone Production and Developmental Timing. Dev. Cell 2016, 37, 558–570. [Google Scholar] [CrossRef]
- Carvalho, M.; Schwudke, D.; Sampaio, J.L.; Palm, W.; Riezman, I.; Dey, G.; Gupta, G.D.; Mayor, S.; Riezman, H.; Shevchenko, A.; et al. Survival Strategies of a Sterol Auxotroph. Development 2010, 137, 3675–3685. [Google Scholar] [CrossRef] [PubMed]
- Niwa, R.; Niwa, Y.S. The Fruit Fly Drosophila Melanogaster as a Model System to Study Cholesterol Metabolism and Homeostasis. Cholesterol 2011, 2011, 176802. [Google Scholar] [CrossRef] [PubMed]
- Bloch, K. The Biological Synthesis of Cholesterol. Science 1965, 150, 19–28. [Google Scholar] [CrossRef]
- CLARK, A.J.; BLOCK, K. The Absence of Sterol Synthesis in Insects. J. Biol. Chem. 1959, 234, 2578–2582. [Google Scholar] [CrossRef]
- Yue, S.; Li, J.; Lee, S.-Y.; Lee, H.J.; Shao, T.; Song, B.; Cheng, L.; Masterson, T.A.; Liu, X.; Ratliff, T.L.; et al. Cholesteryl Ester Accumulation Induced by PTEN Loss and PI3K/AKT Activation Underlies Human Prostate Cancer Aggressiveness. Cell Metab. 2014, 19, 393–406. [Google Scholar] [CrossRef]
- Parra-Peralbo, E.; Culi, J. Drosophila Lipophorin Receptors Mediate the Uptake of Neutral Lipids in Oocytes and Imaginal Disc Cells by an Endocytosis-Independent Mechanism. PLoS Genet. 2011, 7, e1001297. [Google Scholar] [CrossRef] [PubMed]
- Fluegel, M.L.; Parker, T.J.; Pallanck, L.J. Mutations of a Drosophila NPC1 Gene Confer Sterol and Ecdysone Metabolic Defects. Genetics 2006, 172, 185–196. [Google Scholar] [CrossRef]
- Huang, X.; Suyama, K.; Buchanan, J.; Zhu, A.J.; Scott, M.P. A Drosophila Model of the Niemann-Pick Type C Lysosome Storage Disease: Dnpc1a Is Required for Molting and Sterol Homeostasis. Development 2005, 132, 5115–5124. [Google Scholar] [CrossRef]
- Chi, K.-C.; Tsai, W.-C.; Wu, C.-L.; Lin, T.-Y.; Hueng, D.-Y. An Adult Drosophila Glioma Model for Studying Pathometabolic Pathways of Gliomagenesis. Mol. Neurobiol. 2019, 56, 4589–4599. [Google Scholar] [CrossRef]
- Voght, S.P.; Fluegel, M.L.; Andrews, L.A.; Pallanck, L.J. Drosophila NPC1b Promotes an Early Step in Sterol Absorption from the Midgut Epithelium. Cell Metab. 2007, 5, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Horner, M.A.; Pardee, K.; Liu, S.; King-Jones, K.; Lajoie, G.; Edwards, A.; Krause, H.M.; Thummel, C.S. The Drosophila DHR96 Nuclear Receptor Binds Cholesterol and Regulates Cholesterol Homeostasis. Genes Dev. 2009, 23, 2711–2716. [Google Scholar] [CrossRef] [PubMed]
- Bujold, M.; Gopalakrishnan, A.; Nally, E.; King-Jones, K. Nuclear Receptor DHR96 Acts as a Sentinel for Low Cholesterol Concentrations in Drosophila Melanogaster. Mol. Cell. Biol. 2010, 30, 793–805. [Google Scholar] [CrossRef]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative Genomic Profiling of Human Prostate Cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Haenszel, W.; Kurihara, M. Studies of Japanese Migrants. I. Mortality from Cancer and Other Diseases among Japanese in the United States. J. Natl. Cancer Inst. 1968, 40, 43–68. [Google Scholar] [PubMed]
- Mostaghel, E.A.; Solomon, K.R.; Pelton, K.; Freeman, M.R.; Montgomery, R.B. Impact of Circulating Cholesterol Levels on Growth and Intratumoral Androgen Concentration of Prostate Tumors. PLoS ONE 2012, 7, e30062. [Google Scholar] [CrossRef] [PubMed]
- Moon, H.; Ruelcke, J.E.; Choi, E.; Sharpe, L.J.; Nassar, Z.D.; Bielefeldt-Ohmann, H.; Parat, M.-O.; Shah, A.; Francois, M.; Inder, K.L.; et al. Diet-Induced Hypercholesterolemia Promotes Androgen-Independent Prostate Cancer Metastasis via IQGAP1 and Caveolin-1. Oncotarget 2015, 6, 7438–7453. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Huang, L.; Dai, T.; Pei, X.; Xia, L.; Zeng, G.; Ye, M.; Liu, K.; Zeng, F.; Han, W.; et al. SQLE Mediates Metabolic Reprogramming to Promote LN Metastasis in Castration-Resistant Prostate Cancer. Onco. Targets. Ther. 2021, 14, 4285–4295. [Google Scholar] [CrossRef]
- Slattum, G.M.; Rosenblatt, J. Tumour Cell Invasion: An Emerging Role for Basal Epithelial Cell Extrusion. Nat. Rev. Cancer 2014, 14, 495–501. [Google Scholar] [CrossRef]
- Fadul, J.; Rosenblatt, J. The Forces and Fates of Extruding Cells. Curr. Opin. Cell Biol. 2018, 54, 66–71. [Google Scholar] [CrossRef]
- Slattum, G.; Gu, Y.; Sabbadini, R.; Rosenblatt, J. Autophagy in Oncogenic K-Ras Promotes Basal Extrusion of Epithelial Cells by Degrading S1P. Curr. Biol. 2014, 24, 19–28. [Google Scholar] [CrossRef]
- Fadul, J.; Zulueta-Coarasa, T.; Slattum, G.M.; Redd, N.M.; Jin, M.F.; Redd, M.J.; Daetwyler, S.; Hedeen, D.; Huisken, J.; Rosenblatt, J. KRas-Transformed Epithelia Cells Invade and Partially Dedifferentiate by Basal Cell Extrusion. Nat. Commun. 2021, 12, 7180. [Google Scholar] [CrossRef]
- Shirai, T.; Sekai, M.; Kozawa, K.; Sato, N.; Tanimura, N.; Kon, S.; Matsumoto, T.; Murakami, T.; Ito, S.; Tilston-Lunel, A.; et al. Basal Extrusion of Single-Oncogenic Mutant Cells Induces Dome-like Structures with Altered Microenvironments. Cancer Sci. 2022, 113, 3710–3721. [Google Scholar] [CrossRef]
- Shen, J.; Lu, J.; Sui, L.; Wang, D.; Yin, M.; Hoffmann, I.; Legler, A.; Pflugfelder, G.O. The Orthologous Tbx Transcription Factors Omb and TBX2 Induce Epithelial Cell Migration and Extrusion in Vivo without Involvement of Matrix Metalloproteinases. Oncotarget 2014, 5, 11998–12015. [Google Scholar] [CrossRef] [PubMed]
- Hendley, A.M.; Wang, Y.J.; Polireddy, K.; Alsina, J.; Ahmed, I.; Lafaro, K.J.; Zhang, H.; Roy, N.; Savidge, S.G.; Cao, Y.; et al. P120 Catenin Suppresses Basal Epithelial Cell Extrusion in Invasive Pancreatic Neoplasia. Cancer Res. 2016, 76, 3351–3363. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
drosophila accessory gland tumour cells accumulate cholesterol esters. A-H : confocal imaging of representative glands initiated for tumorigenesis. Clonal induction of an activated form of EGFR (EGRFl) induces the formation of intraglandular clones (white arrowheads in B-D and F-H) and of tumours outside the gland (empty arrowheads in B-D and F-H). Muscle cells surrounding the glands are revealed by phalloidin staining of F-actin, yellow in A and E). Cell nuclei are revealed by DAPI staining (blue in A-B and E-H). Clonal cells are revealed by GFP co-expression (green in A-B and E-H, grey for single channel in C and G). A-D : Nile Red staining (magenta in A-B and grey in single channel D) shows an accumulation of neutral lipids specifically in tumour cells. E-H : Bodipy staining (magenta in E-F and grey in single channel H) indicates a strong proportion of cholesterol esters among these neutral lipids. Representative images in (A-H) are from three or more experiments. Scale bars: 50μM.
Figure 1.
drosophila accessory gland tumour cells accumulate cholesterol esters. A-H : confocal imaging of representative glands initiated for tumorigenesis. Clonal induction of an activated form of EGFR (EGRFl) induces the formation of intraglandular clones (white arrowheads in B-D and F-H) and of tumours outside the gland (empty arrowheads in B-D and F-H). Muscle cells surrounding the glands are revealed by phalloidin staining of F-actin, yellow in A and E). Cell nuclei are revealed by DAPI staining (blue in A-B and E-H). Clonal cells are revealed by GFP co-expression (green in A-B and E-H, grey for single channel in C and G). A-D : Nile Red staining (magenta in A-B and grey in single channel D) shows an accumulation of neutral lipids specifically in tumour cells. E-H : Bodipy staining (magenta in E-F and grey in single channel H) indicates a strong proportion of cholesterol esters among these neutral lipids. Representative images in (A-H) are from three or more experiments. Scale bars: 50μM.
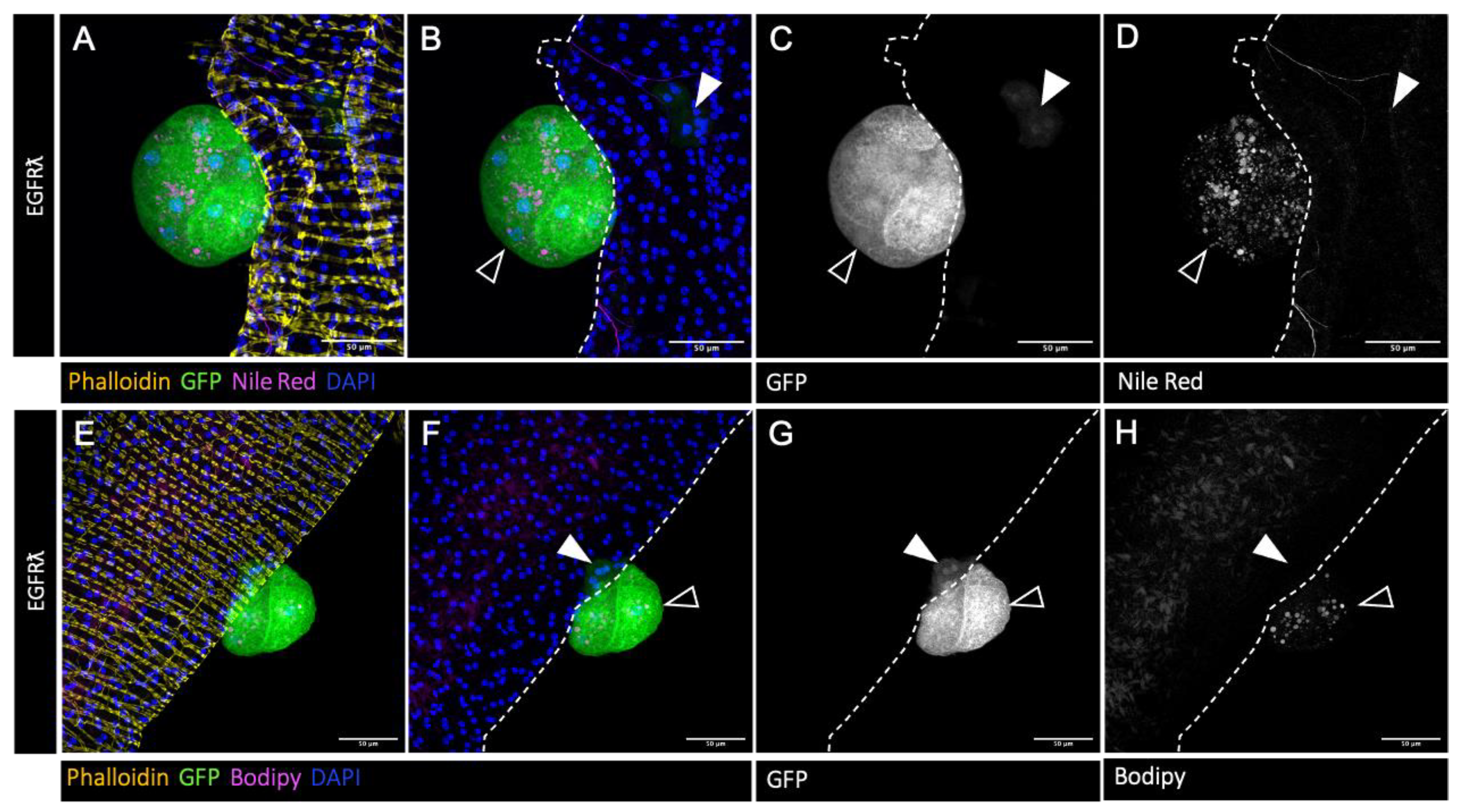
Figure 2.
Tumor-specific downregulation of cholesterol metabolism reduces cholesterol esters accumulation. A-T : confocal imaging of representative glands initiated for tumorigenesis. White arrowheads indicate intra-glandular clones as empty arrowheads indicate tumors outside the gland. Muscle cells surrounding the glands are revealed by phalloidin staining in A, I, Q (yellow). Normal epithelial cells are revealed by coracle staining in E and M (yellow). Cell nuclei are revealed by DAPI staining (blue). Clonal cells are revealed by GFP co-expression (green in the first and second columns, grey in third). Neutral lipids accumulation is revealed by Nile Red staining (magenta in the first and second columns, grey in fourth). A-D : Downregulation of ncp1b, which is expressed specifically in intestinal cells, is used as control RNAi. In this condition, a strong accumulation of neutral lipids is seen specifically in the tumor cells (in A, B and D) as for the EGFRl condition. E-T : downregulation of genes implicated in cholesterol uptake (E-L), intracellular trafficking (M-P) or storage (Q-T) strongly impairs lipid accumulation (compare images in right column to D). Representative images in (A-T) from three or more experiments. Scale bars: 50 μm.
Figure 2.
Tumor-specific downregulation of cholesterol metabolism reduces cholesterol esters accumulation. A-T : confocal imaging of representative glands initiated for tumorigenesis. White arrowheads indicate intra-glandular clones as empty arrowheads indicate tumors outside the gland. Muscle cells surrounding the glands are revealed by phalloidin staining in A, I, Q (yellow). Normal epithelial cells are revealed by coracle staining in E and M (yellow). Cell nuclei are revealed by DAPI staining (blue). Clonal cells are revealed by GFP co-expression (green in the first and second columns, grey in third). Neutral lipids accumulation is revealed by Nile Red staining (magenta in the first and second columns, grey in fourth). A-D : Downregulation of ncp1b, which is expressed specifically in intestinal cells, is used as control RNAi. In this condition, a strong accumulation of neutral lipids is seen specifically in the tumor cells (in A, B and D) as for the EGFRl condition. E-T : downregulation of genes implicated in cholesterol uptake (E-L), intracellular trafficking (M-P) or storage (Q-T) strongly impairs lipid accumulation (compare images in right column to D). Representative images in (A-T) from three or more experiments. Scale bars: 50 μm.
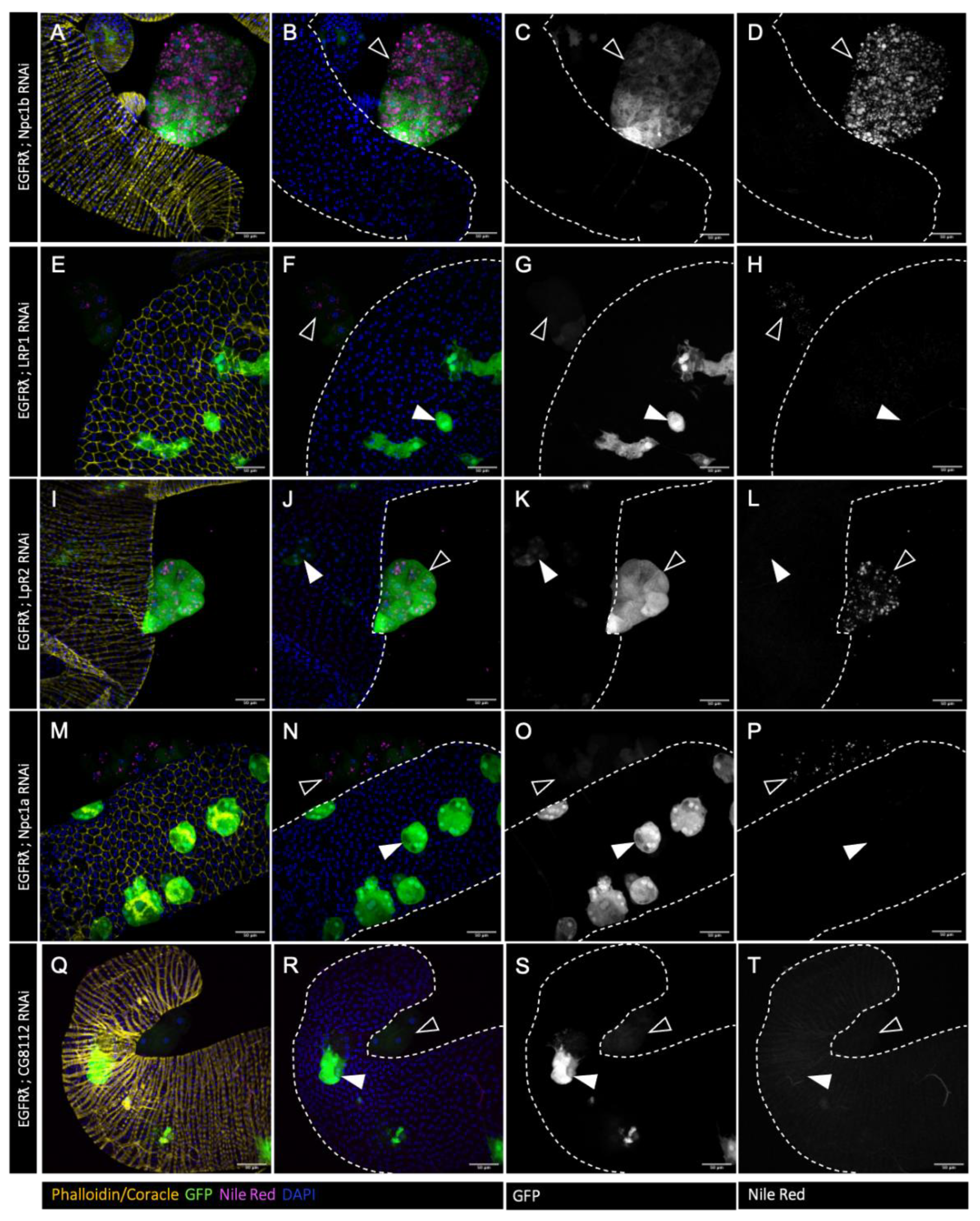
Figure 3.
Tumor-specific downregulation of cholesterol metabolism impairs basal extrusion. A : at dissection, presence of tumor(s) was assessed for all considered genotypes outside the glands for several flies (n) in independent experiments (N). As expected, expression of an RNAi against intestinal cell-specific ncp1b does not induce a change in tumor formation compared to EGFRl condition (right and left columns, respectively). On the contrary, downregulation of genes implicated in cholesterol uptake, intracellular trafficking or storage significantly decrease tumor formation. B-D : Tumor size (B), cell number (C) or tumor cell size remain unchanged when cholesterol uptake, intracellular trafficking or storage are down-regulated (see also Figure S1). (A) Chi2-test; EGFRλ: N=29; LRP1 RNAi: N=9; LpR2 RNAi: N=8; Npc1a RNAi: N=11; CG8112 RNAi: N=5; Npc1b RNAi: N=3 (B-D) Krustal-Wallis tests; EGFRλ: N=11; LRP1 RNAi: N=10; LpR2 RNAi: N=7; Npc1a RNAi: N=8; Npc1b RNAi: N=3. **** p<0.0001; ns: non significant.
Figure 3.
Tumor-specific downregulation of cholesterol metabolism impairs basal extrusion. A : at dissection, presence of tumor(s) was assessed for all considered genotypes outside the glands for several flies (n) in independent experiments (N). As expected, expression of an RNAi against intestinal cell-specific ncp1b does not induce a change in tumor formation compared to EGFRl condition (right and left columns, respectively). On the contrary, downregulation of genes implicated in cholesterol uptake, intracellular trafficking or storage significantly decrease tumor formation. B-D : Tumor size (B), cell number (C) or tumor cell size remain unchanged when cholesterol uptake, intracellular trafficking or storage are down-regulated (see also Figure S1). (A) Chi2-test; EGFRλ: N=29; LRP1 RNAi: N=9; LpR2 RNAi: N=8; Npc1a RNAi: N=11; CG8112 RNAi: N=5; Npc1b RNAi: N=3 (B-D) Krustal-Wallis tests; EGFRλ: N=11; LRP1 RNAi: N=10; LpR2 RNAi: N=7; Npc1a RNAi: N=8; Npc1b RNAi: N=3. **** p<0.0001; ns: non significant.
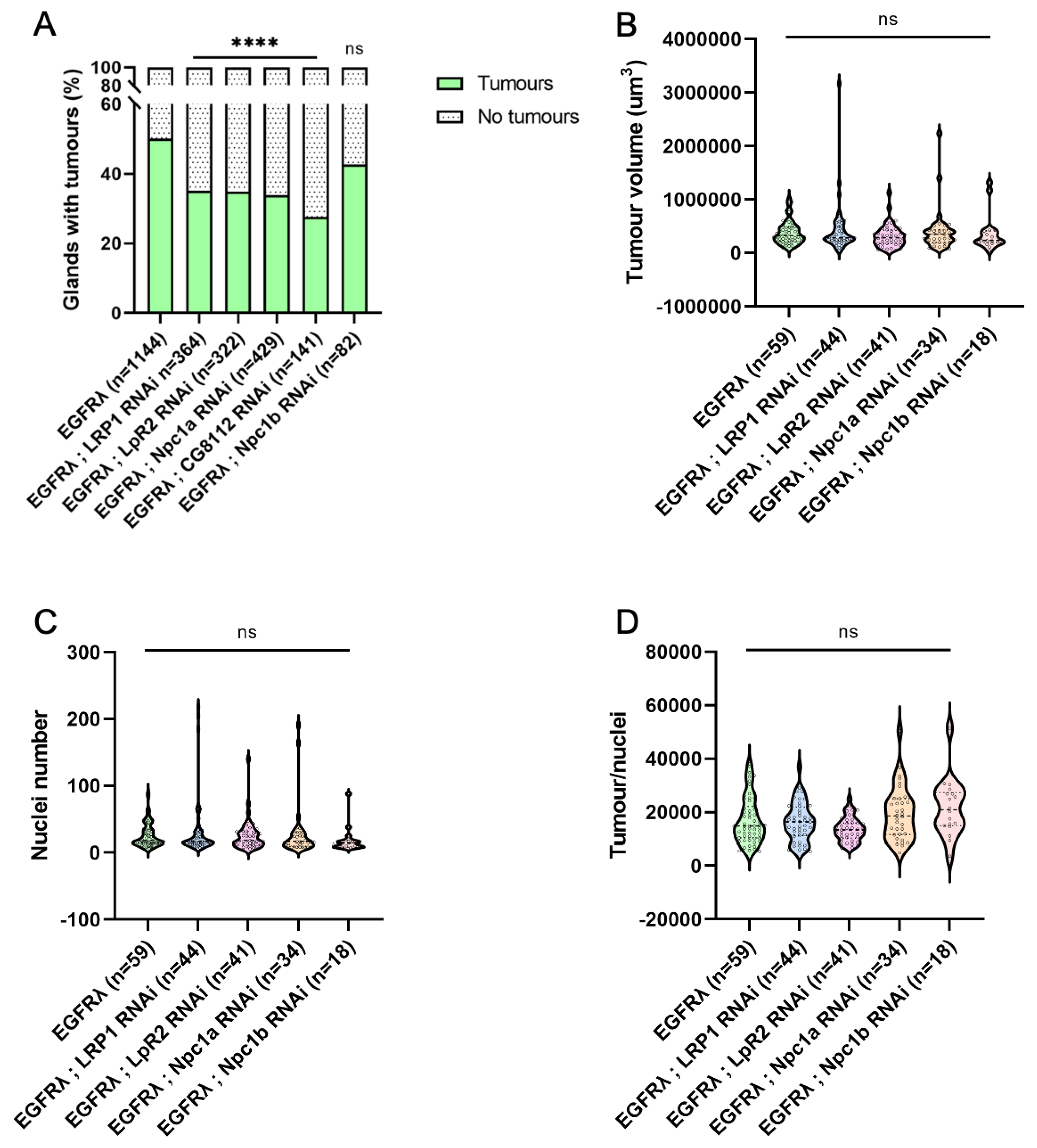
Figure 4.
Further cholesterol metabolism deregulation has no effect on tumour phenotype. A-P : confocal imaging of representative glands initiated for tumorigenesis. White arrowheads indicate intra-glandular clones as empty arrowheads indicate tumours outside the gland. Muscle cells surrounding the glands are revealed by phalloidin staining in A, I, M (yellow). Normal epithelial cells are revealed by coracle staining in E (yellow). Cell nuclei are revealed by DAPI staining (blue). Clonal cells are revealed by GFP co-expression (green in the first and second columns, grey in third). Neutral lipids accumulation is revealed by Nile Red staining (magenta in the first and second columns, grey in fourth). A-D :, tumour control condition with normal diet . In this condition, a strong accumulation of neutral lipids is seen specifically in the tumour cells (in A, B and D). E-H : High cholesterol diet (+ 0.2% cholesterol) does not modify tumour phenotype despite slightly increased neutral lipid accumulation (quantification in Figure 5). I-P : Downregulation of master controller of cholesterol homeostasis DHR96 +/- high cholesterol diet (respectively normal diet in I-L and HCD in M-P) induces no evident changes in tumour phenotypes (quantifications in Figure 5). Representative images in (A-P) from three or more experiments. Scale bars: 50 μm.
Figure 4.
Further cholesterol metabolism deregulation has no effect on tumour phenotype. A-P : confocal imaging of representative glands initiated for tumorigenesis. White arrowheads indicate intra-glandular clones as empty arrowheads indicate tumours outside the gland. Muscle cells surrounding the glands are revealed by phalloidin staining in A, I, M (yellow). Normal epithelial cells are revealed by coracle staining in E (yellow). Cell nuclei are revealed by DAPI staining (blue). Clonal cells are revealed by GFP co-expression (green in the first and second columns, grey in third). Neutral lipids accumulation is revealed by Nile Red staining (magenta in the first and second columns, grey in fourth). A-D :, tumour control condition with normal diet . In this condition, a strong accumulation of neutral lipids is seen specifically in the tumour cells (in A, B and D). E-H : High cholesterol diet (+ 0.2% cholesterol) does not modify tumour phenotype despite slightly increased neutral lipid accumulation (quantification in Figure 5). I-P : Downregulation of master controller of cholesterol homeostasis DHR96 +/- high cholesterol diet (respectively normal diet in I-L and HCD in M-P) induces no evident changes in tumour phenotypes (quantifications in Figure 5). Representative images in (A-P) from three or more experiments. Scale bars: 50 μm.
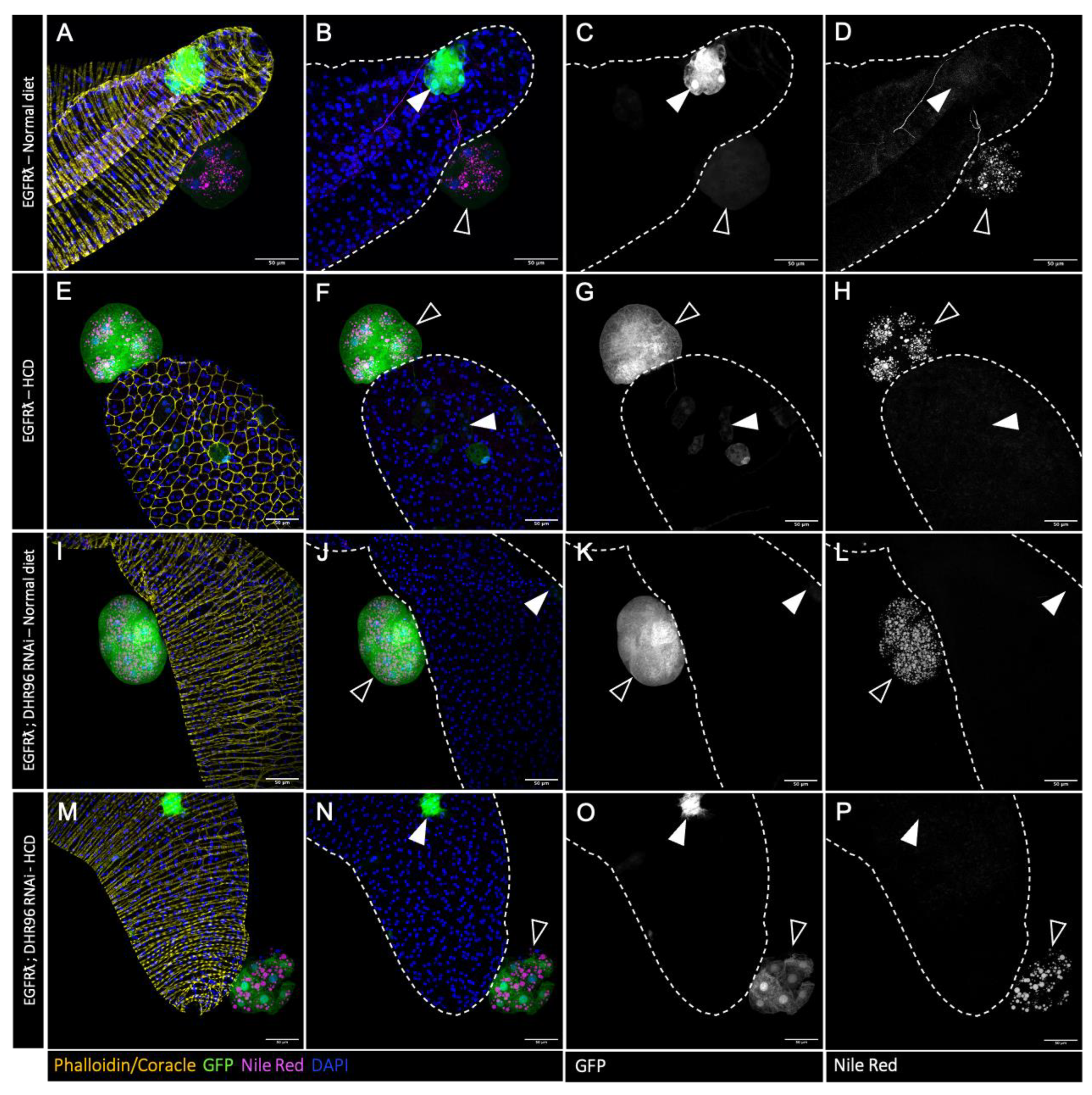
Figure 5.
Forcing further the deregulation of cholesterol metabolism has limited effect on tumor phenotype and no effect on basal extrusion. A : high cholesterol diet (HCD) or DHR96 tumor cell-specific downregulation significantly increase the volume of lipid droplets compared to EGFRl condition. B-C : Tumor size (B) and cell number (C) remain unchanged when flies are fed high cholesterol diet and/or for tumor cells tumor cell-specific downregulation of DHR96. D : at dissection, presence of tumor(s) was assessed for all considered genotypes outside the glands for several flies (n) in several independent experiments. Neither HCD nor DHR96 downregulation induce a change in tumor formation compared to EGFRl condition. (A-C) Krustal-Wallis test; EGFRλ: N=6; EGFRλ – HCD: N=6; DHR96 RNAi: N=4; DHR96 RNAi – HCD: N=3 (D) Chi2-test; EGFRλ: N=9; EGFRλ – HCD: N=10; DHR96 RNAi: N=6; DHR96 RNAi – HCD: N=6* p<0.05; ns: non significant.
Figure 5.
Forcing further the deregulation of cholesterol metabolism has limited effect on tumor phenotype and no effect on basal extrusion. A : high cholesterol diet (HCD) or DHR96 tumor cell-specific downregulation significantly increase the volume of lipid droplets compared to EGFRl condition. B-C : Tumor size (B) and cell number (C) remain unchanged when flies are fed high cholesterol diet and/or for tumor cells tumor cell-specific downregulation of DHR96. D : at dissection, presence of tumor(s) was assessed for all considered genotypes outside the glands for several flies (n) in several independent experiments. Neither HCD nor DHR96 downregulation induce a change in tumor formation compared to EGFRl condition. (A-C) Krustal-Wallis test; EGFRλ: N=6; EGFRλ – HCD: N=6; DHR96 RNAi: N=4; DHR96 RNAi – HCD: N=3 (D) Chi2-test; EGFRλ: N=9; EGFRλ – HCD: N=10; DHR96 RNAi: N=6; DHR96 RNAi – HCD: N=6* p<0.05; ns: non significant.
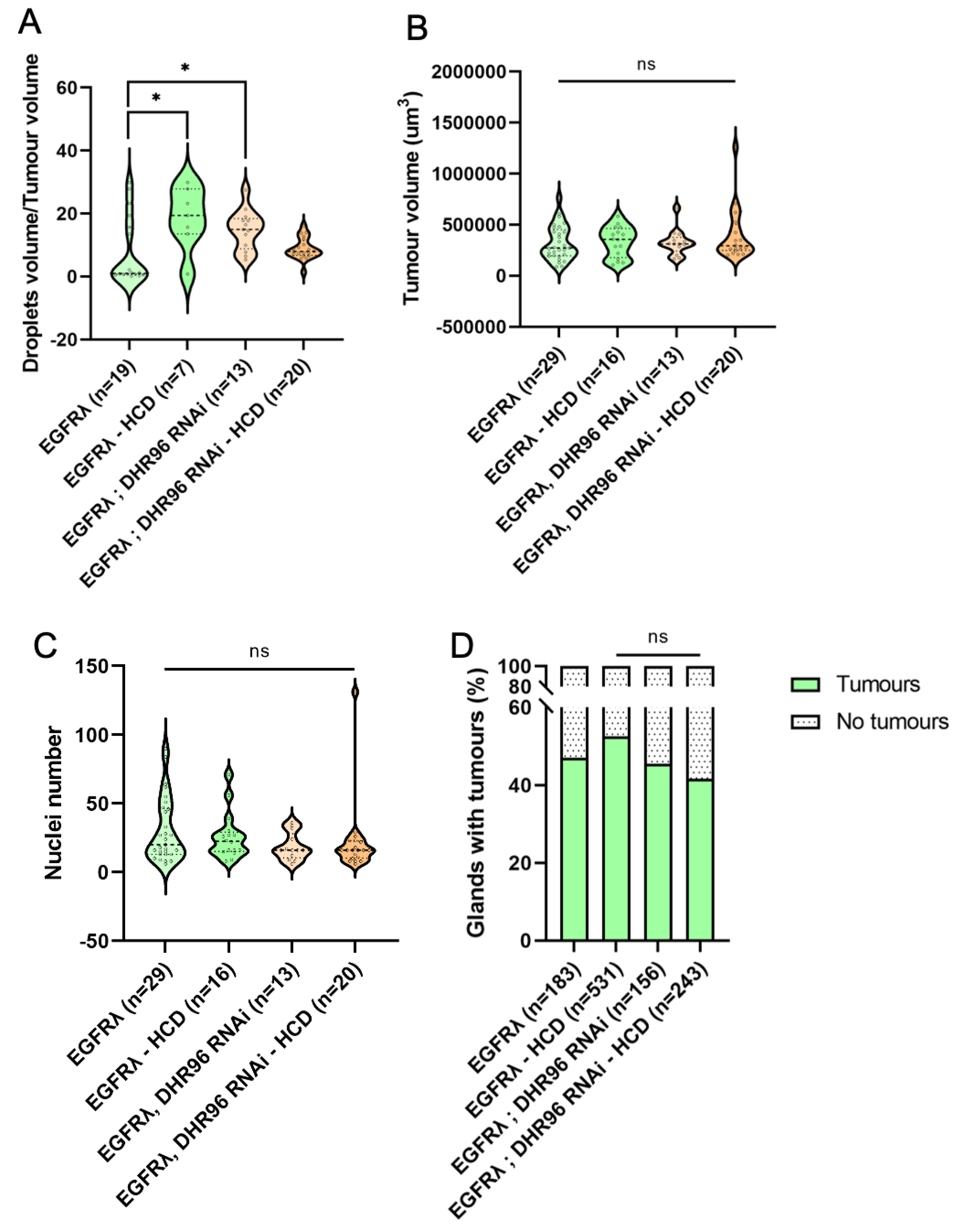
Figure 6.
High cholesterol diet restores tumour cell lipid accumulation but has no impact on tumour phenotype. A-T : confocal imaging of representative glands initiated for tumorigenesis. White arrowheads indicate intra-glandular clones as empty arrowheads indicate tumours outside the gland. Muscle cells surrounding the glands are revealed by phalloidin staining in A, E, I, Q (yellow). Normal epithelial cells are revealed by coracle staining in M (yellow). Cell nuclei are revealed by DAPI staining (blue). Clonal cells are revealed by GFP co-expression (green in the first and second columns, grey in third). Neutral lipids accumulation is revealed by Nile Red staining (magenta in the first and second columns, grey in fourth). Increasing dietary cholesterol restores lipid accumulation in tumours harboring downregulation of cholesterol import (A-H), intracellular trafficking (I-L) or storage (M-P). Representative images in (A-P) from three or more experiments. Scale bars: 50 μm.
Figure 6.
High cholesterol diet restores tumour cell lipid accumulation but has no impact on tumour phenotype. A-T : confocal imaging of representative glands initiated for tumorigenesis. White arrowheads indicate intra-glandular clones as empty arrowheads indicate tumours outside the gland. Muscle cells surrounding the glands are revealed by phalloidin staining in A, E, I, Q (yellow). Normal epithelial cells are revealed by coracle staining in M (yellow). Cell nuclei are revealed by DAPI staining (blue). Clonal cells are revealed by GFP co-expression (green in the first and second columns, grey in third). Neutral lipids accumulation is revealed by Nile Red staining (magenta in the first and second columns, grey in fourth). Increasing dietary cholesterol restores lipid accumulation in tumours harboring downregulation of cholesterol import (A-H), intracellular trafficking (I-L) or storage (M-P). Representative images in (A-P) from three or more experiments. Scale bars: 50 μm.
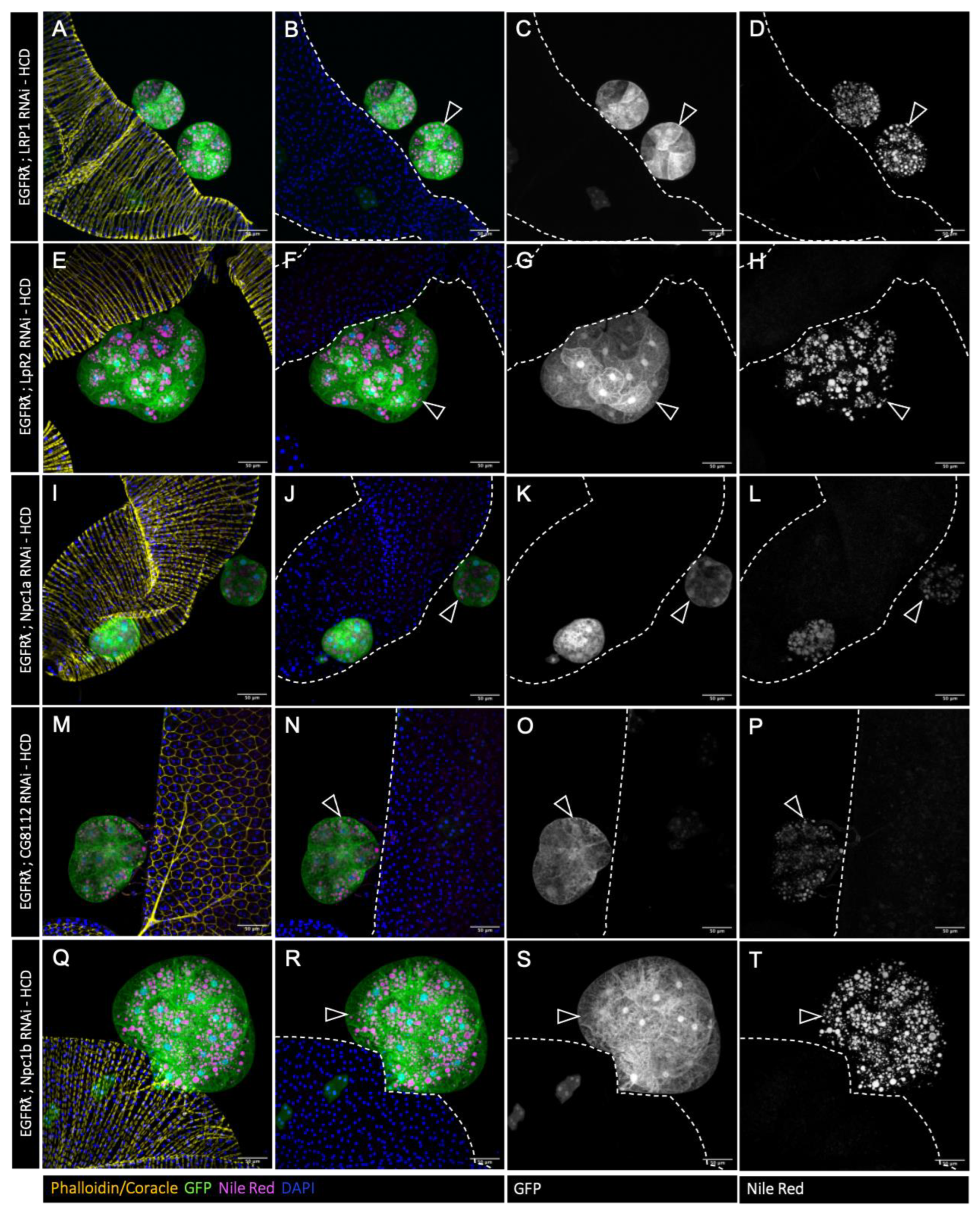
Figure 7.
High cholesterol diet counteracts the effect of decreased cholesterol metabolism on basal extrusion. At dissection, presence of tumour(s) was assessed for all considered genotypes outside the glands for several flies (n) in several independent experiments (N). A-D : Increasing dietary cholesterol significantly increases tumour formation in tumours harbouring downregulation of cholesterol import (A-B), intracellular trafficking (C) or storage (D) (compare the two right columns for each graph). By opposition, these high cholesterol diet conditions become indistinguishable from EGFRl phenotype independently of diet status (compare right column to the two left columns). E : As expected, a diet enriched in cholesterol has no effect on basal extrusion in a control negative condition, Npc1b RNAi. (A-E) Krustal-Wallis tests; EGFRλ: N=29; EGFRλ – HCD: 10; LRP1 RNAi: N=9; LRP1 RNAi – HCD: N=5; LpR2 RNAi: N=8; LpR2 RNAi – HCD: N=4; Npc1a RNAi: N=11; Npc1a RNAi – HCD: N=6; Npc1b RNAi: N=3; Npc1b RNAi – HCD: N=3; CG8112 RNAi: N=5; CG8112 RNAi – HCD: N=3. *p<0.05; **p<0.01; ****p<0.0001; ns: non significant.
Figure 7.
High cholesterol diet counteracts the effect of decreased cholesterol metabolism on basal extrusion. At dissection, presence of tumour(s) was assessed for all considered genotypes outside the glands for several flies (n) in several independent experiments (N). A-D : Increasing dietary cholesterol significantly increases tumour formation in tumours harbouring downregulation of cholesterol import (A-B), intracellular trafficking (C) or storage (D) (compare the two right columns for each graph). By opposition, these high cholesterol diet conditions become indistinguishable from EGFRl phenotype independently of diet status (compare right column to the two left columns). E : As expected, a diet enriched in cholesterol has no effect on basal extrusion in a control negative condition, Npc1b RNAi. (A-E) Krustal-Wallis tests; EGFRλ: N=29; EGFRλ – HCD: 10; LRP1 RNAi: N=9; LRP1 RNAi – HCD: N=5; LpR2 RNAi: N=8; LpR2 RNAi – HCD: N=4; Npc1a RNAi: N=11; Npc1a RNAi – HCD: N=6; Npc1b RNAi: N=3; Npc1b RNAi – HCD: N=3; CG8112 RNAi: N=5; CG8112 RNAi – HCD: N=3. *p<0.05; **p<0.01; ****p<0.0001; ns: non significant.
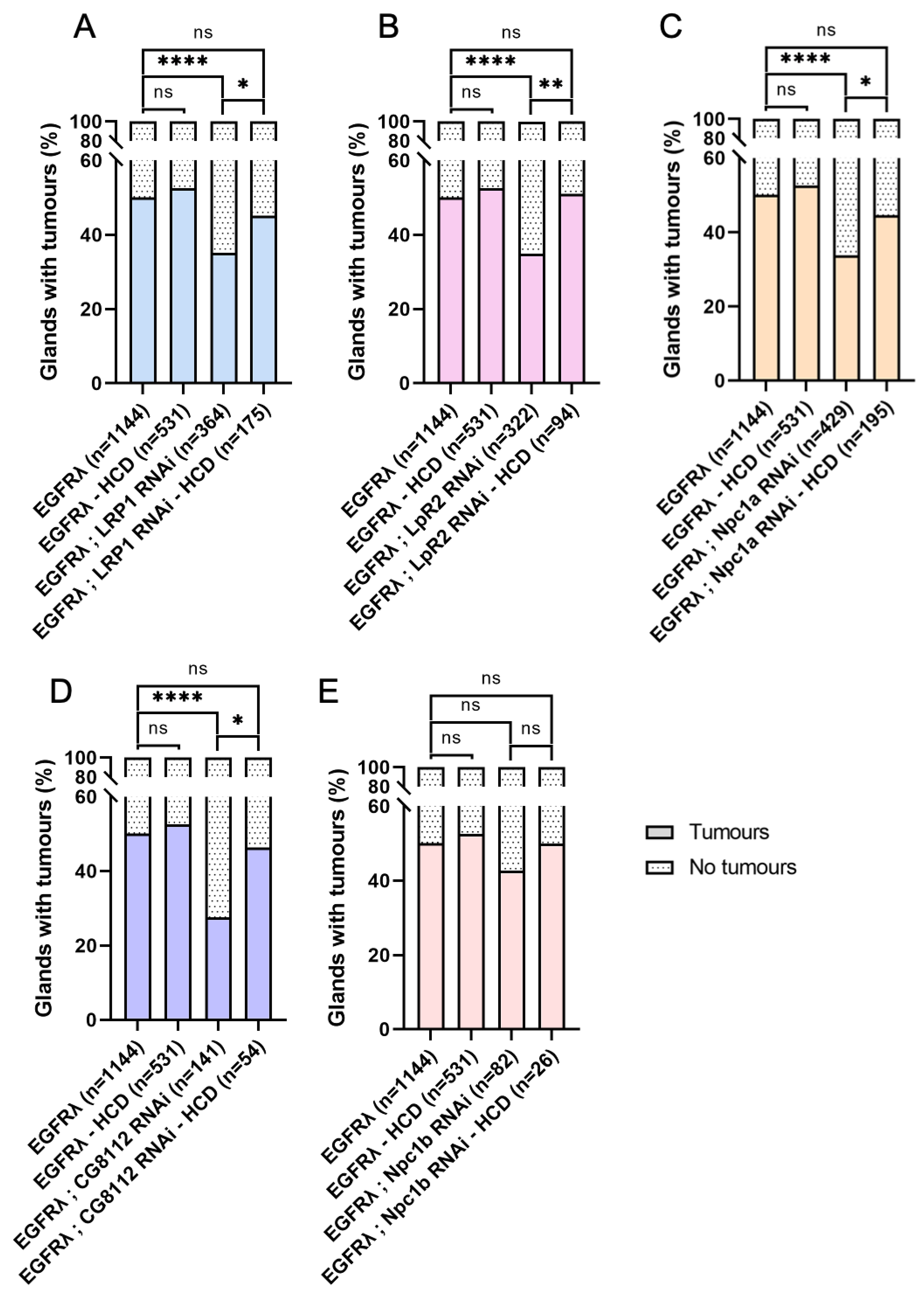
Figure 8.
Cholesterol metabolism is deregulated in primary and metastatic prostate cancer. (A-I) Violin-plots showing mRNA expression data for nine genes in normal prostate tissues (PAN, green), primary prostate tumours (Primary tumours, pink) and metastatic prostate tumours (Metastatic, blue). Expression data were first published by Taylor et al. Unpaired t test; *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001; ; ns: non significant. PAN: N=29; Primary tumours: N=131; Metastatic: N=29.
Figure 8.
Cholesterol metabolism is deregulated in primary and metastatic prostate cancer. (A-I) Violin-plots showing mRNA expression data for nine genes in normal prostate tissues (PAN, green), primary prostate tumours (Primary tumours, pink) and metastatic prostate tumours (Metastatic, blue). Expression data were first published by Taylor et al. Unpaired t test; *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001; ; ns: non significant. PAN: N=29; Primary tumours: N=131; Metastatic: N=29.
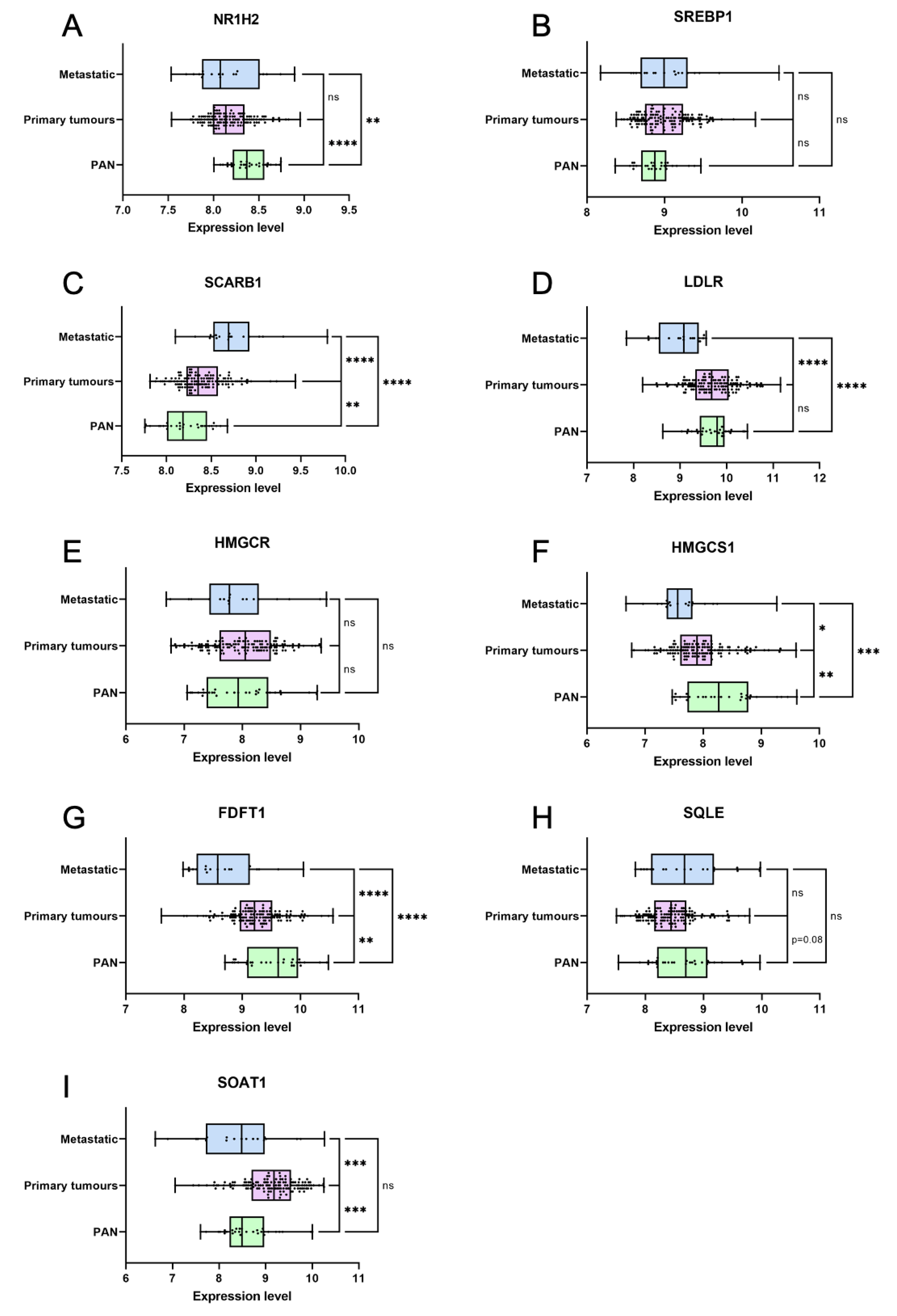
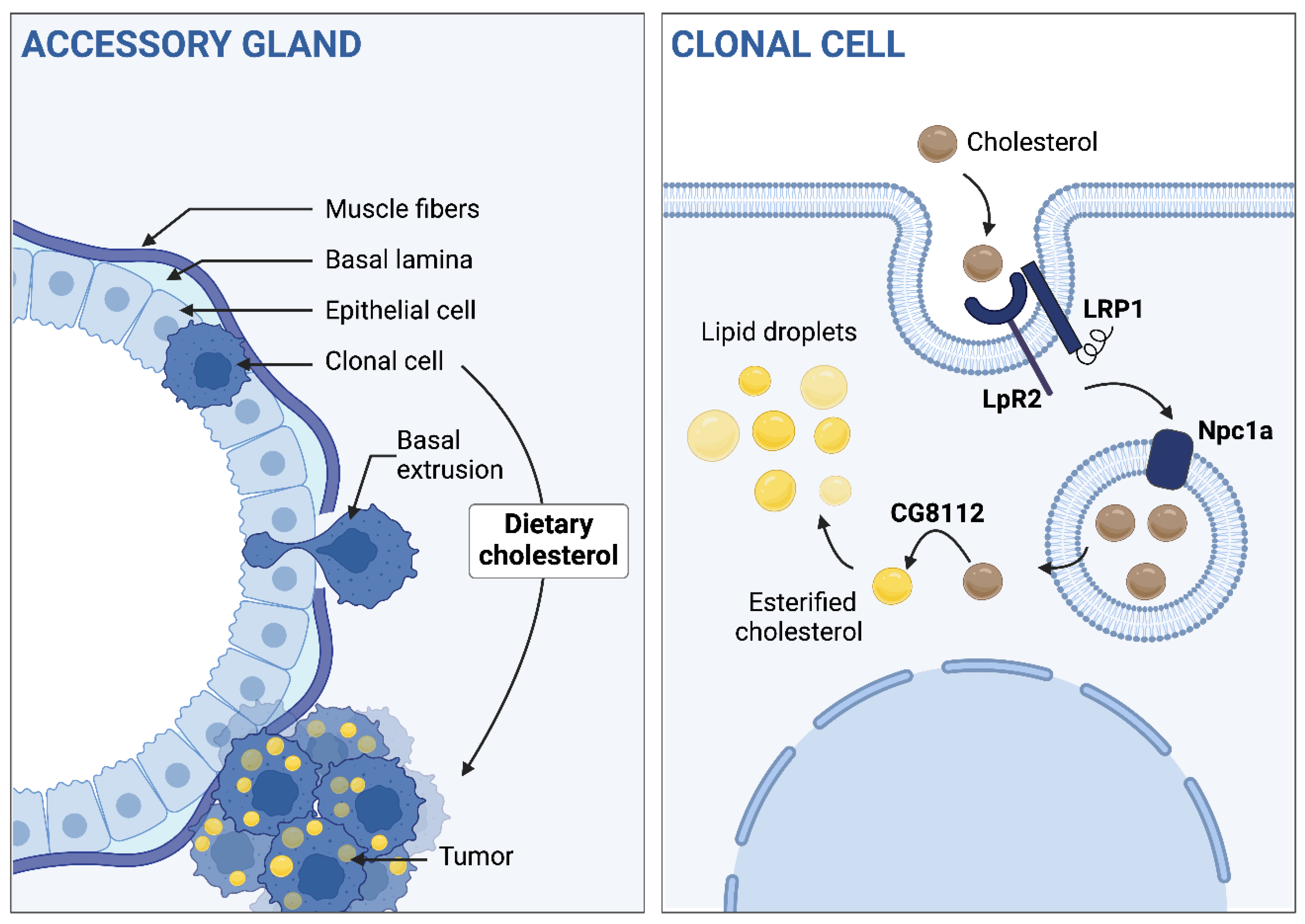
Figure 9.
Dietary cholesterol and its metabolism and storage in situ is involved in basal extrusion. Left part, from top to bottom: after the oncogenic hit, clonal cells undergo a basal extrusion to form tumours outside the gland and these accumulate cholesterol into droplets. Dietary cholesterol promotes this phenomenon. Right part, from top to bottom: In situ cholesterol intake, trafficking and storage drives the dietary cholesterol effect on tumorigenesis.
Figure 9.
Dietary cholesterol and its metabolism and storage in situ is involved in basal extrusion. Left part, from top to bottom: after the oncogenic hit, clonal cells undergo a basal extrusion to form tumours outside the gland and these accumulate cholesterol into droplets. Dietary cholesterol promotes this phenomenon. Right part, from top to bottom: In situ cholesterol intake, trafficking and storage drives the dietary cholesterol effect on tumorigenesis.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



