
Submitted:
10 July 2024
Posted:
11 July 2024
You are already at the latest version
Abstract
Traumatic brain injury (TBI) is an important global clinical issue, requiring not only prevention but also effective treatment. Following TBI, diverse parallel and intertwined pathological mechanisms affecting biochemical, neurochemical, and inflammatory pathways can have severe impact on the patient’s quality of life. The current review summarizes evidence for utility of amantadine in TBI in connection to its mechanism of action. Amantadine combining multiple mechanisms of action may offer both neuroprotective and neuroactivating effects in TBI patients. Indeed, use of amantadine in TBI has been encouraged by several clinical practice guidelines/recommendations. Amantadine is also available as infusion which may be of particular benefit in unconscious patients with TBI, due to immediate delivery to the central nervous system and the possibility of precise dosing. In other situations, orally administered amantadine may be used. There are several questions that remain to be addressed: Can amantadine be effective in disorders of consciousness requiring long-term treatment and in combination with drugs approved for treatment of TBI? Do the observed beneficial effects of amantadine extend to disorders of consciousness due to factors other than TBI? Well controlled clinical studies are warranted to ultimately confirm its utility in the TBI and provide answers these questions.
Keywords:
traumatic brain injury
; clinical
; preclinical
; mechanism of action
; sigma-1
; aromatic amino acids decarboxylase
; GDNF
; NMDA receptors
1. Selected Epidemiological Aspects of Traumatic Brain Injury
Traumatic brain injury (TBI) is one of the leading causes of death and disabilities ranging from paralysis to plethora of psychiatric abnormalities. It is most often caused by vehicle accidents and falls. In the USA, based on Centers for Disease Control and Prevention report for 2014, TBI contributed to nearly 3 million emergency department visits and hospitalizations [1]. Annually, an estimated 200,000 individuals who had sustained TBI need hospitalization. TBI leads to 56,000 deaths and was reported to account for approximately 40% of all deaths from acute injuries in the USA [1,2]. The mortality rate was found to be high (33%) in severe TBI, while it was much lower (2.5%) in moderate TBI [2].
In Europe, TBI incidence amounts to 500 cases per 100,000 population [3]. In a more recent, extensive review including sixty-six studies from European countries, Brazinova et al. reported crude incidence rates ranging 47.3 - 694/100,000 persons/year (country-level studies) and 83.3 - 849/100,000/year (regional-level studies) [4]. Crude mortality rates ranged 9 - 28.10/100,000/year (country-level), and 3.3 - 24.4/100,000/year (regional-level). Similar to the USA, the most common reasons of injury were traffic accidents and falls [4]. Majdan et al. [5] reported occurrence of a total of 17,049 TBI-related deaths (translating into 374,636 years of lost lives, YLLs) in 16 European countries in the year 2013. The pooled age-standardized rate of YLLs per 100,000 people per year was 259.1 [6]. The same research group estimated that in the year 2012, in the European Union (approx. 500 million) there were roughly 57,000 TBI-related deaths and 1,000,000 hospital discharges. At the same time, in the entire Europe (approx. 750 million) approximately 82,000 deaths and about 2,100,000 hospital discharges occurred. The authors concluded that TBI is an important cause of death and hospital admissions in Europe [5]. In summary, even though epidemiological data vary across different geographical regions, TBI remains a very relevant clinical issue deserving a great deal of attention about both prevention measures and treatment all over the world.
2. Pathophysiology of Traumatic Brain Injury and Possible Therapeutic Window for Intervention with Amantadine
There are diverse causes of primary damage in TBI but the mechanisms of subsequent damage underlying the pathology and observed symptoms seem to converge (Figure 1). While there is only limited knowledge about specific causal mechanisms underlying TBI, there is accumulating evidence, that interplay between oxidative stress, excitotoxicity, inflammation, lysosomal and autophagy dysfunction etc. are key elements leading to cell death [7,8,9]. It is not likely to find a treatment halting cell death but more realistic is therapeutic benefit resulting from inhibition of secondary degeneration and/or improving recovery and functional outcome.
According to severity, TBI can be categorized as mild, moderate, and severe [10]. In mild TBI, 30-53% of patients show disability symptoms for at least one year but their life expectancy is typically unchanged [10]. In contrast, moderate to severe TBI is connected with progressive loss of consciousness, as well as with cognitive and neurological impairments [11].
In terms of temporal sequence, both pathomechanisms and symptoms of TBI can be categorized into those which are characteristic for the acute phase (hours), the subacute phase (days to weeks) and the chronic phase (weeks to years). Each of these three phases is characterized by presence of distinct mechanisms and symptoms [9] (Figure 1).
Amantadine can positively influence chronologically different, sometimes cascading damage (Figure 1) and recovery processes taking place after a TBI due to its diverse modes of action. During the initial, acute phase of TBI, the window for a therapeutic intervention is very narrow making a pharmacological therapeutic intervention, e.g., with amantadine virtually impossible. However, the subacute phase, in which additional secondary neurodegenerative processes occur, offers a potential therapeutic window to interrupt some of the ongoing damaging processes; in this phase, beneficial effects of amantadine on TBI-induced pathology might be expected (Figure 1). Also at the chronic stage of TBI, which may involve cognitive deficits, affective alterations, sleep disturbances and aggression [11], amantadine may be useful by providing support of dopaminergic transmission (Figure 1).
3. Damage and Subsequent Dysfunction in Traumatic Brain Injury
The primary damage results from a direct impact of a force that can lead to different, multiple sequelae, including skull fractures, intracranial bleeding (coup mechanism); it can also occur in the contrecoup contusion mechanism, whereby injury takes place at a brain area opposite to the side of the directly impacted area. The initial damage in TBI may also occur due to diffuse axonal injury in which widespread lesions are present in both white and grey matter because of effect of forces associated with rapid acceleration and/or deceleration, (e.g., in traffic accidents or falls). For a thorough review see, e.g., McKee and Daneshvar, 2015 [12]. Due to nature of TBI causes and very short duration of the phase of primary damage, the window for a pharmacological intervention at this stage is very short-lasting and thus extremely challenging.
There are multiple mechanisms possibly involved in a secondary damage in TBI. Cerebral metabolic dysfunction may result directly from the primary injury or occur in course of any secondary processes. This dysfunction can lead to reduced cerebral metabolism (as measured by oxygen and glucose consumption) and to a reduced energetic status of the brain [13].
In terms of cerebral autoregulation, a change in cerebral perfusion pressure leads to either vasoconstriction or vasodilation. When this autoregulation mechanism gets depleted due to a TBI, there is a risk of a secondary ischemia. This can also occur as a response to hypo- or hypercapnia and is referred to as cerebrovascular CO2 reactivity [14]. When the intracerebral pressure increases and reaches the value of the mean systolic blood pressure, the cerebral blood flow decreases. As a result, the systemic blood pressure increases, and the cerebral vessels expand. Consequently, the intracerebral pressure increases even further which is followed by cerebral hypoxia, cerebral oedema (sometimes associated with herniation) [15,16]. In the vasogenic cerebral oedema, reflexive dilation of the brain vessels and a mechanical-functional disturbance of the endothelial wall both lead to a disruption of the blood-brain barrier and an accumulation of a relatively large volume of fluid in the extracellular space. In the cytotoxic (intracellular) brain oedema, it is the altered permeability of the cell membrane that leads to altered reabsorption of osmotically active substances and thus to a change in the cellular osmolality. The associated intracellular water accumulation primarily affects neurons, microglia and astrocytes [17].
Minutes after a TBI, extracellular levels of the excitatory amino acids glutamate and aspartate rise dramatically [18]. This leads to excessive stimulation of the N-methyl-D-aspartate (NMDA) receptors, leading to depolarization of neurons. The increased glutamate outflow results in an increased Na+ and Ca2+ influx into the cell and eventually leads to triggering cell damage mechanisms by Ca2+ overload. This occurs first in neurons, while astrocytes can take up glutamate and convert it into glutamine. The resulting increase in activity of the Na+/K+-ATPase rises the metabolic demand. The magnitude of glutamate release is age-dependent, being more pronounced in the older TBI patients than in the younger ones [19]. The widely described NMDA receptor antagonist properties of amantadine may potentially contribute to the observed beneficial effects of this compound in TBI patients (for details refer to the Section 5.1).
After a TBI, stimulation of the NMDA receptor leads to the release of glutamate and ultimately to intracellular accumulation of Ca2+ in the mitochondria. The most important consequence of the increased Ca2+ load is the formation of a mitochondrial permeability transition pore, which ultimately leads to emptying of the Ca2+ pool into the cytoplasm. In turn, this paves the way for apoptosis [20]. Like for excitotoxity, also for this pathway, the amantadine’s NMDA receptor antagonism may contribute its reported therapeutic effects. It should however be mentioned that pharmacological profile of amantadine extends beyond NMDA receptor blockade; a detailed discussion of putative amantadine targets can be found in the Section 5.
The oxidative stress is caused by the imbalance between the production of free radicals and the body's ability to neutralize their harmful effects through endogenous antioxidant mechanisms. Depletion of the endogenous antioxidants (e.g., superoxide dismutase, glutathione peroxidase, catalase) leads to excessive production of reactive oxygen species and related species (nitric oxide, superoxide, hydrogen peroxide). Free radicals have unpaired electrons and find them in the environment, thus leading to the oxidation of proteins, the cleavage of DNA and the inhibition of the mitochondrial electron transport chain. This in turn leads to inflammatory processes, immediate cell death or triggers delayed apoptotic programs [21]. Indeed, there is some limited preclinical evidence existing for amantadine’s antioxidant properties (cf. Section 5.5).
TBI leads to immunological and inflammatory tissue reactions. Inflammation can cause damage on one hand and promote regeneration on the other. In the acute phase, the direct injury being a consequence of a mechanical impact is accompanied by disruption of blood-brain barrier and inflammation involving release of cytokines, and mobilization of neutrophiles and macrophages [9]. However, during the subacute phase, additional secondary neurodegenerative processes occur which are accompanied by apoptosis and increasing immune response with activation of microglia (promoting phagocytosis and delivering growth factors to the injured tissue), astrocytes, as well as of T and B lymphocytes [9]. Microglia also deliver growth factors to injured brain tissue. This stage offers a potential therapeutic window to interrupt some of the ongoing damaging processes. In this phase, beneficial effects of amantadine on humoral and cellular TBI-induced pathology might be expected (Figure 1).
Both primary and secondary injuries activate the release of cellular mediators (cytokines, prostaglandins, free radicals and complement) [22]. Leukocytes, macrophages, and T-cell lymphocytes infiltrate injured tissue which is degraded in response to these inflammatory processes. Additionally, within hours of injury, pro-inflammatory enzymes, and other mediators (e.g., tumor necrosis factor (TNF), interleukin IL-1-ß und IL-6) are upregulated. Amantadine may exert anti-inflammatory effects mediated through inhibition of microglial activation and inflammatory cytokines such as interferons and TNF , as well as through stimulation of IL production [23,24,25]. All of the aforementioned factors can influence the inflammatory response characteristic for the pathophysiology of TBI.
Neuronal cell death following TBI causes neurological deficits and mortality. Neuronal death phenotypes are categorized based morphological or molecular changes. In necrosis - a passive process - loss of ionic homeostasis, failure of membrane integrity, and organelle and cell swelling take place. On the other hand, Apoptosis is an active, energy-dependent process of condensation and fragmentation of the cytoplasm and the nucleus, leading to decrease in cell volume with preserved structure of the organelle. In some cases, apoptosis and necrosis coexist, constituting an intermediate type of cell death sometimes referred to as aponecrosis. It has been suggested that future neuroprotective strategies need to target multiple pathways to reflect both regional and temporal changes underlying different types of neuronal cell death (for review see [26] and references therein). Indeed, some experimental studies suggest that anti-necrotic, anti-apoptotic, and neuroprotective effects of amantadine could be related to its antioxidant, anti-inflammatory and biochemical mechanisms [27,28,29].
4. Disorders of Consciousness—Recovery Enhancement
Consciousness is thought to comprise arousal (wakefulness, sustained attention, vigilance) and awareness (subjective perceptions, feelings, thoughts) [30,31]. Arousal and vigilance require normal function of the brainstem and the thalamus [32,33,34,35] which are interrelated with the parts of the frontoparietal network known to be impaired in subjects presenting with disturbances of consciousness [36,37,38]. Dopamine is a neurotransmitter most implicated into arousal and consequently also in the TBI; indeed, widespread axonal injury is related to a reduced brain dopamine availability [39,40].
Coma has been described as a pathological state characterized by severe and prolonged dysfunction of vigilance and consciousness [41], and may either occur due to a diffuse insult to both hemispheres (e.g., epileptic seizures, poisoning, drug or alcohol overdose) or due to a focal insult (e.g., stroke or head trauma) [42].
While a subset of comatose patients presents with an extensive or complete recovery of awareness, many others who awaken from the acute comatose state do not show any signs of awareness. If repeated examinations yield no evidence of a sustained, reproducible, purposeful, or voluntary behavioral response to visual, auditory, tactile, or noxious stimuli, a diagnosis of a “unresponsive wakefulness syndrome” is made one month after the injury [43]. Some patients remain in this condition. Others eventually show inconsistent but reproducible signs of awareness, including the ability to follow commands, but they remain unable to communicate interactively. In 2002, the Aspen Neurobehavioral Conference Work Group coined the term “minimally conscious state” (MCS) to describe the condition of such patients, thereby adding a new clinical entity to the spectrum of disorders of consciousness [44]. The MCS diagnosis has been further sub-categorized into MCS minus and MCS plus. The most frequent signs of consciousness in MCS minus patients are visual fixation and pursuit, automatic motor reactions (e.g., scratching, pulling the bed sheet), and localization to noxious stimulation, whereas MCS plus patients can, in addition, follow simple commands, intelligibly verbalize or intentionally communicate [45].
5. Potential Mechanism of Amantadine Effects in Traumatic Brain Injury—NMDA Receptors and Beyond
Recently, we analyzed possible targets of amantadine that could play a role in its observed therapeutic effects based on comparison of its concentrations reached at a putative target in humans following administration of this drug at therapeutic doses and in vitro affinity at this target [50]. The analysis demonstrated that there are several targets such as sigma-1 receptors, aromatic l-amino acids decarboxylase (AADC) and glial cell line-derived neurotrophic factor (GDNF) were found to possibly have stronger involvement in amantadine’s actions than the glutamatergic NMDA receptors. For the purpose of that analysis, we also considered that intracellular concentrations of amantadine in the brain are 10- or 20-times higher than plasma levels in animal and human studies, respectively, due to lysosomal trapping [51,52,53]. Although there are dozens of publications showing in vitro inhibition of NMDA receptors by amantadine, only one of them observed that effect at therapeutic range of concentrations (up to 10 µM) [50].
Below, we provide a short characterization of the most relevant therapeutic targets with evidence supporting their potential utility in TBI treatment.
5.1. NMDA Receptors and Neuroprotection
As discussed in a recent review, there are many functional and binding studies showing inhibition of glutamatergic NMDA receptors in a range from 10 µM to 640 µM [50], however only one of these studies showed this effect at maximal plasma concentrations achieved at therapeutic doses (approximately 10 µM). It cannot however be excluded that an incomplete inhibitory effect on NMDA receptors may be supportive to other mechanisms which are described in the following (see also Figure 1).
Shortly after discovery of NMDA receptors [54,55] and of their high permeability to calcium, their role in acute and chronic brain insult has been postulated [56,57]. However, all clinical trials with NMDA receptor antagonists in stroke or TBI failed [58], likely due to a need of administering these substances at doses which may actually produce a detrimental effect on neuronal recovery. Therefore, NMDA antagonism alone cannot be regarded as viable approach to prevent TBI-induced damage, but it could still support other mechanism if the degree of NMDA receptor blockade remains mild to moderate. We believe that it may be the case for amantadine, which produces only weak effect on NMDA receptors at therapeutic doses (see also [50]).
It has recently been suggested that improved neuroprotective effects can be achieved by selective targeting of extrasynaptic NMDA receptors of so-called “death signaling complex”. These receptors are mainly composed of NR2B subunits and coupled to different signaling pathways than the physiologically more relevant subsynaptic receptors [59,60]. It is however not known whether amantadine has preference for these extrasynaptic receptors.
It is beyond the scope of this review to discuss all studies focusing on effects of NMDA receptor antagonists in animal models of TBI. It should however be mentioned that the majority of studies showed beneficial effects in terms of improvement of structural and/or functional outcomes as reviewed elsewhere in more detail [61,62,63,64].
As mentioned above, the positive preclinical data did not result in a therapeutic use of such compounds, since clinical studies failed to demonstrate their efficacy [58].
5.2. Sigma 1 Receptors and Neuroprotection
Kornhuber and colleagues were the first to describe that amantadine binds to sigma-1 receptors with approximately. 20 µM Ki as evidenced by [3H](+)-pentazocine binding in homogenates of post-mortem human frontal cortex [65]. Even higher affinity was observed in guinea pig or rat brain homogenates [66,67]. Amantadine seems to function as an agonist at sigma-1 receptors [67].
These receptors are located intracellularly on membranes of the endoplasmatic reticulum and mitochondria where they control Ca2+ signaling [68,69,70].
There are many studies indicating involvement of sigma-1 receptors in the function of the dopaminergic system which may have implications for the effect of amantadine on recovery from TBI, in particular for a faster return to the conscious state (Figure 1). Sigma-1 receptor activation enhances tyrosine hydroxylase (TH) activity [71], increases dopamine in vivo in the striatum [72] and decreases DA uptake [73]. Moreover, it has been described that sigma-1 ligands modulate NMDA stimulated dopamine release [74].
Apart from the role in modulation of dopamine transmission, sigma-1 receptors have been associated with neuroprotective activity which has been demonstrated in various models focusing on neuronal insults [75,76,77,78,79,80,81,82]. Studies in animal models of neurodegenerative diseases, reviewed recently by Shi and colleagues [83], indirectly support amantadine’s use in TBI.
How can the neuroprotective effect of sigma-1 agonism be mediated? It has been suggested that upon ligand stimulation, sigma-1 receptor dissociates from the binding immunoglobulin protein on the endoplasmic reticulum (ER) membrane and modulates three sensors of ER stress. These comprise protein kinase RNA-like ER kinase, inositol requiring enzyme 1α, and factor 6 [83]. Similar protective mechanisms occur on mitochondria which play a crucial role in TBI. Change of balance between anti-apoptotic/pro-apoptotic factors and reactive oxygen species are part of these mechanisms.
Sigma-1 receptors have been suggested to exert dual effect on NMDA receptors. They enhance the function of synaptic NMDA receptors responsible for plasticity while they inhibit extrasynaptic NMDA receptors responsible for excitotoxic neuronal death [83].
Several effects such as decrease of ER stress, improvement of mitochondrial function, normalization of calcium homeostasis and inhibition of excitotoxicity could play in concert for recovery from TBI [83].
On top of that, improvement of recovery may be supported by inhibition of microglia mediated inflammation including normalization of imbalance of M1/M2 phenotypes, the subpopulations which have pro- and anti-inflammatory functions, respectively [83,84].
The data on the efficacy of sigma-1 ligands in animal models of TBI are limited. In one study activation of sigma-1 by 2-(4-morpholinethyl)-1-phenylcyclohexanecarboxylate (PRE-084, 10 mg/kg i.p.) given 15 min after TBI, reduced lesion volume, brain edema, neurological severity score, and accelerated body weight recovery [85]. A decrease in microglia activation was also observed.
The activation of sigma-1 receptors is important for the anti-inflammatory effect of amantadine [85,86].
In summary, it may be expected that sigma-1 receptor activation may enhance recovery from TBI (Figure 1) through increase in synaptogenesis and inhibition of inflammation [83,85].
To the best of our knowledge, there have been no clinical trials with selective sigma-1 ligands in TBI.
5.3. Aromatic Amino Acids Decarboxylase and Neuroactivation
Amantadine was demonstrated to increase the activity of AADC, the enzyme responsible for dopamine synthesis [87]. In this way, dopaminergic activity increases, which can have a supportive effect on recovery after TBI, since dysfunctions of the dopaminergic and noradrenergic systems occur here.
In vitro, in pheochromocytoma (PC12) cells, amantadine (at 10 µM) enhances expression of mRNA of AADC by 70% [88]. In an ex vivo study in rats, amantadine (at 40 mg/kg) increased activity of AADC in the striatum (3 fold) and in the substantia nigra (10 fold) one hour after injection [89].
Amantadine (30 mg/kg) administered to rats subjected to 6-hydroxydopamine (6-OHDA) lesions of the dopaminergic system, increases ex vivo L-DOPA conversion in the striatum indicating increased AADC activity [90].
In humans, Deep and colleagues [87] showed that amantadine (100 mg for 3 days) increases activity of AADC up to 27 % in the ventral striatum using 6-[18F]fluoro-L-DOPA (L-DOPA = 3,4-Dihydroxy-L-phenylalanin) as an exogenous AADC substrate.
Enhanced activity and/or increase in concentration leads to an increase in dopamine levels which can be released to the synaptic cleft. In turn, this effect could be clearly supportive for recovery from TBI (Figure 1), in particular for enhancement of recovery from unconsciousness and cognitive performance [91,92,93].
In TBI, benefit of dopamine enhancement is not expected in the initial insult [39,94]. This relates to the fact that excess of dopamine produces oxidative stress, energy deficit and activates inflammation [39,94]. At the same time, dopaminergic neurons are victims of neurotoxicity in the hippocampus and striatum resulting in the impairment of cognitive and motor function, respectively [39,94]. In the chronic phase, this creates a gradually increasing dopaminergic deficit in aforementioned structures. In turn, enhancement of dopaminergic transmission may be particularly useful to enhance and/or increase recovery of cognitive and motor functions. Apart from amantadine, positive effects in TBI have been reported for enhancers of dopaminergic transmission such as amphetamine, methylphenidate, or bromocriptine in preclinical and/or clinical conditions [39,94].
5.4. Glial Cell Line-Derived Neurotrophic Factor and Neuroprotection/Regeneration
The GDNF is a neurotrophin connected with action on dopaminergic neurons. It has been shown to support neuronal morphology of these neurons and to protect against neurotoxicity through increase in pro-survival genes expression and a decrease in pro-apoptosis factors [95]. In C6 glioma cells, amantadine, at a concentration of 5 µM, increases GDNF mRNA [96]. In primary cultures from rat midbrain, amantadine (10-30 µM) increases GDNF mRNA by up to 70% 48 and 72 hours after exposure to mixed cultures of astroglia and microglia [27]. The authors indicate the role of induction of acetylation of histone H3 by inhibiting the histone deacetylase as underlying mechanism [27].
In rats, amantadine (25 mg/kg) increases GDNF in the hippocampus 6 and 24 h after surgery by approx. two-fold as demonstrated by immunohistochemistry and Western blot [97]. Amantadine also improved recovery after post-operative insult. Interestingly, attenuation of learning impairment by amantadine after surgery was inhibited by anti-GDNF antibody [97,98] suggesting this mechanism of action.
In primary hippocampal cultures, GDNF (1 ng/ml) prevented hypoxia-induced functional and structural changes [99]. In rats with TBI, GDNF infused into the lateral ventricle for 7 days (200 ng/day) decreased neuronal loss in CA2 and CA3 hippocampal regions by approx. 50% [100]. Umbilical cord-derived mesenchymal stem cells expressing GDNF and brain-derived neurotrophic factor (BDNF) provided neuroprotection in rats subjected to TBI [101]. Similarly, AdV-GDNF delivery in a TBI model in rats enhanced neuronal survival and induced neuroprotection [102]. Supportive evidence for neuroprotective and /or restorative effects of GDNF results from studies on various models of acute and chronic neurodegenerative diseases as reviewed recently [103,104]. Anti-inflammatory and tissue-protective functions of reactive astrocytes has been suggested to be likely mediated through GDNF [105]. In turn, the role of GDNF among other trophic factors (e.g. nerve growth factor (NGF), BDNF, bFGF, Neurotrophins 3,4,5) has been implied in TBI [106]. In conclusion, action of amantadine on GDNF may be valuable contribution to its therapeutic effect in TBI (Figure 1).
5.5. Other Possible Mechanisms of Action
After chronic (6 weeks) treatment with amantadine in mice, the effectiveness of presynaptically acting CNS stimulants was reduced, while the effect of the dopaminergic agonist apomorphine was enhanced. This was accompanied by an increase in the number of spiroperidol binding to presumably dopamine receptors [107]. Also, anti-inflammatory properties of amantadine may play a role in supporting recovery from TBI. In vitro, amantadine (4 µM) inhibited inflammatory activation of microglia by approximately 25% following lipopolysaccharide (LPS) stimulation [23]. Moreover, at a concentration of 40 µM, amantadine protected neurons in co-culture against LPS-induced toxicity [23]. The same authors reported that in mice, amantadine (10 mg/kg) given for 4 days, inhibits microglia activation and protects against 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridin (MPTP)-induced toxicity at 25 mg/kg [23]. In an in vitro study in human blood, amantadine (1 µM) inhibited production of pro-inflammatory cytokines such as interferon-γ and tumor necrosis factor-α. [24]. Similarly, Wandinger and colleagues [25] reported that in Parkinson patients amantadine corrected decreased interleukine-2 and interferon-γ secretion as measured in blood samples collected from Parkinson’s disease patients. Finally, the blockade of α4β2- and α7-nicotine receptors mediated by amantadine also appears to exert anti-inflammatory effects [108]. Furthermore, amantadine was demonstrated to exert antioxidant-like activity in vitro in the 2,2-Diphenyl-1-picrylhydrazyl (DPPH)-test [28].
It was shown that the expression of phosphodiesterase (PDE) in the hippocampus is altered after TBI. Amantadine’s suggested effect on PDE could thus favorably alter the deficits in synaptic plasticity of the hippocampus and contribute to the improvement of cognitive abilities after TBI. Amantadine inhibits calmodulin-dependent PDE 1 with IC50 of approximately 5 µM, which may increase adenosine 3′,5′-cyclic monophosphate (cAMP) and in turn produce neuroprotective activity [109] and anti-inflammatory properties of amantadine [110]. In another in vitro study, amantadine, at concentration of 6 µM, inhibited PDEs responsible for guanosine 3′,5′-cyclic monophosphate (cGMP) and cAMP degradation by up to 30 and 20%, respectively. This effect was even stronger, i.e., reaching 50%, when analyzed ex vivo in hemiparkinsonian rats rendered dyskinetic with repeated doses of L-DOPA. Moreover, amantadine treatment (40 mg/kg) decreased cGMP in the striatum of the dyskinetic rat brain microdialysates [111]. There is an indication that PDEs may be upregulated in TBI. The effect of amantadine on PDE could thus favorably alter the deficits in synaptic plasticity of the hippocampus and contribute to the improvement of cognitive abilities after TBI. Indeed, PDEs, particularly of group 4 (PDE4), have been suggested as potential target for the treatment of TBI [112,113]. The reduction in PDE-1 is also related to the anti-inflammatory properties of amantadine. This shows an impact on microglia signaling pathways and the ability of PDE-1 inhibitors to prevent or attenuate an excessive inflammatory response from BV2 cells and microglia [110].
6. Preclinical and Clinical Evidence of Amantadine Efficacy in Traumatic Brain Injury
Amantadine was first developed in 1960´s as a treatment against influenza A2 [114,115]. Later, its antiparkinsonian activity was accidentally discovered by Robert Schwab [116]. Therefore, amantadine has been used for several decades in the treatment of influenza infections and Parkinson’s disease.
Amantadine’s efficacy in TBI has been initially suggested by Gualtieri and colleagues based on their clinical observations [117,118]. It is probably not surprising given the fact that neuroprotection by amantadine had previously been suggested in various neurological conditions such as Parkinson’s disease, stroke, or infectious disease [119,120,121,122,123,124].
6.1. Preclinical Studies
A number of studies demonstrated neuroprotective activity of amantadine in various experimental paradigms. The effects observed in these studies can also be pertinent to pathomechanisms of TBI. However, for the sake of keeping the focus on TBI, we refer the reader to our previous extensive review discussing these aspects [50].
Several preclinical studies were performed or have been ongoing, that are specifically evaluating effects of amantadine on different outcome measures in various animal models of TBI. From the therapeutic point of view, studies employing the delayed treatment initiation paradigm are closer to clinical practice as compared to pretreatment. The effect of amantadine may thus be related to improvement of recovery from the insult, enhancement of regeneration processes or effect on neurochemical pathways implicated in TBI symptoms.
One of the earliest studies of amantadine in a TBI model was conducted by Dixon and colleagues in rats. The authors demonstrated that amantadine given for 18 days, starting one day post injury at a dose of 10 mg/kg/day attenuated deficits in water maze learning 14-18 days after injury. The motor tasks and hippocampal histology were not improved [125].
Wang and colleagues investigated effects of amantadine treatment initiated one hour after TBI and subsequently followed by its administration using thrice-daily dose regimen for 16 consecutive days at 15, 45, or 135 mg/kg/day. Only the highest dose improved performance in Morris maze spatial learning paradigm and afforded neuroprotection as observed on the level of the hippocampus. However, the effective dose resulted in serum concentrations of approximately 12 000 ng/ml (corresponding to 80 µM) [126]. Such serum level is far above the therapeutic range of amantadine.
In another study, amantadine, given at 45 or 135 mg/kg three times a day for 28 days following experimental TBI in rats, decreased the neuronal degeneration and apoptosis in the substantia nigra [127]. In addition, amantadine reversed the TBI-related decrease in dopamine in the striatum, decreased depressive-like behavior (as demonstrated in experiments using forced swim test and sucrose preference paradigms) and improved learning deficits [126,127].Noteworthy, even the lower dose of amantadine (45 mg/kg) is expected to exceed clinically relevant plasma concentrations [50].
Bleimeister and colleagues started administration of amantadine at the dose of 20 mg/kg to rats 24 hours after cortical impact injury; the treatment was continued for 19 days. Improvement in motor and learning disabilities were observed. However, amantadine failed to improve structural changes, i.e., volume of cortical lesions, as measured by lesion area in histological slices [128].
Huang and colleagues showed that infusion of amantadine (86.4 mg/kg/day starting five days after insult and continuing for eight weeks) reversed dopamine deficit, decreased motor impairment on rotarod and improved novel object recognition learning test in rats after cerebral cortical fluid percussion injury, the widely used model of brain injury [129].
In yet another study, treatment with amantadine, starting 24 hours after cortical impact injury and continuing for 19 days, partially attenuated motor co-ordination deficit (as measured using beam walking test, on days 1-5) and improved spatial learning (on days 14-19). Interestingly, a statistically significant effect was observed at mid-dose (20 mg/kg/day) but not at 10 or 40 mg/kg/day, suggesting a bell-shaped dose-response relationship [130].
The majority of the aforementioned studies indicate some types of functional improvement by amantadine; however, most authors also report lack of structural improvement with amantadine. The main shortcoming of many of these studies is use of too high doses, that lack therapeutic relevance. However, in general, the preclinical data clearly suggest beneficial effect of amantadine in post-treatment of TBI. This remains in agreement with the available favorable results of clinical studies (see below).
6.2. Clinical Studies
The awakening mechanism associated with amantadine in disturbance of consciousness is related to the enhancement of dopamine in the substantia nigra and in neurotransmission within the mesencephalic limbic and frontal striatum loop system, which are responsible for regulating awakening, activation and attention [131]. This has been confirmed by positron emitted tomography examination [132]. Neuropharmacological therapies are commonly used off-label to enhance arousal and behavioral responsiveness, on the premise that pathological derangements in dopaminergic and noradrenergic neurotransmitter systems can be improved through supplementation. In that context, Amantadine is one of the most commonly used drugs. There have been a multitude of studies analyzing the effects of Amantadine in recent years.
Amantadine has been widely investigated in consciousness disorders, however, the clinical trials are rather heterogenous regarding the studied populations , treatment modalities (e.g., the timing of the initiation of the pharmacological intervention, treatment duration, the dosage), and clinical outcome measures, for review see [133]. Indeed, both neuroprotection and neuroactivation can be envisaged as potential mechanisms underlying amantadine's effects on overall recovery following brain injury. There is a relatively large body of evidence suggesting that amantadine promotes functional amelioration in patients following acute TBI. In the earliest published placebo-controlled randomized controlled trial (RCT) using crossover design, amantadine failed to increase the rate of cognitive recovery in 10 patients moderate to severe TBI [134]. A placebo controlled RCT conducted later showed improvements with amantadine on the Disability Rating Scale (DRS) and cognitive function tests. Furthermore, following switch to amantadine, the placebo-treated patients showed further improvements [40]. Likewise, the most robust and large placebo-controlled RCT (n=184) involving patients 4-16 weeks after severe TBI in the vegetative state or minimally conscious state showed 4-week treatment with amantadine accelerate recovery as measured on the DRS and Coma Recovery Scale-Revised (CRS-R) [131]. The rate of improvement decreased during a 2-week wash-out period in the amantadine more than in placebo group, with no difference in DRS and CRS-R scores at 6 weeks. Rates of adverse effects were similar in both groups [131].
A number of retrospective chart reviews, case-control studies, or case reports in patients with disorders of consciousness remain in concordance with the results of the aforementioned RCTs [118,135,136,137,138,139,140,141,142,143,144,145,146]. Furthermore, amantadine-induced specific metabolic changes in affected brain areas of TBI patients, which were correlated with some clinical improvements [132,138]. In an open-label study effect of amantadine (400 mg) on executive function and activity in pre-frontal cortex was studies in twenty-two subjects pre- and post-12-week treatment. Improvement in executive function were observed and positron emission tomography (PET) data showed increase in left pre-frontal cortex glucose metabolism with significant correlation between these two measures (Kraus et al., 2005). Shafiee et al. compared observed numerical improvement in an acute phase after injury as measured with Glasgow Coma Scale (GCS) and Glasgow Outcome Scale (GOS) with amantadine when compared with zolpidem and placebo groups, but without any significant statistical difference [147]. Very recently, Shimia et al. showed significant improvements compared to placebo on DRS, but not on GOS. The authors themselves acknowledged limitations of their study: small sample size, short duration, absence of a wash-out period, and shortcomings of the GOS for this kind of clinical study [148].
Therapeutic potential of amantadine has also been tested in pediatric TBI patients. In placebo-controlled studies in pediatric population (age range 3-18 years), amantadine was reported to be well tolerated, with adverse effects profile similar to that of placebo [for review see [149]]. Green et al. (2004) evaluated the safety of amantadine in children with TBI, with only 5 of 54 patients experienced side effects, all of which were reversible [150]. Also, a later study investigating the effects of amantadine in pediatric TBI patients found it safe. Despite lack of statistically significant differences in cognition, a cognition-improving potential of amantadine was suggested [151]. In yet another pediatric study – a RCT comparing amantadine to pramipexole in low responsive children and adolescents one month after brain injury – the patients in amantadine group made significant improvements from the baseline on several outcome parameters (Coma/ Near Coma Scale, Western NeuroSensory Stimulation Profile, DRS weekly gains, and Rancho Los Amigos Scale) without any significant side effects [152]. More recently, McMahon et al. performed a randomized placebo-controlled crossover trial in children (n=7). The observed improvements in consciousness parameters were greater with amantadine than with placebo, however, the differences were not found to be significant [153].
Some studies investigated effects of amantadine on neurobehavioral parameters, e.g., irritability, aggression or anger in patients recovering from the TBI in its chronic phase (≥6 months following TBI). Among patients with moderate-severe irritability, amantadine significantly improved the frequency and severity of irritability and aggression and was safe [154]. Amantadine significantly reduced aggression but not anger, in patients with moderate-to-severe aggression [155]. Even though aggression is one of possible sequelae of TBI in children [149], it should be interpreted with caution, since amantadine was reported to increase aggression in pediatric TBI patients [150]. In a recent publication, McLaughlin et al. reported on amantadine use in 234 children and young adults (age range 2 months to 21 years) with TBI during inpatient rehabilitation. Of those, 21% patients (0.9 - 20 years) received amantadine. Almost half of the patients admitted with a disorder of consciousness (median age 11.6 years) were treated with amantadine (dose range 0.7 - 13.5 mg/kg/d, the highest total daily dose was 400 mg/d). Nausea/abdominal discomfort (N=3) and agitation (N=3) were the most commonly reported adverse effects (8 patients; 16%). None of the adverse events were reported as serious [156].
Up to date, there are no comprehensive guidelines for treatment of disorders of consciousness in children and adolescents. Recently, Molteni et al. [157] reviewed the available evidence with the aim to provide a base for development of pediatric guidelines for diagnosis, prognosis, and treatment such disorders. Based on their analysis, amantadine treatment was associated with improvement of consciousness parameters in approximately 55% of cases [157].
It should be mentioned that some studies failed to demonstrate favorable effects of amantadine on various outcome measures in patients with brain injury (e.g., [134] [155,158,159,160,161,162]. Recently, Passman et al. evaluated the efficacy of early amantadine administration on recovery of consciousness after severe TBI in a retrospective analysis of medical records of patients over 11 years [163]. The authors compared the patients receiving amantadine (N = 60) to all other patients (N = 344) with respect to the outcomes on GCSe, GOS-Extended score, length of stay, mortality, recovery of command-following, and days to command following. The authors found no difference between these two groups in terms of mortality, rates of command following, or percentage of patients with severe (3-8) GCS scores at discharge, but also with respect to adverse events. In addition, the amantadine group was less likely to have a favorable recovery, had a longer length of hospital stay and a longer time to command following. The authors underlined a necessity of larger inpatient randomized trials investigating amantadine treatment for severe TBI [163].
In conclusion, there is some published evidence that amantadine improves arousal, attention, concentration, alertness, mobility without compromising safety in comatose patients at different stages following acute brain injury [164,165]. Accordingly, amantadine has been recommended by several clinical practice guidelines related to TBI treatment [117,166,167]. It should also be mentioned that amantadine was classified by American Academy of Neurology (AAN) at the level of evidence B in the recent guidelines for disorders of consciousness [117,166,167,168]. In addition, amantadine may have potential of normalizing behavioral disturbances in patients recovering from TBI [155]. Very recently, an expert panel (INCOG) reviewed evidence published from 2014 and developed updated guidelines for the management of attention in adults. The panel concluded that amantadine may facilitate arousal in comatose or vegetative patients but does not enhance performance on attentional measures over the longer term [169]. New evidence-based German clinical practice guidelines for the neurological rehabilitation of patients with disorders of consciousness have recently become available (Bender et al., 2023). The authors listed TBI among the most common causes of disorders of consciousness and called for use of standardized instruments in research. Mostly based on the results of the placebo-controlled study of [131] they recommended use of escalating doses of amantadine up to a 400 mg daily to treat post-coma vigilance impairment [49].
A detailed overview of selected important clinical studies with amantadine in the indication TBI can be found in the Table 1. The table covers amantadine doses, treatment durations, study designs, descriptions of the treated population, clinical tools (e.g., scales) used, and study results.
7. Non-Traumatic Brain Injury
For the use of amantadine in chronic disorders of consciousness, there is also a recommendation for non-traumatic causes [49]. The authors consider this to be appropriate, since the evidence for efficacy is very good and the risk-benefit ratio speaks in favor of an application trial.
Gao et al. [173] investigated the efficacy of amantadine in non-traumatic cerebral hemorrhage. In their study, 6 out of 12 patients on amantadine regained consciousness within three months. Efficacy was lower for bleeding in the frontal, parietal and temporal lobes than in the thalamus and basal ganglia.
No significant improvement in the recovery rate was noted in the amantadine group, but a reduction in the time to regain consciousness was reported in non-traumatic patients [174].
There is also evidence that amantadine improves attention, concentration, alertness, arousal and mobility in comatose patients at various stages of acute brain injury [164].
8. Differences Between Amantadine Sulphate and Hydrochloride
It should be noted that there are two amantadine salts on the market: amantadine hydrochloride originally introduced by Dupont as Symmetrel® and amantadine sulphate introduced by Merz Pharmaceuticals as PK Merz®. It has been suggested that after oral treatment, the increase in plasma levels after amantadine sulphate (PK Merz®) is more gradual and lasts longer due to slower absorption, which is likely the result of lower solubility [175]. Due to this feature, higher doses of amantadine sulphate (up to 600 mg) have been claimed to be used with lower risk of side effects as opposed to amantadine hydrochloride [175]. Moreover, longer half-life provides a potential advantage of more constant plasma levels by lower treatment frequency.
However, well-controlled clinical studies supporting these observations of differences between amantadine sulphate and hydrochloride are missing. Inspired by this gap, we compared the pharmacokinetics of amantadine sulphate vs. that of amantadine hydrochloride after oral administration [176] of equimolar doses to Sprague Dawley (SD) male rats using 0.5% methylcellulose as a vehicle (N=8 per group) (Table 2, Figure 2, internal report) [176]. Plasma obtained by serial sampling was analyzed for amantadine at 0.5, 1, 1.5, 2, 2.5, 3, 4, 8, 16 and 24 h after administration using liquid chromatography-mass spectrometry (LC/MS).). Indeed, we could demonstrate that amantadine sulphate had delayed plasma half-life (T1/2) and higher area under the curve (Table 2, Figure 2). There was also a trend for delayed Tmax which however failed to reach statistical significance. Cmax values were comparable. It remains to be demonstrated whether these animal data can be translated into clinical findings.
Apart from the differences between amantadine salts after oral administration, it should be noted that intravenous infusions are only available as amantadine sulphate. This form of application has the following potential advantages:
- Possibility of treatment when oral use is not possible or difficult like in unconscious state (e.g. TBI) or swallowing difficulties (e.g. Parkinson´s disease).
- Faster onset of action as compared to oral administration which could offer advantage in e.g. TBI) or in akinetic crisis [175].
- Better monitoring of PK-PD relationship through flexible adjustment of infusion speed
9. Future Research Questions
There is some robust, even though limited evidence that amantadine is effective and safe in treatment of consequences of TBI. Results of the largest, randomized, placebo-controlled clinical trial by Giacino et al. (2012) are further supported by a number of rather heterogenous studies employing different clinical scales and readouts in various populations of patients who had undergone TBI (see Table 1 for additional information). The current state of knowledge found reflection in several guidelines and recommendation papers. There are multiple preclinical publications existing, that suggest a wide array of potential mechanisms by which amantadine may exert its beneficial effects in TBI patients. Future preclinical studies are needed to explore these mechanisms and understand how to employ them in an optimal way in clinical settings. Moreover, further clinical studies are needed to confirm and fully reveal the therapeutic potential of amantadine in patients post TBI. There are still some important clinical questions that are yet to be answered, e.g.:
- -
- What are the effects of amantadine in disorders of consciousness with a therapy duration of more than four weeks?
- -
- How does amantadine work in different disorders of consciousness, especially those with non-traumatic causes?
- -
- What is the interaction of amantadine administered in combination with other drugs (e.g., with cerebrolysin) in patients with impaired consciousness?
10. Conclusions
Considering the poly-pharmacology of amantadine, we believe that the potential of this compound for the treatment of TBI is not fully utilized. Amantadine may offer neuroprotective and neuroactivating benefits. The causes of TBI are diverse in terms of impact magnitude, localization, conditions of the affected person and the age. In turn, diversity of pathological pathways may be present already in the beginning. Moreover, resulting neurodegeneration can have severe impact on the patient’s quality of life and occurs via diverse, parallel mechanisms interacting with each other. This implies that treatment with multiple targets may show better efficacy than those with selectivity for one target. We believe that amantadine may fulfill this expectation and in turn, well controlled clinical studies of amantadine in TBI seem to be warranted.
In the situation when oral treatment is possible, amantadine sulphate salt may show superiority over amantadine hydrochloride due to a slower rate of absorption, and, in turn, a longer duration of action connected with the decreased risk of peak-dose side effects. On the other hand, application of amantadine as an infusion may be of particular benefit in unconscious patients with TBI by whom oral route od administration cannot be utilized. Furthermore, intravenously administered amantadine rapidly appears in the CNS thanks to bypassing absorption from the gastrointestinal tract. Finally, parenteral administration allows for precise dose adjustment based on blood monitoring and/or patients physiological reaction.
The clinical practice seems to support the use of amantadine in TBI, as it is encouraged by several recommendations in different countries (e.g., in Brazil, Canada, France, Germany, USA) for practice guidelines for disorders of consciousness and TBI recovery [117,166,167,168].
Authors contributions: AD & WD contributed equally to all aspects related to concept of this review manuscript as well as its preparation. Specifically, AD prepared Table 1 and most of the clinical part. WD prepared Table 2 and most of the preclinical part as well as description of mechanism of action amantadine. GP, REB, and AS contributed equally to finalizing the manuscript from the stage of first draft by writing corrections and text additions resulting from discussions, in particular for clinical aspects. The manuscript in its present form was read and approved by all authors.
Acknowledgements
The authors would like to thank Andreas Gravius for valuable comments and corrections and Malgorzata Dekundy for concept of graphic presentation of Figure 1 and graphic design.
Conflict of interest: AD and AS are employees of Merz Therapeutics. WD is previous employee of Merz Therapeutics and currently serves as scientific advisor for Merz Therapeutics.
References
- Agarwal, N. Traumatic Brain Injury. Available online: https://www.aans.org/Patients/Neurosurgical-Conditions-and-Treatments/Traumatic-Brain-Injury (accessed on 2020).
- Dawodu, S.T. Traumatic Brain Injury (TBI) - Definition, Epidemiology, Pathophysiology. Available online: https://emedicine.medscape.com/article/326510-overview#showall (accessed on.
- Lingsma, H.F.; Roozenbeek, B.; Steyerberg, E.W.; Murray, G.D.; Maas, A.I. Early prognosis in traumatic brain injury: from prophecies to predictions. Lancet Neurol 2010, 9, 543–554. [Google Scholar] [CrossRef]
- Brazinova, A.; Rehorcikova, V.; Taylor, M.S.; Buckova, V.; Majdan, M.; Psota, M.; Peeters, W.; Feigin, V.; Theadom, A.; Holkovic, L.; et al. Epidemiology of Traumatic Brain Injury in Europe: A Living Systematic Review. Journal of neurotrauma 2021, 38, 1411–1440. [Google Scholar] [CrossRef]
- Majdan, M.; Plancikova, D.; Brazinova, A.; Rusnak, M.; Nieboer, D.; Feigin, V.; Maas, A. Epidemiology of traumatic brain injuries in Europe: a cross-sectional analysis. Lancet Public Health 2016, 1, e76–e83. [Google Scholar] [CrossRef]
- Majdan, M.; Plancikova, D.; Maas, A.; Polinder, S.; Feigin, V.; Theadom, A.; Rusnak, M.; Brazinova, A.; Haagsma, J. Years of life lost due to traumatic brain injury in Europe: A cross-sectional analysis of 16 countries. PLoS Med 2017, 14, e1002331. [Google Scholar] [CrossRef]
- Ray, S.K.; Dixon, C.E.; Banik, N.L. Molecular mechanisms in the pathogenesis of traumatic brain injury. Histol Histopathol 2002, 17, 1137–1152. [Google Scholar] [CrossRef]
- Veenith, T.; Goon, S.; Burnstein, R.M. Molecular mechanisms of traumatic brain injury: the missing link in management. World J Emerg Surg 2009, 4, 7. [Google Scholar] [CrossRef]
- Jarrahi, A.; Braun, M.; Ahluwalia, M.; Gupta, R.V.; Wilson, M.; Munie, S.; Ahluwalia, P.; Vender, J.R.; Vale, F.L.; Dhandapani, K.M.; et al. Revisiting Traumatic Brain Injury: From Molecular Mechanisms to Therapeutic Interventions. Biomedicines 2020, 8. [Google Scholar] [CrossRef]
- Traeger, J.; Hoffman, B.; Misencik, J.; Hoffer, A.; Makii, J. Pharmacologic Treatment of Neurobehavioral Sequelae Following Traumatic Brain Injury. Crit Care Nurs Q 2020, 43, 172–190. [Google Scholar] [CrossRef]
- Dixon, K.J. Pathophysiology of Traumatic Brain Injury. Phys Med Rehabil Clin N Am 2017, 28, 215–225. [Google Scholar] [CrossRef]
- McKee, A.C.; Daneshvar, D.H. The neuropathology of traumatic brain injury. Handb Clin Neurol 2015, 127, 45–66. [Google Scholar] [CrossRef]
- Wu, H.M.; Huang, S.C.; Hattori, N.; Glenn, T.C.; Vespa, P.M.; Yu, C.L.; Hovda, D.A.; Phelps, M.E.; Bergsneider, M. Selective metabolic reduction in gray matter acutely following human traumatic brain injury. Journal of neurotrauma 2004, 21, 149–161. [Google Scholar] [CrossRef]
- Enevoldsen, E.M.; Jensen, F.T. Autoregulation and CO2 responses of cerebral blood flow in patients with acute severe head injury. J Neurosurg 1978, 48, 689–703. [Google Scholar] [CrossRef]
- Brenner, M.; Stein, D.M.; Hu, P.F.; Aarabi, B.; Sheth, K.; Scalea, T.M. Traditional systolic blood pressure targets underestimate hypotension-induced secondary brain injury. J Trauma Acute Care Surg 2012, 72, 1135–1139. [Google Scholar] [CrossRef] [PubMed]
- Stein, D.M.; Lindel, A.L.; Murdock, K.R.; Kufera, J.A.; Menaker, J.; Scalea, T.M. Use of serum biomarkers to predict secondary insults following severe traumatic brain injury. Shock 2012, 37, 563–568. [Google Scholar] [CrossRef]
- Unterberg, A.W.; Stover, J.; Kress, B.; Kiening, K.L. Edema and brain trauma. Neuroscience 2004, 129, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- Bullock, R.; Zauner, A.; Woodward, J.J.; Myseros, J.; Choi, S.C.; Ward, J.D.; Marmarou, A.; Young, H.F. Factors affecting excitatory amino acid release following severe human head injury. J Neurosurg 1998, 89, 507–518. [Google Scholar] [CrossRef] [PubMed]
- Mellergard, P.; Sjogren, F.; Hillman, J. The cerebral extracellular release of glycerol, glutamate, and FGF2 is increased in older patients following severe traumatic brain injury. Journal of neurotrauma 2012, 29, 112–118. [Google Scholar] [CrossRef]
- Lifshitz, J.; Sullivan, P.G.; Hovda, D.A.; Wieloch, T.; McIntosh, T.K. Mitochondrial damage and dysfunction in traumatic brain injury. Mitochondrion 2004, 4, 705–713. [Google Scholar] [CrossRef]
- Shao, C.; Roberts, K.N.; Markesbery, W.R.; Scheff, S.W.; Lovell, M.A. Oxidative stress in head trauma in aging. Free Radic Biol Med 2006, 41, 77–85. [Google Scholar] [CrossRef]
- Lucas, S.M.; Rothwell, N.J.; Gibson, R.M. The role of inflammation in CNS injury and disease. Br J Pharmacol 2006, 147 Suppl 1, S232–240. [Google Scholar] [CrossRef]
- Kim, J.H.; Lee, H.W.; Hwang, J.; Kim, J.; Lee, M.J.; Han, H.S.; Lee, W.H.; Suk, K. Microglia-inhibiting activity of Parkinson's disease drug amantadine. Neurobiol Aging 2012, 33, 2145–2159. [Google Scholar] [CrossRef] [PubMed]
- Kubera, M.; Maes, M.; Budziszewska, B.; Basta-Kaim, A.; Leskiewicz, M.; Grygier, B.; Rogoz, Z.; Lason, W. Inhibitory effects of amantadine on the production of pro-inflammatory cytokines by stimulated in vitro human blood. Pharmacological reports : PR 2009, 61, 1105–1112. [Google Scholar] [CrossRef] [PubMed]
- Wandinger, K.P.; Hagenah, J.M.; Kluter, H.; Rothermundt, M.; Peters, M.; Vieregge, P. Effects of amantadine treatment on in vitro production of interleukin-2 in de-novo patients with idiopathic Parkinson's disease. Journal of Neuroimmunology 1999, 98, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Stoica, B.A.; Faden, A.I. Cell death mechanisms and modulation in traumatic brain injury. Neurotherapeutics 2010, 7, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Ossola, B.; Schendzielorz, N.; Chen, S.H.; Bird, G.S.; Tuominen, R.K.; Mannisto, P.T.; Hong, J.S. Amantadine protects dopamine neurons by a dual action: reducing activation of microglia and inducing expression of GDNF in astroglia [corrected]. Neuropharmacology 2011, 61, 574–582. [Google Scholar] [CrossRef] [PubMed]
- Kranthi, K.; Anand Priya, V.V.M.; Punnagai, K.; Chellathai David, D. A Comparative Free Radical Scavenging Evaluation of Amantadine and Rasagiline. Biomedical & Pharmacology Journal 2019, 12, 1175–1179. [Google Scholar] [CrossRef]
- Wenk, G.L.; Danysz, W.; Roice, D.D. The effects of mitochondrial failure upon cholinergic toxicity in the nucleus basalis. Neuroreport 1996, 7, 1453–1456. [Google Scholar] [CrossRef] [PubMed]
- Posner, J.B.; Saper, C.B.; Plum, F. Diagnosis of stupor and coma; Oxford University Press.: New-York, 2007.
- Zeman, A. Consciousness. Brain 2001, 124, 1263-1289. [CrossRef]
- Buckwalter, J.A.; Parvizi, J.; Morecraft, R.J.; van Hoesen, G.W. Thalamic projections to the posteromedial cortex in the macaque. J Comp Neurol 2008, 507, 1709–1733. [Google Scholar] [CrossRef]
- Lin, J.S. Brain structures and mechanisms involved in the control of cortical activation and wakefulness, with emphasis on the posterior hypothalamus and histaminergic neurons. Sleep Med Rev 2000, 4, 471–503. [Google Scholar] [CrossRef]
- Schiff, N.D. Central thalamic contributions to arousal regulation and neurological disorders of consciousness. Ann N Y Acad Sci 2008, 1129, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Sherman, S.M.; Guillery, R.W. The role of the thalamus in the flow of information to the cortex. Philos Trans R Soc Lond B Biol Sci 2002, 357, 1695–1708. [Google Scholar] [CrossRef] [PubMed]
- Laureys, S. The neural correlate of (un)awareness: lessons from the vegetative state. Trends Cogn Sci 2005, 9, 556–559. [Google Scholar] [CrossRef] [PubMed]
- Laureys, S.; Faymonville, M.E.; Luxen, A.; Lamy, M.; Franck, G.; Maquet, P. Restoration of thalamocortical connectivity after recovery from persistent vegetative state. Lancet 2000, 355, 1790–1791. [Google Scholar] [CrossRef] [PubMed]
- Laureys, S.; Goldman, S.; Phillips, C.; Van Bogaert, P.; Aerts, J.; Luxen, A.; Franck, G.; Maquet, P. Impaired effective cortical connectivity in vegetative state: preliminary investigation using PET. Neuroimage 1999, 9, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Bales, J.W.; Wagner, A.K.; Kline, A.E.; Dixon, C.E. Persistent cognitive dysfunction after traumatic brain injury: A dopamine hypothesis. Neuroscience and biobehavioral reviews 2009, 33, 981–1003. [Google Scholar] [CrossRef] [PubMed]
- Meythaler, J.M.; Brunner, R.C.; Johnson, A.; Novack, T.A. Amantadine to improve neurorecovery in traumatic brain injury-associated diffuse axonal injury: a pilot double-blind randomized trial. J Head Trauma Rehabil 2002, 17, 300–313. [Google Scholar] [CrossRef]
- Plum, F.; Posner, J.B. The diagnosis of stupor and coma. Contemp Neurol Ser 1972, 10, 1–286. [Google Scholar] [PubMed]
- Cooksley, T.; Rose, S.; Holland, M. A systematic approach to the unconscious patient. Clin Med (Lond) 2018, 18, 88–92. [Google Scholar] [CrossRef]
- Laureys, S.; Celesia, G.G.; Cohadon, F.; Lavrijsen, J.; Leon-Carrion, J.; Sannita, W.G.; Sazbon, L.; Schmutzhard, E.; von Wild, K.R.; Zeman, A.; et al. Unresponsive wakefulness syndrome: a new name for the vegetative state or apallic syndrome. BMC Med 2010, 8, 68. [Google Scholar] [CrossRef]
- Giacino, J.T.; Ashwal, S.; Childs, N.; Cranford, R.; Jennett, B.; Katz, D.I.; Kelly, J.P.; Rosenberg, J.H.; Whyte, J.; Zafonte, R.D.; et al. The minimally conscious state: definition and diagnostic criteria. Neurology 2002, 58, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Bruno, M.A.; Vanhaudenhuyse, A.; Thibaut, A.; Moonen, G.; Laureys, S. From unresponsive wakefulness to minimally conscious PLUS and functional locked-in syndromes: recent advances in our understanding of disorders of consciousness. J Neurol 2011, 258, 1373–1384. [Google Scholar] [CrossRef] [PubMed]
- Pichler, G.; Fazekas, F. Cardiopulmonary arrest is the most frequent cause of the unresponsive wakefulness syndrome: A prospective population-based cohort study in Austria. Resuscitation 2016, 103, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Lei, J.; Gao, G.; Feng, J.; Mao, Q.; Jiang, J. Prevalence of persistent vegetative state in patients with severe traumatic brain injury and its trend during the past four decades: A meta-analysis. NeuroRehabilitation 2017, 40, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Bender, A. S3-LL Neurologische Rehabilitation bei Koma und schwerer Bewusstseinsstörung im Erwachsenenalter. DEUTSCHE GESELLSCHAFT FÜR NEUROREHABILITATION E.V. (DGNR) (Hrsgb.), Leitlinien für die Neurorehabilitation. 2022, 1, 1-8.
- Bender, A.; Eifert, B.; Rubi-Fessen, I.; Jox, R.J.; Maurer-Karattup, P.; Muller, F. The Neurological Rehabilitation of Adults With Coma and Disorders of Consciousness. Dtsch Arztebl Int 2023, 120, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Danysz, W.; Dekundy, A.; Scheschonka, A.; Riederer, P. Amantadine: reappraisal of the timeless diamond-target updates and novel therapeutic potentials. J Neural Transm (Vienna) 2021, 128, 127–169. [Google Scholar] [CrossRef] [PubMed]
- Kornhuber, J.; Retz, W.; Riederer, P. Slow accumulation of psychotropic substances in the human brain. Relationship to therapeutic latency of neuroleptic and antidepressant drugs? Journal of neural transmission. Supplementum 1995, 46, 315–323. [Google Scholar] [PubMed]
- Danysz, W.; Gossel, M.; Zajaczkowski, W.; Dill, D.; Quack, G. Are NMDA antagonistic properties relevant for antiparkinsonian-like activity in rats? case of amantadine and memantine. Journal of Neural Transmission. Parkinsons Disease and Dementia Section 1994, 7, 155–166. [Google Scholar] [CrossRef]
- Hesselink, M.B.; DeBoer, B.G.; Breimer, D.D.; Danysz, W. Brain penetration and in vivo recovery of NMDA receptor antagonists amantadine and memantine: A quantitative microdialysis study. Pharmaceutical Research 1999, 16, 637–642. [Google Scholar] [CrossRef]
- Monaghan, D.T.; Yao, D.; Cotman, C. L-[3H]Glutamate binds to kainate-, NMDA- and AMPA- sensitive binding sites: an autoradiographic analysis. Brain Research 1985, 340, 378–383. [Google Scholar] [CrossRef]
- Cotman, C.W.; Iversen, L.L. Excitatory aminio acids in the brain - focus on NMDA receptors. Trends in Neurosciences 1987, 10, 263–265. [Google Scholar] [CrossRef]
- Faden, A.I.; Demediuk, P.; Panter, S.S.; Vink, R. The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science 1989, 244, 798–800. [Google Scholar] [CrossRef] [PubMed]
- Rader, R.K.; T.H., L. Experimental ischemia induces a persistent depolarisation blocked by decreased calcium and NMDA antagonists. Neuroscience Letters 1989, 99, 125-130.
- Ikonomidou, C.; Turski, L. Why did NMDA receptor antagonists fail clinical trials for stroke and traumatic brain injury? Lancet Neurol 2002, 1, 383–386. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, S.; Pouladi, M.A.; Talantova, M.; Yao, D.; Xia, P.; Ehrnhoefer, D.E.; Zaidi, R.; Clemente, A.; Kaul, M.; Graham, R.K.; et al. Balance between synaptic versus extrasynaptic NMDA receptor activity influences inclusions and neurotoxicity of mutant huntingtin. Nat Med 2009, 15, 1407–1413. [Google Scholar] [CrossRef]
- Xia, P.; Chen, H.S.; Zhang, D.; Lipton, S.A. Memantine Preferentially Blocks Extrasynaptic over Synaptic NMDA Receptor Currents in Hippocampal Autapses. J Neurosci 2010, 30, 11246–11250. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, T.K. Novel pharmacologic therapies in the treatment of experimental traumatic brain injury - a review. J Neurotrauma 1993, 10, 215–261. [Google Scholar] [CrossRef]
- Parsons, C.G.; Danysz, W.; Quack, G. Glutamate in CNS Disorders as a target for drug development: an update. Drug News Perspect 1998, 11, 523–569. [Google Scholar] [CrossRef]
- Danysz, W.; Parsons, C.G.; Bresink, I.; Quack, G. Glutamate in CNS disorders - A revived target for drug development. Drug News Perspect 1995, 8, 261–277. [Google Scholar]
- Loane, D.J.; Faden, A.I. Neuroprotection for traumatic brain injury: translational challenges and emerging therapeutic strategies. Trends Pharmacol Sci 2010, 31, 596–604. [Google Scholar] [CrossRef]
- Kornhuber, J.; Schoppmeyer, K.; Riederer, P. Affinity of 1-aminoadamantanes for the sigma binding site in post-mortem human frontal cortex. Neuroscience Letters 1993, 163, 129–131. [Google Scholar] [CrossRef]
- Nguyen, V.H.; Kassiou, M.; Johnston, G.A.R.; Christie, M.J. Comparison of binding parameters of sigma-1 and sigma-2 binding sites in rat and guinea pig brain membranes: novel subtype-selective trishomocubanes. Eur.J.Pharmacol. 1996, 311, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Peeters, M.; Romieu, P.; Maurice, T.; Su, T.P.; Maloteaux, J.M.; Hermans, E. Involvement of the sigma 1 receptor in the modulation of dopaminergic transmission by amantadine. Eur.J Neurosci. 2004, 19, 2212–2220. [Google Scholar] [CrossRef] [PubMed]
- Piechal, A.; Jakimiuk, A.; Mirowska-Guzel, D. Sigma receptors and neurological disorders. Pharmacological reports : PR 2021. [CrossRef]
- Salaciak, K.; Pytka, K. Revisiting the sigma-1 receptor as a biological target to treat affective and cognitive disorders. Neuroscience and biobehavioral reviews 2022, 132, 1114–1136. [Google Scholar] [CrossRef] [PubMed]
- Monnet, F.P. Sigma-1 receptor as regulator of neuronal intracellular Ca2+: clinical and therapeutic relevance. Biology of the cell / under the auspices of the European Cell Biology Organization 2005, 97, 873–883. [Google Scholar] [CrossRef] [PubMed]
- Weiser, S.D.; Patrick, S.L.; Mascarella, S.W.; Downing-Park, J.; Bai, X.; Carroll, F.I.; Walker, J.M.; Patrick, R.L. Stimulation of rat striatal tyrosine hydroxylase activity following intranigral administration of sigma receptor ligands. Eur J Pharmacol 1995, 275, 1–7. [Google Scholar] [CrossRef]
- Gudelsky, G.A. Effects of sigma receptor ligands on the extracellular concentration of dopamine in the striatum and prefrontal cortex of the rat. Eur J Pharmacol 1995, 286, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Thompson, T.L.; Bridges, S.; Miller, C. Modulation of dopamine uptake in rat nucleus accumbens: effect of specific dopamine receptor antagonists and sigma ligands. Neurosci Lett 2001, 312, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Alvear, G.M.; Werling, L.L. sigma1 Receptors in rat striatum regulate NMDA-stimulated [3H]dopamine release via a presynaptic mechanism. Eur J Pharmacol 1995, 294, 713–719. [Google Scholar] [CrossRef]
- Rousseaux, C.G.; Greene, S.F. Sigma receptors [sigmaRs]: biology in normal and diseased states. J Recept Signal Transduct Res 2015, 36, 327–388. [Google Scholar] [CrossRef]
- Francardo, V. Sigma-1 receptor: a potential new target for Parkinson's disease? Neural Regen Res 2014, 9, 1882–1883. [Google Scholar] [CrossRef]
- Mori, T.; Hayashi, T.; Su, T.P. Compromising sigma-1 receptors at the endoplasmic reticulum render cytotoxicity to physiologically relevant concentrations of dopamine in a nuclear factor-kappaB/Bcl-2-dependent mechanism: potential relevance to Parkinson's disease. J Pharmacol Exp Ther 2012, 341, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Decoster, M.A.; Klette, K.L.; Knight, E.S.; Tortella, F.C. sigma receptor-mediated neuroprotection against glutamate toxicity in primary rat neuronal cultures. Brain Research 1995, 671, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Maurice, T.; Lockhart, B.P. Neuroprotective and anti-amnesic potentials of sigma (sigma) receptor ligands. Progress in Neuro - Psychopharmacology & Biological Psychiatry 1997, 21, 69–102. [Google Scholar]
- Mancuso, R.; Olivan, S.; Rando, A.; Casas, C.; Osta, R.; Navarro, X. Sigma-1R agonist improves motor function and motoneuron survival in ALS mice. Neurotherapeutics 2012, 9, 814–826. [Google Scholar] [CrossRef] [PubMed]
- Meunier, J.; Ieni, J.; Maurice, T. The anti-amnesic and neuroprotective effects of donepezil against amyloid beta25-35 peptide-induced toxicity in mice involve an interaction with the sigma1 receptor. Br J Pharmacol 2006, 149, 998–1012. [Google Scholar] [CrossRef] [PubMed]
- Oneill, M.; Caldwell, M.; Earley, B.; Canney, M.; Ohalloran, A.; Kelly, J.; Leonard, B.E.; Junien, J.L. The sigma receptor ligand JO 1784 (igmesine hydrochloride) is neuroprotective in the gerbil model of global cerebral ischaemia. European Journal of Pharmacology 1995, 283, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Chen, F.; Chen, Z.; Yang, W.; Yue, S.; Zhang, J.; Chen, X. Sigma-1 Receptor: A Potential Therapeutic Target for Traumatic Brain Injury. Front Cell Neurosci 2021, 15, 685201. [Google Scholar] [CrossRef] [PubMed]
- Cervero, C.; Blasco, A.; Tarabal, O.; Casanovas, A.; Piedrafita, L.; Navarro, X.; Esquerda, J.E.; Caldero, J. Glial Activation and Central Synapse Loss, but Not Motoneuron Degeneration, Are Prevented by the Sigma-1 Receptor Agonist PRE-084 in the Smn2B/- Mouse Model of Spinal Muscular Atrophy. J Neuropathol Exp Neurol 2018, 77, 577–597. [Google Scholar] [CrossRef]
- Dong, H.; Ma, Y.; Ren, Z.; Xu, B.; Zhang, Y.; Chen, J.; Yang, B. Sigma-1 Receptor Modulates Neuroinflammation After Traumatic Brain Injury. Cell Mol Neurobiol 2016, 36, 639–645. [Google Scholar] [CrossRef]
- Ryskamp, D.A.; Korban, S.; Zhemkov, V.; Kraskovskaya, N.; Bezprozvanny, I. Neuronal Sigma-1 Receptors: Signaling Functions and Protective Roles in Neurodegenerative Diseases. Front Neurosci 2019, 13, 862. [Google Scholar] [CrossRef]
- Deep, P.; Dagher, A.; Sadikot, A.; Gjedde, A.; Cumming, P. Stimulation of dopa decarboxylase activity in striatum of healthy human brain secondary to NMDA receptor antagonism with a low dose of amantadine. Synapse 1999, 34, 313–318. [Google Scholar] [CrossRef]
- Li, X.M.; Juorio, A.V.; Qi, J.; Boulton, A.A. Amantadine increases aromatic L-amino acid decarboxylase mRNA in PC12 cells. Journal of Neuroscience Research 1998, 53, 490–493. [Google Scholar] [CrossRef]
- Fisher, A.; Biggs, C.S.; Starr, M.S. Effects of glutamate antagonists on the activity of aromatic L-amino acid decarboxylase. Amino Acids 1998, 14, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Arai, A.; Kannari, K.; Shen, H.; Maeda, T.; Suda, T.; Matsunaga, M. Amantadine increases L-DOPA-derived extracellular dopamine in the striatum of 6-hydroxydopamine-lesioned rats. Brain research 2003, 972, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Liepert, J. Update on pharmacotherapy for stroke and traumatic brain injury recovery during rehabilitation. Curr Opin Neurol 2016, 29, 700–705. [Google Scholar] [CrossRef] [PubMed]
- Barra, M.E.; Izzy, S.; Sarro-Schwartz, A.; Hirschberg, R.E.; Mazwi, N.; Edlow, B.L. Stimulant Therapy in Acute Traumatic Brain Injury: Prescribing Patterns and Adverse Event Rates at 2 Level 1 Trauma Centers. J Intensive Care Med 2019, 885066619841603. [Google Scholar] [CrossRef] [PubMed]
- Karli, D.C.; Burke, D.T.; Kim, H.J.; Calvanio, R.; Fitzpatrick, M.; Temple, D.; Macneil, M.; Pesez, K.; Lepak, P. Effects of dopaminergic combination therapy for frontal lobe dysfunction in traumatic brain injury rehabilitation. Brain Injury 1999, 13, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Bales, J.W.; Kline, A.E.; Wagner, A.K.; Dixon, C.E. Targeting Dopamine in Acute Traumatic Brain Injury. Open Drug Discov J 2010, 2, 119–128. [Google Scholar] [CrossRef]
- d'Anglemont de Tassigny, X.; Pascual, A.; Lopez-Barneo, J. GDNF-based therapies, GDNF-producing interneurons, and trophic support of the dopaminergic nigrostriatal pathway. Implications for Parkinson's disease. Front Neuroanat 2015, 9, 10. [Google Scholar] [CrossRef]
- Caumont, A.S.; Octave, J.N.; Hermans, E. Amantadine and memantine induce the expression of the glial cell line-derived neurotrophic factor in C6 glioma cells. Neurosci Lett 2006, 394, 196–201. [Google Scholar] [CrossRef]
- Zhang, J.; Tan, H.; Jiang, W.; Zuo, Z. Amantadine alleviates postoperative cognitive dysfunction possibly by increasing glial cell line-derived neurotrophic factor in rats. Anesthesiology 2014, 121, 773–785. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Li, J.; Ni, C.; Zuo, Z. Amantadine Alleviates Postoperative Cognitive Dysfunction Possibly by Preserving Neurotrophic Factor Expression and Dendritic Arborization in the Hippocampus of Old Rodents. Frontiers in Aging Neuroscience 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Mitroshina, E.V.; Mishchenko, T.A.; Shirokova, O.M.; Astrakhanova, T.A.; Loginova, M.M.; Epifanova, E.A.; Babaev, A.A.; Tarabykin, V.S.; Vedunova, M.V. Intracellular Neuroprotective Mechanisms in Neuron-Glial Networks Mediated by Glial Cell Line-Derived Neurotrophic Factor. Oxid Med Cell Longev 2019, 2019, 1036907. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.T.; Rao, V.L.; Sailor, K.A.; Bowen, K.K.; Dempsey, R.J. Protective effects of glial cell line-derived neurotrophic factor on hippocampal neurons after traumatic brain injury in rats. J Neurosurg 2001, 95, 674–679. [Google Scholar] [CrossRef]
- Qi, L.; Xue, X.; Sun, J.; Wu, Q.; Wang, H.; Guo, Y.; Sun, B. The Promising Effects of Transplanted Umbilical Cord Mesenchymal Stem Cells on the Treatment in Traumatic Brain Injury. J Craniofac Surg 2018, 29, 1689–1692. [Google Scholar] [CrossRef] [PubMed]
- Minnich, J.E.; Mann, S.L.; Stock, M.; Stolzenbach, K.A.; Mortell, B.M.; Soderstrom, K.E.; Bohn, M.C.; Kozlowski, D.A. Glial cell line-derived neurotrophic factor (GDNF) gene delivery protects cortical neurons from dying following a traumatic brain injury. Restor Neurol Neurosci 2010, 28, 293–309. [Google Scholar] [CrossRef] [PubMed]
- Bahlakeh, G.; Rahbarghazi, R.; Mohammadnejad, D.; Abedelahi, A.; Karimipour, M. Current knowledge and challenges associated with targeted delivery of neurotrophic factors into the central nervous system: focus on available approaches. Cell Biosci 2021, 11, 181. [Google Scholar] [CrossRef] [PubMed]
- Abe, K. Therapeutic potential of neurotrophic factors and neural stem cells against ischemic brain injury. J Cereb Blood Flow Metab 2000, 20, 1393–1408. [Google Scholar] [CrossRef]
- Linnerbauer, M.; Rothhammer, V. Protective Functions of Reactive Astrocytes Following Central Nervous System Insult. Front Immunol 2020, 11, 573256. [Google Scholar] [CrossRef]
- Lin, P.H.; Kuo, L.T.; Luh, H.T. The Roles of Neurotrophins in Traumatic Brain Injury. Life (Basel) 2021, 12. [Google Scholar] [CrossRef]
- Gianutsos, G.; Chute, S.; Dunn, J.P. Pharmacological changes in dopaminergic systems induced by long term administration of amantadine. European Journal of Pharmacology 1985, 110, 357–361. [Google Scholar] [CrossRef] [PubMed]
- Dineley, K.T.; Pandya, A.A.; Yakel, J.L. Nicotinic ACh receptors as therapeutic targets in CNS disorders. Trends Pharmacol Sci 2015, 36, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Kakkar, R.; Raju, R.V.S.; Rajput, A.H.; Sharma, R.K. Amantadine: An antiparkinsonian agent inhibits bovine brain 60 kDa calmodulin-dependent cyclic nucleotide phosphodiesterase isozyme. Brain Research 1997, 749, 290–294. [Google Scholar] [CrossRef] [PubMed]
- O'Brien, J.J.; O'Callaghan, J.P.; Miller, D.B.; Chalgeri, S.; Wennogle, L.P.; Davis, R.E.; Snyder, G.L.; Hendrick, J.P. Inhibition of calcium-calmodulin-dependent phosphodiesterase (PDE1) suppresses inflammatory responses. Mol Cell Neurosci 2020, 102, 103449. [Google Scholar] [CrossRef] [PubMed]
- Sancesario, G.; Morrone, L.A.; D'Angelo, V.; Castelli, V.; Ferrazzoli, D.; Sica, F.; Martorana, A.; Sorge, R.; Cavaliere, F.; Bernardi, G.; et al. Levodopa-induced dyskinesias are associated with transient down-regulation of cAMP and cGMP in the caudate-putamen of hemiparkinsonian rats: reduced synthesis or increased catabolism? Neurochem Int 2014, 79, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Titus, D.J.; Oliva, A.A.; Wilson, N.M.; Atkins, C.M. Phosphodiesterase inhibitors as therapeutics for traumatic brain injury. Curr Pharm Des 2014, 21, 332–342. [Google Scholar] [CrossRef]
- Wilson, N.M.; Titus, D.J.; Oliva, A.A.; Furones, C.; Atkins, C.M. Traumatic Brain Injury Upregulates Phosphodiesterase Expression in the Hippocampus. Frontiers in Systems Neuroscience 2016, 10. [Google Scholar] [CrossRef] [PubMed]
- Gerzon, K.; Krumkalns, E.V.; Brindle, R.L.; Marshall, F.J.; Root, M.A. The adamantyl group in medicinal agents. I. Hypoglycemic N- arylsulfonyl-N'-adamantylureas. J.Med.Chem. 1963, 6, 760–763. [Google Scholar] [CrossRef] [PubMed]
- Maj, J.; Sowinska, H.; Baran, L.; Sarnek, J. Pharmacological effects of 1,3-dimethyl-5-aminoadamantane, a new adamantane derivative. European Journal of Pharmacology 1974, 26, 9–14. [Google Scholar] [CrossRef]
- Schwab, R.S.; England, A.C., Jr.; Poskanzer, D.C.; Young, R.R. Amantadine in the treatment of Parkinson's disease. JAMA 1969, 208, 1168–1170. [Google Scholar] [CrossRef]
- Butterworth, R.F. Amantadine for the Treatment of Traumatic Brain Injury and its Associated Cognitive and Neurobehavioural Complications. Journal of Pharmacology and Pharmaceutical Research 2020, 3, 1–5. [Google Scholar]
- Gualtieri, T.; Chandler, M.; Coons, T.B.; Brown, L.T. Amantadine: a new clinical profile for traumatic brain injury. Clinical Neuropharmacology 1989, 12, 258–270. [Google Scholar] [CrossRef] [PubMed]
- Uitti, R.J.; Rajput, A.H.; Ahlskog, J.E.; Offord, K.P.; Ho, M.M.; Prasad, M.; Rajput, A.; Basran, P. Amantadine treatment is an independent predictor of improved survival in parkinsonism. Canadian Journal of Neurological Sciences 1993, 20 (Suppl. 4), S235. [Google Scholar]
- Khasanova, D.R.; Saikhunov, M.V.; Kitaeva, E.A.; Khafiz'ianova, R.; Islaamov, R.R.; Demin, T.V. [Amantadine sulfate (PK-Merz) in the treatment of ischemic stroke: a clinical-experimental study]. Zhurnal nevrologii i psikhiatrii imeni S.S. Korsakova / Ministerstvo zdravookhraneniia i meditsinskoi promyshlennosti Rossiiskoi Federatsii, Vserossiiskoe obshchestvo nevrologov [i] Vserossiiskoe obshchestvo psikhiat 2009, 109, 37–43. [Google Scholar]
- Brison, E.; Jacomy, H.; Desforges, M.; Talbot, P.J. Novel treatment with neuroprotective and antiviral properties against a neuroinvasive human respiratory virus. J Virol 2014, 88, 1548–1563. [Google Scholar] [CrossRef]
- Quarato, G.; Scrima, R.; Ripoli, M.; Agriesti, F.; Moradpour, D.; Capitanio, N.; Piccoli, C. Protective role of amantadine in mitochondrial dysfunction and oxidative stress mediated by hepatitis C virus protein expression. Biochem Pharmacol 2014, 89, 545–556. [Google Scholar] [CrossRef]
- Rejdak, K.; Grieb, P. Adamantanes might be protective from COVID-19 in patients with neurological diseases: multiple sclerosis, parkinsonism and cognitive impairment. Mult Scler Relat Disord 2020, 42, 102163. [Google Scholar] [CrossRef] [PubMed]
- Butterworth, R.F. Amantadine, Parkinson’s Disease and COVID-19. Covid perspect res & rev 2020, 2020, 1–6. [Google Scholar]
- Dixon, C.E.; Kraus, M.F.; Kline, A.E.; Ma, X.C.; Yan, H.Q.; Griffith, R.G.; Wolfson, B.M.; Marion, D.W. Amantadine improves water maze performance without affecting motor behavior following traumatic brain injury in rats. Restorative Neurology and Neuroscience 1999, 14, 285–294. [Google Scholar]
- Wang, T.; Huang, X.J.; Van, K.C.; Went, G.T.; Nguyen, J.T.; Lyeth, B.G. Amantadine improves cognitive outcome and increases neuronal survival after fluid percussion traumatic brain injury in rats. Journal of neurotrauma 2014, 31, 370–377. [Google Scholar] [CrossRef]
- Tan, L.; Ge, H.; Tang, J.; Fu, C.; Duanmu, W.; Chen, Y.; Hu, R.; Sui, J.; Liu, X.; Feng, H. Amantadine preserves dopamine level and attenuates depression-like behavior induced by traumatic brain injury in rats. Behavioural brain research 2015, 279, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Bleimeister, I.H.; Wolff, M.; Lam, T.R.; Brooks, D.M.; Patel, R.; Cheng, J.P.; Bondi, C.O.; Kline, A.E. Environmental enrichment and amantadine confer individual but nonadditive enhancements in motor and spatial learning after controlled cortical impact injury. Brain research 2019, 1714, 227–233. [Google Scholar] [CrossRef]
- Huang, E.Y.; Tsui, P.F.; Kuo, T.T.; Tsai, J.J.; Chou, Y.C.; Ma, H.I.; Chiang, Y.H.; Chen, Y.H. Amantadine ameliorates dopamine-releasing deficits and behavioral deficits in rats after fluid percussion injury. PloS one 2014, 9, e86354. [Google Scholar] [CrossRef] [PubMed]
- Okigbo, A.A.; Helkowski, M.S.; Royes, B.J.; Bleimeister, I.H.; Lam, T.R.; Bao, G.C.; Cheng, J.P.; Bondi, C.O.; Kline, A.E. Dose-dependent neurorestorative effects of amantadine after cortical impact injury. Neurosci Lett 2019, 694, 69–73. [Google Scholar] [CrossRef]
- Giacino, J.T.; Whyte, J.; Bagiella, E.; Kalmar, K.; Childs, N.; Khademi, A.; Eifert, B.; Long, D.; Katz, D.I.; Cho, S.; et al. Placebo-controlled trial of amantadine for severe traumatic brain injury. The New England journal of medicine 2012, 366, 819–826. [Google Scholar] [CrossRef]
- Schnakers, C.; Hustinx, R.; Vandewalle, G.; Majerus, S.; Moonen, G.; Boly, M.; Vanhaudenhuyse, A.; Laureys, S. Measuring the effect of amantadine in chronic anoxic minimally conscious state. Journal of neurology, neurosurgery, and psychiatry 2008, 79, 225–227. [Google Scholar] [CrossRef] [PubMed]
- Loggini, A.; Tangonan, R.; El Ammar, F.; Mansour, A.; Goldenberg, F.D.; Kramer, C.L.; Lazaridis, C. The role of amantadine in cognitive recovery early after traumatic brain injury: A systematic review. Clin Neurol Neurosurg 2020, 194, 105815. [Google Scholar] [CrossRef]
- Schneider, W.N.; Drew-Cates, J.; Wong, T.M.; Dombovy, M.L. Cognitive and behavioural efficacy of amantadine in acute traumatic brain injury: An initial double-blind placebo-controlled study. Brain Injury 1999, 13, 863–872. [Google Scholar] [CrossRef]
- Chandler, M.C.; Barnhill, J.L.; Gualtieri, C.T. Amantadine for the agitated head-injury patient. Brain injury : [BI] 1988, 2, 309–311. [Google Scholar] [CrossRef]
- Kraus, M.F.; Maki, P. The combined use of amantadine and l-dopa/carbidopa in the treatment of chronic brain injury. Brain Injury 1997, 11, 455–460. [Google Scholar] [CrossRef]
- Kraus, M.F.; Maki, P.M. Effect of amantadine hydrochloride on symptoms of frontal lobe dysfunction in brain injury: case studies and review. The Journal of neuropsychiatry and clinical neurosciences 1997, 9, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Kraus, M.F.; Smith, G.S.; Butters, M.; Donnell, A.J.; Dixon, E.; Yilong, C.; Marion, D. Effects of the dopaminergic agent and NMDA receptor antagonist amantadine on cognitive function, cerebral glucose metabolism and D2 receptor availability in chronic traumatic brain injury: a study using positron emission tomography (PET). Brain injury : [BI] 2005, 19, 471–479. [Google Scholar] [CrossRef]
- Nickels, J.L.; Schneider, W.N.; M. L., D.; Wong, T.M. Clinical use of amantadine in brain injury rehabilitation. Brain Injury 1994, 8, 709–718. [Google Scholar] [CrossRef]
- Saniova, B.; Drobny, M.; Lehotsky, J.; Sulaj, M.; Schudichova, J. Biochemical and clinical improvement of cytotoxic state by amantadine sulphate. Cell Mol Neurobiol 2006, 26, 1475–1482. [Google Scholar] [CrossRef] [PubMed]
- Zafonte, R.D.; Watanabe, T.; Mann, N.R. Amantadine : a potential treatment for the minimally conscious state. Brain Injury 1998, 12, 617–621. [Google Scholar] [CrossRef]
- Raffaele, R.; Nicoletti, G.; Vecchio, I.; Ruggieri, M.; Malaguarnera, M.; Rampello, L.; Brunetto, M.B.; Nicoletti, F. Use of amantadine in the treatment of the neurobehavioral sequelae after brain injury in elderly patients. Arch Gerontol Geriatr Suppl 2002, 8, 309–312. [Google Scholar] [CrossRef] [PubMed]
- Saniova, B.; Drobny, M.; Kneslova, L.; Minarik, M. The outcome of patients with severe head injuries treated with amantadine sulphate. J Neural Transm (Vienna) 2004, 111, 511–514. [Google Scholar] [CrossRef]
- Whyte, J.; Katz, D.; Long, D.; DiPasquale, M.C.; Polansky, M.; Kalmar, K.; Giacino, J.; Childs, N.; Mercer, W.; Novak, P.; et al. Predictors of outcome in prolonged posttraumatic disorders of consciousness and assessment of medication effects: A multicenter study. Arch Phys Med Rehabil 2005, 86, 453–462. [Google Scholar] [CrossRef]
- Wu, T.S.; Garmel, G.M. Improved neurological function after Amantadine treatment in two patients with brain injury. J Emerg Med 2005, 28, 289–292. [Google Scholar] [CrossRef]
- Zafonte, R.D.; Lexell, J.; Cullen, N. Possible applications for dopaminergic agents following traumatic brain injury: part 2. J Head Trauma Rehabil 2001, 16, 112–116. [Google Scholar] [CrossRef]
- Shafiee, S.; Ehteshami, S.; Moosazadeh, M.; Aghapour, S.; Haddadi, K. Placebo-controlled trial of oral amantadine and zolpidem efficacy on the outcome of patients with acute severe traumatic brain injury and diffuse axonal injury. Caspian J Intern Med 2022, 13, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Shimia, M.; Iranmehr, A.; Valizadeh, A.; Mirzaei, F.; Namvar, M.; Rafiei, E.; Rahimi, A.; Khadivi, A.; Aeinfar, K. A placebo-controlled randomized clinical trial of amantadine hydrochloride for evaluating the functional improvement of patients following severe acute traumatic brain injury. J Neurosurg Sci 2023, 67, 598–604. [Google Scholar] [CrossRef] [PubMed]
- Hosenbocus, S.; Chahal, R. Amantadine: A Review of Use in Child and Adolescent Psychiatry. J Can Acad Child Adolesc Psychiatry 2013, 22, 55–60. [Google Scholar] [PubMed]
- Green, L.B.; Hornyak, J.E.; Hurvitz, E.A. Amantadine in pediatric patients with traumatic brain injury: a retrospective, case-controlled study. Am J Phys Med Rehabil 2004, 83, 893–897. [Google Scholar] [CrossRef]
- Beers, S.R.; Skold, A.; Dixon, C.E.; Adelson, P.D. Neurobehavioral effects of amantadine after pediatric traumatic brain injury: a preliminary report. J Head Trauma Rehabil 2005, 20, 450–463. [Google Scholar] [CrossRef]
- Patrick, P.D.; Blackman, J.A.; Mabry, J.L.; Buck, M.L.; Gurka, M.J.; Conaway, M.R. Dopamine agonist therapy in low-response children following traumatic brain injury. J Child Neurol 2006, 21, 879–885. [Google Scholar] [CrossRef] [PubMed]
- McMahon, M.A.; Vargus-Adams, J.N.; Michaud, L.J.; Bean, J. Effects of amantadine in children with impaired consciousness caused by acquired brain injury: a pilot study. Am J Phys Med Rehabil 2009, 88, 525–532. [Google Scholar] [CrossRef]
- Hammond, F.M.; Bickett, A.K.; Norton, J.H.; Pershad, R. Effectiveness of amantadine hydrochloride in the reduction of chronic traumatic brain injury irritability and aggression. J Head Trauma Rehabil 2014, 29, 391–399. [Google Scholar] [CrossRef]
- Hammond, F.M.; Sherer, M.; Malec, J.F.; Zafonte, R.D.; Whitney, M.; Bell, K.; Dikmen, S.; Bogner, J.; Mysiw, J.; Pershad, R.; et al. Amantadine Effect on Perceptions of Irritability after Traumatic Brain Injury: Results of the Amantadine Irritability Multisite Study. Journal of neurotrauma 2015, 32, 1230–1238. [Google Scholar] [CrossRef]
- McLaughlin, M.J.; Caliendo, E.; Lowder, R.; Watson, W.D.; Kurowski, B.; Baum, K.T.; Blackwell, L.S.; Koterba, C.H.; Hoskinson, K.R.; Tlustos, S.J.; et al. Prescribing Patterns of Amantadine During Pediatric Inpatient Rehabilitation After Traumatic Brain Injury: A Multicentered Retrospective Review From the Pediatric Brain Injury Consortium. J Head Trauma Rehabil 2022, 37, 240–248. [Google Scholar] [CrossRef]
- Molteni, E.; Canas, L.D.S.; Briand, M.M.; Estraneo, A.; Font, C.C.; Formisano, R.; Fufaeva, E.; Gosseries, O.; Howarth, R.A.; Lanteri, P.; et al. Scoping Review on the Diagnosis, Prognosis, and Treatment of Pediatric Disorders of Consciousness. Neurology 2023, 101, e581–e593. [Google Scholar] [CrossRef] [PubMed]
- Hughes, S.; Colantonio, A.; Santaguida, P.L.; Paton, T. Amantadine to enhance readiness for rehabilitation following severe traumatic brain injury. Brain injury : [BI] 2005, 19, 1197–1206. [Google Scholar] [CrossRef] [PubMed]
- Abbasivash, R.; Valizade Hasanloei, M.A.; Kazempour, A.; Mahdkhah, A.; Shaaf Ghoreishi, M.M.; Akhavan Masoumi, G. The Effect of Oral Administration of Amantadine on Neurological Outcome of Patients With Diffuse Axonal Injury in ICU. J Exp Neurosci 2019, 13, 1179069518824851. [Google Scholar] [CrossRef]
- Ghalaenovi, H.; Fattahi, A.; Koohpayehzadeh, J.; Khodadost, M.; Fatahi, N.; Taheri, M.; Azimi, A.; Rohani, S.; Rahatlou, H. The effects of amantadine on traumatic brain injury outcome: a double-blind, randomized, controlled, clinical trial. Brain injury : [BI] 2018, 32, 1050–1055. [Google Scholar] [CrossRef]
- Gramish, J.A.; Kopp, B.J.; Patanwala, A.E. Effect of Amantadine on Agitation in Critically Ill Patients With Traumatic Brain Injury. Clinical neuropharmacology 2017, 40, 212–216. [Google Scholar] [CrossRef]
- Hammond, F.M.; Sherer, M.; Malec, J.F.; Zafonte, R.D.; Dikmen, S.; Bogner, J.; Bell, K.R.; Barber, J.; Temkin, N. Amantadine Did Not Positively Impact Cognition in Chronic Traumatic Brain Injury: A Multi-Site, Randomized, Controlled Trial. Journal of neurotrauma 2018, 35, 2298–2305. [Google Scholar] [CrossRef] [PubMed]
- Passman, J.N.; Cleri, N.A.; Saadon, J.R.; Naddaf, N.; Gilotra, K.; Swarna, S.; Vagal, V.; Zheng, X.; Zhang, J.; Wong, J.; et al. In-Hospital Amantadine Does Not Improve Outcomes After Severe Traumatic Brain Injury: An 11-Year Propensity-Matched Retrospective Analysis. World Neurosurg 2023. [Google Scholar] [CrossRef]
- DeMarchi, R.; Bansal, V.; Hung, A.; Wroblewski, K.; Dua, H.; Sockalingam, S.; Bhalerao, S. Review of awakening agents. Can J Neurol Sci 2005, 32, 4–17. [Google Scholar] [CrossRef]
- Sawyer, E.; Mauro, L.S.; Ohlinger, M.J. Amantadine enhancement of arousal and cognition after traumatic brain injury. Ann Pharmacother 2008, 42, 247–252. [Google Scholar] [CrossRef]
- Anghinah, R.; Amorim, R.L.O.; Paiva, W.S.; Schmidt, M.T.; Ianof, J.N. Traumatic brain injury pharmacological treatment: recommendations. Arq Neuropsiquiatr 2018, 76, 100–103. [Google Scholar] [CrossRef]
- Plantier, D.; Luaute, J. Drugs for behavior disorders after traumatic brain injury: Systematic review and expert consensus leading to French recommendations for good practice. Ann Phys Rehabil Med 2016, 59, 42–57. [Google Scholar] [CrossRef] [PubMed]
- Giacino, J.T.; Katz, D.I.; Schiff, N.D.; Whyte, J.; Ashman, E.J.; Ashwal, S.; Barbano, R.; Hammond, F.M.; Laureys, S.; Ling, G.S.F.; et al. Practice Guideline Update Recommendations Summary: Disorders of Consciousness: Report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology; the American Congress of Rehabilitation Medicine; and the National Institute on Disability, Independent Living, and Rehabilitation Research. Arch Phys Med Rehabil 2018, 99, 1699–1709. [Google Scholar] [CrossRef]
- Ponsford, J.; Velikonja, D.; Janzen, S.; Harnett, A.; McIntyre, A.; Wiseman-Hakes, C.; Togher, L.; Teasell, R.; Kua, A.; Patsakos, E.; et al. INCOG 2.0 Guidelines for Cognitive Rehabilitation Following Traumatic Brain Injury, Part II: Attention and Information Processing Speed. J Head Trauma Rehabil 2023, 38, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Vargus-Adams, J.N.; McMahon, M.A.; Michaud, L.J.; Bean, J.; Vinks, A.A. Pharmacokinetics of amantadine in children with impaired consciousness due to acquired brain injury: preliminary findings using a sparse-sampling technique. PM R 2010, 2, 37–42. [Google Scholar] [CrossRef]
- Reddy, C.C.; Collins, M.; Lovell, M.; Kontos, A.P. Efficacy of amantadine treatment on symptoms and neurocognitive performance among adolescents following sports-related concussion. J Head Trauma Rehabil 2013, 28, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Ghate, P.S.; Bhanage, A.; Sarkar, H.; Katkar, A. Efficacy of Amantadine in Improving Cognitive Dysfunction in Adults with Severe Traumatic Brain Injury in Indian Population: A Pilot Study. Asian J Neurosurg 2018, 13, 647–650. [Google Scholar] [CrossRef]
- Gao, Y.; Zhang, Y.; Li, Z.; Ma, L.; Yang, J. Persistent vegetative state after severe cerebral hemorrhage treated with amantadine: A retrospective controlled study. Medicine (Baltimore) 2020, 99, e21822. [Google Scholar] [CrossRef]
- Avecillas-Chasin, J.M.; Barcia, J.A. Effect of amantadine in minimally conscious state of non-traumatic etiology. Acta Neurochir (Wien) 2014, 156, 1375–1377. [Google Scholar] [CrossRef]
- Danielczyk, W. Twenty-five years of amantadine therapy in Parkinson´s disease. Journal of Neural Transmission 1995, 46 (Suppl.), 399–405. [Google Scholar]
- Wang, H.; Ji, X.; Li, H.; Liang, J. In Vivo DMPK Report - Amantadine. PL_20201211_WD_1; 2021; pp. 1-8.
Figure 1.
Pathophysiology of TBI and possible targets of amantadine. AADC – aromatic l-amino acids decarboxylase; Ca2+ - calcium cation; DA – Dopamine; GDNF – glial cell line-derived neurotrophic factor; NMDA – N-methyl-D-aspartate; TH – tyrosine hydroxylase.
Figure 1.
Pathophysiology of TBI and possible targets of amantadine. AADC – aromatic l-amino acids decarboxylase; Ca2+ - calcium cation; DA – Dopamine; GDNF – glial cell line-derived neurotrophic factor; NMDA – N-methyl-D-aspartate; TH – tyrosine hydroxylase.
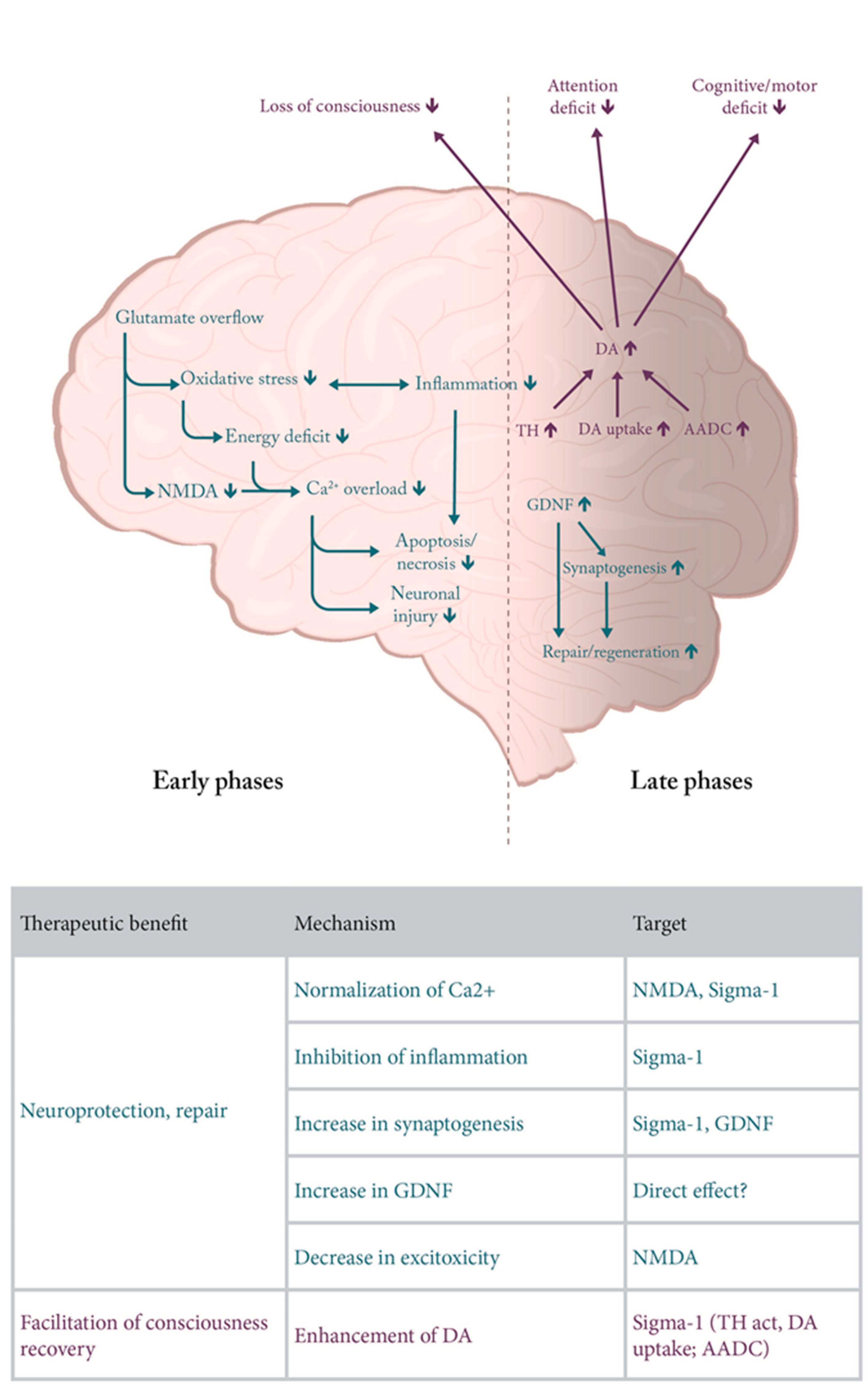
Figure 2.
Comparison of pharmacokinetic of amantadine hydrochloride and amantadine sulphate given orally as suspension in carboxy methyl cellulose (CMC) at equimolar doses (50.00 and 53,36 mg/kg). Sulphate salt shows delayed Tmax and higher area under the curve (see Table 1 for details). Symbols are means of 8 replicates per group [176].
Figure 2.
Comparison of pharmacokinetic of amantadine hydrochloride and amantadine sulphate given orally as suspension in carboxy methyl cellulose (CMC) at equimolar doses (50.00 and 53,36 mg/kg). Sulphate salt shows delayed Tmax and higher area under the curve (see Table 1 for details). Symbols are means of 8 replicates per group [176].
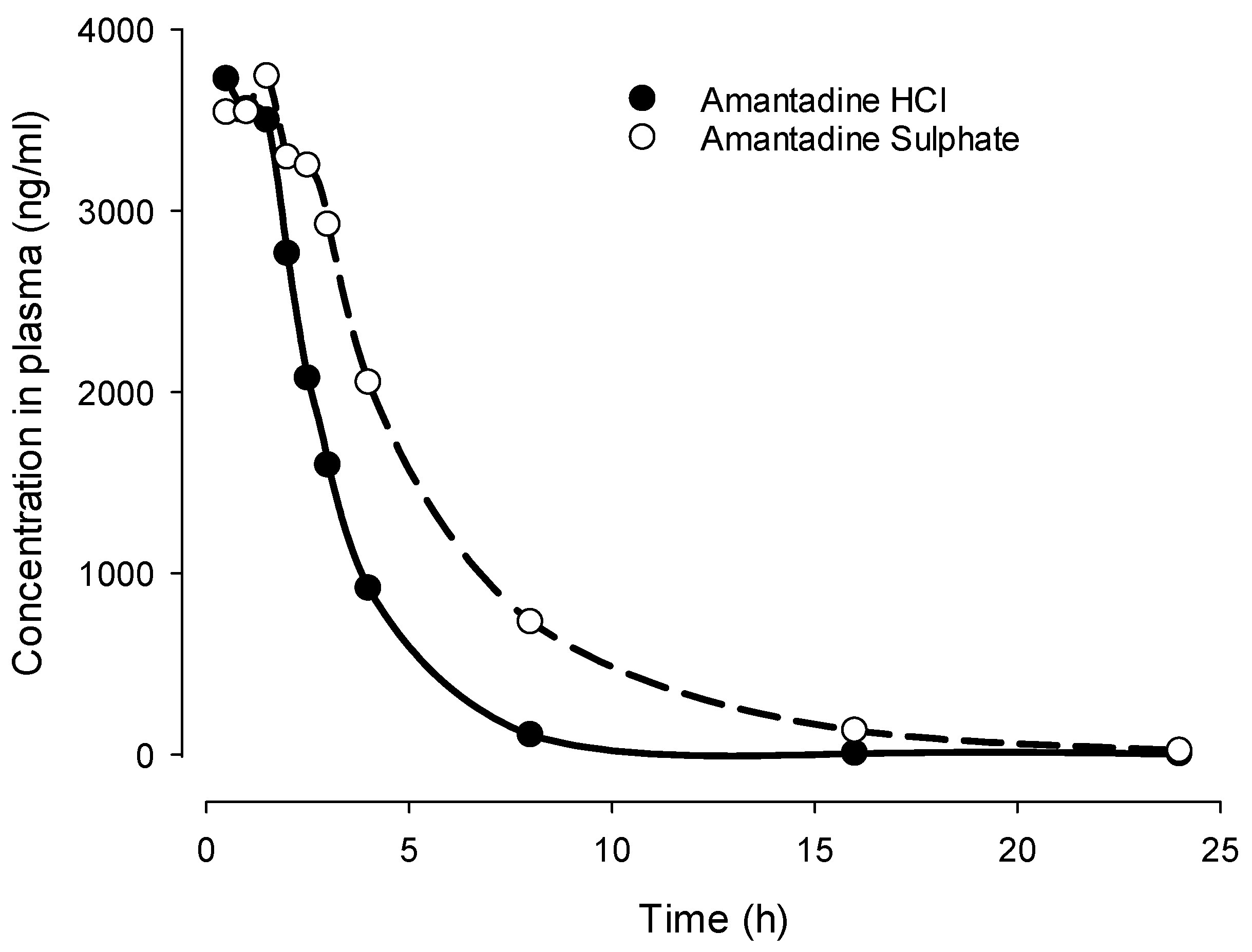

Table 1.
Summary of clinical studies with amantadine for TBI.
| Reference | Dose, treatment duration | Study design, | Clinical measures | Results |
|---|---|---|---|---|
| [139] | 50–200 mg/day BID |
Case Series Acute inpatient rehabilitation following brain injuries. N=12 |
Functional, neurobehavioral and cognitive status (e.g., attention, concentration, alertness, arousal, reaction time, agitation, anxiety) | Improvements in attention and concentration, alertness, arousal, processing time, psychomotor speed, mobility, vocalization, agitation, anxiety, and participation in therapy. |
| [136,137] | 25–400 mg/day | Case Series TBI N=7 |
The Mini-Mental State Examination (MMSE), Test for Severe Impairment; Clock Drawing Test; The Hopkins Verbal Learning Test; Hopkins Attention Screening Test; The Brief Test of Attention; verbal fluency tests; The Trail Making Test; Boston Naming Test | All patients had significant frontal lobe dysfunction from TBI, and 4 were “responders” while 3 were “non-responders” to amantadine treatment, with improvements in alertness, attention, executive function, cognition, speech, behavior, mood, motivation, motor abilities and psychomotor speed, as well as less dyscontrol. |
| [134] | 50-150 mg BID over 2 weeks | RCT, Crossover TBI N=10 2 weeks on AMH, 2 weeks wash out, 2 weeks on placebo |
Neurobehavioural Rating Score (NRS) Orientation, memory, attention, executive Rate of patients’ cognitive recovery |
Amantadine had no effect on the rate of patients’ cognitive recovery. Results limited by small sample size, heterogeneous population, acute time course, and limited study power and high drop-out rate. |
| [40] | 200 mg/day over 6 weeks | RCT, Crossover Acute TBI N=35 6 weeks on AMH, 6 weeks on placebo |
Agitated Behavioural Scale (ABS); MMSE; Disability Rating Scale (DRS); GOS; and Functional Independence Measure (FIM-cog) scale; Galveston Orientation and Amnesia Test (GOAT) | Significant improvements in the MMSE, DRS, GOS, and FIM cognitive scale in both groups of patients recovering from acute TBI during the first 6 weeks of the study, but only in the amantadine-treatment group during the second 6 weeks. However, the groups had similar functional levels after the study had finished. Amantadine was safe in the study population. |
| [142] | up to 150 mg BID | RCT, crossover Brain injuries N=6 |
Attention and concentration, fatigue | Amantadine improved attention and concentration, and reduced fatigue. |
| [150] | 100 mg BID to 400 mg QD | Case Control, Retrospective TBI (pediatric) N = 118 (amantadine N=54) |
Ranchos Los Amigos (RLA) | Amantadine-treated subjects had a greater improvement in their RLA level during their admission. Subjective improvements noted in most patients administered amantadine. Side effects were minimal and resolved when treatment was reduced. |
| [151] | up to 150 mg/d (<10 y/o) or 200 mg/d (>10 y/o) |
RCT (BUT: no placebo) TBI (pediatric subjects) N=27 (amantadine N=17); Only per protocol set analysed: N=13 (amantadine N=9) |
Cognition | Improvements with amantadine in cognitive testing when compared to age- and severity-matched TBI control patients observed in those ≤2 years post injury. The results limited since just per-protocol analysis was used. |
| [158] | 200 mg BID | Retrospective Cohort Severe TBI N=123 (amantadine N=28) |
GCS and somatosensory evoked potentials | Amantadine failed to shorten the time to emerge from coma. |
| [138] | 400 mg/day | RCT, Open label, Crossover TBI N=22 |
Executive function | Amantadine improved performance on executive function tests, correlated with a significant increase in left prefrontal cortex glucose metabolism in the first 6 male subjects enrolled. |
| [144] | Not provided | Cohort TBI N=124 (amantadine N=47) |
DRS | Amantadine significantly improved recovery |
| [152] | 100 mg BID | RCT TBI N=10 (amantadine N=6) |
Coma Near Coma (CNC) scale, DRS, and Western NeuroSensory Stimulation Profile | Weekly rate of change in the CNC scale, DRS, and Western NeuroSensory Stimulation Profile was significantly greater with amantadine or pramipexole than without and slowed 6 weeks after treatment termination). |
| [140] | 200 mg BID (i.v.) | RCT, Open Label Closed head injury N=32 (amantadine N=18) |
GCS, survival, biochemical parameters: glycaemia, malondialdehyde (MDA; marker of lipid peroxidation), beta-carotene, total SH groups | Amantadine-treated patients had reduced MDA and increased Beta-carotene (antioxidant), as well as improved survival, after only 1 week of treatment. |
| [170] | 400 mg/day | RCT, crossover Brain injuries in pediatric population N=7 |
CNC Scale or Coma Recovery Scale – Revised (CRS-R) | Amantadine was well tolerated, but had no significant effect on CNC Scale or CRS-R. |
| [131] | 200 mg BID, 4 weeks | RCT, crossover Post-traumatic disorders of consciousness Patients in the vegetative state or minimally conscious state 4-16 weeks after severe TBI N=184 (amantadine N=87) |
DRS – primary outcome measure CRS-R |
Amantadine accelerated the rate of functional recovery during active treatment. The rate of improvement decreased during a 2-week wash-out period in the amantadine more than in placebo group, with no difference in DRS and CRS-R scores at 6 weeks. Amantadine did not increase the incidence of adverse effects. |
| [171] | 100 mg BID | Case Control, Retrospective Subjects with history of head concussion N= 50 (amantadine N=25) |
Verbal memory, reaction time | After 3–4 weeks, amantadine-treated patients made significantly greater improvements in verbal memory and reaction time, as well as reported fewer persistent post-concussion symptoms, when compared to matched controls (by age, sex, and concussion history). |
| [154] | 100 mg BID, 4 weeks | RCT TBI N=76 (amantadine N=38) |
Neuropsychiatric Inventory - Irritability (NPI-I); Neuropsychiatric Inventory - Aggression (NPI-A) | Among patients with moderate-severe irritability (≥6 months following TBI), 4 weeks of amantadine significantly improved the frequency and severity of irritability and aggression and was safe. |
| [155] | 100 mg BID | RCT TBI N=118 (amantadine N=61) |
Aggression, anger | Among patients (≥6 months post-TBI) with moderate-to-severe aggression, amantadine significantly reduced aggression, with no beneficial effect on anger. |
| [155] | 100 mg BID | RCT TBI N=168 (amantadine N=82) |
NPI | Because of a very large placebo effect, amantadine did not significantly improve irritability (in patients with moderate-severe irritability, who suffered TBI ≥6 months prior to enrollment). |
| [161] | 100 mg BID | Cohort, retrospective TBI N=139 (amantadine N=70) |
Agitation, length of stay in intensive care unit (ICU) | Agitation was significantly more prevalent in the amantadine group. Patients given amantadine had longer ICU lengths of stay and received more opioids. |
| [160] | 100 mg BID | RCT severe TBI N=40 (amantadine N=19) |
GCS | Patients having received amantadine had a faster rate of improvement in their GCS scores during the first week of treatment. No functional differences observed at 6-month follow-up. |
| [172] | 100 mg BID over 4 weeks | Observational severe TBI (at 2 months orally or through enteral feeding tube) |
Full Outline of Unresponsiveness (FOUR) score, DRS, GOS during 4 weeks of treatment and 2 weeks posttreatment was assessed. | Improvement of cognitive function over 4-weeks of treatment interval as shown by significant improvement on FOUR score, DRS, and GOS. Recovery speed slowed down after discontinuation of amantadine. Convulsions (adverse effect) occurred in 8 out of 50 patients (5 discontinued). |
| [162] | 100 mg BID | RCT TBI (at least 6 months prior to enrollment, with moderate-severe irritability) N=119 (amantadine N=59) |
Cognitive battery, irritability | No differences between groups were observed after 60 days of treatment, but the placebo responses were high. Cognitive battery baseline scores for the treatment group were higher, increasing the group’s susceptibility to ceiling effects. At day 28, the mean change for the placebo group was greater (more room for improvement?). |
| [159] | 100 mg BID increased to 200 mg BID within 3 days |
Double-blind placebo-controlled trial Acute TBI (patients admitted to the intensive care unit, ICU) N=66 (amantadine N=33) |
GCS, GOS duration of mechanical ventilation length of hospitalization fatality at the hospital mortality in patients. |
No significant differences between amantadine and placebo on the GCS, GOS, duration of mechanical ventilation and hospitalization, fatality at the hospital. Statistical differences were found on GCS and GOS in discharged and deceased patients. |
| [147] | 200 mg/day | RCT (with parallel placebo and zolpidem groups) Acute severe TBI N=66 (amantadine N=22) |
GCS, GOS | The improvement on GCS and GOS was non-significantly better with amantadine than with zolpidem or placebo. No clinically significant adverse events were observed. |
| [156] | 0.7 - 13.5 mg/kg/d; up to 400 mg/d. | N = 234 children and young adults (2 mo - 21 y) TBI, inpatient rehabilitation. (amantadine N= (21%) patients, 0.9 - 20 years) |
Retrospective review of behavioral descriptions of function based on, e.g., Coma Recovery Scale-Revised (CRS-R) and post-traumatic amnesia (PTA) as measured using, e.g., Children’s Orientation and Amnesia Test | Almost half of the patients admitted with a disorder of consciousness (median age 11.6 years) were treated with amantadine Nausea/abdominal discomfort (N=3) and agitation (N=3) were the most commonly reported adverse effects (8 patients; 16%). None of the adverse events were reported as serious |
| [148] | 100 mg BID for 14 days, then 150 mg BID for 7 days, then 200 mg BID for 21 days |
RCT (triple-blind, placebo-controlled Severe TBI N= 57 (amantadine N=29) |
GOS, DRS | On DRS, change from baseline was significantly (p = 0.015) better with amantadine (10.88 ± 5.24) than with placebo (8.04 ± 4.07). No significant difference between these groups was found for GOS |
| [163] | tbd | Retrospective Severe TBI amantadine N =60 control N=344 |
GCS GOS-Extended Score (GCS-ES) Length of stay Mortality Recovery of command following Days to command following |
No difference between these two groups in terms of mortality, rates of command following, or percentage of patients with severe (3-8) Glasgow Coma Scale scores at discharge. No difference in adverse events. The amantadine group was less likely to have a favorable recovery, had a longer length of hospital stay and a longer time to command following. |
Table 2.
Comparison of pharmacokinetic analysis of amantadine sulphate and amantadine hydrochloride given orally as suspension in CMC at equimolar doses (53.36 and 50.00 mg/kg respectively). Symbols are means of 8 replicates per group [176].
Table 2.
Comparison of pharmacokinetic analysis of amantadine sulphate and amantadine hydrochloride given orally as suspension in CMC at equimolar doses (53.36 and 50.00 mg/kg respectively). Symbols are means of 8 replicates per group [176].
| Equimolar doses | Amantadine Sulphate | Amantadine hydrochloride | Statistical analysis |
|---|---|---|---|
| T½ (h) | 2.07±0.62 | 1.67±0.41 | Student T-test; P=0.001 |
| tmax (h) | 1.43±1.02 | 0.75±0.38 | NS |
| Cmax (ng/l) | 4045±689.35 | 3911±427.11 | NS |
| AUC 0-∞ (ng*h/ml) | 22226.88±4387.05 | 11690±1366.33 | Student T-test; P<0.0001 |
Values are Mean±SD, N=8 per group.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



