
Submitted:
14 May 2024
Posted:
14 May 2024
You are already at the latest version
Abstract
Equine herpesvirus-1 (EHV-1) causes respiratory diseases, abortion, and encephalomyelitis in horses. EHV-1 immediate-early (IE) protein, essential for viral replication, is transactivated by the binding of a multiprotein complex including the open reading frame 12 (ORF12) and some host factors to the IE promoter region. Promoter-associated non-coding RNAs (pancRNAs), which are transcribed from bidirectional promoters, regulate the transcription of neighboring genes in mammals and pathogens. In this study, we identified a novel pancRNA transcribed from across the areas of the 5’-untranslated region and promoter of EHV-1 IE and named it IE pancRNA. IE pancRNA and mRNA were simultaneously expressed in EHV-1-infected RN33B-A68B2M cells. This pancRNA was also transcribed in RK13 and E. Derm cells, which are highly susceptible to EHV-1 infection. Furthermore, IE pancRNA upregulated IE gene expression in the presence of ORF12, and stable expression of IE pancRNA increased the number of EHV-1-infected RN33B-A68B2M cells. These results suggest that IE pancRNAs facilitate EHV-1 proliferation by promoting IE gene expression.
Keywords:
equine herpesvirus-1
; gene expression
; non-coding RNA
1. Introduction
Equine herpesvirus-1 (EHV-1), an alphaherpesvirus belonging to the Herpesviridae family, is distributed worldwide and causes respiratory diseases, abortion, and encephalomyelitis (equine herpesvirus myeloencephalopathy) in horses. EHV-1 is a serious problem in the equine industry and causes significant economic losses. EHV-1 is transmitted via the respiratory pathway. This virus causes latent infections in horses, leading to periodic reactivation as reservoirs of infection for other susceptible horses. Latent infections have been reported in the lymphoid and neural tissues [1,2].
During lytic infection, EHV-1 genes are coordinately regulated in the following phases: immediate-early (IE), early, and late phases [3,4]. IE gene, which maps within each of the two inverted repeat sequences, is the first gene to be expressed in all EHV-1 coding genes [4,5], from which 6.0-kb transcripts are synthesized [4]. EHV-1 mutant lacking the IE gene cannot replicate in cultured cells, and no viral protein synthesis is detected in such cells, suggesting that IE is essential for EHV-1 replication [6]. Transcription of EHV-1 IE gene is activated by the open reading frame 12 (ORF12) protein, a functional homolog of α-transinducing factor (TIF) gene in herpes simplex virus (HSV) type 1 [7,8]. Similar to α-TIF, ORF12 forms a multiprotein complex with host cellular factors, including OCT-1 and HCF, on DNA-binding sites to interact with the IE promoter and enhance its transcription [9]. α-TIF plays important roles in reactivating latent HSV and causing infections [10].
Viral non-coding RNAs (ncRNAs) are vital for viral infections [11,12]. Several types of viral ncRNAs have been reported to affect viral replication [13]. For example, HSV-1 latency-associated transcript, which is processed into several microRNAs (miRNAs) and is the only viral gene expressed during latent infection in neurons, inhibits apoptosis and reduces viral protein expression to maintain latency [14,15,16]. Kaposi’s sarcoma-associated herpes virus polyadenylated nuclear RNA interacts with demethylases and physically binds to promoter regions to mediate viral gene expression [17]. Furthermore, miR-U86, an miRNA encoded by human herpes virus 6A, inhibits viral lytic replication by regulating the IE gene [18]. However, the specific roles of ncRNAs in EHV-1 infection remain unclear.
Promoter-associated ncRNAs (pancRNAs) are ncRNAs transcribed within a few hundred bases of the transcription start sites (TSSs) of protein-coding or non-coding genes [19,20]. pancRNAs affect the promoter region and play an important role in the epigenetic regulation of neighboring protein-coding gene expression [19]. For example, antisense pancRNA expression from the promoter regions of neurofilament light chain (Nefl) and Il17d induces DNA demethylation in the promoter region to promote transcription [21,22]. However, the roles of pancRNAs in viral infections remain poorly understood.
In this study, we aimed to identify novel pancRNAs from the IE promoter region of EHV-1 and analyze their roles in IE gene expression.
2. Materials and Methods
2.1. Cells and Viruses
Here, Rn33B-A68B2M cells previously established and stored in our laboratory [23] were used. RK13 and E. Derm cells were obtained from the American Type Culture Collection (VA, USA). Rn33B-A68B2M cells were cultured in a mixture of Dulbecco’s modified Eagle’s medium (DMEM; Nissui, Tokyo, Japan) and Ham’s F-12 (Nissui) in a 1:1 ratio supplemented with 10% fetal bovine serum (FBS; Biowest, UT, USA) at 32 °C with 5% CO2. For differentiation, Rn33B-A68B2M cells were cultured in a high-glucose DMEM (FUJIFILM Wako Pure Chemical Corporation, Osaka, Japan) and Ham’s F12 in a 1:1 ratio containing 1% bovine serum albumin (FUJIFILM Wako) and N2 supplemented with transferrin (Holo; FUJIFILM Wako) at 37 °C with 5% CO2. RK13 cells were cultured in the Eagle’s minimum essential medium (Nissui) containing 10% FBS at 37 °C with 5% CO2. E. Derm cells were cultured in DMEM containing 10% FBS and MEM non-essential amino acid solution (FUJIFILM Wako) at 37 °C with 5% CO2. EHV-1 strain Ab4 [24] and EHV-1 mutant Ab4-green fluorescent protein (GFP) containing a GFP expression cassette between open reading frames (ORFs) 62 and 63 [25] were used. Stock viruses were propagated in RK13 cells and titrated using a plaque formation assay in RK13 cells.
2.2. Primers and Synthetic dsDNA
All primers used for polymerase chain reaction (PCR) and rapid amplification of cDNA ends (RACE) were synthesized by Eurofins Genomics (Tokyo, Japan). All primers used in this study are listed in Table 1. Long synthetic dsDNA was obtained from GenScript Biotech Corporation (Tokyo, Japan).
2.3. Reverse Transcription-Quantitative PCR (RT-qPCR)
Undifferentiated and differentiated Rn33B-A68B2M cells were cultured at 32 and 37 °C, respectively. Two days after seeding, the cells were infected with EHV-1 Ab4 at a multiplicity of infection (MOI) of 5. After incubation at 37 °C for 1 h, cells were washed with phosphate-buffered saline (PBS) three times and cultured with a growth medium. At 24 h post-infection (p.i.), the cells were collected, and total RNA was extracted using NucleoSpin RNA (Takara, Shiga, Japan) according to the manufacturer’s instructions. Complementary DNA (cDNA) was synthesized using PrimeScript II first-strand cDNA Synthesis Kit (Takara) from 1 μg of total RNA of samples. cDNA (1 μL) was used in a final reaction volume of 5 μL, and the experiments were performed in triplicate using KAPA SYBR FAST qPCR Master Mix (2×) ABI Prism (KAPA Biosystems, MA, USA) and primers for EHV-1 IE promoter_F and EHV-1 IE promoter_R (Table 1) in a Step One Real-Time PCR system (Thermo Fisher Scientific, Waltham, MA, USA). The real-time PCR program was 95 °C for 3 min and 40 cycles of 95 °C for 3 s (s) and 60 °C for 20 s. Rat β-actin was used as an internal control, and amplified by the primers Rat_β-actin_F and Rat_β-actin_R (Table 1). The Ct values of the target genes were normalized by that of internal controls, and then we compared the expression levels of genes using the 2−ΔΔCt values.
2.4. RACE
5’- and 3’- RACE for ncRNA were performed using a SMARTer RACE 5’/3’ Kit (Takara) according to the manufacturer’s instructions. Briefly, undifferentiated Rn33B-A68B2M cells were infected with EHV-1 Ab4 at an MOI of five. At 24 p.i., cells were collected, and total RNA was extracted using NucleoSpin® RNA according to the manufacturer’s instructions.
For 5’ RACE, The 5’-first strand cDNA synthesis was performed with random primers. The PCR reaction was performed with the Universal Primers Mix provided in the kit and 5’ RACE GSP of 5’ RACE Antisense GSP, 5’ RACE Sense GSP-1 or 5’ RACE Sense GSP-2 (Table 1).
For 3’RACE, poly(A) tailing was performed by using Poly (A) Tailing of RNA using E.coli Poly(A) Polymerase (New England Biolabs Japan, Tokyo, Japan) because ncRNA possibly lacks a polyadenylated tail. Reverse transcription was performed with 3’-CDS Primer A provided in the kit. The PCR reaction was performed with the Universal Primers Mix and 3’ RACE GSP of 3’ RACE Antisense GSP, 3’ RACE Sense GSP-1, 3’ RACE Sense GSP-2, 3’ RACE Sense GSP-3 (Table 1). All the amplified products were cloned into a pRACE vector and sequenced.
2.5. DNA Cloning and Preparation of RNA Probes
The cDNA fragment of ncRNA (designated as IE promoter-associated ncRNA; IE pan-cRNA in the Results section) was amplified by PCR using synthetic dsDNA spanning the entire sequence of the ncRNA transcripts as a template. cDNA fragments of EHV-1 IE and rat β-actin were synthesized using reverse transcription PCR. For the EHV-1 IE gene, cDNA was synthesized from total RNA of EHV-1-infected RK13 cells. For the rat β-actin gene, cDNA was synthesized from the total RNA of undifferentiated Rn33B-A68B2M. The primer pairs used for amplification were as follows: for the probe complementary to the 5’ side of IE pancRNA, 5’-GGCGAATTCCTCTTGGCACTCCTTCTTCG-3’ and 5’-ACTCAAGCTTCTTCGAGGTAAGTATCCCCAC-3’; for the probe complementary to the 3’ side of IE pancRNA, 5’-GGCGAATTCCTTTAATGAGATTCAACCGGG-3’ and 5’-ACTCAAGCTTCCGCTCATATGCATAAAGACG-3’; for IE mRNA, 5’-TCGCCGCGATGCTGAAGATG-3’ and 5’-TTCGTCGCTGTCGCTGTCGT-3’; for β-actin, 5’-ACTCAAGCTTAGGCCAACCGTGAAAAGATG-3’ and 5’-GGCGAATTCAGTCTAGGGCAACATAGCAC-3. ’ The PCR products of IE pancRNA and rat β-actin were cloned into the EcoRI-HindIII site of the pGEM-3Z vector (Promega, Madison, WI, USA). The PCR products of IE mRNA were cloned into the pGEM-T vector (Promega).
Antisense RNA probes were prepared using the digoxigenin (DIG) RNA-labeling kit (SP6/T7) (Roche, Basel, Switzerland). To obtain templates for RNA transcription, the plasmid DNA containing the PCR product was linearized with the restriction enzyme HindIII or NotI. Each linearized DNA fragment was labeled by the T7 transcription runoff method by incorporating DIG-11-UTP into single-stranded specific RNA probes. The labeled probes generated from 3 μg of the plasmid DNA were precipitated with ethanol and then dissolved in 50 μL of RNase-free water. The RNA probes were stored at –80 °C.
2.6. Northern Hybridization
Total RNA was extracted using TriPure Isolation Reagent (Roche) from EHV-1-infected or uninfected Rn33B-A68B2M, RK13, and E. Derm cells according to the manufacturer’s instructions. Hybridization was performed using the method described by Shifman and Stein [26] with slight modifications. Briefly, 1 μg of RNA samples were electrophoresed through a 1.5% agarose-2.2M formaldehyde gel and were transferred to nylon membranes (positively charged; Roche). The RNAs were fixed to the membrane by XL-1000 UV crosslinker (Spectronics Corporation, Lincoln, NB, USA) with subsequent baking at 80 °C for 2 h. Membranes were prehybridized in 0.25 M Na2HPO4 (pH 7.2), sodium dodecyl sulfate (SDS) 10%, 1 mM EDTA, and blocking reagent 2% at 68 °C for 3 h. Hybridization was carried out in the same buffer containing 6-8 ng/mL of the DIG-labeled cRNA probe at 68 °C for 15 h. After hybridization, membranes were washed three times for 20 min each in 25 mM Na2HPO4 (pH 7.2), SDS 1%, and 1 mM EDTA at 68 °C. Hybridization signals were visualized using an alkaline phosphatase-conjugated anti-DIG antibody and disodium 3-(4-methoxyspiro{1,2-dioxetane-3,2’-(5’-chloro)tricyclo[3.3.1.13,7]decan}-4-yl) phenyl phosphate (CSPD) chemiluminescent substrate (Roche).
2.7. Expression Vector and Reporter Plasmid Construction
The pcDNA3.1 (+) vector encoding full-length IE pancRNA was generated using GenScript. This plasmid was designated pcDNA-IE pancRNA. EHV-1 ORF12 gene was amplified from EHV-1 HH1 strain genome by PCR using the primers 5’-GGCGAATTCACCATGTGCCTCTTACATATTTC-3’ and 5’-ACTCAAGCTTTTAAATGTCAAACATCTGGT-3. ’ The PCR product was digested with EcoRI and Hind III and cloned into the EcoRI-HindIII site of the pcDNA 3.1 (-) vector (Thermo Fisher Scientific). Flag tag sequences (5’-GACTACAAAGACGATGACGACAAG-3’) were inserted at the 5’ end of cloned ORF12 gene by inverse PCR and subsequent self-ligation. The resulting ORF12 expression vector was designated as pcDNA-ORF12 flag. pUC19 vector containing IE promoter region (from +78 to -1807 of IE gene) [9] at Hind III site was generated by GenScript. This plasmid was digested with HindIII and the gel-purified fragment was cloned into the pGL4.10 [luc2] vector (Promega). To check whether the insert was cloned in the proper direction, the cloned plasmid was digested with EcoRV, which exists at the multi-cloning site of the vector and in the cloned sequences, and the length of the fragment was determined by gel electrophoresis. The resultant plasmid was designated as pGL4-IE promoter-Luc.
2.8. Luciferase Reporter Assay
Undifferentiated Rn33B-A68B2M cells were seeded in a 6-well plate (Greiner Bio-One) and cultured at 32 °C. Two days after seeding, cells (≈70% confluent) were co-transfected with 0.8 μg each of pcDNA-IE pancRNA, pGL4-IE promoter-Luc, and pcDNA-ORF12 flag using Lipofectamine 3000 (Thermo Fisher Scientific) according to manufacturer’s instructions. As a negative control, pcDNA3.1(+) cells were transfected instead of the pcDNA-IE pancRNA. At 24 h after transfection, the cells were lysed with Passive Lysis Buffer (Promega) and assayed for firefly luciferase activity using Luciferase Assay Reagent II (Promega). To evaluate the amount of plasmids incorporated into cells by transfection, genome DNA was extracted from cell pellets in the lysate of reporter assay using Wizard® GenomicDNA Purification Kit (Promega) according to the manufacturer’s instructions. Genomic DNA (10 ng of genome DNA was used for qPCR with the primers Firefly LuciferaseF and Firefly Luciferase_R [27] (Table 1). Serial 10-fold dilutions of the pGL4-IE promoter-Luc plasmid were used to generate a standard curve. The real-time PCR program was 95 °C for 20 s and 40 cycles of 95 °C for 3 s and 60 °C for 20 s. Normalized firefly luciferase activity was calculated by dividing firefly luciferase activity (RLU) by firefly plasmid amount (ng). To examine whether the Renilla luciferase vector could be used as an internal control reporter vector, three Renilla luciferase vectors with different promoters, pGL4.73[hRluc/SV40] vector (Promega), pGL4.74[hRluc/TK] vector (Promega), pGL4.75[hRluc/CMV] vector (Promega), were used. 1.0 μg each of pGL4-IE promoter-Luc and pcDNA-ORF12 flag and 0.1 μg of renilla luciferase vector were transfected and lysed as described above. Renilla luciferase activity was analyzed using the Stop and Glo Reagent (Promega). To evaluate the effects of the ORF12 protein on the expression of Renilla luciferase, cells transfected with vectors, except for pcDNA-ORF12 flag, were assayed.
2.9. Construction of IE pancRNA-Expressing Rn33B-A68B2M Cells
The self-inactivating lentiviral vector construct (pCSII-CMV-MCS-IRES2-Bsd), packing construct (pCAG-HIV-gp), and VSV-G and Rev-repression constructs (pCMV-VSV-G-RSV-Rev) were kindly provided by Dr. Miyoshi (RIKEN Bio Resource Center, Ibaraki, Japan). The DNA fragment containing IE pancRNA was digested from pcDNA-IE pancRNA with NheI and XhoI, and the fragment was cloned into the NheI-XhoI site of the pCSII-CMV-MCS-IRES2-Bsd vector. A recombinant lentiviral vector was generated by transient transfection of 293T cells with a combination of pCAG-HIVgp and pCMV-VSV-G-RSV-Rev. The supernatant containing the lentiviral vector was collected after incubation of the cells at 37 °C for 48 h. Rn33B-A68B2M cells were infected with a lentiviral vector and cultured in the presence of 5 μg/mL blasticidin S (Merck, Darmstadt, Germany). The cells were designated as Rn33B-A68B2M-IE pancRNA cells.
2.10. Flow Cytometry
3.0×105 undifferentiated Rn33B-A68B2M-IE pancRNA cells were seeded in a 6-well plate (Falcon) and cultured at 32 °C. Two days after seeding, cells were infected with EHV-1 Ab4-GFP at an MOI of 0.05. At 24 and 48 h p.i., the cells were harvested, dissociated, and analyzed using a BD FACSVerse system (BD Biosciences, Franklin Lakes, NJ, USA), and the data were analyzed using FACSuite (BD Biosciences). Undifferentiated Rn33B-A68B2M cells were used as negative controls.
2.11. Statistical Analyses
Statistical analyses were conducted using Microsoft Excel (Microsoft Corporation 2017) and R Statistical Software (version 4.0.3; R Core Team 2020). Student’s t-test was used to analyze the differences between two groups, whereas Dunnett’s test was used to analyze the differences among multiple groups. Statistical significance was set at P < 0.05.
3. Results
3.1. Identification of a Novel ncRNA Expressed in the Upstream Region of IE Coding Sequences in EHV-1-Infected Rn33B-A68B2M Cells
We examined whether the ncRNAs are expressed in the EHV-1 IE promoter region of EHV-1-infected cells. Rn33B-A68B2M rat neuronal cell line was used to investigate viral gene expression in both undifferentiated and neuronally differentiated EHV-1-susceptible and -unsusceptible cells [23]. RT-qPCR was performed using primer pairs amplifying a part of the IE promoter (nt. 143841–143943) to detect the RNA expression in both EHV-1-infected undifferentiated and differentiated cells (Figure 1). Expression levels were higher in the undifferentiated cells than in the differentiated cells but were undetectable in the uninfected cells.
3.2. Determination of the 5’- and 3’-Ends of ncRNA via RACE
Next, we performed 5’- and 3’-RACE analyses to determine the full length of IE promoter-associated RNA. We could amplify 5’- and 3’-ends of RNA transcribed in the antisense direction (Figure 2A). Nucleotide sequences of the 5’- and 3’-ends revealed that the antisense RNA was transcribed from nt region 144287–143290 of the EHV-1 genome, which is equivalent to the position from +344 to –654 in the IE TSS. Moreover, 3’-RACE without poly (A) tail in cDNA synthesis resulted in amplification similar to that with 3’-RACE with poly (A) tail (Figure 2B), suggesting that the antisense RNA has a polyadenylated tail. However, we could not amplify the 5’- and 3’- ends of the RNA transcribed in the sense direction.
Antisense RNA predicted to be transcribed from the region overlapping the 5’-untranslated and promoter regions (from +344 to –654) of IE was designated as the IE promoter-associated ncRNA (IE pancRNA).
3.3. IE pancRNA Is Expressed in EHV-1-Infected RN33B-A68B2M, RK13, and E. Derm Cells
The expression and length of IE pancRNA, as expected by RT-qPCR and RACE analyses, respectively, were validated by northern blot hybridization using Rn33B-A68B2M cells. Undifferentiated and differentiated Rn33B-A68B2M cells were infected with EHV-1 Ab4 at an MOI of 5, and total RNA was extracted at 24 h p.i. Uninfected Rn33B-A68B2M cells were used as the negative control. We prepared two strand-specific probes complementary to the 5’ side and 3’ side of IE pancRNA. The IE pancRNA was detected approximately 1.3 kb in length in EHV-1 infected-undifferentiated and differentiated Rn33B-A68B2M cells treated with both probes (Figure 3A, left side, B). Consistent with the qPCR data (Figure 1), the expression levels were higher in undifferentiated cells than in differentiated cells.
Next, we examined whether pancRNA expression preceded or lagged IE gene expression in EHV-1-infected Rn33B-A68B2M cells. Undifferentiated Rn33B-A68B2M cells were infected with EHV-1 at an MOI of 5. RNA was extracted at 0, 12, 14, 16, 18, 20, 22, and 24 h p.i. and hybridized to probes specific for IE pancRNA or IE transcript. Both IE pancRNA and IE transcripts were detected at the same time point, as early as 14 h p. i., by northern blot hybridization (Figure 3C).
To confirm whether IE pancRNA was expressed in other cell lines, northern blot analysis was performed in RK13 and E. Derm cells, which are highly susceptible to EHV-1 infection. RK13 cells were infected with EHV-1 Ab4 at an MOI of 0.5, and RNA was extracted at 12 h p.i. E. Derm cells were infected with EHV-1 at an MOI of 5 and RNA was extracted at 24 h p.i. Uninfected cells were used as negative controls. Similar to Rn33B-A68B2M cells, RK13 and E. Derm cells also showed IE pancRNA expression upon EHV-1 infection (Figure 3A, right side).
We also performed northern hybridization using a probe for the IE promoter-associated sense RNA in Rn33B-A68B2M cells. Weak transcript signals spanning the IE promoter region approximately 6.7 kb in length, were detected in EHV-1 infected undifferentiated and differentiated Rn33B-A68B2M cells (Figure 3D). This may have been caused by the detection of IE mRNA transcribed upstream of a known transcription initiation site.
3.4. IE pancRNA Promotes IE Gene Transcription
To investigate the role of IE pancRNAs in IE gene transcription, a luciferase reporter assay was performed. EHV-1 ORF12 protein was previously reported as a transactivator of IE gene expression [28,29,30]. Control reporter vectors containing constitutive promoters (SV40, HSV-TK, or CMV) were not used to normalize Luc activity as EHV-1 ORF12 significantly activated these promoters (Figure 4A). In the presence of ORF12, cells transfected with the IE pancRNA expression vector exhibited higher RLU than those transfected with the empty control vector (Figure 4B). In the absence of ORF12, RLU was low, regardless of the expression of IE pancRNA. These results suggest that IE pancRNA promotes the transcription of IE via ORF12.
3.5. IE pancRNA Promotes EHV-1 Infection in Rn33B-A68B2M Cells
Finally, effect of IE pancRNA on EHV-1 infection was examined using Rn33B-A68B2M cells stably expressing pancRNA (Rn33B-A68B2M-IE pancRNA). At 24 and 48 h p.i. with GFP-expressing EHV-1, number of GFP-positive cells in the Rn33B-A68B2M-IE pancRNA group was higher than that in the Rn33B-A68B2M group (Figure 5). This suggests that IE pancRNA promotes EHV-1 infection in Rn33B-A68B2M cells.
4. Discussion
In this study, we identified a novel ncRNA, IE pancRNA, transcribed from the EHV-1 IE 5’-untranslated and promoter regions in EHV-1-infected cells. IE pancRNA promoted the transcription of IE gene and promoted EHV-1 infection in Rn33B-A68B2M. Besides the report on latency-associated transcripts in ganglionic neurons and equine leukocytes [31,32], the involvement of ncRNAs in EHV-1 infections remains largely unknown. To the best of our knowledge, this is the first report of a virus-derived lncRNA regulating EHV-1 expression.
PancRNAs are lncRNAs transcribed from bidirectional promoters regulating the expression of protein-coding genes [20]. Recent studies revealed the mechanisms underlying the regulation of gene expression by pancRNAs. pancRNA expression in the promoter region of vimentin induces sequence-specific DNA demethylation in the promoter region to promote transcription [22]. Myoparr, which is expressed in the promoter region of myogenin, is presumed to promote myogenin expression by regulating the association between the transcriptional coactivator and histone acetyltransferase [33]. Here, our study revealed IE pancRNA as a functional pancRNA regulating paired gene transcription. However, the underlying mechanisms, especially those responsible for enhanced transactivation by ORF12, remain unknown. RACE analysis revealed that the 5’-end of IE pancRNA is located downstream of the IE TSS. Although the majority of pancRNAs are transcribed upstream of the TSS of a paired gene, a functional pancRNA transcribed from 1161 bp downstream of the TSS of Nefl in rat PC12 cells causes DNA demethylation in Nefl upstream region [22].
We identified IE pancRNA as a 998 nucleotide transcript (nt. 144287–143290) using RACE. However, an approximately 1.3 kb band was detected in EHV-1-infected cells via northern blotting for IE pancRNA. This discrepancy may be due to the addition of a long (approximately 200–300 bp) poly(A) tail [34]. LncRNAs can have a 5’-cap structure and/or be spliced similar to mRNAs, warranting further analyses on the post-transcriptional modification of IE pancRNA [35].
Here, we demonstrated that IE pancRNA was expressed in the EHV-1-infected RK13 rabbit kidney-derived, E. Derm equine dermal, and Rn33B-A68B2M rat neuronal cell lines. Our findings suggest that IE pancRNA is expressed in various cell types and regulates EHV-1 gene expression during EHV-1 lytic infection.
In conclusion, we identified a pancRNA transcribed from the IE promoter region that enhanced EHV-1 infection. Moreover, our findings suggest the potential involvement of IE pancRNA, which upregulated IE gene transcription, in EHV-1 infection mechanisms in horses. However, further studies are necessary to validate these findings.
Author Contributions
Conceptualization, T.K.; Investigation, M.Maeda and M.Murashita; Formal Analysis, K.A.; Resources, H.F. and T.K.; Writing – Original Draft Preparation, M.Maeda; Writing – Review & Editing, K.A., A.K., and T.K.; Supervision, T.K.; Funding Acquisition, T.K. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by a Grant-in-Aid for Scientific Research (B) (grant number 22H02508) from the Ministry of Education, Culture, Sports, Science, and Technology, Japan (TK).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data generated or analyzed in this study are included in the published article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Slater, J.D.; Borchers, K.; Thackray, A.M.; Field, H.J. The trigeminal ganglion is a location for equine herpesvirus 1 latency and reactivation in the horse. J. Gen. Virol. 1994, 75, 2007–2016. [Google Scholar] [CrossRef] [PubMed]
- Welch, H.M.; Bridges, C.G.; Lyon, A.M.; Griffiths, L.; Edington, N. Latent equid herpesviruses 1 and 4: Detection and distinction using the polymerase chain reaction and co-cultivation from lymphoid tissues. J. Gen. Virol. 1992, 73, 261–268. [Google Scholar] [CrossRef]
- Gray, W.L.; Baumann, R.P.; Robertson, A.T.; Caughman, G.B.; O’Callaghan, D.J.; Staczek, J. Regulation of equine herpesvirus type 1 gene expression: Characterization of immediate early, early, and late transcription. Virology 1987, 158, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Gray, W.L.; Baumann, R.P.; Robertson, A.T.; O’Callaghan, D.J.; Staczek, J. Characterization and mapping of equine herpesvirus type 1 immediate early, early, and late transcripts. Virus Res. 1987, 8, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Grundy, F.J.; Baumann, R.P.; O’Callaghan, D.J. DNA sequence and comparative analyses of the equine herpesvirus type 1 immediate early gene. Virology 1989, 172, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Garko-Buczynski, K.A.; Smith, R.H.; Kim, S.K.; O’Callaghan, D.J. Complementation of a replication-defective mutant of equine herpesvirus type 1 by a cell line expressing the immediate-early protein. Virology 1998, 248, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.B.; Thompson, Y.G.; Feng, X.; Holden, V.R.; O’Callaghan, D.; Caughman, G.B. Structural and antigenic identification of the ORF12 protein (alpha TIF) of equine herpesvirus 1. Virology 1997, 230, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Purewal, A.S.; Smallwood, A.V.; Kaushal, A.; Adegboye, D.; Edington, N. Identification and control of the cis-acting elements of the immediate early gene of equid herpesvirus type 1. J. Gen. Virol. 1992, 73, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Elliott, G.; O’Hare, P. Equine herpesvirus 1 gene 12, the functional homologue of herpes simplex virus VP16, transactivates via octamer sequences in the equine herpesvirus IE gene promoter. Virology 1995, 213, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Fan, D.; Wang, M.; Cheng, A.; Jia, R.; Yang, Q.; Wu, Y.; Zhu, D.; Zhao, X.; Chen, S.; Liu, M.; Zhang, S.; Ou, X.; Mao, S.; Gao, Q.; Sun, D.; Wen, X.; Liu, Y.; Yu, Y.; Zhang, L.; Tian, B.; Pan, L.; Chen, X. The role of VP16 in the life cycle of alphaherpesviruses. Front. Microbiol. 2020, 11, 1910. [Google Scholar] [CrossRef] [PubMed]
- Fortes, P.; Morris, K.V. Long noncoding RNAs in viral infections. Virus Res., 212, 1–11.
- Gottwein, E.; Cullen, B.R. Viral and cellular microRNAs as determinants of viral pathogenesis and immunity. Cell Host Microbe 2008, 3, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, W.; Liu, Z.F. Long non-coding RNAs: Novel players in regulation of immune response upon herpesvirus infection. Front. Immunol. 2018, 9, 761. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Gartner, J.J.; Sethupathy, P.; Hatzigeorgiou, A.G.; Fraser, N.W. Anti-apoptotic function of a microRNA encoded by the HSV-1 latency-associated transcript. Nature 2006, 442, 82–85, (Gupta, A.; Gartner, J.J.; Sethupathy, P.; Hatzigeorgiou, A.G.; Fraser, N.W. Retraction Note: Anti-apoptotic function of a microRNA encoded by the HSV-1 latency-associated transcript. Nature 2008, 451(7178), 600). [Google Scholar] [CrossRef] [PubMed]
- Perng, G.C.; Jones, C.; Ciacci-Zanella, J.; Stone, M.; Henderson, G.; Yukht, A.; Slanina, S.M.; Hofman, F.M.; Ghiasi, H.; Nesburn, A.B.; Wechsler, S.L. Virus-induced neuronal apoptosis blocked by the herpes simplex virus latency-associated transcript. Science 2000, 287, 1500–1503. [Google Scholar] [CrossRef] [PubMed]
- Umbach, J.L.; Kramer, M.F.; Jurak, I.; Karnowski, H.W.; Coen, D.M.; Cullen, B.R. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature 2008, 454, 780–783. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, C.C.; Tarrant-Elorza, M.; Verma, S.; Purushothaman, P.; Pari, G.S. Regulation of viral and cellular gene expression by Kaposi’s sarcoma-associated herpesvirus polyadenylated nuclear RNA. J. Virol. 2013, 87, 5540–5553. [Google Scholar] [CrossRef]
- Nukui, M.; Mori, Y.; Murphy, E.A. A human herpesvirus 6A-encoded microRNA: role in viral lytic replication. J. Virol. 2015, 89, 2615–2627. [Google Scholar] [CrossRef] [PubMed]
- Chellini, L.; Frezza, V.; Paronetto, M.P. Dissecting the transcriptional regulatory networks of promoter-associated noncoding RNAs in development and cancer. J. Exp. Clin. Cancer Res. 2020, 39, 51. [Google Scholar] [CrossRef]
- Uesaka, M.; Nishimura, O.; Go, Y.; Nakashima, K.; Agata, K.; Imamura, T. Bidirectional promoters are the major source of gene activation-associated non-coding RNAs in mammals. BMC Genomics 2014, 15, 35. [Google Scholar] [CrossRef] [PubMed]
- Hamazaki, N.; Uesaka, M.; Nakashima, K.; Agata, K.; Imamura, T. Gene activation-associated long noncoding RNAs function in mouse preimplantation development. Development 2015, 142, 910–920. [Google Scholar] [CrossRef]
- Tomikawa, J.; Shimokawa, H.; Uesaka, M.; Yamamoto, N.; Mori, Y.; Tsukamura, H.; Maeda, K.; Imamura, T. Single-stranded noncoding RNAs mediate local epigenetic alterations at gene promoters in rat cell lines. J. Biol. Chem. 2011, 286, 34788–34799. [Google Scholar] [CrossRef] [PubMed]
- Minato, E.; Kobayashi, A.; Aoshima, K.; Fukushi, H.; Kimura, T. Susceptibility of rat immortalized neuronal cell line Rn33B expressing equine major histocompatibility class 1 to equine herpesvirus-1 infection is differentiation dependent. Microbiol. Immunol. 2020, 64, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Nugent, J.; Birch-Machin, I.; Smith, K.C.; Mumford, J.A.; Swann, Z.; Newton, J.R.; Bowden, R.J.; Allen, G.P.; Davis-Poynter, N. Analysis of equid herpesvirus 1 strain variation reveals a point mutation of the DNA polymerase strongly associated with neuropathogenic versus nonneuropathogenic disease outbreaks. J. Virol. 2006, 80, 4047–4060. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim elS, M.; Pagmajav, O.; Yamaguchi, T.; Matsumura, T.; Fukushi, H. Growth and virulence alterations of equine herpesvirus 1 by insertion of a green fluorescent protein gene in the intergenic region between ORFs 62 and 63. Microbiol. Immunol. 2004, 48, 831–842. [Google Scholar] [CrossRef] [PubMed]
- Shifman, M.I.; Stein, D.G. A reliable and sensitive method for non-radioactive northern blot analysis of nerve growth factor mRNA from brain tissues. J. Neurosci. Methods 1995, 59, 205–208. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; McLaughlin, S.L.; Klinke, D.J. Quantifying spontaneous metastasis in a syngeneic mouse melanoma model using real time PCR. Analyst 2017, 142, 2945–2953. [Google Scholar] [CrossRef] [PubMed]
- Elliott, G.D. The extreme carboxyl terminus of the equine herpesvirus 1 homolog of herpes simplex virus VP16 is essential for immediate-early gene activation. J. Virol. 1994, 68, 4890–4897. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.B.; Thompson, Y.G.; Caughman, G.B. Transcriptional control of the equine herpesvirus 1 immediate early gene. Virology 1993, 197, 788–792. [Google Scholar] [CrossRef] [PubMed]
- Purewal, A.S.; Allsopp, R.; Riggio, M.; Telford, E.A.; Azam, S.; Davison, A.J.; Edington, N. Equid herpesviruses 1 and 4 encode functional homologs of the herpes simplex virus type 1 virion transactivator protein, VP16. Virology 1994, 198, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Baxi, M.K.; Efstathiou, S.; Lawrence, G.; Whalley, J.M.; Slater, J.D.; Field, H.J. The detection of latency-associated transcripts of equine herpesvirus 1 in ganglionic neurons. J. Gen. Virol. 1995, 76(12), 3113–3118. [Google Scholar] [CrossRef] [PubMed]
- Chesters, P.; Allsop, R.; Purewal, A.; Edington, N. Detection of latency-associated transcripts of equid herpesvirus 1 in equine leukocytes but not in trigeminal ganglia. J. Virol. 1997, 71(5), 3437–3443. [Google Scholar] [CrossRef] [PubMed]
- Hitachi, K.; Nakatani, M.; Takasaki, A.; Ouchi, Y.; Uezumi, A.; Ageta, H.; Inagaki, H.; Kurahashi, H.; Tsuchida, K. Myogenin promoter-associated lncRNA Myoparr is essential for myogenic differentiation. EMBO Rep. 2019, 20, e47468. [Google Scholar] [CrossRef] [PubMed]
- Jolles, B.; Aliouat, A.; Stierlé, V.; Salhi, S.; Jean-Jean, O. Translation termination-dependent deadenylation of MYC mRNA in human cells. Oncotarget 2018, 9, 26171–26182. [Google Scholar] [CrossRef] [PubMed]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; Xue, C.; Marinov, G.K.; Khatun, J.; Williams, B.A.; Zaleski, C.; Rozowsky, J.; Röder, M.; Kokocinski, F.; Abdelhamid, R.F.; Alioto, T.; Antoshechkin, I.; Baer, M.T.; Bar, N.S.; Batut, P.; Bell, K.; Bell, I.; Chakrabortty, S.; Chen, X.; Chrast, J.; Curado, J.; Derrien, T.; Drenkow, J.; Dumais, E.; Dumais, J.; Duttagupta, R.; Falconnet, E.; Fastuca, M.; Fejes-Toth, K.; Ferreira, P.; Foissac, S.; Fullwood, M.J.; Gao, H.; Gonzalez, D.; Gordon, A.; HGunawardena, H.; Howald, C.; Jha, S.; Johnson, R.; Kapranov, P.; King, B.; Kingswood, C.; Luo, O.J.; Park, E.; Persaud, K.; Preall, J.B.; Ribeca, P.; Risk, B.; Robyr, D.; Sammeth, M.; Schaffer, L.; See, L.-H.; Shahab, A.; Skancke, J.; Suzuki, A.M.; Takahashi, H.; Tilgner, H.; Trout, D.; Walters, N.; Wang, H.; Wrobel, J.; Yu, Y.; Ruan, X.; Hayashizaki, Y.; Harrow, J.; Gerstein, M.; Hubbard, T.; Reymond, A.; Antonarakis, S.E.; Hannon, G.; Giddings, M.C.; Ruan, Y.; Wold, B.; Carninci, P.; Guigó, R.; Gingeras, T.R. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
RNA expression levels from the promoter region of the immediate-early (IE) gene in equine herpesvirus-1 (EHV-1)-infected Rn33B-A68B2M cells. Undifferentiated and differentiated Rn33B-A68B2M cells were infected with EHV-1 at a multiplicity of infection (MOI) of 5. Total RNA was extracted 24 h post-infection (p.i.). RNA transcribed from the IE promoter region was detected via reverse transcription-quantitative polymerase chain reaction (RT-qPCR). β-actin was used as an internal control. Bar represents the mean with standard deviation from three independent experiments. Student’s t-test.
Figure 1.
RNA expression levels from the promoter region of the immediate-early (IE) gene in equine herpesvirus-1 (EHV-1)-infected Rn33B-A68B2M cells. Undifferentiated and differentiated Rn33B-A68B2M cells were infected with EHV-1 at a multiplicity of infection (MOI) of 5. Total RNA was extracted 24 h post-infection (p.i.). RNA transcribed from the IE promoter region was detected via reverse transcription-quantitative polymerase chain reaction (RT-qPCR). β-actin was used as an internal control. Bar represents the mean with standard deviation from three independent experiments. Student’s t-test.
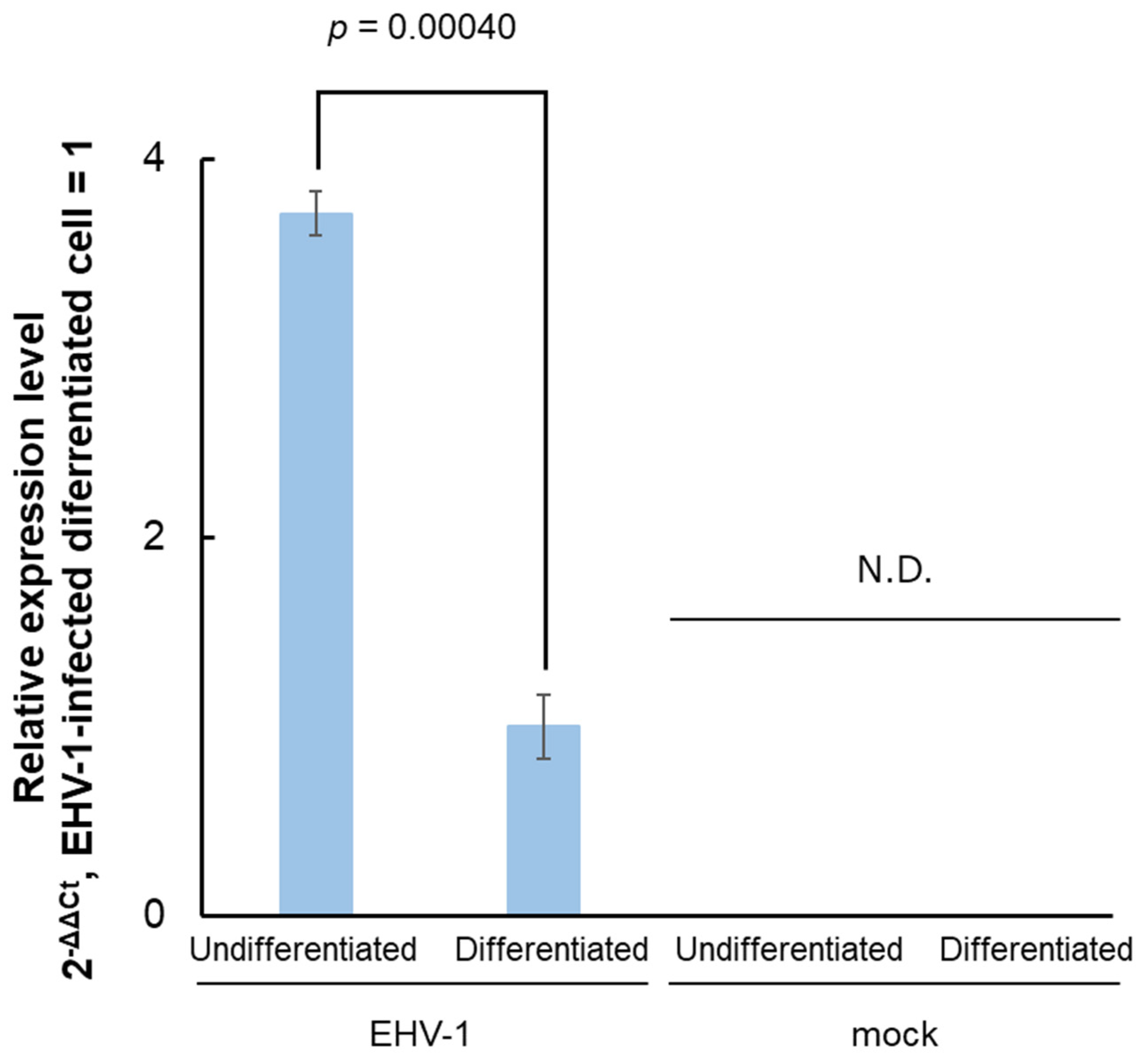
Figure 2.
5’- and 3’-rapid amplification of cDNA ends (RACE) of the IE promoter-associated RNA in EHV-1 infected Rn33B-A68B2M cells. Undifferentiated Rn33B-A68B2M cells were infected with EHV-1 at an MOI of 5. Total RNA was extracted at 24 h p.i. (A) Gel electrophoresis images of PCR products from the 5’- and 3’-RACE assays of ncRNA in the antisense orientation of the IE promoter. (B) Gel electrophoresis images of PCR products from the 3’-RACE assay of ncRNA without the addition of a poly(A) tail.
Figure 2.
5’- and 3’-rapid amplification of cDNA ends (RACE) of the IE promoter-associated RNA in EHV-1 infected Rn33B-A68B2M cells. Undifferentiated Rn33B-A68B2M cells were infected with EHV-1 at an MOI of 5. Total RNA was extracted at 24 h p.i. (A) Gel electrophoresis images of PCR products from the 5’- and 3’-RACE assays of ncRNA in the antisense orientation of the IE promoter. (B) Gel electrophoresis images of PCR products from the 3’-RACE assay of ncRNA without the addition of a poly(A) tail.

Figure 3.
Expression levels of the IE promoter-associated non-coding RNA (pancRNA) in Rn33B-A68B2M, RK13, and E. Derm cells. Cells were infected with EHV-1 at an MOI of 5. Total RNA was extracted from EHV-1- and mock-infected cells at 24 h p.i. (A) Northern blotting with digoxigenin (DIG)-labeled RNA probe that hybridizes to the 5’ region of IE pancRNA. (B) Northern blotting with DIG-labeled RNA probe that hybridizes to the 3’ region of IE pancRNA. (C) Time course of the expression levels of IE pancRNA and IE gene in EHV-1-infected undifferentiated RN33B-A68B2M cells. (D) Northern blotting with DIG-labeled RNA probe that hybridizes to the sense transcript from the IE promoter region.
Figure 3.
Expression levels of the IE promoter-associated non-coding RNA (pancRNA) in Rn33B-A68B2M, RK13, and E. Derm cells. Cells were infected with EHV-1 at an MOI of 5. Total RNA was extracted from EHV-1- and mock-infected cells at 24 h p.i. (A) Northern blotting with digoxigenin (DIG)-labeled RNA probe that hybridizes to the 5’ region of IE pancRNA. (B) Northern blotting with DIG-labeled RNA probe that hybridizes to the 3’ region of IE pancRNA. (C) Time course of the expression levels of IE pancRNA and IE gene in EHV-1-infected undifferentiated RN33B-A68B2M cells. (D) Northern blotting with DIG-labeled RNA probe that hybridizes to the sense transcript from the IE promoter region.
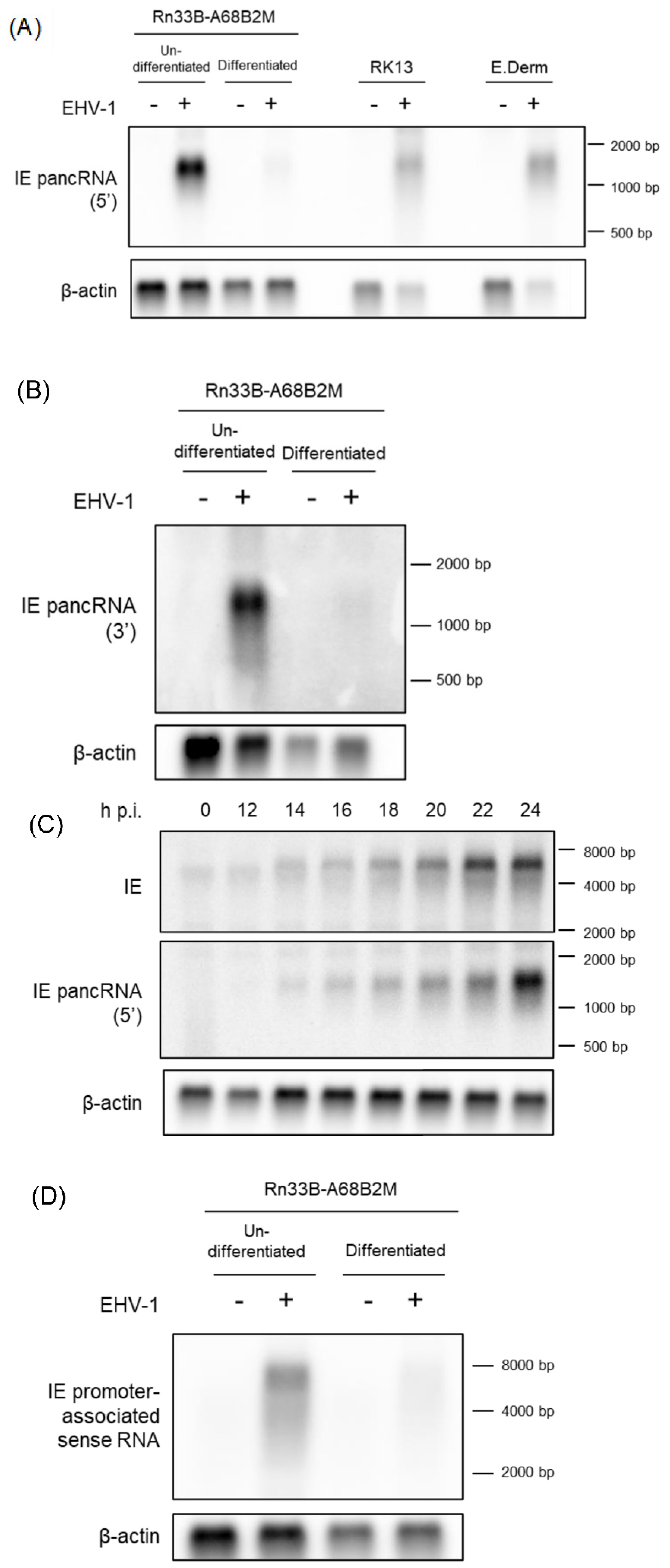
Figure 4.
Effect of IE pancRNA on IE promoter activity in undifferentiated Rn33B-A68B2M cells. Undifferentiated Rn-33B-A68B2M cells were co-transfected with luciferase reporter plasmids containing IE promoter (pGL4-ORF 64 promoter-Luc), IE pancRNA expression vector (pcDNA-IE pancRNA), and ORF12 expression vector (ocDNA-ORF12 flag). At 24 h p.i., cells were lysed and assayed for luciferase activities. (A) Firefly/Renilla dual luciferase assay was performed to verify whether the renilla luciferase vector with Simian virus 40 (pGL4.73[hRluc/SV40] vector), thymidine kinase (pGL4.74[hRluc/TK] vector), or cytomegalovirus (pGL4.75[hRluc/CMV] vector) promoters can be used as an internal control. All samples were analyzed in singlicate. (B) Firefly luciferase assay was used to examine the role of IE pancRNA in IE gene transcription. Normalized firefly luciferase activity was calculated by dividing the firefly luciferase activity (RLU) by the firefly plasmid amount (ng) incorporated into cells via transfection. Bar represents the mean with standard deviation from three independent experiments. Student’s t-test.
Figure 4.
Effect of IE pancRNA on IE promoter activity in undifferentiated Rn33B-A68B2M cells. Undifferentiated Rn-33B-A68B2M cells were co-transfected with luciferase reporter plasmids containing IE promoter (pGL4-ORF 64 promoter-Luc), IE pancRNA expression vector (pcDNA-IE pancRNA), and ORF12 expression vector (ocDNA-ORF12 flag). At 24 h p.i., cells were lysed and assayed for luciferase activities. (A) Firefly/Renilla dual luciferase assay was performed to verify whether the renilla luciferase vector with Simian virus 40 (pGL4.73[hRluc/SV40] vector), thymidine kinase (pGL4.74[hRluc/TK] vector), or cytomegalovirus (pGL4.75[hRluc/CMV] vector) promoters can be used as an internal control. All samples were analyzed in singlicate. (B) Firefly luciferase assay was used to examine the role of IE pancRNA in IE gene transcription. Normalized firefly luciferase activity was calculated by dividing the firefly luciferase activity (RLU) by the firefly plasmid amount (ng) incorporated into cells via transfection. Bar represents the mean with standard deviation from three independent experiments. Student’s t-test.
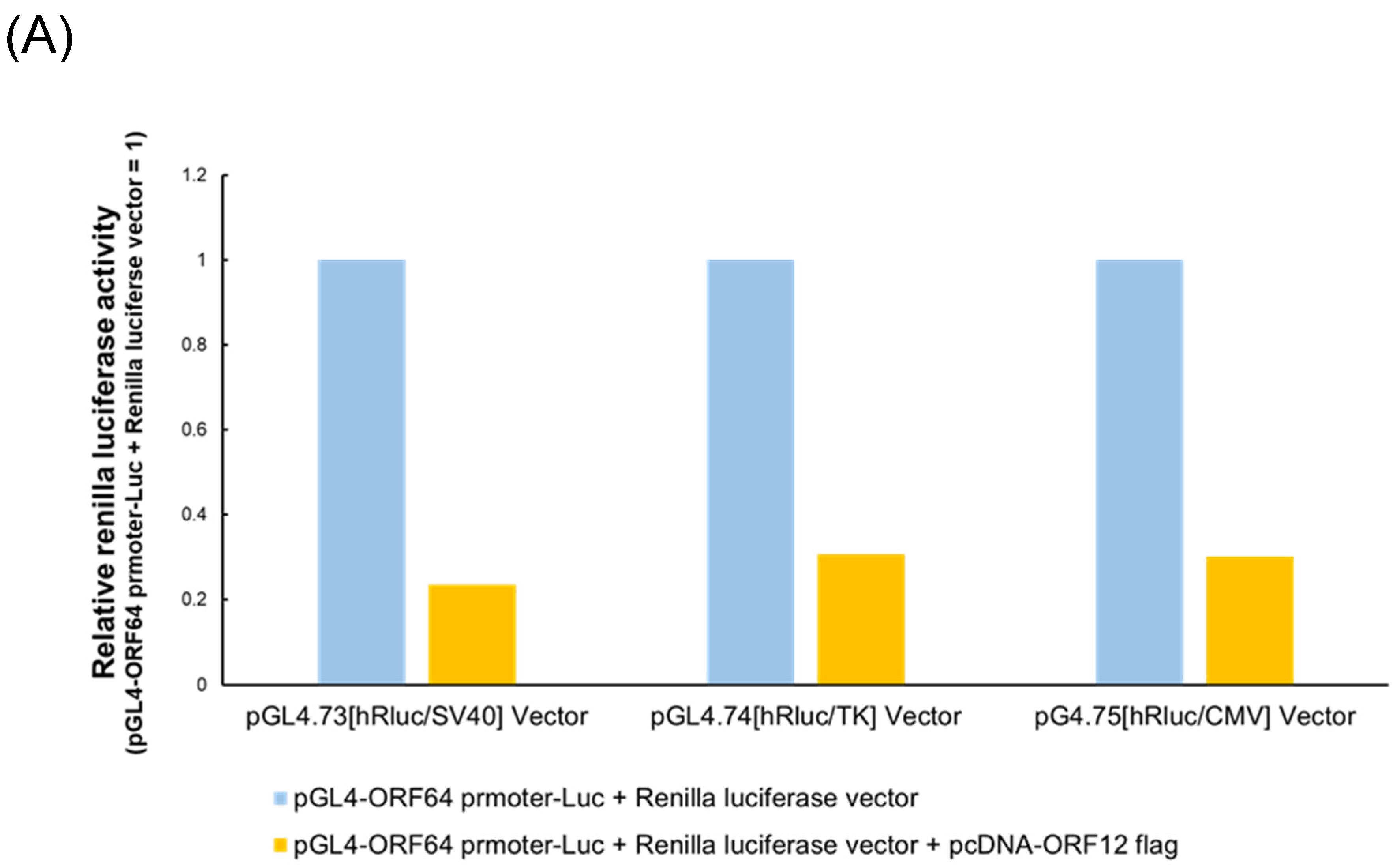
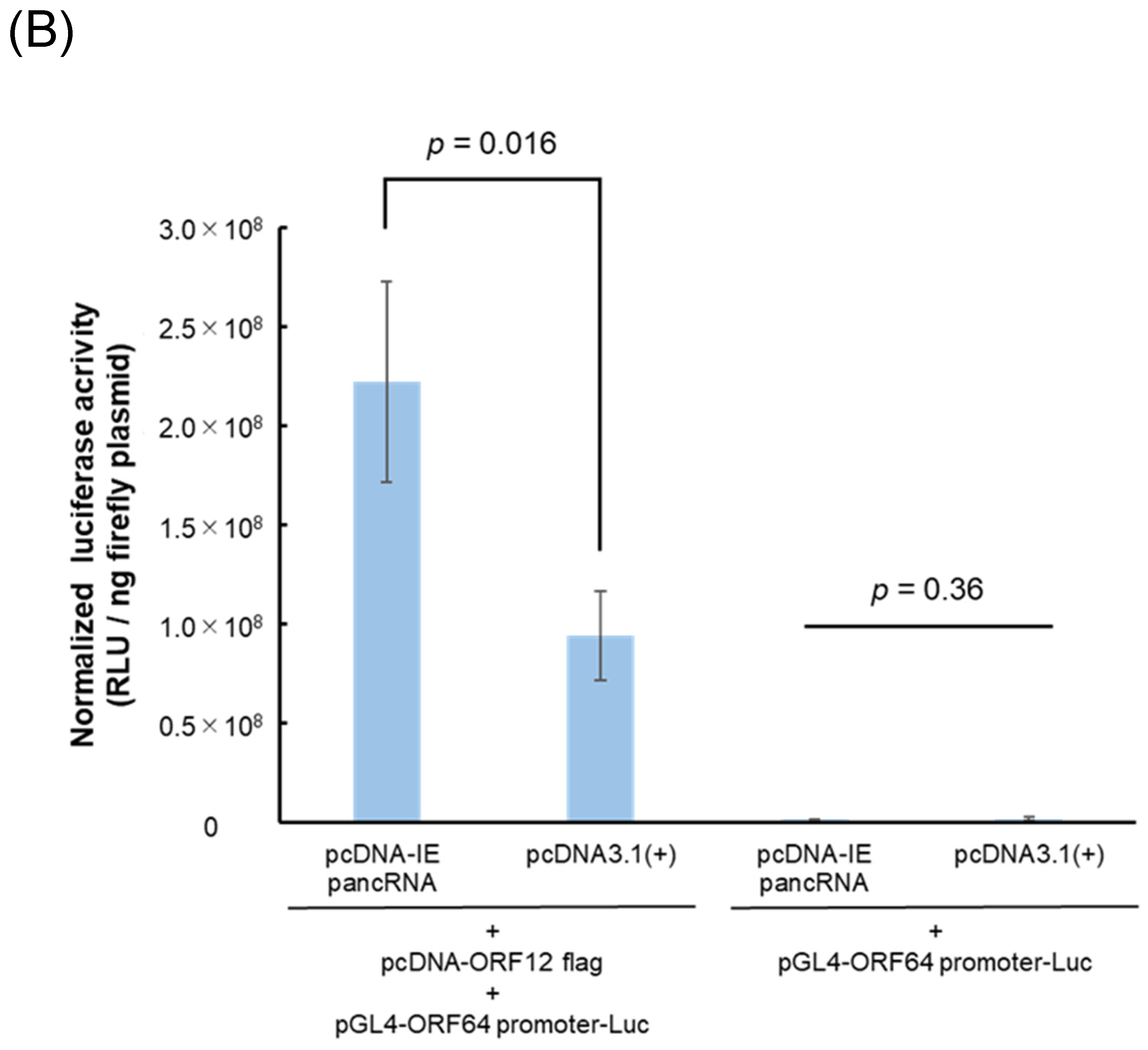
Figure 5.
Effect of IE pancRNA expression on EHV-1 infection in undifferentiated Rn33B-A68B2M cells. Undifferentiated Rn33B-A68B2M cells expressing IE pancRNA (Rn33B-A68B2M-IE pancRNA) and undifferentiated Rn33B-A68B2M cells were infected with Ab4-green fluorescent protein (GFP) at an MOI of 0.05. At 24 and 48 h p.i., the cells were harvested, dissociated, and the number of GFP-positive cells was quantified via flow cytometry. Bar represents the mean with standard deviation from three independent experiments. Student’s t-test.
Figure 5.
Effect of IE pancRNA expression on EHV-1 infection in undifferentiated Rn33B-A68B2M cells. Undifferentiated Rn33B-A68B2M cells expressing IE pancRNA (Rn33B-A68B2M-IE pancRNA) and undifferentiated Rn33B-A68B2M cells were infected with Ab4-green fluorescent protein (GFP) at an MOI of 0.05. At 24 and 48 h p.i., the cells were harvested, dissociated, and the number of GFP-positive cells was quantified via flow cytometry. Bar represents the mean with standard deviation from three independent experiments. Student’s t-test.
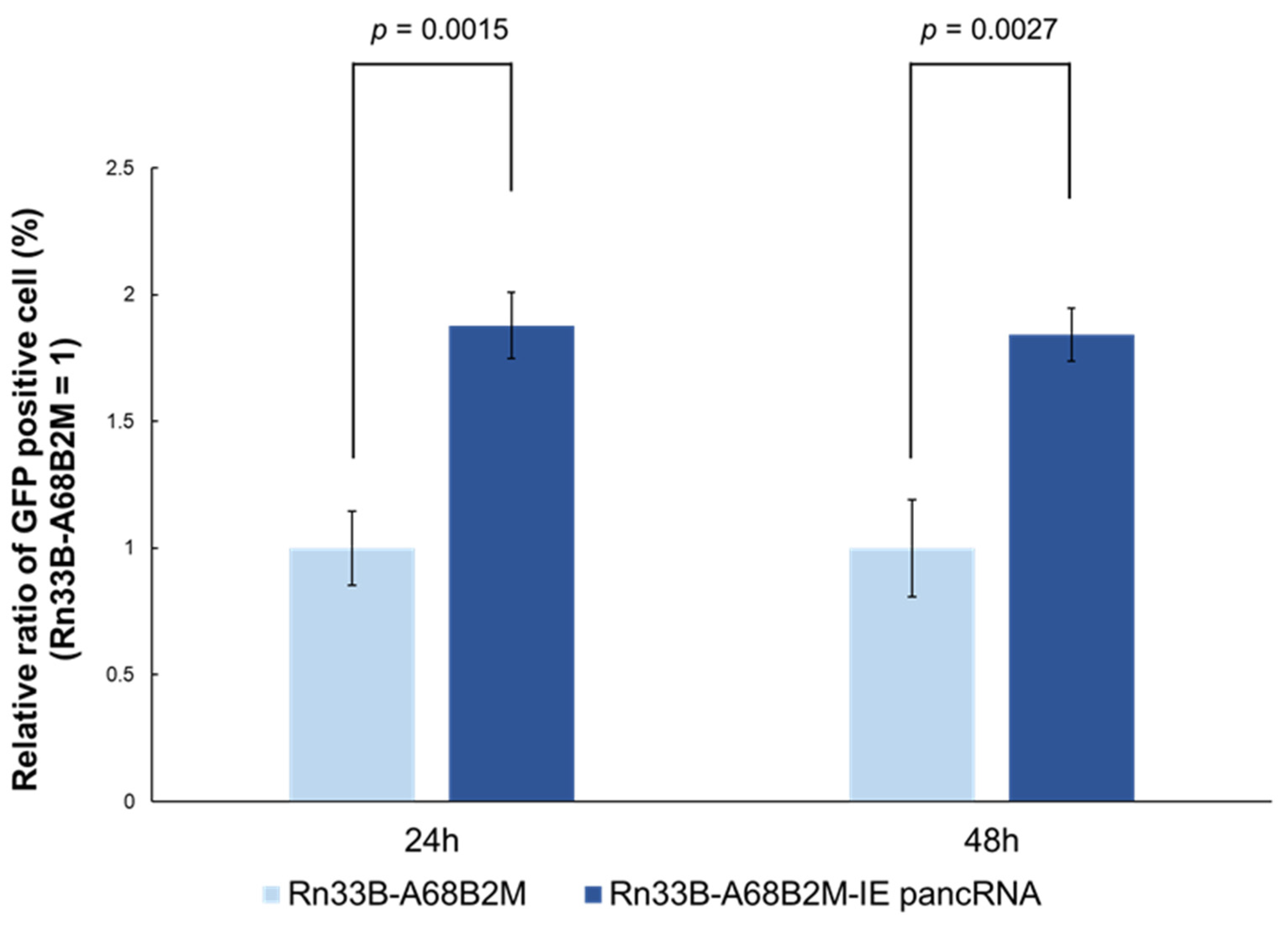

Table 1.
Nucleotide sequences of primers used for reverse transcription-quantitative polymerase chain reaction (RT-qPCR) and rapid amplification of cDNA ends (RACE).
Table 1.
Nucleotide sequences of primers used for reverse transcription-quantitative polymerase chain reaction (RT-qPCR) and rapid amplification of cDNA ends (RACE).
| Primer | Sequence (5’–3’) | |
|---|---|---|
| EHV-1 IE promoter_F | TCAACGGCCAATCACAATCG | (20 bp) |
| EHV-1 IE promoter_R | TACGATGGGTAAGCAACAGGTG | (22 bp) |
| Rat_β-actin_F | AAGTCCCTCACCCTCCCAAAAG | (22 bp) |
| Rat_β-actin_R | AAGCAATGCTGTCACCTTCCC | (21 bp) |
| 5’ RACE Antisense GSP | GATTACGCCAAGCTTCGACACACGGGTTCTAATTGGTTGGAG | (42 bp) |
| 3’ RACE Antisense GSP | GATTACGCCAAGCTTCTACGATGGAGTTTTGCCTTCCCCCTA | (42 bp) |
| 5’ RACE Sense GSP-1 | GATTACGCCAAGCTTTACGATGGAGTTTTGCCTTCCCCCTAGT | (43 bp) |
| 5’ RACE Sense GSP-2 | GATTACGCCAAGCTTCTCCAACCAATTAGAACCCGTGTGTCG | (42 bp) |
| 3’ RACE Sense GSP-1 | GATTACGCCAAGCTTACGACACACGGGTTCTAATTGGTTGGAG | (43 bp) |
| 3’ RACE Sense GSP-2 | GATTACGCCAAGCTTCGCTTCCCTGGGAGGAGACATACGCAAA | (43 bp) |
| 3’ RACE Sense GSP-3 | GATTACGCCAAGCTTCACTAGGGGGAAGGCAAAACTCCATCG | (42 bp) |
| Firefly Luciferase_F | CACCGTCGTATTCGTGAGCA | (20 bp) |
| Firefly Luciferase_R | AGTCGTACTCGTTGAAGCCG | (20 bp) |
Abbreviation: bp, base pair.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



