
Submitted:
08 May 2024
Posted:
08 May 2024
You are already at the latest version
Abstract
Oligodendrocyte progenitor cells (OPCs) represent a subtype of glia giving rise to oligodendrocytes, the myelin forming cells in the central nervous system (CNS). While OPCs are highly proliferating during development, they become relatively quiescent during adulthood, when their fate is strictly influenced by the extracellular context. In traumatic injuries and chronic neurodegenerative conditions including those of autoimmune origin, oligodendrocytes undergo apoptosis, and demyelination starts. Adult OPCs become immediately activated, they migrate at the lesion site and proliferate to replenish the damaged area, but their efficiency is hampered by the presence of a glial scar, a barrier mainly formed by reactive astrocytes, microglia, and the deposition of inhibitory extracellular matrix components. If, on one hand, glial scar limits the lesion spreading, it also blocks tissue regeneration. Therapeutic strategies aimed at reducing astrocyte or microglia activation and shifting them toward a neuroprotective phenotype have been proposed, whereas the role of OPCs has been largely overlooked. In this review, we have considered glial scar from the perspective of OPCs, analysing their behaviour when lesions originate and exploring the potential therapies aimed at sustaining OPCs to efficiently differentiate and promote remyelination.
Keywords:
remyelination
; oligodendrocytes
; glia
; astrocytes
; proteoglycans
; spinal cord injury
; multiple sclerosis
; demyelination
; CNS lesion.
1. Introduction
Responses to injury include a wound-healing structural remodeling of the lesion site and nearby regions culminating in the formation of a glial scar. This process has been observed in a wide range of CNS pathological conditions, including spinal cord injury (SCI), traumatic brain injury (TBI), ischemic stroke and in neurodegenerative diseases, such as multiple sclerosis (MS). The size and features of the glial scar varies in distinct CNS diseases and injuries, based on the different pathological dynamics, location, and severity of the insult. Yet, a fundamental arrangement of the glial scar can be recognized in all cases and includes three main compartments: a fibrotic lesion core encircled by a compact border of a heterogeneous population of glial cells, and, more externally, by an adjacent reactive neural parenchyma [1,2].
The lesion core or fibrotic scar is an area of severe tissue damage - often containing a central cystic cavity - populated by blood-borne cells (i.e., macrophages and lymphocytes), perivascular-derived macrophages and stromal cells. Early after acute injuries, blood-derived cells are recruited to the lesion site by resident astrocytes and microglia – which are the first responders to insults – secreting pro-inflammatory cytokines and chemokines. Then, overtime, the stromal component (i.e., fibroblasts and pericytes) progressively prevails over the blood-derived inflammatory cells and become the dominant cell population of the fibrotic scar, being embedded in a rich deposit of extracellular matrix (ECM) [3,4]. Newly generated astrocytes, oligodendrocyte progenitor cells (OPCs, or NG2 glia) and microglia are instead the main components of the surrounding ring, the proper glial scar [2,5], whereas the adjacent neural parenchyma comprises neurons actively engaged in neurite and synaptic remodelling [6].
Formation of the glial scar has been traditionally interpreted as a protective process, instrumental to confine the inflammatory response and blood-derived cells to the lesion core, preserving the integrity of the healthy nervous tissue and allowing the repair of the blood-brain barrier. Such a protective role has been mainly ascribed to astrocytes and microglia, whose ablation/manipulation leads to increased influx of blood-derived macrophages and fibrotic cells and exacerbated neuronal cell death, axon loss and demyelination after injury [2,7,8]. The role of the other glial cell types - and particularly of oligodendroglia - participating in the formation of the glial scar has been less investigated so far. Yet, an emerging corpus of data starts to be available. In this review, we will specifically focus on OPCs and oligodendrocytes, their behaviour and their molecular alterations in non-permissive inflammatory conditions, and potential therapeutic strategies to restore their functions and to promote lesion resolution.
2. Myelin as a Structural and Trophic “Organ” for Axons
In the CNS, oligodendrocytes are highly specialized cells responsible for the synthesis of myelin, a multilamellar structure characterized by up to 70% in lipid and myelin proteins that enable membrane compaction in a sheath that enwraps single axons thus allowing fast saltatory conduction [9].
The composition of myelin is unique compared to other membranes, with an approximate 2:2:1 ratio for cholesterol:phospholipids:glycolipids [10]. The extremely high cholesterol content in in myelin, that represent 80% of the cholesterol present in the brain, is ensured by both de novo biosynthesis and uptake of cholesterol precursors from other glial cells [11]. Other lipids are synthesized using fatty acids as building blocks. Myelination is a dynamic process that requires continuous maintenance of membranes, thus making oligodendrocytes highly active in both protein and lipid metabolism.
Moreover, in the last decades, it has been clearly demonstrated that oligodendrocytes also provide axons with fuel to enable proper neuronal functions, thus further increasing their energy demand (for review, see Saab and Nave 2017 [12]). Oligodendrocytes can directly transfer glucose, through the GLUT1 transporter; in addition, oligodendrocytes metabolize glucose to pyruvate through glycolysis, convert pyruvate to lactate, and then export lactate via monocarboxylate transporter 1 (MCT1) to the axons. Oligodendrocytes could also play a more indirect trophic role, fuelling axons with lactate produced by astrocytes and imported through gap junctions, rather than synthesising lactate by extensive glycolysis [13]. Via gap junctions, oligodendrocytes also regulate the concentrations of ions, such as Na+, potassium K+, and chloride Cl-, in both intracellular and extracellular spaces in the neuron-oligodendrocyte synapse, thus contributing to the maintenance of the resting membrane potential [14,15,16]. More recently, extracellular vesicles have been described as important components able to shuttle proteins, lipids, nutrients, and non-coding RNAs to axons, and maintain their homeostasis also in nutrient-deprived conditions [17]. Axonal trophic support includes typical fatty acids, such as docosahexaenoic acid, which constitutes a major component of excitable membranes, produced through beta-oxidation by oligodendrocyte peroxisomes in the myelin sheath [18].
3. More than Oligodendrocyte Progenitor Cells
Oligodendrocytes originate from OPCs, a population of highly proliferating glia that represents approximately 5–8% of the total cells in the CNS. OPCs derive from multiple niches in the developing brain and they migrate in both brain and spinal cord, where they will differentiate and contribute to myelination [9]. OPCs can be identified based on their expression of the platelet-derived growth factor receptor α (PDGFRα), the proteoglycan NG2, and the transcription factors SOX10 and OLIG2. In this early stage, OPCs show small capacitance, currents mediated by KV channels, small-medium sized NaV channels, and glutamate receptors [19,20,21]. As OPCs differentiate, they firstly form pre-myelinating oligodendrocytes, down-regulating PDGFRα, NG2, voltage-gated Na+ and Ca2+ channels, while they progressively express the sulfatide O4, the galactocerebroside GalC, and GPR17, a G protein-coupled receptor typical of this intermediate stage and acts as a transient inhibitor of myelination [22]. During their maturation, pre-myelinating oligodendrocytes progressively downregulate GPR17, rewire their glucose and lipid metabolism, produce myelin proteins and lipids that allow the organization of the myelin sheath around axons. This three-stage classification is an oversimplification since OPCs and oligodendrocytes represent a continuum. Recent transcriptomic analysis in oligodendroglial cells isolated from different brain areas in young and adult mice have identified 13 distinct populations that represent different oligodendroglial functional states [23]. This study highlighted that differentiation-committed oligodendrocyte precursors are distinct from OPCs and show lower levels of cell cycle markers while expressing genes involved in migration. One population of newly formed oligodendrocytes was shown to respond to motor learning, while another of vascular and leptomeningeal cells could migrate following vessels. This heterogeneity could be both intrinsic to their developmental origin and acquired by extrinsic cues at their final location. It was demonstrated that OPCs change their differentiation ability and show different responsiveness to growth factors when transplanted into other CNS areas [24]. OPCs are the only glial cell type that receives neuronal synapses, which activity was shown to modulate their proliferation, differentiation, and migration and is important for myelination and remyelination (for review, see Moura et al. 2022 [25] and Habermacher, Angulo and Benamer 2019 [26]). Recently, OPCs have been shown to phagocytose synapses in the developing and mature mouse visual cortex thus contributing to remodelling of neural circuitries [27]. OPCs are extremely plastic and adapt their program in response to a multitude of signals both in physiological and pathological conditions [28].
4. Oligodendroglial Cells Responses to Injury
Myelin and oligodendrocyte loss starts immediately after acute injuries such as SCI or TBI (i.e., within 15 minutes and for 24-48h at the core of the injury), likely due to the combinatorial effects of bleeding, hypoxia, oxidative damage, ATP- and glutamate-mediated excitotoxicity and release of proinflammatory cytokines such as interleukin-1α (IL-1α), tumour necrosis factor-α (TNFα) [6,29] (Figure 1). While neurons and astrocytes do not typically die beyond 24 h post-injury, oligodendrocyte apoptosis is instead protracted up to the subacute (i.e., 3-14 days after injury) and chronic phases, especially in distal degenerating axon tracts after SCI [30]. By impairing axonal conduction and reducing the metabolic supply and ion buffering, such a protracted loss of oligodendrocytes and demyelination contribute to neuronal and circuit dysfunction, leading to post-injury functional impairment. Accordingly, the number of spared axons per se does not always predict functional recovery and, in case of SCI, complete loss of motor and sensory functions frequently occurs also in presence of anatomically incomplete spinal lesions [29].
While demyelination is still ongoing, OPCs are rapidly recruited at the lesion site and set up a robust proliferative response, peaking at around 5-7 dpi in the tissue directly bordering the lesion upon SCI or stroke [29,31,32]. Such OPC expansion can be sustained by both parenchymal OPCs and periventricular neural stem cells [33,34,35], partly as a consequence of the loss of cell-to-cell contact inhibition [36,37], and partly in response to developmental (e.g., fibroblast growth factor - FGF, platelet-derived growth factor-PDGF, WNTs) and proinflammatory factors present in the lesion milieu and in the glial scar [2]. In case of SCI, OPC proliferative response is surprisingly long-lasting, being detected even at chronic stages [29]. OPC proliferative burst is accompanied by a prominent reduction in neuron-to-OPC synaptic connectivity, as assessed by the transient disappearance of glutamatergic inputs onto virtually all proliferating OPCs responding to a demyelinating injury [38].
Newly born OPCs accumulate at the border of the lesion (e.g., at the stroke penumbra) together with astrocytes, whereas reactive microglia enter the margins of the fibrotic scar [39]. Such an OPC confinement depends at least in part by stromal cell-derived signals, as demonstrated by OPC infiltration in the central fibrotic core of the lesion in case of fibroblast experimental ablation [40]. Acutely, OPCs produce fibronectin and laminin that protect axon growth cones from the neuroinflammatory milieu [41]. However, in the long term, glial cells overexpress matrix metalloproteases (MMPs) resulting in the degradation of hyaluronan and other ECM components into low molecular weight fragments that can amplify inflammation [42]. Glial cells, including OPCs, further modify the ECM composition by overexpressing chondroitin sulphate proteoglycans (CSPGs), among which brevican, neurocan, versican, and NG2 are highly represented, and known for contributing to hostile environment to axonal regrowth and neuronal migration [43].
On one hand, most of the newly generated OPCs entrap axon growth-cones and hamper regeneration, on the other hand, they do not differentiate into myelinating oligodendrocytes and do not contribute to remyelination. An increased number of immature OPCs and immature oligodendrocytes was reported at the border of the lesion even at later times after stroke, SCI or cortical stab wound [30,31,44], suggesting a blockade in their progression along the lineage or a local requirement for cells exerting roles other than differentiation into myelinating cells. The first hypothesis is corroborated by the presence of a plethora of environmental signals in the lesion milieu, that operate as inhibitors of OPC differentiation, including proinflammatory cytokines and CSPGs (Figure 1). Accordingly, when plated on CSPGs-coated plates, OPCs show maturation delay, reduced process outgrowth, and impaired migration, suggesting a direct chemorepulsive effect that alters their potential to access to demyelinated sites and the consequent recovery process [45]. Similar repellent effects of CSPGs in oligodendrocyte function were observed in scars of traumatic origin. After SCI in rats, treatments with chondroitinase increased the numbers of OPCs surrounding lesions in the first two weeks post-lesion, and the number of OPCs inside the lesion without altering OPC proliferation and cell death, likely as a consequence of improved OPC migration. Improving OPC migration into lesioned sites was shown to be necessary to enable the increased axonal sprouting and improved recovery observed at the second week after SCI [46,47]. Interestingly, CSPG-induced oligodendrocyte maturation impairment could not be reverted by previously reported pro-remyelinating compounds such as clemastine, benztropine, and miconazole, which suggests that remyelination strongly relies on the extracellular microenvironment [48,49,50]. Furthermore, OPCs have functional receptors for many cytokines such as IL-10, IL-6, IL-4, IL-18, IFN-γ, and TNF-α [51,52,53]. These factors, primarily secreted by astrocytes and microglia, were shown to have a negative impact on OPC maturation in vivo and in vitro [54,55].
OPC or oligodendrocyte transdifferentiation in other cell types may also contribute to such a failure in providing new myelinating elements. Specifically, fate mapping experiments revealed that a fraction (10-25%) of OPCs acquire features of astrocytes (e.g., expression of the glial cell fibrillary protein – GFAP, or Gja1, Aqp4, Kcnq4 mRNAs and ion currents pattern similar to that observed in cortical astrocytes) upon cerebral stroke, cortical stab wound, SCI and experimental autoimmune encephalomyelitis - EAE [56,57,58]. These OPC-derived astrocytes are a transient cell population characterized by a high expression of proliferation and motility markers [56]. As astrocytes have a limited capacity to migrate and amplify compared to OPCs (i.e., astrocyte numbers increase just by 10-20% after acute injuries; [59,60], it has been proposed that OPC-derived astrocytes might temporarily perform the functions of astrocytes (such as regulating extracellular ion and neurotransmitter homeostasis or providing energy supply) in proximity to the lesion [56], and then die or re-enter the oligodendroglia lineage [57]. Unexpectedly, a subpopulation of mature oligodendrocytes has been also shown to activate astrocytic genes and transgress via a transitional precursor phenotype toward the astroglial fate after acute brain injuries [61]. Mechanistically, mature oligodendrocyte-to-astrocyte conversion has been shown to be microglia-dependent and attributed to the activation of the IL-6 signalling [61]. The specific function of this additional population of astrocytes remains to be determined.
In addition, after injury, NG2 cells can trans-differentiate into Schwann cells. Yet, the appearance of NG2 cell-derived Schwann cells is context-dependent (i.e., they are detected following SCI, but not after cortical stab wound injury) and their actual contribution to remyelination remains controversial [62,63].
In agreement with the idea that, upon injury, OPC roles go beyond the generation of new myelinating cells, recent studies have shown that OPCs per se participate in tissue remodelling and healing. Although OPCs were shown to contribute to synapse and axon pruning during normal development, they were also shown to selectively internalize myelin debris through phagocytosis upon injury [27,54,64,65]. Moreover, in the acute phase after brain injury, OPCs release metalloproteinases (e.g., MMP9) which in turn promote blood-brain barrier (BBB) leakage and infiltration of blood-derived cells at lesion site [66]. For instance, ablation of proliferating NG2 glia results in reduced astrocyte hypertrophy and disorganization of the glial scar, which eventually lead to delayed brain wound closure and worse functional outcome [39,67]. Similarly, upon an inflammatory challenge, OPC depletion leads to a profound downregulation of the expression of microglia-specific genes and an exacerbated inflammatory response [68]. Consistently, manipulations of OPCs not resulting in their depletion (e.g., OPC-specific deletion of β-catenin) reduce astrocyte hypertrophy and GFAP expression, and promote microglia/macrophage acquisition of an anti-inflammatory/pro-regenerative phenotype [69]. Such effects likely depend on OPC ability to release cytokines and chemokines and to present antigen when exposed to inflammatory conditions [70]. Both these capabilities are shared by more advanced stages along the oligodendroglia lineage. For instance, immature oligodendrocytes can upregulate the expression of toll-like receptor Tlr3, IL-1β, IFN-β, Ccl2, and Cxcl10 when stimulated with IL-1β [71]. Similarly, the chronic activation of NF-kB, the transcription factor downstream to TNF-α, IL-1 and toll-like receptors [72], leads to exacerbated neuroinflammatory conditions in mature oligodendrocytes [73]. Moreover, recent transcriptomic data allowed the identification of populations of disease-associated mature oligodendrocytes enriched in immune-related and antigen presentation genes (e.g., major histocompatibility complex -MHC- class II) in the brain of mouse models and Alzheimer’s Disease and Multiple Sclerosis patients [54,74,75]. Together, this evidence suggests that upon injury oligodendroglia can actively participate in fuelling inflammation and might have a role in disease development and antigen presentation.
5. Therapeutic Approaches and Targets for Promoting Remyelination and Lesion Resolution
In the field of neurodegenerative disorders characterized by glial scar formation and demyelination, the main goal of treatment is to recover damaged motor and sensory axons, promote remyelination, and heal glial scar. More recently, remyelination is increasingly emerging as an important strategy to protect axons from degeneration after demyelinating injuries. In the last decade, three main approaches have been proposed for improving remyelination: cellular transplantation, enhancement of cell transdifferentiation toward the oligodendroglial fate, and promotion of OPC differentiation by fostering pro-myelinating mechanisms and/or inhibiting pathologically altered mechanisms.
5.1. Cell Replacement Strategies
Cell transplantation emerged as one of the most effective strategies, with the ability to potentially restore functionality and neuronal connections by replacing damaged cells. Grafted cells could provide trophic support for neurons and manipulate the environment within the lesion to facilitate axonal regeneration, promote plasticity and foster endogenous remyelination (Figure 2A) [76]. Sources for cell transplantation are typically multipotent stem cells, including embryonic stem cells (ESCs), neural stem cells (NSCs), neural precursor cells (NPCs), induced pluripotent stem cells (iPSCs), and mesenchymal stem cells (MSCs) [77].
In the context of SCI, several studies have highlighted the relevance of remyelination by graft-derived cells for neurobehavioral recovery after transplantation [78,79]. Following these findings, Nori and colleagues have developed a novel method to directly generate reprogrammed NPCs committed toward the oligodendrogenic fate (oNPCs) [80], to use them in an integrated strategy employing a cross-linked methylcellulose (XMC) hydrogel containing chondroitinase ABC (ChABC), the enzyme responsible for the degradation of the sulphated glycosaminoglycan chains on the CSPGs [81]. Indeed, previous studies have reported therapeutic efficacy for NPCs transplantation in the subacute period, but effective therapies to target the chronic phase of SCI are greatly lacking, due to the unfavourable microenvironment of the chronically injured spinal cord [82,83]. oNPC transplantation in combination with intrathecal injection of XMC-ChABC increased the survival rate of the grafted cells 12 weeks post-treatment. Grafted cells mainly differentiated into mature CC1-positive oligodendrocytes, contributed to the formation of mature myelin sheaths on spared axons and promoted the formation of nodes of Ranvier. More importantly, XMC-ChABC and oNPC combinatorial therapy improved motor function after chronic SCI, compared to the other groups [80], representing a clinically promising option to regenerate the chronically injured spinal cord.
The advent of gene therapy allowed the delivery of stem cells expressing therapeutic genes, to increase the viability of damaged cells and promote regeneration, while also limiting inflammation, demyelination, and astrogliosis. In this context, Park and colleagues recently tested to possibility to increase functional recovery from SCI by transplantation of NPCs overexpressing human arginine decarboxylase (hADC) [77], the enzyme that synthesizes agmatine [84], an endogenous primary amine with neuroprotective effects [85]. Six weeks after SCI, hADC-NPCs transplantation improved neurological outcomes, induced oligodendrocyte differentiation and remyelination, increased neural lineage differentiation, and decreased glial scar formation [77]. Moreover, locomotor and bladder function were both recovered, suggesting that hADC overexpression in NPCs is a potential therapeutic approach for SCI treatment.
NPC-based therapies may represent a valuable option also for the treatment of progressive MS (PMS). In the EAE model, transplanted NPCs showed pathotropic properties, migrating to demyelinating areas and inducing a rescue of the functional impairment in transplanted rodents [86]. Within these areas, OPCs markedly proliferated and differentiated, with many of them being of donor origin and actively remyelinating axons. Furthermore, a significant reduction of astrogliosis and a marked decrease in both extent of demyelination and axonal loss were observed in transplanted animals. NPC transplantation ameliorated EAE in a non-human primate model as well [87]. Very recently, results from a phase I clinical trial, involving 12 severely disabled people with PMS who had already received the available therapies with little or no success, have demonstrated the safety and tolerability of NPC transplantation in humans [88]. This study has also shown a significant reduction in the loss of cerebral tissue, evaluated by magnetic resonance imaging during the subsequent two years, and an amelioration of the CSF profile in the patients who received the higher dose of NPCs [88]. Further clinical studies in a larger cohort of patients are necessary to establish whether NPC transplantation might have a long-lasting bystander effect in PMS patients.
Beyond NPCs and NSCs, oligodendrocytes have been considered suitable therapeutic targets for cell replacement therapy in neurodegenerative disease. In this respect, a recent study evaluated the contribution of the inhibitory microenvironment combining glial scar ablation (GSA) with the transplantation of iPSC-derived ventral spinal neural progenitor (pre-OPCs) [89], a cell population that arise from the motor neuron progenitor domain in the developing neural tube able to generate both motor and interneurons and subsequently OPCs [90]. In SCI, the magnitude of the lesion cavity was significantly decreased when GSA was performed after pre-OPCs injection when compared to GSA-induced-only and pre-OPCs-injected-only groups. Grafted cells survived and differentiated into astrocytes, oligodendrocytes and neurons when transplanted into chronically injured rats. The local environment created by GSA addressed the transplanted cells towards oligodendrocyte lineage differentiation, at the expense of ventrally located V2a interneurons [89]. Altogether, these observations highlight the importance of the crosstalk between transplanted cells and local cues from the host environment to drive the successful integration and differentiation of xenografts.
5.2. Limiting Astrogliosis to Favour Remyelination
Acutely, astrogliosis does limit the spread of inflammation, but, later, it creates a physical and chemical barrier to the regeneration and axon remyelination. Despite many therapies for CNS injuries have sought to remove this barrier by blocking astrogliosis, several studies suggested that, in certain contexts, blocking reactive astrocyte formation impedes functional recovery [91]. Therefore, rather than halting astrogliosis entirely, alternative therapies could target specific factors released by reactive astrocytes (e.g., CSPGs) that inhibit axon regeneration and remyelination (Figure 2B). Astrocytes in the white matter of MS lesions associate with CSPGs, and remyelination tends to be more robust in areas with less reactive astrocytes, lower levels of CSPGs, and a higher number of OPCs. The mechanisms by which CSPGs build a hostile environment for recovery from glial scarring are still unclear. CPSGs can bind and activate different receptors, such as the protein tyrosine phosphatase-sigma receptor family (PTPR) [92], mostly known to regulate synapse formation. When activated by CSPGs, it activates the RhoA-Rock signaling which has been associated to impaired oligodendrocyte maturation and myelination by inhibiting the extension and branching of oligodendrocyte processes [93]. In vitro silencing of PTPRσ reversed the inhibitory effect of CSPGs on oligodendrocyte process outgrowth and myelination. A similar effect on oligodendrocyte process outgrowth and myelination was observed using Y-27632, an inhibitor of ROCK [92]. Inhibiting this pathway using the intracellular sigma peptide was also shown to reduce gliosis and demyelination in the LPC model, and to improve oligodendrocyte remyelination in the optic nerve after LPC treatment [94]. Another recent study in a mouse model of MS [95] described a pharmacological strategy aimed at reducing CSPG deposition into the lesions by administering UCM03025, a new potent selective and reversible inhibitor of monoacylglycerol lipase (MagL), the enzyme that accounts for 2-arachidonoylglycerol (2-AG) degradation. 2-AG accumulation reduced microglia activation, CSPGs levels and astrogliosis around demyelinated lesions in the spinal cord of Theiler’s murine encephalomyelitis virus-infected mice. Moreover, inhibition of 2-AG hydrolysis increased the number of mature oligodendrocytes, leading to remyelination and functional recovery. These data suggest that targeting MAGL represents a promising therapeutic strategy for the treatment of progressive phases of chronic demyelinating diseases and of other pathologies involving white matter injury.
Accumulating evidence show that, in pathological tissues, reactive astrocytes have stem cell properties and multiple differentiation potentials [96], suggesting that some extracellular signals in the pathological microenvironment are involved in the cell fate switching. Thus, directly reprogramming or converting astrocytes to neurons and oligodendrocytes has been investigated as a potential therapeutic strategy that aims at attenuating astrogliosis and facilitating brain and spinal cord remyelination (Figure 2B). A recent study showed that reactive astrocytes can be address toward the oligodendroglial lineage by addition of Neuregulin1 (Nrg1), an extracellular axon-localized factor able to promote oligodendrogenesis and remyelination [97]. Cultured reactive astrocytes treated with Nrg1 generated oligodendrocyte-like cells characterized by expressing specific oligodendroglial markers (i.e, PDGFR-α and O4). In addition, the number of cells co-expressing PDGFR-α or O4 or CNPase and GFAP was significantly increased in the injured spinal cord treated with Nrg1 for 2-4 weeks, compared to vehicle treated mice [98]. More importantly, in the Nrg1-treated group, demyelination was significantly lower compared to the controls, suggesting that Nrg1-promoted astrocyte-to-oligodendrocyte conversion may contribute to enhanced remyelination after SCI [98]. Further characterization of the extracellular factors in the lesion microenvironment will not only help to reveal the molecular mechanism underlying the astrocyte-to-oligodendrocyte fate change, but also aid to identify therapeutic targets for improving the efficiency of lineage conversion.
Conversion of reactive astrocytes to other cell types can also be achieved by somatic reprogramming techniques. A recent study has shown that SCI elicits neurogenic potential of OPCs and that SOX2 is sufficient for neurogenic reprogramming of these cells, which can eventually lead to functional neurogenesis in non-neurogenic adult mouse spinal cord [99]. Although this injury-induced phenotypic reprogramming was found to be transient and incomplete for producing mature neurons, the injection of SOX2-producing viral particles into the surrounding lesion area increased neurogenesis, reduced glial scar volume, and ultimately contributed to functional improvements after SCI [99].
Among the different transcription factors involved in OPC specification and differentiation, SOX10 is one of the most investigated and widely used to produce inducible oligodendrocytes from fibroblasts, iPSCs and NSCs [100,101,102]. SOX10 is also involved in the transdifferentiation of astrocytes to oligodendrocytes occurring in SCI and this conversion can be enhanced by EGF signalling activation [103]. Continuous delivery of EGF into injury area after SCI significantly increased the percentage of SOX10-induced oligodendrocytes, compared to untreated mice, whereas EGFR inhibitor Gefitinib counteracted the synergistic effect of EGF and SOX10. Similarly, Gefitinib reduced the transdifferentiation in the SOX10 only infected tissues as well, suggesting that, in the pathological milieu, EGF participates in the astrocyte-to-oligodendrocyte reprogramming [103].
5.3. Promoting OPC Differentiation
Remyelination is controlled by both intrinsic and extrinsic factors that act either as inhibitors or promoters of OPC differentiation [106,107]. As discussed above, incomplete remyelination reported in MS and in other neurodegenerative disorders, is partly due to the failure of OPCs to differentiate into myelinating oligodendrocytes, rather than to a reduction in the pool of OPCs. Therefore, new therapeutic agents promoting OPC differentiation and remyelination are urgently needed [108]. Molecules able to promote remyelination can be classified based on their nature [109] in small molecules targeting physiological or pathologically altered receptors (e.g., GPCRs, intracellular receptors), endogenously occurring free molecules (e.g., microRNAs, metabolite, peptides), and non-physiologically occurring free molecules (e.g., synthetic compounds, plant-derived compounds) (Figure 2C).

Table 2.
Summary of the studies targeting astrocytes to improve remyelination.
| Pathology | Drug | Mechanism of action | Effects | Reference |
|---|---|---|---|---|
| MS (LPC) | Intracellular sigma peptide | Inhibition of protein tyrosine phosphatase sigma (PTPσ) | Decreased demyelination and astrogliosis. Increased remyelination and functional recovery in optic pathway |
[110] |
| MS (TMEV) | UCM03025 | Inhibition of monoacylglycerol lipase (MagL) | Reduction of microglia activation, astrogliosis and CSPGs levels around demyelinated lesions. Improvement in remyelination and motor functions. |
[95] |
| SCI | Neuregulin1 | Astrocyte reprogramming | Reduction of astrogliosis. Enhancement of remyelination, axonal preservation, locomotor recovery |
[98] |
| SCI | SOX2 | Astrocyte reprogramming | Reduction in glial scar volume. Increased neurogenesis and improvement in motor functions. |
[99] |
| SCI | SOX10 | Astrocyte reprogramming | Increased generation of oligodendrocytes through EGF signalling activation | [103] |
Among the different GPCRs expressed by oligodendrocytes during their differentiation, GPR17 and GPR37 are known for their role in preventing excessive or premature myelination via a cAMP-dependent mechanism. GPR17, whose expression starts in early OPCs, reaches its maximal expression in pre-oligodendrocytes, and must be downregulated to allow terminal maturation [111,112]. Previous studies have shown an increased number of GPR17-expressing cells blocked at immature stages in several models of neurodegenerative diseases [113,114] and human brain pathological specimens [115]. Accordingly, GPR17 conditional KO in oligodendrocytes induced untimely oligodendrocyte maturation, while its overexpression inhibited oligodendrocyte development and myelination. GPR17 has been explored as a potential therapeutic target in remyelination either by inhibiting GPR17 via antagonists or by activating the reserve pool of GPR17-expressing OPCs to promote their maturation [116].
Instead, GPR37 starts to be expressed later, in pre-oligodendrocytes, and its expression is maintained in more mature stages. Like GPR17, GPR37 stimulation reduces cAMP levels, while its inhibition increases cAMP levels and the Epac/Raf/ERK downstream signalling. A recent study found that osteocalcin, a hormone synthesized in osteoblasts, can cross the BBB and interact with GPR37 to inhibit the transition between pre-oligodendrocytes and myelinating oligodendrocytes [117]. GPR37L1 shares structural and functional similarities with GPR37 and plays an important role in brain health and remyelination. Both the receptors are activated by prosaposin [118], a precursor of saposins, which is involved in the metabolism of sphingolipids, a fundamental building block of the myelin sheath. Prosaposin (PSAP) and prosaptides (peptides mimicking the neurotrophic region in prosaposin) have been shown to protect oligodendrocyte cell line and to stimulate myelin lipid synthesis [119,120]. Interestingly, a recent spatial transcriptomic analysis of neurodegeneration in PMS indicated that the interaction between neuronal PSAP and oligodendroglial GPR37L1 is reduced in disease condition and highlighted this receptor as potential novel drug target for progressive MS [121]. Further studies are necessary to clarify the exact role of GPR37/GPR37L1 in remyelination.
Another receptor, called GPR149, recently found highly expressed in OPCs and downregulated during oligodendrocyte differentiation, acts as a negative regulator of myelination and remyelination in the CNS [122]. Despite the signal transduction mechanism and endogenous ligands of GPR149 are still unknown, its inhibitory action on oligodendrocyte differentiation is likely mediated by the activation of ERK1/2 pathway. Similarly to GPR17, genetic deletion of GPR149 in mice resulted in transiently precocious differentiation of oligodendrocytes, enhanced myelination during development and myelin regeneration following cuprizone-induced demyelination [122], suggesting that blocking GPR149 might be an intriguing way to promote myelin repair in demyelinating diseases.
Repurposing approaches, allowed the identification of clobetasol and miconazole, marketed drugs with a history of safe use, as promising remyelinating molecules able to markedly improve motor function, reduce demyelination and improved MBP expression in the spinal cord, when daily-administered in EAE mice starting from the peak of disease [123]. A following repurposing study focused on drugs acting on the nucleocytoplasmic shuttling of the p57kip2 protein, an early cellular process involved in OPC differentiation decision [124]. After evaluation in human primary OPCs, and in organotypic cerebellar slice cultures, the steroid danazol and the anthelmintic parbendazole were selected for in vivo evaluation [125]. Of these, parbendazole increased the number of mature oligodendrocytes and enhanced spontaneous remyelination following cuprizone-induced demyelination, providing a new pharmacological compound with the potential to boost myelin repair processes. Other FDA-approved drugs that showed promising results in demyelinating models are montelukast and clemastine. Montelukast is a CysLT1R and GPR17 antagonist approved as adjuvant therapy for asthma in both children and adults. Montelukast neuroprotective and anti-inflammatory effects have been proven in different models of neurodegenerative diseases, including SCI, MS, stroke, amyotrophic lateral sclerosis, and Huntington’s [126,127,128,129,130]. Clemastine is a first-generation H1 histamine antagonist with anticholinergic properties that has been shown to be effective in promoting myelination in hypoxic mouse brains demyelinating and SCI models. A phase 2a clinical trial in patients with relapsing remitting MS is now evaluating the beneficial effects of clemastine in combination with metformin, a drug which is already used to treat type 2 diabetes that showed remyelinating properties [131,132,133].
In the last decade, also the role of microRNAs in oligodendrocyte development and the consequences of their dysregulation in these cells have been extensively investigated, thus opening the way to exploit miRNA mimics or antago-miR as pharmacological targets [134,135]. Among the numerous miRNAs able to directly influence oligodendrocyte development, miR-219 and miR-338 are the most characterized. These oligodendrocyte-specific miRNAs promote oligodendrocyte differentiation by repressing negative regulators of oligodendrocyte differentiation [136,137]. Administration of exogenous miR-219 and miR-338 promoted myelin regeneration in models of demyelinating diseases [138,139,140,141]. In this context, a recent study evaluated the effects of miR-219/miR-338 delivery on microglia and astrocyte behaviour showing that their overexpression diminished microglial expression of pro-inflammatory cytokines and suppressed astrocyte activation [142]. These results suggest that miR-219/miR-338 CNS delivery represents a promising treatment for axonal remyelination following nerve injuries.
miRNAs can act as inhibitors of oligodendrocyte differentiation. Over-expression of miR-125a-3p in cultured OPCs impaired oligodendrocyte maturation, while administration of an antago-mir promoted maturation through different mechanisms [143]. Expression levels of this miRNA have been found increased following demyelinating injuries, in MS animal models and brain specimens, suggesting its possible involvement in disease pathogenesis. Accordingly, in vivo silencing of miR-125a-3p following lysolecithin-induced demyelination promoted myelin repair [144]. Due to their unique interacting features miRNAs can act at different stages of a process, on different targets and leading to different outcomes. This implies that potential off-target effects can occur in miRNA-based therapy. It is the case of miR-146a, a miRNA that recently showed therapeutic potential for remyelination. Several studies demonstrated that miR-146a was able to increase oligodendrocyte differentiation and remyelination in animal models [145,146,147]. However, further studies indicated that miR-146a-deficient mice demonstrated increased oligodendrocyte numbers and reduced inflammation, demyelination, and axonal loss following cuprizone-induced demyelination [148]. This example highlights the importance of designing stage-specific and cell-specific therapeutic strategies based on miRNA mimics/inhibitors to prevent unwanted off-target effects.
6. Conclusions
Due to the cellular heterogeneity and to the complex dynamics of the lesions in time and space, therapeutic intervention to treat glial scar or scar-associated neuroinflammation still remains a challenge. It is clear that glial scar formation is part of a regenerative attempt and should not be completely blocked. Instead, the inhibitory factors that progressively establish a hostile environment should be dissected to identify more focused therapies. The roles of oligodendrocytes and OPCs have been overlooked in glial scar. Early after injury, oligodendrocyte apoptosis leads to demyelination, whereas OPCs react and migrate toward the damage to replenish and repair. Acutely, they form synapse-like connections with the tips of transected axons and produce ECM components that protect axon growth cones from the neuroinflammatory milieu. However, in the long term, they overexpress matrix metalloproteinases (MMPs) and CSPGs, contributing to the production of aberrant unfavourable ECM components and to the generation of a hostile environment, which in turn impairs long-term axonal regeneration and inhibit OPC differentiation and remyelination. The production of high amounts of CSPGs within the lesions is associated with chemorepulsion and pathological behaviour of OPCs. NG2 is one of the major CSPGs expressed on the surface of early OPCs, whose accumulation at the lesioned areas could take part to a vicious cycle, impeding their own differentiation. Accordingly, depletion of CSPGs was shown to successfully restore OPC differentiation and remyelination in several models of disease.
Fostering remyelination is an unmet need in the context of demyelinating diseases, such as MS. However, since oligodendrocyte dysregulation and demyelination are pathological features of several neurodegenerative diseases or acute conditions, and often they take place in the early phases of neurodegeneration, it is possible to speculate that remyelination would be a potential strategy also in other disorders, trauma, and brain ischemia. It is worth observing that the generation of a favourable microenvironment is a key step to pave the way to remyelination. Accordingly, cell transplantation approaches have shown promising results when coupled to other interventions aimed at reestablishing a favourable microenvironment, targeting CSPG deposition and degradation. These results may explain why previously reported pro-remyelinating compounds have shown poor efficacy in the presence of an unfavourable CSPG-rich environment, suggesting that also these therapeutic approaches should be complemented with other compounds targeting the hostile microenvironment.
Author Contributions
All the authors contributed to conception and writing of the present review. JHCS prepared the figures. DL combined the contributions and revised the manuscript. All authors read, revised, and approved the submitted version.
Funding
The research was supported by Italian Ministry of University and Research (MUR); PRIN—Progetti di Ricerca di Interesse Nazionale (n.2022AJ3FHA); and European Union—NextGenerationEU (PNRR M4C2-Investimento 1.4- CN00000041). The authors acknowledge support from the University of Milan through the APC initiative. EB’s research is supported by the Italian Ministry of Research (PRIN - PNRR 2022 ID: P20225Z3J5, PRIN2022 ID: 20224YJBBP and “Dipartimenti di Eccellenza 2023–2027” to Dept. of Neuroscience “Rita Levi Montalcini”) and by the Italian Telethon Foundation (ID: GMR22T1066).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- O’Shea, T.M.; Burda, J.E.; Sofroniew, M. V. Cell biology of spinal cord injury and repair. J. Clin. Invest. 2017, 127, 3259–3270. [Google Scholar] [CrossRef] [PubMed]
- Adams, K.L.; Gallo, V. The diversity and disparity of the glial scar. Nat. Neurosci. 2018, 21, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Beck, K.D.; Nguyen, H.X.; Galvan, M.D.; Salazar, D.L.; Woodruff, T.M.; Anderson, A.J. Quantitative analysis of cellular inflammation after traumatic spinal cord injury: Evidence for a multiphasic inflammatory response in the acute to chronic environment. Brain 2010, 133, 433–447. [Google Scholar] [CrossRef] [PubMed]
- Dias, D.O.; Kalkitsas, J.; Kelahmetoglu, Y.; Estrada, C.P.; Tatarishvili, J.; Holl, D.; Jansson, L.; Banitalebi, S.; Amiry-Moghaddam, M.; Ernst, A.; et al. Pericyte-derived fibrotic scarring is conserved across diverse central nervous system lesions. Nat. Commun. 2021, 12, 5501. [Google Scholar] [CrossRef] [PubMed]
- Bellver-Landete, V.; Bretheau, F.; Mailhot, B.; Vallières, N.; Lessard, M.; Janelle, M.E.; Vernoux, N.; Tremblay, M.È.; Fuehrmann, T.; Shoichet, M.S.; et al. Microglia are an essential component of the neuroprotective scar that forms after spinal cord injury. Nat. Commun. 2019, 10, 518. [Google Scholar] [CrossRef] [PubMed]
- Shafqat, A.; Albalkhi, I.; Magableh, H.M.; Saleh, T.; Alkattan, K.; Yaqinuddin, A. Tackling the glial scar in spinal cord regeneration: new discoveries and future directions. Front. Cell. Neurosci. 2023, 17. [Google Scholar] [CrossRef]
- Brennan, F.H.; Li, Y.; Wang, C.; Ma, A.; Guo, Q.; Li, Y.; Pukos, N.; Campbell, W.A.; Witcher, K.G.; Guan, Z.; et al. Microglia coordinate cellular interactions during spinal cord repair in mice. Nat. Commun. 2022, 13, 4096. [Google Scholar] [CrossRef]
- Raffaele, S.; Fumagalli, M. Dynamics of Microglia Activation in the Ischemic Brain: Implications for Myelin Repair and Functional Recovery. Front. Cell. Neurosci. 2022, 16. [Google Scholar] [CrossRef]
- Kuhn, S.; Gritti, L.; Crooks, D.; Dombrowski, Y. Oligodendrocytes in Development, Myelin Generation and Beyond. Cells 2019, 8, 1424. [Google Scholar] [CrossRef]
- Montani, L. Lipids in regulating oligodendrocyte structure and function. Semin. Cell Dev. Biol. 2021, 112, 114–122. [Google Scholar] [CrossRef]
- Saher, G.; Brügger, B.; Lappe-Siefke, C.; Möbius, W.; Tozawa, R.; Wehr, M.C.; Wieland, F.; Ishibashi, S.; Nave, K.-A. High cholesterol level is essential for myelin membrane growth. Nat. Neurosci. 2005, 8, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Saab, A.S.; Nave, K.A. Myelin dynamics: protecting and shaping neuronal functions. Curr. Opin. Neurobiol. 2017, 47, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Morrison, B.M.; Li, Y.; Lengacher, S.; Farah, M.H.; Hoffman, P.N.; Liu, Y.; Tsingalia, A.; Jin, L.; Zhang, P.W.; et al. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 2012, 487, 443–448. [Google Scholar] [CrossRef]
- Papanikolaou, M.; Lewis, A.; Butt, A.M. Glial and neuronal expression of the Inward Rectifying Potassium Channel Kir7.1 in the adult mouse brain. J. Anat. 2019, 235, 984–996. [Google Scholar] [CrossRef]
- Duncan, G.J.; Simkins, T.J.; Emery, B. Neuron-Oligodendrocyte Interactions in the Structure and Integrity of Axons. Front. Cell Dev. Biol. 2021, 9. [Google Scholar] [CrossRef]
- Menichella, D.M.; Majdan, M.; Awatramani, R.; Goodenough, D.A.; Sirkowski, E.; Scherer, S.S.; Paul, D.L. Genetic and Physiological Evidence That Oligodendrocyte Gap Junctions Contribute to Spatial Buffering of Potassium Released during Neuronal Activity. J. Neurosci. 2006, 26, 10984. [Google Scholar] [CrossRef] [PubMed]
- Krämer-Albers, E.M.; Werner, H.B. Mechanisms of axonal support by oligodendrocyte-derived extracellular vesicles. Nat. Rev. Neurosci. 2023, 24, 474–486. [Google Scholar] [CrossRef] [PubMed]
- Kassmann, C.M. Myelin peroxisomes - Essential organelles for the maintenance of white matter in the nervous system. Biochimie 2014, 98, 111–118. [Google Scholar] [CrossRef]
- Fletcher, J.L.; Makowiecki, K.; Cullen, C.L.; Young, K.M. Oligodendrogenesis and myelination regulate cortical development, plasticity and circuit function. Semin. Cell Dev. Biol. 2021, 118, 14–23. [Google Scholar] [CrossRef]
- Káradóttir, R.; Attwell, D. Neurotransmitter receptors in the life and death of oligodendrocytes. Neuroscience 2007, 145, 1426–1438. [Google Scholar] [CrossRef]
- Gudz, T.I.; Komuro, H.; Macklin, W.B. Glutamate Stimulates Oligodendrocyte Progenitor Migration Mediated via anαv Integrin/Myelin Proteolipid Protein Complex. J. Neurosci. 2006, 26, 2458. [Google Scholar] [CrossRef]
- Fumagalli, M.; Lecca, D.; Coppolino, G.T.; Parravicini, C.; Abbracchio, M.P. Pharmacological properties and biological functions of the GPR17 receptor, a potential target for neuro-regenerative medicine. Adv. Exp. Med. Biol. 2017, 1051, 169–192. [Google Scholar] [CrossRef]
- Marques, S.; Zeisel, A.; Codeluppi, S.; van Bruggen, D.; Mendanha Falcão, A.; Xiao, L.; Li, H.; Häring, M.; Hochgerner, H.; Romanov, R.A.; et al. Oligodendrocyte heterogeneity in the mouse juvenile and adult central nervous system. Science (80). 2016, 352, 1326–1329. [Google Scholar] [CrossRef]
- Viganò, F.; Möbius, W.; Götz, M.; Dimou, L. Transplantation reveals regional differences in oligodendrocyte differentiation in the adult brain. Nat. Neurosci. 2013, 16, 1370–1372. [Google Scholar] [CrossRef]
- Moura, D.M.S.; Brennan, E.J.; Brock, R.; Cocas, L.A. Neuron to Oligodendrocyte Precursor Cell Synapses: Protagonists in Oligodendrocyte Development and Myelination, and Targets for Therapeutics. Front. Neurosci. 2022, 15. [Google Scholar] [CrossRef]
- Habermacher, C.; Angulo, M.C.; Benamer, N. Glutamate versus GABA in neuron–oligodendroglia communication. Glia 2019, 67, 2092–2106. [Google Scholar] [CrossRef]
- Auguste, Y.S.S.; Ferro, A.; Kahng, J.A.; Xavier, A.M.; Dixon, J.R.; Vrudhula, U.; Nichitiu, A.-S.; Rosado, D.; Wee, T.-L.; Pedmale, U. V.; et al. Publisher Correction: Oligodendrocyte precursor cells engulf synapses during circuit remodeling in mice. Nat. Neurosci. 2022, 25, 1735–1735. [Google Scholar] [CrossRef]
- Lecca, D.; Baron, W.; Butt, A.M. Editorial: Cellular and molecular factors that drive the behavior of oligodendrocyte progenitor cells in physiological conditions and disease. Front. Cell. Neurosci. 2023, 17, 1145627. [Google Scholar] [CrossRef]
- Pukos, N.; Goodus, M.T.; Sahinkaya, F.R.; McTigue, D.M. Myelin status and oligodendrocyte lineage cells over time after spinal cord injury: What do we know and what still needs to be unwrapped? Glia 2019, 67, 2178–2202. [Google Scholar] [CrossRef]
- Pukos, N.; Marion, C.M.; Arnold, W.D.; Noble, B.T.; Popovich, P.G.; McTigue, D.M. Chronic demyelination and myelin repair after spinal cord injury in mice: A potential link for glutamatergic axon activity. Glia 2023, 71, 2096–2116. [Google Scholar] [CrossRef]
- Bonfanti, E.; Gelosa, P.; Fumagalli, M.; Dimou, L.; Viganò, F.; Tremoli, E.; Cimino, M.; Sironi, L.; Abbracchio, M.P. The role of oligodendrocyte precursor cells expressing the GPR17 receptor in brain remodeling after stroke. Cell Death Dis. 2017, 8, e2871–e2871. [Google Scholar] [CrossRef]
- Tripathi, R.; McTigue, D.M. Prominent oligodendrocyte genesis along the border of spinal contusion lesions. Glia 2007, 55, 698–711. [Google Scholar] [CrossRef]
- Butt, A.M.; Rivera, A.D.; Fulton, D.; Azim, K. Targeting the Subventricular Zone to Promote Myelin Repair in the Aging Brain. Cells 2022, 11, 1809. [Google Scholar] [CrossRef] [PubMed]
- Meletis, K.; Barnabé-Heider, F.; Carlén, M.; Evergren, E.; Tomilin, N.; Shupliakov, O.; Frisén, J. Spinal cord injury reveals multilineage differentiation of ependymal cells. PLoS Biol. 2008, 6, 1494–1507. [Google Scholar] [CrossRef] [PubMed]
- Llorens-Bobadilla, E.; Chell, J.M.; Le Merre, P.; Wu, Y.; Zamboni, M.; Bergenstråhle, J.; Stenudd, M.; Sopova, E.; Lundeberg, J.; Shupliakov, O.; et al. A latent lineage potential in resident neural stem cells enables spinal cord repair. Science (80-). 2020, 370. [Google Scholar] [CrossRef] [PubMed]
- Hughes, E.G.; Kang, S.H.; Fukaya, M.; Bergles, D.E. Oligodendrocyte progenitors balance growth with self-repulsion to achieve homeostasis in the adult brain. Nat. Neurosci. 2013, 16, 668–676. [Google Scholar] [CrossRef]
- Barber, H.M.; Ali, M.F.; Kucenas, S. Glial Patchwork: Oligodendrocyte Progenitor Cells and Astrocytes Blanket the Central Nervous System. Front. Cell. Neurosci. 2022, 15. [Google Scholar] [CrossRef]
- Sahel, A.; Ortiz, F.C.; Kerninon, C.; Maldonado, P.P.; Angulo, M.C.; Nait-Oumesmar, B. Alteration of synaptic connectivity of oligodendrocyte precursor cells following demyelination. Front. Cell. Neurosci. 2015, 9, 77. [Google Scholar] [CrossRef]
- Hesp, Z.C.; Yoseph, R.Y.; Suzuki, R.; Jukkola, P.; Wilson, C.; Nishiyama, A.; McTigue, D.M. Proliferating NG2-Cell-Dependent Angiogenesis and Scar Formation Alter Axon Growth and Functional Recovery After Spinal Cord Injury in Mice. J. Neurosci. 2018, 38, 1366–1382. [Google Scholar] [CrossRef]
- Dorrier, C.E.; Aran, D.; Haenelt, E.A.; Sheehy, R.N.; Hoi, K.K.; Pintarić, L.; Chen, Y.; Lizama, C.O.; Cautivo, K.M.; Weiner, G.A.; et al. CNS fibroblasts form a fibrotic scar in response to immune cell infiltration. Nat. Neurosci. 2021 242 2021, 24, 234–244. [Google Scholar] [CrossRef]
- Tran, A.P.; Warren, P.M.; Silver, J. The Biology of Regeneration Failure and Success After Spinal Cord Injury. Physiol. Rev. 2018, 98, 881–917. [Google Scholar] [CrossRef]
- Jiang, D.; Liang, J.; Noble, P.W. Hyaluronan in tissue injury and repair. Annu. Rev. Cell Dev. Biol. 2007, 23, 435–461. [Google Scholar] [CrossRef]
- Quraishe, S.; Forbes, L.H.; Andrews, M.R. The Extracellular Environment of the CNS: Influence on Plasticity, Sprouting, and Axonal Regeneration after Spinal Cord Injury. Neural Plast. 2018, 2018, 1–18. [Google Scholar] [CrossRef]
- Boda, E.; Viganò, F.; Rosa, P.; Fumagalli, M.; Labat-Gest, V.; Tempia, F.; Abbracchio, M.P.; Dimou, L.; Buffo, A. The GPR17 receptor in NG2 expressing cells: Focus on in vivocell maturation and participation in acute trauma and chronic damage. Glia 2011, 59, 1958–1973. [Google Scholar] [CrossRef]
- Lau, L.W.; Keough, M.B.; Haylock-Jacobs, S.; Cua, R.; Döring, A.; Sloka, S.; Stirling, D.P.; Rivest, S.; Yong, V.W. Chondroitin sulfate proteoglycans in demyelinated lesions impair remyelination. Ann. Neurol. 2012, 72, 419–432. [Google Scholar] [CrossRef]
- Siebert, J.R.; Stelzner, D.J.; Osterhout, D.J. Chondroitinase treatment following spinal contusion injury increases migration of oligodendrocyte progenitor cells. Exp. Neurol. 2011, 231, 19–29. [Google Scholar] [CrossRef]
- Siebert, J.R.; Osterhout, D.J. The inhibitory effects of chondroitin sulfate proteoglycans on oligodendrocytes. J. Neurochem. 2011, 119, 176–188. [Google Scholar] [CrossRef]
- Keough, M.B.; Rogers, J.A.; Zhang, P.; Jensen, S.K.; Stephenson, E.L.; Chen, T.; Hurlbert, M.G.; Lau, L.W.; Rawji, K.S.; Plemel, J.R.; et al. An inhibitor of chondroitin sulfate proteoglycan synthesis promotes central nervous system remyelination. Nat. Commun. 2016, 7, 11312. [Google Scholar] [CrossRef]
- Sobel, R.A.; Ahmed, A.S. White Matter Extracellular Matrix Chondroitin Sulfate/Dermatan Sulfate Proteoglycans in Multiple Sclerosis. J. Neuropathol. Exp. Neurol. 2001, 60, 1198–1207. [Google Scholar] [CrossRef] [PubMed]
- Marangon, D.; Caporale, N.; Boccazzi, M.; Abbracchio, M.P.; Testa, G.; Lecca, D. Novel in vitro Experimental Approaches to Study Myelination and Remyelination in the Central Nervous System. Front. Cell. Neurosci. 2021, 15. [Google Scholar] [CrossRef] [PubMed]
- Bonora, M.; De Marchi, E.; Patergnani, S.; Suski, J.M.; Celsi, F.; Bononi, A.; Giorgi, C.; Marchi, S.; Rimessi, A.; Duszyński, J.; et al. Tumor necrosis factor-α impairs oligodendroglial differentiation through a mitochondria-dependent process. Cell Death Differ. 2014, 21, 1198–1208. [Google Scholar] [CrossRef] [PubMed]
- Cannella, B.; Raine, C.S. Multiple Sclerosis: Cytokine Receptors on Oligodendrocytes Predict Innate Regulation. Ann. Neurol. 2004, 55, 46–57. [Google Scholar] [CrossRef]
- Tchélingérian, J.; Monge, M.; Le Saux, F.; Zalc, B.; Jacque, C. Differential Oligodendroglial Expression of the Tumor Necrosis Factor Receptors In Vivo and In Vitro. J. Neurochem. 1995, 65, 2377–2380. [Google Scholar] [CrossRef]
- Falcão, A.M.; van Bruggen, D.; Marques, S.; Meijer, M.; Jäkel, S.; Agirre, E.; Samudyata; Floriddia, E.M.; Vanichkina, D.P.; ffrench-Constant, C.; et al. Disease-specific oligodendrocyte lineage cells arise in multiple sclerosis. Nat. Med. 2019, 24, 1837. [Google Scholar] [CrossRef]
- Zeis, T.; Enz, L.; Schaeren-Wiemers, N. The immunomodulatory oligodendrocyte. Brain Res. 2016, 1641, 139–148. [Google Scholar] [CrossRef]
- Kirdajova, D.; Valihrach, L.; Valny, M.; Kriska, J.; Krocianova, D.; Benesova, S.; Abaffy, P.; Zucha, D.; Klassen, R.; Kolenicova, D.; et al. Transient astrocyte-like NG2 glia subpopulation emerges solely following permanent brain ischemia. Glia 2021, 69, 2658–2681. [Google Scholar] [CrossRef]
- Komitova, M.; Serwanski, D.R.; Richard Lu, Q.; Nishiyama, A. NG2 Cells Are Not a Major Source of Reactive Astrocytes After Neocortical Stab Wound Injury. Glia 2011, 59, 800. [Google Scholar] [CrossRef]
- Hackett, A.R.; Yahn, S.L.; Lyapichev, K.; Dajnoki, A.; Lee, D.H.; Rodriguez, M.; Cammer, N.; Pak, J.; Mehta, S.T.; Bodamer, O.; et al. Injury type-dependent differentiation of NG2 glia into heterogeneous astrocytes. Exp. Neurol. 2018, 308, 72. [Google Scholar] [CrossRef] [PubMed]
- Buffo, A.; Rite, I.; Tripathi, P.; Lepier, A.; Colak, D.; Horn, A.P.; Mori, T.; Götz, M. Origin and progeny of reactive gliosis: A source of multipotent cells in the injured brain. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 3581. [Google Scholar] [CrossRef]
- Bardehle, S.; Krüger, M.; Buggenthin, F.; Schwausch, J.; Ninkovic, J.; Clevers, H.; Snippert, H.J.; Theis, F.J.; Meyer-Luehmann, M.; Bechmann, I.; et al. Live imaging of astrocyte responses to acute injury reveals selective juxtavascular proliferation. Nat. Neurosci. 2013 165 2013, 16, 580–586. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Zhao, N.; Koupourtidou, C.; Fang, L.P.; Schwarz, V.; Caudal, L.C.; Zhao, R.; Hirrlinger, J.; Walz, W.; Bian, S.; et al. In the mouse cortex, oligodendrocytes regain a plastic capacity, transforming into astrocytes after acute injury. Dev. Cell 2023, 58, 1153–1169. [Google Scholar] [CrossRef] [PubMed]
- Duncan, G.J.; Manesh, S.B.; Hilton, B.J.; Assinck, P.; Plemel, J.R.; Tetzlaff, W. The fate and function of oligodendrocyte progenitor cells after traumatic spinal cord injury. Glia 2020, 68, 227–245. [Google Scholar] [CrossRef] [PubMed]
- Assinck, P.; Duncan, G.J.; Plemel, J.R.; Lee, M.J.; Stratton, J.A.; Manesh, S.B.; Liu, J.; Ramer, L.M.; Kang, S.H.; Bergles, D.E.; et al. Myelinogenic Plasticity of Oligodendrocyte Precursor Cells following Spinal Cord Contusion Injury. J. Neurosci. 2017, 37, 8635. [Google Scholar] [CrossRef] [PubMed]
- Hamanaka, G.; Hernández, I.C.; Takase, H.; Ishikawa, H.; Benboujja, F.; Kimura, S.; Fukuda, N.; Guo, S.; Lok, J.; Lo, E.H.; et al. Myelination- and migration-associated genes are downregulated after phagocytosis in cultured oligodendrocyte precursor cells. J. Neurochem. 2023, 167, 571–581. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, J.A.; Elabbady, L.; Collman, F.; Jorstad, N.L.; Bakken, T.E.; Ott, C.; Glatzer, J.; Bleckert, A.A.; Bodor, A.L.; Brittain, D.; et al. Oligodendrocyte precursor cells ingest axons in the mouse neocortex. Proc. Natl. Acad. Sci. U. S. A. 2022, 119. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.H.; Miyamoto, N.; Hayakawa, K.; Pham, L.D.D.; Maki, T.; Ayata, C.; Kim, K.W.; Lo, E.H.; Arai, K. Oligodendrocyte precursors induce early blood-brain barrier opening after white matter injury. J. Clin. Invest. 2013, 123, 782. [Google Scholar] [CrossRef] [PubMed]
- von Streitberg, A.; Jäkel, S.; Eugenin von Bernhardi, J.; Straube, C.; Buggenthin, F.; Marr, C.; Dimou, L. NG2-Glia Transiently Overcome Their Homeostatic Network and Contribute to Wound Closure After Brain Injury. Front. Cell Dev. Biol. 2021, 9. [Google Scholar] [CrossRef]
- Zhang, S.Z.; Wang, Q.Q.; Yang, Q.Q.; Gu, H.Y.; Yin, Y.Q.; Li, Y.D.; Hou, J.C.; Chen, R.; Sun, Q.Q.; Sun, Y.F.; et al. NG2 glia regulate brain innate immunity via TGF-β2/TGFBR2 axis. BMC Med. 2019, 17, 204. [Google Scholar] [CrossRef]
- Rodriguez, E.G.; Wegner, C.; Kreutzfeldt, M.; Neid, K.; Thal, D.R.; Jürgens, T.; Brück, W.; Stadelmann, C.; Merkler, D. Oligodendroglia in cortical multiple sclerosis lesions decrease with disease progression, but regenerate after repeated experimental demyelination. Acta Neuropathol. 2014, 128, 231–246. [Google Scholar] [CrossRef]
- Kirby, L.; Jin, J.; Cardona, J.G.; Smith, M.D.; Martin, K.A.; Wang, J.; Strasburger, H.; Herbst, L.; Alexis, M.; Karnell, J.; et al. Oligodendrocyte precursor cells present antigen and are cytotoxic targets in inflammatory demyelination. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef]
- Boccazzi, M.; Raffaele, S.; Fumagalli, M. Not only myelination: the immune-inflammatory functions of oligodendrocytes. Neural Regen. Res. 2022, 17, 2661. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, T. The Nuclear Factor NF-κB Pathway in Inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1. [Google Scholar] [CrossRef] [PubMed]
- Schlett, J.S.; Mettang, M.; Skaf, A.; Schweizer, P.; Errerd, A.; Mulugeta, E.A.; Hein, T.M.; Tsesmelis, K.; Tsesmelis, M.; Büttner, U.F.G.; et al. NF-κB is a critical mediator of post-mitotic senescence in oligodendrocytes and subsequent white matter loss. Mol. Neurodegener. 2023, 18, 24. [Google Scholar] [CrossRef] [PubMed]
- Kenigsbuch, M.; Bost, P.; Halevi, S.; Chang, Y.; Chen, S.; Ma, Q.; Hajbi, R.; Schwikowski, B.; Bodenmiller, B.; Fu, H.; et al. A shared disease-associated oligodendrocyte signature among multiple CNS pathologies. Nat. Neurosci. 2022, 25, 876. [Google Scholar] [CrossRef]
- Jäkel, S.; Agirre, E.; Mendanha Falcão, A.; van Bruggen, D.; Lee, K.W.; Knuesel, I.; Malhotra, D.; Ffrench-Constant, C.; Williams, A.; Castelo-Branco, G. Altered human oligodendrocyte heterogeneity in multiple sclerosis. Nature 2019, 566, 543–547. [Google Scholar] [CrossRef] [PubMed]
- Assinck, P.; Duncan, G.J.; Hilton, B.J.; Plemel, J.R.; Tetzlaff, W. Cell transplantation therapy for spinal cord injury. Nat. Neurosci. 2017 205 2017, 20, 637–647. [Google Scholar] [CrossRef]
- Park, Y.M.; Kim, J.H.; Lee, J.E. Neural Stem Cells Overexpressing Arginine Decarboxylase Improve Functional Recovery from Spinal Cord Injury in a Mouse Model. Int. J. Mol. Sci. 2022, 23. [Google Scholar] [CrossRef]
- Hawryluk, G.W.J.; Spano, S.; Chew, D.; Wang, S.; Erwin, M.; Chamankhah, M.; Forgione, N.; Fehlings, M.G. An examination of the mechanisms by which neural precursors augment recovery following spinal cord injury: A key role for remyelination. Cell Transplant. 2014, 23, 365–380. [Google Scholar] [CrossRef]
- Nagoshi, N.; Khazaei, M.; Ahlfors, J.E.; Ahuja, C.S.; Nori, S.; Wang, J.; Shibata, S.; Fehlings, M.G. Human Spinal Oligodendrogenic Neural Progenitor Cells Promote Functional Recovery After Spinal Cord Injury by Axonal Remyelination and Tissue Sparing. Stem Cells Transl. Med. 2018, 7, 806–818. [Google Scholar] [CrossRef]
- Nori, S.; Khazaei, M.; Ahuja, C.S.; Yokota, K.; Ahlfors, J.E.; Liu, Y.; Wang, J.; Shibata, S.; Chio, J.; Hettiaratchi, M.H.; et al. Human Oligodendrogenic Neural Progenitor Cells Delivered with Chondroitinase ABC Facilitate Functional Repair of Chronic Spinal Cord Injury. Stem Cell Reports 2018, 11, 1433. [Google Scholar] [CrossRef]
- Takiguchi, M.; Miyashita, K.; Yamazaki, K.; Funakoshi, K. Chondroitinase ABC Administration Facilitates Serotonergic Innervation of Motoneurons in Rats With Complete Spinal Cord Transection. Front. Integr. Neurosci. 2022, 16, 881632. [Google Scholar] [CrossRef]
- Nishimura, S.; Yasuda, A.; Iwai, H.; Takano, M.; Kobayashi, Y.; Nori, S.; Tsuji, O.; Fujiyoshi, K.; Ebise, H.; Toyama, Y.; et al. Time-dependent changes in the microenvironment of injured spinal cord affects the therapeutic potential of neural stem cell transplantation for spinal cord injury. Mol. Brain 2013, 6, 3. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Ahuja, C.S.; Salewski, R.P.; Li, L.; Satkunendrarajah, K.; Nagoshi, N.; Shibata, S.; Fehlings, M.G. Neural stem cell mediated recovery is enhanced by Chondroitinase ABC pretreatment in chronic cervical spinal cord injury. PLoS One 2017, 12, e0182339. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.Y.; Iyo, A.; Piletz, J.E.; Regunathan, S. Expression of human arginine decarboxylase, the biosynthetic enzyme for agmatine. Biochim. Biophys. Acta 2004, 1670, 156. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Gao, L.; Li, T.; Shao, A.; Zhang, J. Neuroprotective Role of Agmatine in Neurological Diseases. Curr. Neuropharmacol. 2018, 16, 1296–1305. [Google Scholar] [CrossRef] [PubMed]
- Pluchino, S.; Quattrini, A.; Brambilla, E.; Gritti, A.; Salani, G.; Dina, G.; Galli, R.; Del Carro, U.; Amadio, S.; Bergami, A.; et al. Injection of adult neurospheres induces recovery in a chronic model of multiple sclerosis. Nature 2003, 422, 688–694. [Google Scholar] [CrossRef] [PubMed]
- Pluchino, S.; Gritti, A.; Blezer, E.; Amadio, S.; Brambilla, E.; Borsellino, G.; Cossetti, C.; Del Carro, U.; Comi, G.; ’T Hart, B.; et al. Human neural stem cells ameliorate autoimmune encephalomyelitis in non-human primates. Ann. Neurol. 2009, 66, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Genchi, A.; Brambilla, E.; Sangalli, F.; Radaelli, M.; Bacigaluppi, M.; Furlan, R.; Andolfo, A.; Drago, D.; Magagnotti, C.; Scotti, G.M.; et al. Neural stem cell transplantation in patients with progressive multiple sclerosis: an open-label, phase 1 study. Nat. Med. 2023, 29, 75–85. [Google Scholar] [CrossRef]
- Patil, N.; Walsh, P.; Carrabre, K.; Holmberg, E.G.; Lavoie, N.; Dutton, J.R.; Parr, A.M. Regionally Specific Human Pre-Oligodendrocyte Progenitor Cells Produce Both Oligodendrocytes and Neurons after Transplantation in a Chronically Injured Spinal Cord Rat Model after Glial Scar Ablation. J. Neurotrauma 2021, 38, 777–788. [Google Scholar] [CrossRef]
- Maire, C.L.; Wegener, A.; Kerninon, C.; Oumesmar, B.N. Gain-of-function of olig transcription factors enhances oligodendrogenesis and myelination. Stem Cells 2010, 28, 1611–1622. [Google Scholar] [CrossRef]
- Pearson, C. A Therapeutic Link between Astrogliosis and Remyelination in a Mouse Model of Multiple Sclerosis. J. Neurosci. 2018, 38, 29. [Google Scholar] [CrossRef] [PubMed]
- Pendleton, J.C.; Shamblott, M.J.; Gary, D.S.; Belegu, V.; Hurtado, A.; Malone, M.L.; McDonald, J.W. Chondroitin sulfate proteoglycans inhibit oligodendrocyte myelination through PTPσ. Exp. Neurol. 2013, 247, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Chen, Z.; Wen, J. The role of RhoA/ROCK pathway in the ischemic stroke-induced neuroinflammation. Biomed. Pharmacother. 2023, 165, 115141. [Google Scholar] [CrossRef] [PubMed]
- Niknam, P.; Raoufy, M.R.; Fathollahi, Y.; Javan, M. Modulating proteoglycan receptor PTPσ using intracellular sigma peptide improves remyelination and functional recovery in mice with demyelinated optic chiasm. Mol. Cell. Neurosci. 2019, 99, 103391. [Google Scholar] [CrossRef] [PubMed]
- Feliú, A.; Del Río, I.B.; Carrillo-Salinas, F.J.; Hernández-Torres, G.; Mestre, L.; Puente, N.; Ortega-Gutiérrez, S.; López-Rodríguez, M.L.; Grandes, P.; Mecha, M.; et al. 2-arachidonoylglycerol reduces proteoglycans and enhances remyelination in a progressive model of demyelination. J. Neurosci. 2017, 37, 8385–8398. [Google Scholar] [CrossRef] [PubMed]
- Shimada, I.S.; LeComte, M.D.; Granger, J.C.; Quinlan, N.J.; Spees, J.L. Self-renewal and differentiation of reactive astrocyte-derived neural stem/progenitor cells isolated from the cortical peri-infarct area after stroke. J. Neurosci. 2012, 32, 7926–7940. [Google Scholar] [CrossRef] [PubMed]
- Kataria, H.; Alizadeh, A.; Shahriary, G.M.; Saboktakin Rizi, S.; Henrie, R.; Santhosh, K.T.; Thliveris, J.A.; Karimi-Abdolrezaee, S. Neuregulin-1 promotes remyelination and fosters a pro-regenerative inflammatory response in focal demyelinating lesions of the spinal cord. Glia 2018, 66, 538–561. [Google Scholar] [CrossRef]
- Ding, Z.; Dai, C.; Zhong, L.; Liu, R.; Gao, W.; Zhang, H.; Yin, Z. Neuregulin-1 converts reactive astrocytes toward oligodendrocyte lineage cells via upregulating the PI3K-AKT-mTOR pathway to repair spinal cord injury. Biomed. Pharmacother. 2021, 134, 111168. [Google Scholar] [CrossRef] [PubMed]
- Tai, W.; Wu, W.; Wang, L.L.; Ni, H.; Chen, C.; Yang, J.; Zang, T.; Zou, Y.; Xu, X.M.; Zhang, C.L. In vivo reprogramming of NG2 glia enables adult neurogenesis and functional recovery following spinal cord injury. Cell Stem Cell 2021, 28, 923. [Google Scholar] [CrossRef]
- Chanoumidou, K.; Hernández-Rodríguez, B.; Windener, F.; Thomas, C.; Stehling, M.; Mozafari, S.; Albrecht, S.; Ottoboni, L.; Antel, J.; Kim, K.P.; et al. One-step Reprogramming of Human Fibroblasts into Oligodendrocyte-like Cells by SOX10, OLIG2, and NKX6.2. Stem Cell Reports 2021, 16, 771–783. [Google Scholar] [CrossRef]
- Langseth, A.J.; Munji, R.N.; Choe, Y.; Huynh, T.; Pozniak, C.D.; Pleasure, S.J. Wnts Influence the Timing and Efficiency of Oligodendrocyte Precursor Cell Generation in the Telencephalon. J. Neurosci. 2010, 30, 13367–13372. [Google Scholar] [CrossRef] [PubMed]
- El Waly, B.; Cayre, M.; Durbec, P. Promoting Myelin Repair through In Vivo Neuroblast Reprogramming. Stem Cell Reports 2018, 10, 1492–1504. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, C.; Li, J.; Xie, L.; Hong, Z.; Zheng, K.; Zhao, X.; Yang, A.; Xu, X.; Tao, H.; et al. EGF signaling promotes the lineage conversion of astrocytes into oligodendrocytes. Mol. Med. 2022, 28, 50. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Chen, W.; Zeng, J.; Tong, W.; Zheng, P. Long noncoding RNA NKILA transferred by astrocyte-derived extracellular vesicles protects against neuronal injury by upregulating NLRX1 through binding to mir-195 in traumatic brain injury. Aging (Albany. NY). 2021, 13, 8127–8145. [Google Scholar] [CrossRef] [PubMed]
- Zare, L.; Baharvand, H.; Javan, M. In vivo conversion of astrocytes to oligodendrocyte lineage cells using chemicals: Targeting gliosis for myelin repair. Regen. Med. 2018, 13, 803–819. [Google Scholar] [CrossRef] [PubMed]
- Zuchero, J.B.; Barres, B.A. Intrinsic and extrinsic control of oligodendrocyte development. Curr. Opin. Neurobiol. 2013, 23, 914. [Google Scholar] [CrossRef] [PubMed]
- Mozafari, S.; Starost, L.; Manot-Saillet, B.; Garcia-Diaz, B.; Xu, Y.K.T.; Roussel, D.; Levy, M.J.F.; Ottoboni, L.; Kim, K.P.; Schöler, H.R.; et al. Multiple sclerosis iPS-derived oligodendroglia conserve their properties to functionally interact with axons and glia in vivo. Sci. Adv. 2020, 6. [Google Scholar] [CrossRef]
- Huntemer-Silveira, A.; Patil, N.; Brickner, M.A.; Parr, A.M. Strategies for Oligodendrocyte and Myelin Repair in Traumatic CNS Injury. Front. Cell. Neurosci. 2021, 14. [Google Scholar] [CrossRef] [PubMed]
- Göttle, P.; Förster, M.; Weyers, V.; Küry, P.; Rejdak, K.; Hartung, H.P.; Kremer, D. An unmet clinical need: Roads to remyelination in MS. Neurol. Res. Pract. 2019, 1, 21. [Google Scholar] [CrossRef]
- Niknam, P.; Raoufy, M.R.; Fathollahi, Y.; Javan, M. Modulating proteoglycan receptor PTPσ using intracellular sigma peptide improves remyelination and functional recovery in mice with demyelinated optic chiasm. Mol. Cell. Neurosci. 2019, 99, 103391. [Google Scholar] [CrossRef]
- Marangon, D.; Audano, M.; Pedretti, S.; Fumagalli, M.; Mitro, N.; Lecca, D.; Caruso, D.; Abbracchio, M.P. Rewiring of Glucose and Lipid Metabolism Induced by G Protein-Coupled Receptor 17 Silencing Enables the Transition of Oligodendrocyte Progenitors to Myelinating Cells. Cells 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Boccazzi, M.; Macchiarulo, G.; Lebon, S.; Janowska, J.; Le Charpentier, T.; Faivre, V.; Hua, J.; Marangon, D.; Lecca, D.; Fumagalli, M.; et al. G protein-coupled receptor 17 is regulated by WNT pathway during oligodendrocyte precursor cell differentiation. Neurobiol. Dis. 2023, 187, 106315. [Google Scholar] [CrossRef] [PubMed]
- Paladini, M.S.; Marangon, D.; Rossetti, A.C.; Guidi, A.; Coppolino, G.T.; Negri, C.; Spero, V.; Abbracchio, M.P.; Lecca, D.; Molteni, R. Prenatal Stress Impairs Spinal Cord Oligodendrocyte Maturation via BDNF Signaling in the Experimental Autoimmune Encephalomyelitis Model of Multiple Sclerosis. Cell. Mol. Neurobiol. 2022, 42, 1225. [Google Scholar] [CrossRef] [PubMed]
- Coppolino, G.T.; Marangon, D.; Negri, C.; Menichetti, G.; Fumagalli, M.; Gelosa, P.; Dimou, L.; Furlan, R.; Lecca, D.; Abbracchio, M.P. Differential local tissue permissiveness influences the final fate of GPR17-expressing oligodendrocyte precursors in two distinct models of demyelination. Glia 2018, 66, 1118–1130. [Google Scholar] [CrossRef] [PubMed]
- Angelini, J.; Marangon, D.; Raffaele, S.; Lecca, D.; Abbracchio, M.P. The Distribution of GPR17-Expressing Cells Correlates with White Matter Inflammation Status in Brain Tissues of Multiple Sclerosis Patients. Int. J. Mol. Sci. 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Parravicini, C.; Lecca, D.; Marangon, D.; Coppolino, G.T.; Daniele, S.; Bonfanti, E.; Fumagalli, M.; Raveglia, L.; Martini, C.; Gianazza, E.; et al. Development of the first in vivo GPR17 ligand through an iterative drug discovery pipeline: A novel disease-modifying strategy for multiple sclerosis. PLoS One 2020, 15, e0231483. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.; Li, H.; Yang, H.; Yang, Q.; Lu, Z.; Wang, L.; Chen, Y.; Li, X. Osteocalcin attenuates oligodendrocyte differentiation and myelination via GPR37 signaling in the mouse brain. Sci. Adv. 2021, 7. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Mosienko, V.; Vaccari Cardoso, B.; Prokudina, D.; Huentelman, M.; Teschemacher, A.G.; Kasparov, S. Glio- and neuro-protection by prosaposin is mediated by orphan G-protein coupled receptors GPR37L1 and GPR37. Glia 2018, 66, 2414–2426. [Google Scholar] [CrossRef]
- Harzer, K.; Hiraiwa, M.; Paton, B.C. Saposins (sap) A and C activate the degradation of galactosylsphingosine. FEBS Lett. 2001, 508, 107–110. [Google Scholar] [CrossRef]
- Hiraiwa, M.; Marie Campana, W.; Martin, B.M.; O’Brien, J.S. Prosaposin receptor: Evidence for a G-protein-associated receptor. Biochem. Biophys. Res. Commun. 1997, 240, 415–418. [Google Scholar] [CrossRef]
- Kaufmann, M.; Schaupp, A.L.; Sun, R.; Coscia, F.; Dendrou, C.A.; Cortes, A.; Kaur, G.; Evans, H.G.; Mollbrink, A.; Navarro, J.F.; et al. Identification of early neurodegenerative pathways in progressive multiple sclerosis. Nat. Neurosci. 2022 257 2022, 25, 944–955. [Google Scholar] [CrossRef]
- Suo, N.; He, B.; Cui, S.; Yang, Y.; Wang, M.; Yuan, Q.; Xie, X. The orphan G protein-coupled receptor <scp>GPR149</scp> is a negative regulator of myelination and remyelination. Glia 2022, 70, 1992–2008. [Google Scholar] [CrossRef]
- Najm, F.J.; Madhavan, M.; Zaremba, A.; Shick, E.; Karl, R.T.; Factor, D.C.; Miller, T.E.; Nevin, Z.S.; Kantor, C.; Sargent, A.; et al. Drug-based modulation of endogenous stem cells promotes functional remyelination in vivo. Nature 2015, 522, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Göttle, P.; Sabo, J.K.; Heinen, A.; Venables, G.; Torres, K.; Tzekova, N.; Parras, C.M.; Kremer, D.; Hartung, H.P.; Cate, H.S.; et al. Oligodendroglial Maturation Is Dependent on Intracellular Protein Shuttling. J. Neurosci. 2015, 35, 906. [Google Scholar] [CrossRef]
- Manousi, A.; Göttle, P.; Reiche, L.; Cui, Q.L.; Healy, L.M.; Akkermann, R.; Gruchot, J.; Schira-Heinen, J.; Antel, J.P.; Hartung, H.P.; et al. Identification of novel myelin repair drugs by modulation of oligodendroglial differentiation competence. EBioMedicine 2021, 65, 103276. [Google Scholar] [CrossRef] [PubMed]
- Bonfanti, E.; Bonifacino, T.; Raffaele, S.; Milanese, M.; Morgante, E.; Bonanno, G.; Abbracchio, M.P.; Fumagalli, M. Abnormal upregulation of gpr17 receptor contributes to oligodendrocyte dysfunction in SOD1G93A mice. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.-L.; Wei, E.-Q.; Zhang, S.-H.; Xu, H.-M.; Chu, L.-S.; Zhang, W.-P.; Zhang, Q.; Chen, Z.; Mei, R.-H.; Zhao, M.-H. Montelukast, a Cysteinyl Leukotriene Receptor-1 Antagonist, Dose- and Time-Dependently Protects against Focal Cerebral Ischemia in Mice. Pharmacology 2005, 73, 31–40. [Google Scholar] [CrossRef]
- Tassan Mazzocco, M.; Murtaj, V.; Martins, D.; Schellino, R.; Coliva, A.; Toninelli, E.; Vercelli, A.; Turkheimer, F.; Belloli, S.; Moresco, R.M. Exploring the neuroprotective effects of montelukast on brain inflammation and metabolism in a rat model of quinolinic acid-induced striatal neurotoxicity. J. Neuroinflammation 2023, 20, 1–17. [Google Scholar] [CrossRef]
- Xiao, Y.; Zhang, Y.; Gao, Y.H.; Zhao, Z.H.; He, J.; Gao, R.; Guo, Y.X.; Wang, L. Bin; Li, X. A targeted extracellular vesicles loaded with montelukast in the treatment of demyelinating diseases. Biochem. Biophys. Res. Commun. 2022, 594, 31–37. [Google Scholar] [CrossRef]
- Han, B.; Zhang, Y.Y.; Ye, Z.Q.; Xiao, Y.; Rasouli, J.; Wu, W.C.; Ye, S.M.; Guo, X.Y.; Zhu, L.; Rostami, A.; et al. Montelukast alleviates inflammation in experimental autoimmune encephalomyelitis by altering Th17 differentiation in a mouse model. Immunology 2021, 163, 185–200. [Google Scholar] [CrossRef]
- Green, A.J.; Gelfand, J.M.; Cree, B.A.; Bevan, C.; Boscardin, W.J.; Mei, F.; Inman, J.; Arnow, S.; Devereux, M.; Abounasr, A.; et al. Clemastine fumarate as a remyelinating therapy for multiple sclerosis (ReBUILD): a randomised, controlled, double-blind, crossover trial. Lancet 2017, 390, 2481–2489. [Google Scholar] [CrossRef] [PubMed]
- Neumann, B.; Baror, R.; Zhao, C.; Segel, M.; Dietmann, S.; Rawji, K.S.; Foerster, S.; McClain, C.R.; Chalut, K.; van Wijngaarden, P.; et al. Metformin Restores CNS Remyelination Capacity by Rejuvenating Aged Stem Cells. Cell Stem Cell 2019, 25, 473. [Google Scholar] [CrossRef] [PubMed]
- De Keersmaecker, A.-V.; Van Doninck, E.; Popescu, V.; Willem, L.; Cambron, M.; Laureys, G.; D’ Haeseleer, M.; Bjerke, M.; Roelant, E.; Lemmerling, M.; et al. A metformin add-on clinical study in multiple sclerosis to evaluate brain remyelination and neurodegeneration (MACSiMiSE-BRAIN): study protocol for a multi-center randomized placebo controlled clinical trial. Front. Immunol. 2024, 15, 1362629. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, J.M.K.; Anderson, R.C.; McDermott, K.W. MicroRNA: Key regulators of oligodendrocyte development and pathobiology. Int. J. Biochem. Cell Biol. 2015, 65, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Ngo, C.; Kothary, R. MicroRNAs in oligodendrocyte development and remyelination. J. Neurochem. 2022, 162, 310–321. [Google Scholar] [CrossRef] [PubMed]
- Dugas, J.C.; Cuellar, T.L.; Scholze, A.; Ason, B.; Ibrahim, A.; Emery, B.; Zamanian, J.L.; Foo, L.C.; McManus, M.T.; Barres, B.A. Dicer1 and miR-219 Are Required for Normal Oligodendrocyte Differentiation and Myelination. Neuron 2010, 65, 597–611. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; He, X.; Han, X.; Yu, Y.; Ye, F.; Chen, Y.; Hoang, T.; Xu, X.; Mi, Q.-S.; Xin, M.; et al. MicroRNA-Mediated Control of Oligodendrocyte Differentiation. Neuron 2010, 65, 612–626. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Moyano, A.L.; Ma, Z.; Deng, Y.; Lin, Y.; Zhao, C.; Zhang, L.; Jiang, M.; He, X.; Ma, Z.; et al. miR-219 Cooperates with miR-338 in Myelination and Promotes Myelin Repair in the CNS. Dev. Cell 2017, 40, 566–582. [Google Scholar] [CrossRef] [PubMed]
- Milbreta, U.; Lin, J.; Pinese, C.; Ong, W.; Chin, J.S.; Shirahama, H.; Mi, R.; Williams, A.; Bechler, M.E.; Wang, J.; et al. Scaffold-Mediated Sustained, Non-viral Delivery of miR-219/miR-338 Promotes CNS Remyelination. Mol. Ther. 2019, 27, 411–423. [Google Scholar] [CrossRef]
- Liu, S.; Ren, C.; Qu, X.; Wu, X.; Dong, F.; Chand, Y.K.; Fan, H.; Yao, R.; Geng, D. miR-219 attenuates demyelination in cuprizone-induced demyelinated mice by regulating monocarboxylate transporter 1. Eur. J. Neurosci. 2017, 45, 249–259. [Google Scholar] [CrossRef]
- Osorio-Querejeta, I.; Carregal-Romero, S.; Ayerdi-Izquierdo, A.; Mäger, I.; Nash, L.A.; Wood, M.; Egimendia, A.; Betanzos, M.; Alberro, A.; Iparraguirre, L.; et al. MiR-219a-5p enriched extracellular vesicles induce OPC differentiation and EAE improvement more efficiently than liposomes and polymeric nanoparticles. Pharmaceutics 2020, 12, 186. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.; Ong, W.; Wang, K.; Wang, M.; Nizetic, D.; Chew, S. Effects of miR-219/miR-338 on microglia and astrocyte behaviors and astrocyte-oligodendrocyte precursor cell interactions. Neural Regen. Res. 2020, 15, 739. [Google Scholar] [CrossRef] [PubMed]
- Marangon, D.; Abbracchio, M.P.; Lecca, D. Pathway-Focused Profiling of Oligodendrocytes Over-Expressing miR-125a-3p Reveals Alteration of Wnt and Cell-to-Cell Signaling. Cell. Mol. Neurobiol. 2021, 41, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Marangon, D.; Boda, E.; Parolisi, R.; Negri, C.; Giorgi, C.; Montarolo, F.; Perga, S.; Bertolotto, A.; Buffo, A.; Abbracchio, M.P.; et al. In vivo silencing of miR-125a-3p promotes myelin repair in models of white matter demyelination. Glia 2020, 68, 2001–2014. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.S.; Chopp, M.; Pan, W.L.; Wang, X.L.; Fan, B.Y.; Zhang, Y.; Kassis, H.; Zhang, R.L.; Zhang, X.M.; Zhang, Z.G. MicroRNA-146a Promotes Oligodendrogenesis in Stroke. Mol. Neurobiol. 2017, 54, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, Z.G.; Lu, M.; Zhang, Y.; Shang, X.; Chopp, M. MiR-146a promotes oligodendrocyte progenitor cell differentiation and enhances remyelination in a model of experimental autoimmune encephalomyelitis. Neurobiol. Dis. 2019, 125, 154–162. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Z.G.; Lu, M.; Wang, X.; Shang, X.; Elias, S.B.; Chopp, M. MiR-146a promotes remyelination in a cuprizone model of demyelinating injury. Neuroscience 2017, 348, 252–263. [Google Scholar] [CrossRef]
- Martin, N.A.; Molnar, V.; Szilagyi, G.T.; Elkjaer, M.L.; Nawrocki, A.; Okarmus, J.; Wlodarczyk, A.; Thygesen, E.K.; Palkovits, M.; Gallyas, F.; et al. Experimental demyelination and axonal loss are reduced in MicroRNA-146a deficient mice. Front. Immunol. 2018, 9, 490. [Google Scholar] [CrossRef]
Figure 1.
Pathological mechanisms contributing to remyelination failure in glial scar. Microglia and astrocytes contribute to the scar microenvironment by secreting pro-inflammatory CTKs, CSPGs and ROS, that inhibit OPC differentiation and induce a disease-associated phenotype in OPCs and mature OLs. Disease-associated OPCs overexpress MMPs, release CSPGs, acquire phagocytic functions, and astrocyte-like phenotype, instead of generating new myelinating OLs. Together, all these pathological mechanisms lead to remyelination failure. OPC = oligodendrocyte precursor cells; OL = oligodendrocyte; CTK = cytokines; CSPG = chondroitin sulphate proteoglycans; ROS = radical oxygen species; MMP = matrix metalloproteinases; MHC = major histocompatibility complex.
Figure 1.
Pathological mechanisms contributing to remyelination failure in glial scar. Microglia and astrocytes contribute to the scar microenvironment by secreting pro-inflammatory CTKs, CSPGs and ROS, that inhibit OPC differentiation and induce a disease-associated phenotype in OPCs and mature OLs. Disease-associated OPCs overexpress MMPs, release CSPGs, acquire phagocytic functions, and astrocyte-like phenotype, instead of generating new myelinating OLs. Together, all these pathological mechanisms lead to remyelination failure. OPC = oligodendrocyte precursor cells; OL = oligodendrocyte; CTK = cytokines; CSPG = chondroitin sulphate proteoglycans; ROS = radical oxygen species; MMP = matrix metalloproteinases; MHC = major histocompatibility complex.
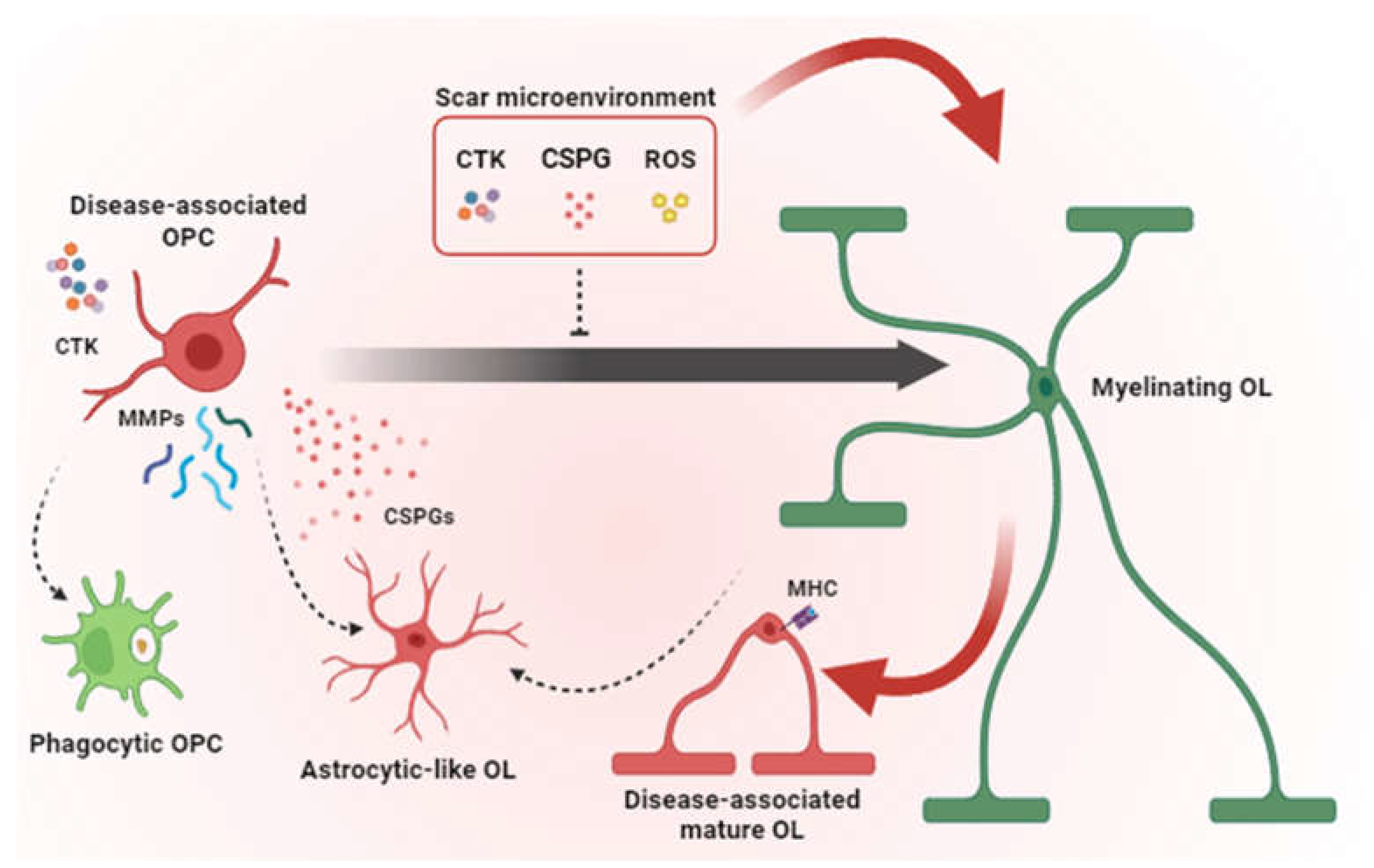
Figure 2.
Therapeutic strategies to promote remyelination and lesion resolution. (A) NPC transplantation has the potential to restore myelination and neuronal connections replacing damaged cells. (B) Modulation of astrocyte reactivity and promotion of their transdifferentiation toward the oligodendroglial fate can reduce the levels of inhibitory factors, promoting axon regeneration and endogenous remyelination. (C) Promotion of OPC differentiation and/or inhibition of pathologically altered mechanisms aim at fostering endogenous remyelination and in turn protect axons from damage. NPC = Neural progenitor cell; OPC = oligodendrocyte precursor cells; OL = oligodendrocyte; CSPG = chondroitin sulphate proteoglycans.
Figure 2.
Therapeutic strategies to promote remyelination and lesion resolution. (A) NPC transplantation has the potential to restore myelination and neuronal connections replacing damaged cells. (B) Modulation of astrocyte reactivity and promotion of their transdifferentiation toward the oligodendroglial fate can reduce the levels of inhibitory factors, promoting axon regeneration and endogenous remyelination. (C) Promotion of OPC differentiation and/or inhibition of pathologically altered mechanisms aim at fostering endogenous remyelination and in turn protect axons from damage. NPC = Neural progenitor cell; OPC = oligodendrocyte precursor cells; OL = oligodendrocyte; CSPG = chondroitin sulphate proteoglycans.
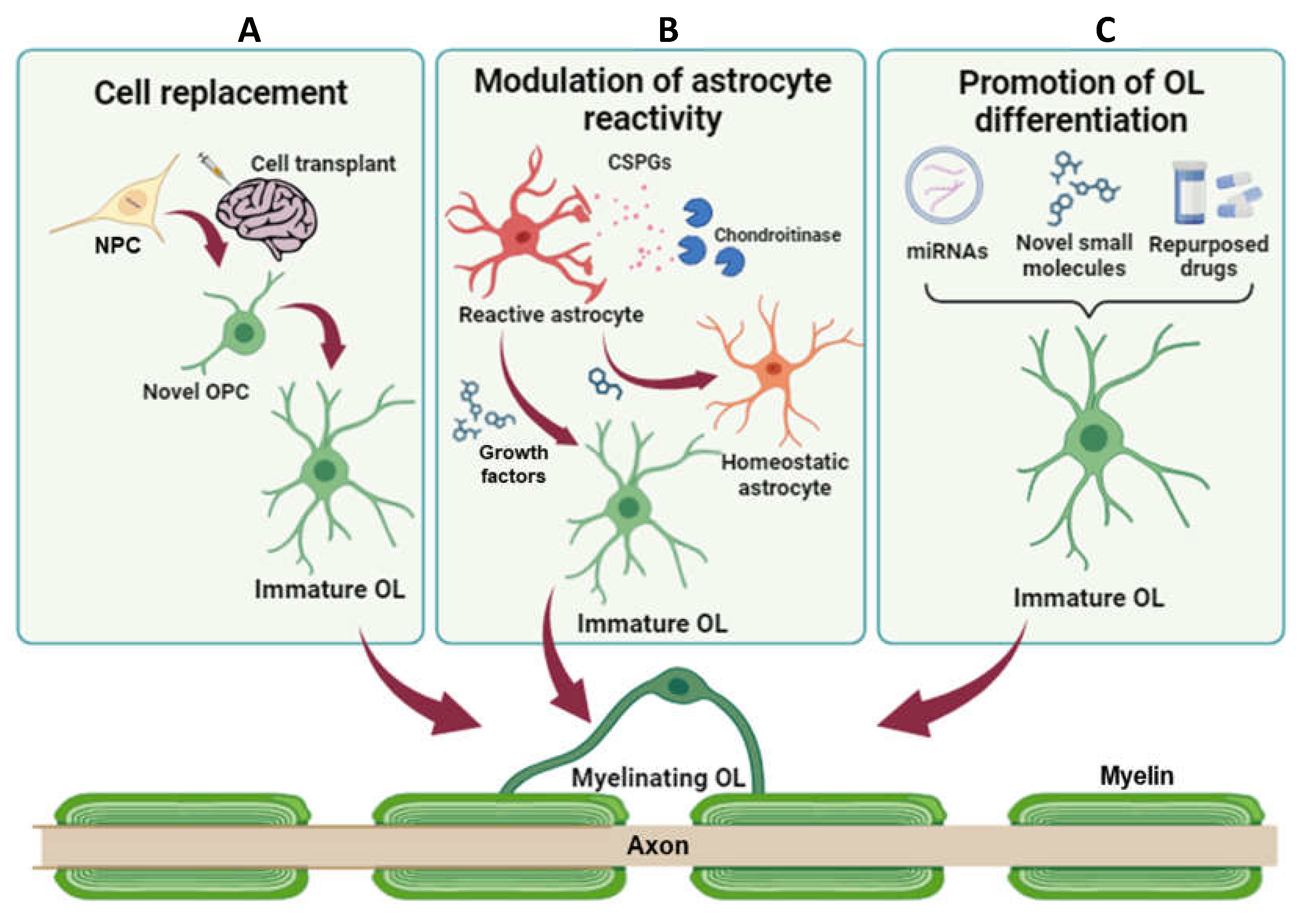
Table 1.
Summary of the studies that applied cell replacement strategies to improve remyelination.
| Model | Cell type | Combination treatment |
Effects | Reference |
|---|---|---|---|---|
| SCI | OPC-committed NPCs | Chondroitinase ABC | Formation of nodes of Ranvier. Improvement in remyelination and motor functions. |
[80] |
| SCI | NPCs | Overexpression of human arginine decarboxylase | Decreased glial scar formation. Increased remyelination and neuronal differentiation. Improvement in neurological outcomes. |
[77] |
| MS (EAE) | NPCs | / | Decreased demyelination and astrogliosis. Increased remyelination and improvement in neurological scores. |
[86] |
| SCI | Pre-OPCs | Glial scar ablation | Generation of new OLs and neurons | [89] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



