
Submitted:
09 May 2024
Posted:
10 May 2024
You are already at the latest version
Abstract
(1) Background: Multiple alterations of cellular metabolism have been documented in experimental studies of autosomal dominant polycystic kidney disease (ADPKD) and are thought to contribute to its pathogenesis. (2) Methods: To elucidate the molecular pathways and transcriptional regulators associated with the metabolic changes of renal cysts in ADPKD, we compared global gene expression data from human PKD1 renal cysts, minimally cystic tissues (MCT) from the same patients, and healthy human kidney cortical tissue samples. (3) Results: We found PKD1 renal cysts displayed the Warburg effect with gene pathway changes favoring increased cellular glucose uptake and lactate production, instead of pyruvate oxidation. Additionally, mitochondrial energy metabolism was globally depressed, associated with downregulation of gene pathways related to fatty acid oxidation (FAO), branched-chain amino acid (BCAA) degradation, the Krebs cycle, and oxidative phosphorylation (OXPHOS) in renal cysts. Activation of mTORC1 and its two target proto-oncogenes, HIF-1α and MYC, was predicted to drive the expression of multiple genes involved in the observed metabolic reprogramming (e.g., GLUT3, HK1/HK2, ALDOA, ENO2, PKM, LDHA/LDHB, MCT4, PDHA1, PDK1/3, MPC1/2, CPT2, BCAT1, NAMPT); indeed, their predicted expression patterns were confirmed by our data. Conversely, we found AMPK inhibition was predicted in renal cysts. AMPK inhibition was associated with decreased expression of PGC-1α, a transcriptional coactivator for transcription factors PPARα, ERRα, and ERRγ, all of which play a critical role in regulating oxidative metabolism and mitochondrial biogenesis. (4) Conclusions: These data provide a comprehensive map of metabolic pathway reprogramming in ADPKD and highlight nodes of regulation that may serve as targets for therapeutic intervention.
Keywords:
ADPKD
; renal cysts
; global gene profiling
; metabolic reprogramming
1. Introduction
ADPKD is the most common hereditary kidney disease worldwide with an estimated cumulative lifetime prevalence of ~1 in 1000 [1]. Progressive increase in cyst number and size results in the distortion of normal kidney architecture and ultimately end-stage renal disease in the majority of patients [2]. Mutations of two genes, PKD1 and PKD2, account for 75-85% and 15-25% of the genetically resolved cases, respectively [3,4,5,6]. Recent advances have led to the discovery of multiple therapeutic targets in preclinical studies of ADPKD. Among them, aberrant mTORC1 activation and increased cAMP signaling in cystic tissues are two highly promising pathogenic mechanisms driving cyst growth in ADPKD [7,8]. Both have been experimentally validated and clinically tested as therapeutic targets [7,9,10,11]. However, only vasopressin V2 receptor inhibition by Tolvaptan, which lowers cystic cellular cAMP, has been found to be effective and safe by clinical trials, and has become the first disease-modifying therapy in ADPKD.
As recently reviewed [12,13,14,15,16,17], multiple experimental studies have highlighted a pathogenic role of metabolic reprogramming in ADPKD. Increased aerobic glycolysis [18] and sirtuin 1 (SIRT1) activity [19], reduced AMPK activity [18,20,21,22], mitochondrial dysfunction [18,23,24,25,26,27,28], enhanced reactive oxygen species (ROS) production [27], oxidative stress [24,27,29,30,31,32,33], lipid peroxidation [30,33], defective FAO [34,35], increased glutamine usage [36,37], and arginine auxotrophy [38] have been observed both in vitro and in vivo in animal models of ADPKD or in the tissues of patients with ADPKD. Importantly, targeting metabolic reprogramming defects in ADPKD has been shown to ameliorate cystic disease progression in rodent and non-rodent models [12,13,14,15,16,17].
Repurposing drugs targeting cellular metabolism for the treatment of ADPKD would bypass much of the cost and time associated with novel drug discovery and development [39,40]. For instance, the reliance of Pkd1 null cells/cystic tissues on glucose for growth and proliferation has led to the use of 2-deoxyglucose as a novel experimental treatment in ADPKD [41,42,43]. Similarly, AMPK is a master metabolic regulator that has been targeted for the treatment of various pathological entities such as obesity, diabetes, inflammation, and cancer [44,45,46,47]. Accumulating evidence suggests that AMPK activation (using metformin, salsalate, 2-deoxyglucose, or diet) may restore mitochondrial function and slow cystogenesis by inhibiting mTORC1 and cystic fibrosis transmembrane conductance regulator (CFTR) in the cystic kidney [16,20,22,41,42,43,48,49,50,51,52]. In animal models, the PPARα agonist fenofibrate enhances FAO and attenuates polycystic kidney and liver disease in mice [35], and inhibitors of glutamine metabolism retard disease progression [37,53]. These preclinical findings demonstrate the pivotal importance of better understanding the interacting metabolic irregularities in ADPKD to identify potential therapeutic targets.
Previously, we performed a systems biology analysis to discover upregulated gene pathways and key transcription factors associated with renal cyst growth in human ADPKD [54]. Of the 637 pathways tested, 212 (128 up- and 84 downregulated) pathways were enriched in renal cysts compared to MCT control. We found that PKD1 renal cysts displayed a rich network of upregulated signaling pathways for mitogenic responses, including receptor tyrosine kinases (e.g., IGF/IGF1R, FGF/FGFR, EGF/EGFR, VEGF/VEGFR), G-protein-coupled receptors, and intracellular cascades involved in calcium, cAMP and mTORC1 signaling [54]. Here we performed the complimentary analysis of gene sets that were downregulated in PKD1 renal cysts, the majority (77/84) of which were found to be involved in metabolic reprogramming. These data support efforts toward novel therapeutics targeting the key regulators of metabolic reprogramming in ADPKD.
2. Results
2.1. Metabolic Pathway Analysis of PKD1 Renal Cysts
We used Gene Set Enrichment Analysis (GSEA) to identify dysregulated signaling pathways [55]. The gene sets in the GSEA Molecular Signatures Database (MSigDB) are highly overlapping; Kyoto Encyclopedia of Genes and Genomes (KEGG) is a collection of manually drawn pathways representing experimental knowledge on metabolism and various other functions of the cell; the best organized part of the KEGG pathway database is metabolism [56]. In order to reduce the redundancy among the enriched gene sets, we performed GSEA on 186 gene sets from the GSEA C2 KEGG pathway database [55]. At a nominal P-value (NOM P-value) ≤ 0.01 with a false discovery rate (FDR) ≤ 0.1, we found 75 pathways were dysregulated (30 up- and 45 downregulated) in the renal cysts (Table 1). Replicating our previous results [54], the upregulated gene sets in PKD1 renal cysts displayed a rich signature of mitogen-mediated proliferation. By contrast, of the 45 downregulated pathways, 39 represented metabolic pathways or their regulators.
Table 1 Size is the total number of genes in a given gene set. NES represents degree of enrichment of the gene set at the top or bottom of the ordered gene list. NOM P-value measures the significance of NES for a gene set by using permutation testing. The FDR is the estimated probability that a set with a given NES represents a false positive finding. Pathways marked with an asterisk (*) can entirely or mostly be found within mitochondria. Detailed descriptions of each pathway can also be found on the GSEA Molecular Signatures Database (MSigDB) website: http://software.broadinstitute.org/gsea/msigdb/genesets.jsp?collection=CP:KEGG.
2.1.1. Human PKD1 Renal Cysts Display the Warburg Effect
Aerobic glycolysis or the ‘Warburg effect’, a hallmark of cancer or proliferative tissues [18,57], has been observed in animal models and human cystic kidney tissues. KEGG pathway analysis identified glycolysis/gluconeogenesis, pentose phosphate pathway (PPP), and pyruvate metabolism as downregulated in human PKD1 renal cysts (Table 1). More detailed analysis revealed that key enzymes of gluconeogenesis were highly downregulated, while glycolytic enzymes were moderately upregulated in renal cysts (Figure 1).
To determine whether renal cysts display the Warburg effect, we first checked changes in the expression of multiple key sequential regulatory points in these pathways. Glucose transporter GLUT3, which plays a major role in the enhanced glucose uptake by many cancer cells [58], was upregulated 40x in renal cysts compared to MCT. In addition, enzymes for irreversible steps of glycolysis were upregulated in renal cysts, including hexokinase 1 and 2 (HK1, 1.7x; HK2, 6.2x), and pyruvate kinase (PKM, 1.6x). Genes encoding enzymes for lactate fermentation and export were also upregulated in renal cysts. Lactate dehydrogenase is a tetrameric enzyme consisting of differing ratios of LDHA and LDHB subunits, with LDHA having a higher affinity for pyruvate, and LDHB having a higher affinity for lactate [58]. The upregulation of LDHA (1.4x) and downregulation of LDHB (-1.8x) in renal cysts suggest increased pyruvate to lactate flux. The carrier that exports glycolysis-derived lactate, MCT4, which is predominantly expressed in glycolytic tissues [58], was upregulated (2.9x) in renal cysts (Figure 1, left). On the other hand, the MPC1 (-1.5x)/MPC2 (-2.1x) heterodimer responsible for transporting pyruvate into the mitochondria for ATP production [59] was downregulated in renal cysts. Concurrently, multiple genes in the mitochondrial pyruvate dehydrogenase complex (PDC) were downregulated in renal cysts. The PDC acts as a rate-limiting enzyme that catalyzes the irreversible conversion of pyruvate into acetyl coenzyme A (acetyl-CoA), providing the primary link between glycolysis and the Krebs cycle [60]. Downregulated genes include PDHA1 (-2.8x), DLAT (-1.6x), and DLD (-1.7x). The activity of the PDC is regulated by the PDHA1 subunit; its phosphorylation by PDH kinases (PDKs) leads to a strong decrease in PDC activity [60]. The upregulation of PDK1 (3.3x) and PDK3 (1.5x) in renal cysts suggests the inhibition of PDC activity, and therefore decreased conversion of pyruvate to acetyl-CoA for oxidative metabolism (Figure 1, bottom).
The PPP branches from glycolysis at the first committed step of glucose metabolism to provide the precursors for nucleotide and amino acid biosynthesis. It is the major source of nicotinamide adenine dinucleotide phosphate (NADPH) for the reduction of glutathione (GSH) and fatty acid biosynthesis [58]. Although the PPP pathway was identified as downregulated, we found G6PD, encoding the rate-limiting enzyme for the irreversible oxidative phase of the PPP, was upregulated (2.1x) in renal cysts. The genes contributing to downregulation of the PPP either encoded enzymes of the reversible non-oxidative phase of the PPP, or shared enzymes involved in glycolysis/gluconeogenesis. Among these reversible enzymes shared by the glycolysis/gluconeogenesis and PPP pathways, we found an isoform switch of aldolases in renal cysts. ALDOA has a high affinity for fructose-1,6-BP and favors glycolysis, whereas ALDOB has a higher affinity for glyceraldehyde3-P and dihydroxyacetone phosphate and favors gluconeogenesis [58]. The observed substantial downregulation of ALDOB (-233x) and upregulation of ALDOA (1.7x) further support increased glycolytic flux in renal cysts. Taken together, these results suggest that instead of fully oxidizing glucose, the PKD1 renal cysts shuttle glucose through aerobic glycolysis and the PPP in order to sustain cell growth and proliferation.
2.1.2. Inhibition of Gluconeogenesis
Gluconeogenesis is the process of generating glucose from non-carbohydrate carbon substrates such as lactate, glycerol and amino acids [61]. The kidney is the only organ other than the liver able to perform gluconeogenesis [62]. Gluconeogenesis and glycolysis share many reversible enzymes. However, gluconeogenesis uses 4 distinct reactions to bypass the 3 metabolically irreversible reactions of glycolysis. The enzymes catalyzing these irreversible reactions are the potential sites for regulatory control [61]. We found that 5 genes encoding the 4 enzymes that catalyze the irreversible reactions of gluconeogenesis were all downregulated in renal cysts, including pyruvate carboxylase (PC, -3.4x), phosphoenolpyruvate carboxykinase (PCK1, -43.4x; PCK2, -3.9x), fructose 1,2-bisphosphatase (FBP1, -7.2x), and glucose 6-phosphate phosphatase (G6PC, -7.3x). Notably, FBP1 is the rate-limiting enzyme during gluconeogenesis. In addition, GLUT2, a glucose transporter normally enriched in the kidney that is responsible for glucose export, was greatly downregulated (-20x) in renal cysts (Figure 1, right). Overall, these data suggest that gluconeogenesis is downregulated in renal cysts.
2.1.3. Downregulation of Mitochondrial Catalytic Pathways in Renal Cysts
In normal cells, mitochondrial acetyl-CoA derived from glycolysis, fatty acids, or BCAAs is fed into the Krebs cycle, followed by OXPHOS for high-efficiency ATP generation [63]. Consistent with defective mitochondrial metabolism in ADPKD, 7 of the 10 most downregulated pathways in cystic tissue occur predominantly in the mitochondria. These included BCAA degradation, pyruvate metabolism, fatty acid metabolism, propanoate metabolism, butanoate metabolism, the Krebs cycle, and OXPHOS (Table 1). Most individual genes within these mitochondrial metabolic pathways were also downregulated in renal cysts (Figure 2a-2d).
BCAAs (i.e., valine, leucine, and isoleucine) are essential amino acids that play a crucial role in activating mTORC1 [64]. BCAA supplementation has been shown to accelerate the ADPKD progression in mice through mTORC1 and MAPK/ERK activation [65]. Among the downregulated pathways, BCAA degradation was identified as the most downregulated pathway with 32 differentially expressed genes (31 down, 1 up) in renal cysts (Figure 2a). The one upregulated gene was BCAT1 (5.3x), which catalyzes the only step in BCAA degradation that occurs outside of the mitochondria, and is the major isoform implicated in cancer growth [64]. In contrast, all 31 downregulated genes encode multiple sequential mitochondrial enzymes in the catabolism of BCAA, suggesting defective mitochondrial BCAA degradation in renal cysts.
Fatty acid metabolism was another highly downregulated pathway, with 26 differentially expressed genes (25 down, 1 up). FAO, which occurs in the mitochondria and peroxisomes, and is the preferred energy source for renal tubular epithelial cells [66]. Of interest, all 25 downregulated genes in this pathway encode enzymes in fatty acid degradation. Notably, CPT2, encoding one of the rate-limiting enzymes for transferring fatty acids into the mitochondria during FAO, was downregulated (-1.6x) in renal cysts. Concurrently, peroxisome metabolism was also identified among the top downregulated pathways in renal cysts (Table 1). On the other hand, CD36, encoding a multifunctional receptor that mediates the binding and cellular uptake of long-chain fatty acids, was greatly upregulated (12.3x), consistent with the upregulation of CD36 in the setting of chronic kidney disease (CKD) [67]. These data suggest increased uptake and reduced catabolism of fatty acids. Together these would cause aberrant intracellular lipid accumulation, which has a demonstrated role in the pathogenesis of kidney injury and fibrosis [34,67,68].
2.1.3. Alteration of GSH Synthesis and GSH-Dependent Antioxidant Response in Renal Cysts
Oxidative damage, as measured by lipid peroxidation [69], has been shown to be greatly elevated in the cystic kidney [30,33], and to drive renal cyst growth by activating the anoctamin 1 (ANO1) [33,70]. Indeed, increased expression of ANO1 (3.7x) was observed in our PKD1 renal cysts. Along with evidence of oxidative damage, we found impairment of the GSH-dependent system, which is critical in antioxidant response [71,72]. Our pathway analysis revealed that GSH metabolism, as well as drug metabolism via multiple enzymes including cytochrome P450, were all downregulated in human PKD1 renal cysts (Table 1).
The kidney salvages circulating GSH through the γ-glutamyl cycle, which breaks down extracellular GSH to provide cysteine, the rate-limiting substrate, for intracellular de novo synthesis of GSH [71,73]. We found multiple genes encoding enzymes in the γ-glutamyl cycle were highly downregulated in renal cysts, including γ-glutamyl transferase (GGT1, -14x), dipeptidase 1 (DPEP1, -32.5x), aminopeptidase N (ANPEP, -12x) and 5-oxoprolinase (OPLAH, -2.6x). Levels of GSH biosynthetic enzymes were also downregulated in renal cysts, including the catalytic subunit of the rate-limiting enzyme glutamyl cysteine ligase (GCLC, -2.5x), glutathione synthetase (GSS, -1.9x), and glutathione reductase (GSR, -2.3x) (Figure 3). Aside from the γ-glutamyl cycle, cysteine can be also produced from extracellular cystine through the xCT antiporter encoded by SLC7A11, which is known to maintain the cysteine pool in many cancer cells [74,75]. We found SLC7A11 was expressed very low levels in MCT, but was upregulated (4.9x) in renal cysts. Cells can also synthesize cysteine de novo from methionine-derived homocysteine using the trans-sulfuration pathway [75]. Expression levels of some enzymes in the trans-sulfuration pathway were reduced, while others were unaltered (Figure 3).
GSH exerts its antioxidant function directly, by interacting with ROS and electrophiles, or by serving as a cofactor for various antioxidant enzymes [69,72]. In renal cysts, we also identified the dysregulation of genes encoding multiple GSH-linked antioxidant enzymes, including superoxide dismutase (SOD1, -1.3x; SOD2, 1.8x), catalase (CAT, -1.6x), and glutathione S-transferase (GSTA1, -58x; GSTA3, -3.5x; GSTK1, -2,2x; GSTM5, 2.8x; GSTO1, 1.9x), glutathione peroxidase (GPX7 (1.9x), GPX8 (4.1x)), glutaredoxin (GLRX (-2.1x)), and peroxiredoxin (PRDX1, -1.2x; PRDX3, -1.7x; PRDX4, 2.8x; PRDX6, 1.4x). Of interest, GSTT1, encoding glutathione S-transferase theta 1, was consistently overexpressed in both MCT (27x) and renal cysts (33x) relative to normal kidneys. Taken together, these results suggest aberrant GSH synthesis and GSH-dependent antioxidant response in PKD1 renal cysts.
2.2. In Silico Prediction of Key Transcriptional Regulators Based on Differentially Expressed Genes
To discover potential transcriptional regulators responsible for metabolic dysregulation in PKD, we applied our differentially expressed genes with at least 1.5x changes (up: 3142; down: 1690) to Upstream Regulator Analysis (URA) in the Ingenuity® Pathway Analysis (IPA®) software. URA predicted 102 activated and 48 inhibited transcriptional regulators with z-scores ≥ 2 or ≤ -2. Overall, there is excellent concordance between our results from the pathway and URA analyses. The top 50 most activated and 48 most inhibited transcriptional regulators in the renal cysts are shown in Table 2. Many of the predicted transcriptional regulators were differentially expressed in renal cysts compared with MCT.
Table 2 The bias-corrected z-score is used to infer the activation states of upstream regulators. It is calculated from the proportions of genes that are differentially regulated in an expected direction based on the known interactions between the regulator and the genes present in the Ingenuity database. z-scores ≥ 2 or ≤ -2 are considered to be either activated or inhibited, respectively. The p-value of overlap is the calculated statistical significance of overlap between genes from the dataset and genes that are known to be regulated by the regulator using Fisher’s exact test. Gene expression direction is not taken into account for this calculation.
The most upregulated transcriptional regulators were associated with the activation of TGFβ, growth factor/receptor tyrosine kinase, Wnt/β-catenin, hypoxic, and immune/inflammatory response pathways in PKD1 renal cysts, consistent with our previous study [54]. In contrast, many of the top inhibited transcriptional regulators were associated with metabolism and development. As expected, URA predicted PKD1 (z-score = -7.8) as the most inhibited protein. Consistent with our previous results [54], hepatocyte nuclear factor family members HNF1α (z-score = -7.3) and HNF4α (z-score = -5), which regulate glucose homeostasis and tissue-specific gene expression, were again predicted to be highly inhibited and both were indeed downregulated in renal cysts. The inhibition of HNF4α also supports experimental work in a Pkd1 mouse model that identified Hnf4a as a key disease modifier [76].
Multifunctional metabolic sensors, including mTORC1, SIRT1, and AMPK, act under a network of cooperative signaling cascades. AMPK is one of the master coordinators of cell energy homeostasis, growth, and metabolism [44,45,46,47]. Of interest, URA predicted the moderate inhibition of AMPKα2 (z-score = -2.1). At the mRNA level, although no significant change was observed in the expression of PRKKA2 (encoding AMPKα2), we found slightly increased expression of PRKAA1 (1.5x, encoding AMPKα1) in human PKD1 renal cysts, consistent with the isoform shift in the catabolic subunit of AMPK from AMPKα2 to AMPKα1 in renal fibrosis [77,78,79]. Other transcriptional regulators that were predicted to be most inhibited in renal cysts included PGC-1α (z-score = -4.9), PPARα (z-score = -3.4) and ERRα (z-score = -3.2). These act under a network of cooperation: AMPK can inhibit mTORC1 and activate PGC-1α, whereas PGC-1α acts as a transcriptional coactivator for PPARα and estrogen-related receptors (e.g., ERRα and ERRγ), which promote the expression of genes in OXPHOS, FAO, the Krebs cycle, and mitochondrial biosynthesis [80,81,82]. Concordantly, the genes encoding PPARα and estrogen-related receptors (PPARGC1A, PPARA, ESRRA and ESRRG) were all downregulated in renal cysts.
3. Discussion
As one of the most metabolically active organs in the body, the kidney has an abundance of mitochondria to provide sufficient energy for waste filtration, salt-water balance, and electrolyte homeostasis [83,84]. Healthy renal tubular epithelial cells rely on FAO and OXPHOS as their main energy source [66]. In ADPKD, there are reductions in mitochondrial biogenesis, OXPHOS, and FAO, with cells instead relying on aerobic glycolysis (the Warburg effect) to produce energy. Concomitantly, there is decreased AMPK and increased mTORC1 activity.
In this study, we found that human PKD1 renal cysts, regardless of their tubular origins, displayed the Warburg effect and had globally depressed mitochondrial oxidative metabolism. Of all pathways involved, mTORC1 and AMPK are two central regulators of energy metabolism, cell growth, and proliferation with opposing effects [44,45,46,47]. mTORC1 integrates signals from growth factors, energy status, oxygen, and amino acid availability to promote anabolic processes and cell growth [44,45,46,47]. mTORC1 also activates two key transcription factors: MYC and HIF-1α [85,86], causing increased expression of genes in aerobic glycolysis (e.g., glucose transporters, glycolytic enzymes) and inhibiting the mitochondrial TCA cycle and OXPHOS. Mitochondrial dysfunction in ADPKD further contributes to reduced FAO and OXPHOS and leads to increased ROS production, causing lipid peroxidation and tissue damage. This is further exacerbated by increased lipid uptake. Activation of ANO1 by lipid peroxidation drives the proliferation and expansion of renal cysts [33,70]. Therefore, restoring mitochondrial homeostasis and function may be beneficial for the treatment of ADPKD.
A target of particular interest is AMPK, a major cellular energy sensor driving catabolic processes which has received a lot of attention as a treatment target in diseases with underlying metabolic perturbations [44,45,46,47]. AMPK is highly expressed in the kidney and is involved in the regulation of a variety of physiological and pathological processes, including ion transport, podocyte function, renal fibrosis, diabetic renal hypertrophy, and polycystic kidney disease [12,13,16,17,87,88,89]. The AMPK molecule is a heterotrimeric complex composed of a catalytic α subunit, and regulatory β and γ subunits, each of which has multiple isoforms (α1/α2, β1/β2, γ1/γ2/γ3) [44,45,46,47]. In renal fibrosis, AMPKα1 plays a deleterious role, whereas AMPKα2 is protective [77,78,79,90]. Fibrosis and inflammation are common findings in ADPKD, and indeed, we found the gene encoding AMPKα1 to be upregulated in human PKD1 renal cysts. Given the protective role of AMPKα2 and deleterious role of AMPKα1 in the kidney, we hypothesize that selective activation of AMPKα2-containing isoforms may have the potential to slow ADPKD progression.
An additional function of AMPK is the regulation of PGC-1α by multiple direct and indirect mechanisms [46,47]. As the master regulator of mitochondrial biogenesis, PGC-1α is a transcriptional coactivator interacting with many transcription factors, including PPARα, ERRα, and ERRγ, to stimulate the expression of genes involved in FAO, OXPHOS, and mitochondrial DNA transcription and replication [80,81,82]. Mitochondrial dysfunction along with decreased PGC-1α activity is a common feature of acute kidney injury (AKI) and CKD, and its pharmaceutical activation has renoprotective effects in both [91,92,93]. PGC-1α is also downregulated in murine and human cystic kidney cells and tissues [27,35,50,52,94]. Thus, increasing PGC-1α expression or activity may be a promising approach to restore mitochondrial metabolism and attenuate injury and fibrosis in ADPKD. As an upstream regulator, activation of AMPK would be one method to achieve this.
Regulators of FAO and OXPHOS, both of which are deficient in ADPKD, that were highlighted by our analysis include PPAR𝛼, ERRα and ERRγ. PPAR𝛼 is the master regulator of lipid metabolism, controlling mitochondrial, peroxisomal and microsomal FAO [95]. Notably, fenofibrate, a PPARα agonist, was found to increase FAO and attenuate cystic kidney and liver disease in Pkd1RC/RC mice [35]. Both ERRα and ERRγ are orphan nuclear receptors that regulate mitochondrial biogenesis and OXPHOS. Genetic ERRα deficiency leads to abnormal mitochondrial morphology and increases susceptibility to cisplatin-induced AKI in mice [96]. In addition to regulating mitochondrial OXPHOS/FAO functions, ERRγ also cooperates with HNF1β to activate the expression of renal reabsorption genes including PKD2; deletion of ERRγ in renal tubular epithelial cells results in renal cysts [97].
In parallel to these metabolic changes, evidence from experimental studies in humans and animals suggests that oxidative stress is increased in ADPKD. The mechanisms underlying oxidative damage remain incompletely understood [24,29,30,31,32,33]. Of interest, GSH depletion with L-buthionine-sulfoximine, a specific inhibitor of γ-glutamylcysteine synthetase, caused a marked aggravation of renal cystic disease in a rat model of ADPKD [29]. Our transcriptome profiling in human cysts revealed defective GSH metabolism and a highly downregulated γ-glutamyl cycle. Consistent with our findings, recent integrated transcriptome and metabolome profiling inPkd1 mutant mouse kidneys also showed strongly decreased expression of GGT1 and DPEP1, and a striking decrease of multiple γ-glutamyl amino acids, which are the direct products of GGT1 [36]. This indicates that the defective γ-glutamyl cycle pathway in ADPKD is strikingly similar between humans and mice. However, although both GGT1 and DPEP1 were found to be greatly inhibited, the levels of cysteine (the direct product of DPEP1), which acts both as a building block for protein translation and as the rate-limiting substrate for GSH synthesis, were not altered, and the levels of GSH were strikingly increased (39x) inPkd1 mutant mouse kidneys [36]. Since GSH is an important ROS scavenger, the increased GSH levels could be considered the main strategy used by renal cysts to overcome ROS stress and prevent oxidative stress-induced cell death.
Our data suggest that Pkd1 mutant cells reprogram their cysteine production to enhance intracellular GSH synthesis through xCT to compensate for the defective γ-glutamyl cycle pathway. The cystine-glutatmate antiporter xCT is upregulated in a variety of cancers for cystine uptake and GSH production. Recent studies revealed that xCT also plays a critical role in the glucose and glutamine dependency of cancer cells, and inhibition of xCT activity is emerging as a promising antiproliferative therapeutic strategy [74,75]. We hypothesize that increased expression of xCT could be an important mechanism of cysteine recruitment for the proliferation of PKD1 renal cysts.
A previous study revealed that NAD+-dependent enzyme SIRT1 to be upregulated and involved in the pathophysiology of a mouse model of ADPKD [19]. Consistent with this, we also found increased expression of SIRT1 (1.4x) in human PKD1 renal cysts. In humans, NAD+ is synthesized via two major pathways: via de novo NAD+ biosynthesis and via the NAD+ salvage pathway. Although we found no definitive enrichment of this pathway, we did observe upregulation of NAMPT (2.9x) and downregulation of QPRT (-12.5x) (Figure 2e), the rate-limiting enzymes in the NAD+ salvage and de novo synthesis pathways, respectively [98]. These data suggest that renal cysts may favor the salvage over the de novo pathway to produce NAD+ for a variety of NAD+-dependent enzymes including SIRT1.
4. Materials and Methods
The tissue samples, RNA extraction, microarray procedure, and bioinformatics analysis used in this study have been described previously (GEO ID: GSE7869) [54]. In brief, by using Affymetrix HG-U133 Plus 2.0 arrays (Affymetrix, Santa Clara, CA), global gene profiling was performed on cysts of different sizes (<1 ml, n = 5; 10–20 ml, n = 5; >50 ml, n = 3) and minimally cystic tissue (MCT, n = 5) from five PKD1 human polycystic kidneys, and normal kidney cortical tissue samples (n=3), respectively.
4.1. Pathway Analysis
We used Gene Set Enrichment Analysis (GSEA) (http://software.broadinstitute.org/gsea/index.jsp) to identify dysregulated signaling and metabolic pathways that may modulate renal cyst growth [55]. Before running GSEA, Affymetrix probe sets were collapsed to one gene level by Partek Genomics Suite 6.6 (Partek Inc., Chesterfield, MO, USA) and t-test statistics scores were used to create a ranked list of genes of the entire data set (in total, 22486 unique genes with gene symbols). GSEA was performed using 186 gene sets from the GSEA C2 KEGG pathway database (MSigDB database v6.2 updated July 2018), which has a comprehensive collection of metabolic pathways. We defined overrepresented pathways by a NOM P-value ≤ 0.01 with an FDR ≤ 10%.
4.2. Upstream Regulator Analysis (URA)
The Upstream Regulator Analysis (URA) feature within the Ingenuity® Pathway Analysis (IPA®, QIAGEN) was utilized to infer potential upstream transcriptional regulators influencing gene expression in our microarray dataset. This analysis uses known relationships documented in the Ingenuity® Knowledge Base based on prior scientific findings of the interactions between transcriptional regulators and their target genes. Specifically, the URA algorithm identifies transcriptional regulators whose known target genes are significantly represented in the dataset and assesses the concordance of the observed gene expression changes (upregulation or downregulation) with the expected effects if these regulators were active in order to predict the transcriptional regulatory networks influencing the observed gene expression patterns. For each potential transcriptional regulator, two statistical measures - an activation z-score and an overlap p-value - are computed. The z-score and bias-corrected z-score are computed to infer the activation states of upstream regulators. An overlap p-value is computed by Fisher's exact test based on significant overlap between genes in the dataset and known targets regulated by the transcriptional regulator [99]. We used Significance Analysis of Microarrays analysis to identify differentially expressed genes with an FDR ≤ 1% [100]. The top differentially expressed genes with a minimum fold-change of ±1.5 (Cyst vs. MCT) were applied to URA to predict the transcriptional regulators. A bias-corrected z-score ≥ 2 (activated) or ≤ -2 (inhibited) was considered significant.
5. Conclusions
In conclusion, the present analysis highlights a complex rewiring of energy metabolism in human PKD1 renal cysts. Metabolism in cysts is directed toward the generation of metabolic intermediates to support cellular proliferation, rather than efficient extraction of ATP through OXPHOS. We have generated a comprehensive map of key metabolic pathways and regulators altered in PKD1 renal cysts (Figure 4). Despite the complexity, redundancy, and crosstalk between these pathways, it is conceivable that therapeutic interventions targeting key points of convergence in intracellular signaling cascades may provide broad renal protective effects in ADPKD. For example, our pathway and transcriptional regulator analyses highlighted the importance of AMPK, PGC-1α, PPARα, ERRα, and ERRγ in regulating metabolic reprogramming in ADPKD. These regulators are all highly expressed in the kidney and form an interconnected network. PGC-1α is downstream of the intensively investigated drug target AMPK, while PPARα, ERRα and ERRγ are the downstream targets of PGC-1α. Their expression and/or activity were reduced in renal cysts, in parallel with reduced expression of genes implicated in mitochondrial biogenesis, FAO and OXPHOS. Interventions and drugs that activate an energy-sensing network consisting of these key transcriptional regulators have the potential to inhibit cyst growth.
Author Contributions
X.S. and Y.P. designed the study; X.S. analyzed the data; X.S. and L.P. produced the figures and drafted the paper; J.S, H.S. and Y.P reviewed and edited the paper. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by a grant from the Canadian Institutes of Health Research (MOP 67084) to Y.P.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Microarray data are available in Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) (ID: GSE7869)
Acknowledgments
We thank the study patients who donated their nephrectomized polycystic kidneys for our research and Dr. Andrei Iliuta for proof-reading our manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
Appendix A
ADPKD (autosomal dominant polycystic kidney disease); FAO (fatty acid oxidation); BCAA (branched-chain amino acid); OXPHOS (oxidative phosphorylation); GSH (glutathione); ROS (reactive oxygen species); GSEA (gene set enrichment analysis); MSigDB (Molecular Signatures Database); KEGG (Kyoto Encyclopedia of Genes and Genomes); NOM P-value (nominal P-value); FDR (false discovery rate); PPP (pentose phosphate pathway); NAPDH (nicotinamide adenine dinucleotide phosphate, reduced); PDC (pyruvate dehydrogenase complex); PDK (PDH kinase); acetyl-CoA (acetyl coenzyme A); BCKA (branched-chain α-keto acid); α-KG (α-ketoglutarate); NAD (nicotinamide adenine dinucleotide); NAM (nicotinamide); NMN (nicotinamide mononucleotide); AA (amino acid); Glu (glutamate); Gln (glutamine); Cys (cysteine); Gly (glycine); Met (methionine); GSSG (glutathione disulfide); MTs (methyltransferases); SAM (S-adenosylmethionine); SAH (S-adenosylhomocysteine); SOD (superoxide dismutase); CAT (catalase); GST (glutathione S-transferase); GPX (glutathione peroxidase); PRDX (peroxiredoxin); NEAA (non-essential amino acid); IPA® (Ingenuity® Pathway Analysis); URA (Upstream Regulator Analysis); NES (normalized enrichment score); AKI (acute kidney injury); CKD (chronic kidney disease); MCT (minimally cystic tissue).
References
- Lanktree, M.B.; Haghighi, A.; Guiard, E.; Iliuta, I.A.; Song, X.; Harris, P.C.; Paterson, A.D.; Pei, Y. Prevalence estimates of polycystic kidney and liver disease by population sequencing. J. Am. Soc. Nephrol. 2018, 29, 2593–2600. [Google Scholar] [CrossRef] [PubMed]
- Grantham, J.J. Clinical practice. Autosomal dominant polycystic kidney disease. The New England journal of medicine 2008, 359, 1477–1485. [Google Scholar] [CrossRef]
- Peters, D.J.; Sandkuijl, L.A. Genetic heterogeneity of polycystic kidney disease in Europe. Contrib. Nephrol. 1992, 97, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Hwang, Y.H.; Conklin, J.; Chan, W.; Roslin, N.M.; Liu, J.; He, N.; Wang, K.; Sundsbak, J.L.; Heyer, C.M.; Haider, M.; et al. Refining genotype-phenotype correlation in autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 2016, 27, 1861–1868. [Google Scholar] [CrossRef] [PubMed]
- Cornec-Le Gall, E.; Audrézet, M.P.; Chen, J.M.; Hourmant, M.; Morin, M.P.; Perrichot, R.; Charasse, C.; Whebe, B.; Renaudineau, E.; Jousset, P.; et al. Type of PKD1 mutation influences renal outcome in ADPKD. J. Am. Soc. Nephrol. 2013, 24, 1006–1013. [Google Scholar] [CrossRef]
- Heyer, C.M.; Sundsbak, J.L.; Abebe, K.Z.; Chapman, A.B.; Torres, V.E.; Grantham, J.J.; Bae, K.T.; Schrier, R.W.; Perrone, R.D.; Braun, W.E.; et al. Predicted mutation strength of nontruncating PKD1 mutations aids genotype-phenotype correlations in autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 2016, 27, 2872–2884. [Google Scholar] [CrossRef]
- Harris, P.C.; Torres, V.E. Genetic mechanisms and signaling pathways in autosomal dominant polycystic kidney disease. J. Clin. Invest. 2014, 124, 2315–2324. [Google Scholar] [CrossRef]
- Bergmann, C.; Guay-Woodford, L.M.; Harris, P.C.; Horie, S.; Peters, D.J.M.; Torres, V.E. Polycystic kidney disease. Nat Rev Dis Primers 2018, 4, 50. [Google Scholar] [CrossRef]
- Shillingford, J.M.; Murcia, N.S.; Larson, C.H.; Low, S.H.; Hedgepeth, R.; Brown, N.; Flask, C.A.; Novick, A.C.; Goldfarb, D.A.; Kramer-Zucker, A.; et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proceedings of the National Academy of Sciences of the United States of America 2006, 103, 5466–5471. [Google Scholar] [CrossRef]
- Shillingford, J.M.; Piontek, K.B.; Germino, G.G.; Weimbs, T. Rapamycin ameliorates PKD resulting from conditional inactivation of Pkd1. J Am Soc Nephrol 2010, 21, 489–497. [Google Scholar] [CrossRef]
- Novalic, Z.; van der Wal, A.M.; Leonhard, W.N.; Koehl, G.; Breuning, M.H.; Geissler, E.K.; de Heer, E.; Peters, D.J. Dose-dependent effects of sirolimus on mTOR signaling and polycystic kidney disease. J. Am. Soc. Nephrol. 2012, 23, 842–853. [Google Scholar] [CrossRef] [PubMed]
- Padovano, V.; Podrini, C.; Boletta, A.; Caplan, M.J. Metabolism and mitochondria in polycystic kidney disease research and therapy. Nat Rev Nephrol 2018, 14, 678–687. [Google Scholar] [CrossRef]
- Menezes, L.F.; Germino, G.G. The pathobiology of polycystic kidney disease from a metabolic viewpoint. Nat. Rev. Nephrol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Podrini, C.; Cassina, L.; Boletta, A. Metabolic reprogramming and the role of mitochondria in polycystic kidney disease. Cell. Signal. 2019, 67, 109495. [Google Scholar] [CrossRef]
- Nowak, K.L.; Hopp, K. Metabolic reprogramming in autosomal dominant polycystic kidney disease: evidence and therapeutic potential. Clin. J. Am. Soc. Nephrol. 2020. [Google Scholar] [CrossRef]
- Song, X.; Tsakiridis, E.; Steinberg, G.R.; Pei, Y. Targeting AMP-activated protein kinase (AMPK) for treatment of autosomal dominant polycystic kidney disease. Cell. Signal. 2020, 73, 109704. [Google Scholar] [CrossRef]
- Iliuta, I.A.; Song, X.; Pickel, L.; Haghighi, A.; Retnakaran, R.; Scholey, J.; Sung, H.K.; Steinberg, G.R.; Pei, Y. Shared pathobiology identifies AMPK as a therapeutic target for obesity and autosomal dominant polycystic kidney disease. Front. Mol. Biosci. 2022, 9, 962933. [Google Scholar] [CrossRef] [PubMed]
- Rowe, I.; Chiaravalli, M.; Mannella, V.; Ulisse, V.; Quilici, G.; Pema, M.; Song, X.W.; Xu, H.; Mari, S.; Qian, F.; et al. Defective glucose metabolism in polycystic kidney disease identifies a new therapeutic strategy. Nature medicine 2013, 19, 488–493. [Google Scholar] [CrossRef]
- Zhou, X.; Fan, L.X.; Sweeney, W.E., Jr.; Denu, J.M.; Avner, E.D.; Li, X. Sirtuin 1 inhibition delays cyst formation in autosomal-dominant polycystic kidney disease. The Journal of clinical investigation 2013, 123, 3084–3098. [Google Scholar] [CrossRef]
- Takiar, V.; Nishio, S.; Seo-Mayer, P.; King, J.D., Jr.; Li, H.; Zhang, L.; Karihaloo, A.; Hallows, K.R.; Somlo, S.; Caplan, M.J. Activating AMP-activated protein kinase (AMPK) slows renal cystogenesis. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 2462–2467. [Google Scholar] [CrossRef]
- Yuajit, C.; Muanprasat, C.; Gallagher, A.R.; Fedeles, S.V.; Kittayaruksakul, S.; Homvisasevongsa, S.; Somlo, S.; Chatsudthipong, V. Steviol retards renal cyst growth through reduction of CFTR expression and inhibition of epithelial cell proliferation in a mouse model of polycystic kidney disease. Biochem. Pharmacol. 2014, 88, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Warner, G.; Hein, K.Z.; Nin, V.; Edwards, M.; Chini, C.C.; Hopp, K.; Harris, P.C.; Torres, V.E.; Chini, E.N. Food restriction ameliorates the development of polycystic kidney disease. J. Am. Soc. Nephrol. 2016, 27, 1437–1447. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.C.; Kurashige, M.; Liu, Y.; Terabayashi, T.; Ishimoto, Y.; Wang, T.; Choudhary, V.; Hobbs, R.; Liu, L.K.; Lee, P.H.; et al. A cleavage product of Polycystin-1 is a mitochondrial matrix protein that affects mitochondria morphology and function when heterologously expressed. Sci. Rep. 2018, 8, 2743. [Google Scholar] [CrossRef]
- Kahveci, A.S.; Barnatan, T.T.; Kahveci, A.; Adrian, A.E.; Arroyo, J.; Eirin, A.; Harris, P.C.; Lerman, A.; Lerman, L.O.; Torres, V.E.; et al. Oxidative stress and mitochondrial abnormalities contribute to decreased endothelial nitric oxide synthase expression and renal disease progression in early experimental polycystic kidney disease. Int. J. Mol. Sci. 2020, 21, 1994. [Google Scholar] [CrossRef]
- Kuo, I.Y.; Brill, A.L.; Lemos, F.O.; Jiang, J.Y.; Falcone, J.L.; Kimmerling, E.P.; Cai, Y.; Dong, K.; Kaplan, D.L.; Wallace, D.P.; et al. Polycystin 2 regulates mitochondrial Ca2+ signaling, bioenergetics, and dynamics through mitofusin 2. Sci. Signal. 2019, 12, eaat7397. [Google Scholar] [CrossRef]
- Cassina, L.; Chiaravalli, M.; Boletta, A. Increased mitochondrial fragmentation in polycystic kidney disease acts as a modifier of disease progression. FASEB J 2020, 34, 6493–6507. [Google Scholar] [CrossRef]
- Ishimoto, Y.; Inagi, R.; Yoshihara, D.; Kugita, M.; Nagao, S.; Shimizu, A.; Takeda, N.; Wake, M.; Honda, K.; Zhou, J.; et al. Mitochondrial abnormality facilitates cyst formation in autosomal dominant polycystic kidney disease. Mol. Cell. Biol. 2017, 37. [Google Scholar] [CrossRef] [PubMed]
- Padovano, V.; Kuo, I.Y.; Stavola, L.K.; Aerni, H.R.; Flaherty, B.J.; Chapin, H.C.; Ma, M.; Somlo, S.; Boletta, A.; Ehrlich, B.E.; et al. The polycystins are modulated by cellular oxygen-sensing pathways and regulate mitochondrial function. Mol. Biol. Cell. 2017, 28, 261–269. [Google Scholar] [CrossRef]
- Torres, V.E.; Bengal, R.J.; Litwiller, R.D.; Wilson, D.M. Aggravation of polycystic kidney disease in Han:SPRD rats by buthionine sulfoximine. J. Am. Soc. Nephrol. 1997, 8, 1283–1291. [Google Scholar] [CrossRef]
- Maser, R.L.; Vassmer, D.; Magenheimer, B.S.; Calvet, J.P. Oxidant stress and reduced antioxidant enzyme protection in polycystic kidney disease. J. Am. Soc. Nephrol. 2002, 13, 991–999. [Google Scholar] [CrossRef]
- Menon, V.; Rudym, D.; Chandra, P.; Miskulin, D.; Perrone, R.; Sarnak, M. Inflammation, oxidative stress, and insulin resistance in polycystic kidney disease. Clin. J. Am. Soc. Nephrol. 2011, 6, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Klawitter, J.; Reed-Gitomer, B.Y.; McFann, K.; Pennington, A.; Abebe, K.Z.; Klepacki, J.; Cadnapaphornchai, M.A.; Brosnahan, G.; Chonchol, M.; Christians, U.; et al. Endothelial dysfunction and oxidative stress in polycystic kidney disease. Am. J. Physio.l Renal Physiol. 2014, 307, F1198–1206. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.; Buchholz, B.; Kraus, A.; Schley, G.; Scholz, J.; Ousingsawat, J.; Kunzelmann, K. Lipid peroxidation drives renal cyst growth in vitro through activation of TMEM16A. J. Am. Soc. Nephrol. 2019, 30, 228–242. [Google Scholar] [CrossRef] [PubMed]
- Menezes, L.F.; Lin, C.C.; Zhou, F.; Germino, G.G. Fatty acid oxidation is impaired in an orthologous mouse model of autosomal dominant polycystic kidney disease. EBioMedicine 2016, 5, 183–192. [Google Scholar] [CrossRef]
- Lakhia, R.; Yheskel, M.; Flaten, A.; Quittner-Strom, E.B.; Holland, W.L.; Patel, V. PPARalpha agonist fenofibrate enhances fatty acid beta-oxidation and attenuates polycystic kidney and liver disease in mice. Am. J. Physiol. Renal Physiol. 2018, 314, F122–F131. [Google Scholar] [CrossRef] [PubMed]
- Podrini, C.; Rowe, I.; Pagliarini, R.; Costa, A.S.H.; Chiaravalli, M.; Di Meo, I.; Kim, H.; Distefano, G.; Tiranti, V.; Qian, F.; et al. Dissection of metabolic reprogramming in polycystic kidney disease reveals coordinated rewiring of bioenergetic pathways. Commun. Biol. 2018, 1, 194. [Google Scholar] [CrossRef] [PubMed]
- Soomro, I.; Sun, Y.; Li, Z.; Diggs, L.; Hatzivassiliou, G.; Thomas, A.G.; Rais, R.; Slusher, B.S.; Somlo, S.; Skolnik, E.Y. Glutamine metabolism via glutaminase 1 in autosomal-dominant polycystic kidney disease. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. [CrossRef]
- Trott, J.F.; Hwang, V.J.; Ishimaru, T.; Chmiel, K.J.; Zhou, J.X.; Shim, K.; Stewart, B.J.; Mahjoub, M.R.; Jen, K.Y.; Barupal, D.K.; et al. Arginine reprogramming in ADPKD results in arginine-dependent cystogenesis. Am. J. Physiol. Renal Physiol. 2018, 315, F1855–F1868. [Google Scholar] [CrossRef] [PubMed]
- Malas, T.B.; Leonhard, W.N.; Bange, H.; Granchi, Z.; Hettne, K.M.; Van Westen, G.J.P.; Price, L.S.; 't Hoen, P.A.C.; Peters, D.J.M. Prioritization of novel ADPKD drug candidates from disease-stage specific gene expression profiles. EBioMedicine 2020, 51, 102585. [Google Scholar] [CrossRef]
- Asawa, R.R.; Danchik, C.; Zahkarov, A.; Chen, Y.; Voss, T.; Jadhav, A.; Wallace, D.P.; Trott, J.F.; Weiss, R.H.; Simeonov, A.; et al. A high-throughput screening platform for Polycystic Kidney Disease (PKD) drug repurposing utilizing murine and human ADPKD cells. Sci. Rep. 2020, 10, 4203. [Google Scholar] [CrossRef]
- Riwanto, M.; Kapoor, S.; Rodriguez, D.; Edenhofer, I.; Segerer, S.; Wuthrich, R.P. Inhibition of aerobic glycolysis attenuates disease progression in polycystic kidney disease. PLoS One 2016, 11, e0146654. [Google Scholar] [CrossRef]
- Chiaravalli, M.; Rowe, I.; Mannella, V.; Quilici, G.; Canu, T.; Bianchi, V.; Gurgone, A.; Antunes, S.; D'Adamo, P.; Esposito, A.; et al. 2-Deoxy-d-Glucose Ameliorates PKD Progression. J Am Soc Nephrol 2016, 27, 1958–1969. [Google Scholar] [CrossRef] [PubMed]
- Lian, X.; Wu, X.; Li, Z.; Zhang, Y.; Song, K.; Cai, G.; Li, Q.; Lin, S.; Chen, X.; Bai, X.Y. The combination of metformin and 2-deoxyglucose significantly inhibits cyst formation in miniature pigs with polycystic kidney disease. Br. J. Pharmacol. 2019, 176, 711–724. [Google Scholar] [CrossRef]
- Hardie, D.G. AMPK: a target for drugs and natural products with effects on both diabetes and cancer. Diabetes 2013, 62, 2164–2172. [Google Scholar] [CrossRef] [PubMed]
- Garcia, D.; Shaw, R.J. AMPK: Mechanisms of cellular energy sensing and restoration of metabolic balance. Mol. Cell 2017, 66, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Herzig, S.; Shaw, R.J. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol 2018, 19, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, G.R.; Carling, D. AMP-activated protein kinase: the current landscape for drug development. Nat. Rev. Drug Discov. 2019. [Google Scholar] [CrossRef]
- Kipp, K.R.; Rezaei, M.; Lin, L.; Dewey, E.C.; Weimbs, T. A mild reduction of food intake slows disease progression in an orthologous mouse model of polycystic kidney disease. Am. J. Physiol. Renal Physiol. 2016, 310, F726–F731. [Google Scholar] [CrossRef] [PubMed]
- Torres, J.A.; Kruger, S.L.; Broderick, C.; Amarlkhagva, T.; Agrawal, S.; Dodam, J.R.; Mrug, M.; Lyons, L.A.; Weimbs, T. Ketosis ameliorates renal cyst growth in polycystic kidney disease. Cell Metab. 2019, 30, 1007–1023. [Google Scholar] [CrossRef] [PubMed]
- Leonhard, W.N.; Song, X.; Kanhai, A.A.; Iliuta, I.A.; Bozovic, A.; Steinberg, G.R.; Peters, D.J.M.; Pei, Y. Salsalate, but not metformin or canagliflozin, slows kidney cyst growth in an adult-onset mouse model of polycystic kidney disease. EBioMedicine 2019, 47, 436–445. [Google Scholar] [CrossRef]
- Pastor-Soler, N.M.; Li, H.; Pham, J.; Rivera, D.; Ho, P.Y.; Mancino, V.; Saitta, B.; Hallows, K.R. Metformin improves relevant disease parameters in an autosomal dominant polycystic kidney disease mouse model. Am J Physiol Renal Physiol 2022, 322, F27–F41. [Google Scholar] [CrossRef]
- Song, X.; Leonhard, W.N.; Kanhai, A.A.; Steinberg, G.R.; Pei, Y.; Peters, D.J.M. Preclinical evaluation of tolvaptan and salsalate combination therapy in a Pkd1-mouse model. Front. Mol. Biosci. 2023, 10, 1058825. [Google Scholar] [CrossRef] [PubMed]
- Flowers, E.M.; Sudderth, J.; Zacharias, L.; Mernaugh, G.; Zent, R.; DeBerardinis, R.J.; Carroll, T.J. Lkb1 deficiency confers glutamine dependency in polycystic kidney disease. Nature communications 2018, 9, 814. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Di Giovanni, V.; He, N.; Wang, K.; Ingram, A.; Rosenblum, N.D.; Pei, Y. Systems biology of autosomal dominant polycystic kidney disease (ADPKD): computational identification of gene expression pathways and integrated regulatory networks. Hum. Mol. Genet. 2009, 18, 2328–2343. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Hay, N. Reprogramming glucose metabolism in cancer: can it be exploited for cancer therapy? Nat. Rev. Cancer 2016, 16, 635–649. [Google Scholar] [CrossRef]
- McCommis, K.S.; Finck, B.N. Mitochondrial pyruvate transport: a historical perspective and future research directions. Biochem. J. 2015, 466, 443–454. [Google Scholar] [CrossRef]
- Patel, M.S.; Nemeria, N.S.; Furey, W.; Jordan, F. The pyruvate dehydrogenase complexes: structure-based function and regulation. J. Biol. Chem. 2014, 289, 16615–16623. [Google Scholar] [CrossRef]
- Scrutton, M.C.; Utter, M.F. Regulation of Glycolysis and Gluconeogenesis in Animal Tissues. Annu Rev Biochem 1968, 37, 249. [Google Scholar] [CrossRef]
- Guder, W.G.; Rupprecht, A. Metabolism of isolated kidney tubules. Independent actions of catecholamines on renal cyclic adenosine 3':5'-monophosphate levels and gluconeogenesis. Eur. J. Biochem. 1975, 52, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Pietrocola, F.; Galluzzi, L.; Bravo-San Pedro, J.M.; Madeo, F.; Kroemer, G. Acetyl coenzyme A: a central metabolite and second messenger. Cell Metab. 2015, 21, 805–821. [Google Scholar] [CrossRef]
- Ananieva, E.A.; Wilkinson, A.C. Branched-chain amino acid metabolism in cancer. Current opinion in clinical nutrition and metabolic care 2018, 21, 64–70. [Google Scholar] [CrossRef]
- Yamamoto, J.; Nishio, S.; Hattanda, F.; Nakazawa, D.; Kimura, T.; Sata, M.; Makita, M.; Ishikawa, Y.; Atsumi, T. Branched-chain amino acids enhance cyst development in autosomal dominant polycystic kidney disease. Kidney Int. 2017, 92, 377–387. [Google Scholar] [CrossRef]
- Kang, H.M.; Ahn, S.H.; Choi, P.; Ko, Y.A.; Han, S.H.; Chinga, F.; Park, A.S.; Tao, J.; Sharma, K.; Pullman, J.; et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nature medicine 2015, 21, 37–46. [Google Scholar] [CrossRef]
- Yang, X.; Okamura, D.M.; Lu, X.; Chen, Y.; Moorhead, J.; Varghese, Z.; Ruan, X.Z. CD36 in chronic kidney disease: novel insights and therapeutic opportunities. Nat. Rev. Nephrol. 2017, 13, 769–781. [Google Scholar] [CrossRef]
- Declèves, A.E.; Zolkipli, Z.; Satriano, J.; Wang, L.; Nakayama, T.; Rogac, M.; Le, T.P.; Nortier, J.L.; Farquhar, M.G.; Naviaux, R.K.; et al. Regulation of lipid accumulation by AMP-activated kinase in high fat diet-induced kidney injury. Kidney Int. 2014, 85, 611–623. [Google Scholar] [CrossRef]
- Kurutas, E.B. The importance of antioxidants which play the role in cellular response against oxidative/nitrosative stress: current state. Nutr. J. 2016, 15, 71. [Google Scholar] [CrossRef]
- Cabrita, I.; Kraus, A.; Scholz, J.K.; Skoczynski, K.; Schreiber, R.; Kunzelmann, K.; Buchholz, B. Cyst growth in ADPKD is prevented by pharmacological and genetic inhibition of TMEM16A in vivo. Nat. Commun. 2020, 11, 4320. [Google Scholar] [CrossRef]
- Deneke, S.M.; Fanburg, B.L. Regulation of cellular glutathione. Am. J. Physiol. 1989, 257, L163–173. [Google Scholar] [CrossRef]
- Lushchak, V.I. Glutathione homeostasis and functions: potential targets for medical interventions. J. Amino Acids 2012, 2012, 736837. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.C. Glutathione synthesis. Biochim. Biophys. Acta 2013, 1830, 3143–3153. [Google Scholar] [CrossRef] [PubMed]
- Koppula, P.; Zhang, Y.; Zhuang, L.; Gan, B. Amino acid transporter SLC7A11/xCT at the crossroads of regulating redox homeostasis and nutrient dependency of cancer. Cancer Commun. (Lond) 2018, 38, 12. [Google Scholar] [CrossRef] [PubMed]
- Combs, J.A.; DeNicola, G.M. The non-essential amino acid cysteine becomes essential for tumor proliferation and survival. Cancers (Basel) 2019, 11, E678. [Google Scholar] [CrossRef] [PubMed]
- Menezes, L.F.; Zhou, F.; Patterson, A.D.; Piontek, K.B.; Krausz, K.W.; Gonzalez, F.J.; Germino, G.G. Network analysis of a Pkd1-mouse model of autosomal dominant polycystic kidney disease identifies HNF4alpha as a disease modifier. PLoS Genet. 2012, 8, e1003053. [Google Scholar] [CrossRef] [PubMed]
- Mia, S.; Federico, G.; Feger, M.; Pakladok, T.; Meissner, A.; Voelkl, J.; Groene, H.J.; Alesutan, I.; Lang, F. Impact of AMP-activated protein kinase α1 deficiency on tissue injury following unilateral ureteral obstruction. PLoS One 2015, 10, e0135235. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jia, L.; Hu, Z.; Entman, M.L.; Mitch, W.E. AMP-activated protein kinase/myocardin-related transcription factor-A signaling regulates fibroblast activation and renal fibrosis. Kidney Int. 2018, 93, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Qiu, S.; Xiao, Z.; Piao, C.; Zhang, J.; Dong, Y.; Cui, W.; Liu, X.; Zhang, Y.; Du, J. AMPKα2 reduces renal epithelial transdifferentiation and inflammation after injury through interaction with CK2β. J. Pathol. 2015, 237, 330–342. [Google Scholar] [CrossRef]
- Cantó, C.; Auwerx, J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. 2009, 20, 98–105. [Google Scholar] [CrossRef]
- Lin, J.; Handschin, C.; Spiegelman, B.M. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005, 1, 361–370. [Google Scholar] [CrossRef]
- Dominy, J.E.; Puigserver, P. Mitochondrial biogenesis through activation of nuclear signaling proteins. Cold Spring Harbor perspectives in biology 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, P.; Schnellmann, R.G. Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 2017, 13, 629–646. [Google Scholar] [CrossRef] [PubMed]
- Cargill, K.; Sims-Lucas, S. Metabolic requirements of the nephron. Pediatr. Nephrol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Stine, Z.E.; Walton, Z.E.; Altman, B.J.; Hsieh, A.L.; Dang, C.V. MYC, metabolism, and cancer. Cancer Discov. 2015, 5, 1024–1039. [Google Scholar] [CrossRef]
- Masoud, G.N.; Li, W. HIF-1α pathway: role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef] [PubMed]
- Hallows, K.R.; Mount, P.F.; Pastor-Soler, N.M.; Power, D.A. Role of the energy sensor AMP-activated protein kinase in renal physiology and disease. Am J Physiol Renal Physiol 2010, 298, F1067–1077. [Google Scholar] [CrossRef] [PubMed]
- Rajani, R.; Pastor-Soler, N.M.; Hallows, K.R. Role of AMP-activated protein kinase in kidney tubular transport, metabolism, and disease. Curr. Opin. Nephrol. Hypertens. 2017, 26, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Glosse, P.; Föller, M. AMP-activated protein kinase (AMPK)-dependent regulation of renal transport. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Wang, S.; Zhang, Y.; Xiao, H. Metformin attenuates renal fibrosis in both AMPKα2-dependent and independent manners. Clin. Exp. Pharmacol. Physiol. 2017, 44, 648–655. [Google Scholar] [CrossRef]
- Lynch, M.R.; Tran, M.T.; Parikh, S.M. PGC1α in the kidney. Am. J. Physiol. Renal Physiol. 2018, 314, F1–F8. [Google Scholar] [CrossRef]
- Fontecha-Barriuso, M.; Martin-Sanchez, D.; Martinez-Moreno, J.M.; Monsalve, M.; Ramos, A.M.; Sanchez-Niño, M.D.; Ruiz-Ortega, M.; Ortiz, A.; Sanz, A.B. The role of PGC-1α and mitochondrial biogenesis in kidney diseases. Biomolecules 2020, 10, 347. [Google Scholar] [CrossRef] [PubMed]
- Chambers, J.M.; Wingert, R.A. PGC-1α in disease: recent renal insights into a versatile metabolic regulator. Cells 2020, 9, 2234. [Google Scholar] [CrossRef] [PubMed]
- Hajarnis, S.; Lakhia, R.; Yheskel, M.; Williams, D.; Sorourian, M.; Liu, X.; Aboudehen, K.; Zhang, S.; Kersjes, K.; Galasso, R.; et al. microRNA-17 family promotes polycystic kidney disease progression through modulation of mitochondrial metabolism. Nat. Commun. 2017, 8, 14395. [Google Scholar] [CrossRef] [PubMed]
- Kersten, S. Integrated physiology and systems biology of PPARα. Mol. Metab. 2014, 3, 354–371. [Google Scholar] [CrossRef] [PubMed]
- Tsushida, K.; Tanabe, K.; Masuda, K.; Tanimura, S.; Miyake, H.; Arata, Y.; Sugiyama, H.; Wada, J. Estrogen-related receptor α is essential for maintaining mitochondrial integrity in cisplatin-induced acute kidney injury. Biochem Biophys Res Commun 2018, 498, 918–924. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Lupino, K.; Wilkins, B.J.; Qiu, C.; Liu, J.; Omura, Y.; Allred, A.L.; McDonald, C.; Susztak, K.; Barish, G.D.; et al. Genomic integration of ERRγ-HNF1β regulates renal bioenergetics and prevents chronic kidney disease. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, E4910–E4919. [Google Scholar] [CrossRef]
- Chiarugi, A.; Dolle, C.; Felici, R.; Ziegler, M. The NAD metabolome--a key determinant of cancer cell biology. Nat. Rev. Cancer 2012, 12, 741–752. [Google Scholar] [CrossRef]
- Kramer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef]
- Tusher, V.G.; Tibshirani, R.; Chu, G. Significance analysis of microarrays applied to the ionizing radiation response. Proceedings of the National Academy of Sciences of the United States of America 2001, 98, 5116–5121. [Google Scholar] [CrossRef]
Figure 1.
Human PKD1 renal cysts display the Warburg effect and increased pentose phosphate pathway (PPP) flux. Schematic summary of the upregulation of glycolysis and PPP (left) and downregulation of gluconeogenesis (right) in PKD1 renal cysts. Upregulated genes are shown in red, and downregulated genes in blue, with mean expression fold changes in brackets. Genes that were not differentially expressed are shown in black. Arrows indicate irreversible enzymatic steps, and bi-directional arrows indicate interconverting reversible reactions determined by substrate concentration. Asterisk* denotes rate-limiting enzymes.
Figure 1.
Human PKD1 renal cysts display the Warburg effect and increased pentose phosphate pathway (PPP) flux. Schematic summary of the upregulation of glycolysis and PPP (left) and downregulation of gluconeogenesis (right) in PKD1 renal cysts. Upregulated genes are shown in red, and downregulated genes in blue, with mean expression fold changes in brackets. Genes that were not differentially expressed are shown in black. Arrows indicate irreversible enzymatic steps, and bi-directional arrows indicate interconverting reversible reactions determined by substrate concentration. Asterisk* denotes rate-limiting enzymes.
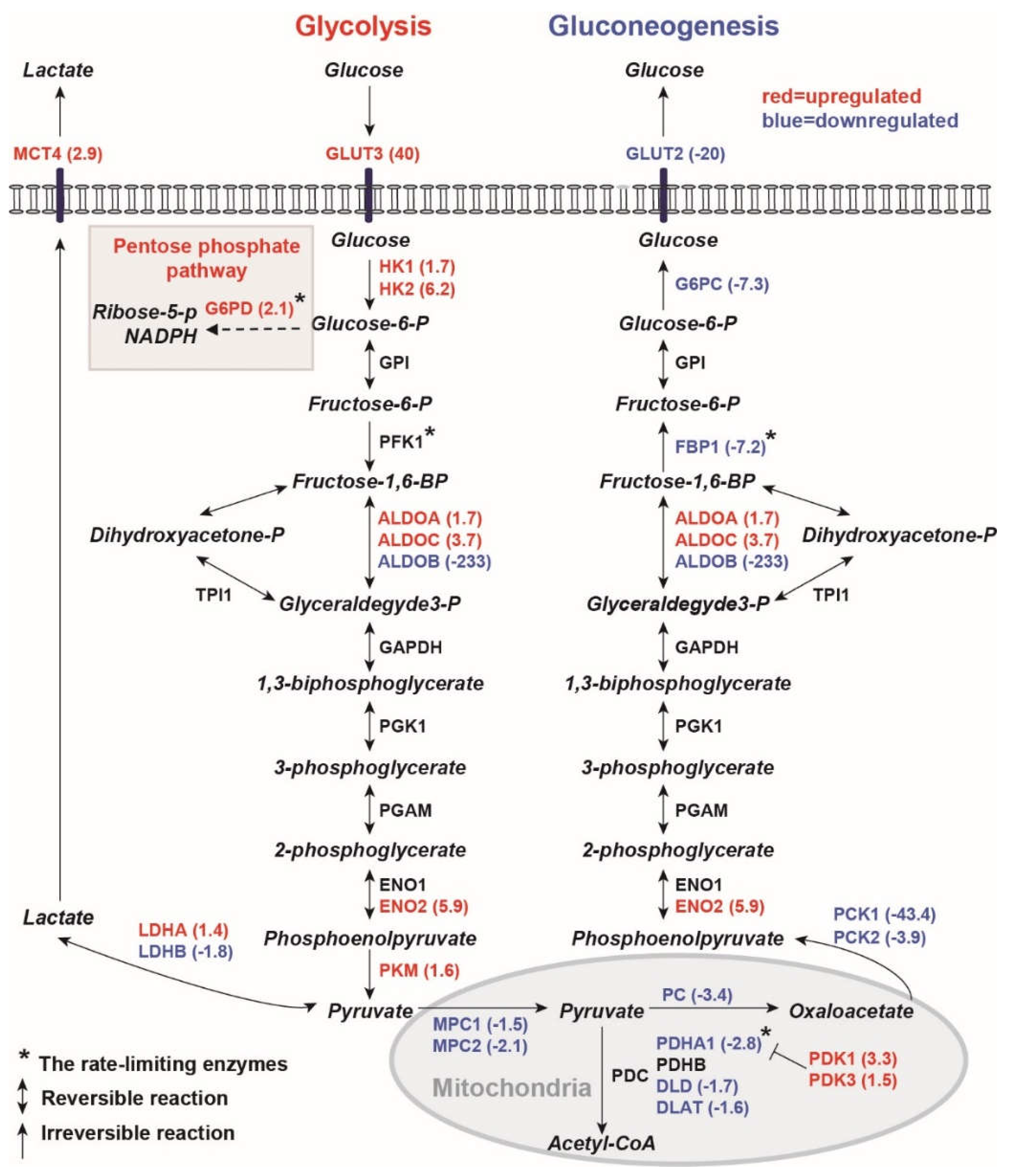
Figure 2.
Metabolic reprogramming in human PKD1 renal cysts. Downregulation of the majority of genes in branched-chain amino acid degradation (a), fatty acid degradation (b), the Krebs cycle (c), and oxidative phosphorylation (d) suggests defective mitochondrial oxidative metabolism in PKD1 renal cysts. (e) Upregulation of NAMPT and downregulation of QPRT suggest renal cysts may favor the salvage over the de novo pathway to produce NAD+. All genes listed in the panels were differentially expressed between the cysts and MCT samples with an FDR ≤ 1%. In the heatmap, each column represents an individual sample, and each row represents the Z-score scaled gene expression levels across all samples; white is the mean Z-score (set to 0), red indicates greater than the mean and blue, less than the mean. Z-scores are computed for individual genes by subtracting the mean and then dividing by the standard deviation.
Figure 2.
Metabolic reprogramming in human PKD1 renal cysts. Downregulation of the majority of genes in branched-chain amino acid degradation (a), fatty acid degradation (b), the Krebs cycle (c), and oxidative phosphorylation (d) suggests defective mitochondrial oxidative metabolism in PKD1 renal cysts. (e) Upregulation of NAMPT and downregulation of QPRT suggest renal cysts may favor the salvage over the de novo pathway to produce NAD+. All genes listed in the panels were differentially expressed between the cysts and MCT samples with an FDR ≤ 1%. In the heatmap, each column represents an individual sample, and each row represents the Z-score scaled gene expression levels across all samples; white is the mean Z-score (set to 0), red indicates greater than the mean and blue, less than the mean. Z-scores are computed for individual genes by subtracting the mean and then dividing by the standard deviation.
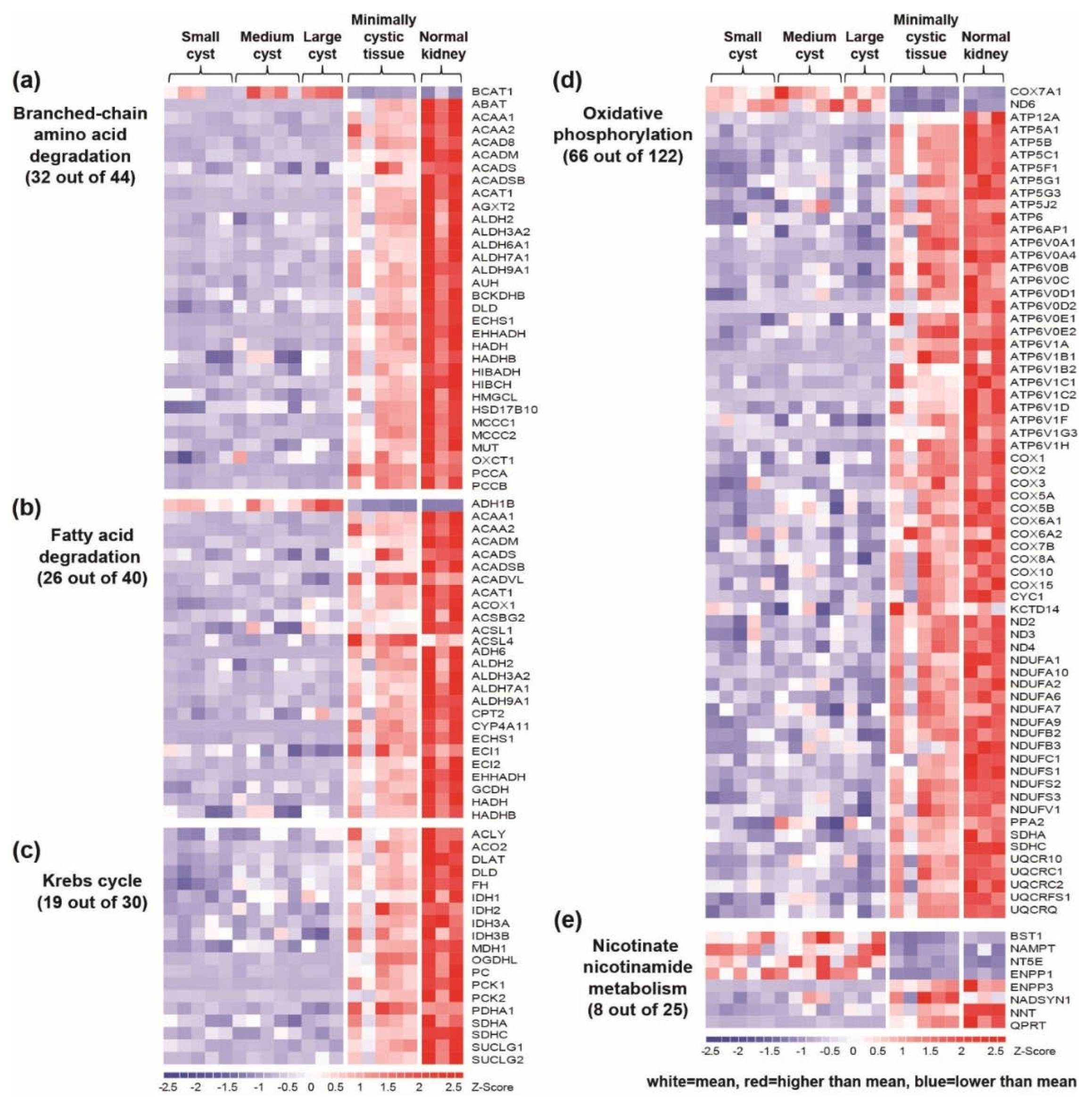
Figure 3.
Rewiring of GSH metabolism in human PKD1 renal cysts. (a) Schematic summary of the downregulation of the γ-glutamyl cycle and upregulation of Na+-independent cystine/glutamate antiporter xCT (encoded by SLC7A11), which may serve as important sources for maintaining the cysteine pool in PKD1 renal cysts. NADPH may be resupplied by the reduction of NADP+ via the pentose phosphate pathway. Upregulated genes are shown in red, and downregulated genes in blue, with mean expression fold-changes in brackets. Genes that were not differentially expressed are shown in black. Asterisk* denotes the rate-limiting enzyme or substrate. (b) Gene expression profiling showing the differentially expressed genes involved in GSH metabolism in PKD1 renal cysts. In the heatmap, each column represents an individual sample, and each row represents the Z-score scaled gene expression levels across all samples; white is the mean Z-score (set to 0), red indicates greater than the mean and blue, less than the mean. Z-scores are computed for individual genes by subtracting the mean and then dividing by the standard deviation. Abbreviations: GSH (glutathione); AA (amino acid); Glu (glutamate); Cys (cysteine); Gly (glycine); Met (methionine); ROS (reactive oxygen species); MTs (methyltransferases); SAM (S-adenosylmethionine); SAH (S-adenosylhomocysteine); GSSG (glutathione disulfide); NAPDH (nicotinamide adenine dinucleotide phosphate, reduced); SOD (superoxide dismutase); CAT (catalase); GST (glutathione S-transferase); GPX (glutathione peroxidase); PRDX (peroxiredoxin).
Figure 3.
Rewiring of GSH metabolism in human PKD1 renal cysts. (a) Schematic summary of the downregulation of the γ-glutamyl cycle and upregulation of Na+-independent cystine/glutamate antiporter xCT (encoded by SLC7A11), which may serve as important sources for maintaining the cysteine pool in PKD1 renal cysts. NADPH may be resupplied by the reduction of NADP+ via the pentose phosphate pathway. Upregulated genes are shown in red, and downregulated genes in blue, with mean expression fold-changes in brackets. Genes that were not differentially expressed are shown in black. Asterisk* denotes the rate-limiting enzyme or substrate. (b) Gene expression profiling showing the differentially expressed genes involved in GSH metabolism in PKD1 renal cysts. In the heatmap, each column represents an individual sample, and each row represents the Z-score scaled gene expression levels across all samples; white is the mean Z-score (set to 0), red indicates greater than the mean and blue, less than the mean. Z-scores are computed for individual genes by subtracting the mean and then dividing by the standard deviation. Abbreviations: GSH (glutathione); AA (amino acid); Glu (glutamate); Cys (cysteine); Gly (glycine); Met (methionine); ROS (reactive oxygen species); MTs (methyltransferases); SAM (S-adenosylmethionine); SAH (S-adenosylhomocysteine); GSSG (glutathione disulfide); NAPDH (nicotinamide adenine dinucleotide phosphate, reduced); SOD (superoxide dismutase); CAT (catalase); GST (glutathione S-transferase); GPX (glutathione peroxidase); PRDX (peroxiredoxin).
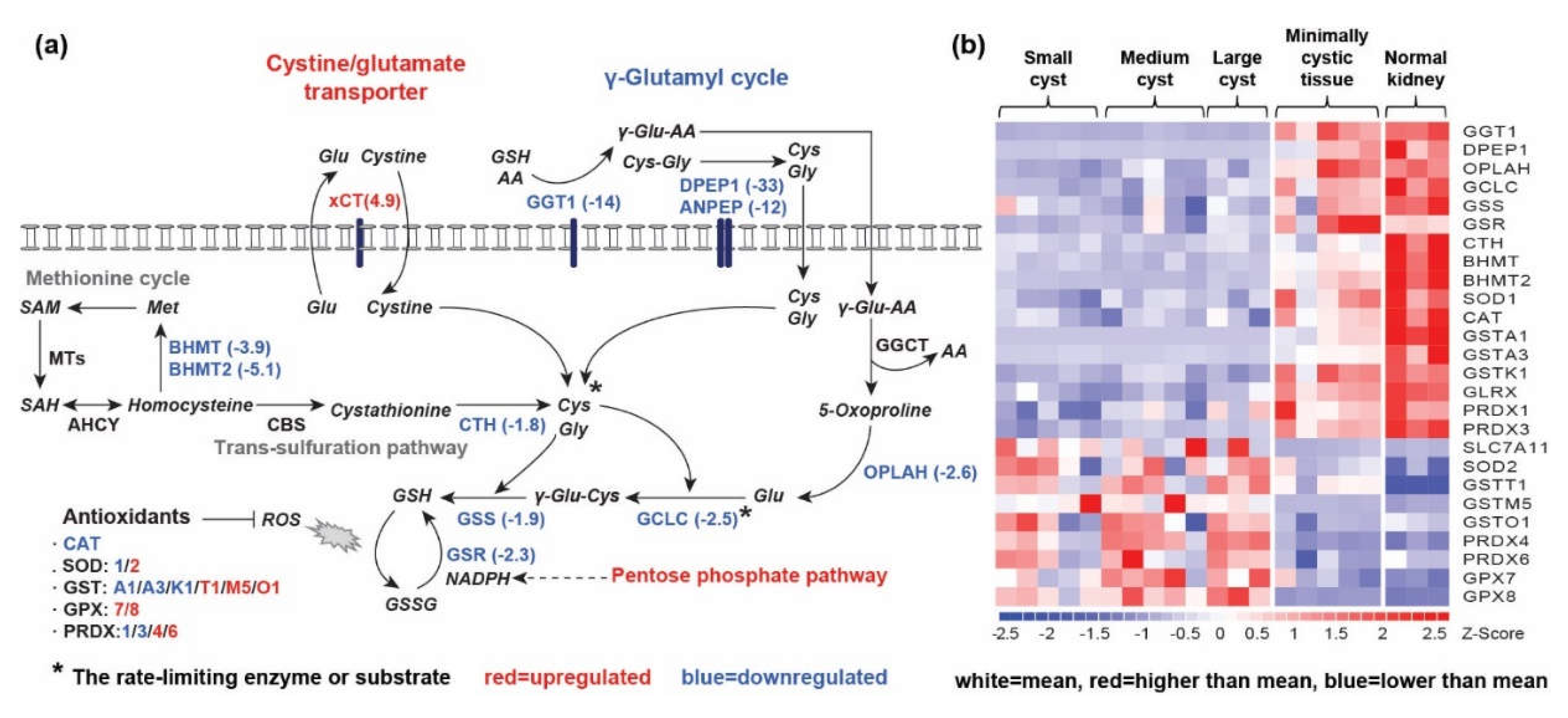
Figure 4.
Schematic summary of interrelationships between growth factors and energy sensing pathways in PKD1 renal cysts. Cysts switch from oxidative metabolism (fatty acid oxidation, branched-chain amino acid degradation, the Krebs cycle, oxidative phosphorylation, and peroxisomal proteins) to aerobic glycolysis to meet their energy needs. The PI3K/Akt pathway is activated upon growth factor/receptor tyrosine kinase stimulation (e.g., IGF1/IGF1R). The mTORC1 pathway integrates signals from growth factor stimulation, amino acid availability, and energy status via AMPK. The oncogenes HIF-1α and MYC together drive the expression of genes promoting aerobic glycolysis and the NAD+ salvage pathway. Upregulated pathways/genes are shown in red, and downregulated pathways/genes in blue, with mean expression fold-changes in brackets. Genes that were not differentially expressed are shown in black. Asterisk * denotes proteins that were predicted to be activated (red) or inhibited (blue) by GSEA or URA. Abbreviations: BCAA (branched-chain amino acid); BCKA (branched-chain α-keto acid); α-KG (α-ketoglutarate); OXPHOS (oxidative phosphorylation); Glu (glutamate); Gln (glutamine); NEAA (non-essential amino acids); ROS (reactive oxygen species); NAD (nicotinamide adenine dinucleotide); NAM (nicotinamide); NMN (nicotinamide mononucleotide).
Figure 4.
Schematic summary of interrelationships between growth factors and energy sensing pathways in PKD1 renal cysts. Cysts switch from oxidative metabolism (fatty acid oxidation, branched-chain amino acid degradation, the Krebs cycle, oxidative phosphorylation, and peroxisomal proteins) to aerobic glycolysis to meet their energy needs. The PI3K/Akt pathway is activated upon growth factor/receptor tyrosine kinase stimulation (e.g., IGF1/IGF1R). The mTORC1 pathway integrates signals from growth factor stimulation, amino acid availability, and energy status via AMPK. The oncogenes HIF-1α and MYC together drive the expression of genes promoting aerobic glycolysis and the NAD+ salvage pathway. Upregulated pathways/genes are shown in red, and downregulated pathways/genes in blue, with mean expression fold-changes in brackets. Genes that were not differentially expressed are shown in black. Asterisk * denotes proteins that were predicted to be activated (red) or inhibited (blue) by GSEA or URA. Abbreviations: BCAA (branched-chain amino acid); BCKA (branched-chain α-keto acid); α-KG (α-ketoglutarate); OXPHOS (oxidative phosphorylation); Glu (glutamate); Gln (glutamine); NEAA (non-essential amino acids); ROS (reactive oxygen species); NAD (nicotinamide adenine dinucleotide); NAM (nicotinamide); NMN (nicotinamide mononucleotide).
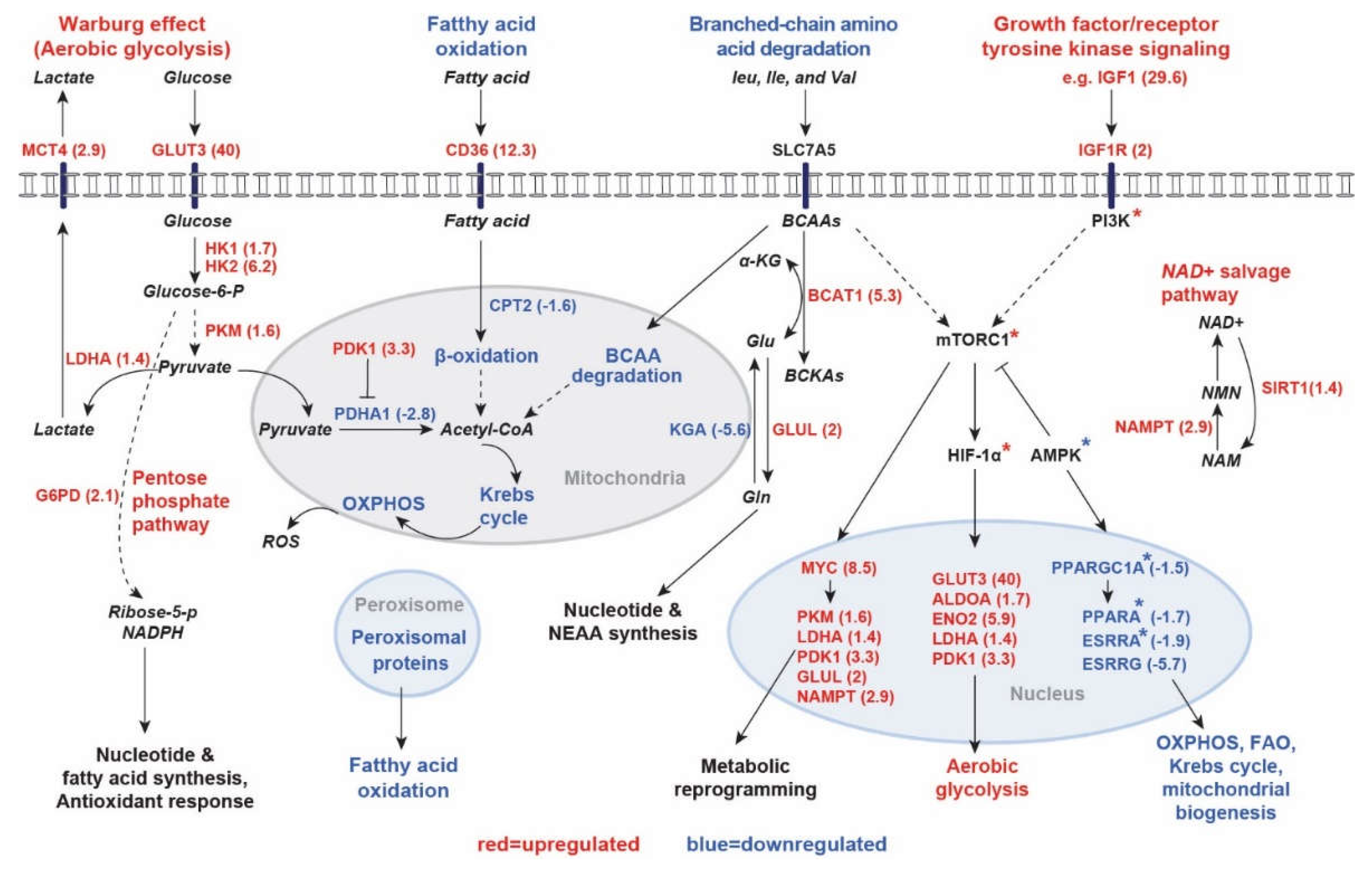

Table 1.
Dysregulated KEGG pathways (n=75) in PKD1 renal cysts (NOM p≤0.01 and FDR≤0.1).
| Upregulated (n=30) | SIZE | NES | NOM p-val | FDR q-val | Rank by NES |
|---|---|---|---|---|---|
| RIBOSOME | 81 | 2.95 | 0.00 | 0.000 | 1 |
| TGF_BETA_SIGNALING_PATHWAY | 84 | 2.44 | 0.00 | 0.000 | 2 |
| SPLICEOSOME | 118 | 2.36 | 0.00 | 0.000 | 3 |
| WNT_SIGNALING_PATHWAY | 145 | 2.19 | 0.00 | 0.000 | 4 |
| NUCLEOTIDE_EXCISION_REPAIR | 43 | 2.00 | 0.00 | 0.002 | 5 |
| FOCAL_ADHESION | 197 | 1.86 | 0.00 | 0.013 | 6 |
| BASAL_CELL_CARCINOMA | 53 | 1.85 | 0.00 | 0.012 | 7 |
| PATHWAYS_IN_CANCER | 320 | 1.85 | 0.00 | 0.011 | 8 |
| ECM_RECEPTOR_INTERACTION | 82 | 1.80 | 0.00 | 0.018 | 9 |
| PATHOGENIC_ESCHERICHIA_COLI_INFECTION | 54 | 1.79 | 0.00 | 0.018 | 10 |
| COLORECTAL_CANCER | 62 | 1.78 | 0.00 | 0.018 | 11 |
| ACUTE_MYELOID_LEUKEMIA | 56 | 1.76 | 0.00 | 0.020 | 12 |
| UBIQUITIN_MEDIATED_PROTEOLYSIS | 132 | 1.76 | 0.00 | 0.020 | 13 |
| MELANOMA | 71 | 1.70 | 0.01 | 0.030 | 14 |
| PROSTATE_CANCER | 89 | 1.70 | 0.00 | 0.028 | 15 |
| RENAL_CELL_CARCINOMA | 69 | 1.68 | 0.01 | 0.032 | 16 |
| MELANOGENESIS | 98 | 1.67 | 0.00 | 0.032 | 17 |
| CELL_CYCLE | 123 | 1.66 | 0.00 | 0.035 | 18 |
| OOCYTE_MEIOSIS | 109 | 1.64 | 0.00 | 0.040 | 19 |
| CHRONIC_MYELOID_LEUKEMIA | 72 | 1.64 | 0.00 | 0.040 | 20 |
| CYTOSOLIC_DNA_SENSING_PATHWAY | 54 | 1.63 | 0.00 | 0.040 | 21 |
| NOD_LIKE_RECEPTOR_SIGNALING_PATHWAY | 61 | 1.63 | 0.01 | 0.039 | 22 |
| REGULATION_OF_ACTIN_CYTOSKELETON | 210 | 1.63 | 0.00 | 0.038 | 23 |
| AXON_GUIDANCE | 128 | 1.62 | 0.00 | 0.037 | 24 |
| JAK_STAT_SIGNALING_PATHWAY | 150 | 1.62 | 0.00 | 0.037 | 25 |
| VIRAL_MYOCARDITIS | 67 | 1.61 | 0.01 | 0.039 | 26 |
| DILATED_CARDIOMYOPATHY | 89 | 1.58 | 0.01 | 0.045 | 27 |
| HYPERTROPHIC_CARDIOMYOPATHY_HCM | 82 | 1.57 | 0.00 | 0.048 | 28 |
| MAPK_SIGNALING_PATHWAY | 261 | 1.57 | 0.00 | 0.047 | 29 |
| CHEMOKINE_SIGNALING_PATHWAY | 180 | 1.47 | 0.01 | 0.082 | 30 |
| Downregulated (n=45) | SIZE | NES | NOM p-val | FDR q-val | Rank by NES |
| VALINE_LEUCINE_AND_ISOLEUCINE_DEGRADATION* | 44 | -2.99 | 0.00 | 0.000 | 1 |
| PROPANOATE_METABOLISM* | 32 | -2.90 | 0.00 | 0.000 | 2 |
| OXIDATIVE_PHOSPHORYLATION* | 122 | -2.72 | 0.00 | 0.000 | 3 |
| BUTANOATE_METABOLISM* | 33 | -2.71 | 0.00 | 0.000 | 4 |
| PYRUVATE_METABOLISM* | 39 | -2.62 | 0.00 | 0.000 | 5 |
| PEROXISOME | 77 | -2.61 | 0.00 | 0.000 | 6 |
| FATTY_ACID_METABOLISM* | 40 | -2.60 | 0.00 | 0.000 | 7 |
| PROXIMAL_TUBULE_BICARBONATE_RECLAMATION | 23 | -2.42 | 0.00 | 0.000 | 8 |
| CITRATE_CYCLE_TCA_CYCLE* | 30 | -2.41 | 0.00 | 0.000 | 9 |
| ARGININE_AND_PROLINE_METABOLISM | 49 | -2.39 | 0.00 | 0.000 | 10 |
| BETA_ALANINE_METABOLISM | 22 | -2.39 | 0.00 | 0.000 | 11 |
| ASCORBATE_AND_ALDARATE_METABOLISM | 14 | -2.32 | 0.00 | 0.000 | 12 |
| GLYCINE_SERINE_AND_THREONINE_METABOLISM | 30 | -2.29 | 0.00 | 0.000 | 13 |
| RENIN_ANGIOTENSIN_SYSTEM | 17 | -2.24 | 0.00 | 0.000 | 14 |
| PPAR_SIGNALING_PATHWAY | 67 | -2.19 | 0.00 | 0.000 | 15 |
| LYSINE_DEGRADATION | 41 | -2.17 | 0.00 | 0.000 | 16 |
| GLYCOLYSIS_GLUCONEOGENESIS | 60 | -2.15 | 0.00 | 0.000 | 17 |
| DRUG_METABOLISM_OTHER_ENZYMES | 39 | -2.15 | 0.00 | 0.000 | 18 |
| ALANINE_ASPARTATE_AND_GLUTAMATE_METABOLISM | 32 | -2.12 | 0.00 | 0.000 | 19 |
| FRUCTOSE_AND_MANNOSE_METABOLISM | 34 | -2.08 | 0.00 | 0.001 | 20 |
| MATURITY_ONSET_DIABETES_OF_THE_YOUNG | 24 | -1.99 | 0.00 | 0.002 | 21 |
| FOLATE_BIOSYNTHESIS | 11 | -1.97 | 0.00 | 0.002 | 22 |
| RETINOL_METABOLISM | 47 | -1.95 | 0.00 | 0.002 | 23 |
| TRYPTOPHAN_METABOLISM | 39 | -1.95 | 0.00 | 0.002 | 24 |
| TERPENOID_BACKBONE_BIOSYNTHESIS | 15 | -1.94 | 0.01 | 0.002 | 25 |
| PARKINSONS_DISEASE | 118 | -1.94 | 0.00 | 0.002 | 26 |
| PENTOSE_AND_GLUCURONATE_INTERCONVERSIONS | 16 | -1.94 | 0.00 | 0.002 | 27 |
| GLYCEROLIPID_METABOLISM | 42 | -1.94 | 0.00 | 0.002 | 28 |
| DRUG_METABOLISM_CYTOCHROME_P450 | 59 | -1.94 | 0.00 | 0.002 | 29 |
| LYSOSOME | 117 | -1.91 | 0.00 | 0.003 | 30 |
| HISTIDINE_METABOLISM | 28 | -1.90 | 0.00 | 0.003 | 31 |
| HUNTINGTONS_DISEASE | 174 | -1.88 | 0.00 | 0.003 | 32 |
| LIMONENE_AND_PINENE_DEGRADATION | 10 | -1.88 | 0.00 | 0.003 | 33 |
| METABOLISM_OF_XENOBIOTICS_BY_CYTOCHROME_P450 | 57 | -1.85 | 0.00 | 0.004 | 34 |
| VIBRIO_CHOLERAE_INFECTION | 53 | -1.82 | 0.00 | 0.006 | 35 |
| ALZHEIMERS_DISEASE | 158 | -1.82 | 0.00 | 0.006 | 36 |
| ARACHIDONIC_ACID_METABOLISM | 52 | -1.81 | 0.00 | 0.006 | 37 |
| STARCH_AND_SUCROSE_METABOLISM | 36 | -1.81 | 0.00 | 0.006 | 38 |
| PANTOTHENATE_AND_COA_BIOSYNTHESIS | 16 | -1.78 | 0.00 | 0.008 | 39 |
| PHENYLALANINE_METABOLISM | 18 | -1.75 | 0.01 | 0.010 | 40 |
| PENTOSE_PHOSPHATE_PATHWAY | 26 | -1.74 | 0.00 | 0.011 | 41 |
| TYROSINE_METABOLISM | 42 | -1.74 | 0.01 | 0.011 | 42 |
| STEROID_HORMONE_BIOSYNTHESIS | 43 | -1.71 | 0.00 | 0.013 | 43 |
| GLUTATHIONE_METABOLISM | 47 | -1.70 | 0.00 | 0.014 | 44 |
| PORPHYRIN_AND_CHLOROPHYLL_METABOLISM | 29 | -1.66 | 0.01 | 0.019 | 45 |
Table 2.
In silico prediction of top activated (n=50) and inhibited upstream regulators (n=48) in PKD1 renal cysts.
Table 2.
In silico prediction of top activated (n=50) and inhibited upstream regulators (n=48) in PKD1 renal cysts.
| Upstream regulator | Molecule type | Predicted Activation State | z-score | p-value of overlap |
|---|---|---|---|---|
|
Activated (z-score ≥ 2) |
||||
| TGFB1 | growth factor | Activated | 5.99 | 4.73E-18 |
| NUPR1 | transcription regulator | Activated | 5.91 | 1.32E-01 |
| Tgf beta | growth factor | Activated | 4.50 | 2.81E-05 |
| IL1B | cytokine | Activated | 4.39 | 2.34E-07 |
| IL6 | cytokine | Activated | 3.82 | 3.60E-02 |
| NR0B2 | ligand-dependent nuclear receptor | Activated | 3.82 | 2.01E-03 |
| SMAD4 | transcription regulator | Activated | 3.68 | 1.13E-03 |
| TGFBR2 | kinase | Activated | 3.67 | 9.54E-05 |
| Vegf | growth factor | Activated | 3.59 | 1.71E-04 |
| WNT1 | cytokine | Activated | 3.59 | 1.97E-05 |
| TGFB3 | growth factor | Activated | 3.51 | 1.45E-09 |
| F2 | peptidase | Activated | 3.49 | 1.03E-05 |
| TNF | cytokine | Activated | 3.48 | 1.54E-11 |
| TGFA | growth factor | Activated | 3.33 | 2.24E-01 |
| LDL | complex | Activated | 3.32 | 1.99E-01 |
| IL17A | cytokine | Activated | 3.18 | 8.81E-02 |
| SRF | transcription regulator | Activated | 3.17 | 2.43E-02 |
| IL1A | cytokine | Activated | 3.15 | 1.02E-02 |
| EDN1 | cytokine | Activated | 3.12 | 7.12E-02 |
| MKL1 | transcription regulator | Activated | 3.10 | 5.94E-02 |
| STAT4 | transcription regulator | Activated | 3.04 | 2.04E-07 |
| SMAD3 | transcription regulator | Activated | 3.01 | 6.54E-04 |
| EGF | growth factor | Activated | 2.94 | 3.73E-05 |
| P38 MAPK | mitogen-activated protein kinase | Activated | 2.82 | 4.94E-03 |
| CSF3 | cytokine | Activated | 2.82 | 5.17E-01 |
| FOXL2 | transcription regulator | Activated | 2.77 | 4.34E-01 |
| MTPN | transcription regulator | Activated | 2.75 | 1.05E-04 |
| IFNG | cytokine | Activated | 2.71 | 3.54E-05 |
| IGF2BP1 | translation regulator | Activated | 2.71 | 3.43E-05 |
| HTT | transcription regulator | Activated | 2.68 | 4.33E-05 |
| TGFBR1 | kinase | Activated | 2.67 | 2.17E-05 |
| HGF | growth factor | Activated | 2.66 | 1.37E-04 |
| C5 | cytokine | Activated | 2.66 | 1.00E+00 |
| STAT3 | transcription regulator | Activated | 2.62 | 5.35E-02 |
| OSM | cytokine | Activated | 2.59 | 3.47E-09 |
| F7 | peptidase | Activated | 2.59 | 1.65E-05 |
| CYP1B1 | enzyme | Activated | 2.56 | 2.38E-04 |
| Cg | complex | Activated | 2.56 | 3.21E-08 |
| IRF8 | transcription regulator | Activated | 2.55 | 1.00E+00 |
| MAP2K1/2 | MEK/ERK | Activated | 2.55 | 6.69E-03 |
| HIF1A | transcription regulator | Activated | 2.55 | 7.28E-05 |
| GDF9 | growth factor | Activated | 2.55 | 5.25E-03 |
| SMAD2 | transcription regulator | Activated | 2.53 | 1.00E+00 |
| NRG1 | growth factor | Activated | 2.53 | 1.74E-02 |
| CTNNB1 | transcription regulator | Activated | 2.50 | 9.74E-11 |
| MAP3K1 | kinase | Activated | 2.49 | 1.32E-01 |
| CSF1 | cytokine | Activated | 2.48 | 4.49E-01 |
| PDGF BB | complex | Activated | 2.47 | 9.83E-17 |
| SRC | kinase | Activated | 2.45 | 8.64E-04 |
| ADAM17 | peptidase | Activated | 2.43 | 2.63E-01 |
|
Inhibited (z-score ≤ -2) |
Molecule type | Predicted Activation State | z-score | p-value of overlap |
| PKD1 | ion channel | Inhibited | -7.82 | 3.64E-27 |
| HNF1A | transcription regulator | Inhibited | -7.29 | 2.31E-06 |
| LHX1 | transcription regulator | Inhibited | -7.07 | 2.01E-14 |
| PXR ligand-PXR-Retinoic acid-RXR | complex | Inhibited | -5.34 | 8.61E-04 |
| HNF4A | transcription regulator | Inhibited | -4.97 | 1.10E-08 |
| PPARGC1A | transcription regulator | Inhibited | -4.88 | 1.51E-02 |
| INSR | kinase | Inhibited | -4.54 | 3.69E-03 |
| Alpha catenin | group | Inhibited | -4.16 | 1.24E-07 |
| Ncoa-Nr1i2-Rxra | complex | Inhibited | -4.11 | 2.81E-04 |
| CAR ligand-CAR-Retinoic acid-RXR | complex | Inhibited | -4.07 | 3.38E-03 |
| Ncoa-Nr1i3-Rxra | complex | Inhibited | -3.68 | 1.01E-02 |
| HNF4 dimer | complex | Inhibited | -3.64 | 1.01E-02 |
| AHR | ligand-dependent nuclear receptor | Inhibited | -3.51 | 4.40E-12 |
| WISP2 | growth factor | Inhibited | -3.45 | 5.90E-03 |
| PPARA | ligand-dependent nuclear receptor | Inhibited | -3.44 | 1.33E-03 |
| FOXA2 | transcription regulator | Inhibited | -3.41 | 1.86E-01 |
| estrogen receptor | group | Inhibited | -3.25 | 3.21E-09 |
| ESRRA | ligand-dependent nuclear receptor | Inhibited | -3.23 | 1.36E-01 |
| SOX2 | transcription regulator | Inhibited | -3.22 | 5.04E-04 |
| NKX2-1 | transcription regulator | Inhibited | -3.20 | 8.61E-03 |
| SGK1 | kinase | Inhibited | -3.16 | 2.07E-01 |
| FOXA3 | transcription regulator | Inhibited | -3.15 | 7.92E-03 |
| DICER1 | enzyme | Inhibited | -2.97 | 3.94E-03 |
| POU3F3 | transcription regulator | Inhibited | -2.91 | 3.95E-02 |
| RXRA | ligand-dependent nuclear receptor | Inhibited | -2.86 | 7.54E-04 |
| FOXI1 | transcription regulator | Inhibited | -2.84 | 1.52E-03 |
| SMAD7 | transcription regulator | Inhibited | -2.82 | 3.44E-07 |
| FGF21 | growth factor | Inhibited | -2.71 | 1.25E-01 |
| Immunoglobulin | complex | Inhibited | -2.67 | 3.73E-01 |
| ALDH1A2 | enzyme | Inhibited | -2.67 | 1.73E-04 |
| KRAS | enzyme | Inhibited | -2.63 | 8.58E-08 |
| NR4A3 | ligand-dependent nuclear receptor | Inhibited | -2.58 | 3.87E-01 |
| DKK1 | growth factor | Inhibited | -2.40 | 1.29E-03 |
| MAX | transcription regulator | Inhibited | -2.35 | 1.42E-02 |
| KLF2 | transcription regulator | Inhibited | -2.32 | 3.91E-02 |
| CFTR | ion channel | Inhibited | -2.32 | 2.12E-03 |
| GSK3B | kinase | Inhibited | -2.29 | 3.75E-01 |
| NOG | growth factor | Inhibited | -2.29 | 2.88E-01 |
| PPIF | enzyme | Inhibited | -2.24 | 5.10E-01 |
| SPDEF | transcription regulator | Inhibited | -2.20 | 2.17E-05 |
| DACH1 | transcription regulator | Inhibited | -2.15 | 3.50E-03 |
| AMPKα2 | kinase | Inhibited | -2.13 | 2.97E-01 |
| PTPN1 | phosphatase | Inhibited | -2.10 | 1.00E+00 |
| SPTLC2 | enzyme | Inhibited | -2.10 | 3.88E-02 |
| Laminin | complex | Inhibited | -2.07 | 5.25E-04 |
| INHA | growth factor | Inhibited | -2.05 | 1.92E-08 |
| KDM1A | enzyme | Inhibited | -2.03 | 1.00E+00 |
| ERP29 | transporter | Inhibited | -2.00 | 1.48E-01 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



