
Submitted:
16 May 2024
Posted:
16 May 2024
You are already at the latest version
Abstract
Human immunodeficiency virus (HIV) and malaria, caused by infection with Plasmodium spp., are endemic in similar geographical locations. As a result, there is high potential for HIV/Plasmodium co-infection, which increases the pathology of both diseases. However, the immunological mechanisms underlying the exacerbated disease pathology observed in co-infected individuals are poorly understood. Here, we used the rhesus macaque (RM) model to characterize the immunopathogenic impact of Plasmodium fragile co-infection during antiretroviral therapy (ART)-treated simian immunodeficiency virus (SIV)-infection. We observed that P. fragile co-infection resulted in parasitemia and anemia, as well as persistently detectable viral loads (VL) and decreased absolute CD4+ T-cell counts despite daily ART treatment. Notably, P. fragile co-infection was associated with increased levels of inflammatory cytokines linked with neutrophil function, including monocyte chemoattractant protein 1 (MCP-1). P. fragile co-infection was associated with increased levels of neutrophil elastase, a plasma marker of neutrophil extracellular trap (NET) formation, but significant decreases in markers of neutrophil degranulation, potentially indicating a shift in neutrophil functionality during co-infection. Finally, we characterized levels of plasma markers of gastrointestinal (GI) barrier permeability and microbial translocation and observed significant correlations between indicators of GI dysfunction, clinical markers of SIV and Plasmodium infection, and neutrophil frequency and function. Taken together, these data indicate that neutrophil-driven inflammation and GI dysfunction may underlie heightened SIV/P. fragile co-infection pathogenesis.
Keywords:
NHP
; P. fragile
; SIV
; malaria
; neutrophils
; co-infection
; immunology
Introduction
Human immunodeficiency virus (HIV) and malaria, caused by Plasmodium parasites, are two of the world’s most devastating infections. In 2022, over 39 million people were living with HIV (PWH) [1], and there were over 247 million cases of malaria [2]. Despite effective tools and treatments, challenges in prevention and eradication of both infections remain. Indeed, consistent use of antiretrovirals (ART) allows for sustained viral suppression, improving health and quality of life of PWH [3]. However, ART does not eliminate the viral reservoir and plasma viremia rapidly rebounds post-treatment interruption [4,5]. Similarly, antimalarial drugs can prevent and cure Plasmodium infection, but emergence of drug-resistance undermines control efforts and contributes to increased morbidity and mortality [6,7]. For both, the complexities of the infectious agents, combined with an incomplete knowledge of the immunological mechanisms underlying pathogenesis, hampers identification of immune correlates of protection and development of fully efficacious prophylactic vaccines.
HIV and malaria endemicity are geographically overlapped, creating high potential for co-infection. A meta-analysis of studies conducted between 1991 and 2018 found that co-incidence of HIV and severe malaria, or presence of peripheral parasitemia associated with fatal outcomes, was 43% [8]. Prior work has demonstrated reciprocal antagonistic effects that results in increased transmission of both HIV and malaria. For example, Plasmodium increases HIV VL both in vitro and in ART-naïve PWH [9,10,11]. High VLs correlate with increased HIV transmission, suggesting that malaria co-infection of PWH could enhance HIV transmission risk [12,13,14]. Similarly, clinical malaria prevalence, malaria infection severity, and malaria-associated mortality rates are increased in ART-naïve PWH [15,16,17,18]. Additionally, previous in vitro and ex vivo studies have indicated that co-infection with both HIV and Plasmodium exacerbates disease pathogenesis. For example, in vitro infection of monocyte-derived macrophages with a laboratory strain of HIV-1 resulted in inhibited phagocytic capability and cytokine production in response to stimulation with opsonized trophozoites from a laboratory-adapted strain of P. falciparum [19]. Moreover, HIV-infected children in Malawi with cerebral malaria infection had higher rates and more rapid progression to mortality, greater parasite loads in brain and spleen, and greater brain accumulation of monocytes and platelets in the brain, as compared to children without HIV [20,21]. Notably, uncontrolled inflammation underlies disease pathogenesis in separate HIV and malaria infection [22,23]. In PWH, increased inflammation associates with viral persistence, disruptions in intestinal homeostasis, and increased risk of co-infection with other pathogens [24]. Likewise, a pro-inflammatory environment associates with severe malaria and increased malaria-associated mortality [23,25]. Importantly, the impact of Plasmodium co-infection on ART efficacy in PWH, and the link between inflammatory responses and disease pathology during co-infection, has not yet been defined [26].
Neutrophils are granulocytes that constitute up to 70% of all circulating leukocytes [27] and aid in host defense through: 1) exocytosis of anti-microbial molecule-containing granules; 2) phagocytosis and destruction of microbes in phagosomes; and 3) formation of neutrophil extracellular traps (NETs), DNA decorated with granule contents that aid pathogen clearance [28,29,30,31]. Conversely, dysregulated neutrophil activation causes uncontrolled inflammation and collateral host tissue damage [32]. For example, although NET formation associates with protection against HIV infection and impaired replication in vitro [33], increased neutrophil activation was linked with adverse clinical outcomes in PWH [34,35,36] and ART-treated PWH exhibited impaired neutrophil phagocytosis and increased neutrophil apoptosis, as compared to uninfected controls [37,38]. Additionally, increased neutrophil infiltration into the gastrointestinal (GI) mucosa in ART-treated PWH and macaques with chronic simian immunodeficiency virus (SIV) infection associated with loss of GI epithelial barrier integrity and elevated microbial translocation [39,40,41], both of which associate with chronic inflammation, morbidity, and mortality in PWH [42,43,44]. Likewise, neutrophil phagocytosis aids in Plasmodium clearance [45,46,47] and NET formation in children associated with parasite killing [48]. However, NET formation also associated with increased inflammation and severe malaria [49,50,51], and excessive neutrophil degranulation contributed to risk of severe malaria (48, 52, 53). Importantly, the role of neutrophils in disease pathogenesis during HIV/Plasmodium co-infection has not been established.
SIV infection induces pathologies similar to progressive HIV, including high peak and chronic plasma VLs and CD4+ T-cell depletion [54]. Additionally, like P. falciparum, which causes most human malaria cases worldwide [2], P. fragile is capable of endothelial adherence, tissue sequestration, and antigenic variation in rhesus macaques (RMs) [55]. SIV/P. fragile co-infection in RMs mimics HIV/P. falciparum co-infection in humans, including increased SIV VL and innate immune dysfunction in ART-naïve SIV/P. fragile co-infected versus singly infected RMs [56,57]. However, SIV/P. fragile co-infection has not been characterized in the context of ART. In this pilot study, we sought to define the impact of P. fragile co-infection on ART-treated SIV infection. We hypothesized that P. fragile co-infection would result in exacerbated SIV pathology that associated with neutrophil dysfunction despite ART. To test this hypothesis, we infected four adult RMs with SIVmac239, followed by ART-initiation, and P. fragile co-infection, and longitudinally monitored clinical and immune markers.
Methods and Materials:
Study Animals and Approval
Four adult (aged 6–12 years) male Indian-origin RMs were housed and cared for at the Tulane National Primate Research Center (TNPRC) under an Institutional Animal Care and Use Committee (IACUC; Office of Laboratory Animal Welfare Assurance Number A4499-01) approved protocol (P0477-3564). Animal housing, care, and procedures were performed in Association for Assessment and Accreditation of Laboratory Animal Care accredited facilities (AAALAC Number 000594), compliant with United States Department of Agriculture regulations, including the Animal Welfare Act (9 CFR) and the Animal Care Policy Manual, with guidelines established by the National Research Council in the Guide for the Care and Use of Laboratory Animals and the Weatherall Report. All animals were naïve for both SIV and Plasmodium prior to study assignment. In addition, animals were negative for MHC class I alleles associated with SIV control, including Mamu-A*01, Mamu-B*08, and Mamu-B*17 [58,59,60]. Animals were singly housed indoors under climate-controlled conditions, a 12-hour light/12-hour dark cycle and were monitored daily to ensure welfare. Abnormalities were recorded and reported to a veterinarian. Water was available ad libitum and animals were fed commercial monkey chow (Purina LabDiet; PMI Nutrition International, Richmond, IN), supplemented with fruits, vegetables, and foraging treats as a part of the TNPRC environmental enrichment program. At week 2 post-SIV infection (p.i.), one animal (LN07) received a topical antibiotic for a surface wound. At weeks 10 and 13 p.i., all RMs received Kefzol (6.25mg/kg) during surgical procedures. At week 14 p.i. one animal (LE96) received a blood transfusion. At weeks 14 (LC40) and 16 p.i. (LC40, LE96), two animals received a dose of the antibiotic Excede (200mg/ml). Procedures were performed under direction of TNPRC veterinarians. Anesthesia was used in accordance with TNPRC policy and the Weatherall Report. Euthanasia at study endpoint was performed using methods consistent with recommendations of the American Veterinary Medical Association and per the recommendations of the IACUC.
SIV Inoculation, Monitoring, and ART Treatment
RMs were intravenously inoculated with 50 TCID50 SIVmac239 [61]. Plasma VLs were monitored via RT-qPCR [62]. Starting at week 8 p.i. and continuing until endpoint, RMs received daily ART, administered subcutaneously, consisting of tenofovir disoproxil fumarate (TDF; 5.1mg/kg), emtricitabine (FTC; 30mg/kg; both from Gilead, Foster City, CA), and dolutegravir (DTG; 2.5mg/kg; ViiV Healthcare, London, England, UK), formulated in Kleptose (15% in 0.1 N NaOH, Roquette, Lestrem, France), a formulation selected for effectiveness in suppressing SIV replication in RMs [63].
P. fragile Inoculation, Monitoring, and Anti-Malarial Treatment
RMs were intravenously inoculated with 20x10^6 P. fragile-infected erythrocytes (Sri Lanka strain) [64,65,66]. Briefly, cryopreserved erythrocytes were thawed and resuspended in 12% NaCl (Thermo Fisher Scientific, Waltham, MA) for 5min at room temperature (RT). Next, 1.6% NaCl was added dropwise, followed by centrifugation for 10min, RT, 1400 revolutions per minute (RPM). The pellet was resuspended in 0.9% NaCl and 2% dextrose (Thermo Fisher Scientific), centrifuged for 10min, RT, 1400 RPM, and resuspended in 0.9% of NaCl for inoculation. Anemia was monitored via hematocrit (HCT), calculated as the ratio of erythrocytes to total blood volume. Parasitemia was monitored via Giemsa staining of thin blood smears collected from sedated animals via venipuncture or tail sticks from non-sedated animals using positive reinforcement, three days a week starting in Week 12, delineated as week A, B, or C. Smears were fixed in methanol for 5min, followed by staining in 5% Giemsa solution (pH=7.2) for 45-60min and washed in distilled water. Parasitemia was calculated as the average number of parasitized erythrocytes among all erythrocytes in 10 randomized fields of view. Post week 14A p.i., RMs received anti-malarial drugs via oral gavage consisting of one administration of quinine sulfate (150mg; Archway Apothecary, NDC: 51927-1588-00), followed by four daily administrations of chloroquine (20mg/kg; Health Warehouse, NDC: 64980-0177-50).
Sample Collection and Processing
EDTA and serum gel vacutainer tubes (Starstedt, Newton, NC) were used to collect peripheral blood. Complete blood counts (CBCs) were performed using EDTA blood on a Sysmex XN-1000v (Sysmex, Kobe, Hyogo, Japan). Blood chemistry was performed using fresh serum and C-reactive protein (CRP) levels were quantified using frozen serum on a Beckman AU480 (Beckman, Brea, CA). For experimental procedures, EDTA blood was centrifuged for 10min, 2000 RPM, RT to isolate plasma, which was stored at -80°C. After plasma removal, whole blood was reconstituted with PBS. Aliquots of 250µL of blood-PBS were set aside for flow cytometric staining. Peripheral blood mononuclear cells (PBMCs) were isolated from remaining blood via density-gradient centrifugation using Ficoll-Paque Plus (Sigma-Aldrich, St. Louis, MO) and Accuspin tubes (Sigma-Aldrich). PBMCs were cryopreserved in freezing media (5mls dimethyl sulfoxide (DMSO) [Sigma-Aldrich] + 45mls heat inactivated fetal bovine serum [Thermo Fisher Scientific]) and stored in liquid nitrogen.
Flow Cytometry
Multicolor flow cytometry was performed on whole blood using RM cross-reactive monoclonal antibodies. Samples were first stained with a Live/Dead Fixable Aqua dead cell stain (Thermo Fisher Scientific), then treated with Fc block (BD Biosciences, Franklin Lakes, NJ). Extracellular staining was performed using predetermined fluorochrome conjugated antibody concentrations (Supplemental Table S1), followed by red blood cell lysis using 1x FACS lysing solution (BD Biosciences). Cells were fixed, permeabilized (CytoFix/Perm Kit, BD Biosciences), then intracellularly stained (Supplemental Table S1 and Supplemental Figure S1).
Phagocytosis was evaluated using E. coli bioparticles conjugated to a dye that fluoresces in acidic environments (pHrodo Red E. coli Bioparticles Phagocytosis Kit for Flow Cytometry; Thermo Fisher Scientific). Briefly, pHrodo bioparticles were incubated with plasma from healthy RMs (1:3 plasma:pHrodo ratio) for 30min to allow for bioparticle opsonization. Opsonized pHrodo bioparticles were added to 250µL whole blood aliquots for 2hrs at 37°C, followed by surface staining (Supplemental Figure S2).
All samples were fixed with 1% paraformaldehyde and held at 4°C until acquisition on a BD LSRFortessa using FACSDiva software (v9.0). Single-color controls were acquired in every experiment as compensation. Analysis was performed using FlowJo (v10). In all analysis, individual cell subsets with less than 100 parental gate events were not included in downstream analysis due to the inability to ensure adequate fluorescence separation.
CD4+ T lymphocyte kinetics were monitored by flow cytometric evaluation of absolute counts. Briefly, 50μL whole blood were surface stained (Supplemental Table S2), and incubated for 20min, RT, in the dark. Red blood cells were lysed with 1X BD FACS Lysing Solution for 30-45 minutes. The sample was mixed and volumetrically analyzed on a Miltenyi MACSQuant 16 (Miltenyi, Bergisch Gladbach, Germany).
Detection of Plasma Markers of Neutrophil Function, GI Mucosal Barrier Integrity, and Microbial Translocation
Commercially available enzyme-linked immunosorbent assay (ELISA) kits were used to quantify plasma levels of neutrophil granule components, including myeloperoxidase (MPO; Abcam, Cambridge, UK), cathepsin G (MyBiosource, San Diego, CA), proteinase 3 (PR3; MyBiosource); biomarkers of NET formation, including citrullinated histone 3 (CitH3; Cayman Chemicals, Ann Arbor, MI), and neutrophil elastase (NE; LSBio, Lynwood, WA); markers of GI barrier permeability, including Zonulin-1 (MyBiosource) and intestinal fatty acid binding protein (IFAB-P; Novus Biologicals, Centennial, CO); and surrogate markers of microbial translocation, including lipopolysaccharide (LPS) binding protein (LBP; Novus Biologicals) and soluble CD14 (sCD14) ELISA (ThermoFisher), as per the manufacturers’ recommended protocols.
Detection of Plasma Markers of Systemic Inflammation
A BioLegend LEGENDplex™ NHP Inflammation Panel (13-plex) with V-bottom plate was used to quantify plasma levels of inflammation markers IL-6, IL-10, CXCL10 (IP-10), IL-1β, IL-12p40, IL-17A, IFN-β, IL-23, TNF-α, IFN-γ, GM-CSF, CXCL8 (IL-8), CCL2 (MCP-1). Plasma samples were run in duplicate at a 1:4 dilution, as per manufacturer recommendations. Samples were acquired on a Miltenyi MACSQuant 16 in a 96 well plate format. Data were analyzed with the BioLegend LEGENDplex™ online analysis software against a standard curve.
Data and Statistical Analysis
We used the absolute number of neutrophils/µL blood to calculate the number of neutrophils positive for pHrodo bioparticles to characterize phagocytosis (phagocytic score, or the number of neutrophils capable of phagocytosis) [67]. Phagocytic index, representing phagocytic proficiency, was calculated by multiplying the phagocytic score by the Mean Fluorescence Intensity of pHrodo positive neutrophils [68].
Statistical significance between all timepoints was calculated using a mixed-effects analysis of variance with the Geisser-Greenhouse correction. Multiple comparisons between all timepoints were performed using Tukey’s multiple comparisons tests based on individual variances for each comparison. In all figures, multiplicity adjusted significant P values are shown above horizontal black bars. Multivariate analysis of variance (MANOVA) approaches were applied to identify potential relationships between various neutrophil measures, clinical signs of both SIV and P. fragile infection, peripheral markers of inflammation, and peripheral markers of GI dysfunction. The MANOVA approach allowed us to account for temporal dependence, as previously described [69]. MANOVA was used to model 13 parameters (VL, anemia, absolute CD4+ count, peripheral neutrophils, plasma zonulin, sCD4, I-FABP, LBP, NE, cathepsin G, CitH3, IP-10, MCP-1, and CRP) against animal number. Partial correlation coefficients were generated based on the MANOVA error terms adjusted for animal effects. Statistical analyses were performed using GraphPad Prism (Version 10; GraphPad Software, San Diego, CA) and Statistical Analysis System (SAS, Version 9.4; Cary, NC). All reported P values were multiplicity-adjusted and values of <0.05 were considered significant. The JoinPoint Regression Program (NIH, V5.0.2, Bethesda, MD) was utilized to assess longitudinal trends in VLs.
Results:
Experimental Design
Following baseline (BL) sampling, RMs (n=4) were intravenously inoculated with SIVmac239 (Figure 1). At week 8 post-SIV infection (p.i.), RMs initiated ART, which continued until euthanasia at week 20 p.i.. For simplicity, all time points in our analyses are reported as post-SIV infection. At week 12 p.i., RMs were intravenously inoculated with P. fragile. At week 14 p.i., RMs surpassed our treatment threshold of 0.5% parasitemia and received antimalarial drugs via oral gavage. Physical exams, peripheral blood collections, CBCs, and serum chemistries were conducted throughout the study.
P. fragile co-Infection of ART-Treated SIV+ RMs Results in Clinical Signs of Malaria
Parasitemia and anemia are clinical hallmarks of malaria [70,71]. Following P. fragile co-infection, parasitemia was assessed tri-weekly (A, B, C). RMs reached peak parasitemia by week 14A p.i. and had undetectable parasitemia following antimalarial treatment (P=0.0429 for week 14 p.i. versus all other timepoints; Figure 2A). All RMs experienced mild to severe anemia between weeks 14-15 p.i., coinciding with peak parasitemia (Figure 2B). Percent hematocrit (% HCT) was significantly lower at week 14 p.i. compared to weeks 10, 12, and 13 p.i. (P=0.0367, 0.0391, and 0.049, respectively; Figure 2B). Additionally, % HCT was significantly lower at week 15 p.i. compared to BL and weeks 2, 6, 19, and 20 p.i. (P=0.0122, 0.0155, 0.0448, 0.0286, and 0.0366, respectively; Figure 2B). RMs remained mildly anemic at week 17 p.i., demonstrated by significantly lower % HCT compared to week 8 p.i. (P=0.0493; Figure 2B). By week 18 p.i., all RMs were non-anemic and had significantly greater % HCT at week 19 p.i. as compared to week 17 p.i. (P=0.0485; Figure 2B). Taken together, P. fragile co-infection of ART-treated SIV+ RMs induced significant parasitemia and anemia, hallmarks of clinical malaria.
P. fragile co-Infection Results in Clinical Signs of SIV Infection Despite Daily ART
Uncontrolled VL and decreased CD4+ T-cell counts within a few weeks following infection are hallmarks of pathogenic HIV/SIV [72,73,74,75]. Treatment with ART has been shown to suppress viral replication and restore CD4+ T-cell counts in SIV-infected RMs even at end stage disease [63,76]. Here, we observed that peak viremia in RMs inoculated with SIVmac239 occurred by week 3 p.i. (median=1.160 x107 copies/ml; Figure 2C). Lower VLs were observed following ART, with 2/4 RMs (LC40 and LE96) becoming undetectable by week 12 p.i. (Figure 2C). Following P. fragile inoculation, all RMs maintained VLs above the limit of detection from weeks 13-17 p.i.(Figure 2C). Longitudinal assessment by JoinPoint regression demonstrated that following P. fragile co-infection, the VL of all 4 RM remained unchanged, indicating viral persistence (Supplemental Figure 3). Between weeks 18-20 p.i., one RM (LE96) had transiently detectable VLs, two RMs became undetectable by weeks 19 (LN07) and 20 p.i. (LC40), with the remaining RM (JF97) remaining incompletely suppressed. Absolute CD4+T-cell counts were significantly lower at week 3 p.i. compared to BL (P=0.0414) and remained low until ART initiation at week 8 p.i. (Figure 2D). Following P. fragile inoculation, CD4+ T-cell counts were significantly increased at week 13 p.i. compared to week 3 p.i. (P=0.0397; Figure 2D). By week 17 p.i. following antimalarial treatment, CD4+ T-cell counts returned to pre-ART levels (Figure 2D). These data suggest that co-infection of ART-treated SIV+ RMs with P. fragile results in persistently detectable VLs and fluctuations in CD4+ T cell counts.
Increased Levels of Inflammatory Markers Associated with Neutrophil Function during ART-Treated SIV/P. Fragile co-Infection
CRP is an acute phase plasma marker of inflammation that has been shown to be elevated in ART-naïve PWH and is associated with HIV disease progression even despite ART [77,78,79,80]. Additionally, elevated CRP has been observed in individuals with complicated and uncomplicated malaria and can be used as a biomarker for monitoring of malaria severity [81,82]. Here, we observed that serum CRP was significantly increased at week 14 p.i. as compared to BL and weeks 2, 6, 8, and 10 p.i. (P=0.0455 for all), as well as compared to weeks 12, 15, and 20 p.i. (P=0.0435, 0.0424, and 0.0428, respectively; Figure 3A). These data suggest that there is an increase in systemic inflammation following P. fragile co-infection of ART-treated, SIV+ RMs that coincides with peak parasitemia.
Both HIV and Plasmodium infection result in elevated levels of pro-inflammatory cytokines and chemokines [23,83]. Here, we observed some longitudinal and inter-animal variation in plasma levels of interleukin (IL)-8 and interferon gamma-induced protein 10 (IP-10), but no statistically significant changes in these analytes throughout P. fragile co-infection of ART-treated SIV+ RMs (Figure 3B and C). Plasma monocyte chemoattractant protein-1 (MCP-1) was significantly increased at week 14 p.i. compared to BL, and weeks 16 and 20 p.i. (P=0.0223, 0.0406, and 0.0412, respectively; Figure 3D). All other inflammatory analytes remained unchanged (Supplementary Figure 4). These findings suggest that neutrophil-associated chemokines are altered after P. fragile co-infection of ART-treated, SIV+ RMs.
Stable Peripheral Neutrophil Frequencies and Apoptosis during ART-Treated SIV/P. fragile co-Infection
We next characterized peripheral neutrophil dynamics and apoptosis via flow cytometry. Neutrophils were identified as viable, CD45+ HLA-DR- CD11b+ CD66abce+ CD14+ CD49d- cells, as previously described (Supplemental Figure S1) [84,85,86,87,88,89]. As previously demonstrated [90], total peripheral neutrophil frequency was consistent throughout acute SIV and ART-treatment as compared to BL (Figure 4A). Neutrophil frequencies were significantly decreased following anti-malarial treatment as compared to the start of anti-malarial treatment (week 17 vs. 14 p.i., P=0.0136), followed by stable neutrophil frequencies until study endpoint in week 20 p.i. (Figure 4A). Percentages of apoptotic peripheral neutrophils (Caspase3+) fluctuated across all timepoints (Figure 4B). These data indicate that neutrophil frequencies decrease following clearance of P. fragile, while neutrophil apoptosis is unchanged throughout co-infection.
Minimal Disruption in Neutrophil Phagocytosis during ART-Treated SIV/P. Fragile co-Infection
Impaired phagocytosis has been observed in separate HIV and Plasmodium infection [37,38,45,46,47]. Here, we calculated peripheral blood neutrophil phagocytic score (capability) and index (proficiency) throughout ART-treated SIV/P. fragile co-infection. Although inter-animal variations in neutrophil phagocytic score (Figure 5A) and index (Figure 5B) were observed, no statistically significant changes in either phagocytosis parameter were detected over time.
Decreased Plasma Levels of Neutrophil Granule Components during ART-Treated SIV/P. fragile co-Infection
Plasma levels of MPO, PR3, and CATG, three extracellular neutrophil degranulation secreted components [91,92], were quantified throughout ART-treated SIV/P. fragile co-infection. Consistent with prior work [93], plasma MPO levels were elevated in all four RMs at week 2 p.i., but no significant differences were observed throughout SIV infection, ART-treatment and P. fragile co-infection (Figure 6A). Likewise, plasma levels of PR3 were stable throughout acute SIV infection, ART-treatment, and P. fragile co-infection (Figure 6B). Finally, a statistically significant decrease in CATG was observed at week 14 p.i., compared to BL and weeks 6 and 10 p.i. (P=0.032, 0.0038, and 0.0331, respectively), followed by a return to BL levels following antimalarial treatment (Figure 6C). These data indicate that P. fragile co-infection of ART-treated SIV+ RMs resulted in lowered plasma levels CATG.
Increased Plasma Biomarker of NET Formation during ART-Treated SIV/P. Fragile co-Infection
Excessive NET formation contributes to inflammation in separate HIV and Plasmodium infection (33, 48, 52, 53). We assessed plasma levels of NE and CitH3, biomarkers of NET formation [94], throughout ART-treated SIV/P. fragile co-infection. Plasma levels of NE were significantly increased at week 14 p.i. compared to BL and weeks 2, 10, 12, and 13 p.i. (P=0.0354, 0.0237, 0.0450, 0.0406, and 0.0066, respectively; Figure 7A), but significantly reduced at weeks 16 and 20 p.i. as compared to week 14 p.i. (P=0.0137 and 0.0096, respectively) and week 15 p.i. (P=0.0359 and 0.0216, respectively; Figure 7A). Plasma NE was significantly lower at week 20 p.i. compared to week 16 p.i. (P=0.0341; Figure 7A). Plasma levels of CitH3 were elevated in all four RMs at week 14 p.i., but no significant differences were observed throughout SIV infection, ART-treatment, and P. fragile co-infection (Figure 7B). These findings suggest that P. fragile co-infection of ART-treated SIV+ RMs resulted in increased expression of NE, a marker of NET formation.
Increased Plasma Markers of Gut Permeability and Microbial Translocation during ART-Treated SIV/P. Fragile co-Infection
Loss of GI epithelial barrier integrity leads to microbial translocation in both HIV and Plasmodium infection [39,43,44,95]. Here, we examined plasma levels of zonulin, a protein that modulates tight junctions [96], I-FABP, a circulating biomarker of intestinal injury [97,98], sCD14, which is released from monocytes upon lipopolysaccharide (LPS) stimulation, and LBP, which assists in LPS recognition by interacting with LPS receptors [99]. Inter-animal variation in plasma zonulin levels were observed throughout the study (Figure 8A). Plasma I-FABP was significantly increased at week 14 p.i. compared to week 2 p.i. (P=0.0436; Figure 8B), sCD14 was significantly greater at week 16 p.i. compared to week 6 p.i. (P=0.0377; Figure 8C) and LBP was significantly increased at week 14 p.i. compared to weeks 2, 15 and 20 p.i. (P=0.0441, 0.0305 and 0.0027, respectively; Figure 8D). These findings indicate that P. fragile co-infection may exacerbate GI epithelial barrier disruption, resulting in microbial translocation, in ART-treated SIV+ RMs.
Discussion:
Here, we characterized the impact of P. fragile co-infection on ART-treated, SIV+ RMs. Pathogenic SIV infection in RMs has been well-characterized, with RMs exhibiting uncontrolled VLs and decreased CD4+ T-cell counts within two weeks following SIV infection, similar to pathogenic HIV infection in humans [73,74,75]. Additionally, previous work has shown that ART-initiation during SIV infection results in decreased VLs within two weeks of starting treatment [63] and that ART treatment rapidly restores CD4+ T-cells and T-cell functionality, even at end stage disease [76]. Consistent with these prior findings, all four RMs in our study exhibited elevated VLs and depleted CD4+ T-cell counts during acute SIV infection, followed by decreased VLs and elevated CD4+ T-cell counts post ART-initiation, indicating that all RMs followed the expected progression of acute SIV infection and response to suppressive therapy.
Following P. fragile co-infection, SIV-infected RMs maintained detectable VLs and decreased CD4+ T-cell counts for several weeks. Although SIV/P. fragile co-infection has been examined previously [56,57], to the best of our knowledge, this is the first assessment of SIV/P. fragile co-infection during ART. Our observation of detectable VLs during P. fragile co-infection despite ART agrees with prior reports noting detectable VLs during co-infection of ART-naïve RMs [56]. Additionally, Plasmodium co-infection resulted in increased HIV replication sans ART in vitro and in vivo in humans (9-11, 100-102). Of note, one RM (JF97) had persistently high VLs despite ART, as well as higher levels of inflammatory cytokines and chemokines. Previous work has shown associations between host genetics, including expression of particular MHC alleles, and high VLs and rapid disease progression in both humans [103] and macaques [104,105]. Recent work suggests an association between Mamu-B*012 and high VLs in RM, but only with specific KIR alleles [104]. Here, one animal (LE96) did express Mamu-B*012, but KIR genotyping was not performed and this animal did not exhibit exceptionally high VLs. In addition, there were no marked differences in clinical history, such as values reported in weekly CBCs or blood chemistries, or physical exams, between JF97 and the other three RMs assessed here. Additional work is needed to understand JF97’s inability to virally control despite ART. Importantly, there is no expected interaction between the ART regimen (TDF/FTC/DTG) and anti-malarial drugs (chloroquine and quinine sulfate) used, indicating that drug-drug interactions are unlikely to be the cause of persistent SIV VLs despite ART treatment [106].
Systemic inflammation is a hallmark of HIV infection, even with consistent ART [24]. Indeed, CRP, an acute-phase marker of inflammation, has been shown to be elevated in ART-naïve and ART-treated PWH [77,78,79,80]. Additionally, increased CRP has been used as a biomarker of malaria severity [81,82]. Here, we observed increased levels of serum CRP that coincided with peak parasitemia in ART-treated, SIV+ RMs, indicating increased inflammation during co-infection. We also observed that serum CRP was significantly positively correlated with not just neutrophil frequency, but also markers of NE and CitH3, biomarkers of NET formation [94]. Notably, neutrophil-associated inflammation contributes to pathogenesis during separate HIV and Plasmodium infection (50, 84, 107-109). Prior work has also identified links between residual viral replication during ART and uncontrolled inflammation [110,111]. Thus, neutrophil-associated inflammation could constitute a mechanism underlying continued SIV replication during P. fragile co-infection despite ART. Here, ART-treated SIV/P. fragile co-infection resulted in increased MCP-1, a potent monocyte chemoattractant produced by many cells including neutrophils [112]. Additionally, we observed significant correlations between plasma MCP-1 with NE and CitH3. Previously, increased MCP-1 was associated with NET release in individuals with myocardial infarction, which in turn stimulated further MCP-1 production [113]. Taken together, increased MCP-1 production during ART-treated SIVP. fragile co-infection could result from increased NET formation [114], and these processes may cooperatively contribute to heightened inflammation, allowing for persistent viral replication. Supporting this, plasma MCP-1 was correlated with SIV VL, indicating a potential association between neutrophil-mediated inflammation and SIV reactivation during P. fragile co-infection.
Notably, although neutrophil frequency was minimally altered throughout ART-treated SIV/P. fragile co-infection, a significant shift in NE, a peripheral marker of neutrophil function, was detected. We identified that the increase in this marker of NET formation was significantly inversely correlated with anemia, a clinical marker of Plasmodium infection. Conversely, we noted decreased expression of the neutrophil degranulation marker, CATG, and unchanged neutrophil phagocytosis. Neutrophil selection between defense mechanisms appears to be size-dependent; phagocytosis of smaller microbes inhibits NET release but inhibition of phagocytosis due to microbe size prompts NETosis [115]. Notably, previous studies have shown that both opsonized and non-opsonized monocyte/macrophage phagocytosis of P. falciparum-infected erythrocytes are impaired in PWH in vitro and in vivo [115,116,117], although non-opsonized parasite phagocytosis was restored after 6 months of ART [117]. Our data could indicate that an insufficient phagocytic response during SIV/P. fragile co-infection skews neutrophils towards NET formation, providing a potential mechanism by which neutrophils contribute to systemic inflammation during SIV/P. fragile co-infection.
GI dysfunction is a major pathogenic process in separate HIV and malaria infection (42-44, 118). Plasmodium parasite sequestration in the GI tract causes barrier permeability [95,119], while HIV-associated GI mucosal dysfunction is linked with loss of barrier integrity and elevated microbial translocation [43,99]. Our data indicating that SIV VL is positively correlated with plasma zonulin levels is in agreement with this. Importantly, GI neutrophil infiltration and survival has previously been correlated with HIV-associated GI mucosal dysfunction [39,40,41], thus GI neutrophil activity in response to parasite sequestration could further exacerbate SIV-associated GI dysfunction. Our data indicating that plasma markers of GI barrier permeability (plasma sCD14, iFABP, LBP) were associated peripheral markers of NET formation (plasma NE and CitH3) in ART-treated SIV/P. fragile co-infected RMs supports this. Moreover, our observation of an inverse correlation between sCD14, LBP, and anemia, a hallmark of clinical malaria infection further supports a potential relationship between malaria-induced GI dysfunction, possibly mediated by GI neutrophil infiltration and inflammation, during ART-treated SIV co-infection. A caveat of these data is that they are currently limited to plasma markers of neutrophil function, GI barrier integrity, and microbial translocation. Future studies will focus on assessing the relationship between these factors in mucosal tissues to fully define mechanistic relationships between neutrophil-associated GI dysfunction and SIV/malaria co-infection pathogenesis.
A major strength of our work is the utilization of an NHP model which mimics HIV and P. falciparum infection (54, 55, 64, 65). Additionally, our longitudinal assessments provided an opportunity to identify how Plasmodium co-infection could influence SIV pathology in the setting of viral suppression. A caveat of our study is the short duration of ART and lack of complete viral suppression for an extended period prior to P. fragile co-infection. Additional work characterizing P. fragile co-infection in RMs with long-term SIV suppression is needed. Another caveat to this study is the usage of chloroquine and quinine sulfate as our anti-malarial treatment. Both quinine sulfate and chloroquine have been shown to influence various immune parameters, including NET formation and phagocytosis [120,121,122,123,124]. Given that our data indicate that neutrophil phagocytosis was unchanged throughout anti-malarial drug treatment, and the significant increase we observed in NE occurred prior to quinine sulfate and chloroquine administration, the use of these drugs in our study likely did not impact our observations on the effects of P. fragile co-infection on neutrophil responses in ART-treated SIV-infected RMs. Nonetheless, we cannot completely discount the possibility that quinine sulfate and/or chloroquine administration may have affected these parameters, and future studies should include anti-malarial drug treatment only groups to control for this possibility. Previous work has found that although neutrophil frequency and function are unchanged during the pre-erythrocytic stage of Plasmodium infection, total leukocytes are significantly increased [125]. Future studies that incorporate Plasmodium transmission through infected mosquito bites will allow for assessment of the pre-erythrocytic stage. Another limitation is the small number of RMs used, particularly since inter-animal variation was observed in some parameters, which may have contributed to the lack of statistical significance observed at some time points. Additional work with more animals, with the addition of SIV-only, P. fragile-only, and anti-malaria treatment only control groups, will be necessary to fully characterize the kinetics and impact of co-infection, particularly in critical tissues, including the GI mucosa.
In summary, ART-treated SIV/P. fragile co-infected RMs displayed clinical signs of SIV and malaria, which were associated with shifts in neutrophil function and increased markers of GI mucosal dysfunction. These observations could have implications for HIV and malaria co-endemic areas; Plasmodium co-infection in PWH may lead to viral reactivation, creating a scenario in which rates of HIV transmission are sustained even despite widespread use and adherence to ART. Indeed, PWH who have viral loads greater than 1000 copies/ml, regardless of ART, are at increased risk of transmitting HIV [126]. It is important to note, however, that little work has been done to examine whether sustained SIV VL correlates with increased risk of SIV transmission, thus additional work will be needed to extend our observations using an NHP model to ART-treated PWH with Plasmodium co-infection. Moreover, our observations of a link between neutrophil function with clinical signs of SIV/P. fragile and GI dysfunction highlight the need for additional research to define the role of this cellular subset during co-infection and supports the rationale for examining the potential for neutrophil-targeted interventions to reduce the burden of HIV and malaria, separately and in the context of co-infection.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org
Author Contributions
SMN performed and analyzed experiments and prepared the manuscript. HCG, JEP and SLF assisted with tissue processing, flow cytometry and ELISA/LegendPLEX. KG, NJM, and PAM assisted with viral inoculum selection, preparation, and administration. LAR performed MHC genotyping. KDP provided the original P. fragile stock used to propagate the parasite stock used in these studies. MV, KD, NG and MJM assisted with flow cytometry acquisition and expertise. CT, MBB, and CA supported qPCR for viral load assessments. CM and ME supported parasitemia assessments. BG provided veterinary support for animals. JGS provided statistical support. RVB, AK, THM, and BLR assisted with data interpretation and paper preparation. JAM conceived of the study and oversaw the planning and direction of the project, including data interpretation and writing of the paper. All authors read and approved the final manuscript.
Data Availability
All data are available withing the manuscript or in its supplemental materials.
Acknowledgements
We thank all the veterinary staff at the Tulane National Primate Research Center (TNPRC) for their aid with this animal study. We thank Gilead and ViiV for their generous contribution of ART drugs for these studies. Flow Cytometry analysis and expertise were provided by the Flow Cytometry Core, Research Resource Identifier (RRID) SCR_024611. The Flow Cytometry Core is funded by TNPRC base grant OD011104 from NIH. Blood chemistry and CBC were provided the Clinical Pathology Cre (RRID: SCR_024609). The Pathogen Detection and Quantification Core provided viral load data and analysis (RRID: SSCR_024614). Parasitemia data and analysis was provided by the Vector-Borne Infectious Disease and Diagnostic Parasitology Core (RRID: SSC_024680). Virus, virus preparation, and inoculation were given and performed by the Virus Characterization, Isolation, Production, and Sequencing Core (SCR_024679) This work was supported by R21OD031435 to JAM. Research reported in this publication was additionally supported in part by TNPRC (RRID:SCR_008167) NIH core grants P51OD011104, U42OD010568, and U42OD024282. The funders had no direct role in study design, data collection, analysis, or preparation and publication of the manuscript.
Conflict of Interest
The authors declare no conflict of interest.
References
- WHO. 2022. HIV data and statistics 2022, on World Health Organization. Accessed 11/5/23.
- WHO. 2023. World malaria report 2023, on World Health Organization. https://www.who.int/publications/i/item/9789240086173. Accessed 12/12/23.
- Tseng A, Seet J, Phillips EJ. 2015. The evolution of three decades of antiretroviral therapy: challenges, triumphs and the promise of the future. Br J Clin Pharmacol 79:182-94.
- Finzi D, Blankson J, Siliciano JD, Margolick JB, Chadwick K, Pierson T, Smith K, Lisziewicz J, Lori F, Flexner C, Quinn TC, Chaisson RE, Rosenberg E, Walker B, Gange S, Gallant J, Siliciano RF. 1999. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat Med 5:512-7.
- Holkmann Olsen C, Mocroft A, Kirk O, Vella S, Blaxhult A, Clumeck N, Fisher M, Katlama C, Phillips AN, Lundgren JD. 2007. Interruption of combination antiretroviral therapy and risk of clinical disease progression to AIDS or death. HIV Med 8:96-104.
- Menard D, Dondorp A. 2017. Antimalarial Drug Resistance: A Threat to Malaria Elimination. Cold Spring Harb Perspect Med 7.
- Balikagala B, Fukuda N, Ikeda M, Katuro OT, Tachibana SI, Yamauchi M, Opio W, Emoto S, Anywar DA, Kimura E, Palacpac NMQ, Odongo-Aginya EI, Ogwang M, Horii T, Mita T. 2021. Evidence of Artemisinin-Resistant Malaria in Africa. N Engl J Med 385:1163-1171.
- Mahittikorn A, Kotepui KU, De Jesus Milanez G, Masangkay FR, Kotepui M. 2021. A meta-analysis on the prevalence and characteristics of severe malaria in patients with Plasmodium spp. and HIV co-infection. Sci Rep 11:16655.
- Froebel K, Howard W, Schafer JR, Howie F, Whitworth J, Kaleebu P, Brown AL, Riley E. 2004. Activation by malaria antigens renders mononuclear cells susceptible to HIV infection and re-activates replication of endogenous HIV in cells from HIV-infected adults. Parasite Immunol 26:213-7.
- Hoffman IF, Jere CS, Taylor TE, Munthali P, Dyer JR, Wirima JJ, Rogerson SJ, Kumwenda N, Eron JJ, Fiscus SA, Chakraborty H, Taha TE, Cohen MS, Molyneux ME. 1999. The effect of Plasmodium falciparum malaria on HIV-1 RNA blood plasma concentration. Aids 13:487-94.
- Kublin JG, Patnaik P, Jere CS, Miller WC, Hoffman IF, Chimbiya N, Pendame R, Taylor TE, Molyneux ME. 2005. Effect of Plasmodium falciparum malaria on concentration of HIV-1-RNA in the blood of adults in rural Malawi: a prospective cohort study. Lancet 365:233-40.
- Abu-Raddad LJ, Patnaik P, Kublin JG. 2006. Dual infection with HIV and malaria fuels the spread of both diseases in sub-Saharan Africa. Science 314:1603-6.
- Cuadros DF, Branscum AJ, Crowley PH. 2011. HIV-malaria co-infection: effects of malaria on the prevalence of HIV in East sub-Saharan Africa. Int J Epidemiol 40:931-9.
- Quinn TC, Wawer MJ, Sewankambo N, Serwadda D, Li C, Wabwire-Mangen F, Meehan MO, Lutalo T, Gray RH. 2000. Viral load and heterosexual transmission of human immunodeficiency virus type 1. Rakai Project Study Group. N Engl J Med 342:921-9.
- Berg A, Patel S, Aukrust P, David C, Gonca M, Berg ES, Dalen I, Langeland N. 2014. Increased severity and mortality in adults co-infected with malaria and HIV in Maputo, Mozambique: a prospective cross-sectional study. PLoS One 9:e88257.
- Beyene HB, Tadesse M, Disassa H, Beyene MB. 2017. Concurrent Plasmodium infection, anemia and their correlates among newly diagnosed people living with HIV/AIDS in Northern Ethiopia. Acta Trop 169:8-13.
- Ojurongbe O, Oyeniran OA, Alli OA, Taiwo SS, Ojurongbe TA, Olowe AO, Opaleye OO, Adeyeba OA. 2014. Prevalence of Plasmodium falciparum Parasitaemia and Its Correlation with Haematological Parameters among HIV-Positive Individuals in Nigeria. J Trop Med 2014:161284.
- Tay SC, Badu K, Mensah AA, Gbedema SY. 2015. The prevalence of malaria among HIV seropositive individuals and the impact of the co- infection on their hemoglobin levels. Ann Clin Microbiol Antimicrob 14:10.
- Ludlow LE, Zhou J, Tippett E, Cheng WJ, Hasang W, Rogerson SJ, Jaworowski A. 2012. HIV-1 inhibits phagocytosis and inflammatory cytokine responses of human monocyte-derived macrophages to P. falciparum infected erythrocytes. PLoS One 7:e32102.
- Hochman SE, Madaline TF, Wassmer SC, Mbale E, Choi N, Seydel KB, Whitten RO, Varughese J, Grau GE, Kamiza S, Molyneux ME, Taylor TE, Lee S, Milner DA, Jr., Kim K. 2015. Fatal Pediatric Cerebral Malaria Is Associated with Intravascular Monocytes and Platelets That Are Increased with HIV Coinfection. mBio 6:e01390-15.
- Joice R, Frantzreb C, Pradham A, Seydel KB, Kamiza S, Wirth DF, Duraisingh MT, Molyneux ME, Taylor TE, Marti M, Milner DA, Jr. 2016. Evidence for spleen dysfunction in malaria-HIV co-infection in a subset of pediatric patients. Mod Pathol 29:381-90.
- Deeks SG, Tracy R, Douek DC. 2013. Systemic effects of inflammation on health during chronic HIV infection. Immunity 39:633-45.
- Popa GL, Popa MI. 2021. Recent Advances in Understanding the Inflammatory Response in Malaria: A Review of the Dual Role of Cytokines. J Immunol Res 2021:7785180.
- Lv T, Cao W, Li T. 2021. HIV-Related Immune Activation and Inflammation: Current Understanding and Strategies. J Immunol Res 2021:7316456.
- Dobbs KR, Crabtree JN, Dent AE. 2020. Innate immunity to malaria-The role of monocytes. Immunol Rev 293:8-24.
- Kwenti TE. 2018. Malaria and HIV coinfection in sub-Saharan Africa: prevalence, impact, and treatment strategies. Res Rep Trop Med 9:123-136.
- Nathan C. 2006. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol 6:173-82.
- Borregaard N, Cowland JB. 1997. Granules of the human neutrophilic polymorphonuclear leukocyte. Blood 89:3503-21.
- Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A. 2004. Neutrophil extracellular traps kill bacteria. Science 303:1532-5.
- Häger M, Cowland JB, Borregaard N. 2010. Neutrophil granules in health and disease. J Intern Med 268:25-34.
- Schönrich G, Raftery MJ. 2016. Neutrophil Extracellular Traps Go Viral. Front Immunol 7:366.
- Burn GL, Foti A, Marsman G, Patel DF, Zychlinsky A. 2021. The Neutrophil. Immunity 54:1377-1391.
- Saitoh T, Komano J, Saitoh Y, Misawa T, Takahama M, Kozaki T, Uehata T, Iwasaki H, Omori H, Yamaoka S, Yamamoto N, Akira S. 2012. Neutrophil extracellular traps mediate a host defense response to human immunodeficiency virus-1. Cell Host Microbe 12:109-16.
- Bowers NL, Helton ES, Huijbregts RP, Goepfert PA, Heath SL, Hel Z. 2014. Immune suppression by neutrophils in HIV-1 infection: role of PD-L1/PD-1 pathway. PLoS Pathog 10:e1003993.
- Elbim C, Prevot MH, Bouscarat F, Franzini E, Chollet-Martin S, Hakim J, Gougerot-Pocidalo MA. 1994. Polymorphonuclear neutrophils from human immunodeficiency virus-infected patients show enhanced activation, diminished fMLP-induced L-selectin shedding, and an impaired oxidative burst after cytokine priming. Blood 84:2759-66.
- Ramsuran V, Kulkarni H, He W, Mlisana K, Wright EJ, Werner L, Castiblanco J, Dhanda R, Le T, Dolan MJ, Guan W, Weiss RA, Clark RA, Karim SS, Ahuja SK, Ndung'u T. 2011. Duffy-null-associated low neutrophil counts influence HIV-1 susceptibility in high-risk South African black women. Clin Infect Dis 52:1248-56.
- Hadad N, Levy R, Schlaeffer F, Riesenberg K. 2007. Direct effect of human immunodeficiency virus protease inhibitors on neutrophil function and apoptosis via calpain inhibition. Clin Vaccine Immunol 14:1515-21.
- Roilides E, Venzon D, Pizzo PA, Rubin M. 1990. Effects of antiretroviral dideoxynucleosides on polymorphonuclear leukocyte function. Antimicrob Agents Chemother 34:1672-7.
- Estes JD, Harris LD, Klatt NR, Tabb B, Pittaluga S, Paiardini M, Barclay GR, Smedley J, Pung R, Oliveira KM, Hirsch VM, Silvestri G, Douek DC, Miller CJ, Haase AT, Lifson J, Brenchley JM. 2010. Damaged intestinal epithelial integrity linked to microbial translocation in pathogenic simian immunodeficiency virus infections. PLoS Pathog 6:e1001052.
- Hensley-McBain T, Wu MC, Manuzak JA, Cheu RK, Gustin A, Driscoll CB, Zevin AS, Miller CJ, Coronado E, Smith E, Chang J, Gale M, Jr., Somsouk M, Burgener AD, Hunt PW, Hope TJ, Collier AC, Klatt NR. 2019. Increased mucosal neutrophil survival is associated with altered microbiota in HIV infection. PLoS Pathog 15:e1007672.
- Somsouk M, Estes JD, Deleage C, Dunham RM, Albright R, Inadomi JM, Martin JN, Deeks SG, McCune JM, Hunt PW. 2015. Gut epithelial barrier and systemic inflammation during chronic HIV infection. Aids 29:43-51.
- Hunt PW, Sinclair E, Rodriguez B, Shive C, Clagett B, Funderburg N, Robinson J, Huang Y, Epling L, Martin JN, Deeks SG, Meinert CL, Van Natta ML, Jabs DA, Lederman MM. 2014. Gut epithelial barrier dysfunction and innate immune activation predict mortality in treated HIV infection. J Infect Dis 210:1228-38.
- Klatt NR, Funderburg NT, Brenchley JM. 2013. Microbial translocation, immune activation, and HIV disease. Trends Microbiol 21:6-13.
- Sandler NG, Wand H, Roque A, Law M, Nason MC, Nixon DE, Pedersen C, Ruxrungtham K, Lewin SR, Emery S, Neaton JD, Brenchley JM, Deeks SG, Sereti I, Douek DC. 2011. Plasma levels of soluble CD14 independently predict mortality in HIV infection. J Infect Dis 203:780-90.
- Feng G, Wines BD, Kurtovic L, Chan JA, Boeuf P, Mollard V, Cozijnsen A, Drew DR, Center RJ, Marshall DL, Chishimba S, McFadden GI, Dent AE, Chelimo K, Boyle MJ, Kazura JW, Hogarth PM, Beeson JG. 2021. Mechanisms and targets of Fcγ-receptor mediated immunity to malaria sporozoites. Nat Commun 12:1742.
- Kumaratilake LM, Ferrante A. 2000. Opsonization and phagocytosis of Plasmodium falciparum merozoites measured by flow cytometry. Clin Diagn Lab Immunol 7:9-13.
- Sun T, Chakrabarti C. 1985. Schizonts, merozoites, and phagocytosis in falciparum malaria. Ann Clin Lab Sci 15:465-9.
- Tannous S, Ghanem E. 2018. A bite to fight: front-line innate immune defenses against malaria parasites. Pathog Glob Health 112:1-12.
- Baker VS, Imade GE, Molta NB, Tawde P, Pam SD, Obadofin MO, Sagay SA, Egah DZ, Iya D, Afolabi BB, Baker M, Ford K, Ford R, Roux KH, Keller TC, 3rd. 2008. Cytokine-associated neutrophil extracellular traps and antinuclear antibodies in Plasmodium falciparum infected children under six years of age. Malar J 7:41.
- Knackstedt SL, Georgiadou A, Apel F, Abu-Abed U, Moxon CA, Cunnington AJ, Raupach B, Cunningham D, Langhorne J, Krüger R, Barrera V, Harding SP, Berg A, Patel S, Otterdal K, Mordmüller B, Schwarzer E, Brinkmann V, Zychlinsky A, Amulic B. 2019. Neutrophil extracellular traps drive inflammatory pathogenesis in malaria. Sci Immunol 4.
- Rodrigues DAS, Prestes EB, Gama AMS, Silva LS, Pinheiro AAS, Ribeiro JMC, Campos RMP, Pimentel-Coelho PM, De Souza HS, Dicko A, Duffy PE, Fried M, Francischetti IMB, Saraiva EM, Paula-Neto HA, Bozza MT. 2020. CXCR4 and MIF are required for neutrophil extracellular trap release triggered by Plasmodium-infected erythrocytes. PLoS Pathog 16:e1008230.
- Kho S, Minigo G, Andries B, Leonardo L, Prayoga P, Poespoprodjo JR, Kenangalem E, Price RN, Woodberry T, Anstey NM, Yeo TW. 2019. Circulating Neutrophil Extracellular Traps and Neutrophil Activation Are Increased in Proportion to Disease Severity in Human Malaria. J Infect Dis 219:1994-2004.
- Lee HJ, Georgiadou A, Walther M, Nwakanma D, Stewart LB, Levin M, Otto TD, Conway DJ, Coin LJ, Cunnington AJ. 2018. Integrated pathogen load and dual transcriptome analysis of systemic host-pathogen interactions in severe malaria. Sci Transl Med 10.
- Gardner MB CM, Luciw PA.. 2004. AIDS and other manifestations of HIV infection, 4th ed ed. Raven Press, New York.
- Coatney GR CW, Contacos PG. 1971. The primate malarias. US National Institute of Allergy and Infectious Diseases, Washington DC.
- Trott KA, Chau JY, Hudgens MG, Fine J, Mfalila CK, Tarara RP, Collins WE, Sullivan J, Luckhart S, Abel K. 2011. Evidence for an increased risk of transmission of simian immunodeficiency virus and malaria in a rhesus macaque coinfection model. J Virol 85:11655-63.
- Trott KA, Richardson A, Hudgens MA, Abel K. 2013. Immune activation and regulation in simian immunodeficiency virus-Plasmodium fragile-coinfected rhesus macaques. J Virol 87:9523-37.
- Loffredo JT, Maxwell J, Qi Y, Glidden CE, Borchardt GJ, Soma T, Bean AT, Beal DR, Wilson NA, Rehrauer WM, Lifson JD, Carrington M, Watkins DI. 2007. Mamu-B*08-positive macaques control simian immunodeficiency virus replication. J Virol 81:8827-32.
- Mothé BR, Weinfurter J, Wang C, Rehrauer W, Wilson N, Allen TM, Allison DB, Watkins DI. 2003. Expression of the major histocompatibility complex class I molecule Mamu-A*01 is associated with control of simian immunodeficiency virus SIVmac239 replication. J Virol 77:2736-40.
- Yant LJ, Friedrich TC, Johnson RC, May GE, Maness NJ, Enz AM, Lifson JD, O'Connor DH, Carrington M, Watkins DI. 2006. The high-frequency major histocompatibility complex class I allele Mamu-B*17 is associated with control of simian immunodeficiency virus SIVmac239 replication. J Virol 80:5074-7.
- Del Prete GQ, Scarlotta M, Newman L, Reid C, Parodi LM, Roser JD, Oswald K, Marx PA, Miller CJ, Desrosiers RC, Barouch DH, Pal R, Piatak M, Jr., Chertova E, Giavedoni LD, O'Connor DH, Lifson JD, Keele BF. 2013. Comparative characterization of transfection- and infection-derived simian immunodeficiency virus challenge stocks for in vivo nonhuman primate studies. J Virol 87:4584-95.
- Monjure CJ, Tatum CD, Panganiban AT, Arainga M, Traina-Dorge V, Marx PA, Jr., Didier ES. 2014. Optimization of PCR for quantification of simian immunodeficiency virus genomic RNA in plasma of rhesus macaques (Macaca mulatta) using armored RNA. J Med Primatol 43:31-43.
- Del Prete GQ, Smedley J, Macallister R, Jones GS, Li B, Hattersley J, Zheng J, Piatak M, Jr., Keele BF, Hesselgesser J, Geleziunas R, Lifson JD. 2016. Short Communication: Comparative Evaluation of Coformulated Injectable Combination Antiretroviral Therapy Regimens in Simian Immunodeficiency Virus-Infected Rhesus Macaques. AIDS Res Hum Retroviruses 32:163-8.
- Collins WE, Warren M, Sullivan JS, Galland GG, Strobert E, Nace D, Williams A, Williams T, Barnwell JW. 2006. Studies on sporozoite-induced and chronic infections with Plasmodium fragile in Macaca mulatta and New World monkeys. J Parasitol 92:1019-26.
- Dissanaike A, Nelson P, Garnham P. 1965. Two new malaria parasites plasmodium cynomolgi ceylonensis subsp. nov. and plasmodium fragile sp. nov. from monkeys in Ceylon.
- Moll K KA, Scherf A, Wahlgren M. 2013. Methods in Malaria Researcb, 6th ed ed. EviMalaR, Glasgow, UK & Manassas VA, USA.
- Karsten CB, Mehta N, Shin SA, Diefenbach TJ, Slein MD, Karpinski W, Irvine EB, Broge T, Suscovich TJ, Alter G. 2019. A versatile high-throughput assay to characterize antibody-mediated neutrophil phagocytosis. J Immunol Methods 471:46-56.
- Butcher SK, Chahal H, Nayak L, Sinclair A, Henriquez NV, Sapey E, O'Mahony D, Lord JM. 2001. Senescence in innate immune responses: reduced neutrophil phagocytic capacity and CD16 expression in elderly humans. J Leukoc Biol 70:881-6.
- Christensen R. 2013. Plane Answers to Complex Questions, 4 ed. Springer New York, NY, New York, New York.
- Trampuz A, Jereb M, Muzlovic I, Prabhu RM. 2003. Clinical review: Severe malaria. Crit Care 7:315-23.
- White NJ. 2018. Anaemia and malaria. Malar J 17:371.
- Schacker TW, Hughes JP, Shea T, Coombs RW, Corey L. 1998. Biological and virologic characteristics of primary HIV infection. Ann Intern Med 128:613-20.
- Brenchley JM, Paiardini M. 2011. Immunodeficiency lentiviral infections in natural and non-natural hosts. Blood 118:847-54.
- Estes JD, Wong SW, Brenchley JM. 2018. Nonhuman primate models of human viral infections. Nat Rev Immunol 18:390-404.
- Silvestri G. 2008. AIDS pathogenesis: a tale of two monkeys. J Med Primatol 37 Suppl 2:6-12.
- Okoye AA, Picker LJ. 2013. CD4(+) T-cell depletion in HIV infection: mechanisms of immunological failure. Immunol Rev 254:54-64.
- Lau B, Sharrett AR, Kingsley LA, Post W, Palella FJ, Visscher B, Gange SJ. 2006. C-reactive protein is a marker for human immunodeficiency virus disease progression. Arch Intern Med 166:64-70.
- Mabhida SE, McHiza ZJ, Mokgalaboni K, Hanser S, Choshi J, Mokoena H, Ziqubu K, Masilela C, Nkambule BB, Ndwandwe DE, Kengne AP, Dludla PV. 2024. High-sensitivity C-reactive protein among people living with HIV on highly active antiretroviral therapy: a systemic review and meta-analysis. BMC Infect Dis 24:160.
- Redd AD, Eaton KP, Kong X, Laeyendecker O, Lutalo T, Wawer MJ, Gray RH, Serwadda D, Quinn TC. 2010. C-reactive protein levels increase during HIV-1 disease progression in Rakai, Uganda, despite the absence of microbial translocation. J Acquir Immune Defic Syndr 54:556-9.
- Shivakoti R, Yang WT, Berendes S, Mwelase N, Kanyama C, Pillay S, Samaneka W, Santos B, Poongulali S, Tripathy S, Riviere C, Lama JR, Cardoso SW, Sugandhavesa P, Balagopal A, Gupte N, Semba RD, Campbell TB, Bollinger RC, Gupta A. 2016. Persistently Elevated C-Reactive Protein Level in the First Year of Antiretroviral Therapy, Despite Virologic Suppression, Is Associated With HIV Disease Progression in Resource-Constrained Settings. J Infect Dis 213:1074-8.
- Hashmi F, Aqeel S, Zuberi UF, Khan W. 2023. A systematic review and meta-analysis of inflammatory biomarkers associated with malaria infection and disease severity. Cytokine 169:156305.
- Wilairatana P, Mahannop P, Tussato T, Hayeedoloh IM, Boonhok R, Klangbud WK, Mala W, Kotepui KU, Kotepui M. 2021. C-reactive protein as an early biomarker for malaria infection and monitoring of malaria severity: a meta-analysis. Sci Rep 11:22033.
- Breen EC. 2002. Pro- and anti-inflammatory cytokines in human immunodeficiency virus infection and acquired immunodeficiency syndrome. Pharmacol Ther 95:295-304.
- Hensley-McBain T, Klatt NR. 2018. The Dual Role of Neutrophils in HIV Infection. Curr HIV/AIDS Rep 15:1-10.
- Jones R, Manickam C, Ram DR, Kroll K, Hueber B, Woolley G, Shah SV, Smith S, Varner V, Reeves RK. 2021. Systemic and mucosal mobilization of granulocyte subsets during lentiviral infection. Immunology 164:348-357.
- Lin A, Liang F, Thompson EA, Vono M, Ols S, Lindgren G, Hassett K, Salter H, Ciaramella G, Loré K. 2018. Rhesus Macaque Myeloid-Derived Suppressor Cells Demonstrate T Cell Inhibitory Functions and Are Transiently Increased after Vaccination. J Immunol 200:286-294.
- Rogers KA, Scinicariello F, Attanasio R. 2006. IgG Fc receptor III homologues in nonhuman primate species: genetic characterization and ligand interactions. J Immunol 177:3848-56.
- Musich T, Rahman MA, Mohanram V, Miller-Novak L, Demberg T, Venzon DJ, Felber BK, Franchini G, Pavlakis GN, Robert-Guroff M. 2018. Neutrophil Vaccination Dynamics and Their Capacity To Mediate B Cell Help in Rhesus Macaques. J Immunol 201:2287-2302.
- Vono M, Lin A, Norrby-Teglund A, Koup RA, Liang F, Loré K. 2017. Neutrophils acquire the capacity for antigen presentation to memory CD4(+) T cells in vitro and ex vivo. Blood 129:1991-2001.
- Hensley-McBain T, Berard AR, Manuzak JA, Miller CJ, Zevin AS, Polacino P, Gile J, Agricola B, Cameron M, Hu SL, Estes JD, Reeves RK, Smedley J, Keele BF, Burgener AD, Klatt NR. 2018. Intestinal damage precedes mucosal immune dysfunction in SIV infection. Mucosal Immunol 11:1429-1440.
- Korkmaz B, Horwitz MS, Jenne DE, Gauthier F. 2010. Neutrophil elastase, proteinase 3, and cathepsin G as therapeutic targets in human diseases. Pharmacol Rev 62:726-59.
- Segal AW. 2005. How neutrophils kill microbes. Annu Rev Immunol 23:197-223.
- Klebanoff SJ, Coombs RW. 1992. Viricidal effect of polymorphonuclear leukocytes on human immunodeficiency virus-1. Role of the myeloperoxidase system. J Clin Invest 89:2014-7.
- Remijsen Q, Kuijpers TW, Wirawan E, Lippens S, Vandenabeele P, Vanden Berghe T. 2011. Dying for a cause: NETosis, mechanisms behind an antimicrobial cell death modality. Cell Death Differ 18:581-8.
- Wilairatana P, Meddings JB, Ho M, Vannaphan S, Looareesuwan S. 1997. Increased gastrointestinal permeability in patients with Plasmodium falciparum malaria. Clin Infect Dis 24:430-5.
- Tripathi A, Lammers KM, Goldblum S, Shea-Donohue T, Netzel-Arnett S, Buzza MS, Antalis TM, Vogel SN, Zhao A, Yang S, Arrietta MC, Meddings JB, Fasano A. 2009. Identification of human zonulin, a physiological modulator of tight junctions, as prehaptoglobin-2. Proc Natl Acad Sci U S A 106:16799-804.
- Kanda T, Fujii H, Tani T, Murakami H, Suda T, Sakai Y, Ono T, Hatakeyama K. 1996. Intestinal fatty acid-binding protein is a useful diagnostic marker for mesenteric infarction in humans. Gastroenterology 110:339-43.
- Pelsers MM, Hermens WT, Glatz JF. 2005. Fatty acid-binding proteins as plasma markers of tissue injury. Clin Chim Acta 352:15-35.
- Brenchley JM, Douek DC. 2012. Microbial translocation across the GI tract. Annu Rev Immunol 30:149-73.
- Ariyoshi K, Schim van der Loeff M, Berry N, Jaffar S, Whittle H. 1999. Plasma HIV viral load in relation to season and to Plasmodium falciparum parasitaemia. Aids 13:1145-6.
- Pisell TL, Hoffman IF, Jere CS, Ballard SB, Molyneux ME, Butera ST, Lawn SD. 2002. Immune activation and induction of HIV-1 replication within CD14 macrophages during acute Plasmodium falciparum malaria coinfection. Aids 16:1503-9.
- Xiao L, Owen SM, Rudolph DL, Lal RB, Lal AA. 1998. Plasmodium falciparum antigen-induced human immunodeficiency virus type 1 replication is mediated through induction of tumor necrosis factor-alpha. J Infect Dis 177:437-45.
- Sharma G, Kaur G, Mehra N. 2011. Genetic correlates influencing immunopathogenesis of HIV infection. Indian J Med Res 134:749-68.
- Albrecht C, Malzahn D, Brameier M, Hermes M, Ansari AA, Walter L. 2014. Progression to AIDS in SIV-Infected Rhesus Macaques is Associated with Distinct KIR and MHC class I Polymorphisms and NK Cell Dysfunction. Front Immunol 5:600.
- Evans DT, Jing P, Allen TM, O'Connor DH, Horton H, Venham JE, Piekarczyk M, Dzuris J, Dykhuzen M, Mitchen J, Rudersdorf RA, Pauza CD, Sette A, Bontrop RE, DeMars R, Watkins DI. 2000. Definition of five new simian immunodeficiency virus cytotoxic T-lymphocyte epitopes and their restricting major histocompatibility complex class I molecules: evidence for an influence on disease progression. J Virol 74:7400-10.
- Liverpool Uo. HIV Drug Interactions, on University of Liverpool. https://www.hiv-druginteractions.org/checker. Accessed November 6th, 2023.
- Aitken EH, Alemu A, Rogerson SJ. 2018. Neutrophils and Malaria. Front Immunol 9:3005.
- Babatunde KA, Adenuga OF. 2022. Neutrophils in malaria: A double-edged sword role. Front Immunol 13:922377.
- Gabali AM, Anzinger JJ, Spear GT, Thomas LL. 2004. Activation by inflammatory stimuli increases neutrophil binding of human immunodeficiency virus type 1 and subsequent infection of lymphocytes. J Virol 78:10833-6.
- Massanella M, Fromentin R, Chomont N. 2016. Residual inflammation and viral reservoirs: alliance against an HIV cure. Curr Opin HIV AIDS 11:234-41.
- Mazzuti L, Turriziani O, Mezzaroma I. 2023. The Many Faces of Immune Activation in HIV-1 Infection: A Multifactorial Interconnection. Biomedicines 11.
- Yoshimura T, Takahashi M. 2007. IFN-gamma-mediated survival enables human neutrophils to produce MCP-1/CCL2 in response to activation by TLR ligands. J Immunol 179:1942-9.
- Hofbauer TM, Ondracek AS, Mangold A, Scherz T, Nechvile J, Seidl V, Brostjan C, Lang IM. 2020. Neutrophil Extracellular Traps Induce MCP-1 at the Culprit Site in ST-Segment Elevation Myocardial Infarction. Front Cell Dev Biol 8:564169.
- Cahilog Z, Zhao H, Wu L, Alam A, Eguchi S, Weng H, Ma D. 2020. The Role of Neutrophil NETosis in Organ Injury: Novel Inflammatory Cell Death Mechanisms. Inflammation 43:2021-2032.
- Branzk N, Lubojemska A, Hardison SE, Wang Q, Gutierrez MG, Brown GD, Papayannopoulos V. 2014. Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat Immunol 15:1017-25.
- Ataíde R, Mwapasa V, Molyneux ME, Meshnick SR, Rogerson SJ. 2011. Antibodies that induce phagocytosis of malaria infected erythrocytes: effect of HIV infection and correlation with clinical outcomes. PLoS One 6:e22491.
- Serghides L, Finney CA, Ayi K, Loutfy M, Kain KC. 2015. Chronic HIV infection impairs nonopsonic phagocytosis of malaria parasites. J Acquir Immune Defic Syndr 68:128-32.
- Donnelly E, de Water JV, Luckhart S. 2021. Malaria-induced bacteremia as a consequence of multiple parasite survival strategies. Curr Res Microb Sci 2:100036.
- Olsson RA, Johnston EH. 1969. Histopathologic changes and small-bowel absorption in falciparum malaria. Am J Trop Med Hyg 18:355-9.
- Boone BA, Murthy P, Miller-Ocuin J, Doerfler WR, Ellis JT, Liang X, Ross MA, Wallace CT, Sperry JL, Lotze MT, Neal MD, Zeh HJ, 3rd. 2018. Chloroquine reduces hypercoagulability in pancreatic cancer through inhibition of neutrophil extracellular traps. BMC Cancer 18:678.
- Boone BA, Orlichenko L, Schapiro NE, Loughran P, Gianfrate GC, Ellis JT, Singhi AD, Kang R, Tang D, Lotze MT, Zeh HJ. 2015. The receptor for advanced glycation end products (RAGE) enhances autophagy and neutrophil extracellular traps in pancreatic cancer. Cancer Gene Ther 22:326-34.
- Labro MT, Babin-Chevaye C. 1988. Effects of amodiaquine, chloroquine, and mefloquine on human polymorphonuclear neutrophil function in vitro. Antimicrob Agents Chemother 32:1124-30.
- Murthy P, Singhi AD, Ross MA, Loughran P, Paragomi P, Papachristou GI, Whitcomb DC, Zureikat AH, Lotze MT, Zeh Iii HJ, Boone BA. 2019. Enhanced Neutrophil Extracellular Trap Formation in Acute Pancreatitis Contributes to Disease Severity and Is Reduced by Chloroquine. Front Immunol 10:28.
- Liles NW, Page EE, Liles AL, Vesely SK, Raskob GE, George JN. 2016. Diversity and severity of adverse reactions to quinine: A systematic review. Am J Hematol 91:461-6.
- van Wolfswinkel ME, Langenberg MCC, Wammes LJ, Sauerwein RW, Koelewijn R, Hermsen CC, van Hellemond JJ, van Genderen PJ. 2017. Changes in total and differential leukocyte counts during the clinically silent liver phase in a controlled human malaria infection in malaria-naïve Dutch volunteers. Malar J 16:457.
- Broyles LN, Luo R, Boeras D, Vojnov L. 2023. The risk of sexual transmission of HIV in individuals with low-level HIV viraemia: a systematic review. Lancet 402:464-471.
Figure 1.
Experimental Timeline. Experimental timeline depicting sample collection from adult male rhesus macaques (RM, n=4). RMs were inoculated with SIVmac239, TCID50=50, intravenously (i.v.) at week 0. Daily antiretroviral treatment (ART) was given subcutaneously, began at week 8, and continued until the end of the study (TDF/FTC/DTG; 5.1/30/2.5 mg/kg). RMs were inoculated with Plasmodium fragile, 20x106 infected erythrocytes, via i.v.. Anti-malaria treatment occurred over one week, at week 14, and consisted of one oral gavage of quinine sulfate (150mg) followed by four daily oral gavages of chloroquine (20mg/kg). .
Figure 1.
Experimental Timeline. Experimental timeline depicting sample collection from adult male rhesus macaques (RM, n=4). RMs were inoculated with SIVmac239, TCID50=50, intravenously (i.v.) at week 0. Daily antiretroviral treatment (ART) was given subcutaneously, began at week 8, and continued until the end of the study (TDF/FTC/DTG; 5.1/30/2.5 mg/kg). RMs were inoculated with Plasmodium fragile, 20x106 infected erythrocytes, via i.v.. Anti-malaria treatment occurred over one week, at week 14, and consisted of one oral gavage of quinine sulfate (150mg) followed by four daily oral gavages of chloroquine (20mg/kg). .
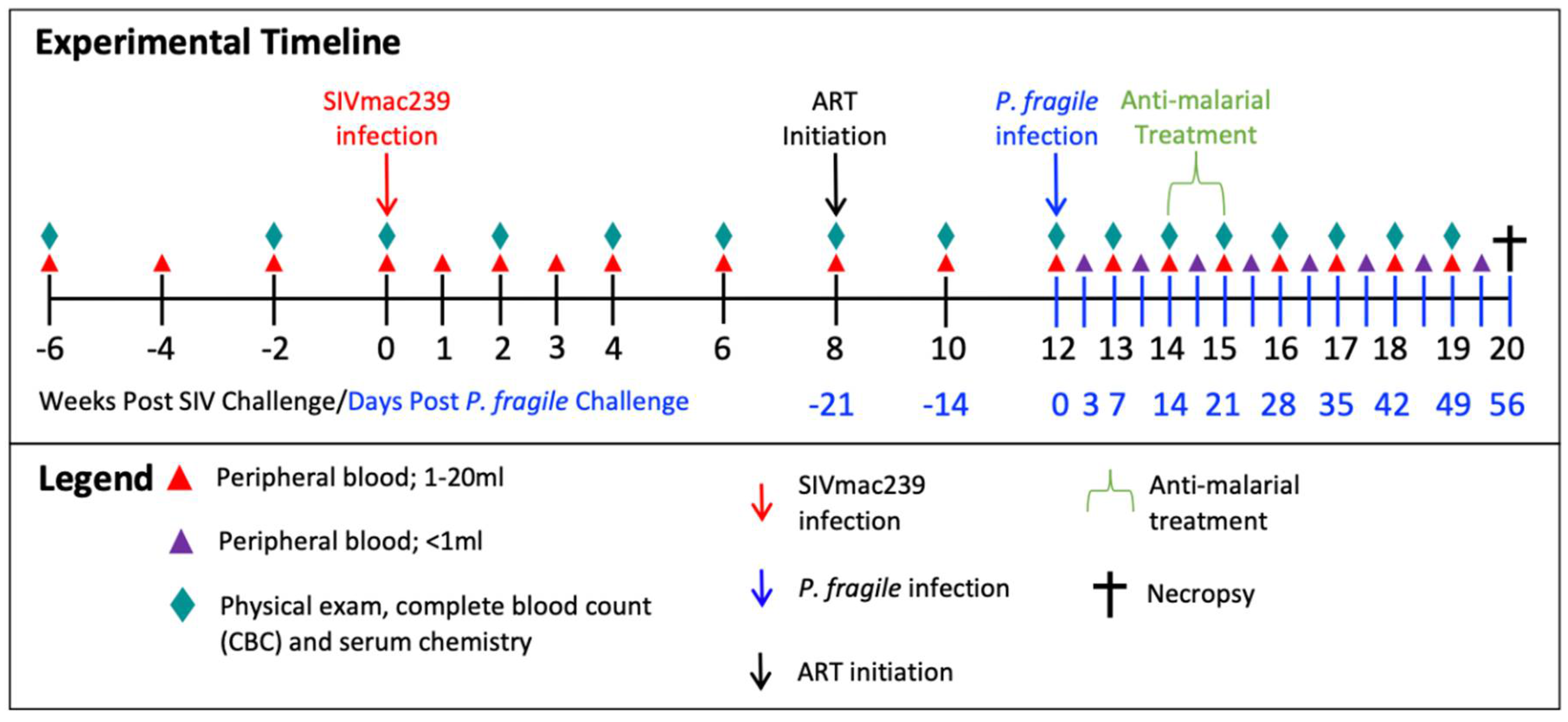
Figure 2.
P. fragile co-infection results in clinical signs of SIV infection despite persistent, daily ART. Peripheral P. fragile parasitemia, anemia, SIVmac239 peripheral viral load (VL), and blood CD4+ T cell count were assessed in adult male rhesus macaques (RMs; n=4). A) Following P. fragile inoculation at week 12 post-SIV infection (p.i.), parasitemia was assessed tri-weekly, indicated as week p.i. A, B, and C. % Parasitemia was assessed via Giemsa staining of thin blood smears and was defined as the percentage of erythrocytes infected by a parasite among all erythrocytes. B) Anemia was assessed by characterizing % hematocrit, defined as the ratio of red blood cells to total blood. C) Plasma VL (RNA copies/ml plasma) was assessed by qPCR. D) Absolute number of CD4+ T cells per µL of blood was assessed via flow cytometry. In all panels, each RM is represented by a different symbol and color. Baseline (BL) is an average of data collected at weeks -6, -4, -2, and 0 p.i.. Inoculation with SIVmac239 at week 0 p.i. is indicated by a purple dashed arrow. Inoculation with P. fragile at week 12 p.i. is indicated by a blue dashed arrow. Antiretroviral therapy (ART) was initiated at week 8 p.i., indicated by the dark grey bar. Anti-malarial treatment occurred throughout week 14 p.i., indicated by the light grey bar. Statistical significance between all time points was calculated using a mixed-effects analysis with the Geisser-Greenhouse correction and a Tukey’s multiple comparisons test, with individual variances computed for each comparison. Multiplicity adjusted significant P values are shown above horizontal black bars.
Figure 2.
P. fragile co-infection results in clinical signs of SIV infection despite persistent, daily ART. Peripheral P. fragile parasitemia, anemia, SIVmac239 peripheral viral load (VL), and blood CD4+ T cell count were assessed in adult male rhesus macaques (RMs; n=4). A) Following P. fragile inoculation at week 12 post-SIV infection (p.i.), parasitemia was assessed tri-weekly, indicated as week p.i. A, B, and C. % Parasitemia was assessed via Giemsa staining of thin blood smears and was defined as the percentage of erythrocytes infected by a parasite among all erythrocytes. B) Anemia was assessed by characterizing % hematocrit, defined as the ratio of red blood cells to total blood. C) Plasma VL (RNA copies/ml plasma) was assessed by qPCR. D) Absolute number of CD4+ T cells per µL of blood was assessed via flow cytometry. In all panels, each RM is represented by a different symbol and color. Baseline (BL) is an average of data collected at weeks -6, -4, -2, and 0 p.i.. Inoculation with SIVmac239 at week 0 p.i. is indicated by a purple dashed arrow. Inoculation with P. fragile at week 12 p.i. is indicated by a blue dashed arrow. Antiretroviral therapy (ART) was initiated at week 8 p.i., indicated by the dark grey bar. Anti-malarial treatment occurred throughout week 14 p.i., indicated by the light grey bar. Statistical significance between all time points was calculated using a mixed-effects analysis with the Geisser-Greenhouse correction and a Tukey’s multiple comparisons test, with individual variances computed for each comparison. Multiplicity adjusted significant P values are shown above horizontal black bars.
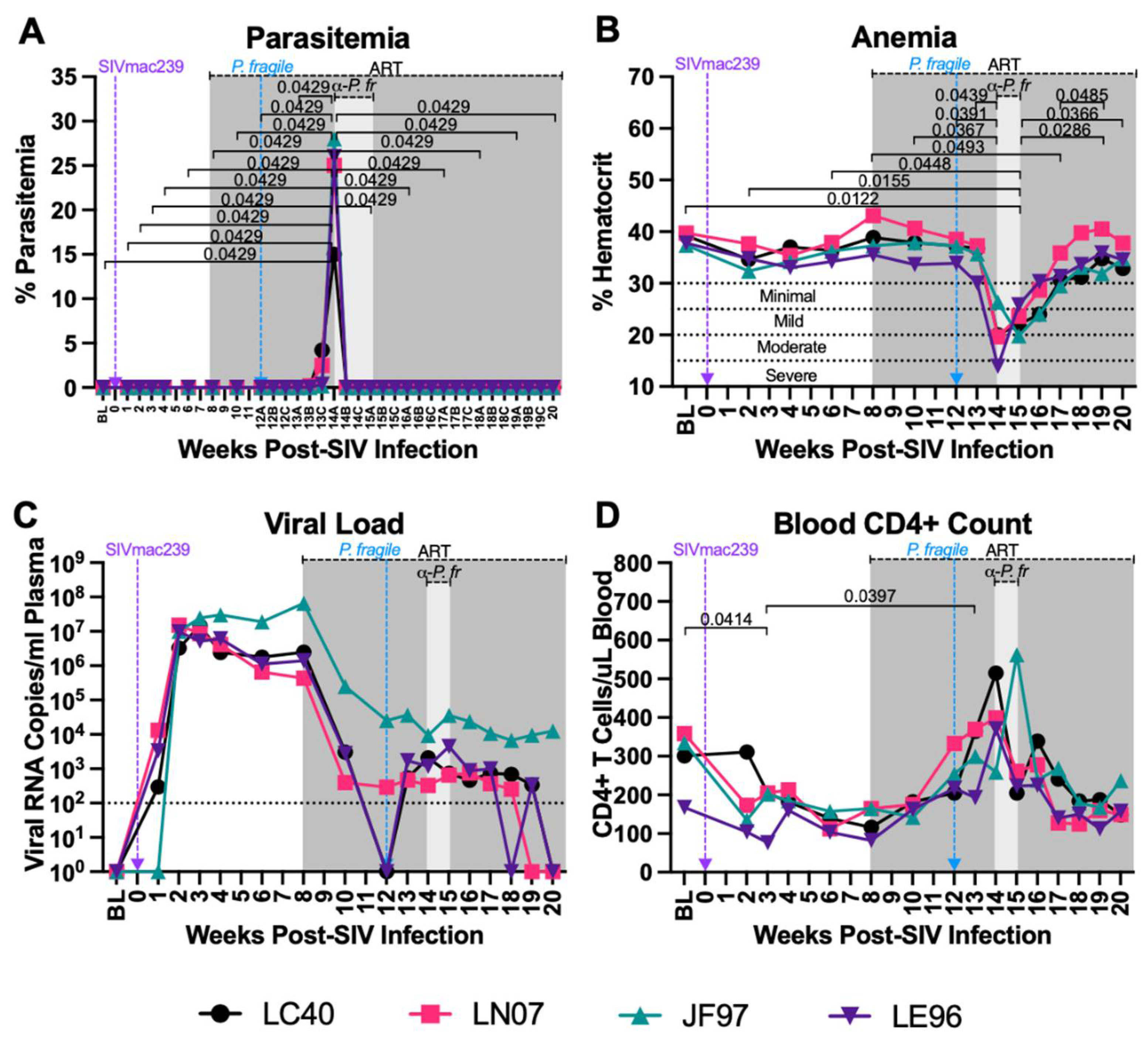
Figure 3.
Variable levels of inflammatory markers throughout P. fragile co-infection of ART-treated SIVmac239-infected rhesus macaques. CRP, cytokine and chemokine levels were measured throughout P. fragile co-infection of ART-treated SIVmac239-infected rhesus macaques (RMs; n=4). A) CRP levels were measured in serum by a Beckman au480. B-D) IL-8 (B), IP-10 (C), and MCP-1 (D) levels were measured in plasma by LegendPlex. In all panels, each RM is represented by a different symbol and color. Baseline (BL) is an average of data collected at weeks -6, -2, and 0 post-SIV infection (p.i.). Inoculation with SIVmac239 at week 0 p.i. is indicated by a purple dashed arrow. Inoculation with P. fragile at week 12 p.i. is indicated by a blue dashed arrow. Antiretroviral therapy (ART) was initiated at week 8 p.i., indicated by the dark grey bar. Anti-malarial treatment occurred throughout week 14 p.i., indicated by the light grey bar. Statistical significance between all time points was calculated using a mixed-effects analysis with the Geisser-Greenhouse correction and a Tukey’s multiple comparisons test, with individual variances computed for each comparison. Multiplicity adjusted significant P values are shown above horizontal black bars.
Figure 3.
Variable levels of inflammatory markers throughout P. fragile co-infection of ART-treated SIVmac239-infected rhesus macaques. CRP, cytokine and chemokine levels were measured throughout P. fragile co-infection of ART-treated SIVmac239-infected rhesus macaques (RMs; n=4). A) CRP levels were measured in serum by a Beckman au480. B-D) IL-8 (B), IP-10 (C), and MCP-1 (D) levels were measured in plasma by LegendPlex. In all panels, each RM is represented by a different symbol and color. Baseline (BL) is an average of data collected at weeks -6, -2, and 0 post-SIV infection (p.i.). Inoculation with SIVmac239 at week 0 p.i. is indicated by a purple dashed arrow. Inoculation with P. fragile at week 12 p.i. is indicated by a blue dashed arrow. Antiretroviral therapy (ART) was initiated at week 8 p.i., indicated by the dark grey bar. Anti-malarial treatment occurred throughout week 14 p.i., indicated by the light grey bar. Statistical significance between all time points was calculated using a mixed-effects analysis with the Geisser-Greenhouse correction and a Tukey’s multiple comparisons test, with individual variances computed for each comparison. Multiplicity adjusted significant P values are shown above horizontal black bars.
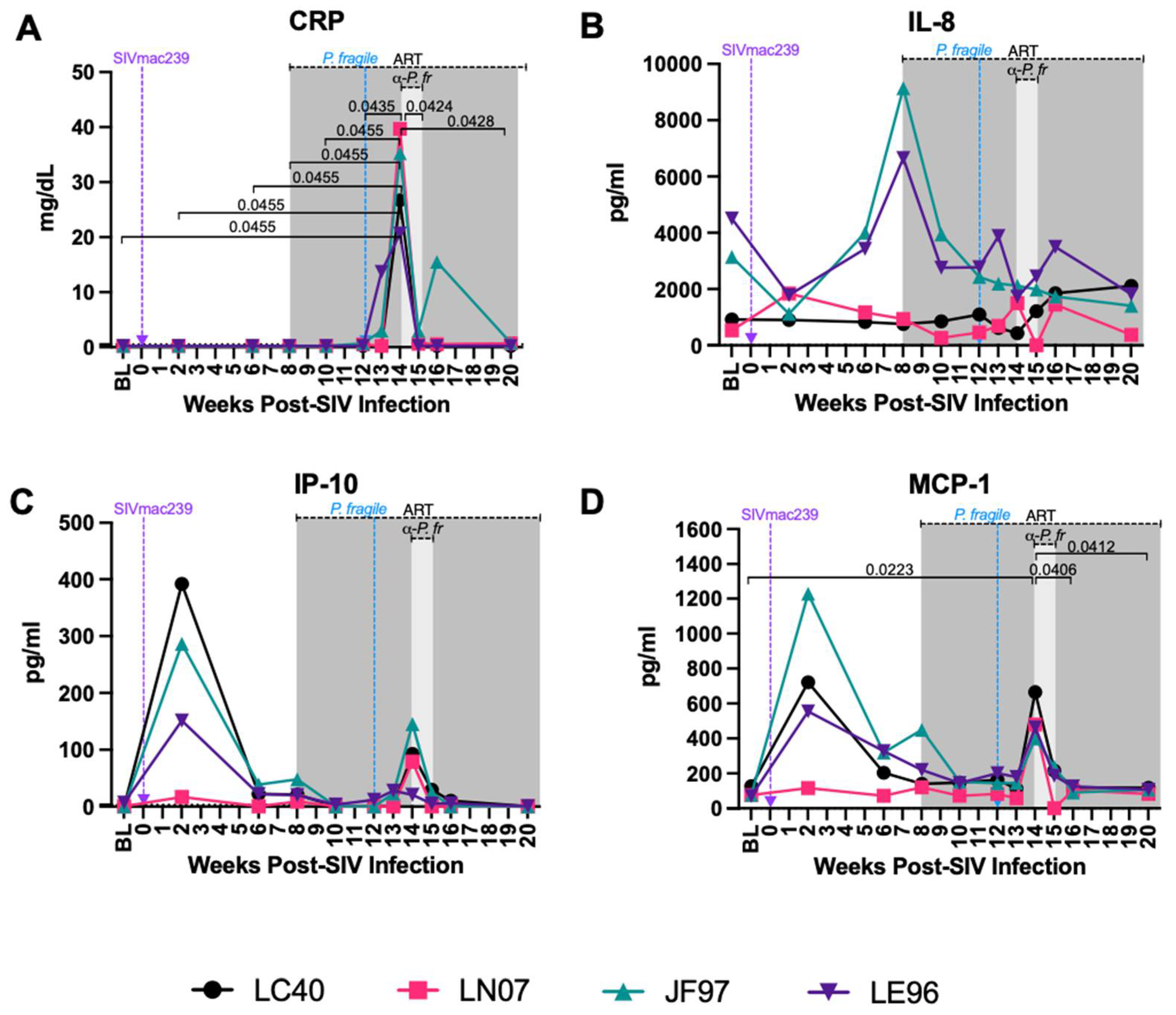
Figure 4.
Minimal disruption of peripheral neutrophil frequencies and percentage of neutrophils undergoing apoptosis during P. fragile co-infection of ART-treated SIVmac239-infected rhesus macaques. Total neutrophil frequencies and frequencies of neutrophils undergoing apoptosis were assessed in whole blood before and after P. fragile co-infection of ART-treated SIVmac239-infected rhesus macaques (RMs; n=4) by flow cytometry. A) Neutrophil (HLA-DR-CD11b+CD66abce+CD14+) frequency of live CD45+ cells were assessed throughout co-infection. B) The frequency of neutrophils undergoing apoptosis (caspase3+) was assessed throughout co-infection. In both panels, each RM is represented by a different symbol and color. Baseline (BL) is an average of data collected at weeks -6, -4, -2, and 0 post-SIV infection (p.i.). Inoculation with SIVmac239 at week 0 p.i. is indicated by a purple dashed arrow. Inoculation with P. fragile at week 12 p.i. is indicated by a blue dashed arrow. Antiretroviral therapy (ART) was initiated at week 8 p.i., indicated by the dark grey bar. Anti-malarial treatment occurred throughout week 14 p.i., indicated by the light grey bar. Statistical significance between all time points was calculated using a mixed-effects analysis with the Geisser-Greenhouse correction and a Tukey’s multiple comparisons test, with individual variances computed for each comparison. Multiplicity adjusted significant P values are shown above horizontal black bars.
Figure 4.
Minimal disruption of peripheral neutrophil frequencies and percentage of neutrophils undergoing apoptosis during P. fragile co-infection of ART-treated SIVmac239-infected rhesus macaques. Total neutrophil frequencies and frequencies of neutrophils undergoing apoptosis were assessed in whole blood before and after P. fragile co-infection of ART-treated SIVmac239-infected rhesus macaques (RMs; n=4) by flow cytometry. A) Neutrophil (HLA-DR-CD11b+CD66abce+CD14+) frequency of live CD45+ cells were assessed throughout co-infection. B) The frequency of neutrophils undergoing apoptosis (caspase3+) was assessed throughout co-infection. In both panels, each RM is represented by a different symbol and color. Baseline (BL) is an average of data collected at weeks -6, -4, -2, and 0 post-SIV infection (p.i.). Inoculation with SIVmac239 at week 0 p.i. is indicated by a purple dashed arrow. Inoculation with P. fragile at week 12 p.i. is indicated by a blue dashed arrow. Antiretroviral therapy (ART) was initiated at week 8 p.i., indicated by the dark grey bar. Anti-malarial treatment occurred throughout week 14 p.i., indicated by the light grey bar. Statistical significance between all time points was calculated using a mixed-effects analysis with the Geisser-Greenhouse correction and a Tukey’s multiple comparisons test, with individual variances computed for each comparison. Multiplicity adjusted significant P values are shown above horizontal black bars.
Figure 5.
Nominal alterations in neutrophil phagocytosis during P. fragile co-infection of ART-treated SIVmac239-infected rhesus macaques. The frequency of phagocytic neutrophils and neutrophil phagocytic capacity was assessed in whole blood throughout P. fragile co-infection of ART-treated SIVmac239-infected rhesus macaques (RMs; n=4) by flow cytometry. Phagocytosis was determined by assessing uptake of pHrodo Red E. coli Bioparticles. A) Neutrophil phagocytic score was calculated by multiplying the absolute number of neutrophils/μL of whole blood by the percentage of neutrophils positive for uptake of pHrodo bioparticles. B) Phagocytic index was calculated by multiplying the phagocytic score in A by the Mean Fluorescence Intensity (MFI) of pHrodo positive neutrophils. In both panels, each RM is represented by a different symbol and color. Baseline (BL) is an average of data collected at weeks -6, -4, -2, and 0 post-SIV infection (p.i.). Inoculation with SIVmac239 at week 0 p.i. is indicated by a purple dashed arrow. Inoculation with P. fragile at week 12 p.i. is indicated by a blue dashed arrow. Antiretroviral therapy (ART) was initiated at week 8 p.i., indicated by the dark grey bar. Anti-malarial treatment occurred throughout week 14 p.i., indicated by the light grey bar. Statistical significance between all time points was calculated using a mixed-effects analysis with the Geisser-Greenhouse correction and a Tukey’s multiple comparisons test, with individual variances computed for each comparison. Multiplicity adjusted significant P values are shown above horizontal black bars.
Figure 5.
Nominal alterations in neutrophil phagocytosis during P. fragile co-infection of ART-treated SIVmac239-infected rhesus macaques. The frequency of phagocytic neutrophils and neutrophil phagocytic capacity was assessed in whole blood throughout P. fragile co-infection of ART-treated SIVmac239-infected rhesus macaques (RMs; n=4) by flow cytometry. Phagocytosis was determined by assessing uptake of pHrodo Red E. coli Bioparticles. A) Neutrophil phagocytic score was calculated by multiplying the absolute number of neutrophils/μL of whole blood by the percentage of neutrophils positive for uptake of pHrodo bioparticles. B) Phagocytic index was calculated by multiplying the phagocytic score in A by the Mean Fluorescence Intensity (MFI) of pHrodo positive neutrophils. In both panels, each RM is represented by a different symbol and color. Baseline (BL) is an average of data collected at weeks -6, -4, -2, and 0 post-SIV infection (p.i.). Inoculation with SIVmac239 at week 0 p.i. is indicated by a purple dashed arrow. Inoculation with P. fragile at week 12 p.i. is indicated by a blue dashed arrow. Antiretroviral therapy (ART) was initiated at week 8 p.i., indicated by the dark grey bar. Anti-malarial treatment occurred throughout week 14 p.i., indicated by the light grey bar. Statistical significance between all time points was calculated using a mixed-effects analysis with the Geisser-Greenhouse correction and a Tukey’s multiple comparisons test, with individual variances computed for each comparison. Multiplicity adjusted significant P values are shown above horizontal black bars.
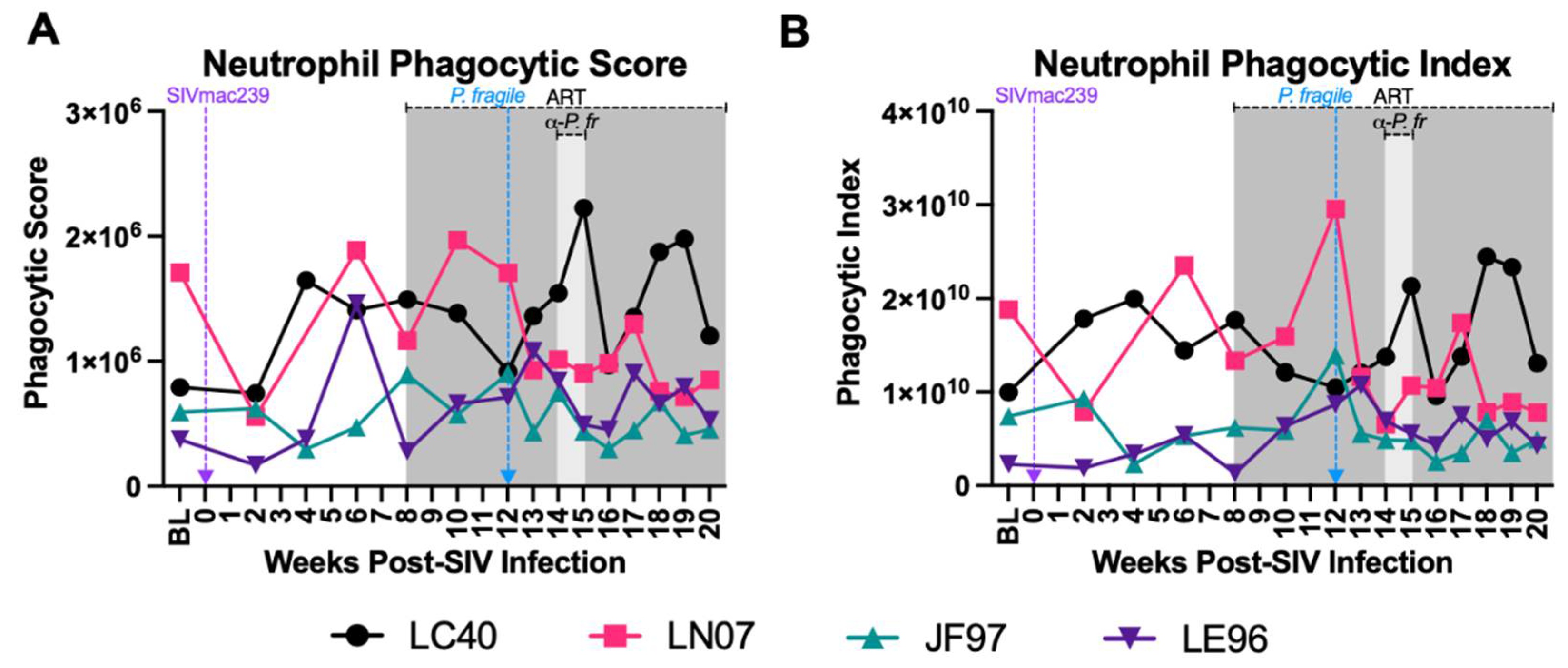
Figure 6.
Decreased plasma levels of neutrophil degranulation markers during P. fragile co-infection of ART-treated SIVmac239-infected rhesus macaques. Products of neutrophil degranulation were measured throughout P. fragile co-infection of ART-treated SIVmac239-infected rhesus macaques (RMs; n=4) via ELISA. A-B) Myeloperoxidase (A), Proteinase 3 (B), and Cathepsin G (C) levels were measured in plasma by ELISA. In all panels, each RM is represented by a different symbol and color. Baseline (BL) is an average of data collected at weeks -6, -2, and 0 post-SIV infection (p.i.). Inoculation with SIVmac239 at week 0 p.i. is indicated by a purple dashed arrow. Inoculation with P. fragile at week 12 p.i. is indicated by a blue dashed arrow. Antiretroviral therapy (ART) was initiated at week 8 p.i., indicated by the dark grey bar. Anti-malarial treatment occurred throughout week 14 p.i., indicated by the light grey bar. Statistical significance between all time points was calculated using a mixed-effects analysis with the Geisser-Greenhouse correction and a Tukey’s multiple comparisons test, with individual variances computed for each comparison. Multiplicity adjusted significant P values are shown above horizontal black bars.
Figure 6.
Decreased plasma levels of neutrophil degranulation markers during P. fragile co-infection of ART-treated SIVmac239-infected rhesus macaques. Products of neutrophil degranulation were measured throughout P. fragile co-infection of ART-treated SIVmac239-infected rhesus macaques (RMs; n=4) via ELISA. A-B) Myeloperoxidase (A), Proteinase 3 (B), and Cathepsin G (C) levels were measured in plasma by ELISA. In all panels, each RM is represented by a different symbol and color. Baseline (BL) is an average of data collected at weeks -6, -2, and 0 post-SIV infection (p.i.). Inoculation with SIVmac239 at week 0 p.i. is indicated by a purple dashed arrow. Inoculation with P. fragile at week 12 p.i. is indicated by a blue dashed arrow. Antiretroviral therapy (ART) was initiated at week 8 p.i., indicated by the dark grey bar. Anti-malarial treatment occurred throughout week 14 p.i., indicated by the light grey bar. Statistical significance between all time points was calculated using a mixed-effects analysis with the Geisser-Greenhouse correction and a Tukey’s multiple comparisons test, with individual variances computed for each comparison. Multiplicity adjusted significant P values are shown above horizontal black bars.
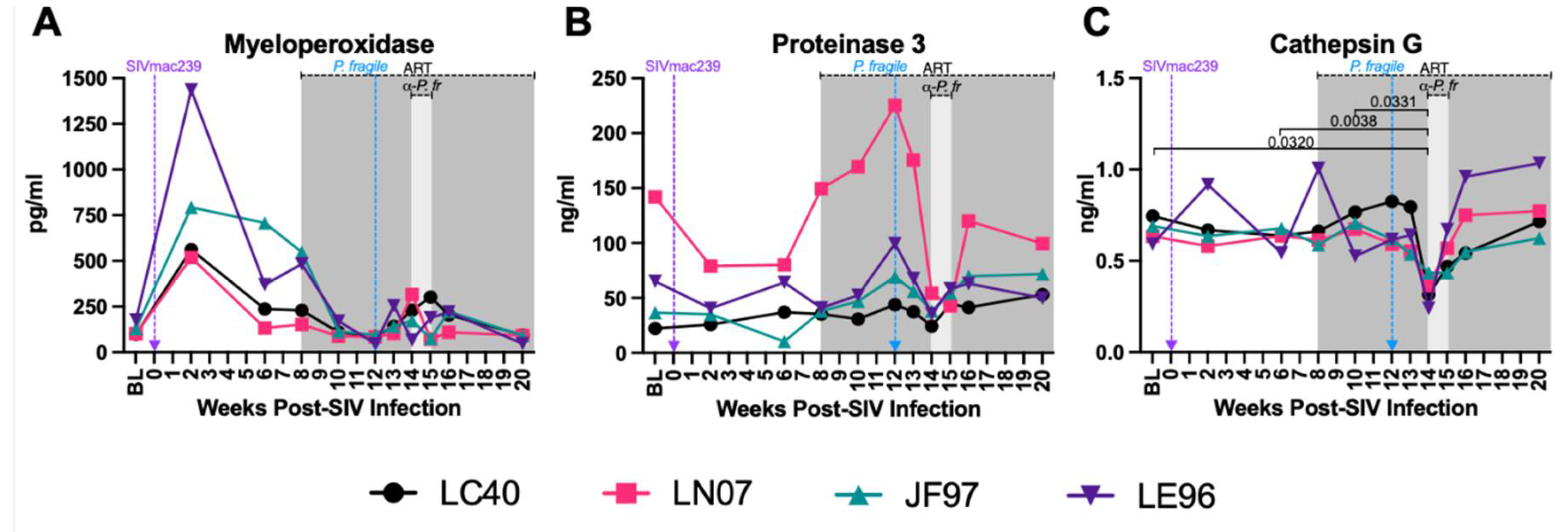
Figure 7.
Increased plasma levels of neutrophil extracellular trap markers during P. fragile co-infection of ART-treated SIVmac239-infected rhesus macaques. Markers of neutrophil extracellular trap formation were measured throughout P. fragile co-infection of ART-treated SIVmac239-infected rhesus macaques (RMs; n=4) via ELISA. A) Neutrophil elastase and B) Citrullinated histone 3 levels were measured in plasma by ELISA. In both panels, each RM is represented by a different symbol and color. Baseline (BL) is an average of data collected at weeks -6, -2, and 0 post-SIV infection (p.i.). Inoculation with SIVmac239 at week 0 p.i. is indicated by a purple dashed arrow. Inoculation with P. fragile at week 12 p.i. is indicated by a blue dashed arrow. Antiretroviral therapy (ART) was initiated at week 8 p.i., indicated by the dark grey bar. Anti-malarial treatment occurred throughout week 14 p.i., indicated by the light grey bar. Statistical significance between all time points was calculated using a mixed-effects analysis with the Geisser-Greenhouse correction and a Tukey’s multiple comparisons test, with individual variances computed for each comparison. Multiplicity adjusted significant P values are shown above horizontal black bars.
Figure 7.
Increased plasma levels of neutrophil extracellular trap markers during P. fragile co-infection of ART-treated SIVmac239-infected rhesus macaques. Markers of neutrophil extracellular trap formation were measured throughout P. fragile co-infection of ART-treated SIVmac239-infected rhesus macaques (RMs; n=4) via ELISA. A) Neutrophil elastase and B) Citrullinated histone 3 levels were measured in plasma by ELISA. In both panels, each RM is represented by a different symbol and color. Baseline (BL) is an average of data collected at weeks -6, -2, and 0 post-SIV infection (p.i.). Inoculation with SIVmac239 at week 0 p.i. is indicated by a purple dashed arrow. Inoculation with P. fragile at week 12 p.i. is indicated by a blue dashed arrow. Antiretroviral therapy (ART) was initiated at week 8 p.i., indicated by the dark grey bar. Anti-malarial treatment occurred throughout week 14 p.i., indicated by the light grey bar. Statistical significance between all time points was calculated using a mixed-effects analysis with the Geisser-Greenhouse correction and a Tukey’s multiple comparisons test, with individual variances computed for each comparison. Multiplicity adjusted significant P values are shown above horizontal black bars.
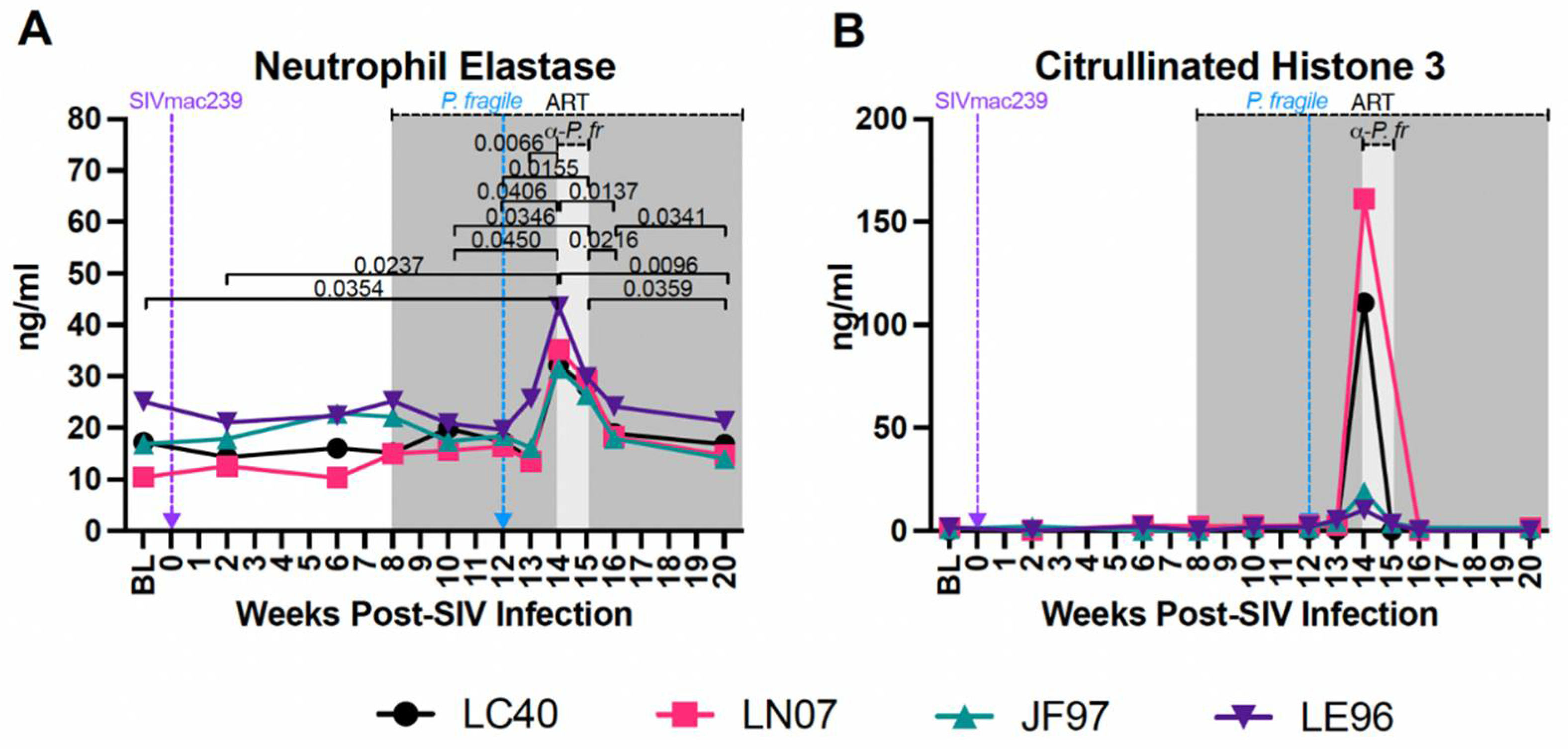
Figure 8.
Increased levels of microbial translocation and gastrointestinal (GI) barrier permeability markers P. fragile co-infection of ART-treated SIVmac239-infected rhesus macaques. Markers of microbial translocation and GI barrier permeability were measured throughout P. fragile co-infection of ART-treated SIVmac239-infected rhesus macaques (RMs; n=4) via ELISA. A-D) Zonulin (A), Intestinal fatty-acid binding protein (I-FABP; B), Soluble CD14 (sCD14; C), and LPS binding protein (LBP; D) levels were measured in plasma by ELISA. In all panels, each RM is represented by a different symbol and color. Baseline (BL) is an average of data collected at weeks -6, -2, and 0 post-SIV infection (p.i.). Inoculation with SIVmac239 at week 0 p.i. is indicated by a purple dashed arrow. Inoculation with P. fragile at week 12 p.i. is indicated by a blue dashed arrow. Antiretroviral therapy (ART) was initiated at week 8 p.i., indicated by the dark grey bar. Anti-malarial treatment occurred throughout week 14 p.i., indicated by the light grey bar. Statistical significance between all time points was calculated using a mixed-effects analysis with the Geisser-Greenhouse correction and a Tukey’s multiple comparisons test, with individual variances computed for each comparison. Multiplicity adjusted significant P values are shown above horizontal black bars.
Figure 8.
Increased levels of microbial translocation and gastrointestinal (GI) barrier permeability markers P. fragile co-infection of ART-treated SIVmac239-infected rhesus macaques. Markers of microbial translocation and GI barrier permeability were measured throughout P. fragile co-infection of ART-treated SIVmac239-infected rhesus macaques (RMs; n=4) via ELISA. A-D) Zonulin (A), Intestinal fatty-acid binding protein (I-FABP; B), Soluble CD14 (sCD14; C), and LPS binding protein (LBP; D) levels were measured in plasma by ELISA. In all panels, each RM is represented by a different symbol and color. Baseline (BL) is an average of data collected at weeks -6, -2, and 0 post-SIV infection (p.i.). Inoculation with SIVmac239 at week 0 p.i. is indicated by a purple dashed arrow. Inoculation with P. fragile at week 12 p.i. is indicated by a blue dashed arrow. Antiretroviral therapy (ART) was initiated at week 8 p.i., indicated by the dark grey bar. Anti-malarial treatment occurred throughout week 14 p.i., indicated by the light grey bar. Statistical significance between all time points was calculated using a mixed-effects analysis with the Geisser-Greenhouse correction and a Tukey’s multiple comparisons test, with individual variances computed for each comparison. Multiplicity adjusted significant P values are shown above horizontal black bars.
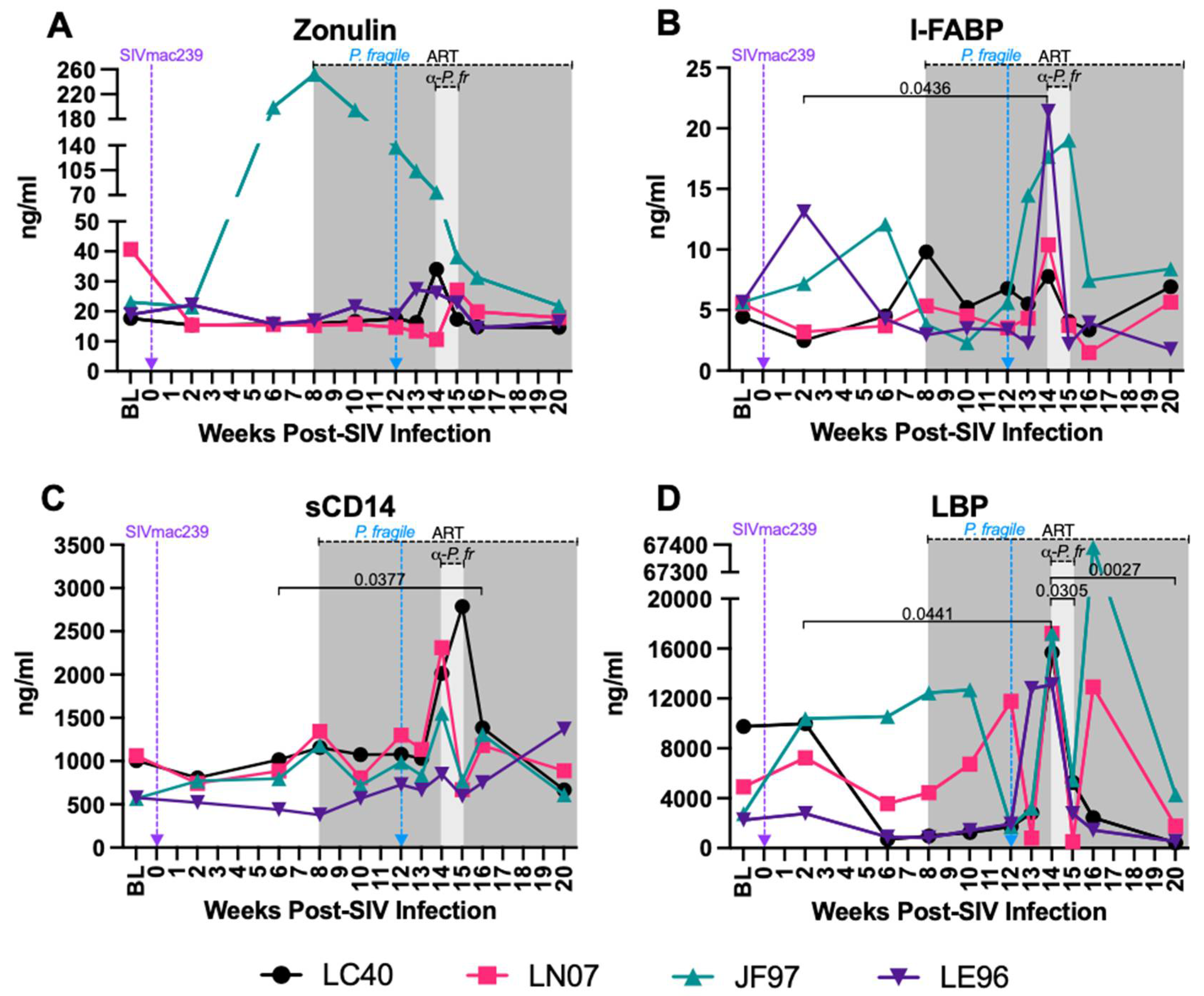
Figure 9.
Multivariate ANOVA (MANOVA) reveals significant correlations between clinical markers of SIV and malaria infection, as well as neutrophil frequency, function, and peripheral markers of GI dysfunction. Pearson’s partial correlation coefficients were generated using a MANOVA for 13 different parameters (VL, anemia, absolute CD4+ count, peripheral neutrophils, plasma zonulin, sCD4, I-FABP, LBP, NE, cathepsin G, CitH3, IP-10, MCP-1, and CRP). The correlation coefficients were adjusted against animal number. Boxes highlighted in light green represent positive correlations trending towards significance (0.05<p<0.07) and boxes highlighted dark green or red represent statistically significant positive or negative correlations, respectively (p<0.05).
Figure 9.
Multivariate ANOVA (MANOVA) reveals significant correlations between clinical markers of SIV and malaria infection, as well as neutrophil frequency, function, and peripheral markers of GI dysfunction. Pearson’s partial correlation coefficients were generated using a MANOVA for 13 different parameters (VL, anemia, absolute CD4+ count, peripheral neutrophils, plasma zonulin, sCD4, I-FABP, LBP, NE, cathepsin G, CitH3, IP-10, MCP-1, and CRP). The correlation coefficients were adjusted against animal number. Boxes highlighted in light green represent positive correlations trending towards significance (0.05<p<0.07) and boxes highlighted dark green or red represent statistically significant positive or negative correlations, respectively (p<0.05).
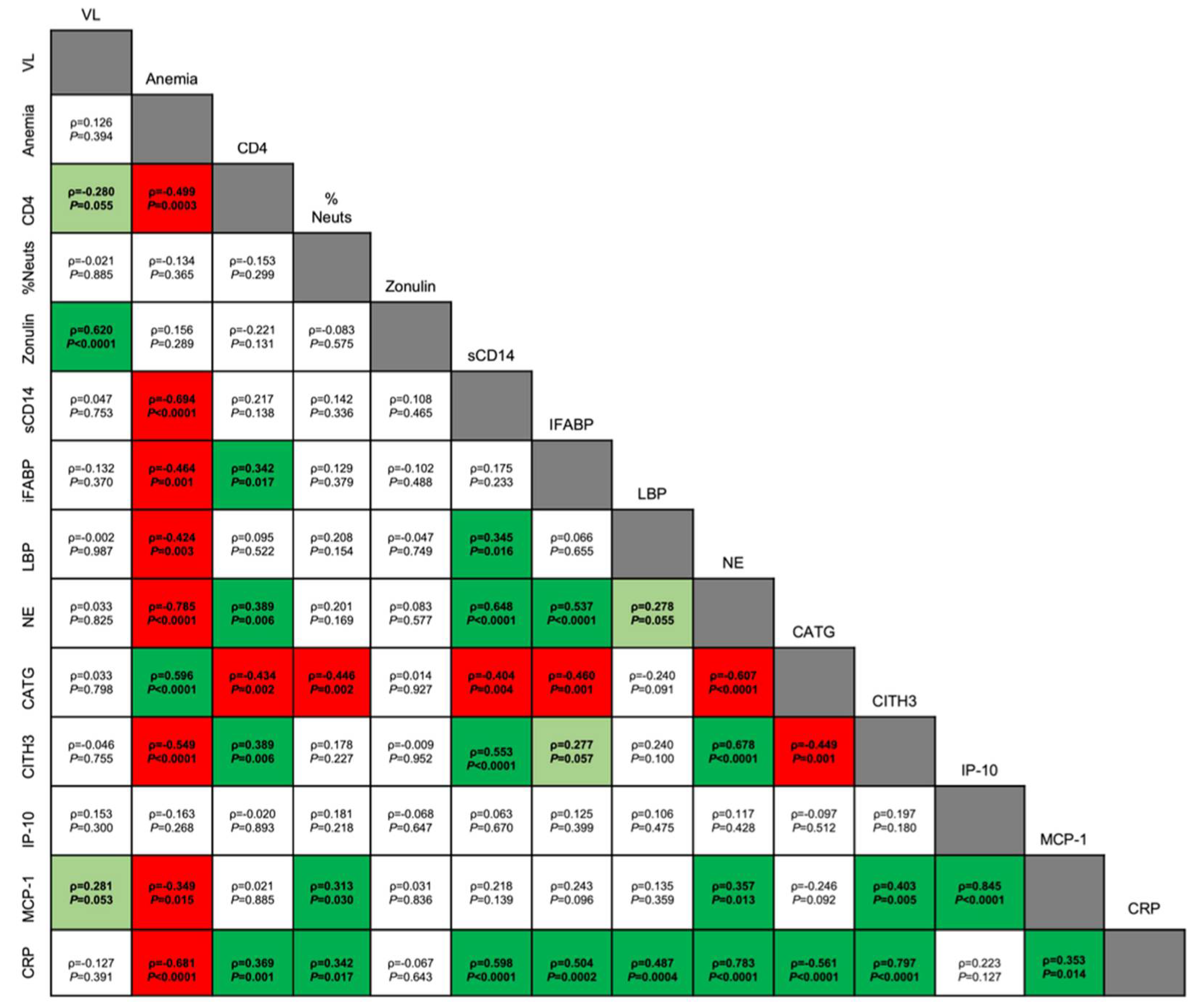
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



