
Submitted:
14 May 2024
Posted:
15 May 2024
You are already at the latest version
Abstract
Skeletal muscle contraction evokes numerous genetic and biochemical alterations that underpin the beneficial effects of exercise. The present study aimed to elucidate the mechanism and significance for contraction-evoked antioxidant adaptation in C2C12 myotubes using electrical pulse stimulation (EPS). As NQO1 and GSTA2 are the top antioxidants upregulated by Nrf2, dose-response and time-course experiments of EPS were performed to optimize the parameter at 10 volts for 4 days by evaluating their protein expression. Western blot analysis demonstrated that EPS-cells expressed significantly higher Nrf2 and a broad array of downstream antioxidant enzymes involved in multiple antioxidant systems. These effects were completely abolished by pretreatment with a ROS scavenger, N-acetylcysteine. MitoSOX-Red, CM-H2DCFDA, and EPR spectroscopy revealed a significantly higher ROS level in mitochondria and cytosol in EPS-cells compared to non-stimulated cells. Seahorse and Oroboros revealed that EPS significantly increased maximal mitochondrial oxygen consumption rate, along with upregulated protein expression of mitochondrial complexes I/V, mitofusin-1, and mitochondrial fission factor. A post-stimulation time-course experiment demonstrated that upregulated NQO1 and GSTA2 last at least 24 hours following the cessation of EPS, whereas elevated ROS declines immediately. These finding suggest an antioxidant preconditioning effect in the EPS-cells. A cell viability study using the CCK-8 assay suggested that EPS-cells displayed 11- and 36-fold higher survival rate compared to control-cells in response to 2 and 4 mM H2O2 treatment, respectively. In summary, we found that EPS upregulated a large group of antioxidant enzymes in C2C12 myotubes via a contraction-mitochondrial-ROS-Nrf2 pathway. This antioxidant adaptation protects cells against oxidative stress-associated cytotoxicity.
Keywords:
Electrical Pulse Stimulation
; Mitochondria
; ROS
; Nrf2
; Antioxidant Preconditioning
; Cellular Protection.
1. Introduction
Skeletal muscle (SkM) is a highly plastic tissue capable of functional and structural remodeling in response to numerous physiological stimuli. Particularly during endurance training, skeletal myocytes undergo various challenges from neuronal, hormonal, mechanical, and metabolic stressors, leading to genetic and biochemical adaptations that ultimately determine the beneficial phenotypes evoked by chronic exercise [1].
A predominant stressor on contracting muscle fibers is reactive oxygen species (ROS). ROS at high levels can react with biomolecules to oxidize proteins, lipids, and nucleic acids. This results in cell damage and organ dysfunction, a pathological redox state referred to as “oxidative stress”, which underlies the pathogenesis of numerous diseases. Indeed, in skeletal muscle, it is well-known that excessive ROS contribute to muscle fatigue during exhaustive exercise [2], doxorubicin (DOX)-induced muscle wasting [3], and age-related progressive loss of muscle mass and strength [4]. On the other hand, ROS at low- to moderate- levels function as essential intracellular signaling molecules, playing a crucial role in many biological processes under normal conditions. Physiological ROS can induce posttranslational modifications (PTMs) on specific target proteins by a reversible oxidation of sulfur groups and metal centers [5], therefore altering protein activity, localization, and interactions that result in orchestration of various biological events in cells and organs, such as cell proliferation, differentiation, migration, and angiogenesis [6]. Physiological ROS or “normal redox signaling” is essential to maintain normal metabolism and function of cells and organs [7]. Indeed, studies have demonstrated that selective depletion of ROS from normal skeletal muscle decreases force generation8. Accordingly, there is an optimal ROS concentration in skeletal myocytes which determines the maximal force generation during contraction. Deviations of redox status away from this point, either via an increase or decrease in ROS concentration, will impair muscle contractility forming a classic bell-shaped hormesis dose-response curve [9].
While there is no clear boundary of ROS levels to induce oxidative stress and function as physiological redox signaling, there is no doubt that these free radicals can activate intracellular antioxidant defense under both conditions via compensatory mechanisms to upregulate enzymatic and non-enzymatic antioxidants. This enhanced antioxidant mechanisms not only supports the cells to overcome instant oxidative stress, but also provides cells with an extended antioxidant capability to counteract subsequent similar insults, a phenomenon we term “antioxidant preconditioning”. This process has been proposed as a foundation of redox adaptation of skeletal muscle to endurance exercise [9].
A well-known sensor-effector apparatus in response to a redox disturbance is the Keap1-Nrf2 system. Keap1 acts as a ROS sensor and Nrf2 repressor while Nrf2 is responsible for activating multiple antioxidant genes thus playing a critical role in maintaining intracellular redox homeostasis [10]. Employing muscle-specific Nrf2 deletion and overexpression transgenic mouse models, iMS-Nrf2flox/flox and iMS-Keap1flox/flox, we previously identified over 200 cytoprotective and regulative proteins governed by Nrf2 in skeletal muscle, which are involved in several intracellular signaling pathways. These include antioxidant defense and mitochondrial function [11]. We further demonstrated that these Nrf2-orchestrated molecular networks contribute to chronic exercise-induced enhancement of contractility and muscle antioxidant adaptatioin [12]. However, it remains to be determined if short-term endurance training (STET) can evoke similar beneficial effects on skeletal myocytes to protect the cells against oxidative stress-associated injury.
In the present study, we used electrical pulse stimulation (EPS) on differentiated C2C12 myotubes, a validated in vitro and widely used acute muscle preparation [13, 14] to address the hypothesis that a short-term contraction can evoke an antioxidant preconditioning in skeletal myocytes via a mitochondrial-ROS-Nrf2 pathway. Furthermore, we provide evidence to demonstrate that this antioxidant adaptation protects cells against H2O2-induced toxicity.
2. Materials and Methods
2.1. Cell Culture
The C2C12 cell line (CRL-1722, ATCC, Manassas, VA) was used in these experiments. Cells were seeded in a 6-well plate at ~50,000 or 100,000 cells per well and grown for one day in complete media (CpM) (Dulbecco's Modification of Eagle's Medium (DMEM; Sigma-Aldrich) supplemented with 10% fetal bovine serum (FBS; Gibco/ThermoFisher Scientific) and 1% penicillin/streptomycin (P/S)). After ~24 hours, the media was changed from CpM to differentiation media (DM) (DMEM supplemented with 5% horse serum (HS; Gibco/ThermoFisher Scientific) and 1% P/S), where the cells differentiated from myoblasts to myotubes after four days in culture at 37 ˚C, 5% CO2, 20% O2, and 95% humidity. On the fourth day, the media was replaced by fresh DM and the cells underwent electrical pulse stimulation (EPS).
2.2. Electrical Pulse Stimulation (EPS) and Cell Collection
Electrical current of EPS was delivered to the C2C12 myotubes via stimulation electrodes designed and fabricated in our lab. These electrodes were built on the lid of standard 6-well plates, similar to what has previously been reported15. On the first day of EPS, the regular 6-well plate lids were replaced by the EPS-lids containing the electrodes, which were connected to a single channel pulse generator (A310 Accupulse, WPI, Sarasota, FL). The EPS lids were cleaned before and after each study using the following protocol: The EPS chamber was flooded with 70% ethanol for 10 minutes. The electrodes and EPS lid were washed with ~3 mL of sterile PBS. Finally, the EPS lid was exposed to UV light for 10 – 30 minutes in the cell-culture laminar flow hood. The C2C12 myotubes underwent EPS at 10 v, 50 Hz, 10 ms, 0.3 sec/3 sec for 1h/day for 4 days. At the end of EPS, the cells were rinsed with 200 µL of sterile PBS, followed by trypsinizing with 200 µL of 0.05% trypsin for 2 minutes in 37 ºC. The cells were then collected and transferred to 1.5 mL EP tubes, which were stored in -80 ºC for future analysis.
2.3. Western Blotting
2.3.1. Sample Preparation (Protein Extraction).
Cells were defrosted from -80 ºC, incubated on ice in 100 µL of 100:1 RIPA:PIC for 15 minutes, and then homogenized utilizing a sonicator. The soluble proteins of cell lysate were then extracted by centrifugation for 20 minutes at 13,800 x g and 4 ºC. Protein concentration was measured via BSA assay and a Multiskan Sky Microplate Spectrometer. Samples were then diluted to 2 µg/µL with RIPA. Equal volume of 2X SDS was added to each sample to further dilute the samples to 1 µg/µL and 1X SDS. Samples were then boiled in a 70 ºC water bath for 2 minutes and immediately returned to ice.
2.3.2. Gel Electrophoresis.
The protein samples (15 μg in 15 μL/each sample) were loaded on a precast polyacrylamide gel (NW00105BOX, Invitrogen) along with 2 μL of Invitrogen MagicMark™ Western Standard and 4 μL of Bio Rad Precision Plus Protein Dual Color Standards in separate wells. Electrophoresis was performed using Mini Gel Tank and Blot Module Set (NW2000, Invitrogen), initially at 70 V, for approximately 15 minutes, and then 120 V, until the desired separation had been reached.
2.3.3. Membrane transfer (iBlot2).
The fractionated protein on the gel was electrically transferred onto a nitrocellulose membrane by employing Thermo Fischer Scientific iBlot2 (IB21001, Thermo Fisher Scientific) with the preset program P0 (20 V for 1 minute, then 23 V for 4 minutes, followed by 25 V for 2 minutes). Membranes were gently rinsed with Milli-Q water and then stained with Ponceau S for 5 minutes to visualize total protein. Once ideal bands of total protein were obtained, Ponceau S on the membrane was removed by rinsing it in 10 mL of 1X phosphate-buffered saline-Tween 20 solution (PBST) for 5 minutes.
2.3.4. Immunodetection (Antibodies)
The membranes were then blocked for 30 minutes in 5% milk in 1X PBST at room temperature; this was followed by two 5-minute washes in 10 mL of 1X PBST at room temperature. The membranes were then incubated in primary antibody in 4% BSA overnight at 4 °C. The primary antibodies used in the present experiment were purchased from Abcam (ab-), ABclonal (A-), Santa Cruz Biotechnology (sc-), and Proteintech (-AB/-ig), including NQO1 (ab80588), GSTA2 (ab232833), GSTA4 (ab231601), GPX1 (ab22604), PRX Pathway (TRX, TXNRD1, and PRX1; ab184868), total OXPHOS (ab110413), calmodulin 1/2/3 (A4885), MFN1 (A9880), MFF (A8700), DRP1 (A2586), FIS1 (A21527), SOD1 (sc-8637), SOD2 (sc-30080), catalase (sc-50508), MyoG (sc-576), Nrf2 (A0674 and 16396-1-AP), and mTOR (66888-1-ig). The following day, membranes were washed three times for 5 minutes in 10 mL of 1X PBST; this was followed by incubation in anti-rabbit IgG horseradish peroxidase secondary antibody (1:5000) in 5% milk in 1X PBST for 30 minutes at room temperature. After this, the membrane was again washed three times for 5 minutes in 10 mL of 1X PBST. Membranes were developed in chemiluminescence detection reagent (SuperSignal West Dura or SuperSignal Femto; ThermoScientific). The films were scanned using the G:Box Syngene and Genesys. The intensity of the bands was quantified using the ImageJ software (NIH) and normalized for loading using Ponceau S staining.
2.4. ROS Measurements and NAC Application
2.4.1. Electron Paramagnetic Resonance (EPR) Spectroscopy
C2C12 myoblasts were seeded in six-well plates (100,000 cells/well) followed by differentiation and EPS processes, as described above. After the final EPS, the old DM was replaced with fresh DM, and the cells were processed for EPR spectroscopy analysis as described below at their specified time points (0, 3, 12, or 24 hours after EPS, or NS).
The media was removed from the wells, and the cells were then washed twice with 1 mL of KDD buffer (Krebs-HEPES buffer [pH 7.4]; 99 mM NaCl, 4.69 mM KCl, 2.5 mM CaCl2, 1.2 mM MgSO4, 25 mM NaHCO3, 1.03 mM KH2PO4, 5.6 mM d-glucose, 20 mM HEPES, 5 μM diethyldithiocarbamic acid sodium salt [DETC], 25 μM deferoxamine). Then, the cells were mixed with 200 μM cell-permeable ROS-sensitive spin probe 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethyl pyrrolidine (CMH; Noxygen Science Transfer and Diagnostics, Elzach, Germany) and incubated for 1 hour and 15 minutes at 37 °C. After incubation, 900 µL of KDD buffer was removed from the wells, and the cells were gently scraped into the remaining 100 µL. 50 µL of the cell suspension was pipetted into an EPR glass capillary tube, and inserted into a Bruker e-scan EPR spectrometer with the following settings: field sweep width, 60.0 G; microwave frequency, 9.75 kHz; microwave power, 21.90 mW; modulation amplitude, 2.37 G; conversion time, 10.24 ms; time constant, 40.96 ms. The remaining cell suspension was utilized to count the number of cells to normalize the EPR spectrum amplitude.
2.4.2. Confocal Imaging - MitoSOX Red (MSR, Mitochondrial ROS Indicator) and CM-H2DCFDA (Cytoplasmic ROS Indicator)
C2C12 myoblasts were seeded in six-well plates (5,000 cells/well) containing polylysine coated slides for cell growth, followed by differentiation and EPS processes, as described above. Immediately, after the final day of EPS, the well was washed with 1 mL of HBSS, and the coverslips were then transferred to a 24-well plate, where 2 mL of warm 1 µM MSR working solution or 1 mL of 5 µM CM-H2DCFDA was added, followed by incubation in at 37 °C and 5% CO2 for 30 minutes. The cover slips were then transferred into clean black boxes and gently washed 3 times with warm HBSS. Cells on the coverslips were fixed with cold 4% paraformaldehyde (PFA) for 10 minutes, washed 3 times with PBS, and then mounted onto a glass slide using mounting medium (sc-24941 with DAPI) and clear nail polish. The fluorescence was analyzed via confocal microscopy at the following wavelengths: Mitochondrial ROS: Ex 488 nm/Em 550 – 650 nm, Total ROS: Ex 488 nm/Em 520 nm, and Cellular Nuclear: Ex 360 nm/Ex 460 nm.
2.4.3. N-Acetylcysteine (NAC) Treatment
Three six-well plates were seeded at 100,000 cells per well and underwent differentiation as described above. On the fourth day of differentiation, 40.80 mg of NAC was dissolved in 1 mL of complete media, which was then further diluted to a final working concentration of 5 mM NAC in 50 mL of differentiation media. The pH of this media was neutralized to ~7.5 with 0.5 M NaOH, and then filtered through a 0.2 µm syringe filter. After 15 hours pretreatment with this differentiation media containing 5 mM NAC, the cells underwent EPS for 1 hour at 50 Hz, 10 V, 10 ms, and 0.3 sec/3 sec. 8 hours after EPS, the cells underwent a second treatment of NAC following the protocol outlined above. This was repeated daily for four days. After the fourth, and final day of EPS, the cells were collected as described above.
2.5. Mitochondrial Function Assays
2.5.1. Seahorse – Mito Stress Test
After undergoing 4 days EPS, the cells were collected as described above and replated in 96-well plates at 10,000 cells per well in 100 μL differentiation media (DM). The sensor cartridge was hydrated in calibration buffer at 37 °C in a non-CO2 incubator overnight. Sterile filtered 10 mM glucose, 2 mM glutamate, and 1 mM pyruvate were added to the Seahorse media, whose pH was then corrected to ~7.4 with 1 M HCl or 0.5 M NaOH. Next, 80 μL of DM and was removed from each well of the XF96-well plate, followed by washing with 100 μL of Seahorse media. 160 μL of fresh Seahorse media was added to each well, bringing the final volume to 180 μL. The cells were incubated in the Seahorse media in a non-CO2 incubator for 1 hour. Working solutions of Oligomycin, FCCP, and Rotenone/Antimycin A were made and then added to the injection ports of the sensor cartridge in the respective order. The plate was run on a 96-well plate Seahorse instrument using Wave programming. The Seahorse XF Mito Stress Test Report Generator was used to retrieve the data for later analysis.
2.5.2. Oroboros– High-Resolution Respirometry
After undergoing 4 days of EPS or no-stimulation (controls), approximately 300,000 cells suspended in DM were centrifuged at 1500 x g for 5 minutes, and the cell pellet was resuspended in 2 mL of mitochondrial respiration medium [MiR05, containing 0.5 mM EGTA, 3 mM MgCl2, 60mM lactobionic acid, 20 mM taurine, 10 mM KH2PO4, 20 mM HEPES, 110 mM D-Sucrose, 1 g/L BSA, pH 7.1] in a high-resolution respirometer (Oxygraph-2k, Oroboros, Innsbruck, Austria) to measure mitochondrial respiratory function. The chamber block maintained a constant temperature of 37 °C and magnetic stir bars mixed the solution continuously at 750 RPM. The chambers were hyper-oxygenated to 300 μM O2 via syringe with pure oxygen from an Oxia device (Oxia, Innsbruck, Austria). After the O2 respiration rate stabilized, the cells were permeabilized with digitonin (4.05 μM) for 20 minutes. After permeabilization, substrates were administered in the following order: 1) malate (2 mM) & glutamate (10 mM), 2) adenosine diphosphate (ADP, 5mM), 3) succinate (10 mM), 4) cytochrome c (10 μM), 5) rotenone (0.5 μM), 5) oligomycin (5 nM), 6) antimycin-A (2.5 μM), and 7) ascorbate (2 mM) & N,N,N’,N’-tetramethyl-p-phenylenediamine dihydrochloride (TMPD, 0.5 mM)16, 17. Mitochondrial complexes were assessed as follows: 1) complex I state 2 (LEAK respiration) was assessed after malate & glutamate, 2) complex I state 3 was assessed after malate & glutamate + ADP, 3) complex I+II state 3 was assessed after malate & glutamate + ADP + succinate, 4) membrane integrity was evaluated after the addition of cytochrome c, 5) complex II state 3 was assessed after the addition of rotenone, and 6) complex IV respiration was assessed after ascorbate & TMPD17. The respiration rate for each substrate or inhibitor was recorded after O2 flux reached a stable value (~5 min for substrates and ~10 min for inhibitors). The O2 respiration rates were measured as picomoles of O2 per second per mL and were converted to attomoles of O2 per second per cell (amol·s-1·cell).
2.6. H2O2 Treatment and CCK-8 Assay
After undergoing 4-day EPS, the cells were collected as described above and replated in 96-well plates at 5,000 cells per well in 100 μL CpM. The cells were allowed to adhere to the plate overnight. After cells had adhered to the plate, the CpM was removed and replaced with 100 μL of PBS in CpM (control) or 2 or 4 mM H2O2 in CpM. Cells were incubated in H2O2 at 5% CO2 and 37 °C for 5 hours. After 5 hours, 10 μL of CCK-8 was added to each well. Cells were incubated in CCK-8 at 5% CO2 and 37 °C for 1 hour. The absorbance was measured by a Multiskan Sky Microplate Spectrometer at 450 nm to evaluate cell viability.
2.7. Statistical Analyses
Data are expressed as means ± SD. A t test was used for analyzing the differences in protein expression between EPS and control group using GraphPad Prism 8 software. A two-way ANOVA was used to analyze the EPS-NAC data shown in panels C and D of Figure 4. A P value of < 0.05 was taken as indicative of statistical significance..
3. Results
3.1. EPS Evokes Voltage- and Time- Dependent Upregulation of NQO1 and GSTA2 Proteins
The basic EPS parameters (10 ms, 50 Hz, 0.3 s/3 sec) were adopted from our previous experiments in mice, where EPS was employed to induce in situ tetanic contraction for evaluating SkM fatigue development and force generation of mice with chronic heart failure [18], Nrf2 gene manipulation [11], and chronic exercise training [12]. The frequency of EPS was set as 50 Hz because mouse motor unit output impulses fall within the range of 30 - 80 Hz [19]. To optimize the EPS suitable for the present study in cultured cells, we investigated protein expression of NQO1 and GSTA2, two primary targets of Nrf2 in SkM [11], in response to the EPS administration at different voltages (2.5 – 10 V; Panel A of Figure 1) and different durations (1 – 6 days; Panel B of Figure 1). Clearly, both NQO1 and GSTA2 were upregulated by EPS in a voltage- and duration- dependent manner, with a minor response difference between these two proteins. As shown in Panel A, 10 V-EPS evoked a maximal expression in both, whereas 2.5 V-EPS upregulated the expression only in GSTA2 but not in NQO1. As shown in Panel B, NQO1 displayed maximal response to EPS on day 4, while GSTA2’s maximal expression existed on day 5. Based on these results, we chose the EPS parameters of 10 ms, 50 Hz, 0.3 s/3 sec, 10 V, and 1 hr/day for 4 days for all of the following experiments.
3.2. EPS Increases Nrf2 Protein Content in C2C12 Myotubes
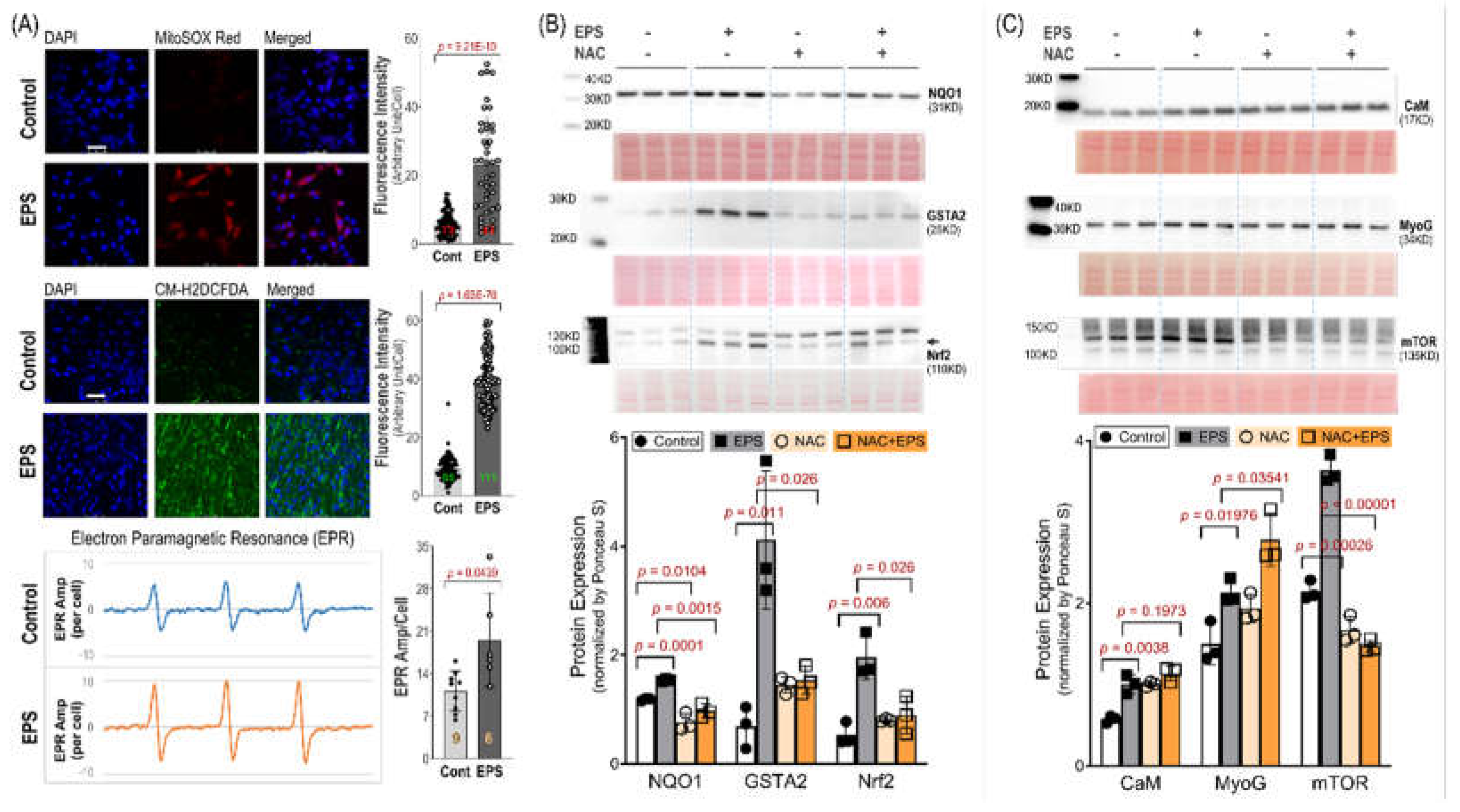
In two previous experiments from our laboratory, we employed the ab13550 antibody (AbCam) to probe Nrf2 protein in mouse SkM lysate and showed a single specific band at 110 kDa, which was significantly upregulated by administrating the Nrf2 activator curcumin [20] or by deleting the Keap1 gene [11]. Unfortunately, the current lot of ab13550 did not detect a Nrf2 band in mouse muscle tissue nor in C2C12 myotubes. We therefore tested seven other Nrf2 antibodies provided by different vendors, including AbCam (ab62352), Proteintech (16396-1-AP, 66504-1-lg), ABclonal (A0674, A1244, A11159), and Invitrogen (PA5-67520). Among these antibodies, we found that two antibodies, A0674 and 16396-1-AP, detected a specific Nrf2 band at 110 kDa, whose intensity was significantly higher in EPS-cells than in control cells, as shown in Figure 2. Interestingly, in the blot of 16396-1-AP antibody we found an additional Nrf2 band at 66 kDa, which was not changed by EPS, as shown in Panel B. The different blots of these two antibodies may reflect a different immunogen used to make the antibodies where the NP_006155.2 was used for A0674 (ABclonal) and the AG9489 (Proteintech) for the 16396-1-A. Since the predicted molecular weight of Nrf2 based on its amino acid sequence is ~66 kDa, we think that the band at ~66 kDa represents a non-modified and non-activated Nrf2 protein, whereas the band in ~110 kDa is the biologically relevant and activated Nrf2, whose molecular weight is higher due to post-translational modifications (PTMs) such as acetylation, phosphorylation, ubiquitination, and SUMOylation [21]. The observation and interpretation of the Nrf2 blots showing two bands have been discussed by two laboratories in the field [22, 23]. Although the current data clearly showed an increased Nrf2 protein content in whole cell lysates of EPS-C2C12 myotubes, the underlying molecular mechanisms remain to be elucidated. We postulated that both activation of the Nrf2 gene (NFE2L2) and reduction of proteasome-mediated Nrf2 protein degradation contribute to this increased Nrf2 protein content with the latter mechanism being the predominant. This conclusion is based on the data shown in Figure 4 where we found that EPS dramatically elevated intracellular ROS, the principal mediator to dissociate Nrf2 from Keap1, and that pretreatment with the ROS scavenger, N-acetylcysteine (NAC), completely abolished the EPS-induced increase in Nrf2 protein.
3.3. EPS Upregulates Multiple Antioxidant Proteins
After demonstrating an increased Nrf2 protein in EPS-cells, we then evaluated Nrf2 downstream target proteins that constitute cellular antioxidant defenses. We found that, in the EPS-cells, a large group of proteins associated with multiple antioxidant systems were significantly upregulated (Figure 3). These proteins include the components of the 1st line antioxidant system (SOD1, SOD2, Cat, and GPX1; Panel A), the thioredoxin antioxidant system (Trx and Prx1; Panel B), and the glutathione antioxidant system (GSTA2, GSTA4, and NQO1; Panel C). Among these evaluated Nrf2 target proteins, NQO1, GSTA2, and GSTA4 were the top three most upregulated proteins following EPS, which were 3-, 12-, and 7-fold higher in EPS-cells than control-cells, respectively (right subpanel of Panel C). These data suggest that EPS can activate multiple antioxidant systems in C2C12 cells, where the glutathione system is the most mobilized.
3.4. EPS-Evoked Activation of Nrf2/Antioxidant System Relies on ROS
To investigate the role of ROS in EPS-evoked activation of Nrf2/antioxidant systems, we employed three techniques to measure ROS levels and used a ROS scavenger, N-acetylcysteine (NAC). As can be seen in the MitoSOX-Red, CM-H2DCFDA, and EPR spectroscopy data shown in Panel A of Figure 4, EPS-cells exhibited a significantly higher fluorescence intensity and EPR spectrum amplitude compared to control-cells. These data indicate a significantly high ROS level in mitochondria, cytosol, and whole cells of the C2C12 myotubes that received stimulation and demonstrate an increased ROS generation from mitochondria of contracting muscle. Panel B of Figure 4 shows that EPS-induced upregulation of Nrf2/antioxidant enzymes was completely abolished by NAC pretreatment (EPS+NAC), suggesting that the increased ROS is the exclusive mediator of the Nrf2/antioxidant upregulation induced by EPS. Interestingly, we found that NQO1 expression was significantly lower in the cells treated with NAC alone compared to cells without any treatment, suggesting that the basal expression of this antioxidant enzyme relies on physiological levels of ROS. Panel C of Figure 4 shows the expression of three non-antioxidant proteins, where NAC had no effects on EPS-induced upregulation of CaM and MyoG but did abolish the mTOR response.
Figure 4.
ROS level and its role in EPS-induced protein expression. (A): ROS levels in mitochondria (MitoSOX-Red), cytosol (CM-H2DCFDA), and whole cells (EPR). Left subpanels: representative confocal images (scale bar: 50 um) and EPR spectra. Right subpanels: quantitative fluorescence intensity and EPR results. The red, green, and orange numbers within the bars indicate cell and sample numbers. (B) and (C): Protein expression of Nrf2/antioxidant enzymes and non-antioxidant proteins. Data are expressed as mean ± SD; n = 3/each group. NAC: N-acetylcysteine.
Figure 4.
ROS level and its role in EPS-induced protein expression. (A): ROS levels in mitochondria (MitoSOX-Red), cytosol (CM-H2DCFDA), and whole cells (EPR). Left subpanels: representative confocal images (scale bar: 50 um) and EPR spectra. Right subpanels: quantitative fluorescence intensity and EPR results. The red, green, and orange numbers within the bars indicate cell and sample numbers. (B) and (C): Protein expression of Nrf2/antioxidant enzymes and non-antioxidant proteins. Data are expressed as mean ± SD; n = 3/each group. NAC: N-acetylcysteine.
3.5. EPS Enhances Mitochondrial Oxidative Phosphorylation And Dynamics
Given mitochondria are one of the major sources of ROS in skeletal myocytes during contraction, we examined the effects of EPS on oxidative phosphorylation (OXPHOS) complex protein expression, oxygen consumption rate, and protein expression of four factors associated with mitochondrial dynamics. We found that EPS significantly upregulated CI, the main site of ROS production24, and CV, the ATP synthase25, whereas CII, CIII, and CIV expression were left unchanged (Panel A, Figure 5). Correspondingly, FCCP-mediated uncoupled maximal oxygen consumption rate was significantly increased in EPS-cells compared to control cells (Panel B, Figure 5). Furthermore, ADP-stimulated respiration (state 3) was increased in EPS compared to non-stimulated cells through complex I, complex II, and complex I+II (Panel C, Figure 5). In addition, complex IV respiration was elevated and complex I state 2 respiration was unchanged in EPS compared to non-stimulated cells (Panel C, Figure 5). Overall, these data suggest that EPS can mediate robust increases in OXPHOS capacity in C2C12 myotubes by upregulating CI and CV expression. Mitochondria are highly dynamic cytoplasmic organelles. During contraction, the mitochondrial integrity and homeostasis in SkM have to be maintained through continual fusion and fission26, 27. Therefore, we evaluated the expression of four proteins associated with mitochondrial dynamics and found that EPS upregulated MFN1 and DRP1 but had no effect on MFF and FIS1 (Panel D, Figure 5).
3.6. EPS Evokes Antioxidant Preconditioning to Protect Cells Against H2O2-Induced Injury
To determine how long antioxidant proteins can remain upregulated after stopping stimulation, we carried out an experiment to evaluate the time course of NQO1 and GSTA2 protein expression in samples harvested at 0, 3, 6, 12, 24 hours after EPS (Panel A of Figure 6). We did not find a significant difference in protein expression between samples at 0, 3, 6, and 12 hours. However, protein expression at the 24-hour sample was significantly lower than that in other EPS samples but still significantly higher than that in the non-stimulated control sample. These data suggest that EPS-enhanced antioxidant enzyme expression can last at least 24 hours. On the other hand, EPR spectroscopy showed a significant increase in ROS immediately after EPS (i.e., 0 hour), which rapidly declined in the 3 – 24 hour samples to levels below that in the non-EPS sample (Panel B, Figure 6). These data suggest that the upregulated endogenous antioxidants by EPS provide C2C12 myotubes with excess antioxidant capacity, a status we term “antioxidant preconditioning”, which can protect the cells against subsequent oxidative stressor-induced injury. As can be seen in Panel C of Figure 6, the survival rates of the EPS-cells exposed to 2 mM and 4 mM H2O2 were 77.8% and 54.3% of PBS-NS control group, about 11- and 36-fold higher than that of H2O2-NS-cells whose survival rates were 7.2% and 1.5%, respectively.
4. Discussion
Using EPS on cultured C2C12 myotubes to investigate the effects of contraction on the intracellular antioxidant system in SkM, we showed that EPS for up to 4 days induced an obvious time- and dose- dependent upregulation of NQO1 and GSTA2 protein expression. We found that EPS markedly activated Nrf2 and multiple antioxidant systems, including the first line antioxidant system, the thioredoxin system, and the glutathione system by upregulating a large group of antioxidant enzymes and antioxidant-associated proteins. Among these systems, the glutathione system displayed the largest response to EPS, with approximately a 10-fold increase in GSTA2 and GSTA4 protein expression (Panel C, Figure 3). We further demonstrated that EPS-mediated increase in ROS played a critical role in the activation of Nrf2/antioxidant systems, since EPS-mediated enhancements in antioxidant capacity were abolished when the C2C12 myotubes were pre-treated with the ROS scavenger, NAC (Panel B, Figure 4).
Importantly, our data, of substantially elevated mitochondrial-derived superoxide after EPS, supports the view that mitochondria may be one of the major sources of ROS and responsible for the activation of Nrf2/antioxidant pathways (Panel A, Figure 4). Furthermore, our data suggest that EPS-induced mitochondrial adaptations may promote EPS-induced augmentation in antioxidant capacity. Of note, complex IV respiration was significantly increased after 4 days of EPS, compared to non-stimulated cells (Panels B and C, Figure 5). Furthermore, ADP-stimulated respiration (state 3) through complex I, complex II, and complex I+II, were all elevated after 4-days of EPS (Panel C, Figure 5). In addition, these adaptations were paralleled by increased protein expression of complexes I, the main mitochondrial site for ROS production [28], and V (Panel A, Figure 5), and upregulated fission/fusion factors (Panel D, Figure 5). Altogether, these data indicate that EPS leads to substantial adaptations to mitochondrial function and elevations in OXPHOS capacity, which is consistent with known adaptations in human SkM mitochondria after aerobic training [29, 30]. Importantly, the stepwise elevation in OXPHOS capacity due to repeated EPS exposure may also lead to elevations in mitochondrial ROS production during high-intensity EPS [31-34], which may provide an incremental stimulus necessary to induce cumulative increases in antioxidant capacity through Nrf2 as observed in the present study (Panel A, Figure 1). Furthermore, our data support the notion that ROS is transiently elevated during EPS (Panel B, Figure 6) while antioxidant capacity remains elevated up to 24 hours after EPS (Panel A, Figure 6), indicating an overcompensation of antioxidant synthesis and the development of an antioxidant reserve. Importantly, we demonstrated that “antioxidant preconditioning” induced by EPS protects C2C12 myotubules from future oxidative injury, since EPS-cells were more resistant to H2O2-induced cytotoxicity than non-stimulated control cells. Overall, our data indicate that mitochondrial ROS during EPS activates Nrf2/antioxidant pathways and may lead to an antioxidant reserve that protects cells from future oxidative injury.
C2C12 is a frequently used cell line to investigate the physiology and pathology of skeletal myocytes. After exposure to a differential media, C2C12 are transformed from primitive myoblasts into mature myotubes, which have the primary characteristics of rodent SkM, including the ability to contract and generate force. Indeed, it has been demonstrated that EPS evokes actual contraction of C2C12-myotubes, making this cell line a convenient in vitro model suitable to investigate the physiological significance and molecular responses of skeletal myocytes to stimulated exercise [14]. A single-bout of EPS at 1 Hz for 1, 3, or 6 hour duration has been demonstrated to upregulate Nrf2/antioxidant system in C2C12 myotubes [35]. We employed EPS at 50 Hz and 1 hour per day for 4 consecutive days in the present experiment, which may better simulate SkM response to chronic short-term exercise. Importantly, our time-course data clearly show a significantly higher protein expression of NQO1 and GSTA2 in 4-day-EPS-cells compared to those that undergo 1 day of EPS. These findings suggest cumulative effects of repeated contraction on the activation of the muscle antioxidant system and differential responses of SkM to a single bout of exercise compared to multiple sets of a short-term exercise training.
Intracellular redox homeostasis is crucial for maintaining normal muscle structure and function. However, this balance tends to be disturbed during exercise due to increased ROS production by membrane NADPH oxidases and mitochondria where the oxidative phosphorylation process increases dramatically to meet the increased ATP demand. As shown in Panel A of Figure 4, we detected a profound increase in ROS in mitochondria, cytosol, and whole cells receiving 4 days of EPS. The exact mechanisms responsible for this increased ROS remain to be determined. However, given the data in Panels A and B of Figure 5, a significant upregulation of complexes I/V and increased mitochondrial oxygen consumption rate were detected. We believe that mitochondria are, at least, one of the major sources for the increased ROS in these EPS cells [36].
To preserve redox homeostasis in response to oxidative stress, intracellular antioxidant defenses must be mobilized to remove the excessive ROS. Antioxidant protein expression is primarily orchestrated by the Nrf2-Keap1 system, a thiol-based sensor-effector apparatus, where ROS oxidizes three key cysteine residues (Cys151, Cys273, and Cys288) on the Keap1 molecule, resulting in dissociation of Nrf2 from Keap1 and translocation from cytosol to the nucleus. In the nucleus, Nrf2 binds to antioxidant response elements (AREs) on DNA to upregulate hundreds of proteins involved in antioxidant defense, anti-inflammation, detoxification, and metabolism [10]. This ROS-trigger and Nrf2-Keap1-dependent activation of antioxidant defense in SkM has been characterized by our laboratory employing transgenic mouse models with muscle-specific Nrf2/Keap1 deficiency, iMS-Nrf2flox/flox and iMS-Keap1flox/flox [11], and in a chronic exercise training model in C57BL/6 mice [12]. In the present study using 4 days of EPS on C2C12 myotubes, we describe a useful in vitro model whose Nrf2/antioxidant system can be activated to a similar level as observed in animal models. This is particularly true for the expression of NQO1 and GSTA2 proteins.
One of the most important findings from the present study, we believe, is the prolonged elevated protein expression of antioxidant enzymes for up to 24 hours after ceasing 4-day-EPS (Panel A, Figure 6). This phenomenon suggests potential cumulative effects on protein expression induced by consecutive multiple sets of EPS when the interval of any two chronologically adjacent stimulations is less than 24 hours. This cumulative effect may represent a mechanism for the gradual increased protein content of NQO1 and GSTA2 observed in the time course experiment shown in Panel B of Figure 1. Translation of this in vitro experimental data into a strategy to optimize long-term exercise points to the significance of intervals between two sets of exercise-induced muscle antioxidant adaptation. While the molecular mechanisms underlying this prolonged antioxidant protein overexpression remain to be elucidated, we postulate that they are involved in an extended alteration of DNA transcription, protein translation, and/or post-translational modifications.
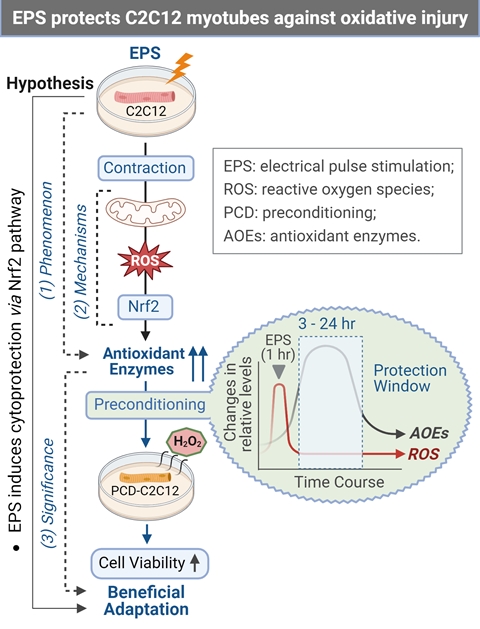
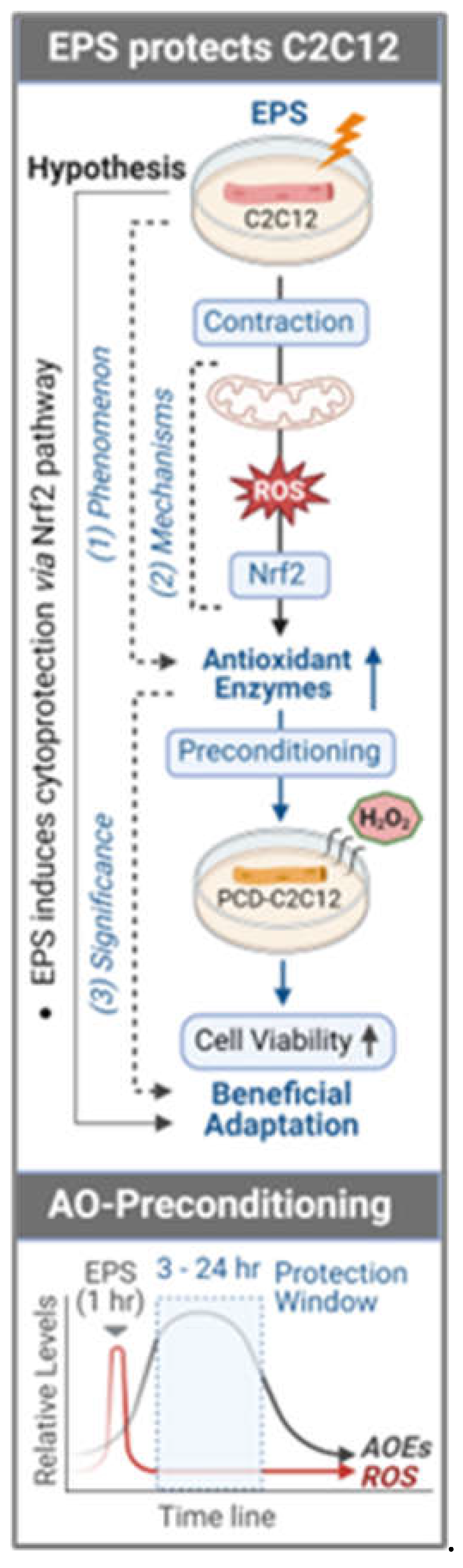
In contrast to the expression of antioxidant enzymes, EPS-induced increase in ROS did not last more than 3 hours after ceasing stimulation (Panel B, Figure 6). This chronological dissociation between antioxidant enzymes and ROS tilts the redox balance to the antioxidant side and confers cells with excess and chronic antioxidant capacity, a state we term as “antioxidant preconditioning” (Panel B, Figure 7), thus protecting cells against subsequent oxidative injury. Indeed, as the data shows in Panel C of Figure 6, EPS-cells have 11- and 36-fold higher viability than control-cells when they are exposed to the medium containing 2 mM and 4 mM H2O2.
5. Conclusions
By employing C2C12 myotubes, this study provides evidence to demonstrate that short-term contraction can protect skeletal myocytes against subsequent oxidative stress-induced cytotoxicity likely via ROS-dependent Nrf2-evoked activation of intracellular antioxidant mechanisms (Panel A, Figure 7), which provides novel insight in the area of SkM redox signaling responses to exercise [36].
Author Contributions
Conceptualization, and L.G. and S.P.P.; Methodology, Data curation, Visualization, Investigation: S.P.P.; A.B.; C.A.; M.F.A.; W.B.; J.J.; V.V.P.; L.Y.; T.B.; Writing—original draft preparation, S.P.P.; L.G.; Writing—review and editing, H.I.Z.; M.C.Z.; S.Y.P.; Supervision, L.G. and S.P.P.; Project administration, L.G. and S.P.P.; Funding acquisition, L.G.. All authors have read and agreed to the published version of the manuscript.”
Funding
This research was funded by National Institutes of Health (NIH) grant R01HL160820 awarded to LG. IHZ was supported, in part, by the Theodore F. Hubbard Foundation. The EPR spectroscopy data was collected in the UNMC EPR Spectroscopy Core, which was established with support, in part, from an NIH Centers of Biomedical Research Excellence (COBRE) grant (1P30GM103335) awarded to the University of Nebraska's Redox Biology Center.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data are contained within the article.
Acknowledgments
The authors thank the Seahorse Core Facility of University of Nebraska Medical Center, Dr. Kelly Stauch and Dr. Andrew Trease, for professional assistance in measurement of oxygen consumption rate. The graphical summary was created with BioRender.com.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Egan B and Sharples, AP. Molecular responses to acute exercise and their relevance for adaptations in skeletal muscle to exercise training. Physiol Rev. 2023;103:2057-2170.
- Reid, MB. Free radicals and muscle fatigue: Of ROS, canaries, and the IOC. Free Radic Biol Med. 2008;44:169-79.
- Hiensch AE, Bolam KA, Mijwel S, Jeneson JA, Huitema AD, Kranenburg O, Van der Wall E, Rundqvist H, Wengstrom Y and May AMJAp. Doxorubicin-induced skeletal muscle atrophy: elucidating the underlying molecular pathways. 2020;229:e13400.
- Fulle S, Protasi F, Di Tano G, Pietrangelo T, Beltramin A, Boncompagni S, Vecchiet L and Fanò GJEg. The contribution of reactive oxygen species to sarcopenia and muscle ageing. 2004;39:17-24.
- Sinenko SA, Starkova TY, Kuzmin AA and Tomilin AN. Physiological Signaling Functions of Reactive Oxygen Species in Stem Cells: From Flies to Man. Front Cell Dev Biol. 2021;9:714370.
- Sies H, Berndt C and Jones DP. Oxidative Stress. Annu Rev Biochem. 2017;86:715-748.
- Sies H and Jones, DP. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat Rev Mol Cell Biol. 2020;21:363-383.
- Reid MB, Khawli FA and Moody MR. Reactive oxygen in skeletal muscle. III. Contractility of unfatigued muscle. J Appl Physiol (1985). 1993;75:1081-7.
- Powers SK, Deminice R, Ozdemir M, Yoshihara T, Bomkamp MP and Hyatt H. Exercise-induced oxidative stress: Friend or foe? J Sport Health Sci. 2020;9:415-425.
- Yamamoto M, Kensler TW and Motohashi H. The KEAP1-NRF2 System: a Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol Rev. 2018;98:1169-1203.
- Gao L, Kumar V, Vellichirammal NN, Park SY, Rudebush TL, Yu L, Son WM, Pekas EJ, Wafi AM, Hong J, Xiao P, Guda C, Wang HJ, Schultz HD and Zucker IH. Functional, proteomic and bioinformatic analyses of Nrf2- and Keap1- null skeletal muscle. J Physiol. 2020;598:5427-5451.
- Bhat A, Abu R, Jagadesan S, Vellichirammal NN, Pendyala VV, Yu L, Rudebush TL, Guda C, Zucker IH, Kumar V and Gao L. Quantitative Proteomics Identifies Novel Nrf2-Mediated Adaptative Signaling Pathways in Skeletal Muscle Following Exercise Training. Antioxidants (Basel). 2023;12.
- Lautaoja JH, Turner DC, Sharples AP, Kivela R, Pekkala S, Hulmi JJ and Yla-Outinen L. Mimicking exercise in vitro: effects of myotube contractions and mechanical stretch on omics. Am J Physiol Cell Physiol. 2023;324:C886-C892.
- Manabe Y, Miyatake S, Takagi M, Nakamura M, Okeda A, Nakano T, Hirshman MF, Goodyear LJ and Fujii NL. Characterization of an acute muscle contraction model using cultured C2C12 myotubes. PLoS One. 2012;7:e52592.
- Mobini S, Leppik L and Barker JH. Direct current electrical stimulation chamber for treating cells in vitro. Biotechniques. 2016;60:95-8.
- Robinson MM, Sather BK, Burney ER, Ehrlicher SE, Stierwalt HD, Franco MC and Newsom SA. Robust intrinsic differences in mitochondrial respiration and H(2)O(2) emission between L6 and C2C12 cells. Am J Physiol Cell Physiol. 2019;317:C339-C347.
- Park SY, Pekas EJ, Anderson CP, Kambis TN, Mishra PK, Schieber MN, Wooden TK, Thompson JR, Kim KS and Pipinos, II. Impaired microcirculatory function, mitochondrial respiration, and oxygen utilization in skeletal muscle of claudicating patients with peripheral artery disease. Am J Physiol Heart Circ Physiol. 2022;322:H867-H879.
- Wafi AM, Yu L, Gao L and Zucker IH. Exercise training upregulates Nrf2 protein in the rostral ventrolateral medulla of mice with heart failure. J Appl Physiol (1985). 2019;127:1349-1359.
- Manuel M, Chardon M, Tysseling V and Heckman CJP. Scaling of motor output, from mouse to humans. 2019;34:5-13.
- Wafi AM, Hong J, Rudebush TL, Yu L, Hackfort B, Wang H, Schultz HD, Zucker IH and Gao L. Curcumin improves exercise performance of mice with coronary artery ligation-induced HFrEF: Nrf2 and antioxidant mechanisms in skeletal muscle. J Appl Physiol (1985). 2019;126:477-486.
- Walters TS, McIntosh DJ, Ingram SM, Tillery L, Motley ED, Arinze IJ and Misra S. SUMO-Modification of Human Nrf2 at K(110) and K(533) Regulates Its Nucleocytoplasmic Localization, Stability and Transcriptional Activity. Cell Physiol Biochem. 2021;55:141-159.
- Lau A, Tian W, Whitman SA and Zhang DD. The predicted molecular weight of Nrf2: it is what it is not. Antioxid Redox Signal. 2013;18:91-3.
- Kopacz A, Rojo AI, Patibandla C, Lastra-Martinez D, Piechota-Polanczyk A, Kloska D, Jozkowicz A, Sutherland C, Cuadrado A and Grochot-Przeczek A. Overlooked and valuable facts to know in the NRF2/KEAP1 field. Free Radic Biol Med. 2022;192:37-49.
- Zhao RZ, Jiang S, Zhang L and Yu ZB. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int J Mol Med. 2019;44:3-15.
- Long Q, Yang K and Yang Q. Regulation of mitochondrial ATP synthase in cardiac pathophysiology. Am J Cardiovasc Dis. 2015;5:19-32.
- Kitaoka Y, Ogasawara R, Tamura Y, Fujita S, Hatta HJAP, Nutrition, and Metabolism. Effect of electrical stimulation-induced resistance exercise on mitochondrial fission and fusion proteins in rat skeletal muscle. 2015;40:1137-1142.
- Drake JC, Wilson RJ and Yan Z. Molecular mechanisms for mitochondrial adaptation to exercise training in skeletal muscle. FASEB J. 2016;30:13-22.
- Brand MD, Affourtit C, Esteves TC, Green K, Lambert AJ, Miwa S, Pakay JL, Parker NJFRB and Medicine. Mitochondrial superoxide: production, biological effects, and activation of uncoupling proteins. 2004;37:755-767.
- Granata C, Oliveira RS, Little JP, Renner K and Bishop DJ. Mitochondrial adaptations to high-volume exercise training are rapidly reversed after a reduction in training volume in human skeletal muscle. FASEB J. 2016;30:3413-3423.
- Greggio C, Jha P, Kulkarni SS, Lagarrigue S, Broskey NT, Boutant M, Wang X, Conde Alonso S, Ofori E, Auwerx J, Canto C and Amati F. Enhanced Respiratory Chain Supercomplex Formation in Response to Exercise in Human Skeletal Muscle. Cell Metab. 2017;25:301-311.
- Layec G, Blain GM, Rossman MJ, Park SY, Hart CR, Trinity JD, Gifford JR, Sidhu SK, Weavil JC, Hureau TJ, Amann M and Richardson RS. Acute High-Intensity Exercise Impairs Skeletal Muscle Respiratory Capacity. Med Sci Sports Exerc. 2018;50:2409-2417.
- Tonkonogi M, Walsh B, Svensson M and Sahlin K. Mitochondrial function and antioxidative defence in human muscle: effects of endurance training and oxidative stress. J Physiol. 2000;528 Pt 2:379-88.
- Chandwaney R, Leichtweis S, Leeuwenburgh C and Ji LLJA. Oxidative stress and mitochondrial function in skeletal muscle: effects of aging and exercise training. 1998;21:109-117.
- Sahlin K, Shabalina IG, Mattsson CM, Bakkman L, Fernstrom M, Rozhdestvenskaya Z, Enqvist JK, Nedergaard J, Ekblom B and Tonkonogi M. Ultraendurance exercise increases the production of reactive oxygen species in isolated mitochondria from human skeletal muscle. J Appl Physiol (1985). 2010;108:780-7.
- Horie M, Warabi E, Komine S, Oh S and Shoda J. Cytoprotective Role of Nrf2 in Electrical Pulse Stimulated C2C12 Myotube. PLoS One. 2015;10:e0144835.
- Powers SK and Schrager, M. Redox signaling regulates skeletal muscle remodeling in response to exercise and prolonged inactivity. Redox Biol. 2022;54:102374.
Figure 1.
NQO1 and GSTA2 protein expression in C2C12 myotubes receiving EPS at different voltages (A) and durations (B). Red arrows indicate the target bands. Data are expressed as mean ± SD; n = 4 - 6/each group. NS, non-stimulated control.
Figure 1.
NQO1 and GSTA2 protein expression in C2C12 myotubes receiving EPS at different voltages (A) and durations (B). Red arrows indicate the target bands. Data are expressed as mean ± SD; n = 4 - 6/each group. NS, non-stimulated control.
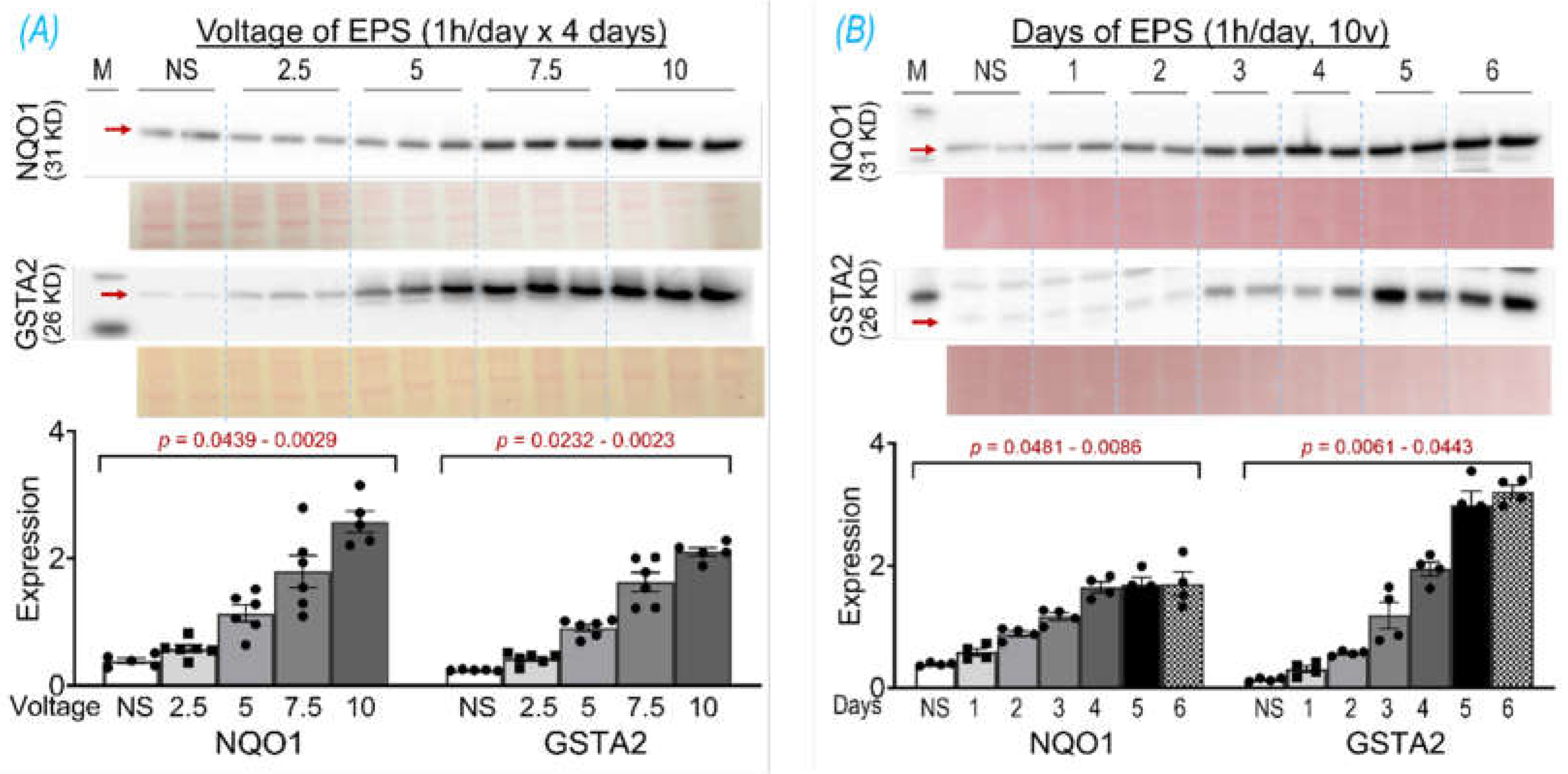
Figure 2.
Protein expression of Nrf2 in the C2C12 myotubes receiving EPS, detected by using two different antibodies: A, A0674 (ABclonal); B, 16396-1-AP (Proteintech). Data are expressed as mean ± SD; n = 3/each group.
Figure 2.
Protein expression of Nrf2 in the C2C12 myotubes receiving EPS, detected by using two different antibodies: A, A0674 (ABclonal); B, 16396-1-AP (Proteintech). Data are expressed as mean ± SD; n = 3/each group.
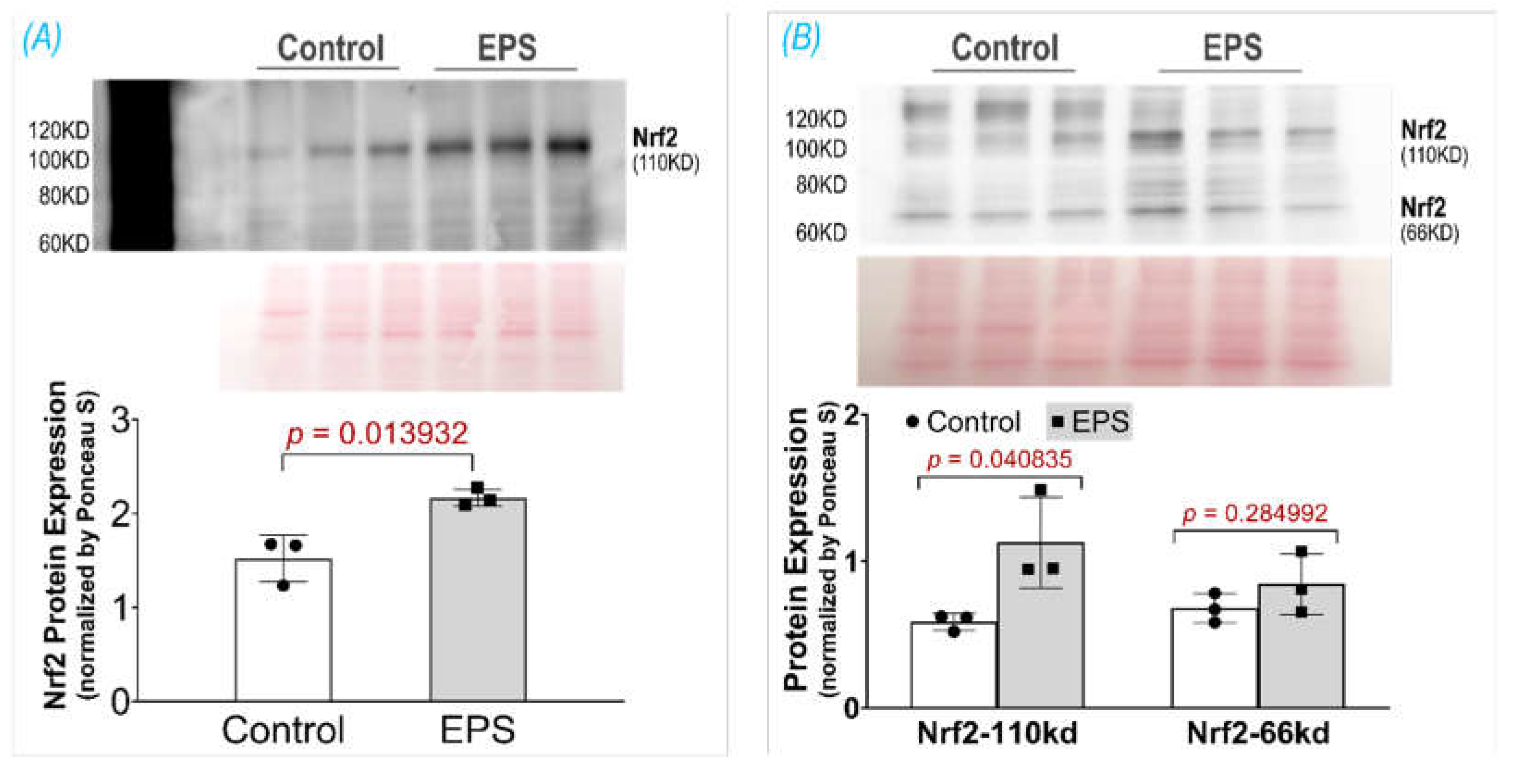
Figure 3.
Protein expression of key antioxidant components in three antioxidant systems: the 1st line antixodant system (A), thioredoxin antioxidant system (B), and glutathione antioxidant system (C). Middle subpanel of (C) shows raw data; Right subpanel of (C) is relative data normalized by the expression in the control group. Data are expressed as mean ± SD; n = 3/each group.
Figure 3.
Protein expression of key antioxidant components in three antioxidant systems: the 1st line antixodant system (A), thioredoxin antioxidant system (B), and glutathione antioxidant system (C). Middle subpanel of (C) shows raw data; Right subpanel of (C) is relative data normalized by the expression in the control group. Data are expressed as mean ± SD; n = 3/each group.
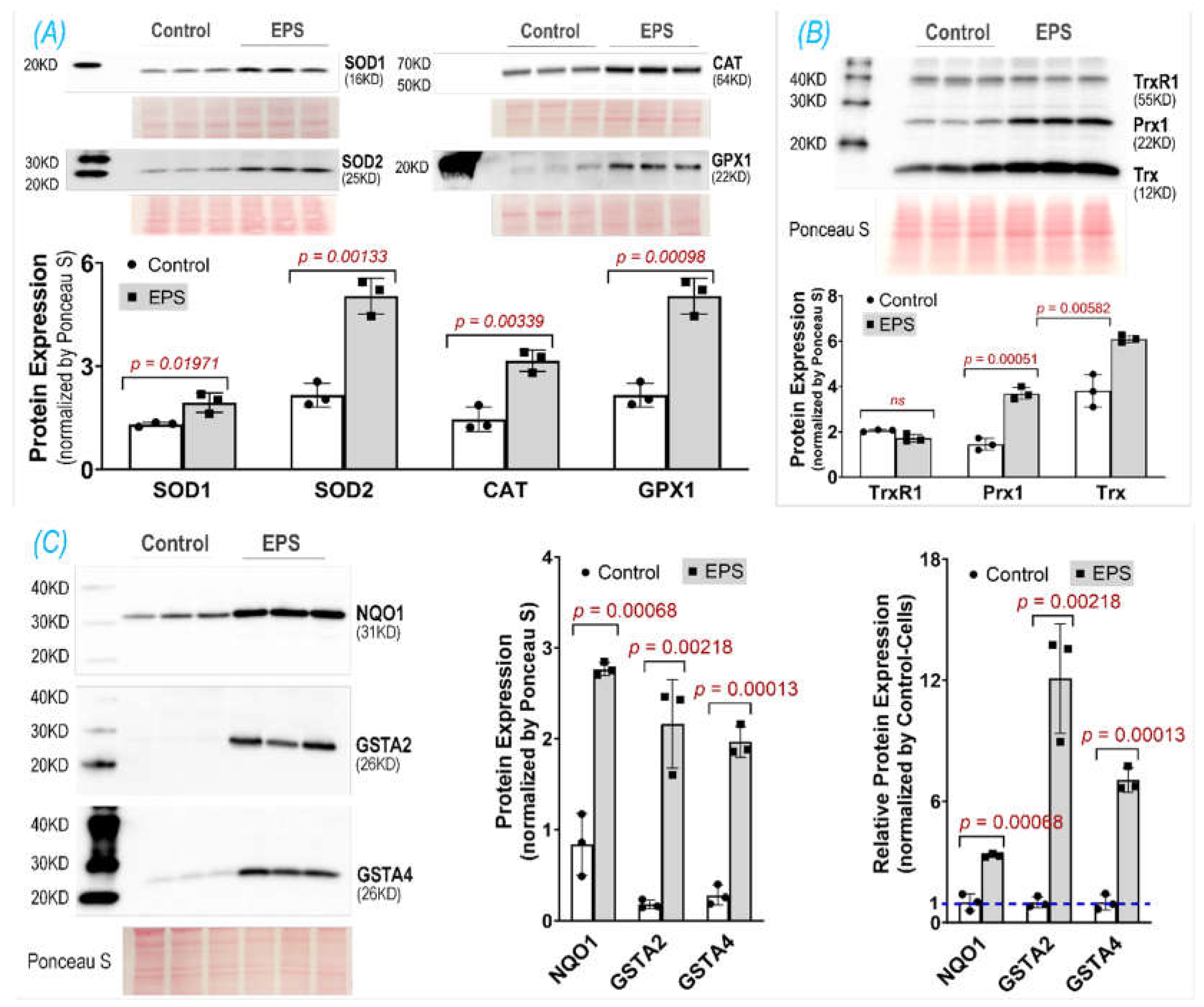
Figure 5.
Mitochondrial protein express and function. (A) Protein expression of respiratory complexes I-V. n = 3/group. (B) Oxygen comsuption rate measured by Seahorse. n = 46 in NS group and 46 in EPS group. (C) Oxygen consumption rates measured by Oroboros. n = 6/group. (D) Protein expression of Fission/Fusion factors. n = 3/group. All data are expressed as mean ± SD.
Figure 5.
Mitochondrial protein express and function. (A) Protein expression of respiratory complexes I-V. n = 3/group. (B) Oxygen comsuption rate measured by Seahorse. n = 46 in NS group and 46 in EPS group. (C) Oxygen consumption rates measured by Oroboros. n = 6/group. (D) Protein expression of Fission/Fusion factors. n = 3/group. All data are expressed as mean ± SD.
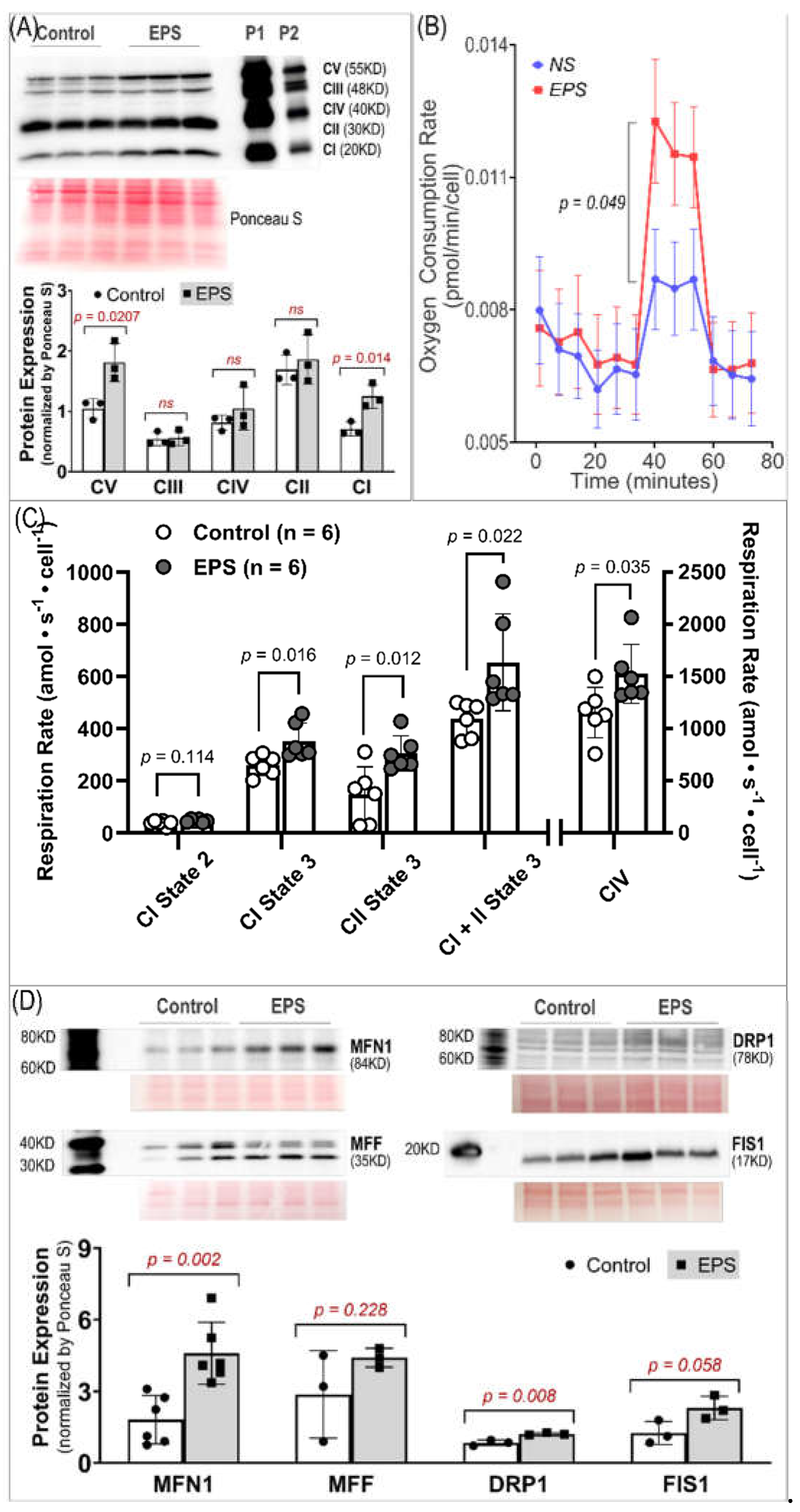
Figure 6.
EPS induced antioxidant preconditioning and protective effects. (A) Time course of NQO1 and GSTA2 expression; (B) Time course of ROS levels; (C) Cell viability assay using CCK-8 shown as raw data of OD 450 nm absorbance (upper panel) and normalized viability by NS-PBS as percentage (lower panel). Data are expressed as mean ± SD; n = 3/group in A, 12-21/group in B, and 23-24/group in C. NS, non-stimulation.
Figure 6.
EPS induced antioxidant preconditioning and protective effects. (A) Time course of NQO1 and GSTA2 expression; (B) Time course of ROS levels; (C) Cell viability assay using CCK-8 shown as raw data of OD 450 nm absorbance (upper panel) and normalized viability by NS-PBS as percentage (lower panel). Data are expressed as mean ± SD; n = 3/group in A, 12-21/group in B, and 23-24/group in C. NS, non-stimulation.
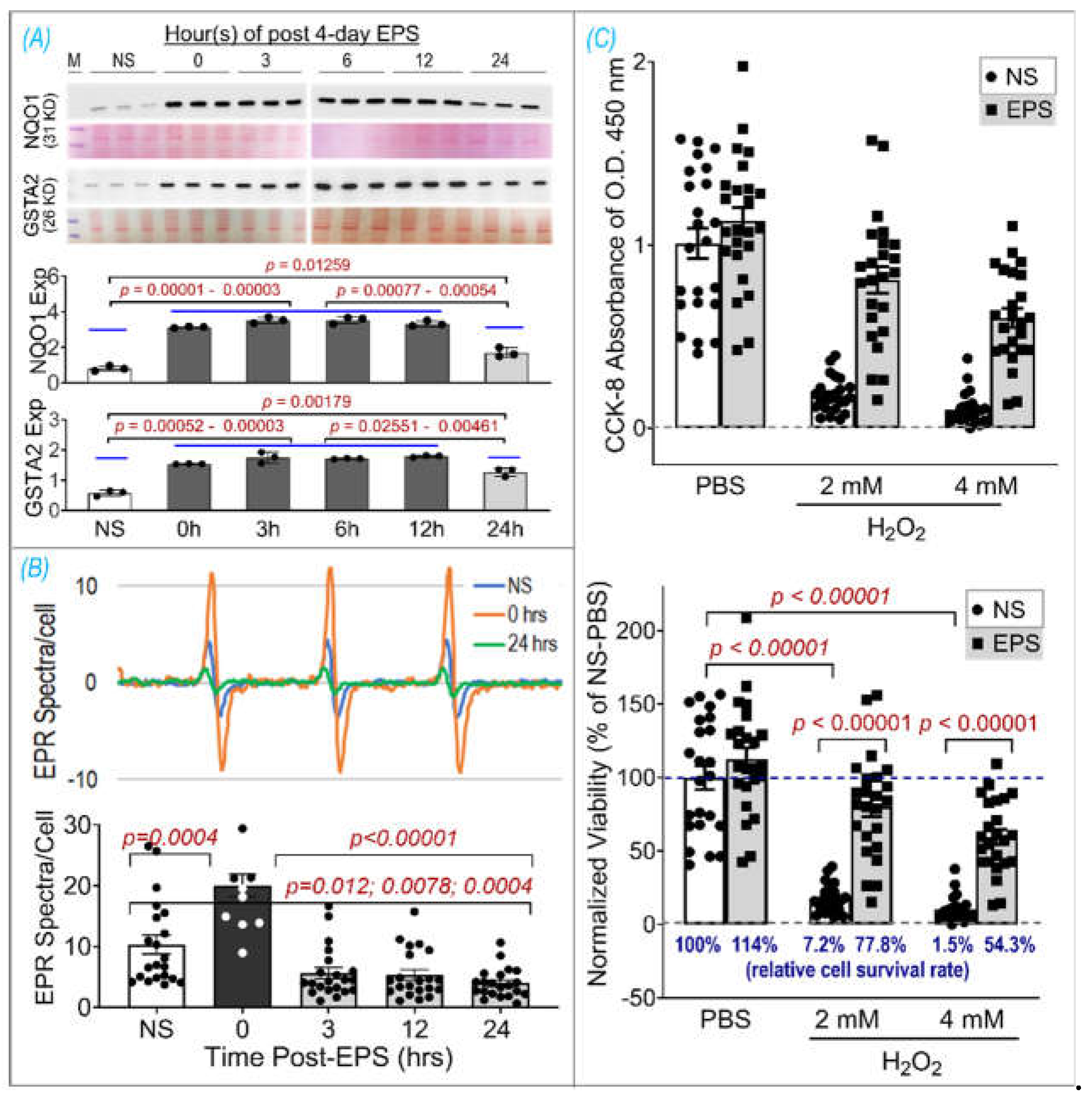
Figure 7.
Graphical Summary (A) and Antioxidant Preconditioning (B). AOEs-Antioxidant (AO) Enzymes (E); PCD-Preconditioning.
Figure 7.
Graphical Summary (A) and Antioxidant Preconditioning (B). AOEs-Antioxidant (AO) Enzymes (E); PCD-Preconditioning.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



