
Submitted:
20 May 2024
Posted:
21 May 2024
You are already at the latest version
Abstract
. Background. Hypoxic-ischemic brain injury is one primary cause of long-term neurological disability, morbidity, and death worldwide. The decrease in blood flow and oxygen concentration leads to insufficient nutrient supply to the brain, energy depletion, increased free radical generation, and mitochondrial dysfunction. Perinatal asphyxia — the oxygen supply suspension near birth-time — causes hypoxic-ischemic brain injury and is a major risk factor for neurodevelopmental damage. In pathological scenarios, raloxifene, a selective estrogen receptor modulator, has shown neuroprotective effects. Purpose. To examine whether raloxifene showed neuroprotection in an oxygen-glucose deprivation/reoxygenation astrocyte cell model. Methods. T98G cells in culture were treated with a glucose-free DMEM medium and incubated at 37ºC in a hypoxia chamber with 1% O2 for 3, 6, 12, and 24 hours. Cultures were supplemented with raloxifene 1, 10, and 100 nM during both glucose and oxygen deprivation and reoxygenation periods. Results. Raloxifene 100 nM and 10 nM improved cell survival — 65.34% and 70.56% — respectively compared to the control cell groups. Mitochondrial membrane potential was preserved by 58.9% 10 nM raloxifene and 81.57% 100 nM raloxifene cotreatment. Raloxifene cotreatment reduced superoxide production by 72.72% and peroxide production by 57%. Mitochondrial mass was preserved by 47.4%, 75.5%, and 89% in T98G cells exposed to 6-hour oxygen-glucose deprivation followed by 3, 6, and 9 hours of reoxygenation, respectively. Conclusions. Raloxifene improved cell survival and mitochondrial membrane potential, and reduced lipid peroxidation and reactive oxygen species (ROS) production, suggesting a direct effect on mitochondria. Raloxifene protected the oxygen-glucose-deprived astrocyte cells used to mimic hypoxic-ischemic brain injury in this study.
Keywords:
raloxifene
; neuroprotection: astrocytes: glucose-deprivation
; oxygen deprivation
; hypoxic-ischemic brain injury
1. Introduction
Perinatal asphyxia (PA) is a clinical syndrome characterized by oxygen supply shortage around birth time (Herrera-Marschitz et al., 2011). It may be the result of a variety of factors, including umbilical cord compression, alterations in gas exchange in the placenta, and fetal pulmonary failure, resulting in multiorgan oxygen (hypoxia) and/or hypoperfusion (ischemia) during the perinatal period (Galeano et al., 2013).
PA affects not only glial and neural circuits in the corpus striatum and the hippocampus (Herrera et al., 2018; Udovin et al., 2020) and increases presynaptic terminal buttons density and striatal and cortical dendritic spines (G. E. Saraceno et al., 2016, 2018). It also leads to necrosis and apoptosis in these areas and the cortex (Perez-Lobos et al., 2017), and astrogliosis in the striatum and cerebellum (Barkhuizen et al., 2017; Udovin et al., 2020). It disrupts the dopaminergic, GABAergic, glutamatergic, and redox systems (Lespay-Rebolledo et al., 2018, 2019; Perez-Lobos et al., 2017), leading to oxidative stress (Lespay-Rebolledo et al., 2018) and protein aggregation with ubiquitination (G. E. Saraceno et al., 2016), among other deleterious effects.
Mortality caused by PA has decreased in the last decades, while morbidity has increased (Holubiec et al., 2018). Many physical and mental disorders can be associated with PA (Herrera-Marschitz et al., 2014), including cognitive deficits (Ahearne, 2016; Barkhuizen et al., 2017; Galeano et al., 2015), seizures and/or epilepsy (Johne et al., 2021), schizophrenia (Pugliese et al., 2019), autism spectrum disorders (Modabbernia et al., 2016), attention-deficit/hyperactivity (Perna & Cooper, 2012), and neurodegenerative disorders (Toro-Urrego et al., 2020).
Glucose is the primary energy source for the adult brain, with neurons having the highest energy demand among cells (Detka et al., 2015). From glucose metabolism, the brain gets precursors for neurotransmitter synthesis, ATP for physiological functions, and neuronal and non-neuronal cell maintenance (Mergenthaler et al., 2013). However, astrocytes and other brain cells are crucial for brain glucose metabolism (Chen et al., 2022).
Astrocytes make up nearly 25% of brain volume, and their role in multiple processes has been studied over the past 20 years, including supporting the nervous system, maintaining the cerebral microenvironment for proper function, and regulating cerebral blood flow, which is crucial for optimal neuronal function. One of their most important functions is their involvement in brain metabolism. On one hand, astrocytes take up glucose from the blood vessels and provide energy metabolites to neurons (Guillamón-Vivancos et al., 2015). Through the astrocyte-neuron lactate shuttle (ANLS), astrocytes supply neurons with lactate, which serves as a substrate for the citric acid cycle to meet their energy demands (Fuller et al., 2010).
Neurons are very sensitive to energy deficits. Changes in glucose metabolism have been linked to cell death pathways and autophagy, increasing the risk of various central nervous system disorders. Different studies have shown how these alterations are associated with various neurodegenerative disorders’ progression, including Alzheimer’s disease, Parkinson’s disease, amyotrophic lateral sclerosis, neuroglycopenia, and Huntington’s disease, among others (Mergenthaler et al., 2013; Sifat et al., 2022). Reduced oxidation levels observed in post-ischemic brain tissues suggest these alterations may contribute to cerebrovascular and neurodegenerative abnormalities, including stroke (Sifat et al., 2022).
Therefore, there is considerable research focused on the prevention or reversal of neuronal damage caused by hypoglycemia. Steroids, such as estrogens and progesterone, have shown the ability to modulate neuronal activity and provide neuroprotective effects against toxicity and neurodegeneration in vivo studies (Nematipour et al., 2020; Wang et al., 2017). Neurons and glial cells have high-affinity steroid receptors, highlighting their importance in brain activity (Bove & Chitnis, 2014; Graham et al., 2008; Nematipour et al., 2020).
It has been described that the protective response of astrocytes can be modulated by neurosteroids, which regulate and modulate the expression of many genes involved in the development, connectivity, and survival of the nervous system in both glial and neuronal cells (Ávila Rodriguez et al., 2014; Barreto et al., 2011). Neurosteroids are promising molecules that provide neuroprotection due to their ability to modify the brain’s response to injury, modulating different processes — reactive gliosis, reactive oxygen species (ROS) production, and dysregulation of mitochondrial calcium levels, among others — involved in neurological conditions (Guennoun et al., 2015; Rodríguez-Rodríguez et al., 2014). Therefore, we examined whether raloxifene showed neuroprotection in an oxygen-glucose deprivation/reoxygenation astrocyte cell model.
2. Materials and Methods
2.1. T98G Cell Cultures
T98G cell line was used as an astrocytic cell model system (ATCC CRL-1690) (Avila-Rodriguez et al., 2016; de Joannon et al., 2000; Stein, 1979). Cells were kept under exponential growth in Dulbecco's modified Eagle's medium (DMEM) (LONZA, Walkersville, USA), containing 10% fetal bovine serum (FBS, LONZA, Walkersville, USA), and 10 U penicillin/10 μg streptomycin/25 ng amphotericin (LONZA, Walkersville, USA). The medium was changed three times a week. Cultures were incubated at 37°C in a humidified atmosphere containing 5% carbon dioxide and 95% oxygen. Cells were seeded in 96-well plates for cell death measurement, 12-well plates for flow cytometry determinations and 24-well plates for tetra-methyl rhodamine methyl ester (TMRM) and fluorescence measurements and microphotographs.
2.2. Drug Treatments
To determine raloxifene concentrations to be tested on T98G cells, dose-response curves were performed. Cells were trypsinized for 3 minutes at 37ºC and DMEM medium was added for trypsin inactivation; the cells were transferred to a 15 mL centrifuge tube and centrifuged at 1700 rpm for 5 minutes. The supernatant was discarded, and the cell pellet was resuspended in 1 mL of medium. Around 100,000 cells/well were seeded in a 24-well plate with a final volume of 500 μL/ well. The plate was incubated at 37ºC and 5% CO2 until reaching 80% confluence. Next, the cell medium was replaced with serum-free DMEM and incubated for 12 hours. Once this time was completed, the cells were treated with a DMEM medium without glucose supplemented with raloxifene at concentrations of 1, 10, and 100 nM during the metabolic insult hours of glucose and oxygen deprivation, as well as during the reoxygenation period. Neurosteroid solutions were prepared by diluting a 100 μM stock drug solution in DMSO in a glucose-free DMEM medium to achieve a concentration of 0.0001% of the vehicle.
2.3. Oxygen and Glucose Deprivation
To induce oxygen-glucose deprivation, T98G cells were first washed three times with a glucose-free DMEM solution and then treated with glucose-free DMEM and incubated at 37ºC in a hypoxia chamber (Stemcell) with 1% O2 for 3, 6, 12, and 24 hours. Control cultures were treated with a DMEM medium under normoxic conditions for the same incubation times. Then, the hypoxic cells were subjected to reoxygenation under normoxic conditions with both oxygen and glucose.
2.4. Cell Viability Assessment
Cell viability was assessed using the MTT (3- (4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium) assay (Sigma, St Louis, Missouri, USA). Cells were seeded in 96-well plates in DMEM culture medium containing 10% fetal bovine serum at a seeding density of 10,000 cells per well and incubated for 2-3 days until reaching confluence. Subsequently, cells were treated according to different experimental schemes. Viability was assessed after oxygen and glucose deprivation/ reoxygenation (OGD/R) by adding an MTT solution at a final concentration of 5 mg/ml, for 4 hours at 37°C. Cells were then lysed by the addition of dimethyl sulfoxide (DMSO). The resulting blue formazan product was measured using a plate reader at 595 nm (spectrophotometer). The values were normalized to the control cultures without oxygen and glucose deprivation, which were considered as 100% cell survival. Each assay was performed with a minimum of 18 replicated wells for each condition, with three replicates per experiment. Cell morphology was qualitatively analyzed using a phase contrast microscope to evaluate cell shape changes after OGD/R insult in T98G cells (Fluorescence NIKON Eclipse Ti-E PFS).
2.5. Mitochondrial Membrane Potential Determination
Mitochondrial membrane potential was measured by flow cytometry using Tetramethyl Rhodamine Methyl Ester (TMRM). TMRM is a fluorescent cationic probe that permeates the cell and is taken up by active mitochondria (Scaduto & Grotyohann, 1999). Following treatment with neurosteroids and OGD/reoxygenation, T98G cells were incubated with a 500 nM TMRM solution in the absence of light at 37°C for 20 minutes. After the incubation period, the probe was removed, and the cells were washed with 1X PBS three times to remove residual TMRM. Cells treated with 10 nM of the uncoupling protonophore carbonyl cyanide m-chlorophenyl hydrazone (CCCP, Sigma) were used as mitochondrial oxidative phosphorylation inhibition experimental control. CCCP dissipates the mitochondrial membrane potential, providing a baseline for analysis. The quantitative analysis was evaluated by flow cytometry (Becton Dickinson FACS Calibur cytometer), and the cells were observed using fluorescence microscopy, and photomicrographs were taken. The experiment was performed in quintuplicate.
2.6. Reactive Oxygen Species (ROS) Production Determination
Hydrogen peroxide (H2O2) and superoxide production in cells exposed to OGD was measured using 2',7'-Dichlorofluorescein Diacetate (DCFDA) 1 nM and Dihydroethidium (DHE), respectively. Cells were incubated with the compounds at 37°C for 30 minutes in the dark, washed with 1X PBS, and trypsinized for flow cytometry analysis (Becton Dickinson FACS Calibur cytometer). Each assay was performed with six replicates for each condition and three repetitions. The cells were observed using fluorescence microscopy, and photomicrographs were taken. The experiment was performed in quintuplicate.
2.7. Mitochondrial Mass Determination
Mitochondrial mass was determined using Acridine Orange Nonyl (NAO) and quantitative analysis by flow cytometry and fluorescence microphotographs. After completion of the OGD insult and reoxygenation time, cells were washed three times with 1X PBS. Then, cells were trypsinized and transferred to centrifuge tubes with fetal bovine serum to inactivate the enzyme. The cells were centrifuged at 4500 rpm for 5 minutes and resuspended in a solution containing the light-protected NAO compound. Qualitative analysis was also performed by flow cytometry analysis (Becton Dickinson FACS Calibur cytometer). Cells were fixed with 4% paraformaldehyde (PFA) as described previously (Toro-Urrego et al., 2016) and stained with NAO. The cells were observed using fluorescence microscopy, and photomicrographs were taken. The experiment was performed in quintuplicate.
2.8. Statistical Analysis
The Kolmogorov-Smirnov test and Levene’s test were used to test data normal distribution and homogeneity of variance, respectively. Data were examined by analysis of variance, followed by the post hoc Dunnet’s test for between-group comparisons and Tukey’s test for multiple comparisons. Data are expressed as the mean ± SEM. A statistically significant difference was set at p < 0.05.
3. Results
3.1. Cell Viability
The MTT assay was used to examine the effect of raloxifene on OGD/R-exposed T98G cells’ viability. Figure 1A shows cell viability when cells were co-treated with OGD and raloxifene at different concentrations (during injury). Cells were treated with raloxifene since they were subjected to oxygen and glucose deprivation until the end of the reoxygenation period. Cells exposed to OGD for 6 hours followed by reoxygenation for 3 hours showed 65.34% (p = 0.0021) and 70.56% (p < 0.0001) recovery with100 nM and 10 nM of raloxifene, respectively compared with the untreated control cells (Figure 1A).
In this model, cell viability assessed with MTT decreased from the early hours of OGD/reoxygenation, progressively decreasing from 3 hours to 24 hours of OGD/ 24-hour reoxygenation (data not shown). Approximately 50% of the damaged cells died after 24-hour OGD/24-hour reoxygenation (data not shown).
3.2. Cell Morphology
Cells’ viability and morphology are directly related. We performed a qualitative analysis using phase contrast microscope to evaluate cell shape changes after OGD/R insult in T98G cells (Figure 1B-D). OGD/R insult resulted in cellular shrinkage, vacuolization, and a decrease in the number of processes (Figure 1C). Co-treatment with raloxifene decreased cell degeneration (Figure 1D), showing astrocytic-like morphology comparable to control cells (Figure 1B).
3.3. Raloxifene Reduced Superoxide Production in OGD-Exposed T98G Cells
Flow cytometry analysis using dihydroethidium (DHE) was used to measure the effect of treatment with raloxifene on superoxide anion (O2•–) production in OGD-exposed cells. Dihydroethidium (DHE) is a cell-permeable compound that interacts with the superoxide anion O2•– to form ethidium, which binds to nucleic acids and emits bright red fluorescence (Tarpey et al., 2004).
Figure 2 shows OGD/R effect on superoxide (O2•–) production in T98G cells with and without raloxifene. OGD/R increased O2•– production compared with control cells (p<0.0001). The OGD/R-raloxifene group showed decreased O2•– production compared with the OGD/R group (p<0.0001). Qualitative analysis was performed on DHE-stained cells. Fluorescence microphotographs confirmed the results obtained by flow cytometry (Figure 2C-E. The OGD/R group showed a large increase in fluorescence intensity compared with control cells (Figure 2D,C). Raloxifene treatment largely decreased fluorescence intensity compared with OGD/R cells (Figure 2E,C). This suggests that raloxifene reduced reactive oxygen species (ROS) production and oxidative stress in this OGD/R model.
We measured hydrogen peroxide production to confirm reactive oxygen species (ROS) production decrease by raloxifene in OGD/R-exposed T98G cells, using another dye. DCFDA (Dichlorofluorescein diacetate) is an organic dye of the fluorescein family that emits fluorescence after oxidation by hydrogen peroxide and other ROS (Cristóvão et al., 2009). Using DCFDA, the mean fluorescence intensity was measured in cell culture microphotographs (Figure 3).
3.4. Raloxifene Effect on Mitochondrial Membrane Potential Loss in Reperfused OGD-Exposed T98G Cells
To evaluate the effect of raloxifene on mitochondrial membrane potential (Δψm), a quantitative analysis was performed using flow cytometry (Figure 4A). Δψm was measured using the lipophilic cation tetramethylrhodamine methyl ester (TMRM), a dye that changes its intensity in response to Δψm changes (Scaduto and Grotyohann, 1999). Figure 4 shows OGD/R and raloxifene effects on T98G cells’ Δψm. Mitochondrial membrane potential loss (p= 0.0006) was observed after 6 hours of OGD followed by 3 hours of reoxygenation and was attenuated by 10 nM raloxifene treatment (p = 0.0180) compared with untreated OGD/R-exposed cells (Figure 4A). Fluorescence microphotographs showed a similar pattern with decreased fluorescence staining after OGD/R exposure and intensity recovery after raloxifene treatment (Figure 4C-E).
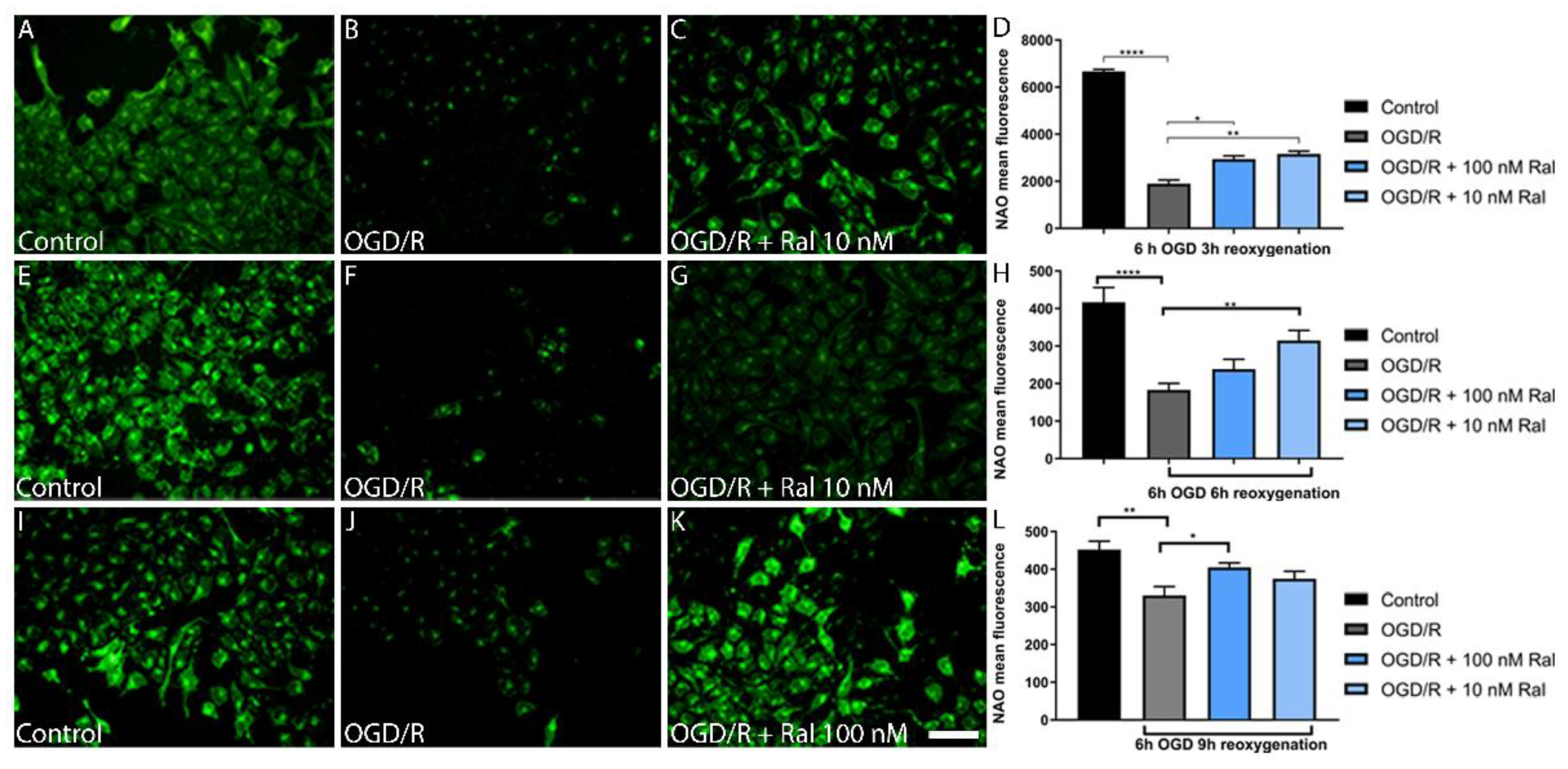
3.5. Raloxifene Attenuated Mitochondrial Mass Reduction in OGD/R-Exposed T98G Cells
To determine the OGD/R effect on mitochondrial mass, a quantitative analysis was performed using nonyl acridine orange (NAO) and flow cytometry (Figure 5). NAO is a marker of the mitochondrial membrane surface and measures mitochondrial lipid peroxidation by detecting cardiolipin oxidation (Oliva et al., 2011).
OGD/R resulted in mitochondrial mass reduction in OGD/R-exposed T98G cells compared with control cells (p <0.0001) exposed to 6h OGD and 3 h reoxygenation (Figure 5D). This change was counteracted by cotreatment with 100 nM (p=0.0349) and 10 nM (p=0.0038) raloxifene, which preserved mitochondrial mass (Figure 5D). Fluorescence microphotographs confirmed a similar pattern using the NAO dye. OGD/R exposure decreased fluorescence intensity compared with control cells, while treatment with raloxifene preserved fluorescence intensity (Figure 5 A, B, and C).
To understand lipid peroxidation dynamics, mitochondrial mass changes were measured after 6 hours of OGD using fluorescence microphotographs, followed by different reoxygenation periods (Figure 5D, H, L). Figure 5 shows intensity loss in OGD/R-exposed cells and raloxifene corrective effect at different reoxygenation times between 6 hours (Figure 5H) and 9 hours (Figure 5L).
Qualitative studies measuring fluorescence intensity in the microphotographs confirmed as observed (Figure 5). 10Nm (p=0.0073) and 100 Nm (p= 0.0468) raloxifene attenuated mitochondrial mass loss at different reoxygenation times (6 and 9 h respectively) in 6-hour OGD-exposed cells (Figure 5h,L).
4. Discussion
The study focused on characterizing the astrocytic T98G-OGD/R model. Careful observation of the events occurring during OGD rendered noteworthy evidence on cell viability, lipid peroxidation, reactive species production, and mitochondrial function.
In vitro experimentation is crucial due to the methodological limitations of in vivo models (Toro-Urrego et al., 2019). Therefore, the choice of an in vitro model that meets the specific requirements of the parameters to be evaluated is of paramount importance. Models using hypoxia-simulating agents are among the in vitro approaches. These are based on producing, at the molecular level, the effects caused by low oxygen concentrations, primarily those based on the expression of hypoxia-inducible factor 1α (HIF1A) (Toro-Urrego et al., 2019). However, they do not consider removing glucose, the most important energy substrate in cerebral energy metabolism.
Previous studies have shown that estrogen activity in astrocytes contributes to its neuroprotective effects (Dhandapani & Brann, 2007; Spence et al., 2011; Wang et al., 2014). Raloxifene protective effects have been reported in various models of brain injury: schizophrenia, excitotoxic neuronal death (Karki et al., 2014) glucose deprivation (Vesga-Jiménez et al., 2019), among others Raloxifene is a neurosteroid that can modify various metabolic and genomic responses in the brain and has shown neuroprotective effects in different pathologies (Felgel-Fernholz et al., 2023; Karki et al., 2014; Vesga-Jiménez et al., 2019). Neuroprotective effects have been observed in murine models of common carotid artery ligation, where neurogenesis in the ipsilateral subventricular zone has been observed in rats treated with raloxifene (Khan et al., 2015). Raloxifene treatment has shown neuroprotective effects, mitigating cell death. 100 nM of raloxifene has shown protective effects in metabolic insults of cerebral injury in astrocytic models with T98G cells (Vesga-Jiménez et al., 2019). The findings in this study agree with previous observations. Cotreatment with 100 nM raloxifene reduced OGD-exposed cell death after reoxygenation (Figure 1). Different molecular mechanisms may mediate the reduction in cell death (Mergenthaler et al., 2013; Nematipour et al., 2020; Thornton & Hagberg, 2015; H. F. Wang et al., 2018; Wassink et al., 2014; Yao et al., 2007), which may explain the results observed in the study.
In astrocytic models, raloxifene has been found to regulate glutamate transporters (GLAST and GLT-1), improving cell survival (Karki et al., 2014). Noteworthy, its protective effects might involve the expression of Bcl-2, an anti-apoptotic gene (Bourque et al., 2014). Through non-genomic mechanisms, raloxifene can activate survival proteins like MAPk, PI3K/Akt, Src, and CREB, which are related to anti-apoptotic metabolic pathways and neuroprotection (Acaz-Fonseca et al., 2016; Bourque et al., 2014; Karki et al., 2014).
Changes in cell morphology have been associated with cell viability, in particular, the state in which the cell is found. Cells exhibit different characteristics during apoptosis, including nuclear envelope disintegration, cytoplasmic condensation, and surface reduction (Xiong et al., 2006; Ziegler & Groscurth, 2004), parameters mostly affecting cell morphology. These changes align with the findings in this study (Figure 1B-D), where raloxifene decreases morphological alterations caused by glucose and oxygen deprivation, showing a potential reduction in apoptotic cellular processes.
In our model, superoxide and hydrogen peroxide production was detected from 6 hours of OGD/R injury onwards (Figure 2 and Figure 3). In physiological conditions, these species are produced in regulated concentrations and are transformed into less reactive species through energy-dependent mechanisms (Bertero & Maack, 2018; De Nicolo et al., 2023; Görlach et al., 2015; Ryan et al., 2023). Energy shortage hinders the transformation of these species into a less reactive form, triggering damage to cellular machinery. Therefore, this parameter is a potential indicator of damage that, if reversed, could explain neuroprotective effects. ROS production depends on Δψm (Murphy, 2009; Zorov et al., 2014). Maintaining an optimal mitochondrial membrane potential (maximum 140 mV) prevents ROS formation and exhaustively uses ATP production capacity.
Reactive oxygen species can be generated at low Δψm levels under certain conditions. Yet, ROS are produced in excess when mitochondrial membrane potential levels are high and Δψm exceeds 140 mV (membrane hyperpolarization). In these conditions, ROS production in mitochondrial respiratory complexes I (NADH: ubiquinone oxidoreductase) and III (ubiquinol–cytochrome c oxidoreductase) increases exponentially (Fang et al., 2022) and an increase in oxidative stress could generate significant morphological changes. This corresponds to the results presented in this study, where an exponential increase in superoxide anion production at hour 6 of OGD/R (Figure 2A) and H2O2 at hour 6 (Figure 3D) is observed. These periods match the highest peaks of mitochondrial membrane potential production in glucose deprivation insults in T98G cells, as reported in previous studies (Ávila Rodriguez et al., 2014; Toro-Urrego et al., 2016). This phenomenon is attributed to the mitochondria’s need to adapt to the cell’s energy needs, regardless of the deleterious increase in mitochondrial membrane potential. The cell enters mitochondrial apoptotic characterized by excessive calcium release, Δψm hyperpolarization, and exacerbated ROS production (Kadenbach et al., 2004; Z. Zhang et al., 2023).
The collected data revealed a significant alteration in mitochondrial potential at 6 hours of OGD and 3 hours of reoxygenation (Figure 4). This loss of potential coincides with the loss of viability described earlier. In different models, this potential has been found to increase to a peak within the first few hours of damage and then decrease gradually until a complete loss, matching cell death time (Ávila Rodriguez et al., 2014; Toro-Urrego et al., 2016; Vesga-Jiménez et al., 2019).
While hydrogen peroxide is fairly less reactive, it can form different reactive hydroxyl radicals in the presence of iron ions, starting lipid peroxidation cascades in the cell membrane. This oxidative stress is a major problem in neurodegenerative diseases and one of their major causes (Stockwell et al., 2017; Z. Zhang et al., 2023). Neurosteroids have shown the ability to attenuate these oxidative stress processes, decreasing ROS levels, in injuries caused by oxidative stress in particular (Hidalgo-Lanussa et al., 2018; Stark et al., 2015). This aligns with the results reported by Capani et al. (2001). In a perinatal asphyxia model, they found increased H2O2 production — likely related to an increase in the dynamics of superoxide transfer and transformation — preventing the harmful accumulation of this more dangerous species. Superoxide species conversion to less reactive forms like hydrogen peroxide and water, helps cells achieve a more stable physiological state during injury (Stark et al., 2015; Yuan et al., 2016). However, this ROS increase, like membrane hyperpolarization, is not sustained throughout the injury period. As cellular energy levels have decreased because of cellular energy shortage, the cell has shifted to other energy sources like fatty acid oxidation or glycogenolysis, in the early hours of insult (Korenic et al., 2014), which may explain the changes in ROS production in this study.
The mitochondrial role in cell death stands on the control of energy metabolism, ROS production, and the release of apoptotic factors into the cytoplasm. Cytochrome C is the most prominent pro-apoptotic factor (Toro-Urrego et al., 2020), and mitochondrial apoptosis-inducing proteins like endonuclease G, Smac/DIABLO, and Omi/HtrA have also been described to play a significant role in apoptosis regulation. These pro-apoptotic factors are not released necessarily through mitochondrial permeability transition pores (MPT). This suggests that changes in Δψm are directly related to cellular necrosis and apoptosis (Jacobson & Duchen, 2002). Our results showed raloxifene preserved mitochondrial membrane potential in cells exposed to oxygen and glucose deprivation, in agreement with Vesga-Jiménez et al. (2019) that reported raloxifene protected astrocytes from oxidative stress.
One of the most critical targets is cardiolipin, a phospholipid found in the inner mitochondrial membrane. It is crucial for the insertion into the membrane and the function of cytochrome C, cytochrome C oxidase, and other phosphorylation complexes. It is required for the optimal functioning of complex I (NADH: ubiquinone reductase), complex III (NADH: ubiquinone cytochrome C oxidoreductase), complex IV (cytochrome C oxidase), and complex V (ATP synthase). Changes in phospholipid structure can lead to mitochondrial dysfunction, as the integrity of cardiolipin depends on it. However, its high content of fatty acids makes it susceptible to ROS-induced damage (Anthonymuthu et al., 2016; Vähäheikkilä et al., 2018; J. Zhang & Shi, 2022). Considering the above, the findings in this study may help understand the reasons behind the mitochondrial dysfunction observed.
Figure 5 shows how OGD/R affected T98G cells’ mitochondrial mass at different insult-exposure times. Raloxifene preserved cells from mitochondrial mass loss, disclosing another edge of its neuroprotective effects. The decrease in ROS production has been found to mediate this effect (J. Zhang & Shi, 2022).
In sum, understanding whether and how OGD/R damage contributes to disease could explain why hypoxic-ischemic brain injury is a complex concurrence of simultaneous processes, and how it may be approached to enhance the therapeutic effects of neuroprotective treatments. The outcome of this part of the study shows that raloxifene exerted neuroprotective effects on T98G astrocytic cells exposed to hypoxia-reoxygenation injury. The survival of these cells in the OGD/R model suggests astrocytes might participate in the protective effects of these neurosteroids in hypoxia-ischemia injury. Further studies will determine if the present findings may be extrapolated to an in vivo model.
Author Contributions
Conceptualization, NTU, JPL and FC; methodology, NTU, JPL and SF; formal analysis, NTU, JPL and SF; investigation NTU, JPL and SF; writing—original draft preparation, NTU, JPL and SF; writing—review and editing, MO-L.; visualization, MO-L; supervision, MO-L and FC; funding acquisition, FC. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded Consejo Nacional de Investigaciones Científicas y Técnicas. (CONICET) (UBACYT 2014- 2017), PIP CONICET (2015-2018), and PICT- ANPCYT 2018-2021.
Institutional Review Board Statement
Not applicable as the study involved no humans or animals.
Informed Consent Statement
Not applicable as the study involved no humans.
Data Availability Statement
Research data are available and shared in this study.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Acaz-Fonseca, E., Avila-Rodriguez, M., Garcia-Segura, L. M., & Barreto, G. E. (2016). Regulation of astroglia by gonadal steroid hormones under physiological and pathological conditions. Progress in Neurobiology, 144, 5–26. [CrossRef] [PubMed]
- Anthonymuthu, T. S., Kenny, E. M., & Bayır, H. (2016). Therapies targeting lipid peroxidation in traumatic brain injury. Brain Research, 1640(Pt A), 57–76. [CrossRef] [PubMed]
- Avila-Rodriguez, M., Garcia-Segura, L. M., Hidalgo-lanussa, O., Baez, E., Gonzalez, J., & Barreto, G. E. (2016). Tibolone protects astrocytic cells from glucose deprivation through a mechanism involving estrogen receptor beta and the upregulation of neuroglobin expression. Molecular and Cellular Endocrinology, 433, 35–46. [CrossRef] [PubMed]
- Ávila Rodriguez, M., Garcia-Segura, L. M., Cabezas, R., Torrente, D., Capani, F., Gonzalez, J., & Barreto, G. E. (2014). Tibolone protects T98G cells from glucose deprivation. PART B, 294–303. https://pubmed.ncbi.nlm.nih.gov/25086299/.
- Bertero, E., & Maack, C. (2018). Calcium signaling and reactive oxygen species in Mitochondria. Circulation Research, 122(10), 1460–1478. [CrossRef]
- Bourque, M., Morissette, M., & Di Paolo, T. (2014). Raloxifene activates G protein-coupled estrogen receptor 1/Akt signaling to protect dopamine neurons in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mice. Neurobiology of Aging, 35(10), 2347–2356. [CrossRef] [PubMed]
- Capani, F., Loidl, C. F., Aguirre, F., Piehl, L., Facorro, G., Hager, A., De Paoli, T., Farach, H., & Pecci-Saavedra, J. (2001). Changes in reactive oxygen species (ROS) production in rat brain during global perinatal asphyxia: an ESR study. Brain Research, 914(1–2), 204–207. [CrossRef] [PubMed]
- Cristóvão, A. C., Choi, D. H., Baltazar, G., Beal, M. F., & Kim, Y. S. (2009). The Role of NADPH Oxidase 1–Derived Reactive Oxygen Species in Paraquat-Mediated Dopaminergic Cell Death. Antioxidants & Redox Signaling, 11(9), 2105. [CrossRef] [PubMed]
- de Joannon, A. C., Mancini, F., Landolfi, C., Soldo, L., Leta, A., Ruggieri, A., Mangano, G., Polenzani, L., Pinza, M., & Milanese, C. (2000). Adenosine triphosphate affects interleukin -1β release by T98G glioblastoma cells through a purinoceptor-independent mechanism. Neuroscience Letters, 285(3), 218–222. [CrossRef] [PubMed]
- De Nicolo, B., Cataldi-Stagetti, E., Diquigiovanni, C., & Bonora, E. (2023). Calcium and Reactive Oxygen Species Signaling Interplays in Cardiac Physiology and Pathologies. Antioxidants (Basel, Switzerland), 12(2), 353. [CrossRef] [PubMed]
- Dhandapani, K. M., & Brann, D. W. (2007). Role of astrocytes in estrogen-mediated neuroprotection. Experimental Gerontology, 42(1–2), 70–75. [CrossRef]
- Fang, M., Xia, F., Chen, Y., Shen, Y., Ma, L., You, C., Tao, C., & Hu, X. (2022). Role of Eryptosis in Hemorrhagic Stroke. Frontiers in Molecular Neuroscience, 15. [CrossRef] [PubMed]
- Felgel-Farnholz, V., Hlusicka, E. B., Edemann-Callesen, H., Garthe, A., Winter, C., & Hadar, R. (2023). Adolescent raloxifene treatment in females prevents cognitive deficits in a neurodevelopmental rodent model of schizophrenia. Behavioural Brain Research, 441. [CrossRef] [PubMed]
- Görlach, A., Bertram, K., Hudecova, S., & Krizanova, O. (2015). Calcium and ROS: A mutual interplay. Redox Biology, 6, 260–271. [CrossRef] [PubMed]
- Hidalgo-Lanussa, O., Ávila-Rodriguez, M., Baez-Jurado, E., Zamudio, J., Echeverria, V., Garcia-Segura, L. M., & Barreto, G. E. (2018). Tibolone Reduces Oxidative Damage and Inflammation in Microglia Stimulated with Palmitic Acid through Mechanisms Involving Estrogen Receptor Beta. Molecular Neurobiology, 55(7), 5462–5477. [CrossRef]
- Jacobson, J., & Duchen, M. R. (2002). Mitochondrial oxidative stress and cell death in astrocytes--requirement for stored Ca2+ and sustained opening of the permeability transition pore. Journal of Cell Science, 115(Pt 6), 1175–1188. [CrossRef]
- Kadenbach, B., Arnold, S., Lee, I., & Hüttemann, M. (2004). The possible role of cytochrome c oxidase in stress-induced apoptosis and degenerative diseases. Biochimica et Biophysica Acta (BBA) - Bioenergetics, 1655(1–3), 400–408. [CrossRef]
- Karki, P., Webb, A., Zerguine, A., Choi, J., Son, D. S., & Lee, E. (2014). Mechanism of raloxifene-induced upregulation of glutamate transporters in rat primary astrocytes. GLIA, 62(8), 1270–1283. [CrossRef] [PubMed]
- Khan, M. M., Wakade, C., De Sevilla, L., & Brann, D. W. (2015). Selective estrogen receptor modulators (SERMs) enhance neurogenesis and spine density following focal cerebral ischemia. 146, 38–47. [CrossRef]
- Korenic, A., Boltze, J., Deten, A., Peters, M., Andjus, P., & Radenovic, L. (2014). Astrocytic mitochondrial membrane hyperpolarization following extended oxygen and glucose deprivation. PloS One, 9(2). [CrossRef]
- Mergenthaler, P., Lindauer, U., Dienel, G. A., & Meisel, A. (2013). Sugar for the brain: the role of glucose in physiological and pathological brain function. Trends in Neurosciences, 36(10). [CrossRef]
- Murphy, M. P. (2009). How mitochondria produce reactive oxygen species. Biochemical Journal, 417(1), 1–13. [CrossRef]
- Nematipour, S., Vahidinia, Z., Nejati, M., Naderian, H., Beyer, C., & Tameh, A. A. (2020). Estrogen and progesterone attenuate glutamate neurotoxicity via regulation of EAAT3 and GLT-1 in a rat model of ischemic stroke. Iranian Journal of Basic Medical Sciences, 23(10), 1346–1352. [CrossRef]
- Ramos, E., Patiño, P., Reiter, R. J., Gil-Martín, E., Marco-Contelles, J., Parada, E., los Rios, C. de, Romero, A., & Egea, J. (2017). Ischemic brain injury: New insights on the protective role of melatonin. Free Radical Biology & Medicine, 104, 32–53. [CrossRef]
- Ryan, A. K., Rich, W., & Reilly, M. A. (2023). Oxidative stress in the brain and retina after traumatic injury. Frontiers in Neuroscience, 17. [CrossRef]
- Scaduto, R. C., & Grotyohann, L. W. (1999). Measurement of mitochondrial membrane potential using fluorescent rhodamine derivatives. Biophysical Journal, 76(1 Pt 1), 469. [CrossRef]
- Spence, R. D., Hamby, M. E., Umeda, E., Itoh, N., Du, S., Wisdom, A. J., Cao, Y., Bondar, G., Lama, J., Ao, Y., Sandoval, F., Suriany, S., Sofroniew, M. V., & Voskuhl, R. R. (2011). Neuroprotection mediated through estrogen receptor-alpha in astrocytes. Proceedings of the National Academy of Sciences of the United States of America, 108(21), 8867–8872. [CrossRef]
- Stark, J., Varbiro, S., Sipos, M., Tulassay, Z., Sara, L., Adler, I., Dinya, E., Magyar, Z., Szekacs, B., Marczell, I., Kloosterboer, H. J., Racz, K., & Bekesi, G. (2015). Antioxidant effect of the active metabolites of tibolone. 31(1), 31–35. [CrossRef]
- Stein, G. H. (1979). T98G: an anchorage-independent human tumor cell line that exhibits stationary phase G1 arrest in vitro. Journal of Cellular Physiology, 99(1), 43–54. [CrossRef]
- Stockwell, B. R., Friedmann Angeli, J. P., Bayir, H., Bush, A. I., Conrad, M., Dixon, S. J., Fulda, S., Gascón, S., Hatzios, S. K., Kagan, V. E., Noel, K., Jiang, X., Linkermann, A., Murphy, M. E., Overholtzer, M., Oyagi, A., Pagnussat, G. C., Park, J., Ran, Q., … Zhang, D. D. (2017). Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell, 171(2), 273–285. [CrossRef]
- Tarpey, M. M., Wink, D. A., & Grisham, M. B. (2004). Methods for detection of reactive metabolites of oxygen and nitrogen: in vitro and in vivo considerations. American Journal of Physiology. Regulatory, Integrative and Comparative Physiology, 286(3). [CrossRef]
- Thornton, C., & Hagberg, H. (2015). Role of mitochondria in apoptotic and necroptotic cell death in the developing brain. Clinica Chimica Acta; International Journal of Clinical Chemistry, 451(Pt A), 35–38. [CrossRef]
- Toro-Urrego, N., Avila-Rodriguez, M., Herrera, M. I., Aguilar, A., Udovin, L., Luaces, J. P., Toro-Urrego, N., Avila-Rodriguez, M., Herrera, M. I., Aguilar, A., Udovin, L., & Luaces, J. P. (2020). Neuroactive Steroids in Hypoxic–Ischemic Brain Injury: Overview and Future Directions. Neuroprotection - New Approaches and Prospects. [CrossRef]
- Toro-Urrego, N., Garcia-Segura, L. M., Echeverria, V., & Barreto, G. E. (2016). Testosterone Protects Mitochondrial Function and Regulates Neuroglobin Expression in Astrocytic Cells Exposed to Glucose Deprivation. Frontiers in Aging Neuroscience, 8(JUN), 152. [CrossRef]
- Toro-Urrego, N., Vesga-Jiménez, D. J. D. J., Herrera, M. I. M. I., Luaces, J. P. J. P., & Capani, F. (2019). Neuroprotective Role of Hypothermia in Hypoxic-ischemic Brain Injury: Combined Therapies using Estrogen. Current Neuropharmacology, 17(9), 874. [CrossRef]
- Vähäheikkilä, M., Peltomaa, T., Róg, T., Vazdar, M., Pöyry, S., & Vattulainen, I. (2018). How cardiolipin peroxidation alters the properties of the inner mitochondrial membrane? Chemistry and Physics of Lipids, 214, 15–23. [CrossRef]
- Vesga-Jiménez, D. J., Hidalgo-Lanussa, O., Baez-Jurado, E., Echeverria, V., Ashraf, G. M., Sahebkar, A., & Barreto, G. E. (2019). Raloxifene attenuates oxidative stress and preserves mitochondrial function in astrocytic cells upon glucose deprivation. 234(3), 2051–2057. [CrossRef]
- Wang, C., Jie, C., & Dai, X. (2014). Possible roles of astrocytes in estrogen neuroprotection during cerebral ischemia. Reviews in the Neurosciences, 25(2), 255–268. [CrossRef]
- Wang, H. F., Wang, Z. Q., Ding, Y., Piao, M. H., Feng, C. S., Chi, G. F., Luo, Y. N., & Ge, P. F. (2018). Endoplasmic reticulum stress regulates oxygen-glucose deprivation-induced parthanatos in human SH-SY5Y cells via improvement of intracellular ROS. CNS Neuroscience and Therapeutics, 24(1), 29–38. [CrossRef]
- Wassink, G., Gunn, E. R., Drury, P. P., Bennet, L., & Gunn, A. J. (2014). The mechanisms and treatment of asphyxial encephalopathy. 8(8 FEB), 40. https://pubmed.ncbi.nlm.nih.gov/24578682/.
- Xiong, J., Dai, W., Chen, L., Liu, G., Liu, M., Zhang, Z., & Xiao, H. (2006). New method for studying the relationship between morphological parameters and cell viability. 6150, 828–832. [CrossRef]
- Yao, M., Nguyen, T. V. v., & Pike, C. J. (2007). Estrogen Regulates Bcl-w and Bim Expression: Role in Protection against β-Amyloid Peptide-Induced Neuronal Death. Journal of Neuroscience, 27(6), 1422–1433. [CrossRef]
- Yuan, L., Dietrich, A. K., Ziegler, Y. S., & Nardulli, A. M. (2016). 17β-Estradiol alters oxidative damage and oxidative stress response protein expression in the mouse mammary gland. Molecular and Cellular Endocrinology, 426, 11–21. [CrossRef]
- Zhang, J., & Shi, Y. (2022). In Search of the Holy Grail: Toward a Unified Hypothesis on Mitochondrial Dysfunction in Age-Related Diseases. Cells, 11(12). [CrossRef]
- Zhang, Z., Huang, Q., Zhao, D., Lian, F., Li, X., & Qi, W. (2023). The impact of oxidative stress-induced mitochondrial dysfunction on diabetic microvascular complications. Frontiers in Endocrinology, 14, 1112363. [CrossRef]
- Ziegler, U., & Groscurth, P. (2004). Morphological features of cell death. News in Physiological Sciences, 19(3), 124–128. [CrossRef]
- Zorov, D. B., Juhaszova, M., & Sollott, S. J. (2014). Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol Rev, 94, 909–950. [CrossRef]
Figure 1.
Raloxifene decreases OGD-induced cell death. A) T98G cells were treated with different concentrations of raloxifene during 6 h of OGD and 3 h of reoxygenation, and cell viability was assessed by MTT assay. Data are represented as the mean ± SEM of 4 independent experiments. Control (101.99 ± 1.85); OGD/R (52.59 ± 2.02); OGD/R + 100nM raloxifene (65.34 ± 2.03); OGD/R + 10nM raloxifene (70.56 ± 2.36) *P<0.005. B-D) Raloxifene diminishes morphological alterations induced by oxygen-glucose deprivation/reoxygenation. Representative microphotographs showing the morphology of cells exposed to B) DMEM, C) OGD/R, and D) OGD/R + Ral 100 nM. Scale bar 50 µm.
Figure 1.
Raloxifene decreases OGD-induced cell death. A) T98G cells were treated with different concentrations of raloxifene during 6 h of OGD and 3 h of reoxygenation, and cell viability was assessed by MTT assay. Data are represented as the mean ± SEM of 4 independent experiments. Control (101.99 ± 1.85); OGD/R (52.59 ± 2.02); OGD/R + 100nM raloxifene (65.34 ± 2.03); OGD/R + 10nM raloxifene (70.56 ± 2.36) *P<0.005. B-D) Raloxifene diminishes morphological alterations induced by oxygen-glucose deprivation/reoxygenation. Representative microphotographs showing the morphology of cells exposed to B) DMEM, C) OGD/R, and D) OGD/R + Ral 100 nM. Scale bar 50 µm.
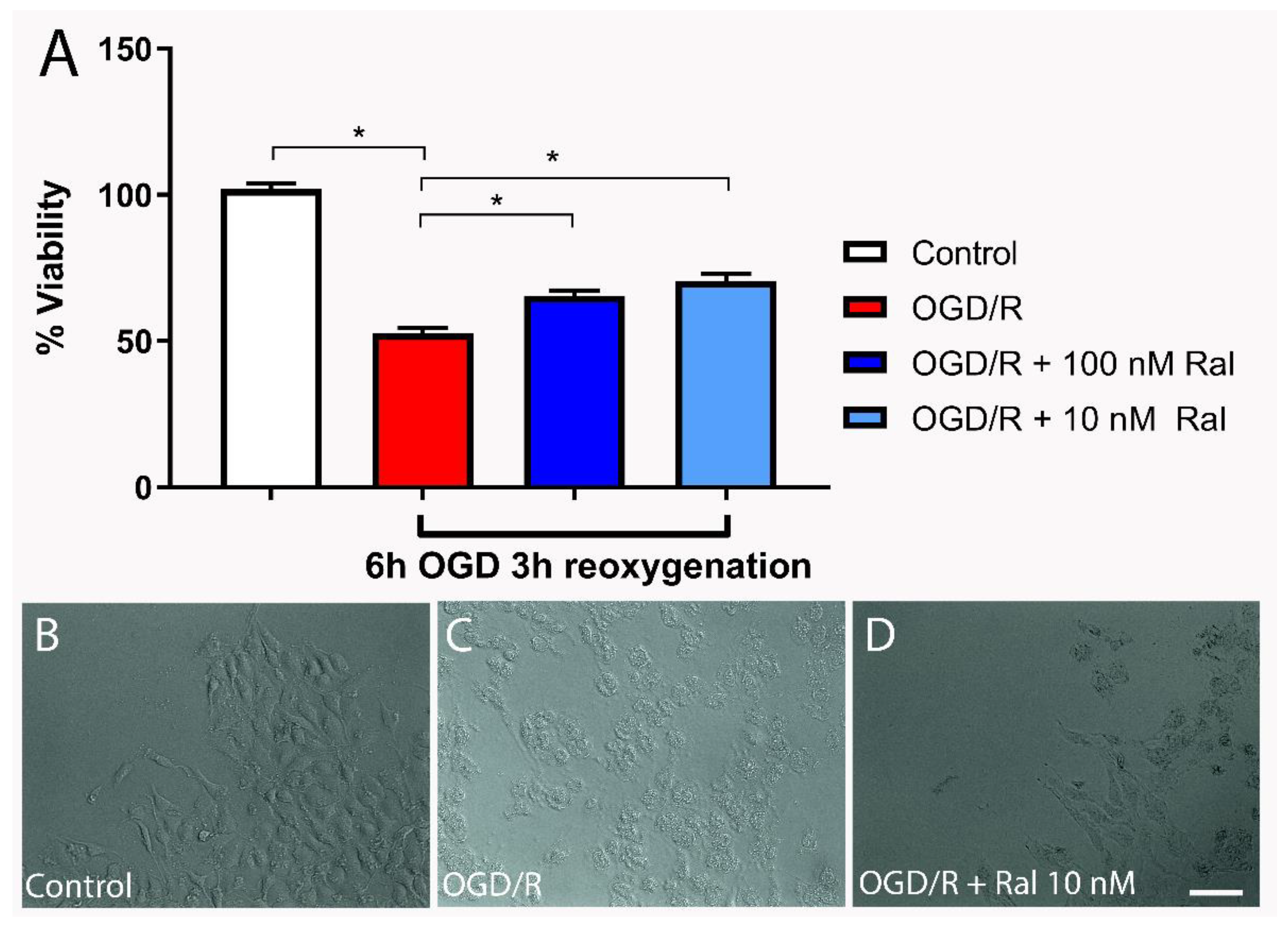
Figure 2.
Raloxifene reduces superoxide production at 6 h of OGD and 3 h of reoxygenation. A) Mean fluorescence values of dihydroethidium (DHE) intensity and B) representative flow cytometry plots are shown. Data are represented as the mean ± SEM of 5 independent experiments. Control (5028.58 ± 424.80); OGD/R (39510.50 ± 1718.64); OGD/R + 100nM raloxifene (28734.70 ± 1856.91); OGD/R + 10nM raloxifene (28538.67 ± 1017.65) ***P<0.0001. C-E) Representative fluorescence micrographs of dihydroethidium (DHE) staining in T98G cells exposed to C) DMEM, D) OGD/R, and E) OGD/R + Ral 100 nM with 6 h of OGD and 3 h of reoxygenation. Scale bar 50 µm.
Figure 2.
Raloxifene reduces superoxide production at 6 h of OGD and 3 h of reoxygenation. A) Mean fluorescence values of dihydroethidium (DHE) intensity and B) representative flow cytometry plots are shown. Data are represented as the mean ± SEM of 5 independent experiments. Control (5028.58 ± 424.80); OGD/R (39510.50 ± 1718.64); OGD/R + 100nM raloxifene (28734.70 ± 1856.91); OGD/R + 10nM raloxifene (28538.67 ± 1017.65) ***P<0.0001. C-E) Representative fluorescence micrographs of dihydroethidium (DHE) staining in T98G cells exposed to C) DMEM, D) OGD/R, and E) OGD/R + Ral 100 nM with 6 h of OGD and 3 h of reoxygenation. Scale bar 50 µm.
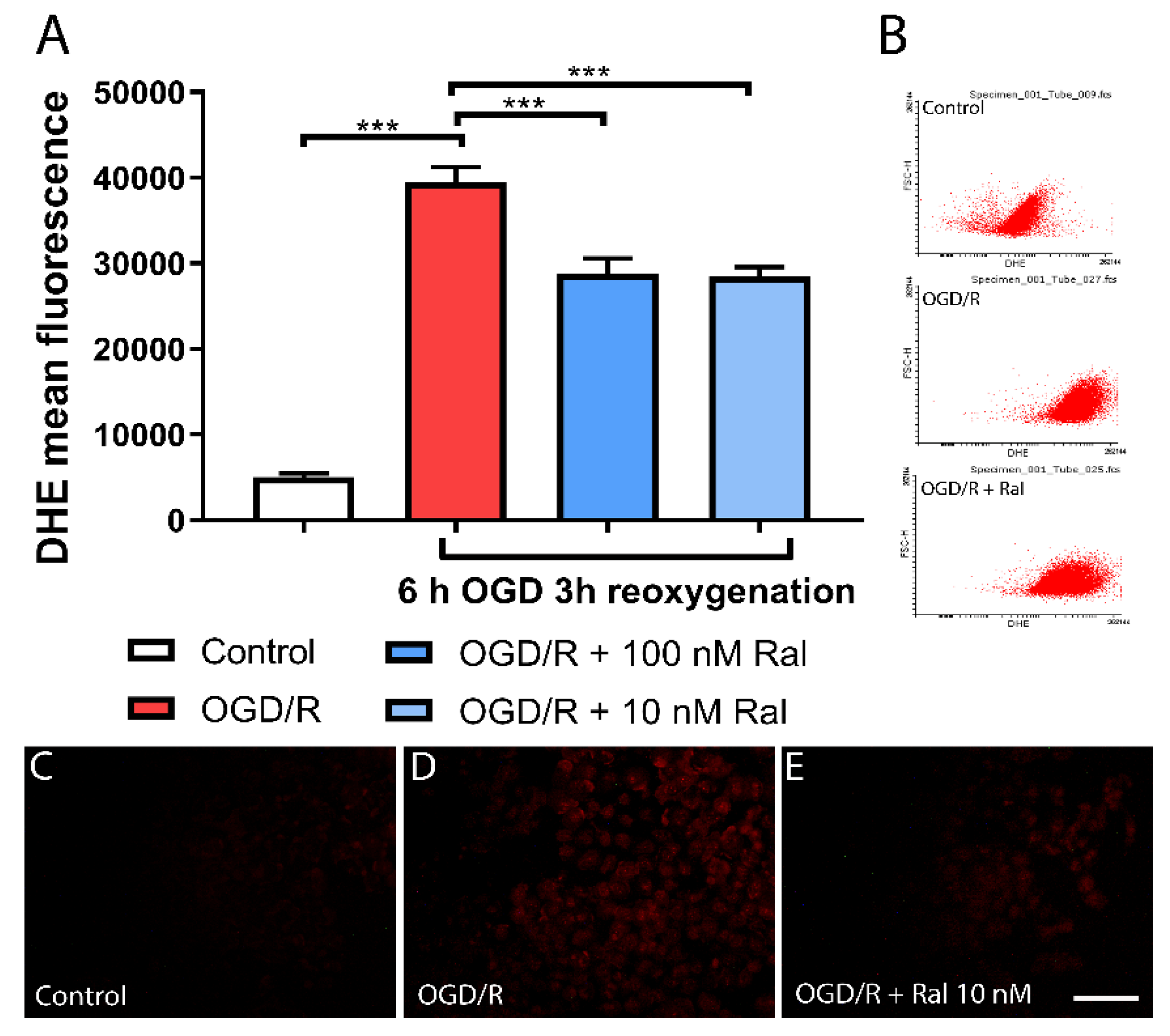
Figure 3.
Raloxifene reduces peroxide production at 6 h of OGD and 6 h of reoxygenation. The Figure shows the representative fluorescence microphotographs of 2′,7′-Dichlorofluorescin Diacetate (DCFDA) staining of T98G cells exposed to A) DMEM, B) OGD/R, OGD/R + Ral 100nM with 6 h of OGD and 6 h of reoxygenation, and D) the mean fluorescence values of DCFDA intensity measured by flow cytometry. Data are represented as the mean ± SEM of 5 independent experiments. Control (55.51 ± 1.03); OGD/R (131.00 ± 4.01); OGD/R + 100nM raloxifene (75.15 ± 6.60) ****P<0.0001. Scale bar 50 µm.
Figure 3.
Raloxifene reduces peroxide production at 6 h of OGD and 6 h of reoxygenation. The Figure shows the representative fluorescence microphotographs of 2′,7′-Dichlorofluorescin Diacetate (DCFDA) staining of T98G cells exposed to A) DMEM, B) OGD/R, OGD/R + Ral 100nM with 6 h of OGD and 6 h of reoxygenation, and D) the mean fluorescence values of DCFDA intensity measured by flow cytometry. Data are represented as the mean ± SEM of 5 independent experiments. Control (55.51 ± 1.03); OGD/R (131.00 ± 4.01); OGD/R + 100nM raloxifene (75.15 ± 6.60) ****P<0.0001. Scale bar 50 µm.
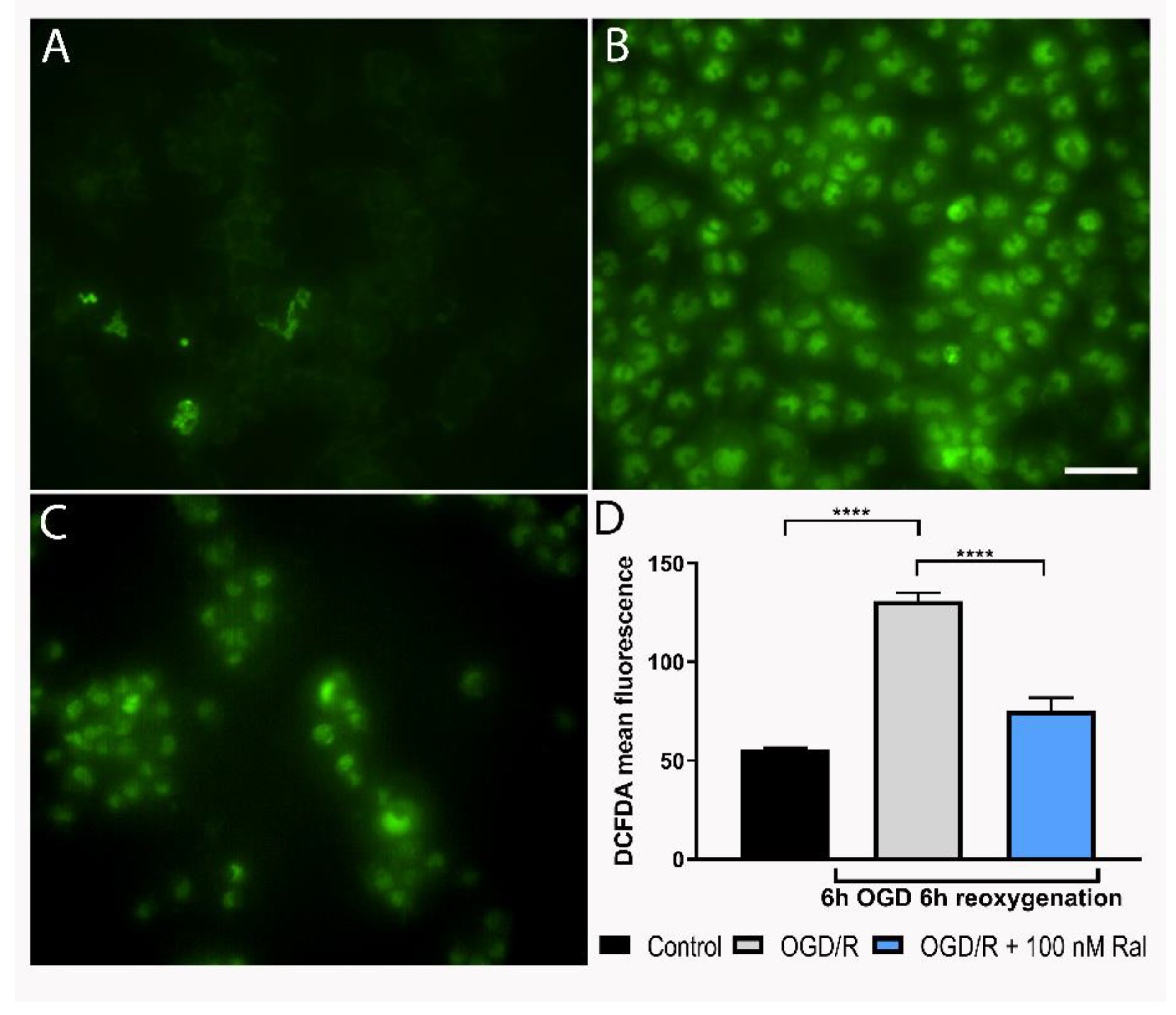
Figure 4.
Raloxifene attenuates mitochondrial membrane potential loss at 6 h of OGD and 3 h of reoxygenation. A) The Figure shows the mean fluorescence values and B) the representative flow cytometry plots of tetramethylrhodamine methyl ester (TMRM) intensity. Data are represented as the mean ± SEM of 5 independent experiments. Control (102.60 ± 7.43); OGD/R (9.86 ± 2.88); OGD/R + 100nM raloxifene (60.44 ± 14.92); OGD/R + 10nM raloxifene (83.70 ± 16.84); Carbonyl cyanide 3-chlorophenylhydrazone (cccp) (24.58 ± 13.58) ***P<0.0001 C-E). Representative fluorescence micrographs of tetra-methyl rhodamine methyl ester (TMRM) staining in T98G cells exposed to C) DMEM, D) OGD/R, and E) OGD/R + Ral 100 nM with 6 h of OGD and 3 h of reoxygenation. Scale bar 50 µm.
Figure 4.
Raloxifene attenuates mitochondrial membrane potential loss at 6 h of OGD and 3 h of reoxygenation. A) The Figure shows the mean fluorescence values and B) the representative flow cytometry plots of tetramethylrhodamine methyl ester (TMRM) intensity. Data are represented as the mean ± SEM of 5 independent experiments. Control (102.60 ± 7.43); OGD/R (9.86 ± 2.88); OGD/R + 100nM raloxifene (60.44 ± 14.92); OGD/R + 10nM raloxifene (83.70 ± 16.84); Carbonyl cyanide 3-chlorophenylhydrazone (cccp) (24.58 ± 13.58) ***P<0.0001 C-E). Representative fluorescence micrographs of tetra-methyl rhodamine methyl ester (TMRM) staining in T98G cells exposed to C) DMEM, D) OGD/R, and E) OGD/R + Ral 100 nM with 6 h of OGD and 3 h of reoxygenation. Scale bar 50 µm.
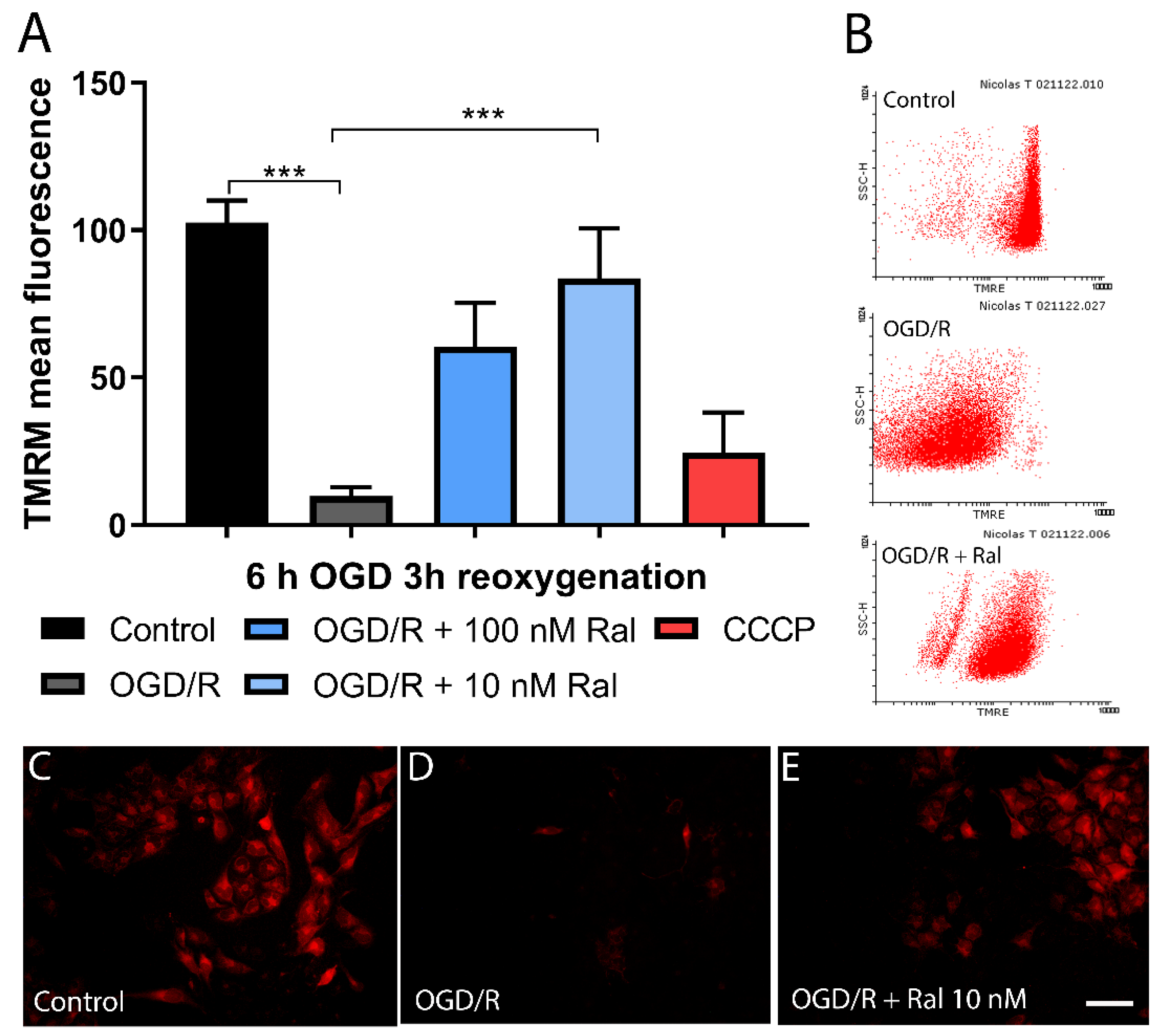
Figure 5.
Raloxifene preserves mitochondrial mass in T98G cells exposed to 6 hours of OGD and 3 hours of reoxygenation. The Figure shows the mitochondrial mass in T98G cells exposed to 6 hours of oxygen-glucose deprivation (OGD) to 3 h (A-D), 6 h (E-H) and 9 h (I-L) of reoxygenation. The representative microphotographs of acridine orange (NAO) fluorescence in T98G astrocytic cells exposed to A) DMEM, B) OGD/R, and C) OGD/R + Ral 100 nM with 3 hours of reoxygenation. D) Mean fluorescence values of NAO intensity in this period of insult. Data are represented as the mean ± SEM of 5 independent experiments. Control (6671.00 ± 86.18); OGD/R (1903.00 ± 155.30); OGD/R + 100nM raloxifene (2940.00 ± 142.90); OGD/R + 10nM raloxifene (3163.00 ± 119.80). E) DMEM, F) OGD/R, and G) OGD/R + Ral 100 nM with 6 hours of reoxygenation. H) Mean fluorescence values of NAO intensity in this period of insult. Data are represented as the mean ± SEM of 5 independent experiments. Control (416.7.00 ± 39.47); OGD/R (183.1 ± 17.70); OGD + 100nM raloxifene (238.4 ± 26.43); OGD + 10nM raloxifene (314.6 ± 27.45) I) DMEM, J) OGD/R, and K) OGD/R + Ral 10 nM with 9 hours of reoxygenation. L) Mean fluorescence values of NAO intensity in this period of insult. Data are represented as the mean ± SEM of 5 independent experiments. Control (452.2 ± 22.28); OGD/R (330.4 ± 23.45); OGD/R + 100nM raloxifene (404.7 ± 12.34); OGD/R + 10nM raloxifene (374.6 ± 19.78). *P<0.005. Scale bar 50 µm.
Figure 5.
Raloxifene preserves mitochondrial mass in T98G cells exposed to 6 hours of OGD and 3 hours of reoxygenation. The Figure shows the mitochondrial mass in T98G cells exposed to 6 hours of oxygen-glucose deprivation (OGD) to 3 h (A-D), 6 h (E-H) and 9 h (I-L) of reoxygenation. The representative microphotographs of acridine orange (NAO) fluorescence in T98G astrocytic cells exposed to A) DMEM, B) OGD/R, and C) OGD/R + Ral 100 nM with 3 hours of reoxygenation. D) Mean fluorescence values of NAO intensity in this period of insult. Data are represented as the mean ± SEM of 5 independent experiments. Control (6671.00 ± 86.18); OGD/R (1903.00 ± 155.30); OGD/R + 100nM raloxifene (2940.00 ± 142.90); OGD/R + 10nM raloxifene (3163.00 ± 119.80). E) DMEM, F) OGD/R, and G) OGD/R + Ral 100 nM with 6 hours of reoxygenation. H) Mean fluorescence values of NAO intensity in this period of insult. Data are represented as the mean ± SEM of 5 independent experiments. Control (416.7.00 ± 39.47); OGD/R (183.1 ± 17.70); OGD + 100nM raloxifene (238.4 ± 26.43); OGD + 10nM raloxifene (314.6 ± 27.45) I) DMEM, J) OGD/R, and K) OGD/R + Ral 10 nM with 9 hours of reoxygenation. L) Mean fluorescence values of NAO intensity in this period of insult. Data are represented as the mean ± SEM of 5 independent experiments. Control (452.2 ± 22.28); OGD/R (330.4 ± 23.45); OGD/R + 100nM raloxifene (404.7 ± 12.34); OGD/R + 10nM raloxifene (374.6 ± 19.78). *P<0.005. Scale bar 50 µm.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



