
Submitted:
24 May 2024
Posted:
27 May 2024
You are already at the latest version
Abstract
Presynaptic Ca2+-influx through voltage-gated Ca2+ channels (VGCC) is a key signal for synaptic vesicle release. Synaptic neurexins can partially determine the strength of transmission by regulating VGCCs. However, it is unknown whether neurexins modulate Ca2+-influx via all VGCC subtypes similarly. Here, we performed live cell imaging of synaptic boutons from primary hippocampal neurons with a Ca2+-indicator. We used the expression of inactive and active Cre recombinase to compare control to conditional knockout neurons lacking either all or selected neurexin variants. We found that reduced total presynaptic Ca2+-transients caused by the deletion of all neurexins were primarily due to the reduced contribution of P/Q-type VGCCs. The deletion of neurexin1α alone also reduced the total presynaptic Ca2+-influx but increased Ca2+-influx via N-type VGCCs. Moreover, we tested whether the decrease of Ca2+-influx induced by activation of cannabinoid receptor 1 (CB1-receptor) is modulated by neurexins. Unlike earlier observations emphasizing a role for β-neurexins, we found that the decrease of presynaptic Ca2+-transients induced by CB1-receptor activation depended more strongly on the presence of α-neurexins in hippocampal neurons. Together, our results suggest that neurexins have unique roles in the modulation of presynaptic Ca2+-influx through VGCC subtypes and that different neurexin variants may affect specific VGCCs.
Keywords:
Neurexin
; Calcium Channel Subtypes
; Presynapse
; Endocannabinoid System
; Genetically Encoded Calcium Indicator
1. Introduction
Synaptic transmission is a fundamental step in neuronal communication and the main place for neuromodulation. In presynaptic boutons, the opening of high threshold voltage-gated calcium channels (VGCCs) is a central step in the action potential-driven transmitter release [1,2]. Synaptic strength and synchronous release depend on the subtype, number, activity, and topography of VGCCs [3,4]. Action potential-triggered vesicle release mainly depends on Cav2.1 (P/Q-type) and Cav2.2 (N-type) VGCCs as determined by postsynaptic excitatory postsynaptic currents (EPSCs) [5,6], but in some synapses, only P/Q type VGCCs seem to be relevant for fast synaptic vesicle release [7]. Additional Ca2+ influx at the presynapse employs Cav2.3 (R-type) and Cav1.2/3 (L-type) channels [8,9], although the latter is believed to have a limited impact on vesicle release and, thus, eIPSC amplitude [1,7,10]. The high-voltage activated Cav2 channels show faster activation and inactivation making them suitable for fast transmission of neuronal action potential activity, whereas Cav1 channels are primarily involved in slower processes like hormone secretion and Ca2+ signaling to gene transcription [11]. Accordingly, biochemical and functional studies have identified numerous molecular interactions between VGCC subunits and various partners that serve, for example, to couple Ca2+ channels to the release machinery (reviewed in [11,12]). Interestingly, these interactions of VGCC subunits include not only intracellular pathways but also crosstalk to extracellular or cell surface molecules [13,14,15,16,17,18,19,20].
We discovered many years ago that neurexins (Nx), a polymorphic family of synaptic cell surface molecules [21,22], are involved in the regulation of VGCC-dependent neurotransmitter release from excitatory and inhibitory synapses [23]. Neurexins are encoded by three genes in vertebrates, each of which contains independent promoters that drive transcription of longer α-Nx and shorter β-Nx. A truncated γ-isoform is transcribed in neurexin-1 [24] and more variants arise from up to six conserved splice sites [25,26]. Extracellularly, α-Nx proteins mostly comprise six laminin-Nx-sex-hormone-binding (LNS) domains with interspersed epidermal growth factor (EGF)-like repeats. Shorter β-Nx differ by expressing a β-specific, 37-residue-long N-terminal domain before splicing into the last (sixth) LNS domain of the respective gene [21,22]. Since LNS6 and subsequent sequences are identical in α- and β-Nx, they share properties such as a C-terminal PDZ recognition motif required for intracellular trafficking [27,28], a heparan sulfate glycan moiety [29], and physiological ectodomain cleavage [30]. α- and β-Nx also share binding partners such as neuroligins [31,32,33], leucine-rich repeat transmembrane neuronal proteins (LRRTMs) [34,35,36], α -dystroglycan [37,38], latrophilins [39], and cerebellins [40,41].
The functional link between Nx and VGCCs was initially observed in a constitutive deletion mouse model (knockout) of all α-Nx [23,42,43], and later confirmed in conditional knockout neurons lacking all β-Nx [44,45]. Surprisingly, investigations of conditional knockout neurons lacking all Nx variants detected reduced total Ca2+ transients only in somatostatin- but not parvalbumin-positive interneurons of the medial prefrontal cortex [46], and failed to see reduced Ca2+ influx into the parvalbumin-positive excitatory calyx of Held synapses in the brainstem [47]. A possible explanation of this discrepancy might be that the functional link between Nx and VGCCs involves specific combinations of Nx variants and VGCC subtypes which may differ between brain regions and subpopulations of synapses. In support, we found recently that the reduced Ca2+ influx into boutons of excitatory hippocampal neurons in α-Nx triple knockout mice predominantly involved Cav2.1 (P/Q-type) VGCCs [17] and could be rescued by overexpression of the Nx1α variant which is abundant in hippocampal neurons [48].
To further explore this important aspect in our current study, we compared directly in the same model system whether and how deletions of one or all Nx isoforms can affect different synaptic VGCC subtypes. Therefore, we generated a conditional Nx1α knockout mouse model and compared presynaptic Ca2+ influx in primary hippocampal cultures of control to conditional knockout neurons lacking either the single Nx1α variant (Nx1α cKO, created for this study) or all Nx isoforms (Nx123 cKO [46,47]). We particularly focused on how the deletions affect single action potential-evoked Ca2+ influx through different VGCC subtypes, using transfected synGCaMP7b [49] as Ca2+ indicator and pharmacological isolation by sequential addition of subtype-specific blockers [9] which together allowed quantification even at the level of individual presynaptic boutons. We report here that Nx variants likely alter the contribution of most VGCC subtypes to presynaptic Ca2+ transients, including P/Q-type (CaV2.1), N-type (CaV2.1), L-type (CaV1.2/3) and R-type (CaV2.3) channels. Strikingly, the deletions of a single Nx1α or all Nx variants resulted in a different pattern of VGCC subtypes affected. These findings may indicate that Nx variants modulate Ca2+ influx in a partially overlapping, partially unique way, depending on the actual presence and/or relative amount of Nx variants and VGCC subtypes in a particular synapse population or even in individual terminals.
2. Materials and Methods
2.1. Animals
Mice of either sex were used for neuronal cultures derived from timed-pregnant females at E17. Animal experiments were performed at the University of Münster following government regulations for animal welfare and approved by the Landesamt für Natur, Umwelt und Verbraucherschutz (LANUV, NRW, Germany), license numbers 84-02.05.20.11.209 and 84-02.04.2015.A423.
Three mouse models were used in this study, Nx1α cKO, Nx123 cKO, and β-Nx cKO. Nx123 cKO and β-Nx cKO were characterized and reported earlier [44,45,46]. The conditional knockout model for Nx1α (Nx1α cKO) is reported here for the first time. Briefly, a targeting vector was cloned to introduce loxP sites on either side of the first coding exon of the Nx1 gene based on mouse genomic clones. This exon is the largest of all the exons of the gene and codes for the signal peptide as well as the first LNS domain and EGF domain. It was expected that the deletion of this exon would lead to a complete loss of functional protein from the synapses because the same exon was deleted to generate a conventional knockout of Nx1α [50] and led to the loss of the complete protein [23,50]. 5’ and 3’ loxP sites were inserted along with selection markers (Figure 1 B) and the targeting vector was electroporated into ES cells. Homologous recombination was identified by Southern blotting and PCRs, positive ES cell clones were microinjected into blastocysts. Chimeric mice were generated and germline transmission was monitored again by Southern blotting and PCRs, as described before [23,50]. Homozygous KI mice are viable and can be kept on a homozygous background.
2.2. Neuronal Cell Culture
Dissociated primary neurons were prepared in Hank’s Balanced Salt Solution (HBSS) from hippocampi as described [28,45]. Briefly, cell suspensions obtained after 0.25% trypsin and trituration were plated onto 18 mm glass coverslips coated with poly-L-lysine (Sigma) at a density of 40,000 cells/coverslip. After 4 h at 37°C in plating medium (MEM, 10% horse serum, 0.6% glucose, 1 mM sodium pyruvate), coverslips were inverted onto a 70 - 80% confluent monolayer of astrocytes grown in 12-well plates (Falcon) and incubated in Neurobasal medium supplemented with B27, 0.5 mM glutamine, and 12.5 mM glutamate. After 3 days, media were refreshed with Neurobasal medium supplemented with B27, 0.5 mM glutamine, and 5 mM AraC. Cultures were maintained at 37°C in a humidified incubator with an atmosphere of 95% air and 5% CO2. Neurons were transfected at day-in-vitro (DIV) 11 using lipofectamine (Thermo Fisher Scientific, Waltham, MA, USA), and experiments were performed between DIV 17 and DIV 21.
For induction of the conditional knockout of Nx genes marked with loxP sites, neuronal cultures were infected at DIV 4 with lentivirus by adding 100 µl per well of viral supernatant that was made as described earlier [45]. In short, recombinant lentiviral particles were produced in HEK293 cells and the supernatant was collected and snap-frozen (-80°C). Lentivirus contained EGFP fused to active Cre recombinase (Cre), or to an inactive mutated Cre recombinase (Cremut) [45,51], or the same vector with Cre recombinase deleted (ΔCre) [52].
2.3. Ca2+ Imaging
For Ca2+ imaging of synaptic boutons using a genetically encoded indicator, we generated the expression plasmid synGCaMP7b by fusing GCaMP7b [49] to synaptophysin, driven by a human synapsin promotor as described and characterized earlier for synGCaMP6f [9,17,45].
To determine presynaptic Ca2+ influx, primary neurons were transfected at DIV 11 with synGCaMP7b (see above), and co-transfected with pMH4-SYN-tdimer2-RFP (RFP, T. Oertner, Hamburg, Germany) for better identification of neuronal morphology. As only a few neurons were transfected by lipofectamine (about 30 – 50 per coverslip) it was possible to observe areas where only presynaptic boutons were loaded with synGCaMP7b (Figure 2 A,B). Six to eight days post-transfection, neurons growing on glass coverslips were placed in a recording chamber mounted to an inverted microscope (Observer.A1, Zeiss, Oberkochen, Germany) in 2 ml bath solution (temperature 32°C), containing (in mM): NaCl 145, KCl 3, MgCl2 1, CaCl2 2, glucose 11, HEPES 10; pH 7.3 adjusted with NaOH; to suppress postsynaptic signaling, 10 μM 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), 25 μM D, L-2-amino-5-phosphonovaleric acid (AP5), and 10 μM bicuculline were added. All chemicals were obtained from Sigma (St. Louis, MO, USA), except Ca2+ channel blockers (Alomone Labs, Jerusalem, Israel). A stimulation electrode, built by two platinum wires of 10 mm length in 10 mm distance was positioned with a micromanipulator (MPC-200, Sutter Instrument, Novato, CA, USA) and neurons were stimulated with 50 Hz trains of 1, 3, or 10 current pulses (1 ms, 55 mA). Ca2+ transients were visualized and recorded (20 ms exposure time, frame rate 50 Hz, binning 2: 0.46 μm per pixel) with a CMOS camera (Orca Flash4.0, Hamamatsu, Japan), a LED-light source (SpectraX, Lumencor, Beaverton, OR, USA) using the green channel (excitation at 470 ± 20 nm) or red channel (640 ± 20 nm) and controlled by VisiView software (Visitron Systems, Puchheim, Germany). As a standard, 20 frames were recorded before the stimulus train was triggered. For stimulation with one AP, four individual recordings with 10 s time intervals were averaged frame by frame to improve the signal-to-noise ratio. Ca2+ channel antagonists were added by direct application into the recording chamber 3 min prior next stimulation, ω-agatoxin IVA (0.1 μM, P/Q-Type; Alomone Labs), ω-conotoxin GVIA (2 μM, N-Type; Alomone Labs), nifedipine (20 μM, L-Type; Sigma-Aldrich) and SNX-482 (0.5 μM, R-Type; Alomone Labs). Due to the chemical instability of 2-AG (2 μM, CB1-receptor agonist; Avantilipids) in aqueous solutions [53], 1 μl aliquots of a 4 mM 2-AG in DMSO were prepared and frozen at -20°C. Immediately before use, 200 μl of bath solution was added and inserted into the recording chamber.
2.4. Data Analysis
Data analysis of imaging recordings of Ca2+ transients was done with Fiji/ImageJ (National Institute of Health, MA, USA) and IgorPro (Wavemetrics, Lake Oswego, Oregon) or MATLAB R2020b (The MathWorks Inc.). Active boutons were identified by the increase of fluorescence (ΔF) after stimulation with a train of 3 AP. 60-100 regions of interest (ROIs) per measurement were drawn around active boutons using the plugin ‘‘Time Series Analyzer V3’’ with an AutoROI diameter of 8 pixels (Figure 2 C). To quantify fluorescence changes in individual boutons, we first applied the commonly used [54] ‘‘Subtract Background...’’ tool of ImageJ (employing a ‘‘rolling ball’’ algorithm with a radius of 20 pixels ~ 10 μm), to remove the background signal deriving from faint autofluorescence and the dark current of the camera. For each ROI and each frame, the mean of the four pixels with the strongest fluorescence was calculated using a self-made macro. The area of four pixels (0.85 mm2) corresponds to the size of a normal bouton and the restriction to the four brightest pixels avoids the problem of the relevance of the ROI size to the area of increased fluorescence. Further calculations used IgorPro/MATLAB to average for each ROI the value of frame 10–20 as a control value (F0), changes were calculated as the change of fluorescence intensity (Fstim – F0 = ΔF) divided by the control (ΔF/F0) for each ROI. Single AP responses were analyzed after averaging four consecutive recordings already within ImageJ, and for analysis of the individual amplitudes, the traces were binomial Gaussian smoothed (coefficient 3) to improve the signal-to-noise ratio.
To quantify the relative change by application of the CB1-receptor agonist 2-AG, the relative change in synGCaMP7b ΔF/F0 was calculated for each bouton as described in the formula below:
For the analysis of individual presynaptic boutons, only boutons were included with an amplitude larger than 0.12 ΔF/F0 (three times the noise level) before treatment to allow reliable quantification of minor reduction.
2.5. Quantification and Statistical Analysis
Statistical tests were performed in Prism (GraphPad Prism 6.0d, GraphPad Software Inc.). If data showed normal distribution, Student’s t-test was used to compare two groups and ANOVA for multiple comparisons with post hoc Turkey’s multiple-comparison test. In case the criteria for normal distribution were not fulfilled, the corresponding non-parametric tests were used, e.g. Kruskal-Wallis test followed by Dunn’s test. The data are represented as mean ± SEM or boxplot with 25-75 percentile and significance level indicated by asterisks (*p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001). Further information on statistical details can be found in the figure legends. The experiments were not randomized, and investigators were only partially blinded during experiments and analyses. For outlier identification (see figure legend), the ROUT-method [55] was performed in GraphPad Prism with Q = 1. In addition, the number of examined neurons and boutons are shown in the form of boutons/neurons or as a single number indicating the number of neurons in all figures.
3. Results
3.1. Nx1α is the Prominent Nx Variant in Cultured Primary Hippocampal Neurons
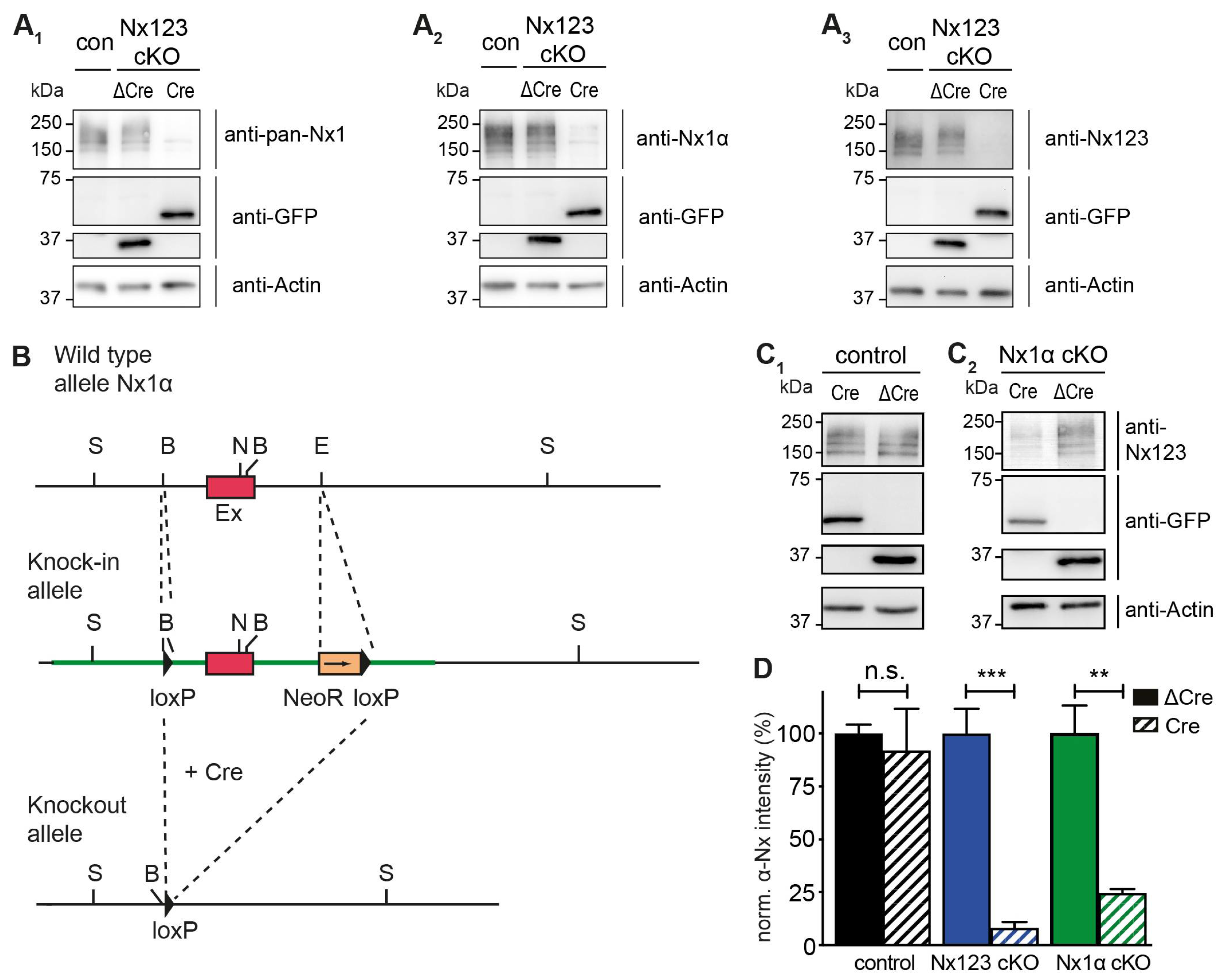
Cultured primary hippocampal neurons from Nx123 conditional knockout (cKO) mice [46] were transduced by lentivirus particles expressing active Cre- or inactive ΔCre-recombinase fused to EGFP at DIV4. Neurons were tested for α-Nx expression at DIV18 by immunoblotting and compared to neurons that contained no floxed α-Nx variants (Figure 1 A). All three antibodies tested (Figure 1 A1-A3) revealed the presence of α-Nx in wild-type and ΔCre-transduced neurons, and strongly reduced protein levels in Cre-transduced Nx123 cKO neurons, indicating an effective deletion of α-Nx. This finding is consistent with the reduced mRNA levels in the same mouse model [46] and the efficient removal of β-Nx protein upon Cre recombination in a related line [45]. To compare these Nx123 cKO neurons to neurons lacking only the single Nx1α variant, which is prominently expressed in the hippocampus [48], a knock-in/ conditional knockout mouse line of Nx1α was generated, in which the first coding exon of the neurexin-1 gene is flanked by loxP sites (Figure 1 B). In hippocampal neurons cultured from these Nx1α cKO mice, Cre recombinase expression also efficiently depleted the α-Nx signal on immunoblots in comparison to cultures infected by ΔCre-expressing lentivirus (Figure 1 C2) in contrast to neurons that contained no floxed α-Nxs (Figure 1 C1). The strong reduction of the signal indicated an effective deletion of Nx1α. Moreover, since we used an antibody that recognizes all α-Nx variants this finding confirms the substantial contribution of the Nx1α isoform to the overall α-Nx protein pool, consistent with earlier mRNA data [48]. The strong reduction of the signal also indicates that the deletion of Nx1α is likely not compensated by other α-Nx isoforms. Quantification of the Nx signal intensities on repeated immunoblots, normalized to the respective control (ΔCre) value (Figure 1 D), confirmed the predominance of Nx1α in the cultured hippocampal neurons because deletion of the single variant reduced the α-Nx signal already by 75% to 25 ± 2% of control (Nx1α cKO Cre), while deletion of all α-Nx reduced the signal to 8 ± 3% (Nx123 cKO Cre).
Figure 1.
Conditional deletion of the single Nx1α variant. (A) Immunoblots of Nx in Nx123 cKO cells tested with 3 different antibodies: pan-Nx1 (A1, Millipore #AB161-I), Nx1α (A2, Frontier Institute #AB_2571817) and Nx123 (A3, SySy #175003). Con, mouse line without floxed α-Nx. (B) Wild type allele of the 5‘ end of the Nx1α gene including the first coding exon (indicated in red) is illustrated. After successful homologous recombination of the wild-type allele with the targeting vector (not depicted), the knock-in allele that resulted is indicated. The 5’ loxP site is introduced via the BamH1 (‘B’) site upstream of the first coding exon. Downstream of the first coding exon and at the EcoR1 site (‘E’), the 3' loxP site and NeoR (Neomycin resistance) gene are inserted (blunt end cloning). Via the addition of a Cre-recombinase, the knock-in allele is converted into the knockout allele. The region between the loxP sites of the Nx1α gene including the first coding exon is excised. Further restriction sites: S = Spel, N = Nhel. (C) Immunoblots of control neurons without floxed α-Nx and Nx1α cKO neurons were probed with anti-Nx123 (SySy #175003). (D) Quantification of αNx normalized to ΔCre condition (100%) for control neurons, Nx123 cKO neurons, and Nx1α cKO neurons. Data are based on n independent immunoblot experiments like in A3 and C (control: 3, 2; Nx123: 4, 4; Nx1α: 3, 3); columns were compared with an unpaired t-test. n.s. = non-significant: p > 0.05, ** p < 0.01, ***p < 0.001.
Figure 1.
Conditional deletion of the single Nx1α variant. (A) Immunoblots of Nx in Nx123 cKO cells tested with 3 different antibodies: pan-Nx1 (A1, Millipore #AB161-I), Nx1α (A2, Frontier Institute #AB_2571817) and Nx123 (A3, SySy #175003). Con, mouse line without floxed α-Nx. (B) Wild type allele of the 5‘ end of the Nx1α gene including the first coding exon (indicated in red) is illustrated. After successful homologous recombination of the wild-type allele with the targeting vector (not depicted), the knock-in allele that resulted is indicated. The 5’ loxP site is introduced via the BamH1 (‘B’) site upstream of the first coding exon. Downstream of the first coding exon and at the EcoR1 site (‘E’), the 3' loxP site and NeoR (Neomycin resistance) gene are inserted (blunt end cloning). Via the addition of a Cre-recombinase, the knock-in allele is converted into the knockout allele. The region between the loxP sites of the Nx1α gene including the first coding exon is excised. Further restriction sites: S = Spel, N = Nhel. (C) Immunoblots of control neurons without floxed α-Nx and Nx1α cKO neurons were probed with anti-Nx123 (SySy #175003). (D) Quantification of αNx normalized to ΔCre condition (100%) for control neurons, Nx123 cKO neurons, and Nx1α cKO neurons. Data are based on n independent immunoblot experiments like in A3 and C (control: 3, 2; Nx123: 4, 4; Nx1α: 3, 3); columns were compared with an unpaired t-test. n.s. = non-significant: p > 0.05, ** p < 0.01, ***p < 0.001.
3.2. Deleting the Single Nx1α Variant is Sufficient to Reduce the Total Presynaptic Ca2+ Influx
To analyze single action potential-driven presynaptic Ca2+ influx, we measured Ca2+ transients using the genetically encoded Ca2+ indicator synGCaMP7b (Figure 2 A-C). We then compared control neurons (Cremut in Figure 2 D, E) to Nx deficient neurons (Cre in Figure 2 D, E). In hippocampal neurons lacking all Nx variants, the maximum amplitude of Ca2+ transients was 0.33 ± 0.02 ΔF/F0 (Figure 2 F, Nx123 cKO Cre) compared to 0.43 ± 0.02 ΔF/F0 in neurons with normal Nx expression (Figure 2 F, Nx123 cKO Cremut; p < 0.0001, unpaired t-test). This corresponds to a reduction by 23.3% in Nx123 cKO Cre compared to Nx123 cKO Cremut neurons, which is compatible with the slightly smaller reduction of total Ca2+ transients (18.5%) we found earlier in constitutive KO neurons that lack all α-Nx [17].
Figure 2.
Presynaptic Ca2+ transients recorded from individual active boutons with synGCaMP7b. (A) Example picture of fluorescence intensity of synGCaMP7b before stimulation (left, F0, shown in magenta), representing the baseline fluorescence; fluorescence intensity changes after stimulation with 3 AP, isolated by subtraction (middle, ΔF, shown in green). The green fluorescence dots lighting up indicate active boutons. Both images merged represent the effect image (right) that allows the identification of active boutons that are not disturbed by high baseline fluorescence of other sources like Cre-EGFP-fluorescent cell nuclei (asterisk). (B) Enlarged perspective (yellow box in A), showing the change in fluorescence (ΔF, green) as well as the cell process morphology indicated by co-transfected RFP (red). (C) ROIs (red circles) were placed on active boutons for the quantification of presynaptic Ca2+ transients. (D) Averaged synGCaMP7b fluorescence changes from Nx123 cKO neurons with Nx (Cremut, n = 14 cells/1045 boutons) or without all Nx variants (dashed line, Cre, 13/916) show Ca2+ transients following a single AP stimulation. (E) Neurons lacking only Nx1α (Cre, 8/681) and equivalent controls (Cremut, 12/1074) showed comparable fluorescence alterations as those seen in N123 cKO. (F) Comparing peak values of Ca2+ transients (mean ± SEM) in Cremut and Cre cells from both mouse lines in response to a single AP stimulation. Nx123 cKO: Cremut 48 cells/3964 boutons, Cre 58/4582, and Nx1α cKO: Cremut 21/1672, Cre 25/2051. The mean values of all boutons of a single cell are shown as dots and used for statistics. Columns were compared with an unpaired t-test. *: p < 0.05, **** p < 0.0001.
Figure 2.
Presynaptic Ca2+ transients recorded from individual active boutons with synGCaMP7b. (A) Example picture of fluorescence intensity of synGCaMP7b before stimulation (left, F0, shown in magenta), representing the baseline fluorescence; fluorescence intensity changes after stimulation with 3 AP, isolated by subtraction (middle, ΔF, shown in green). The green fluorescence dots lighting up indicate active boutons. Both images merged represent the effect image (right) that allows the identification of active boutons that are not disturbed by high baseline fluorescence of other sources like Cre-EGFP-fluorescent cell nuclei (asterisk). (B) Enlarged perspective (yellow box in A), showing the change in fluorescence (ΔF, green) as well as the cell process morphology indicated by co-transfected RFP (red). (C) ROIs (red circles) were placed on active boutons for the quantification of presynaptic Ca2+ transients. (D) Averaged synGCaMP7b fluorescence changes from Nx123 cKO neurons with Nx (Cremut, n = 14 cells/1045 boutons) or without all Nx variants (dashed line, Cre, 13/916) show Ca2+ transients following a single AP stimulation. (E) Neurons lacking only Nx1α (Cre, 8/681) and equivalent controls (Cremut, 12/1074) showed comparable fluorescence alterations as those seen in N123 cKO. (F) Comparing peak values of Ca2+ transients (mean ± SEM) in Cremut and Cre cells from both mouse lines in response to a single AP stimulation. Nx123 cKO: Cremut 48 cells/3964 boutons, Cre 58/4582, and Nx1α cKO: Cremut 21/1672, Cre 25/2051. The mean values of all boutons of a single cell are shown as dots and used for statistics. Columns were compared with an unpaired t-test. *: p < 0.05, **** p < 0.0001.
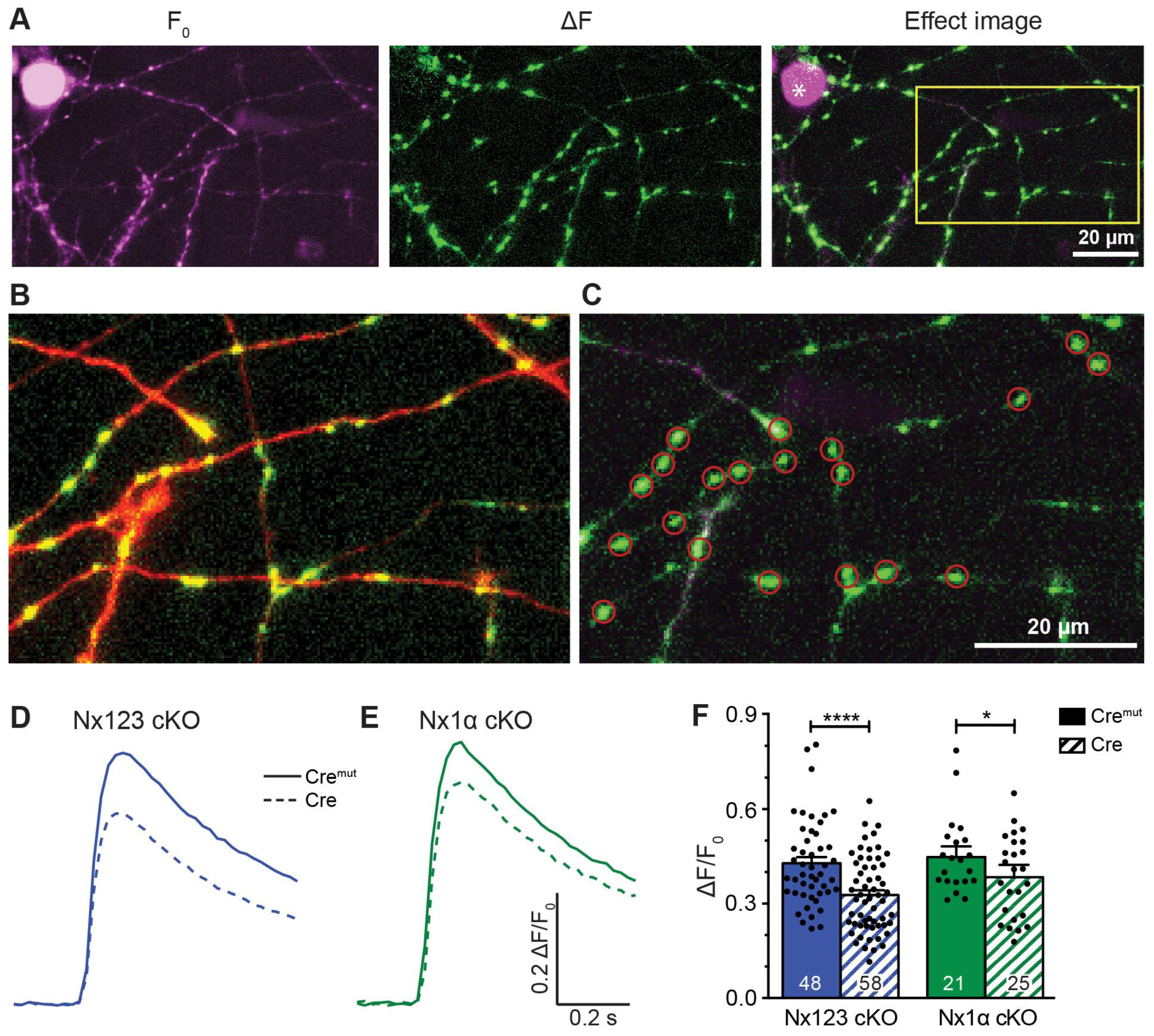
These results suggest that the α-Nx variants are predominantly responsible for regulating the total presynaptic Ca2+ influx. To test if deletion of the single Nx1α variant was already sufficient to reduce the total presynaptic Ca2+ influx, we performed the same experiment using neurons from the newly generated Nx1α cKO mouse line (Figure 2 E, F). We found that again the maximum amplitude of Ca2+ transients was smaller in Cre transduced neurons compared to the control (Nx1α cKO, Cremut: 0.458 ± 0.026 vs. Cre: 0.380 ± 0.025; p < 0.05; Figure 2 F). This reduction of Ca2+ transients by 17.0% in neurons lacking only α-Nx is in line with the strong expression of the Nx1α isoform in hippocampal neurons [48] and constitutive deletion of Nx1α has been shown before to cause functional and behavioral deficits [56,57,58]. These data may indicate that the lack of Nx1α is so fundamental that other α-Nx variants, for example, Nx3α [48], cannot fully compensate for the loss at the level of overall Ca2+ influx, emphasizing the role of Nx1α for the presence and function of presynaptic VGCC. The question arises, however, if all VGCC subtypes contribute equally to the reduction of overall Ca2+ influx, or if the individual VGCC subtypes (P/Q, N, L, or R) contribute disproportionally to the effect.
3.3. Deletion of All Nx Predominantly Reduced Ca2+ Influx through P/Q-Type VGCC
The total Ca2+ influx into presynaptic terminals in response to single action potentials is composed of contributions from different VGCC subtypes, which can be inhibited by specific blockers. In our experiments, we blocked P/Q-type channels by 0.1 μM ω-agatoxin IVA, N-type channels by 2 μM ω-conotoxin GVIA, L-type channels by 20 μM nifedipine, and R-type channels by 0.5 μM SNX-482. Sequential administration of these blockers was used to isolate pharmacologically Ca2+ influx through individual subtypes, which we characterized before in our cell culture model [9]. In that previous study, we observed that sequential addition of the different VGCC blockers caused a reduction in Ca2+ influx after almost every addition, indicating a broad mixture of P/Q-type, N-type, L-type, and R-type VGCCs in presynaptic boutons of primary hippocampal neurons. We, therefore, applied the protocol of sequential blocker administration on Nx123 cKO neurons transduced by active Cre and inactive Cremut recombinase to dissect if deletion of all Nx affected the presynaptic VGCC subtype composition or induced a proportional reduction of all subtypes.
We found in control neurons that P/Q-type VGCCs contributed most to Ca2+ transients, followed by L-type and N-type channels (Figure 3 A, Cremut). The contribution of R-type channels isolated by SNX-482 in normal boutons was so small that a reliable quantification in comparison to noise was hardly possible. The small Ca2+ transient that is still visible in the presence of all blockers is likely explained by some SNX-482 insensitive R-type channels [5,59,60]. More importantly, the reduced total presynaptic Ca2+ transients in neurons lacking all Nx reported above (Figure 2) were mainly due to a substantial reduction of P/Q-type channel activity (Figure 3 A, Cre) and, additionally, the portion of N-type channels is moderately smaller, whereas some SNX-482 sensitive R-type channels could be identified here. The L-type channels seemed not affected by the loss of Nx. The strong impact of Nx on P/Q-type channels and the Nx indifference of L-type channels are visible in a direct comparison of the digitally isolated transients as shown in Figure 3 B. The absolute Ca2+ influx through the different VGCC subtypes was quantified in more detail and compared between Nx-expressing (Cremut) and Nx-deficient (Cre) neurons. The isolated Ca2+ transients passing through P/Q-type channels (Nx123 Cremut: 0.17 ± 0.04 ΔF/F0) were reduced almost by half when all Nx were missing (Nx123 Cre: 0.09 ± 0.03 ΔF/F0). Also the N-type Ca2+ channel transients were reduced in Nx deficient neurons albeit at a lower level (Nx123 Cremut: 0.080 ± 0.014 ΔF/F0 vs. Nx123 Cre: 0.057 ± 0.010 ΔF/F0), whereas the absolute contribution of L-type channels seems not affected by a lack of Nx (Nx123 Cremut: 0.148 ± 0.034 ΔF/F0 vs. Nx123 Cre: 0.143 ± 0.028 ΔF/F0). For the R-type Ca2+ channel, we observed no R-type transient in the presence of Nx, whereas in absence of Nx a small SNX-482 sensitive R-type transient was present (Nx123 Cre: 0.032 ± 0.009 ΔF/F0).
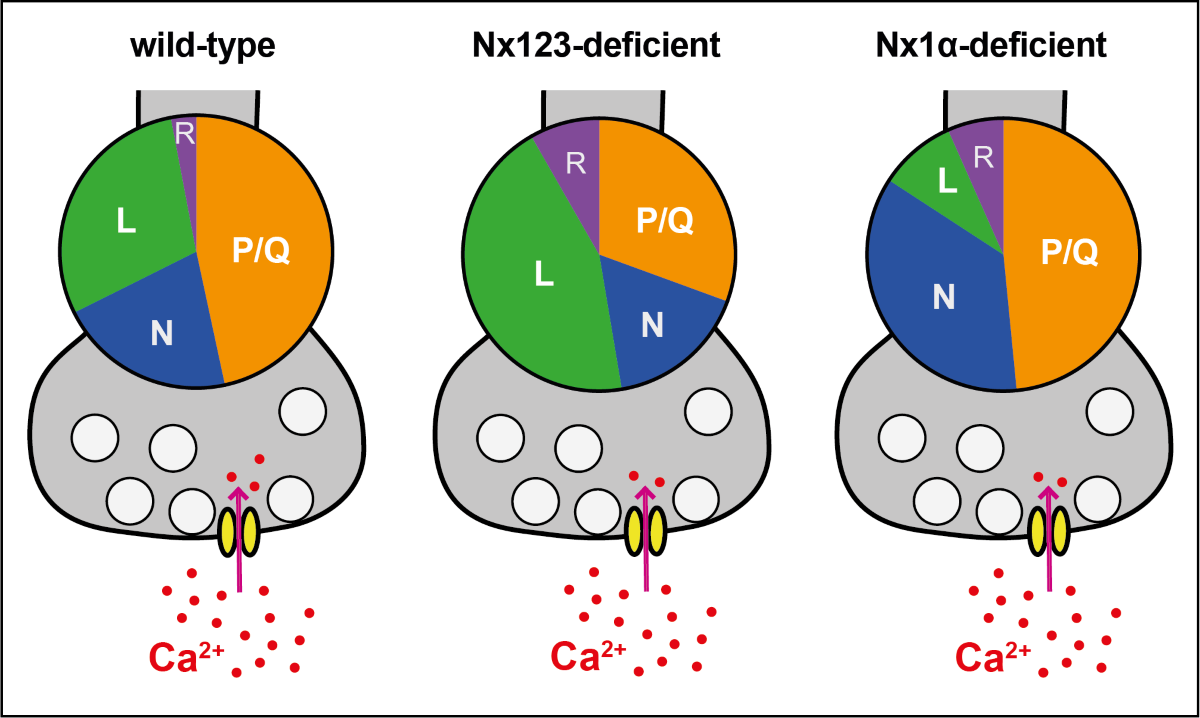
To compare the relative proportion of Ca2+ influx through the different VGCC subtypes, we calculated the share of VGCC subtypes to the total Ca2+ influx for individual boutons of Nx-expressing (Cremut) and Nx-deficient (Cre) neurons (Figure 3 C). As a consequence of the different total presynaptic Ca2+ influx of control and cKO boutons, the absolute Ca2+ influx through a particular VGCC subtype differs from their relative contribution. For example, the almost equal absolute Ca2+ influx via L-type channels (Figure 3 B, green traces) corresponds to a larger relative contribution of L-type channels in N123 cKO (Figure 3 C, green bars) as the total Ca2+ transient is smaller in N123 cKO. 41.2 ± 1.6% of the total Ca2+ influx passed through the P/Q-type Ca2+ channel in control neurons, compared to 26.6 ± 1.5% in Nx-deficient cells (p < 0.0001; Kruskal-Wallis test; Figure 3 C). In contrast to P/Q-type, the part relative to the total Ca2+ transient of the L-type Ca2+ channel influx was larger in Nx deficient cells (Nx123 Cremut: 29.9 ± 1.3% vs. Nx123 Cre: 38.6 ± 1.5%, p < 0.0001) without an increase in absolute amount (see Figure 3 B) as the total Ca2+ transient was smaller in the Nx deficient neurons. A lack of Nx had only minor impact on the relative portion of N-type channels (Nx123 Cremut: 17.8 ± 0.9% vs. Nx123 Cre: 14.6 ± 0.9%, p = 0.1337). A small increase in Nx-deficient presynapses could be observed for R-type (Nx123 Cremut: 1.6 ± 0.5% vs. Nx123 Cre: 7.2 ± 0.7%, p = 0.0028). Taken together, the VGCC subtype with the largest relative contribution shifted from P/Q-type channels in control conditions to the L-type channels in neurons lacking all Nx.
The improved signal-noise ratio of the recordings with GCaMP7b allowed even an evaluation of VGCC subtype contribution not only on cellular level but in individual synaptic boutons. In these recordings we observed a broad heterogeneity in the VGCC subtype contribution of individual synaptic boutons within the same neuron. For each bouton, the relative contributions of P/Q-, N-, and L-type channels to Ca2+ transients were calculated and the frequency distribution was plotted as a histogram (Figure 3 D-F). The analysis for P/Q-type part in individual presynaptic boutons showed many boutons with a P/Q-type contribution of about 60-80% in control conditions, but only a few boutons with this amount of P/Q-type Ca2+ influx in neurons lacking Nx indicated by a clear left-shift in the histogram with a maximum around 20% contribution of P/Q-type in synaptic boutons lacking Nx (Figure 3 D). N- or L-type channels had a maximum at 10-30% of Ca2+ influx in boutons with normal Nx levels, but the contribution reached above 90% within some boutons, indicating that in some individual boutons the Ca2+ transients were driven almost completely by only one of these types of VGCC. Regarding the deletion of all Nx variants, an altered distribution was observed. In Nx deficient boutons N- and P/Q-type revealed a similar contribution with a peak in 10-30%. In terms of L-type, the contribution was more scattered, with most boutons having a contribution of about 50-60%. Consequently, it appeared that in Nx123 KO neurons a shift took place in the opposite direction for P/Q-type (Figure 3 D) and L-type (Figure 3 F) channels. For P/Q-type, the distribution decreased compared to the control (left shift, Figure 3 D), but for L-type it increased (right shift, Figure 3 F). For N-type channels, the distribution remained almost equal (Figure 3 E). Thus, the presynaptic Ca2+ transients in neurons lacking Nx are not only smaller as described already earlier [17,46], but also reveal a shift from vesicle release-supporting VGCC subtypes P/Q- and N-type channels to Ca2+ channels that are primarily involved in slower processes like hormone secretion and Ca2+ signaling to gene transcription [11]. Both effects contribute to the weakening of synaptic transmission in neurons lacking Nx [17,23,43,46].
3.4. Deletion of the Single Nx1α Variant Altered the Pattern of VGCC Subtype Contribution to Presynaptic Ca2+ Influx
Our measurements of total presynaptic Ca2+ transients revealed a smaller, but significant impairment in neurons lacking only one Nx variant, Nx1α (Figure 2 E, F). To investigate a possible specific impact of Nx1α on VGCC subtype distribution, which may differ from the changes caused by a lack of all Nx variants, we compared Cremut and Cre-transduced neurons from the new Nx1α cKO mouse line on presynaptic VGCC subtype composition. Blocking the P/Q-type channel with ω-agatoxin IVA in these neurons induced an equal reduction in Ca2+ influx in control and Nx1α deficient neurons (Figure 4 A). Thus, the isolated P/Q-type Ca2+ transient is not affected by Nx1α, and remains approximately at the same level (Nx1α Cremut: 0.220 ± 0.045 vs. Nx1α Cre: 0.202 ± 0.40; Figure 4 B). In contrast, the addition of ω-conotoxin GVIA resulted in a higher reduction of the Ca2+ influx in Nx1α depleted cells and thus larger isolated Ca2+ influx through the N-type channels (Nx1α Cremut: 0.104 ± 0.023 vs. Nx1α Cre: 0.158 ± 0.034; Figure 4 B). After addition of nifedipine, a higher Ca2+ influx remained in the Nx1α depleted cells. Thus, the absolute influx through the L-type channel was reduced (Nx1α Cremut: 0.146 ± 0.040 vs. Nx1α Cre: 0.069 ± 0.020; Figure 4 B). The minor R-type transient remained almost unchanged hardly above the noise level.
Evaluation of the relative portion of the different VGCC subtypes revealed a comparable result (Figure 4 C). The cKO of Nx1α did not change the relative part of the P/Q-type channel (Nx1α Cremut: 43.2 ± 1.3% vs. Nx1α Cre: 42.3 ± 1.4%, p > 0.9999). In contrast, a shift in the relative VGCC subtype contribution was seen in the N- and L-type channel contributions. The N-type portion significantly increased from 20.2 ± 0.8% (Nx1α Cremut) to 31.2 ± 1.2% (Nx1α Cre; p < 0.0001), in contrast, the L-type portion decreased from 23.3 ± 1.0% (Nx1α Cremut) to 8.0 ± 0.7% (Nx1α Cre; p < 0.0001). No significant changes were found in the portion of R-type (p > 0.9999; Figure 4 C). To sum up, the N-type contribution increased whereas the L-type contribution decreased in neurons lacking Nx1α.
Again we used the possibility to evaluate the VGCC contributions for each synaptic bouton individually and plotted a frequency histogram for P/Q-, N-, and L-type Ca2+ channels (Figure 4 D-F). In a direct comparison between control cells and Nx1α depleted cells, it appears that the Nx1α deletion led to a higher influx through the N-type channel as the peak at 10%, meaning only minor amounts of N-type channels in this bouton, in control cells disappeared in neurons without Nx1α and the number of boutons with a N-type contribution above 30% was always moderately higher (Figure 4 Ε). The distribution of L-type channel transients in single boutons showed that more boutons with a larger influx through the L-type channel existed under control conditions and in the absence of Nx1α more boutons were represented with almost zero Ca2+ influx through the L-type channel (Figure 4 F). To sum up, in neurons lacking Nx1α the N-type contribution increased whereas the L-type contribution decreased and the P/Q-type was not affected and, thus, a lack of the single Nx variant Nx1α led to significantly more vesicle-release supporting N-type channels, which may compensate the effect of reduced total Ca2+ influx on the vesicle release.
In combination, it shows that Nx have an impact on the combination of presynaptic VGCC subtypes, but beyond this, it seems that individual Nx subtypes have correlations to special VGCC subtypes. The Nx1α, which is prominently expressed in hippocampal neurons [48], is supporting L-type Ca2+ channels but seems to dampen the N-type Ca2+ channels, whereas the full Nx KO shows that all Nx in concert promote the activity of P/Q- and N-type Ca2+ channels and, thus, presynaptic vesicle release.
3.5. Deletions of Nx also Affect the Endocannabinoid Receptor-Dependent Modulation of Presynaptic Ca2+ Influx
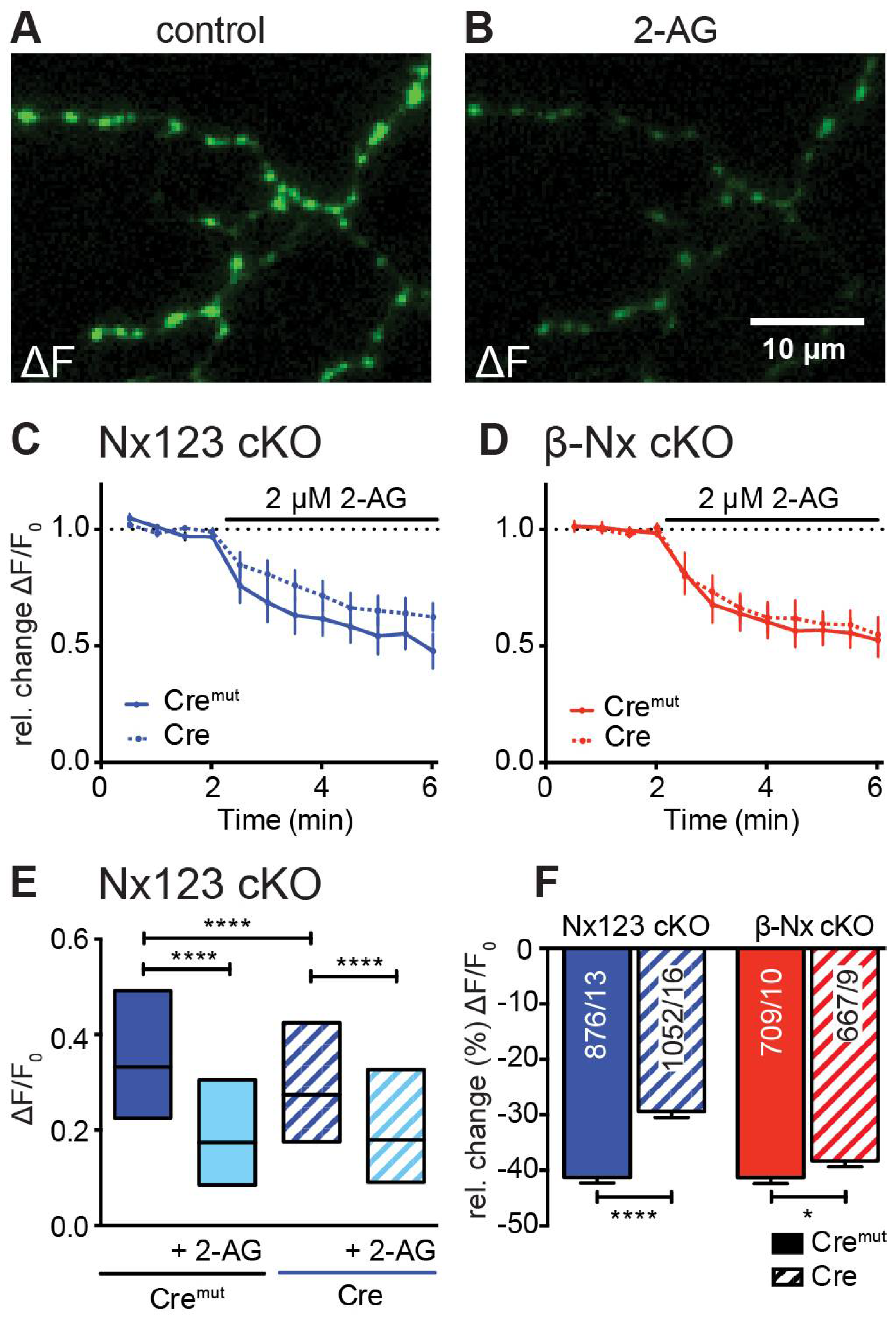
Presynaptic VGCCs are modulated by a wealth of metabotropic receptors including the cannabinoid receptor CB1 [61,62]. Strikingly, the endocannabinoid receptor system was recently shown to be modulated by neurexins [44]. Endocannabinoids such as 2-arachidonoylglycerol (2-AG) are lipid-based neurotransmitters that bind to CB1R [63] and thereby allow the retrograde adaptation of synaptic activity [64,65,66,67]. This modulatory process is regulated postsynaptically by on-demand synthesis and degradation of endocannabinoids [68]. Here, we tested the idea that the decrease of presynaptic Ca2+ transients induced by the CB1-receptor agonist 2-AG depends on Nx. Direct measurements of AP-driven presynaptic Ca2+ transients and subsequent activation of the CB1-receptor with 2-AG resulted in a significant reduction in Ca2+ influx in both, control and Nx deficient neurons as well as in β-Nx deficient neurons (Figure 5). This reduction of the presynaptic Ca2+ influx by 2-AG was larger in control neurons (Nx123 cKO Cremut: 0.37 ± 0.01; with 2-AG: 0.22 ± 0.01) compared to Nx deficient neurons (Nx123 cKO Cre: 0.33 ± 0.01; with 2-AG: 0.24 ± 0.01; Figure 5 E). The relative change (%) by the endocannabinoid 2-AG was significantly lower in the Nx deficient neurons (Cremut -41.3% ± 1.0% vs. Cre -29.4% ± 1.1%, p < 0.0001; Figure 5 F, blue columns), indicating a modulatory role of Nx in the CB1-receptor signaling cascade. In neurons lacking only β-Nx, the relative change by the addition of 2-AG was much weaker, but still significant (Cremut: -41.3% ± 1.1% vs. Cre: -38.3% ± 1.0%; p = 0.047; Figure 5 F, red columns). These data suggest a dominant role of α-Nx for the Nx-related impact on retrograde endocannabinoid signaling extending an earlier study describing a dependence solely on β-Nx [44].
4. Discussion
The present study revealed an unexpectedly complex modulation of presynaptic Ca2+ influx by Nx based on a comparison of Ca2+ transients through specific VGCC subtypes. In hippocampal neurons of Nx123 cKO mice, the presynaptic Ca2+ influx was reduced upon conditional knockout of all Nx variants (Figure 2 D,F). Interestingly, this reduction was stronger than that previously seen in neurons lacking all α-Nx but not β-Nx [17] or in neurons lacking all β-Nx but not α-Nx [44,45]. But even our novel deletion of the ASD candidate gene Nx1α alone induced a reduced total presynaptic Ca2+ influx (Figure 2 E,F), suggesting that already the lack of a single Nx variant affects synaptic efficiency. Obviously, the removal of more and more Nx variants gradually induces a stronger reduction of total presynaptic Ca2+ influx. While these data confirm our initial hypothesis of a general dose effect of Nx on synaptic function [23], we surprisingly found that deletions of Nx may produce different and complex patterns of affected VGCC subtypes.
In the complete Nx123 cKO, the reduced total presynaptic Ca2+ influx was mainly due to a reduced influx through P/Q-type channels (Figure 3). This is in line with similar data from hippocampal neurons of a constitutive knockout of all α-Nx variants [17] and from an analysis of the calyx of Held synapses, in which Nx were shown to be crucial for clustering of P/Q-type channels in the active zone [47]. In addition, our investigation of Nx123 cKO produced a tendency toward the reduction of N-type channel contribution while increasing the contribution of L-type and R-type VGCCs. Since this shift would imply a change from channels directly coupled to vesicle release to channel subtypes with a mere supportive role in fast synaptic release [1,6], the transition away from P/Q- and N-type to L- and R-type likely predicts a more dramatic influence on synaptic release than the moderate reduction of the Ca2+ transients suggest. In fact, a large release defect has been previously described with a reduction of postsynaptic EPSCs in αTKO neurons (lacking all α-Nx) by more than 50% compared to controls [42].
A different pattern of VGCC modulation was seen in the case of the single Nx1α cKO. The deletion of Nx1α alone did not change the Ca2+ influx through the P/Q-type channel, but, unexpectedly, elevated the contribution of N-type channels. Since the total Ca2+ influx was moderately reduced in this deletion model, the increased Ca2+ influx through N-type channels was likely compensated by a reduced L-type channel contribution (Figure 4). As P/Q-type and N-type VGCCs are the main Ca2+ channels for presynaptic transmitter release and deletion of Nx1α induces a shift in the relative contribution from L-type to N-type channels, transmitter release in Nx1α-deficient synapses should be normal or the probability of release increased, unlike in the complete deletion of Nx. Thus, the overall organization of the presynaptic active zone and clustering of P/Q-type channels observed earlier in the Calyx of Held synapse in absence of all Nx [47] most likely does not depend on the Nx1α variant because Nx1α knockout did not affect P/Q-type channel-driven Ca2+ influx as shown here in hippocampal neurons. However, this finding is in contrast to observations in neurons from a constitutive knockout of all α-Nx variants, in which overexpressed Nx1α partially rescued the amount of Ca2+ influx through P/Q-type channels [17]. This discrepancy indicates that Nx1α is not alone responsible for the modulation of the P/Q-type channel [17], but that other α-Nx variants can compensate for the deletion, e.g. in concert with α2δ auxiliary subunits of VGCCs. Together, these results are consistent with the view that Nx regulate presynaptic Ca2+ influx and that individual Nx variants may have partially overlapping, partially non-redundant effects on the distribution or function of different VGCC subtypes.
To further explore the possibility that α-Nx are also involved in additional signaling pathways targeting presynaptic VGCCs as suggested previously for the GABAB receptor pathway [43,69], we investigated retrograde signaling via the endocannabinoid system (ECS). In this retrograde pathway, postsynaptically synthesized endocannabinoids (e.g., 2-AG or AEA) diffuse to the presynapses to stimulate presynaptic CB1-receptor, inhibiting the activity of VGCCs [64,65]. We therefore compared neurons containing or lacking Nx and revealed a lower relative CB1-receptor effect by 2-AG in the absence of all Nx variants. This is in line with an increased tonic endocannabinoid signaling as it has been proposed for reduced β-Nx levels according to a study of the ECS in β-Nx-deficient neurons [44]. Our current results now suggest that the role of α-Nx in this regulation may even be stronger than β-Nx, based on a direct comparison of neurons lacking all Nx123 cKO versus β-Nx cKO neurons. While these results present an important extension of the role of Nx in regulating presynaptic VGCCs, the measured changes in presynaptic Ca2+ transients could not elucidate the precise mechanism of how Nx modulate the ECS. However, at least three hypotheses are conceivable. First, a postsynaptic regulation of 2-AG synthesis as postulated in [44] is possible since Nx engages in transsynaptic interactions and can cluster receptors in the postsynaptic membrane, for example, AMPAR [48,70,71] and GABAAR [72]. Naturally, such a potential postsynaptic influence on 2-AG synthesis could hardly be attributed to β-Nx alone as α-Nx share the same binding partners, supporting our observation here. This scenario would imply that α-Nx have an additional effect on VGCCs by modulating the ECS postsynaptically. Second, Nx could modulate the effect of the ECS system via the presynaptic organization of VGCCs and/or the localization of CB1-receptors. In support, it was shown that α-Nx is presumably involved in the overall organization at the active zone [47], and an altered distribution or activity of either CB1-receptors or the VGCC subtypes themselves may explain the effect of Nx reported here. Third, since the activity of the CB1 receptor is regulated by on-demand production and degradation of 2-AG [68], it cannot be ruled out that Nx might influence presynaptic 2-AG degradation as an additional alternative. Future research will have to distinguish between these possibilities.
5. Conclusion
Neurexins are key players in synapse organization and were found here to be pivotal for the contribution of particular Ca2+ channel subtypes to presynaptic Ca2+ influx into boutons of cultured hippocampal neurons. The lack of all Nx isoforms weakened the contribution of the P/Q-type channels that are normally responsible for fast synaptic vesicle release and elevated the amount of Ca2+ influx via L-type channels. In contrast, the deletion of the single Nx1α variant alone promoted influx through N-type channels at the expense of L-type channels. Our data indicate a complex interplay of different Nx variants with several subtypes of synaptic Ca2+ channel. This complex interplay may include the modulation of the endocannabinoid system by α-Nx that also impact synaptic Ca2+ channels.
Author Contributions
Conceptualization, M.M. and J.B.; methodology, M.M., J.B. and C.R.; software, J.B. and I.K.; formal analysis, J.B., I.K., C.R., D.R. and M.A.; investigation, I.K., J.B., D.R., C.R. and M.A.; resources, M.M.; data curation, J.B. and C.R.; writing—original draft preparation, J.B., I.K. and M.M.; writing—review and editing, M.M.; visualization, J.B., I.K. and C.R.; supervision, M.M.; project administration, M.M.; funding acquisition, M.M. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by grants of the Deutsche Forschungsgemeinschaft SFB1348 TPA03 and DFG MI 479/10-1 (to M.M.).
Institutional Review Board Statement
Animal experiments were performed at the University of Münster following government regulations for animal welfare and approved by the Landesamt für Natur, Umwelt und Verbraucherschutz (LANUV, NRW, Germany), license numbers 84-02.05.20.11.209 and 84-02.04.2015.A423.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors on request.
Acknowledgments
The authors thank I. Wolff and K. Kerkhoff for excellent technical support.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Catterall, W. A. Structure and Regulation of Voltage-Gated Ca2+-Channels. Annu Rev Cell Dev Biol 2000, 16, 521–55. [Google Scholar] [CrossRef] [PubMed]
- Dolphin, A. C.; Lee, A. Presynaptic calcium channels: specialized control of synaptic neurotransmitter release. Nat Rev Neurosci 2020, 21, 213–229. [Google Scholar] [CrossRef] [PubMed]
- Sheng, J.; He, L.; Zheng, H.; Xue, L.; Luo, F.; Shin, W.; Sun, T.; Kuner, T.; Yue, D. T.; Wu, L. G. Calcium-channel number critically influences synaptic strength and plasticity at the active zone. Nat Neurosci 2012, 15, 998–1006. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Harada, H.; Kamasawa, N.; Matsui, K.; Rothman, J. S.; Shigemoto, R.; Silver, R. A.; DiGregorio, D. A.; Takahashi, T. Nanoscale distribution of presynaptic Ca(2+) channels and its impact on vesicular release during development. Neuron 2015, 85, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Bischofberger, J.; Jonas, P. Differential gating and recruitment of P/Q-, N-, and R-type Ca2+ channels in hippocampal mossy fiber boutons. J Neurosci 2007, 27, 13420–9. [Google Scholar] [CrossRef]
- Cao, Y. Q.; Tsien, R. W. Different relationship of N- and P/Q-type Ca2+ channels to channel-interacting slots in controlling neurotransmission at cultured hippocampal synapses. J Neurosci 2010, 30, 4536–46. [Google Scholar] [CrossRef]
- Chen, J. J.; Kaufmann, W. A.; Chen, C.; Arai, I.; Kim, O.; Shigemoto, R.; Jonas, P. Developmental transformation of Ca2+ channel-vesicle nanotopography at a central GABAergic synapse. Neuron 2024, 112, 755–771. [Google Scholar] [CrossRef]
- Helton, T. D.; Xu, W.; Lipscombe, D. Neuronal L-type calcium channels open quickly and are inhibited slowly. J Neurosci 2005, 25, 10247–51. [Google Scholar] [CrossRef]
- Brockhaus, J.; Bruggen, B.; Missler, M. Imaging and Analysis of Presynaptic Calcium Influx in Cultured Neurons Using synGCaMP6f. Front Synaptic Neurosci 2019, 11, 12. [Google Scholar] [CrossRef]
- Dunlap, K.; Luebke, J. I.; Turner, T. J. Exocytotic Ca2+ channels in mammalian central neurons. Trends Neurosci. 1995, 18, 89–98. [Google Scholar] [CrossRef]
- Nanou, E.; Catterall, W. A. Calcium Channels, Synaptic Plasticity, and Neuropsychiatric Disease. Neuron 2018, 98, 466–481. [Google Scholar] [CrossRef] [PubMed]
- Mochida, S. Presynaptic Calcium Channels. Int J Mol Sci 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Eroglu, C.; Allen, N. J.; Susman, M. W.; O'Rourke, N. A.; Park, C. Y.; Ozkan, E.; Chakraborty, C.; Mulinyawe, S. B.; Annis, D. S.; Huberman, A. D.; Green, E. M.; Lawler, J.; Dolmetsch, R.; Garcia, K. C.; Smith, S. J.; Luo, Z. D.; Rosenthal, A.; Mosher, D. F.; Barres, B. A. Gabapentin receptor alpha2delta-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell 2009, 139, 380–92. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Yu, Y. P.; Zhou, C. Y.; Li, K. W.; Wang, D.; Chang, E.; Kim, D. S.; Vo, B.; Zhang, X.; Gong, N.; Sharp, K.; Steward, O.; Vitko, I.; Perez-Reyes, E.; Eroglu, C.; Barres, B.; Zaucke, F.; Feng, G.; Luo, Z. D. Central Mechanisms Mediating Thrombospondin-4-induced Pain States. J Biol Chem 2016, 291, 13335–48. [Google Scholar] [CrossRef] [PubMed]
- Tong, X. J.; Lopez-Soto, E. J.; Li, L.; Liu, H.; Nedelcu, D.; Lipscombe, D.; Hu, Z.; Kaplan, J. M. Retrograde Synaptic Inhibition Is Mediated by alpha-Neurexin Binding to the alpha2delta Subunits of N-Type Calcium Channels. Neuron 2017, 95, 326–340 e5. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Fehlhaber, K. E.; Sarria, I.; Cao, Y.; Ingram, N. T.; Guerrero-Given, D.; Throesch, B.; Baldwin, K.; Kamasawa, N.; Ohtsuka, T.; Sampath, A. P.; Martemyanov, K. A. The Auxiliary Calcium Channel Subunit alpha2delta4 Is Required for Axonal Elaboration, Synaptic Transmission, and Wiring of Rod Photoreceptors. Neuron 2017, 93, 1359–1374 e6. [Google Scholar] [CrossRef] [PubMed]
- Brockhaus, J.; Schreitmuller, M.; Repetto, D.; Klatt, O.; Reissner, C.; Elmslie, K.; Heine, M.; Missler, M. alpha-Neurexins Together with alpha2delta-1 Auxiliary Subunits Regulate Ca(2+) Influx through Cav2.1 Channels. J Neurosci 2018, 38, 8277–8294. [Google Scholar] [CrossRef]
- Dahimene, S.; Page, K. M.; Kadurin, I.; Ferron, L.; Ho, D. Y.; Powell, G. T.; Pratt, W. S.; Wilson, S. W.; Dolphin, A. C. The alpha(2)delta-like Protein Cachd1 Increases N-type Calcium Currents and Cell Surface Expression and Competes with alpha(2)delta-1. Cell Rep 2018, 25, 1610–1621 e5. [Google Scholar] [CrossRef] [PubMed]
- Risher, W. C.; Kim, N.; Koh, S.; Choi, J. E.; Mitev, P.; Spence, E. F.; Pilaz, L. J.; Wang, D.; Feng, G.; Silver, D. L.; Soderling, S. H.; Yin, H. H.; Eroglu, C. Thrombospondin receptor alpha2delta-1 promotes synaptogenesis and spinogenesis via postsynaptic Rac1. J Cell Biol 2018, 217, 3747–3765. [Google Scholar] [CrossRef]
- Geisler, S.; Schopf, C. L.; Stanika, R.; Kalb, M.; Campiglio, M.; Repetto, D.; Traxler, L.; Missler, M.; Obermair, G. J. Presynaptic alpha(2)delta-2 Calcium Channel Subunits Regulate Postsynaptic GABA(A) Receptor Abundance and Axonal Wiring. J Neurosci 2019, 39, 2581–2605. [Google Scholar] [CrossRef]
- Reissner, C.; Runkel, F.; Missler, M. Neurexins. Genome Biol 2013, 14. [Google Scholar] [CrossRef] [PubMed]
- Südhof, T. C. Synaptic Neurexin Complexes: A Molecular Code for the Logic of Neural Circuits. Cell 2017, 171, 745–769. [Google Scholar] [CrossRef]
- Missler, M.; Zhang, W.; Rohlmann, A.; Kattenstroth, G.; Hammer, R. E.; Gottmann, K.; Südhof, T. C. Alpha-neurexins couple Ca2+ channels to synaptic vesicle exocytosis. Nature 2003, 423, 939–948. [Google Scholar] [CrossRef]
- Sterky, F. H.; Trotter, J. H.; Lee, S. J.; Recktenwald, C. V.; Du, X.; Zhou, B.; Zhou, P.; Schwenk, J.; Fakler, B.; Sudhof, T. C. Carbonic anhydrase-related protein CA10 is an evolutionarily conserved pan-neurexin ligand. Proc Natl Acad Sci U S A 2017, 114, E1253–E1262. [Google Scholar] [CrossRef] [PubMed]
- Schreiner, D.; Nguyen, T. M.; Russo, G.; Heber, S.; Patrignani, A.; Ahrne, E.; Scheiffele, P. Targeted combinatorial alternative splicing generates brain region-specific repertoires of neurexins. Neuron 2014, 84, 386–98. [Google Scholar] [CrossRef] [PubMed]
- Treutlein, B.; Gokce, O.; Quake, S. R.; Sudhof, T. C. Cartography of neurexin alternative splicing mapped by single-molecule long-read mRNA sequencing. Proc Natl Acad Sci U S A 2014, 111, E1291–9. [Google Scholar] [CrossRef]
- Fairless, R.; Masius, H.; Rohlmann, A.; Heupel, K.; Ahmad, M.; Reissner, C.; Dresbach, T.; Missler, M. Polarized targeting of neurexins to synapses is regulated by their C-terminal sequences. J Neurosci 2008, 28, 12969–81. [Google Scholar] [CrossRef]
- Neupert, C.; Schneider, R.; Klatt, O.; Reissner, C.; Repetto, D.; Biermann, B.; Niesmann, K.; Missler, M.; Heine, M. Regulated Dynamic Trafficking of Neurexins Inside and Outside of Synaptic Terminals. J Neurosci 2015, 35, 13629–47. [Google Scholar] [CrossRef]
- Zhang, P.; Lu, H.; Peixoto, R. T.; Pines, M. K.; Ge, Y.; Oku, S.; Siddiqui, T. J.; Xie, Y.; Wu, W.; Archer-Hartmann, S.; Yoshida, K.; Tanaka, K. F.; Aricescu, A. R.; Azadi, P.; Gordon, M. D.; Sabatini, B. L.; Wong, R. O. L.; Craig, A. M. Heparan Sulfate Organizes Neuronal Synapses through Neurexin Partnerships. Cell 2018, 174, 1450–1464 e23. [Google Scholar] [CrossRef]
- Trotter, J. H.; Hao, J.; Maxeiner, S.; Tsetsenis, T.; Liu, Z.; Zhuang, X.; Sudhof, T. C. Synaptic neurexin-1 assembles into dynamically regulated active zone nanoclusters. J Cell Biol 2019, 218, 2677–2698. [Google Scholar] [CrossRef]
- Ichtchenko, K.; Hata, Y.; Nguyen, T.; Ullrich, B.; Missler, M.; Moomaw, C.; Südhof, T. C. , Neuroligin 1: a splice site-specific ligand for beta-neurexins. Cell 1995, 81, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Boucard, A. A.; Chubykin, A. A.; Comoletti, D.; Taylor, P.; Sudhof, T. C. A splice code for trans-synaptic cell adhesion mediated by binding of neuroligin 1 to alpha- and beta-neurexins. Neuron 2005, 48, 229–36. [Google Scholar] [CrossRef]
- Reissner, C.; Klose, M.; Fairless, R.; Missler, M. Mutational analysis of the neurexin/neuroligin complex reveals essential and regulatory components. Proc Natl Acad Sci U S A 2008, 105, 15124–9. [Google Scholar] [CrossRef]
- de Wit, J.; Sylwestrak, E.; O'Sullivan, M. L.; Otto, S.; Tiglio, K.; Savas, J. N.; Yates, J. R., 3rd; Comoletti, D.; Taylor, P.; Ghosh, A. LRRTM2 interacts with Neurexin1 and regulates excitatory synapse formation. Neuron 2009, 64, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.; Fuccillo, M. V.; Malenka, R. C.; Sudhof, T. C. LRRTM2 functions as a neurexin ligand in promoting excitatory synapse formation. Neuron 2009, 64, 791–8. [Google Scholar] [CrossRef]
- Siddiqui, T. J.; Pancaroglu, R.; Kang, Y.; Rooyakkers, A.; Craig, A. M. LRRTMs and neuroligins bind neurexins with a differential code to cooperate in glutamate synapse development. J Neurosci 2010, 30, 7495–506. [Google Scholar] [CrossRef]
- Sugita, S.; Saito, F.; Tang, J.; Satz, J.; Campbell, K.; Sudhof, T. C. A stoichiometric complex of neurexins and dystroglycan in brain. J Cell Biol 2001, 154, 435–45. [Google Scholar] [CrossRef]
- Reissner, C.; Stahn, J.; Breuer, D.; Klose, M.; Pohlentz, G.; Mormann, M.; Missler, M. Dystroglycan binding to alpha-neurexin competes with neurexophilin-1 and neuroligin in the brain. J Biol Chem 2014, 289, 27585–603. [Google Scholar] [CrossRef] [PubMed]
- Boucard, A. A.; Ko, J.; Sudhof, T. C. High affinity neurexin binding to cell adhesion G-protein-coupled receptor CIRL1/latrophilin-1 produces an intercellular adhesion complex. J Biol Chem 2012, 287, 9399–413. [Google Scholar] [CrossRef]
- Uemura, T.; Lee, S. J.; Yasumura, M.; Takeuchi, T.; Yoshida, T.; Ra, M.; Taguchi, R.; Sakimura, K.; Mishina, M. Trans-synaptic interaction of GluRdelta2 and Neurexin through Cbln1 mediates synapse formation in the cerebellum. Cell 2010, 141, 1068–79. [Google Scholar] [CrossRef]
- Matsuda, K.; Yuzaki, M. Cbln family proteins promote synapse formation by regulating distinct neurexin signaling pathways in various brain regions. Eur J Neurosci 2011, 33, 1447–61. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Rohlmann, A.; Sargsyan, V.; Aramuni, G.; Hammer, R. E.; Sudhof, T. C.; Missler, M. Extracellular domains of alpha-neurexins participate in regulating synaptic transmission by selectively affecting N- and P/Q-type Ca2+ channels. J Neurosci 2005, 25, 4330–42. [Google Scholar] [CrossRef] [PubMed]
- Dudanova, I.; Sedej, S.; Ahmad, M.; Masius, H.; Sargsyan, V.; Zhang, W.; Riedel, D.; Angenstein, F.; Schild, D.; Rupnik, M.; Missler, M. Important contribution of alpha-neurexins to Ca2+-triggered exocytosis of secretory granules. J Neurosci 2006, 26, 10599–613. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G. R.; Aoto, J.; Tabuchi, K.; Foldy, C.; Covy, J.; Yee, A. X.; Wu, D.; Lee, S. J.; Chen, L.; Malenka, R. C.; Sudhof, T. C. beta-Neurexins Control Neural Circuits by Regulating Synaptic Endocannabinoid Signaling. Cell 2015, 162, 593–606. [Google Scholar] [CrossRef] [PubMed]
- Klatt, O.; Repetto, D.; Brockhaus, J.; Reissner, C.; El Khallouqi, A.; Rohlmann, A.; Heine, M.; Missler, M. Endogenous beta-neurexins on axons and within synapses show regulated dynamic behavior. Cell Rep 2021, 35, 109266. [Google Scholar] [CrossRef]
- Chen, L. Y.; Jiang, M.; Zhang, B.; Gokce, O.; Sudhof, T. C. Conditional Deletion of All Neurexins Defines Diversity of Essential Synaptic Organizer Functions for Neurexins. Neuron 2017, 94, 611–625 e4. [Google Scholar] [CrossRef] [PubMed]
- Luo, F.; Sclip, A.; Jiang, M.; Sudhof, T. C. , Neurexins cluster Ca(2+) channels within the presynaptic active zone. EMBO J 2020, 39, e103208. [Google Scholar] [CrossRef] [PubMed]
- Aoto, J.; Martinelli, D. C.; Malenka, R. C.; Tabuchi, K.; Sudhof, T. C. Presynaptic neurexin-3 alternative splicing trans-synaptically controls postsynaptic AMPA receptor trafficking. Cell 2013, 154, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Dana, H.; Sun, Y.; Mohar, B.; Hulse, B. K.; Kerlin, A. M.; Hasseman, J. P.; Tsegaye, G.; Tsang, A.; Wong, A.; Patel, R.; Macklin, J. J.; Chen, Y.; Konnerth, A.; Jayaraman, V.; Looger, L. L.; Schreiter, E. R.; Svoboda, K.; Kim, D. S. High-performance calcium sensors for imaging activity in neuronal populations and microcompartments. Nat Methods 2019, 16, 649–657. [Google Scholar] [CrossRef]
- Geppert, M.; Khvotchev, M.; Krasnoperov, V.; Goda, Y.; Missler, M.; Hammer, R. E.; Ichtchenko, K.; Petrenko, A. G.; Sudhof, T. C. Neurexin I alpha is a major alpha-latrotoxin receptor that cooperates in alpha-latrotoxin action. J Biol Chem 1998, 273, 1705–10. [Google Scholar] [CrossRef]
- Eroshenko, N.; Church, G. M. Mutants of Cre recombinase with improved accuracy. Nat Commun 2013, 4, 2509. [Google Scholar] [CrossRef] [PubMed]
- de Jong, A. P.; Schmitz, S. K.; Toonen, R. F.; Verhage, M. Dendritic position is a major determinant of presynaptic strength. J Cell Biol 2012, 197, 327–37. [Google Scholar] [CrossRef] [PubMed]
- Docs, K.; Meszar, Z.; Gonda, S.; Kiss-Szikszai, A.; Hollo, K.; Antal, M.; Hegyi, Z. The Ratio of 2-AG to Its Isomer 1-AG as an Intrinsic Fine Tuning Mechanism of CB1 Receptor Activation. Front Cell Neurosci 2017, 11, 39. [Google Scholar] [CrossRef] [PubMed]
- Iwabuchi, S.; Kakazu, Y.; Koh, J. Y.; Harata, N. C. Evaluation of the effectiveness of Gaussian filtering in distinguishing punctate synaptic signals from background noise during image analysis. J Neurosci Methods 2014, 223, 92–113. [Google Scholar] [CrossRef] [PubMed]
- Motulsky, H. J.; Brown, R. E. Detecting outliers when fitting data with nonlinear regression - a new method based on robust nonlinear regression and the false discovery rate. BMC Bioinformatics 2006, 7, 123. [Google Scholar] [CrossRef] [PubMed]
- Etherton, M. R.; Blaiss, C. A.; Powell, C. M.; Südhof, T. C. Mouse neurexin-1alpha deletion causes correlated electrophysiological and behavioral changes consistent with cognitive impairments. Proc Natl Acad Sci U S A 2009, 106, 17998–8003. [Google Scholar] [CrossRef] [PubMed]
- Grayton, H. M.; Missler, M.; Collier, D. A.; Fernandes, C. Altered social behaviours in neurexin 1alpha knockout mice resemble core symptoms in neurodevelopmental disorders. PLoS One 2013, 8, e67114. [Google Scholar] [CrossRef] [PubMed]
- Dachtler, J.; Ivorra, J. L.; Rowland, T. E.; Lever, C.; Rodgers, R. J.; Clapcote, S. J. Heterozygous deletion of alpha-neurexin I or alpha-neurexin II results in behaviors relevant to autism and schizophrenia. Behav Neurosci 2015, 129, 765–76. [Google Scholar] [CrossRef] [PubMed]
- Tottene, A.; Volsen, S.; Pietrobon, D. alpha(1E) subunits form the pore of three cerebellar R-type calcium channels with different pharmacological and permeation properties. J Neurosci 2000, 20, 171–178. [Google Scholar] [CrossRef]
- Metz, A. E.; Jarsky, T.; Martina, M.; Spruston, N. R-type calcium channels contribute to afterdepolarization and bursting in hippocampal CA1 pyramidal neurons. J Neurosci 2005, 25, 5763–73. [Google Scholar] [CrossRef]
- Twitchell, W.; Brown, S.; Mackie, K. Cannabinoids inhibit N- and P/Q-type calcium channels in cultured rat hippocampal neurons. J Neurophysiol 1997, 78, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R. I.; Nicoll, R. A. Endocannabinoid signaling in the brain. Science 2002, 296, 678–82. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, T.; Waku, K. 2-Arachidonoylglycerol and the cannabinoid receptors. Chem Phys Lipids 2000, 108, 89–106. [Google Scholar] [CrossRef] [PubMed]
- Diana, M. A.; Marty, A. Endocannabinoid-mediated short-term synaptic plasticity: depolarization-induced suppression of inhibition (DSI) and depolarization-induced suppression of excitation (DSE). Br J Pharmacol 2004, 142, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Kreitzer AC, R. W. Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron 2001, 29, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Diana, M. A.; Levenes, C.; Mackie, K.; Marty, A. Short-term retrograde inhibition of GABAergic synaptic currents in rat Purkinje cells is mediated by endogenous cannabinoids. J Neurosci 2002, 22, 200–8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Linden, D. J. Neuromodulation at single presynaptic boutons of cerebellar parallel fibers is determined by bouton size and basal action potential-evoked Ca transient amplitude. J Neurosci 2009, 29, 15586–94. [Google Scholar] [CrossRef] [PubMed]
- Blankman, J. L.; Simon, G. M.; Cravatt, B. F. A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem Biol 2007, 14, 1347–56. [Google Scholar] [CrossRef] [PubMed]
- Luo, F.; Sclip, A.; Merrill, S.; Sudhof, T. C. Neurexins regulate presynaptic GABA(B)-receptors at central synapses. Nat Commun 2021, 12, 2380. [Google Scholar] [CrossRef]
- Heine, M.; Thoumine, O.; Mondin, M.; Tessier, B.; Giannone, G.; Choquet, D. Activity-independent and subunit-specific recruitment of functional AMPA receptors at neurexin/neuroligin contacts. Proc Natl Acad Sci U S A 2008, 105, 20947–52. [Google Scholar] [CrossRef]
- Aoto, J.; Foldy, C.; Ilcus, S. M.; Tabuchi, K.; Sudhof, T. C. Distinct circuit-dependent functions of presynaptic neurexin-3 at GABAergic and glutamatergic synapses. Nat Neurosci 2015, 18, 997–1007. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Zhang, X.; Dobie, F.; Wu, H.; Craig, A. M. , Induction of GABAergic postsynaptic differentiation by alpha-neurexins. J Biol Chem 2008, 283, 2323–34. [Google Scholar] [CrossRef] [PubMed]
Figure 3.
Deletion of all Nx variants decreased presynaptic Ca2+ influx primarily via P/Q-type VGCC. Pharmacologically isolated VGCC subtype contribution to the Ca2+ influx measured during single AP stimulation in Nx123 cKO neurons with synGCaMP7b by sequential addition of specific blockers: ω-agatoxin IVA (AgTX, 0.1 μM; P/Q-type); ω-conotoxin GVIA (CTX, 2 μM; N-type); nifedipine (Nif, 20 μM; L-type); SNX-482 (SNX, 0.5 μM; R-type). (A) Averaged traces of control neurons (Cremut, 12 cells/869 boutons, left) and neurons lacking all neurexin variants (Cre, 13/916, right), colors indicate traces after subsequent application of subtype-specific blockers as depicted. Thus, the area in dimmed colors above the traces indicates the amount of Ca2+ influx sensitive to the given blocker. (B) Ca2+ transients that reflect Ca2+ influx through the given VGCC subtypes are isolated by subtraction from the traces in A, comparing Nx123 cKO Cremut (continuous lines) and Nx123 cKO Cre (dashed lines). (C) Mean ± SEM of relative VGCC subtype contribution (% of control) calculated for each bouton (ROI, relative to total Ca2+ influx) in Nx123 cKO neurons. The number of examined boutons/cells is shown in the P/Q columns and applies to all VGCC subtypes. Columns were compared with Kruskal-Wallis test, n.s.: p > 0.05, **: p < 0.01, ****: p < 0.0001. (D) Each presynaptic bouton’s P/Q-type contribution was determined, and the spreading is depicted in a histogram that contrasts the distribution of Nx123 cKO neurons with and without Nx. The data show that without Nx, the number of boutons with more than 50% P/Q-type Ca2+ influx is almost lost (Cremut, 584/12; Cre, 501/13). The same analysis is shown in (E) for the N-type and (F) for the L-type portion in individual boutons.
Figure 3.
Deletion of all Nx variants decreased presynaptic Ca2+ influx primarily via P/Q-type VGCC. Pharmacologically isolated VGCC subtype contribution to the Ca2+ influx measured during single AP stimulation in Nx123 cKO neurons with synGCaMP7b by sequential addition of specific blockers: ω-agatoxin IVA (AgTX, 0.1 μM; P/Q-type); ω-conotoxin GVIA (CTX, 2 μM; N-type); nifedipine (Nif, 20 μM; L-type); SNX-482 (SNX, 0.5 μM; R-type). (A) Averaged traces of control neurons (Cremut, 12 cells/869 boutons, left) and neurons lacking all neurexin variants (Cre, 13/916, right), colors indicate traces after subsequent application of subtype-specific blockers as depicted. Thus, the area in dimmed colors above the traces indicates the amount of Ca2+ influx sensitive to the given blocker. (B) Ca2+ transients that reflect Ca2+ influx through the given VGCC subtypes are isolated by subtraction from the traces in A, comparing Nx123 cKO Cremut (continuous lines) and Nx123 cKO Cre (dashed lines). (C) Mean ± SEM of relative VGCC subtype contribution (% of control) calculated for each bouton (ROI, relative to total Ca2+ influx) in Nx123 cKO neurons. The number of examined boutons/cells is shown in the P/Q columns and applies to all VGCC subtypes. Columns were compared with Kruskal-Wallis test, n.s.: p > 0.05, **: p < 0.01, ****: p < 0.0001. (D) Each presynaptic bouton’s P/Q-type contribution was determined, and the spreading is depicted in a histogram that contrasts the distribution of Nx123 cKO neurons with and without Nx. The data show that without Nx, the number of boutons with more than 50% P/Q-type Ca2+ influx is almost lost (Cremut, 584/12; Cre, 501/13). The same analysis is shown in (E) for the N-type and (F) for the L-type portion in individual boutons.
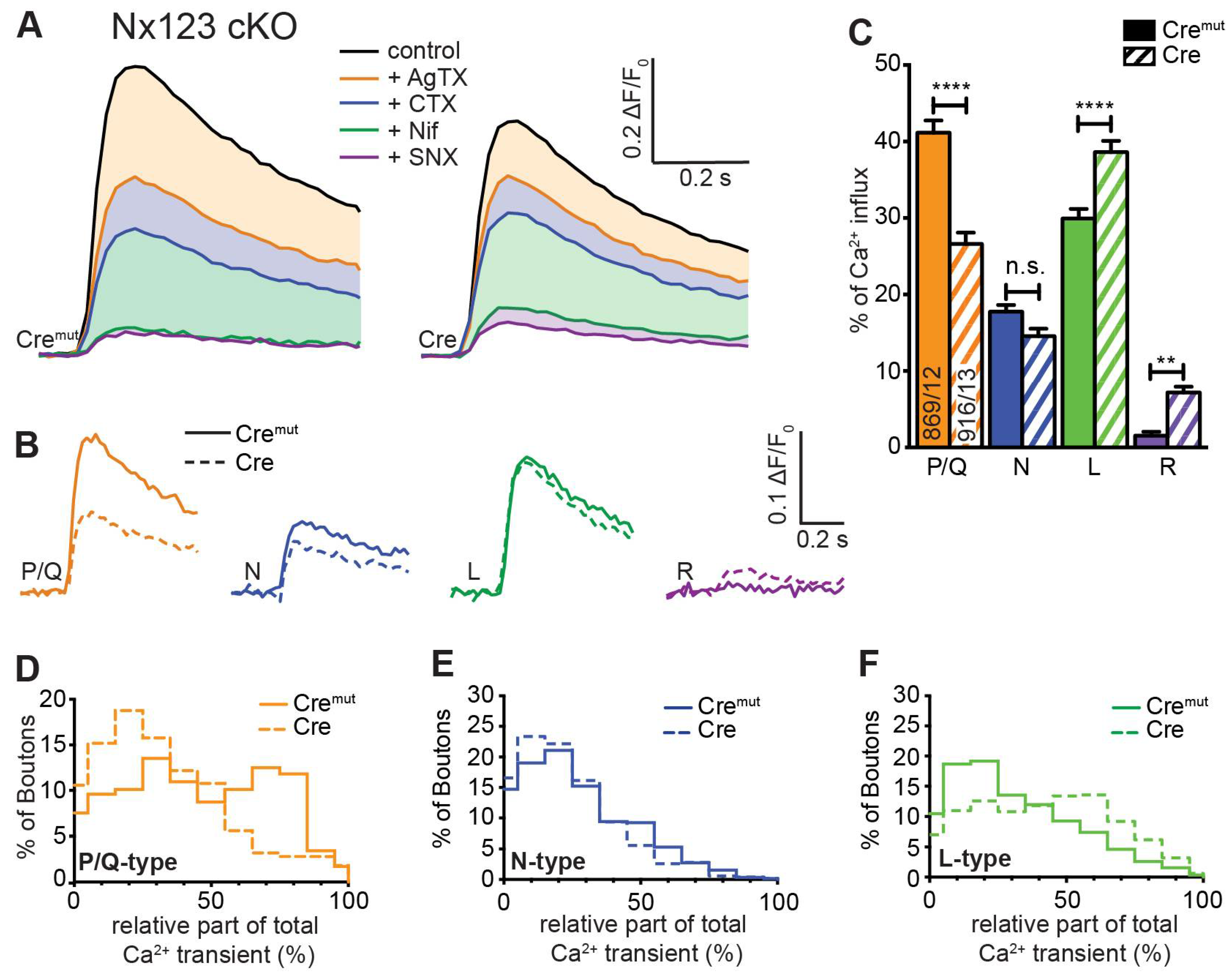
Figure 4.
Single Nx1α deletion changed the VGCC subtype contribution to presynaptic Ca2+ influx. VGCC subtype contribution on Ca2+ influx after single AP stimulation was measured in Nx1α cKO neurons with synGCaMP7b by sequential addition of specific blockers as described in Figure 3. (A) Averaged traces of control neurons (Nx1α cKO Cremut, 13 cells/1154 boutons; left) and neurons lacking only Nx1α (Cre, 11/956, right), colors indicate traces after subsequent application of subtype-specific blockers as depicted. Thus, the area in dimmed colors above the traces indicates the amount of Ca2+ influx sensitive to the given blocker. (B) Ca2+ transients that specifically reflect Ca2+ influx through the sequentially blocked VGCC subtypes were isolated by subtraction from the traces in A and compared between Nx1α cKO Cremut (continuous lines) and Nx1α cKO Cre (dashed lines). (C) Mean ± SEM of relative Ca2+ contribution (% of control) per VGCC subtype calculated for individual boutons (ROIs) in Nx1α cKO neurons. The number of examined boutons/cells is shown in the P/Q columns and applies to all VGCC subtypes. Columns were compared with Kruskal-Wallis test, n.s.: p > 0.05, **: p < 0.01, ****: p < 0.0001. (D) The P/Q-type portion of Ca2+ transients was calculated for each synaptic bouton and the spreading is shown in a histogram comparing the variability in neurons with and without Nx1α (Cremut, 755/13; Cre, 552/11), in (E) for the N-type and (F) for L-type.
Figure 4.
Single Nx1α deletion changed the VGCC subtype contribution to presynaptic Ca2+ influx. VGCC subtype contribution on Ca2+ influx after single AP stimulation was measured in Nx1α cKO neurons with synGCaMP7b by sequential addition of specific blockers as described in Figure 3. (A) Averaged traces of control neurons (Nx1α cKO Cremut, 13 cells/1154 boutons; left) and neurons lacking only Nx1α (Cre, 11/956, right), colors indicate traces after subsequent application of subtype-specific blockers as depicted. Thus, the area in dimmed colors above the traces indicates the amount of Ca2+ influx sensitive to the given blocker. (B) Ca2+ transients that specifically reflect Ca2+ influx through the sequentially blocked VGCC subtypes were isolated by subtraction from the traces in A and compared between Nx1α cKO Cremut (continuous lines) and Nx1α cKO Cre (dashed lines). (C) Mean ± SEM of relative Ca2+ contribution (% of control) per VGCC subtype calculated for individual boutons (ROIs) in Nx1α cKO neurons. The number of examined boutons/cells is shown in the P/Q columns and applies to all VGCC subtypes. Columns were compared with Kruskal-Wallis test, n.s.: p > 0.05, **: p < 0.01, ****: p < 0.0001. (D) The P/Q-type portion of Ca2+ transients was calculated for each synaptic bouton and the spreading is shown in a histogram comparing the variability in neurons with and without Nx1α (Cremut, 755/13; Cre, 552/11), in (E) for the N-type and (F) for L-type.
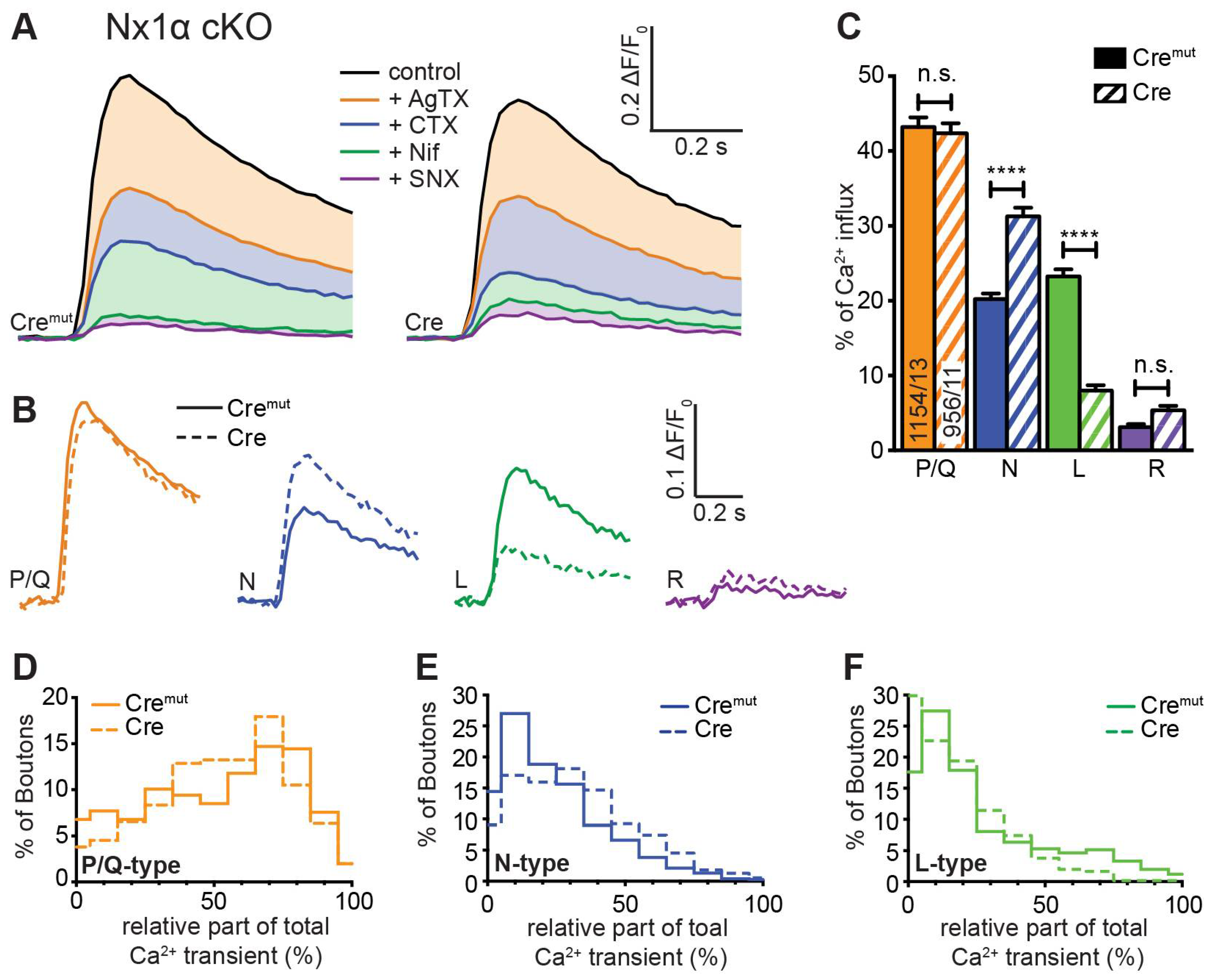
Figure 5.
Endocannabinoid-evoked CB1 receptor activation reduces presynaptic Ca2+ transients in an Nx-dependent manner. (A) Several presynaptic boutons of a synGCaMP7b transfected Nx123 cKO Cre neuron are shown in an exemplary ΔF image during 3 AP stimulation. (B) The identical presynaptic boutons as in A after 5 min of CB1-receptor activation with 2 µM 2-AG, again during a 3 AP stimulation. (C) Repetitive stimulation (1 AP every 30 s) shows a reduction of Ca2+ transients in response to the application of 2-AG, averaged (mean ± SEM) from neurons of Nx123 Cremut (13 neurons, continuous line) and Nx123 Cre (16, dotted line), displayed as relative changes normalized to the mean of four stimulations before 2-AG application. (D) Similar recordings as in C for β-Nx cKO Cremut (10, continuous line) and β-Nx cKO Cre (9, dotted line). (E) Boxplot (quartiles and median) of Ca2+ ΔF/F0 for Nx123 Cremut (960 ROIs/13 cells) and Nx123 Cre (1168/16), respectively, before and after 2-AG application. (F) Relative change (%) of presynaptic Ca2+ transients by activation of CB1-receptor with 2-AG. Cells were measured under both conditions (control and 5 min of 2-AG) and reduction was calculated for each bouton separately, plotted as mean ± SEM in Nx123 cKO (blue) and β-Nx cKO (red) neurons (Cremut and Cre). Outliers were detected and removed with ROUT-method (Q=1) and columns were compared with an unpaired t-test, * p < 0.05 , **** p < 0.0001; numbers (included ROIs/cells) are given in the columns.
Figure 5.
Endocannabinoid-evoked CB1 receptor activation reduces presynaptic Ca2+ transients in an Nx-dependent manner. (A) Several presynaptic boutons of a synGCaMP7b transfected Nx123 cKO Cre neuron are shown in an exemplary ΔF image during 3 AP stimulation. (B) The identical presynaptic boutons as in A after 5 min of CB1-receptor activation with 2 µM 2-AG, again during a 3 AP stimulation. (C) Repetitive stimulation (1 AP every 30 s) shows a reduction of Ca2+ transients in response to the application of 2-AG, averaged (mean ± SEM) from neurons of Nx123 Cremut (13 neurons, continuous line) and Nx123 Cre (16, dotted line), displayed as relative changes normalized to the mean of four stimulations before 2-AG application. (D) Similar recordings as in C for β-Nx cKO Cremut (10, continuous line) and β-Nx cKO Cre (9, dotted line). (E) Boxplot (quartiles and median) of Ca2+ ΔF/F0 for Nx123 Cremut (960 ROIs/13 cells) and Nx123 Cre (1168/16), respectively, before and after 2-AG application. (F) Relative change (%) of presynaptic Ca2+ transients by activation of CB1-receptor with 2-AG. Cells were measured under both conditions (control and 5 min of 2-AG) and reduction was calculated for each bouton separately, plotted as mean ± SEM in Nx123 cKO (blue) and β-Nx cKO (red) neurons (Cremut and Cre). Outliers were detected and removed with ROUT-method (Q=1) and columns were compared with an unpaired t-test, * p < 0.05 , **** p < 0.0001; numbers (included ROIs/cells) are given in the columns.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



