
Submitted:
16 May 2024
Posted:
17 May 2024
You are already at the latest version
Abstract
Among nonsteroidal anti-inflammatory drugs, ibuprofen, diclofenac, and celecoxib have been frequently used in multimodal analgesia. Recent studies challenge the conventional theory that they exhibit activity and toxicity by acting on cyclooxygenase selectively. We compared their membrane interactions that may be associated with analgesic and gastrointestinal toxic effects. Biomimetic membranes suspended in buffers of different pH were prepared with 1-palmitoyl-2-oleoylphosphatidylcholine, sphingomyelin, and cholesterol to mimic neuronal membranes and with 1,2-dipalmitoylphosphatidylcholine to mimic gastrointestinal mucosae. The membrane interactivity was determined by measuring fluorescence polarization. At pH 7.4, the drugs interacted with neuro-mimetic membranes to decrease membrane fluidity at pharmacokinetically-relevant 0.5–100 μM. Celecoxib was most potent, followed by ibuprofen and diclofenac. At pH 4.0 and 2.5, however, the drugs increased the fluidity of 1,2-dipalmitoylphosphatidylcholine membranes at 0.1–1 mM corresponding to gastroduodenal lumen concentrations after administration. Their membrane fluidization was greater at gastric pH 2.5 than at duodenal pH 4.0. Low-micromolar ibuprofen, diclofenac, and celecoxib structure-specifically decrease neuronal membrane fluidity, which hypothetically could affect signal transmission of nociceptive sensory neurons. Under gastroduodenal acidic conditions, high-micromolar ibuprofen, diclofenac, and celecoxib induce fluidity increases of membranous phosphatidylcholines that are hypothetically associated with gastrointestinal toxic effects, which would enhance acid permeability of protective mucosal membranes.
Keywords:
ibuprofen
; diclofenac
; celecoxib
; membrane interaction
; lipid composition
; medium pH
; drug concentration
; analgesic activity
; gastrointestinal toxicity
1. Introduction
Nonsteroidal anti-inflammatory drugs are one of the most common over-the-counter and prescribed medicines for relieving pain and suppressing inflammation. In addition to their use as a popular anti-inflammatory analgesic, ibuprofen (IBU), diclofenac (DIC), and celecoxib (CEL) (Figure 1) have frequently been applied to multimodal analgesia for the purpose of potentiating analgesic effects and sparing opioids [1,2]. These drugs exhibit the analgesic activity when used properly, but otherwise the gastrointestinal toxicity [3,4,5]. Their pharmacological and toxicological effects have been primarily explained by relating to inhibition of cyclooxygenase (COX), which consists of constitutive COX-1 responsible for physiological functions and inducible COX-2 upregulated by various pathological conditions [4,6]. However, recent findings do not necessarily support the conventional theory that nonsteroidal anti-inflammatory drugs selectively act on COX-constituting proteins to exert beneficial effects by inhibiting COX-2 and adverse effects by inhibiting COX-1. They include experimental results indicating that selective COX-1 inhibition does not cause gastric damage to preserve mucosal integrity [7] but inhibition of both COX isozymes is required to induce gastric injuries [8] and that gastrointestinal ulceration occurs independently of a prostanoid metabolic pathway [9]. Involvement of causative factors other than COX mediation has been suggested for the mechanisms of gastrointestinal damage [10].
In sensory neurons, the axonal signal transmission and the membrane-bound receptor, ion channel, and enzyme activity depend on neuronal membrane fluidity. Physicochemical membrane modifiers modulate nociceptive signaling and pain transduction [11]. Membrane-active agents could affect inflammatory pain signaling by altering fluidity or elasticity of lipid bilayer membranes [12]. IBU, DIC, and CEL have amphiphilic structures, which enable them to interact with lipid bilayers.
Even if the enzyme inhibition underlies the effects of nonsteroidal anti-inflammatory drugs, their interactions with monotopic membrane enzyme COX take place in membrane lipid bilayers. Drug-induced changes in physicochemical membrane property modulate the functions of biomembranes and membrane proteins [13]. In addition to the direct effects on neuronal lipid bilayer membranes, IBU, DIC, and CEL may influence the activity of COX through alteration of the membrane lipid environment for COX.
Membrane fluidity has been referred to as a determinant for the integrity of biomembranes and the function of membrane-imbedded proteins [14,15]. Typical nonsteroidal anti-inflammatory drugs have been suggested to interact with biological and artificial membranes [16,17]. A considerable number of studies reported that they consequently decrease membrane fluidity [18,19,20], whereas other studies, increase membrane fluidity [21,22,23]. Such opposing membrane effects can be attributed to experimental conditions that are different in membrane lipid composition, reaction medium pH, and used drug concentration.
In the present study, we compared the interactions of IBU, DIC, and CEL with biomimetic lipid membranes (neuronal membranes and gastrointestinal protective mucosae) by varying lipid composition, medium pH, and drug concentration to associate their membrane interactivity with analgesic and gastrointestinal toxic effects.
2. Materials and Methods
2.1. Chemicals
IBU, DIC, CEL, and reference drugs such as aspirin (ASP), indomethacin (IND), and loxoprofen (LOX) were obtained from Wako Pure Chemicals (Osaka, Japan). 1,2-Dipalmitoylphosphatidylcholine (DPPC), 1-palmitoyl-2-oleoylphosphatidylcholine (POPC), and porcine brain sphingomyelin (SM) were purchased from Avanti Polar Lipids (Alabaster, AL, USA), cholesterol from Wako Pure Chemicals (Osaka, Japan), and diphenyl-1,3,5-hexatriene (DPH) from Molecular Probes (Eugene, OR, USA). 4-(2-Hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), acetate, and phosphate buffers were prepared to contain 125 mM NaCl and 25 mM KCl by using reagent products (Wako Pure Chemicals). Dimethyl sulfoxide (DMSO) of spectroscopic grade, ethanol of spectroscopic grade, and water of liquid chromatographic grade, all of which were used for preparing reagent solutions, were obtained from Kishida (Osaka, Japan). All other chemicals were of the highest analytical grade available commercially.
2.2. Preparation of Biomimetic Membranes
DPH-labelled biomimetic membranes were prepared by an ethanol injection method [24], in which a lipid and DPH solution was injected to an excess of buffer for producing unilamellar vesicles. In brief, the dry film of phospholipids and cholesterol was dissolved with an ethanolic solution of DPH. An aliquot (250 μL) of the resulting solution (total lipids of 10 mM and DPH of 50 μM) was rapidly injected four times into 199 mL of 10 mM HEPES buffer (pH 7.4), 50 mM acetate buffer (pH 4.0), or 20 mM phosphate buffer (pH 2.5) under stirring above the phase transition temperatures of phospholipids. The membrane lipid composition was 45 mol% POPC, 10 mol% SM, and 45 mol% cholesterol for neuro-mimetic membranes [20,24] and 100 mol% DPPC for gastrointestinal protective mucosa-mimetic membranes [25].
2.3. Determination of Membrane Interactivity
The tested drugs dissolved in DMSO were added to the membrane preparations so that final concentrations were 0.5 μM to 1.0 mM for IBU, 0.5–200 μM for DIC, 0.5–200 μM for CEL, and 25–200 μM for ASP. The concentration of DMSO was adjusted to be less than 0.5% (v/v) of the total volume so as not to affect the fluidity of intact membranes. Control experiments were conducted by adding an equivalent volume of DMSO. After reactions at 37 °C for 45 min, DPH fluorescence polarization was measured at 360 nm for excitation wavelength and 430 nm for emission wavelength by an FP-777 spectrofluorometer (Japan Spectroscopic Cooperation; Tokyo, Japan) equipped with a polarizer (Shimadzu; Kyoto, Japan). Polarization values were calculated as reported previously [24]. Compared with controls, an increase and a decrease of fluorescence polarization indicates a decrease and an increase of membrane fluidity, respectively. When comparing the membrane interactivity between different conditions, the polarization changes (%) relative to control polarization values were used because the polarization values of intact membranes vary depending on lipid composition and medium pH.
2.4. Statistical Analysis
The data were statistically analyzed by one-way ANOVA with a Bonferroni post-hoc comparison using SPSS version 22 (IBM Corporation; Chicago, IL, USA). All results are expressed as means ± SEM (n = 8 for each experiment), and values of *p < 0.05 and **p < 0.01 were considered statistically significant.
3. Results
3.1. Interactions with Neuro-Mimetic Membranes
When subjected to the reactions with biomimetic membrane in media of pH 7.4, IBU, DIC, CEL, and ASP interacted with neuro-mimetic membranes to decrease membrane fluidity with the potency depending on drug structures as shown by DPH polarization increases in Table 1. At 50 μM for each, CEL was most potent, followed by IBU, DIC, and ASP. IND also showed polarization increases at 50–200 μM. However, IND was excluded from the comparative assessment because the possibility that its natural fluorescence affected polarization analysis cannot be ruled out.
In contrast to the membrane fluidity-decreasing effects at pH 7.4, IBU of 100–500 μM increased the fluidity of neuro-mimetic membranes at pH 4.0 (Table 1), suggesting the pH-dependent drug and membrane interactions.
3.2. Interactions with DPPC Membranes
The tested drugs pH-dependently interacted with DPPC membranes as shown in Table 2. At pH 7.4, IBU, DIC, CEL, and ASP decreased membrane fluidity. Although LOX changed DPH polarization, it was excluded from the comparative assessment because LOX is a prodrug that is converted into an active metabolite after absorption.
Under acidic conditions, IBU, DIC, CEL and ASP increased the fluidity of DPPC membranes with increasing concentrations. The relative potency to increase DPPC membrane fluidity was CEL > DIC > IBU at 100 μM for each. Membrane effects of IBU (≥ 200 μM), DIC (≥ 100 μM), and CEL (≥ 50 μM) at pH 2.5 were greater than those at pH 4.0 (pH 4.0 vs pH 2.5, p < 0.01 for all). DIC and ASP acted on DPPC membranes more potently to increase membrane fluidity at pH 2.5 compared with at pH 4.0.
3.3. Membrane Interactivity Depending on Lipid Composition and Medium pH
IBU, DIC, CEL, and ASP interacted with both neuro-mimetic membranes and DPPC membranes at pH 7.4 to decrease membrane fluidity, but with greater potency for neuro-mimetic membranes (Figure 2a) than for DPPC membranes (Figure 2b). At 50 μM for each, effects of IBU, DIC, and CEL on neuro-mimetic membranes were 2.6, 2.0, and 1.2 times greater than those on DPPC membranes, respectively (neuro-mimetic vs DPPC, p < 0.01 for all).
The membrane interactivity of IBU depended on medium pH as shown in Figure 3. At pH 7.4 (Figure 3a), 50–100 μM IBU preferentially interacted with neuro-mimetic membranes to induce greater decreases in membrane fluidity than DPPC membranes, whereas at pH 4.0 (Figure 3b), 100–500 μM IBU preferentially interacted with DPPC membranes to induce greater increases in membrane fluidity than neuro-mimetic membranes (neuro-mimetic vs DPPC, p < 0.01 for all).
IBU, DIC, CEL, and ASP interacted with DPPC membranes at pH 7.4, 4.0, and 2.5 to change membrane fluidity differently as shown in Figure 4. Although their relatively low concentrations decreased DPPC membrane fluidity at pH 7.4 (Figure 4a), their relatively high concentrations increased DPPC membrane fluidity at pH 4.0 (Figure 4b) and pH 2.5 (Figure 4c). Membrane effects at pH 2.5 of 500 μM IBU, 200 μM DIC, and 100 μM CEL were 1.5, 1.5, and 1.3 times greater than those at pH 4.0, respectively (pH 2.5 vs pH 4.0, p < 0.01 for all).
4. Discussion
While phospholipids and cholesterol are the major components in biomembranes, sphingolipids are abundant in neuronal membranes and play an important role in the signaling process [26] and the pain perception of nociceptive sensory neurons [27]. In particular, SM is preset at high concentrations for the structural and functional significance. We prepared neuro-mimetic membranes with POPC and cholesterol plus SM. IBU, DIC, and CEL were more effective in interacting with such biomimetic membranes containing SM than the membranes consisting of DPPC alone. The membrane interactions of the tested drugs were so dependent on lipid composition, medium pH, and drug concentration that they decreased the fluidity of membranes consisting of phospholipids and cholesterol at pH 7.4 at low-micromolar concentrations, whereas increased the fluidity of membranes consisting of a single phospholipid component at both acidic pH and higher concentrations, but decreased at pH 7.4. Compared with the membranes prepared with 3.6 mol% SM and phosphatidylcholine/cholesterol of the equimolar ratio [20], the present neuro-mimetic membranes containing 10 mol% SM produced greater decreases in membrane fluidity by interacting with IBU, suggesting that SM is responsible for characterizing the drug and neuronal membrane interactions.
When IBU tablet of 400 mg and 800 mg were administered to human subjects, the maximum plasma concentration (Cmax) reached 147 and 305 μM, respectively [28]. DIC showed Cmax of 3.6–7.5 μM after administration of 50-mg tablet [29]. Oral administration of 100-mg CEL or 200-mg CEL-tramadol co-crystal (112 mg CEL and 88 mg tramadol) to healthy subjects resulted in Cmax of CEL ranging from 0.7 to 1.4 μM [30]. Intravenous infusion of 400 and 800 mg Caldolor (IBU injection) for 5–60 min produced Cmax of 190–582 μM for IBU [28] and a single dose of intravenous hydroxypropyl-β-cyclodextrin/DIC of 37.5 mg, Cmax of 24–73 μM for DIC [31]. At concentrations corresponding to these pharmacokinetic parameters, IBU, DIC, and CEL commonly decreased the fluidity of neuro-mimetic membranes at pH 7.4, but with different potencies. Since membrane fluidity regulates signal transmission and neuronal functions in sensory neurons, its alteration is presumed to disturb nociceptive signaling and affect inflammatory pain [11]. Some pain relievers act on lipid bilayer membranes and modify membrane fluidity in addition to acting on channels relevant to pain perception [12]. IBU, DIC, and CEL of pharmacokinetically-relevant low-micromolar concentrations induce structure-specific decreases in neuro-mimetic membrane fluidity, which hypothetically could affect signal transmission of nociceptive sensory neurons, possibly resulting in analgesia.
5-Lipoxygenase, which binds to nuclear membranes for activation, preferentially interacts with fluid (fluidity-increased) membranes, but not with rigid (fluidity-decreased) membranes [14]. Similarly, monotopic membrane enzyme COX may be activated in biomembranes with the relatively high fluidity. The activity of membrane-associated enzymes is considered to be enhanced when the membrane domains have higher fluidity, whereas reduced by decreasing membrane fluidity. We could hypothesize that membrane-rigidifying drugs inhibit COX indirectly through alteration of the membrane lipid environment optimal for COX activity. CEL and DIC are highly and intermediately selective for COX-2, respectively, but IBU is not selective for COX-2, whereas ASP has high selectivity for COX-1. Lucio et al. [32] compared the effects of drugs on lipid bilayers and revealed that COX-2 selective inhibitors change membrane fluidity, but not COX-1 inhibitors. In the present study, the relative membrane-interacting potency was CEL >> IBU >> ASP, suggesting the possibility that the membrane interactivity of drugs may correlate with their selectivity for COX-2. Although the membrane effect of DIC was comparable to or slightly weaker than that of IBU, higher affinity of DIC for COX-2 would enhance its COX-2 selectivity. According to Ki (inhibitory constant) values of CEL, DIC, and IBU, COX-2 binding affinity of DIC is almost similar to CEL but much higher than IBU. A relationship between membrane interactivity and COX-2 selectivity needs to be verified by further studies.
Gastric lesions caused by nonsteroidal anti-inflammatory drugs may be related to their physicochemical properties such as pKa (acid dissociation constant) as well as their COX-2/COX-1 selectivity [33]. For the purpose of investigating the gastrointestinal toxic effects independent of COX inhibition, Lichtenberger et al. [15,34] conducted a series of studies to assess the property of nonsteroidal anti-inflammatory drugs to attenuate the hydrophobic protective barrier of gastrointestinal mucosae. Gastrointestinal tracts are protected against luminal acids by the linings, which are constituted of phospholipids (rich in phosphatidylcholine) monolayers at the interface between mucus gel and luminal fluid, and of phospholipid (primarily phosphatidylcholine) bilayer membranes of epithelial cells [15]. In human gastroduodenal mucosae, the most abundant species of phosphatidylcholine were identified as 16:0/18:1, 16:0/18:2, and 16:0/20:4 [25].
Koenigsknecht et al. [35] administered 800-mg IBU tablet to human subjects to determine pH and IBU in gastrointestinal fluids. They revealed that gastric and duodenal pH are 2.3 and 4–5, respectively, and that intragastric and intraduodenal IBU concentrations are 439 μM after 15 min and 400–900 μM after 3–7 hours, respectively. Hens et al. [36] followed up changes in pH of gastrointestinal fluids for 7 hours after administration of 800-mg IBU tablet. In the fasting state, gastric and duodenal pH were 1.1–7.5 and 1.7–7.6, respectively.
By reference to previous studies [15,35,36], we assessed the effects of CEL, DIC, and IBU on DPPC (16:0/16:0) membranes as protective mucosae at pH 2.5 and pH 4.0 reflecting the gastroduodenal environments and at high-micromolar concentrations corresponding to drug concentrations in the gastroduodenal lumen after administration. The tested drugs increased the fluidity of DPPC membranes at pH 2.5 more significantly than pH 4.0 with the relative potency being CEL > DIC > IBU. These drugs are efficiently ionized at pH being > pKa, but not at pH being < pKa, so that they are very likely to be present in a nonionized form. Nonionized molecules are considered to effectively interact with phospholipid membranes. The greater membrane interactivity at gastric pH 2.5 than at duodenal pH 4.0 is consistent with the relative gastrointestinal toxicity that the incidence of gastric ulcers is higher than that of duodenal ulcers. Pereira-Leite et al. [37] investigated the effects of DIC on 1,2-dimyristoylphosphatidylcholine liposomal membranes at pH 3–5. The neutral form of DIC displayed greater affinity for phospholipid bilayers to modulate the bilayer structures more effectively than the anionic form. They suggested that nonionized DIC-induced changes in membranous phospholipids at low pH constitute a topical mechanism of the gastric toxicity. Increasing membrane fluidity should increase permeability of the membrane lipid bilayers, which may exert adverse effects on gastroduodenal protective mucosae.
Meta-analysis of gastrointestinal complications indicated that the risk of DIC is 2–3 times higher than IBU [38]. The membrane interaction-mediated gastrointestinal effect of CEL may have a longer time course as CEL causes gastrointestinal injuries by oral administration for a long term. While nonsteroidal anti-inflammatory drugs also adversely affect the cardiovascular system, the drug and membrane interactions depending on lipid composition, drug concentration, and drug structure were recently associated with cardiotoxic effects [39].
5. Conclusions
Drugs frequently used in multimodal analgesia interact with biomimetic membranes depending on lipid composition, medium pH, and drug concentration. As a result of membrane interactions, IBU, DIC, and CEL of pharmacokinetically-relevant low-micromolar concentrations structure-specifically increase the fluidity of neuro-mimetic membranes at the physiological pH, which hypothetically could affect signal transmission of nociceptive sensory neurons, possibly resulting in analgesic effects. Under gastroduodenal acidic conditions, however, IBU, DIC, and CEL of high-micromolar concentrations corresponding to intragastric and intraduodenal concentrations after oral administration induce fluidity increases of membranous phosphatidylcholines that are hypothetically associated with gastrointestinal toxic effects, which would enhance acid permeability of protective mucosal membranes, possibly causing damage of gastroduodenal tracts.
Author Contributions
Author 1 (MM) performed the experiments and statistically analyzed the data. Author 2 (HT) designed and conducted the study. Both MM and HT wrote and reviewed the manuscript.
Funding
The present study was supported by JSPS KAKENHI grant number 20K10152 and JSPS KAKENHI grant number 24K12106.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable because the present study includes neither human nor animal experiments.
Data Availability Statement
All data generated or analyzed during the present study are included in this article. The data supporting the findings are kept at the affiliation of author 1 (MM) and are available on request.
Conflicts of Interest
The authors have no conflict of interest to declare.
References
- Young, A.; Buvanendran, A. Recent advances in multimodal analgesia. Anesthesiol. Clin. 2012, 30, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Brubaker, L.; Kendall, L.; Reina, E. Multimodal analgesia: A systematic review of local NSAIDs for non-ophthalmologic postoperative pain management. Int. J. Surg. 2016, 32, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Rainsford, K.D. Ibuprofen: pharmacology, efficacy and safety. Inflammopharmacology 2009, 17, 275–342. [Google Scholar] [CrossRef] [PubMed]
- Harirforoosh, S.; Asghar, W.; Jamali, F. Adverse effects of nonsteroidal antiinflammatory drugs: an update of gastrointestinal, cardiovascular and renal complications. J. Pharm. Pharm. Sci. 2013, 16, 821–847. [Google Scholar] [CrossRef] [PubMed]
- Cooper, C.; Chapurlat, R.; Al-Daghri, N.; Herrero-Beaumont, G.; Bruyère, O.; Rannou, F.; Roth, R.; Uebelhart, D.; Reginster, J.Y. Safety of oral non-selective non-steroidal anti-inflammatory drugs in osteoarthritis: What does the literature say? Drugs Aging 2019, 36 (Suppl 1), 15–24. [Google Scholar] [CrossRef] [PubMed]
- Bindu, S.; Mazumder, S.; Bandyopadhyay, U. Non-steroidal anti-inflammatory drugs (NSAIDs) and organ damage: A current perspective. Biochem. Pharmacol. 2020, 180, 114147. [Google Scholar] [CrossRef] [PubMed]
- Perrone, M.G.; Lofrumento, D.D.; Vitale, P.; De Nuccio, F.; La Pesa, V.; Panella, A.; Calvello, R.; Cianciulli, A.; Panaro, M.A.; Scilimati, A. Selective cyclooxygenase-1 inhibition by p6 and gastrotoxicity: Preliminary investigation. Pharmacology 2015, 95, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Wallace, J.L.; McKnight, W.; Reuter, B.K.; Vergnolle, N. NSAID-induced gastric damage in rats: Requirement for inhibition of both cyclooxygenase 1 and 2. Gastroenterology 2000, 119, 706–714. [Google Scholar] [CrossRef]
- Lim, Y.J.; Dial, E.J.; Lichtenberger, L.M. Advent of novel phosphatidylcholine-associated nonsteroidal anti-inflammatory drugs with improved gastrointestinal safety. Gut Liver 2013, 7, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Bjarnason, I.; Scarpignato, C.; Holmgren, E.; Olszewski, M.; Rainsford, K.D.; Lanas, A. Mechanisms of damage to the gastrointestinal tract from nonsteroidal anti-inflammatory drugs. Gastroenterology 2018, 154, 500–514. [Google Scholar] [CrossRef]
- Wagner, K.; Vito, S.; Inceoglu, B.; Hammock, B.D. The role of long chain fatty acids and their epoxide metabolites in nociceptive signaling. Prostaglandins Other Lipid Mediat. 2014, 113-115, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Matta, J.A.; Miyares, R.L.; Ahern, G.P. TRPV1 is a novel target for omega-3 polyunsaturated fatty acids. J. Physiol. 2007, 578 (Pt 2), 397–411. [Google Scholar] [CrossRef]
- Escribá, P.V.; González-Ros, J.M.; Goñi, F.M.; Kinnunen, P.K.; Vigh, L.; Sánchez-Magraner, L.; Fernández, A.M.; Busquets, X.; Horváth, I.; Barceló-Coblijn, G. Membranes: A meeting point for lipids, proteins and therapies. J. Cell. Mol. Med. 2008, 12, 829–875. [Google Scholar] [CrossRef] [PubMed]
- Pande, A.H.; Qin, S.; Tatulian, S.A. Membrane fluidity is a key modulator of membrane binding, insertion, and activity of 5-lipoxygenase. Biophys. J. 2005, 88, 4084–4094. [Google Scholar] [CrossRef] [PubMed]
- Lichtenberger, L.M.; Zhou, Y.; Jayaraman, V.; Doyen, J.R.; O’Neil, R.G.; Dial, E.J.; Volk, D.E.; Gorenstein, D.G.; Boggara, M.B.; Krishnamoorti, R. Insight into NSAID-induced membrane alterations, pathogenesis and therapeutics: characterization of interaction of NSAIDs with phosphatidylcholine. Biochim. Biophys. Acta 2012, 1821, 994–1002. [Google Scholar] [CrossRef] [PubMed]
- Pereira-Leite, C.; Nunes, C.; Reis, S. Interaction of nonsteroidal anti-inflammatory drugs with membranes: in vitro assessment and relevance for their biological actions. Prog. Lipid Res. 2013, 52, 571–584. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, H.; Mizogami, M. Membrane interactivity of non-steroidal anti-inflammatory drugs: A literature review. J. Adv. Med. Med. Res. 2019, 31, 1–30. [Google Scholar] [CrossRef]
- Giraud, M.N.; Motta, C.; Romero, J.J.; Bommelaer, G.; Lichtenberger, L.M. Interaction of indomethacin and naproxen with gastric surface-active phospholipids: a possible mechanism for the gastric toxicity of nonsteroidal anti-inflammatory drugs (NSAIDs). Biochem. Pharmacol. 1999, 57, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Suwalsky, M.; Manrique, M.; Villena, F.; Sotomayor, C.P. Structural effects in vitro of the anti-inflammatory drug diclofenac on human erythrocytes and molecular models of cell membranes. Biophys. Chem. 2009, 141, 34–40. [Google Scholar] [CrossRef]
- Tsuchiya, H.; Mizogami, M. Discrimination of stereoisomers by their enantioselective interactions with chiral cholesterol-containing membranes. Molecules 2017, 23, 49. [Google Scholar] [CrossRef]
- Nunes, C.; Lopes, D.; Pinheiro, M.; Pereira-Leite, C.; Reis, S. In vitro assessment of NSAIDs-membrane interactions: significance for pharmacological actions. Pharm. Res. 2013, 30, 2097–2107. [Google Scholar] [CrossRef] [PubMed]
- Manrique-Moreno, M.; Heinbockel, L.; Suwalsky, M.; Garidel, P.; Brandenburg, K. Biophysical study of the non-steroidal anti-inflammatory drugs (NSAID) ibuprofen, naproxen and diclofenac with phosphatidylserine bilayer membranes. Biochim. Biophys. Acta 2016, 1858, 2123–2131. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, E.; Soares, T.B.; Gonçalves, H.; Bernstorff, S.; Real Oliveira, M.E.C.D.; Lopes, C.M.; Lúcio, M. A molecular biophysical approach to diclofenac topical gastrointestinal damage. Int. J. Mol. Sci. 2018, 19, 3411. [Google Scholar] [CrossRef]
- Mizogami, M.; Tsuchiya, H. Acetaminophen has lipid composition-dependent membrane interactivity that could be related to nephrotoxicity but not to analgesic activity and hepatotoxicity. Med. Princ. Pract. 2022, 31, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Ehehalt, R.; Wagenblast, J.; Erben, G.; Lehmann, W.D.; Hinz, U.; Merle, U.; Stremmel, W. Phosphatidylcholine and lysophosphatidylcholine in intestinal mucus of ulcerative colitis patients. A quantitative approach by nanoElectrospray-tandem mass spectrometry. Scand. J. Gastroenterol. 2004, 39, 737–742. [Google Scholar] [CrossRef] [PubMed]
- Grassi, S.; Chiricozzi, E.; Mauri, L.; Sonnino, S.; Prinetti, A. Sphingolipids and neuronal degeneration in lysosomal storage disorders. J. Neurochem. 2019, 148, 600–611. [Google Scholar] [CrossRef] [PubMed]
- Sántha, P.; Dobos, I.; Kis, G.; Jancsó, G. Role of gangliosides in peripheral pain mechanisms. Int. J. Mol. Sci. 2020, 21, 1005. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.S.; Voss, B. Pharmacokinetics of intravenous ibuprofen: Implications of time of infusion in the treatment of pain and fever. Drugs 2012, 72, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Altman, R.; Bosch, B.; Brune, K.; Patrignani, P.; Young, C. Advances in NSAID development: Evolution of diclofenac products using pharmaceutical technology. Drugs 2015, 75, 859–877. [Google Scholar] [CrossRef]
- Cebrecos, J.; Carlson, J.D.; Encina, G.; Lahjou, M.; Sans, A.; Sust, M.; Vaqué, A.; Morte, A.; Gascón, N.; Plata-Salamán, C. Celecoxib-tramadol co-crystal: A randomized 4-way crossover comparative bioavailability study. Clin. Ther. 2021, 43, 1051–1065. [Google Scholar] [CrossRef]
- Hamilton, D.A.; Ernst, C.C.; Kramer, W.G.; Madden, D.; Lang, E.; Liao, E.; Lacouture, P.G.; Ramaiya, A.; Carr, D.B. Pharmacokinetics of diclofenac and hydroxypropyl-β-cyclodextrin (HPβCD) following administration of injectable HPβCD-diclofenac in subjects with mild to moderate renal insufficiency or mild hepatic impairment. Clin. Pharmacol. Drug Dev. 2018, 7, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Lúcio, M.; Ferreira, H.; Lima, J.L.; Reis, S. Interactions between oxicams and membrane bilayers: An explanation for their different COX selectivity. Med. Chem. 2006, 2, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Bjarnason, I.; Scarpignato, C.; Takeuchi, K.; Rainsford, K.D. Determinants of the short-term gastric damage caused by NSAIDs in man. Aliment. Pharmacol. Ther. 2007, 26, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Lichtenberger, L.M.; Barron, M.; Marathi, U. Association of phosphatidylcholine and NSAIDs as a novel strategy to reduce gastrointestinal toxicity. Drugs Today 2009, 45, 877–890. [Google Scholar] [CrossRef] [PubMed]
- Koenigsknecht, M.J.; Baker, J.R.; Wen, B.; Frances, A.; Zhang, H.; Yu, A.; Zhao, T.; Tsume, Y.; Pai, M.P.; Bleske, B.E.; et al. In vivo dissolution and systemic absorption of immediate release ibuprofen in human gastrointestinal tract under fed and fasted conditions. Mol. Pharm. 2017, 14, 4295–4304. [Google Scholar] [CrossRef] [PubMed]
- Hens, B.; Tsume, Y.; Bermejo, M.; Paixao, P.; Koenigsknecht, M.J.; Baker, J.R.; Hasler, W.L.; Lionberger, R.; Fan, J.; Dickens, J.; et al. Low buffer capacity and alternating motility along the human gastrointestinal tract: Implications for in vivo dissolution and absorption of ionizable drugs. Mol. Pharm. 2017, 14, 4281–4294. [Google Scholar] [CrossRef] [PubMed]
- Pereira-Leite, C.; Jamal, S.K.; Almeida, J.P.; Coutinho, A.; Prieto, M.; Cuccovia, I.M.; Nunes, C.; Reis, S. Neutral diclofenac causes remarkable changes in phosphatidylcholine bilayers: Relevance for gastric toxicity mechanisms. Mol. Pharmacol. 2020, 97, 295–303. [Google Scholar] [CrossRef]
- Henry, D.; Lim, L.L.; Garcia Rodriguez, L.A.; Perez Gutthann, S.; Carson, J.L.; Griffin, M.; Savage, R.; Logan, R.; Moride, Y.; Hawkey, C.; et al. Variability in risk of gastrointestinal complications with individual non-steroidal anti-inflammatory drugs: Results of a collaborative meta-analysis. BMJ. 1996, 312, 1563–1566. [Google Scholar] [CrossRef]
- Pereira-Leite, C.; Figueiredo, M.; Burdach, K.; Nunes, C.; Reis, S. Unraveling the role of drug-lipid interactions in NSAIDs-induced cardiotoxicity. Membranes 2021, 11, 24. [Google Scholar] [CrossRef]
Figure 1.
Drugs frequently used in multimodal analgesia.
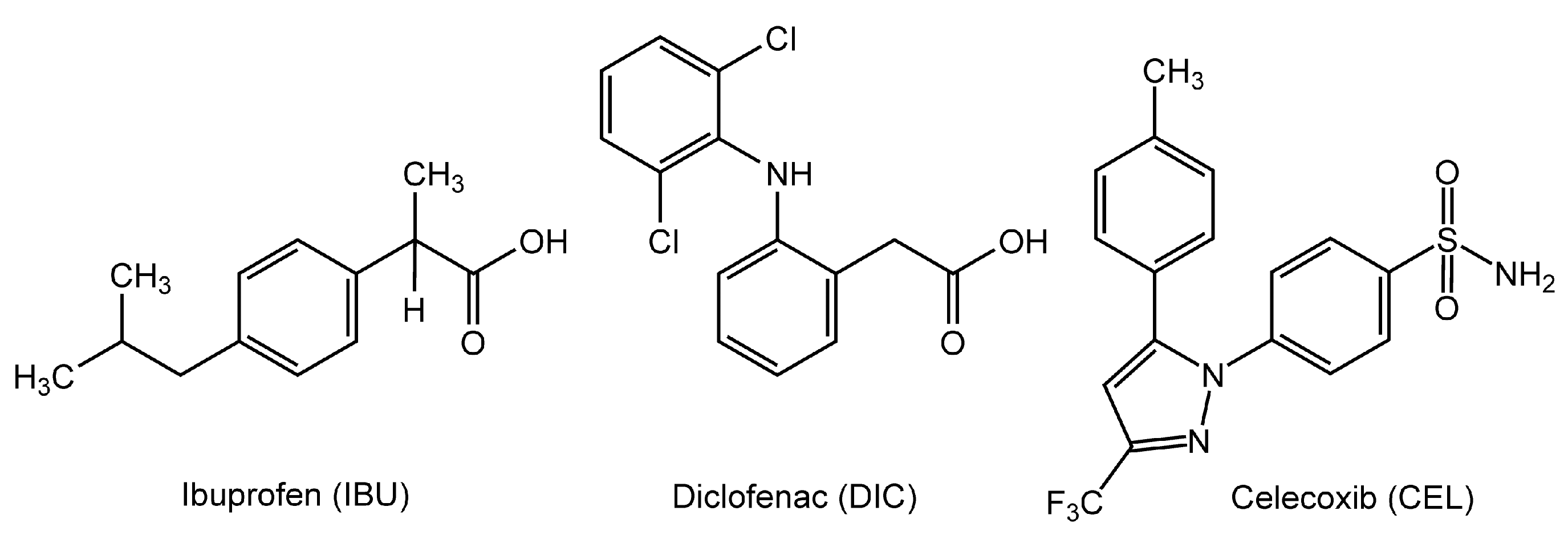
Figure 2.
Interactions of drugs at pH 7.4 with neuro-mimetic membranes (a) and 1,2-dipalmitoylphosphatidylcholine membranes (b). Values are means ± SEM (n = 8). *p < 0.05 and **p < 0.01 compared with controls. IBU, ibuprofen; DIC, diclofenac; CEL, celecoxib; ASP, aspirin.
Figure 2.
Interactions of drugs at pH 7.4 with neuro-mimetic membranes (a) and 1,2-dipalmitoylphosphatidylcholine membranes (b). Values are means ± SEM (n = 8). *p < 0.05 and **p < 0.01 compared with controls. IBU, ibuprofen; DIC, diclofenac; CEL, celecoxib; ASP, aspirin.
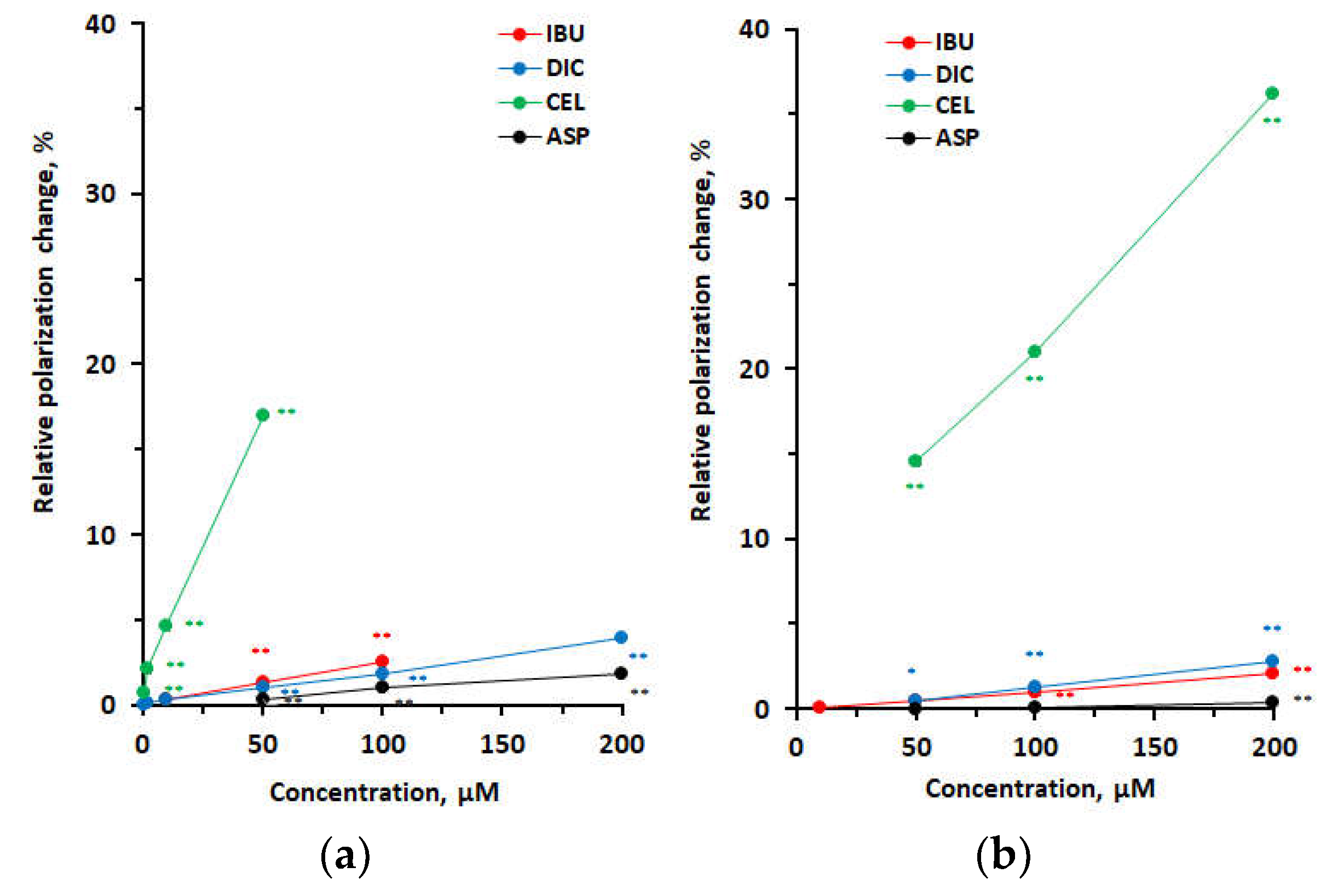
Figure 3.
Interactions of ibuprofen with neuro-mimetic membranes and 1,2-dipalmitoylphosphatidylcholine membranes at pH 7.4 (a) and pH 4.0 (b). Values are means ± SEM (n = 8). *p < 0.05 and **p < 0.01 compared with controls. DPPC, 1,2-dipalmitoylphosphatidylcholine.
Figure 3.
Interactions of ibuprofen with neuro-mimetic membranes and 1,2-dipalmitoylphosphatidylcholine membranes at pH 7.4 (a) and pH 4.0 (b). Values are means ± SEM (n = 8). *p < 0.05 and **p < 0.01 compared with controls. DPPC, 1,2-dipalmitoylphosphatidylcholine.
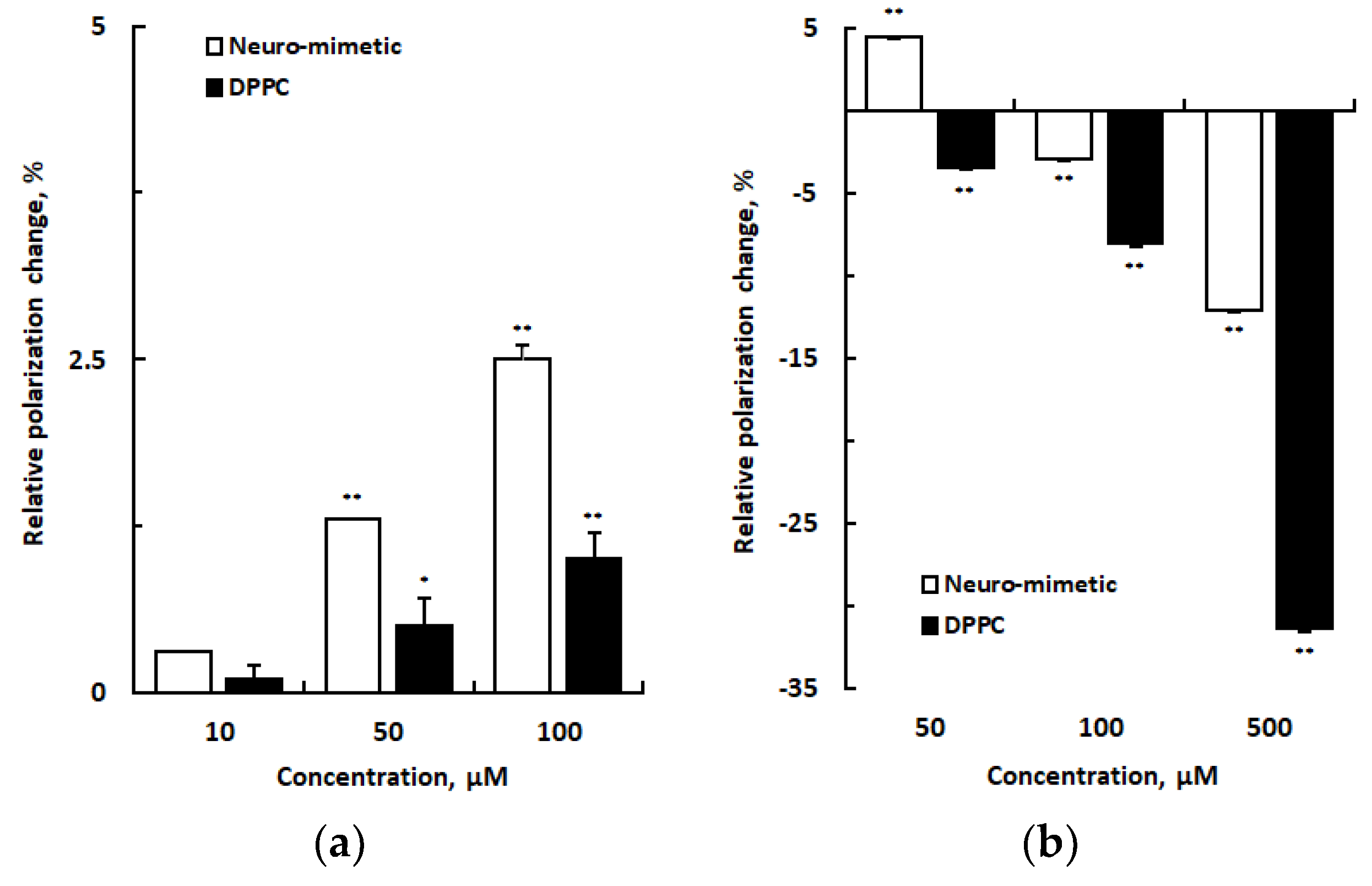
Figure 4.
Interactions of drugs with 1,2-dipalmitoylphosphatidylcholine membranes at pH 7.4 (a), pH 4.0 (b), and pH 2.5 (c). Values are means ± SEM (n = 8). *p < 0.05 and **p < 0.01 compared with controls. IBU, ibuprofen; DIC, diclofenac; CEL, celecoxib; ASP, aspirin.
Figure 4.
Interactions of drugs with 1,2-dipalmitoylphosphatidylcholine membranes at pH 7.4 (a), pH 4.0 (b), and pH 2.5 (c). Values are means ± SEM (n = 8). *p < 0.05 and **p < 0.01 compared with controls. IBU, ibuprofen; DIC, diclofenac; CEL, celecoxib; ASP, aspirin.

Table 1.
Interactions of drugs with neuro-mimetic membranes at different pH.
| Polarization change | |||||
| pH 7.4 | pH 4.0 | ||||
| Concentration (μM) | IBU | DIC | CEL | ASP | IBU |
| 0.5 | 0.0000 ± 0.0003 | 0.0000 ± 0.0002 | 0.0018 ± 0.0002** | ||
| 2 | 0.0002 ± 0.0002 | 0.0002 ± 0.0004 | 0.0054 ± 0.0004** | ||
| 10 | 0.0009 ± 0.0001 | 0.0006 ± 0.0001 | 0.0121 ± 0.0002** | ||
| 50 | 0.0034 ± 0.0000** | 0.0027 ± 0.0001** | 0.0445 ± 0.0003** | 0.0009 ± 0.0000** | 0.0098 ± 0.0003** |
| 100 | 0.0065 ± 0.0003** | 0.0047 ± 0.0000** | 0.0026 ± 0.0000** | –0.0067 ± 0.0002** | |
| 200 | 0.0102 ± 0.0002** | 0.0046 ± 0.0001** | |||
| 500 | –0.0270 ± 0.0002** | ||||
Values are means ± SEM (n = 8). **p < 0.01 compared with controls. IBU, ibuprofen; DIC, diclofenac; CEL, celecoxib; ASP, aspirin.
Table 2.
Interactions of drugs with DPPC membranes at different pH.
| Polarization change | ||||
| Concentration (μM) | IBU | DIC | CEL | ASP |
| pH 7.4 | ||||
| 50 | 0.0010 ± 0.0002* | 0.0010 ± 0.0000* | 0.0283 ± 0.0003** | 0.0000 ± 0.0001 |
| 100 | 0.0018 ± 0.0003** | 0.0025 ± 0.0002** | 0.0407 ± 0.0009** | 0.0002 ± 0.0001 |
| 200 | 0.0040 ± 0.0002** | 0.0054 ± 0.0003** | 0.0703 ± 0.0002** | 0.0008 ± 0.0000** |
| pH 4.0 | ||||
| 50 | –0.0059 ± 0.0002** | –0.0095 ± 0.0005** | –0.0350 ± 0.0007** | 0.0098 ± 0.0003** |
| 100 | –0.0134 ± 0.0004** | –0.0310 ± 0.0008** | –0.0570 ± 0.0005** | |
| 200 | –0.0398 ± 0.0004** | –0.0469 ± 0.0002** | –0.0150 ± 0.0002** | |
| 500 | –0.0518 ± 0.0004** | |||
| 1 mM | –0.0562 ± 0.0005** | |||
| pH 2.5 | ||||
| 25 | –0.0252 ± 0.0004** | –0.0369 ± 0.0004** | –0.0163 ± 0.0006** | |
| 50 | 0.0006 ± 0.0000 | –0.0832 ± 0.0004** | –0.0634 ± 0.0006** | |
| 100 | –0.0059 ± 0.0003** | –0.0920 ± 0.0004** | –0.1028 ± 0.0005** | |
| 200 | –0.0916 ± 0.0005** | –0.1029 ± 0.0006** | ||
| 500 | –0.1081 ± 0.0003** | |||
| 1 mM | –0.1119 ± 0.0005** | |||
Values are means ± SEM (n = 8). *p < 0.05 and **p < 0.01 compared with controls. IBU, ibuprofen; DIC, diclofenac; CEL, celecoxib; ASP, aspirin.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



