
Submitted:
08 May 2024
Posted:
08 May 2024
You are already at the latest version
Abstract
Lifestyle interventions can prevent type 2 diabetes (T2DM). However, some in-dividuals do not experience anticipated improvements despite weight loss. Bi-omarkers to identify such individuals at early stages are lacking. IGF-1 and IGFBP-1 were shown to predict T2DM onset in prediabetes. We assessed if these markers also predict the success of lifestyle interventions, thereby possibly guid-ing personalized strategies.
We analyzed fasting serum levels of IGF-1, IGFBP-1 and IGFBP-2 in relation to changes in metabolic and anthropometric parameters, including intrahepatic li-pids (IHL) and visceral adipose tissue (VAT) volume measured by MRI, in 345 high-risk prediabetic participants (54% female; aged 36-80 years). Participants were enrolled in three randomized dietary intervention trials and assessed both at baseline and one year post-intervention. Statistical analyses were performed using IBM SPSS Statistics (version 28), significance set at p
Keywords:
IGF-axis
; lifestyle intervention
; intervention response
; prediabetes
1. Introduction
Obesity and its metabolic sequelae are increasing worldwide and are the primary causes of the most prevalent diseases of industrialized countries linked to the metabolic syndrome. Lifestyle approaches aiming to prevent diabetes by moderate weight loss, healthy diet and increased physical activity have proven highly successful due to improvements in insulin sensitivity and insulin secretion [1]. However, in these trials some study participants did not show the expected improvements despite significant weight loss and reduction of liver fat [2]. These individuals may require more intense programs aiming at lifestyle factors or might profit from early pharmacological interventions. To date, there is a lack of biomarkers for identifying these individuals at early time points.
The insulin-like growth factor (IGF) system has evolutionarily been separated from the insulin system in vertebrates to regulate growth and food related metabolism with greater flexibility but remained closely intertwined in the insulin/IGF-system [3,4]. This system is closely linked to life expectancy, from worms to mammals, such that a reduced function prolongs life which probably serves to survive unfavorable periods of famine and shortness [5]. In short lived mammals, a deficiency of the growth hormone IGF-axis induces longevity, while this is less clear in long-lived species. Nevertheless, growth hormone (GH) and IGF-1 deficiency is associated with reduced rates of cancer, type 2 diabetes, and diabetic complications in humans, while increased levels of IGF-1 have been linked to the incidence of some types of cancer [5,6].
However, the IGF-system is also associated with regenerative processes; low function predicted sarcopenia [7], cardiovascular risk [8], cognitive dysfunction [9] and type 2 diabetes in epidemiological [10] and randomized controlled studies [11]. This discrepancy possibly relates to the complexity of the insulin/IGF-1 system with its regulation by age, nutrition and behavioral components resulting from epigenetics against the background of a strong genetic disposition [12].
Although circulating IGF-1 is a hepatokine, produced in response to pulses of GH release from the pituitary gland and mediating most of its actions, it is closely linked to insulin release through the regulation of IGF-binding proteins (IGFBP) [13,14]. Circulating levels of IGFBPs determine the bioactivity of IGF-1 by binding 99% of IGF-1 [3,13,15]. IGFBP-1 and IGFBP-2 were shown to be closely related to circulating insulin and insulin sensitivity [13,16,17]. Circulating IGFBP-1 is produced by the liver and suppressed by portal levels of insulin [18]. Acute increases of insulin after meals decrease IGFBP-1 by up to 60% and thereby increase free, biologically active IGF-1 [18-20]. Chronically increased levels of insulin decrease circulating IGFBP-1 which correlates closely with whole body and hepatic insulin resistance as well as with metabolic dysfunction–associated steatotic liver disease (MASLD) [14,18]. IGFBP-2 is also related to insulin resistance but regulated more slowly by insulin [16]. IGF-1 and the IGF-binding proteins regulate visceral and subcutaneous fat depots and exert significant effects on hepatic fat content [3,13-15,20,21].
The IGF-system shows significant inheritance and additionally appears to be regulated by epigenetic factors programmed by obesity, diet and physical activity, thus individual metabolic conditions [12,22].
Further, in a preceding study, we found low IGF-1 and high IGFBP-1 to be predictive for the incidence of T2DM in a prediabetic cohort with high risk for the development of diabetes. This phenomenon is most likely attributable to impaired beta-cell function, possibly explaining a non-responsiveness to lifestyle interventions [11].
Consequently, we hypothesized that components of the IGF-system might be related to the success of metabolic improvements in lifestyle studies; and may allow prediction of responsiveness to intervention studies. We, therefore, investigated the role of the IGF-system in predicting the success of lifestyle interventions in people with impaired glucose metabolism, analyzing the same above-mentioned high-risk prediabetic cohort.
2. Results
2.1. Baseline Characteristics
Most participants of the studies displayed characteristics of the metabolic syndrome, including abdominal obesity and impaired glucose metabolism at baseline (Table 1). At baseline, absolute levels of IGF-1 showed a wide spread between individuals and correlated negatively with fasting glucose, waist-hip ratio (WHR) and VAT (Visceral adipose tissue, measured via magnetic resonance imaging, MRI), but not with IHL (Intrahepatic lipid content, measured via magnetic resonance spectroscopy, MRS), or indices of insulin sensitivity or insulin secretion, as already reported elsewhere [11]. IGFBP-1 similarly showed a wide variation and correlated significantly not only inversely with constitutional markers as BMI, WHR, VAT and IHL but also positively with indices of glucose sensitivity and secretion [11]. IGFBP-2 displayed modest negative correlations with anthropometric markers (BMI, VAT, IHL) [11].
2.2. Responses to Lifestyle Interventions
Lifestyle intervention led to highly significant improvements of anthropometric and metabolic parameters of the participants, as already reported elsewhere [11]. However, this was not reflected by major changes of IGF-1 or the binding proteins, which showed no changes except for a statistically significant, but small increase of IGFBP-1 from 2.1 to 2.2 µg/l, despite their significant correlations with metabolic parameters [11].
We, therefor,e tested whether higher or lower baseline levels of IGF-1 and IGFBP-1 might associate with responses to lifestyle interventions, and compared changes in individuals with levels above or below the medians.
2.3. Responses to Lifestyle Interventions within Median Subgroups of IGF-1 and IGFBP-1 Baseline Levels 1
The stratification of individuals by baseline IGF-1 and IGFBP-1 medians (median subgroups) revealed highly significant differences between groups in terms of changes in IGF-1 and IGFBP-1 levels, as well as changes in metabolic parameters during the intervention.
Participants with baseline IGF-1 levels above the median showed a highly significant decrease in IGF-1 levels, whereas individuals with baseline levels below the median showed increased IGF-1 levels (Table 2a).
Concerning IGFBP-1 subgroups, individuals with levels below the median showed a significant increase, whereas individuals with levels above the median showed a significant decrease in IGFBP-1 levels (Table 2b).
Moreover, individuals with lower IGFBP-1 concentrations to baseline exhibited a significant increase in IGF-1 levels, whereas those with higher IGFBP-1 levels significantly decreased in IGF-1 (Table 2b).
2.4. Differential Response to Lifestyle Interventions Depending on Baseline Levels of IGF-1 and IGFBP-1
The differential change of IGF-1 levels between subgroups below and above the median led to a significant between-group-difference in change of IGF-1 between groups (Table 3a), which persisted after adjusting for change in BMI (MD= 32.6 µg/L; 95%-CI: [23.5; 41.7], F(1, 339)= 49.4, p= <.001, partial η2= .127; note: homogeneity of regression slopes not given for the interaction term subgroup x change in BMI; homogeneity of variances not given).
Regarding intervention-induced metabolic changes, individuals within the subgroup with supra-median levels of IGF-1 mostly showed improved profiles compared to those with lower IGF-1 levels to baseline, despite similar reductions in body weight (Tables 2a and 3a). Specifically, while both groups experienced significant reductions in VAT and IHL, these reductions were significantly greater in the subgroup with supra-median levels. Regarding glucose metabolism, fasting glucose and 2h glucose improved significantly in both groups but fasting insulin and HOMA-IR only improved significantly in the subgroup with supra-median levels. Fasting insulin and Matsuda index showed a significantly greater improvement in this group.
Further, changes in IGFBP-1 levels showed significant between-group differences when comparing the two subgroups of IGFBP-1 baseline levels (Table 3b).
Both IGFBP-1 subgroups showed overall metabolic improvements upon intervention with regard to BMI, total and visceral fat volume, IHL, fasting and 2h-glucose as well as fasting insulin levels, insulin sensitivity and secretion (Table 2b). Except for fasting glucose levels, each of these improvements were more pronounced within the group with lower IGFBP-1 levels at baseline. Significantly greater improvements were observed for change in IHL (Table 3b).
3. Discussion
It is well established that IGF-1 and IGFBP-1 are highly heritable and correlate with anthropometric and metabolic parameters beyond inheritance [22,23]. Here, we show that responses of IGF-1 and IGFBP-1 to lifestyle interventions depended on baseline expression levels. Moreover, the baseline levels predicted the ability to respond to lifestyle changes, and thereby appear to determine the success of lifestyle interventions.
Baseline levels of IGF-1 vary widely between individuals, primarily due to inheritance [12,24] and due to parameters of glucose and insulin metabolism [23]. Although caloric and primarily protein restriction reduce IGF-1 [25], previous studies did not observe significant changes of IGF-1 upon lifestyle interventions and weight loss at 1 or 6 years [26], nor did they report a decrease of IGF-1 [27]. Unexpectedly, upon moderate weight loss, we observed highly significant increases of IGF-1 in people with low levels at baseline, while IGF-1 decreased in participants with initially high levels. Due to the wide spread of baseline levels, the absolute values were still lower in the subgroup with sub-median levels, and higher in the group with supra-median levels after the intervention (Figure A2); possibly due to the strong inheritance, which was estimated at 63% in twin studies [12,24]. Higher levels of IGF-1 are associated with reduced risk of type 2 diabetes in cross-sectional [10] and prospective [28,29] epidemiological studies, but also with increased risk in a Mendelian Randomization study [30].
The observed changes in IGF-1and IGFBP-1 were relatively small. The subgroups with levels above and below the median were closely clustered together and significantly above zero, excluding the possibility of a floor or ceiling effect. Despite this proximity of the subgroups, there was an observable tendency for high IGF-1 levels trending downward, and low IGF-1 levels trending upward. A similar pattern was observed with IGFBP-1. This may potentially represent a simple regression to the mean. However, the contrary argues that the split between the subgroups below and above median levels was associated with significant metabolic consequences, which is an intriguing and novel aspect.
Our data show that higher levels of IGF-1 predisposed to significantly greater improvements of intrahepatic lipids and of visceral fat volume, markers which are strongly associated with the metabolic syndrome, insulin resistance and diabetes risk, despite comparable weight loss. In addition, fasting insulin decreased only in individuals with higher IGF-1, indicating that the group with low levels was unable to improve insulin sensitivity despite weight loss and significant reductions of VAT and IHL. IGF-1, thus, may have determined the capacity for metabolic recompensation in this high-risk group. Although levels of IGF-1 are primarily determined by inheritance, protein intake increases IGF-1 while other foods have minor effects. We monitored food intake and did not observe food dependent effects on IGF-1 in our study, which did not specifically involve high protein intake.
Given that our study was done in prediabetic cohorts with higher risk for progression, it may not be translatable to people without metabolic impairments. However, higher levels of IGF-1 at baseline were also associated with reduced risk of developing T2DM in our study [11], supporting a protective effect of IGF-1 when undergoing lifestyle intervention.
In earlier studies, higher levels of IGFBP-1 are generally associated with better insulin sensitivity and insulin secretion, while low levels are prospectively associated with T2DM and IGT [15,17]. IGFBP-1 is acutely and chronically inhibited by portal insulin levels and, therefore, low levels closely reflect hepatic fat content and hepatic insulin resistance [18]. In our cohorts, we observed similar inverse associations of IGFBP-1 with IHL, VAT, hepatic, and whole-body insulin resistance, reflecting extensive metabolic impairment. It might, therefore, seem unexpected that low IGFBP-1 levels associated with considerably greater improvements of anthropometric and metabolic responses to the lifestyle intervention, despite similar reductions of body weight. One may argue that greater improvements were due to the greater initial impairments, but higher IGFBP-1 also labelled a group with reduced capacity for improvement. This phenomenon was also observed in earlier studies on individuals with prediabetes, which showed that patients with combined IFG-IGT – a prediabetes subtype with most prominent alterations throughout the entire metabolism – respond more effectively to lifestyle intervention than individuals with isolated IGT [31].
In fact, the prediabetic group differed from the high-risk groups identified in cross-sectional or prospective observational studies with regard to IGFBP-1: according to a Swedish study, an increase of IGFBP-1 was observed in prediabetic individuals as they approached overt type 2 diabetes [19,20]. This appears to relate to the progression of hepatic insulin resistance, which reduces the suppression of the hepatokine IGFBP-1 relative to circulating insulin levels [21]. Further, the progressive beta-cell dysfunction reflected by impaired glucose tolerance appears to contribute to this phenotype. Notably, in our present study, participants with higher IGFBP-1 showed smaller reductions of 2h glucose values, and only one quarter of the reduction of fasting insulin compared to the subgroup with sub-median levels. Higher IGFBP-1 thus labels the group that is unable to improve beta-cell function upon reductions of body weight, visceral and hepatic fat content. Accordingly, high IGFBP-1 was also shown to identify prediabetic people who are unresponsive to standard lifestyle interventions [11].
Mechanistically, this phenomenon described above may be attributed to the antagonism of IGF-1 activity by IGFBP-1, which is particularly pronounced in the interstitial and pericellular environment [13,15,21]. IGF-1 was shown to cooperate with insulin in maintaining beta-cell function in adult animals, while its developmental function was negligible [32,33]. The selective deletion of beta-cell IGF-receptors primarily led to impaired glucose sensing rather than loss of beta-cell mass in mice [32]. This appears to translate to humans, as suggested in our present study, by the protective effects of higher IGF-1 and lower IGFBP-1 leading to increased biologically active IGF-1. In addition, our findings indicate that higher activity of the IGF-system appears to support − in context of intervention − loss of ectopic fat stores, as shown by the greater reductions of visceral and hepatic fat in this study.
In mice with diet-induced obesity, overexpression of IGFBP-1 improves insulin sensitivity [34]. In humans, weight loss, reductions of hepatic fat and hepatic insulin resistance and consequently of reduced circulating insulin are associated with increases of IGFBP-1 [35], which we also observed in our study in patients with low IGFBP-1 at baseline.
Taken together, the IGF-1 system in metabolism represents a complex interplay that certainly requires further investigation. Our here presented novel findings suggest that IGF-1 and IGFBP-1 may serve as serological biomarkers to predict lifestyle responses - which, to our knowledge, would be the first of their kind.
4. Materials and Methods
4.1. Project Design, Participants
For the analysis, we used data from three German lifestyle intervention studies: The Prediabetes Lifestyle Intervention Study (PLIS), the Diabetes Nutrition Algorithm-Prediabetes Trial (DiNA-P), and the concluded Optimal Fiber Trial (OptiFiT), all three focusing on lifestyle interventions for individuals with prediabetes, being at high risk of developing type 2 diabetes. High-risk criteria included reduced insulin sensitivity together with presence of MASLD and/ or reduced insulin secretion (PLIS, DiNA-P) or impaired glucose tolerance (OptiFiT).
Data for this analysis cover the first year of intervention of all three studies.
PLIS, a multicenter study initiated in 2013 at eight sites in Germany, is part of the national research association, the German Center for Diabetes Research (DZD) [2]. DiNA-P, designed in parallel with PLIS, was intended to offer equivalent data on an alternative dietary intervention and constitutes an independent trial (refer to clinicaltrials.gov: NCT02609243). Our present analysis includes 135 PLIS participants from the University Hospital Carl Gustav Carus of the Technical University Dresden and 116 DiNA-P participants from sites in Nuthetal and Berlin.
The OptiFiT study was conducted between March 2010 and October 2014 in Berlin and Nuthetal [36].Our analysis included data from 94 participants who completed the first year of intervention.
Primary goal of each study was metabolic improvement and moderate weight loss through lifestyle modification. We assessed changes after a one-year intervention period of each study. Thus, the ultimate cohort comprises 345 participants, from whom fasting levels of IGF-1, IGFBP-1, and IGFBP-2 at both baseline and after 12 months were collectively available.
4.2. Interventions
In PLIS and DiNA-P, participants followed a hypo- to isocaloric diet based on low fat intake, as per 2018 recommendations from the German Nutrition Society (< 30 kcal% fat, <10 kcal% saturated fatty acids, >15 g/1000 kcal fiber/day) for 12 months. They received personalized dietary counseling in 8 or 16 sessions of equal duration, depending on randomization. At DiNA-P, there was an additional three-week comparison between reduced carbohydrate or fat intake, while otherwise maintaining similarity to the PLI study (refer to clinicaltrials.gov; NCT02609243). In both trials, long-term follow-up extended beyond the initial 1-year intervention.
The OptiFiT study focused on insoluble cereal fiber intake's effects on glycemic metabolism in individuals with impaired glucose tolerance (IGT). Participants underwent random assignment to either cereal fiber or placebo supplementation for a duration of 2 years. Both groups engaged in a structured 1-year lifestyle program, adapted from the PREvention of DIAbetes Self-management (PREDIAS, [37]). Details on the study design are published elsewhere[36].
Nutrient and energy intake were monitored via dietary records throughout the studies. All participants were mandated to achieve a certain level of daily physical activity, monitored through a combination of questionnaires and technical devices.
Ethical committees approved the study protocols for all three trials, which also adhered to Good Clinical Practice principles and the Declaration of Helsinki. Before enrollment, all participants provided written informed consent and underwent comprehensive medical evaluations, including history, physical exams, and routine blood and urine tests. At the study’s outset, participants had no evidence of severe chronic diseases, including metabolic, cardiovascular, lung, gastrointestinal, autoimmune diseases or cancer.
4.3. Sample Collection, Anthropometric and Metabolic Assessment
In each study, participants underwent a baseline assessment, which included medical examinations, fasting blood draws, an oral glucose tolerance test (oGTT), anthropometric measurements and magnetic resonance (MR) examination, along with the provision of food records and activity meters. These assessments were repeated 1-year after enrollment into the respective study. Notably, within the OptiFiT cohort under analysis here, MR examination was undergone of only 16 participants.
Measurements of body weight, height, and circumferences were taken with participants wearing light clothing and no shoes. Fat volumes were assessed using magnetic resonance imaging (MRI), while hepatic fat storage was detected using MR spectroscopy (1H-MRS) following a previously published protocol [38]. MR scans were evaluated in a blinded manner by a medical physicist (JM).
Both fasting blood samples and oral glucose tolerance tests (oGTT) using 75 g of glucose provided the basis for the determination of glucose homeostasis, insulin sensitivity, and insulin secretion. In the PLIS and DiNA-P, blood samples after glucose load were collected at minutes 0, 30, 60, 90, and 120. In OptiFiT, capillary blood for determination of glucose levels and whole blood for insulin measurements were drawn at minutes 0, 60 and 120 after glucose load, respectively. Acquired blood samples were either analyzed immediately or stored at -80°C.
We used HOMA-IR and the Matsuda index [39,40] as standard surrogate parameters for insulin resistance (IR). The hepatic insulin resistance (HIRI) was estimated using a formula developed by Abdul-Ghani et al. [41]. Insulin secretion capacity was approximated using the modified Insulinogenic Index (IGI) according to Seltzer [42] and the Disposition Index-2 (DI, [43]). For calculation of oGTT-based indices, only participants with complete data sets for respective required timepoints were analyzed.
4.4. Laboratory Analyses
Glucose and insulin levels, along with routine laboratory safety parameters, were measured using established standard methods (for insulin, ELISA by Mercodia®, Uppsala Sweden, was used in DiNA-P and OptiFiT an chemiluminescent immunoassay by Siemens Healthcare GmbH, Erlangen Germany, in PLIS).
For the measurement of fasting levels of IGF-1, IGFBP-1, and IGFBP-2, we used commercially available ELISA assays (Mediagnost®, Reutlingen, Germany), previously validated by our research group [44], following manufacturer’s instructions (intra- and interassay coefficients of variation IGF-1: 5.8% and 8.6%, IGFBP-1: 6.5% and 6.1, IGFBP-2: both <10%). The measurement was performed by technical assistants in a blinded manner.
4.5. Statistics
We analyzed data of 345 participants having 1-year follow-up data available.
The data are presented as means with standard deviation (SD) or as median with interquartile rane (IQR), depending on the distribution of the data.
Within-group differences were assessed using student’s paired t-test (one-tailed) in case of normal distribution or using Wilcoxon-Signed-Rank test in case of skewed data.
Between-group differences were evaluated using Welch test (one-tailed testing) in case of normally distributed parameters, regardless of homogeneity of variance, with this following a recommendation by Rasch, Kubinger, and Moder [45]. Differences between groups of non-normally distributed data were tested via Mann-Whitney-U test. Mean difference (MD) between groups was indicated as mean difference between the subgroup below the median and the subgroup above the median.
We used ANCOVA models to test for between-group differences between two independent groups, when we controlled for one or more variables. We used Bonferroni correction to adjust for multiple comparisons. We asssessed homogeneity of regression slopes by testing the interaction terms between covariates and the group variable. We indicated, if not given; here, the analysis must be considered with caution. Using Levene’s test (based on median), we assessed homogeneity of variances. In case, these were not given, we acknowledged it, but assumed the robustness of ANCOVA models due to roughly equal group sizes.
A two-sided p-value of <.05 was considered as statistically significant. The analyses were conducted using IBM® SPSS®, Version 28 (SPSS Inc, Chicago, IL).
5. Conclusions
In conclusion, our study proposes that baseline expression levels of IGF-1 and IGFBP-1 play a role in determining responses to lifestyle intervention, with higher levels of IGF-1 predisposing to greater impairment at baseline, but also greater interventional improvements in metabolic risk markers despite similar weight loss. Conversely, low levels of IGFBP-1 are associated with greater improvements in response to lifestyle interventions. These associations are seen in individuals with preexisting impairment of glucose metabolism. Mechanistically, the antagonistic relationship between IGF-1 and IGFBP-1 might form the basis for these associations.
Understanding these relationships might help to identify individuals who may require more intensive interventions early on. As our data's applicability is limited to prediabetic high-risk groups, further research is warranted to validate these findings in broader populations.
Author Contributions
Conceptualization, Nina Meyer, Stefan Kabisch and Andreas Pfeiffer; Data curation, Nina Meyer, Stefan Kabisch and Martin Osterhoff; Formal analysis, Nina Meyer, Stefan Kabisch and Andreas Pfeiffer; Funding acquisition, Stefan Kabisch and Andreas Pfeiffer; Investigation, Nina Meyer, Stefan Kabisch, Ulrike Dambeck, Caroline Honsek, Margrit Kemper, Christiana Gerbracht, Ayman Arafat, Andreas Birkenfeld, Peter Schwarz , Jürgen Machann and Martin Osterhoff; Methodology, Nina Meyer, Stefan Kabisch and Andreas Pfeiffer; Project administration, Andreas Pfeiffer; Resources, Andreas Birkenfeld, Peter Schwarz and Andreas Pfeiffer; Supervision, Stefan Kabisch and Andreas Pfeiffer; Validation, Nina Meyer, Stefan Kabisch and Andreas Pfeiffer; Visualization, Nina Meyer; Writing – original draft, Nina Meyer and Andreas Pfeiffer; Writing – review & editing, Nina Meyer, Stefan Kabisch, Ulrike Dambeck, Caroline Honsek, Margrit Kemper, Christiana Gerbracht, Ayman Arafat, Andreas Birkenfeld, Peter Schwarz , Jürgen Machann, Martin Osterhoff and Andreas Pfeiffer.
Funding
PLIS and DiNA-P were financially backed by the German Center for Diabetes Research (DZD) in Neuherberg, Germany. DiNA-P additionally received material support from the California Walnut Commission in Folsom, CA, USA. OptiFiT received support from the German Diabetes Foundation (Deutsche Diabetes Stiftung. DDS) in Düsseldorf, Germany, and J. Rettenmaier & Soehne GmbH + Co KG in Holzmuehle, Germany, which provided the supplements.
Institutional Review Board Statement
The studies were conducted in accordance with the Declaration of Helsinki, and approved by the respective Ethics Committee (PLIS: Ethics Committee of the Faculty of Medicine of the Eberhard Karls University of Tübingen, 055/2012BO1, 07.03.2012, and Ethics Committee of the Charité and Ethics Committee of the University of Potsdam, session 18/34, 06.05.2013; DiNA-P: Ethics Committee of the Charité and of the University of Potsdam, session 17/34, 06.05.2013; OptiFiT: Ethics Committee at the University of Potsdam: EA4/129/09).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The data presented in this study are available on request from the corresponding author. (the data are not publicly available due to privacy restrictions.).
Acknowledgments
In this section, you can acknowledge any support given which is not covered by the author contribution or funding sections. This may include administrative and technical support, or donations in kind (e.g., materials used for experiments).
Conflicts of Interest
Stefan Kabisch received grants by the German Center for Diabetes Research (DZD), the German Diabetes Association, the Almond Board of California, the California Walnut Commission, the Wilhelm-Doerenkamp-Foundation, J. Rettenmaier & Söhne and Beneo Südzucker as well as personal reimbursements by Lilly Germany, Sanofi, Berlin Chemie, Boehringer-Ingelheim and the JuZo-Akademie. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Appendix A
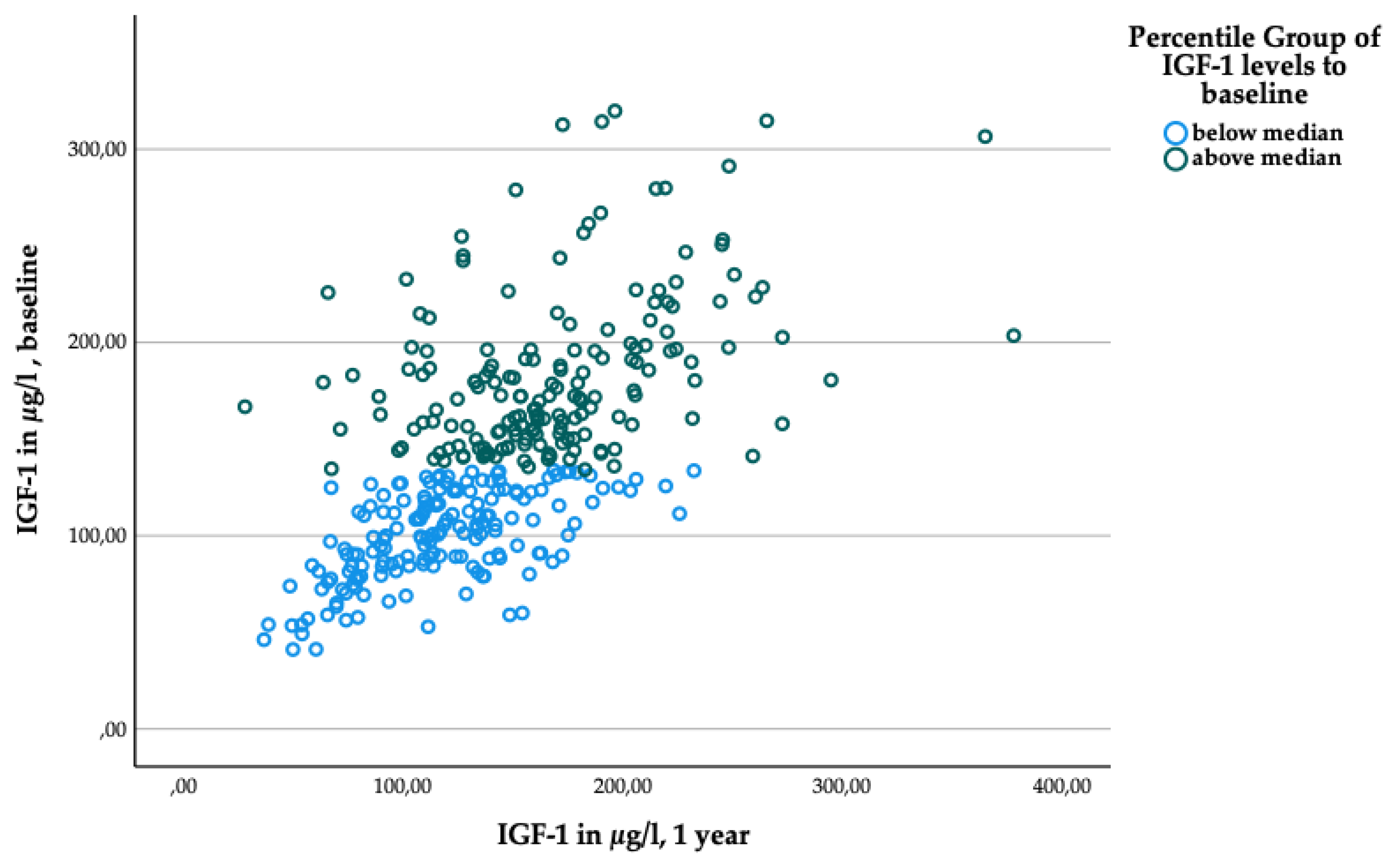
Figure A2.
Scatter plot for IGF-1 levels at baseline and one year, grouped according to IGF-1 baseline levels. Blue dots: participants with sub-median levels. Green dots: participants with super-median levels.
Figure A2.
Scatter plot for IGF-1 levels at baseline and one year, grouped according to IGF-1 baseline levels. Blue dots: participants with sub-median levels. Green dots: participants with super-median levels.
References
- Uusitupa, M.; Khan, T.A.; Viguiliouk, E.; Kahleova, H.; Rivellese, A.A.; Hermansen, K.; Pfeiffer, A.; Thanopoulou, A.; Salas-Salvado, J.; Schwab, U.; Sievenpiper, J.L. Prevention of Type 2 Diabetes by Lifestyle Changes: A Systematic Review and Meta-Analysis. Nutrients 2019, 11. [Google Scholar] [CrossRef]
- Fritsche, A.; Wagner, R.; Heni, M.; Kantartzis, K.; Machann, J.; Schick, F.; Lehmann, R.; Peter, A.; Dannecker, C.; Fritsche, L.; et al. Different Effects of Lifestyle Intervention in High- and Low-Risk Prediabetes: Results of the Randomized Controlled Prediabetes Lifestyle Intervention Study (PLIS). Diabetes 2021, 70, db210526–db210526. [Google Scholar] [CrossRef] [PubMed]
- LeRoith, D.; Yakar, S. Mechanisms of disease: metabolic effects of growth hormone and insulin-like growth factor 1. Nat Clin Pract Endocrinol Metab 2007, 3, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Partridge, L.; Alic, N.; Bjedov, I.; Piper, M.D. Ageing in Drosophila: the role of the insulin/Igf and TOR signalling network. Exp Gerontol 2011, 46, 376–381. [Google Scholar] [CrossRef]
- Fontana, L.; Partridge, L.; Longo, V.D. Extending healthy life span--from yeast to humans. Science 2010, 328, 321–326. [Google Scholar] [CrossRef]
- Milman, S.; Huffman, D.M.; Barzilai, N. The Somatotropic Axis in Human Aging: Framework for the Current State of Knowledge and Future Research. Cell Metabolism 2016, 23, 980–989. [Google Scholar] [CrossRef]
- Sandri, M.; Barberi, L.; Bijlsma, A.Y.; Blaauw, B.; Dyar, K.A.; Milan, G.; Mammucari, C.; Meskers, C.G.; Pallafacchina, G.; Paoli, A.; et al. Signalling pathways regulating muscle mass in ageing skeletal muscle: the role of the IGF1-Akt-mTOR-FoxO pathway. Biogerontology 2013, 14, 303–323. [Google Scholar] [CrossRef] [PubMed]
- Colao, A. The GH-IGF-I axis and the cardiovascular system: clinical implications. Clin Endocrinol (Oxf) 2008, 69, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Broughton, S.; Partridge, L. Insulin/IGF-like signalling, the central nervous system and aging. Biochem J 2009, 418, 1–12. [Google Scholar] [CrossRef]
- Teppala, S.; Shankar, A. Association between serum IGF-1 and diabetes among U.S. adults. Diabetes Care 2010, 33, 2257–2259. [Google Scholar] [CrossRef]
- Meyer, N.M.T.; Kabisch, S.; Dambeck, U.; Honsek, C.; Kemper, M.; Gerbracht, C.; Arafat, A.M.; Birkenfeld, A.L.; Schwarz, P.E.H.; Machann, J.; et al. Low IGF-1 and high IGFBP-1 predict diabetes onset in prediabetic patients. Eur J Endocrinol 2022. [Google Scholar] [CrossRef] [PubMed]
- Kao, P.C.; Matheny, A.P., Jr.; Lang, C.A. Insulin-like growth factor-I comparisons in healthy twin children. J Clin Endocrinol Metab 1994, 78, 310–312. [Google Scholar] [CrossRef] [PubMed]
- Clemmons, D.R. Role of IGF Binding Proteins in Regulating Metabolism. Trends Endocrinol Metab 2016, 27, 375–391. [Google Scholar] [CrossRef] [PubMed]
- Firth, S.M.; Baxter, R.C. Cellular actions of the insulin-like growth factor binding proteins. Endocr Rev 2002, 23, 824–854. [Google Scholar] [CrossRef] [PubMed]
- Haywood, N.J.; Slater, T.A.; Matthews, C.J.; Wheatcroft, S.B. The insulin like growth factor and binding protein family: Novel therapeutic targets in obesity & diabetes. Mol Metab 2019, 19, 86–96. [Google Scholar] [CrossRef]
- Arafat, A.M.; Weickert, M.O.; Frystyk, J.; Spranger, J.; Schofl, C.; Mohlig, M.; Pfeiffer, A.F. The Role of Insulin-Like Growth Factor (IGF) Binding Protein-2 in the Insulin-Mediated Decrease in IGF-I Bioactivity. J Clin Endocrinol Metab 2009, 94, 5093–5101. [Google Scholar] [CrossRef] [PubMed]
- Petersson, U.; Ostgren, C.J.; Brudin, L.; Brismar, K.; Nilsson, P.M. Low levels of insulin-like growth-factor-binding protein-1 (IGFBP-1) are prospectively associated with the incidence of type 2 diabetes and impaired glucose tolerance (IGT): the Soderakra Cardiovascular Risk Factor Study. Diabetes Metab 2009, 35, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Kotronen, A.; Lewitt, M.; Hall, K.; Brismar, K.; Yki-Jarvinen, H. Insulin-like growth factor binding protein 1 as a novel specific marker of hepatic insulin sensitivity. J Clin Endocrinol Metab 2008, 93, 4867–4872. [Google Scholar] [CrossRef]
- Lewitt, M.S.; Hilding, A.; Ostenson, C.G.; Efendic, S.; Brismar, K.; Hall, K. Insulin-like growth factor-binding protein-1 in the prediction and development of type 2 diabetes in middle-aged Swedish men. Diabetologia 2008, 51, 1135–1145. [Google Scholar] [CrossRef]
- Lewitt, M.S.; Hilding, A.; Brismar, K.; Efendic, S.; Ostenson, C.G.; Hall, K. IGF-binding protein 1 and abdominal obesity in the development of type 2 diabetes in women. European Journal of Endocrinology 2010, 163, 233–242. [Google Scholar] [CrossRef]
- Lewitt, M.S. The Role of the Growth Hormone/Insulin-Like Growth Factor System in Visceral Adiposity. Biochem Insights 2017, 10, 1178626417703995. [Google Scholar] [CrossRef]
- Teumer, A.; Qi, Q.; Nethander, M.; Aschard, H.; Bandinelli, S.; Beekman, M.; Berndt, S.I.; Bidlingmaier, M.; Broer, L.; Group, C.L.W.; et al. Genomewide meta-analysis identifies loci associated with IGF-I and IGFBP-3 levels with impact on age-related traits. Aging Cell 2016, 15, 811–824. [Google Scholar] [CrossRef]
- Hong, Y.; Brismar, K.; Hall, K.; Pedersen, N.L.; de Faire, U. Associations between insulin-like growth factor-I (IGF-I), IGF-binding protein-1, insulin and other metabolic measures after controlling for genetic influences: results from middle-aged and elderly monozygotic twins. J Endocrinol 1997, 153, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Pedersen, N.L.; Brismar, K.; Hall, K.; de Faire, U. Quantitative genetic analyses of insulin-like growth factor I (IGF-I), IGF-binding protein-1, and insulin levels in middle-aged and elderly twins. J Clin Endocrinol Metab 1996, 81, 1791–1797. [Google Scholar] [CrossRef] [PubMed]
- Thissen, J.P.; Ketelslegers, J.M.; Underwood, L.E. Nutritional regulation of the insulin-like growth factors. Endocr Rev 1994, 15, 80–101. [Google Scholar] [CrossRef]
- Fontana, L.; Weiss, E.P.; Villareal, D.T.; Klein, S.; Holloszy, J.O. Long-term effects of calorie or protein restriction on serum IGF-1 and IGFBP-3 concentration in humans. Aging Cell 2008, 7, 681–687. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.; Brandhorst, S.; Shelehchi, M.; Mirzaei, H.; Cheng, C.W.; Budniak, J.; Groshen, S.; Mack, W.J.; Guen, E.; Di Biase, S.; et al. Fasting-mimicking diet and markers/risk factors for aging, diabetes, cancer, and cardiovascular disease. Sci Transl Med 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, M.S.; Heald, A.H.; Gibson, J.M.; Cruickshank, J.K.; Dunger, D.B.; Wareham, N.J. Circulating concentrations of insulin-like growth factor-I and development of glucose intolerance: a prospective observational study. Lancet 2002, 359, 1740–1745. [Google Scholar] [CrossRef]
- Rajpathak, S.N.; He, M.; Sun, Q.; Kaplan, R.C.; Muzumdar, R.; Rohan, T.E.; Gunter, M.J.; Pollak, M.; Kim, M.; Pessin, J.E.; et al. Insulin-like growth factor axis and risk of type 2 diabetes in women. Diabetes 2012, 61, 2248–2254. [Google Scholar] [CrossRef]
- Larsson, S.C.; Michaelsson, K.; Burgess, S. IGF-1 and cardiometabolic diseases: a Mendelian randomisation study. Diabetologia 2020, 63, 1775–1782. [Google Scholar] [CrossRef]
- Saito, T.; Watanabe, M.; Nishida, J.; Izumi, T.; Omura, M.; Takagi, T.; Fukunaga, R.; Bandai, Y.; Tajima, N.; Nakamura, Y.; Ito, M. Lifestyle modification and prevention of type 2 diabetes in overweight Japanese with impaired fasting glucose levels: a randomized controlled trial. Archives of internal medicine 2011, 171, 1352–1360. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, R.N.; Holzenberger, M.; Shih, D.Q.; Ozcan, U.; Stoffel, M.; Magnuson, M.A.; Kahn, C.R. beta-cell-specific deletion of the Igf1 receptor leads to hyperinsulinemia and glucose intolerance but does not alter beta-cell mass. Nat Genet 2002, 31, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Ueki, K.; Okada, T.; Hu, J.; Liew, C.W.; Assmann, A.; Dahlgren, G.M.; Peters, J.L.; Shackman, J.G.; Zhang, M.; Artner, I.; et al. Total insulin and IGF-I resistance in pancreatic beta cells causes overt diabetes. Nat Genet 2006, 38, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Rajwani, A.; Ezzat, V.; Smith, J.; Yuldasheva, N.Y.; Duncan, E.R.; Gage, M.; Cubbon, R.M.; Kahn, M.B.; Imrie, H.; Abbas, A.; et al. Increasing Circulating IGFBP1 Levels Improves Insulin Sensitivity, Promotes Nitric Oxide Production, Lowers Blood Pressure, and Protects Against Atherosclerosis. Diabetes 2012, 61, 915–924. [Google Scholar] [CrossRef] [PubMed]
- Telgenkamp, I.; Kusters, Y.H.A.M.; Schalkwijk, C.G.; Houben, A.J.H.M.; Kooi, M.E.; Lindeboom, L.; Bons, J.A.P.; Schaper, N.C.; Joris, P.J.; Plat, J.; et al. Contribution of Liver Fat to Weight Loss-Induced Changes in Serum Hepatokines: A Randomized Controlled Trial. The Journal of clinical endocrinology and metabolism 2019, 104, 2719–2727. [Google Scholar] [CrossRef] [PubMed]
- Honsek, C.; Kabisch, S.; Kemper, M.; Gerbracht, C.; Arafat, A.M.; Birkenfeld, A.L.; Dambeck, U.; Osterhoff, M.A.; Weickert, M.O.; Pfeiffer, A.F.H. Fibre supplementation for the prevention of type 2 diabetes and improvement of glucose metabolism: the randomised controlled Optimal Fibre Trial (OptiFiT). Diabetologia 2018, 61, 1295–1305. [Google Scholar] [CrossRef] [PubMed]
- Kulzer, B.; Hermanns, N.; Gorges, D.; Schwarz, P.; Haak, T. Prevention of diabetes self-management program (PREDIAS): effects on weight, metabolic risk factors, and behavioral outcomes. Diabetes care 2009, 32, 1143–1146. [Google Scholar] [CrossRef] [PubMed]
- Machann, J.; Thamer, C.; Schnoedt, B.; Stefan, N.; Haring, H.U.; Claussen, C.D.; Fritsche, A.; Schick, F. Hepatic lipid accumulation in healthy subjects: A comparative study using spectral fat-selective MRI and volume-localized 1H-MR spectroscopy. Magnetic Resonance in Medicine 2006, 55, 913–917. [Google Scholar] [CrossRef]
- Matthews, D.R.; Hosker, J.P.; Rudenski, A.S.; Naylor, B.A.; Treacher, D.F.; Turner, R.C. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985, 28, 412–419. [Google Scholar] [CrossRef]
- Matsuda, M.; DeFronzo, R.A. Insulin sensitivity indices obtained from oral glucose tolerance testing: comparison with the euglycemic insulin clamp. Diabetes care 1999, 22, 1462–1470. [Google Scholar] [CrossRef]
- Abdul-Ghani, M.A.; Matsuda, M.; Balas, B.; DeFronzo, R.A. Muscle and liver insulin resistance indexes derived from the oral glucose tolerance test. Diabetes Care 2007, 30, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Seltzer, H.S.; Allen, E.W.; Herron, A.L.; Brennan, M.T. Insulin secretion in response to glycemic stimulus: relation of delayed initial release to carbohydrate intolerance in mild diabetes mellitus. The Journal of clinical investigation 1967, 46, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Retnakaran, R.; Shen, S.; Hanley, A.J.; Vuksan, V.; Hamilton, J.K.; Zinman, B. Hyperbolic relationship between insulin secretion and sensitivity on oral glucose tolerance test. Obesity 2008, 16, 1901–1907. [Google Scholar] [CrossRef] [PubMed]
- Schüler, R.; Markova, M.; Osterhoff, M.A.; Arafat, A.; Pivovarova, O.; Machann, J.; Hierholzer, J.; Hornemann, S.; Rohn, S.; Pfeiffer, A.F.H. Similar dietary regulation of IGF-1- and IGF-binding proteins by animal and plant protein in subjects with type 2 diabetes. European Journal of Nutrition 2021. [Google Scholar] [CrossRef]
- Rasch, D.; Kubinger, K.D.; Moder, K. The two-sample t test: Pre-testing its assumptions does not pay off. Statistical Papers 2011, 52, 219–231. [Google Scholar] [CrossRef]

Table 1.
Baseline characteristics of the cohort.
| Parameters | Value | N |
| Women (%) | 54.0 | 186 |
| Age (years) | 62.7 ± 8.7 | 345 |
| Study allocation | ||
| PLIS (%) | 39.1 | 135 |
| DiNA-P (%) | 33.6 | 116 |
| OptiFiT (%) | 27.2 | 94 |
| IGF-1 (µg/L) | 141.8 ± 53.7 | 345 |
| IGFBP-1 (µg/L) | 2.1 [ 1.4; 4.1] | 345 |
| IGFBP-2 (µg/L) | 259.1 [134.2; 422.6] | 345 |
| BMI (kg/m²) | 30.9 ± 5.4 | 345 |
| Present overweight (%) | 38.0 | 132 |
| Present obesity (%) | 50.7 | 175 |
| Grade I (%) | 29.3 | 101 |
| Grade II (%) | 15.1 | 52 |
| Grade III (%) | 6.4 | 22 |
| WHR (cm/cm) | 0.93 ± 0.09 | 341 |
| Body fat content-BIA [%] | 34.7 ± 8.5 | 312 |
| VAT-MRI (l) | 5.5 ± 2.4 | 225 |
| IHL-MRS (%-abs.) | 7.0 [3.0; 14.4] | 231 |
| Present MASLD (%) | 39.4 | 136 |
| Fasting glucose (mmol/L) | 5.7 ± 0.7 | 345 |
| 2-h glucose (mmol/L) | 8.2 ± 1.6 | 345 |
| Fasting insulin (pmol/L) | 73.4 [51.7; 105.5] | 337 |
| Present IFG + NGT (%) | 31.9 | 110 |
| Present NFG + IGT (%) | 31.6 | 109 |
| Present IFG + IGT (%) | 36.5 | 126 |
| HOMA-IR | 2.6 [1.7; 3.8] | 337 |
| Matsuda Index | 2.6 [1.8; 3.5] | 238 |
| HIRI | 37.2 [30.6; 44.4] | 242 |
| IGI | 11.7 [7.5; 21.2] | 242 |
| DI | 30.9 [21.6: 43.6] | 238 |
Data are shown as mean ± SD (normally distributed variables), as median [IQR] (non-normally distributed variables) or as proportions (%). PLIS: Prediabetes Lifestyle Intervention Study. DiNA-P: Diabetes Nutrition Algorithm- Prediabetes. OptiFiT: Optimal Fibre Trial. BMI Body mass index. WHR Waist-hip ratio. VAT Visceral adipose tissue. BIA Bioelectrical impedance analysis. MRI Magnetic resonance imaging. IHL Intrahepatic lipid content. MRS Magnetic resonance spectroscopy. abs absolute. MASLD Metabolic Dysfunction-associated Steatotic Liver Disease. IFG Impaired Fasting Glucose. NGT Normal Glucose Tolerance. IGT Impaired Glucose Tolerance. HOMA Homeostatic model assessment. IR Insulin Resistance. HIRI Hepatic insulin resistance index (Abdul-Ghani). IGI Insulinogenic Index (Seltzer). DI Disposition Index-2. IGFBP1/-2: Insulin-like Growth Factor Binding Protein-1/-2.
Table 2.
a) and b). IGF-1, IGFBP-1, IGFBP-1 and metabolic parameters at baseline and after 1 year, respectively, in association with a) IGF-1 baseline levels, b) IGFBP-1 baseline levels.
Table 2.
a) and b). IGF-1, IGFBP-1, IGFBP-1 and metabolic parameters at baseline and after 1 year, respectively, in association with a) IGF-1 baseline levels, b) IGFBP-1 baseline levels.
| Parameters | baseline | 1 year | n | p | d / r | baseline | 1 year | n | p | d / r |
|---|---|---|---|---|---|---|---|---|---|---|
| (a) | ||||||||||
| IGF-1 < 134.2 µg/L | IGF-1 > 134.2 µg/L | |||||||||
| IGF-1 [µg/L] | 99.9 ± 23.3 | 117.2 ± 38.8 | 172 | <.001 | -.56 | 183.5 ± 41.5 | 168.7 ± 51.0 | 173 | <.001 | .29 |
| IGFBP-1 [µg/L] | 2.2 [1.2; 4.4] | 2.5 [1.3; 4.5] | 172 | .460 | -.06 | 2.1 [.9; 3.7] | 1.9 [1.2; 4.0] | 173 | .015 | -.18 |
| IGFBP-2 [µg/L] | 269.6 [148.1; 453.6] | 271.7 [162.0; 431.9] | 170 | .290 | -.08 | 251.5 [133.9; 385.2] | 250.7 [164.8; 427.7] | 172 | .057 | -.14 |
| Body Mass Index [kg/m²] | 30.8 ± 5.2 | 29.9 ± 5.1 | 171 | <.001 | .51 | 31.1 ± 5.6 | 29.9 ± 5.4 | 171 | <.001 | .65 |
| Waist-to-hip ratio [cm/cm] | 0.94 ± 0.09 | 0.92 ± 0.09 | 166 | .011 | .18 | 0.93 ± 0.09 | 0.93 ± 0.09 | 166 | .359 | .03 |
| Body fat content-BIA [%] | 35.1 ± 8.6 | 34.0 ± 9.0 | 145 | <.001 | .34 | 34.2 ± 8.5 | 33.1 ± 9.1 | 147 | .002 | .25 |
| Visceral fat volume-MRI [l] | 5.6 ± 2.5 | 5.2 ± 2.4 | 111 | <.001 | .43 | 5.6 ± 2.3 | 5.0 ± 2.1 | 86 | <.001 | .71 |
| Intrahepatic Lipid Content -MRS [%-abs.] | 7.0 [3.0; 14.7] | 4.4 [2.3; 8.9] | 113 | <.001 | -.41 | 7.2 [3.0; 14.2] | 3.1 [1.1; 7.1] | 89 | <.001 | -.68 |
| Fasting glucose [mmol/L] | 5.8 ± .7 | 5.6 ± .8 | 164 | <.001 | .34 | 5.7 ± .7 | 5.5 ± .7 | 157 | <.001 | .31 |
| 2-h glucose [mmol/L] | 8.3 ± 1.5 | 7.6 ± 1.9 | 164 | <.001 | .36 | 8.1 ± 1.6 | 7.3 ± 2.0 | 157 | <.001 | .46 |
| Fasting insulin [pmol/L] | 79.7 [55.8; 108.2] | 77.8 [54.9; 111.2] | 170 | .239 | -.09 | 66.0 [49.6; 99.7] | 61.5 [44.1;88.3] | 165 | <.001 | -.34 |
| HOMA-IR | 3.0 [1.9; 3.9] | 2.7 [1.7; 3.9] | 170 | .051 | -.15 | 2.4 [1.6; 3.7] | 2.0 [1.4; 3.1] | 164 | <.001 | -.36 |
| Matsuda Index | 2.5 [1.8; 3.3] | 2.9 [2.0; 4.3] | 122 | <.001 | -.34 | 2.8 [1.9; 3.6] | 3.6 [2.5; 5.0] | 112 | <.001 | -.50 |
| HIRI | 37.5 [30.8; 45.3] | 34.2 [29.8; 42.0] | 127 | .003 | -.26 | 36.7 [30.0;42.6] | 33.5 [27.2; 39.3] | 114 | <.001 | -.41 |
| IGI | 11.7 [7.3; 21.2] | 12.4 [7.8; 19.8] | 127 | .273 | -.10 | 11.6 [7.5; 19.2] | 11.2 [7.0; 17.0] | 114 | .232 | -.11 |
| DI | 28.2 [19.5; 43.6] | 34.3 [21.4; 63.1] | 122 | <.001 | -.36 | 33.6 [22.9; 44.5] | 38.7 [25.0; 68.0] | 112 | <.001 | -.31 |
| b | ||||||||||
| IGFBP-1 < 2.13 µg/L | IGFBP-1 > 2.13 µg/L | |||||||||
| IGF-1 [µg/L] | 141.5 ± 48.5 | 150.5 ± 52.5 | 172 | .002 | -.23 | 142.1 ± 58.5 | 135.6 ± 50.6 | 173 | .043 | .13 |
| IGFBP-1 [µg/L] | 1.0 [.7 ; 1.5] | 1.5 [.9; 2.2] | 172 | <.001 | -.53 | 4.1 [2.8; 6.8] | 3.9 [2.3; 5.6] | 173 | .045 | -.15 |
| IGFBP-2 [µg/L] | 223.6 [119.5; 369.2] | 237.4 [141.2; 352.5] | 172 | .080 | -.13 | 310.2 [175.4; 463.2] | 319.5 [190.2; 515.7] | 170 | .179 | -.10 |
| Body Mass Index [kg/m²] | 31.8 ± 5.0 | 30.7 ± 4.8 | 171 | <.001 | .68 | 30.0 ± 5.7 | 29.1 ± 5.6 | 171 | <.001 | .49 |
| Waist-to-hip ratio [cm/cm] | 0.94 ± 0.08 | 0.93 ± 0.08 | 165 | .035 | .14 | 0.93 ± 0.10 | 0.92 ± 0.09 | 167 | .096 | .10 |
| Body fat content-BIA [%] | 35.5 ± 8.1 | 34.3 ± 8.8 | 149 | <.001 | .35 | 33.6 ± 9.1 | 32.7 ± 9.6 | 143 | .003 | .24 |
| Visceral fat volume-MRI [l] | 6.00 ± 2.1 | 5.5 ± 2.1 | 106 | <.001 | .64 | 5.1 ± 2.7 | 4.7 ± 2.3 | 91 | <.001 | .46 |
| Intrahepatic Lipid Content -MRS [%-abs.] | 9.4 [5.1; 17.1] | 5.3 [2.4; 10.5] | 110 | <.001 | -.55 | 4.1 [1.5; 9.2] | 2.5 [.7; 6.5] | 92 | <.001 | -.50 |
| Fasting glucose [mmol/L] | 5.8 ± .6 | 5.6 ± .7 | 159 | <.001 | .31 | 5.7 ± .7 | 5.5 ± .8 | 162 | <.001 | .34 |
| 2-h glucose [mmol/L] | 8.2 ± 1.5 | 7.3 ± 2.0 | 159 | <.001 | .47 | 8.3 ± 1.6 | 7.6 ± 2.0 | 162 | <.001 | .35 |
| Fasting insulin [pmol/L] | 82.0 [59.3; 115.3] | 74.3 [55.5; 111.1] | 165 | .002 | -.24 | 64.2 [43.2 98.0] | 62.8 [44.1; 87.6] | 170 | .019 | -.18 |
| HOMA-IR | 3.0 [2.1; 4.1] | 2.7 [1.8; 3.9] | 165 | <.001 | -.28 | 2.3 [1.5; 3.4] | 2.0 [1.3; 3.1] | 169 | .003 | -.23 |
| Matsuda Index | 2.4 [1.7; 3.2] | 2.8 [2.0;4.1] | 128 | <.001 | -.53 | 2.9 [2.2; 4.6] | 3.7 [2.4; 5.6] | 106 | .002 | -.30 |
| HIRI | 38.3 [32.8; 45.5] | 35.8 [31.3; 42.3] | 133 | <.001 | -.37 | 34.9 [27.9; 40.9] | 31.2 [25.8; 38.6] | 108 | .003 | -.29 |
| IGI | 13.7 [8.9; 23.5] | 15.2 [8.8; 19.8] | 133 | .560 | -.05 | 8.5 [5.7; 15.7] | 9.9 [6.0; 15.9] | 108 | .434 | -.08 |
| DI | 32.9 [22.1; 46.1] | 38.2 [22.6; 65.5] | 128 | <.001 | -.31 | 28.4 [19.5; 39.2] | 33.8 [23.1; 63.4] | 106 | <.001 | -.3 |
Data are shown as mean ± SD (normally distributed variables) or as median [IQR] (non-normally distributed variables). Within-group differences of normally distributed variables were tested via student’s t-test (one-tailed) and of non-normally distributed parameters via Wilcoxon Signed-Rank Test. p for within-group difference, respectively. Significant p-values are bolded. Effect sizes are given as d= Cohen’s d for parametric testing, or as Pearson’s correlation coefficient r for non-parametric testing. Abbreviations: IGF-1 Growth Factor 1. Insulin-like IGFBP1/-2: Insulin-like Growth Factor Binding Protein-1/-2. BMI Body mass index. WHR Waist-hip ratio. BIA Bioelectrical impedance analysis. MRI Magnetic resonance imaging. MRS Magnetic resonance spectroscopy. abs absolute. HOMA Homeostatic model assessment. IR Insulin Resistance. HIRI Hepatic insulin resistance index (Abdul-Ghani). IGI Insulinogenic Index (Seltzer). DI Disposition Index-2.
Table 3.
a) and b). Changes of metabolic parameters over time in association with a) IGF-1 baseline levels and b) IGFBP-1 baseline levels.
Table 3.
a) and b). Changes of metabolic parameters over time in association with a) IGF-1 baseline levels and b) IGFBP-1 baseline levels.
| Parameters | Mean Difference | 95% CI | p | d / r |
|---|---|---|---|---|
| (a) | ||||
| Subgroups of IGF-1 baseline levels: below vs. above the median | ||||
| ∆ IGF-1 [µg/L] | 32.09 | [23.06; 41.12] | <.001 | .75 |
| ∆ IGFBP-1[µg/L] | -0.06 | [-0.88; 0.76] | .396a | -.05 |
| ∆ IGFBP-2 [µg/L] | -17.90 | [-57.96; 22.16] | .422 a | -.04 |
| ∆ Body Mass Index [kg/m²] | 0.31 | [-0.06; 0.68] | .053 | .18 |
| ∆ Waist-to-hip ratio [cm/cm] | -0.01 | [-0.03; 0.00] | .046 | -.19 |
| ∆ Body fat content-BIA [%] | -0.13 | [-0.99; 0.74] | .386 | -.03 |
| ∆ Visceral fat volume-MRI [l] | 0.24 | [0.00; 0.48] | .027 | .28 |
| ∆ Intrahepatic Lipid Content -MRS [%-abs.] | 1.75 | [0.06; 3.43] | .011 a | -.18 |
| ∆ Fasting glucose [mmol/L] | -0.03 | [-0.15; 0.09] | .321 | -.05 |
| ∆ 2-h glucose [mmol/L] | 0.06 | [-0.35; 0.46] | .394 | .03 |
| ∆ Fasting insulin [pmol/L] | 11.31 | [-5.12; 27.74] | .031 a | -.12 |
| ∆ HOMA-IR | 0.36 | [-0.25; 0.96] | .086 a | -.09 |
| ∆ Matsuda Index | -0.45 | [-0.98; -0.09] | .019 a | -.15 |
| ∆ HIRI | 1.16 | [-1.02; 3.35] | .232 a | -.08 |
| ∆ IGI | 5.11 | [-0.48; 10.70] | .118 a | -.10 |
| ∆ DI | 7.59 | [-6.71; 21.89] | .679 a | -.03 |
| (b) | ||||
| Subgroups of IGFBP-1 baseline levels: below vs. above the median | ||||
| ∆ IGF-1 [µg/L] | 15.49 | [5.97; 25.00] | <.001 | .34 |
| ∆ IGFBP-1 [µg/L] | 1.79 | [0.99; 2.58] | <.001 a | -.22 |
| ∆ IGFBP-2 [µg/L] | -3.60 | [-43.78; 36.58] | .430 a | .00 |
| ∆ Body Mass Index [kg/m²] | -0.17 | [-0.54; 0.20] | .183 | -.10 |
| ∆ Waist-to-hip ratio [cm/cm] | 0.00 | [-0.01; 0.02] | .465 | .01 |
| ∆ Body fat content-BIA [%] | -0.26 | [-1.13; 0.61] | .276 | -.07 |
| ∆ Visceral fat volume-MRI [l] | -0.11 | [-0.36; 0.14] | .193 | -.13 |
| ∆ Intrahepatic Lipid Content -MRS [%-abs.] | -1.28 | [-2.95; 0.38] | .049 a | -.14 |
| ∆ Fasting glucose [mmol/L] | 0.05 | [-0.08; 0.17] | .221 | .08 |
| ∆ 2-h glucose [mmol/L] | -0.12 | [-0.53; 0.28] | .275 | -.06 |
| ∆ Fasting insulin [pmol/L] | -6.11 | [-22.58; 10.35] | .642 a | -.03 |
| ∆ HOMA-IR | -0.22 | [-0.82; 0.39] | .703 a | -.02 |
| ∆ Matsuda Index | 0.27 | [-0.29; 0.83] | .484 a | -.05 |
| ∆ HIRI | -0.60 | [-2.82; 1.62] | .785 a | -.02 |
| ∆ IGI | 1.72 | [-3.75; 7.19] | .375 a | -.06 |
| ∆ DI | 4.07 | [-9.74; 17.89] | .786 a | -.02 |
Between-group differences of normally distributed variables were tested via Welch t-test (one-tailed) and of non-normally distributed parameters via Mann-Whitney-U Test. a non-parametric testing. p for within-group difference, respectively. Significant p-values are bolded. Effect sizes are given as d= Cohen’s d for parametric testing, or as Pearson’s correlation coefficient r for non-parametric testing. ∆= Delta. Abbreviations: IGF-1 Growth Factor 1. Insulin-like IGFBP1/-2: Insulin-like Growth Factor Binding Protein-1/-2. BMI Body mass index. WHR Waist-hip ratio. BIA Bioelectrical impedance analysis. MRI Magnetic resonance imaging. MRS Magnetic resonance spectroscopy. abs absolute. HOMA Homeostatic model assessment. IR Insulin Resistance. HIRI Hepatic insulin resistance index (Abdul-Ghani). IGI Insulinogenic Index (Seltzer). DI Disposition Index-2.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



