
Submitted:
08 May 2024
Posted:
09 May 2024
You are already at the latest version
Abstract
Introduction: Lipoyl transferase 2 is involved in biosynthesis of the lipoate. Lipoate is the cofactor for the glycine cleavage system and four dehydrogenase enzymes. Biallelic variants in LIPT2 causing severe neonatal encephalopathy was first described in 2017. Methods: Clinical data was collected by retrospective chart review after obtaining consent from parents. The pathogenicity of these variants was further delineated using a yeast model. The YEp352-LIPT2 plasmid was used as a template to generate the two patient variants using QuickChange Lightning Site-Directed Mutagenesis Kit. Results: Patient was a 15-month-old female who presented at 1 month with dystonia, developmental delay and feeding difficulties. MR Brain showed cortical malformations including colpocephaly, polymicrogyria and heterotopia. Patient had elevations in lactate (6.1 mmol/L) and glycine. Exome sequencing showed 2 variants of uncertain significance in trans in the LIPT2 gene: c.346 G>T and c.418C>T. Patient was started on lipoic acid, thiamine and COQ10. Yeast complementation experiments indicate that both patient mutation variants result in diminished function versions of the LIPT2 protein. Conclusion: We report the fourth case of LIPT2-related disorder. Proband shared significant overlap with previous patients, however had a distinct movement disorder and brain malformations which have not been previously described. Unlike most neurometabolic disorders where dystonia develops later after metabolic stroke in basal ganglia, LIPT2- related disorder seems unique due to early onset of dystonia due to energy deficit in the developing brain. Lipoic acid supplementation has not led to significant clinical improvement. Analyses in yeast indicate that novel variants are deleterious but have retained some functionality.
Keywords:
LIPT2
; mitochondrial disorder
; dystonia
; lipoic acid biosynthesis disorder
1. Introduction
Lipoic acid is an essential co-factor for five enzyme complexes in the mitochondria: the glycine cleavage system (GCS) and the four alpha-ketoacid dehydrogenases - pyruvate dehydrogenase (pyruvate metabolism), α-oxoglutarate dehydrogenase (Krebs cycle), branched chain α-oxoacid dehydrogenase (leucine, isoleucine, and valine catabolism), and 2-oxoadipate dehydrogenase (lysine and tryptophan catabolism). Production of lipoic acid involves synthesis of fatty acid (up to eight carbon atoms) in the mitochondria followed by three lipoic acid specific enzymes – lipoyltransferase 1 (LIPT1), lipoyl(octanoyl) transferase 2 (LIPT2) and lipoic acid synthase (LIAS). LIPT2 transfers an octanoyl moiety from acyl carrier protein to lipoylate the GCS H protein [1,2,3] Biallelic variants in LIPT1 and LIAS are known to be causative of severe neurodevelopmental phenotypes. Lipoyltransferase 1 deficiency (OMIM 616299) is associated with lactic acidosis, global developmental delay and Leigh-like findings on Brain MRI. [4,5] Pathogenic variants in the LIAS gene are associated with a rare subtype of pyruvate dehydrogenase deficiency (OMIM 607031) characterized by seizures, developmental delay, lactic acidosis and hyperglycinemia. [6,7] Bi-allelic pathogenic variants in LIPT2 as a cause of a distinct neurometabolic disorder (OMIM 61768, Encephalopathy, neonatal severe, with lactic acidosis and brain abnormalities) was first discovered in 2017. [8] The three patients reported in that index paper had severe developmental delay, spastic quadriplegia, varied MRI abnormalities and lactic acidosis. We report the fourth case with this ultra-rare disorder, with novel variants. Functional study using heterologous complementation approach in Baker’s yeast (Saccharomyces cerevisiae) supported their pathogenicity. Based on our data, LIPT2-related disorder is a unique neurodevelopmental disorder characterized by neonatal-onset severe generalized dystonia, spectrum of cortical malformations and lactic acidosis with or without abnormal plasma amino acid and urine organic acid profiles.
2. Materials and Methods
2.1. Clinical and Molecular Analysis
Clinical, neuroradiological and biochemical data were collected by retrospective chart review after obtaining written informed consent from patient’s parents. This case report did not meet the U.S. Department of Health and Human Services Common Rule definition of human subjects’ research per 45 CFR 46.102(d) and thus was not reviewed by an Institutional Review Board (IRB). Selection of testing modality and laboratory occurred at the discretion of the clinical team. Clinical exome sequencing was performed by GeneDx. Variant annotation and analysis were performed using company’s custom-developed analysis tool.
2.2. Functional Study Using Yeast Model
The YEp352-LIPT2 plasmid described in previous publication was used as a template to generate the two patient variants using QuickChange Lightning Site-Directed Mutagenesis Kit (Agilent Technologies, Cedar Creek, Texas, USA, lot:0006708228) according to manufacturer’s instructions. [3] The mutagenesis primers are listed in Table 1. The plasmid constructs and mutations were verified by sequencing in the Biocenter Oulu Sequencing Center.
2.3. LIPT2 Sequence Alignment
The multiple sequence alignment was generated using the free multiple sequence alignment function on the Uniprot Website (https://www.uniprot.org/align). This service uses the Clustal Omega algorithm. [9] Amino acid sequences for human (NP_001138341.1), Mouse (NP_080286.2), Chicken (XP_040514800.1), Alligator (XP_059579098.1), Xenopus tropicalis (NP_001072237.1), Zebrafish (NP_001035082.1) and yeast (QHB10390.1, not shown) LIPT2/Lip2 used in the alignment were retrieved from the NIH National Library of Medicine NCBI protein database (https://www.ncbi.nlm.nih.gov/)
2.4. LIPT2 Protein Structure Predictions
AlphaFold predictions of Human (A6NK58) and Xenopus tropicalis (Q0VFH3) LIPT2 structure were analyzed on the AlphaFold Protein Structure Database website (https://alphafold.ebi.ac.uk/). [10]
3. Results
3.1. Clinical Case Report
The patient is currently a 15-month-old female with global developmental delay, generalized dystonia, feeding difficulties, failure to thrive, and lactic acidosis. She was born to a non-consanguineous couple of Vietnamese & Chinese ancestry after an uneventful pregnancy. She started having episodes of back arching at 3 weeks of life. These were initially attributed to gastroesophageal reflux. However, the back arching episodes worsened progressively. The episodes were captured on EEG with no electrographic correlation and a normal background. MR Brain showed cortical malformations including ventricular colpocephaly, polymicrogyria and possible subependymal heterotopia. (Figure 1). She was referred to combined multidisciplinary Movement Disorder-Neurogenetics Clinic. Upon further phenotyping, her movement disorder was characterized as severe generalized dystonia. On physical examination, she had back and neck arching in an opisthotonic posture. Her neck was rotated to the right and she had frequent repetitive tongue protrusion. She had dystonic posturing in all four extremities. She had frequent exacerbations of dystonia throughout the day. At 15 months of age, she was starting to roll over but was not sitting or standing. She was grabbing objects and babbling but had no words. Cardiological evaluation was notable for a stable ventricular septal defect. On ophthalmological evaluation, she was noted to have no evidence of optic atrophy or retinal abnormalities. The patient was started on a vitamin cocktail with alpha lipoic acid, thiamine, COQ10, and riboflavin. Her dystonia had a suboptimal response to baclofen, trihexyphenidyl, clonazepam, and gabapentin.
On initial laboratory tests at 10 months of age, patient had lactic acidosis as high as 6.1 mmol/L (reference range (RR) 1.0-3.3 mmol/L). Plasma amino acid profile was grossly normal without any elevations in glycine, proline or alanine. Repeat lactic acid at 15 months after starting mitochondrial supplements, showed a lactate of 3.7 mmol/L. Repeat plasma amino acid profile showed elevations in glycine at 326 μmol/L (RR 87-309 μmol/L) and valine at 368 μmol/L (RR 52-268 μmol/L).
3.2. Molecular Analysis
Two variants were identified in the LIPT2 gene: maternally inherited variant, c.346 G>T (p.V116L) and paternally inherited variant, c.418C>T (p.P140S). The c.346G>T variant is a missense variant, which is predicted to be deleterious and appears at extremely low frequency in population databases. The c.418C>T variant is also missense, absent from population databases, though some computational models do not predict deleterious effect. Based on ACMG criteria, these variants were initially classified as variant of uncertain significance (VUS).
3.3. Functional Study Using Yeast Model System
Yeast (Saccharomyces cerevisiae) knocked out for LIP2, the homolog of LIPT2 in humans, are respiratory deficient, lack lipoylated proteins and are unable to grow on media containing only a non-fermentable carbon source like glycerol, although perfectly capable of growth on fermentable substrates like glucose. [3] Growth of the yeast LIP2 deletion strain (Δlip2 strain) on non-fermentable glycerol media can be rescued by transformation with a yeast plasmid expressing human LIPT2, and lipoylation is partially restored in these strains (Figure 2). [3] We introduced the c.346 G>T (p.V116I) and c.418C>T (p.P140S) mutations into the LIPT2 yeast expression construct and tested the ability of these variants to rescue the yeast Δlip2 strain growth and lipoylation phenotypes. When the Δlip2 strain was transformed with the plasmids expressing the patient mutation variants, growth rescue was noticeable weaker for either construct compared to yeast cells expressing the wild-type variant (Figure 2). Likewise, lipoylation in yeast strains expressing the mutant variants was partially restored but clearly reduced compared to lipoylation rescue in yeast cells relying on wild-type LIPT2. This data is consistent with compromised function of the c.346 G>T (p.V116I) and c.418C>T (p.P140S) patient variants (Figure 2).
4. Discussion
We report the fourth case of LIPT2-related disorder caused by novel biallelic variants presenting with infantile-onset dystonia without acute metabolic crises. LIPT2 is involved in integration of the lipoate moiety into the enzymes for which it is the cofactor: the GCS and four dehydrogenase enzymes as discussed above. [1,2] Biallelic variants in LIPT2 as a cause of severe neonatal encephalopathy has been described in a single manuscript, by Habarou et a.l in 2017. [8] Our proband shared major phenotypic overlap with previous patients, however had a distinct movement disorder phenotype and cortical malformations which have not been previously described. Unlike most neurometabolic and mitochondrial disorders where dystonia develops later in the course of disease after metabolic stroke in the basal ganglia, LIPT2-related disorder is unique due to infantile onset of dystonia which could be attributed to severe energy deficit in the developing central nervous system. Supplementation with lipoic acid did not lead to substantial clinical improvement similar to previous reported in other lipoic acid biosynthesis defects. This is likely because exogenous lipoic acid is not used as an enzymatic cofactor.
When comparing our proband to patients reported in the index paper, there are significant clinical commonalities. [Table 2] Our proband is the first Asian patient with LIPT2-related disorder. Index paper described one patient (P1) with mixed ethnicity and two German siblings. Our patient had severe dystonia and developmental delay similar to individual P1 described in the Habarou paper. She had moderate lactic acidosis (up to 6.1) similar to individuals P1, P2 and P3 described in the original cohort. Muscle biopsy in individual P3 from the index paper showed evidence of variable diameters in muscle fibers and abnormal glycogen deposition, although individual respiratory complexes had normal activity. [8] P1 was alive at the time of publication with no episodes of metabolic crisis but P2 and P3 had succumbed in their first year of lives. Our patient was alive at 15 months with severe dystonia and developmental delay but had had no major metabolic crisis or evidence of cardio-respiratory compromise. Biochemical testing in our patient showed elevated glycine in plasma amino acid analysis. Since H protein is a part of GCS, elevated plasma glycine has been reported in some patients. We were unable to obtain urine organic acid analysis, but previous patients had elevated lactate, branched-chain keto acids, alpha-ketoglutarate, 2-ketoadipic and 2-hydroxyadipic acids given the defects in lipoic acid dependent enzymes. These findings, although nonspecific, could be a clue for lipoic acid biosynthesis defect. All the patients reported to date had been diagnosed through exome sequencing. The LIPT2 gene has not been included in most commercial multi-gene panels for dystonia/ movement disorders at the time of writing this report. Maintaining a high index of suspicion in cases of infantile dystonia and lactic acidosis and early ordering of exome/ genome sequencing will be crucial to diagnose more patients and elucidate the disease spectrum.
Hyperkinetic movement disorders such as dystonia, choreoathetosis and tremors can be seen in a myriad of neurometabolic disorders. This list includes but is not limited to 1. disorders of nitrogen containing compounds (such as organic acidemias, urea cycle disorders, dopa responsive dystonia), 2. disorders of Vitamins and Minerals (such as PKAN-related disorder, Wilson disease, biotin-thiamine responsive basal Ganglia Disease), 3. disorders of carbohydrate metabolism (such as glucose transporter 1 deficiency), 4. disorders of mitochondrial metabolism, 5. storage disorders (such as Nieman-Pick Type C, galactosialodosis) and 6. congenital disorders of glycosylation. The pathogenesis of movement disorder in neurometabolic conditions generally involves an acute injury (e.g., due to metabolic strokes) or progressive dysfunction (e.g., due to abnormal deposition) of the basal ganglia circuitry. [11,12] Consequently, movement disorders manifest either acutely following metabolic crises or evolve gradually over time. Our patient, as well as P1 in the index paper, presented with very early onset generalized dystonia without metabolic crises or acute basal ganglia injury in the brain MRI. The etiology of the dystonia in this context is unclear. We hypothesize this is related to severe defects in energy metabolism leading to abnormal development of basal ganglia circuitry. Profound energy deficits during critical periods of development could also underlie neuronal migration anomalies, as seen in our patient and P3 in the index paper. The pathognomic imaging signature in several mitochondrial disorders includes asymmetric DWI changes involving parieto-occipital lobe (stroke-like episodes), T2 hyperintensities in bilateral basal ganglia (Leigh syndrome) as well as ponto-cerebellar hypoplasia. [13,14] Structural malformations are atypical but have increasingly been reported. For instance, ventriculomegaly and agenesis of corpus callosum have been reported in pyruvate dehydrogenase deficiency and TPK deficiency. [15,16] Based on MRI findings in our proband and previously reported patients, LIPT2-related disorder also seems to encompass these congenital brain malformations. Our patient had polymicrogyria and heterotopia and previous cases were noted to have supratentorial ventriculomegaly, cystic changes and bi-frontal white matter changes. This observation suggests that lipoic acid is a critical molecule supporting several key biochemical pathways in the developing brain in utero and defects in its biosynthesis can lead to a malformative progressive encephalopathy.
The yeast complementation experiments indicate that both patient variants result in diminished function versions of the LIPT2 protein, with the p.V116L change in the protein yielding more debilitating results than p.P140S. Expression of human LIPT2 protein variants is controlled by the carbon source responsive CTA1 promoter in the complemented yeast strains. We tested complementation under two different conditions. In the first case, the pre-culturing of the cells was done on media containing glucose as the carbon source, where expression of the human transgene variants is low from the CTA1 promoter. Under these conditions, complementation of the lip2 deletion mutation in yeast by the wild type LIPT2 variant is poor, and even worse by both patient variants (Figure 2A). This is also reflected by the levels of lipoylated proteins detectable on a western blot. Wild type LIPT2 restores lipoylation of both the Lat1 and Kgd2 E2 subunits of yeast pyruvate dehydrogenase and α-ketoglutarate dehydrogenase, albeit at reduced levels. However, neither of the patient LIPT2 variants is able to restore Lat1 lipoylation, and the lipoylation level is much weaker overall (Figure 2C). If the cultures of complemented yeast strains are grown on glycerol, where transcription form the CTA1 promoter is much higher, a different picture emerges. Growth of yeast strains complemented by either wild type LIPT2 and LIPT2 p.P140S is near wild type growth, while complementation by the p.V116L -variant is much poorer (Figure 2B). Also here, the restoration of protein lipoylation on western blots reflects the growth assay results: LIPT2 as well as LIPT2p.P140S restore lipoylation to wild type levels, while Lipt2p.V116L only weakly restores the lipoylation of Kgd2. These data indicate that neither of the patient variants is fully functional, but the p.V116L alteration is much more detrimental to LIPT2 function. At first sight this observation appears counterintuitive. A change from Valine to Leucine is considered a conserved change, replacing one hydrophobic residue with another hydrophobic residue of only slightly larger size. In contrast, it would be expected that changing the hydrophobic amino acid Proline, with its unique side chain bonding with the main chain and resulting in diminished flexibility, with the polar and more flexible amino acid Serine would have more drastic consequences. However, alignment of LIPT2 protein sequences reveal that the V116 residue appears to be universally conserved (Figure 2), even in yeast (not shown), while P140 is not even strictly conserved in vertebrates. Instead of Proline, the Xenopus tropicalis (Western clawfrog) and zebrafish LIPT2 variants harbor a Glutamine or a Serine residue in this position (133 in both cases), respectively. Furthermore, a three amino acid gap can be found in both the Xenopus and zebrafish proteins close to the 133 position (Figure 2). To our knowledge, no crystal structure has been obtained for LIPT2. We turned to the AlphaFold structure database (https://alphafold.ebi.ac.uk/) for a plausible prediction of LIPT2 structures and identification of the putative locations of residues V116 and P140 in humans compared to the corresponding predicted structure of the Xenopus laevis LIPT2 that does not harbor a conserved Proline. AlphaFold shows V116 and the corresponding V112 of the clawfrog protein residing in an α-helix in both the predicted human LIPT2 structure (A6NK58) and the predicted Xenopus LIPT2 structure (Q0VFH3). In contrast, P140 of human LIPT2 and the corresponding Q133 in the Xenopus homolog are proposed to sit in a loop region. It is plausible that such a loop structure of the protein is more tolerant in accepting drastic changes than the α-helix harboring V116/112. It is especially poignant that a Proline to Serine replacement, like in one of the patient variants, can be found in the amino acid 133 position in the zebrafish structure. The partial loss of function of the human mutant protein therefore probably needs to be ascribed to the local amino acid sequence context. Our analyses in yeast indicate that both patient variants are defective but have retained some functionality which supports viability. However, neither protein appears to be sufficiently active to provide the level of lipoylation required in healthy humans.
The management of LIPT2-related disorder is largely supportive and there are no disease modifying ‘cures’. [8] These patients need a multi-disciplinary team consisting of pediatric movement disorder specialists, medical geneticists, physical and occupational therapists, and nutritionists. Annual surveillance with cardiology, ophthalmology, and audiology is recommended. Oral baclofen, clonazepam and Artane were being used in our patient with limited effect. Botulinum toxin injections or baclofen pump can be trialed in refractory cases. Data regarding Deep Brain Stimulation (DBS) in mitochondrial disorders is limited. [12] Our patient was started on a vitamin cocktail consisting of alpha lipoic acid, thiamine, COQ10 and riboflavin. Exogenous supplementation of alpha lipoic acid does not seem to treat or reverse neurological symptoms. Since lipoic acid is a co-factor to the alpha-ketoacid dehydrogenase enzymes, which use thiamine pyrophosphate as an additional cofactor, thiamine can be postulated to have a beneficial effect. Since starting the supplements, our patient had not had any metabolic crisis. Patients should be provided an emergency letter enumerating the need for high calorie/ dextrose containing fluids during acute stressors such as fever, infections, fasting and surgery as well as other standard precautions for mitochondrial diseases. Depakote, propofol and lactated ringers should be avoided at all costs.
5. Conclusions
Our report adds to the growing body of evidence regarding LIPT2- related disorder, an ultra-rare pan-ethnic neurometabolic disorder characterized by infantile onset encephalopathy and dystonia, cortical malformations and lactic acidosis. Our analyses in yeast indicate that both novel variants seen in our patient are deleterious but have retained some functionality which supports viability. The distinctive clinical and imaging features in our patient, in conjunction with previously reported patients, contribute to a deeper understanding of the phenotypic variability and natural history of LIPT2- related disorder. Collaborative care models involving pediatric movement disorder specialists, medical geneticists, nutritionists and physical therapists is essential for optimizing management pathways. Exogenous supplementation of lipoic acid and other vitamins may not treat existing neurological symptoms but may help to reduce frequency and degree of lactic acidosis. Longitudinal studies tracking the outcomes in diagnosed individuals as well as basic science research are required to refine prognostication and treatment strategies for this rare and heterogenous disorder.
Acknowledgments
We thank Cecilia Bouska, CGC for her work with the family and assistance with obtaining genetic testing.
References
- Mayr JA, Feichtinger RG, Tort F, Ribes A, Sperl W. Lipoic acid biosynthesis defects. J Inherit Metab Dis. 2014 Jul;37(4):553-63.
- Booker, SJ. Unraveling the pathway of lipoic acid biosynthesis. Chem Biol. 2004 Jan;11(1):10-2.
- Pietikäinen LP, Rahman MT, Hiltunen JK, Dieckmann CL, Kastaniotis AJ. Genetic dissection of the mitochondrial lipoylation pathway in yeast. BMC Biol. 2021 Jan 25;19(1):14.
- Soreze Y, Boutron A, Habarou F, Barnerias C, Nonnenmacher L, Delpech H, Mamoune A, Chrétien D, Hubert L, Bole-Feysot C, Nitschke P, Correia I, Sardet C, Boddaert N, Hamel Y, Delahodde A, Ottolenghi C, de Lonlay P. Mutations in human lipoyltransferase gene LIPT1 cause a Leigh disease with secondary deficiency for pyruvate and alpha-ketoglutarate dehydrogenase. Orphanet J Rare Dis. 2013 Dec 17;8:192.
- Tort F, Ferrer-Cortès X, Thió M, Navarro-Sastre A, Matalonga L, Quintana E, Bujan N, Arias A, García-Villoria J, Acquaviva C, Vianey-Saban C, Artuch R, García-Cazorla À, Briones P, Ribes A. Mutations in the lipoyltransferase LIPT1 gene cause a fatal disease associated with a specific lipoylation defect of the 2-ketoacid dehydrogenase complexes. Hum Mol Genet. 2014 Apr 1;23(7):1907-15.
- Baker PR 2nd, Friederich MW, Swanson MA, Shaikh T, Bhattacharya K, Scharer GH, Aicher J, Creadon-Swindell G, Geiger E, MacLean KN, Lee WT, Deshpande C, Freckmann ML, Shih LY, Wasserstein M, Rasmussen MB, Lund AM, Procopis P, Cameron JM, Robinson BH, Brown GK, Brown RM, Compton AG, Dieckmann CL, Collard R, Coughlin CR 2nd, Spector E, Wempe MF, Van Hove JL. Variant non ketotic hyperglycinemia is caused by mutations in LIAS, BOLA3 and the novel gene GLRX5. Brain. 2014 Feb;137(Pt 2):366-79.
- Wongkittichote P, Chhay C, Zerafati-Jahromi G, Weisenberg JL, Mian A, Jensen LT, Grange DK. Novel LIAS variants in a patient with epilepsy and profound developmental disabilities. Mol Genet Metab. 2023 Mar;138(3):107373.
- Habarou F, Hamel Y, Haack TB, Feichtinger RG, Lebigot E, Marquardt I, Busiah K, Laroche C, Madrange M, Grisel C, Pontoizeau C, Eisermann M, Boutron A, Chrétien D, Chadefaux-Vekemans B, Barouki R, Bole-Feysot C, Nitschke P, Goudin N, Boddaert N, Nemazanyy I, Delahodde A, Kölker S, Rodenburg RJ, Korenke GC, Meitinger T, Strom TM, Prokisch H, Rotig A, Ottolenghi C, Mayr JA, de Lonlay P. Biallelic Mutations in LIPT2 Cause a Mitochondrial Lipoylation Defect Associated with Severe Neonatal Encephalopathy. Am J Hum Genet. 2017 Aug 3;101(2):283-290.
- Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD, Higgins DG. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011 Oct 11;7:539.
- Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A, Bridgland A, Meyer C, Kohl SAA, Ballard AJ, Cowie A, Romera-Paredes B, Nikolov S, Jain R, Adler J, Back T, Petersen S, Reiman D, Clancy E, Zielinski M, Steinegger M, Pacholska M, Berghammer T, Bodenstein S, Silver D, Vinyals O, Senior AW, Kavukcuoglu K, Kohli P, Hassabis D. Highly accurate protein structure prediction with AlphaFold. Nature. 2021 Aug;596(7873):583-589.
- Ortigoza-Escobar JD. A Proposed Diagnostic Algorithm for Inborn Errors of Metabolism Presenting With Movements Disorders. Front Neurol. 2020 Nov 13;11:582160.
- Zea Vera A, Gropman AL. Surgical treatment of movement disorders in neurometabolic conditions. Front Neurol. 2023 Jun 2;14:1205339.
- Gropman AL. Neuroimaging in mitochondrial disorders. Neurotherapeutics. 2013 Apr;10(2):273-85.
- Set KK, Sen K, Huq AHM, Agarwal R. Mitochondrial Disorders of the Nervous System: A Review. Clin Pediatr (Phila). 2019 Apr;58(4):381-394.
- Brown GK. Congenital brain malformations in mitochondrial disease. J Inherit Metab Dis. 2005;28(3):393-401.
- Ah Mew N, Loewenstein JB, Kadom N, Lichter-Konecki U, Gropman AL, Martin JM, Vanderver A. MRI features of 4 female patients with pyruvate dehydrogenase E1 alpha deficiency. Pediatr Neurol. 2011 Jul;45(1):57-9.
Figure 1.
MRI Brain showed ortical malformations including ventricular colpocephaly, polymicrogyria and possible subependymal heterotopia.
Figure 1.
MRI Brain showed ortical malformations including ventricular colpocephaly, polymicrogyria and possible subependymal heterotopia.
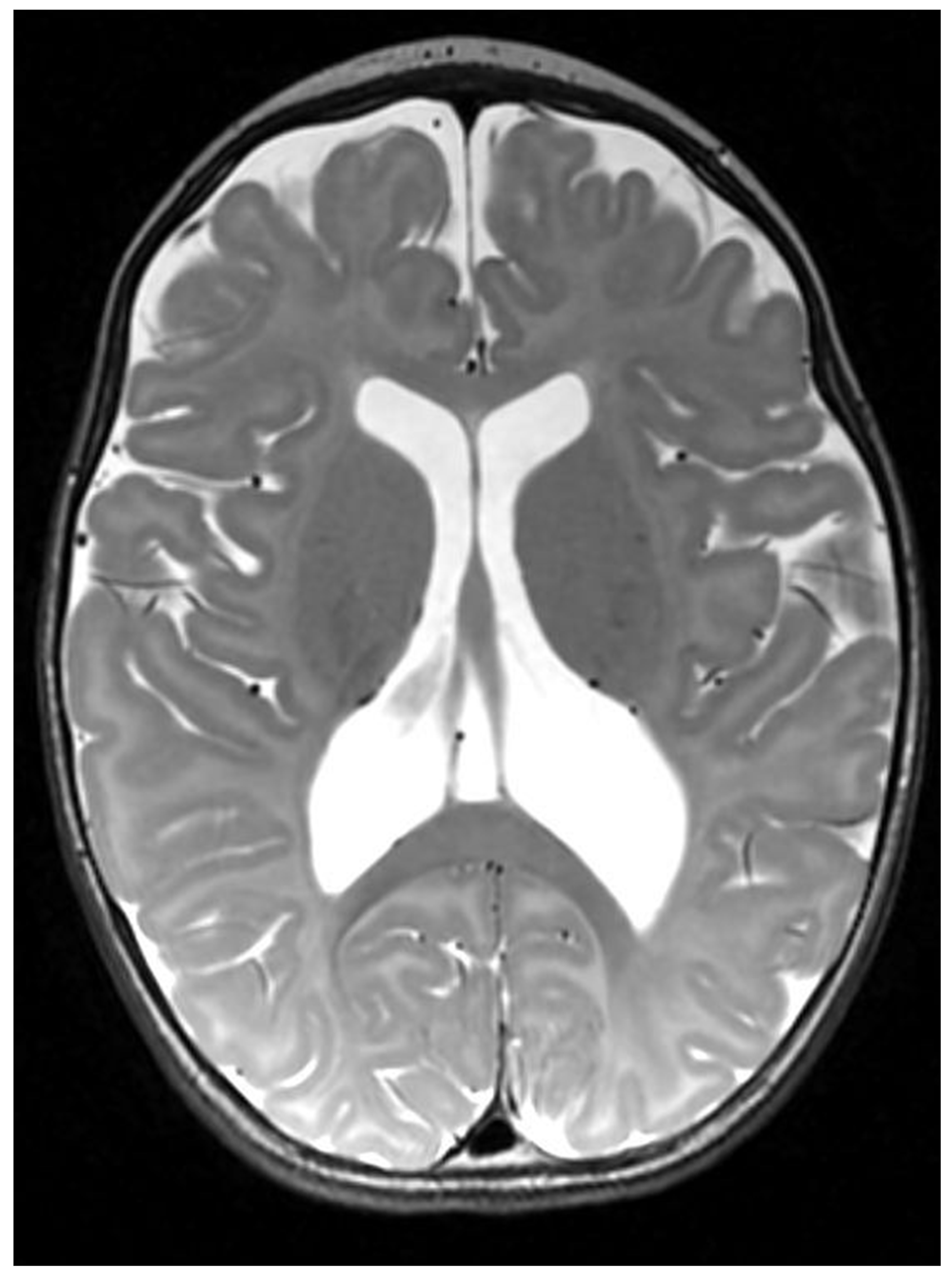
Figure 2.
Growth assay and protein lipoylation investigation for complementation of Δlip2 yeast strains by human wildtype LIPT2 and mutated LIPT2. All plasmids transformed into Δlip2 yeast strain are based on the YEp352 yeast episomal plasmid backbone. A. Growth analysis of wildtype+YEp352, Δlip2+Yep352, Δlip2+YEp352LIPT2, Δlip2+YEp352LIPT2V116L and Δlip2+YEp352LIPT2P140S. Cells were grown for 16 hours in SCD-URA medium, cells were then inoculated to OD600 0.1 in SCD-URA medium and cells were grown for 4-5 hours. OD600 was adjusted to 0.5 and undiluted, 1:10, 1:100 and 1:1000 dilutions were spotted on SCD-URA/YPD media as a general growth control and on SCG-URA media. Cells were incubated at +30°C for 4 days. B. Cells were grown for 16 hours in SCG-URA medium complemented with 0,5% glucose, cells were then inoculated to OD600 0.1 in SCG-URA medium complemented with 0,05% glucose and cells were grown for 6 hours. OD600 was adjusted to 0.5 and undiluted, 1:10, 1:100 and 1:1000 dilutions were spotted on SCD-URA/YPD media as a general growth control and on YPG media. Cells were incubated at +30°C for 4 days. C. Western blot analysis of cell extracts of WT+YEp352, Δlip2+YEp352, Δlip2+YEp352LIPT2, Δlip2+YEp352LIPT2V116L and Δlip2+Yep352LIPT2P140S with anti-LA. Actin and Ponceau were used as loading controls. Cells were grown for 16 hours in SCD-URA medium and then inoculated in YPG medium for 24 hours. D. Cells were grown for 16 hours in SCG-URA medium complemented with 0.5% glucose and then inoculated in SCG-URA medium complemented with 0.05% glucose for 24 hours.
Figure 2.
Growth assay and protein lipoylation investigation for complementation of Δlip2 yeast strains by human wildtype LIPT2 and mutated LIPT2. All plasmids transformed into Δlip2 yeast strain are based on the YEp352 yeast episomal plasmid backbone. A. Growth analysis of wildtype+YEp352, Δlip2+Yep352, Δlip2+YEp352LIPT2, Δlip2+YEp352LIPT2V116L and Δlip2+YEp352LIPT2P140S. Cells were grown for 16 hours in SCD-URA medium, cells were then inoculated to OD600 0.1 in SCD-URA medium and cells were grown for 4-5 hours. OD600 was adjusted to 0.5 and undiluted, 1:10, 1:100 and 1:1000 dilutions were spotted on SCD-URA/YPD media as a general growth control and on SCG-URA media. Cells were incubated at +30°C for 4 days. B. Cells were grown for 16 hours in SCG-URA medium complemented with 0,5% glucose, cells were then inoculated to OD600 0.1 in SCG-URA medium complemented with 0,05% glucose and cells were grown for 6 hours. OD600 was adjusted to 0.5 and undiluted, 1:10, 1:100 and 1:1000 dilutions were spotted on SCD-URA/YPD media as a general growth control and on YPG media. Cells were incubated at +30°C for 4 days. C. Western blot analysis of cell extracts of WT+YEp352, Δlip2+YEp352, Δlip2+YEp352LIPT2, Δlip2+YEp352LIPT2V116L and Δlip2+Yep352LIPT2P140S with anti-LA. Actin and Ponceau were used as loading controls. Cells were grown for 16 hours in SCD-URA medium and then inoculated in YPG medium for 24 hours. D. Cells were grown for 16 hours in SCG-URA medium complemented with 0.5% glucose and then inoculated in SCG-URA medium complemented with 0.05% glucose for 24 hours.
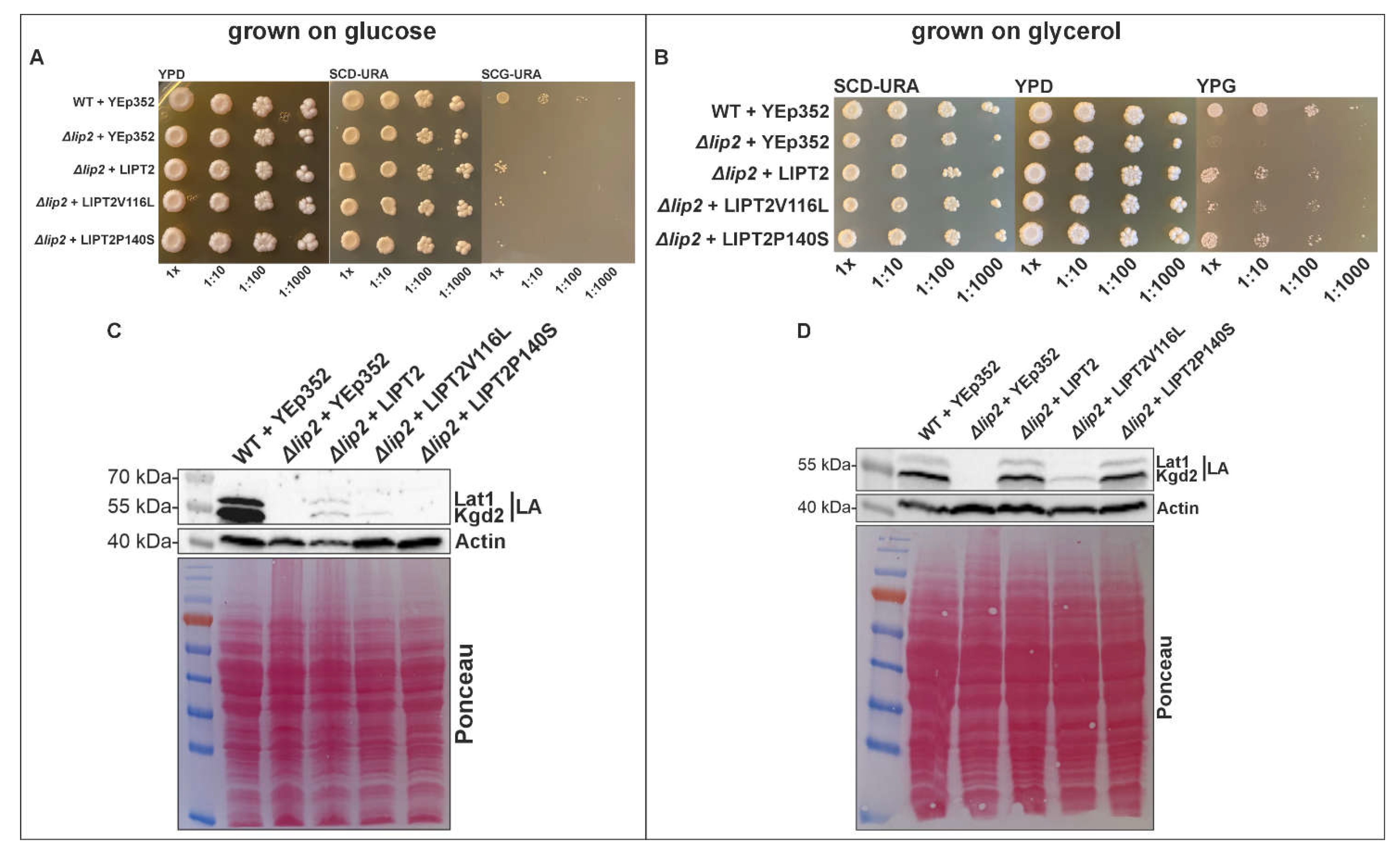

Table 1.
Primers used for site-directed mutagenesis.
| Name of the primer | Sequence 5´-3´ |
|---|---|
| LIPT2_G346T_Forward | CGCTTGCGCATGCACTTAGCGTCGCTGG |
| LIPT2_G346T_Reverse | CCAGCGACGCTAAGTGCATGCGCAAGCG |
| LIPT2_C418T_Forward | CGCGCGGCCCTCGCCCTACACTGGC |
| LIPT2_C418T_Reverse | GCCAGTGTAGGGCGAGGGCCGCGCG |
| Patient 1 (Habaro et al.) | Patient 2 (Habaro et al.) | Patient 3 (Habaro et al.) | Proband | |
|---|---|---|---|---|
| Ethnicity | French/Ivory Coast | German | German | Chinese/Vietnamese |
| Clinical Presentation | Truncal hypotonia, spastic tetraplegia and dystonia, epileptic encephalopathy | Severe hypotonia, developmental delay | Severe hypotonia, developmental delay, epilepsy | Severe generalized dystonia, global developmental delay, feeding difficulties and failure to thrive |
| MRI Findings | Supra-tentorial cortical atrophy with ventricular dilatation, bi-frontal white matter abnormalities, and delayed myelination | Enlarged lateral ventricles and formation of cysts in the cortex and white matter of the whole cerebral structures | Periventricular cystic changes | Cortical malformations including ventricular colpocephaly, polymicrogyria and possible subependymal heterotopia |
| Biochemical characteristics | Hyperlactatemia with a high lactate/pyruvate ratio moderate hyperglycinemia, increased alanine and decreased branched-chain amino acids |
Lactic acidosis up to 17 mmol/L at birth | Lactic acidosis 3.7 mmol/L at birth and increased to values between 8 and 10 Elevation of alanine and proline, moderate increase of glycine, and decrease of branched-chain amino acids |
Lactic acidosis upto 6.1 mmol/L, normal plasma amino acid |
| Molecular Variants | c.89T>C and c.377T>G in trans | c.314T>G (p.Leu105Arg) and c.377T>G (p.Leu126Arg) in trans | c.314T>G (p.Leu105Arg) and c.377T>G (p.Leu126Arg) in trans | c.346 G>T (p.V116L) and c.418C>T (p.P140S) in trans |
| Outcome | Alive with no episodes of metabolic decompensation | Died within first month of life | Died within first month of life | Alive with severe dystonia but no episodes of metabolic decompensation |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



