
Submitted:
20 May 2024
Posted:
21 May 2024
You are already at the latest version
Abstract
Long-range HNCO NMR spectra for proteins show crosspeaks due to 1JNC, 2JNC, 3JNCg and h3JNC couplings. The h3JNC couplings are transmitted through hydrogen bonds and are correlated to hydrogen bond lengths. We collected long-range HNCO data at a series of temperatures for four protein states. P22i and CUS-3i are six-stranded beta-barrel I-domains from phages P22 and CUS-3 that share less than 40% sequence identity. The cis and trans states of the C-terminal domain from pore-forming toxin hemolysin ΙΙ (HlyIIC) arise from isomerization of a single G404-P405 peptide bond. For P22i and CUS-3i the hydrogen bonds detected by NMR agree with those observed in the corresponding domains from cryoEM structures of the two phages. Hydrogen bond lengths derived from the h3JNC couplings, however, are poorly conserved between the distantly related CUS-3i and P22i domains and show differences even between the cis and trans states of HlyIIC. This is consistent with hydrogen bond lengths being determined by local differences in structure rather than the overall folding topology. With increasing temperature hydrogen bonds typically show an apparent increase in length, which has been attributed to protein thermal expansion. Some hydrogen bonds are invariant with temperature, however, while others show apparent decreases in length suggesting they become stabilized with increasing temperature. Considering the data for the three proteins in this study and previously published data for ubiquitin and GB3, lowered protein folding stability and cooperativity correlates with a larger range of temperature responses for hydrogen bonds. This suggests a partial uncoupling of hydrogen bond energetics from global unfolding cooperativity as protein stability decreases.
Keywords:
protein folding
; m-value
; chi-1 angle temperature dependence
; structure conservation
; NMR structure
1. Introduction
Hydrogen bonds (H-bonds) are critical components of protein secondary and tertiary structure. A main driver of protein secondary structure is that backbone H-bonds compensate for the partially polar character of the protein mainchain when it crosses through the hydrophobic core of a protein [1]. H-bonds involving sidechains and/or solvent play additional roles in protein structure specificity, stability, and function. Individually, a H-bond confers only about 1-2 kcal/mol to the stability of the protein, the weakness of the interaction allowing it to be easily formed and broken under physiological conditions [2,3,4]. However, the tens to hundreds of H-bonds in a typical protein, together with other non-covalent interactions, provide the enthalpic stabilization of the structure that counteracts the loss of conformational entropy accompanying protein folding. The degree to which individual H-bonds contribute additively to protein stability or as part of a cooperative network of noncovalent interactions remains an open question [5,6,7].
Initially, H-bonds were viewed as an electrostatic interaction between a hydrogen bonded to an electronegative donor atom and a second electronegative acceptor atom [3,8]. In the 1930’s Pauling suggested that H-bonds have partial covalent character (first edition of [2]). This was finally confirmed in the late 1990’s by NMR detection of J-couplings transmitted through H-bonds [9,10,11,12,13,14] and Compton X-ray scattering anisotropy in ice [3,15]. In proteins, direct NMR investigations of H-bond couplings are usually carried out with the long-range HNCO (lrHNCO) experiment that measures a three-bond h3JNC’ through-hydrogen-bond scalar coupling between amide 15N and carbonyl 13C’ nuclei [9]. The h3JNC’ couplings are small with values typically >1 Hz, requiring high-sensitivity and usually necessitating perdeuteration of proteins larger than ~10 KDa [13]. However, the h3JNC’ couplings provide unambiguous identification of H-bond donors and acceptors, without requiring knowledge about the rest of the molecular structure. This contrasts with hydrogen exchange protection studies [16] that identify the H-bond donor but not the acceptor. Thus, our lab and others have found through-H-bond couplings extremely useful restraints in protein NMR structure calculations [17,18,19,20,21,22,23].
Another interesting but lesser explored application of h3JNC’ couplings is to investigate how structure varies with conditions – a unique strength of NMR in structural biology. Arguably h3JNC’ couplings because of their short distance range are less complicated by averaging and dynamics than other types of NMR restraints such as RDCs, dihedral restraints, and NOEs (that can also be confounded by spin diffusion). The detection of a h3JNC’ coupling unambiguously establishes that an H-bond between 2.5 and 3.5 Å is present, with a relatively sensitive and straightforward dependence on backbone H-bond length [24,25]. By contrast, side-chain H-bonds are seen infrequently in lrHNCO experiments, possibly due to their increased flexibility, and have a more complex dependence on H-bond length [24]. Studies on the variability of H-bonds have investigated how h3JNC’ couplings respond to pressure [26], kosmotropic solvents like trifluoroethanol (TFE) [27], and ligand binding [28].
To date there have been only two studies of the temperature dependence of h3JNC’ couplings, for the proteins ubiquitin [29] and GB3 [30]. Both proteins showed a small weakening of h3JNC’ couplings with increasing temperatures that was interpreted in terms of lengthening of N•••O H-bond distances due to the thermal volume expansion of the proteins [29]. In ubiquitin, a few individual residues showed different behavior than the average H-bond expansion, characterized by H-bonds that became stronger or were invariant with temperature. These exceptions were attributed to localized partially autonomous folding regions at the ends of regular secondary structure that became more stable with increasing temperature [29]. Both the ubiquitin and G3 proteins are small (< 8.5 KDa), have the same protein folding motif, and are very stable proteins (Tm ≥ 80 oC). We therefore wanted to investigate how generalizable the temperature dependence of H-bonds is to proteins with other structural motifs and stabilities. Since we were interested in the conservation of H-bonds between proteins with similar structures [25], we selected two pairs of structures for these studies (Figure 1). P22i and CUS-3i are autonomously folding “insertion domains” form the coat proteins of the related bacteriophages P22 and CUS-3 [21]. The two domains have 40% sequence identity and the same overall 6-strand b-barrel fold, although there are structural differences in the lengths of b-strand elements and intervening loops [21]. The cis and trans states of the C-terminal domain of hemolysin II (HlyIIC) are two slightly different structural forms of the same protein sequence brought about by isomerization of the G404-P405 peptide bond. The HlyIIC domain has a pseudo-barrel fold comprised of five beta strands and two a-helices [19]. Since the cis and trans forms are in slow exchange on the NMR timescale, the two closely related structures which differ mainly in the position of the proline-bearing loop, give separate NMR signals for about half of the residues in the protein [31]. The four protein states considered in this work were used to probe the conservation of H-bonds and their dependence on temperature in distantly and closely related protein structures.
2. Results and Discussion
2.1. lrHNCO Experiments Detect H-bond 3hJNC’ and Sidechain 3JNCg Scalar Couplings
Figure 2 shows representative data from lrHNCO experiments obtained at 5 different temperatures for the protein HlyIIC. In Figure 2A, 15N planes near 116.5 ppm are superposed for the different temperatures. In this portion of the spectrum, none of the six residues have resolvable signals from the cis and trans states of the protein since they are far away in the structure from the site of isomerization, P405. However, separate cis and trans signals are observed for K355 in Figure 2B. Four types of correlations are observed in the lrHNCO spectra (Figure 2A).
The first type of crosspeak, is an ~11 Hz’ 1JNC coupling connecting the amide nitrogen with the carbonyl of the preceding residue [33], exemplified for M349, F384, V390. These couplings are detected in standard HNCO experiments but are suppressed in the lrHNCO experiment by setting the N to C’ dephasing time 2T to a value of 133 ms corresponding to 2/(1JNC) [9]. For comparison a value of ~16 ms corresponding to 1/(2*(1JNC)) is used in the standard HNCO experiment [34]. Despite being suppressed, strong 1JNC’ couplings can nevertheless persist in the lrHNCO experiment (Figure 2). The 1JNC’ couplings are correlated with the strengths of H-bonds [33,35,36].
The second type of crosspeak h3JNC’ is the three-bond through-Hbond coupling between the N atom of the H-bond donor and the C’ of the acceptor (Figure 2), that the lrHNCO experiment is intended to detect [9]. The h3JNC’ coupling constants in units of Hz were calculated from cross-peak volumes in H-bond (VHB) and reference (Vref) versions of long-range HNCO experiments according to the formula:
where Nref and NHB were the number of scans per FID in the two experiments, and T is the 66 msec delay for the N to C’ INEPT refocusing period used to detect H-bond couplings [9,25]. The h3JNC’ couplings are typically less than 1 Hz, with smaller values for H-bonds in a-helices than b-sheets [13]. It has been empirically established that the sizes of the h3JNC’ couplings are inversely correlated with N-to-O distances across H-bonds [24,25] as:
assuming that the h3JNC’ coupling constant depends only on the N-O distance [24].
h3JNC’ = ([(VHB/Vref)/(Nref/NHB)]1/2)/(2pT)
rNO = 2.75 - 0.25*(ln | h3JNC’ |)
The third type of crosspeak that occurs in lrHNCO spectra is the intraresidue two-bond 2JNC’ coupling between the N and the C’ atoms within the same residue [9,25]. These couplings are not seen in standard HNCO experiments, but their small sizes of 1.5 to 0 Hz are comparable to the h3JNC’ couplings, so that both can appear in lrHNCO experiments [9,25]. The size of the 2JNC’ coupling depends on the angle between sequentially adjacent peptide groups [37].
Finally, a set of relatively strong crosspeaks are seen in the lrHNCO spectra from the backbone N to the sidechain carbonyl Cg atoms of Asp and Asn residues, for example D336, N339, and N398 in Figure 2A. These correlations are due to 3JNCg couplings with sizes between 0 and 3 Hz. The 3JNCg couplings depend on the c1 dihedral angles of Asp/Asn residues defined by the atoms N-Ca-Cb-Cg [38,39]. The 3JNCg coupling constant has the largest values near 3 Hz for the trans conformation (c1 = 180o), decreasing to near zero for the gauche conformations (c1 ± 180o). Motional averaging of the c1 angle is expected to give 3JNCg values near 1.4 Hz. To characterize the 3JNCg couplings we supplemented complete mainchain assignments for the four protein states with side-chain assignments for carbonyl containing Asp, Asn, Glu, and Gln using 3D HNCO and the sidechain-HCACO experiment [40]. These side-chain assignments are given in Tables S1-S3. The h3JNC’ and 3JNCg coupling constants determined from lrHNCO experiments at various temperatures are summarized in Tables S4-S5. The d(rNO)/dT slopes defining the temperature dependence of mainchain H-bond distances calculated from h3JNC’ coupling constants are given in Tables S6-S7, and Tables S8-S9 compare H-bonds detected by NMR for CUS-3i and P22i with those from the cryoEM structures of the corresponding phages.
2.2. Agreement of H-Bonds Measured by NMR and cryoEM
None of the four protein states studied have X-ray structures, however, there are cryoEM structures for phages CUS-3 (PDB 8SKG, resolution of 2.8 Å) and P22 (PDB 5UU5 and 8I1V, resolutions of 3.3 and 2.6 Å, respectively). We compared the H-bonds detected in lrHNCO experiments in this work to the H-bonds in the I-domains of the cryoEM phage structures.
The NMR and cryoEM data are in good agreement for CUS-3i with 31 of the 48 mainchain H-bonds (65%) seen by both NMR and cryoEM (Table S8). There were an additional 15 main-chain H-bonds in the cryoEM structure for which h3JNC’ couplings were not detected in the lrHNCO experiment. Two of these could not be identified due to NMR spectral overlap, one is at the dynamic N-terminus of CUS-3i that is free in the NMR fragment but covalently attached to the coat protein in the cryoEM phage structure. An additional 10 H-bonds in the cryoEM structure are in turns connecting residues separated by less than three sequence position. These are probably an artefact of overly close contacts in the cryoEM structure, since the H-bonds have marginal <DHA (donor-H•••acceptor) angles near 120o that would probably not qualify them as true H-bonds based on energetic considerations [12,13,41]. Only three genuine H-bonds disagreed between the NMR and cryoEM data. Two H-bonds G261(N)-A259(O) and R291(N)-S303(O) were observed in the lrHNCO spectra but were not present in the cryoEM structure, with both at the end of regular b-sheet secondary structure. The Q288(N)-V273(O) H-bond in the cryoEM structure was not observed by NMR.
For P22i, the H-bonds detected in lrHNCO experiments are also consistent with those in the cryoEM structures determined at 2.6 Å-resolution [42] and an earlier 3.3 Å-resolution structure [43]. A complicating factor for comparison is that two long D- (a.a.239-254) and S-loops (a.a. 281-291) are disordered when the P22i domain is studied in isolation by solution NMR [32], but become involved in H-bonded b-sheet structure that stabilizes the icosahedral capsid when the P22i domain is studied in the context of the intact P22 phage structure determined by cryoEM [42,43]. For the 2.6 Å resolution cryoEM P22 structure 25 of 37 (68%) mainchain H-bonds are detected by NMR, and 25 of 32 (78%) for the lower 3.3 Å-resolution structure. Conversely, excluding the D- and S-loops, 27 of 30 (90 %) mainchain H-bonds detected by NMR for P22i are seen in one of the corresponding domains from the cryoEM structures of phage P22. As with Cus-3, the differences between the H-bonds detected by NMR and cryoEM for P22i are largely due to differences in dynamics, H-bonds that cannot be detected by NMR due to spectral overlap, and H-bonds with marginal <DHA angles in the cryoEM structures that often involve turns shorter than 4 residues.
By comparison, H-bonds involving sidechains show much poorer agreement between the NMR and cryoEM data. In CUS-3i, only two N-H•••O=C H-bonds involving sidechains were detected in the lrHNCO experiments and only one of these is observed in the cryoEM structure. Conversely, six of the sidechain-involving H-bonds in the cryoEM structure do not give detectable h3JNC’ couplings. For P22i, none of the four sidechain H-bonds observed by NMR are detected in the 2.6 Å-resolution 8I1V cryoEM structure, and only one is seen in the 3.3 Å-resolution 5UU5 structure. Most of the sidechain H-bonds in the cryoEM structures are not seen in the lrHNCO NMR experiments, and there is also poor internal agreement for the side-chain H-bonds between the two cryoEM structures of P22i (Table S9). H-bonds involving sidechains are typically more difficult to detect via h3JNC’ couplings than their backbone counterparts, possibly to due to their more dynamic character [24].
We next investigated the agreement of mainchain H-bond distances calculated from h3JNC’ couplings according to Eq. 2 [24] with the corresponding N-to-O distances across H-bonds in the cryoEM structures. We did not observe a correlation between distances calculated from NMR and cryoEM structures similar to those observed with X-ray structures [24,25], probably due to the more limited resolutions of the cryoEM structures. Nevertheless, when examining the RMS differences in N-to-O distances across H-bonds calculated from h3JNC’ couplings and cryoEM, these were 0.13 Å for the 2.8 Å-resolution CUS-3 structure (31 H-bonds), 0.21 Å for the 2.6 Å-resolution P22 structure (21 H-bonds), and 0.16 Å for the 3.3 Å-resolution P22 structure (25 H-bonds). Thus, the H-bond distances from h3JNC’ couplings agree with those from the cryoEM structures on average within about 0.1 to 0.2 Å.
The temperature dependence of H-bonds d(NO)/dT obtained for the proteins in this work are as large as 0.01 to 0.02 Å /oK, although for the more stable ubiquitin the values are smaller, on average 0.0005 Å /oK (see below). To determine cryoEM structures, samples prepared at physiological temperatures are vitrified by plunging them into cryogens such as liquid ethane. Vitrification necessitates a rapid cooling rate between 105 to 108 oK/s-1 to bring samples to a typical temperature of ~80 oK for cryoEM data collection [44]. The H-bond distances obtained from h3JNC’ couplings and cryoEM structures agree within 0.2 Å, even though the data collection temperatures for the two methods differ by more than 200 oK. This suggests the cryoEM samples are trapped by rapid vitrification in conformations similar to those present at physiological temperatures.
2.3. H-Bonds and Their Temperature Dependence Are Poorly Conserved between Related Proteins Structures
To obtain information on the temperature dependence of H-bonds we recorded lrHNCO and reference HNCO spectra for the three proteins studied in this work at five or six different temperatures. h3JNC’ coupling constants were calculated from peak volumes in the H-bond and reference spectra according to Eq. 1 [9,25]. The resulting coupling constants were used to calculate N-O distances across H-bonds according to Eq. 2 [24]. The derived individual H-bond distances are shown as a function of temperature in Figure 3. H-bond distances for ubiquitin, calculated the same way from published h3JNC’ coupling constants [29] are also included for reference. The H-bond distances show three types of behaviors. Most of the distances increase with temperature, indicated by the red symbols. Some of the H-bonds become shorter with increasing temperature, indicated by the green symbols. The last class of H-bonds with slopes smaller than the uncertainty of the slope and R-values < 0.6 indicated with “X” symbols, were assigned as temperature invariant within experimental uncertainty. The proportions of different kinds of H-bond temperature responses vary among the different proteins studied (Figure 3).
In addition to the main-chain H-bonds we also looked at the temperature dependence of the sidechain 3JNCg couplings that to our knowledge has not been described before. Of 18 Asp/Asn sidechains analyzed in four protein states, six (33%) showed decreases in 3JNCg couplings with increasing temperature and the rest were invariant within experimental uncertainty (Table S4, S5). Most of the six residues that experienced a decrease in the coupling constant with temperature had a large sidechain 3JNCg above 2 Hz at low temperature characteristic of a trans conformation (c1 = 180o), that decreased towards the ~1.4 Hz limit expected for c1 dihedrals undergoing conformational averaging [39].
For the analysis of main-chain H-bonds we first looked at conservation between related structures. The phage I-domains CUS-3i and P22i share 40% sequence identity and the same 6-strand b-barrel folding motif but have differences in secondary structure elements, differences in loop dynamics, as well as markedly different surface electrostatics [21]. The cryoEM structures of the two proteins (PDB codes 8SKG, 5UU5) align with an RMSD of 0.9 Å, allowing for the comparison of 12 structurally equivalent H-bonds after best-fit superposition. The rNO distances (Eq. 2) for equivalent H-bonds show only a moderate correlation between the two proteins (R-value = 0.76, p = 0.0038), that could be due to the proteins sharing similar secondary structures and the fact that H-bonds are shorter in b-sheets. The d(rNO)/dT slopes describing the changes in H-bond distances with temperature are not significantly correlated between the two proteins (R-value = -0.50, p = 0.093).
We next examined the cis and trans states of HlyIIC related by isomerization about the G404-P405 peptide bond. The two states are in slow exchange on the NMR timescale, giving separate NMR signals for about half of the residues in the protein [45]. The main difference between the structures of the two states is in the orientation of the loop between helix a2 and strand b5 that harbors P405 [31]. We were able to resolve and analyze ten structurally equivalent H-bonds in the cis and trans states. The H-bond distances at 307 oK are moderately correlated (R-value = 0.69, p = 0.026). However, in the same 3D lrHNCO spectrum where the cis and trans state can be analyzed simultaneously, three of 28 H-bonds in the cis state are not seen in the trans state (N377-Y406, G404-N352, E408-F375). The three H-bonds involve residues near the P405 site of isomerization that are lost in the trans state due to the structural differences accompanying isomerization. The d(rNO)/dT slopes for the ten structurally equivalent H-bonds are only moderately correlated between the cis and trans states (R-value = 0.80, p = 0.0059). In several cases N350-Q353, T357-S346, V381-I374, I397-I393 the H-bonds show markedly different d(rNO)/dT slopes between the two states.
The relatively weak conservation of H-bond distances and temperature responses between closely similar protein structures such as the cis and trans states of HlyIIC, suggest that the length and temperature dependence of an H-bond is determined mostly by short-range interactions in its immediate vicinity and less by the overall protein fold. This is further supported by the observation that three H-bonds are lost near P405 in the trans compared to the cis state of HlyIIC due to the conformational differences localized to the loop bearing the proline.
2.4. The Variability of H-Bond Temperature Responses Is Inversely Correlated with Global Folding Stability
Data on the stabilities of the three proteins used to carry out temperature dependent H-bond studies in this work, together with ubiquitin from a previously published study [29] are given in Table 1. A plot summarizing the variability in d(rNO)/dT slopes is shown in Figure 4A. The proteins in Figure 4A are arranged in order of increasing stability to unfolding from left to right (Table 1). The average d(rNO)/dT slope near (10.7 ± 4.6) x 10-4 Å/K is similar for all the proteins. This average probably reflects the thermal volume expansion coefficient, an intrinsic property that has a conserved value of ~5.2 x10-4 1/K for a variety of proteins [29]. The spread in d(rNO)/dT slopes, however, increases with decreasing stability to unfolding. For the moderately stable protein HlyIIC, the range of d(rNO)/dT slopes between 0.019 and -0.022 Å/K is more than 10-fold larger than for the stable protein ubiquitin where d(rNO)/dT slopes vary between 0.0015 and -0.0004 Å/K. This is illustrated in Figure 4B where the standard deviation of the d(rNO)/dT slopes is correlated with the ∆G0unf and the m-values obtained for the three proteins from equilibrium unfolding experiments (Table 1). Studies of the temperature dependence of H-bond h3JNC couplings were done for the additional protein GB3 [30] but are not included in Figure 4 because data for the couplings of individual H-bonds were not available. Nevertheless, the range of d(h3JNC)/dT slopes between 0.001 and -0.0003 Hz/K for GB3 [30] are very similar to those for ubiquitin [29]. The GB3 protein has a similar fold to ubiquitin [46], and a high thermal stability estimated to be ≥ 90 oC [47,48]. Therefore, the data for GB3 also qualitatively supports that d(rNO)/dT slopes are more uniform for stable proteins.
The ∆G0unf parameter is the change in the Gibbs free energy for unfolding, measured in equilibrium denaturation experiments, and reflects the stability of the folded compared to the unfolded state. The m-value is a descriptor of the slope or steepness of the unfolding transition [53,54,55]. There are two interpretations of the m-value. The first, is that it describes the change in solvent accessible surface between the folded and unfolded state [53,54]. The second is that it describes the cooperativity of the unfolding transition [55,56]. The two interpretations are largely equivalent, in as much as cooperative all-or-none unfolding transitions lead to a large change in accessible surface area whereas non-cooperative partial unfolding leads to a smaller change in accessible surface area. It is well established that for most proteins ∆G0unf and the m-value for unfolding are highly correlated [50,54]. The m-values tend to be small for poorly structured proteins such as molten globules, and large for stable proteins [55,57]. The ∆G0unf and m-values are also highly correlated for the four proteins considered in Table 1 and Figure 4 (R-value = 0.991, p = 0.0088).
The correlation between increased variability in H-bond d(rNO)/dT slopes and lower m-values (Figure 4B) is consistent with the interpretation of the latter in terms of cooperativity. For the highly cooperative and stable protein ubiquitin all H-bonds show nearly the same small 5 x10-4 Å/K expansion that coincides with the value for the thermal volume expansion of the protein. For the least stable proteins CUS-3 and HlyIIC with the smallest m-values indicative of diminished unfolding cooperativity, the range of d(rNO)/dT slopes is about 10-fold larger and includes an increasing proportion of H-bonds with negative slopes, shown in green in Figure 3. Most of the H-bonds with negative d(rNO)/dT slopes occur in irregular or a-helical secondary structure, or at the edges corresponding to the first or last residues of b-strands. Few occur in the middle of b-strands, except F295(N)-I273(O) in strand b4 of P22i and several H-bonds connecting strands b3-b4-b5 of CUS3-i. The segment b3-b4-b5 in CUS-3i behaves as an autonomously folding sub-domain under acid denaturing conditions, retaining b-sheet structure while the rest of the protein is unfolded (A.T.A, unpublished observations).
Residues with negative d(rNO)/dT slopes tend to correspond to the some of the shortest H-bonds in the proteins HlyIIC, CUS-3i and P22i (green in Figure 3). This suggests these H-bonds are energetically favorable but opposed by strain arising from the remainder of non-covalent interactions stabilizing the structure. In weekly cooperative proteins, as the structure becomes increasingly dynamic at higher temperature, the strain due to rigid side-chain packing could become abated allowing some of the H-bonds to move towards their energetic optimum. By contrast in a highly cooperative stable systems like ubiquitin nearly all H-bonds experience the same increase driven by thermal expansion, with the few exceptions of H-bonds in the b-strand near the N-terminus that are invariant or become more stable at higher temperature [29].
The H-bonds may not be experiencing changes in length with temperature but changes in the ratio of conformers that have the H-bonds formed or broken. These conformers are in fast exchange on the NMR timescale, so that the measured h3JNC’ coupling will be a population-weighted average. When a-helical structure of the RNaseA S-peptide is stabilized by increasing concentrations of the kosmotropic solvent TFE, the h3JNC’ coupling constants show apparent increases suggestive of decreasing H-bond distances [27]. However, circular dichroism ellipticity becomes more negative [58] and the NMR Ca secondary chemical shifts increase [27]. Both observations are more consistent with an increasing fraction of molecules adopting H-bonded a-helical conformers than with a shortening of pre-existing H-bonds. Similarly, in proteins with low m-values indicative of reduced unfolding cooperativity, the responses of H-bonds to changes in temperature will be more varied if the populations of individual sub-structures become uncoupled from the unfolding of the overall global structure.
3. Materials and Methods
3.1. Samples
Double-labeled 13C/15N and triple-labeled 13C/15N/2H samples of wild type HlyIIC [19], P22i [59], and CUS-3i [60], were prepared as described previously. For long-range HNCO experiments to measure H-bonds, triple-labeled 13C/15N/2H proteins in 90% H2O/10% D2O were used with the following sample conditions: 1.0 mM HlyIIC in 20 mM NaH2PO4 with 1mM EDTA and 1 mM PMSF at pH 6.6; 1.7 mM P22i in 20 mM NaH2PO4 at pH 6.1, 1.6 mM CUS-3i in 20 mM NaH2PO4 at pH 6.3. For sidechain-HCACO experiments to obtain Asp/Asn and Glu/Gln assignments, double-labeled 13C/15N samples were lyophilized and re-dissolved in 99.96% D2O with the following sample conditions: 1 mM HlyIIC at pH* 6.0, 3.8 mM P22i at pH* 6.1, 0.7 mM CUS-3i at pH* 6.3, where pH* is the pH measured in D2O with a glass pH electrode.
3.2. NMR Data Acquisition and Analysis
Experiments were done on 600 MHz Bruker Avance and 600 MHz Varian Inova spectrometers, both equipped with cryogenic probes. For the Bruker instrument we modified the BEST TROSY HNCO H-bond experiment [61] from the Bruker IBS library (pulse sequence BT_HNCO_hbonds) to work on Topsin 2.1. At each temperature we obtained long-range HNCO (d23 = 66 ms) and reference HNCO (d24 = 16.5 ms) versions of the experiments to quantify h3JNC’ couplings [9,25,27]. For HlyIIC we recorded 3D spectra on the Bruker 600 MHz spectrometer at temperatures of 285, 290, 295, 301, and 307 oK, with 32(t1,C’) x 16(t2,N) x 1024(t3,H) complex points and acquisition times of 13, 9, and 114 ms in the t1, t2, and t3 dimensions. Total experiment times were on the order of 46 and 4 h for the H-bond and reference spectra, respectively. For CUS-3i we recorded 3D spectra on the Bruker 600 MHz spectrometer at temperatures of 274, 286, 295, 298, and 305 oK, with 64(t1,C) x 16(t2,N) x 1024(t3,H) complex points and acquisition times of 26, 10, and 107 ms in the t1, t2, and t3 dimensions. Total experiment times were about 54 and 8 h for each of the H-bond and reference spectra, respectively. Since P22i had the best dispersion of the three proteins we recorded 2D versions of the TROSY-HNCO experiment (pulse sequence best_trosy_hbonds) on the Varian 600 MHz spectrometer at temperatures of 274, 282, 290, 298, 307, and 314 oK. For the reference experiment we modified the Varian best_trosy_hbonds pulse sequence to shift the 13C’ 180o-pulses by 16.5 ms with respect to the 15N 180o-pulses in the INEPT steps as described in the literature [9]. The 2D data sets on P22i were recorded with 64 (t1,C) x 512 (t2,H) complex points with acquisition times of 14 (t1) and 107 ms (t2). Total acquisition times were 11 h for the H-bond and 1 h for the reference experiments. NMR sample temperatures were calibrated using 100% methanol (T < 300 oK) and 100% ethylene glycol (T ≥ 300 oK) standards, as described in the Bruker VT-calibration manual.
Sidechain carbonyl and amide resonances were assigned from 3D HNCO and HCACO experiments. The 3D HCACO experiment (Bruker pulse sequence hcacogp3d) was modified for sidechains as described in the literature [40], namely the aliphatic 13C center was placed at ~39 ppm, the carbonyl 13C center at ~177 ppm, and the delay 1/[4(JHC)] (called d4 in the hcacogp3d pulse sequence) was set to 1.8 ms for methylene protons rather than 3.3 ms in the standard experiment. The 3D HCACO spectrum for HlyIIC was recorded at a sample temperature of 37 oC on a 500 MHz instrument with 50(t1,C) x 32(t2,C’) x 1024(t3,H) complex points, and acquisition times of 20, 12, and 122 ms in the t1, t2, and t3 dimensions, respectively. The total acquisition time was 34 h. The 3D HCACO spectrum for CUS-3i was recorded at a temperature of 25 oC on a 500 MHz magnet with 16(t1,C) x 11(t2,C’) x 1024(t3,H) complex points, and acquisition times of 4 (t1), 3 (t2), and 157 (t3) ms; total 30 h. The 3D HCACO for P22i was at 37 oC on a 600 MHz spectrometer with 48(t1,C) x 32(t2,C’) x 1024(t3,H) complex points, and acquisition times of 29 (t1), 13 (t2), and 122 (t3) ms; total 64 h.
NMR spectra were processed with iNMR 6.3 (Mestrelab Research, Santiago de Compostela, Spain) and analyzed with CcpNmr Analysis version 2.5.2 [62]. Published assignments were used to analyze the NMR data for cis HlyIIC (BMRB 19461), trans HlyIIC (BMRB 19462), CUS-3i (BMRB 25263) and P22i (BMRB 18566).
h3JNC’ coupling constants were calculated according to Eq. 1. To estimate uncertainties in h3JNC’ values experiments were replicated on two separate samples at one temperature for each protein: HlyIIC (307 oK), CUS-3i (298 oK), and P22i (307 oK). The RMS differences between the duplicate h3JNC’ values were 0.10, 0.06 and 0.04 Hz for HlyIIC, CUS-3i, and P22i, respectively. H-bond distances were calculated using the empirical relationship (Eq. 2) established by Bax and co-workers [24].
3.3. Structure Analysis
To correlate NMR with structural parameters we used the segment corresponding to CUS-3i (Chain A of the asymmetric unit, residues 223-337) from the 2.8 Å-resolution cryoEM structure of phage CUS-3 (Richard Whitehead, Carolyn Teschke, and A.T.A, in preparation; PDB accession code 8SKG, coordinates are available from the corresponding author until release). For P22i we used the 3.3 Å-resolution cryoEM structure of phage P22 (PDB 5UU5, chain E of asymmetric unit, residues 223-345 [43]), and the 2.6 Å-resolution cryoEM structure of phage P22 (PDB 8I1V, chain A of asymmetric unit, residues 223-345 [42]). Interatomic distances were calculated with the “get_distance” command of PyMol (Schrödinger software, New York). H-bonds were calculated with the program HBPLUS v.3.06 [63].
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, A.T.A; methodology, A.T.A. and A.J.D; formal analysis, A.T.A; resources, A.T.A; writing – original draft preparation, A.T.A; writing – reviewing and editing, A.T.A. and A.J.D. All authors have read and agreed to the published version of the manuscript.
Acknowledgments
We thank Richard Whitehead III and Carol Teschke for the coordinates of the cryoEM CUS-3 phage structure. We thank Anne Kaplan and Rich Olson for samples of HlyIIC, and Carol Teschke and Therese Tripler for samples of CUS-3i and P22i. A.T.A. thanks Prof. Mei Hong and the Hong lab for the use of their solution 600 MHz instrument during his sabbatical. The NMR experiments used equipment at the MIT-Harvard Center for Magnetic Resonance, which is supported by the P41 grant GM132079. M.H. is partially supported by NIH grant AG059661. A.J.D. was partially supported by an NIH fellowship F31AG069418.
Conflict of Interest
The authors declare no potential conflict of interest.
Abbreviations
2D, 3D, one-, two, three-dimensional; cryoEM, cryogenic electron microscopy; CUS-3i, insertion domain from the coat protein of phage CUS-3; FID, free induction decay, H-bond, hydrogen bond; HlyIIC, hemolysin II C-terminal domain; INEPT, insensitive nuclei enhancement by polarization transfer; lrHNCO, long-range HNCO experiment, NMR, nuclear magnetic resonance; P22i, insertion domain from the coat protein of phage P22; RMS, root-mean-square, RMSD, root-mean-square deviation; TFE, trifluoroethanol; TROSY, transverse relaxation optimized spectroscopy.
References
- Branden, C.; Tooze, J. Introduction to Protein Structure, 2nd ed.; Garland Science: New York, 1998. [Google Scholar]
- Pauling, L. The hydrogen bond. In The Nature of the Chemical Bond, 3rd ed.; Pauling, L., Ed.; Cornell University Press: New York, NY, USA, 1960; pp. 449–504. [Google Scholar]
- Martin, T.W.; Derewenda, Z.S. The name is bond--H bond. Nat Struct Biol 1999, 6, 403–406. [Google Scholar] [CrossRef]
- Pace, C.N.; Fu, H.; Lee Fryar, K.; Landua, J.; Trevino, S.R.; Schell, D.; Thurlkill, R.L.; Imura, S.; Scholtz, J.M.; Gajiwala, K.; et al. Contribution of hydrogen bonds to protein stability. Protein Sci 2014, 23, 652–661. [Google Scholar] [CrossRef] [PubMed]
- Boresch, S.; Archontis, G.; Karplus, M. Free energy simulations: the meaning of the individual contributions from a component analysis. Proteins 1994, 20, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Brady, G.P.; Szabo, A.; Sharp, K.A. On the decomposition of free energies. J Mol Biol 1996, 263, 123–125. [Google Scholar] [CrossRef]
- Mark, A.E.; van Gunsteren, W.F. Decomposition of the free energy of a system in terms of specific interactions. Implications for theoretical and experimental studies. J Mol Biol 1994, 240, 167–176. [Google Scholar] [CrossRef]
- Pauling, L. The Shared-Electron Chemical Bond. Proc Natl Acad Sci USA 1928, 14, 359–362. [Google Scholar] [CrossRef]
- Cordier, F.; Grzesiek, S. Direct observation of hydrogen bonds in proteins by interresidue 3HJNC' scalar couplings. J Am Chem Soc 1999, 121, 1601–1602. [Google Scholar] [CrossRef]
- Cordier, F.; Rogowski, M.; Grzesiek, S.; Bax, A. Observation of through-hydrogen-bond 2hJHC' in a perdeuterated protein. J Magn Reson 1999, 140, 510–512. [Google Scholar] [CrossRef]
- Cornilescu, G.; Hu, J.S.; Bax, A. Identification of the hydrogen bonding network in a protein by scalar couplings. J Am Chem Soc 1999, 121, 2949–2950. [Google Scholar] [CrossRef]
- Dingley, A.J.; Grzesiek, S. Direct Observation of Hydrogen Bonds in Nucleic Acid Base Pairs by Internucleotide 2JNN Couplings. J Am Chem Soc 1998, 120, 8293–8297. [Google Scholar] [CrossRef]
- Grzesiek, S.; Cordier, F.; Jaravine, V.A.; Barfield, M. Insights into biomolecular hydrogen bonds from hydrogen bond scalar couplings. Prog. Nucl. Magn. Reson. Spectrosc. 2004, 45, 275–300. [Google Scholar] [CrossRef]
- Pervushin, K.; Ono, A.; Fernandez, C.; Szyperski, T.; Kainosho, M.; Wuthrich, K. NMR scalar couplings across Watson-Crick base pair hydrogen bonds in DNA observed by transverse relaxation-optimized spectroscopy. Proc Natl Acad Sci U S A 1998, 95, 14147–14151. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, E.D.; Shukla, A.; Platzman, P.M.; Hamann, D.R.; Barbiellini, B.; Tulk, C.A. Covalency of the Hydrogen Bond in Ice: A Direct X-Ray Measurement. Phys. Rev. Lett. 1999, 82, 600–603. [Google Scholar] [CrossRef]
- Englander, S.W.; Sosnick, T.R.; Englander, J.J.; Mayne, L. Mechanisms and uses of hydrogen exchange. Curr Opin Struct Biol 1996, 6, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Alexandrescu, A.T. Strategy for supplementing structure calculations using limited data with hydrophobic distance restraints. Proteins 2004, 56, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Sheftic, S.R.; Garcia, P.P.; White, E.; Robinson, V.L.; Gage, D.J.; Alexandrescu, A.T. Nuclear magnetic resonance structure and dynamics of the response regulator Sma0114 from Sinorhizobium meliloti. Biochemistry 2012, 51, 6932–6941. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, A.R.; Kaus, K.; De, S.; Olson, R.; Alexandrescu, A.T. NMR structure of the Bacillus cereus hemolysin II C-terminal domain reveals a novel fold. Sci Rep 2017, 7, 3277. [Google Scholar] [CrossRef] [PubMed]
- Newcomer, R.L.; Schrad, J.R.; Gilcrease, E.B.; Casjens, S.R.; Feig, M.; Teschke, C.M.; Alexandrescu, A.T.; Parent, K.N. The phage L capsid decoration protein has a novel OB-fold and an unusual capsid binding strategy. Elife 2019, 8. [Google Scholar] [CrossRef]
- Tripler, T.N.; Kaplan, A.R.; Alexandrescu, A.T.; Teschke, C.M. Conservation and Divergence of the I-Domain Inserted into the Ubiquitous HK97 Coat Protein Fold in P22-Like Bacteriophages. J Virol 2019, 93. [Google Scholar] [CrossRef]
- Kanelis, V.; Rotin, D.; Forman-Kay, J.D. Solution structure of a Nedd4 WW domain-ENaC peptide complex. Nat Struct Biol 2001, 8, 407–412. [Google Scholar] [CrossRef]
- Jee, J.G.; Ikegami, T.; Hashimoto, M.; Kawabata, T.; Ikeguchi, M.; Watanabe, T.; Shirakawa, M. Solution structure of the fibronectin type III domain from Bacillus circulans WL-12 chitinase A1. J Biol Chem 2002, 277, 1388–1397. [Google Scholar] [CrossRef] [PubMed]
- Cornilescu, G.; Ramirez, B.E.; Frank, M.K.; Clore, G.M.; Gronenborn, A.M.; Bax, A. Correlation between 3hJNC' and hydrogen-bond length in proteins. J Am Chem Soc 1999, 121, 6275–6279. [Google Scholar] [CrossRef]
- Alexandrescu, A.T.; Snyder, D.R.; Abildgaard, F. NMR of hydrogen bonding in cold-shock protein A and an analysis of the influence of crystallographic resolution on comparisons of hydrogen bond lengths. Protein Sci 2001, 10, 1856–1868. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yamada, H.; Akasaka, K.; Gronenborn, A.M. Pressure alters electronic orbital overlap in hydrogen bonds. J Biomol NMR 2000, 18, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Jaravine, V.A.; Alexandrescu, A.T.; Grzesiek, S. Observation of the closing of individual hydrogen bonds during TFE-induced helix formation in a peptide. Protein Sci 2001, 10, 943–950. [Google Scholar] [CrossRef] [PubMed]
- Cordier, F.; Wang, C.; Grzesiek, S.; Nicholson, L.K. Ligand-induced strain in hydrogen bonds of the c-Src SH3 domain detected by NMR. J Mol Biol 2000, 304, 497–505. [Google Scholar] [CrossRef]
- Cordier, F.; Grzesiek, S. Temperature-dependence of protein hydrogen bond properties as studied by high-resolution NMR. J Mol Biol 2002, 317, 739–752. [Google Scholar] [CrossRef]
- Hong, J.; Jing, Q.; Yao, L. The protein amide (1)H(N) chemical shift temperature coefficient reflects thermal expansion of the N-H...O=C hydrogen bond. J Biomol NMR 2013, 55, 71–78. [Google Scholar] [CrossRef]
- Kaplan, A.R.; Olson, R.; Alexandrescu, A.T. Protein yoga: Conformational versatility of the Hemolysin II C-terminal domain detailed by NMR structures for multiple states. Protein Sci 2021, 30, 990–1005. [Google Scholar] [CrossRef]
- Rizzo, A.A.; Suhanovsky, M.M.; Baker, M.L.; Fraser, L.C.; Jones, L.M.; Rempel, D.L.; Gross, M.L.; Chiu, W.; Alexandrescu, A.T.; Teschke, C.M. Multiple functional roles of the accessory I-domain of bacteriophage P22 coat protein revealed by NMR structure and CryoEM modeling. Structure 2014, 22, 830–841. [Google Scholar] [CrossRef]
- Juranic, N.; Ilich, P.K.; Macura, S. Hydrogen Bonding Networks in Proteins As Revealed by the Amide 1JNC' Coupling Constant. J Am Chem Soc 1995, 117, 405–410. [Google Scholar] [CrossRef]
- Kay, L.E.; Ikura, M.; Tschudin, R.; Bax, A. Three-dimensional triple-resonance NMR Spectroscopy of isotopically enriched proteins. 1990. J Magn Reson 2011, 213, 423–441. [Google Scholar] [CrossRef] [PubMed]
- Juranic, N.; Moncrieffe, M.C.; Likic, V.A.; Prendergast, F.G.; Macura, S. Structural dependencies of h3JNC' scalar coupling in protein H-bond chains. J Am Chem Soc 2002, 124, 14221–14226. [Google Scholar] [CrossRef] [PubMed]
- Juranic, N.; Macura, S. Correlations among (1)J(NC)' and (h3)J(NC)' coupling constants in the hydrogen-bonding network of human ubiquitin. J Am Chem Soc 2001, 123, 4099–4100. [Google Scholar] [CrossRef] [PubMed]
- Juranic, N.; Dannenberg, J.J.; Cornilescu, G.; Salvador, P.; Atanasova, E.; Ahn, H.C.; Macura, S.; Markley, J.L.; Prendergast, F.G. Structural dependencies of protein backbone 2JNC' couplings. Protein Sci 2008, 17, 768–776. [Google Scholar] [CrossRef]
- Benirschke, R.C.; Thompson, J.R.; Nomine, Y.; Wasielewski, E.; Juranic, N.; Macura, S.; Hatakeyama, S.; Nakayama, K.I.; Botuyan, M.V.; Mer, G. Molecular basis for the association of human E4B U box ubiquitin ligase with E2-conjugating enzymes UbcH5c and Ubc4. Structure 2010, 18, 955–965. [Google Scholar] [CrossRef] [PubMed]
- Juranic, N.; Atanasova, E.; Moncrieffe, M.C.; Prendergast, F.G.; Macura, S. Calcium-binding proteins afford calibration of dihedral-angle dependence of 3J(NC(gamma)) coupling constant in aspartate and asparagine residues. J Magn Reson 2005, 175, 222–225. [Google Scholar] [CrossRef]
- Yamazaki, T.; Nicholson, L.K.; Torchia, D.A.; Wingfield, P.; Stahl, S.J.; Kaufman, J.D.; Eyermann, C.J.; Hodge, C.N.; Lam, P.Y.S.; Ru, Y.; et al. NMR and X-ray evidence that the HFV protease catalytic aspartyl groups are protonated in the complex formed by the protease and a non-peptide cyclic urea-based inhibitor. J Am Chem Soc 1994, 116, 10791–10792. [Google Scholar] [CrossRef]
- Wood, P.A.; Allen, F.H.; Pidock, E. Hydrogen-bond directionality at the donor H atom—analysis of interaction energies and database statistics. CrystEngComm 2009, 11, 1563–1571. [Google Scholar] [CrossRef]
- Xiao, H.; Zhou, J.; Yang, F.; Liu, Z.; Song, J.; Chen, W.; Liu, H.; Cheng, L. Assembly and Capsid Expansion Mechanism of Bacteriophage P22 Revealed by High-Resolution Cryo-EM Structures. Viruses 2023, 15. [Google Scholar] [CrossRef]
- Hryc, C.F.; Chen, D.H.; Afonine, P.V.; Jakana, J.; Wang, Z.; Haase-Pettingell, C.; Jiang, W.; Adams, P.D.; King, J.A.; Schmid, M.F.; et al. Accurate model annotation of a near-atomic resolution cryo-EM map. Proc Natl Acad Sci USA 2017, 114, 3103–3108. [Google Scholar] [CrossRef]
- Weissenberger, G.; Henderikx, R.J.M.; Peters, P.J. Understanding the invisible hands of sample preparation for cryo-EM. Nat Methods 2021, 18, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, A.R.; Maciejewski, M.W.; Olson, R.; Alexandrescu, A.T. NMR assignments for the cis and trans forms of the hemolysin II C-terminal domain. Biomol. NMR Assign. 2014, 8, 419–423. [Google Scholar] [CrossRef]
- Kraulis, P.J. Similarity of protein G and ubiquitin. Science 1991, 254, 581–582. [Google Scholar] [CrossRef] [PubMed]
- Alexander, P.; Fahnestock, S.; Lee, T.; Orban, J.; Bryan, P. Thermodynamic analysis of the folding of the streptococcal protein G IgG-binding domains B1 and B2: why small proteins tend to have high denaturation temperatures. Biochemistry 1992, 31, 3597–3603. [Google Scholar] [CrossRef] [PubMed]
- Granata, D.; Camilloni, C.; Vendruscolo, M.; Laio, A. Characterization of the free-energy landscapes of proteins by NMR-guided metadynamics. Proc Natl Acad Sci USA 2013, 110, 6817–6822. [Google Scholar] [CrossRef] [PubMed]
- Tripler, T.N. Conservation and Divergence between P22-like Bacteriophages Coat Protein’s I-Domains and Procapsid-like Particles. Doctoral Dissertation, University of Connecticut, Storrs, CT, USA, 2019. [Google Scholar]
- Newcomer, R.L.; Fraser, L.C.R.; Teschke, C.M.; Alexandrescu, A.T. Mechanism of Protein Denaturation: Partial Unfolding of the P22 Coat Protein I-Domain by Urea Binding. Biophysical journal 2015, 109, 2666–2677. [Google Scholar] [CrossRef] [PubMed]
- Surana, P.; Das, R. Observing a late folding intermediate of Ubiquitin at atomic resolution by NMR. Protein Sci 2016, 25, 1438–1450. [Google Scholar] [CrossRef]
- Wintrode, P.L.; Makhatadze, G.I.; Privalov, P.L. Thermodynamics of ubiquitin unfolding. Proteins 1994, 18, 246–253. [Google Scholar] [CrossRef]
- Myers, J.K.; Pace, C.N.; Scholtz, J.M. Denaturant m values and heat capacity changes: relation to changes in accessible surface areas of protein unfolding. Protein Sci 1995, 4, 2138–2148. [Google Scholar] [CrossRef]
- Shortle, D. Staphylococcal nuclease: a showcase of m-value effects. Adv Protein Chem 1995, 46, 217–247. [Google Scholar] [CrossRef] [PubMed]
- Alexandrescu, A.T.; Jaravine, V.A.; Dames, S.A.; Lamour, F.P. NMR hydrogen exchange of the OB-fold protein LysN as a function of denaturant: the most conserved elements of structure are the most stable to unfolding. J Mol Biol 1999, 289, 1041–1054. [Google Scholar] [CrossRef] [PubMed]
- Carra, J.H.; Privalov, P.L. Thermodynamics of denaturation of staphylococcal nuclease mutants: an intermediate state in protein folding. FASEB J 1996, 10, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.J.; Alexandrescu, A.T.; Pitkeathly, M.; Dobson, C.M. Solution structure of a peptide fragment of human alpha-lactalbumin in trifluoroethanol: a model for local structure in the molten globule. Structure 1994, 2, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Nelson, J.W.; Kallenbach, N.R. Stabilization of the ribonuclease S-peptide alpha-helix by trifluoroethanol. Proteins 1986, 1, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, A.A.; Fraser, L.C.; Sheftic, S.R.; Suhanovsky, M.M.; Teschke, C.M.; Alexandrescu, A.T. NMR assignments for the telokin-like domain of bacteriophage P22 coat protein. Biomolecular NMR assignments 2013, 7, 257–260. [Google Scholar] [CrossRef] [PubMed]
- Tripler, T.N.; Maciejewski, M.W.; Teschke, C.M.; Alexandrescu, A.T. NMR assignments for the insertion domain of bacteriophage CUS-3 coat protein. Biomolecular NMR assignments, 1007. [Google Scholar] [CrossRef]
- Favier, A.; Brutscher, B. Recovering lost magnetization: polarization enhancement in biomolecular NMR. J Biomol NMR 2011, 49, 9–15. [Google Scholar] [CrossRef]
- Vranken, W.F.; Boucher, W.; Stevens, T.J.; Fogh, R.H.; Pajon, A.; Llinas, M.; Ulrich, E.L.; Markley, J.L.; Ionides, J.; Laue, E.D. The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins 2005, 59, 687–696. [Google Scholar] [CrossRef]
- McDonald, I.K.; Thornton, J.M. Satisfying hydrogen bonding potential in proteins. J Mol Biol 1994, 238, 777–793. [Google Scholar] [CrossRef]
Figure 1.
Solution structures of the four protein states studied in this work. (A) Cartoons of the NMR conformer closest to the ensemble average, shown with a color ramp from the N (blue) to the C-terminus (red). For HlyIIC, the largest difference between the cis and trans structures [31] is in the loop between helix a2 (orange) and strand b5 (red). The D- and S-loops in the P22i structure are dynamically disordered [32]. In Cus3i, the segment corresponding to the P22i loop becomes structured to form an extension of the b1-b2 hairpin [21]. (B) NMR backbone ensembles of the four protein states. Backbone H-bonds identified with the PyMol program are shown with dotted red lines.
Figure 1.
Solution structures of the four protein states studied in this work. (A) Cartoons of the NMR conformer closest to the ensemble average, shown with a color ramp from the N (blue) to the C-terminus (red). For HlyIIC, the largest difference between the cis and trans structures [31] is in the loop between helix a2 (orange) and strand b5 (red). The D- and S-loops in the P22i structure are dynamically disordered [32]. In Cus3i, the segment corresponding to the P22i loop becomes structured to form an extension of the b1-b2 hairpin [21]. (B) NMR backbone ensembles of the four protein states. Backbone H-bonds identified with the PyMol program are shown with dotted red lines.
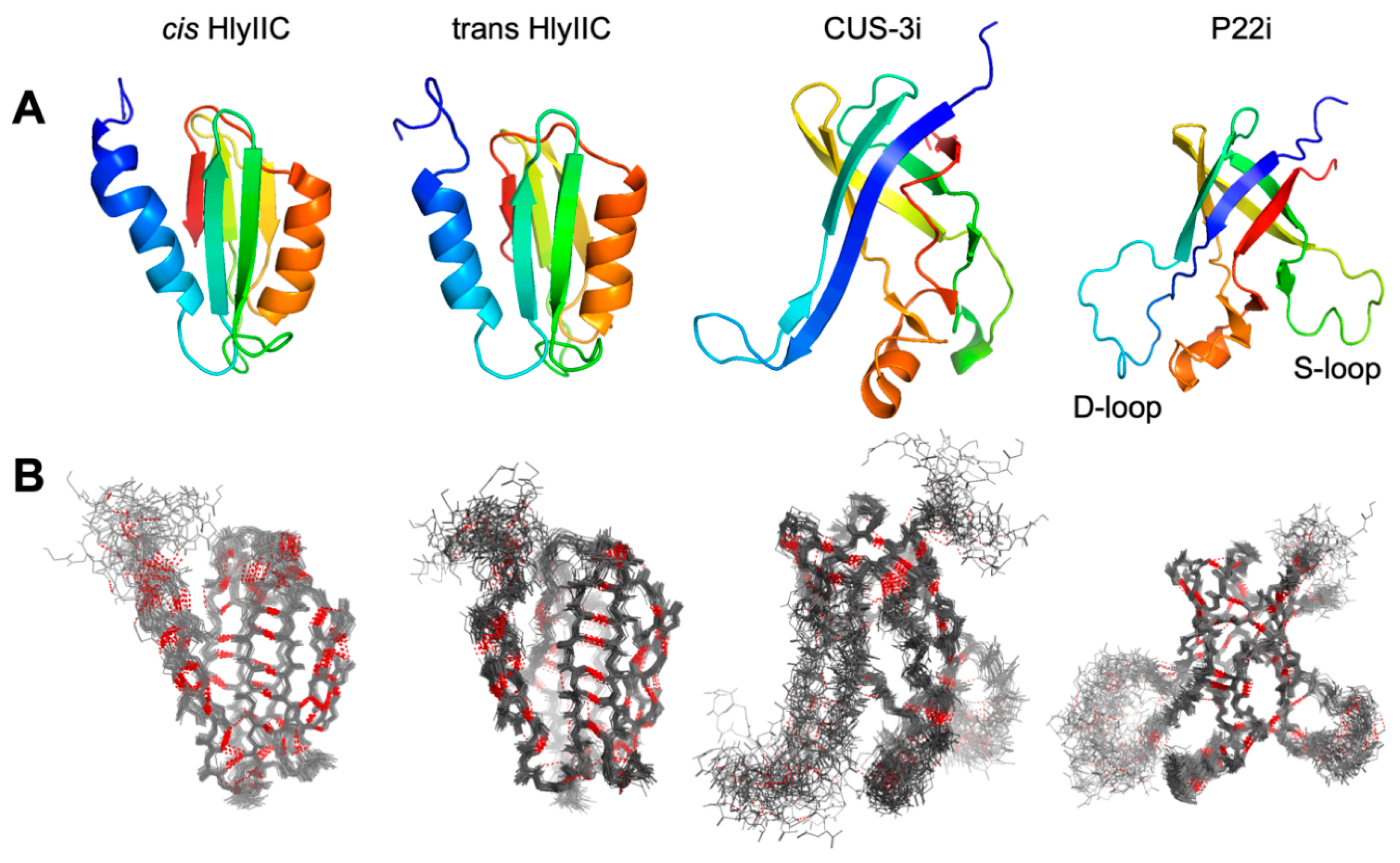
Figure 2.
Superposed 15N planes from lrHNCO spectra for HlyIIC at five temperatures. (A) 15N plane at 116.6 ppm illustrating the various types of correlations observed: 1JNC’, h3JNC’, and 3JNCg. Two weak intraresidue 2JNC’ crosspeaks are also present in this plane but at contour levels lower than shown (gray boxes near 8.5 ppm). (B) 1H-15N HSQC of HlyIIC highlighting crosspeaks due to trans (left) and cis (right) states of the protein. The expansion shows superpositions of 3D lrHNCO and reference HNCO planes at 15N 128.4 ppm for five different temperatures, using the same coloring scheme for contour levels as in A. At each temperature a pair of 1HN resonances is observed making it possible to investigate the temperature dependence of h3JNC’ couplings for K355 in both the cis and trans states. There were ten residues in HlyIIC for which separate mainchain h3JNC’ couplings could be resolved from the cis and trans states and fifteen for which NMR signals from the two conformations were unresolved (Table S4, S6).
Figure 2.
Superposed 15N planes from lrHNCO spectra for HlyIIC at five temperatures. (A) 15N plane at 116.6 ppm illustrating the various types of correlations observed: 1JNC’, h3JNC’, and 3JNCg. Two weak intraresidue 2JNC’ crosspeaks are also present in this plane but at contour levels lower than shown (gray boxes near 8.5 ppm). (B) 1H-15N HSQC of HlyIIC highlighting crosspeaks due to trans (left) and cis (right) states of the protein. The expansion shows superpositions of 3D lrHNCO and reference HNCO planes at 15N 128.4 ppm for five different temperatures, using the same coloring scheme for contour levels as in A. At each temperature a pair of 1HN resonances is observed making it possible to investigate the temperature dependence of h3JNC’ couplings for K355 in both the cis and trans states. There were ten residues in HlyIIC for which separate mainchain h3JNC’ couplings could be resolved from the cis and trans states and fifteen for which NMR signals from the two conformations were unresolved (Table S4, S6).
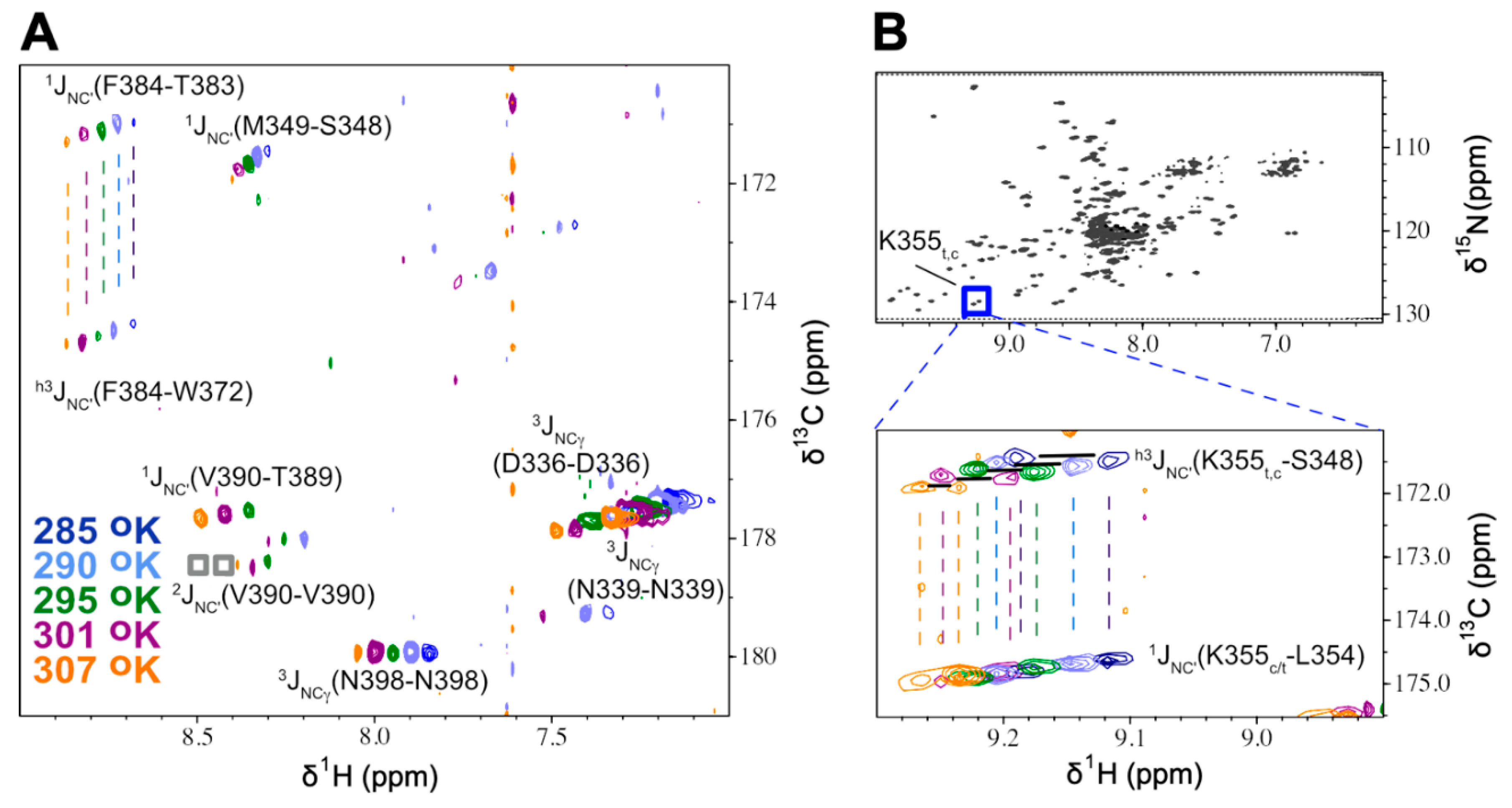
Figure 3.
Temperature dependence of mainchain H-bond distances derived from h3JNC’ couplings. (A) HlyIIC cis state (left to right 285, 290, 295, 301, 307 oK), (B) HlyIIC trans state (same temperatures as cis), (C) CUS-3i (274, 286, 295, 298, 305 oK), (D) P22i (274, 282, 290, 298, 307, 314 oK), (E) ubiquitin (278, 298, 318, 328, 338 oK). The data for ubiquitin is derived from a previously published paper [29]. Each H-bond is labeled as donor(N)-acceptor(O). H-bonds that show an increase with temperature, decrease, or no change within uncertainty are shown with red, green, and gray “X” symbols, respectively. Linear fits are shown for all H-bonds but in some cases the lines are obscured by the data points. For Cus3i and P22i, H-bonds labeled blue are structurally equivalent after superposition of the structures. N.P. indicates 3 H-bonds that are present in the cis but not trans state of HlyIIC.
Figure 3.
Temperature dependence of mainchain H-bond distances derived from h3JNC’ couplings. (A) HlyIIC cis state (left to right 285, 290, 295, 301, 307 oK), (B) HlyIIC trans state (same temperatures as cis), (C) CUS-3i (274, 286, 295, 298, 305 oK), (D) P22i (274, 282, 290, 298, 307, 314 oK), (E) ubiquitin (278, 298, 318, 328, 338 oK). The data for ubiquitin is derived from a previously published paper [29]. Each H-bond is labeled as donor(N)-acceptor(O). H-bonds that show an increase with temperature, decrease, or no change within uncertainty are shown with red, green, and gray “X” symbols, respectively. Linear fits are shown for all H-bonds but in some cases the lines are obscured by the data points. For Cus3i and P22i, H-bonds labeled blue are structurally equivalent after superposition of the structures. N.P. indicates 3 H-bonds that are present in the cis but not trans state of HlyIIC.
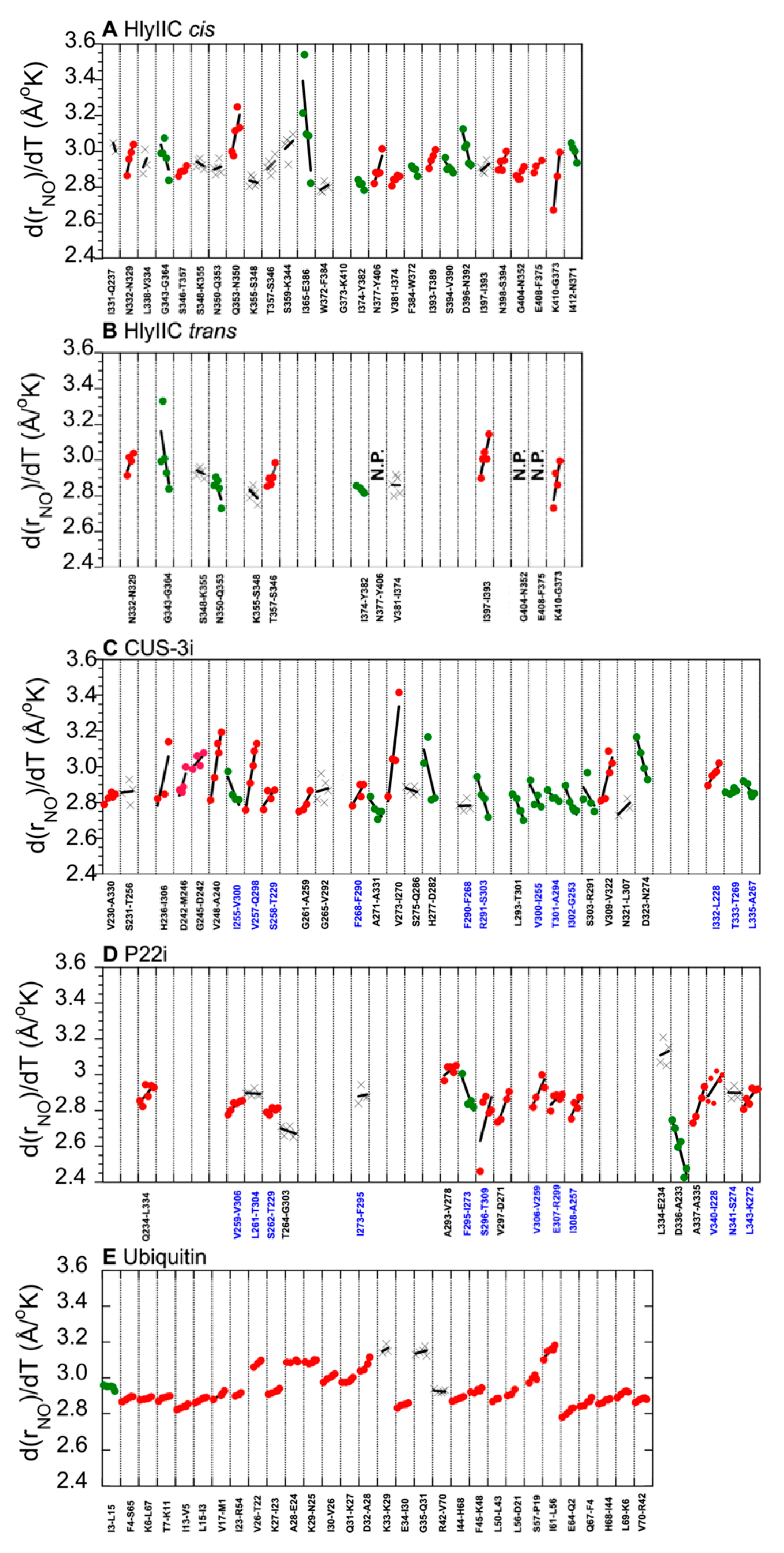
Figure 4.
Variability in the temperature dependence of H-bonds as a function of folding stability. (A) Distribution of d(rNO)/dT slopes in four proteins arranged in order of increasing stability to unfolding (least stable right, most stable left). (B) Correlation between the standard deviation in d(rNO)/dT slopes and parameters related to protein stability: ∆G0unf and m-values (from Table 1).
Figure 4.
Variability in the temperature dependence of H-bonds as a function of folding stability. (A) Distribution of d(rNO)/dT slopes in four proteins arranged in order of increasing stability to unfolding (least stable right, most stable left). (B) Correlation between the standard deviation in d(rNO)/dT slopes and parameters related to protein stability: ∆G0unf and m-values (from Table 1).
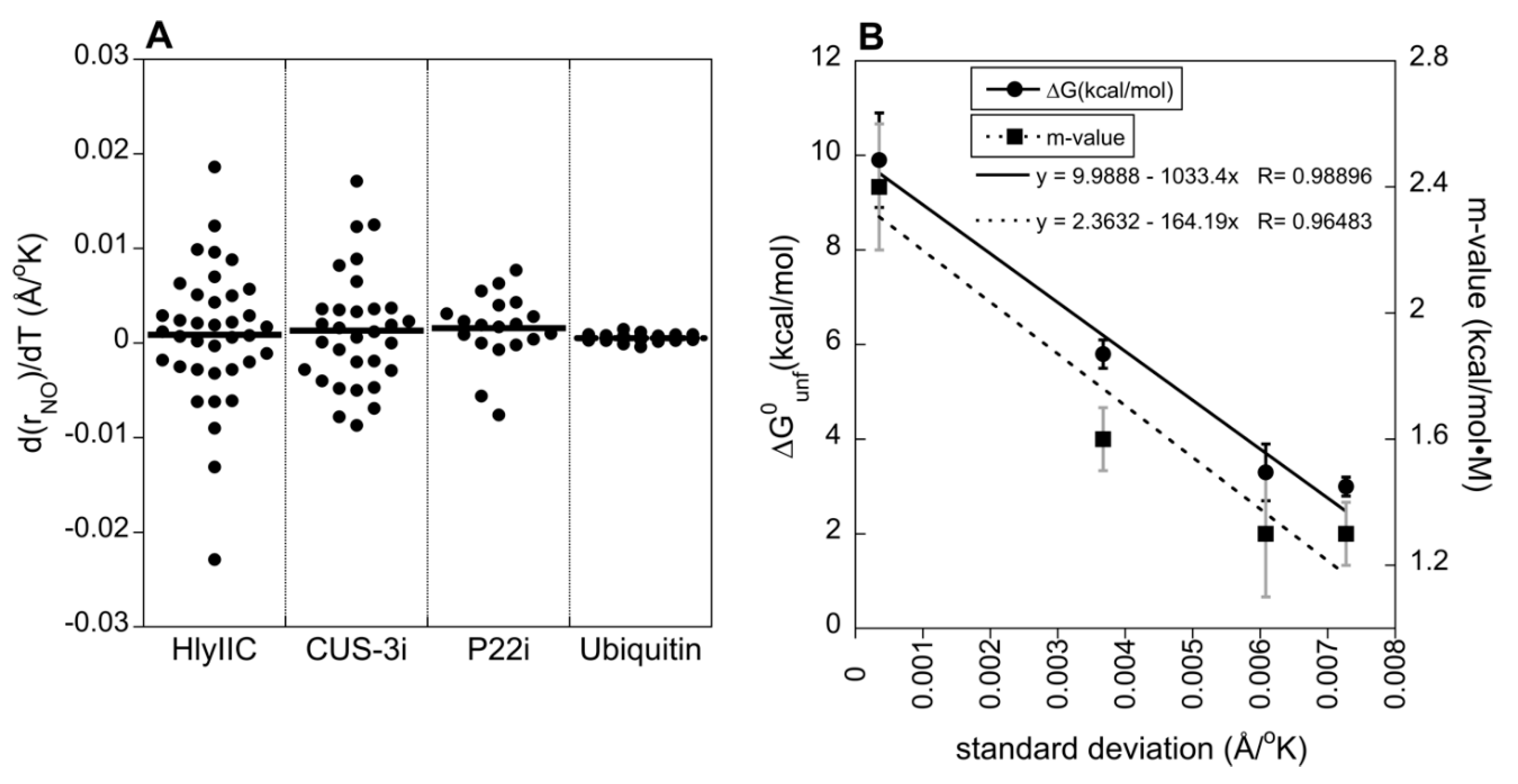

Table 1.
Folding stability information on proteins used for lrHNCO studiesa.
| Protein | ∆G0unf (kcal/mol) |
m-value (kcal/mol•M) |
Tmelt (oC) |
References |
|---|---|---|---|---|
| HlyIIC | 3.0 ± 0.2 | 1.3 ± 0.1 | 52 | [19,31] |
| CUS-3i | 3.3 ± 0.6 | 1.3 ± 0.2 | 48 | [49] |
| P22 | 5.8 ± 0.3 | 1.6 ± 0.1 | 54 | [50] |
| ubiquitin | 9.9 ± 1.0 | 2.4 ± 0.2 | > 90 | [51,52] |
| GB3 | ~ 6 to 7 | N.D. | ≥ 90 | [47,48] |
aThe ∆G0unf and m values are from equilibrium denaturation experiments using urea, except for ubiquitin where guanidine chloride was used as a denaturant, and GB3 where values were estimated from the highly homologous GB1 and GB2 domains [47,48]. The Tmelt values are midpoints for thermal unfolding.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



