
Submitted:
13 May 2024
Posted:
14 May 2024
You are already at the latest version
Abstract
Cigarette smoke (CS) is a major driver of many respiratory diseases including chronic obstructive pulmonary disease (COPD) and non-small cell lung cancer (NSCLC). Tobacco causes oxidative stress, impaired phagocytosis of alveolar macrophages (AMs), and alterations in gene expression in the lungs of smokers. MicroRNAs (miRNAs) are small non-coding RNAs that influence several bi-ological processes and interfere with several regulatory pathways. The purpose of this study was to assess the effect of active CS on miRNAs expression in AMs obtained from bronchoalveolar lavage (BAL) of ever- or never-smoker subjects and patients with COPD or NSCLC. BAL specimens were collected from 43 sex-matched subjects to determine the expression of has-miR-34a-5p, 17-5p, 16-5p, 106a-5p, 223-5p, and 20a-5p before and after in vitro CS exposure by RT-PCR. In addition, bioin-formatic analysis of miRNAs target genes linked to inflammation was performed. Distinct and common miRNA expression profiles were identified in response to CS, suggesting their possible role in smoking-related diseases. It is noteworthy that, following exposure to CS, the expression levels of hsa-miR-34a-5p and 17-5p in both ever- or never-smokers, 106a-5p in never-smokers and 20a-5p in ever-smokers, shifted towards those found in individuals with COPD, suggesting them like a risk factor in developing this lung condition. Moreover, we identified miRNA targets involved in the immune system or AMs property regulation using in silico analysis. In conclusion, our study iden-tified miRNA signatures in AMs exposed to CS, indicating that CS is an important driver of epige-netic changes that contribute to the onset of various lung diseases.
Keywords:
microRNAs
; alveolar macrophages
; cigarette smoke
; COPD
; lung cancer
1. Introduction
Cigarette smoke (CS) is the leading cause of preventable deaths worldwide and is commonly considered a major driver of many respiratory diseases [1]. Many epidemiological studies have established a high burden of diseases resulting from smoking, including non-small cells lung cancer (NSCLC) and chronic obstructive pulmonary disease (COPD) [2,3]. COPD is an heterogenous disease characterized by progressive deterioration of lung function over time and is generally associated with lung inflammation triggered by harmful particles or gases [4,5,6,7,8,9,10]. COPD and lung cancer, beyond a common etiology, are closely linked conditions, and patients with COPD have twice the risk of cancer diagnosis [12,13,14,15,16]. In the lungs of smokers, tobacco promotes oxidative stress and systemic /local inflammation, which is characterized by the upregulation of circulating inflammatory cells and release of inflammatory mediators [17,18,19]. In addition, CS has been shown to lead to genetic and molecular impairments, which may increase the chance of mutations and lung carcinogenesis [20]. Moreover, it is becoming increasingly evident that the development of COPD or NSCLC phenotypes in response to harmful agents is regulated by both the innate and adaptive immune systems. [20,21,22]. Alveolar macrophages (AMs) are essential effector cells that play a key role in the innate lung immune system by performing pathogen clearance, recruiting other immune cells, phagocyting and processing inhaled environmental particles, and producing pro-inflammatory mediators [23,24,25,26,27]. Smoking causes AMs impairment in phagocytosis and responses to pathogens, compromising their protection from noxious agents [28,29,30]. Importantly, AMs gene expression can be altered in response to environmental exposure, leading to epigenetic changes such as DNA methylation, covalent histone modifications, and microRNAs (miRNA) expression [31,32,33]. miRNAs are small non-coding endogenous RNA molecules capable of modulating gene expression by binding their target mRNAs at 3’ end and, leading to gene silencing through mRNA cleavage or translational repression. miRNAs influence most biological processes and interfere with several regulatory networks, thereby coordinating gene expression under pathological conditions [34,35,36]. Indeed, aberrant miRNA expression appears to be a signature of human diseases, including tumors and inflammatory lung diseases [37,38]. In our previous study, we reported that has-miR-34a-5p, 17-5p, 16-5p, 106a-5p, 223-5p, and 20a-5p expression profiles in AMs are dysregulated in NSCLC, COPD and ever- or never-smoker controls, suggesting their potential role as an index of the smoking-related disease microenvironment [39]. Notably, all selected miRNAs have been shown to influence processes related to inflammation, carcinogenesis or immunity, which are closely linked to CS [40,41,42,43,44,45,46]. However, despite the known association between CS and lung diseases, little is known about the effect of active smoking on the expression levels of miRNAs in AMs, and how it affects the identification of potential candidate miRNAs as biomarkers of pulmonary conditions. Therefore, to further assess the role of CS in the regulation of AMs miRNA expression, we evaluated the levels of the above mentioned-miRNAs in AMs of bronchoalveolar lavage (BAL) from ever- or never-smoker controls and patients with COPD or NSCLC before and after CS exposure.
2. Materials and Methods
2.1. Ethics Statement
This study belongs to a cross-sectional nonpharmacological clinical study recorded at clinicaltrials.gov (NCT04654104) and all procedures and protocols described were approved by the local Ethics Committee “Calabria Centro”. The criteria of the Institutional Review Board/Human Subjects Research Committee, the Declaration of Helsinki, and the Guidelines for Good Clinical Practice were followed and, all patients or legal guardians signed an informed consent form prior of the beginning of the study.
2.2. Study Population
We enrolled 43 individuals who were equally distributed in terms of age (≥18 years) and sex, at the “Mater Domini” Hospital in Catanzaro, Italy. All participants underwent spirometry in compliance to international guidelines as well as bronchoscopy and BAL for suspected pulmonary neoplasia [47]. Samples that were not employed for histopathological purposes or in our previous research were used in the current study 8 [39]. Based on the clinical data and the pathological diagnosis obtained after bronchoscopy, we divided the enrolled subjects into:1) healthy never-smoker control (“HNS”; n = 9); 2) healthy ever-smokers control (“HS”; n = 11); 3) smokers with Global Initiative for Obstructive Lung Diseases (GOLD) stage 1–4 (“COPD,” n =11); 4) non-small cell lung cancer (“NSCLC”; n=12). The main clinical and pathological characteristics of the cohorts are reported in our previous study and are available in the online version, at https://doi.org/10.3390/biomedicines12051050. In summary, those who had lung infections, extrapulmonary tumors, airflow obstruction other than COPD, autoimmune disorders, or who did not sign the informed consent form were excluded. All enrolled subjects were smokers except for HNS group; specifically, HS were 8 current and 3 former smokers, COPD were 11 current smokers and NSCLC were equally distributed between current and ex-smokers. Within each group, the subjects were comparable in terms of age, sex, and lung cancer histology. Indeed, only those with NSCLC were enrolled among the subject’s presenting cancer. The most frequent comorbidities were hypertension (p < 0.05), and the most used drugs were bronchodilators (p < 0.0001).
2.3. Bronchoalveolar Lavage and AMs Extraction
After obtaining informed consent, the subjects underwent standard flexible bronchoscopy for clinical indications [47]. Premedication and local anesthesia were followed by BAL with 200 ml of sterile isotonic saline solution (37 °C) in the right middle lobe. Specifically, BAL was obtained by instilling 50 ml up to four times, as previously reported [39]. The samples were filtered through sterile gauze and centrifuged at 400 g for 10 min at 4 C° to pellet cellular material. The cells were washed, resuspended in buffer phosphate saline (PBS), and counted in a Bürker chamber. The cell yield was determined as the total cells /total volume obtained for each saline installation. Then, cell viability was determined by Trypan blue exclusion assay, and differential cell count was performed with QUICK-DIFF staining; at least 100 cells were counted.
2.4. Preparation of CS Extract
CS extract was prepared as previously described bubbling ten Red Marlboro cigarettes (Phillip Morris; Cracow, Poland) without a filter through 250 ml of serum-free RPMI with a customized vacuum pump apparatus. The obtained suspension was adjusted to pH 7.4 and filtered through a 0.20 µm pore filter to remove bacteria and large particles [19].
2.5. TH-P1 Culture and Cytotoxicity Assay
Macrophages from acute monocytic leukemia (THP-1) ware used as a pilot model to establish the exact dose (2%, 5%, or 10%) of CS that was able to affect cell viability at 24h using the Thiazolyl Blue Tetrazolium Bromide solution (MTT) assay. THP-1 cells (ATCCR TIB-202TM), purchased from the American Type Culture Collection (Manassas, Virginia, USA), were maintained at 2 × 105 cells/ml in RPMI-1640 medium containing 10% FBS and 2 mM L-glutamine, 200 U/ml penicillin, and 200 mg/ml streptomycin. To obtain a macrophages-like phenotype, THP-1 cells were treated with 100 ng/ml phorbol 12-myristate 13-acetate (PMA, Sigma-Aldrich) for two days. The cells were then incubated with fresh medium for one day to allow cell recovery and exposed to 2%, 5%, or 10% CS medium for 24h. Following 24 h MTT was added and incubated for 4 h to perform the proliferation assay. MTT is a colorimetric method that allow to assess the mitochondrial reductive function as an indicator of growth inhibition. After 4 h, DMSO was added to measure the absorbance at 595 nm using a microplate reader.
2.6. AMs Culture and CS Exposure
BAL cell pellets were suspended in RPMI-1640 medium supplemented with 10% FBS, 2 mM L-glutamine, 200 U/ml penicillin, and 200 mg/ml streptomycin. The cell suspension was added at 0.5 × 106 cells/mL to a 75 tissue culture flask and maintained at 37 °C in a 5% CO2 humidified milieu for 2 h to allow AMs adherence. Lymphocytes, red blood cells and other non-adherent cells were removed by washing several times with PBS. AMs purity, as determined by morphology, was greater than 95%. The AMs were then exposed to 10% CS for 24 h, based on THP-1 treatments results.
2.7. Biochemistry Assays and Real Time PCR (RT-PCR)
The extraction of miRNAs in AMs obtained from BAL was carried out through the miRNeasy mini kit and RNA was eluted at a volume of 15 µL, as previously described 37). RNA degradation was assessed using a qubit RNA Integrity and Quality (IQ) assay (catalog number Q33222) with a Qubit 4 fluorometer (serial number 2322618032114). The expression levels of has-miR-34a-5p, 17-5p, 16-5p, 106a-5p 223-5p and 20a-5p were determined using TaqMan™ Advanced miRNA Assay RT-PCR, following Thermo Fisher Scientific procedures (Waltham, MA, USA), with U6 snRNA as the housekeeping miRNA as previously described [39]. Nine biological replicates for the HNS group, eleven for HS, eleven for COPD, and twelve for NSCLC were analyzed, and all samples were run in triplicate; after the achievement of the RT-PCR, the cycle threshold (Ct) of the reactions was determined. Data from all RT–PCR experiments and miRNA expression was analyzed applying the comparative and normalizing to the endogenous miRNA control 2ˆ-DDCt method, where DCt = CtmiRNA – Ct housekeeping miRNA, whereas the relative differences in expression was determined with DDCt = DCt HS/COPD/NSCLC (with or without CS) − DCt HNS.
2.8. In Silico Prediction of hsa-miRs Target Genes
mRNA targets of has-miR-34a-5p, 17-5p, 16-5p, 106a-5p 223-5p and 20a-5p that are linked to inflammation or AMs properties were analyzed in silico using DIANA Tools (http://diana.imis.athena-innovation.gr/DianaTools/index.php) and miRTargetLink 2.0 (https://ccb-compute.cs.uni-saarland.de/mirtargetlink2) databases. Only genes that were already validated experimentally were selected and potential biochemical pathways were checked using the GeneCard database (https://www.genecards.org/).
2.9. Statistical Analysis
Unless specified, all data are expressed as mean ± standard deviation (SD). The ordinary one-way ANOVA test followed by Dunnet Multiple Comparison Test (for MTT assay) and Tukey Multiple comparison test (for miRNAs analysis) with a single pooled variance, was used to assess the differences between the groups. Nominal (sex, age, comorbidity, or treatment) and categorical variables were considered and the correlation between clinical data was calculated using one-way ANOVA followed by Tukey Multiple Comparison Test. GraphPad software (version 9.1.0) was used for statistical analyses (GraphPad Software, San Diego, CA, USA). Differences were considered statistically significant at p < 0.05.
3. Results
3.1. CS Effect on TH-P1 Viability
To determine the dose of CS for subsequent analysis, we used THP-1 macrophage cell line. Treatment of THP-1 cells with CS for 24h significantly affect cell viability, which peaked at 10% CS (p <0.0001) (Figure 1). Therefore, 24h 10% CS was used to perform AMs exposure.
3.2. miRNA Expression Levels
hsa-miR-34a-5p, 17-5p and 16-5p
We assessed miRNA signatures in each pathological condition (NSCLC and COPD), smoking habit (HS), and control group (HNS) before and after exposure to 10% CS for 24h. The effects of CS in vitro stimulation on hsa-miR-34a-5p, 17-5p, and 16-5p expression in BAL AMs in vitro are shown in Figure 2, Figure 3 and Figure 4. Following stimulation with 10% CS for 24 h, we observed a significant increase in hsa-miR-34a-5p (p < 0.01), 17-5p (p < 0.001), and 16-5p (p < 0.001) expression in AMs obtained from HNS group (Figures 2A–4A). Interestingly, acute in vitro CS stimulation also led to significant positive modulation of hsa-miR-34a-5p (p < 0.001), 17-5p (p < 0.001), and 16-5p (p < 0.001) expression in AMs from HS (Figures 2A–4B). In contrast, CS stimulation of COPD and NSCLC AMs did not affect hsa-miR expression (Figures 2C,D–4C,D). This could suggest that acute CS stimulation is sufficient to affect hsa-miR expression exclusively in AMs from subjects without preexisting lung diseases.
hsa-miR-106a-5p
Following in vitro exposure to 10% CS for 24 h, we observed a significant increase in hsa-miR-106a-5p expression in HNS AMs (p < 0.01) (Figure 5A) and in AMs from the COPD group (p < 0.05) (Figure 5C). Interestingly, CS led to the opposite trend in AMs from HS (p < 0.01) (Figure 5B) while it did not affect hsa-miR-106a-5p expression in AMs obtained from patients with NSCLC (Figure 5D).
hsa-miR-223-5p and 20a-5p
RT-PCR results showed that CS significantly decreased hsa-miR-223-5p (p < 0.001) and 20a-5p (p < 0.01) expression in HNS AMs (Figures 6A and 7A). In contrast, in vitro CS stimulation in COPD AMs induced both hsa-miR-223-5p (p < 0.01) and 20a-5p (p < 0.05) upregulation (Figure 6C and 7C). Moreover, CS affected only hsa-miR-20a-5p expression in AMs from HS, leading to a significant positive modulation (p < 0.0001) (Figure 7B), while did not further impact hsa-miR expression in AMs from individuals with NSCLC (Figure 6D and 7D).
3.3. In Silico Identification of Target mRNAs
The relationship between miRNAs and lung response to CS, was assessed by in silico analysis of sequence similarity between miRNAs and different mRNAs. Experimentally validated target genes were compared in two different databases of which miR target Link 2.0 was chosen as it had a higher number of validated genes for each miRNA. We analyzed all selected miRNA targets, focusing on those that are common to several miRNA and implicated in the cellular pathways regulating inflammation or AMs properties. Abbreviations, names of the selected genes, methods, and validation tissues are listed in Table 2. The results showed that the miRNA may be involved in regulation of several inflammation driver genes, such as BCL2, LAMTOR, MCL1, SOCS5 or VEGFA, among others. However, other target genes linked to apoptosis or cytokines production were found, as shown in Table 3. Given that several authors have experimentally confirmed all genes, miRNA might modulate these targets in a coordinated or individual manner, affecting several hallmarks of lung response to CS.

Table 1.
Bioinformatics tools for in silico analysis. Number of validated genes for each miRNAanalyzed in miR Target Link 2.0 and Diana Tools databases.
Table 1.
Bioinformatics tools for in silico analysis. Number of validated genes for each miRNAanalyzed in miR Target Link 2.0 and Diana Tools databases.
| Number of Target Genes | ||
|---|---|---|
| miRNA | miR Target Link 2.0 | DIANA Tools |
| hsa-miR-223-5p | 551 | 10 |
| hsa-miR-16-5p | 2.279 | 455 |
| hsa-miR-20a-5p | 1.659 | 611 |
| hsa-miR-17-5p | 1.817 | 136 |
| hsa-miR-34a-5p | 968 | 324 |
| hsa-miR-106a-5p | 1.166 | 435 |
4. Discussion
In this study, we assessed the effect of in vitro CS exposure on the regulation of miRNA expression in AMs, the main participants in the development of smoking-related conditions such as COPD and NSCLC. Tobacco promotes oxidative stress, systemic inflammation, and local inflammation in the lungs of smokers, leading to innate and adaptive immune system impairments [17,18,19]. In addition, CS leads to genetic and molecular impairments, which may increase the chance of mutations and lung carcinogenesis [20]. Given the high prevalence of smoking-related diseases, the search for new biomarkers has prompted in-depth epidemiological studies [48,49]. Several authors attempted to identify miRNA signature of CS and evidence for their causal role in smoking-related inflammation. Willinger et al. profiled 283 miRNAs and found six associated with serum levels of C-reactive protein, interleukin-6 and pulmonary function [50]. In our previous study, we reported that has-miR-34a-5p, 17-5p, 16-5p, 223-5p, 20a-5p, and 106a-5p expression profiles in AMs were dysregulated in NSCLC, COPD and ever- or never-smoker controls, suggesting their possible role as an index of smoking-associated conditions [39]. To further investigate the effect of active smoking on the expression levels of these miRNAs, we analyzed the changes in their expression before and after in vitro CS exposure in the above mentioned groups. This profiling was carried out in AMs recovered from BAL, a precious biological sample that is highly representative of the pulmonary microenvironment [51]. First, we identified that never-smokers AMs in vitro stimulated with 10% CS for 24h results in two specific trends, leading to hsa-miR-34a-5p, 17-5p, 16-5p, and 106a-5p upregulation and negative modulation of hsa-miR-223-5p and 20a-5p levels. In contrast, in vitro CS exposure in AMs obtained from individuals chronically exposed in vivo to CS such as ever smokers or with pre-existing lung conditions such as COPD or NSCLC variably affects miRNAs. In the parenchyma of COPD patients, a key hallmark of alveolar epithelial and endothelial cells is apoptosis and, as one of the most important risk factors for COPD, CS can initiate apoptosis in several cell types, including macrophages [52]. In this context, Long et al. reported that Notch-1 receptor protein, a transmembrane receptor which has been implicated in cell proliferation and apoptosis control, is lower expressed in primary lung microvascular endothelial cells (HPMECs) treated with CS, with the upregulation of hsa-miR-34a-5p [53]. In addition, Zeng et al. showed that exposure of BEAS-2B cells to CS increased the expression of hsa-miR-34a-5p and senescence-associated pro-inflammatory cytokines such as IL-1β, IL-6, IL-8, and TNF-α) in a dose-dependent manner [54]. Furthermore, in our previous study, we reported a significant positive modulation of hsa-miR-34a-5p in the tissue and AMs of COPD subjects compared to healthy never-smoker controls [55]. Consistent with these data, we observed after in vitro exposure to CS a significant positive modulation of hsa-miR-34a-5p in the AMs of healthy individuals towards COPD level, providing evidence of the role of CS in COPD-like dysregulation. Interestingly, in vitro CS exposure equally affected hsa-miR-34a-5p expression in AMs obtained from HS suggesting the potent effect of acute CS stimulation. CS can directly damage epithelial cells, the first barrier for the respiratory tract, and cause infiltration of immune cells in the lungs, including AMs [56]. Leukocyte signal-regulatory protein-α (SIRPα), a member of the immunoglobulin superfamily, modulates many aspects of the inflammatory response to noxious agents, including immune cell activation, chemotaxis, and phagocytosis [57]. In this regard, Zhu et al. showed that upregulation of hsa-miR-17-5p by lipopolysaccharide (LPS) in macrophages is the mechanism underlying LPS-induced SIRPα reduction and AMs activation [58]. This was consistent with our finding of higher hsa-miR-17-5p levels in AMs of never-smokers and ever-smokers following acute in vitro CS treatment, supporting the importance of CS in the mechanisms underlying AMs impairment in lung diseases. The innate immune system represents the first line of host defense against harmful particles or bacterial infections through phagocytosis by resident macrophages [59]. One of the most important features of this process is the activation of TLRs immune receptors and the release of a variety of toxic products, including reactive oxygen species (ROS) such as NO, hydrogen peroxide, and superoxide anions [60]. Moon et al. reported that bacterial LPS enhanced the level of has-miR-16-5p in bone marrow–derived macrophages, resulting in decreased phagocytosis and the generation of mitochondrial ROS [61]. Accordingly, our findings showed a positive modulation of has-miR-16-5p in never-smokers and ever-smokers AMs following CS treatment suggesting the ability of CS exposure to modulate the lung inflammatory response. Although a few studies have investigated the hsa-miR-106a-5p expression patterns associated with CS and chronic lung diseases, Liu et al. reported that it dramatically inhibited the activation of autophagy induced by M. tuberculosis in human THP-1 macrophages [62]. Indeed, CS impairs AMs autophagy playing an important role in COPD [63]. Moreover, Sharma et al. reported that hsa-miR-106a-5p negatively regulates IL-10 expression with an increase in proinflammatory cytokines in in vitro and in vivo model of airway inflammation [64]. This was in line with our findings of increased has-miR-106a-5p in AMs of both never-smokers and COPD subjects following in vitro CS exposure. However, the regulatory effects of has-miR-106a-5p in CS-related diseases are not fully understood, making its role controversial, as suggested by its reduction in AMs from ever-smokers after in vitro CS treatment. Our findings highlight that in vitro CS stimulation of AMs obtained from never-smokers results in negative modulation of hsa-miR-223-5p and 20a-5p levels. Several authors have described hsa-miR-223-5p role in macrophage differentiation, neutrophil recruitment, and pro-inflammatory responses, which are key features of lung inflammation and remodeling [65]. Interestingly, in never-smokers AMs, CS resulted in the modulation of hsa-miR-223-5p to levels comparable to those observed in individuals with smoking-related conditions. Consistent with our data, Schembri et al. reported lower has-miR-223-5p levels in bronchial epithelial cells from current smokers than in those from never-smokers [66]. Furthermore, it is important to point out that in COPD AMs, the expression of this miRNA is increased following exposure to CS, indicating a unique function for acute CS in COPD microenvironment. In fact, acute CS exposure can induce chemotactic factors in the lungs, stimulate AMs, and lead to neutrophil influx, which can require at least six months to normalize completely [67]. In this context, Roffel et al. detected higher levels of has-miR-223-5p in the lung tissue of COPD patients, assuming that it could be associated with impaired lung function and higher neutrophil counts [68]. As for hsa-miR-223-5p, CS led to hsa-miR-20a-5p downregulation in AMs obtained from never-smokers. Importantly, our data showed that ever-smokers and patients with COPD shared increased levels of this miRNA after exposure to CS. Specifically, exposure to CS in ever-smokers increases levels towards those reported in COPD, highlighting the close link between CS and the development of a COPD-like phenotype. hsa-miR-20a-5p has been shown to regulate AMs inflammatory responses by targeting SIRPα [58]. Moreover, Liu et al. reported higher hsa-miR-20a-5p levels in children with pneumonia and in lung cells exposed to LPS, highlighting its role in inflammation through activation of the NF-κB signaling pathway [69]. However, given its role in controlling cellular networks, such as the PI3K/Akt axis, the regulatory effects of CS on its expression cannot be generalized, making a more in-depth analysis necessary to explain our results in AMs from never-smokers [44]. Finally, in vitro CS exposure did not influence the expression of any miRNAs in AMs from subjects with NSCLC. In our previous study the same trend was seen for the Programmed death-ligand 1 (PD-L1) mRNA expression. Indeed, we reported that after CS exposure, PD-L1 mRNA expression was increased in AMs derived from never-smoker subjects but not in NSCLC patients, suggesting an overwhelm effect of cancer on acute CS exposure [70]. It is important to note that the intensity of the reaction against immunogenic antigens produced in response to CS varies across a wide range of disease manifestations highlighting the crucial role of immune responses in regulating the development of distinct phenotypes (such as COPD or NSCLC) in response to CS [71,72]. In this context, dysregulation of miRNAs could reflect phenotype switching or the onset of different lung manifestations, underlining the prominent, but not exclusive, role of CS. Indeed, miRNA expression profiles can be influenced by other environmental factors, which can further modulate the correlation between the expression of miRNAs and mRNA targets in response to CS [73]. Finally, to assess the potential interconnection of miRNAs in lung response to CS, we performed in silico prediction of hsa-miR target genes. Bioinformatic results revealed that the miRNAs analyzed may potentially be involved in the regulation of several inflammation driver genes, such as ATG14, BCL2, CPT1A, FOXC1, HAS2, HSPA1A, LAMTOR1, MCL1, MFN2, SCAMP5, SENP1, SOCS5, TGFBR2 and VEGFA, which are involved in immune system regulatory pathways [74,75,76,77,78,79,80,81,82,83,84,85,86,87]. Since several authors have experimentally confirmed all miRNA-regulated genes, miRNAs may modulate these targets combined or individually, affecting different hallmarks of lung response to CS.
5. Conclusions
This study, even if preliminary, indicates that CS is an important driver of epigenetic changes that contribute to the onset of various lung diseases. Moreover, we demonstrated that the effects of acute CS on miRNA expression levels could differ between never-smokers and subjects who were already chronically exposed to CS, such as HS, or had pre-existing lung diseases, such as COPD. It is noteworthy that, following exposure to CS, the expression levels of hsa-miR-34a-5p and 17-5p in both ever- or never-smokers, 106a-5p in never-smokers and 20a-5p in ever-smokers, shifted towards those found in individuals with COPD, suggesting them like a risk factor in developing this lung condition. A potential limitation of our study was the small sample size used for miRNA analysis, which did not allow subanalysis according to COPD severity. However, our data could be of clinical relevance and lead to future studies involving larger populations, allowing to better understand the networks involved in the pathogenesis of smoking-related diseases.
Author Contributions
Conceptualization, Davida Mirra, Giuseppe Spaziano, Luca Gallelli and Bruno D’Agostino; Data curation, Renata Esposito, Giuseppe Spaziano, Antonio Squillante, Luca Gallelli and Erika Cione; Formal analysis, Davida Mirra, Francesca Panico, Maddalena Falciani, Fiorentina Roviezzo and Erika Cione; Funding acquisition, Concetta Rafaniello; Investigation, Davida Mirra and Fiorentina Roviezzo; Methodology, Davida Mirra, Renata Esposito, Giuseppe Spaziano, Maddalena Falciani and Erika Cione; Project administration, Giuseppe Spaziano, Luca Gallelli and Bruno D’Agostino; Resources, Antonio Squillante, Maddalena Falciani and Luca Gallelli; Software, Renata Esposito, Giuseppe Spaziano, Francesca Panico, Antonio Squillante, Maddalena Falciani and Erika Cione; Supervision, Giuseppe Spaziano, Luca Gallelli and Bruno D’Agostino; Validation, Renata Esposito, Giuseppe Spaziano, Francesca Panico and Antonio Squillante; Visualization, Giuseppe Spaziano; Writing – original draft, Davida Mirra and Erika Cione; Writing – review & editing, Davida Mirra, Giuseppe Spaziano, Luca Gallelli and Bruno D’Agostino.
Funding
This research received no funding.
Institutional Review Board Statement
This study was part of a clinical trial recorded at clini-caltrials.gov (NCT04654104) and was approved by the local ethics committee “Calabria Centro” (n. 361 del 22 Ottobre 2020). This work was conducted in compliance with the Institutional Re-view Board/Human Subjects Research Committee requirements, the Declaration of Helsinki, and the Guidelines for Good Clinical Practice criteria.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- GBD 2019 Tobacco Collaborators (2021). Spatial, temporal, and demographic patterns in prevalence of smoking tobacco use and attributable disease burden in 204 countries and territories, 1990-2019: a systematic analysis from the Global Burden of Disease Study. Lancet (London, England), 2019, 397, 2337–2360. [Google Scholar] [CrossRef]
- Bade, B.C.; Dela Cruz, C.S. Lung cancer 2020: Epidemiology, etiology, and prevention. Clin. Chest Med. 2020, 41, 1–24. [Google Scholar] [CrossRef]
- Shaykhiev, R.; Krause, A.; Salit, J.; Strulovici-Barel, Y.; Harvey, B.G.; O’Connor, T. P.; Crystal, R. G. Smoking-dependent reprogramming of alveolar macrophage polarization: implication for pathogenesis of chronic obstructive pulmonary disease. Journal of immunology 2009, 183, 2867–2883. [Google Scholar] [CrossRef] [PubMed]
- Pauwels, R. A.; Rabe, K. F. Burden and Clinical Features of Chronic Obstructive Pulmonary Disease (COPD). The Lancet 2004, 364, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Kotlyarov, S. The Role of Smoking in the Mechanisms of Development of Chronic Obstructive Pulmonary Disease and Atherosclerosis. IJMS 2023, 24, 8725. [Google Scholar] [CrossRef] [PubMed]
- Urbanek, K.; De Angelis, A.; Spaziano, G.; Piegari, E.; Matteis, M.; Cappetta, D.; Esposito, G.; Russo, R.; Tartaglione, G.; De Palma, R.; Rossi, F.; D’Agostino, B. Intratracheal Administration of Mesenchymal Stem Cells Modulates Tachykinin System, Suppresses Airway Remodeling and Reduces Airway Hyperresponsiveness in an Animal Model. PLoS ONE 2016, 11, e0158746. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, B.; Advenier, C.; De Palma, R.; Gallelli, L.; Marrocco, G.; Abbate, G. F.; Rossi, F. The Involvement of Sensory Neuropeptides in Airway Hyper-Responsiveness in Rabbits Sensitized and Challenged to Parietaria Judaica: Sensory Neuropeptides in Airway Hyper-Responsiveness. Clinical & Experimental Allergy 2002, 32, 472–479. [Google Scholar] [CrossRef] [PubMed]
- Roviezzo, F.; Sorrentino, R.; Terlizzi, M.; Riemma, M. A.; Iacono, V. M.; Rossi, A.; Spaziano, G.; Pinto, A.; D’Agostino, B.; Cirino, G. Toll-Like Receptor 4 Is Essential for the Expression of Sphingosine-1-Phosphate-Dependent Asthma-Like Disease in Mice. Frontiers in immunology 2017, 8, 1336. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, B.; Marrocco, G.; De Nardo, M.; Calò, G.; Guerrini, R.; Gallelli, L.; Advenier, C.; Rossi, F. Activation of the Nociceptin/Orphanin FQ Receptor Reduces Bronchoconstriction and Microvascular Leakage in a Rabbit Model of Gastroesophageal Reflux: N/OFQ Effects in the Airways in a GER Animal Model. British Journal of Pharmacology 2005, 144, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Rouget, C.; Cui, Y. Y.; D’Agostino, B.; Faisy, C.; Naline, E.; Bardou, M.; Advenier, C. Nociceptin Inhibits Airway Microvascular Leakage Induced by HCl Intra-Oesophageal Instillation: Nociceptin and Gastro-Oesophageal Reflux. British Journal of Pharmacology 2004, 141, 1077–1083. [Google Scholar] [CrossRef]
- Gallelli, L.; D’Agostino, B.; Marrocco, G.; De Rosa, G.; Filippelli, W.; Rossi, F.; Advenier, C. Role of Tachykinins in the Bronchoconstriction Induced by HCl Intraesophageal Instillation in the Rabbit. Life Sciences 2003, 72, 1135–1142. [Google Scholar] [CrossRef] [PubMed]
- Young, R. P.; Hopkins, R. J. Link between COPD and Lung Cancer. Respiratory Medicine 2010, 104, 758–759. [Google Scholar] [CrossRef] [PubMed]
- Schetter, A. J.; Heegaard, N. H. H.; Harris, C. C. Inflammation and Cancer: Interweaving MicroRNA, Free Radical, Cytokine and P53 Pathways. Carcinogenesis 2010, 31, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Caramori, G.; Adcock, I. M.; Casolari, P.; Ito, K.; Jazrawi, E.; Tsaprouni, L.; Villetti, G.; Civelli, M.; Carnini, C.; Chung, K. F.; Barnes, P. J.; Papi, A. Unbalanced Oxidant-Induced DNA Damage and Repair in COPD: A Link towards Lung Cancer. Thorax 2011, 66, 521–527. [Google Scholar] [CrossRef]
- Schaible, A. M.; Filosa, R.; Temml, V.; Krauth, V.; Matteis, M.; Peduto, A.; Bruno, F.; Luderer, S.; Roviezzo, F.; Di Mola, A.; de Rosa, M.; D’Agostino, B.; Weinigel, C.; Barz, D.; Koeberle, A.; Pergola, C.; Schuster, D.; Werz, O. Elucidation of the Molecular Mechanism and the Efficacy in Vivo of a Novel 1,4-Benzoquinone That Inhibits 5-Lipoxygenase. Br J Pharmacol 2014, 171, 2399–2412. [Google Scholar] [CrossRef]
- Schaible, A. M.; Filosa, R.; Krauth, V.; Temml, V.; Pace, S.; Garscha, U.; Liening, S.; Weinigel, C.; Rummler, S.; Schieferdecker, S.; Nett, M.; Peduto, A.; Collarile, S.; Scuotto, M.; Roviezzo, F.; Spaziano, G.; de Rosa, M.; Stuppner, H.; Schuster, D.; D’Agostino, B.; Werz, O. The 5-Lipoxygenase Inhibitor RF-22c Potently Suppresses Leukotriene Biosynthesis in Cellulo and Blocks Bronchoconstriction and Inflammation in Vivo. Biochemical Pharmacology 2016, 112, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Yanbaeva, D.G.; Dentener, M.A.; Creutzberg, E.C.; Wesseling, G.; Wouters, E.F.M. Systemic effects of smoking. Chest 2007, 131, 1557–1566. [Google Scholar] [CrossRef]
- Jensen, E. J.; Pedersen, B.; Frederiksen, R.; Dahl, R. Prospective study on the effect of smoking and nicotine substitution on leucocyte blood counts and relation between blood leucocytes and lung function. Thorax, 1998, 53, 784–789. [CrossRef] [PubMed]
- Kono, Y.; Colley, T.; Masako, To.; Papaioannou, A. I.; Mercado, N.; Baker, J. R.; To, Y.; Abe, S.; Haruki, K.; Ito, K.; Barnes, P. J. Cigarette smoke-induced impairment of autophagy in macrophages increases galectin-8 and inflammation. Scientific reports, 2021, 11, 335. [CrossRef]
- Hecht, S.S. Lung carcinogenesis by tobacco smoke. International journal of cancer, 2012, 131, 2724–2732. [CrossRef]
- Mark, N. M.; Kargl, J.; Busch, S. E.; Yang, G. H. Y.; Metz, H. E.; Zhang, H.; Hubbard, J. J.; Pipavath, S. N. J.; Madtes, D. K.; Houghton, A. M. Chronic Obstructive Pulmonary Disease Alters Immune Cell Composition and Immune Checkpoint Inhibitor Efficacy in Non–Small Cell Lung Cancer. Am J Respir Crit Care Med 2018, 197, 325–336. [Google Scholar] [CrossRef]
- Punturieri, A.; Szabo, E.; Croxton, T. L.; Shapiro, S. D.; Dubinett, S. M. Lung Cancer and Chronic Obstructive Pulmonary Disease: Needs and Opportunities for Integrated Research. JNCI Journal of the National Cancer Institute 2009, 101, 554–559. [Google Scholar] [CrossRef]
- sbv IMPROVER project team (in alphabetical order), Boue, S.; Fields, B.; Hoeng, J.; Park, J.; Peitsch, M. C.; Schlage, W. K.; Talikka, M.; Challenge Best Performers (in alphabetical order), Binenbaum, I.; Bondarenko, V.; Bulgakov, O. V.; Cherkasova, V.; Diaz-Diaz, N.; Fedorova, L.; Guryanova, S.; Guzova, J.; Igorevna Koroleva, G.; Kozhemyakina, E.; Kumar, R.; Zelikman, M. Enhancement of COPD biological networks using a web-based collaboration interface. F1000Research 2015, 4, 32. [CrossRef]
- Hautamaki, R.D.; Kobayashi, D.K.; Senior, R.M.; Shapiro, S.D. Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science (New York, N.Y.), 1997, 277, 2002–2004. [CrossRef]
- Shapiro, S.D. COPD unwound. The New England journal of medicine, 2005, 352, 2016–2019. [CrossRef]
- Shapiro, S.D.; Ingenito, E.P. The pathogenesis of chronic obstructive pulmonary disease: advances in the past 100 years. American journal of respiratory cell and molecular biology, 2005, 32, 367–372. [CrossRef]
- Hunninghake, G. M.; Cho, M. H.; Tesfaigzi, Y.; Soto-Quiros, M. E.; Avila, L.; Lasky-Su, J.; Stidley, C.; Melén, E.; Söderhäll, C.; Hallberg, J.; Kull, I.; Kere, J.; Svartengren, M.; Pershagen, G.; Wickman, M.; Lange, C.; Demeo, D. L.; Hersh, C. P.; Klanderman, B. J.; Raby, B. A.; Celedón, J. C. MMP12, lung function, and COPD in high-risk populations. The New England journal of medicine, 2009, 361, 2599–2608. [CrossRef]
- Bitterman, P. B.; Rennard, S. I.; Hunninghake, G. W.; Crystal, R. G. Human alveolar macrophage growth factor for fibroblasts. Regulation and partial characterization. The Journal of clinical investigation, 1982, 70, 806–822. [CrossRef]
- Chen, H.; Cowan, M. J.; Hasday, J. D.; Vogel, S. N.; Medvedev, A. E. Tobacco smoking inhibits expression of proinflammatory cytokines and activation of IL-1R-associated kinase, p38, and NF-kappaB in alveolar macrophages stimulated with TLR2 and TLR4 agonists. Journal of immunology (Baltimore, Md.: 1950), 2007, 179, 6097–6106. [CrossRef]
- Hodge, S.; Hodge, G.; Ahern, J.; Jersmann, H.; Holmes, M.; Reynolds, P. N. Smoking alters alveolar macrophage recognition and phagocytic ability: implications in chronic obstructive pulmonary disease. American journal of respiratory cell and molecular biology, 2007, 37, 748–755. [CrossRef]
- Stämpfli, M. R.; Anderson, G. P. How cigarette smoke skews immune responses to promote infection, lung disease and cancer. Nature reviews. Immunology, 2009, 9, 377–384. [CrossRef]
- Heguy, A.; O’Connor, T. P.; Luettich, K.; Worgall, S.; Cieciuch, A.; Harvey, B. G.; Hackett, N. R.; Crystal, R. G. Gene expression profiling of human alveolar macrophages of phenotypically normal smokers and nonsmokers reveals a previously unrecognized subset of genes modulated by cigarette smoking. Journal of molecular medicine (Berlin, Germany), 2006, 84, 318–328. [CrossRef]
- Shaykhiev, R.; Krause, A.; Salit, J.; Strulovici-Barel, Y.; Harvey, B. G.; O’Connor, T. P.; Crystal, R. G. Smoking-dependent reprogramming of alveolar macrophage polarization: implication for pathogenesis of chronic obstructive pulmonary disease. Journal of immunology (Baltimore, Md.: 1950), 2009, 183, 2867–2883. [CrossRef]
- Woodruff, P. G.; Koth, L. L.; Yang, Y. H.; Rodriguez, M. W.; Favoreto, S.; Dolganov, G. M.; Paquet, A. C.; Erle, D. J. (2005). A distinctive alveolar macrophage activation state induced by cigarette smoking. American journal of respiratory and critical care medicine, 2005, 172, 1383–1392. [CrossRef]
- Lu, L.-F.; Liston, A. MicroRNA in the Immune System, MicroRNA as an Immune System. Immunology 2009, 127, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Iannone, F.; Montesanto, A.; Cione, E.; Crocco, P.; Caroleo, M. C.; Dato, S.; Rose, G.; Passarino, G. Expression Patterns of Muscle-Specific MiR-133b and MiR-206 Correlate with Nutritional Status and Sarcopenia. Nutrients 2020, 12, 297. [Google Scholar] [CrossRef] [PubMed]
- Cannataro, R.; Caroleo, M. C.; Fazio, A.; La Torre, C.; Plastina, P.; Gallelli, L.; Lauria, G.; Cione, E. Ketogenic Diet and MicroRNAs Linked to Antioxidant Biochemical Homeostasis. Antioxidants 2019, 8, 269. [Google Scholar] [CrossRef] [PubMed]
- Molina-Pinelo, S.; Pastor, M. D.; Suarez, R.; Romero-Romero, B.; Gonzalez De la Pena, M.; Salinas, A.; Garcia-Carbonero, R.; De Miguel, M. J.; Rodriguez-Panadero, F.; Carnero, A.; Paz-Ares, L. MicroRNA Clusters: Dysregulation in Lung Adenocarcinoma and COPD. European Respiratory Journal 2014, 43, 1740–1749. [Google Scholar] [CrossRef] [PubMed]
- Mirra, D.; Cione, E.; Spaziano, G.; Esposito, R.; Sorgenti, M.; Granato, E.; Cerqua, I.; Muraca, L.; Iovino, P.; Gallelli, L.; D’Agostino, B. Circulating MicroRNAs Expression Profile in Lung Inflammation: A Preliminary Study. JCM 2022, 11, 5446. [Google Scholar] [CrossRef]
- Mirra, D.; Esposito, R.; Spaziano, G.; Sportiello, L.; Panico, F.; Squillante, A.; Falciani, M.; Cerqua, I.; Gallelli, L.; Cione, E.; et al. MicroRNA Monitoring in Human Alveolar Macrophages from Patients with Smoking-Related Lung Diseases: A Preliminary Study. Biomedicines 2024, 12, 1050. [Google Scholar] [CrossRef]
- Zhang, L.; Liao, Y.; Tang, L. MicroRNA-34 Family: A Potential Tumor Suppressor and Therapeutic Candidate in Cancer. J Exp Clin Cancer Res 2019, 38, 53. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y. MiR-223-5p Suppresses OTX1 to Mediate Malignant Progression of Lung Squamous Cell Carcinoma Cells. Computational and Mathematical Methods in Medicine 2021, 2021, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Luo, Y.; Lin, M.; Peng, X.; Liu, M.; Wang, Y.; Li, S.; Yang, D.; Yang, Z. Serum Exosomal miR -16-5p Functions as a Tumor Inhibitor and a New Biomarker for PD-L1 Inhibitor-dependent Immunotherapy in Lung Adenocarcinoma by Regulating PD-L1 Expression. Cancer Medicine 2022, 11, 2627–2643. [Google Scholar] [CrossRef]
- Ye, T.; Changyu, S.; Limeng, Z.; Yuan, P. Clinical Significance of MiRNA - 106a in Non-Small Cell Lung Cancer Patients Who Received Cisplatin Combined with Gemcitabine Chemotherapy. Cancer Biology & Medicine 2018, 15, 157. [Google Scholar] [CrossRef]
- Han, J.; Hu, J.; Sun, F.; Bian, H.; Tang, B.; Fang, X. MicroRNA-20a-5p Suppresses Tumor Angiogenesis of Non-Small Cell Lung Cancer through RRM2-Mediated PI3K/Akt Signaling Pathway. Mol Cell Biochem 2021, 476, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, Y.; Qi, P.; Ma, Z. Biology of MiR-17-92 Cluster and Its Progress in Lung Cancer. Int. J. Med. Sci. 2018, 15, 1443–1448. [Google Scholar] [CrossRef] [PubMed]
- Sweat, Y.; Ries, R. J.; Sweat, M.; Su, D.; Shao, F.; Eliason, S.; Amendt, B. A. MiR-17 Acts as a Tumor Suppressor by Negatively Regulating the MiR-17-92 Cluster. Molecular Therapy - Nucleic Acids 2021, 26, 1148–1158. [Google Scholar] [CrossRef] [PubMed]
- Sokolowski, J.W., Jr.; Burgher, L.W.; Jones, F.L., Jr.; Patterson, J.R.; Selecky, P.A. Guidelines for fiberoptic bronchoscopy in adults. American Thoracic Society guidelines. Medical Section of the American Lung Association. Am. Rev. Respir. Dis. 1987, 136, 1066. [Google Scholar] [CrossRef] [PubMed]
- de Torres, J.P.; Marín, J.M.; Casanova, C.; Cote, C.; Carrizo, S.; Cordoba-Lanus, E.; Baz-Dávila, R.; Zulueta, J.J.; Aguirre-Jaime, A.; Saetta, M.; et al. Lung Cancer in Patients with Chronic Obstructive Pulmonary Disease: Incidence and Predicting Factors. Am. J. Respir. Crit. Care Med. 2011, 184, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Park, H.Y.; Kang, D.; Shin, S.H.; Yoo, K.-H.; Rhee, C.K.; Suh, G.Y.; Kim, H.; Shim, Y.M.; Guallar, E.; Cho, J.; et al. Chronic Obstructive Pulmonary Disease and Lung Cancer Incidence in Never Smokers: A Cohort Study. Thorax 2020, 75, 506–509. [Google Scholar] [CrossRef] [PubMed]
- Willinger, C. M.; Rong, J.; Tanriverdi, K.; Courchesne, P. L.; Huan, T.; Wasserman, G. A.; Lin, H.; Dupuis, J.; Joehanes, R.; Jones, M. R.; Chen, G.; Benjamin, E. J.; O’Connor, G. T.; Mizgerd, J. P.; Freedman, J. E.; Larson, M. G.; & Levy, D. MicroRNA Signature of Cigarette Smoking and Evidence for a Putative Causal Role of MicroRNAs in Smoking-Related Inflammation and Target Organ Damage. Circulation. Cardiovascular genetics 2017, 10, e001678. [CrossRef] [PubMed]
- Lofdahl, J. M. Bronchoalveolar Lavage in COPD: Fluid Recovery Correlates with the Degree of Emphysema. European Respiratory Journal 2005, 25, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Aoshiba, K.; Tamaoki, J.; Nagai, A. Acute cigarette smoke exposure induces apoptosis of alveolar macrophages. Am J Physiol Lung Cell Mol Physiol 2001, 281, L1392–401. [Google Scholar] [CrossRef]
- Long, Y. J.; Liu, X. P.; Chen, S. S.; Zong, D. D.; Chen, Y.; Chen, P. miR-34a is involved in CSE-induced apoptosis of human pulmonary microvascular endothelial cells by targeting Notch-1 receptor protein. Respiratory research 2018, 19, 21. [Google Scholar] [CrossRef]
- Zeng, X. L.; Yang, X. N.; Liu, X. J. Resveratrol attenuates cigarette smoke extract induced cellular senescence in human airway epithelial cells by regulating the miR-34a/SIRT1/NF-κB pathway. Medicine 2022, 101, e31944. [Google Scholar] [CrossRef] [PubMed]
- Mirra, D.; Esposito, R.; Spaziano, G.; La Torre, C.; Vocca, C.; Tallarico, M.; Cione, E.; Gallelli, L.; D’Agostino, B. Lung MicroRNAs Expression in Lung Cancer and COPD: A Preliminary Study. Biomedicines 2023, 11, 736. [Google Scholar] [CrossRef] [PubMed]
- Danov, O.; Wolff, M.; Bartel, S.; Böhlen, S.; Obernolte, H.; Wronski, S.; Jonigk, D.; Hammer, B.; Kovacevic, D.; Reuter, S.; Krauss-Etschmann, S.; Sewald, K. Cigarette Smoke Affects Dendritic Cell Populations, Epithelial Barrier Function, and the Immune Response to Viral Infection With H1N1. Front. Med. 2020, 7, 571003. [Google Scholar] [CrossRef] [PubMed]
- Barclay, A. N.; Brown, M. H. The SIRP Family of Receptors and Immune Regulation. Nat Rev Immunol 2006, 6, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Pan, C.; Li, L.; Bian, Z.; Lv, Z.; Shi, L.; Zhang, J.; Li, D.; Gu, H.; Zhang, C.-Y.; Liu, Y.; Zen, K. MicroRNA-17/20a/106a Modulate Macrophage Inflammatory Responses through Targeting Signal-Regulatory Protein α. Journal of Allergy and Clinical Immunology 2013, 132, 426–436. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Pamer, E. G. Monocyte recruitment during infection and inflammation. Nat. Rev. Immunol 2011, 11, 762–774. [Google Scholar] [CrossRef]
- Lee, C.C.; Avalos, A. M.; Ploegh, H. L. Accessory molecules for Tolllike receptors and their function. Nat. Rev. Immunol.
- Moon, H. G.; Yang, J.; Zheng, Y.; Jin, Y. miR-15a/16 regulates macrophage phagocytosis after bacterial infection. Journal of immunology 2014, (Baltimore, Md.: 1950), 193, 4558–4567. [CrossRef]
- Liu, K.; Hong, D.; Zhang, F.; Li, X.; He, M.; Han, X.; Zhang, G.; Xu, G.; Stonehouse, N. J.; Jiang, Z.; An, W.; Guo, L. MicroRNA-106a Inhibits Autophagy Process and Antimicrobial Responses by Targeting ULK1, ATG7, and ATG16L1 During Mycobacterial Infection. Frontiers in immunology 2021, 11, 610021. [Google Scholar] [CrossRef] [PubMed]
- Vij, N.; Chandramani-Shivalingappa, P.; Van Westphal, C.; Hole, R. Cigarette smoke-induced autophagy impairment accelerates lung aging, COPD-emphysema exacerbations and pathogenesis. Am J Physiol Cell Physiol 2018, 314, C73-C87. [CrossRef]
- Sharma, A.; Kumar, M.; Ahmad, T.; Mabalirajan, U.; Aich, J.; Agrawal, A.; Ghosh, B. Antagonism of Mmu-Mir-106a Attenuates Asthma Features in Allergic Murine Model. Journal of Applied Physiology 2012, 113, 459–464. [Google Scholar] [CrossRef] [PubMed]
- Roffel, M. P.; Bracke, K. R.; Heijink, I. H.; Maes, T. MiR-223: A Key Regulator in the Innate Immune Response in Asthma and COPD. Front. Med. 2020, 7, 196. [Google Scholar] [CrossRef]
- Schembri, F.; Sridhar, S.; Perdomo, C.; Gustafson, A. M.; Zhang, X.; Ergun, A.; Lu, J.; Liu, G.; Zhang, X.; Bowers, J.; Vaziri, C.; Ott, K.; Sensinger, K.; Collins, J. J.; Brody, J. S.; Getts, R.; Lenburg, M. E.; Spira, A. MicroRNAs as Modulators of Smoking-Induced Gene Expression Changes in Human Airway Epithelium. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 2319–2324. [Google Scholar] [CrossRef]
- Lugg, S. T.; Scott, A.; Parekh, D.; Naidu, B.; Thickett, D. R. Cigarette smoke exposure and alveolar macrophages: mechanisms for lung disease. Thorax 2022, 77, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Roffel, M. P.; Maes, T.; Brandsma, C. A.; van den Berge, M.; Vanaudenaerde, B. M.; Joos, G. F.; Brusselle, G. G.; Heijink, I. H.; Bracke, K. R. MiR-223 is increased in lungs of patients with COPD and modulates cigarette smoke-induced pulmonary inflammation. American journal of physiology. Lung cellular and molecular physiology 2021, 321, L1091–L1104. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Yu, H.; Guo, Q. MicroRNA-20a promotes inflammation via the nuclear factor-κB signaling pathway in pediatric pneumonia. Molecular medicine reports 2018, 17, 612–617. [Google Scholar] [CrossRef] [PubMed]
- Polverino, F.; Mirra, D.; Yang, C. X.; Esposito, R.; Spaziano, G.; Rojas-Quintero, J.; Sgambato, M.; Piegari, E.; Cozzolino, A.; Cione, E.; Gallelli, L.; Capuozzo, A.; Santoriello, C.; Berrino, L.; de-Torres, J. P.; Hackett, T. L.; Polverino, M.; D’Agostino, B. Similar Programmed Death Ligand 1 (PD-L1) Expression Profile in Patients with Mild COPD and Lung Cancer. Sci Rep 2022, 12, 22402. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-H.; Goswami, S.; Grudo, A.; Song, L.; Bandi, V.; Goodnight-White, S.; Green, L.; Hacken-Bitar, J.; Huh, J.; Bakaeen, F.; Coxson, H. O.; Cogswell, S.; Storness-Bliss, C.; Corry, D. B.; Kheradmand, F. Antielastin Autoimmunity in Tobacco Smoking–Induced Emphysema. Nat Med 2007, 13, 567–569. [Google Scholar] [CrossRef] [PubMed]
- Polverino, F.; Laucho-Contreras, M.; Rojas Quintero, J.; Divo, M.; Pinto-Plata, V.; Sholl, L.; de-Torres, J. P.; Celli, B. R.; Owen, C. A. Increased Expression of A Proliferation-Inducing Ligand (APRIL) in Lung Leukocytes and Alveolar Epithelial Cells in COPD Patients with Non Small Cell Lung Cancer: A Possible Link between COPD and Lung Cancer? Multidiscip Respir Med 2016, 11, 17. [Google Scholar] [CrossRef] [PubMed]
- Qiu, C.; Chen, G.; Cui, Q. Towards the Understanding of MicroRNA and Environmental Factor Interactions and Their Relationships to Human Diseases. Sci Rep 2012, 2, 318. [Google Scholar] [CrossRef] [PubMed]
- Ryter, S. W.; Choi, A. M. ; Autophagy in the lung. Proceedings of the American Thoracic Society 2010, 7, 13–21. [Google Scholar] [CrossRef]
- Zeng, H.; Li, T.; He, X.; Cai, S.; Luo, H.; Chen, P.; Chen, Y. Oxidative stress mediates the apoptosis and epigenetic modification of the Bcl-2 promoter via DNMT1 in a cigarette smoke-induced emphysema model. Respiratory research 2020, 21, 229. [Google Scholar] [CrossRef]
- Li, L.; Zhang, Y.; Gong, J.; Yang, G.; Zhi, S.; Ren, D.; Zhao, H. Cpt1a alleviates cigarette smoke-induced chronic obstructive pulmonary disease. Experimental and therapeutic medicine 2022, 25, 54. [Google Scholar] [CrossRef]
- Xia, S.; Qu, J.; Jia, H.; He, W.; Li, J.; Zhao, L.; Mao, M.; Zhao, Y. Overexpression of Forkhead box C1 attenuates oxidative stress, inflammation and apoptosis in chronic obstructive pulmonary disease. Life sciences 2019, 216, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Dentener, M. A.; Vernooy, J. H.; Hendriks, S.; Wouters, E. F. Enhanced levels of hyaluronan in lungs of patients with COPD: relationship with lung function and local inflammation. Thorax 2005, 60, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Hlapčić, I.; Hulina-Tomašković, A.; Grdić Rajković, M.; Popović-Grle, S.; Vukić Dugac, A.; Rumora, L. Association of Plasma Heat Shock Protein 70 with Disease Severity, Smoking and Lung Function of Patients with Chronic Obstructive Pulmonary Disease. Journal of clinical medicine 2020, 9, 3097. [Google Scholar] [CrossRef]
- Hayama, Y.; Kimura, T.; Takeda, Y.; Nada, S.; Koyama, S.; Takamatsu, H.; Kang, S.; Ito, D.; Maeda, Y.; Nishide, M.; Nojima, S.; Sarashina-Kida, H.; Hosokawa, T.; Kinehara, Y.; Kato, Y.; Nakatani, T.; Nakanishi, Y.; Tsuda, T.; Koba, T.; Okada, M.; Kumanogoh, A. Lysosomal Protein Lamtor1 Controls Innate Immune Responses via Nuclear Translocation of Transcription Factor EB. Journal of immunology 2018, (Baltimore, Md.: 1950), 200, 3790–3800. [CrossRef]
- Bewley, M. A.; Preston, J. A.; Mohasin, M.; Marriott, H. M.; Budd, R. C.; Swales, J.; Collini, P.; Greaves, D. R.; Craig, R. W.; Brightling, C. E.; Donnelly, L. E.; Barnes, P. J.; Singh, D.; Shapiro, S. D.; Whyte, M. K. B.; Dockrell, D. H. Impaired Mitochondrial Microbicidal Responses in Chronic Obstructive Pulmonary Disease Macrophages. American journal of respiratory and critical care medicine 2017, 196, 845–855. [Google Scholar] [CrossRef] [PubMed]
- Giordano, L.; Gregory, A. D.; Pérez Verdaguer, M.; Ware, S. A.; Harvey, H.; DeVallance, E.; Brzoska, T.; Sundd, P.; Zhang, Y.; Sciurba, F. C.; Shapiro, S. D.; Kaufman, B. A. Extracellular Release of Mitochondrial DNA: Triggered by Cigarette Smoke and Detected in COPD. Cells 2022, 11, 369. [Google Scholar] [CrossRef]
- Murray, R. Z.; Stow, J. L. Cytokine Secretion in Macrophages: SNAREs, Rabs, and Membrane Trafficking. Frontiers in immunology, 2014, 5, 538. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Li, D.; Zhan, W.; He, K.; Yang, H. (2020). Downregulation of SENP1 suppresses LPS-induced macrophage inflammation by elevating Sp3 SUMOylation and disturbing Sp3-NF-κB interaction. American journal of translational research 2020, 12, 7439–7448. [Google Scholar]
- Diao, X.; Zhou, J.; Wang, S.; Ma, X. Upregulation of miR-132 contributes to the pathophysiology of COPD via targeting SOCS5. Experimental and molecular pathology, 2018, 105, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Dutta, R. K.; Chinnapaiyan, S.; Rasmussen, L.; Raju, S. V.; Unwalla, H. J. (2019). A Neutralizing Aptamer to TGFBR2 and miR-145 Antagonism Rescue Cigarette Smoke- and TGF-β-Mediated CFTR Expression. Molecular therapy: the journal of the American Society of Gene Therapy, 2019 27, 442–455. [CrossRef]
- Michaud, S. E.; Dussault, S.; Groleau, J.; Haddad, P.; Rivard, A. Cigarette smoke exposure impairs VEGF-induced endothelial cell migration: role of NO and reactive oxygen species. Journal of molecular and cellular cardiology 2006, 41, 275–284. [Google Scholar] [CrossRef]
Figure 1.
THP-1 cells viability after CS exposure. THP-1 cells were treated with CS at 24h for the indicated concentration, and the cell viability was assessed by MTT assay. Cell viability is shown as absorbance at 595 nm. All samples were run in triplicate, and results are shown as means ± SD. The statistical tests used in these analyses were one-way analysis of variance followed by Dunnet Multiple Comparison Test. *** p < 0.001, **** p < 0.0001.
Figure 1.
THP-1 cells viability after CS exposure. THP-1 cells were treated with CS at 24h for the indicated concentration, and the cell viability was assessed by MTT assay. Cell viability is shown as absorbance at 595 nm. All samples were run in triplicate, and results are shown as means ± SD. The statistical tests used in these analyses were one-way analysis of variance followed by Dunnet Multiple Comparison Test. *** p < 0.001, **** p < 0.0001.
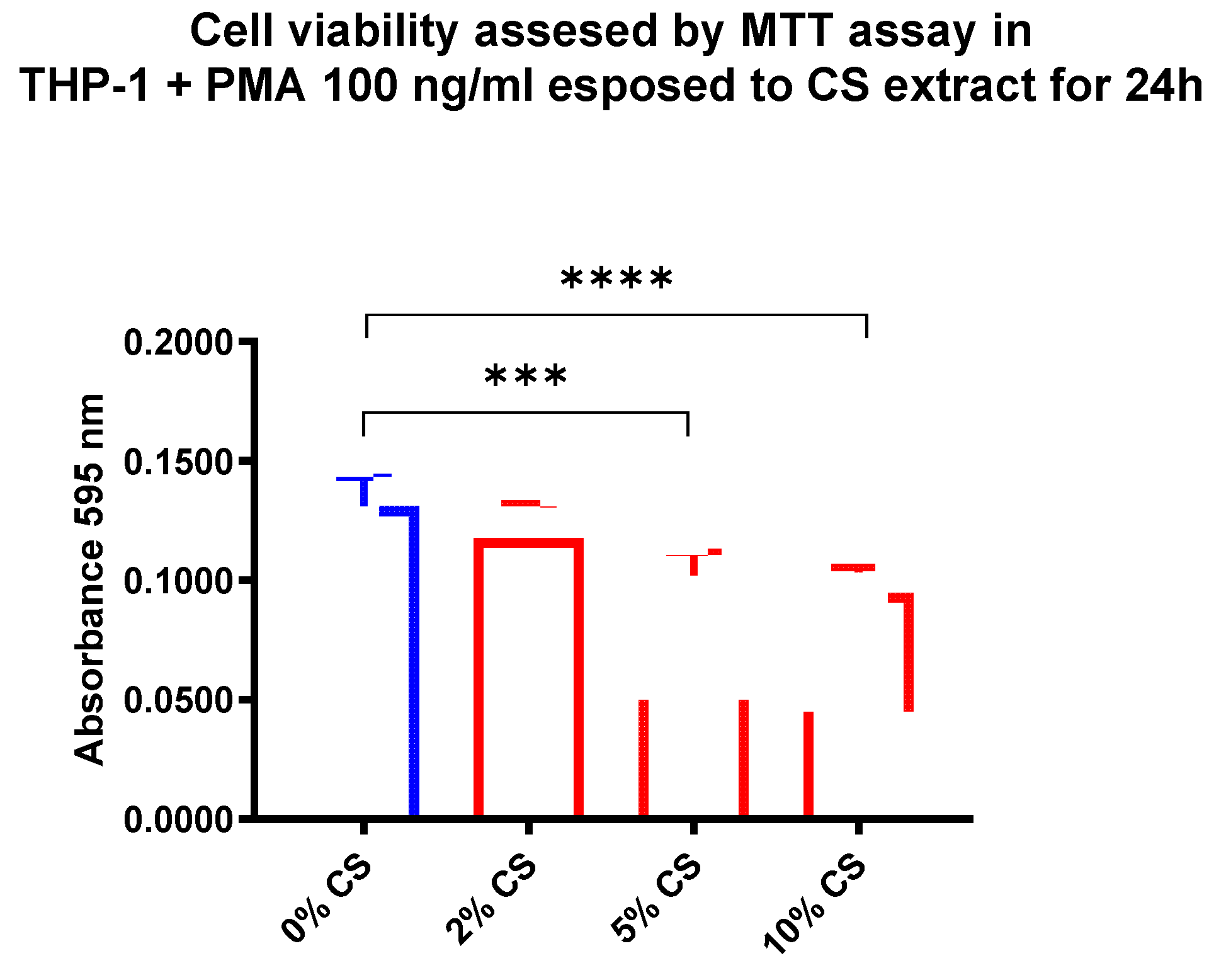
Figure 2.
Analysis of hsa-miR-34a-5p AMs expression levels in HNS (2A; biological replicates n = 9), HS (2B; biological replicates n = 11), COPD (2C; biological replicates n = 11) and NSCLC (2D; biological replicates n = 12) before and after 10% CS at 24 h. AMs from BAL all groups were treated with CS at 10% for 24 h and the expression of hsa-miR-34a-5p was assessed by real-time RT-PCR. All samples were run in triplicate, and results are shown as means ± SD. The statistical tests used in these analyses were one-way analysis of variance followed by Tukey Multiple Comparison Test. ** p < 0.01, **** p < 0.0001.
Figure 2.
Analysis of hsa-miR-34a-5p AMs expression levels in HNS (2A; biological replicates n = 9), HS (2B; biological replicates n = 11), COPD (2C; biological replicates n = 11) and NSCLC (2D; biological replicates n = 12) before and after 10% CS at 24 h. AMs from BAL all groups were treated with CS at 10% for 24 h and the expression of hsa-miR-34a-5p was assessed by real-time RT-PCR. All samples were run in triplicate, and results are shown as means ± SD. The statistical tests used in these analyses were one-way analysis of variance followed by Tukey Multiple Comparison Test. ** p < 0.01, **** p < 0.0001.
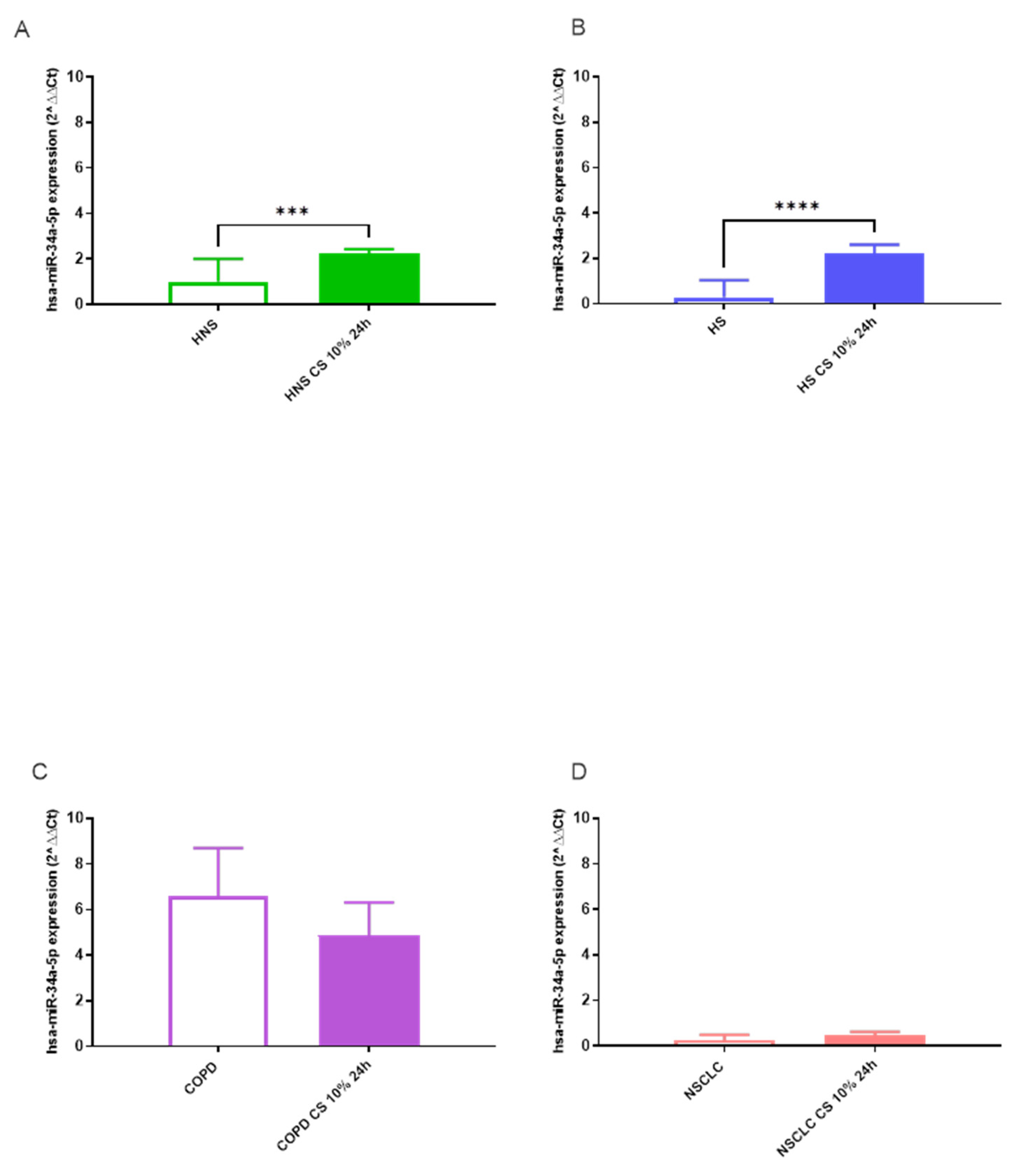
Figure 3.
Analysis of hsa-miR-17-5p AMs expression levels in HNS (2A; biological replicates n = 9), HS (2B; biological replicates n = 11), COPD (2C; biological replicates n = 11) and NSCLC (2D; biological replicates n = 12) before and after 10% CS at 24 h. AMs from BAL all groups were treated with CS at 10% for 24 h and the expression of hsa-miR-17-5p was assessed by real-time RT-PCR. All samples were run in triplicate, and results are shown as means ± SD. The statistical tests used in these analyses were one-way analysis of variance followed by Tukey Multiple Comparison Test. *** p < 0.001, **** p < 0.0001.
Figure 3.
Analysis of hsa-miR-17-5p AMs expression levels in HNS (2A; biological replicates n = 9), HS (2B; biological replicates n = 11), COPD (2C; biological replicates n = 11) and NSCLC (2D; biological replicates n = 12) before and after 10% CS at 24 h. AMs from BAL all groups were treated with CS at 10% for 24 h and the expression of hsa-miR-17-5p was assessed by real-time RT-PCR. All samples were run in triplicate, and results are shown as means ± SD. The statistical tests used in these analyses were one-way analysis of variance followed by Tukey Multiple Comparison Test. *** p < 0.001, **** p < 0.0001.
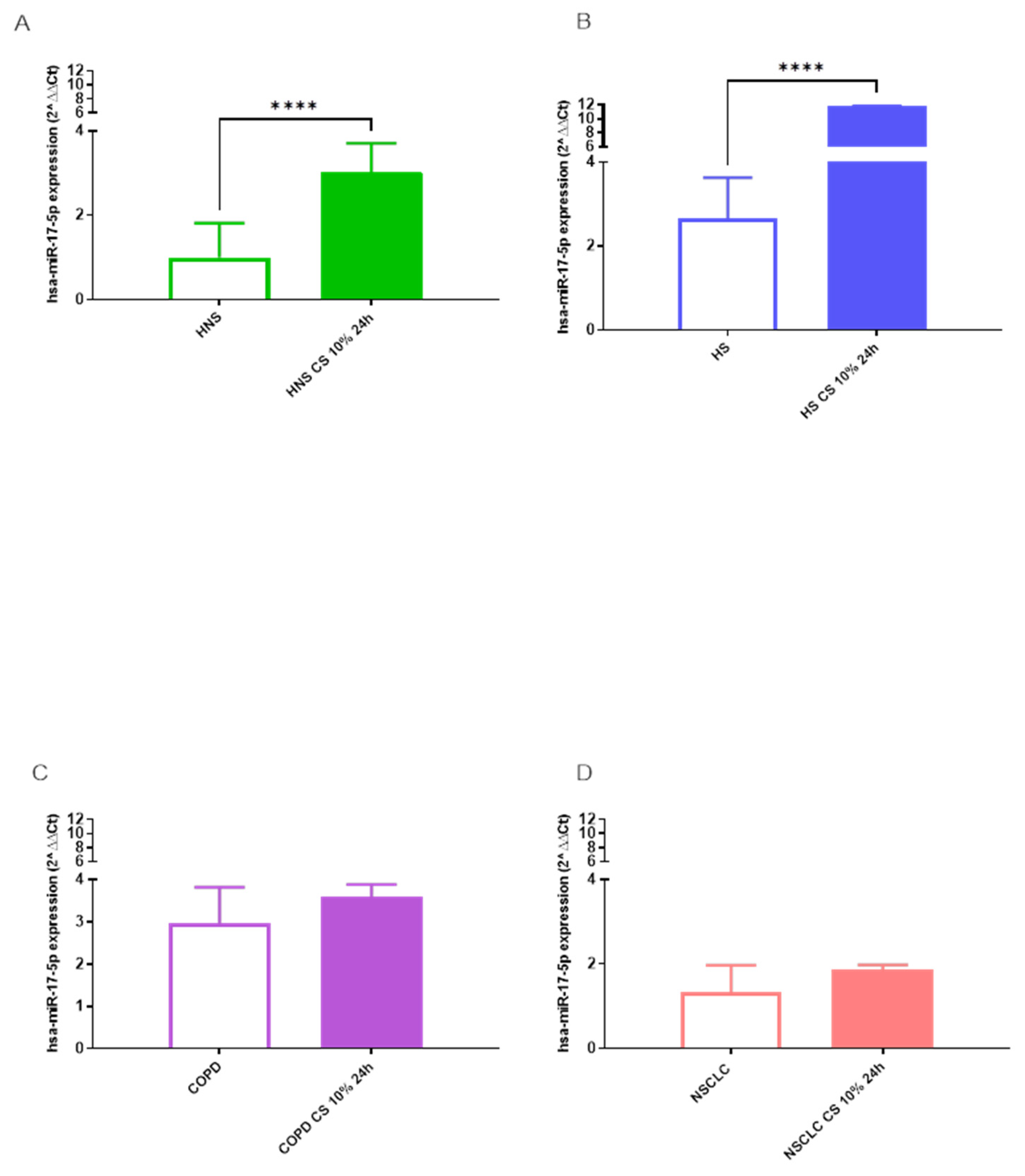
Figure 4.
Analysis of hsa-miR-16-5p AMs expression levels in HNS (2A; biological replicates n = 9), HS (2B; biological replicates n = 11), COPD (2C; biological replicates n = 11) and NSCLC (2D; biological replicates n = 12) before and after 10% CS at 24 h. AMs from BAL all groups were treated with CS at 10% for 24 h and the expression of hsa-miR-16-5p was assessed by real-time RT-PCR. All samples were run in triplicate, and results are shown as means ± SD. The statistical tests used in these analyses were one-way analysis of variance followed by Tukey Multiple Comparison Test. *** p < 0.001, **** p < 0.0001.
Figure 4.
Analysis of hsa-miR-16-5p AMs expression levels in HNS (2A; biological replicates n = 9), HS (2B; biological replicates n = 11), COPD (2C; biological replicates n = 11) and NSCLC (2D; biological replicates n = 12) before and after 10% CS at 24 h. AMs from BAL all groups were treated with CS at 10% for 24 h and the expression of hsa-miR-16-5p was assessed by real-time RT-PCR. All samples were run in triplicate, and results are shown as means ± SD. The statistical tests used in these analyses were one-way analysis of variance followed by Tukey Multiple Comparison Test. *** p < 0.001, **** p < 0.0001.
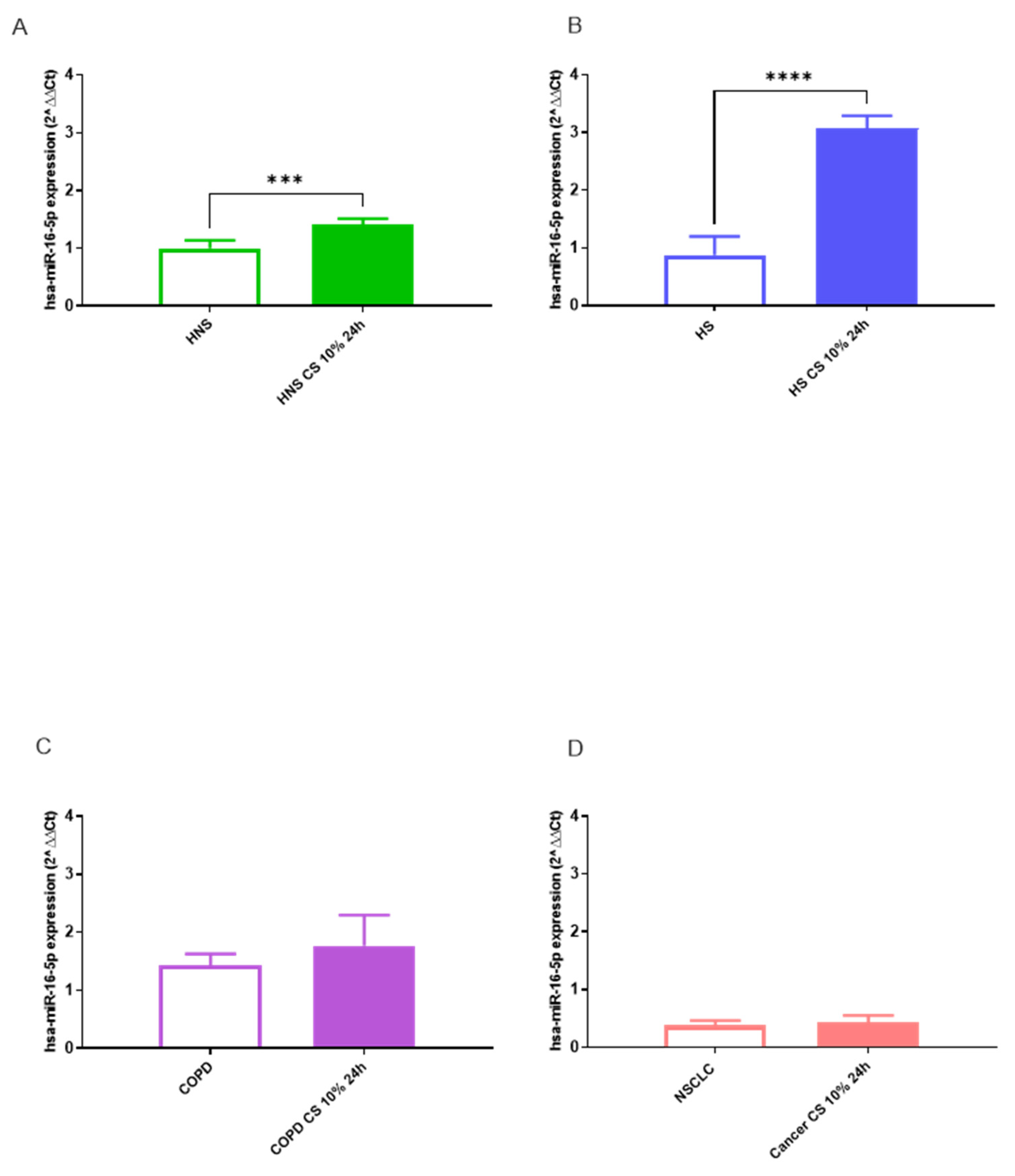
Figure 5.
Analysis of hsa-miR-106a-5p AMs expression levels in HNS (2A; biological replicates n = 9), HS (2B; biological replicates n = 11), COPD (2C; biological replicates n = 11) and NSCLC (2D; biological replicates n = 12) and after 10% CS at 24 h. AMs from BAL all groups were treated with CS at 10% for 24 h and the expression of hsa-miR-106a-5p was assessed by real-time RT-PCR. All samples were run in triplicate, and results are shown as means ± SD. The statistical tests used in these analyses were one-way analysis of variance followed by Tukey Multiple Comparison Test. * p < 0.05, ** p < 0.01.
Figure 5.
Analysis of hsa-miR-106a-5p AMs expression levels in HNS (2A; biological replicates n = 9), HS (2B; biological replicates n = 11), COPD (2C; biological replicates n = 11) and NSCLC (2D; biological replicates n = 12) and after 10% CS at 24 h. AMs from BAL all groups were treated with CS at 10% for 24 h and the expression of hsa-miR-106a-5p was assessed by real-time RT-PCR. All samples were run in triplicate, and results are shown as means ± SD. The statistical tests used in these analyses were one-way analysis of variance followed by Tukey Multiple Comparison Test. * p < 0.05, ** p < 0.01.
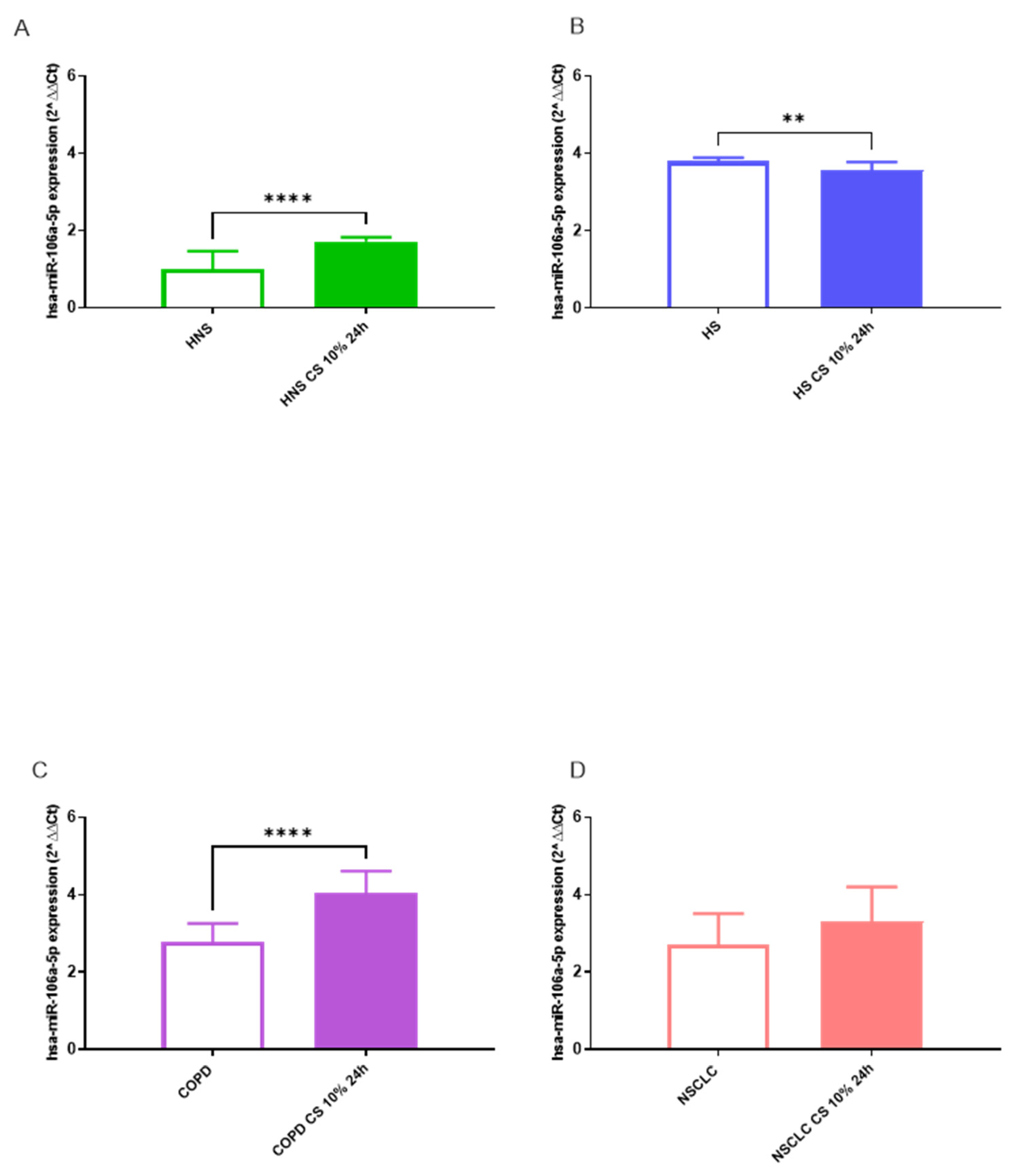
Figure 6.
Analysis of hsa-miR-223-5p AMs expression levels in HNS (2A; biological replicates n = 9), HS (2B; biological replicates n = 11), COPD (2C; biological replicates n = 11) and NSCLC (2D; biological replicates n = 12) before and after 10% CS at 24 h. AMs from BAL all groups were treated with CS at 10% for 24 h and the expression of hsa-miR-223-5p was assessed by real-time RT-PCR. All samples were run in triplicate, and results are shown as means ± SD. The statistical tests used in these analyses were one-way analysis of variance followed by Tukey Multiple Comparison Test. ** p < 0.01, *** p < 0.001.
Figure 6.
Analysis of hsa-miR-223-5p AMs expression levels in HNS (2A; biological replicates n = 9), HS (2B; biological replicates n = 11), COPD (2C; biological replicates n = 11) and NSCLC (2D; biological replicates n = 12) before and after 10% CS at 24 h. AMs from BAL all groups were treated with CS at 10% for 24 h and the expression of hsa-miR-223-5p was assessed by real-time RT-PCR. All samples were run in triplicate, and results are shown as means ± SD. The statistical tests used in these analyses were one-way analysis of variance followed by Tukey Multiple Comparison Test. ** p < 0.01, *** p < 0.001.

Figure 7.
Analysis of hsa-miR-20a-5p AMs expression levels in HNS (2A; biological replicates n = 9), HS (2B; biological replicates n = 11), COPD (2C; biological replicates n = 11) and NSCLC (2D; biological replicates n = 12) before and after 10% CS at 24 h. AMs from BAL all groups were treated with CS at 10% for 24 h and the expression of hsa-miR-20a-5p was assessed by real-time RT-PCR. All samples were run in triplicate, and results are shown as means ± SD. The statistical tests used in these analyses were one-way analysis of variance followed by Tukey Multiple Comparison Test. * p < 0.05, ** p < 0.01, **** p < 0.0001.
Figure 7.
Analysis of hsa-miR-20a-5p AMs expression levels in HNS (2A; biological replicates n = 9), HS (2B; biological replicates n = 11), COPD (2C; biological replicates n = 11) and NSCLC (2D; biological replicates n = 12) before and after 10% CS at 24 h. AMs from BAL all groups were treated with CS at 10% for 24 h and the expression of hsa-miR-20a-5p was assessed by real-time RT-PCR. All samples were run in triplicate, and results are shown as means ± SD. The statistical tests used in these analyses were one-way analysis of variance followed by Tukey Multiple Comparison Test. * p < 0.05, ** p < 0.01, **** p < 0.0001.
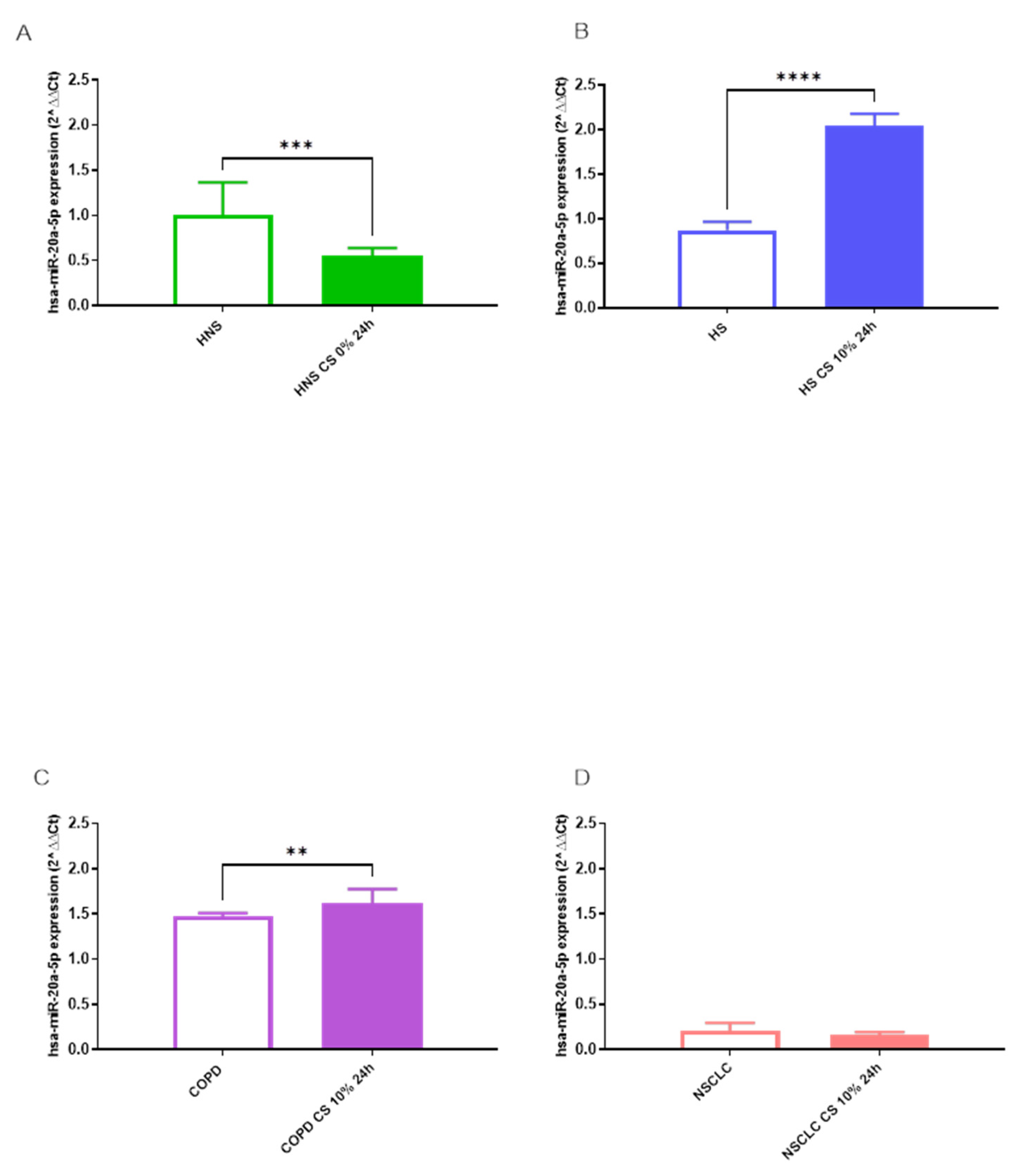
Table 2.
Abbreviations, gene names, methods, and tissue on which the miRNA selected were validated targets from miR Target Link 2.0.
Table 2.
Abbreviations, gene names, methods, and tissue on which the miRNA selected were validated targets from miR Target Link 2.0.
| Abbreviation | Gene Name | Methods | Tissues | References (PMID) |
|---|---|---|---|---|
| ATG14 | Autophagy Related 14 | Sequencing, HITS-CLIP |
Embryonic kidney cells, B cells |
20371350 22473208 |
| BCL2 | BCL2 Apoptosis Regulator | Luciferase reporter assay, qRT-PCR, Western blot, Proteomics analysis, Immunohistochemistry, Microarray, Sequencing, HITS-CLIP, Immunoblot, Immunoprecipitaion | Cervix cells, gastric cells, bone cells, marrow cells, spleen, liver, kidney, lymph node, tracheal/bronchial epithelial cells, breast cells, ovary cells, human embryonic kidney cells, B cells, mesothelial cell, glioma cells |
17877811 18449891 18362358 17351108 17707831 20643754 20876285 19269153 16166262 19903841 20371350 23907579 22473208 24148817 25435430 26397135 26722459 |
| CPT1A | Carnitine Palmitoyltransferase 1A | Proteomics HITS-CLIP |
Cervix cells, neuronal cells |
18668040 23313552 |
| FOXC1 | Forkhead Box C1 | PAR-CLIP | Human embryonic kidney cells | 21572407 |
| HAS2 | Hyaluronan Synthase 2 | HITS-CLIP | B-cell | 22927820 |
| HSPA1A | Heat Shock Protein Family A (Hsp70) Member 1A | Microarray pSILAC, Proteomics, PAR-CLIP | Leukemic cells, cervix cells, human embryonic stem cells |
18362358 18668040 22012620 |
| LAMTOR1 | Late Endosomal/Lysosomal Adaptor, MAPK And MTOR Activator 1 | Proteomics, PAR-CLIP |
Cervix cells, brain tissue human embryonic kidney cells, B cells |
18668040 24398324 23446348 20371350 |
| MCL1 | MCL1 Apoptosis Regulator, BCL2 Family Member | HITS-CLIP, microarray, Immunohistochemistry, Luciferase reporter assay, qRT-PCR, Western blot, PCR array | Human embryonic kidney cells, leukemic cells, liver | 22473208 18362358 23594563 28097098 |
| MFN2 | Mitofusin 2 | Proteomics, luciferase reporter assay, western blot, CLASH | Breast cells, lungs, human embryonic kidney cells | 18668040 27640178 23622248 |
| SCAMP5 | Secretory Carrier Membrane Protein 5 | HITS-CLIP | B cells | 22473208 22473208 |
| SENP1 | SUMO Specific Peptidase 1 | HITS-CLIP, PAR-CLIP |
Human embryonic kidney cells | 22473208 20371350 21572407 |
| SOCS5 | Suppressor Of Cytokine Signaling 5 | CLASH, PAR-CLIP |
Human embryonic kidney cells, peripheral blood mononuclear cells, macrophages, brain tissue |
23622248 23592263 23446348 |
| TGFBR2 | Transforming Growth Factor Beta Receptor 2 | Immunoblot, Luciferase reporter assay, Microarray, qRT-PCR, Western blot HITS-CLIP PAR-CLIP Immunohistochemistry, In situ hybridization |
Colorectal cancer cells, umbilical cord blood cell, B cells, human embryonic stem cells, human embryonic kidney cells, B cells, epithelial cells of the small and large intestines, esophageal cells |
20940405 19435428 22473208 22012620 21572407 20371350 27080303 27508097 26729221 |
| VEGFA | Vascular Endothelial Growth Factor A | ELISA, Luciferase reporter assay | Kidney cells | 18320040 |
Table 3.
miRNA gene interaction and possible biochemical pathways involved.
| Biochemical Pathways | miRNA | Validated target genes |
|---|---|---|
| Autophagy- adaptive immune response regulation | hsa-miR-16-5p hsa-miR-20a-5p hsa-miR-17-5p |
ATG14 |
| Apoptosis- ROS production | hsa-miR-16-5p hsa-miR-17-5p hsa-miR-20a-5p hsa-miR-34a-5p |
BCL2 |
| Apoptosis- inflammatory response regulation | hsa-miR-16-5p hsa-miR-20a-5p hsa-miR-17-5p hsa-miR-106a-5p |
CPT1A |
| Oxidative stress-inflammation responses- cell apoptosis | hsa-miR-20a-5p hsa-miR-17-5p hsa-miR-223-5p |
FOXC1 |
| Cytokines, chemokines, and matrix metalloproteinase production | hsa-miR-20a-5p hsa-miR-17-5p hsa-miR-106a-5p |
HAS2 |
| Protein folding- prevention of protein aggregation - apoptosis | hsa-miR-16-5p hsa-miR-34a-5p hsa-miR-223-5p |
HSPA1A |
| Macrophages polarization-innate immune response regulation | hsa-miR-16-5p hsa-miR-20a-5p hsa-miR-17-5p |
LAMTOR1 |
| Apoptosis- bacterial clearance | hsa-miR-16-5p hsa-miR-17-5p hsa-miR-20a-5p hsa-miR-34a-5p |
MCL1 |
| Mitochondrial fusion- mitochondrial membranes regulation | hsa-miR-16-5p hsa-miR-17-5p hsa-miR-34a-5p hsa-miR-106a-5p |
MFN2 |
| TNF secretory pathway | hsa-miR-16-5p hsa-miR-20a-5p hsa-miR-17-5p |
SCAMP5 |
| Cytokines secretion- NF-κB pathway | hsa-miR-20a-5p hsa-miR-16-5p hsa-miR-34a-5p hsa-miR-223-5p |
SENP1 |
| EGFR signaling pathway | hsa-miR-16-5p hsa-miR-20a-5p hsa-miR-17-5p |
SOCS5 |
| TGF-β signaling pathway | hsa-miR-20a-5p hsa-miR-17-5p hsa-miR-34a-5p |
TGFBR2 |
| VEGF pathway- ROS generation- Akt/eNOS/NO pathway | hsa-miR-16-5p hsa-miR-20a-5p hsa-miR-17-5p |
VEGFA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



