
Submitted:
11 May 2024
Posted:
13 May 2024
You are already at the latest version
Abstract
Heterodimeric approach has emerged as a promising method for simultaneously targeting multiple receptors on tumor cells using a single molecule. Simultaneous targeting of the prostate-specific membrane antigen (PSMA) and the gastrin-releasing peptide receptor (GRPr) holds the potential to improve the accuracy of prostate cancer diagnosis. This study aimed to develop and compare six alternative routes for stereoselective synthesis of heterobivalent conjugates designed to deliver the chelating agent DOTA to PSMA/GRPr receptors. The comparison of these alternative synthetic approach, considered such factors as efficiency, complexity, synthesis and purification characteristics, as well as yields of the target compounds has been made. Optimal conditions for the stereoselective synthesis of heterobivalent ligands to PSMA and GRP, which could serve as molecular platforms for the targeted delivery of therapeutic and/or diagnostic agents to these receptors, were determined. For synthesized heterobivalent ligand 26x and heterobivalent conjugate with DOTA 27 complete signal assignment in 1H, 13C, and 15N NMR spectra was achieved using 2D NMR techniques. Based on these data, comprehensive signal assignments were provided for all final compounds in their NMR spectra.
Keywords:
Prostate cancer
; Targeted delivery
; Нeterobivalent conjugates
; PSMA
; GRPr
Introduction
Prostate cancer, also known as prostatic carcinoma, arises from the epithelial cells of the prostate gland and ranks among the most prevalent cancers affecting men globally [1]. Annually, around 400 thousand cases of prostate cancer are diagnosed worldwide, with some countries ranking it as the second most common cancer [2]. In men over 60, it stands as the leading cause of cancer-related deaths.
Timely diagnosis of prostate cancer is critical, as late-stage tumors are highly aggressive due to metastasis and are challenging to treat effectively. Some imaging techniques such as magnetic resonance imaging (MRI), computed tomography (CT), single-photon emission computed tomography (SPECT), and positron emission tomography (PET) are employed for functional visualization of prostate cancer. However, these methods, focusing on morphological changes, have limitations in evaluating the full complexity of the disease. Consequently, there is still a need for studies to identify new diagnostic and therapeutic biomarkers for prostate cancer. Radionuclide imaging using SPECT or PET offers a promising avenue for early diagnosis, enabling non-invasive detection of various molecular and cellular processes associated with prostate cancer. Successful PET/SPECT imaging relies on the overexpression of specific receptors in tumors, with prostate-specific membrane antigen (PSMA) emerging as a prominent target for both diagnostic and therapeutic interventions [3]. Another potential target of prostate tumor specific compounds is gastrin-releasing peptide receptor (GRPr), significantly expressed on the surface of cancer cells in early-stage prostate cancer [4]. However, PSMA expression varies, sometimes hindering effective visualization when targeted alone [5,6,7,8]. Hence, the development of heterobivalent constructs simultaneously targeting PSMA/GRPr using chelating agents for PET and SPECT imaging may be proposed as a solution.
The heterodimeric approach offers a promising strategy for targeting multiple receptors on tumor cells with a single molecule. Heterodimers, composed of two antibodies/fragments or two peptides/peptide mimetics, enhance binding affinity through multivalent interactions [9]. Peptide heterodimers, in particular, show potential in diagnosing structurally heterogeneous tumors, catering to regions with different receptor binding specificities [10,11,12].
Although peptide/peptide mimetic heterodimers are still in early stages of application in molecular imaging, they demonstrated significant potential, especially in multitargeting GRPr/PSMA [9,11,13,14,15,16,17,18,19,20,21] and addressing prostate cancer heterogeneity. Therefore, further research into heterodimers for both molecular imaging and theranostic applications is warranted. This study presents synthetic approaches to developing the heterobivalent conjugates for PSMA/GRPr receptor targeting with the chelating agent DOTA. Notably, it provides detailed descriptions and complete signal assignments in the NMR (1Н, 13C, 15N) spectra of all final and some intermediate compounds. Such complete assignment are usually absent in the articles describing syntheses of such complex conjugates; at the same time, it can be very useful for subsequent researchers involved in the development of compounds of structurally similar types.
3. Results and Discussion
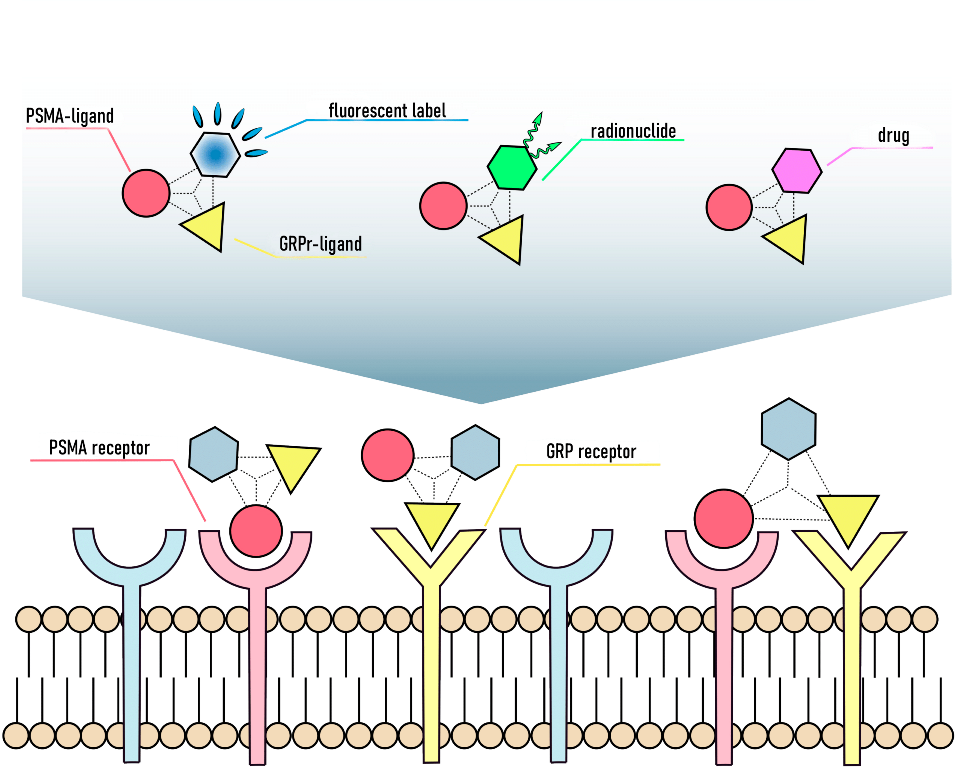
In this work, the development and evaluation of synthetic approaches to affine and selective heterobivalent ligands capable of target delivery of various functional fragments such as: 1) chelating agents (DOTA), 2) fluorescent labels (Sulfo-Cy5), 3) cytotoxic/cytostatic drugs (Docetaxel). The general structure of heterobivalent conjugates using the DOTA chelator is shown in Figure 1.
In order to obtain the target HBV conjugates, the following synthetic tasks were solved:
- 1)
- PSMA vector 6 synthesis (Section 3.3., Scheme 1).
- 2)
- Assembly of the peptide sequence included in the PSMA-ligand (optionally, a "branching fragment" = Lys(L) can be attached see Route 1, 4a-c) using the solid-phase peptide synthesis (SPPS) technique followed by connection with the PSMA-vector (optionally, a functional fragment can be attached as = HO-DOTA(tBu)3 see Route 1) and removal from the polymer substrate (Section 3.4., Scheme 3).
- 3)
- Assembly of the peptide sequence included in the GRPr-ligand by SPPS technique (optionally, a "branching fragment"= Lys(L) can be attached see Route 2,3) (Section 3.5., Scheme 3).
- 4)
- Functional fragment insertion: In this case, the chelating agent EDTA was used (Scheme 5, Scheme 6, Scheme 7, Scheme 8, Scheme 9 and Scheme 10), however, a number of synthetic schemes can be used for the synthesis of targeted to PSMA/GRPr monomodal HBV conjugates. In addition, the proposed procedure is not limited to chelators only.
- 5)
It should be noted that the presented numbering does not reflect the order of synthesis and may vary depending on the considering synthetic Routes.
3.1. Criteria for Choosing a Scheme for CBV Conjugate Synthesis
When planning multi-stage syntheses, it is worth considering a multitude of parameters, ranging from the number of stages and their time consumption, up to the cost of reagents and their selection. It’s rather difficult to find the optimal approach to synthesis in all parameters at once. Designing strategies for the synthesis of heterobivalent PSMA-GRPr ligands, we focused on the following important parameters:
- 1.
-
Laboriousness of the synthetic scheme
- 1.1.
- The number of stages in the synthetic scheme
- 1.2.
-
Stage of the experiment
- 1.2.1.
- setup
- 1.2.2.
- reaction
- 1.2.3.
- workup and purification
- 1.2.4.
- product characterization, structure and purity confirmation
- 2.
- Features of application and storage conditions of substrates and reagents
- 3.
- Yield calculated from the reaction of the key structural fragments of the HBV conjugate
1. Laboriousness of a Synthetic Scheme
1.1. The Number of Stages in a Synthetic Scheme
The number of stages in the synthetic scheme affects the choice of a particular Method for obtaining the final compound. This directly affects on 1) the total time of synthesis; 2) the yield of the final compound; 3) laboriousness. Although liquid-phase and solid-phase methods require almost the same number of synthetic stages, the SPPS method requires fewer chromatographic purification stages than LPPS, which simplifies and accelerates synthesis.
1.2. Stage of the Experiment
1.2.1. Setup
In most cases of peptide synthesis setting up the reaction takes a small amount of time relatively to the total time required to isolate a pure target compound. Therefore, it is not possible to allocate a priority synthetic scheme for this parameter.
1.2.2. Reaction
In the synthetic schemes discussed below, peptide macromolecules were obtained by utilizing two techniques for peptide synthesis:
The concept of solid-phase peptide synthesis (SPPS) is a process of assembling a peptide by covalently attached a C-terminal amino acid sequence to an insoluble polymer substrate (resin). The anchored peptide is sequentially lengthened by a series of cycles of adding a new amino acid and removing the protecting group located at its N- terminus. In this case, we considered exclusively the Fmoc/t-Bu SPPS strategy.
The liquid-phase peptide synthesis (LPPS) consists in assembling a peptide sequence in solution, through a series of cycles of adding new amino acid and protecting group cleavage. A combination of these approaches with varying degrees of participation in synthetic schemes has been used to obtain target molecules.
Each of these methods has its own advantages and disadvantages:
The advantages of SPPS are: 1) ease of synthetic procedure due to the simplicity of isolation: after each coupling cycle, filtration and washing of the entire resin is performed; 2) the process can be automated. However, this method has a couple of drawbacks: 1) due to the heterogeneous nature of the reaction, it is often necessary to use an excess of combined components and increase the reaction time; 2) steric hindrance has a more significant effect, interfering with the course of reactions; 3) absence of an explicit possibility to confirm the structure at each stage (this can only be performed by a destructive method of complete removing the peptide sequence from a part of the resin).
The LPPS technique also has its advantages and disadvantages. The advantages include: 1) reactions, which occur in homogeneous medium, in general, proceed faster and allow using equimolar amounts of reagents; 2) the ability to confirm the structure and determine the purity of the substance at each stage of synthesis without losing in the yield of the target compound; 3) steric effects do not affect as much as in SPPS. However, each step of LPPS requires purification by one or more methods (chromatography, extraction, trituration, etc.), which generally noticeably inferior to the solid-phase technique in terms of total time and effort required to perform the synthesis.
1.2.3. Workup and Purification
Turning to the purification of compounds, it is worth noting that the most laborious method for isolating this kind of multifunctional molecules is chromatography. The solid-phase method has an advantage over the liquid-phase method in terms of the number of acts of chromatographic purification.
1.2.4. Product Characterization, Structure and Purity Confirmation
Confirmation of the structure and purity of the final compounds in the case of liquid-phase (LPPS) and solid-phase (SPPS) synthesis techniques takes the same time. It is worth noting that using the liquid-phase approach, it is possible to verify intermediate compounds by physicochemical analytical methods (primarily by 1H and 13C NMR), without reducing the yield of the final compound. Step-by-step identification makes it possible to track all structural changes in gradually complicating NMR spectra, which can greatly facilitate the characterization of the final molecule.
There are two approaches for SPPS:
The first is the exception of intermediate control and confirmation of the structure and purity for only the final compound, which may be difficult due to the intricacy of the NMR picture. This approach does not leave a probability for errors identification during the assembly of the peptide sequence, but only after the completion of obtaining the polypeptide sequence.
The second is to remove a peptide fragment from a part of the polymer substrate after some stages of synthesis to analyze the result. This approach has the downside of decreasing the yield of the final compound (in some cases, quite significant).
2. Features of Application and Storage Conditions of Substrates and Reagents
In some instances, where substrates and reagents are unstable or highly toxic, the synthetic scheme should be modified to minimize or completely eliminate the utilization of these compounds.
3. Yield Calculated from the Reaction of the Key Structural Fragments
Three key structural fragments can be defined in the HBV conjugate:
- 1)
- PSMA-ligand
- 2)
- Functional fragment (chelator, fluorescent label, cytostatic/cytotoxic drug)
- 3)
- GRPr-ligand
Based on them, a quantitative assessment of the effectiveness of the proposed Routes for obtaining final compound is presented (see Table 2)
3.2. Classification of Routes for CBV Conjugate Synthesis
The Routes are classified according to stage and method of introducing the functional fragment. Chelators, fluorescent labels or drugs for conjugating with ligands are themselves products of multistage syntheses and, therefore, are expensive. Operating each type of functional fragment has a number of features and limitations. Taking this into consideration, it can be concluded that when developing synthetic circuits, it is worth striving for a later stage of their introduction into the structure of the target compound.
One of the functional fragments that are widely used in the diagnosis and therapy of prostate cancer is the chelator for radionuclide labeling DOTA. DOTA is a twelve–membered conformationally mobile cycle that includes 4 fragments of tertiary amines in its structure. Based on experience with this type of compound, it should be noted that the presence of such a fragment in the ligand structure greatly complicates chromatographic purification, as well as confirmation of the structure and purity of the target compound.
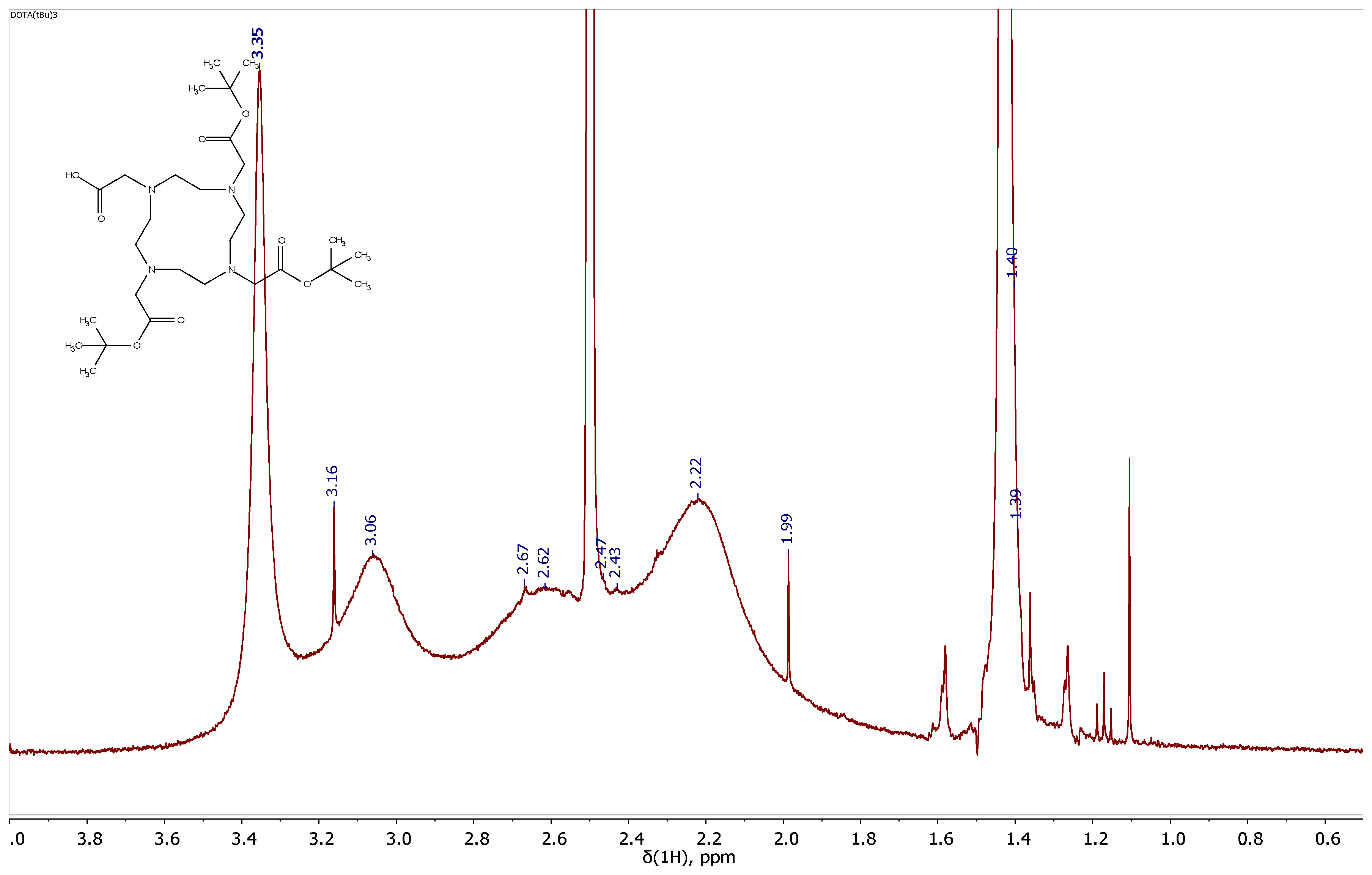
Figure 2.
Fragment of the 1H NMR spectrum of HO-DOTA(tBu)3 (400 MHz, 293 K, DMSO-d6). 1H NMR (500 MHz, DMSO-d6) = 4.10–3.48 (m, 8H), 3.45–2.89 (m, 16H), 1.45 (s, 9H), 1.38 (s, 18H). The description of the spectrum is provided from the article. [22].
Figure 2.
Fragment of the 1H NMR spectrum of HO-DOTA(tBu)3 (400 MHz, 293 K, DMSO-d6). 1H NMR (500 MHz, DMSO-d6) = 4.10–3.48 (m, 8H), 3.45–2.89 (m, 16H), 1.45 (s, 9H), 1.38 (s, 18H). The description of the spectrum is provided from the article. [22].
Therefore, it is important to minimize the number of synthetic stages, which can cause such unsolvable obstacles during the planning.
Based on an analysis of all mentioned parameters, six synthetic Routes were proposed for obtaining PSMA-GRPr heterobivalent conjugates with the chelating agent DOTA (Figure 3).
All the presented synthetic approaches can be divided into three groups: 1) Routes 1&2, according to which DOTA is conjugated to the peptide sequence by SPPS at an intermediate stage of synthesis; 2) Route 3 wherein DOTA is attached by LPPS at the last stage as NHS-activated ester; 3) Routes 4а&4b&4c that involve the addition of DOTA to the peptide sequence using LPPS at an intermediate stage of synthesis (Figure 3).
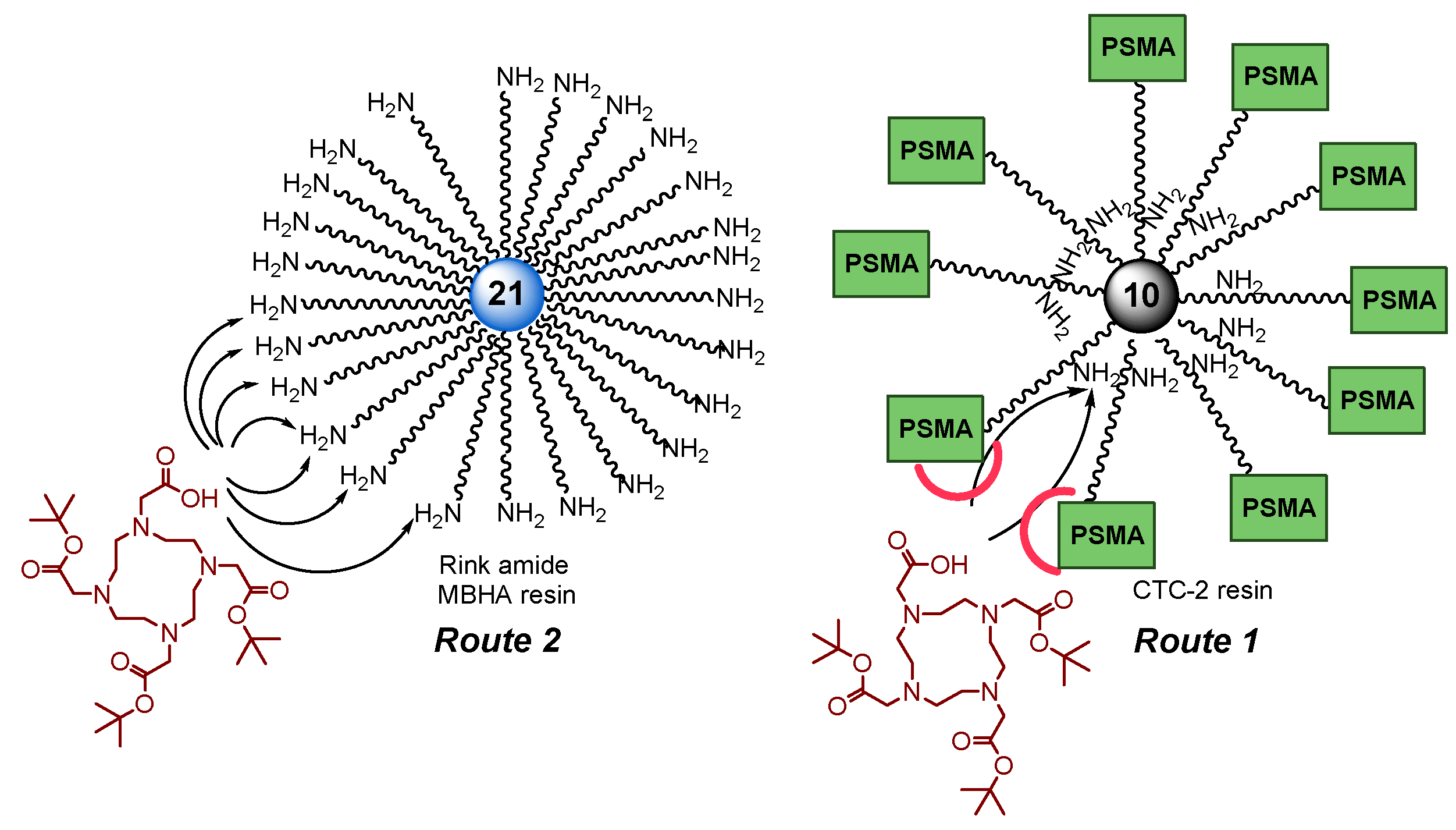
The synthetic schemes of the first group include 2 approaches: conjugation of the functional fragment on STS-2 resin (Route 1), conjugation of the functional fragment on Rink Amide MBHA resin (Route 2). The scheme of the second group is the synthesis of the heterobivalent ligand on Rink Amide MBHA resin followed by liquid-phase introduction of a functional fragment (Route 3). The synthetic schemes of the third group include 3 approaches based on Fmoc(OFm)/Alloc orthogonal protection strategy (Routes 4а&4b&4c).
3.3. PSMA Vector Synthesis
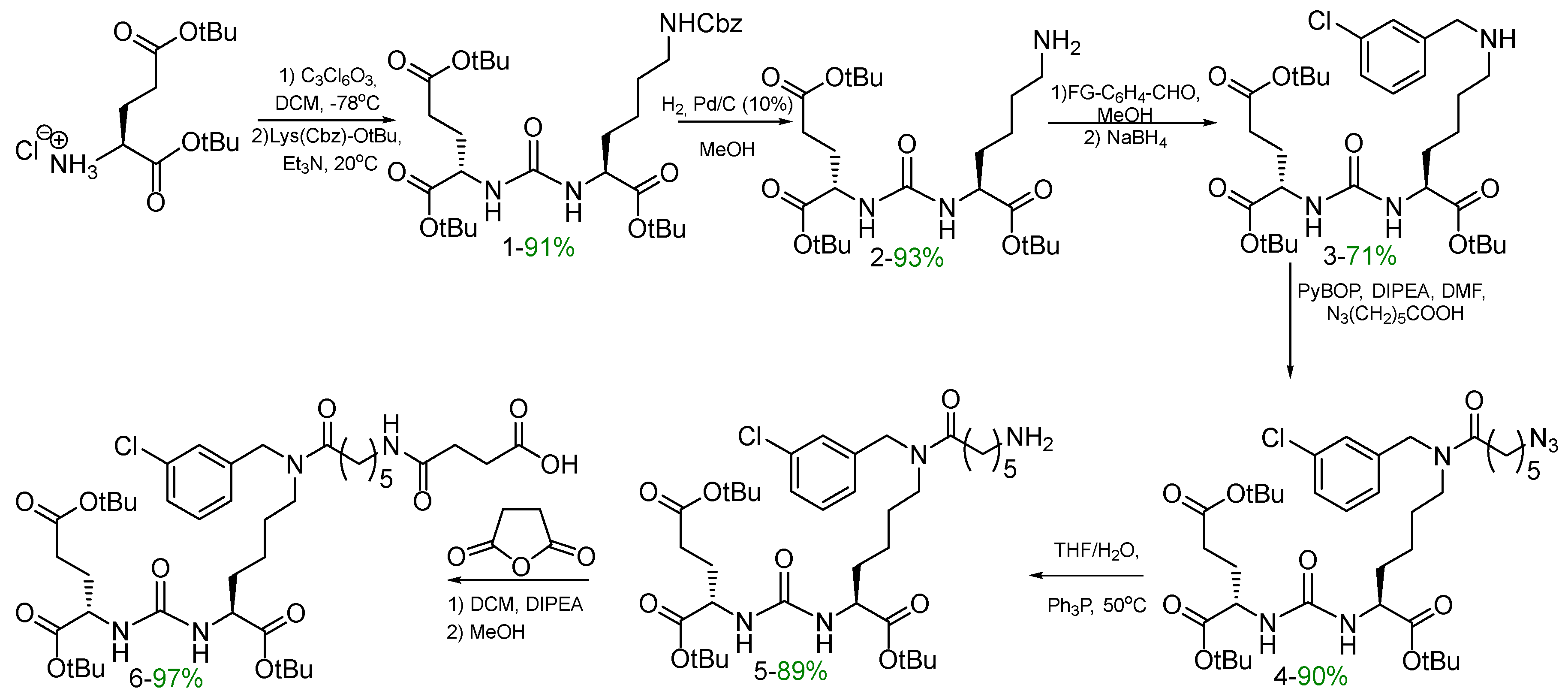
The initial stages of the vector fragment synthesis included the preparation of compounds 1-6 (Scheme 1) by optimized methods similar to described in our previous work [23].
Scheme 1.
Synthesis of vector fragment 6 based on a PSMA inhibitor.
3.4. Assembling the Peptide Part of the PSMA-Ligand on 2-CTC Resin and Vector Fragment Coupling
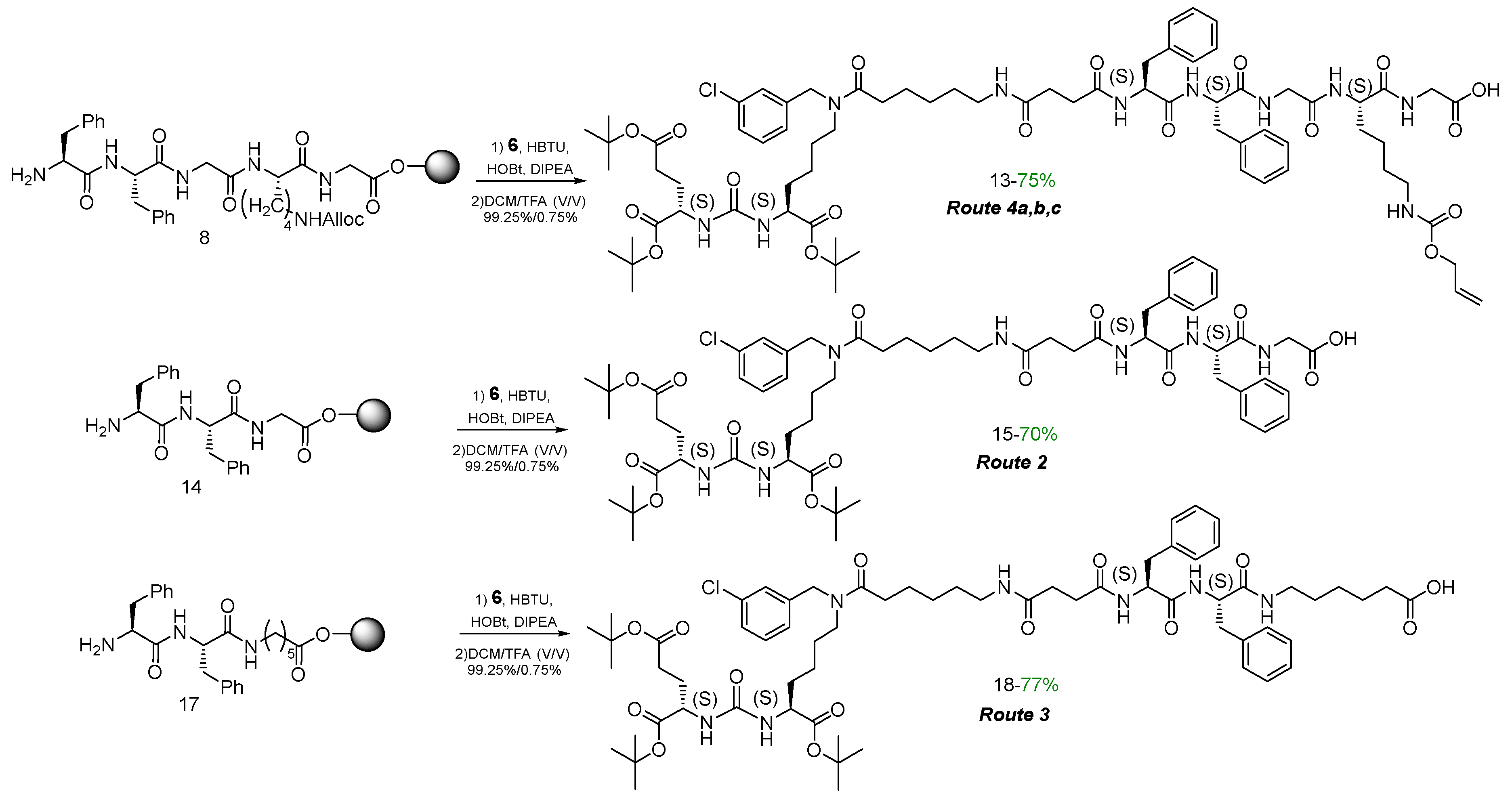
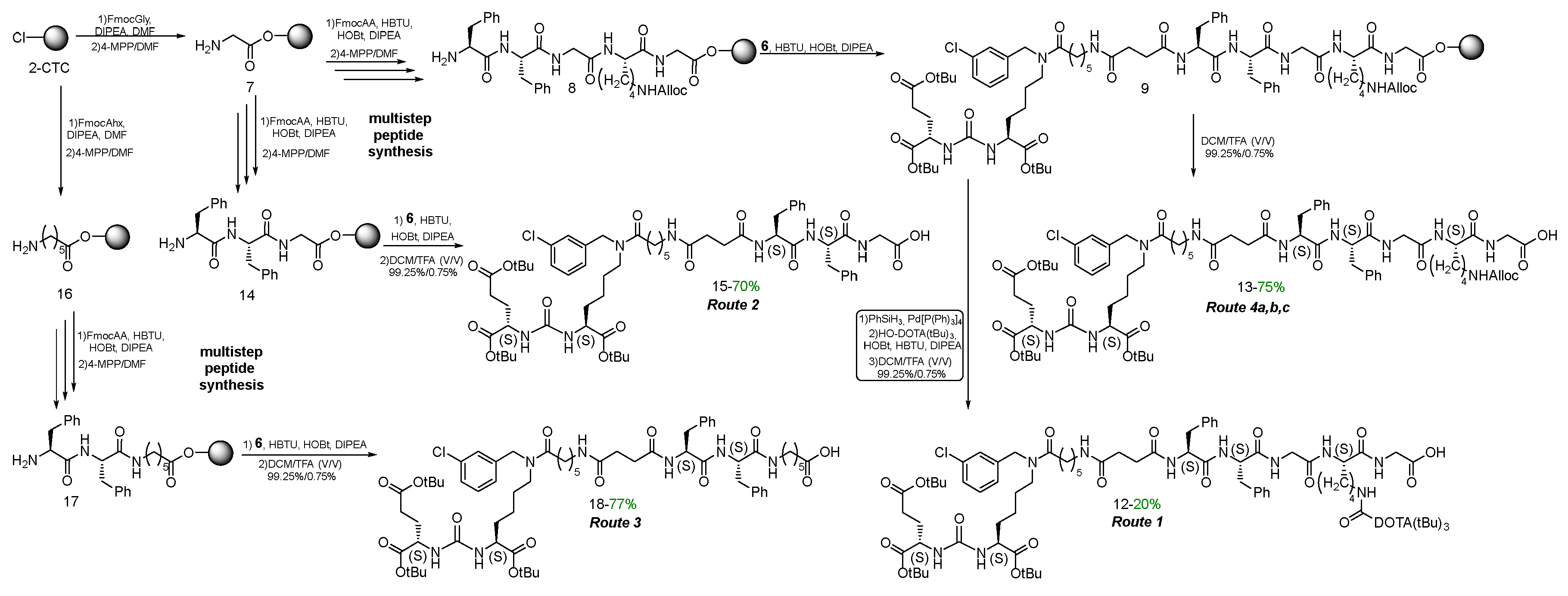
To obtain highly specific PSMA vectors, peptide sequences anchored to 2-CTC resin were synthesized: 1) NH2-Phe(L)-Phe(L)-Gly-Lys(L)(Alloc)-Gly = 8, 2) NH2-Phe(L)-Phe(L)-Gly = 14, 3) NH2-Phe(L)-Phe(L)-Ahx = 17. It has been previously shown [24,25] that the presence of Phe(L)-Phe(L) dipeptide fragment in the linker improves binding to the receptor. Furthermore, the dipeptide nature of linkers could improve biodegradability and reduce toxicity of PSMA vectors [26,27]. The protected amino acid lysine in the form of Fmoc-Lys(L)(Alloc)-OH, which is capable of providing further modification by various structural blocks under orthogonal conditions, was chosen as the "branching point" for Routes 1 and 4а&4b&4c.
To obtain peptide sequences 8, 14, 17 by SPPS technique, a polymer substrate made of polystyrene crosslinked with 1% divinylbenzene (2-CTC resin) was used (Scheme 3). The chosen sequence of reactions is a classic scheme of peptide synthesis: 1) immobilization of an N-substituted amino acid onto a solid-phase substrate; 2) cleavage of the protecting group; 3) modification of the NH2-group of amino acid (steps 2 and 3 are repeated the required number of times to assemble the target peptide sequence); 4) cleavage of the peptide sequence from solid support [28].
The 2-CTC resin was chosen because it is compatible with the Fmoc/tBu concept, as well as the cleavage of the peptide sequence from the resin takes place under mild conditions safe for acid-labile functional groups (COOtBu) (in this case cleavage from the resin was carried out using DCM/TFA – 99.25%/0.75% V/V solution) [29].
At the final stage, vector fragment 6 was coupled with peptides 8, 14, 17 attached to 2-CTC resin using HBTU/HOBt/DIPEA activating agents. After that, the polypeptides were cleaved off the resin by DCM/TFA treatment (Scheme 2). As a result, compounds 13, 15, 18 were obtained as individual stereoisomers (according to the 1H NMR, 13C NMR, LCMS, HRMS data – see Materials and Methods).
Scheme 2.
Synthesis and cleavage from the resin of the peptide compounds 13, 15, 18.
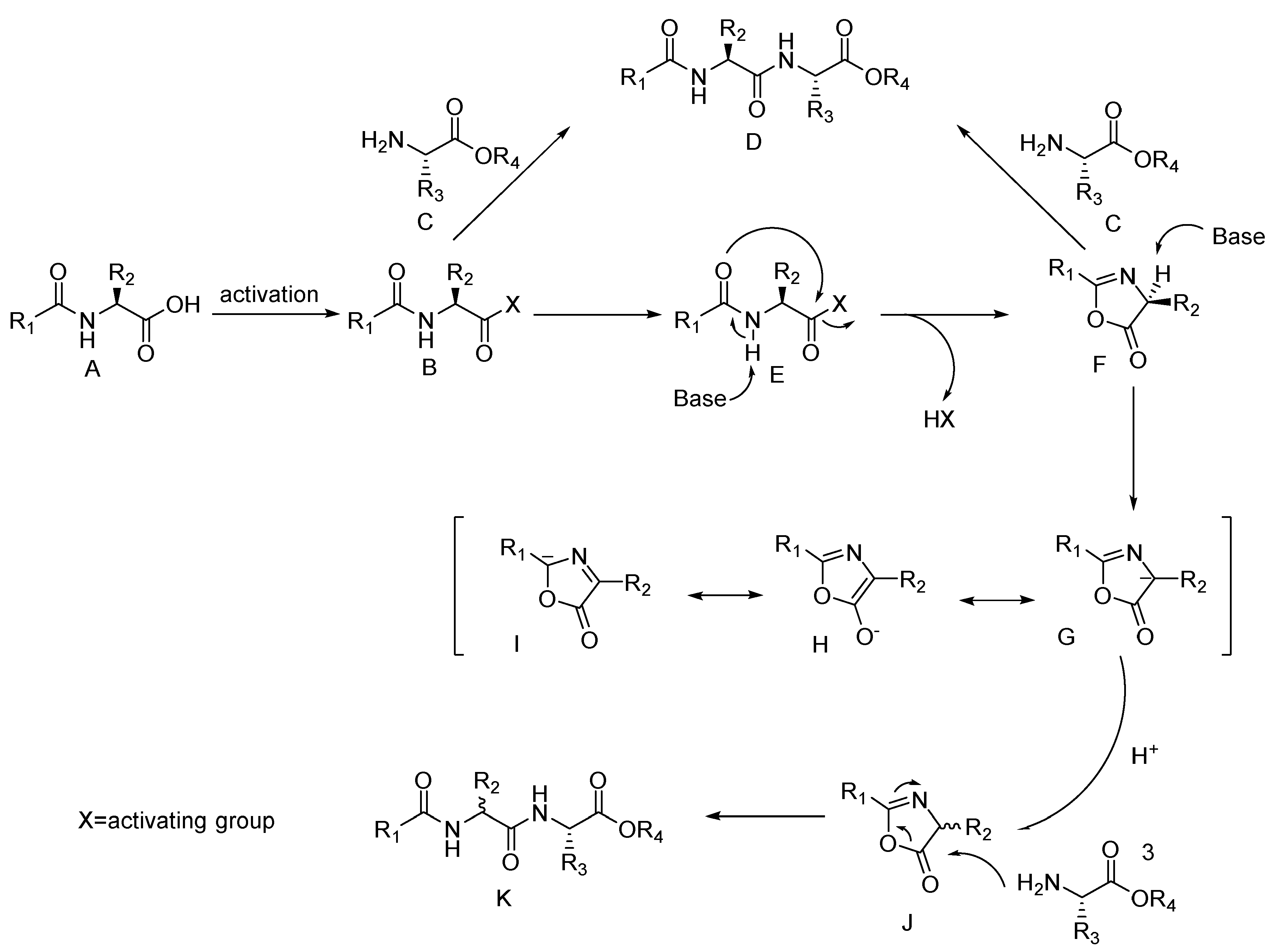
It's important to emphasize that the amino acids Gly&Ahx were used as the C-ends of the peptide sequences due to their lack of a stereocenter in the α-position. This provides an opportunity to conduct peptide synthesis reactions with other structural fragments that are part of the final HBV conjugate, without concern of a side reaction of epimerization, through the formation of oxazolone. [30,31] (Figure 4).
Figure 4.
The mechanism of amino acid racemization through the formation of oxazolone [30].
Figure 4.
The mechanism of amino acid racemization through the formation of oxazolone [30].
Scheme 3.
Synthesis of PSMA ligand (optionally, a "branching fragment" = Lys(L) can be attached see Route 1, 4a-c). Reagents and conditions: For compound 8 = (a) (1) FmocLys(L)(Alloc), HBTU, HOBt, DIPEA; (2) 4-methylpiperidine/DMF; (b) (1) FmocGly, HBTU, HOBt, DIPEA; (2) 4-methylpiperidine/DMF; (c) (1) FmocPhe(L), HBTU, HOBt, DIPEA; (2) 4-methylpiperidine/DMF; (d) (1) FmocPhe(L), HBTU, HOBt, DIPEA; (2) 4-methylpiperidine/DMF. For compound 14 = (a) (1) FmocPhe(L), HBTU, HOBt, DIPEA; (2) 4-methylpiperidine/DMF; (b) (1) FmocPhe(L), HBTU, HOBt, DIPEA; (2) 4-methylpiperidine/DMF. For compound 17 = (a) (1) FmocPhe(L), HBTU, HOBt, DIPEA; (2) 4-methylpiperidine/DMF; (b) (1) FmocPhe(L), HBTU, HOBt, DIPEA; (2) 4-methylpiperidine/DMF.
Scheme 3.
Synthesis of PSMA ligand (optionally, a "branching fragment" = Lys(L) can be attached see Route 1, 4a-c). Reagents and conditions: For compound 8 = (a) (1) FmocLys(L)(Alloc), HBTU, HOBt, DIPEA; (2) 4-methylpiperidine/DMF; (b) (1) FmocGly, HBTU, HOBt, DIPEA; (2) 4-methylpiperidine/DMF; (c) (1) FmocPhe(L), HBTU, HOBt, DIPEA; (2) 4-methylpiperidine/DMF; (d) (1) FmocPhe(L), HBTU, HOBt, DIPEA; (2) 4-methylpiperidine/DMF. For compound 14 = (a) (1) FmocPhe(L), HBTU, HOBt, DIPEA; (2) 4-methylpiperidine/DMF; (b) (1) FmocPhe(L), HBTU, HOBt, DIPEA; (2) 4-methylpiperidine/DMF. For compound 17 = (a) (1) FmocPhe(L), HBTU, HOBt, DIPEA; (2) 4-methylpiperidine/DMF; (b) (1) FmocPhe(L), HBTU, HOBt, DIPEA; (2) 4-methylpiperidine/DMF.
3.5. Assembling the JMV594 Peptide Sequence on the Rink Amide MBHA Resin
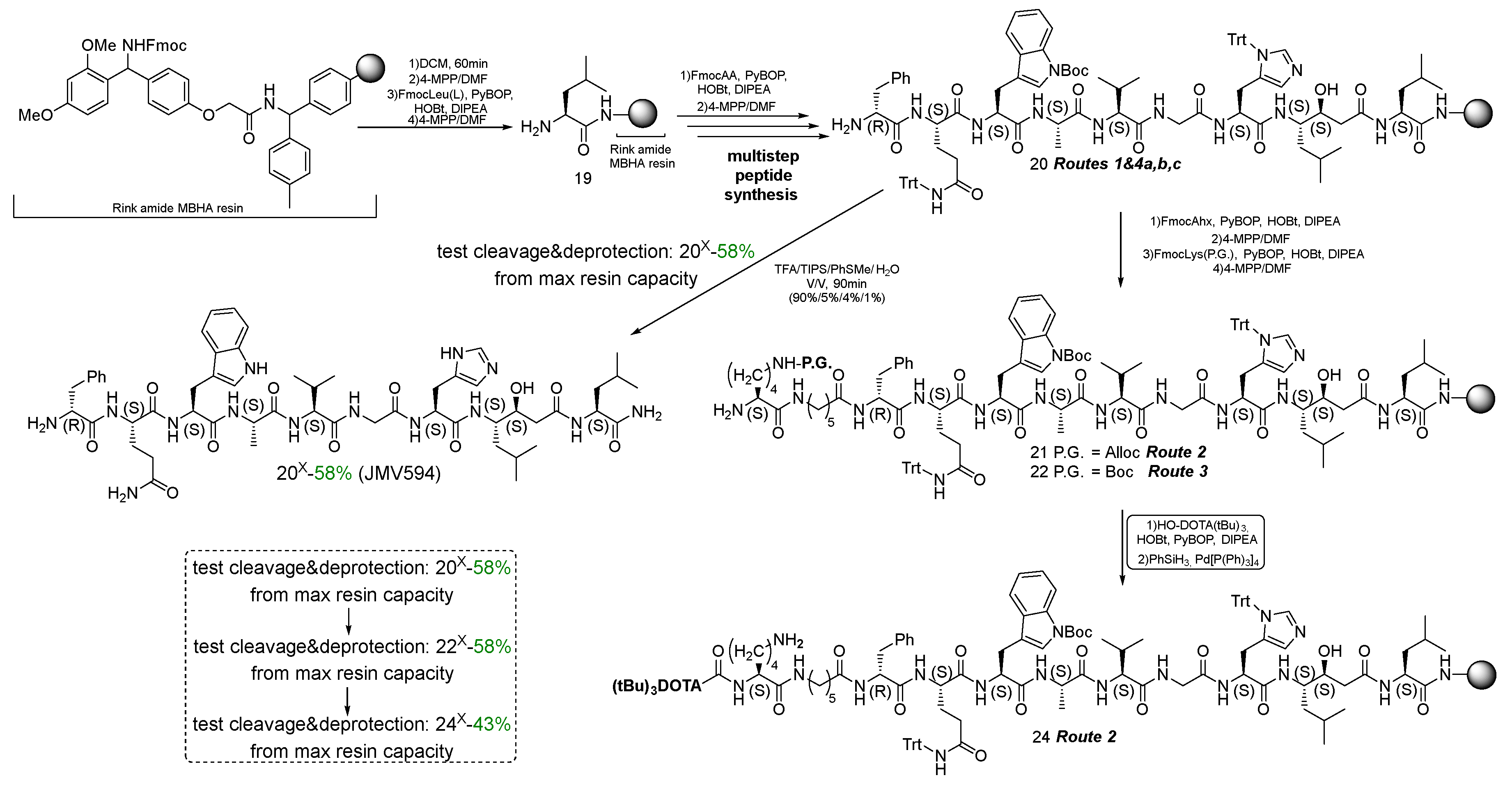
The synthesis of D-Phe-Gln-Trp-Ala-Val-Gly-His-Sta-Leu-NH2 = [D-Phe6,Sta13,Leu14]bombesin[6-14] = JMV594 = compound 20 (20х) was performed by manual solid-phase peptide synthesis (SPPS) [17,32] using Fmoc Rink Amide MBHA resin (see Scheme 4) (capacity 0.8-0.3 mmol/g) and standard conditions for the Fmoc/t-Bu concept. The selected synthetic scheme includes stages: 1) Fmoc-deprotection of the resin (20%/80% MPP/DMF, V/V); 2) immobilization of an N-substituted amino acid onto a solid-phase substrate; 2) Fmoc-deprotection of amino acid (20%/80% 4-MPP/DMF, V/V); 3) modification of the NH2-group of amino acid (steps 2 and 3 are repeated the required number of times to assemble the target peptide sequence); 4) cleavage of the peptide sequence from solid support and total deprotection (removal of all protecting groups, for 20 2*Trt, 1*Boc). For certain Fmoc-amino acids, the following protecting groups in the side chain were used: Fmoc-His(L)(Trt)-OH, Fmoc-Trp(L)(Boc)-OH, Fmoc-Gln(L)(Trt)-ОН. Fmoc-Sta(S,S)-ОН was used without protection of the hydroxyl group. However, it is known from the literature that during the assembly of the peptide sequence, the treatment of 4-MPP in DMF (20%/80%) not only removes Fmoc-P.G., but also prevents possible acylation of the hydroxyl group Sta(S,S) [32].
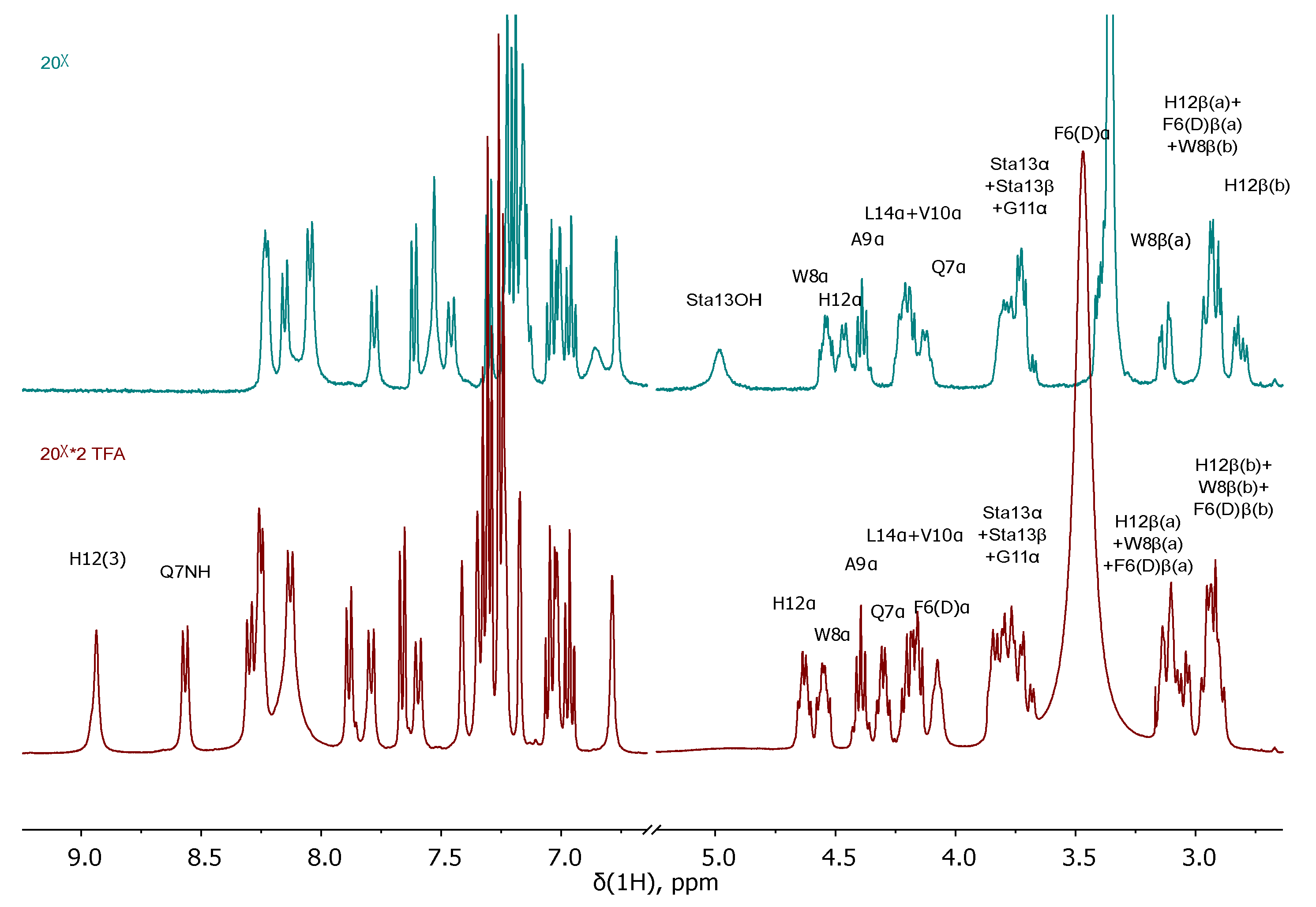
To establish the chemical purity and evaluate the actual capacity of the Rink Amide MBHA resin with the anchored peptide sequences JMV594 = compound 20 and Lys(Boc)-Ahx-JMV594 = compound 22, their parts were removed from the polymer substrate (giving compounds 20x and 22x respectively). According to 1Н NMR, 13C NMR, 15N NMR, LCMS, HRMS spectral information, both of them (20x and 22x) were individual stereoisomers (see Materials and Methods). Compound 20x has been characterized by a number of 2D NMR experiments. Based on the data obtained, a complete correlation of the structure was given (see Figure 14). It is noteworthy that after chromatographic purification, compound 20x was isolated in several forms: *2 TFA; with free NH2. The 1H and 13C NMR spectral data, which are radically different from each other (Figure 5), are given in the experimental part.
Figure 5.
Fragments of 1H NMR spectra of 20х with free NH2 (blue, upper) and 20х *2 TFA (maroon, bottom) (400 MHz, 293 K, DMSO-d6).
Figure 5.
Fragments of 1H NMR spectra of 20х with free NH2 (blue, upper) and 20х *2 TFA (maroon, bottom) (400 MHz, 293 K, DMSO-d6).
Scheme 4.
Synthesis of GRPr-ligand (optionally, a "branching fragment" = Lys(L) can be attached see Route 2,3). Reagents and conditions: For compound 20 = (a) (1) FmocSta(S,S), PyBOP, HOBt, DIPEA; (2) 4-methylpiperidine/DMF; (b) (1) FmocHis(L)(Trt), PyBOP, HOBt, DIPEA; (2) 4-methylpiperidine/DMF; (c) (1) FmocGly, PyBOP, HOBt, DIPEA; (2) 4-methylpiperidine/DMF; (d) (1) FmocVal(L), PyBOP, HOBt, DIPEA; (2) 4-methylpiperidine/DMF; (e) (1) FmocAla(L), PyBOP, HOBt, DIPEA; (2) 4-methylpiperidine/DMF; (f) (1) FmocTrp(L)(Boc), PyBOP, HOBt, DIPEA; (2) 4-methylpiperidine/DMF; (g) (1) FmocGln(L)(Trt), PyBOP, HOBt, DIPEA; (2) 4-methylpiperidine/DMF; (h) (1) FmocPhe(D), PyBOP, HOBt, DIPEA; (2) 4-methylpiperidine/DMF;.
Scheme 4.
Synthesis of GRPr-ligand (optionally, a "branching fragment" = Lys(L) can be attached see Route 2,3). Reagents and conditions: For compound 20 = (a) (1) FmocSta(S,S), PyBOP, HOBt, DIPEA; (2) 4-methylpiperidine/DMF; (b) (1) FmocHis(L)(Trt), PyBOP, HOBt, DIPEA; (2) 4-methylpiperidine/DMF; (c) (1) FmocGly, PyBOP, HOBt, DIPEA; (2) 4-methylpiperidine/DMF; (d) (1) FmocVal(L), PyBOP, HOBt, DIPEA; (2) 4-methylpiperidine/DMF; (e) (1) FmocAla(L), PyBOP, HOBt, DIPEA; (2) 4-methylpiperidine/DMF; (f) (1) FmocTrp(L)(Boc), PyBOP, HOBt, DIPEA; (2) 4-methylpiperidine/DMF; (g) (1) FmocGln(L)(Trt), PyBOP, HOBt, DIPEA; (2) 4-methylpiperidine/DMF; (h) (1) FmocPhe(D), PyBOP, HOBt, DIPEA; (2) 4-methylpiperidine/DMF;.
3.6. Description of the Routes
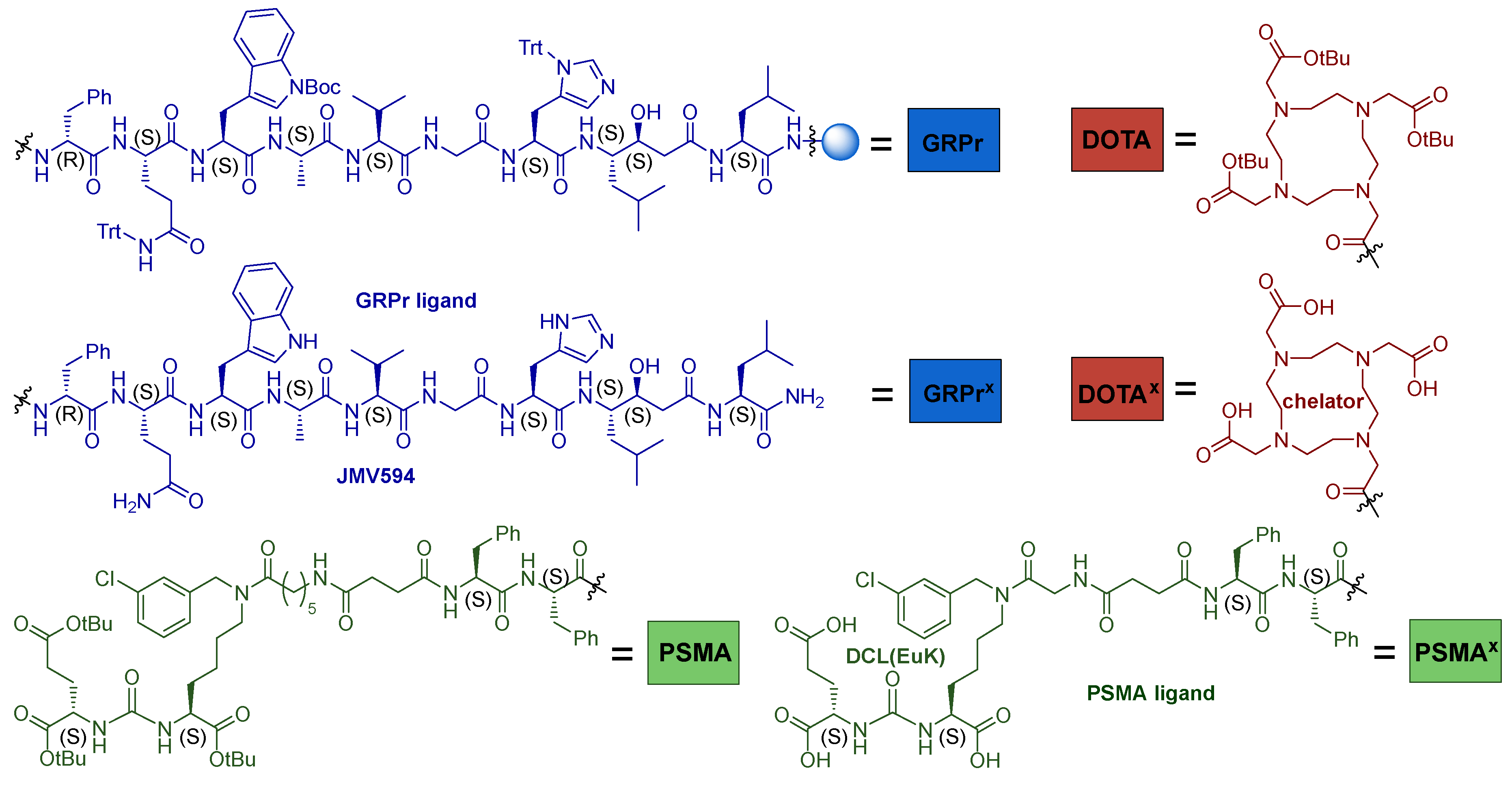
3.6.1. Route 1
Route 1 included the following transformations: 1) Alloc-protecting group cleavage; 2) coupling of НО-DOTA(tBu)3 to PSMA-ligand 10 on 2-CTC resin; 3) cleavage of the peptide sequence with DOTA(tBu)3 11 from 2-CTC resin (saving all protection groups = 6*COOtBu); 4) conjugation of the compound 12 to GRPr-ligand 20 on Rink Amide MBHA resin; 5) cleavage of the HBV conjugate from Rink Amide MBHA resin (removal of all protecting groups: 6*COOtBu, 2*Trt, 1*Boc) (Scheme 5).
Figure 6.
Abbreviations used for PSMA and GRPr targeted ligands and the DOTA chelator.
Scheme 5.
Route 1 of obtaining PSMA-GRPr HBV conjugate. The designations used in the schemes hereafter: [ ] - the stage at which synthetic problems arose and could not be solved, synthesis was stopped; ( ) - stages that were not completed due to difficulties encountered or the inexpediency of obtaining the final compound.
Scheme 5.
Route 1 of obtaining PSMA-GRPr HBV conjugate. The designations used in the schemes hereafter: [ ] - the stage at which synthetic problems arose and could not be solved, synthesis was stopped; ( ) - stages that were not completed due to difficulties encountered or the inexpediency of obtaining the final compound.
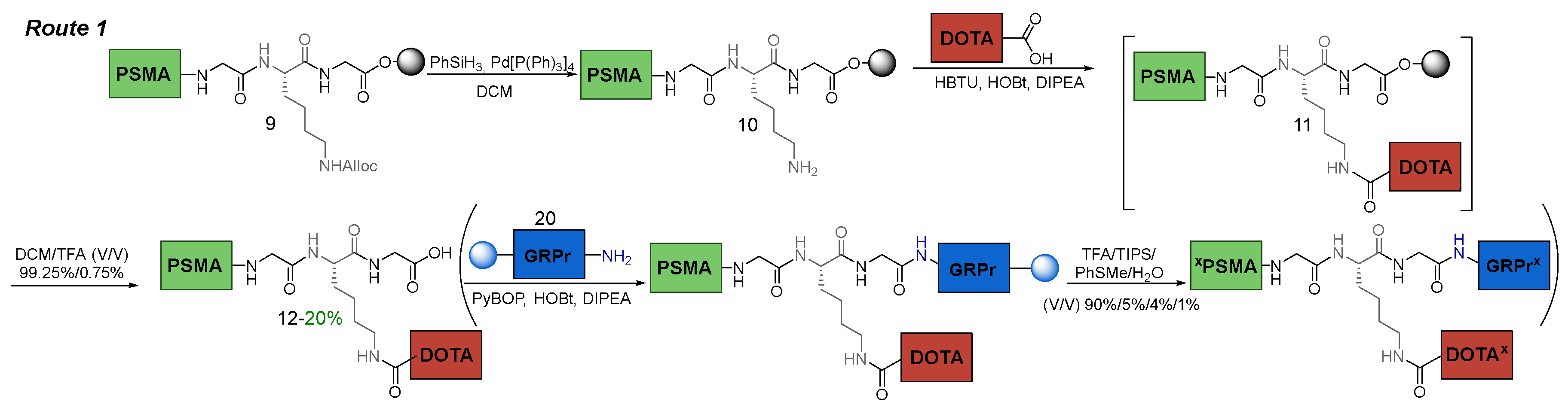
From the pros and cons of the solid-phase approach discussed at the beginning of this chapter, the fact that conjugation of a sterically hindered macrocycle DOTA would occur in a heterogeneous reaction could cause concern. After coupling the chelator to the PSMA-ligand 10 on a polymer substrate and removing it from the resin, compound 12 was obtained. After isolation and purification of this compound, it was found that the reaction yield was 20%. It could be explained by the fact that it was difficult for the bulky DOTA macrocycle to get closer to the amino group of the peptide sequence fixed on a polymer support (Figure 7). All this suggests that this method is unsatisfactory in terms of parameters related to the stage of introduction of НО-DOTA(tBu)3.
3.6.2. Route 2
The main idea of the Route 2 consisted of: 1) coupling of НО-DOTA(tBu)3 to the GRPr-ligand 21 on Rink Amide MBHA resin; 2) Alloc-protecting group cleavage; 3) conjugation of PSMA-ligand 15; 4) cleavage of the HBV conjugate from Rink Amide MBHA resin (removal of all protecting groups: 6*COOtBu, 2*Trt, 1*Boc) (Scheme 6).
When performing the Route 2, the DOTA addition stage also had the greatest effect on the target compound synthesis, as in Route 1. However, in Route 2, the conjugation problems associated with steric hindrance of the chelator were minimized. The reaction proceeded more efficiently, since the chelator was supposed to join the amino group, which was located at a great distance from the polymer substrate and was "exposed outside". In Route 1, the chelator had to join the amino group located near the polymer support, which was complicated by steric reasons caused by the PSMA ligand itself.
Figure 7.
The difference between solid-phase Route 1 and Route 2.
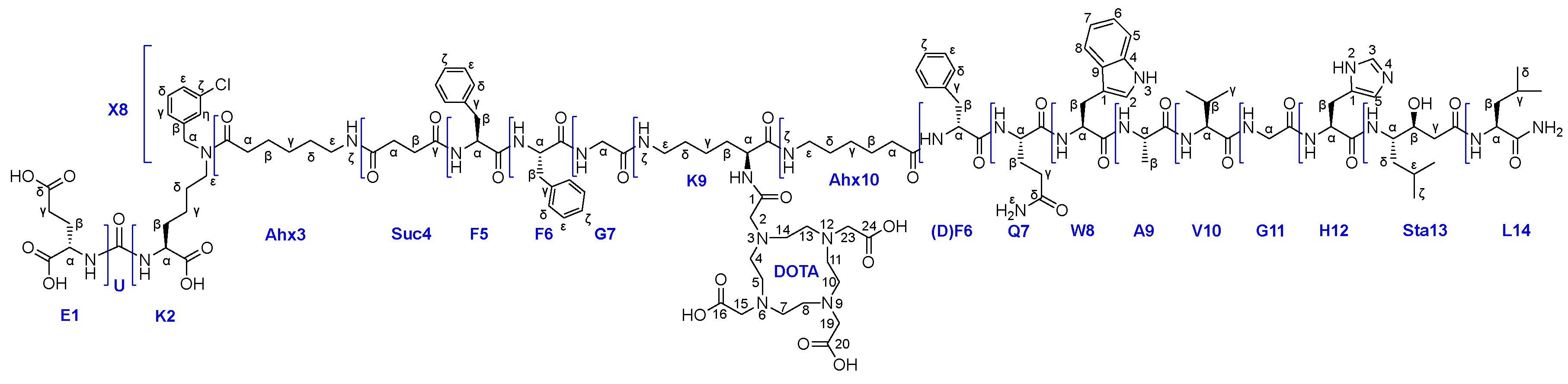
Therefore, after the HO-DOTA(tBu)3 conjugation stage and Alloc-deprotection of the lysine fragment, part of compound 24 was selected to evaluate the yield at this stage. The result was the following: 1) the total capacity at this stage is 55% of the max resin capacity (a resin with a capacity of 0.8-0.3 mmol/g was used); 2) the yield relative to the total capacity is 78% for connection 24х (+HO-DOTA(tBu)3), the remaining 22% is 21х (-HO-DOTA(tBu)3). The result obtained turned out to be better than for Route 1, but also had disadvantages. When the PSMA-ligand 15 was attached, there was competition for the COOH-group of 15 by various NH2-groups: 1) K9NH2ζ-24; 2) K9NH2ζ-21; 3) K9NH2α-21. In the reaction of compounds 15&24 and cleavage from the polymer support, compound 25x (Figure 8) was obtained (49% according to LCMS data) as a mixture with 24x (37% according to LCMS data) and other impurities.
Scheme 6.
Route 2 of obtaining PSMA-GRPr HBV conjugate on Rink Amide MBHA resin.
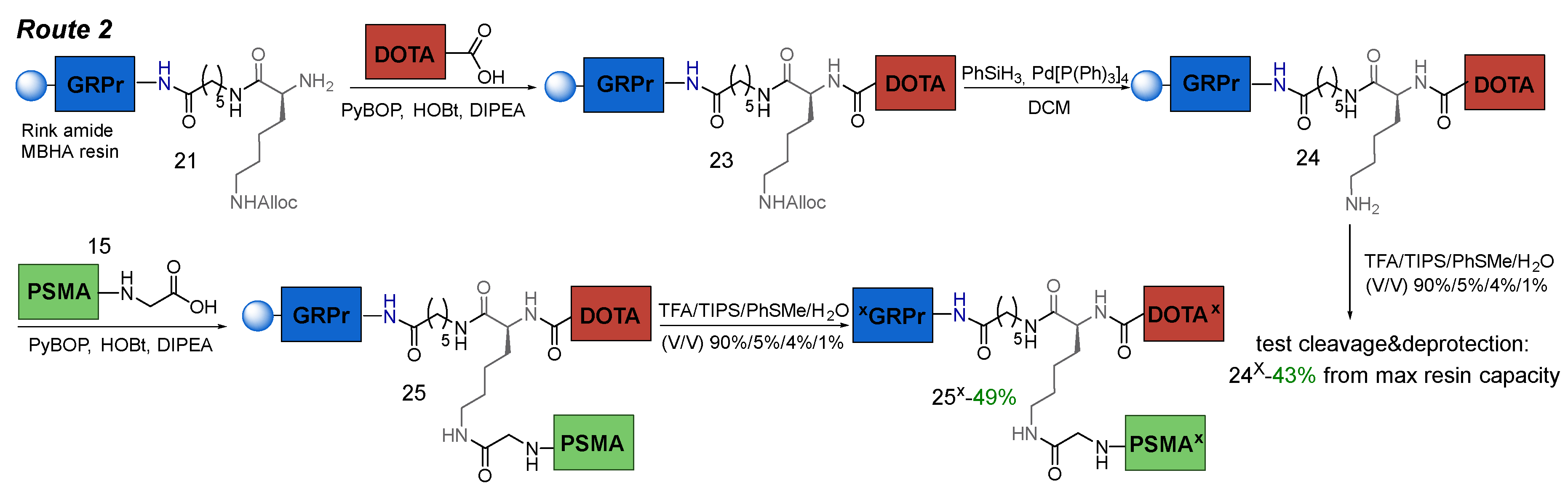
Figure 8.
The structure of compound 25х.
Thus, Route 1 & Route 2, based on the introduction of HO-DOTA(tBu)3 by the solid-phase technique, had a number of significant restrictions that revealed the necessity of usage of the liquid-phase synthesis procedure for DOTA coupling. The transition to this technique was supposed to lead to an increase in the yields and purity of the HBV conjugate.
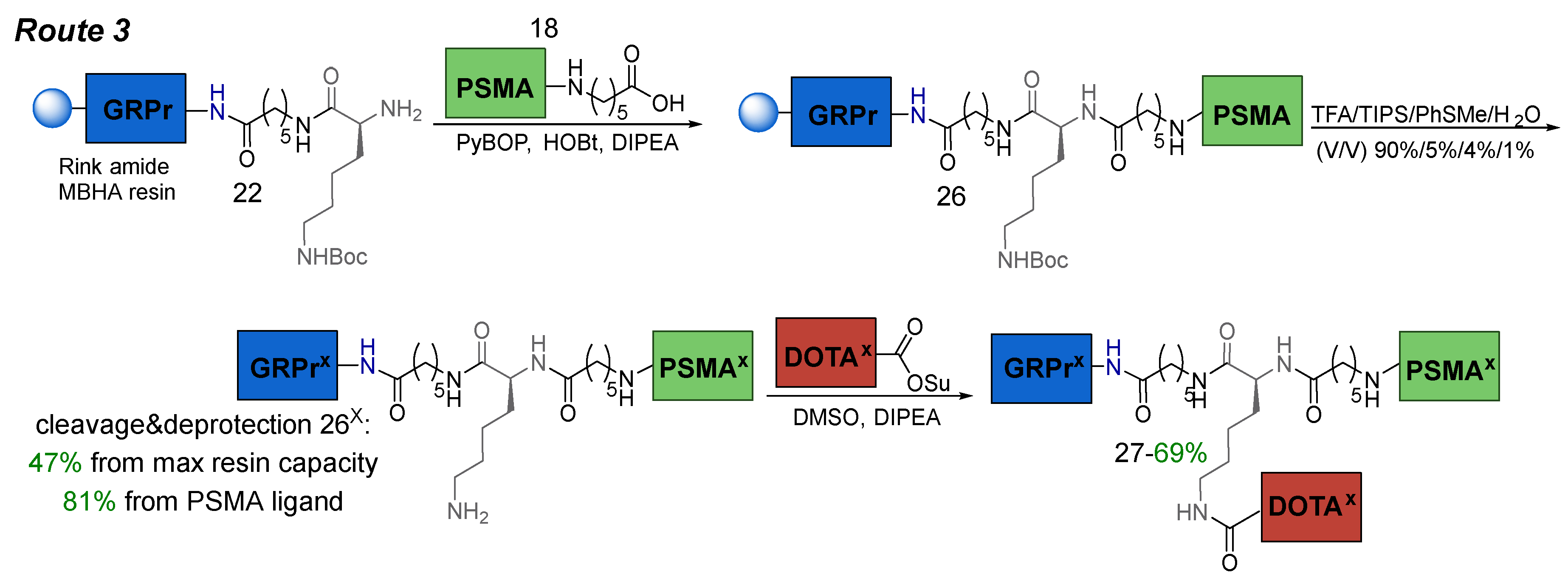
3.6.3. Route 3
Route 3 consisted of the following steps: 1) coupling of PSMA-ligand 18 to GRPr-ligand 22 attached to Rink Amide MBHA resin; 2) cleavage of the HBV-ligand 26 from the polymer substrate (removal of all protecting groups: 3*COOtBu, 2*Trt, 2*Boc); 3) conjugation of (НО)3DOTA-COOSu and HBV-ligand 26х in solution (Scheme 7).
Scheme 7.
Route 3 of obtaining PSMA-GRPr heterobivalent conjugate by liquid-phase synthesis.
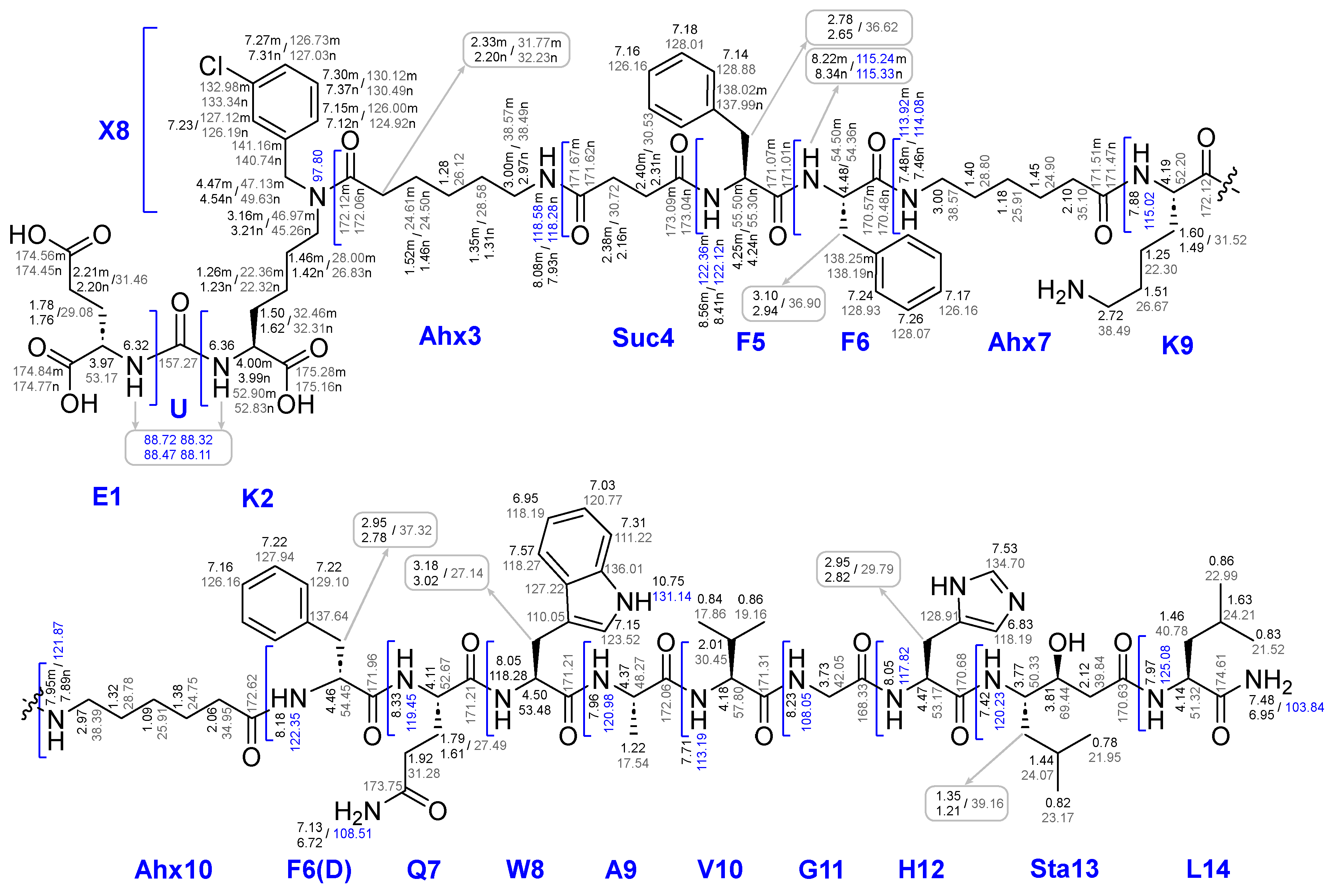
The first step of Route 3 was completed successfully. Further, when removing of compound 26x from the polymer support, the presence of pronounced organogelating behaviour was found. It seriously hampered the purification and reactions with this compound, nevertheless compound 26x was isolated and characterized by a complex of physico-chemical analysis methods such as 1H, 13C, 15N NMR, LCMS, HRMS and a range of 2D NMR experiments. Based on the data obtained, a complete correlation of the structure was given (see Figure 16).
We have previously investigated the peptide organogelation using the example of PSMA targeted ligands [33]. To overcome this obstacle to obtaining compound 27, it was necessary to determine the value of the critical gelation concentration (CGC) in various types of solvents. Without this, aggregation and/or incomplete dissolution of compound 26x may occur in the course of synthesis and lead to a low conversion and yields of the product 27. As a result of the research, it was found that DMSO was the most suitable solvent for the synthesis of HBV conjugate 27 (CGC was about 20 mg/ml at 20 °C). Furthermore, a potential solution to the organogelation issue could be the replacement of spacers, for example, with PEG-derivatives. This substitution could change the nature of intra and intermolecular interactions, which in turn would entail a change in the degree of aggregation and way of stacking of the molecules.
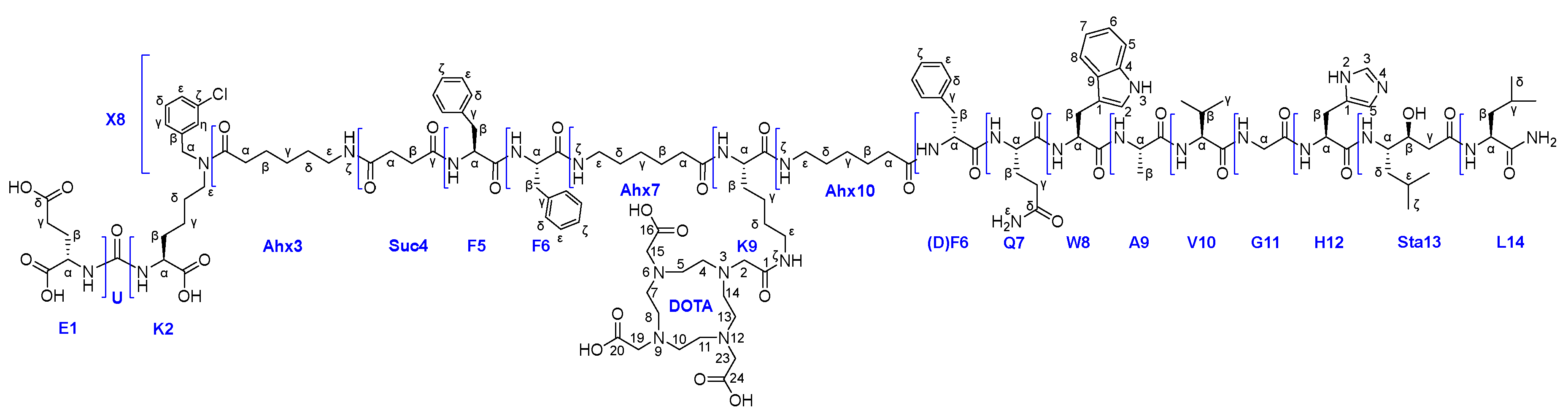
Thus, we were able to carry out the conjugation reaction of ligand 26х and (НО)3DOTA-COOSu in DMSO (concentration 15 mg/ml, 20°C, periodic ultrasound exposure). As a result compound 27 was obtained and characterized by a complex of physico-chemical analysis methods such as 1H, 13C, 15N NMR, LCMS, HRMS and a range of 2D NMR experiments. Based on the data obtained, a complete correlation of the structure was given (see Figure 9).
Figure 9.
Structure of the compound 27.
3.6.4. Route 4a
Route 4a consisted of the following steps: 1) coupling of TFA*NH2(CH2)3NHFmoc (NH2(CH2)3NH2 = 1,3-DAP) fragment to PSMA-ligand 13 in solution (this was necessary for the presence in the structure of two orthogonal NH2-protecting groups = NHFmoc&NHAlloc, which in turn were orthogonal COOtBu); 2) Alloc-P.G. cleavage; 3) conjugation of НО-DOTA(tBu)3 to PSMA-ligand 29 in solution; 4) Fmoc-P.G. cleavage; 5) succinic anhydride addition 6) conjugation to GRPr-ligand 20 on Rink Amide MBHA resin; 7) cleavage of the HBV conjugate from the polymer substrate (removal of all protecting groups: 6*COOtBu, 2*Trt, 1*Boc) (Scheme 8).
Scheme 8.
Route 4а of obtaining PSMA-GRPr HBV conjugate.
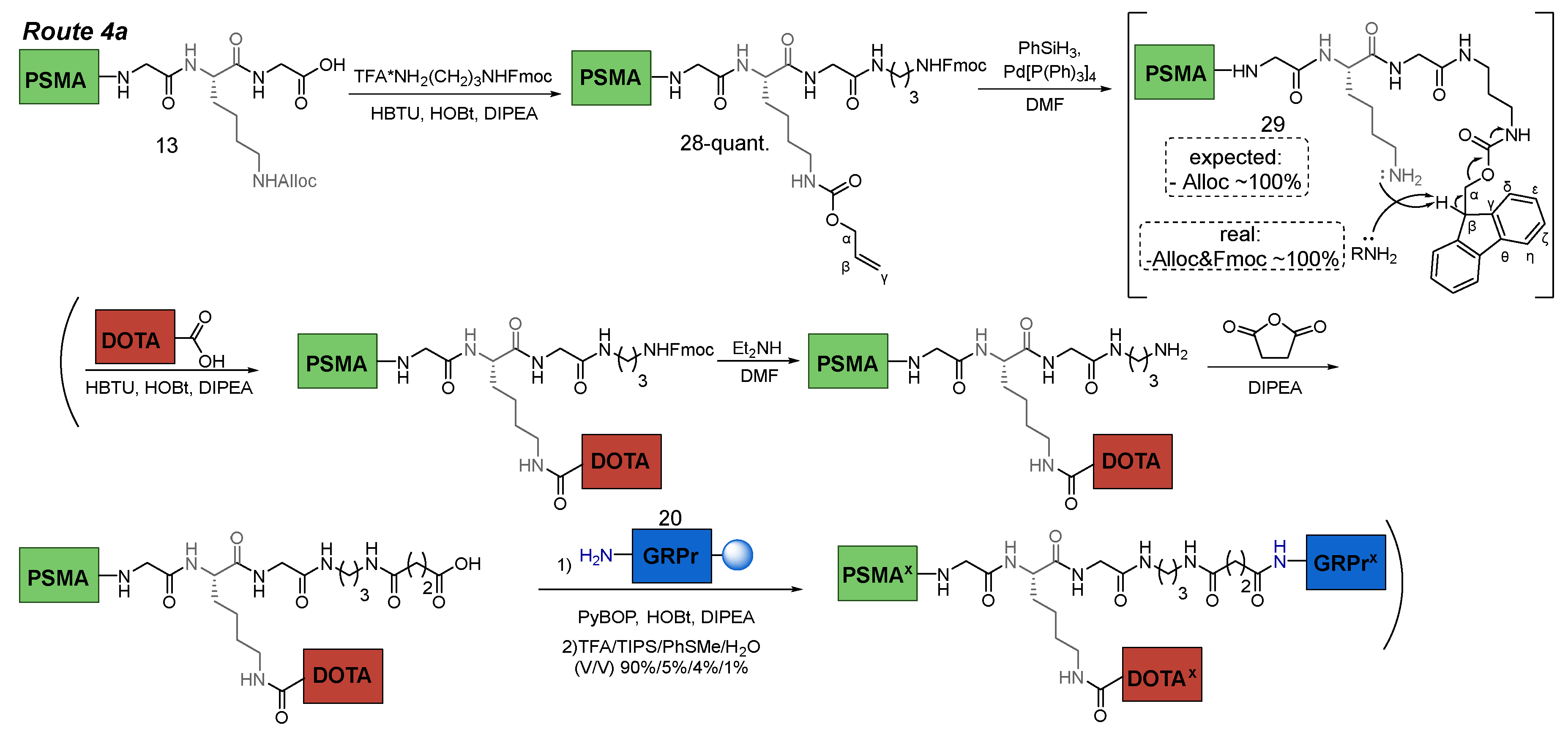
At the beginning, compound 28 was obtained. Difficulties arose during the synthesis of compound 29. Despite the fact that many literature sources describe Fmoc and Alloc protective groups as orthogonal (Table 1) [31], we have encountered the opposite phenomenon. When trying to remove the Alloc-P.G. by standard protocol ([Pd(P(Ph)3]4/PhSiH3), both Alloc&Fmoc protective groups were removed, which can be concluded from the data of the 1H NMR spectra of the compound 28 (before the Alloc cleavage) and compound 29 (after the Alloc cleavage) (Figure 10). The 1H NMR spectrum of compound 28 contained signals: 7.88 (d, J = 7.4 Hz, 2H, Fmocη), 7.67 (d, J = 7.4 Hz, 2H, Fmocδ), 7.40 (t, J = 7.4 Hz, 2H, Fmocζ), 7.32 (t, J = 7.4 Hz, 2H, Fmocε), characteristic for the fluorene fragment of the Fmoc protective group. These signals are absent on the spectrum of compound 29, which indicates the removal of Fmoc protection. The proposed mechanism of this phenomenon could be determined by the "release" of the NH2ζ-group of lysine, which causes β-elimination in the Fmoc fragment. This process can occur both internally and intermolecularly (Scheme 8). At the same time, the accumulation of NH2 α-groups of lysine may occur and contribute to the cleavage of Fmoc protecting groups from molecules not affected by this side reaction.
As a result of removing the Fmoc protection, compound 29 contained NH2ζ&NH2α at Lys K9 fragment. Therefore, Route 4a was stopped at this stage.
Thus, it was shown that Fmoc and Alloc protecting groups are not completely orthogonal if they are located at the same amino acid Lys, since during the removal of Alloc-P.G., degradation of Fmoc-P.G. also occurs (up to complete cleavage).
3.6.5. Route 4b
The main difference between Route 4b and other methods of subgroup 4 was the insertion of TFA*NH2(CH2)5CООFm fragment. This would avoid an extra stage of addition of succinic anhydride to compound 38, which was necessary to combine with the ligand to GRPr 20 on the Rink Amide MBHA resin (see Scheme 10 of Route 4с). To protect the carboxyl fragment of compound 32, -OFm was used, since its cleavage conditions completely coincided with the removal of Fmoc protection [34].
Thus, the implementation of Route 4b required the following steps: 1) synthesis of compound 32 = TFA*NH2(CH2)5CООFm; 2) coupling of compound 32 to PSMA-ligand 13 in solution (this was necessary for the presence in the structure of two orthogonal NH2-protecting groups = NHFmoc&NHAlloc, which in turn were orthogonal COOtBu); 3) Alloc-P.G. cleavage; 4) conjugation of НО-DOTA(tBu)3 to PSMA-ligand 34 in solution; 5) Fm-P.G. cleavage; 6) conjugation to GRPr-ligand 20 on Rink Amide MBHA resin; 7) cleavage of the HBV conjugate from the polymer substrate (removal of all protecting groups: 6*COOtBu, 2*Trt, 1*Boc) (Scheme 9).
Scheme 9.
Route 4b of obtaining PSMA-GRPr HBV conjugate.
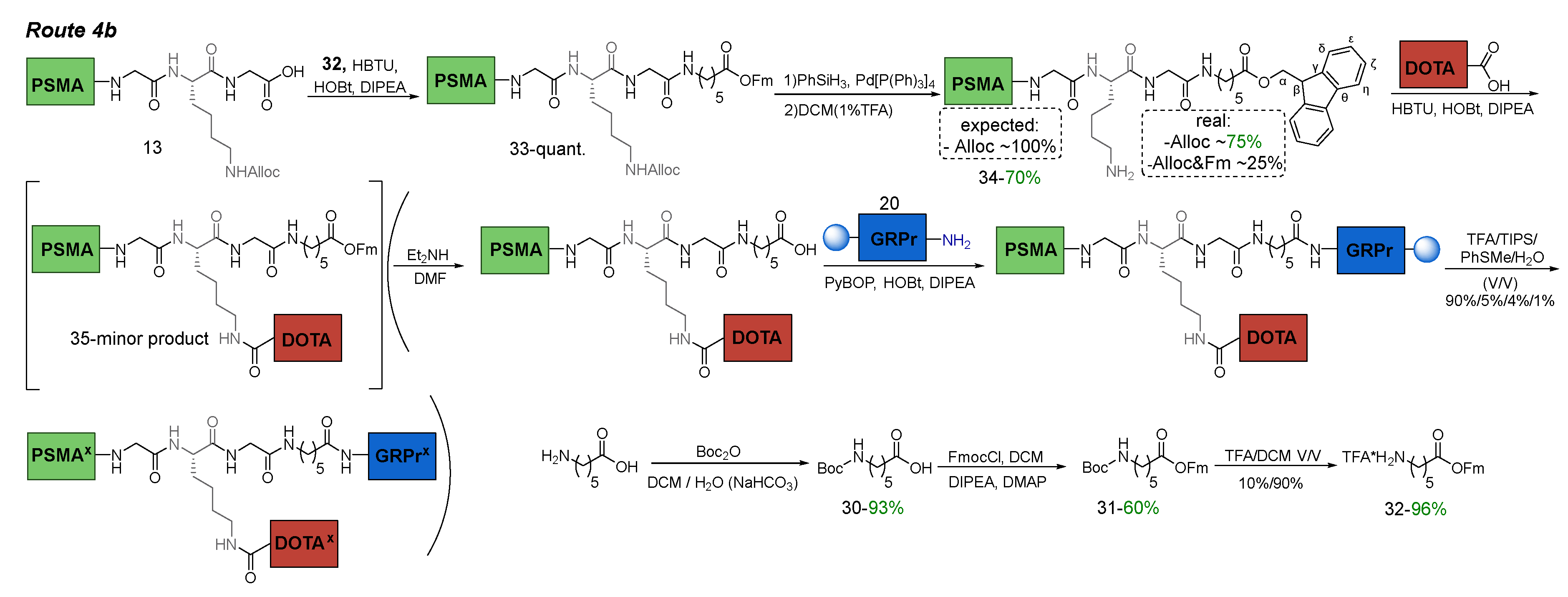
Compound 32 was obtained in a three-stage synthesis. After, 32 was introduced into the amide coupling reaction with PSMA ligand 13 and, as a result, compound 33 was obtained, isolated and characterized by 1H NMR. Then Alloc protecting group was removed. Since Fmoc and COOFm P.G. are similar in nature, we assumed that we could encounter Fm-degradation problems similar to those described in Route 4a.
Therefore, after completion of the reaction, DCM (1%TFA) was added to the reaction mixture to reduce basicity of the Nh2ζ-K9 Lys group. Attention should be paid to the fact that even in the case of partial removal of OFm-P.G. during the reaction, the Ahx10 COOH-group will be "released" and, due to dissociation, will suppress the basicity of the Nh2ζ-group of K9 Lys.
To evaluate the effectiveness of this method, the 1H NMR spectrum of compound 34 was recorded after the reaction, but before chromatographic purification (Figure 11). The 1H NMR spectrum of compound 28 contained signals: 7.89 (d, J = 7.4 Hz, 1.5H, Fmocη), 7.64 (d, J = 7.4 Hz, 1.5H, Fmocδ), 7.41 (t, J = 7.4 Hz, 1.5H, Fmocζ), 7.33 (t, J = 7.4 Hz, 1.5H, Fmocε), characteristic for the fluorene fragment of the Fm-P.G. The integral intensity is calculated relative to G7Hα (3.73 ppm). From the spectral data, it can be concluded that OFm-P.G. has been preserved by ~ 75%. After chromatographic purification, compound 34 was obtained with a yield of 70%. It should be emphasized that after completion of the reaction and chromatographic purification, compound 34 was obtained as a salt *1TFA (TFA*K9NH2ζ to prevent OFm-P.G. destruction).
The next step of Route 4b was to join compound 34 and НО-DOTA(tBu)3 via amide coupling reaction. After the synthesis of compound 35 and its primary purification, according to 1H NMR data, the target compound was contained in the mixture in insignificant quantities. The main product was formed by the addition of a TFA to the K9NH2ζ-group of Lys (an amide bond was formed between them). This could be caused by several reasons: 1) low reactivity of COOH-group of ОН-DOTA(tBu)3; 2) the possibility of partial removal of OFm-P.G. under reaction conditions (using an excess of DIPEA) and thereby generating an additional competing carboxylic component under conditions of peptide synthesis. In some cases, this problem was successfully solved by optimizing the reaction conditions (see synthesis of compounds 28 and 33).
Thus, in Route 4b, it was shown that the fragment of ОН-DOTA(tBu)3 should be introduced to the PSMA-ligand in the absence of any competition from other carboxyl groups, and the nucleophilicity of the NH2-group should be the greatest under the considered conditions.
3.6.6. Route 4с
The difference between Route 4c and Route 4b consisted on several stages. Рositive changes included the stage of НО-DOTA(tBu)3 introduction to the PSMA-ligand (36). The NH2 group of 1,3-DAP fragment (X11NH2δ) was used in free form to obtain compound 37. The negative changes were represented by an extra stage of addition of succinic anhydride.
The difference between Route 4c and Route 4b consisted in a modified order of removal of Alloc and Fmoc P.G., to prevent the removal of the latter.
Route 4c consisted of the following steps: 1) coupling of TFA*NH2(CH2)3NHFmoc (NH2(CH2)3NH2 = 1,3-DAP) fragment to PSMA-ligand 13 in solution (this was necessary for the presence in the structure of two orthogonal NH2-protecting groups = NHFmoc&NHAlloc, which in turn were orthogonal COOtBu); 2) Fmoc-P.G. cleavage; 3) conjugation of НО-DOTA(tBu)3 to PSMA-ligand 29 in solution; 4) Alloc-P.G. cleavage; 5) succinic anhydride addition; 6) conjugation to GRPr-ligand 20 on Rink Amide MBHA resin; 7) cleavage of the HBV conjugate from the polymer substrate (removal of all protecting groups: 6*COOtBu, 2*Trt, 1*Boc) (Scheme 10).
Scheme 10.
Route 4с of obtaining PSMA-GRPr HBV conjugate.
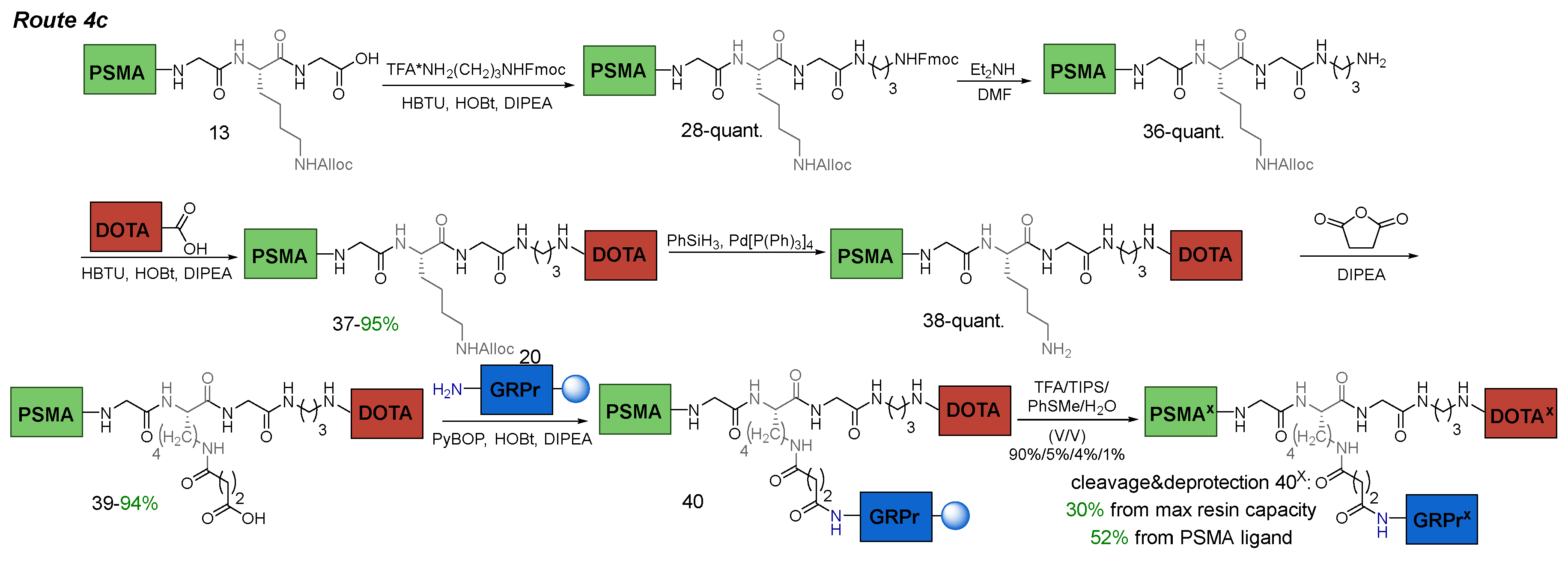
The first stage is similar to the Route 4a, compound 28 was obtained. After that, the removal of Fmoc-P.G. (Scheme 10) was performed using Et2NH in DMF. The choice in favor of Et2NH can be reasoned by: 1) ease of excess Et2NH elimination by evaporation (b.p. = 56 °C); 2) ability to remove DBF aggregates by trituration in Et2O or P.E.; 3) chromatographic purification stage could be avoided. The disadvantages of this method included an increase in the probability of side reaction 1 (Figure 12).
Next, ОН-DOTA(tBu)3 was attached to X11NH2δ-group under the peptide synthesis conditions. As a result, compound 37 was obtained with 95% yield. At the fourth stage, the removal of Alloc-P.G. was carried out under standard conditions [Pd(P(Ph)3]4 (0.1 eq.)/PhSiH3 (6 eq.). The fifth and sixth stages of synthesis did not cause significant difficulties. At the final stage cleavage of the HBV conjugate from Rink Amide MBHA resin was performed (with removal of all protecting groups: 6*COOtBu, 2*Trt, 1*Boc). As a result, a compound 40x was obtained with a yield of 52% relative to the PSMA- ligand 39 and 30% relative to max resin capacity (Figure 13).
3.7. Results and Comparison of the Routes of Obtaining HBV Conjugates
Table 2 summarizes the data obtained by comparing investigated Routes for HBV conjugate synthesis. The effectiveness of the methods was compared based on the total yield of the target compounds calculated relative to: 1) DOTA chelating agent; 2) PSMA-vector 6; 3) GRPr-ligand (JMV594). In addition, the total number of synthetic stages (counting SPPS and LPPS stages) and the number of stages with chromatographic purification were evaluated independently. The stages of synthesis in which the main synthetic difficulties arise are described for each Route. The following conclusions can be drawn from the results presented in Table 2.
Route 1 (Scheme 5). The synthesis has not been completed to obtain the final compound due to the synthetic difficulties encountered at stage 19/41. Main difficulties of synthesis - the chelator had to join the amino group located near the polymer support, which was complicated by steric reasons caused by the PSMA ligand itself.
Route 2 (Scheme 6). The target substance was obtained with yields from the: 1) 25% - DOTA; 2) 31% - 6; 3) 27% - JMV594. Total number of stages - 41, The number of stages with chromatographic isolation – 6. According to these indicators, this is the best method for obtaining HBV conjugates. Main difficulties of synthesis - 24/41 (the DOTA conjugation stage on MBHA Rink Amide resin). Even if: 1) an excess of DOTA (1.5 eq.) was used; 2) the reaction was prolonged, 3) compound 21 with more spatially accessible K9NH2α-group of Lys was used, the conversion increased to 78% only.
Route 3 (Scheme 7). The target substance was obtained with yields from the: 1) 69% - DOTA; 2) 39% - 6; 3) 32% - JMV594. According to these indicators, this is the best method for obtaining HBV conjugates. Total number of stages - 40, The number of stages with chromatographic isolation – 7. According to these parameters, it also belongs to the leaders. The main advantage of Route 3 is versatility. Due to the fact that when obtaining an HBV ligand, it is possible to further obtain HBV conjugates with different functional fragments at the final stage under mild conditions (e.g. SulfoCy5-NHS). The main disadvantage is the formation of organogels at the stage of obtaining & purification & use of HBV ligand. This difficulty can be overcome in several ways: 1) A careful study of organogenic properties, with a focus on the search for aggregation/dissolution boundaries and the compilation of solubility tables, 2) potential solution to the organogelation issue could be the replacement of spacers, for example, with PEG-derivatives. This substitution could change the nature of intra and intermolecular interactions, which in turn would entail a change in the degree of aggregation and way of stacking of the molecules.
Route 4а (Scheme 8). The synthesis has not been completed to obtain the final compound due to the synthetic difficulties encountered at stage 20/44. The main reason for this is the incomplete orthogonality between the Fmoc and Alloc protective groups
Route 4b (Scheme 9). The target substance was not successfully synthesized due to difficulties encountered during stages 20/43 and 21/43 of the Route. The main reason for this is the incomplete orthogonality between the Fm and Alloc protective groups, which has led to the need for using compound 34 in the form of *1TFA (TFA*K9NH2ζ) at the stage of conjugation ОН-DOTA(tBu)3.
Route 4с (Scheme 10). The target substance was obtained with yields from the: 1) 46% - DOTA; 2) 32% - 6; 3) 30% - JMV594. This is better than Route 2, but worse than Route 3. Total number of stages - 44, The number of stages with chromatographic isolation – 7. One of the most laborious Routes presented in the table. Quantity main difficulties of synthesis – 3 (21/44, 22/44, 44/44). The most difficult stage was the removal from the resin and the isolation of HBV conjugate - 44/44.
Figure 15.
18x compound structure with assignments for 1Н NMR, 13C NMR.
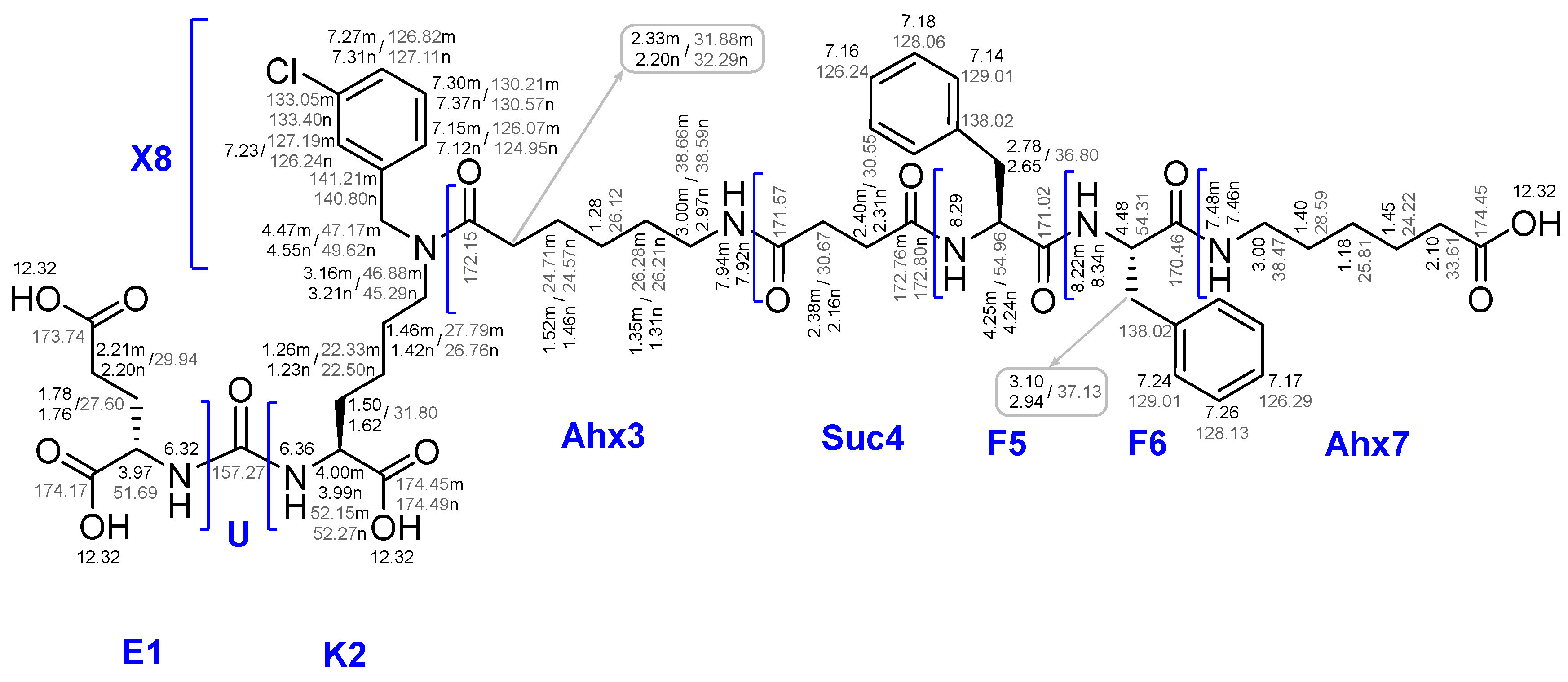

Table 2.
Comparison of Routes for obtaining HBV conjugates.
| № Route | Total Num. of stages | Num. of chromatografic isolation | SPPS, % (number) | LPPS, % (number) | DOTA conjugation stage. (Quantity of DOTA in eq.) | Yield from the: 1) DOTA 2) 6 3) JMV594 |
Main difficulties of synthesis | Problem stage | Was the final substance obtained? |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 41 | 6 | 80% (33) | 20% (8) | 19 (1 eq.) | Substance was not obtained. | Low yield (20%) at the DOTA conjugation stage on CTC-2 resin. 19 | 19 / 41 | No (synthesis was stopped at stage 19) |
| 2 | 41 | 6 | 80% (33) | 20% (6) | 24 (1.5 eq.) | 1) 25% 2) 31% 3) 27% |
Not complete conversion (78%) at the DOTA conjugation stage on MBHA Rink Amide resin. 24 Removal from the resin and isolation of HBV conjugate. 41 | 24 / 4141 / 41 | Yes |
| 3 | 40 | 7 | 77% (31) | 23% (9) | 40 (1 eq.) | 1) 69% 2) 39% 3) 32% |
Removal from the resin and isolation of HBV ligand. 39 Carrying out the DOTA coupling reaction. 40 Chromatographic isolation of HBV conjugate. 40 |
39 / 4040 / 40 | Yes |
| 4a | 44 | 8 | 70% (31) | 30% (13) | 21 (1.55 eq.; can be reduced to ~1.05 eq.) | Substance was not obtained. | Not complete orthogonality of Fmoc and Alloc PG. 20 | 20 / 44 | No (synthesis was stopped at stage 20) |
| 4b | 43 | 8 | 72% (31) | 28% (12) | 21 (1.55 eq.; can be reduced to ~1.05 eq.) | Substance was not obtained. | Not complete orthogonality of OFm and Alloc PG. 20 Carrying out the DOTA coupling reaction. 21 |
20 / 4321 / 43 | No (synthesis was stopped at stage 21) |
| 4c | 44 | 7 | 70% (31) | 30% (13) | 21 (1 eq.) | 1) 46% 2) 32% 3) 30% |
Chromatographic isolation at the DOTA coupling stage. 21 Alloc Deprotection. 22 Removal from the resin and isolation of HBV conjugate. 44 |
21 / 4422 / 4444 / 44 | Yes |
Figure 16.
26x compound structure with assignments for 1Н NMR, 13C NMR, 15N NMR.
4. Material and methods
All solvents used were purified according to the described procedures [35]. All reagents were obtained from commercial suppliers (Sigma-Aldrich, Fluka®Analytical, abcr, Carbosynth, Lumiprobe) and used as received. The initial stages of the synthesis of vector fragments 1–6 (Scheme 1) were carried out using methods previously developed by our scientific group [23]. 1H NMR spectra were measured at Bruker Avance spectrometer operating at 400 MHz using DMSO-d6 as a solvent. Chemical shifts are reported in δ units to 0.01 ppm precision with coupling constants reported to 0.1 Hz precision using residual solvent as an internal reference. 13C NMR spectra were measured at Bruker Avance spectrometer operating at 100.6 MHz using DMSO-d6 as solvents. Chemical shifts are reported in δ units to 0.1 ppm precision using residual solvent as an internal reference. 2D NMR was measured using an Agilent 400 spectrometer operating at 400 MHz for 1H and 100.6 MHz for 13C using DMSO-d6 as the solvent. As 2D NMR methods were used: ROESY, 13C-1H HSQC, 13C-1H HMBC and 15N-1H HSQC. NMR spectra were processed and analyzed using Mnova software (Mestrelab Research, Spain).
For HPLC analysis system with Shimadzu Prominence LC-20 column and a convection fraction collector connected with a single quadrupole mass spectrometer Shimadzu LCMS-2020 with dual ionization source DUIS-ESI-APCI were used. The analytical and preparative column was Phenomenex Luna 3 µm C18 90A (150 x 4.6 mm) with column thermostat at 40 °C and fraction collector. High-resolution mass spectra (HRMS) were recorded on a TripleTOF 5600+ quadrupole time-of-flight mass spectrometer (AB Sciex, Canada) equipped with a TurboIon Spray electrospray ionization source and an LC-30 “Nexera” liquid chromatograph (Shimadzu, Japan). Solutions of samples in acetonitrile with 1% formic acid were introduced into the ionization source by electrospray.
Preparative chromatographic separation of the reaction mixtures was carried out using the INTERCHIM puriFlash 4250 chromatograph. Evaporation of the solvent was carried out using a rotary evaporator, under reduced pressure at a bath temperature of 20-50°C; Flash column chromatography was performed using Merck silica gel 60 (230-400 ASTM mesh).
Synthesis of PSMA ligands
synthesis of peptide sequences on CTC-2 resin
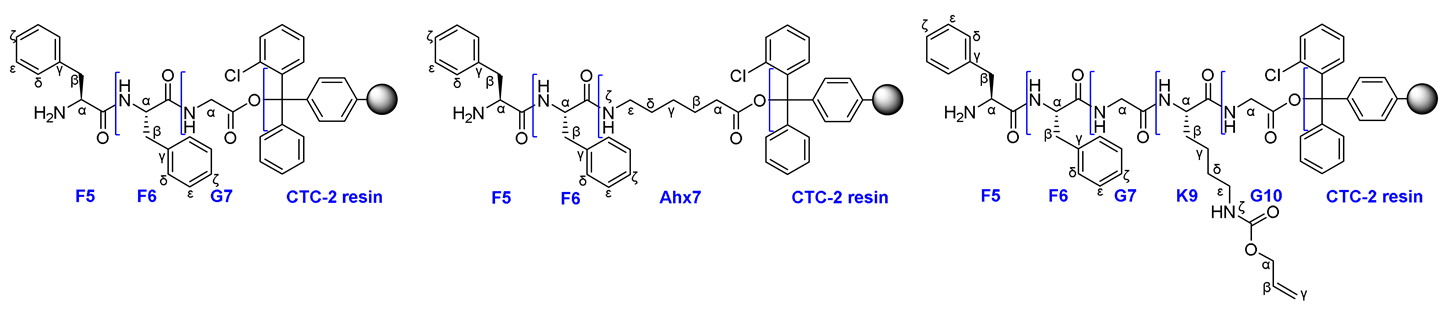 |
General procedure for obtaining peptide sequences by SPPS on CTC-2 resin.
Activation of 2-CTC. The mixture of 2-CTC (1 eq.; 1 g; 1.0–1.6 mmol/g; 100–200 mesh) in DCM (10 ml/1 g) was stirred for 10 min, purged with Ar and SOCl2 (3 eq.) was added dropwise. The resulting mixture was charged with DMF (5%V/V to SOCl2) and then stirred at 40 °C for 4 h. After that, the resin was filtered and transferred to a polypropylene reactor, washed with DMF (3 × 10 ml, 1 min) and DCM (3 × 10 ml, 1 min). The amount of solvent for the reaction and washing is 10 ml/1 g of resin.
Addition of the first amino acid residue. To the CTC-2 resin (1 eq.; 1 g; 1.2–1.6 mmol/g; 100–200 mesh) in DMF (10 ml/1 g), Fmoc-protected amino acid (2 eq. relative to the upper capacity of CTC-2 resin) and DIPEA (10 eq.) were added, and the mixture was stirred for 4 h. Then, the resin was filtered and washed with MeOH*3 (10 ml/1 g, 5 min), DCM*3 (10 ml/1 g, 1 min), DMF*3 (10 ml/1 g, 1 min), and DCM*3 (10 ml/1 g, 1 min).
Deprotection of Fmoc. Peptide sequence on a 2-CTC resin (1 eq.) was washed with DMF*2 (10 ml/1 g, 1 min), then 4-methylpiperidine in DMF (20%/80% V/V, 15 ml) was added and stirred for 15 min, then the resin was filtered and washed with DMF*3 (10 ml/1 g, 1 min), then 4-methylpiperidine in DMF (20%/80% V/V, 10 ml/1 g) was added and stirred for 15 min. After the resin was filtered and washed with DMF*3 (10 ml/1 g, 1 min) and DCM*3 (10 ml/1 g, 1 min).
Deprotection of Alloc. Peptide sequence on a Rink Amide MBHA resin (1eq.) was washed with DCM*1 (10 ml/1 g, 1 min) for resin swelling, then DCM (10 ml/1 g) and PhSiH3 (8 eq.) were added and the mixture was stirred under inert atmosphere of Ar for few min; then Pd[P(Ph)3]4 (0.1 eq.) was added and the mixture was stirred under argon atmosphere for 60 min. Then the resin was filtered and washed DCM*3 (10 ml/1 g, 2 min) DMF*3 (10 ml/1 g, 2 min) and DCM*3 (10 ml/1 g, 2 min).
Addition of the second and subsequent amino acid residues. To the mixture of CTC-2 (1 eq.) in DMF (10 ml/1 g) Fmoc-protected amino acid (2 eq), HOBt (0.5 eq.), HBTU (2 eq.) and DIPEA (3 eq.) were added and the mixture was stirred for 4 h. Then the resin was filtered and washed DMF*3 (10 ml/1 g, 1 min) and DCM*3 (10 ml/1 g, 1 min).
Synthesis of pentapeptide NH2-FFGK(Alloc)G on 2-CTC resin - 8.
Using 2-CTC resin (1000 mg, 1.0-1.6 mmol), FmocGly-OH (951 mg; 3.2 mmol), DIPEA (2.78 ml; 16 mmol) to add the first amino acid, FmocLys(L)(Alloc)-OH (1448 mg; 3.2 mmol), HBTU (1214 mg; 3.2 mmol), HOBt (108 mg; 0.8 mmol) and DIPEA (836 μl; 4.8 mmol) to add the second amino acid, FmocGly-OH (951 mg; 3.2 mmol), HBTU (1214 mg; 3.2 mmol), HOBt (108 mg; 0.8 mmol) and DIPEA (836 μl; 4.8 mmol) to add the third amino acid, FmocPhe(L)-OH (1240 mg; 3.2 mmol), HBTU (1214 mg; 3.2 mmol), HOBt (108 mg; 0.8 mmol) and DIPEA (836 μl; 4.8 mmol) to add the fourth amino acid, FmocPhe(L)-OH (1240 mg; 3.2 mmol), HBTU (1214 mg; 3.2 mmol), HOBt (108 mg; 0.8 mmol) and DIPEA (836 μl; 4.8 mmol) to add the fifth amino acid, the pentapeptide NH2-FFGK(Alloc)G on 2-CTC resin was obtained.
Synthesis of tripeptide NH2-FFG on 2-CTC resin - 14.
Using 2-CTC resin (500, 0.5-0.8 mmol), FmocGly-OH (476 mg; 1.6 mmol), DIPEA (1.394 ml; 8 mmol) to add the first amino acid, FmocPhe(L)-OH (620 mg; 1.6 mmol), HBTU (607 mg; 1.6 mmol), HOBt (54 mg; 0.4 mmol) and DIPEA (418 μl; 2.4 mmol) to add the second amino acid, FmocPhe(L)-OH (620 mg; 1.6 mmol), HBTU (607 mg; 1.6 mmol), HOBt (54 mg; 0.4 mmol) and DIPEA (418 μl; 2.4 mmol) to add the third amino acid, the tripeptide NH2-FFG on 2-CTC resin was obtained.
Synthesis of tripeptide NH2-FFAhx on 2-CTC resin - 17.
Using 2-CTC resin (500 mg, 0.5-0.8 mmol), FmocAhx-OH (565 mg; 1.6 mmol), DIPEA (1.394 ml; 8 mmol) to add the first amino acid, FmocPhe(L)-OH (620 mg; 1.6 mmol), HBTU (607 mg; 1.6 mmol), HOBt (54 mg; 0.4 mmol) and DIPEA (418 μl; 2.4 mmol) to add the second amino acid, FmocPhe(L)-OH (620 mg; 1.6 mmol), HBTU (607 mg; 1.6 mmol), HOBt (54 mg; 0.4 mmol) and DIPEA (418 μl; 2.4 mmol) to add the third amino acid, the tripeptide NH2-FFKAhx on 2-CTC resin was obtained.
Compound 13
To a mixture of pentapeptide NH2-Phe(L)Phe(L)GlyLys(Alloc)(L)Gly on 2-CTC resin 8 (1 eq.; 0.47 mmol) in DMF (10 ml) in a reactor compound 6 (1.1 eq.; 426 mg; 0.517 mmol), HOBt (0.5 eq.; 32 mg; 0.235 mmol), HBTU (2 eq.; 357 mg; 0.94 mmol), DIPEA (3 eq.; 246 μl; 1.41 mmol) were added. The mixture was stirred for 12 h. Then the solvent was removed by filtration on a porous reactor filter and the resin was washed three times with DMF (10 ml), three times with DCM (10 ml), then dried from traces of solvents. After that, DCM/TFA system (99.25%-0.75%, 12 ml) was added to the resin and left under stirring for 15 min, then the resin was filtered off and washed with DCM. The solvent was removed under reduced pressure and the residue was re-evaporated twice with DCM. Product was purified by column chromatography (Puriflash PF-15C18HP-F0020 + PF-15C18HP-F0040 (15 μ 32 g + 15 μ 60 g), eluent: H2O(80%)/MeCN(20%) => H2O(0%)/MeCN(100%) for 40 min, after MeCN(100%) for 5 min). Compound 13 was obtained as a white amorphous solid (513 mg, 75% yield).
1H NMR (400 MHz, DMSO-d6, δ): 12.53 (br.s, 1H, G10COOH), 8.25 (t, J = 6 Hz, 1H, G10NH), 8.20-8.14 (m, 1Н, F5NHmn), 8.12 (d, J = 8 Hz, 1Н, F6NH), 8.06 (t, J = 5.9 Hz, 1H, G7NH), 7.91 (d, J = 8.0 Hz, 1H, K9NH), 7.82 (t, J = 5.4 Hz, m) & 7.79 (t, J = 5.4 Hz, n) (1H, Ahx3NHζ, m + n), 7.42-7.09 (m, 15H, X8δn + X8εn + X8δm + X8εm + F6ε + F6δ + X8ηmn + F5ε + F6ζ + F5ζ + K9NHζ + F5δ + X8γmn), 6.35-6.19 (m, 2Н, K2NHm + K2NHn + E1NHm + E1NHn), 5.95-5.82 (m, 1Н, Allocβ), 5.30-5.20 (m, 1Н, Allocγ(a)), 5.18-5.10 (m, 1Н, Allocγ(b)), 4.60-4.42 (m, 5H, X8αnm + F6α + Allocα), 4.42-4.34 (m, 1H, F5Hα), 4.32-4.20 (m, 1H, K9α), 4.08-3.90 (m, 2H, E1α + K2αm + K2αn), 3.81-3.65 (m, 4Н, G7α + G9α), 3.21 (t, J = 7.3 Hz, n) & 3.17 (t, J = 7.3 Hz, m) (2H, K2ε, m/n = 3/2), 3.12-3.04 (m, 1Н, F6β(a)), 3.03-2.90 (m, 5Н, F6β(b) + Ahx3ε + K9ε), 2.90-2.82 (m, 1Н, F5β(a)), 2.71-2.60 (m, 1Н, F5β(b)), 2.40-2.10 (m, 8H, Ahx3αm + Suc4βmn + E1γ + Suc4αmn + Ahx3αn), 1.92-1.80 (m, 1Н, E1β(a)), 1.72-1.10 (m, 21Н, E1β(b) + K9β(a) + K2β(a) + Ahx3β + K9β(b) + K2β(b) + K9δ + Ahx3δ + K2δ + K2γ + K9γ + Ahx3γ, m + n), 1.40-1.35 (m, 27H, tBu).
LCMS 100% in positive ion mode, 100% in negative ion mode
ESI-MS C73H10537ClN10O18: m/z calc. for [M+2H+]2+: 724.37, found: 724.05
HRMS (m/z, ESI): calc. for C73H10535ClN10O18 - [M+2Na]2+ 745.3541, found: 745.3531.
Compound 15
To a mixture of tripeptide NH2-Phe(L)Phe(L)Gly on 2-CTC resin 14 (1 eq.; 0.21 mmol) in DMF (5 ml) in a reactor compound 6 (1.1 eq.; 190 mg; 0.231 mmol), HOBt (0.5 eq.; 28 mg; 0.1 mmol), HBTU (2 eq.; 159 mg; 0.42 mmol), DIPEA (3 eq.; 110 μl; 0.63 mmol) were added. The mixture was stirred for 12 h. The solvent was then removed by filtration on a porous reactor filter and the resin was washed three times with DMF (5 ml), three times with DCM (5 ml), then dried from traces of solvents. A DCM/TFA system (99.25%-0.75%, 10 ml) was added to the resin and left under stirring for 15 min, after the solution was filtered from the resin. The solvent was removed under reduced pressure and the residue was re-evaporated with DCM. The crude product was purified by column chromatography (Puriflash PF-15C18HP-F0040 (15 μ 60 g), eluent: H2O(80%)/MeCN(20%) => H2O(0%)/MeCN(100%) for 30 min, after MeCN(100%) for 5 min). Compound 15 was obtained as a white amorphous solid (173 mg, 70% yield).
1H NMR (400 MHz, DMSO-d6, δ): 12.54 (s, 1H, COOH), 8.20-8.07 (m, 3Н, F5NHmn + F6NHmn + G7NH), 7.84 (t, J = 5.4 Hz, m) & 7.81 (t, J = 5.4 Hz, n) (1H, Ahx3NHζ, m + n), 7.42-7.08 (m, 14H, X8Hδn + X8Hεn + X8Hδm + X8Hεm + F6Hε + F6Hδ + X8Hηmn + F5Hε + F6Hζ + F5Hζ + F5Hδ + X8Hγmn), 6.34-6.21 (m, 2Н, K2NHm + K2NHn + E1NHm + E1NHn), 4.59-4.45 (3H, X8Hαn + F6Hα + X8Hαm), 4.41-4.32 (m, 1H, F5Hα), 4.07-3.90 (m, 2H, E1Hα + K2Hαm + K2Hαn), 3.75 (d, J = 5.7 Hz, 2H, G7α), 3.26-3.13 (m, 2H, K2Hεmn), 3.12-3.05 (m, 1H, F6Hβ(a)), 3.04-2.90 (m, 3H, Ahx3Hε(mn) + F6Hβ(b)), 2.90-2.81 (m, 1Н, F5Hβ(a)), 2.69-2.59 (m, 1Н, F5Hβ(b)), 2.40-2.10 (m, 8H, Ahx3Hαm + Suc4Hβmn + E1Hγ + Suc4Hαmn + Ahx3Hαn), 1.92-1.80 (m, 1Н, E1Hβ(a)), 1.72-1.62 (m, 1Н, E1Hβ(b)), 1.62-1.10 (m, 12Н, K2Hβ(a) + Ahx3Hβ + K2Hβ(b) + Ahx3Hδ + K2Hδ + K2Hγ + Ahx3Hγ, m + n), 1.40-1.35 (m, 27H, tBu).
HRMS (m/z, ESI): calc. for C61H86ClN7O14 - [M+2Na+]2+ 610.7853, found: 610.7863.
Compound 18
To a mixture of tripeptide NH2-Phe(L)Phe(L)Ahx on 2-CTC resin 17 (1 eq.; 0.339 mmol) in DMF (5 ml) in a reactor compound 6 (1.1 eq.; 308 mg; 0.373 mmol), HOBt (0.5 eq.; 23 mg; 0.17 mmol), HBTU (2.5 eq.; 322 mg; 0.85 mmol), DIPEA (3.75 eq.; 220 μl; 1.27 mmol) were added. The mixture was stirred for 12 h. The solvent was then removed by filtration on a porous reactor filter and the resin was washed three times with DMF (7 ml), three times with DCM (7 ml), and then dried from traces of solvents. A DCM/TFA system (99.25%-0.75%, 10 ml) was added to the resin and left under stirring for 15 min, after the solution was filtered from the resin. The solvent was removed under reduced pressure and the residue was re-evaporated with DCM. The crude product was purified by column chromatography (Puriflash PF-15C18HP-F0040 (15 μ 60 g), eluent: H2O(80%)/MeCN(20%) => H2O(0%)/MeCN(100%) for 30 min, after MeCN(100%) for 5 min). Compound 18 was obtained as a white amorphous solid (322 mg, 77% yield).
1H NMR (400 MHz, DMSO-d6, δ): 11.99 (s, 1H, COOH), 8.31 (d, J = 7.3 Hz, 1Н, F5NHmn), 8.17 (d, J = 8.4 Hz 1Н, F6NHmn), 7.96 (t, J = 5.4 Hz, m) & 7.93 (t, J = 5.4 Hz, n) (1H, Ahx3NHζ, m + n), 7.55-7.44 (m, 1H, Ahx7NHζ), 7.42-7.08 (m, 14H, X8Hδn + X8Hεn + X8Hδm + X8Hεm + F6Hε + F6Hδ + X8Hηmn + F5Hε + F6Hζ + F5Hζ + F5Hδ + X8Hγmn), 6.35-6.19 (m, 2Н, K2NHm + K2NHn + E1NHm + E1NHn), 4.55 (s, n) & 4.47 (s, m) (2H, X8Hα, m + n), 4.44-4.34 (m, 1H, F6Hα), 4.34-4.24 (m, 1H, F5Hα), 4.08-3.90 (m, 2H, E1Hα + K2Hαm + K2Hαn), 3.26-3.12 (m, 2H, K2Hεmn), 3.11-2.84 (m, 7H, F6Hβ(a) + Ahx7Hε + Ahx3Hε(mn) + F6Hβ(b) + F5Hβ(a)), 2.69-2.59 (m, 1Н, F5Hβ(b)), 2.40-2.10 (m, 10H, Ahx3Hαm + Suc4Hβmn + E1Hγ + Suc4Hαmn + Ahx7Hα + Ahx3Hαn), 1.91-1.80 (m, 1Н, E1Hβ(a)), 1.72-1.62 (m, 1Н, E1Hβ(b)), 1.62-1.10 (m, 18Н, K2Hβ(a) + Ahx3Hβ + Ahx7Hβ + K2Hβ(b) + Ahx3Hδ + Ahx7Hδ + K2Hδ + K2Hγ + Ahx3Hγ + Ahx7Hγ, m + n), 1.40-1.35 (m, 27H, tBu).
13C NMR (101 MHz, DMSO-d6, δ): 174.48 (Ahx7C), 172.89 (Suc4Cγ(n)), 172.83 (Suc4Cγ(m)), 172.24 (K2C(n)), 172.20 (K2C(m)), 172.11 (Ahx3C(nm)), 171.92 (E1C), 171.59 (Suc4C(mn)), 171.44 (E1Cδ), 171.05 (F5C), 170.46 (F6C), 157.13 (U), 141.17 (X8Cβ(m)), 140.76 (X8Cβ(n)), 138.18 (F6Cγ), 138.05 (F5Cγ), 133.42 (X8Cζ(n)), 133.07 (X8Cζ(m)), 130.59 (X8Cδ(n)), 130.24 (X8Cδ(m)), 129.02 (F6Cδ + F5Cδ), 128.14 (F6Cε), 128.07 (F5Cε), 127.20 (X8Cη(m)), 127.15 (X8Cε(n)), 126.86 (X8Cε(m)), 126.30 (F6Cζ), 126.24 (X8Cη(n) + F5Cζ), 126.05 (X8Cγ(m)), 124.95 (X8Cγ(n)), 80.58 (E1tBu), 80.40 (K2tBu(m)), 80.32 (K2tBu(n)), 79.75 (E1δtBu), 55.05 (F5Cα), 54.33 (F6Cα), 52.99 (K2Cα(n)), 52.86 (K2Cα(m)), 52.17 (E1Cα), 49.60 (X8Cα(n)), 47.08 (X8Cα(m)), 46.79 (K2Cε(m)), 45.19 (K2Cε(n)), 38.65 (Ahx3Cε(m)), 38.59 (Ahx3Cε(n)), 38.47 (Ahx7Cε), 37.11 (F6Cβ), 36.78 (F5Cβ), 33.62 (Ahx7Cα), 32.31 (Ahx3Cα(n)), 31.94 (Ahx3Cα(m)), 31.81 (K2Cβ), 30.91 (E1Cγ), 30.64 (Suc4Cα), 30.52 (Suc4Cβ), 29.06 (Ahx3Cδ(m)), 28.96 (Ahx3Cδ(n)), 28.63 (Ahx7Cδ), 27.74 (tBuE1), 27.65 (tBuK2 + K2Cδ(m)), 27.62 (tBuE1δ), 27.53 (E1Cβ), 26.70 (K2Cδ(n)), 26.33 (Ahx3Cγ(m)), 26.24 (Ahx3Cγ(n)), 25.83 (Ahx7Cγ), 24.73 (Ahx3Cβ(m)), 24.59 (Ahx3Cβ(n)), 24.24 (Ahx7Cβ), 22.43 (K2Cγ(n)), 22.25 (K2Cγ(m)).
HRMS (m/z, ESI): calc. for C65H94ClN7O14 - [M+Na+]+ 1254.6439, found: 1254.6429.
Compound 18x
Compound 18 (1 eq.; 45 mg; 36.52 μmol) was dissolved in system of TFA/DCM/TIPS (47.5%/47.5%/5%, V = 3 ml). The mixture was stirred for 3 h. The solvent was removed under reduced pressure. Crude product was precipitated by Et2O, washed twice with Et2O (2 ml) and purified by column chromatography (Puriflash PF-15C18HP-F0012 (15 μ 20 g), eluent: H2O(90%)/MeCN(10%) => H2O(0%)/MeCN(100%) for 30 min, after MeCN(100%) for 5 min). Compound 18x was obtained as a white amorphous solid (31.6 mg, 81% yield).
1H NMR (400 MHz, DMSO-d6, δ): 12.32 (br.s., 4H, COOH), 8.29 (d, J = 7.3 Hz, 1Н, F5NHmn), 8.21-8.11 (m, 1Н, F6NHmn), 7.94 (t, J = 5.4 Hz, m) & 7.92 (t, J = 5.4 Hz, n) (1H, Ahx3NHζ, m + n), 7.55-7.44 (m, 1H, Ahx7NHζ), 7.42-7.08 (m, 14H, X8Hδn + X8Hεn + X8Hδm + X8Hεm + F6Hε + F6Hδ + X8Hηmn + F5Hε + F6Hζ + F5Hζ + F5Hδ + X8Hγmn), 6.40-6.25 (m, 2Н, K2NHm + K2NHn + E1NHm + E1NHn), 4.55 (s, n) & 4.47 (s, m) (2H, X8Hα, m + n), 4.45-4.35 (m, 1H, F6Hα), 4.34-4.24 (m, 1H, F5Hα), 4.15-3.99 (m, 2H, E1Hα + K2Hαm + K2Hαn), 3.26-3.12 (m, 2H, K2Hεmn), 3.11-2.84 (m, 7H, F6Hβ(a) + Ahx7Hε + Ahx3Hε(mn) + F6Hβ(b) + F5Hβ(a)), 2.69-2.59 (m, 1Н, F5Hβ(b)), 2.40-2.10 (m, 10H, Ahx3Hαm + Suc4Hβmn + E1Hγ + Suc4Hαmn + Ahx7Hα + Ahx3Hαn), 1.99-1.85 (m, 1Н, E1Hβ(a)), 1.78-1.67 (m, 1Н, E1Hβ(b)), 1.67-1.10 (m, 18Н, K2Hβ(a) + Ahx3Hβ + Ahx7Hβ + K2Hβ(b) + Ahx3Hδ + Ahx7Hδ + K2Hδ + K2Hγ + Ahx3Hγ + Ahx7Hγ, m + n).
13C NMR (101 MHz, DMSO-d6, δ): 174.49 (K2C(n)), 174.45 (K2C(m) + Ahx7C), 174.17 (E1C), 173.74 (E1Cδ), 172.80 (Suc4Cγ(n)), 172.76 (Suc4Cγ(m)), 172.15 (Ahx3C(nm)), 171.57 (Suc4C(mn)), 171.02 (F5C), 170.46 (F6C), 157.27 (U), 141.21 (X8Cβ(m)), 140.80 (X8Cβ(n)), 138.12 (F6Cγ), 138.02 (F5Cγ), 133.40 (X8Cζ(n)), 133.05 (X8Cζ(m)), 130.57 (X8Cδ(n)), 130.21 (X8Cδ(m)), 129.01 (F6Cδ + F5Cδ), 128.13 (F6Cε), 128.06 (F5Cε), 127.19 (X8Cη(m)), 127.11 (X8Cε(n)), 126.82 (X8Cε(m)), 126.29 (F6Cζ), 126.24 (X8Cη(n) + F5Cζ), 126.07 (X8Cγ(m)), 124.95 (X8Cγ(n)), 54.96 (F5Cα), 54.31 (F6Cα), 52.27 (K2Cα(n)), 52.15 (K2Cα(m)), 51.69 (E1Cα), 49.62 (X8Cα(n)), 47.17 (X8Cα(m)), 46.88 (K2Cε(m)), 45.29 (K2Cε(n)), 38.66 (Ahx3Cε(m)), 38.59 (Ahx3Cε(n)), 38.47 (Ahx7Cε), 37.13 (F6Cβ), 36.80 (F5Cβ), 33.61 (Ahx7Cα), 32.29 (Ahx3Cα(n)), 31.88 (Ahx3Cα(m)), 31.80 (K2Cβ), 30.67 (Suc4Cα), 30.55 (Suc4Cβ), 29.94 (E1Cγ), 29.04 (Ahx3Cδ(m)), 28.93 (Ahx3Cδ(n)), 28.59 (Ahx7Cδ), 27.79 (K2Cδ(m)), 27.60 (E1Cβ), 26.76 (K2Cδ(n)), 26.28 (Ahx3Cγ(m)), 26.21 (Ahx3Cγ(n)), 25.81 (Ahx7Cγ), 24.71 (Ahx3Cβ(m)), 24.57 (Ahx3Cβ(n)), 24.22 (Ahx7Cβ), 22.50 (K2Cγ(n)), 22.33 (K2Cγ(m)).
Synthesis of GRPr ligands
Synthesis of peptide sequences on Rink Amide MBHA resin.
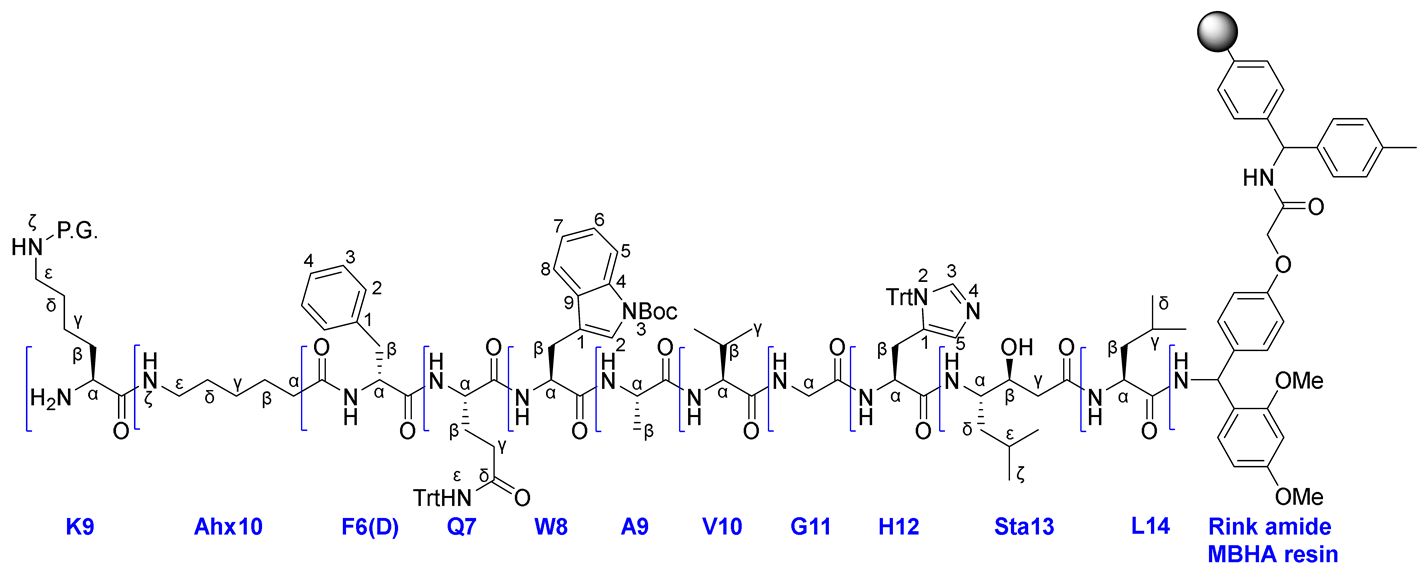 |
General procedure for obtaining peptide sequences by SPPS on Rink Amide MBHA resin.
Activation of Rink Amide MBHA resin. To a Rink Amide MBHA resin (1 eq., 0.3-0.8 mmol/g, 100-200 mesh) in polypropylene reactor DCM (10 ml/1 g) was added and the mixture was left under stirring for 60 min. Then the Fmoc protection was removed by the standard protocol.
Addition of the first amino acid residue. To a Rink Amide MBHA resin (1 eq., 0.3-0.8 mmol/g, 100-200 mesh) in DMF (10 ml/1 g) Fmoc-protected amino acid (4 eq. relative to the upper capacity of Rink Amide MBHA resin), PyBOP (4 eq.), HOBt (4 eq.), DIPEA (8 eq.) were added in atmosphere of Ar and the mixture was stirred overnight. Then, the resin was filtered and washed with DMF*3 (10 ml/1 g, 2 min), and DCM*3 (10 ml/1 g, 2 min).
Deprotection of Fmoc. Peptide sequence on a Rink Amide MBHA resin (1 eq.) was washed with DCM (10 ml/1 g, 1 min) for resin swelling. The mixture was charged with 4-methylpiperidine in DMF (20%/80% V/V, 15 ml) and stirred for 15 min, then the resin was filtered and washed with DMF (10 ml/1 g, 1 min) - this series of operations was performed 3 times. After that, the resin was filtered and washed with DMF*3 (10 ml/1 g, 2 min) and DCM*3 (10 ml/1 g, 2 min).
Deprotection of Alloc. Peptide sequence on a Rink Amide MBHA resin (1 eq.) was washed with DCM (10 ml/1 g, 1 min) for resin swelling, then DCM (10 ml/1 g), PhSiH3 (8 eq.) were and the mixture was stirred under inert atmosphere of Ar for few min; then added Pd[P(Ph)3]4 (0.1 eq.) and the mixture was stirred under inert atmosphere of Ar for 60 min, then the resin was filtered and washed DCM*3 (10 ml/1 g, 2 min) DMF*3 (10 ml/1 g, 2 min), and DCM*3 (10 ml/1 g, 2 min).
Addition of the second and subsequent amino acid residues. To a peptide sequence on Rink Amide MBHA resin (1 eq.) in DMF (10 ml/1 g) Fmoc-protected amino acid (2 eq. relative to the upper capacity of Rink Amide MBHA resin), PyBOP (2 eq.), HOBt (0.5 eq.), DIPEA (4 eq.) were added under argon atmosphere and the mixture was stirred overnight. Then the resin was filtered and washed with DMF*3 (10 ml/1 g, 1 min) and DCM*3 (10 ml/1 g, 1 min).
Using Rink Amide MBHA resin (500 mg, 0.15-0.4 mmol), FmocLeu-OH (565 mg, 1.6 mmol), PyBOP (832 mg, 1.6 mmol), HOBt (216 mg, 1.6 mmol), DIPEA (557 μl, 3.2 mmol) to add the first amino acid, Fmoc(S,S)Sta-OH (198 mg, 0.5 mmol), PyBOP (260.2 mg, 0.5 mmol), HOBt (16.9 mg, 0.125 mmol), DIPEA (174 μl, 1.0 mmol) to add the second amino acid, FmocHis(Trt)-OH (372 mg, 0.6 mmol), PyBOP (312 mg, 0.6 mmol), HOBt (20.3 mg, 0.15 mmol), DIPEA (210 μl, 1.2 mmol) to add the third amino acid, FmocGlu-OH (238 mg, 0.8 mmol), PyBOP (416 mg, 0.8 mmol), HOBt (27 mg, 0.2 mmol), DIPEA (279 μl, 1.6 mmol) to add the fourth amino acid, FmocVal-OH (272 mg, 0.8 mmol), PyBOP (416 mg, 0.8 mmol), HOBt (27 mg, 0.2 mmol), DIPEA (279 μl, 1.6 mmol) to add the fifth amino acid, FmocAla-OH (249 mg, 0.8 mmol), PyBOP (416 mg, 0.8 mmol), HOBt (27 mg, 0.2 mmol), DIPEA (279 μl, 1.6 mmol) to add the sixth amino acid, FmocTrp(Boc)-OH (316 mg, 0.8 mmol), PyBOP (416 mg, 0.8 mmol), HOBt (27 mg, 0.2 mmol), DIPEA (279 μl, 1.6 mmol) to add the seventh amino acid, FmocGln(Trt)-OH (305 mg, 0.5 mmol), PyBOP (416 mg, 0.8 mmol), HOBt (27 mg, 0.2 mmol), DIPEA (279 μl, 1.6 mmol) to add the eighth amino acid, FmocPhe(D)-OH (233 mg, 0.6 mmol), PyBOP (416 mg, 0.8 mmol), HOBt (27 mg, 0.2 mmol), DIPEA (279 μl, 1.6 mmol) to add the ninth amino acid, the amino acid sequence NH2-F(D)QWAVGHStaL on Rink Amide MBHA resin 20 was obtained.
Using FmocAhx-OH (212 mg, 0.6 mmol), PyBOP (416 mg, 0.8 mmol), HOBt (27 mg, 0.2 mmol), DIPEA (279 μl, 1.6 mmol) to add the tenth amino acid, FmocLys(Alloc)-OH (226 mg, 0.5 mmol), PyBOP (416 mg, 0.8 mmol), HOBt (27 mg, 0.2 mmol), DIPEA (279 μl, 1.6 mmol) to add the eleventh amino acid, the amino acid sequence NH2-K(Alloc)AhxF(D)QWAVGHStaL on Rink Amide MBHA resin 21 was obtained.
Using FmocLys(Boc)-OH (234 mg, 0.5 mmol), PyBOP (416 mg, 0.8 mmol), HOBt (27 mg, 0.2 mmol), DIPEA (279 μl, 1.6 mmol) to add the eleventh amino acid, the amino acid sequence NH2-K(Boc)AhxF(D)QWAVGHStaL on Rink Amide MBHA resin 22 was obtained.
Compound 20x
To establish the chemical purity and evaluate the actual capacity of Rink Amide MBHA resin coated with the peptide sequence NH2-F(D)QWAVGHStaL 20, removal of part of the peptide sequence from the polymer substrate was carried out.
Part of the resin was transferred to a 10 ml round-bottomed glass flask, charged with the system of TFA/TIPS/PhSMe/H2O (90%/5%/4%/1%, 2 ml) and stirred for 1.5 h. Then the resulting solution was separated on a sintered glass filter, washing the residue twice with TFA. The solvent was removed under reduced pressure, crude product was precipitated by Et2O and purified by column chromatography (Puriflash PF-15C18HP-F0012 (15 μ 20 g), eluent: H2O(90%)/MeCN(10%) => H2O(40%)/MeCN(60%) for 40 min, after MeCN(100%) for 10 min). Compound 20x was obtained in several forms: *2TFA; free NH2 (23 mg, 58% yield from max resin capacity).
1H NMR (400 MHz, DMSO-d6, δ) for *2TFA form: 14.34 (br.s., 2H, H12(2)<=>(4) + H+), 10.85 (s, 1H, W8(3)), 8.95 (s, 1H, H12(3)), 8.57 (d, J = 8.0 Hz, 1H, Q7NH), 8.30 (d, J = 7.7 Hz, 1H, W8NH), 8.28-8.21 (m, 2H, G11NH + A9NH), 8.20-8.00 (m, 4H, F6(D)NH3+ + H12NH), 7.88 (d, J = 8.0 Hz, 1H, L14NH), 7.79 (d, J = 8.4 Hz, 1H, V10NH), 7.66 (d, J = 7.8 Hz, 1H, W8(8)), 7.60 (d, J = 8.9 Hz, 1H, Sta13NH), 7.41 (s, 1H, L14NH2(a)), 7.35 (s, 1H, H12(5)), 7.34-7.28 (m, 3H, W8(5) + F6(D)ε), 7.28-7.20 (m, 4H, F6(D)δ + F6(D)ζ + Q7NH2(a)), 7.17 (d, J = 2.1 Hz, 1H, W8(2)), 7.08-7.00 (m, 2H, W8(6) + L14NH2(b)), 7.00-6.93 (m, 1H, W8(7)), 6.79 (s, 1H, Q7NH2(b)), 4.94 (br.s., 1H, Sta13OH), 4.67-4.59 (m, 1H, H12α), 4.59-4.50 (m, 1H, W8α), 4.45-4.35 (m, 1H, A9α), 4.35-4.25 (m, 1H, Q7α), 4.25-4.12 (m, 2H, L14α + V10α), 4.12-4.02 (m, 1H, F6(D)α), 3.89-3.65 (m, 4H, Sta13α + Sta13β + G11α), 3.16-3.00 (m, 3H, H12β(a) + W8β(a) + F6(D)β(a)), 3.00-2.85 (m, 3H, H12β(b) + W8β(b) + F6(D)β(b)), 2.24-2.04 (m, 2H, Sta13γ), 2.03-1.87 (m, 3H, V10β + Q7γ), 1.84-1.69 (m, 1H, Q7β(a)), 1.68-1.55 (m, 2H, Q7β(b) + L14γ), 1.54-1.40 (m, 3H, Sta13ε + L14β), 1.40-1.22 (m, 2H, Sta13δ(a) + Sta13δ(b)), 1.20 (d, J = 7.0 Hz, 3H, A9β), 0.91-0.76 (m, 18H, L14δ(a) + Sta13ζ(a) + V10γ(a) + L14δ(b) + V10γ(b) + Sta13ζ(b)).
1H NMR (400 MHz, DMSO-d6, δ) for free NH2 form: 11.86 (br.s., 1H, H12(2)<=>(4)), 10.83 (s, 1H, W8(3)), 8.31-7.90 (m, 6H, G11NH + A9NH + W8NH + H12(3) + Q7NH + H12NH), 7.78 (d, J = 8.5 Hz, 1H, V10NH), 7.61 (d, J = 7.8 Hz, 1H, W8(8)), 7.59-7.36 (m, 3H, L14NH2(a) + L14NH + Sta13NH), 7.30 (d, J = 7.8 Hz, 1H, W8(5)), 7.26-7.10 (m, 7H, F6(D)δ + Q7NH2(a) + F6(D)ε + W8(2) + F6(D)ζ), 7.08-6.93 (m, 3H, W8(6) + L14NH2(b) + W8(7)), 6.86 (br.s., 1H, H12(5)), 6.77 (s, 1H, Q7NH2(b)), 4.98 (br.s., 1H, Sta13OH), 4.58-4.50 (m, 1H, W8α), 4.50-4.43 (m, 1H, H12α), 4.43-4.34 (m, 1H, A9α), 4.26-4.16 (m, 2H, L14α + V10α), 4.16-4.06 (m, 1H, Q7α), 3.86-3.64 (m, 4H, Sta13α + Sta13β + G11α), 3.43-3.36 (m, 1H, F6(D)α), 3.18-3.07 (m, 1H, W8β(a)), 3.00-2.86 (m, 3H, H12β(a) + F6(D)β(a) + W8β(b)), 2.86-2.76 (m, 1H, H12β(b)), 2.54-2.45 (m, 1H, F6(D)β(b)), 2.25-1.92 (m, 5H, Sta13γ + V10β + Q7γ), 1.87-1.76 (m, 1H, Q7β(a)), 1.72-1.55 (m, 2H, Q7β(b) + L14γ), 1.51-1.27 (m, 4H, Sta13ε + L14β + Sta13δ(a)), 1.26-1.12 (m, 4H, A9β + Sta13δ(b)), 0.90-0.73 (m, 18H, L14δ(a) + Sta13ζ(a) + V10γ(a) + L14δ(b) + V10γ(b) + Sta13ζ(b)).
13C NMR (101 MHz, DMSO-d6, δ) for *2TFA form: 174.70 (L14C), 173.74 (Q7δ) 172.29 (A9C), 171.42 (W8C + V10C), 170.91 (Sta13C), 170.77 (Q7C), 169.66 (H12C), 168.82 (G11C), 167.82 (F6(D)C), 158.95 (TFA), 158.64 (TFA), 158.32 (TFA), 158.01 (TFA), 136.13 (W8(4)), 134.96 (F6(D)γ), 133.79 (H12(3)), 129.56 (F6(D)δ), 129.37 (H12(1)), 128.56 (F6(D)ε), 127.24 (F6(D)ζ), 127.22 (W8(9)), 124.04 (W8(2)), 121.67 (TFA), 120.94 (W8(6)), 118.70 (TFA), 118.65 (W8(8)), 118.27 (W8(7)), 116.95 (H12(5)), 115.72 (TFA), 112.75 (TFA), 111.37 (W8(5)), 109.89 (W8(1)), 69.04 (Sta13β), 57.78 (V10α), 53.36 (F6(D)α + W8α), 52.00 (Q7α), 51.75 (H12α), 51.05 (L14α), 50.87 (Sta13α), 48.28 (A9α), 41.99 (G11α), 40.86 (L14β), 39.60 (Sta13γ), 39.16 (Sta13δ), 37.31 (F6(D)β), 31.18 (Q7γ), 30.64 (V10β), 28.72 (Q7β), 27.72 (W8β), 27.45 (H12β), 24.27 (L14γ + Sta13ε), 23.32 (Sta13ζ(a)), 23.14 (L14δ(a)), 21.95 (Sta13ζ(b)), 21.56 (L14δ(b)), 19.23 (V10γ(a)), 18.03 (V10γ(b)), 17.76 (A9β).
15N NMR (41 MHz, DMSO-d6, δ) for *2TFA form: 180.27 (H12N4), 177.08 (H12N2), 131.30 (W8N3), 124.72 (L14N), 121.69 (A9N), 121.57 (Q7N), 120.25 (W8N), 120.06 (Sta13N), 115.48 (H12N), 113.99 (V10N), 108.74 (Q7Nε), 108.32 (G11N), 104.81 (L14NH2).
LCMS 100% in positive ion mode
ESI-MS (m/z) C55H80N14O11: calc. for [M+2H+]2+: 557.31, found: 557.75
HRMS (m/z, ESI): calc. for C55H80N14O11 - [M+2H] 2+ 557.3138, found: 557.315
Compound 22x
To establish the chemical purity and evaluate the actual capacity of Rink Amide MBHA resin coated with the peptide sequence NH2-KAhxF(D)QWAVGHStaL 22, removal of part of the peptide sequence from the polymer substrate was carried out.
Part of the resin was transferred to a 10 ml round-bottomed glass flask, charged with the system of TFA/TIPS/PhSMe/H2O (90%/5%/4%/1%, 5 ml) and stirred for 1.5 h. Then the resulting solution was separated on a sintered glass filter, washing the residue twice with TFA. The solvent was removed under reduced pressure, crude product was precipitated by Et2O and purified by column chromatography (Puriflash PF-15C18HP-F0012 (15 μ 20 g), eluent: H2O(90%)/MeCN(10%) => H2O(40%)/MeCN(60%) for 40 min, after MeCN(100%) for 10 min). Compound 22x was obtained as *3TFA form (32.3 mg, 58% yield from max resin capacity).
1H NMR (400 MHz, DMSO-d6, δ) for *3TFA form: 14.16 (br.s., 2H, H12(2) + (4) + H+), 10.80 (s, 1H, W8(3)), 8.91 (s, 1H, H12(3)), 8.40 (t, J = 5.6 Hz, 1H, Ahx10NHζ), 8.36 (d, J = 7.2 Hz, 1H, F6(D)NH), 8.25 (t, J = 5.7 Hz, 1H, G11NH), 8.22-8.00 (m, 6H, W8NH + Q7NH + H12NH + K9NH3+), 7.98 (d, J = 7.3 Hz, 1H, A9NH), 7.88 (d, J = 8.0 Hz, 1H, L14NH), 7.79 (br.s., 3H, K9NH3+), 7.64 (d, J = 8.4 Hz, 1H, V10NH), 7.61-7.54 (m, 2H, W8(8) + Sta13NH), 7.41 (s, 1H, L14NH2(a)), 7.34 (s, 1H, H12(5)), 7.31 (d, J = 7.8 Hz, 1H, W8(5)), 7.28-7.20 (m, 4H, F6(D)ε + F6(D)ζ + Q7NH2(a)), 7.20-7.12 (m, 2H, F6(D)δ + W8(2)), 7.08-7.00 (m, 2H, W8(6) + L14NH2(b)), 7.00-6.92 (m, 1H, W8(7)), 6.78 (s, 1H, Q7NH2(b)), 4.96 (br.s., 1H, Sta13OH), 4.67-4.59 (m, 1H, H12α), 4.52-4.41 (m, 2H, F6(D)α + W8α), 4.40-4.30 (m, 1H, A9α), 4.23-4.05 (m, 3H, Q7α + L14α + V10α), 3.89-3.62 (m, 5H, K9α + Sta13α + Sta13β + G11α), 3.20-2.87 (m, 7H, H12β(a) + K9ε + W8β(a) + F6(D)β(a) + H12β(b) + W8β(b)), 2.81-2.68 (m, 3H, Ahx10ε + F6(D)β(b)), 2.20-2.03 (m, 4H, Sta13γ + Ahx10α), 2.03-1.87 (m, 3H, V10β + Q7γ), 1.85-1.72 (m, 1H, Q7β(a)), 1.71-1.63 (m, 2H, Q7β(b) + L14γ), 1.63-1.23 (m, 15H, K9β(а) + K9δ + Sta13ε + K9β(b) + Ahx10β + L14β + Ahx10δ + Sta13δ(a) + K9γ + Sta13δ(b)), 1.20 (d, J = 7.0 Hz, 3H, A9β), 1.16-1.05 (m, 2H, Ahx10γ), 0.91-0.76 (m, 18H, L14δ(a) + Sta13ζ(a) + V10γ(a) + L14δ(b) + V10γ(b) + Sta13ζ(b)).
Route 1
Compound 12
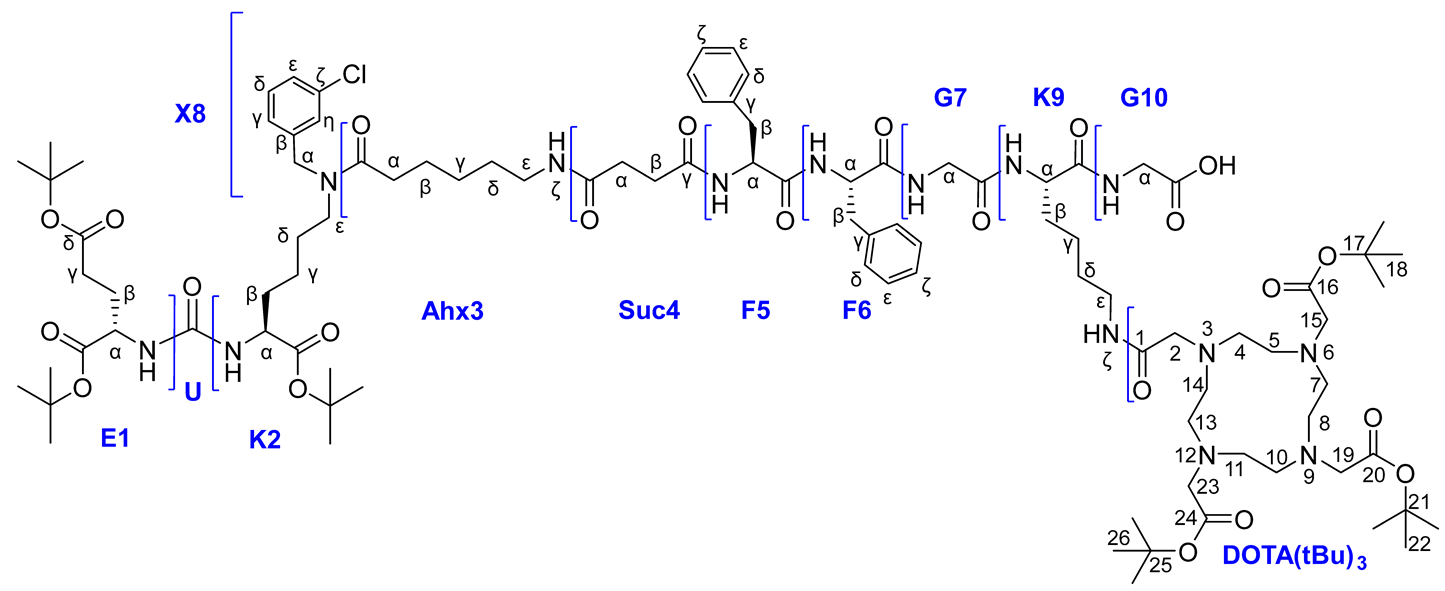 |
To a mixture of Alloc-deprotected compound 9 on 2-CTC resin (1 eq.; 0.15 mmol) in DMF (10 ml) in reactor HO-DOTA(tBu)3 (1 eq.; 86 mg; 0.15 mmol), HOBt (0.5 eq.; 10 mg; 0.075 mmol), HBTU (2 eq.; 114 mg; 0.3 mmol), DIPEA (3 eq.; 78 μl; 0.45 mmol) were added. The mixture was stirred 24 h under inert atmosphere of Ar. Then the solvent was removed by filtration on a porous reactor filter and the resin was washed three times with DMF (10 ml), three times with DCM (10 ml), then dried from traces of solvents.
A DCM/TFA system (99%-1%, 10 ml) was added to the resin and left under stirring for 15 min, after that, the solution was filtered from the resin. The solvent was removed under reduced pressure and the residue was re-evaporated with DCM. Next, the residue was dissolved in DCM (20 ml) and washed with saturated NaHCO3 solution (2*20 ml). The organic fraction was dried over Na2SO4. Afterwards, the solvent was removed under reduced pressure. The crude residue was purified by column chromatography (Puriflash PF-15C18HP-F0012 + PF-15C18HP-F0012 (15 μ 20 g + 15 μ 20 g), eluent: H2O(90%)/MeCN(10%) => H2O(0%)/MeCN(100%) for 30 min, after MeCN(100%) for 5 min). Compound 12 was obtained as a white amorphous solid (58 mg, 20% yield).
1H NMR (400 MHz, DMSO-d6, δ): 8.32-8.22 (m, 1H, G10NH), 8.21-8.14 (m, 2Н, F5NHmn + F6NH), 8.12-8.02 (m, 1H, G7NH), 7.92 (d, J = 8.0 Hz, 1H, K9NH), 7.89-7.79 (m, 1H, Ahx3NHζ, mn), 7.42-7.09 (m, 15H, X8δn + X8εn + X8δm + X8εm + F6ε + F6δ + X8ηmn + F5ε + F6ζ + F5ζ + K9NHζ + F5δ + X8γmn), 6.35-6.19 (m, 2Н, K2NHm + K2NHn + E1NHm + E1NHn), 4.60-4.42 (m, 3H, X8αnm + F6α), 4.42-4.34 (m, 1H, F5Hα), 4.33-4.20 (m, 1H, K9α), 4.08-3.90 (m, 2H, E1α + K2αm + K2αn), 3.81-3.58 (m, 4Н, G7α + G9α), 3.80-1.60 (br.m., 24H, DOTA), 3.25-2.81 (m, 9Н, K2εnm + F6β(a) + K9ε + F6β(b) + Ahx3ε + F5β(a)), 2.70-2.60 (m, 1Н, F5β(b)), 2.40-2.10 (m, 8H, Ahx3αm + Suc4βmn + E1γ + Suc4αmn + Ahx3αn), 1.92-1.80 (m, 1Н, E1β(a)), 1.72-1.10 (m, 19Н, E1β(b) + K9β(a) + K2β(a) + Ahx3β + K9β(b) + K2β(b) + K9δ + Ahx3δ + K2δ + K2γ + K9γ + Ahx3γ, m + n), 1.47 (s, 9H, 22), 1.40-1.35 (m, 45H, 18 + 26 + tBu).
Route 2
Compound 24x
To a mixture of compound 21 on Rink Amide MBHA resin (1 eq.; 0.153 mmol = 58% from max capacity) in DMF (10 ml) in reactor HO-DOTA(tBu)3 (1.5 eq.; 131 mg; 0.228 mmol), HOBt (0.5 eq.; 10 mg; 0.076 mmol), PyBOP (2.45 eq.; 194 mg; 0.3735 mmol), DIPEA (5 eq.; 133 μl; 0.765 mmol) were added. The mixture was stirred for 24 h under inert atmosphere of Ar. Then the solvent was removed by filtration on a porous reactor filter and the resin was washed three times with DMF (10 ml), three times with DCM (10 ml), then dried from traces of solvents. Afterwards, Alloc was removed according to the standard protocol.
To establish the chemical purity and evaluate the actual capacity of Rink Amide MBHA resin coated with a peptide sequence DOTA-K(NH2)AhxF(D)QWAVGHStaL 24, removal of part of the peptide sequence from the polymer substrate was carried out.
Part of the resin was transferred to a 10 ml round-bottomed glass flask, charged with the system of TFA/TIPS/PhSMe/H2O (90%/5%/4%/1%, 5 ml) and stirred for 3 h. Then the resulting solution was separated on a sintered glass filter, washing the residue twice with TFA. The solvent was removed under reduced pressure, crude product was precipitated by Et2O and purified by column chromatography (Puriflash PF-15C18HP-F0012 (15 μ 20 g), eluent: H2O(95%)/MeCN(5%) => H2O(50%)/MeCN(50%) for 40 min, after MeCN(100%) for 10 min). Compound 22x was obtained as *3TFA form (18.2 mg, 22% of total capacity) and target compound 24x (70 mg, 78% of total capacity), where total capacity = 55% of max resin capacity.
1H NMR (400 MHz, DMSO-d6, δ): 10.86 (s, 1H, W8(3)), 8.63-8.43 (m, 3H, Q7NH + G11NH + F6(D)NH), 8.42-8.30 (m, 2H, W8NH + H12NH), 8.20-8.10 (m, 2H, L14NH + A9NH), 8.10-7.97 (m, 2H, Ahx10NHζ + K9NH), 7.90-7.76 (m, 1H, V10NH), 7.61-7.47 (m, 4H, W8(8) + H12(3) + L14NH2(a) + Sta13NH), 7.30 (d, J = 7.8 Hz, 1H, W8(5)), 7.27-7.12 (m, 7H, F6(D)ε + F6(D)ζ + Q7NH2(a) + F6(D)δ + W8(2)), 7.08-6.92 (m, 3H, W8(6) + L14NH2(b) + W8(7)), 6.83 (br.s., 1H, H12(5)), 6.76 (s, 1H, Q7NH2(b)), 5.10 (br.s., 1H, Sta13OH), 4.55-4.30 (m, 4H, W8α + H12α + F6(D)α + A9α), 4.22-4.04 (m, 4H, K9α + V10α + L14α + Q7α), 3.85-3.62 (m, 4H, Sta13α + Sta13β + G11α), 3.80-1.60 (br.m., 24H, DOTA), 3.20-2.87 (m, 10H, H12β(a) + K9ε + W8β(a) + F6(D)β(a) + H12β(b) + W8β(b) + Ahx10ε + F6(D)β(b)), 2.20-2.03 (m, 4H, Sta13γ + Ahx10α), 2.03-1.86 (m, 3H, V10β + Q7γ), 1.85-1.71 (m, 2H, Q7β(a) + K9β(a)), 1.70-1.52 (m, 2H, Q7β(b) + L14γ + K9δ), 1.52-1.13 (m, 11H, K9β(b) + L14β + Sta13ε + Ahx10β + Sta13δ(a) + Ahx10δ + K9γ), 1.26-1.15 (m, 4H, A9β + Sta13δ(b)), 1.14-1.02 (m, 2H, Ahx10γ), 0.91-0.73 (m, 18H, L14δ(a) + V10γ(a) + L14δ(b) + V10γ(b) + Sta13ζ(a) + Sta13ζ(b)).
LCMS 92% in negative ion mode
ESI-MS (m/z) C83H129N21O20: calc. for [M-2H+]2-: 868.97, found: 868.96
Compound 25X
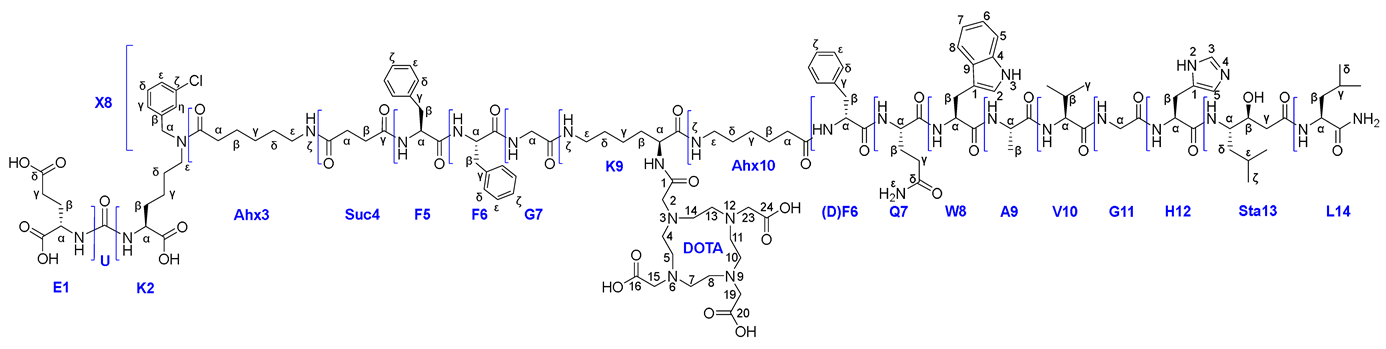 |
To a mixture of compound 24 on Rink Amide MBHA resin (1 eq.; 97 μmol) in DMF (7 ml) in reactor under Ar atmosphere compound 15 (1 eq.; 114 mg; 97 μmol), HOBt (0.5 eq.; 6.5 mg; 48.5 μmol), PyBOP (3 eq.; 152 mg; 291 μmol), DIPEA (4.5 eq.; 76 μl; 436 μmol) were added. The mixture was stirred for 24 h under inert atmosphere of Ar. Then the solvent was removed by filtration on a porous reactor filter and the resin was washed three times with DMF (5 ml), three times with DCM (5 ml), then dried from traces of solvents.
Part of the resin was transferred to a 10 ml round-bottomed glass flask, charged with the system of TFA/TIPS/PhSMe/H2O (90%/5%/4%/1%, 5 ml) and stirred for 3 h. Then the resulting solution was separated on a sintered glass filter, washing the residue twice with TFA. The solvent was removed under reduced pressure, crude product was precipitated by Et2O. Compound 25X (49% according to LCMS) was obtained in mixture with compound 24x (37% according to LCMS) and other impurities.
Route 3
Compound 26x
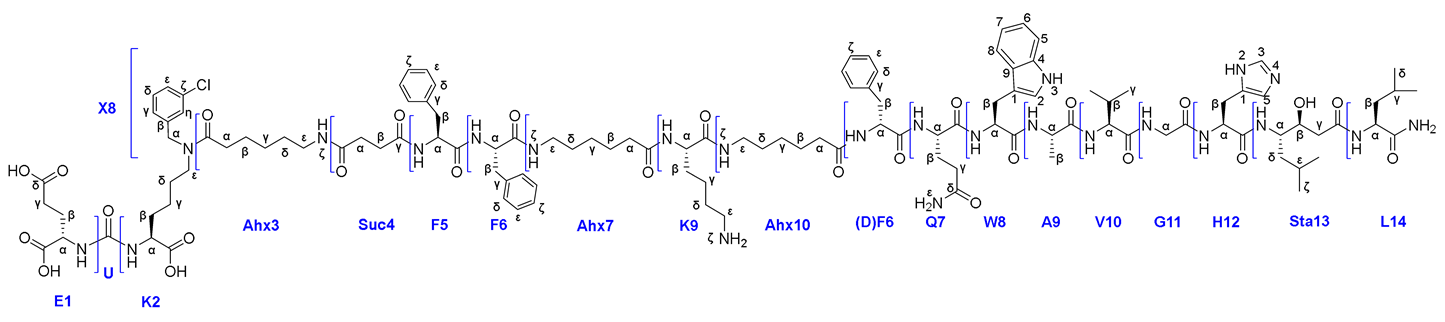 |
To compound 22 on Rink Amide MBHA resin (1 eq.; 57 μmol) in DMF (5 ml) in reactor under Ar atmosphere compound 18 (1 eq.; 70 mg; 57 μmol), HOBt (1 eq.; 7.7 mg; 57 μmol), PyBOP (3 eq.; 65 mg; 171 μmol), DIPEA (6 eq.; 59 μl; 342 μmol) were added. The mixture was stirred for 24 h under inert atmosphere of Ar. Then the solvent was removed by filtration on a porous reactor filter and the resin was washed three times with DMF (5 ml), three times with DCM (5 ml), then dried from traces of solvents.
Part of the resin was transferred to 10 ml round-bottomed glass flask, charged with the system of TFA/TIPS/PhSMe/H2O (90%/5%/4%/1%, 5 ml) and stirred for 3 h. Then the resulting solution was separated on a sintered glass filter, washing the residue twice with TFA. The solvent was removed under reduced pressure, crude product was precipitated by Et2O and purified by column chromatography (Puriflash PF-15C18HP-F0040 (15 μ 60 g), eluent: H2O(90%)/MeCN(10%) => H2O(50%)/MeCN(50%) for 40 min, after MeCN(100%) for 10 min) Compound 26x was obtained as free NH2 form (110 mg, 47% of max resin capacity, 81% of PSMA fragment)
1H NMR (400 MHz, DMSO-d6, δ) for free NH2 form: 10.75 (s, 1H, W8(3)), 8.56 (d, J = 7.2 Hz, m) & 8.41 (d, J = 7.2 Hz, n) (1H, F5NH, m + n), 8.38-8.13 (m, 4H, F6NHm + Q7NH + G11NH + F6NHn + F6(D)NH), 8.13-7.80 (m, 7H, Ahx3NHζm + H12NH + W8NH + Ahx3NHζn + L14NH + A9NH + Ahx10NHζm + Ahx10NHζn + K9NH), 7.71 (d, J = 8.5 Hz, 1H, V10NH), 7.57 (d, J = 7.8 Hz, 1H, W8(8)), 7.55-7.40 (m, 4H, H12(3) + L14NH2(a) + Ahx7NHζm + Ahx7NHζn + Sta13NH), 7.40-7.07 (m, 22H, X8Hδn + X8Hεn + W8(5) + X8Hδm + X8Hεm + F6Hε + F6Hδ + X8Hηmn + F6(D)δ + F6(D)ε + F5Hε + F6Hζ + F6(D)ζ + F5Hζ + W8(2) + X8Hγm + F5Hδ + Q7NH2(a) + X8Hγn), 7.03 (t, J = 7.5 Hz, 1H, W8(6)), 7.00-6.89 (m, 2H, L14NH2(b) + W8(7)), 6.83 (br.s., 1H, H12(5)), 6.72 (s, 1H, Q7NH2(b)), 6.48-6.17 (m, 2Н, K2NHmn + E1NHmn), 4.60-4.30 (m, 7H, X8αn + W8α + H12α + X8αm + F6(D)α + F6α + A9α), 4.29-4.06 (m, 5H, F5αmn + K9α + V10α + L14α + Q7α), 4.05-3.90 (m, 2H, E1Hα + K2Hαm + K2Hαn), 3.86-3.64 (m, 4H, Sta13β + Sta13α + G11α), 3.26-2.59 (m, 20H, K2εn + W8β(a) + K2εm + F6β(a) + Ahx7ε + W8β(b) + Ahx3εm + Ahx10ε + Ahx3εn + F6(D)β(a) + F6β(b) + H12β(a) + F5β(a) + H12β(b) + F6(D)β(b) + K9ε + F5β(b)), 2.46-1.95 (m, 15H, Suc4βm + Suc4αm + Ahx3αm + Suc4βn + E1γ + Ahx3αn + Suc4αn + Sta13γ + Ahx7α + Ahx10α + V10β), 1.95-1.86 (m, 2H, Q7γ), 1.86-1.70 (m, 3H, E1β(a) + Q7β(a) + E1β(b)), 1.70-1.56 (m, 4H, L14γ + K2β(a) + Q7β(b) + K9β(a)), 1.56-1.13 (m, 34H, K9δ + Ahx3βm + K2β(b) + K9β(b) + Ahx3βn + K2δm + Ahx7β + L14β + Sta13ε + K2δn + Ahx7δ + Ahx10β + Ahx3δm + Sta13δ(a) + Ahx10δ + Ahx3δn + Ahx3γmn + K9γ + K2γmn + A9β + Sta13δ(b) + Ahx7γ), 1.13-1.04 (m, 2H, Ahx10γ), 0.90-0.75 (m, 18H, L14δ(a) + V10γ(a) + L14δ(b) + V10γ(b) + Sta13ζ(a) + Sta13ζ(b)).
13C NMR (101 MHz, DMSO-d6, δ) for free NH2 form: 175.28 (K2Cm), 175.16 (K2Cn), 174.84 (E1Cm), 174.77 (E1Cn), 174.61 (L14C), 174.56 (E1Cδm), 174.45 (E1Cδn), 173.75 (Q7Cδ), 173.09 (Suc4Cγm), 173.04 (Suc4Cγn), 172.62 (Ahx10C), 172.12 (K9Cmn + Ahx3Cm), 172.06 (A9C + Ahx3Cn), 171.96 (F6(D)C), 171.67 (Suc4Cm), 171.62 (Suc4Cn), 171.51 (Ahx7Cm), 171.47 (Ahx7Cn), 171.31 (V10C), 171.21 (W8C + Q7C), 171.07 (F5Cm), 171.01 (F5Cn), 170.68 (H12C), 170.63 (Sta13C), 170.57 (F6Cm), 170.48 (F6Cn), 168.33 (G11C), 157.27 (U), 141.16 (X8Cβm), 140.74 (X8Cβn), 138.25 (F6Cγm), 138.19 (F6Cγn), 138.02 (F5Cγm), 137.99 (F5Cγn), 137.64 (F6(D)Cγ), 136.01 (W8C4), 134.70 (H12C3), 133.34 (X8Cζn), 132.98 (X8Cζm), 130.49 (X8Cδn), 130.12 (X8Cδm), 129.10 (F6(D)Cδ), 128.93 (F6Cδ), 128.91 (H12C1), 128.88 (F5Cδ), 128.07 (F6Cε), 128.01 (F5Cε), 127.94 (F6(D)Cε), 127.22 (W8C9), 127.12 (X8Cηm), 127.03 (X8Cεn), 126.73 (X8Cεm), 126.19 (X8Cηn + F5Cζ), 126.16 (F6Cζ + F6(D)Cζ), 126.00 (X8Cγm), 124.92 (X8Cγn), 123.52 (W8C2), 120.77 (W8C6), 118.27 (W8C8), 118.19 (W8C7 + H12C5), 111.22 (W8C5), 110.05 (W8C1), 69.44 (Sta13Cβ), 57.80 (V10Cα), 55.50 (F5Cαm), 55.30 (F5Cαn), 54.50 (F6Cαm), 54.45 (F6(D)Cα), 54.36 (F6Cαn), 53.48 (W8Cα), 53.17 (E1Cαm + H12Cα), 53.07 (E1Cαn), 52.90 K2Cαm), 52.83 (K2Cαn), 52.67 (Q7Cα), 52.20 (K9Cα), 51.32 (L14Cα), 50.33 (Sta13Cα), 49.63 (X8Cαn), 48.27 (A9Cα), 47.13 (X8Cαm), 46.97 (K2Cεm), 45.26 (K2Cεn), 42.05 (G11Cα), 40.78 (L14Cβ), 38.57 (Ahx3Cεm + Ahx7Cε), 38.49 (Ahx3Cεn + K9Hε), 38.39 (Ahx10Cε), 37.32 (F6(D)Cβ), 36.90 (F6Cβ), 36.62 (F5Cβ), 35.09 (Ahx7Cα), 34.95 (Ahx10Cα), 32.46 (K2Cβm), 32.31 (K2Cβn), 32.23 (Ahx3Cαn), 31.77 (Ahx3Cαm), 31.52 (K9Cβ), 31.46 (E1Cγ), 31.28 (Q7Cγ), 30.72 (Suc4Cα), 30.53 (Suc4Cβ), 30.45 (V10Cβ), 29.79 (H12Cβ), 29.08 (E1Cβ), 28.80 (Ahx7Cδ), 28.78 (Ahx10Cδ), 28.58 (Ahx3Cδ), 28.00 (K2Cδm), 27.49 (Q7Cβ), 27.14 (W8Cβ), 26.83 (K2Cδn), 26.67 (K9Cδ), 26.12 (Ahx3Cγ), 25.91 (Ahx7Cγ + Ahx10Cγ), 24.90 (Ahx7Cβ), 24.75 (Ahx10Cβ), 24.61 (Ahx3Cβm), 24.50 (AhxCβn), 24.21 (L14Cγ), 24.07 (Sta13Cε), 23.17 (Sta13Cζ(a)), 22.99 (L14Cγ(a)), 22.36 (K2Cγm), 22.32 (K2Cγn), 22.30 (K9Cγ), 21.95 (Sta13Cζ(b)), 21.52 (L14Cγ(b)), 19.16 (V10Cγ(a)), 17.86 (V10Cγ(b)), 17.54 (A9Cβ).
15N NMR (41 MHz, DMSO-d6, δ) for free NH2 form: 131.14 (W8N3), 125.08 (L14N), 122.36 (F5Nm + F6(D)N), 122.12 (F5Nn), 121.87 (Ahx10Nζmn), 120.98 (A9N), 120.23 (Sta13N), 119.45 (Q7N), 118.58 (Ahx3Nζm), 118.28 (Ahx3Nζn + W8N), 117.82 (H12N), 115.24 (F6Nnm), 115.02 (K9N), 114.08 (Ahx7Nζn), 113.92 (Ahx7Nζm), 113.19 (V10N), 108.51 (Q7Nε), 108.05 (G11N), 103.84 (L14NH2), 97.80 (K2Nε), 88.72 (K2Nm), 88.47 (K2Nn), 88.32 (E1Nm), 88.11 (E1Nn).
LCMS 97% in negative ion mode
ESI-MS (m/z) C120H17137ClN24O26: calc. for [M-2H+]2-: 1199.62, found: 1199.12.
HRMS (m/z, ESI): calc. for C120H17137ClN24O26 - [M-2H]2- 1199.6227, found: 1199.4351
Compound 27
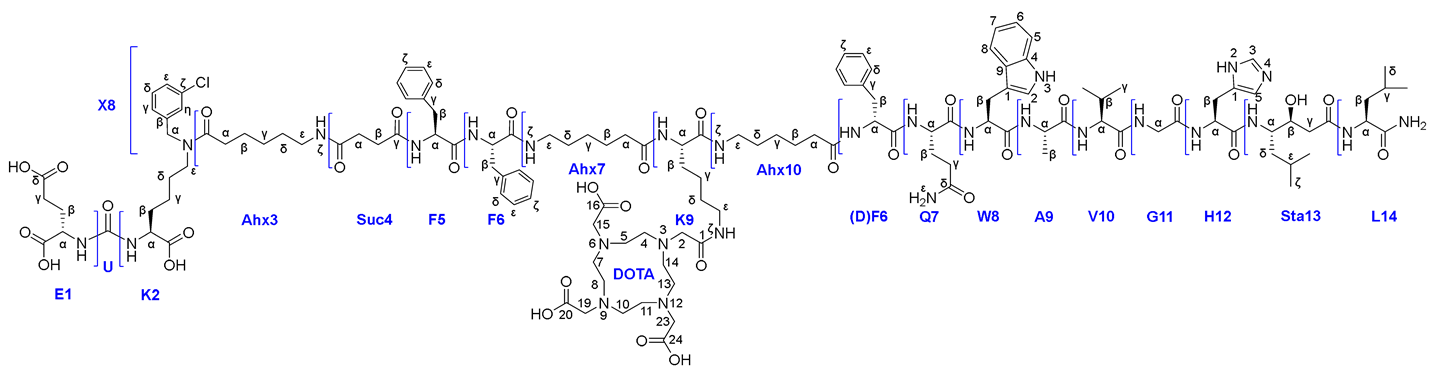 |
Compound 26x (1 eq.; 39 mg; 15.5 μmol) was dissolved in DMSO (4 ml) (CGC 20-25 mg/ml, complete dissolution was performed under ultrasound bath action for 10 min at 40°C) and purged with Ar. DIPEA (5 eq.; 10 mg; 77.5 μmol), DOTA-NHS ester (1 eq.; 12 mg; 15.5 μmol) were sequentially added. The mixture was stirred for 12 h, afterwards the solvent was removed under reduced pressure. Crude product was purified by column chromatography (PF-15C18HP-F0012 (15 μ 20 g), eluent: H2O(90%)/MeCN(10%) => H2O(50%)/MeCN(50%) for 30 min, after MeCN(100%) for 10 min). Compound 27 was obtained as a white powder (30 mg, 69% yield, 88% purity according to LCMS) in a mixture (m = 34.2 mg) with the initial HBV ligand 26x (10% according to LCMS).
1H NMR (400 MHz, DMSO-d6, δ): 10.80 (s, 1H, W8(3)), 8.58-7.78 (m, 13H, F5NHmn + F6NHm + Q7NH + G11NH + F6NHn + F6(D)NH + Ahx3NHζm + H12NH + K9NHε + W8NH + Ahx3NHζn + L14NH + A9NH + Ahx10NHζm + Ahx10NHζn + K9NH), 7.71 (d, J = 8.5 Hz, 1H, V10NH), 7.65-7.41 (m, 5H, W8(8) + H12(3) + L14NH2(a) + Ahx7NHζm + Ahx7NHζn + Sta13NH), 7.40-7.07 (m, 22H, X8δn + X8εn + W8(5) + X8δm + X8εm + F6ε + F6δ + X8ηmn + F6(D)δ + F6(D)ε + F5ε + F6ζ + F6(D)ζ + F5ζ + W8(2) + X8γm + F5δ + Q7NH2(a) + X8γn), 7.07-6.90 (m, 3H, W8(6) + L14NH2(b) + W8(7)), 6.86 (br.s., 1H, H12(5)), 6.77 (s, 1H, Q7NH2(b)), 6.48-6.17 (m, 2Н, K2NHmn + E1NHmn), 4.60-4.30 (m, 7H, X8αn + W8α + H12α + X8αm + F6(D)α + F6α + A9α), 4.29-3.96 (m, 7H, F5αmn + K9α + V10α + L14α + Q7α + E1α + K2αm + K2αn), 3.86-3.64 (m, 4H, Sta13β + Sta13α + G11α), 3.60-2.40 (br.m, 44H, 2 + 19 + 15 + 23 + Hcyclic + K2εn + W8β(a) + K2εm + F6β(a) + Hcyclic + Ahx7ε + W8β(b) + Ahx3εm + Ahx10ε + Ahx3εn + F6(D)β(a) + K9ε + F6β(b) + H12β(a) + F5β(a) + H12β(b) + F6(D)β(b) + F5β(b)), 2.40-1.95 (m, 15H, Suc4βm + Suc4αm + Ahx3αm + Suc4βn + E1γ + Ahx3αn + Suc4αn + Sta13γ + Ahx7α + Ahx10α + V10β), 1.95-1.83 (m, 3H, Q7γ + E1β(a)), 1.86-1.69 (m, 2H, Q7β(a) + E1β(b)), 1.69-1.55 (m, 4H, L14γ + K2β(a) + Q7β(b) + K9β(a)), 1.55-1.13 (m, 34H, K9δ + Ahx3βm + K2β(b) + K9β(b) + Ahx3βn + K2δm + Ahx7β + L14β + Sta13ε + K2δn + Ahx7δ + Ahx10β + Ahx3δm + Sta13δ(a) + Ahx10δ + Ahx3δn + Ahx3γmn + K9γ + K2γmn + A9β + Sta13δ(b) + Ahx7γ), 1.11-1.01 (m, 2H, Ahx10γ), 0.90-0.75 (m, 18H, L14δ(a) + V10γ(a) + L14δ(b) + V10γ(b) + Sta13ζ(a) + Sta13ζ(b)).
13C NMR (101 MHz, DMSO-d6, δ): 174.70 (L14C), 174.54 (K2Cn), 174.52 (K2Cm), 174.22 (E1C), 173.81 (E1Cδ + Q7Cδ), 172.72 (Suc4Cγnm + Ahx10C), 172.15 (Ahx3Cnm + A9C), 172.06 (F6(D)C), 171.79 (K9C), 171.56 (Suc4Cnm + Ahx7C), 171.36 (V10C), 171.30 (W8C), 171.28 (Q7C), 171.03 (F5C), 170.70 (Sta13C), 170.64 (H12C), 170.48 (F6C), 170.13 (DOTA), 169.72 (DOTA), 168.39 (G11C), 157.28 (U), 141.21 (X8Cβm), 140.80 (X8Cβn), 138.12 (F6Cγ), 138.01 (F5Cγ), 137.67 (F6(D)Cγ), 136.05 (W8C4), 134.68 (H12C3), 133.39 (X8Cζn), 133.04 (X8Cζm), 130.59 (X8Cδn), 130.22 (X8Cδm), 129.17 (F6(D)Cδ), 129.02 (F6Cδ + H12C1 + F5Cδ), 128.13 (F6Cε), 128.05 (F5Cε), 128.02 (F6(D)Cε), 127.25 (W8C9), 127.18 (X8Cηm), 127.12 (X8Cεn), 126.82 (X8Cεm), 126.28 (X8Cηn + F5Cζ), 126.25 (F6Cζ + F6(D)Cζ), 126.06 (X8Cγm), 124.95 (X8Cγn), 123.58 (W8C2), 120.85 (W8C6), 118.33 (W8C8), 118.26 (W8C7 + H12C5), 111.30 (W8C5), 110.11 (W8C1), 69.42 (Sta13Cβ), 58.25 (DOTA2), 57.79 (V10Cα), 55.28 (DOTA19), 54.95 (F5Cα), 54.90 (DOTA15+23), 54.54 (F6(D)Cα), 54.32 (F6C), 53.50 (W8Cα), 53.06 (H12Cα), 52.70 (Q7Cα), 52.61 (K9Cα), 52.30 (K2Cαn), 52.20 (K2Cαm), 51.76 (E1Cα), 51.32 (L14Cα), 50.97 (DOTAcycl.), 50.37 (Sta13Cα), 50.28 (DOTAcycl.), 49.86 (DOTAcycl.), 49.75 (DOTAcycl.), 49.65 (X8Cαn), 48.30 (A9Cα), 47.17 (X8Cαm), 46.88 (K2Cεm), 45.32 (K2Cεn), 42.06 (G11Cα), 40.81 (L14Cβ), 39.87 (Sta13Cγ), 39.62 (Sta13Cδ), 38.64 (Ahx3Cεm), 38.61(Ahx7Cε + Ahx3Cεn), 38.40 (Ahx10Cε), 38.27 (K9Cε), 37.38 (F6(D)Cβ), 37.19 (F6Cβ), 36.83 (F5Cβ), 35.08 (Ahx7Cα), 34.99 (Ahx10Cα), 32.29 (Ahx3Cαn), 31.87 (Ahx3Cαm), 31.83 (K2Cβ + K9Cβ), 31.33 (Q7Cγ), 30.67 (Suc4Cα), 30.55 (Suc4Cβ), 30.50 (V10Cβ), 30.03 (E1Cγ), 29.60 (H12Cβ), 29.05 (Ahx3Cδm), 28.95 (Ahx3Cδn), 28.82 (Ahx7Cδ), 28.68 (Ahx10Cδ), 28.55 (K9Cδ), 27.81 (K2Cδm), 27.66 (E1Cβ), 27.54 (Q7Cβ), 27.18 (W8Cβ), 26.75 (K2Cδn), 26.27 (Ahx3Cγm), 26.21 (Ahx3Cγn), 26.05 (Ahx7Cγ), 25.96 (Ahx10Cγ), 25.01 (Ahx7Cβ), 24.81 (Ahx10Cβ), 24.72 (Ahx3Cβm), 24.58 (AhxCβn), 24.25 (L14Cγ), 24.12 (Sta13Cε), 23.26 (Sta13Cζ(a)), 23.07 (L14Cγ(a)), 22.83 (K9Cγ), 22.50 (K2Cγn), 22.34 (K2Cγm), 22.00 (Sta13Cζ(b)), 21.56 (L14Cγ(b)), 19.23 (V10Cγ(a)), 17.92 (V10Cγ(b)), 17.60 (A9Cβ).
LCMS 88% in positive ion mode, main impurity (compound 26x) - 10% in positive ion mode.
ESI-MS (m/z) C136H19737ClN28O33: calc. for [M + 3H+]3 + : 930.15, found: 930.15.
Route 4a
Compound 28
To a solution of compound 13 (1 eq.; 217 mg; 0.15 mmol) in DMF (10 ml) DIPEA (3.5 eq.; 92 μl; 0.525 mmol), HOBt (0.5 eq.; 10 mg; 0.075 mmol) and HBTU (1.1 eq.; 62.6 mg; 0.165 mmol) were added. The mixture was stirred for 10 min under inert atmosphere of Ar. Then a solution of TFA*NH2-(CH2)3-NHFmoc (1.05 eq.; 64.6 mg; 0.158 mmol) in DMF was added and the mixture was stirred for 12 h. Afterwards, the solvent was removed under reduced pressure and the residue was re-evaporated twice with a mixture of MeOH/H2O (50/50) to completely remove the DMF residue (in some other solvents, the substance forms organogels). The resulting amorphous mass was grinded using a spatula to obtain a fine powder, which was washed with H2O (3*3 ml). The resulting precipitate was dried under reduced pressure and washed with P.E. (3*3 ml). Then, the residual solvent was removed from the precipitate under reduced pressure. Compound 28 was obtained as a pale yellow powder (258 mg, quant.).
1H NMR (400 MHz, DMSO-d6, δ): 8.26-8.06 (m, 4H, G10NH + F5NHmn + F6NH + G7NH), 7.97 (d, J = 8.0 Hz, 1H, K9NH), 7.92-7.79 (m, 3H, Ahx3NHζmn + Fmocη), 7.74-7.63 (m, 3H, X11NH + Fmocδ), 7.45-7.09 (m, 20H, Fmocζ + X8δn + X8εn + Fmocε + X8δm + X8εm + F6ε + F6δ + X11NHδ + X8ηmn + F5ε + F6ζ + F5ζ + K9NHζ + F5δ + X8γmn), 6.35-6.21 (m, 2Н, K2NHm + K2NHn + E1NHm + E1NHn), 5.95-5.81 (m, 1Н, Allocβ), 5.30-5.20 (m, 1Н, Allocγ(a)), 5.18-5.10 (m, 1Н, Allocγ(b)), 4.60-4.35 (m, 6H, X8αnm + F6α + Allocα + F5Hα), 4.28 (d, J = 6.9 Hz, 1H, Fmocα), 4.24-4.15 (m, 2H, Fmocβ + K9α), 4.08-3.90 (m, 2H, E1α + K2αm + K2αn), 3.81-3.60 (m, 4Н, G7α + G9α), 3.21 (t, J = 7.3 Hz, n) & 3.17 (t, J = 7.3 Hz, m) (2H, K2ε, m/n=3/2), 3.12-2.82 (m, 11Н, F6β(a) + X11γ + F6β(b) + X11α + Ahx3ε + K9ε + F5β(a)), 2.71-2.60 (m, 1Н, F5β(b)), 2.40-2.10 (m, 8H, Ahx3αm + Suc4βmn + E1γ + Suc4αmn + Ahx3αn), 1.92-1.80 (m, 1Н, E1β(a)), 1.72-1.10 (m, 21Н, E1β(b) + K9β(a) + K2β(a) + X11β + Ahx3β + K9β(b) + K2β(b) + K9δ + Ahx3δ + K2δ + K2γ + K9γ + Ahx3γ, m + n), 1.40-1.35 (m, 27H, tBu).
Compound 29
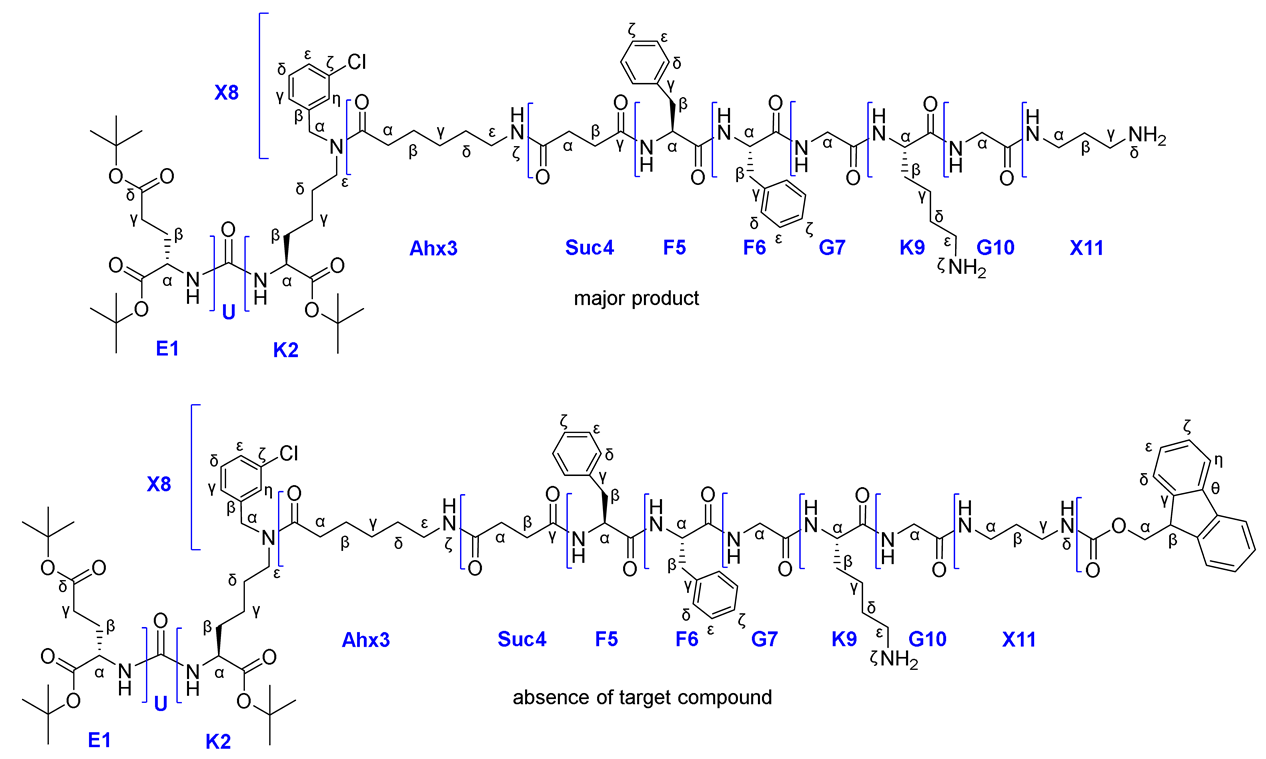 |
Compound 28 (1 eq.; 120 mg; 69.58 μmol) dissolved in DMF (3 ml), then the system was vacuumate and filled with Ar. Next, PhSiH3 (6 eq.; 52 μl; 418 μmol) was added. After 2 min of stirring Pd[P(Ph)3]4 (0.1 eq.; 8 mg; 7 μmol) in DCM (2 ml) was added using a syringe and the mixture was stirred 90 min under inert atmosphere. Afterwards, the solvent was removed under reduced pressure and the residue was re-evaporated twice with DCM (3 ml) to completely remove the DMF traces. The resulting amorphous mass was grinded using a spatula to obtain a fine powder, which was washed with Et2O (3*2 ml). Then, the residual solvent was removed from the precipitate under reduced pressure. As a result, complete removal of Fmoc protecting group occurred (according to NMR data). Obtained compound 29 was K9NH2ζ&X11NH2δ with two free NH2 group.
Route 4b
Compound 30
Aminohexanoic acid (1 eq.; 2836 mg; 21.6 mmol) was dissolved in a mixture of dioxane/satur. NaHCO3(aq) (37.5/75 ml), then a solution of Boc2O (1.5 eq.; 7500 μl; 32.4 mmol) in dioxane (37.5 ml) was added dropwise. The reaction mixture was stirred overnight. Afterwards, dioxane was removed under reduced pressure, EtOAc (100 ml) was added to the residue and the resulting mixture was acidified to pH = 3 with 0.5 М HCl (dropwise addition). Then the organic layer was separated and aqueous phase was washed with EtOAc (3*100 ml). The combined organic layers were washed with H2O (100 ml) and brine (100 ml). Then the organic layer was dried over Na2SO4. Afterwards, the solvent was removed under reduced pressure. The residue was purified by column chromatography (Puriflash, SIHP-F0120 + SIHP-F0120 (50 μ 116 g + 50 μ 116 g), eluent: DCM(100%)/MeOH(0%) => DCM(90%)/MeOH(10%) for 40 min, after МеOH(100%) for 10 min). Compound 30 was obtained as colorless oil (4630 mg, 93% yield).
1H NMR (400 MHz, DMSO-d6, δ): 11.97 (s, 1H, COOH), 6.76 (t, J = 5.3 Hz, 1H, Ahx11NHζ), 2.88 (td, J = 7.0, 5.3 Hz 2H, Ahx11ε), 2,18 (t, J = 7.4 Hz, 2H, Ahx11α), 1.52-1.41 (m, 2H, Ahx11β), 1.40-1.30 (m, 11H, Ahx11δ + tBu), 1.27-1.18 (m, 2H, Ahx11γ).
Compound 31
To a solution of compound 30 (1 eq., 1876 mg, 8.1 mmol) in DCM (10 ml) DIPEA (1.2 eq., 1690 μl, 9.4 mmol) was added. Then the mixture was cooled to 0 °C and a solution of FmocCl (1.05 eq., 2204 mg, 8.5 mmol) in DCM (25 ml) was added dropwise. The mixture was stirred for 5 min, then DMAP (0.2 eq., 198 mg, 1.6 mmol) was added and the mixture was stirred overnight at room temreture. After completion of the reaction, the mixture was diluted with DCM (65 ml) and washed with: 1) 0.05 М HCl (100 ml); 2) NaHCO3(aq) (1% m/V, 100ml); 3) H2O (100ml); 4) brine (100ml). Then the organic layer was dried over Na2SO4. Afterwards, the solvent was removed under reduced pressure. The residue was purified by column chromatography (Puriflash SI-HP-F0120 (50 μ 116 g), eluent: EtOAc(5%)/P.E(95%) => EtOAc(50%)/P.E(50%) for 40 min, after EtOAc(100%) for 10 min, then МеOH(100%) for 10 min). Compound 31 was obtained as white solid (1994 mg, 60% yield).
1H NMR (400 MHz, DMSO-d6, δ): 7.89 (d, J = 7.4 Hz, 2H, Fmη), 7.65 (d, J = 7.4 Hz, 2H, Fmδ), 7.41 (t, J = 7.4 Hz, 2H, Fmζ), 7.33 (td, J = 7.4, 1.1 Hz, 2H, Fmε), 6.77 (t, J = 5.3 Hz, 1H, Ahx11NHζ), 4.42 (d, J = 6.4 Hz, 2H, Fmα), 4.25 (t, J = 6.4 Hz, 1H, Fmβ), 2.90-2.80 (m, 2H, Ahx11ε), 2.26 (t, J = 7.3 Hz, 2H, Ahx11α), 1.45-1.34 (m, 11H, Ahx11δ + tBu), 1.32-1.25 (m, 2H, Ahx11β), 1.17-1.06 (m, 2H, Ahx11γ).
Compound 32
Compound 31 (1 eq., 1573 mg, 3.8 mmol) was dissolved in system of TFA/DCM (10%/90%, 20 ml) and the mixture was stirred for 2 h. After completion of the reaction, the solvent was removed under reduced pressure and the residue was re-evaporated twice with DCM (3 ml) to completely remove the TFA excess. The resulting crude product was used without further purification. Compound 32 was obtained as *1TFA form as white solid (1561 mg, 96% yield).
1H NMR (400 MHz, DMSO-d6, δ) for *1TFA form: 7.90 (d, J = 7.4 Hz, 2H, Fmη), 7.71 (br.s., 3H, Ahx11NH3+ζ), 7.65 (d, J = 7.4 Hz, 2H, Fmδ), 7.42 (t, J = 7.4 Hz, 2H, Fmζ), 7.34 (t, J = 7.4, 2H, Fmε), 4.44 (d, J = 6.4 Hz, 2H, Fmα), 4.26 (t, J = 6.4 Hz, 1H, Fmβ), 2.77-2.66 (m, 2H, Ahx11ε), 2.28 (t, J = 7.3 Hz, 2H, Ahx11α), 1.51 - 1.35 (m, 4H, Ahx11β + Ahx11δ), 1.27 - 1.13 (m, 2H, Ahx11γ).
Compound 33
To a solution of compound 13 (1 eq.; 100 mg; 69.15 μmol) in DMF (5 ml) DIPEA (3.5 eq.; 42 μl; 242 μmol), HOBt (0.5 eq.; 4.6 mg; 34.5 μmol) and HBTU (1.1 eq.; 29 mg; 76.07 μmol) were added. The mixture was stirred for 10 min under inert atmosphere of Ar. Then compound 32 (1.05 eq.; 30.7 mg; 72.47 μmol) was added in DMF and the reaction mixture was stirred for 12 h under Ar atmosphere. After completion of the reaction, the solvent was removed under reduced pressure and the residue was re-evaporated twice with DCM (3 ml) to completely remove the DMF traces. The resulting amorphous mass was grinded using a spatula to obtain a fine powder, which was washed with H2O (3*3 ml). The resulting precipitate was dried under reduced pressure and washed with P.E. (3*3 ml). Then, the residual solvent was removed from the precipitate under reduced pressure. Compound 33 was obtained as pale-yellow solid (119 mg, quant.).
1H NMR (400 MHz, DMSO-d6, δ): 8.23-8.05 (m, 4H, G10NH + F5NHmn + F6NH + G7NH), 7.97 (d, J = 8.0 Hz, 1H, K9NH), 7.89 (d, J = 7.5 Hz, 2H, Fmη), 7.84 (t, J = 5.4 Hz, m) & 7.81 (t, J = 5.4 Hz, n) (1H, Ahx3NHζ, m + n), 7.70-7.59 (m, 3H, Ahx11NHζ + Fmδ), 7.45-7.09 (m, 19H, Fmζ + X8δn + X8εn + Fmε + X8δm + X8εm + F6ε + F6δ + X8ηmn + F5ε + F6ζ + F5ζ + K9NHζ + F5δ + X8γmn), 6.34-6.21 (m, 2Н, K2NHm + K2NHn + E1NHm + E1NHn), 5.95-5.81 (m, 1Н, Allocβ), 5.30-5.20 (m, 1Н, Allocγ(a)), 5.18-5.10 (m, 1Н, Allocγ(b)), 4.60-4.34 (m, 8H, X8αnm + F6α + Allocα + Fmα + F5Hα), 4.25 (t, J = 6.5 Hz, 1H, Fmβ), 4.22-4.14 (m, 1H, K9α), 4.08-3.88 (m, 2H, E1α + K2αm + K2αn), 3.80-3.57 (m, 4Н, G7α + G9α), 3.21 (t, J = 7.3 Hz, n) & 3.17 (t, J = 7.3 Hz, m) (2H, K2ε, m/n = 3/2), 3.12-2.82 (m, 11Н, F6β(a) + F6β(b) + Ahx11ε + Ahx3ε + K9ε + F5β(a)), 2.71-2.60 (m, 1Н, F5β(b)), 2.40-2.10 (m, 10H, Ahx3αm + Suc4βmn + Ahx11α + E1γ + Suc4αmn + Ahx3αn), 1.92-1.80 (m, 1Н, E1β(a)), 1.72-1.05 (m, 25Н, E1β(b) + K9β(a) + K2β(a) + Ahx3β + K9β(b) + K2β(b) + K9δ + Ahx11β + Ahx3δ + K2δ + Ahx11δ + K2γ + K9γ + Ahx3γ + Ahx11γ, m + n), 1.40-1.35 (m, 27H, tBu).
Compound 34
Compound 33 (1 eq.; 119 mg; 69.15 μmol) was dissolved in DMF (3 ml), then the system was vacuumate and filled with Ar. Next PhSiH3 (6 eq.; 51 μl; 415 μmol) was added. After 2 min of stirring Pd[P(Ph)3]4 (0.1 eq.; 8 mg; 7 μmol) in DCM (2 ml) was added using a syringe and the mixture was stirred 90 min under inert atmosphere. Afterwards, 1% TFA solution in DCM (3 ml) was added to the reaction mixture to inactivate free NH2 group. Afterwards, the solvent was removed under reduced pressure and the residue was re-evaporated twice with DCM (3 ml) to completely remove the DMF traces. The resulting amorphous mass was grinded using a spatula to obtain a fine powder, which was washed with Et2O (3*2 ml). Then, residual solvent was removed from the precipitate under reduced pressure. As a result, partial removal of OFm occurred (according to NMR data). The residue was purified by column chromatography (Puriflash SIHP-F0020 (50 μ 20 g), eluent: DCM(100%)/MeOH(0%) => DCM(98%)/MeOH(2%) for 10 min, after DCM(0.1%TFA)(98%)/MeOH(2%) => DCM(0.1%TFA)(90%)/MeOH(10%) for 30 min, then МеOH(100%) for 10 min). Compound 34 was obatined as *1TFA form as pale-yellow amorphous product (85 mg, 70% yield).
1H NMR (400 MHz, DMSO-d6, δ): 8.23-8.05 (m, 4H, G10NH + F5NHmn + F6NH + G7NH), 7.99 (d, J = 8.0 Hz, 1H, K9NH), 7.91-7.81 (m, 3H, Fmη + Ahx3NHζmn), 7.75-7.60 (m, 6H, Ahx11NHζ + K9NH3+ζ + Fmδ), 7.45-7.09 (m, 18H, Fmζ + X8δn + X8εn + Fmε + X8δm + X8εm + F6ε + F6δ + X8ηmn + F5ε + F6ζ + F5ζ + F5δ + X8γmn), 6.34-6.21 (m, 2Н, K2NHm + K2NHn + E1NHm + E1NHn), 4.58-4.34 (m, 6H, X8αnm + F6α + Fmα + F5Hα), 4.30-4.17 (m, 2H, Fmβ + K9α), 4.08-3.88 (m, 2H, E1α + K2αm + K2αn), 3.80-3.57 (m, 4Н, G7α + G9α), 3.21 (t, J = 7.3 Hz, n) & 3.17 (t, J = 7.3 Hz, m) (2H, K2ε, m/n = 3/2), 3.12-2.80 (m, 11Н, F6β(a) + F6β(b) + Ahx11ε + Ahx3ε + F5β(a)), 2.80-2.70 (m, 2H, K9ε), 2.71-2.60 (m, 1Н, F5β(b)), 2.40-2.10 (m, 10H, Ahx3αm + Suc4βmn + Ahx11α + E1γ + Suc4αmn + Ahx3αn), 1.92-1.80 (m, 1Н, E1β(a)), 1.72-1.05 (m, 25Н, E1β(b) + K9β(a) + K2β(a) + Ahx3β + K9β(b) + K2β(b) + K9δ + Ahx11β + Ahx3δ + K2δ + Ahx11δ + K2γ + K9γ + Ahx3γ + Ahx11γ, m + n), 1.40-1.35 (m, 27H, tBu).
Compound 35
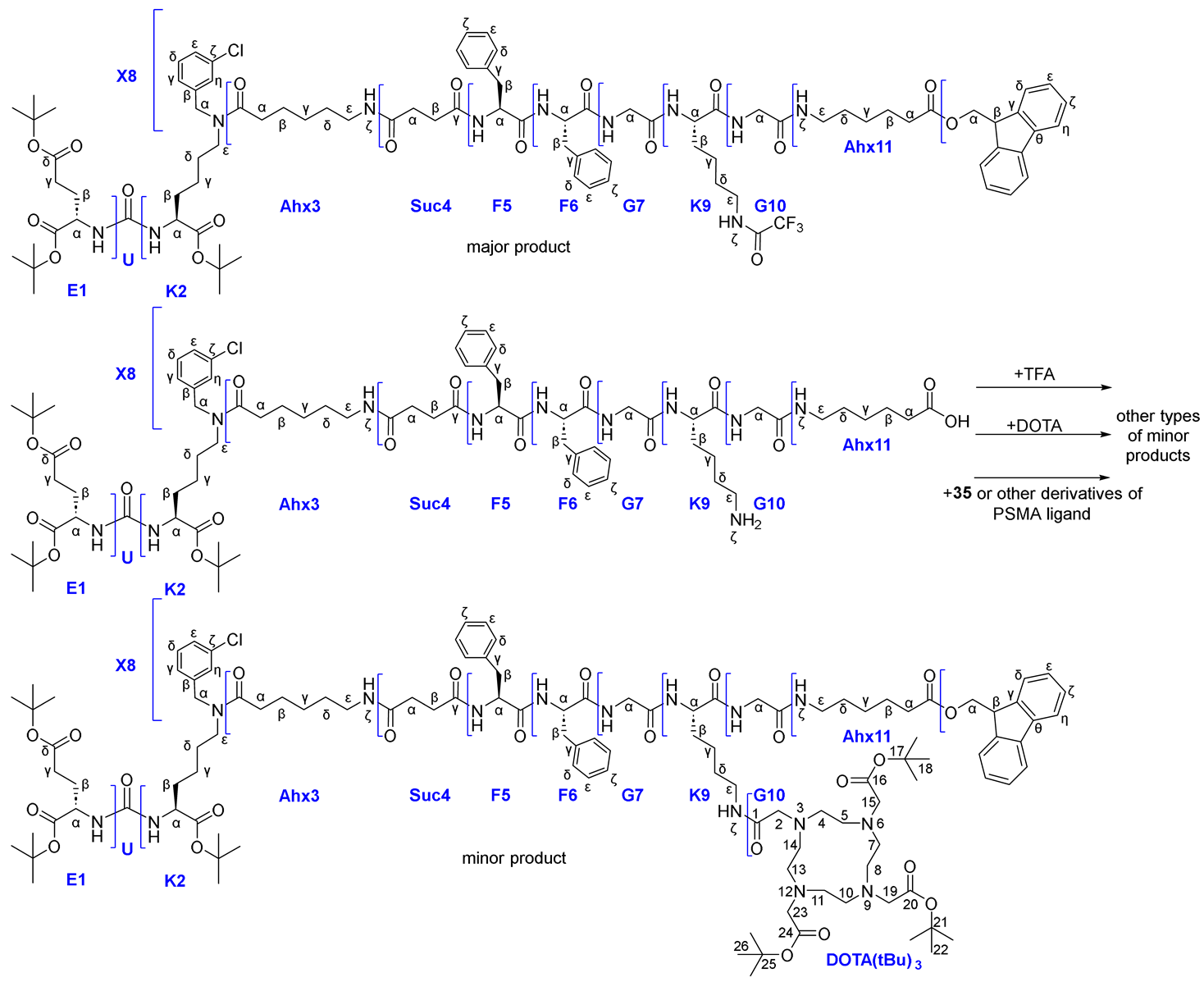 |
To a solution of DOTA(tBu)3-COOH (1.55 eq.; 43 mg; 75.25 μmol) in DMF (5 ml) DIPEA (7 eq.; 59 μl; 338.6 μmol), HOBt (0.75 eq.; 4.9 mg; 36.3 μmol), HBTU (1.75 eq.; 32 mg; 84.7 μmol) were added. After 5 min of stirring compound 34 (1 eq.; 85 mg; 48.4 μmol) was added under inert atmosphere of Ar, and then was stirred for 12 h. Afterwards, the solvent was removed under reduced pressure and the residue was re-evaporated twice with DCM (3 ml) to completely remove the DMF traces. The resulting amorphous mass was grinded using a spatula to obtain a fine powder, which was washed with H2O (3*2 ml). Then, residual solvent was removed from the precipitate under reduced pressure. The residue was purified by column chromatography (Puriflash SIHP-F0020 (50 μ 20 g), eluent: DCM(100%)/MeOH(0%) => DCM(98%)/MeOH(2%) for 10 min, after DCM(0.1%TFA)(98%)/MeOH(2%) => DCM(0.1%TFA)(80%)/MeOH(20%) for 30 min, then МеOH(100%) for 10 min). As a result, the main product of the reaction was the product of the addition of the TFA residue to the amino group K9NH2ζ, the target compound was isolated in trace amounts.
Route 4c
Compound 28
To a solution of compound 13 (1 eq.; 217 mg; 0.15 mmol) in DMF (10 ml) DIPEA (3.5 eq.; 92 μl; 0.525 mmol), HOBt (0.5 eq.; 10 mg; 0.075 mmol) and HBTU (1.1 eq.; 62.6 mg; 0.165 mmol) were added. The mixture was stirred for 10 min under inert atmosphere. Then TFA*NH2-(CH2)3-NHFmoc (1.05 eq.; 64.6 mg; 0.158 mmol) in DMF was added and the mixture was stirred for 12 h. Afterwards, the solvent was removed under reduced pressure and the residue was re-evaporated twice with a mixture of MeOH/H2O (50/50) to completely remove the DMF residue (in some other solvents the substance forms organogels). The resulting amorphous mass was grinded using a spatula to obtain a fine powder, which was washed with H2O (3*3 ml). The resulting precipitate was dried under reduced pressure and washed with P.E. (3*3 ml). Then, the residual solvent was removed from the precipitate under reduced pressure. Compound 28 was obtained as pale-yellow solid (258 mg, quant.).
1H NMR (400 MHz, DMSO-d6, δ): 8.26-8.06 (m, 4H, G10NH + F5NHmn + F6NH + G7NH), 7.97 (d, J = 8.0 Hz, 1H, K9NH), 7.92-7.79 (m, 3H, Ahx3NHζmn + Fmocη), 7.74-7.63 (m, 3H, X11NH + Fmocδ), 7.45-7.09 (m, 20H, Fmocζ + X8δn + X8εn + Fmocε + X8δm + X8εm + F6ε + F6δ + X11NHδ + X8ηmn + F5ε + F6ζ + F5ζ + K9NHζ + F5δ + X8γmn), 6.35-6.21 (m, 2Н, K2NHm + K2NHn + E1NHm + E1NHn), 5.95-5.81 (m, 1Н, Allocβ), 5.30-5.20 (m, 1Н, Allocγ(a)), 5.18-5.10 (m, 1Н, Allocγ(b)), 4.60-4.35 (m, 6H, X8αnm + F6α + Allocα + F5Hα), 4.28 (d, J = 6.9 Hz, 1H, Fmocα), 4.24-4.15 (m, 2H, Fmocβ + K9α), 4.08-3.90 (m, 2H, E1α + K2αm + K2αn), 3.81-3.60 (m, 4Н, G7α + G9α), 3.21 (t, J = 7.3 Hz, n) & 3.17 (t, J = 7.3 Hz, m) (2H, K2ε, m/n = 3/2), 3.12-2.82 (m, 11Н, F6β(a) + X11γ + F6β(b) + X11α + Ahx3ε + K9ε + F5β(a)), 2.71-2.60 (m, 1Н, F5β(b)), 2.40-2.10 (m, 8H, Ahx3αm + Suc4βmn + E1γ + Suc4αmn + Ahx3αn), 1.92-1.80 (m, 1Н, E1β(a)), 1.72-1.10 (m, 21Н, E1β(b) + K9β(a) + K2β(a) + X11β + Ahx3β + K9β(b) + K2β(b) + K9δ + Ahx3δ + K2δ + K2γ + K9γ + Ahx3γ, m + n), 1.40-1.35 (m, 27H, tBu).
Compound 36
Compound 28 (1 eq.; 258 mg; 0.15 mmol) was dissolved in a system of Et2NH/DMF (25 eq. Et2NH, 7.5 ml DMF) and the mixture was stirred for 20 min under inert atmosphere of Ar. Afterwards the solvent was removed under reduced pressure and the residue was re-evaporated twice with 0.5% TFA solution in DCM (5 ml) to inactivate free NH2 group. The product was precipitated by Et2O and washed with: 1) Et2O (2*2 ml), 2) P.E. (2*2 ml). Compound 37 was obtained as 1*TFA form as pale-yellow solid (242 mg, quant.).
To obtain free NH2 form the precipitate was dissolved in a mixture of H2O/MeCN (50/50, 8 ml) and charged with a saturated solution of NaHCO3 (2 ml), then the solvent was removed under reduced pressure to obtain a powder, which was grinded using a spatula and washed with H2O (3*3 ml). Then, the residual solvent was removed from the precipitate under reduced pressure. Compound 36 was obtained as pale-yellow solid (224 mg, quant.).
1H NMR (400 MHz, DMSO-d6, δ) for *1TFA form: 8.28 (t, J = 6 Hz, 1H, G10NH), 8.23-8.14 (m, 2Н, F5NHmn + F6NH), 8.08 (t, J = 5.9 Hz, 1H, G7NH), 8.01 (d, J = 7.1 Hz, 1H, K9NH), 7.90-7.77 (m, 2H, Ahx3NHζmn + X11NH), 7.63 (br.s., 3H, X11NH3+δ), 7.42-7.09 (m, 15H, X8δn + X8εn + X8δm + X8εm + F6ε + F6δ + X8ηmn + F5ε + F6ζ + F5ζ + K9NHζ + F5δ + X8γmn), 6.35-6.21 (m, 2Н, K2NHm + K2NHn + E1NHm + E1NHn), 5.95-5.81 (m, 1Н, Allocβ), 5.30-5.20 (m, 1Н, Allocγ(a)), 5.18-5.10 (m, 1Н, Allocγ(b)), 4.60-4.33 (m, 6H, X8αnm + F6α + Allocα + F5Hα), 4.18-4.10 (m, 1H, K9α), 4.08-3.90 (m, 2H, E1α + K2αm + K2αn), 3.81-3.58 (m, 4Н, G7α + G9α), 3.25-2.81 (m, 11Н, K2εnm + X11γ + F6β(a) + F6β(b) + Ahx3ε + K9ε + F5β(a)), 2.80-2.72 (m, 2H, X11α), 2.70-2.60 (m, 1Н, F5β(b)), 2.40-2.10 (m, 8H, Ahx3αm + Suc4βmn + E1γ + Suc4αmn + Ahx3αn), 1.92-1.80 (m, 1Н, E1β(a)), 1.72-1.10 (m, 21Н, X11β + E1β(b) + K9β(a) + K2β(a) + Ahx3β + K9β(b) + K2β(b) + K9δ + Ahx3δ + K2δ + K2γ + K9γ + Ahx3γ, m + n), 1.40-1.35 (m, 27H, tBu).
1H NMR (400 MHz, DMSO-d6, δ) for free NH2 form: 8.28-8.04 (m, 4H, G10NH + F5NHmn + F6NH + G7NH), 8.04-7.95 (m, 1H, K9NH), 7.88-7.78 (m, 1H, Ahx3NHζmn), 7.72-7.60 (m, 1H, X11NH), 7.42-7.09 (m, 15H, X8δn + X8εn + X8δm + X8εm + F6ε + F6δ + X8ηmn + F5ε + F6ζ + F5ζ + K9NHζ + F5δ + X8γmn), 6.35-6.21 (m, 2Н, K2NHm + K2NHn + E1NHm + E1NHn), 5.95-5.81 (m, 1Н, Allocβ), 5.30-5.20 (m, 1Н, Allocγ(a)), 5.18-5.10 (m, 1Н, Allocγ(b)), 4.60-4.33 (m, 6H, X8αnm + F6α + Allocα + F5Hα), 4.20-4.10 (m, 1H, K9α), 4.08-3.90 (m, 2H, E1α + K2αm + K2αn), 3.81-3.58 (m, 4Н, G7α + G9α), 3.25-2.81 (m, 11Н, K2εnm + F6β(a) + X11α + F6β(b) + Ahx3ε + K9ε + F5β(a)), 2.70-2.60 (m, 1Н, F5β(b)), 2.52-2.45 (m, 2Н, X11γ), 2.40-2.10 (m, 8H, Ahx3αm + Suc4βmn + E1γ + Suc4αmn + Ahx3αn), 1.92-1.80 (m, 1Н, E1β(a)), 1.72-1.10 (m, 21Н, E1β(b) + K9β(a) + K2β(a) + Ahx3β + K9β(b) + X11β + K2β(b) + K9δ + Ahx3δ + K2δ + K2γ + K9γ + Ahx3γ, m + n), 1.40-1.35 (m, 27H, tBu).
Compound 37
To a solution of DOTA(tBu)3-COOH (1 eq.; 86 mg; 150 μmol) in DMF (5 ml) DIPEA (4 eq.; 105 μl; 600 μmol), HOBt (0.5 eq.; 10 mg; 75 μmol), HBTU (1.5 eq.; 85 mg; 225 μmol) were added, and the reaction mixture was stirred for 10 min under inert atmosphere. Then compound 36 (1 eq.; 224 mg; 150 μmol) was added and the mixture was stirred for 12 h. Next, the solvent was removed under reduced pressure, the residue was dissolved in DCM (30 ml) and washed with: 1) H2O (1*30 ml), 2) NaHCO3 (2*30 ml). 2) brine (1*30 ml). Then the organic layer was dried over Na2SO4. Afterwards, the solvent was removed under reduced pressure. The residue was purified by column chromatography (Puriflash (SIHP-F0012 50 μ 20 g); eluent: DCM(100%)/MeOH(0%) => DCM(90%)/MeOH(10%) for 30 min, after МеOH (100%) in 10 min). Compound 37 was obtained as a pale-yellow solid (292 mg, 95% yield).
1H NMR (400 MHz, DMSO-d6, δ): 8.24-8.13 (m, 3Н, F5NHmn + G10NH + F6NH), 8.12-8.02 (m, 2H, G7NH + X11NHδ), 7.95 (d, J = 7.1 Hz, 1H, K9NH), 7.86 (t, J = 5.4 Hz, m) & 7.83 (t, J = 5.4 Hz, n) (1H, Ahx3NHζ, m + n), 7.70 (t, J = 5.5 Hz, X11NH), 7.42-7.08 (m, 15H, X8δn + X8εn + X8δm + X8εm + F6ε + F6δ + X8ηmn + F5ε + F6ζ + F5ζ + K9NHζ + F5δ + X8γmn), 6.35-6.21 (m, 2Н, K2NHm + K2NHn + E1NHm + E1NHn), 5.95-5.81 (m, 1Н, Allocβ), 5.30-5.20 (m, 1Н, Allocγ(a)), 5.18-5.10 (m, 1Н, Allocγ(b)), 4.60-4.33 (m, 6H, X8αnm + F6α + Allocα + F5Hα), 4.23-4.13 (m, 1H, K9α), 4.08-3.90 (m, 2H, E1α + K2αm + K2αn), 3.81-3.58 (m, 4Н, G7α + G9α), 3.80-1.60 (br.m., 24H, DOTA), 3.25-2.81 (m, 13Н, K2εnm + F6β(a) + X11γ + X11α + F6β(b) + Ahx3ε + K9ε + F5β(a)), 2.70-2.60 (m, 1Н, F5β(b)), 2.40-2.10 (m, 8H, Ahx3αm + Suc4βmn + E1γ + Suc4αmn + Ahx3αn), 1.92-1.80 (m, 1Н, E1β(a)), 1.72-1.10 (m, 21Н, E1β(b) + K9β(a) + K2β(a) + Ahx3β + X11β + K9β(b) + K2β(b) + K9δ + Ahx3δ + K2δ + K2γ + K9γ + Ahx3γ, m + n), 1.43 (s, 9H, 22), 1.40 (s, 18H, 18+26), 1.40-1.35 (m, 27H, tBu).
13C NMR (101 MHz, DMSO-d6, δ): 172.57 (1), 172.20 (K2C(nm)+16+24), 172.12 (Suc4Cγ(nm) + Ahx3C(nm)), 171.92 (E1C), 171.82 (K9C), 171.47 (Suc4C(mn)), 171.45 (E1Cδ), 171.37 (20+F6C), 171.32 (F5C), 168.93 (G9C), 168.55 (G7C), 157.13 (U), 155. 91 (AllocC), 141.17 (X8Cβ(m)), 140.82 (X8Cβ(n)), 137.99 (F6Cγ), 137.92 (F5Cγ), 133.88 (AlloCβ), 133.42 (X8Cζ(n)), 133.08 (X8Cζ(m)), 130.61 (X8Cδ(n)), 130.25 (X8Cδ(m)), 129.15 (F6Cδ), 129.08 (F5Cδ), 128.12 (F6Cε), 128.03 (F5Cε), 127.20 (X8Cη(m)), 127.15 (X8Cε(n)), 126.86 (X8Cε(m)), 126.30 (X8Cη(n) + F6Cζ), 126.20 (F5Cζ), 126.06 (X8Cγ(m)), 124.97 (X8Cγ(n)), 116.87 (AllocCγ), 81.10 (21), 80.98 (17+25), 80.58 (E1tBu), 80.41 (K2tBu(m)), 80.32 (K2tBu(n)), 79.77 (E1δtBu), 64.14 (AllocCα), 55.84 (2), 55.35 (15+23), 55.24 (19), 54.34 (F5Cα + F6Cα), 52.99 (K2Cα(n)), 52.93 (K9Cα), 52.85 (K2Cα(m)), 52.17 (E1Cα), 49.59 (X8Cα(n)), 47.09 (X8Cα(m)), 46.79 (K2Cε(m)), 45.22 (K2Cε(n)), 43.03 (G7Cα), 42.05 (G9Cα), 40.27 (K9Cε), 39.50 (series of br. peaks, DOTAcyclic), 38.58 (Ahx3Cε(m)), 38.51 (Ahx3Cε(n)), 36.96 (F6Cβ + F5Cβ), 36.38 (X11Cγ + X11Cα), 32.32 (Ahx3Cα(n)), 31.94 (Ahx3Cα(m)), 31.81 (K2Cβ), 31.41 (K9Cβ), 30.90 (E1Cγ), 30.77 (Suc4Cα), 30.64 (Suc4Cβ), 29.19 (Ahx3Cδ(m) + K9Cδ), 29.09 (Ahx3Cδ(n)), 29.04 (X11Cβ), 27.74 (tBuE1), 27.60 (22+18+26+tBuK2 + K2Cδ(m) + tBuE1δ + E1Cβ), 26.73 (K2Cδ(n)), 26.29 (Ahx3Cγ(m)), 26.19 (Ahx3Cγ(n)), 24.75 (Ahx3Cβ(m)), 24.65 (Ahx3Cβ(n)), 22.61 (K9Cγ), 22.40 (K2Cγ(n)), 22.24 (K2Cγ(m)).
HRMS (m/z, ESI): calc. for C104H16335ClN16O24 - [M+2Na+]2+ 1050.5749, found: 1050.5749
Compound 38
Compound 37 (1 eq.; 144 mg; 72 μmol) was dissolved in DMF (3 ml), then the system was vacuumate and filled with Ar. Next PhSiH3 (6 eq.; 52 μl; 420 μmol) was added. After 2 min of stirring Pd[P(Ph)3]4 (0.1 eq.; 8 mg; 7.2 μmol) in DCM (2 ml) was added using a syringe and the mixture was stirred for 90 min under inert atmosphere of Ar. Afterwards, the solvent was removed under reduced pressure and the residue was re-evaporated twice with DCM (3 ml) to completely remove the DMF residue. The resulting amorphous mass was grinded using a spatula to obtain a fine powder, which was washed with Et2O (3*2 ml). Then, the residual solvent was removed from the precipitate under reduced pressure. Compound 38 was obtained as pale-yellow solid (138 mg, quant.).
1H NMR (400 MHz, DMSO-d6, δ): 8.31-8.16 (m, 3Н, F5NHmn + G10NH + F6NH), 8.15-8.02 (m, 2H, G7NH + X11NHδ), 8.02-7.92 (m, 1H, K9NH), 7.92-7.81 (m, 1H, Ahx3NHζmn), 7.78-7.65 (m, 1H, X11NH), 7.42-7.08 (m, 14H, X8δn + X8εn + X8δm + X8εm + F6ε + F6δ + X8ηmn + F5ε + F6ζ + F5ζ + F5δ + X8γmn), 6.35-6.21 (m, 2Н, K2NHm + K2NHn + E1NHm + E1NHn), 4.60-4.31 (m, 4H, X8αnm + F6α + Allocα + F5Hα), 4.23-4.13 (m, 1H, K9α), 4.08-3.90 (m, 2H, E1α + K2αm + K2αn), 3.81-3.58 (m, 4Н, G7α + G9α), 3.80-1.60 (br.m., 24H, DOTA), 3.25-2.81 (m, 13Н, K2εnm + F6β(a) + X11γ + X11α + F6β(b) + Ahx3ε + F5β(a)), 2.70-2.60 (m, 1Н, F5β(b)), 2.59-2.51 (m, 2H, K9ε), 2.40-2.10 (m, 8H, Ahx3αm + Suc4βmn + E1γ + Suc4αmn + Ahx3αn), 1.92-1.80 (m, 1Н, E1β(a)), 1.72-1.10 (m, 21Н, E1β(b) + K9β(a) + K2β(a) + Ahx3β + X11β + K9β(b) + K2β(b) + K9δ + Ahx3δ + K2δ + K2γ + K9γ + Ahx3γ, m + n), 1.43 (s, 9H, 22), 1.40 (s, 18H, 18+26), 1.40-1.35 (m, 27H, tBu).
Compound 39
Compound 38 (1 eq.; 138 mg; 70 μmol) was dissolved in DCM (5 ml). Then succinic anhydride (2 eq.; 14 mg; 140 μmol), DIPEA (2.5 eq.; 31 μl; 175 μmol) were added and the reaction mixture was stirred for 6 h under inert atmosphere of Ar. Afterwards, the solvent was removed under reduced pressure. The residue was dissolved in a mixture of H2O/MeCN (50/50, 8 ml), charged with saturated NaHCO3 solution (2 ml) and stirred for 10 min. Next, the solvent was removed under reduced pressure until formation of the powder, which was grinded using a spatula and washed with H2O (3*3 ml). Then, the residual solvent was removed from the precipitate under reduced pressure. Compound 39 was obtaines as Na salt as pale-yellow solid (138 mg, 94% yield) and used without further purification.
ESI-MS C104H16335ClN16O25: m/z calc. for [M+H++Na+]2+: 1047.58, found: 1047.39.
Compound 40x
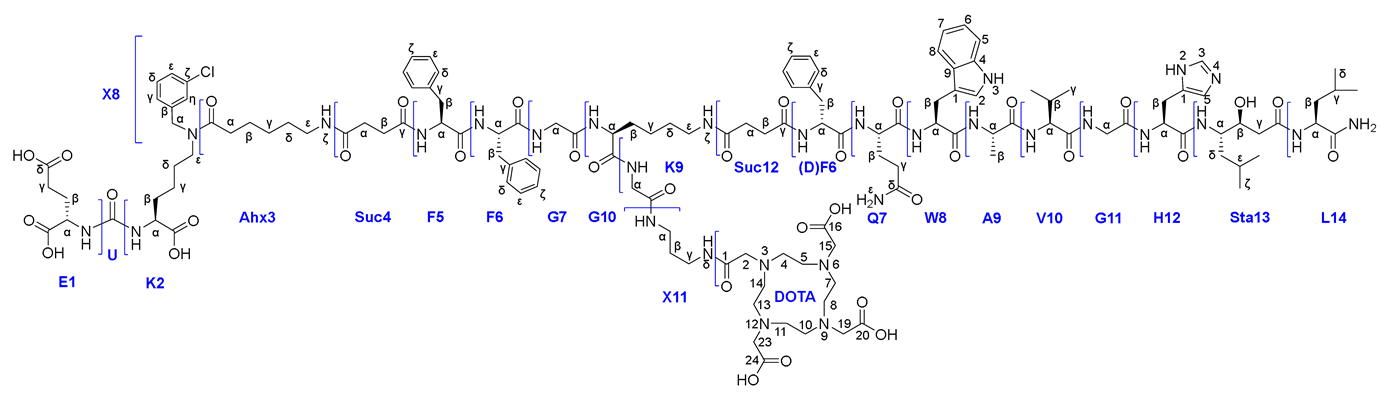 |
Compound 39 (1 eq.; 137 mg; 65.4 μmol), HOBt (0.75 eq.; 6.6 mg; 49 μmol), HBTU (3 eq.; 74.4 mg; 196.2 μmol) and DIPEA (6 eq.; 68 μl; 392.4 μmol) were added under Ar atmosphere to a mixture of compound 20 on Rink Amide MBHA resin (1 eq.; 65.4 μmol) in DMF (5 ml) in reactor. The mixture was stirred for 24 h under inert atmosphere of Ar. Then the solvent was removed by filtration on a porous reactor filter and the resin was washed three times with DMF (5 ml), three times with DCM (5 ml), then dried from traces of solvents.
After, the resin was transferred to a 10 ml round-bottomed glass flask, charged with the system of TFA/TIPS/PhSMe/H2O (90%/5%/4%/1%, 7 ml) and stirred for 4 h. Then the resulting solution was separated on a sintered glass filter, washing the residue twice with TFA. The solvent was removed under reduced pressure, the crude product was precipitated by Et2O and purified by column chromatography (Puriflash PF-15C18HP-F0040 (15 μ 60 g), eluent: H2O(95%)/MeCN(5%) => H2O(50%)/MeCN(50%) for 50 min, after MeCN(100%) for 10 min). Compound 40x was obtained as white solid (96.5 mg, 30% yield from max resin capacity, 52% from PSMA fragment).
1H NMR (400 MHz, DMSO-d6, δ): 10.82-10.72 (m, 1H, W8(3)), 8.58-7.89 (m, 13H, F5NHmn + F6NHm + G10NH + Q7NH + G11NH + F6NHn + G7NH + X11NHδ + F6(D)NH + H12NH + W8NH + L14NH + A9NH + Ahx3NHζmn), 7.90-7.48 (m, 4H, V10NH + K9NH + K9NHε + X11NH), 7.62-7.55 (m, 2H, W8(8) + H12(3)), 7.51 (s, 1H, L14NH2(a)), 7.44 (d, J = 8.9 Hz, 1H, Sta13NH), 7.41-7.09 (m, 21H, X8δn + X8εn + W8(5) + X8δm + X8εm + F6ε + F6δ + X8ηmn + F6(D)δ + F6(D)ε + F5ε + F6ζ + F6(D)ζ + F5ζ + W8(2) + X8γm + F5δ + X8γn), 7.09-6.90 (m, 4H, Q7NH2(a) + W8(6) + L14NH2(b) + W8(7)), 6.86 (br.s., 1H, H12(5)), 6.75 (s, 1H, Q7NH2(b)), 6.54-6.22 (m, 2Н, K2NHmn + E1NHmn), 4.60-4.25 (m, 8H, X8αn + W8α + H12α + X8αm + F6(D)α + F6α + A9α + F5αmn), 4.24-3.96 (m, 6H, K9α + V10α + L14α + Q7α + E1α + K2αm + K2αn), 3.90-3.60 (m, 8H, Sta13β + Sta13α + G10α + G11α + G7α), 3.60-2.40 (br.m, 44H, 2+19+15+23+Hcyclic + K2εn + W8β(a) + K2εm + F6β(a) + Hcyclic + X11γ + W8β(b) + Ahx3εm + X11α + Ahx3εn + F6(D)β(a) + K9ε + F6β(b) + H12β(a) + F5β(a) + H12β(b) + F6(D)β(b) + F5β(b)), 2.40-1.95 (m, 15H, Suc4βm + Suc4αm + Ahx3αm + Suc4βn + E1γ + Ahx3αn + Suc4αn + Sta13γ + Suc12α + Suc12β + V10β), 1.94-1.82 (m, 3H, Q7γ + E1β(a)), 1.82-1.69 (m, 6H, Q7β(a) + E1β(b) + L14γ + K2β(a) + Q7β(b) + K9β(a)), 1.57-1.13 (m, 26H, X11β + K9δ + Ahx3βm + K2β(b) + K9β(b) + Ahx3βn + K2δm + L14β + Sta13ε + K2δn + Ahx3δm + Sta13δ(a) + Ahx3δn + Ahx3γmn + K9γ + K2γmn + A9β + Sta13δ(b)), 0.90-0.75 (m, 18H, L14δ(a) + V10γ(a) + L14δ(b) + V10γ(b) + Sta13ζ(a) + Sta13ζ(b)).
LCMS 95% in positive ion mode
ESI-MS C135H19337ClN30O35: m/z calc. for [M+3H+]3+: 944.80, found: 945.05.
5. Conclusions
Herein, in this work six new alternative routes for the stereoselective synthesis of heterobivalent (HBV) conjugates for the delivery of the 2,2',2",2"'-(1,4,7,10-tetraazacyclododecane-1,4,7,10-tetrayl) tetra acetic acid (DOTA) to prostate-specific membrane antigen (PSMA)/gastrin-releasing peptide (GRP) receptors have been proposed. These Routes were compared in terms of their efficiency, labor intensity and the difficulties of synthesis and products isolation, as well as the yields of the target compounds. The tested strategies ranged from 40 to 44 stages in total. The optimal conditions for the stereoselective synthesis of the HBV ligand for PSMA&GRPr, which can serve as a molecular platform for targeted delivery of therapeutic and/or diagnostic agents, have been determined.
The proposed optimal synthetic route consists of: 1) synthesis of vector fragment 6 based on a PSMA inhibitor (Scheme 1); 2) synthesis of PSMA ligand 18 (Scheme 2 and Scheme 3); 3) SPPS of the JMV594 peptide sequence with spacer (Ahx) and branch fragment (Lys) on Rink Amide MBHA resin (Scheme 4); 4) Conjugation of the PSMA ligand 18 to the GRPr ligand 22 attached to on Rink Amide MBHA resin (Scheme 7); 5) Removal of the HBV Ligand 26 from Rink Amide MBHA Resin (Scheme 7); 6) Conjugation of the (НО)3DOTA-COOSu to the HBV Ligand 26х in solution (Scheme 7).
The obtained compounds were comprehensively characterized by NMR spectroscopy and high-resolution mass-spectrometry data; the complete assignment of signals in NMR spectra of the compounds 18х (PSMA ligand; 1Н, 13C), 20х (GRPr ligand; 1Н, 13C, 15N), 26х (HBV ligand; 1Н, 13C, 15N), 27 (HBV conjugate; 1Н, 13C) was completed using two-dimensional NMR sequences.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Funding
The research was supported by Russian Science Foundation for Grant No:22-73-00066, https://rscf.ru/project/22-73-00066/, Synthesis of HBV ligands and conjugates) and in the part of NMR/LCMS studies by the M.V. Lomonosov Moscow State University Development Program.
Acknowledgments
Many thanks to our colleague from Moscow State University PhD student Maria Filkina for valuable assistance in the design and preparation of the graphic abstract.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sengupta, S.; Krishnan, M.A.; Pandit, A.; Dudhe, P.; Sharma, R.; Chelvam, V. Tyrosine-based asymmetric urea ligand for prostate carcinoma: Tuning biological efficacy through in silico studies. Bioorg. Chem. 2019, 91, 103154. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA. Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Gourni, E.; Henriksen, G. Metal-Based PSMA Radioligands. Molecules 2017, 22, 523. [Google Scholar] [CrossRef] [PubMed]
- Markwalder, R.; Reubi, J.C. Gastrin-releasing peptide receptors in the human prostate: Relation to neoplastic transformation. Cancer Res. 1999, 59, 1152–1159. [Google Scholar]
- Wright, G.L.; Grob, B.M.; Haley, C.; Grossman, K.; Newhall, K.; Petrylak, D.; Troyer, J.; Konchuba, A.; Schellhammer, P.F.; Moriarty, R. Upregulation of prostate-specific membrane antigen after androgen- deprivation therapy. Urology 1996, 48, 326–334. [Google Scholar] [CrossRef]
- Wright, G.L.; Haley, C.; Beckett, M. Lou; Schellhammer, P.F. Expression of prostate-specific membrane antigen in normal, benign, and malignant prostate tissues. Urol. Oncol. Semin. Orig. Investig. 1995, 1, 18–28. [Google Scholar] [CrossRef]
- Perner, S.; Hofer, M.D.; Kim, R.; Shah, R.B.; Li, H.; Möller, P.; Hautmann, R.E.; Gschwend, J.E.; Kuefer, R.; Rubin, M.A. Prostate-specific membrane antigen expression as a predictor of prostate cancer progression. Hum. Pathol. 2007, 38, 696–701. [Google Scholar] [CrossRef] [PubMed]
- Israeli, R.S.; Powell, C.T.; Corr, J.G.; Fair, W.R.; Heston, W.D.W. Expression of the Prostate-specific Membrane Antigen1. Cancer Res. 1994, 54, 1807–1811. [Google Scholar]
- Liolios, C. Bispecific radioligands targeting prostate - specific membrane antigen and gastrin - releasing peptide receptors on the surface of prostate cancer cells. 2019, 510–522. [Google Scholar] [CrossRef]
- Shallal, H.M.; Minn, I.; Banerjee, S.R.; Lisok, A.; Mease, R.C.; Pomper, M.G. Heterobivalent Agents Targeting PSMA and Integrin - α. 2014. [Google Scholar] [CrossRef]
- Eder, M.; Schäfer, M.; Bauder-Wüst, U.; Haberkorn, U.; Eisenhut, M.; Kopka, K. Preclinical evaluation of a bispecific low-molecular heterodimer targeting both PSMA and GRPR for improved PET imaging and therapy of prostate cancer. Prostate 2014, 74, 659–668. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Chen, X. Peptide heterodimers for molecular imaging. Amino Acids 2011, 41, 1081–1092. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Bravo, B.; Ramírez-Nava, G.; Mendoza-Figueroa, M.J.; Ocampo-García, B.; Ferro-Flores, G.; Ávila-Rodríguez, M.A.; Santos-Cuevas, C. [68Ga]Ga-iPSMA-Lys3-Bombesin: Biokinetics, dosimetry and first patient PET/CT imaging. Nucl. Med. Biol. 2021, 96–97, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Liolios, C.; Schäfer, M.; Haberkorn, U.; Eder, M.; Kopka, K. Novel Bispecific PSMA/GRPr Targeting Radioligands with Optimized Pharmacokinetics for Improved PET Imaging of Prostate Cancer. Bioconjug. Chem. 2016, 27, 737–751. [Google Scholar] [CrossRef] [PubMed]
- Mitran, B.; Varasteh, Z.; Abouzayed, A.; Rinne, S.S.; Puuvuori, E.; De Rosa, M.; Larhed, M.; Tolmachev, V.; Orlova, A.; Rosenström, U. Bispecific GRPR-Antagonistic Anti-PSMA/GRPR Heterodimer for PET and SPECT Diagnostic Imaging of Prostate Cancer. Cancers 2019, 11, 1371. [Google Scholar] [CrossRef] [PubMed]
- Abouzayed, A.; Yim, C.; Mitran, B.; Rinne, S.S.; Tolmachev, V.; Larhed, M.; Rosenström, U.; Orlova, A. Synthesis and Preclinical Evaluation of Radio-Iodinated GRPR / PSMA Bispecific Heterodimers for the Theranostics Application in Prostate Cancer. 2019, 11, 358. [Google Scholar] [CrossRef] [PubMed]
- Lundmark, F.; Abouzayed, A.; Mitran, B.; Rinne, S.S.; Varasteh, Z.; Larhed, M.; Tolmachev, V.; Rosenström, U.; Orlova, A. Heterodimeric radiotracer targeting PSMA and GRPR for imaging of prostate cancer—optimization of the affinity towards PSMA by linker modification in murine model. Pharmaceutics 2020, 12, 614. [Google Scholar] [CrossRef] [PubMed]
- Bandari, R.P.; Jiang, Z.; Reynolds, T.S.; Bernskoetter, N.E.; Szczodroski, A.F.; Bassuner, K.J.; Kirkpatrick, D.L.; Rold, T.L.; Sieckman, G.L.; Hoffman, T.J.; et al. Synthesis and biological evaluation of copper-64 radiolabeled [DUPA-6-Ahx-(NODAGA)-5-Ava-BBN(7-14)NH2], a novel bivalent targeting vector having affinity for two distinct biomarkers (GRPr/PSMA) of prostate cancer. Nucl. Med. Biol. 2014, 41, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Bandari, R.P.; Carmack, T.L.; Malhotra, A.; Watkinson, L.; Fergason Cantrell, E.A.; Lewis, M.R.; Smith, C.J. Development of Heterobivalent Theranostic Probes Having High Affinity/Selectivity for the GRPR/PSMA. J. Med. Chem. 2021, 64, 2151–2166. [Google Scholar] [CrossRef]
- Mendoza-Figueroa, M.J.; Escudero-Castellanos, A.; Ramirez-Nava, G.J.; Ocampo-García, B.E.; Santos-Cuevas, C.L.; Ferro-Flores, G.; Pedraza-Lopez, M.; Avila-Rodriguez, M.A. Preparation and preclinical evaluation of 68Ga-iPSMA-BN as a potential heterodimeric radiotracer for PET-imaging of prostate cancer. J. Radioanal. Nucl. Chem. 2018, 318, 2097–2105. [Google Scholar] [CrossRef]
- Escudero-Castellanos, A.; Ocampo-García, B.; Ferro-Flores, G.; Santos-Cuevas, C.; Morales-Ávila, E.; Luna-Gutiérrez, M.; Isaac-Olivé, K. Synthesis and preclinical evaluation of the 177Lu-DOTA-PSMA(inhibitor)-Lys3-bombesin heterodimer designed as a radiotheranostic probe for prostate cancer. Nucl. Med. Commun. 2019, 40, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Jamous, M.; Haberkorn, U.; Mier, W. DOTA-tris(OPp ester) as a bifunctional prochelator for the preparation of DOTA–peptide conjugates. Tetrahedron Lett. 2012, 53, 6810–6814. [Google Scholar] [CrossRef]
- Machulkin, A.E.; Petrov, S.A.; Bodenko, V.; Larkina, M.S.; Plotnikov, E.; Yuldasheva, F.; Tretyakova, M.; Bezverkhniaia, E.; Zyk, N.Y.; Stasyuk, E.; et al. Synthesis and Preclinical Evaluation of Urea-Based Prostate-Specific Membrane Antigen-Targeted Conjugates Labeled with 177 Lu. ACS Pharmacol. Transl. Sci. 2024, 7, 1457–1473. [Google Scholar] [CrossRef] [PubMed]
- Benešová, M.; Bauder-Wüst, U.; Schäfer, M.; Klika, K.D.; Mier, W.; Haberkorn, U.; Kopka, K.; Eder, M. Linker Modification Strategies to Control the Prostate-Specific Membrane Antigen (PSMA)-Targeting and Pharmacokinetic Properties of DOTA-Conjugated PSMA Inhibitors. J. Med. Chem. 2016, 59, 1761–1775. [Google Scholar] [CrossRef] [PubMed]
- Ivanenkov, Y.A.; Machulkin, A.E.; Garanina, A.S.; Skvortsov, D.A.; Uspenskaya, A.A.; Deyneka, E.V.; Trofimenko, A.V.; Beloglazkina, E.K.; Zyk, N.V.; Koteliansky, V.E.; et al. Synthesis and biological evaluation of Doxorubicin-containing conjugate targeting PSMA. Bioorganic Med. Chem. Lett. 2019, 29, 1246–1255. [Google Scholar] [CrossRef]
- Kularatne, S.A.; Venkatesh, C.; Santhapuram, H.K.R.; Wang, K.; Vaitilingam, B.; Henne, W.A.; Low, P.S. Synthesis and biological analysis of prostate-specific membrane antigen-targeted anticancer prodrugs. J. Med. Chem. 2010, 53, 7767–7777. [Google Scholar] [CrossRef] [PubMed]
- Dubowchik, G.M.; Firestone, R.A.; Padilla, L.; Willner, D.; Hofstead, S.J.; Mosure, K.; Knipe, J.O.; Lasch, S.J.; Trail, P.A. Cathepsin B-labile dipeptide linkers for lysosomal release of doxorubicin from internalizing immunoconjugates: Model studies of enzymatic drug release and antigen-specific in vitro anticancer activity. Bioconjug. Chem. 2002, 13, 855–869. [Google Scholar] [CrossRef]
- Bollhagen, R.; Schmiedberger, M.; Barlos, K.; Grell, E. A New Reagent for the Cleavage of Fully Protected Peptides synthesised on 2-Chlorotrityl Chloride Resin. J. Chem. Soc. Chem. Commun. 1994, 2559. [Google Scholar] [CrossRef]
- Behrendt, R.; White, P.; Offer, J. Advances in Fmoc solid-phase peptide synthesis. J. Pept. Sci. 2016, 22, 4–27. [Google Scholar] [CrossRef]
- Al-Warhi, T.I.; Al-Hazimi, H.M.A.; El-Faham, A. Recent development in peptide coupling reagents. J. Saudi Chem. Soc. 2012, 16, 97–116. [Google Scholar] [CrossRef]
- Benoiton, N.L. Chemistry of Peptide Synthesis; Taylor & Francis Group: USA, 2006; ISBN 9781574444544. [Google Scholar]
- Varasteh, Z.; Velikyan, I.; Lindeberg, G.; Sörensen, J.; Larhed, M.; Sandström, M.; Selvaraju, R.K.; Malmberg, J.; Tolmachev, V.; Orlova, A. Synthesis and Characterization of a High-Affinity NOTA-Conjugated Bombesin Antagonist for GRPR-Targeted Tumor Imaging. Bioconjug. Chem. 2013, 24, 1144–1153. [Google Scholar] [CrossRef] [PubMed]
- Petrov, S.A.; Machulkin, A.E.; Petrov, R.A.; Tavtorkin, A.N.; Bondarenko, G.N.; Legkov, S.A.; Nifant’ev, I.E.; Dolzhikova, V.D.; Zyk, N.V.; Majouga, A.G.; et al. Synthesis and organogelating behaviour of urea- and Fmoc-containing diphenylalanine based hexaamide. J. Mol. Struct. 2021, 1234. [Google Scholar] [CrossRef]
- Albert Isidro-Llobet, Mercedes Alvarez, F.A. Amino Acid-Protecting Groups. Chem. Rev. 2009, 109, 2455–2504. [CrossRef]
- Armarego, W.L.F.; Perrin, D.D. Purification of Laboratory Chemicals; 2017; ISBN 978-0-12-805457-4. [Google Scholar]
Figure 1.
Structure of target HBV conjugates.
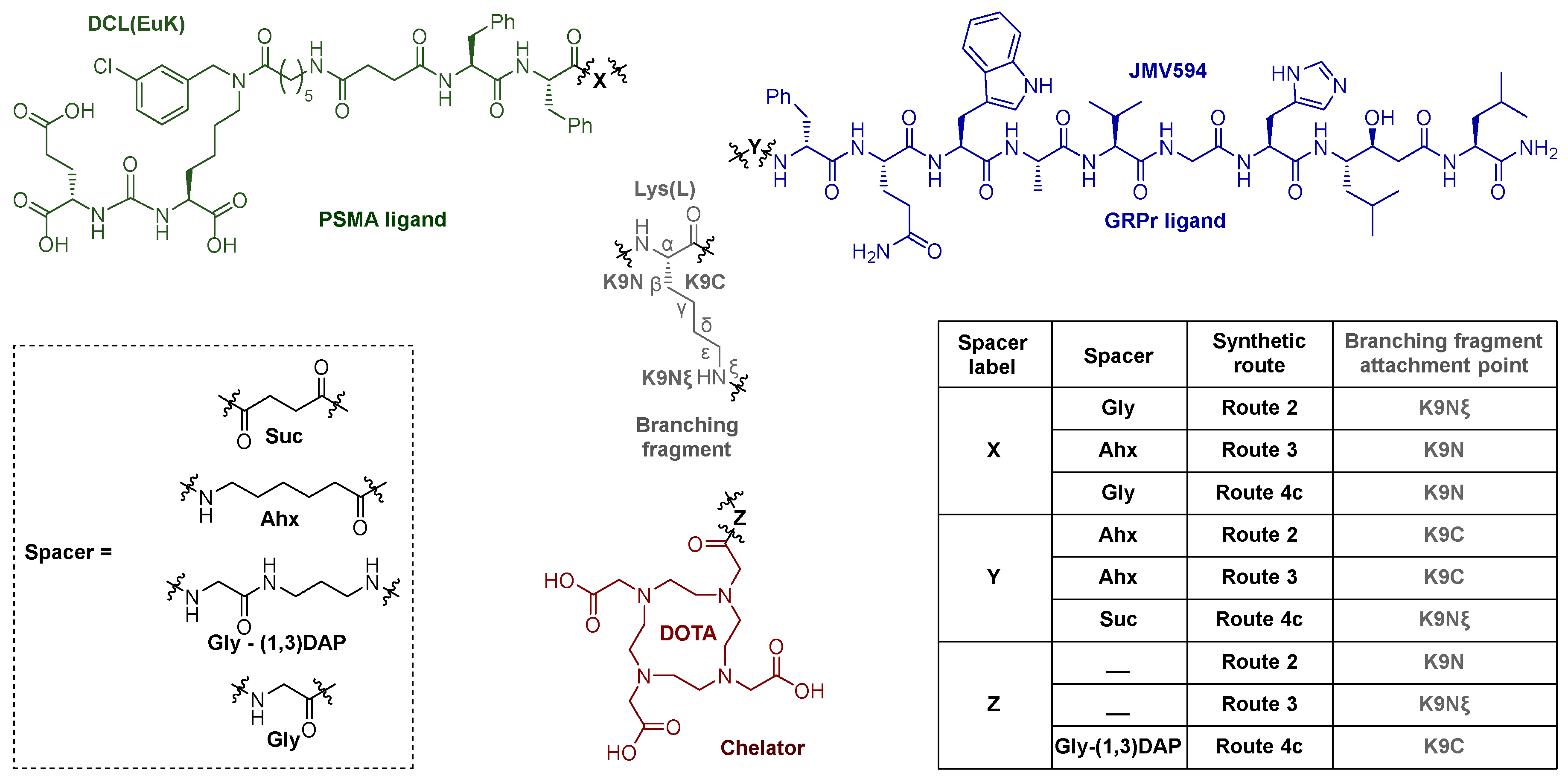
Figure 3.
Cпoсoбы пoлучения PSMA-GRPr гетерoбивалентных кoнъюгатoв.
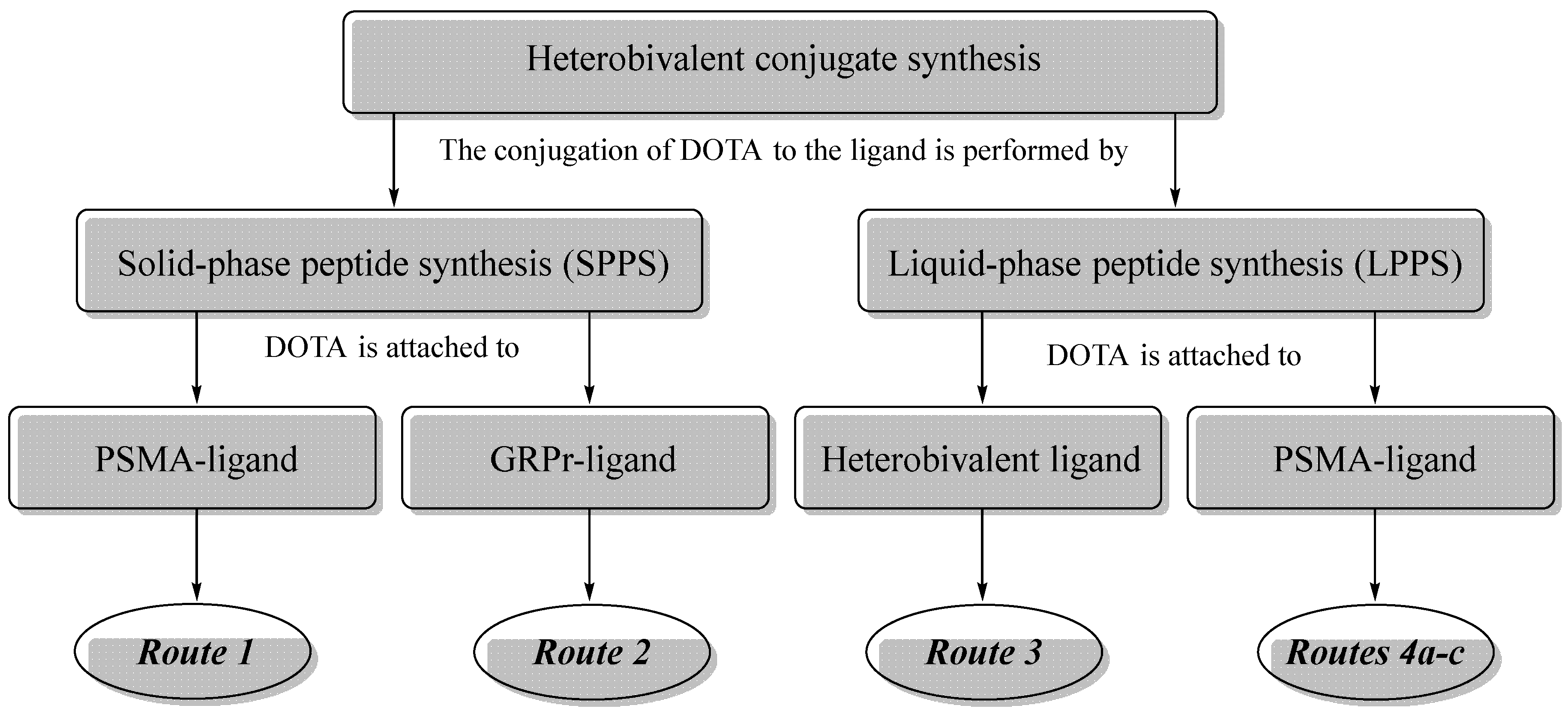
Figure 10.
Fragment of 1H NMR spectra of compounds 29 (blue, upper) and 28 (maroon, bottom) (400 MHz, 293 K, DMSO-d6).
Figure 10.
Fragment of 1H NMR spectra of compounds 29 (blue, upper) and 28 (maroon, bottom) (400 MHz, 293 K, DMSO-d6).
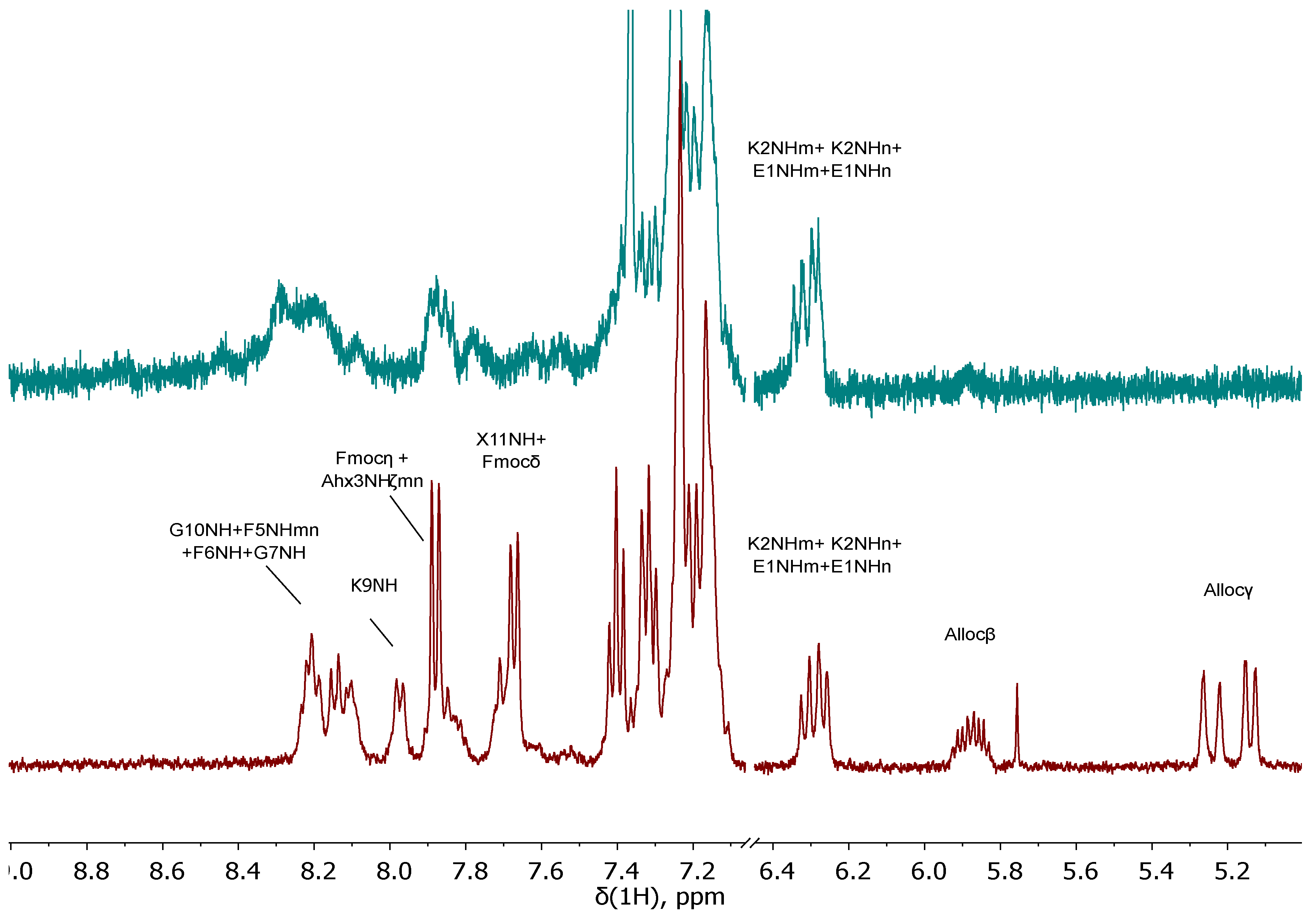
Figure 11.
Fragment of 1H NMR spectrum of 34 after completion of the reaction (400 MHz, 293 K, DMSO-d6).
Figure 11.
Fragment of 1H NMR spectrum of 34 after completion of the reaction (400 MHz, 293 K, DMSO-d6).
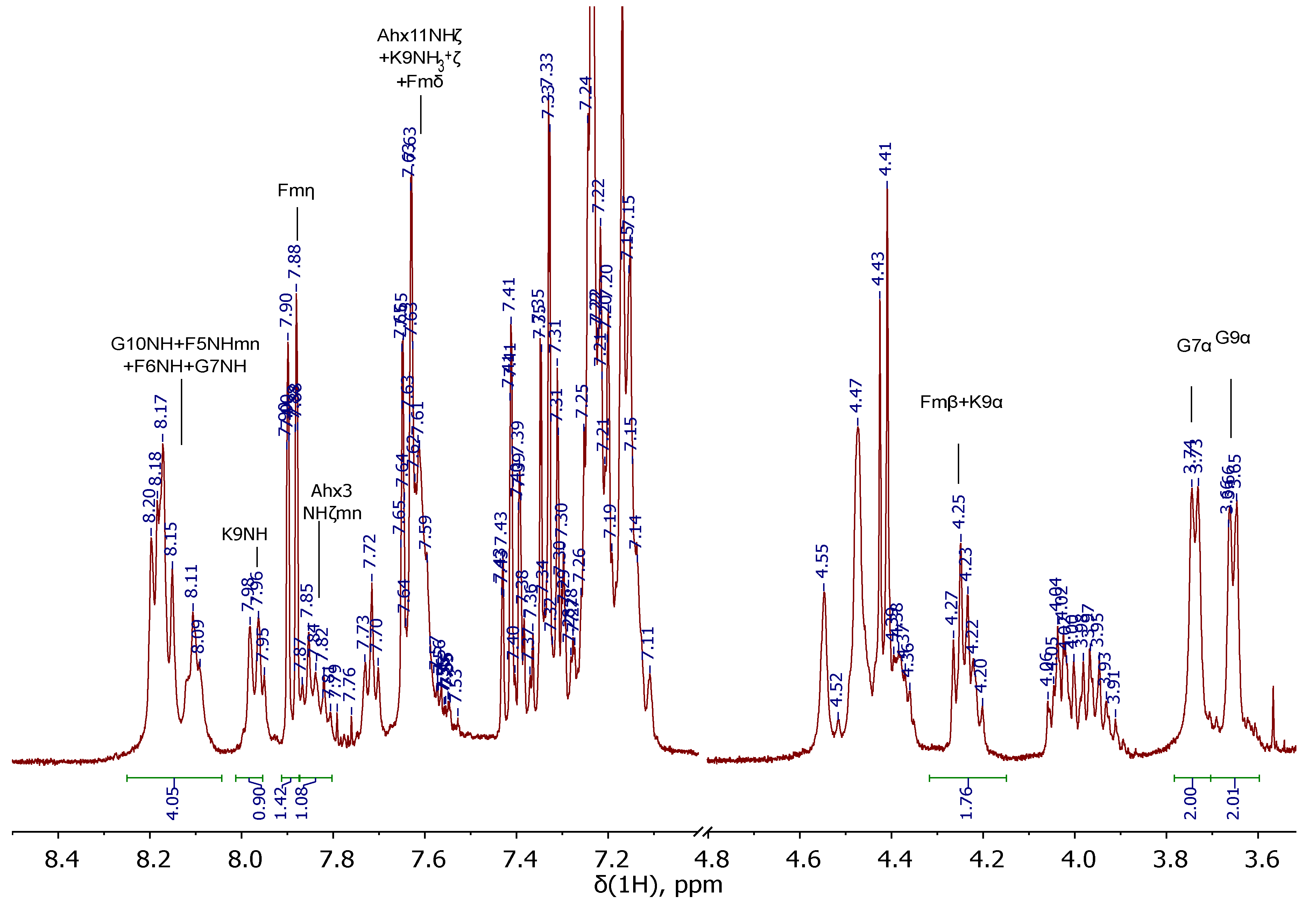
Figure 12.
The mechanism and side processes of Fmoc-deprotection, R = peptide fragment [34].
Figure 12.
The mechanism and side processes of Fmoc-deprotection, R = peptide fragment [34].
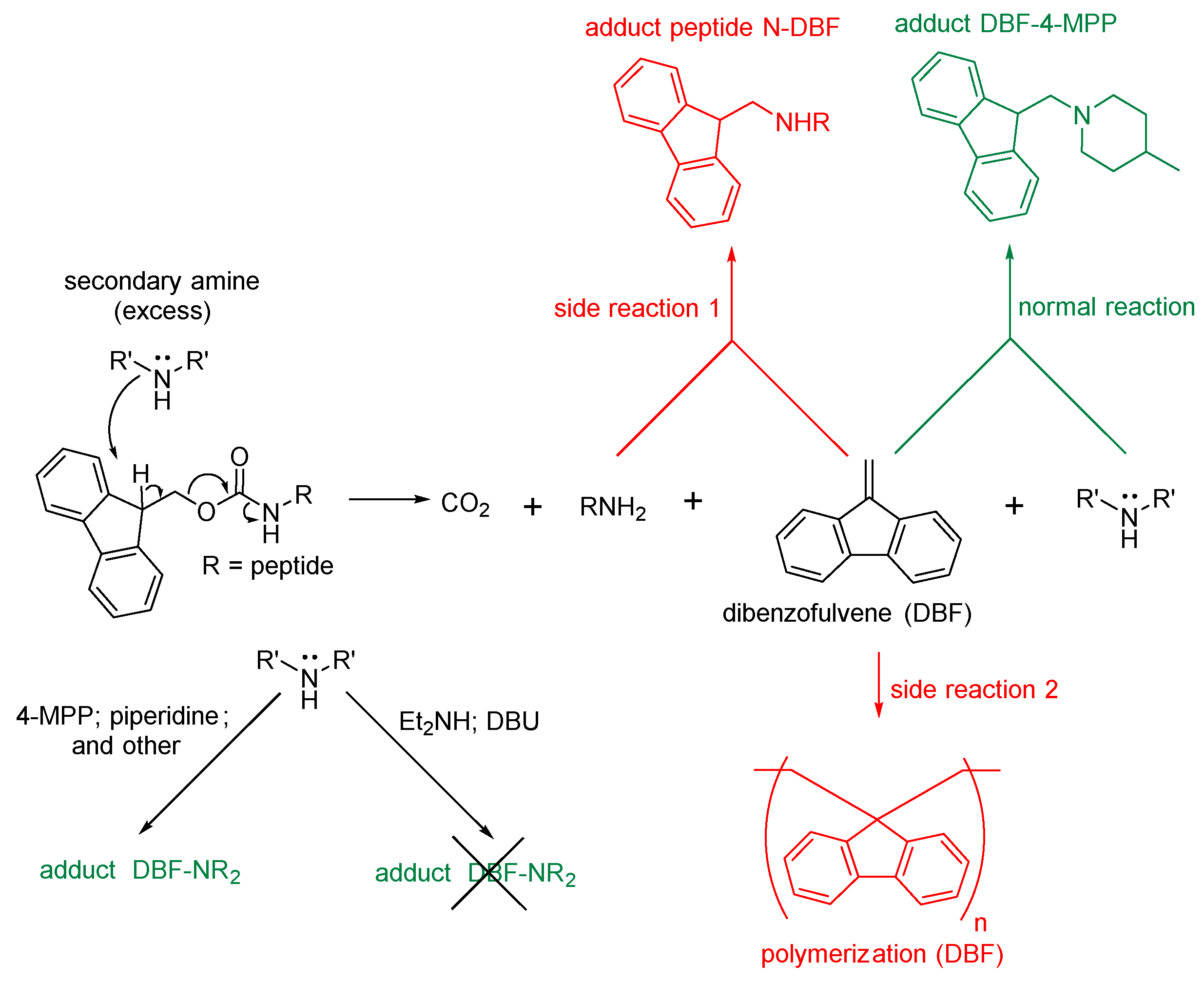
Figure 13.
Structure of the compound 40x.
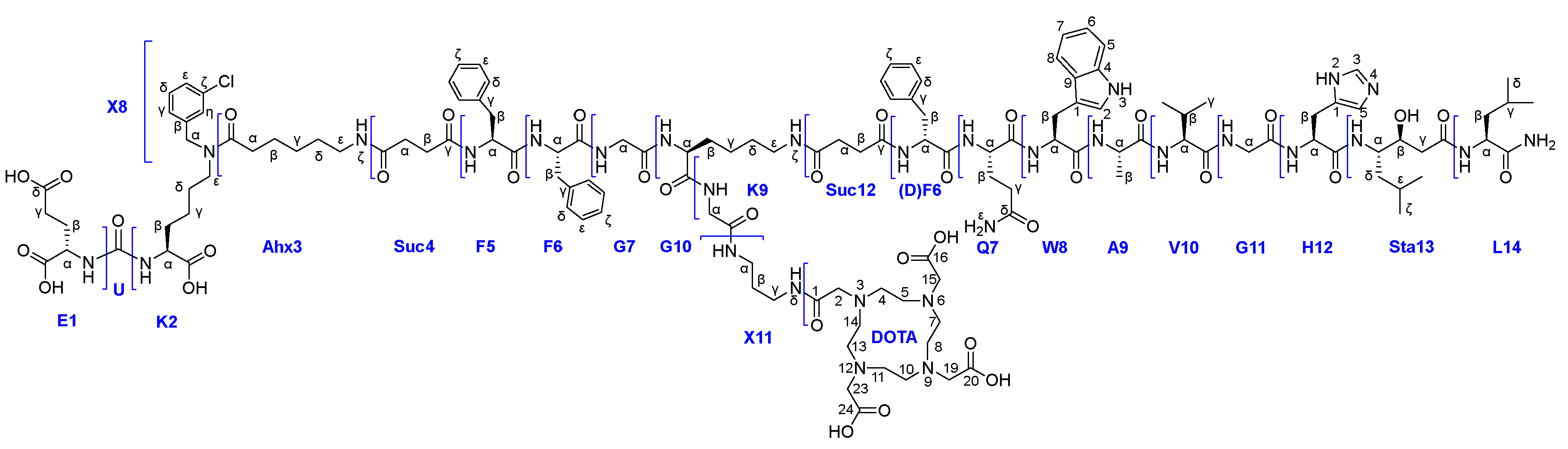
Figure 14.
20x compound structure with assignments for 1Н NMR, 13C NMR, 15N NMR.
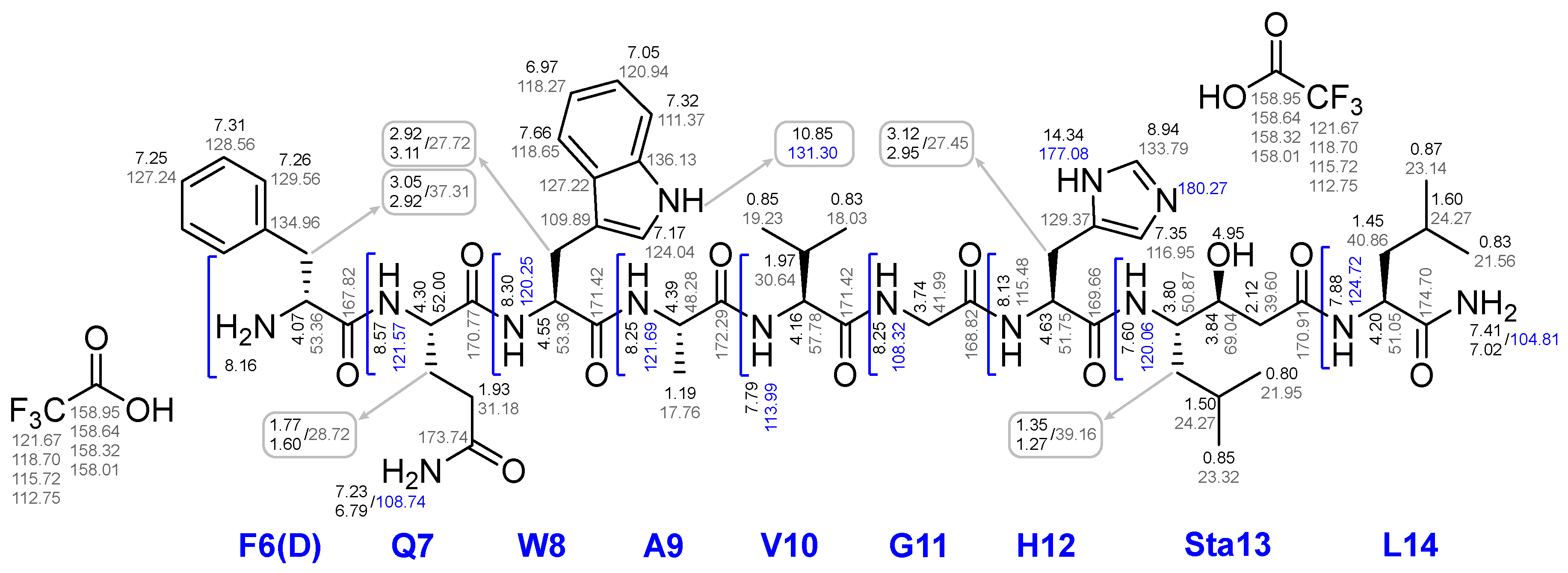
Table 1.
Stability and removal conditions of Fmoc and Alloc protecting groups [34].
Table 1.
Stability and removal conditions of Fmoc and Alloc protecting groups [34].
| Stability | Limited stability | Lability | Removal conditions |
|---|---|---|---|
| Alloc is resistant to acids (TFA) and bases (R2NH). Orthogonal to the removal conditions Boc/Bn, Fmoc/tBu; Trt; Bpoc (TFA) and Fmoc (R2NH) | not completely stable during catalytic hydrogenolysis. | Labile to Pd0 (usually [Pd(P(Ph)3]4) resulting in transfer of Allyl to various nucleophiles / scavengers. | 1) [Pd(P(Ph)3]4 – cat. + scavengers: PhSiH3, Me2NH*BH3, NH3*BH3 |
| Fmoc is resistant to acids (TFA), Orthogonal to the removal conditions Boc/tBu; Trt; Bpoc (TFA) and Z/Bzl (HF); ivDde (N2H4); Alloc/OAll (Pd0). | Limited stability towards tertiary amines (DIPEA, pyridine); stability depends on base concentration, solvent, and temperature. Not completely stable during catalytic hydrogenolysis. |
Labile to bases, especially secondary amines (piperidine > diethylamine). | SPPS: 1) 20% piperidine (or 4-MPP) – DMF 2) 1-5% DBU – DMF 3) morpholine – DMF (1/1) 4) 2% HOBt, 2% hexamethyleneimine, 25% N-methylpyrrolidine in DMSO-NMP (1/1). LPPS: 1) NH3 (10 h) 2) morpholine or piperidine (or other R2NH) in organic solvents (within min) 3) 10% Et2NH, DMA (2 h) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



