
Submitted:
20 May 2024
Posted:
21 May 2024
You are already at the latest version
Abstract
Alzheimer's disease (AD), a progressive neurodegenerative disorder, manifests through dysregulation of brain function and subsequent loss of bodily control, attributed to β-amyloid plaque deposition and TAU protein hyperphosphorylation and aggregation, leading to neuronal death. Concurrently, similar cannabinoids to the ones derived from Cannabis sativa are present in the endocannabinoid system, acting through receptors CB1R and CB2R and other related receptors such as Trpv-1 and GPR-55, and are being extensively investigated for AD therapy. Given the limited efficacy and adverse effects of current available treatments, alternative approaches are crucial. Therefore, this review aims to identify effective natural and synthetic cannabinoids and elucidate their beneficial actions for AD treatment. PubMed and Scopus databases were queried (2014–2024) using keywords such as "Alzheimer's disease" and "cannabinoids". The majority of natural (THC, CBD, AEA, etc.) and synthetic (JWH-133, WIN55,212-2, CP55-940, etc.) cannabinoids included, showed promise in improving memory, cognition, and behavioral symptoms, potentially via pathways involving antioxidant effects of selective CB1R agonists (such as the BDNF/TrkB/Akt pathway) and immunomodulatory effects of selective CB2R agonists (TLR4/NF-κB p65 pathway). Combining anticholinesterase properties with cannabinoid moiety may enhance therapeutic responses, addressing cholinergic deficits of AD brains. Thus, the positive outcomes of the majority of studies discussed supports further advancing cannabinoids in clinical trials for AD treatment.
Keywords:
Cannabinoids
; Alzheimer’s disease
; Cannabis sativa
; CB1R
; CB2R
; Trpv-1
; GPR-55
1. Introduction
Alzheimer's disease (AD) is a neurodegenerative disorder of acquired dementia, which progressively destroys a significant part of the brain's neuronal network. This disease belongs to the broad spectrum of dementia, with the latest data classifying it as a "neurocognitive disorder resulting from Alzheimer's disease" [1]. Today, AD is responsible for over 50% of dementia diagnoses, associated with dysfunctions in memory, cognitive and motor skills, speech, senses, visuospatial abilities and attentional focus [2]. These symptoms bring about behavior alterations in AD patients, which in turn will lead to serious impairment of their social and/or professional abilities and lives. Consequentially, 3 main stages of disease progression are created; the early (preclinical) which is devoid of symptoms (and can be diagnosed by the analysis of mainly protein biomarkers), the intermediate where the first symptoms appear in the form of mild cognitive impairment, and the final stage leading to the conclusive diagnosis of AD [3].
Alzheimer's disease is characterized by two key neuroanatomical changes: senile neuritic plaques and neurofibrillary tangles, resulting from abnormal protein formations. Neuritic plaques form due to the abnormal deposition of beta-amyloid (Aβ) protein, particularly Aβ oligomers, known for their neurotoxicity. These plaques disrupt memory and learning processes, with synapses being initial targets [4,5].
Genetic factors play a significant role, with overexpression of the amyloid precursor protein (APP) gene linked to the disease, notably seen in trisomy 21 (Down syndrome) patients. Mutations in the APP gene lead to increased production of amyloid-β proteins, contributing to early-onset familial AD [4,6]. The pathogenesis of AD involves two metabolic pathways of APP: the non-amyloidogenic and amyloidogenic pathways. Imbalance towards the latter leads to increased production of insoluble Aβ monomers, promoting plaque formation. Dysfunctions in Aβ clearance enzymes, such as apolipoprotein E (apoE), further exacerbate this imbalance [7]. Microglial and other cells attracted to the area may cause further plaque formation from the amorphous fragments of degenerated neuronal cells, subsequently releasing pro-inflammatory molecules and inducing oxidative stress [8,9]. Aβ aggregation triggers neurotoxic phenomena, including hyperphosphorylation of the TAU protein and disruption of calcium homeostasis, intensifying neurotoxic effects [7,10]. Hyperphosphorylated TAU protein, enhanced by decreased expression of certain phosphatases, aberrant activation of kinases such as glycogen synthase kinase-3 beta (GSK-3β), chronic stress and other diseases [11,12], forms insoluble filaments (paired helical filaments, PHFs), leading to neurofibrillary tangles (NFTs) and neuronal atrophy [13]. Brain changes in AD include temporal lobe degeneration, especially in the hippocampus, affecting memory formation. Progressive neurodegeneration results in cortical atrophy, particularly in posterior regions. Anatomical changes manifest as cerebral sulci enlargement and lateral ventricle expansion [14,15].
The most common therapeutic methods currently available include a few basic classes of drugs. These classes mainly include acetylcholinesterase (AChE) inhibitors such as rivastigmine, donepezil and galantamine, as well as inhibitors of activated NMDA receptors, such as memantine [16]. AChE inhibitors enhance cholinergic neurotransmission, which is considered to have a key role in memory and learning functions, while memantine mitigates excitotoxicity, which has been associated with neuronal death, thereby normalizing glutamate neurotransmission [17]. Brexpiprazole belongs to atypical antipsychotics and is the only approved drug against the characteristic agitation symptoms of AD, such as akathisia, extreme verbal and physical behavior, repetitive movements, etc. [18]. These pharmacotherapies may offer temporary relief from AD symptoms. However, none of them provide a cure, as they cannot halt the progression of the disease, instead providing only symptomatic treatment [16].
The U.S. Food and Drug Administration (FDA) has already approved the use of two disease-modifying immunotherapies, lecanemab and aducanumab, which are anti-amyloid monoclonal antibodies that minimize the total number of Aβ plaques and seem promising for curing cognitive impairment if taken in the early stages of AD. Nevertheless, they are accompanied by severe adverse effects, including brain swelling and bleeding, severe allergic reactions, etc. [19]. Therefore, alternative treatment options are urgently needed.
2. Endocannabinoid System and Its Association with Alzheimer’s Disease
Cannabinoids are the main components that can be isolated from hemp or marijuana, i.e., from the dried leaves and flowers of the plant species Cannabis sativa [20]. This plant product is considered one of the most widely used illicit drugs worldwide [21] and contains two characteristic cannabinoids, tetrahydrocannabinol (THC) and cannabidiol (CBD). Its consumption is followed by psychotropic effects, due to its action on the central nervous system (CNS), for which THC is responsible. CBD lacks psychotropic properties and is mainly responsible for the relaxing effects of marijuana [22]. Cannabinoids show affinity with receptors in the central and peripheral nervous system (PNS). These receptors, in combination with cannabinoids that occur naturally in the body (endocannabinoids), make up the endocannabinoid system (ECS). In particular, CB1 (CB1R) and CB2 (CB2R) receptors are reported, with CB1Rs normally found in CNS regions such as hippocampus, cerebellum and basal ganglia neurons, as well as in tissues of the PNS such as the heart, liver and cells of the immune system. CB2Rs mainly exist in peripheral tissues, such as cells of the immune system, including monocytes, macrophages, B-cells and T-cells. Regarding the nervous system, CB2Rs are mainly located in microglia, in response to various damaging conditions associated with local inflammation, while their expression is relatively limited in some neurons too [23,24]. CB1R and CB2R belong to the class of G-protein-coupled receptors (GPCR), whose role is the inhibitory regulation of neurotransmitters such as noradrenaline, dopamine, serotonin and acetylcholine (ACh) [25,26], with CB1Rs being solely responsible for the psychotropic effects of cannabinoids.
The ECS plays a crucial physiological role, encompassing functions like inflammation regulation, promotion of apoptotic processes, stimulation of neurogenesis, and display of antioxidant properties [27,28]. Anandamide (AEA), 2-arachidonylglycerol (2-AG), and 2-arachidonyl glyceryl ether or nolantine ether (2-AGE) are the endocannabinoids constituting this system, functioning as lipid retrograde neurotransmitters [29]. These substances participate in a myriad of biological processes, governing essential functions including memory, learning, neuronal development, emotional regulation, sleep, temperature control, pain modulation, appetite regulation, hormonal balance, and immune system regulation, including inflammation [30].
During the progression of AD, there is a gradual dysfunction of the ECS [28]. This is marked by alterations in the levels and/or expression areas of both CB1R and CB2R. Initially, CB1R expression increases in the frontal cortex and hippocampus during the early stages of AD, but decreases progressively over time. Conversely, CB2R expression becomes exclusive to microglial cells and elevates notably, likely due to intense neuroinflammation, in the later stages of AD [25]. At the same time, the levels of AEA in cortical areas decrease, directly correlating with cognitive decline [29]. Tang et al. [31] investigated the relationship between CBRs and AD-related neuroinflammation in SAMP8 mice. Their research revealed that overexpression of microRNA-139 (miR-139), observed in the hippocampus of AD mice, impaired spatial memory, object recognition, fear response, and reactions to pro-inflammatory stimuli by inhibition of intercellular adhesion molecule 1 (ICAM-1) and cluster of differentiation 40 (CD40), and reduction of interleukin-6 (IL-6) and tumor necrosis factor-a (TNF-a). This effect was attributed to the regulation of CB2R gene expression by miRNAs, indicating the involvement of CB2-mediated neuroinflammatory processes in AD's neurotoxic effects.
Animal experiments have revealed the protective role of CB1Rs against AD-related pathologies [25,32]. These receptors are pivotal in fundamental brain functions such as cognition, memory, emotion, motor control, hunger, and pain sensation [33], as well as in regulating energy balance and metabolism [34]. Conversely, microglial cells' actions significantly contribute to the development of amyloid plaques and neurofibrils through inflammation induction. Overexpression of CB2R in these cells underscores its crucial role in limiting neuroinflammation [35]. This highlights the extensive involvement of CBRs in neurodegenerative disease-linked biological processes, emphasizing their importance in addressing AD pathology [30]. Thus, despite the known adverse effects of cannabinoids on various bodily systems, including the central nervous, respiratory, cardiovascular, and skeletal systems, recent research has explored their potential in combating diseases like epilepsy, psychotic episodes, Parkinson's disease, anxiety disorders, depression, and Alzheimer's disease.
As science continues to advance, it both addresses existing questions and raises new ones, leading to ongoing research. Despite the rapid pace of scientific inquiry, gaps in knowledge persist, often due to limited study or understanding. These gaps are particularly evident in the search for effective treatments for diseases like AD. Consequently, there's a growing need to explore alternative therapeutic approaches, leveraging the expanding use of medicinal cannabis and deepening understanding of the ECS. The association between CBRs and cognitive functions has prompted a shift in scientific focus towards identifying new cannabinoids and optimizing known ones for Alzheimer's treatment. Therefore, this review aimed to synthesize a wide range of studies of the last decade, highlighting promising CBR agonist molecules and their mechanisms of action in reversing AD symptoms. Ultimately, the goal was to contribute to the development of innovative therapeutic models and enrich the scientific literature in this field.
3. Therapeutic Potential of Cannabinoids in Alzheimer’s Disease
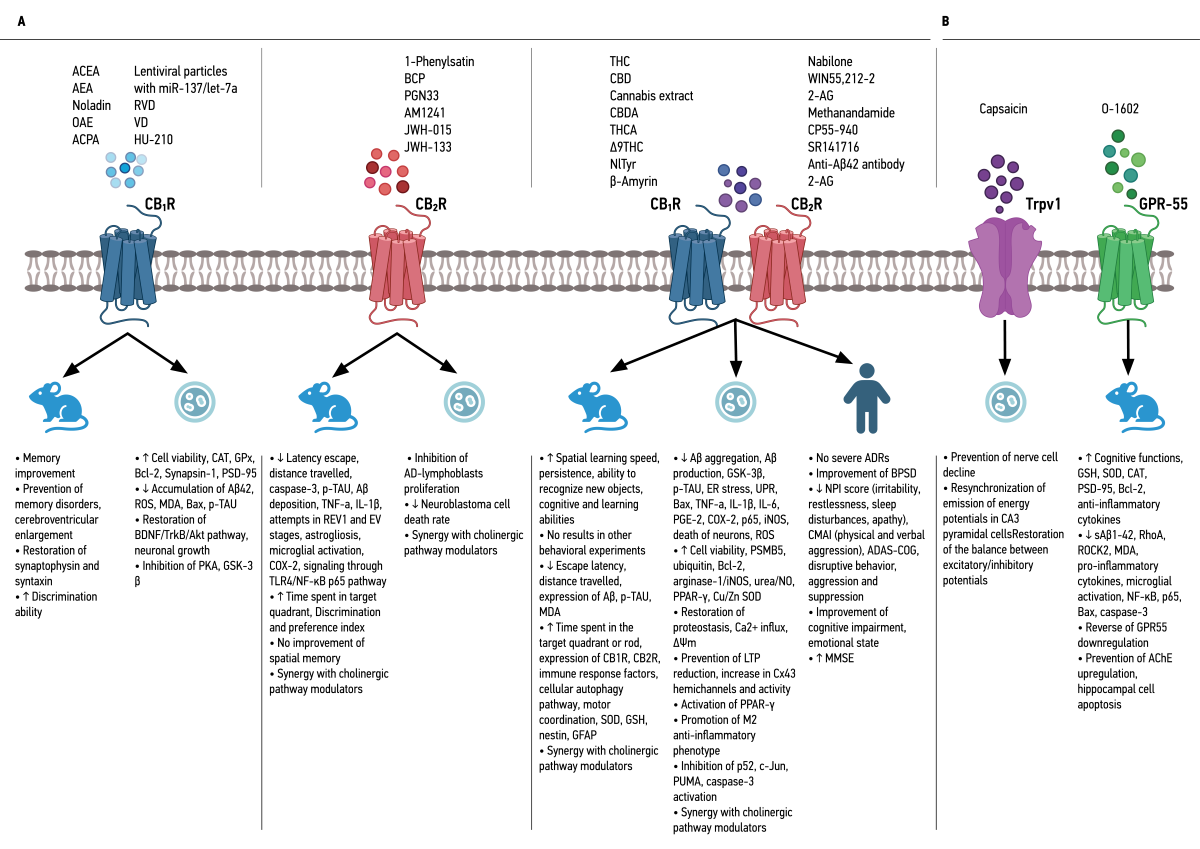
3.1. Selective Agonists of Cannabinoid Receptor 1 (CB1R)
The research by Crunfli et al. [36] explored the therapeutic potential of the CB1R agonist arachidonyl-2'-chloroethylamide (ACEA) against sporadic AD, which was explored through various in vitro in neuro-2a neuroblastoma cells and in vivo experiments in adult male Wistar rats, following streptozotocin (STZ) infusion. It was observed that intraperitoneal (i.p.) treatment with 3 mg/kg ACEA restored cognitive impairment, improving both short-term (33.07%) and long-term (44.94%) memory in comparison to STZ-treated rats. It was also shown that the STZ+ACEA group showed a significant increase in the levels of insulin receptors (92.13%) and antiapoptotic proteins Bcl-2 (110.4%), while at the same time a significant decrease in the activity of protein kinase B (Akt) (≈61%) and extracellular signal-regulated kinase (ERK) (144.9%) was observed. Finally, administration of ACEA to STZ-treated cells increased their viability by 34.18%±10.93%.
In their study, Moreira-Silva et al. [37] investigated the impact of intracerebroventricular (i.c.v.) injection of AEA (100 ng) in a rat model induced with STZ, simulating sporadic AD dementia. Cognitive performance was evaluated using the Novel Object Recognition (NOR) test and the escape latency index in the elevated plus maze (EPM) test. AEA demonstrated a preventive effect against STZ-induced impairments in recognition and non-associative emotional memory. Moreover, STZ-induced cerebroventricular enlargement was mitigated by AEA administration. Notably, key components of synaptic transmission, such as synaptophysin and syntaxin, which were reduced by STZ, showed a reversal after AEA treatment.
Khavandi et al. [38] explored the impact of specific endocannabinoids—AEA, noladin, and O-arachidonylethanolamine (OAE)—on the accumulation and toxicity of Aβ42. Employing in vitro techniques with mouse hippocampal HT22 cells and human CB1R-expressing hamster ovary CHO cells, they found significant inhibition of Aβ42 accumulation by AEA (93.3%), noladin (72.9%), and arachidonic acid (AA) (94.5%) at a concentration of 10μM. Moreover, they demonstrated the capacity of AEA (10μM), noladin (10μM), OAE (1μM, 10μM), and AA (5μM, 10μM) to enhance the viability of HT22 cells via CB1R agonism, with only AEA showing no efficacy in CHO cells at concentrations of 1μM, 5μM, and 10μM.
In their study, Hosseininia et al. [39] investigated the impact of chronic corticolimbic microinjection of the selective CB1R agonist arachidonylcyclopropylamide (ACPA; 10 ng/0.5 μL) and lentiviral particles containing miRNA-137 (miR-137) or miR-let-7a on memory-impaired animal models exposed to i.c.v. STZ. Following ACPA microinjection, the step-through latency of STZ rats in the passive avoidance (PA) test significantly increased compared to controls, indicating enhanced memory function in various brain regions including the hippocampal CA1 region (≈90%), central amygdala (CeA) (≈75%), and medial prefrontal cortex (mPFC) (≈95%). Notably, this effect highlighted the cannabinoid's action across different injection sites. Additionally, the expression levels of the MAGL gene, directly linked to the ECS, were observed to decrease (resulting in increased endocannabinoids) in all targeted regions of STZ rat brains receiving miRNA-137 or -let-7a-carrying lentivirus particles, effectively reversing the amnesic effects of STZ.
In another study, Zhang et al. [40] explored the effects of CB1R peptide agonists (m)RVD-hemopressin (RVD) and (m)VD-hemopressin (VD), on memory in Aβ1-42-lesioned mice. Through i.c.v. injection of RVD or VD (5 nmol), they found restoration of Aβ1-42-induced memory impairment, with mice displaying significantly higher discrimination indices in both the NOR test and the Object Location Recognition (OLR) task. Building upon their prior research, the same group [41] conducted a similar experiment, this time using a mouse model of neurodegeneration affected by an i.p. injection of scopolamine. The results resembled those of the previous study, with RVD and VD (1, 2.5, 5 nmol) demonstrating a dose-dependent restoration of memory function in the NOR and OLR tests, as expected. In an attempt to elucidate the mechanism underlying VD's role in memory restoration, they revealed that VD countered the Aβ1-42-induced elevation of reactive oxidant species (ROS) and malondialdehyde (MDA) in mouse hippocampal neurons. Concurrently, there was an increase in the levels of antioxidant enzymes such as catalase (CAT) and glutathione peroxidase (GPx). Moreover, VD administration resulted in a decrease in the expression of the pro-apoptotic protein Bax, alongside an increase in the expression of the anti-apoptotic protein Bcl-2, shedding light on its apoptotic regulatory mechanisms [42]. In their latest attempt to elucidate the mechanism of action of RVD [43], they observed similar mechanisms of action, this time in HT22 cells treated with scopolamine. Notably, RVD prevented the dysfunction of the brain-derived neurotrophic factor (BDNF)/Tropomyosin receptor kinase B (TrkB)/protein kinase B (Akt) signaling pathway, concurrently boosting the expression of synapsin-1 and postsynaptic density protein 95 (PSD-95) proteins. Adding to these findings, prior research [44] showcased RVD's efficacy in mitigating Aβ1-42-induced TAU protein phosphorylation by inhibiting protein kinase A (PKA) and GSK-3β enzyme activity, as well as curtailing neuronal growth in SH-SY5Y cells. These actions of both RVD and VD, facilitated through CB1R activation, position them as promising candidates for therapeutic interventions aimed at mitigating the characteristic pathogenesis of AD.
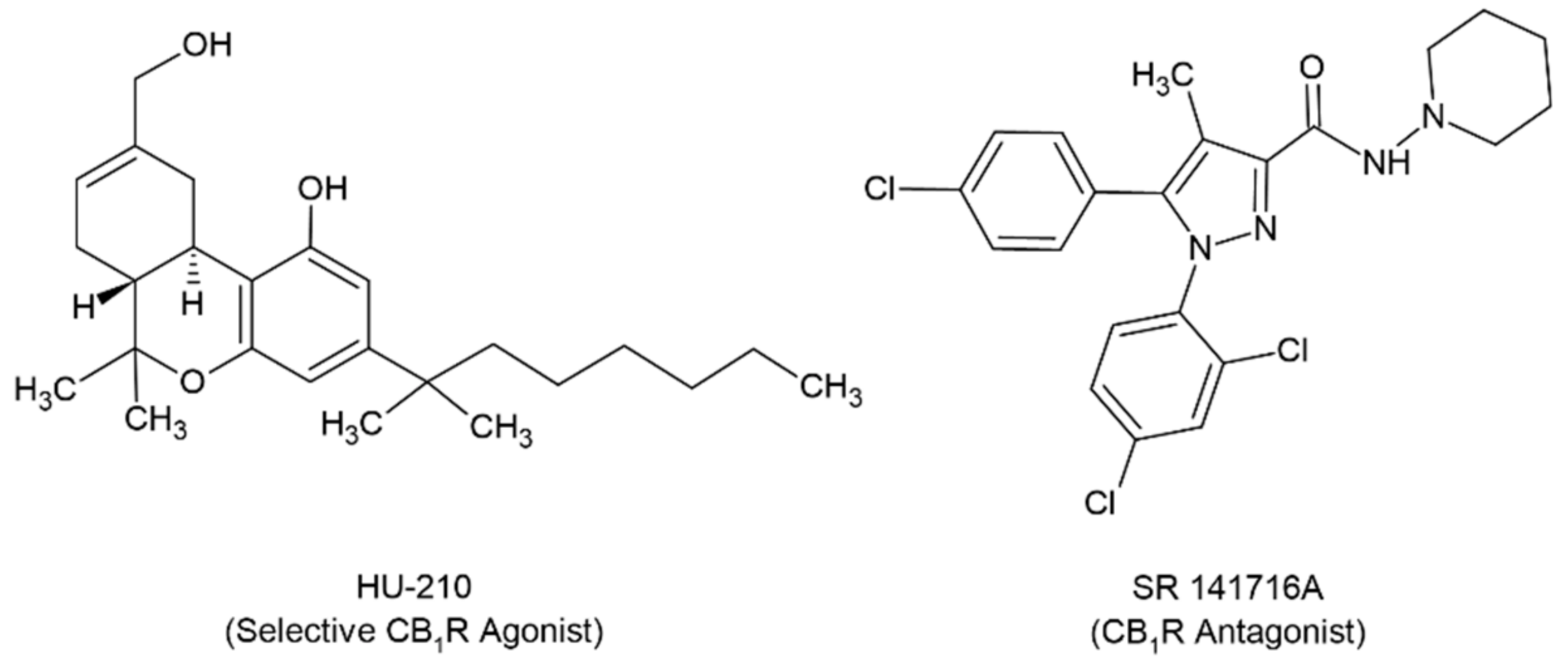
Finally, the research conducted by Velikova et al. [45] focused on examining the role of CB1R in memory and learning processes. Through their study, they administered either the CB1R agonist HU-210 (5 μg/day), or the CB1R antagonist SR 141716A (3 μg/day), via i.c.v. injection to olfactory bulbectomy rats (OBX) (Scheme 1). Their findings revealed that HU-210 improved memory function in OBX rats, whereas SR 141716A exacerbated the memory deficits induced by bulbectomy, as evidenced by active/passive avoidance tests. This underscores the direct involvement of the CB1R in memory function in OBX rats, serving as a model simulating AD symptomatology.
The main outcomes of the studies involving selective CB1R agonists are summarized in Table 1.
3.2. Selective Agonists of Cannabinoid Receptor 2 (CB2R)
The study conducted by Jayant et al. [46] explored the impact of the CB2R agonist, 1-phenylsatin, in a mouse model of NA induced by either STZ or aluminum chloride (AlCl3) + D-galactose (D-Gal). Oral administration of 20mg/kg 1-phenylsatin restored escape latency and time spent in the target quadrant to normal levels more effectively than donepezil in the Morris Water Maze (MWM) test. In the attentional set shifting test, 1-phenylsatin reduced efforts in the reversal 1 (REV1) and extra-dimensional (EV) stages, while also mitigating biochemical (oxidative stress, AChE activity, etc.) and structural (accumulation of Aβ plaques) brain lesions.
Cheng et al. [47] conducted a study to investigate the anti-inflammatory effects of the CB2R agonist β-caryophyllene (BCP) in APP/PS1 mice, focusing on its action via the PPARγ pathway. Following oral administration of 48mg/kg BCP, significant improvements in escape latency and distance traveled were observed in the MWM test starting from the 3rd day. Moreover, the time spent in the target quadrant closely resembled that of healthy mice. BCP administration also dose-dependently reduced Aβ deposition in the cerebral cortex and hippocampus. Additionally, the study measured reduced levels of astrogliosis, microglial activation, cyclooxygenase-2 (COX-2), and of the expression of pro-inflammatory cytokines TNF-a and IL-1β. Overall, there was restoration of spatial memory and cognitive functions by BCP.
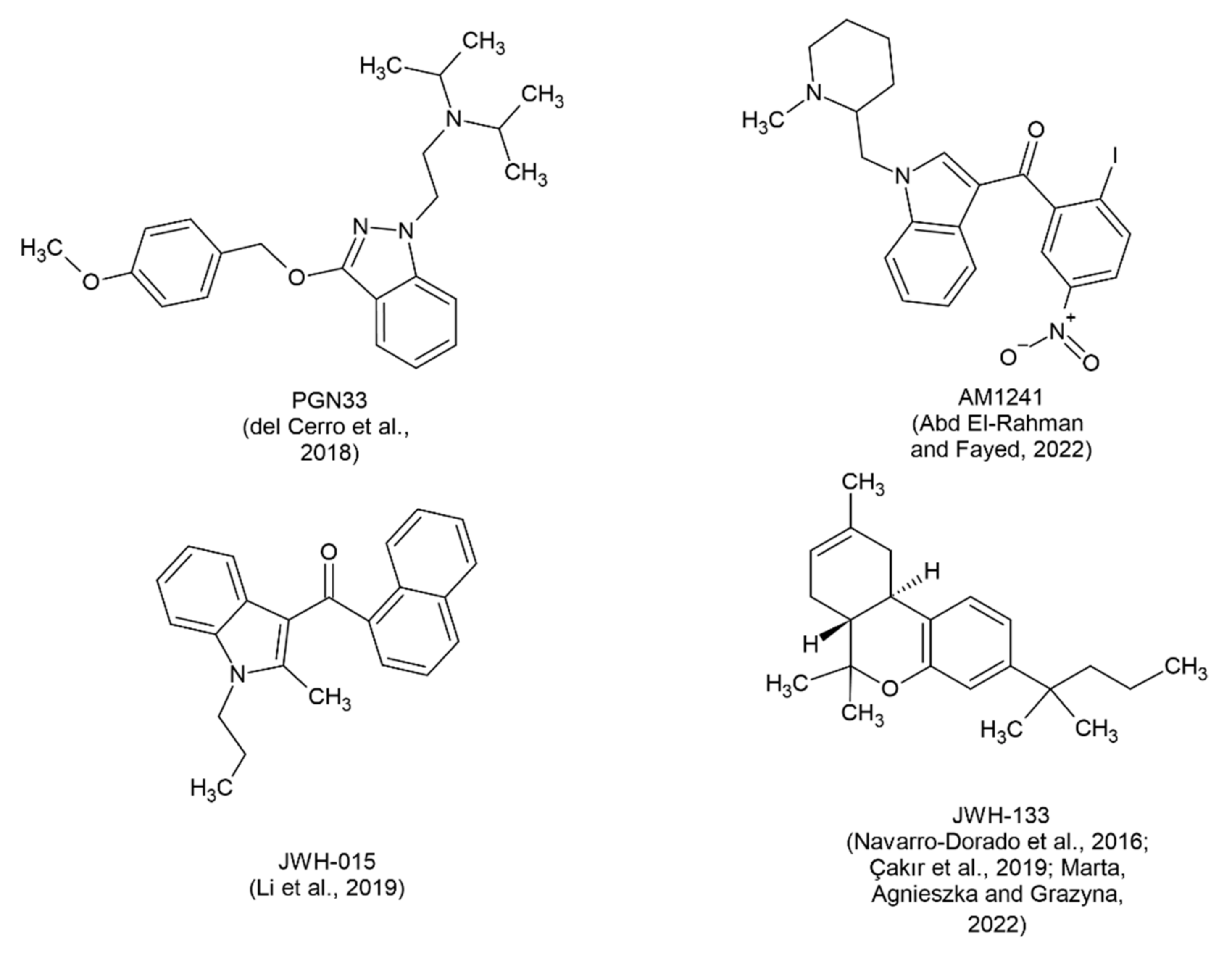
Del Cerro et al. [48] demonstrated the effectiveness of the innovative CB2R agonist, PGN33 (Scheme 2), in influencing the viability of lymphoblasts isolated from late-onset AD patients. PGN33 administration at varying doses (2.5nM, 5nM, 7.5nM, 10nM) dose-dependently decreased the viability of NA-lymphoblasts, limiting their uncontrolled proliferation with a potency similar to that of the agonist JWH-133. This mechanism is achieved by impeding Ca2+/CaM-dependent activation of phosphoinositide 3-kinase (PI3K)/Akt signaling, which typically triggers cell cycle activation. Additionally, PGN33 demonstrated efficacy in mitigating Aβ-induced death of SH-SY5Y neuroblastoma cells.
Abd El-Rahman and Fayed [49] aimed to investigate the potential protective effects and mechanisms of action of the CB2R agonist AM1241 (Scheme 2) in alleviating cognitive and learning deficits in a model of AD in female rats (oophorectomy + D-gal). Administration of AM1241 led to a dose-dependent increase in the discrimination index [0.66±0.02 and 0.95±0.04 for 3 and 6 mg (i.p.), respectively] as well as the preference index (1.06±0.05 at 6mg) compared to the AD-model values in the NOR test. Concurrently, 6mg AM1241 significantly reduced the escape latency of the rats compared to the AD-model (10.38±0.95 vs. 39.08±3.52) in the MWM test. These improvements were accompanied by a decrease in inflammatory signaling via the toll-like receptor 4 (TLR4)/nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) p65 pathway, evidenced by downregulation of the adapter protein myeloid differentiation primary response 88 (Myd88) and enhancement of cAMP response element-binding protein (CREB)/ brain-derived neurotrophic factor (BDNF) expression. Additionally, AM1241 inhibited astrocyte and microglial cell activation, highlighting the importance of CB2R activation in AD treatment.
Li et al. [50] investigated the effects of CB2R activation via i.p. administration of 0.5 mg/kg of the selective CB2R agonist JWH-015 (Scheme 2) in transgenic APP/PS1 mice. Following the NOR and MWM tests, CB2R activation was found to enhance novel object recognition but did not ameliorate the impaired hippocampus-dependent spatial memory in mice. Additionally, using ionized calcium-binding adaptor molecule 1 (Iba1) immunofluorescence and real-time PCR techniques, the researchers observed that the agonist prevented the activation of cortical microglia while simultaneously promoting their conversion from M1 to M2 phenotype, demonstrating the immunoprotective effect of activated CB2R. Moreover, Golgi staining revealed improvements in dendritic complexity in the cortex of APP/PS1 mice.
The present study conducted by Çakır et al. [51] describes an experiment utilizing a rat model with hyperphosphorylated TAU induced by okadaic acid (OKA) administration, wherein the CB2R agonist JWH-133 (0.2 mg/kg, i.p.) (Scheme 2) was administered. During the 1st to 4th day of the MWM test, reductions were observed in both escape latency (85sec - 45sec vs. 95sec - 35sec) and distance traveled (1500cm - 900cm vs. 1550cm - 600cm) in the OKA and OKA+JWH-133 groups, respectively. Additionally, decreased levels of caspase-3, phosphorylated TAU, Aβ, TNF-a, and IL-1β were observed in the cerebral cortex and hippocampus of the OKA+JWH-133 group compared to the OKA group. Overall, the agonist JWH-133 prevented the decline of spatial memory, limited the inflammatory response, and consequently reduced neuronal apoptosis, positioning it as a promising novel therapeutic agent.
The main outcomes of the studies involving selective CB2R agonists are summarized in Table 2.
3.3. Agonists of Cannabinoid Receptor 2 (CB2R) Associated with Cholinergic Pathways
In their investigation, Marta et al. [52] explored the relationship between cholinergic pathways and the CB2R agonist JWH-133, as well as their impact on mouse memory following scopolamine administration. Utilizing PA tests in mice, they demonstrated that combining a suboptimal dose of JWH-133 (0.25 mg/kg) with a suboptimal dose of a cholinergic receptor agonist (nicotine, 0.05 mg/kg) significantly improved cognitive performance in mice. Likewise, the co-administration of JWH-133 with an effective dose of a cholinergic receptor antagonist (scopolamine, 1 mg/kg) mitigated the cognitive impairment induced by the antagonist. These findings suggest a significant involvement of CB2R activation in memory processes associated with cholinergic pathways.
Montanari et al. [53] outlined a procedure for synthesizing 2-arylbenzofuran derivatives, examining their interactions with cholinesterases and cannabinoid receptors. Among these derivatives, compound 8 (5 μM) (Scheme 3) emerged as a promising candidate for a comprehensive treatment approach. It not only acted as a butyrylcholinesterase (BChE) inhibitor, addressing cholinergic impairment, but also displayed neuroprotective properties against Aβ1-42 oligomers. Furthermore, it exhibited potent and selective agonism toward CB2R, offering immunoregulatory effects by prompting a shift in microglial cells from the inflammatory (M1) to the neuroprotective (M2) phenotype. Conversely, compound 10 (5 μM) (Scheme 3) demonstrated robust immunomodulatory activity as an inverse agonist of CB2R, suggesting its potential as a foundational model for the development of other immunomodulatory pharmaceutical agents.
Spatz et al. [54] focused on synthesizing and investigating hybrid molecules acting as CB2R agonists and BChE antagonists. Among the compounds they synthesized, 15d (with IC50 BChE= 0.62 µM, EC50 CB2R= 244 nM) and 21d (with IC50 BChE= 0.15 µM, EC50 CB2R= 1.3 µM) (Scheme 3) demonstrated considerable promise. These compounds exhibited CB2R-dependent immunomodulatory effects, particularly by attenuating the inflammatory M1 phenotype in lipopolysaccharide (LPS)-treated N9 microglial cells. Moreover, when administered i.p. to mice challenged with Aβ25–35 oligomers, compound 15d (at doses of 0.3–3 mg/kg/day) effectively prevented learning impairments in both the spontaneous alternation Y-maze and PA tests. The dual action of these substances, coupled with their ability to cross the blood-brain barrier (BBB), suggests their potential therapeutic utility against AD.
In their study, Scheiner et al. [55] attempted to merge the therapeutic benefits of AChE inhibitors with the neuroprotective properties of CB2R agonists by designing hybrid synthetic analogs of tacrine and a selective CB2R agonist. Among these compounds, 3e, 4a, and 8 (Scheme 3) exhibited neuroprotective effects in a cellular model of neuronal oxidative stress, with compound 8 (at 5 µM) proving to be the most potent, while compound 4a displayed toxicity at doses exceeding 5 µM. Subsequently, in AD mouse models, administration of compounds 3e (at 0.3 mg/kg), 4a (at 1 mg/kg), and 8 (at 0.3 mg/kg) effectively prevented Aβ25-35 infusion-induced memory impairments and learning deficits. Importantly, these compounds demonstrated significantly greater efficacy compared to the parent molecules, with the ability to penetrate the BBB.
The main outcomes of the studies involving compounds targeting CB2R and cholinergic pathways are summarized in Table 3.
3.4. Non-Selective Agonists of Cannabinoid Receptors 1 and 2 (CB1R and CB2R)
The study conducted by Cao et al. [56] explored the potential therapeutic benefits of THC in N2a/AβPPswe cells. Through a series of experiments, they established a time-dependent (6h, 24h, 48h) and dose-dependent (0.25nM, 2.5nM, 25nM, 250nM, 2500nM) relationship between THC administration and the reduction of Aβ40 levels. This reduction occurred alongside the inhibition of Aβ40 aggregation, all without compromising the normal immune response. Additionally, repeated THC administration over 24 h led to further decreases in Aβ protein production. Furthermore, a dose-dependent correlation emerged between THC and the reduction of GSK-3β levels, as well as phosphorylated TAU.
In their study, Gugliandolo et al. [57] investigated the impact of pretreating SH-SY5Y cells with 20μM Δ8-THC before exposure to retinoic acid and subsequent treatment with Aβ1-42. They observed that pretreatment with Δ8-THC elevated cell viability from approximately 75% to about 87%, without inducing any cytotoxic effects upon direct administration. Further analysis revealed that this effect was partly attributed to the attenuation of endoplasmic reticulum (ER) stress induced by Aβ1-42. Specifically, Δ8-THC restored proteostasis by upregulating the expression of proteasome subunits (PSMB5) and ubiquitin, while concurrently suppressing the unfolded protein response (UPR). Additionally, a reduction in Bax protein levels accompanied by an increase in Bcl-2 levels was noted.
Due to the promising outcomes observed in prior studies involving THC, a recent randomized controlled trial [58] focused on the oral administration of THC (1.5 mg thrice daily) or a placebo over a span of 21 days. This trial involved 50 patients with dementia, including AD, and accompanying neuropsychiatric symptoms (NPS). Despite earlier encouraging findings, this study revealed no statistically significant disparities in both the total Neuropsychiatric Inventory (NPI) score and its various subscales (such as agitation/aggression and aberrant motor behavior) between the THC and placebo cohorts (mean difference in NPI total = 3.2, 95% CI -0.8 to 1.9). Similar trends were observed across other assessment metrics like the Cohen-Mansfield Agitation Inventory (CMAI), Quality of Life-Alzheimer's Disease, and the Barthel Index. Additionally, no severe adverse effects attributable to the treatment were documented. Hence, while THC administration was well-tolerated, its therapeutic efficacy for managing dementia-related NPS did not demonstrate discernible benefits.
In their specific research, Coles et al. [59] conducted a series of experiments in female APPxPS1 mice. They demonstrated that the chronic daily administration of a moderate dose (5mg/kg, i.p.) of CBD could have beneficial effects in AD. These experiments aimed to observe potential improvements in AD symptoms such as anxiety, disinterest in exploration, hyperactivity, cognitive and motor dysfunction, as well as sensorimotor impairment. Despite encountering unsuccessful outcomes in the majority of experiments, CBD administration seemed to restore both spatial learning speed and perseveration, along with the ability to recognize novel objects.
In this study [60], CBD administration was explored as a potential safeguard for synaptic plasticity in slices from the CA1 region of the hippocampus in C57Bl/6 mice. The researchers utilized hippocampal long-term potentiation (LTP), a marker of synaptic strength limited by Aβ, to assess the impact. Treating hippocampal slices with CBD before Aβ injection seemed to prevent the decline of LTP, restoring it to levels comparable to the control group. Through the use of various antagonists, it was determined that CBD's specific therapeutic effect is attributed to the activation of the PPAR-γ receptor, with the CB1R not directly involved.
In the study conducted by Amini and Abdolmaleki [61], the efficacy of nano-chitosan-coated CBD administration was evaluated in AD-model rats to assess its impact on learning and memory. Results from the MWM test indicated a significant decrease in both escape latency and distance traveled. Notably, rats treated with 120mg/kg CBD spent more time on the exit platform. Concurrently, there was an observed increase in the expression of CB1R and CB2R in the hippocampus. These findings underscore the potential of coating CBD with nano-chitosan to enhance memory and learning processes.
Hao and Feng [62] focused on uncovering the mechanisms behind CBD's effects on AD by analyzing gene expression data from RNA sequencing experiments conducted on APP/PS1 mice following cannabinoid administration. Their analysis of differentially expressed genes (DEGs) revealed an up-regulation of factors involved in both the immune response and the cellular autophagy pathway. This suggests that CBD's therapeutic actions stem from two primary mechanisms: firstly, by curbing neuroinflammation through bolstering the immune response, and secondly, by impeding AD's pathological processes through the induction of cellular recycling mechanisms.
Building upon the encouraging outcomes mentioned earlier, Alexandri et al. [63] conducted the present study to assess the efficacy of CBD compared to standard treatment over a span of 6 months for managing dementia-related NPS. The administration of 3% CBD resulted in a significant reduction in the NPI index, indicating an improvement in the behavioral and psychological symptoms of dementia (BPSD) among the 20 patients involved. Conversely, classical treatment showed limited or no effects during this timeframe. These findings suggest that CBD might offer a more effective and safer alternative for treating BPSD, although larger clinical studies are warranted to validate these results.
In a limited-sized cohort study [64], the administration of cannabis extract dissolved in oil (22% THC, 0.5% CBD, maximum dose 1ml/day) was investigated in 30 patients diagnosed with moderate to severe AD. The findings from the NPI questionnaire revealed a noteworthy improvement in key behavioral issues, including restlessness, irritability, sleep disturbances, and apathy. Moreover, both physical and verbal aggression behaviors exhibited notable reductions, as evidenced by the results of the CMAI questionnaire. Remarkably, following the administration of the cannabis extract, 45% of patients achieved a Mini Mental State Examination (MMSE) score indicative of mild to moderate cognitive improvement (15-17). Despite these promising results, the study's limited sample size underscores the necessity for larger-scale investigations to ascertain the efficacy of cannabis in the symptomatic treatment of AD.
In the specific case report by Ruver-Martins et al. [28], the focus is on a 75-year-old patient diagnosed with mild AD, presenting with primary symptoms of memory impairment and spatiotemporal disorientation. Over a period of 22 months, the patient underwent treatment with small doses of cannabis extract, characterized by a THC:CBD ratio of 8:1. Throughout this treatment duration, the patient underwent evaluation using the MMSE and the Alzheimer's Disease Assessment Scale-Cognitive Subscale (ADAS-COG), revealing significant improvement, as evidenced by notable increases and decreases in respective scores. The doses of the cannabis extracts varied over the months, with a dosage of 500 μg being the most frequently administered. Remarkably, the treatment led to marked improvement in symptoms, corroborated both by the test results and the patient's subjective experience. The patient opted to continue the treatment, maintaining stable test results (MMSE → 24, ADAS-COG → 10).
In their study, Kim et al. [65] explored the impact of intrahippocampal injection of cannabidiol acid (CBDA, 6µM, 3 µL) and tetrahydrocannabinolic acid (THCA, 12µM, 3 µL) in Aβ1-42-injected ICR mice. Notably, mice exhibited a significant improvement in escape latency during day 4 of the MWM test when administered either THCA (≈35 s) or CBDA (≈25 s), compared to the Aβ group (≈45 s). Additionally, both cannabinoids demonstrated an equivalent increase in the discrimination index in the NOR test. Furthermore, there was a noteworthy reduction in the levels of Aβ polymers (Aβ group: 202%, CBDA: 66%, THCA: 81%, vs. control) and p-TAU (Aβ group: 160%, CBDA: 116%, THCA: 105%, vs. control) in the hippocampus of mice. These findings suggest that cannabinoids achieved restoration of cognitive functions, coupled with neuroprotective properties and ability to penetrate the BBB.
In another case report [66], a 69-year-old woman diagnosed with AD presented severe NPS including depression and paranoid perceptions. From 2008 to 2019, her condition was regularly monitored, during which she underwent treatment with six different psychotropic medications, yielding no improvement. In the final two years of observation, the patient was prescribed dronabinol (Δ9-THC) drops at a dosage ranging from 4.9mg to 6.7mg per day. Remarkably, this intervention enabled the simultaneous discontinuation of three out of the six previously administered psychotropic drugs. Consequently, an improvement in the patient's emotional state was noted, alongside a reduction in disruptive behavior, aggression, and sedation, with no adverse effects reported.
In their study, Long et al. [67] administered the AEA analog, N-linoleyltyrosine (NlTyr), to APP/PS1 mice. Notably, on the 7th day of the Rotarod test (RRT), mice treated with NlTyr exhibited increased time spent on the rod (control: 106.6 ± 3.7 s, APP/PS1: 66.25 ± 7.29 s, NlTyr 60 mg/kg: 101.75 ± 5.56 s), indicating a restoration of motor coordination. Concurrently, NlTyr-treated mice displayed improved cognitive and learning abilities, as evidenced by a return to normal escape latency values on the 6th day of the MWM test (control: 42.38 ± 7.73 s, APP/PS1: 53.00 ± 4.41 s, NlTyr 60 mg/kg: 43.13 ± 5.41 s) and enhanced time spent in the target quadrant. Moreover, through the induction of cannabinoid receptor-mediated autophagy, NlTyr effectively reduced Aβ42 levels in the hippocampal CA1 region (APP/PS1: 26.33 ± 8.19 ng/g, NlTyr 30 mg/kg: 15 ± 1.63 ng/g), thereby mitigating neuronal injury.
In another study [68], the objective was to explore the potential anti-inflammatory effects of the cannabinoid agonist β-amyrin in rat microglial cells treated with LPS/interferon-γ (IFN-γ). Results revealed that β-amyrin, at concentrations ranging from 4 to 16 µM, not only enhanced cell survival rates without inducing any toxicity but also elicited a significant reduction in the levels and expression of pro-inflammatory cytokines such as TNF-a, IL-1β, IL-6, and prostaglandin E2 (PGE-2). Additionally, it downregulated the expression of COX-2. Interestingly, β-amyrin also modulated the gene expression ratio of arginase-1/inducible nitric oxide synthase (iNOS) and the urea/nitric oxide (NO) ratio, indicating a shift towards an M2 (anti-inflammatory) state in microglial cells.
In their randomized double-blind crossover clinical trial, Herrmann et al. [69] showcased the efficacy of nabilone, a synthetic cannabinoid derivative (administered at 1-2mg daily for 6 weeks), in addressing restlessness observed in patients with moderate to severe AD, despite standard treatments, when compared with a placebo. Utilizing various assessment tools including the CMAI (with a reduction of b = -4.0), the Neuropsychiatric Inventory-Nursing Home (NPI-NH) (with a reduction of b = -4.6), the Mini-Nutritional Assessment Short-Form (MNA-SF) (with a reduction of b = -1.7), and the Standardized Mini-Mental State Examination (sMMSE) (with an increase of b = +1.1), they observed the superiority of nabilone over placebo. However longer trials are definitely needed.
In a different study [70], the effect of the synthetic CBR agonist WIN 55,212-2 (Scheme 4) on astrocytes from the cerebral cortex of rats was investigated, particularly when exposed to the toxic peptide Aβ1-42. Remarkably, pre-treatment of the cells with 10µM WIN 55,212-2 significantly enhanced their viability while preventing the Aβ1-42-induced surge in proinflammatory cytokines IL-1β and TNF-a. Concurrently, the application of this agonist notably diminished the expression of the p65 protein and inflammatory proteins COX-2 and iNOS, while increasing the expression of the transcription factor PPAR-γ and the antioxidant enzyme Cu/Zn superoxide dismutase (SOD).
In their study, Mahdi et al. [71] investigated the therapeutic potential of WIN55,212-2 at varying doses (0.5, 1, 2mg/kg) in an AD rat model, induced by injection of aluminum chloride (AlCl3) and D-galactose (D-Gal). Notably, on days 4-5, statistically significant differences were observed in escape latency and time spent in the target quadrant among the different groups in the MWM test. Biochemical analyses of their brains revealed a significant reduction in MDA levels, along with a restoration of the antioxidant molecules glutathione (GSH) and SOD to higher levels. Additionally, administration of WIN55,212-2 mitigated cellular abnormalities in the hippocampus of rats by promoting the production of the proteins nestin and glial fibrillary acidic protein (GFAP), which serve as markers of neurogenesis.
In their study, Gajardo-Gómez et al. [72] investigated the potential of single administrations (5μM) of synthetic and endogenous cannabinoid agonists, including WIN-55,212-2, 2-AG, and methanandamide, to mitigate neuronal death triggered by the opening of astroglial Cx43 hemichannels—an event linked to the excitotoxic release of ATP and glutamate. Their findings revealed that these agonists effectively prevented the increase in the number of surface Cx43 hemichannels, thereby significantly reducing their activity in both astroglia and hippocampal pyramidal cells incubated with Aβ25-35. Moreover, the same agonists demonstrated a notable decrease in the secretion of glutamate (from 200 to 24, 25 and 27 pmol/mg, respectively) and ATP (from 86 to 12, 13 and 14 pmol/mg, respectively) in pyramidal cells, resulting in reduced rates of pyramidal neuron death. These actions were primarily mediated by CB1R.
In the study conducted by Soto-Mercado et al. [73], the action of the non-selective CBR agonist, CP55-940 (Scheme 4), in PSEN1 E280A cells—a model of familial AD—was elucidated. CP55-940 demonstrated the capability to inhibit both intracellular sAβPPβf aggregation and TAU phosphorylation, while restoring mitochondrial membrane potential (ΔΨm) to normal levels, particularly when used in combination with the CB1R inverse agonist, SR141716. Simultaneously, this combination effectively curtailed the formation of ROS and suppressed the activation of transcription factors p53 and c-Jun, as well as the expression of p53 upregulated modulator of apoptosis (PUMA) protein and caspase-3, which are markers of apoptosis. However, the combination failed to reverse the dysfunction of ACh-induced Ca2+ influx, crucial for neuronal function, which was only achieved after co-administration of an anti-Aβ42 antibody. Thus, the synergistic approach of combining cannabinoids with CB1R inverse agonists and anti-Aβ42 antibodies holds promise as a decisive therapeutic strategy for treating familial AD.
The main outcomes of the studies involving non-selective agonists of CB1R and CB2R are summarized in Table 4.
3.5. Non-Selective Agonists of Cannabinoid Receptors 1 and 2 (CB1R and CB2R) Related to Cholinergic Pathways
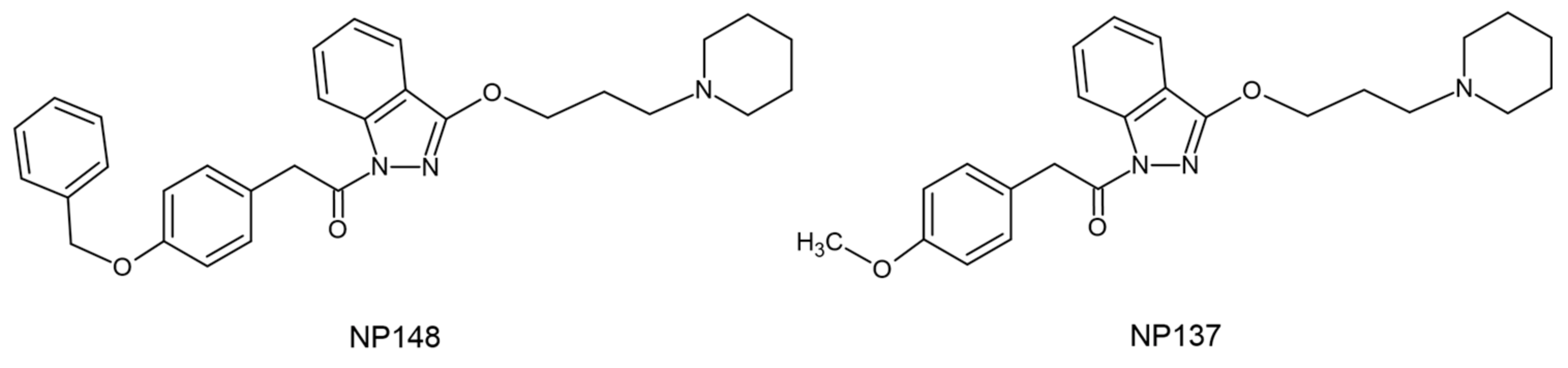
Nuñez-Borque et al. [74] conducted a study to examine the effects of two CBR agonists, NP137 and NP148 (Scheme 5), in both immortalized lymphocytes from patients with delayed onset AD and TgAPP mice. These agonists demonstrated inhibitory effects on β-secretase-1 (BACE-1) and BACE-1/BChE. Initially, the study revealed a significant attenuation of Aβ-induced cell death in neural cortical cells following pretreatment with NP137 or NP148 (2.5 and 5 µM). Moreover, through the MWM test, long-term administration of NP137 (1 mg/kg/day orally) to TgAPP mice effectively restored their cognitive functions, evidenced by a reduction in escape latency comparable to that of the control group. Additionally, the addition of NP137 (5 µM) was found to attenuate the increased proliferative activity of AD cells and normalize ERK1/2 phosphorylation and p21 content in AD lymphoblasts.
Scheme 5.
Chemical structures of the non-selective CB1R and CB2R agonists associated with cholinergic pathways synthesized by [74].
Scheme 5.
Chemical structures of the non-selective CB1R and CB2R agonists associated with cholinergic pathways synthesized by [74].
3.6. Molecules That Act through Pathways Related to the Endocannabinoid System
In the study conducted by Balleza-Tapia et al. [75], the focus was on investigating the potential restorative effects of activating the ionotropic cannabinoid receptor Trpv-1 [76], in counteracting the Aβ-induced impairment of gamma oscillations in hippocampal cells. Through receptor activation using capsaicin (10μM), the study observed a significant prevention of neuronal degeneration. This intervention also reversed the desynchronization of action potential emission in CA3 pyramidal cells and restored the balance between excitatory and inhibitory potential emissions.
In their study, Xiang et al. [77] investigated the impact of G protein-coupled receptor 55 (GPR55) activation [78] using the agonist O-1602 (Scheme 6) on murine neurotoxicity induced by Aβ1-42 administration. Through the MWM and NOR tests, they demonstrated that i.c.v. injection of O-1602 (at doses of 2.0 or 4.0 μg/mouse) mitigated cognitive impairment by reducing the levels of soluble Aβ1-42 in the hippocampus and frontal cortex. Notably, the administration of the agonist reversed the downregulation of the GPR55 and concurrently led to a decrease in Ras homolog family member A (RhoA) and Rho associated coiled-coil containing protein kinase 2 (ROCK2) proteins.
In their subsequent research [79], the same team explored the activity of O-1602 in a murine model of STZ-induced neurotoxicity. Building upon their previous findings, they observed not only a further decrease in BACE1 activity but also a reduction in oxidative stress markers (such as decreased MDA and increased levels of GSH, SOD, and CAT). Additionally, the administration of this agonist not only ameliorated synaptic dysfunction by up-regulating the PSD-95 protein, thereby improving synaptic plasticity, but also mitigated the levels of pro-inflammatory cytokines and reduced microglial activation. Moreover, the study highlighted O-1602's ability to prevent the STZ-induced up-regulation of AChE.
In a complementary study conducted by Wang et al. [80], the same agonist was employed in LPS-challenged ICR mice to investigate its impact on induced cognitive impairment. The novel findings of this research included, among other results, the attenuation of not only the expression of NF-kB p65 and Bax protein but also the activity of caspase-3. Conversely, there was an increase in the expression of both Bcl-2 and anti-inflammatory cytokines. Additionally, a notable contribution of O-1602 to the reduction of hippocampal cell apoptosis was observed through TUNEL-positive cells.
The main outcomes of the studies involving non-selective agonists of CB1R and CB2R associated with cholinergic pathways and molecules that act through pathways related to the ECS are summarized in Table 5.
3.7. Combination Studies of Agonists of Different Classes
The study conducted by Elmazoglu et al. [81] aimed to explore the potential of a range of cannabinoid agents (ranging from 1 to 1000nM) in a primary rat hippocampal neuron model of toxic hyperglycemia and Aβ1-42 treatment. The objective was to enhance cell viability by activating nuclear factor erythroid 2-related factor 2 (Nrf2) and reducing oxidative stress and inflammation. Among the tested agents, URB597 [a fatty acid amide hydrolase (FAAH) inhibitor, Scheme 7] emerged as the most effective in promoting cell survival and suppressing ROS formation. Subsequently, synthetic cannabinoids WIN55,212-2 and CP55-940, followed by endocannabinoids 2-AG and AEA, exhibited efficacy in this regard. Additionally, URB597, particularly in combination with AEA, demonstrated effectiveness in limiting Aβ aggregation. Furthermore, all tested agents induced an increase in antioxidant enzymes including SOD, CAT, GPx, and glutaredoxin (GRx), along with Nrf2, to mitigate inflammation.
Navarro-Dorado et al. [82] explored the alterations in vascular function within the AD brain, examining the effects of prolonged oral administration (at 0.2mg/kg/day) of synthetic cannabinoids WIN55,212-2 and JWH-133 in TgAPP mice. Both agonists were found to normalize the elevated levels of collagen IV positive vessels in the frontal cortex, thereby reducing collagen IV vascular density. While the dilator effect of cannabinoids was limited in the aortic valve of TgAPP mice compared to controls, their administration effectively prevented Aβ-induced desensitization of the vasodilatory action of ACh.
Finally, another study [83] investigated the neuroprotective potential of 11 non-psychoactive cannabinoids using a preclinical drug screening platform for AD. They conducted various assays in HT22 or MC65 cells after inducing C99 production, which represented proteotoxicity, loss of trophic factors and energy, and oxidative stress. Additionally, they examined cannabinoids for their ability to reduce accumulated Aβ. Notably, Δ9-THC (EC50= 100nM) and Δ8-THC (EC50= 85nM) demonstrated efficacy in preventing Aβ toxicity. Furthermore, cannabinoids were assessed for their capacity to suppress the pro-inflammatory response of microglial cells to LPS, with only CBD, dimethyl cannabidiol, cannabigerolic acid, and Δ9-THC exhibiting EC50 values <10 µM in this regard. Importantly, this study revealed that the neuroprotection offered by the tested cannabinoids is independent of the activation of CB1R and CB2R, as none of the cells in the study expressed them.
The main outcomes of the combination studies with agonists from different classes are summarized in Table 6.
4. Discussion
The analysis began with a focus on CB1R selective agonists, which included ACEA, AEA, Noladin, OAE, RVD, VD, and HU-210. Across various studies [36,38,42,43,44], these cannabinoids consistently demonstrated positive effects. Specifically, ACEA, AEA, Noladin, OAE, RVD, and VD were noted to partially or completely restore cell viability. Notably, RVD and VD also increased antioxidant enzymes GPx and CAT, as highlighted in relevant studies [42,43]. This led to a reduction in oxidative stress through the BDNF/TrkB/Akt pathway. In an attempt to enhance memory function, co-administration of ACPA and miRNA-137/-let-7a lentiviral particles was explored, resulting in increased endocannabinoids via induction of the MAGL gene [39]. Additionally, ACEA, AEA, and HU-210 showed significant memory function improvement in AD-mice across several studies [36,37,40,41,45], emphasizing the direct correlation between CB1R and memory function, as underscored in [45], consistent with other findings.
Similarly, studies on CB2R selective agonists consistently demonstrated a direct correlation between receptor activation and memory recovery. Experiments utilizing the MWM and NOR tests [46,47,49,51] supported this correlation. Notably, a decrease in pro-inflammatory cytokines such as TNF-a, IL-1β and COX-2, and apoptotic enzymes such as caspase-3, was observed [47,51], further supported by [48], who found that PGN33 reduced the death rate of Aβ-challenged neuroblastoma cells. These findings suggest that CB2R agonists possess anti-inflammatory and immunomodulatory properties possibly via suppression of the TLR4/NF-κB p65 pathway [49].
Expanding on these findings, the positive association of CB2R with memory, particularly involving cholinergic pathways was demonstrated [52]. This association was corroborated by several other studies [53,54,55], who studied synthetic molecules combining CB2R agonist function with AChE/BChE inhibition. These compounds showed effective prevention of memory impairments, neuroprotection and cholinergic repair without psychotropic effects. Cholinergic pathways were also investigated using non-selective agonists NP137 and NP148, showing promising results in in vitro and in vivo conditions [74].
Moving to non-selective CB1R/CB2R agonists, this category includes natural cannabinoids such as THC, CBD, THCA/CBDA, and various Cannabis extracts. THC studies yielded conflicting results, with some showing reductions in Aβ40 production and TAU phosphorylation possibly through mitigating ER stress [56,57], while others found no significant difference compared to controls, possibly due to methodological considerations [58]. To account for the observed Hawthorne or in-study effect [84], an individually randomized crossover study design may prove beneficial. CBD testing on the other hand, consistently demonstrated promising results, effectively reversing a wide spectrum of traits associated with AD [59,60,61,62,63]. The therapeutic actions of CBD and other cannabinoids partially stem from the activation of the PPAR-γ receptor [47,60,70], further emphasizing the need for its more extensive association with AD in future studies. Limited-sized studies [28,64,66] further supported this notion, showing significant cognitive improvement and emotional stabilization in AD patients after oral cannabis extract or dronabinol administration.
Equally noteworthy were the findings concerning the synthetic, non-selective cannabinoid nabilone from a controlled clinical trial [69], which showcased its effectiveness in combating NPS and restoring cognitive functions, advocating for its broader therapeutic utilization, even though sedation should be considered. Another synthetic cannabinoid of interest, WIN-55,212-2, exhibited anti-inflammatory and neuroprotective effects attributed to the modulation of Cx43 hemichannel activity in hippocampal astroglial cells [72], as well as the augmentation of antioxidant enzymes and neurogenic proteins [71], alongside the reduction of pro-inflammatory cytokines such as TNF-a and IL-1β. PPAR-γ upregulation further corroborated its implication in the mechanism of action of cannabinoids [70]. Notably, co-administration of WIN-55,212-2 with the selective CB2R agonist JWH-133 proved effective in mitigating cerebrovascular dysfunction associated with microvascular alterations in AD [82]. Additionally, the agonist CP55-940 demonstrated promising activity in cellular models of familial AD, particularly when co-administered with an anti-Aβ42 antibody, presenting a novel therapeutic approach [73], mirroring the efficacy on cell viability of WIN-55,212-2 in models of toxic hyperglycemia and Aβ1-42 [81].
The last class of compounds discussed included molecules that do not directly interact with CBRs, but act on receptors indirectly involved in the ECS. One such receptor is Trpv-1, which is considered by many researchers to be an ionotropic CBR, due to its ability to be activated by endocannabinoids such as AEA [76]. Here, its activation by capsaicin restored the balance of excitatory/inhibitory potentials emission, thereby preventing the decline of hippocampal neurons [75]. Another case of such a receptor is that of GPR-55, which has also been proposed as a candidate CBR [85]. The synthetic receptor agonist O-1602 [77,79,80], exerts its neuroprotective effect through inhibition of the RhoA/ROCK2 pathway, simultaneously exhibiting both anti-inflammatory (reduction of pro-inflammatory cytokines) and anti-apoptotic (reduction of NF-kB p65, caspase-3) activity. At the same time, it improved synaptic plasticity by upregulating PSD-95, while also being able to regulate cholinergic system dysfunction in AD. The activation of these receptors has shown very promising results, paving the way for future trials around their therapeutic applications in AD and indicating a broader spectrum of targets for intervention.
5. Conclusions and Future directions
As understood from the above, cannabinoids exhibit efficacy in reversing several of the manifestations of AD. The number of included studies, both in laboratory settings and clinical trials, provides a rather solid foundation for drawing reliable conclusions. The involvement of several cellular pathways as well as cannabinoid receptors in their mechanism of action holds promise for AD treatment, requiring further investigation. This investigation should also consider the synergistic effect between cannabinoids and other therapeutic methods, such as anti-Aβ42 antibodies and anticholinesterase agents, which have shown promising results. Given the current challenge of treating AD, the therapeutic potential of cannabinoids presents a new focus for research in this field.
However, several key considerations must be addressed to advance the translation of preclinical findings into clinically meaningful outcomes. The following future directions outline crucial areas of focus for researchers, clinicians, and regulatory agencies:
1. Even though our review article thoroughly presented the key findings of each included study aiming at elucidating cannabinoid’s mechanism of action in AD, further exploration of the molecular mechanisms underlying these beneficial effects is imperative. Understanding specific pathways involved in neuroprotection, neuroinflammation modulation, and amyloid plaque reduction will facilitate the development of targeted therapeutic strategies.
2. Given their diverse physicochemical characteristics, standardization of cannabinoid formulations is essential to ensure consistency in dosing and efficacy across studies and clinical trials. Addressing variability in cannabinoid composition, purity, and delivery methods is paramount for reliable and reproducible results.
3. Based on divergent observations in the limited clinical trials and case reports discussed, the optimization of dosage and treatment regimens based on preclinical and early clinical data is necessary. Conducting dose-ranging studies will help identify the most effective and safe doses for AD patients, considering individual variability and disease progression. Apart from that, well-designed, placebo-controlled clinical trials with sufficient statistical power are urgently needed to evaluate the efficacy and safety of cannabinoid-based interventions in AD. Consideration of patient selection criteria, outcome measures, trial duration, and follow-up assessments is essential for robust clinical evidence.
4. Rigorous safety assessments are crucial to address concerns regarding potential adverse effects and long-term consequences of cannabinoid use in AD. Comprehensive evaluation of cognitive, psychiatric, and addictive risks is essential for patient safety.
5. As discussed throughout this article, cannabinoids exhibit synergistic effects with other therapeutic agents that affect cholinergic neurotransmission by inhibiting AChE or BChE or by inhibiting BACE-1 or FAAH, among others. These combinations may enhance treatment outcomes in AD thereby reducing the required doses of both agents. Additionally, cannabinoids should be explored along NMDA receptor antagonists in preclinical and clinical settings.
6. Last but not least, addressing misconceptions and stigma surrounding cannabinoid use is crucial for fostering acceptance and support for cannabinoid-based therapies in AD. Education, public awareness campaigns, and destigmatization efforts are essential to garnering broader societal acceptance.
In conclusion, while the beneficial effects of cannabinoids in AD are promising, careful consideration of future directions is imperative to ensure responsible and evidence-based advancement. By addressing mechanistic insights, standardization, safety concerns, clinical trial design, ethical considerations, translational challenges, and public perception, the field can progress towards realizing the therapeutic potential of cannabinoids for AD patients.
Author Contributions
Conceptualization, M.K.; methodology, P.T. and C.A.; data curation, P.T. and C.A.; writing—original draft preparation, P.T., C.A.; writing—review and editing, M.K.; supervision, M.K. All authors have read and agreed to the published version of the manuscript.
Funding
This work received no external funding.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data sharing is not applicable.
Acknowledgments
The authors would like to thank Athanasios Metaxas, who supported this study with his valuable comments and feedback on the original drafted manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders : DSM-5. 5th ed. American Psychiatric Association; 2013.
- Dunne, T.E. Alzheimer’s Disease: An Overview. Encyclopedia of Mental Health 2016. [CrossRef]
- Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s and Dementia. 2011;7(3):280-292. [CrossRef]
- Takahashi RH, Nagao T, Gouras GK. Plaque formation and the intraneuronal accumulation of β-amyloid in Alzheimer’s disease. Pathol Int. 2017;67(4):185-193. [CrossRef]
- Kim T, Vidal GS, Djurisic M, et al. Human LilrB2 is a β-amyloid receptor and its murine homolog PirB regulates synaptic plasticity in an Alzheimer’s model. Science (1979). 2013;341(6152):1399-1404. [CrossRef]
- Selkoe DJ. THE GENETICS AND MOLECULAR PATHOLOGY OF ALZHEIMER’S DISEASE: Roles of Amyloid and the Presenilins. Neurol Clin. 2000;18(4):903-921. [CrossRef]
- de Paula V de JR, Guimarães FM, Diniz BS, Forlenza OV. Neurobiological pathways to Alzheimer’s disease: Amyloid-beta, TAU protein or both? Dement Neuropsychol. 2009;3(3):188-194. [CrossRef]
- Meyer-Luehmann M, Spires-Jones TL, Prada C, et al. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer’s disease. Nature. 2008;451(7179):720-724. [CrossRef]
- Masters CL, Bateman R, Blennow K, Rowe CC, Sperling RA, Cummings JL. Alzheimer’s disease. Nat Rev Dis Primers. 2015;1. [CrossRef]
- Rhein V, Eckert A. Effects of Alzheimer’s amyloid-beta and tau protein on mitochondrial function - Role of glucose metabolism and insulin signalling. Arch Physiol Biochem. 2007;113(3):131-141. [CrossRef]
- Wagner U, Utton M, Gallo JM, Miller CCJ. Cellular phosphorylation of tau by GSK-3β influences tau binding to microtubules and microtubule organisation. J Cell Sci. 1996;109(6):1537-1543.
- Gao Y, Tan L, Yu JT, Tan L. Tau in Alzheimer’s Disease: Mechanisms and Therapeutic Strategies. Curr Alzheimer Res. 2018;15(3):283-300. [CrossRef]
- Sheppard O, Coleman M. Alzheimer’s Disease: Etiology, Neuropathology and Pathogenesis. In: Alzheimer’s Disease: Drug Discovery. Exon Publications; 2020:1-22. [CrossRef]
- Gajardo-Gómez R, Labra VC, Maturana CJ, et al. Cannabinoids prevent the amyloid β-induced activation of astroglial hemichannels: A neuroprotective mechanism. Glia. 2017;65(1):122-137. [CrossRef]
- Deture MA, Dickson DW. The neuropathological diagnosis of Alzheimer’s disease. Mol Neurodegener. 2019;14(1). [CrossRef]
- Wang JF, Chen S, Gao XD. Research Progress of Alzheimer’s Disease Targets and Related Drugs | 阿尔兹海默症靶点及相关药物研究进展. Chinese Journal of Pharmaceutical Biotechnology. 2021;28(3):323-330. [CrossRef]
- Clark MA, Harvey RA, Finkel R, Rey JA, Whalen K. Pharmacology. 5th ed. Lippincott Williams & Wilkins; 2011.
- Caraci F, Santagati M, Caruso G, et al. New antipsychotic drugs for the treatment of agitation and psychosis in Alzheimer’s disease: Focus on brexpiprazole and pimavanserin. F1000Res. 2020;9. [CrossRef]
- Shi M, Chu F, Zhu F, Zhu J. Impact of Anti-amyloid-β Monoclonal Antibodies on the Pathology and Clinical Profile of Alzheimer’s Disease: A Focus on Aducanumab and Lecanemab. Front Aging Neurosci. 2022;14. [CrossRef]
- Huestis MA. Human cannabinoid pharmacokinetics. Chem Biodivers. 2007;4(8):1770-1804. [CrossRef]
- Boadu O, Gombolay GY, Caviness VS, El Saleeby CM. Intoxication From Accidental Marijuana Ingestion in Pediatric Patients: What May Lie Ahead. Pediatr Emerg Care. 2020;36(6):e349-e354. [CrossRef]
- Oberbarnscheidt T, Miller NS. Pharmacology of Marijuana. Published online 2016. [CrossRef]
- Ronan PJ, Wongngamnit N, Beresford TP. Molecular Mechanisms of Cannabis Signaling in the Brain. Prog Mol Biol Transl Sci. 2016;137:123-147. [CrossRef]
- Ruver-Martins AC, Bicca MA, de Araujo FS, et al. Cannabinoid extract in microdoses ameliorates mnemonic and nonmnemonic Alzheimer’s disease symptoms: A case report. J Med Case Rep. 2022;16(1):277. [CrossRef]
- Li S, Huang Y, Yu L, Ji X, Wu J. Impact of the Cannabinoid System in Alzheimer’s Disease. Curr Neuropharmacol. 2023;21(3):715-726. [CrossRef]
- Lafaye G, Karila L, Blecha L, Benyamina A. Cannabis, cannabinoids, and health. Dialogues Clin Neurosci. 2017;19(3):309-316. [CrossRef]
- Iuvone T, Esposito G, Esposito R, Santamaria R, Di Rosa M, Izzo AA. Neuroprotective effect of cannabidiol, a non-psychoactive component from Cannabis sativa, on beta-amyloid-induced toxicity in PC12 cells. J Neurochem. 2004;89(1):134-141. [CrossRef]
- Ruver-Martins AC, Bicca MA, de Araujo FS, et al. Cannabinoid extract in microdoses ameliorates mnemonic and nonmnemonic Alzheimer’s disease symptoms: a case report. J Med Case Rep. 2022;16(1):277. [CrossRef]
- Monteiro KLC, Dos Santos Alcântara MG, de Aquino TM, da Silva-Júnior EF. Cannabinoid pharmacology and its therapeutic uses in Alzheimer’s disease. Neural Regen Res. 2021;16(5):990-991. [CrossRef]
- Páez JA, Campillo NE. Innovative Therapeutic Potential of Cannabinoid Receptors as Targets in Alzheimer’s disease and Less Well-Known Diseases. Curr Med Chem. 2019;26(18):3300-3340. Available online: https://mc04.manuscriptcentral.com/crmchttps://mc04.manuscriptcentral.com/crmc.
- Tang Y, Bao JS, Su JH, Huang W. MicroRNA-139 modulates Alzheimer’s-associated pathogenesis in SAMP8 mice by targeting cannabinoid receptor type 2. Genetics and Molecular Research. 2017;16(1). [CrossRef]
- Aso E, Andrés-Benito P, Ferrer I. Genetic deletion of CB1 cannabinoid receptors exacerbates the Alzheimer-like symptoms in a transgenic animal model. Biochem Pharmacol. 2018;157:210-216. [CrossRef]
- Campillo NE, Páez JA. Cannabinoid system in neurodegeneration: New perspectives in Alzheimer’s disease. Mini Rev Med Chem. 2009;9(5):539-559. [CrossRef]
- Silvestri C, Di Marzo V. The endocannabinoid system in energy homeostasis and the etiopathology of metabolic disorders. Cell Metab. 2013;17(4):475-490. [CrossRef]
- Komorowska-Müller JA, Schmöle AC. CB2 receptor in microglia: The guardian of self-control. Int J Mol Sci. 2021;22(1):1-27. [CrossRef]
- Crunfli F, Vrechi TA, Costa AP, Torrão AS. Cannabinoid Receptor Type 1 Agonist ACEA Improves Cognitive Deficit on STZ-Induced Neurotoxicity Through Apoptosis Pathway and NO Modulation. Neurotox Res. 2019;35(3):516-529. [CrossRef]
- Moreira-Silva D, Carrettiero DC, Oliveira ASA, et al. Anandamide effects in a streptozotocin-induced Alzheimer’s disease-like sporadic dementia in rats. Front Neurosci. 2018;12(SEP). [CrossRef]
- Khavandi M, Rao PPN, Beazely MA. Differential Effects of Endocannabinoids on Amyloid-Beta Aggregation and Toxicity. Int J Mol Sci. 2023;24(2). [CrossRef]
- Hosseininia M, Rostami F, Delphi L, Ghasemzadeh Z, Kouhkan F, Rezayof A. Memory impairment was ameliorated by corticolimbic microinjections of arachidonylcyclopropylamide (ACPA) and miRNA-regulated lentiviral particles in a streptozotocin-induced Alzheimer’s rat model. Exp Neurol. 2023;370:114560. [CrossRef]
- Zhang R san, He Z, Jin W dong, Wang R. Effects of the cannabinoid 1 receptor peptide ligands hemopressin, (m)RVD-hemopressin(α) and (m)VD-hemopressin(α) on memory in novel object and object location recognition tasks in normal young and Aβ 1–42 -treated mice. Neurobiol Learn Mem. 2016;134:264-274. [CrossRef]
- Zhang R, Lao K, Lu B, et al. (m)RVD-hemopressin (α) and (m)VD-hemopressin (α) improve the memory-impairing effect of scopolamine in novel object and object location recognition tasks in mice. Peptides (NY). 2021;136:170442. [CrossRef]
- Zhang R, Zheng Y, Hu F, et al. Effect of (m)VD-hemopressin against Aβ1-42-induced oxidative stress and apoptosis in mouse hippocampal neurons. Peptides (NY). 2020;124:170185. [CrossRef]
- Zhang R, He X, Cheng J, et al. (m) RVD-hemopressin (α) Ameliorated Oxidative Stress, Apoptosis and Damage to the BDNF/TrkB/Akt Pathway Induced by Scopolamine in HT22 Cells. Neurotox Res. 2023;41(6):627-637. [CrossRef]
- Zhang R, Luan J, Hu F, et al. Effect of (m)RVD-hemopressin against Aβ1-42-induced apoptosis and inhibition of neurite outgrowth in SH-SY5Y cells. Neuropeptides. 2020;81:102044. [CrossRef]
- Velikova M, Doncheva D, Tashev R. Subchronic effects of ligands of cannabinoid receptors on learning and memory processes of olfactory bulbectomized rats. Acta Neurobiol Exp (Wars). 2020;80(3):286-296. [CrossRef]
- Jayant S, Sharma BM, Bansal R, Sharma B. Pharmacological benefits of selective modulation of cannabinoid receptor type 2 (CB2) in experimental Alzheimer’s disease. Pharmacol Biochem Behav. 2016;140:39-50. [CrossRef]
- Cheng Y, Dong Z, Liu S. β-Caryophyllene Ameliorates the Alzheimer-Like Phenotype in APP/PS1 Mice through CB2 Receptor Activation and the PPARγ Pathway. Pharmacology. 2014;94(1-2):1-12. [CrossRef]
- del Cerro P, Alquézar C, Bartolomé F, et al. Activation of the Cannabinoid Type 2 Receptor by a Novel Indazole Derivative Normalizes the Survival Pattern of Lymphoblasts from Patients with Late-Onset Alzheimer’s Disease. CNS Drugs. 2018;32(6):579-591. [CrossRef]
- Abd El-Rahman SS, Fayed HM. Improved cognition impairment by activating cannabinoid receptor type 2: Modulating CREB/BDNF expression and impeding TLR-4/ NFκBp65/M1 microglia signaling pathway in D-galactose-injected ovariectomized rats. PLoS ONE. 2022;17(3 3). [CrossRef]
- Li C, Shi J, Wang B, Li J, Jia H. CB2 cannabinoid receptor agonist ameliorates novel object recognition but not spatial memory in transgenic APP/PS1 mice. Neurosci Lett. 2019;707:134286. [CrossRef]
- Çakır M, Tekin S, Doğanyiğit Z, et al. Cannabinoid type 2 receptor agonist JWH-133, attenuates Okadaic acid induced spatial memory impairment and neurodegeneration in rats. Life Sci. 2019;217:25-33. [CrossRef]
- Marta KS, Agnieszka D, Grazyna B. The Influence of CB2-Receptor Ligands on the Memory-Related Responses in Connection with Cholinergic Pathways in Mice in the Passive Avoidance Test. Molecules. 2022;27(13). [CrossRef]
- Montanari S, Mahmoud AM, Pruccoli L, et al. Discovery of novel benzofuran-based compounds with neuroprotective and immunomodulatory properties for Alzheimer’s disease treatment. Eur J Med Chem. 2019;178:243-258. [CrossRef]
- Spatz P, Steinmüller SAM, Tutov A, et al. Dual-Acting Small Molecules: Subtype-Selective Cannabinoid Receptor 2 Agonist/Butyrylcholinesterase Inhibitor Hybrids Show Neuroprotection in an Alzheimer’s Disease Mouse Model. J Med Chem. 2023;66(9):6414-6435. [CrossRef]
- Scheiner M, Dolles D, Gunesch S, et al. Dual-Acting Cholinesterase–Human Cannabinoid Receptor 2 Ligands Show Pronounced Neuroprotection in Vitro and Overadditive and Disease-Modifying Neuroprotective Effects in Vivo. J Med Chem. 2019;62(20):9078-9102. [CrossRef]
- Cao C, Li Y, Liu H, et al. The potential therapeutic effects of THC on Alzheimer’s disease. Journal of Alzheimer’s Disease. 2014;42(3):973-984. [CrossRef]
- Gugliandolo A, Blando S, Salamone S, et al. Δ8-THC Protects against Amyloid Beta Toxicity Modulating ER Stress In Vitro: A Transcriptomic Analysis. Int J Mol Sci. 2023;24(7):6598. [CrossRef]
- van den Elsen GAH, Ahmed AIA, Verkes RJ, et al. Tetrahydrocannabinol for neuropsychiatric symptoms in dementia. Neurology. 2015;84(23):2338-2346. [CrossRef]
- Coles M, Watt G, Kreilaus F, Karl T. Medium-Dose Chronic Cannabidiol Treatment Reverses Object Recognition Memory Deficits of APPSwe/PS1ΔE9 Transgenic Female Mice. Front Pharmacol. 2020;11. [CrossRef]
- Hughes B, Herron CE. Cannabidiol Reverses Deficits in Hippocampal LTP in a Model of Alzheimer’s Disease. Neurochem Res. 2019;44(3):703-713. [CrossRef]
- Amini M, Abdolmaleki Z. The Effect of Cannabidiol Coated by Nano-Chitosan on Learning and Memory, Hippocampal CB1 and CB2 Levels, and Amyloid Plaques in an Alzheimer’s Disease Rat Model. Neuropsychobiology. 2022;81(3):171-183. [CrossRef]
- Hao F, Feng Y. Cannabidiol (CBD) enhanced the hippocampal immune response and autophagy of APP/PS1 Alzheimer’s mice uncovered by RNA-seq. Life Sci. 2021;264:118624. [CrossRef]
- Alexandri F, Papadopoulou L, Tsolaki A, Papantoniou G, Athanasiadis L, Tsolaki M. The Effect of Cannabidiol 3% on Neuropsychiatric Symptoms in Dementia – Six-Month Follow-Up. Clin Gerontol. Published online May 8, 2023:1-8. [CrossRef]
- Palmieri B, Vadalà M. Oral THC: CBD cannabis extract in main symptoms of Alzheimer disease: Agitation and weight loss. Clin Ter. 2023;174(1):53-60. [CrossRef]
- Kim J, Choi P, Park YT, Kim T, Ham J, Kim JC. The Cannabinoids, CBDA and THCA, Rescue Memory Deficits and Reduce Amyloid-Beta and Tau Pathology in an Alzheimer’s Disease-like Mouse Model. Int J Mol Sci. 2023;24(7). [CrossRef]
- Defrancesco M, Hofer A. Cannabinoid as Beneficial Replacement Therapy for Psychotropics to Treat Neuropsychiatric Symptoms in Severe Alzheimer’s Dementia: A Clinical Case Report. Front Psychiatry. 2020;11. [CrossRef]
- Long C mei, Zheng Q xue, Zhou Y, et al. N-linoleyltyrosine exerts neuroprotective effects in APP/PS1 transgenic mice via cannabinoid receptor-mediated autophagy. J Pharmacol Sci. 2021;147(4):315-324. [CrossRef]
- Askari VR, Fereydouni N, Baradaran Rahimi V, et al. β-Amyrin, the cannabinoid receptors agonist, abrogates mice brain microglial cells inflammation induced by lipopolysaccharide/interferon-γ and regulates Mφ1/Mφ2 balances. Biomedicine & Pharmacotherapy. 2018;101:438-446. [CrossRef]
- Herrmann N, Ruthirakuhan M, Gallagher D, et al. Randomized Placebo-Controlled Trial of Nabilone for Agitation in Alzheimer’s Disease. The American Journal of Geriatric Psychiatry. 2019;27(11):1161-1173. [CrossRef]
- Aguirre-Rueda D, Guerra-Ojeda S, Aldasoro M, et al. WIN 55,212-2, agonist of cannabinoid receptors, prevents amyloid β<inf>1-42</inf> effects on astrocytes in primary culture. PLoS ONE. 2015;10(4). [CrossRef]
- Mahdi O, Chiroma SM, Baharuldin MTH, et al. Win55,212-2 attenuates cognitive impairments in alcl<inf>3</inf> + d-galactose-induced alzheimer’s disease rats by enhancing neurogenesis and reversing oxidative stress. Biomedicines. 2021;9(9). [CrossRef]
- Gajardo-Gómez R, Labra VC, Maturana CJ, et al. Cannabinoids prevent the amyloid β-induced activation of astroglial hemichannels: A neuroprotective mechanism. Glia. 2017;65(1):122-137. [CrossRef]
- Soto-Mercado V, Mendivil-Perez M, Jimenez-Del-Rio M, Velez-Pardo C. Multi-Target Effects of the Cannabinoid CP55940 on Familial Alzheimer’s Disease PSEN1 E280A Cholinergic-Like Neurons: Role of CB1 Receptor. Journal of Alzheimer’s Disease. 2021;82(s1):S359-S378. [CrossRef]
- Nuñez-Borque E, González-Naranjo P, Bartolomé F, et al. Targeting Cannabinoid Receptor Activation and BACE-1 Activity Counteracts TgAPP Mice Memory Impairment and Alzheimer’s Disease Lymphoblast Alterations. Mol Neurobiol. 2020;57(4):1938-1951. [CrossRef]
- Balleza-Tapia H, Crux S, Andrade-Talavera Y, et al. TrpV1 receptor activation rescues neuronal function and network gamma oscillations from Aβ-induced impairment in mouse hippocampus in vitro. Elife. 2018;7. [CrossRef]
- Muller C, Morales P, Reggio PH. Cannabinoid Ligands Targeting TRP Channels. Front Mol Neurosci. 2019;11. [CrossRef]
- Xiang X, Wang X, Jin S, et al. Activation of GPR55 attenuates cognitive impairment and neurotoxicity in a mouse model of Alzheimer’s disease induced by Aβ1–42 through inhibiting RhoA/ROCK2 pathway. Prog Neuropsychopharmacol Biol Psychiatry. 2022;112:110423. [CrossRef]
- Ryberg E, Larsson N, Sjögren S, et al. The orphan receptor GPR55 is a novel cannabinoid receptor. Br J Pharmacol. 2007;152(7):1092-1101. [CrossRef]
- Xiang X, Wang X, Wu Y, et al. Activation of GPR55 attenuates cognitive impairment, oxidative stress, neuroinflammation, and synaptic dysfunction in a streptozotocin-induced Alzheimer’s mouse model. Pharmacol Biochem Behav. 2022;214:173340. [CrossRef]
- Wang X, Xiang X, Hu J, et al. Pharmacological Activation of GPR55 Improved Cognitive Impairment Induced by Lipopolysaccharide in Mice. Journal of Molecular Neuroscience. 2022;72(8):1656-1669. [CrossRef]
- Elmazoglu Z, Rangel-López E, Medina-Campos ON, et al. Cannabinoid-profiled agents improve cell survival via reduction of oxidative stress and inflammation, and Nrf2 activation in a toxic model combining hyperglycemia+Aβ1-42 peptide in rat hippocampal neurons. Neurochem Int. 2020;140:104817. [CrossRef]
- Navarro-Dorado J, Villalba N, Prieto D, et al. Vascular dysfunction in a transgenic model of Alzheimer’s disease: Effects of CB1R and CB2R cannabinoid agonists. Front Neurosci. 2016;10(SEP). [CrossRef]
- Schubert D, Kepchia D, Liang Z, Dargusch R, Goldberg J, Maher P. Efficacy of Cannabinoids in a Pre-Clinical Drug-Screening Platform for Alzheimer’s Disease. Mol Neurobiol. 2019;56(11):7719-7730. [CrossRef]
- Brannigan A, Zwerman W. The real “Hawthorne effect.” Society. 2001;38(2):55-60. [CrossRef]
- Oyagawa CRM, Grimsey NL. Cannabinoid receptor CB1 and CB2 interacting proteins: Techniques, progress and perspectives. In: ; 2021:83-132. [CrossRef]
Scheme 1.
Chemical structures of HU-210 and SR 141716A. Adapted from [45].
Scheme 1.
Chemical structures of HU-210 and SR 141716A. Adapted from [45].
Scheme 2.
Chemical structures of new selective CB2R agonists.
Scheme 3.
Chemical structures of novel compounds targeting CB2R and cholinergic pathways.
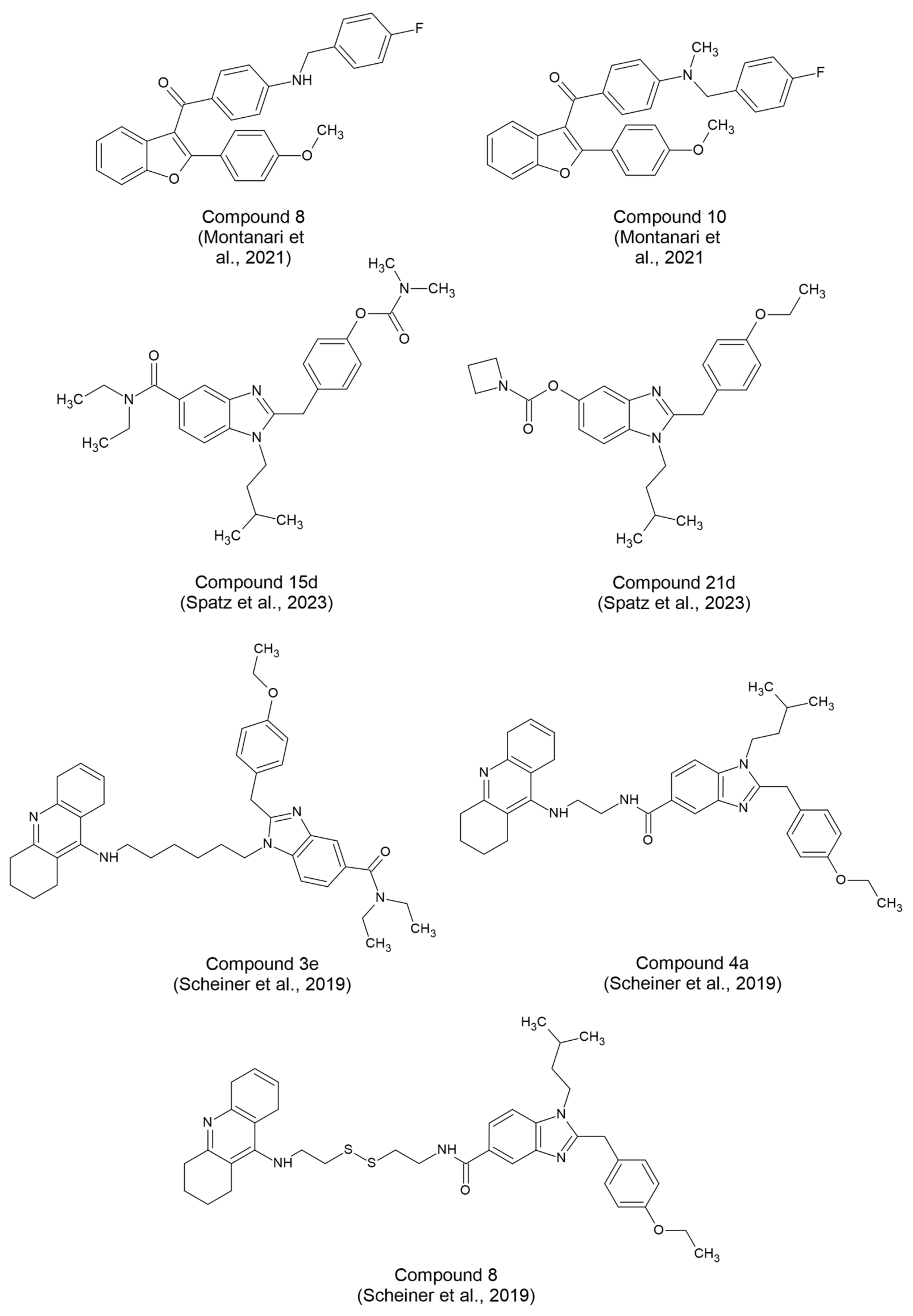
Scheme 4.
Chemical structures of non-selective CB1R and CB2R agonists.
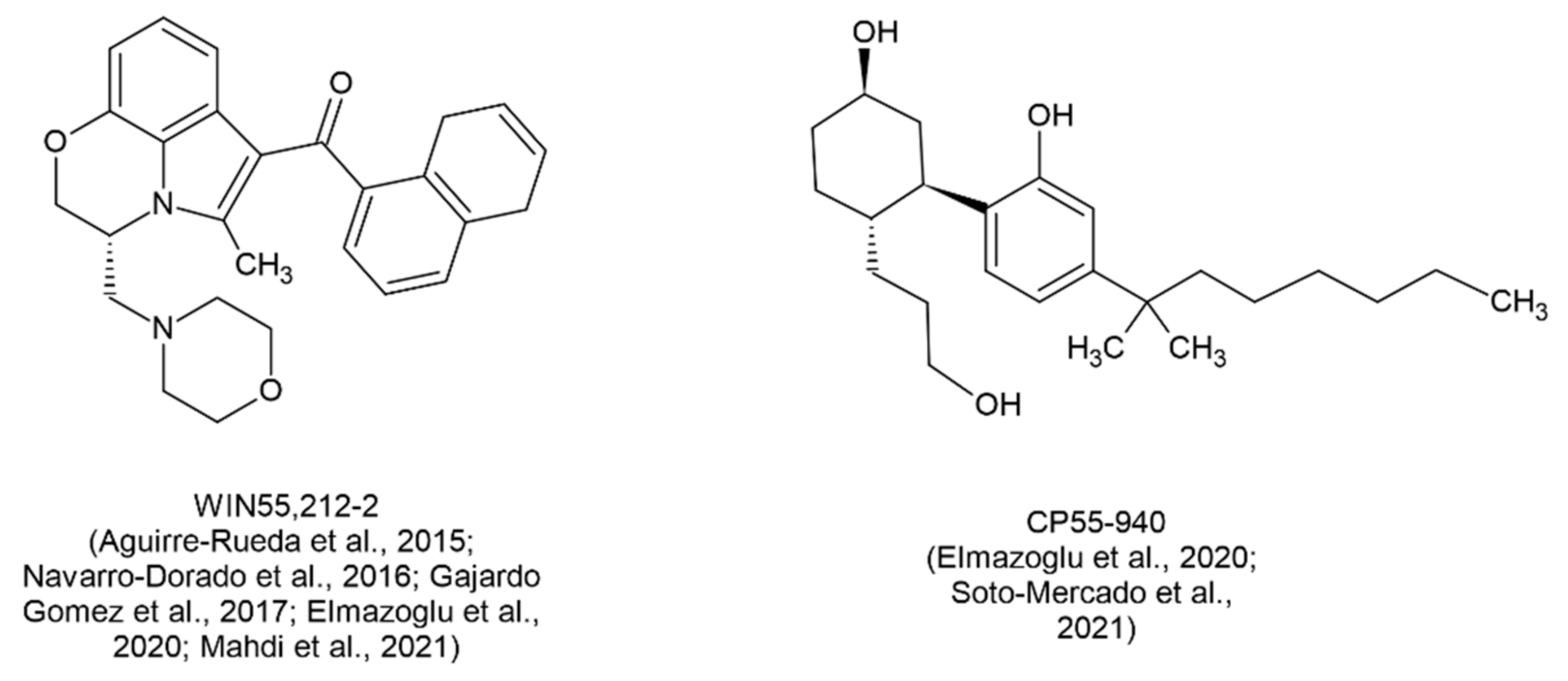
Scheme 6.
Chemical structure of O-1602.
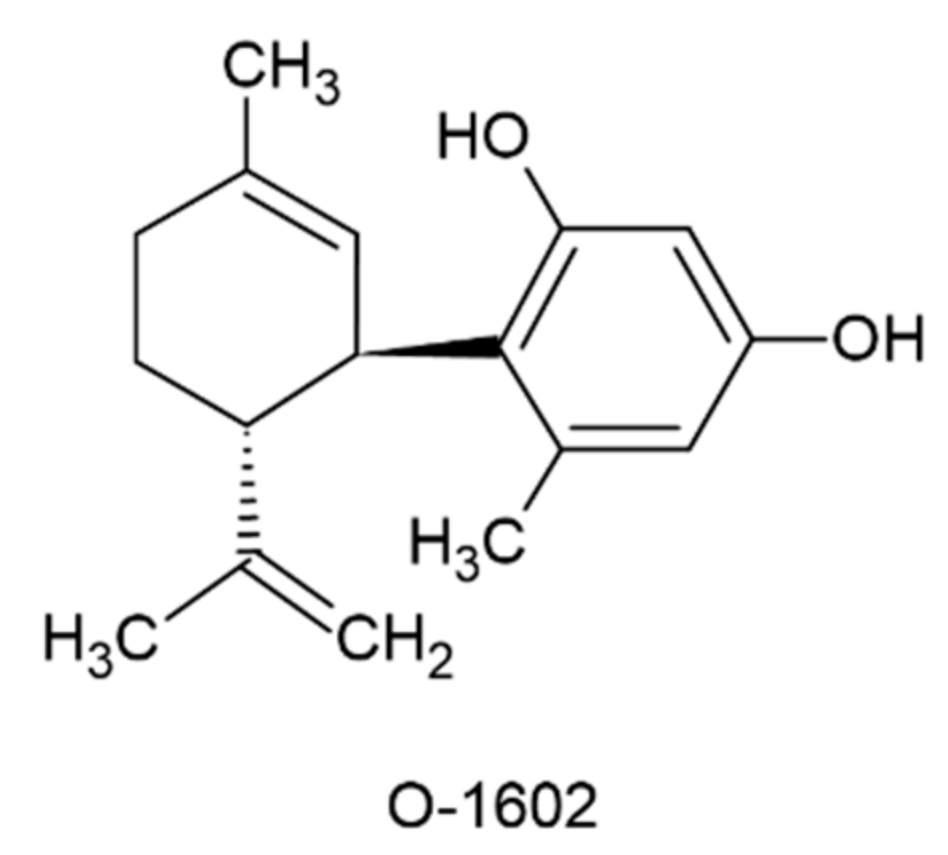
Scheme 7.
Chemical structure of URB597.
Figure 1.
Summary of the Effects of Cannabinoid Receptor Agonists in In Vitro and In Vivo Models, and in Patients with Alzheimer's Disease. A. Effects of CB1R and CB2R agonists in various Alzheimer's disease models. B. Effects of TRPV-1 and GPR-55 agonists in Alzheimer's disease models. Abbreviations: Aβ: amyloid beta; ACh: acetylcholine; AChE: acetylcholinesterase; ACEA: arachi-donyl-2'-chloroethylamide; ACPA: arachidonylcyclopropylamide; AD: Alzheimer’s disease; ADAS-COG: Alzheimer's Disease Assessment Scale-Cognitive Subscale; ADR: adverse drug reaction; AEA: anandamide; 2-AG: 2-arachidonylglycerol; Akt: protein kinase B; AlCl3: aluminum choloride; BACE-1: β-site amyloid precursor protein cleaving enzyme; Bax: Bcl-2 associated X protein; BChE: butyr-ylcholinesterase; Bcl-2: B cell leukemia/lymphoma 2 protein; BCP: β-caryophyllene; BDNF: brain-derived neurotrophic factor; BPSD: behavioral and psychological symptoms of dementia; CAT: catalase; CBC: cannabichromene; CBD: cannabidiol; CBDA: cannabidiol acid; CBDV: cannabidivarin; CBG: can-nabigerol; CBGA: cannabigerolic acid; CBN: cannabinol; CB1R: cannabinoid receptor 1; CB2R: can-nabinoid receptor 2; CeA: central amygdala; CMAI: Cohen-Mansfield agitation inventory; Col IV: col-lagen IV; COX-2: cyclooxygenase-2; DEGs: differentially expressed genes; D-Gal: D-galactosidase; DMCBD: dimethyl cannabidiol; ΔΨm: mitochondrial membrane potential; e.o.d.: every other day; EPM: elevated plus maze; ER: endoplasmic reticulum; ERK1/2: extracellular signal-regulated kinase 1/2; EV: extra-dimensional; FAAH: fatty acid amide hydrolase; GFAP: glial fibrillary acidic protein; GPx: gluta-thione peroxidase; GPR-55: G-protein coupled receptor 55; GRx: glutaredoxin; GSH: glutathione; GSK-3β: glycogen synthase kinase-3 beta; i.c.v.: intracerebroventricular; IFN-γ: interferon γ; IL-1β: inter-leukin-1β; IL-6: interleukin-6; iNOS: inducible nitric oxide synthase; i.p.: intraperitoneally; LPS: lipopoly-saccharide; LTP: long-term potentiation; MAGL: monoglyceride lipase; MDA: malondialdehyde; miR: microRNA; MMSE: mini mental state examination; MNA-SF: mini-nutritional assessment short-form; MCBN: cannabinol methyl ether; mPFC: medial prefrontal cortex; MWM: Morris water maze; NF-κB: nuclear factor-κB; NlTyr: N-linoleyltyrosine; NO: nitric oxide; NOR: novel object recognition; NPI: neuro-psychiatric inventory; NPI-NH: neuropsychiatric inventory-nursing home; NPS: neuropsychiatric symp-toms; Nrf2: nuclear factor erythroid 2-related factor 2; OAE: O-arachidonylethanolamine; OBX: olfacto-ry bulbectomy; o.d.: once daily; OKA: okadaic acid; OLR: object location recognition; PA: passive avoid-ance; PGE-2: prostaglandin E2; PKA: protein kinase A; p.o.: per os; PPAR-γ: peroxisome prolifera-tor-activated receptor-γ; PSMB5: proteasome subunit beta type-5; p-TAU: phosphorylated TAU; PSEN1: presenilin 1; PSD-95: postsynaptic density protein 95; PUMA: p53 upregulated modulator of apoptosis; REV1: reversal 1; RhoA: Ras homolog family member A; ROCK2: Rho associated coiled-coil containing protein kinase 2; ROS: reactive oxygen species; RRT: rotarod test; RVD: (m)RVD-hemopressin; s.c.: subcutaneously; sMMSE: standardized mini mental state examination; SOD: superoxide dismutase; STZ: streptozotocin; THC: tetrahydrocannabinol; THCA: tetrahydrocannabinolic acid; TLR-4: toll-like receptor 4; TNF-a: tumor necrosis factor-a; TrkB: tropomyosin receptor kinase B; Trpv-1: transient recep-tor potential cation channel subfamily V member 1; TUNEL: terminal deoxynucleotidyl transferase dUTP nick end labeling; UPR: unfolded protein response; VD: (m)VD-hemopressin.
Figure 1.
Summary of the Effects of Cannabinoid Receptor Agonists in In Vitro and In Vivo Models, and in Patients with Alzheimer's Disease. A. Effects of CB1R and CB2R agonists in various Alzheimer's disease models. B. Effects of TRPV-1 and GPR-55 agonists in Alzheimer's disease models. Abbreviations: Aβ: amyloid beta; ACh: acetylcholine; AChE: acetylcholinesterase; ACEA: arachi-donyl-2'-chloroethylamide; ACPA: arachidonylcyclopropylamide; AD: Alzheimer’s disease; ADAS-COG: Alzheimer's Disease Assessment Scale-Cognitive Subscale; ADR: adverse drug reaction; AEA: anandamide; 2-AG: 2-arachidonylglycerol; Akt: protein kinase B; AlCl3: aluminum choloride; BACE-1: β-site amyloid precursor protein cleaving enzyme; Bax: Bcl-2 associated X protein; BChE: butyr-ylcholinesterase; Bcl-2: B cell leukemia/lymphoma 2 protein; BCP: β-caryophyllene; BDNF: brain-derived neurotrophic factor; BPSD: behavioral and psychological symptoms of dementia; CAT: catalase; CBC: cannabichromene; CBD: cannabidiol; CBDA: cannabidiol acid; CBDV: cannabidivarin; CBG: can-nabigerol; CBGA: cannabigerolic acid; CBN: cannabinol; CB1R: cannabinoid receptor 1; CB2R: can-nabinoid receptor 2; CeA: central amygdala; CMAI: Cohen-Mansfield agitation inventory; Col IV: col-lagen IV; COX-2: cyclooxygenase-2; DEGs: differentially expressed genes; D-Gal: D-galactosidase; DMCBD: dimethyl cannabidiol; ΔΨm: mitochondrial membrane potential; e.o.d.: every other day; EPM: elevated plus maze; ER: endoplasmic reticulum; ERK1/2: extracellular signal-regulated kinase 1/2; EV: extra-dimensional; FAAH: fatty acid amide hydrolase; GFAP: glial fibrillary acidic protein; GPx: gluta-thione peroxidase; GPR-55: G-protein coupled receptor 55; GRx: glutaredoxin; GSH: glutathione; GSK-3β: glycogen synthase kinase-3 beta; i.c.v.: intracerebroventricular; IFN-γ: interferon γ; IL-1β: inter-leukin-1β; IL-6: interleukin-6; iNOS: inducible nitric oxide synthase; i.p.: intraperitoneally; LPS: lipopoly-saccharide; LTP: long-term potentiation; MAGL: monoglyceride lipase; MDA: malondialdehyde; miR: microRNA; MMSE: mini mental state examination; MNA-SF: mini-nutritional assessment short-form; MCBN: cannabinol methyl ether; mPFC: medial prefrontal cortex; MWM: Morris water maze; NF-κB: nuclear factor-κB; NlTyr: N-linoleyltyrosine; NO: nitric oxide; NOR: novel object recognition; NPI: neuro-psychiatric inventory; NPI-NH: neuropsychiatric inventory-nursing home; NPS: neuropsychiatric symp-toms; Nrf2: nuclear factor erythroid 2-related factor 2; OAE: O-arachidonylethanolamine; OBX: olfacto-ry bulbectomy; o.d.: once daily; OKA: okadaic acid; OLR: object location recognition; PA: passive avoid-ance; PGE-2: prostaglandin E2; PKA: protein kinase A; p.o.: per os; PPAR-γ: peroxisome prolifera-tor-activated receptor-γ; PSMB5: proteasome subunit beta type-5; p-TAU: phosphorylated TAU; PSEN1: presenilin 1; PSD-95: postsynaptic density protein 95; PUMA: p53 upregulated modulator of apoptosis; REV1: reversal 1; RhoA: Ras homolog family member A; ROCK2: Rho associated coiled-coil containing protein kinase 2; ROS: reactive oxygen species; RRT: rotarod test; RVD: (m)RVD-hemopressin; s.c.: subcutaneously; sMMSE: standardized mini mental state examination; SOD: superoxide dismutase; STZ: streptozotocin; THC: tetrahydrocannabinol; THCA: tetrahydrocannabinolic acid; TLR-4: toll-like receptor 4; TNF-a: tumor necrosis factor-a; TrkB: tropomyosin receptor kinase B; Trpv-1: transient recep-tor potential cation channel subfamily V member 1; TUNEL: terminal deoxynucleotidyl transferase dUTP nick end labeling; UPR: unfolded protein response; VD: (m)VD-hemopressin.
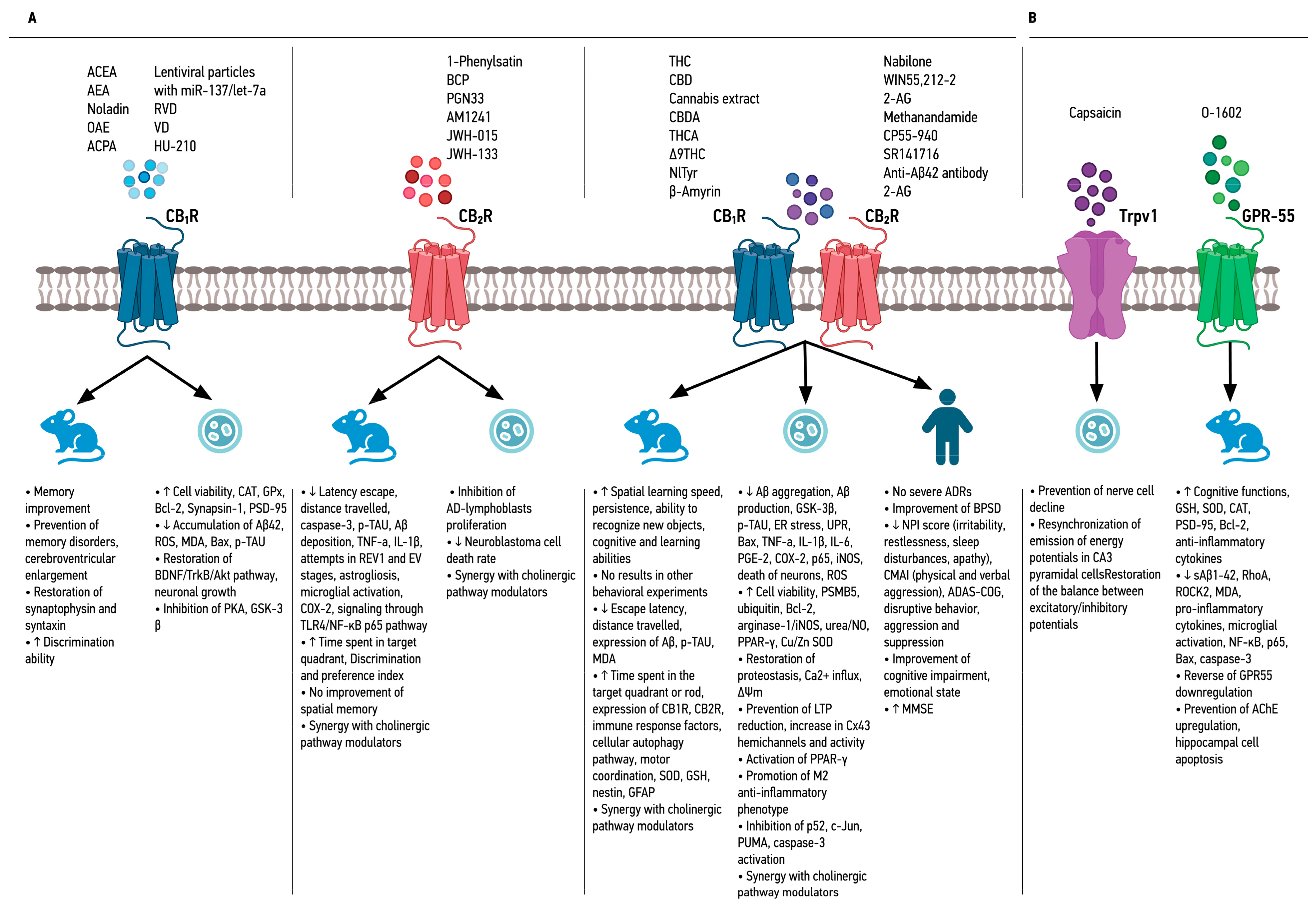

Table 1.
Main results from the studies with selective CB1R agonists.
| Reference | Cannabinoid | Dosage | Major results | Model used | |
|---|---|---|---|---|---|
| Crunfli et al., 2019 [36] | ACEA | 3 mg/kg i.p. |
|
In vitro and in vivo experiments in AD models with STZ | |
| Moreira-Silva et al., 2018 [37] | AEA | 100 ng i.c.v. |
|
In vivo experiments in Wistar rats with STZ injection | |
| Khavandi, Rao and Beazely, 2023 [38] | AΕA | 10 μΜ |
|
In vitro experiments in mouse hippocampal HT22 cells and hamster ovary CHO cells expressing human CB1R | |
| Noladin | 10 μΜ |
|
|||
| OAΕ | 1 μM 10 μΜ |
|
|||
| Hosseininia et al., 2023 [39] | ACPA | 10 ng/0.5 μL (corticolimbic microinjection) |
|
In vivo experiments in Wistar rats after i.c.v. STZ | |
| Lentiviral particles with miR-137 or miR-let-7a | 0.5 μL/rat e.o.d. | ||||
| Zhang et al., 2016 [40] | RVD & VD | 1 nmol 2.5 nmol 5 nmol, i.c.v./i.p. |
|
In vivo experiments in mice infected with Aβ1→42 | |
| Zhang et al., 2021 [41] |
|
In vivo experiments in mice after i.p. scopolamine | |||
| Zhang et al., 2020a [42] | VD |
|
In vitro experiments on hippocampal neurons from mice infected with Aβ1→42 | ||
| Zhang et al., 2023 [43] | RVD |
|
In vitro experiments in HT22 cells treated with scopolamine | ||
| Zhang et al., 2020b [44] |
|
In vitro experiments in SH-SY5Y cells infected with Aβ1→42 | |||
| Velikova, Doncheva and Tashev, 2020 [45] | HU-210 | 5 μg/day i.c.v. |
|
Direct correlation of CB1R with memory function (active and passive avoidance tests) | In vivo experiments in OBX rats |
| SR 141716A (CB1R Antagonist) | 3 μg/day i.c.v. |
|
|||
Abbreviations: Aβ: amyloid beta; ACEA: arachidonyl-2'-chloroethylamide; ACPA: arachidonylcyclopropylamide; AD: Alzheimer’s disease; AEA: anandamide; Akt: protein kinase B; Bax: Bcl-2 associated X protein; Bcl-2: B cell leukemia/lymphoma 2 protein; BDNF: brain-derived neurotrophic factor; CAT: catalase; CB1R: cannabinoid receptor 1; CeA: central amygdala; e.o.d.: every other day; EPM: elevated plus maze; GPx: glutathione peroxidase; GSK-3β: glycogen synthase kinase-3 beta; i.c.v.: intracerebroventricular; i.p.: intraperitoneally; MAGL: monoglyceride lipase; MDA: malondialdehyde; miR: microRNA; mPFC: medial prefrontal cortex; NOR: novel object recognition; OAE: O-arachidonylethanolamine; OBX: olfactory bulbectomy; OLR: object location recognition; PKA: protein kinase A; PSD-95: postsynaptic density protein 95; ROS: reactive oxygen species; RVD: (m)RVD-hemopressin; STZ: streptozotocin; TrkB: tropomyosin receptor kinase B; VD: (m)VD-hemopressin.
Table 2.
Main results from the studies with selective CB2R agonists.
| Reference | Cannabinoid | Dosage | Major results | Model used | |
|---|---|---|---|---|---|
| Jayant et al., 2016 [46] | 1-Phenylsatin | 20 mg/kg p.o. | MWM test |
|
In vivo experiments in rats exposed to STZ or AlCl3 + D-Gal |
| Attentional set shifting test |
|
||||
| Cheng, Dong and Liu, 2014 [47] | BCP | 48 mg/kg p.o. |
|
In vivo experiments in APP/PS1 mice | |
| del Cerro et al., 2018 [48] | PGN33 | 2.5 nM 5 nM 7.5 nM 10 nM |
|
In vitro experiments in lymphoblasts isolated from AD patients and in Aβ-treated SH-SY5Y cells | |
| Abd El-Rahman and Fayed, 2022 [49] | AM1241 | 3 mg 6 mg i.p. |
|
In vivo experiments in model AD female rats, treated with D-Gal after ovariectomy | |
| Li et al., 2019 [50] | JWH-015 | 0.5 mg/kg i.p. |
|
In vivo experiments in transgenic APP/PS1 mice | |
| Çakır et al., 2019 [51] | JWH-133 | 0.2 mg/kg i.p. |
|
In vivo experiments in a rat model with hyperphosphorylated TAU from OKA administration | |
Abbreviations: Aβ: amyloid beta; AD: Alzheimer’s disease; AlCl3: aluminum choloride; BCP: β-caryophyllene; COX-2: cyclooxygenase-2; D-Gal: D-galactosidase; EV: extra-dimensional; IL-1β: interleukin-1β; i.p.: intraperitoneally; MWM: Morris water maze; NF-κB: nuclear factor-κB; NOR: novel object recognition; OKA: okadaic acid; p.o.: per os; p-TAU: phosphorylated TAU; REV1: reversal 1; TLR-4: toll-like receptor 4; TNF-a: tumor necrosis factor-a.
Table 3.
Main results from the studies with CB2R and cholinergic pathways modulators.
| Reference | Cannabinoid | Dosage | Major results | Model used |
|---|---|---|---|---|
| Marta, Agnieszka and Grazyna, 2022 [52] | JWH-133 | 0.25 mg/kg i.p. |
|
In vivo experiments in Swiss mice after scopolamine administration |
| Nicotine (Cholinergic agonist) |
0.05 mg/kg s.c. | |||
| Montanari et al., 2021 [53] | Compound 8 (CB2R agonist, BChE inhibitor) | 5 μΜ |
|
In vitro experiments in SH-SY5Y cells treated with Aβ1-42 |
| Compound 10 (CB2R inverse agonist) | 5 μΜ |
|
||
| Spatz et al., 2023 [54] | Compound 15d (CB2R agonist, BChE inhibitor) |
0.3-3 mg/kg/day i.p. |
|
In vitro experiments in LPS-treated N9 microglial cells and in vivo experiments in mice challenged with Aβ25-35 oligomers |
| Scheiner et al., 2019 [55] | Compound 3e (CB2R agonist, AChE inhibitor) | 0.3 mg/kg i.p., o.d. |
|
In vitro experiments in cellular model of neuronal oxidative stress in N9 microglial cells and in vivo experiments in Aβ25-35 injected AD mice |
| Compound 4a (CB2R agonist, AChE inhibitor) | 1 mg/kg i.p., o.d. | |||
| Compound 8 (CB2R agonist, AChE inhibitor) | 0.3 mg/kg i.p., o.d. |
Abbreviations: Aβ: amyloid beta; AChE: acetylcholinesterase; AD: Alzheimer’s disease; BChE: butyrylcholinesterase; CB2R: cannabinoid receptor 2; i.p.: intraperitoneally; LPS: lipopolysaccharide; o.d.: once daily; PA: passive avoidance; s.c.: subcutaneously.
Table 4.
Main results from the studies with non-selective CB2R and CB2R agonists.
| Reference | Cannabinoid | Dosage | Major results | Model used |
|---|---|---|---|---|
| Cao et al., 2014 [56] | THC | 0.25 nM – 2.5 μΜ every 6h or 24h or 48h |
|
In vitro experiments in N2a/AβPPswe cells |
| Gugliandolo et al., 2023 [57] | THC | 20 μΜ |
|
In vitro experiments in SH-SY5Y cells treated with Aβ1-42 |
| van den Elsen et al., 2015 [58] | THC | 1.5 mg/3 times/day |
|
Randomized controlled trial in 50 patients with dementia |
| Coles et al., 2020 [59] | CBD | 5 mg/kg i.p. |
|
In vivo experiments in female APP/PS1 mice |
| Hughes and Herron, 2019 [60] | CBD | 10 μΜ |
|
In vitro experiments in sections from the CA1 hippocampus region of C57Bl/6 mice after Aβ treatment |
| Amini and Abdolmaleki, 2022 [61] | CBD with nano-chitosan coating | 120 mg/kg p.o. |
|
In vivo experiments in rat AD model after injection of Aβ1-42 |
| Hao and Feng, 2021 [62] | CBD | 5 mg/kg/day i.p. |
|
In vivo study in APP/PS1 mice through analysis of DEGs |
| Alexandri et al., 2023 [63] | CBD oil drops | 3% p.o. for 6 months |
|
Comparative study in 20 patients with dementia between 3% CBD and usual treatment |
| Palmieri and Vadalà, 2023 [64] | Cannabins extract in oil | 1 mL/day (22% THC, 0.5% CBD) p.o. |
|
Limited-size cohort study in 30 patients with moderate-to-severe AD |
| Ruver-Martins et al., 2022 [28] | Cannabis extract | Microdoses (most often 500 µg p.o.) of 8:1 THC:CBD extract for 22 months |
|
Case report on a 75-year-old patient with mild AD (memory impairment, spatiotemporal disorientation) |
| Kim et al., 2023 [65] | CBDA | 6 μΜ, 3 μL (intrahippocampal injection) |
|
In vivo experiments in ICR mice after injection of Aβ1-42 |
| THCA | 12 μΜ, 3 μL (intrahippocampal injection) | |||
| Defrancesco and Hofer, 2020 [66] | Dronabinol drops (Δ9THC) | 4.9 mg – 6.7 mg/day p.o. |
|
Case report on a 69-year-old AD patient with severe NPS (depression, paranoid perception) |
| Long et al., 2021 [67] | NlTyr | 60 mg/kg p.o. |
|
In vivo experiments in AD APP/PS1 mouse model |
| Askari et al., 2018 [68] | β-Amyrin | 4-16 μΜ |
|
In vitro experiments in rat microglial cells treated with LPS/IFN-γ |
| Herrmann et al., 2019 [69] | Nabilone | 1-2 mg/day p.o. |
|
Randomized, double-blind, crossover clinical trial in 39 patients with moderate to severe AD |
| Aguirre-Rueda et al., 2015 [70] | WIN55,212-2 | 10 μΜ |
|
In vitro experiments on rat cortical astrocytes treated with Aβ |
| Mahdi et al., 2021 [71] | WIN55,212-2 | 0.5 mg/kg 1 mg/kg 2 mg/kg |
|
In vivo experiments in a Wistar rat model after administration of AlCl3 + D-Gal |
| Gajardo-Gómez et al., 2017 [72] | WIN55,212-2 | 5 μΜ |
|
In vitro experiments on cells from rat hippocampal slices treated with Aβ25-35 |
| 2-AG | ||||
| Methanandamide | ||||
| Soto-Mercado et al., 2021 [73] | CP55-940 | 1 μΜ |
|
In vitro experiments in PSEN1 E280A cells (familial AD model) |
| SR141716 (CB1R inverse agonist) | ||||
| Anti-Aβ42 antibody | ||||
| 2-AG | ||||
| CP55-940 | ||||
| WIN55,212-2 | ||||
| URB597 (FAAH inhibitor) |
Abbreviations: Aβ: amyloid beta; AD: Alzheimer’s disease; ADAS-COG: Alzheimer's Disease Assessment Scale-Cognitive Subscale; ADR: adverse drug reaction; 2-AG: 2-arachidonylglycerol; AlCl3: aluminum choloride; Bax: Bcl-2 associated X protein; Bcl-2: B cell leukemia/lymphoma 2 protein; BPSD: behavioral and psychological symptoms of dementia; CBD: cannabidiol; CBDA: cannabidiol acid; CB1R: cannabinoid receptor 1; CB2R: cannabinoid receptor 2; CMAI: Cohen-Mansfield agitation inventory; COX-2: cyclooxygenase-2; DEGs: differentially expressed genes; D-Gal: D-galactosidase; ΔΨm: mitochondrial membrane potential; ER: endoplasmic reticulum; FAAH: fatty acid amide hydrolase; GFAP: glial fibrillary acidic protein; GSH: glutathione; GSK-3β: glycogen synthase kinase-3 beta; IFN-γ: interferon γ; IL-1β: interleukin-1β; IL-6: interleukin-6; iNOS: inducible nitric oxide synthase; i.p.: intraperitoneally; LPS: lipopolysaccharide; LTP: long-term potentiation; MDA: malondialdehyde; MMSE: mini mental state examination; MNA-SF: mini-nutritional assessment short-form; MWM: Morris water maze; NlTyr: N-linoleyltyrosine; NO: nitric oxide; NOR: novel object recognition; NPI: neuropsychiatric inventory; NPI-NH: neuropsychiatric inventory-nursing home; NPS: neuropsychiatric symptoms; PGE-2: prostaglandin E2; p.o.: per os; PPAR-γ: peroxisome proliferator-activated receptor-γ; PSMB5: proteasome subunit beta type-5; p-TAU: phosphorylated TAU; PSEN1: presenilin 1; PUMA: p53 upregulated modulator of apoptosis; ROS: reactive oxygen species; RRT: rotarod test; sMMSE: standardized mini mental state examination; SOD: superoxide dismutase; THC: tetrahydrocannabinol; THCA: tetrahydrocannabinolic acid; TNF-a: tumor necrosis factor-a; UPR: unfolded protein response.
Table 5.
Main results from the studies with non-selective agonists of CB1R and CB2R associated with cholinergic pathways and molecules that act through pathways related to the ECS.
Table 5.
Main results from the studies with non-selective agonists of CB1R and CB2R associated with cholinergic pathways and molecules that act through pathways related to the ECS.
| Reference | Cannabinoid | Dosage | Major results | Model used |
|---|---|---|---|---|
| Non-selective agonists of CB1R and CB2R associated with cholinergic pathways | ||||
| Nuñez-Borque et al., 2020 [74] | NP137 | 2.5 μΜ/ 5 μΜ/ 1 mg/kg/day p.o. |
In vitro
|
In vitro experiments in immortalized lymphocytes of patients with delayed AD and in vivo experiments in TgAPP mice |
| NP148 | 5 μΜ | |||
| Modulator molecules that act through pathways related to the endocannabinoid system | ||||
| Balleza-Tapia et al., 2018 [75] | Capsaicin (Trpv-1 agonst) | 10 μΜ |
|
In vitro experiments in Aβ-treated hippocampal cells |
| Xiang et al., 2022a [77] | O-1602 (GPR-55 agonist) | 2 μg/mouse or 4 μg/mouse i.c.v. |
|
In vivo experiments in mice affected by Aβ1-42 |
| Xiang et al., 2022b [79] |
|
In vivo experiments in mice affected by STZ | ||
| Wang et al., 2022 [80] |
|
In vivo experiments in mice affected by LPS | ||
Abbreviations: Aβ: amyloid beta; AChE: acetylcholinesterase; AD: Alzheimer’s disease; BACE-1: β-site amyloid precursor protein cleaving enzyme; Bax: Bcl-2 associated X protein; BChE: butyrylcholinesterase; Bcl-2: B cell leukemia/lymphoma 2 protein; CAT: catalase; ERK1/2: extracellular signal-regulated kinase 1/2; GPR-55: G-protein coupled receptor 55; GSH: glutathione; i.c.v.: intracerebroventricular; LPS: lipopolysaccharide; MDA: malondialdehyde; MWM: Morris water maze; NF-κB: nuclear factor-κB; NOR: novel object recognition; p.o.: per os; PSD-95: postsynaptic density protein 95; RhoA: Ras homolog family member A; ROCK2: Rho associated coiled-coil containing protein kinase 2; SOD: superoxide dismutase; STZ: streptozotocin; Trpv-1: transient receptor potential cation channel subfamily V member 1; TUNEL: terminal deoxynucleotidyl transferase dUTP nick end labeling.
Table 6.
Main results from the combination studies with agonists belonging to different classes.
| Reference | Cannabinoid | Dosage | Major results | Model used |
|---|---|---|---|---|
| Elmazoglu et al., 2020 [81] | AEA | 1-1000 μΜ |
|
In vitro experiments in rat hippocampal neurons - model of combined toxic hyperglycemia and Aβ1-42 |
| 2-AG | ||||
| CP55-940 | ||||
| WIN55,212-2 | ||||
| URB597 (FAAH inhibitor) | ||||
| Navarro-Dorado et al., 2016 [82] | WIN55,212-2 | 0.2 mg/kg/day p.o. |
Combined action
|
In vivo experiments in TgAPP AD mice |
| JWH-133 | ||||
| Schubert et al., 2019 [83] | Δ8THC | 250 nΜ – 10 μM |
|
In vitro experiments in HT22 and MC65 cells after induction of C99 production |
| Δ9THC | ||||
| Δ9THCA | ||||
| CBD | ||||
| CBDA | ||||
| DMCBD | ||||
| CBDV | ||||
| CBG | ||||
| CBGA | ||||
| CBC | ||||
| CBN | ||||
| MCBN |
Abbreviations: Aβ: amyloid beta; ACh: acetylcholine; AD: Alzheimer’s disease; AEA: anandamide; 2-AG: 2-arachidonylglycerol; CAT: catalase; CBC: cannabichromene; CBD: cannabidiol; CBDA: cannabidiol acid; CBDV: cannabidivarin; CBG: cannabigerol; CBGA: cannabigerolic acid; CBN: cannabinol; CB1R: cannabinoid receptor 1; CB2R: cannabinoid receptor 2; Col IV: collagen IV; DMCBD: dimethyl cannabidiol; FAAH: fatty acid amide hydrolase; GPx: glutathione peroxidase; GRx: glutaredoxin; LPS: lipopolysaccharide; MCBN: cannabinol methyl ether; Nrf2: nuclear factor erythroid 2-related factor 2; p.o.: per os; ROS: reactive oxygen species; SOD: superoxide dismutase; THC: tetrahydrocannabinol; THCA: tetrahydrocannabinolic acid.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



