Submitted:
11 May 2024
Posted:
13 May 2024
You are already at the latest version
Abstract
Congenital heart disease (CHD) is the most commonly detected congenital anomaly and affects up to 1 % of all live-born neonates. Current guidelines support the use of Chromosomal Microarray Analysis (CMA) and Next Generation Sequencing (NGS) as diagnostic approaches to identify genetic causes. The aim of our study was to evaluate the diagnostic yield of CMA and NGS in a cohort of neonates with both isolated and syndromic CHD. The present study included 188 infants under 28 days of age with abnormal echocardiography findings hospitalized at the Department of Neonatology, UMC Ljubljana, between January 2014 and December 2023. Phenotypic data were obtained for each infant by retrospective medical chart review. We established the genetic diagnosis of 22 distinct syndromes in 17% of neonates. The most common genetic diagnoses were 22q11.2 microdeletion (22.7%) and CHARGE syndromes (22.7%), Noonan syndrome (9.1%), and Williams syndrome (9.1%). In addition, we detected variants of uncertain significance in 4.8% of neonates. Timely genetic diagnosis is important for the detection of syndrome-related comorbidities, prognosis, reproductive genetic risks and, when appropriate, genetic testing of other family members.
Keywords:
congenital heart disease
; chromosomal microarray analysis
; next-generation sequencing
1. Introduction
Congenital heart defects (CHD) are structural anomalies of the heart and great vessels resulting from errors in early embryogenesis. They are the most commonly detected congenital anomaly affecting 0.8–1% of all live-born infants [1] and present a major cause of morbidity and mortality in infancy [2].
The etiology of CHD is deemed to be multifactorial, an interplay of genetic and environmental factors. CHDs can be caused by environmental exposure to teratogens such as drugs, viral infections, and maternal conditions such as obesity and diabetes [3]. Genetic influences are supported by observed relatively high recurrence risk in families and well-established association of CHD with chromosomal abnormalities [4]; however, the cause of the disease remains unexplained in up to 60% of CHD patients [5]. Genetic cause elucidation is also challenging because of CHD's genetic and phenotypic heterogeneity [6]. With current diagnostic methods mainly comprising cytogenetic techniques (karyotyping and copy number variant platforms) and next-generation sequencing (NGS), we can reach a genetic diagnosis in 18–36% of CHD patients [7]. The overall yield of genetic testing depends on the type of congenital heart malformation, the presence of extracardiac congenital anomalies, and the genetic test modality [7,8].
The value of genetic testing in the setting of CHD lies in the defining etiology and consequently ending diagnostic odyssey for patients and families, possible detection of additional health problems associated with genetic diagnosis, prognostic information for clinical outcomes, genetic reproductive risks for the family, and genetic testing of additional family members when appropriate [9,10].
The aim of this study was to assess the diagnostic yield of genetic testing in the clinical evaluation of neonates with diagnosed CHD who were hospitalized at the Department of Neonatology, Division of Paediatrics, University Medical Centre (UMC) Ljubljana.
2. Materials and Methods
2.1. Patient Selection
In the present study, we enrolled neonates under 28 days with abnormal findings on echocardiography who were hospitalized in the Department of Neonatology, Division of Paediatrics, UMC Ljubljana between January 2014 and December 2023. Phenotypic data was obtained through retrospective chart review for each neonate. Neonates with isolated hemodynamically insignificant patent ductus arteriosus (PDA), isolated hemodynamically insignificant patent foramen ovale (PFO), cardiomyopathies, vasculopathies, and neonates exposed to known environmental teratogenic factors were excluded. We also excluded neonates with prenatally or perinatally detected common trisomies, namely trisomy 13, 18, and 21. Neonates were assigned into one of two groups based on the presence of isolated CHD or additional extracardiac anomalies. Neonates with isolated CHD were subdivided into three groups based on the complexity of congenital heart malformation according to Botto classification (simple, association, and complex). Chromosomal microarray analysis (CMA) was performed as a first-tier genetic test in all neonates with CHD, except in 11 neonatal patients in whom high possibility of monogenic disorder based on the phenotype was detected, and next-generation sequencing (NGS) was performed as a first-tier genetic test instead. In most neonatal CHD patients, however, NGS was performed in neonates with normal CMA results and clinical suspicion of monogenic disease. Informed consent was obtained from the parents of the neonates in accordance with guidelines established by the institutional review boards at their primary site of care.
2.2. Genetic and Bioinformatic Analysis
2.2.1. CMA and Classification of Results
According to the manufacturer's protocol, the DNA was isolated from peripheral blood samples using the Qiagen Mini kit (Qiagen, Valencia, CA). The quality and concentration parameters of the DNA were measured on a NanoDrop 2000c spectrophotometer (Thermo Fisher Scientific Inc.) and a Qubit 2.0 fluorometer (Life Technologies Inc.). Following the sample extraction, the DNA was processed according to the Agilent protocol (Version 8.0 December 2019) using commercially available male and female genomic DNA (Agilent Technologies, Human Reference DNA, Male and Female) as a reference DNA. The Agilent SurePrint G3 Unrestricted CGH 4x180K microarrays were used, which provide a practical average resolution of 50 kb. The array images were acquired using the Agilent laser scanner G2565CA. The image files were quantified using the Agilent Feature extraction software for Cytogenomics 5.3 and analyzed with the Agilent Cytogenomics 5.3 software (Agilent Technologies). Called copy number variants (CNV) were aligned with known aberrations in publicly available databases—ClinGen (http://dbsearch.clinicalgenome.org/search/), DECIPHER (Database of Chromosomal Imbalance and Phenotype in Humans using Ensembl Resources https://decipher.sanger ac.uk/), Database of Genomic Variants (DGV) (http://dgv.tcag.ca/dgv/app/home), as well as with the in-house database of detected variants and their clinical significance, ascertained by the trained analysts. All called CNVs were classified according to ACMG Standards and Guidelines [11].
2.2.2. Next-Generation Sequencing and Variant Interpretation
NGS was performed on the isolated DNA samples at the Clinical Institute for Special Laboratory Diagnostics, University Children's Hospital, UMC Ljubljana, and/or the Clinical Institute of Genomic Medicine, UMC Ljubljana.
We prepared NGS libraries according to standard Illumina protocols (Illumina DNA Prep with Enrichment). Sequencing was performed using Illumina instruments (NovaSeq 6000, NextSeq 550, or MiSeq). We processed, mapped, and aligned raw-read data and identified variants using standard bioinformatics pipelines used at the Clinical Institute for Special Laboratory Diagnostics, University Children's Hospital, UMC Ljubljana, or at the Center for Mendelian Genomics, CIGM, UMC Ljubljana. The interpretation of sequence variants was based on the American College of Medical Genetics and Genomics/Association for Molecular Pathology (ACMG/AMP) Standards and Guidelines [12].
3. Results
The present study included 188 neonates diagnosed with CHD who underwent genetic testing when hospitalized at the Department of Neonatology, Division of Paediatrics, UMC Ljubljana. The cohort comprised 36% of neonates with isolated CHD and 64% of neonates with CHD and additional extracardiac congenital anomalies. Neonates clinically diagnosed with isolated CHD were assigned to one of the three groups according to Botto classification; 15% of neonates were diagnosed with simple isolated CHD, 10% with an association, and 11% with complex CHD (Table 1). CMA was performed as a first-tier test in 94% of neonates; in 6% of neonates, NGS was performed instead due to the high phenotype specificity of a monogenic genetic cause. In two cases of abnormal CMA results, karyotype and FISH were employed to delineate chromosomal aberrations further (Figure 1).
We established the genetic diagnosis of 22 distinct genetic syndromes in 17% of neonates. Genetic cause of CHD was identified in 24.8% of neonates with CHD and additional extra-cardiac anomaly and 3% of neonates with isolated CHD. For neonates with isolated CHD, the diagnosis was made in 5% of complex isolated CHD and 5.6% of associations, and none of the patients with simple isolated CHD. The diagnosis was reached by CMA in 11% of neonates. The most common microdeletion syndromes were 22q11.2 microdeletion syndrome (22.7%) and Williams syndrome (9.1%) (Table 2). By using NGS either sequentially to CMA or as a first-tier genetic test, the diagnosis was reached in 7.4% of neonates with CHD. The most common monogenic conditions identified were CHARGE syndrome (22.7%) and Noonan syndrome (9.1%) (Table 2).
In one patient, we have established a dual genetic diagnosis of 17q12 microduplication syndrome and Weaver syndrome. The phenotype present has characteristics of both conditions. Phenotypes and genetic diagnosis are described in detail in Table 2. Additionally, we detected variants of uncertain significance in 4.8% of neonates with CHD (Table 3).
4. Discussion
In a cohort of 188 neonates with CHD, we identified a genetic cause in 17% of patients. As all phenotypic features of genetic diseases are not always fully present at birth, but may only become apparent during the course of the child's development (e.g. global developmental delay), neonatal CHD patients present a diagnostic challenge that differs from paediatric or adult patients. The general recommendation for clinical genetic testing in CHD includes CMA as a first-tier and exome sequencing as a second-tier genetic test [13,14]. In this paper, we report on a clinical experience of using the recommended protocol in a cohort of neonates in the Slovenian National Tertiary Centre.
The diagnostic yield of CMA in our experience was 11%. The most frequently detected copy number variants associated with syndromic CHDs were those present in 22q11.2 microdeletion syndrome and Williams syndromes. Reported diagnostic yields in other studies varied considerably among different subgroups of CHDs [13,15,16].
The most frequently detected monogenic causes of syndromic CHD were Noonan and CHARGE syndromes. Exome and whole genome sequencing are increasingly used in research, but also in the clinical setting. However, diagnostic rates still differ across the studies and tested phenotypes [17,18,19]. In addition, rapid WGS demonstrated the yield in 27% of individuals with CHD, leading to changes in clinical management in 62% of patients with diagnostic results [20].
The incremental yield of whole genome sequencing over QF-PCR and CMA was estimated to be 26% for the cohort of congenital anomalies patients, with no significant yield increase over exome sequencing [21].
Interestingly, a dual genetic diagnosis was established in one patient in our cohort with a combination of clinical signs of both 17q12 microduplication syndrome and Weaver syndrome, highlighting the complexity of making a genetic diagnosis.
We found a genetic diagnosis in 3% of neonates with isolated CHD. It was estimated that 13.4% of infants with isolated CHD with identifiable genetic causes would have been missed if genetic testing had not been offered [22]. Although the yield of genetic testing in newborns with isolated CHD is relatively low, it is still important to offer them genetic testing because of the clinical benefit of molecular diagnosis. Timely diagnosis provides information about the prognosis and enables us to organise better surveillance for patients. It also provides us with information about the risk of recurrence in future pregnancies of the parents.
5. Conclusions
In a cohort of 188 neonates with CHD, we were able to identify a genetic cause in 17% of patients using CMA and NGS. Timely genetic diagnosis is important for the detection of syndrome-related comorbidities, prognosis, reproductive genetic risks and, when appropriate, genetic testing of other family members.
Author Contributions
Conceptualization G.N. and A.P.; methodology, L.L, A.M. and M. D., formal analysis, L.L, A.M., K.W. and M. D.; data curation, A.P and G.N.; writing—original draft preparation, A.P.; writing—review and editing, A.P., K.W., S.B, G.N. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Medical Ethics Committee of the Republic of Slovenia (protocol code: 0120-234/2019/9, date of approval: 19.09.2019).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The data presented in this study are available on request from the corresponding author due to the reasons regarding privacy of the patients.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Lalani, S.R. Other Genomic Disorders and Congenital Heart Disease. American J of Med Genetics Pt C 2020, 184, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Diab, N.S.; Barish, S.; Dong, W.; Zhao, S.; Allington, G.; Yu, X.; Kahle, K.T.; Brueckner, M.; Jin, S.C. Molecular Genetics and Complex Inheritance of Congenital Heart Disease. Genes 2021, 12, 1020. [Google Scholar] [CrossRef] [PubMed]
- Suluba, E.; Shuwei, L.; Xia, Q.; Mwanga, A. Congenital Heart Diseases: Genetics, Non-Inherited Risk Factors, and Signaling Pathways. Egypt J Med Hum Genet 2020, 21, 11. [Google Scholar] [CrossRef]
- Fotiou, E.; Williams, S.; Martin-Geary, A.; Robertson, D.L.; Tenin, G.; Hentges, K.E.; Keavney, B. Integration of Large-Scale Genomic Data Sources With Evolutionary History Reveals Novel Genetic Loci for Congenital Heart Disease. Circ: Genomic and Precision Medicine 2019, 12, e002694. [Google Scholar] [CrossRef] [PubMed]
- Page, D.J.; Miossec, M.J.; Williams, S.G.; Monaghan, R.M.; Fotiou, E.; Cordell, H.J.; Sutcliffe, L.; Topf, A.; Bourgey, M.; Bourque, G.; et al. Whole Exome Sequencing Reveals the Major Genetic Contributors to Nonsyndromic Tetralogy of Fallot. Circ Res 2019, 124, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Shabana, N.; Shahid, S.U.; Irfan, U. Genetic Contribution to Congenital Heart Disease (CHD). Pediatr Cardiol 2020, 41, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Geddes, G.C.; Basel, D.; Frommelt, P.; Kinney, A.; Earing, M. Genetic Testing Protocol Reduces Costs and Increases Rate of Genetic Diagnosis in Infants with Congenital Heart Disease. Pediatr Cardiol 2017, 38, 1465–1470. [Google Scholar] [CrossRef]
- Ahrens-Nicklas, R.C.; Khan, S.; Garbarini, J.; Woyciechowski, S.; D’Alessandro, L.; Zackai, E.H.; Deardorff, M.A.; Goldmuntz, E. Utility of Genetic Evaluation in Infants with Congenital Heart Defects Admitted to the Cardiac Intensive Care Unit. American J of Med Genetics Pt A 2016, 170, 3090–3097. [Google Scholar] [CrossRef]
- Roos-Hesselink, J.W.; Kerstjens-Frederikse, W.S.; Meijboom, F.J.; Pieper, P.G. Inheritance of Congenital Heart Disease. Neth Heart J 2005, 13, 88–91. [Google Scholar]
- Pierpont, M.E.; Basson, C.T.; Benson, D.W.; Gelb, B.D.; Giglia, T.M.; Goldmuntz, E.; McGee, G.; Sable, C.A.; Srivastava, D.; Webb, C.L. Genetic Basis for Congenital Heart Defects: Current Knowledge: A Scientific Statement From the American Heart Association Congenital Cardiac Defects Committee, Council on Cardiovascular Disease in the Young: Endorsed by the American Academy of Pediatrics. Circulation 2007, 115, 3015–3038. [Google Scholar] [CrossRef]
- Kearney, H.M.; Thorland, E.C.; Brown, K.K.; Quintero-Rivera, F.; South, S.T. American College of Medical Genetics Standards and Guidelines for Interpretation and Reporting of Postnatal Constitutional Copy Number Variants. Genetics in Medicine 2011, 13, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Li, R.; Fu, F.; Pan, M.; Han, J.; Yang, X.; Zhang, Y.; Li, F.; Liao, C. Chromosome Microarray Analysis in the Investigation of Children with Congenital Heart Disease. BMC Pediatr 2017, 17, 117. [Google Scholar] [CrossRef] [PubMed]
- Jerves, T.; Beaton, A.; Kruszka, P. The Genetic Workup for Structural Congenital Heart Disease. American J of Med Genetics Pt C 2020, 184, 178–186. [Google Scholar] [CrossRef]
- Geng, J.; Picker, J.; Zheng, Z.; Zhang, X.; Wang, J.; Hisama, F.; Brown, D.W.; Mullen, M.P.; Harris, D.; Stoler, J.; et al. Chromosome Microarray Testing for Patients with Congenital Heart Defects Reveals Novel Disease Causing Loci and High Diagnostic Yield. BMC Genomics 2014, 15, 1127. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, R.; Fu, F.; Huang, R.; Li, D.; Liao, C. Prenatal Genetic Diagnosis Associated with Fetal Ventricular Septal Defect: An Assessment Based on Chromosomal Microarray Analysis and Exome Sequencing. Front. Genet. 2023, 14, 1260995. [Google Scholar] [CrossRef] [PubMed]
- Slavotinek, A.M.; Thompson, M.L.; Martin, L.J.; Gelb, B.D. Diagnostic Yield after Next-Generation Sequencing in Pediatric Cardiovascular Disease. Human Genetics and Genomics Advances 2024, 5, 100286. [Google Scholar] [CrossRef]
- Li, R.; Fu, F.; Yu, Q.; Wang, D.; Jing, X.; Zhang, Y.; Li, F.; Li, F.; Han, J.; Pan, M.; et al. Prenatal Exome Sequencing in Fetuses with Congenital Heart Defects. Clinical Genetics 2020, 98, 215–230. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, E.E.; Findley, T.O.; Hu, R.; Khazal, Z.S.H.; Signorello, R.; Dash, C.; D’Gama, A.M.; Feldman, H.A.; Agrawal, P.B.; Wojcik, M.H.; et al. Genomic Testing and Molecular Diagnosis among Infants with Congenital Heart Disease in the Neonatal Intensive Care Unit. J Perinatol 2024. [Google Scholar] [CrossRef] [PubMed]
- Hays, T.; Hernan, R.; Disco, M.; Griffin, E.L.; Goldshtrom, N.; Vargas, D.; Krishnamurthy, G.; Bomback, M.; Rehman, A.U.; Wilson, A.T.; et al. Implementation of Rapid Genome Sequencing for Critically Ill Infants With Complex Congenital Heart Disease. Circ: Genomic and Precision Medicine 2023, 16, 415–420. [Google Scholar] [CrossRef]
- Shreeve, N.; Sproule, C.; Choy, K.W.; Dong, Z.; Gajewska-Knapik, K.; Kilby, M.D.; Mone, F. Incremental Yield of Whole-genome Sequencing over Chromosomal Microarray Analysis and Exome Sequencing for Congenital Anomalies in Prenatal Period and Infancy: Systematic Review and Meta-analysis. Ultrasound in Obstet & Gyne 2024, 63, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Helm, B.M.; Ware, S.M. Clinical Decision Analysis of Genetic Evaluation and Testing in 1013 Intensive Care Unit Infants with Congenital Heart Defects Supports Universal Genetic Testing. Genes 2024, 15, 505. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Venn diagram showing the distribution of genetic testing modalities applied in the cohort of neonates with CHD.
Figure 1.
Venn diagram showing the distribution of genetic testing modalities applied in the cohort of neonates with CHD.
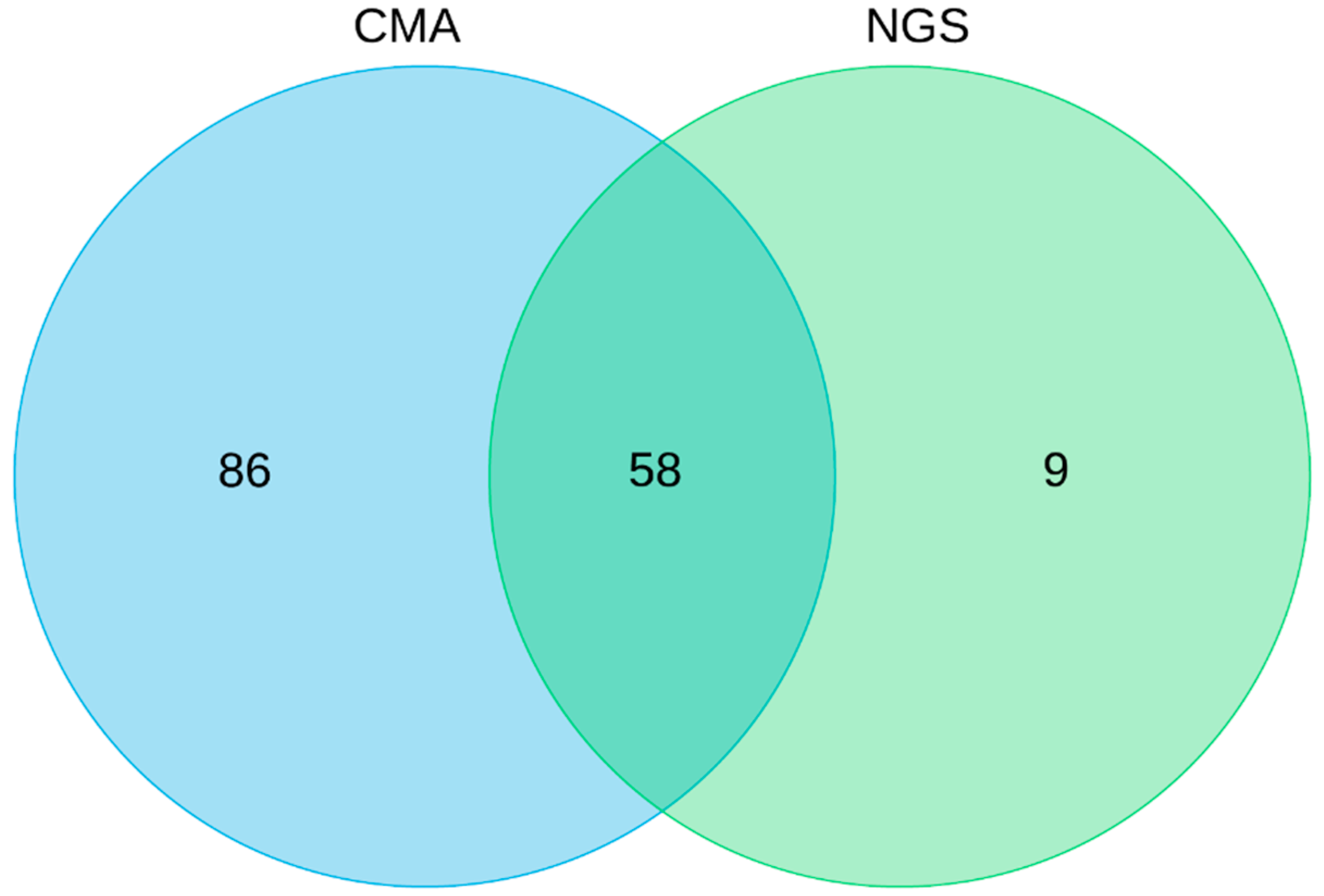

Table 1.
Patients divided into groups according to the Botto classification of congenital heart defect (CHD).
Table 1.
Patients divided into groups according to the Botto classification of congenital heart defect (CHD).
| Category | N of neonates (%) | |
|---|---|---|
| Isolated CHD | Simple | 29 (15) |
| Association | 18 (10) | |
| Complex | 20 (11) | |
| CHD with extracardiac defect | 121 (64) | |
Table 2.
Neonates with congenital heart disease and detected genetic abnormalities.
| N | Congenital heart disease | Extracardiac defects | Results of genetic diagnostics | Geneticlassification | Syndrome |
|---|---|---|---|---|---|
| 1 | sASD | kidney anomaly | arr[hg19] 7q11.23(72,766,313-74,133,332)x1 | P | Williams syndrome |
| 2 | SVAS + stenosis of both pulmonary arteries | / | arr[hg19] 7q11.23(73,474,931-73,493,893)×1 | P | Williams syndrome |
| 3 | mVSD, BAV | hypotonia, hypoplasia of the corpus callosum, feeding difficulties, cryptorchidism, dysmorphic facies | 46,XY, del(8)(p23.3p23.3),dup(8)(p12p23)dn | P | 8p inverted duplication/deletion syndrome |
| 4 | VSD | congenital hydronephrosis, dysmorphic facies | arr[GRCh37] 10q26.13q26.3(126528827-135061104)×1 | P | 10q26 deletion syndrome |
| 5 | VSD, ASD | dysmorphic facies | arr[GRCh37] 22q11.21(21081260-21431174)×1 | P | 22q11.2 microdeletion syndrome |
| 6 | pmVSD | coloboma of irises, hypotonia, anorectal anomaly, feeding difficulty | arr[GRCh37]11q23.3q25(119369472-134904063)x1 | P | Jacobsen syndrome |
| 7 | TGA, ASD, PDA | LGA | arr[GRCh37] 17q12(35149908-36214026)×3, mat | P | 17q12 microduplication syndrome |
| EZH2(NM_004456.5): c.2051G>A | P | Weaver syndrome | |||
| 8 | aortic valve stenosis, BAV, sASD | / | 47,XY,+mar.ish der(22)(pter->q11.21::p12->pter)(acro-p++,SE14/22+,CEP22+,N25+) | P | Cat eye syndrome |
| 9 | ToF, ASD | dysmorphic facies | arr[GRCh37] 1q21.1q21.2(146618988-149224043)×3 dn | P | 1q21.1 microduplication syndrome |
| 10 | VSD | hypocalcemia, dysmorphic facies | arr[GRCh37] 22q11.21(18912870-21431174)×1 dn | P | 22q11.2 microdeletion syndrome |
| 11 | ToF | EA/TEF | CHD7(NM_017780):c.5405-8C>G | P | CHARGE syndrome |
| 12 | pmVSD, multiple ASDs, PFO | SGA, palatoschisis, dysmorphic facies, proximal placement of thumb, pes calcaneovalgus | arr[GRCh37] 18q21.31q23(55111850-78002264)×1 | P | 18q deletion syndrome |
| 13 | VSD, ASD | renal cysts | arr[GRCh37] 17p11.2(16842163-20217777)×1 | P | Smith-Magenis syndrome |
| 14 | pmVSD, truncus arteriosus, | hypothyroidism | arr[GRCh37] 22q11.21(18917796_21431174)×1, | P | 22q11.2 microdeletion syndrome |
| 15 | VSD, ASD | dysmorphic facies | arr[GRCh37] 22q11.21(18917796_21431174)×1, | P | 22q11.2 microdeletion syndrome |
| 16 | VSD, ASD | dysmorphic facies | arr[GRCh37] 1q21.1q21.2(146618988_147826074)×1 dn | P | 1q21.1 microdeletion syndrome |
| 17 | mVSD, sASD, hypoplastic aortic arch | dysmorphic facies | arr[GRCh37] 22q11.21(18917796_21431174)×1, | P | 22q11.2 microdeletion syndrome |
| 18 | ASD, PDA | dysmorphic facies | arr[GRCh37] 16p13.11(15126709_16292235)×3 | P | 16p13. 11 microdeletion syndrome |
| arr[GRCh37] 9q34.13(134077089_134401452)×3 | VUS | ||||
| 19 | ASD | hypotonia, hydronephrosis | arr[GRCh37] 16p13.11(15126709_16292235)×3 | P | 16p13.11 microduplication syndrome |
| 20 | stenosis of aortic valve, BAV | dysmorphic features | arr(X)x1[0.8] | P | mosaic Turner syndrome |
| 21 | valvular pumonary stenosis, SVPS | dysmorphic facies, macrosomia, unilateral cryptorchidism, aplasia cutis | PTPN11(NM_002834.5):c.923A>G | P | Noonan syndrome |
| 22 | sASD | hypotonia, hypoplasia of the corpus callosum, dysmorphic features, palatoschisis, glossoschissis, hypermobility of joints, clinodactyly of 5th fingers | OFD1(NM_003611.3):c.1193_1196del | LP | Orofaciodigital syndrome I |
| 23 | AVSD | coloboma of iris, facial nerve palsy, mixed hearing loss, hypotonia dysmorphic features, feeding difficulties | CHD7(NM_017780.4):c.4353+1G>A | P | CHARGE syndrome |
| 24 | sASD, BAV, PDA | dysmorphic facies, palatoschisis, widely spaced nipples, barrel chest, hypermobility of joints, clinodactyly of 5th fingers | KMT2D(NM_003482.4):c.4364dup | P | Kabuki syndrome |
| 25 | sASD, cleft mitral valve with mild MVR, PDA | dysmorphic facies, chorioretinal coloboma, vocal cord paresis, feeding difficulties, hearing loss | CHD7(NM_017780.4):c.3655C>T | P | CHARGE syndrome |
| 26 | ToF | brachycephaly, ptosis of right eyelid, coloboma of optic nerve papilla, gnatoschisis, choanal atresia, feeding difficulties, unilateral renal agenesis, dysmorphic features, hockey-stick palmar crease, partial 2-3 toe syndactyly, hypotonia, hearing loss | CHD7(NM_017780.3):c.4203_4204delTA | P | CHARGE syndrome |
| 27 | sASD, aortic valve stenosis, BAV | AMC, dynamic upper airway obstruction, ptosis of right eyelid, cryptorchidism, bilateral congenital hip dislocation, clubfoot, fibromatosis colli | CHRNG(NM_005199.5):c.753_754del | P | Multiple pterygium syndrome – Escobar type |
| CHRNG(NM_005199.5):c.250G>A | LP | ||||
| 28 | sASD, PPS, PDA | dysmorphic facies, direct hyperbilirubinemia | JAG1(NM_000214.3):c.2122_2125del | P | Alagille syndrome |
| 29 | pulmonary valve stenosis, PDA, PFO | dysmorphic facies, LGA, renal cyst | PTPN11(NM_002834.5):c.922A>G | P | Noonan syndrome |
| 30 | pulmonary valve stenosis, BAV, bicuspid pulmonary valve, PFO | dysmorphic facies, bilateral coloboma of iris, macula and papilla, horseshoe kidney, ankyloglossia | CHD7(NM_017780.4):c.6292C>T | P | CHARGE syndrome |
| 31 | pmVSD | hypotonia, abnormal cortical gyration, feeding difficulties, dysmorphic facies, single palmar crease | SMARCA4(NM_003072.5):c.4114C>T | LP | Coffin-Siris syndrome 4 |
| 32 | left atrial isomerism | heterotaxy, polysplenia | DNAAF3(NM_001256715.2):c.73_82del | LP | Ciliary dyskinesia, primary, 2 |
* ACC – agenesia of the corpus callosum, AMC – arthrogryposis multiplex congenita, AVSD – atrioventricular septal defect, BAV – bicuspid aortic valve, CoA – coarctation of aorta, dn – de novo, DORV – double outlet right ventricle, EA/TEF – esophageal atresia/tracheoesophageal fistula, LP – likely pathogenic variant, mat – maternally inherited, MVR – mitral valve regurgitation, mVSD – muscular VSD, P – pathogenic variant, PAH – pulmonary artery hypertension, PA-VSD – pulmonary atresia with ventricular septal defect, PDA – patent ductus arteriosus, PFO – patent foramen ovale, pmVSD – perimembranous VSD, PPS – peripheral pulmonary stenosis, sASD – ASD secundum, SGA – small for gestational age, SVAS – supravalvular aortic stenosis, ToF – tetralogy of Fallot, VSD – ventricular septal defect, VUS – variant of uncertain significance, SVPS – supravalvular pulmonary stenosis.
Table 3.
Neonates with congenital heart disease and detected variant of uncertain significance (VUS).
Table 3.
Neonates with congenital heart disease and detected variant of uncertain significance (VUS).
| N | Congenital heart disease | Extracardiac defects | Results of genetic diagnostics | Genetic classification |
|---|---|---|---|---|
| 1 | ASD, PDA | Partial ACC, feeding difficulties, dysmorphic features, occipital subcutaneous vascular malformation | arr[GRCh37] 15q25.2q25.3(85,149,690-85,666,309)x1 | VUS |
| 2 | CoA, hypoplastic distal aortic arch, BAV, pmVSD, ASD, PDA | hypotonia, hypocalcemia, dysmorphic facies | 9p21.2(25713810-26334159)×1 | VUS |
| 3 | ASD, pmVSD | arr[GRCh37] 2q32.3(196614799_196837193)×1dn | VUS | |
| arr[GRCh37] 8p23.2(2470592_4801373)×3 mat | VUS | |||
| 4* | ASD, VSD | hypotonia, HCC, moderate ventriculomegaly, dysmorphic facies, hypoplasia of distal phalanx of fifth finger | arr[GRCh37] Xp22.2(14325345_14757768)×2 mat | VUS |
| 5 | CoA, HLHS | arr[GRCh37] 2q24.2q24.3(163517375_164167131)×3 pat | VUS | |
| 6 | CoA, HLHS | hypotonia | arr[GRCh37] 16q24.1(85002353_85508509)×1 dn | VUS |
| 7 | ToF | coloboma of iris, dysmorphic features | NOTCH1(NM_017617.5):c.3190G>A | VUS |
| 8 | ASD | EA/TEF, annular pancreas, horseshoe kidney, extrarenal pelvis, spina bifida occulta, billiary ducts anomaly | ZNF462(NM_021224.6):c.6334C>T | VUS |
| 9 | CoA | polydactyly, hypospadias, SGA | TLL1(NM_012464.5):c.283G>A (pat) | VUS |
ACC – agenesia of the corpus callosum, AMC – arthrogryposis multiplex congenita, ASD – atrial septal defect, AVSD – atrioventricular septal defect, BAV – bicuspid aortic valve, CoA – coarctation of aorta, DORV – double outlet right ventricle, EA/TEF – esophageal atresia/tracheoesophageal fistula, HCC - hypoplasia of the corpus callosum, HLHS – hypoplastic left heart syndrome, mVSD – muscular VSD, PDA – patent ductus arteriosus, PFO – patent foramen ovale, pmVSD – perimembranous VSD, SGA – small for gestational age, ToF – tetralogy of Fallot, VSD – ventricular septal defect, *WES trio noninformative.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



